
Catalytic applications of a versatile magnetically separable Fe-Mo nanocatalyst
8
Green Chemistry PAPER Cite this: Green Chem., 2013, 15, 682 Received 20th November 2012, Accepted 16th January 2013 DOI: 10.1039/c3gc36844k www.rsc.org/greenchem Catalytic applications of a versatile magnetically separable Fe–Mo (Nanocat-Fe–Mo) nanocatalyst† Manoj B. Gawande,* a Paula S. Branco,* a Isabel D. Nogueira, b C. Amjad A. Ghumman, c Nenad Bundaleski, c Adérito Santos, c Orlando M. N. D. Teodoro c and Rafael Luque d A novel nano-Fe 3 O 4 –MoO 3 (Nanocat-Fe–Mo) catalyst was prepared via simple wet impregnation and characterized by several techniques. The synthesized Nanocat-Fe–Mo was found to be a highly active and efficient catalyst in the oxidation of benzyl alcohol and a series of examples of important organic reac- tions including A3 couplings, hydrogenations and hydrations. The Nanocat-Fe–Mo could be reused and recycled up to 7 times without any significant yield loss. Introduction Nanoparticles have been attracting a great deal of attention related to their special properties as compared to bulk metal counterparts, with multidisciplinary applications in areas including drug discovery, 1 energy storage, 2 electronic devices, 3 and heterogeneous catalysis. 4 Magnetic nanocatalysts recently emerged as highly promising and versatile nanoentities com- bining high surface areas and excellent catalytic activities (similar to those of homogeneous catalysts) with simple prep- aration protocols from inexpensive precursors. 5 Ferrite/magne- tite based nanocatalysts have been extensively used in recent years for various important organic reactions. 5a,6 Examples of such chemistries include cross-coupling reac- tions (using ferrite-immobilized palladium complexes and/or palladium nanoparticles), 7,8 kinetic resolutions 9 (e.g. 1,2- diphenylethane-1-2-diol using magnetic Co/C nanoparticles), magnetite nanoparticles-supported 4-N,N-dialkylaminopyri- dine catalyst, 10 asymmetric hydrogenations of aromatic ketones, 11 selective oxidation reactions, 12 and many others. In addition, various magnetite-supported catalysts have been reported in a range of different chemistries to those high- lighted before. 13 For a comprehensive review on magnetic separable systems and their applications in catalysis, readers are kindly referred to the recent work by Polshettiwar et al. 14 However, the stabilization of metal nanoparticles and/or organoligands over the surface of ferrite/nanomaterials in the absence of linkers or ligands is a significant challenge for organic chemists from the catalytic viewpoint (i.e. leaching). In recent years, only a few reports have been found on the development of stable and highly active metals deposited on the surface of magnetite nanomaterials. Selected examples include impregnated copper on magnetite in the synthesis of propargylamines and addition reactions, 15 a Pd-magnetite for carbonylative Sonogashira coupling reactions, 16 a ferrite sup- ported-Ru catalyst in liquid-phase oxidations and reduc- tions 13m as well as N-alkylation of amines, sulfonamides, sulfinamides, and nitroarenes. 13d Recently, Glorius and co- workers have reported a magnetite–Pd–NHC nanocatalyst for asymmetric α-arylation of ketones with aryl halides. 17 In view of these premises, a remarkable scope and demand for such types of magnetically separable nanocatalysts in organic chemistry and green chemistry is envisaged. In con- tinuation of our research endeavours on sustainable protocols, heterogeneous catalysis and nanomaterials, 18–20 we report herein the design of a novel and highly active magnetite sup- ported Mo nanocatalyst (1) in a range of chemistries by means of a simple wet impregnation method (Scheme 1). Experimental All commercial reagents were used as received unless other- wise mentioned. For analytical and preparative thin-layer chromatography, Merck, 0.2 mm and 0.5 mm Kieselgel GF 254 precoated plates were used, respectively. The spots were visual- ized using UV light. † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3gc36844k a REQUIMTE, Department of Chemistry, Faculty of Science and Technology, New University of Lisbon, Quinta da Torre, 2829-516 Caparica, Lisbon, Portugal. E-mail: [email protected], [email protected]; Fax: +351 21 2948550; Tel: +351 96 4223243, +351 21 2948300 b Instituto de Ciência e Engenharia de Materiais e Superfícies, IST, Lisbon, Portugal c Centre for Physics and Technological Research (CeFITec), Departamento de Física da Faculdade de Ciências Tecnologia (FCT), Universidade Nova de Lisboa, 2829-516 Caparica, Portugal d Departamento de Quimica Organica, Universidad de Cordoba, Edif. Marie Curie, Ctra Nnal IVa, Km 396, 14014 Cordoba, Spain 682 | Green Chem., 2013, 15, 682–689 This journal is © The Royal Society of Chemistry 2013 Downloaded on 25 February 2013 Published on 16 January 2013 on http://pubs.rsc.org | doi:10.1039/C3GC36844K View Article Online View Journal | View Issue
Transcript of Catalytic applications of a versatile magnetically separable Fe-Mo nanocatalyst

Green Chemistry
PAPER
Cite this: Green Chem., 2013, 15, 682
Received 20th November 2012,Accepted 16th January 2013
DOI: 10.1039/c3gc36844k
www.rsc.org/greenchem
Catalytic applications of a versatile magneticallyseparable Fe–Mo (Nanocat-Fe–Mo) nanocatalyst†
Manoj B. Gawande,*a Paula S. Branco,*a Isabel D. Nogueira,b C. Amjad A. Ghumman,c
Nenad Bundaleski,c Adérito Santos,c Orlando M. N. D. Teodoroc and Rafael Luqued
A novel nano-Fe3O4–MoO3 (Nanocat-Fe–Mo) catalyst was prepared via simple wet impregnation and
characterized by several techniques. The synthesized Nanocat-Fe–Mo was found to be a highly active and
efficient catalyst in the oxidation of benzyl alcohol and a series of examples of important organic reac-
tions including A3 couplings, hydrogenations and hydrations. The Nanocat-Fe–Mo could be reused and
recycled up to 7 times without any significant yield loss.
