
Brain injury activates microglia that induce neural stem cell proliferation ex vivo and promote...
11
BRAIN INJURY ACTIVATES MICROGLIA THAT INDUCE NEURAL STEM CELL PROLIFERATION EX VIVO AND PROMOTE DIFFERENTIATION OF NEUROSPHERE-DERIVED CELLS INTO NEURONS AND OLIGODENDROCYTES T. DEIERBORG,* L. ROYBON, A. R. INACIO, J. PESIC AND P. BRUNDIN Neuronal Survival Unit, Department of Experimental Medical Science, Wallenberg Neuroscience Center, Lund University, BMC A10, 22184 Lund, Sweden Abstract—Brain damage, such as ischemic stroke, enhances proliferation of neural stem/progenitor cells (NSPCs) in the subventricular zone (SVZ). To date, no reliable in vitro sys- tems, which can be used to unravel the potential mechanisms underlying this lesion-induced effect, have been established. Here, we developed an ex vivo method to investigate how the proliferation of NSPCs changes over time after experimental stroke or excitotoxic striatal lesion in the adult rat brain by studying the effects of microglial cells derived from an in- jured brain on NSPCs. We isolated NSPCs from the SVZ of brains with lesions and analyzed their growth and differenti- ation when cultured as neurospheres. We found that NSPCs isolated from the brains 1–2 weeks following injury consis- tently generated more and larger neurospheres than those harvested from naive brains. We attributed these effects to the presence of microglial cells in NSPC cultures that origi- nated from injured brains. We suggest that the effects are due to released factors because we observed increased prolifer- ation of NSPCs isolated from non-injured brains when they were exposed to conditioned medium from cultures contain- ing microglial cells derived from injured brains. Furthermore, we found that NSPCs derived from injured brains were more likely to differentiate into neurons and oligodendrocytes than astrocytes. Our ex vivo system reliably mimics what is ob- served in vivo following brain injury. It constitutes a powerful tool that could be used to identify factors that promote NSPC proliferation and differentiation in response to injury-induced activation of microglial cells, by using tools such as proteom- ics and gene array technology. © 2010 IBRO. Published by Elsevier Ltd. All rights reserved. Key words: ischemia, neurogenesis, SVZ, neural progenitor, stroke, microglia. In the adult human brain, newborn neurons arise in the subgranular zone (SGZ) in the dentate gyrus of the hippocampus and the subventricular zone (SVZ) lining the lateral ventricles (Eriksson et al., 1998; Curtis et al., 2007). Brain damage, such as experimental stroke (Arvidsson et al., 2002) or intrastriatal injection of the excitotoxin quinolinic acid (QA; Tattersfield et al., 2004) enhances the proliferation of neural stem/progenitor cells (NSPCs) that are located in the SVZ or the ependymal cell layer (Carlen et al., 2009). Understanding the cellular mechanisms that regulate the proliferation and differentiation of NSPCs fol- lowing brain injury is important, as these cells have been suggested to contribute to endogenous brain repair and can constitute a potential source of donor tissue for cell transplantation therapies (Deierborg et al., 2008; Burns et al., 2009). In the hippocampal SGZ, microglial cells have been reported to impede lesion-induced neurogenesis (Monje et al., 2003). However, recent data suggest that microglia have diverse functions in relation to neurogenesis. For example, microglia activated by endotoxins inhibit neuro- genesis in vivo. Conversely, when activated by cytokines interferon (IFN)-g/ interleukin (IL)-4, they enhance neuro- nal differentiation (Butovsky et al., 2006). Moreover, micro- glia activated by lipopolysaccharide (LPS) for 24 h (acute activation) release pro-inflammatory cytokines, in contrast to chronically activated microglia that adopt an anti-inflam- matory phenotype (Cacci et al., 2008). In light of the above findings, it is valuable to define how different durations of activation of microglia, in clini- cally relevant models of brain injury, affect the proliferation and the fate of NSPCs. Therefore, we developed an ex vivo system to study the effect of activated microglia on NSPCs. We