Introduction
Nanoparticles have been attracting a great deal of attentionrelated to their special properties as compared to bulk metalcounterparts, with multidisciplinary applications in areasincluding drug discovery,1 energy storage,2 electronic devices,3
and heterogeneous catalysis.4 Magnetic nanocatalysts recentlyemerged as highly promising and versatile nanoentities com-bining high surface areas and excellent catalytic activities(similar to those of homogeneous catalysts) with simple prep-aration protocols from inexpensive precursors.5 Ferrite/magne-tite based nanocatalysts have been extensively used in recentyears for various important organic reactions.5a,6
Examples of such chemistries include cross-coupling reac-tions (using ferrite-immobilized palladium complexes and/orpalladium nanoparticles),7,8 kinetic resolutions9 (e.g. 1,2-diphenylethane-1-2-diol using magnetic Co/C nanoparticles),magnetite nanoparticles-supported 4-N,N-dialkylaminopyri-dine catalyst,10 asymmetric hydrogenations of aromaticketones,11 selective oxidation reactions,12 and many others. Inaddition, various magnetite-supported catalysts have beenreported in a range of different chemistries to those high-lighted before.13 For a comprehensive review on magnetic
separable systems and their applications in catalysis, readersare kindly referred to the recent work by Polshettiwar et al.14
However, the stabilization of metal nanoparticles and/ororganoligands over the surface of ferrite/nanomaterials in theabsence of linkers or ligands is a significant challenge fororganic chemists from the catalytic viewpoint (i.e. leaching). Inrecent years, only a few reports have been found on thedevelopment of stable and highly active metals deposited onthe surface of magnetite nanomaterials. Selected examplesinclude impregnated copper on magnetite in the synthesis ofpropargylamines and addition reactions,15 a Pd-magnetite forcarbonylative Sonogashira coupling reactions,16 a ferrite sup-ported-Ru catalyst in liquid-phase oxidations and reduc-tions13m as well as N-alkylation of amines, sulfonamides,sulfinamides, and nitroarenes.13d Recently, Glorius and co-workers have reported a magnetite–Pd–NHC nanocatalyst forasymmetric α-arylation of ketones with aryl halides.17
In view of these premises, a remarkable scope and demandfor such types of magnetically separable nanocatalysts inorganic chemistry and green chemistry is envisaged. In con-tinuation of our research endeavours on sustainable protocols,heterogeneous catalysis and nanomaterials,18–20 we reportherein the design of a novel and highly active magnetite sup-ported Mo nanocatalyst (1) in a range of chemistries by meansof a simple wet impregnation method (Scheme 1).
Experimental
All commercial reagents were used as received unless other-wise mentioned. For analytical and preparative thin-layerchromatography, Merck, 0.2 mm and 0.5 mm Kieselgel GF 254precoated plates were used, respectively. The spots were visual-ized using UV light.
†Electronic supplementary information (ESI) available. See DOI:10.1039/c3gc36844k
aREQUIMTE, Department of Chemistry, Faculty of Science and Technology,
New University of Lisbon, Quinta da Torre, 2829-516 Caparica, Lisbon, Portugal.
E-mail: [email protected], [email protected]; Fax: +351 21 2948550;
Tel: +351 96 4223243, +351 21 2948300bInstituto de Ciência e Engenharia de Materiais e Superfícies, IST, Lisbon, PortugalcCentre for Physics and Technological Research (CeFITec), Departamento de Física da
Faculdade de Ciências Tecnologia (FCT), Universidade Nova de Lisboa, 2829-516
Caparica, PortugaldDepartamento de Quimica Organica, Universidad de Cordoba, Edif. Marie Curie,
Ctra Nnal IVa, Km 396, 14014 Cordoba, Spain
682 | Green Chem., 2013, 15, 682–689 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4K
View Article OnlineView Journal | View Issue

The X-ray powder diffraction pattern was obtained using aconventional powder diffractometer RIGAKU, model: Mini-Flex™ II benchtop X-ray Diffractometer; and an X-ray tube: Cu-Kα (30 kV/15 mA) radiation operating in Bragg–Brentano (θ/2θ)geometry. (Sample preparation: grinding when needed andcompression in the sample holder with a flat glass. Thesample area in the sample holder is about 2 cm2.) Trans-mission electron microscopy (TEM) experiments were per-formed on a Hitachi H8100 microscope, with a ThermoNoranlight elements EDS detector and a CCD camera for imageacquisition. The Fe3O4–MoO3 fine powder was placed on acarbon stub and the images were recorded at 5–15 kV using anLFD detector under low vacuum.
Elemental analysis was done by using an ICP-AES (induc-tively coupled plasma-atomic emission spectrometer) using aHoriba Jobin-Yvon, France, Ultima model equipped with a40.68 MHz RF generator, a Czerny–Turner monochromatorwith 1.00 m (sequential), an autosampler AS500 and a CMA(concomitant metals analyzer).
Scanning electron microscopy images were acquired usinga JEOL JSM7001F FEG-SEM. Elemental analysis was performedusing a light elements EDS detector from Oxford. The Fe3O4–
MoO3 powder was spread on a double-sided carbon tape andanalyzed using 25 kV acceleration voltage.
For SIMS (secondary ion mass spectrometry), positive andnegative secondary ion spectra were collected in the massrange of 0–100 m/z (T = 10 min) with an upgraded VG IonexIX23LS TOF-SIMS set-up based on the Poschenrieder design. Afocused liquid Ga+ gun in pulsed mode (6 kHz) was used as asource of the analytical ions. A beam current in dc mode at 14 keVwas ca. 15 nA with a raster size of 300 × 300 μm2. Sample potentialwas 5 kV. Vacuum during the experiments was maintained in therange of (2–3) × 10−9 mbar in the analytical chamber.
XPS measurements were performed on a VSW XPS systemwith the Class 100 energy analyzer being a part of an exper-imental setup assembled for surface investigation. The spectrawere taken in fixed analyzer transmission mode with the apass energy of 22 eV, i.e. FAT 22. Fe3O4–MoO3 fine powder wasprepared for XPS by pressing on an In (Indium) plate as amatrix in order to reduce the charging problems and provide-ing mechanical support. For the energy axis calibration, Ag(110) and polycrystalline Au samples (previously cleaned byion sputtering) were used. Proton NMR spectra were recordedon a Bruker, 400, 5 mm probe at 400 MHz. 1H shifts arereported relative to internal TMS.
Preparation of ferrites20a–c
The FeCl3·6H2O (5.4 g) and urea (3.6 g) were dissolved in water(200 mL) at 85 to 90 °C for 2 h. The solution turned to a browncolor. To the resultant reaction mixture cooled to room temp-erature was added FeSO4·7H2O (2.8 g) and then 0.1 M NaOHuntil pH 10. The molar ratio Fe(III) to Fe(II) in the above systemwas nearly 2.00. The obtained hydroxides were treated by ultra-sound in the sealed flask at 30 to 35 °C for 30 min. Afterageing for 5 h, the obtained black powder of Fe3O4 waswashed, and dried under vacuum.
Preparation of Nanocat-Fe–Mo MNPs20a–b
Ferrite magnetic nanoparticles Fe3O4 (2 g) and ammoniummolybdate (NH4)6Mo7O24·4H2O (0.147 mg to get 1–2% of Mo)were stirred at room temperature in aqueous solution (50 mL)for 1 h. After impregnation, the suspension was adjusted topH 12 by adding sodium hydroxide (1.0 M) and stirred for20 h. The solid was washed with distilled water (5 × 10 mL).The obtained metal precursors were reduced by adding 0.2 MNaBH4 water solution dropwise under gentle stirring in an ice-water bath for 30 min until no bubbles were observed in thesolution. The resulting Nanocat-Fe–Mo MNPs were kept underultrasonication for 10 min and then washed with distilledwater and subsequently with ethanol and dried under vacuumat 60 °C for 24 h. The Mo content was determined by ICP-AESand it was found to be 0.48%.
Catalytic experiments
Oxidation of benzyl alcohol (2) to benzaldehyde (3). Amixture of benzyl alcohol 2 (1 mmol), TBHP (1 mL, 5 M solu-tion in decane), and Nanocat-Fe–Mo (100 mg, 0.5 mol%) wasstirred at 80 °C for 7 h. After completion of the reaction [moni-tored by TLC; hexane–EtOAc (4 : 1)], 5 mL of ethyl acetate wereadded and the catalyst removed by simple decantation. Theorganic solvent was evaporated under vacuum and the crudeproduct was purified by column chromatography on silica gelor preparative chromatography using n-hexane and ethylacetate as an eluent.
Reduction of 3-chloronitrobenzene (4) to 3-chloroaniline(5). 3-Chloronitro benzene 4 (1 mmol), isopropyl alcohol(3 mL), KOH (1.5 mmol) and Nanocat-Fe–Mo (100 mg, 0.5 mol%)were stirred at 80 °C for 4 h. After completion of the reaction[monitored by TLC; hexane–EtOAc (7 : 3)], the catalyst wasseparated magnetically. The resultant product was extracted
Scheme 1 Synthesis of Nanocat-Fe–Mo (1).
Green Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 682–689 | 683
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online

with ethyl acetate and repeatedly washed with water (5 to 7times) to remove KOH. Then, the organic solvent was evapo-rated in vacuo. The aniline 5 was purified by column chromato-graphy on silica gel using n-hexane and ethyl acetate as aneluent.
Syntheses of the Mannich-type adduct (9a–c). To a mixtureof aldehyde 6 (1 mmol), amine (1.2 mmol) and phenylacety-lene 8 (1.5 mmol) in toluene (2 mL) was added the catalystNanocat-Fe–Mo (50 mg, 0.25 mol%). The reaction mixture wasstirred at 100 °C for 15 h. The progress of the reaction wasmonitored by TLC (hexane–EtOAc = 7 : 3). After completion ofthe reaction, Nanocat-Fe–Mo was separated magnetically. Thefiltrate containing the reaction mixture was concentratedunder reduced pressure to remove the toluene. To the residuewater (5 mL) was added and then extracted with EtOAc (3 ×5 mL). The combined organic layers were washed with brinesolution (10 mL) and then dried over anhydrous Na2SO4. Eva-poration of the solvent under reduced pressure yielded thecrude product, which was then purified by column chromato-graphy using hexane–ethyl acetate as an eluent to afford thepure product.