stimulated microglia by inducing ischemic or excitotoxic lesions in the brain of rats, and examined their effect on NSPCs. We found that microglia harvested dur- ing the first 1–2 weeks following brain injury exert profound effects, mediated via soluble factor(s), on the proliferation of NSPCs grown in vitro. The effect resembles the injury- induced enhacement of neurogenesis previously observed in vivo (Thored et al., 2009). These results suggest that the ex vivo model we have developed is highly relevant to the in vivo situation and can be further used to clarify mecha- *Corresponding author. Tel: 46-46-2229827; fax: 46-46-2220531. E-mail address: [email protected] (T. Deierborg). Abbreviations: ANOVA, analysis of variance; bFGF, basic fibroblast growth factor; BrdU, 5-bromodeoxyuridine; CNPase, 2=,3=-cyclic nucleotide 3=-phosphodiesterase(oligodendrocyte specific marker); DAPI, 4=,6, diamidino-2-phenylindole; DMEM:HAMS-F12, Dulbecco’s Modified Eagle’s Medium and Ham’s Nutrient Mixtures F-12; EGF, epidermal growth factor; GFAP, glial fibrillary acidic protein (astrocyte specific marker); Iba1, ionized calcium binding adaptor molecule 1(macrophages/microglia specific marker); IFN, interferon; IL, interleu- kin; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion; MHC, major histocompatibility complex; NSPCs, neural stem/progen- itor cells; OX42/CD11b, single-pass type-1 chain 1136 aa glycopro- tein(microglia/macrophage-specific marker; clone of antibody that de- tects cell surface antigen-cluster of differentiation 11b, anti-comple- ment receptor 3); PBS, phosphate-buffered saline; QA, quinolinic acid; SVZ, subventricular zone. Neuroscience 171 (2010) 1386 –1396 0306-4522/10 $ - see front matter © 2010 IBRO. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.neuroscience.2010.09.045 1386
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Brain injury activates microglia that induce neural stem cell proliferation ex vivo and promote...

BSDN
TA
NWL
ApstuHpssjbaithtntawiwlastpaiE
*EAgnDMes1kMittmS
Neuroscience 171 (2010) 1386–1396
0d
RAIN INJURY ACTIVATES MICROGLIA THAT INDUCE NEURALTEM CELL PROLIFERATION EX VIVO AND PROMOTEIFFERENTIATION OF NEUROSPHERE-DERIVED CELLS INTO
EURONS AND OLIGODENDROCYTESKs
Isht2
eqpaerlscta
rahegingatm
hcavNeeieoiie
. DEIERBORG,* L. ROYBON, A. R. INACIO, J. PESICND P. BRUNDIN
euronal Survival Unit, Department of Experimental Medical Science,allenberg Neuroscience Center, Lund University, BMC A10, 22184
und, Sweden
bstract—Brain damage, such as ischemic stroke, enhancesroliferation of neural stem/progenitor cells (NSPCs) in theubventricular zone (SVZ). To date, no reliable in vitro sys-ems, which can be used to unravel the potential mechanismsnderlying this lesion-induced effect, have been established.ere, we developed an ex vivo method to investigate how theroliferation of NSPCs changes over time after experimentaltroke or excitotoxic striatal lesion in the adult rat brain bytudying the effects of microglial cells derived from an in-ured brain on NSPCs. We isolated NSPCs from the SVZ ofrains with lesions and analyzed their growth and differenti-tion when cultured as neurospheres. We found that NSPCssolated from the brains 1–2 weeks following injury consis-ently generated more and larger neurospheres than thosearvested from naive brains. We attributed these effects tohe presence of microglial cells in NSPC cultures that origi-ated from injured brains. We suggest that the effects are dueo released factors because we observed increased prolifer-tion of NSPCs isolated from non-injured brains when theyere exposed to conditioned medium from cultures contain-
ng microglial cells derived from injured brains. Furthermore,e found that NSPCs derived from injured brains were more
ikely to differentiate into neurons and oligodendrocytes thanstrocytes. Our ex vivo system reliably mimics what is ob-erved in vivo following brain injury. It constitutes a powerfulool that could be used to identify factors that promote NSPCroliferation and differentiation in response to injury-inducedctivation of microglial cells, by using tools such as proteom-
cs and gene array technology. © 2010 IBRO. Published bylsevier Ltd. All rights reserved.