Hydration of benzonitrile (11). A mixture of benzonitrile 10(1 mmol), 3 mL water and Nanocat-Fe–Mo (50 mg, 0.25 mol%)was added to a Teflon-sealed screw-cap culture tube andstirred at 100 °C for 3 h. The reaction was monitored by TLC(EtOAc–hexane 3 : 7). After completion of the reaction, the reac-tion mixture turned clear and catalyst was deposited on themagnetic needle, which was easily removed from the reactionmixture using an external magnet. After separation of the cata-lyst, the clear liquid was cooled slowly and a pure crystal ofbenzamide was obtained which can be isolated from the watermedium by filtration.
Three component reaction (15a–c). A mixture of aldehyde12 (2 mmol), malonitrile 13 (2.2 mmol), 3,3-dimethyl-1,3-cyclo-hexanedione 14 (2 mmol), and Nanocat-Fe–Mo (50 mg,0.25 mol% Mo) in ethanol (3 mL) was stirred at room tempera-ture until the reaction mixture solidified. After completion ofthe reaction (TLC; EtOAc–hexane 4 : 6), the solid product wasdissolved in hot ethanol (2 mL), and the catalyst removed bysimple decantation. The solvent was removed under reducedpressure and the residue recrystallized from hot ethanol toyield the desired products 15a–c.
Results and discussion
The Nanocat-Fe–Mo was characterized by X-ray diffraction(XRD), inductive coupled plasma-atomic emission spec-troscopy (ICP-AES), transmission electron microscopy (TEM),X-ray photoelectron spectroscopy (XPS), field-emission gunscanning electron microscopy and electron dispersive spec-trometry (FEG-SEM-EDS) as well as secondary ion mass spec-trometry (SIMS). The Mo content in the nanocatalyst wasdetermined to be 0.48 wt% by ICP-AES analysis.
XRD spectra of Fe3O4 and Nanocat-Fe–Mo nanoparticlesdepicted in Fig. 1 showed that the crystallite size of the
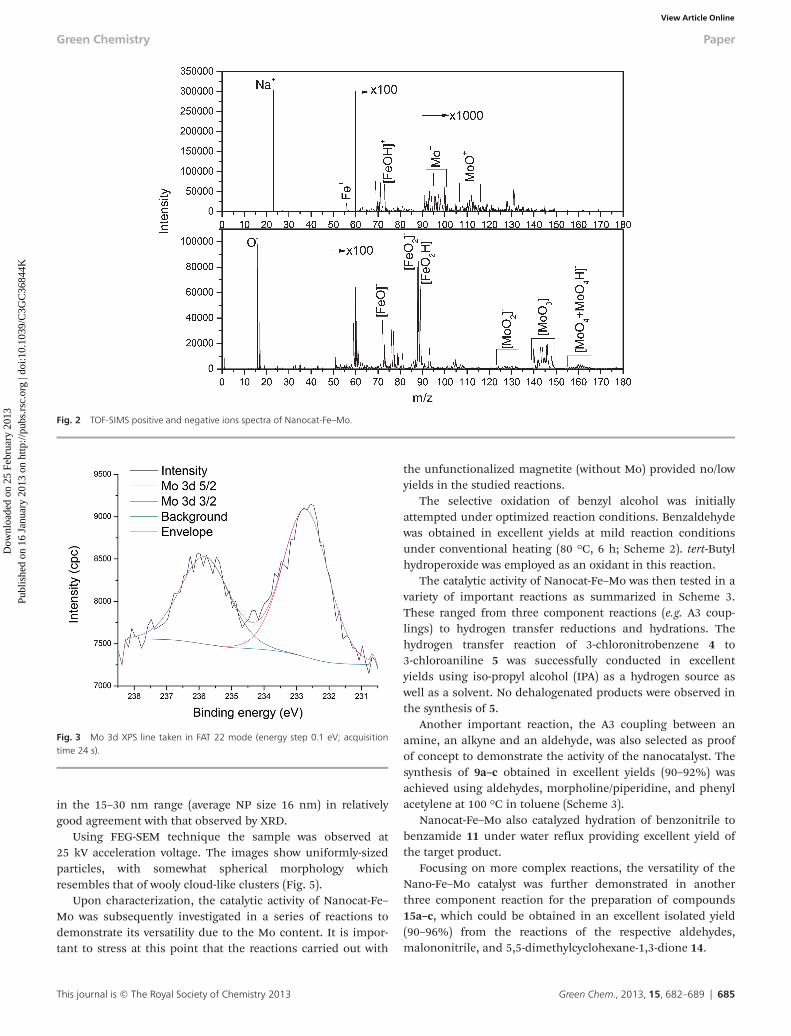
Nanocat-Fe–Mo MNPs was ca. 20 nm (determined by theDebye–Scherrer equation). Due to a low percentage of Mo(0.48 wt%), no diffraction lines corresponding to the observedMo oxides using alternative techniques including XPS andTOF-SIMS could be detected by XRD.
In order to confirm the incorporation of Mo on the ferrite,the Nanocat-Fe–Mo was analyzed by SIMS (secondary ioni-zation mass spectrometry). The sample was analyzed in bothpositive and negative ion mode. TOF-SIMS spectra are shownin Fig. 2. The major ions detected in positive mode corres-ponded to Na (m/z = 23), Fe (56), Ga (69, 71), FeOH (73), FeONa(95), Mo (92, 94, 95, 96, 98, 100), and MoO (108–116, atrespective m/z). Mo and MoO in positive mode were interferingwith other ions including FeONa, Fe2 etc.
Comparatively, the major ions detected in negative ionmode corresponded to FeO (72), FeOH (73), FeO2 (88), FeO2H(89), MoO2, MoO3, MoO4 and interfering MoO4H peaks.
In any case, the oxidation state and nature of the Mospecies in the final nanocatalyst were eventually confirmed byX-ray photoelectron spectroscopy (XPS). Surface compositionof the Nanocat-Fe–Mo powder was determined from thecharacteristic XPS peak intensities of Mo, Fe, O and C (i.e.Mo 3d, Fe 2p, O 1s and C 1s, respectively). Oxygen appeared tobe the most abundant element in the nanocatalyst (52.9%)followed by carbon (32.3%), iron (10.2%) and molybdenum(0.4%). Mo content found by XPS was found to be in goodagreement with that measured by ICP-AES, which indicatesthat most Mo in the final nanocatalyst is present on thesurface of the nanoferrite. The characteristic peaks of molyb-denum (Mo 3d) are presented in Fig. 3. The two peaks (3d5/2and 3d3/2) have intensity ratio 61 : 39 (being close to a theoreti-cal 60 : 40), with FWHM at about 1.68 eV. The position of the3d5/2 line is 232.7 eV, which perfectly matches the MoO3 phaseas confirmed in the literature.21
Transmission electron microscopy (TEM) experiments ofNanocat-Fe–Mo exhibited uniform-sized magnetic nanoparti-cles with almost spherical morphology (Fig. 4). NP sizes were
Fig. 1 X-ray diffraction patterns of Nano-Fe–Mo catalyst as compared to theparent magnetite material.
Paper Green Chemistry
684 | Green Chem., 2013, 15, 682–689 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online
in the 15–30 nm range (average NP size 16 nm) in relativelygood agreement with that observed by XRD.
Using FEG-SEM technique the sample was observed at25 kV acceleration voltage. The images show uniformly-sizedparticles, with somewhat spherical morphology whichresembles that of wooly cloud-like clusters (Fig. 5).
Upon characterization, the catalytic activity of Nanocat-Fe–Mo was subsequently investigated in a series of reactions todemonstrate its versatility due to the Mo content. It is impor-tant to stress at this point that the reactions carried out with
the unfunctionalized magnetite (without Mo) provided no/lowyields in the studied reactions.
The selective oxidation of benzyl alcohol was initiallyattempted under optimized reaction conditions. Benzaldehydewas obtained in excellent yields at mild reaction conditionsunder conventional heating (80 °C, 6 h; Scheme 2). tert-Butylhydroperoxide was employed as an oxidant in this reaction.
The catalytic activity of Nanocat-Fe–Mo was then tested in avariety of important reactions as summarized in Scheme 3.These ranged from three component reactions (e.g. A3 coup-lings) to hydrogen transfer reductions and hydrations. Thehydrogen transfer reaction of 3-chloronitrobenzene 4 to3-chloroaniline 5 was successfully conducted in excellentyields using iso-propyl alcohol (IPA) as a hydrogen source aswell as a solvent. No dehalogenated products were observed inthe synthesis of 5.
Another important reaction, the A3 coupling between anamine, an alkyne and an aldehyde, was also selected as proofof concept to demonstrate the activity of the nanocatalyst. Thesynthesis of 9a–c obtained in excellent yields (90–92%) wasachieved using aldehydes, morpholine/piperidine, and phenylacetylene at 100 °C in toluene (Scheme 3).
Nanocat-Fe–Mo also catalyzed hydration of benzonitrile tobenzamide 11 under water reflux providing excellent yield ofthe target product.
Focusing on more complex reactions, the versatility of theNano-Fe–Mo catalyst was further demonstrated in anotherthree component reaction for the preparation of compounds15a–c, which could be obtained in an excellent isolated yield(90–96%) from the reactions of the respective aldehydes,malononitrile, and 5,5-dimethylcyclohexane-1,3-dione 14.
Fig. 2 TOF-SIMS positive and negative ions spectra of Nanocat-Fe–Mo.
Fig. 3 Mo 3d XPS line taken in FAT 22 mode (energy step 0.1 eV; acquisitiontime 24 s).
Green Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 682–689 | 685
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online

The applicability of the Mo-based catalyst in redox chem-istries (both oxidations and reductions) has been frequentlyreported in the literature.22 The advantage of the Nano-Fe–Mocatalyst is the reusability of the catalyst as well as its facilerecovery and reutilisation from the practicality viewpoint.Extended applications of the herein reported in MCR andhydration reactions can be rationalized by the presence ofLewis acid sites in Nano-Fe–Mo (detected qualitatively, results
Fig. 4 TEM image of (a) Fe3O4 at 200 nm, (b) Nanocat-Fe–Mo at 200 nm and (c) 500 nm. (d) Histogram of Nanocat-Fe–Mo.