Corresponding author. Tel: �46-46-2229827; fax: �46-46-2220531.-mail address: [email protected] (T. Deierborg).bbreviations: ANOVA, analysis of variance; bFGF, basic fibroblastrowth factor; BrdU, 5-bromodeoxyuridine; CNPase, 2=,3=-cyclicucleotide 3=-phosphodiesterase(oligodendrocyte specific marker);API, 4=,6, diamidino-2-phenylindole; DMEM:HAMS-F12, Dulbecco’sodified Eagle’s Medium and Ham’s Nutrient Mixtures F-12; EGF,pidermal growth factor; GFAP, glial fibrillary acidic protein (astrocytepecific marker); Iba1, ionized calcium binding adaptor molecule(macrophages/microglia specific marker); IFN, interferon; IL, interleu-in; LPS, lipopolysaccharide; MCAO, middle cerebral artery occlusion;HC, major histocompatibility complex; NSPCs, neural stem/progen-
tor cells; OX42/CD11b, single-pass type-1� chain 1136 aa glycopro-ein(microglia/macrophage-specific marker; clone of antibody that de-ects cell surface antigen-cluster of differentiation 11b, anti-comple-
ient receptor 3); PBS, phosphate-buffered saline; QA, quinolinic acid;VZ, subventricular zone.
306-4522/10 $ - see front matter © 2010 IBRO. Published by Elsevier Ltd. All rightoi:10.1016/j.neuroscience.2010.09.045
1386
ey words: ischemia, neurogenesis, SVZ, neural progenitor,troke, microglia.
n the adult human brain, newborn neurons arise in theubgranular zone (SGZ) in the dentate gyrus of theippocampus and the subventricular zone (SVZ) lining
he lateral ventricles (Eriksson et al., 1998; Curtis et al.,007).
Brain damage, such as experimental stroke (Arvidssont al., 2002) or intrastriatal injection of the excitotoxinuinolinic acid (QA; Tattersfield et al., 2004) enhances theroliferation of neural stem/progenitor cells (NSPCs) thatre located in the SVZ or the ependymal cell layer (Carlent al., 2009). Understanding the cellular mechanisms thategulate the proliferation and differentiation of NSPCs fol-owing brain injury is important, as these cells have beenuggested to contribute to endogenous brain repair andan constitute a potential source of donor tissue for cellransplantation therapies (Deierborg et al., 2008; Burns etl., 2009).
In the hippocampal SGZ, microglial cells have beeneported to impede lesion-induced neurogenesis (Monje etl., 2003). However, recent data suggest that microgliaave diverse functions in relation to neurogenesis. Forxample, microglia activated by endotoxins inhibit neuro-enesis in vivo. Conversely, when activated by cytokines
nterferon (IFN)-g/ interleukin (IL)-4, they enhance neuro-al differentiation (Butovsky et al., 2006). Moreover, micro-lia activated by lipopolysaccharide (LPS) for 24 h (acutectivation) release pro-inflammatory cytokines, in contrasto chronically activated microglia that adopt an anti-inflam-atory phenotype (Cacci et al., 2008).
In light of the above findings, it is valuable to defineow different durations of activation of microglia, in clini-ally relevant models of brain injury, affect the proliferationnd the fate of NSPCs. Therefore, we developed an exivo system to study the effect of activated microglia onSPCs. We stimulated microglia by inducing ischemic orxcitotoxic lesions in the brain of rats, and examined theirffect on NSPCs. We found that microglia harvested dur-
ng the first 1–2 weeks following brain injury exert profoundffects, mediated via soluble factor(s), on the proliferationf NSPCs grown in vitro. The effect resembles the injury-
nduced enhacement of neurogenesis previously observedn vivo (Thored et al., 2009). These results suggest that thex vivo model we have developed is highly relevant to the
n vivo situation and can be further used to clarify mecha-s reserved.