Fig. 5 SEM images of Nanocat-Fe–Mo nanoparticles.
Scheme 2 Nanocat-Fe–Mo catalyzed selective oxidation of benzyl alcohol.
Paper Green Chemistry
686 | Green Chem., 2013, 15, 682–689 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online
not shown). In terms of coupling chemistries, functionalisedmagnetites have also been recently reported to be active inhomocoupling reactions of benzeneboronic acids.23
Having proved the activity and versatility of the synthesizednanocatalyst in the selected chemistries, its stability was sub-sequently tested by recycling and reuse of the catalyst in theproposed reactions.
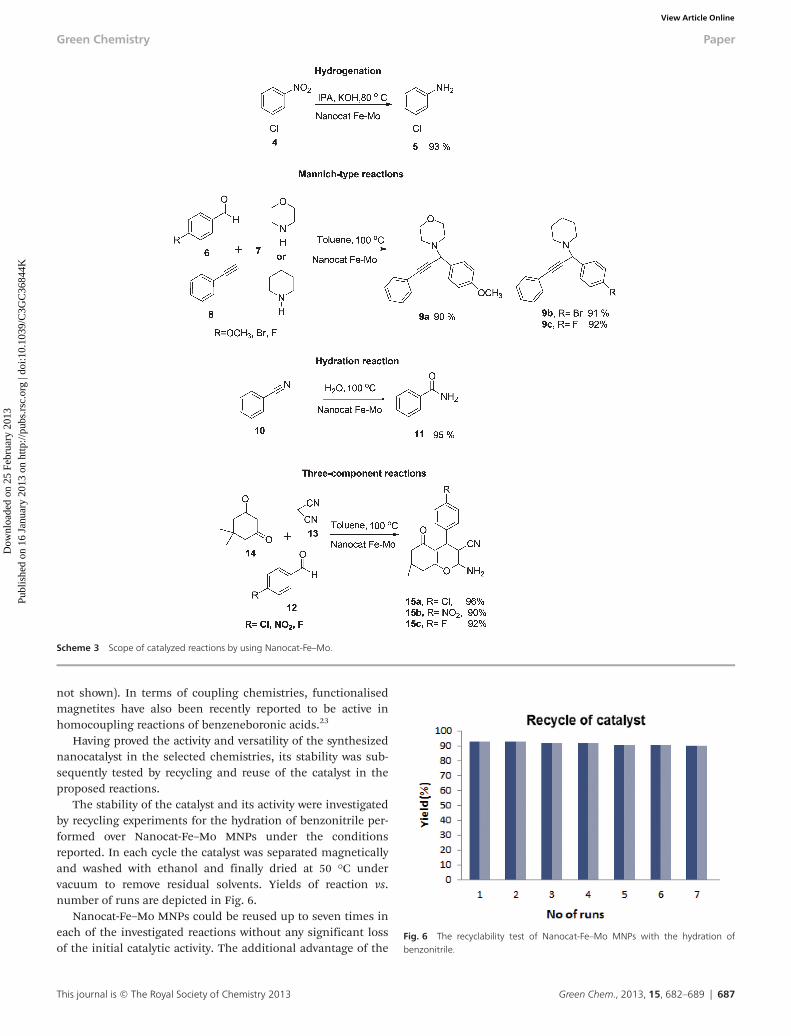
The stability of the catalyst and its activity were investigatedby recycling experiments for the hydration of benzonitrile per-formed over Nanocat-Fe–Mo MNPs under the conditionsreported. In each cycle the catalyst was separated magneticallyand washed with ethanol and finally dried at 50 °C undervacuum to remove residual solvents. Yields of reaction vs.number of runs are depicted in Fig. 6.
Nanocat-Fe–Mo MNPs could be reused up to seven times ineach of the investigated reactions without any significant lossof the initial catalytic activity. The additional advantage of the
Scheme 3 Scope of catalyzed reactions by using Nanocat-Fe–Mo.
Fig. 6 The recyclability test of Nanocat-Fe–Mo MNPs with the hydration ofbenzonitrile.
Green Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 682–689 | 687
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online

proposed system relates to its ease of separation by means of asimple magnet, which could be done at each cycle. Standardleaching experiments (using the hot filtration method andICP-AES) provided no detectable quantities of Fe or Mo in thefiltrate. No conversion evolution was observed in the absenceof catalyst (even after 12 h, hydration of benzonitrile underwater reflux).
Conclusions
A novel, highly active and versatile Nanocat-Fe–Mo catalyst wassynthesized from inexpensive precursors using a simple pre-cipitation/impregnation procedure. The nanomaterial wasfound to be active and highly selective in a range of chosenchemistries including oxidations, transfer hydrogenations,hydrations and three component reactions. Selected processessuccessfully provided target products in excellent isolatedyields. The magnetically separable system was also very stableunder the investigated conditions, preserving almost intact itsinitial activity up to seven uses regardless of the type ofselected reaction. In view of the stability and versatility of thecurrent system, further investigations to expand the scope ofthe nanocatalyst to more substrates and chemistries are under-way in our laboratories.
Acknowledgements
This work has been supported by Fundação para a Ciência e aTecnologia through grant no. PEst-C/EQB/LA0006/2011. ManojB. Gawande also thanks the PRAXIS program for the award ofa research fellowship SFRH/BPD/64934/2009. We acknowledgealso funding from the Portuguese research Grant Pest-OE/FIS/UI0068/2011 through FCT-MEC.
References
1 (a) R. R. Arvizo, S. Bhattacharyya, R. A. Kudgus, K. Giri,R. Bhattacharya and P. Mukherjee, Chem. Soc. Rev., 2012,41, 2943–2970; (b) G. Spada, E. Gavini, M. Cossu, G. Rassuand P. Giunchedi, Nanotechnology, 2012, 23; (c) T. L. Doaneand C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911.
2 N. A. Frey, S. Peng, K. Cheng and S. H. Sun, Chem. Soc.Rev., 2009, 38, 2532–2542.
3 V. Berry and R. F. Saraf, Angew. Chem., Int. Ed., 2005, 44,6668–6673.
4 (a) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int.Ed., 2005, 44, 7852–7872; (b) M. A. Newton, Chem. Soc. Rev.,2008, 37, 2644–2657; (c) A. Schatz, T. R. Long, R. N. Grass,W. J. Stark, P. R. Hanson and O. Reiser, Adv. Funct. Mater.,2010, 20, 4323–4328; (d) M. B. Gawande, P. S. Branco,K. Parghi, J. J. Shrikhande, R. K. Pandey,C. A. A. Ghumman, N. Bundaleski, O. Teodoro andR. V. Jayaram, Catal. Sci. Technol., 2011, 1, 1653–1664;
(e) M. B. Gawande, S. N. Shelke, A. Rathi, P. S. Branco andR. K. Pandey, Appl. Organomet. Chem., 2012, 26, 395–400.
5 (a) A. Schatz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010,16, 8950–8967; (b) S. Roy and M. A. Pericas, Org. Biomol.Chem., 2009, 7, 2669–2677.
6 S. E. Garcia-Garrido, J. Francos, V. Cadierno, J. M. Bassetand V. Polshettiwar, ChemSusChem, 2011, 4, 104–111.
7 P. D. Stevens, G. F. Li, J. D. Fan, M. Yen and Y. Gao, Chem.Commun., 2005, 4435–4437.
8 Z. Yinghuai, S. C. Peng, A. Emi, S. Zhenshun, Monalisa andR. A. Remp, Adv. Synth. Catal., 2007, 349, 1917.
9 A. Schatz, R. N. Grass, Q. Kainz, W. J. Stark and O. Reiser,Chem. Mater., 2010, 22, 305–310.
10 C. O’Dalaigh, S. A. Corr, Y. Gun’ko and S. J. Connon,Angew. Chem., Int. Ed., 2007, 46, 4329–4332.
11 A. G. Hu, G. T. Yee and W. B. Lin, J. Am. Chem. Soc., 2005,127, 12486–12487.
12 F. Shi, M. K. Tse, M. M. Pohl, A. Bruckner, S. M. Zhang andM. Beller, Angew. Chem., Int. Ed., 2007, 46, 8866–8868.
13 (a) C. L. Wu, X. K. Fu and S. Li, Eur. J. Org. Chem., 2011,1291–1299; (b) M. Kawamura and K. Sato, Chem. Commun.,2006, 4718–4719; (c) A. H. Lu, E. L. Salabas and F. Schuth,Angew. Chem., Int. Ed., 2007, 46, 1222–1244; (d) R. Cano,D. J. Ramon and M. Yus, J. Org. Chem., 2011, 76,5547–5557; (e) R. Cano, D. J. Ramon and M. Yus, Tetra-hedron, 2011, 67, 5432–5436; (f ) G. Chouhan, D. S. Wangand H. Alper, Chem. Commun., 2007, 4809–4811;(g) H. M. R. Gardimalla, D. Mandal, P. D. Stevens, M. Yenand Y. Gao, Chem. Commun., 2005, 4432–4434;(h) O. Gleeson, R. Tekoriute, Y. K. Gun’ko and S. J. Connon,Chem.–Eur. J., 2009, 15, 5669–5673; (i) F. M. Koehler,M. Rossier, M. Waelle, E. K. Athanassiou, L. K. Limbach,R. N. Grass, D. Gunther and W. J. Stark, Chem. Commun.,2009, 4862–4864; ( j) C. W. Lim and I. S. Lee, Nano Today,2010, 5, 412–434; (k) D. Rosario-Amorin, X. Wang,M. Gaboyard, R. Clerac, S. Nlate and K. Heuze, Chem.–Eur.J., 2009, 15, 12636–12643; (l) M. Shokouhimehr, Y. Z. Piao,J. Kim, Y. J. Jang and T. Hyeon, Angew. Chem., Int. Ed.,2007, 46, 7039–7043; (m) M. Kotani, T. Koike,K. Yamaguchi and N. Mizuno, Green Chem., 2006, 8,735–741; (n) F. Mi, X. T. Chen, Y. W. Ma, S. T. Yin,F. L. Yuan and H. Zhang, Chem. Commun., 2011, 47,12804–12806; (o) F. Niu, L. Zhang, S. Z. Luo andW. G. Song, Chem. Commun., 2010, 46, 1109–1111;(p) L. Xu, Y. Ma, Y. Zhang, Z. Jiang and W. Huang, J. Am.Chem. Soc., 2009, 131, 16366–16367; (q) R. B. N. Baig andR. S. Varma, Green Chem., 2012, 14, 625–632.
14 V. Polshettiwar, R. Luque, A. Fihri, H. B. Zhu, M. Bouhraraand J. M. Bassett, Chem. Rev., 2011, 111, 3036–3075;M. B. Gawande, P. S. Branco and R. S. Varma, Chem. Soc.Rev., 2013, DOI: 10.1039/c3cs35480f.
15 (a) M. J. Aliaga, D. J. Ramon and M. Yus, Org. Biomol.Chem., 2010, 8, 43–46; (b) R. Cano, D. J. Ramon andM. Yus, J. Org. Chem., 2010, 75, 3458–3460; (c) R. Cano,M. Yus and D. J. Ramon, Tetrahedron, 2012, 68,1393–1400.
Paper Green Chemistry
688 | Green Chem., 2013, 15, 682–689 This journal is © The Royal Society of Chemistry 2013
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online

16 J. Liu, X. Peng, W. Sun, Y. Zhao and C. Xia, Org. Lett., 2008,10, 3933–3936.
17 K. V. S. Ranganath, J. Kloesges, A. H. Schafer andF. Glorius, Angew. Chem., Int. Ed., 2010, 49, 7786–7789.
18 (a) P. S. Branco, V. P. Raje, J. Dourado and J. Gordo, Org.Biomol. Chem., 2010, 8, 2968–2974; (b) M. B. Gawande andP. S. Branco, Green Chem., 2011, 13, 3355–3359.
19 (a) E. J. Garcia-Suarez, A. M. Balu, M. Tristany, A. B. Garcia,K. Philippot and R. Luque, Green Chem., 2012, 14,1434–1439; (b) V. L. Budarin, J. H. Clark, R. Luque,D. J. Macquarrie and R. J. White, Green Chem., 2008, 10,382–387; (c) M. B. Gawande, R. K. Pandey andR. V. Jayaram, Catal. Sci. Technol., 2012, 2, 1113–1125;(d) C. Han, R. Luque and D. D. Dionysiou, Chem. Commun.,2012, 48, 1860–1862; (e) A. Pineda, A. M. Balu,J. M. Campelo, A. A. Romero, D. Carmona, F. Balas,J. Santamaria and R. Luque, ChemSusChem, 2011, 4,1561–1565; (f ) S. N. Shelke, G. R. Mhaske,V. D. B. Bonifácio and M. B. Gawande, Bioorg. Med. Chem.Lett., 2012, 22, 5727–5730; (g) M. B. Gawande, H. Guo,A. K. Rathi, P. S. Branco, Y. Chen, R. S. Varma andD. L. Peng, RSC Adv., 2013, 3, 1050–1054.
20 (a) M. B. Gawande, A. K. Rathi, P. S. Branco, I. D. Nogueira,A. Velhinho, J. J. Shrikhande, U. U. Indulkar, R. V. Jayaram,C. A. A. Ghumman, N. Bundaleski and O. M. N.D. Teodoro, Chem.–Eur. J., 2012, 18, 12628–12632;
(b) M. B. Gawande, A. Velhinho, I. D. Nogueira,C. A. A. Ghumman, O. Teodoro and P. S. Branco, RSC Adv.,2012, 2, 6144–6149; (c) M. B. Gawande, A. Rathi,I. D. Nogueira, C. A. A. Ghumman, N. Bundaleski, O. M. N.D. Teodoro and P. S. Branco, ChemPlusChem, 2012, 77,865–871; (d) A. M. Balu, B. Baruwati, E. Serrano, J. Cot,J. Garcia-Martinez, R. S. Varma and R. Luque, Green Chem.,2011, 13, 2750–2758; (e) F. Rajabi, N. Karimi, M. R. Saidi,A. Primo, R. S. Varma and R. Luque, Adv. Synth. Catal.,2012, 354, 1707–1711; (f ) M. B. Gawande, A. K. Rathi,P. S. Branco, T. M. Potewar, A. Velhinho, I. D. Nogueira,A. Tolstogouzov, C. Amjad, A. Ghumman and O. M. N. D.Teodoro, RSC Adv., 2013, 3, 1050–1053.
21 (a) Th. Weber, J. C. Muijsers, J. H. M. C. van Wolput,C. P. J. Verhagen and J. W. Niemantsverdriet, J. Phys.Chem., 1996, 100, 14144–14150; (b) D. O. Scanlon,G. W. Watson, D. J. Payne, G. R. Atkinson, R. G. Egdell andD. S. L. Law, J. Phys. Chem. C, 2010, 114, 4636–4645.
22 (a) K. Routray, W. Zhou, C. J. Kiely, W. Grünert andI. E. Wachs, J. Catal., 2010, 275, 84–98; (b) S. Shylesh,J. Schweizer, S. Demeshko, V. Schunemann, S. Ernst andW. R. Thiel, Adv. Synth. Catal., 2009, 351, 1789–1795;(c) N. Perret, X. Wang, L. Delannoy, C. Potvin, C. Louis andM. A. Keane, J. Catal., 2012, 286, 172–183.
23 R. Luque, B. Baruwati and R. S. Varma, Green Chem., 2010,12, 1540–1543.
Green Chemistry Paper
This journal is © The Royal Society of Chemistry 2013 Green Chem., 2013, 15, 682–689 | 689
Dow
nloa
ded
on 2
5 Fe
brua
ry 2
013
Publ
ishe
d on
16
Janu
ary
2013
on
http
://pu
bs.r
sc.o
rg |
doi:1
0.10
39/C
3GC
3684
4KView Article Online




