nN
A
Wawhle
E
Wslaaaunitioi2
Eq
WaiPPdidtbta
N
WpScdwlsTasub(SSmfmp
sdi
N
Alcdcp
I
Ft5
ctwibbrm31fiptJ11trfu
Na
Udflicrflo(admlamew
C
Caosg
T. Deierborg et al. / Neuroscience 171 (2010) 1386–1396 1387
isms underlying the effects of activated microglia onSPCs in the injured brain.
EXPERIMENTAL PROCEDURES
nimals
e used male Sprague–Dawley rats (B&K Stockholm, Sweden),ged 10–12 weeks at the start of the experiment. All experimentsere approved by the Malmö-Lund Ethical Committee. Animalsad free access to food and water and were housed under a 12-h
ight/12-h dark cycle. We used a minimal number of animals andfforts were done to avoid suffering.
xperimental model of stroke
e induced experimental stroke, focal brain ischemia, by tran-ient middle cerebral artery occlusion (tMCAO) using an estab-ished protocol (Memezawa et al., 1992). Briefly, the rats werenesthetized with halothane (1.5%, Astra Zeneca, Sweden) andn intraluminal filament was inserted into the internal carotid arterynd placed at the origin of the MCA. An occlusion time of 2 h wassed to induce both striatal and cortical damage. We assessedeurological symptoms after a 2 h recovery period. The neurolog-
cal symptoms were assessed using two behavioral tests: con-ralateral rotation/circling behavior and contralateral fore-/hindpawmpairment (lack of paw withdrawal when hanging over the edgef a table). A combination of these two tests can predict stroke-
njury affecting both the striatum and the cortex (Rickhag et al.,008).
xcitotoxic lesion to the striatum by injection ofuinolinic acid (QA)
e lesioned the striatum by an intracerebral injection of QA, usingn established protocol (Nakao and Brundin, 1997). Briefly, we
nitiated anesthesia by i.p. injection of Fentanyl (50 �g/mL,haramlink, Sweden) / Domitor (1 mg/mL Medetomidine, Orionharma, Finland); proportions: 2 ml Fentanyl�0.1 ml Domitor;ose: 6.3 ml/kg rat, further placed in a stereotaxic frame. We
nduced a large striatal lesion by an injection of 120 nmol of QAissolved in 1 �L of 0.1 M phosphate-buffered saline (PBS) into
he right striatum (rostral 1.2 mm/lateral 2.8 mm/ventral 4.5 mm toregma). The anesthesia was discontinued by a s.c. administra-ion of Antisedan (1 ml/kg) and Temgesic (1.7 ml/kg) was used asn analgesic.
eurosphere generation
e generated neurospheres using a standard protocol as describedreviously (Svendsen et al., 1998; Roybon et al., 2009). Briefly, theVZ was isolated from the ipsilateral side 7 days after a middleerebral artery occlusion (MCAO) or QA lesion, except for the studyesigned to study temporal features of microglial activation (Fig. 3)here the animals were sacrificed 1, 2, 3 and 4 weeks after QA
esion. We dissected out the ipsilateral SVZ from coronal sectionstarting at 1 mm caudal to the optic cross and ending 5 mm rostrally.he tissue was treated with trypsin/deoxyribonuclease (0.1/0.05%)nd was mechanically dissociated to generate a single-cell suspen-ion. The cells were plated at a density of 200,000 cells/mL inncoated T25 flasks in 7 ml of proliferation medium containing Dul-ecco’s Modified Eagle’s Medium and Ham’s Nutrient Mixtures F-12DMEM:HAMS-F12) (3:1), B27 (2%; Invitrogen-Gibco, Stockholm,weden), heparin (5 �g/mL), penicillin:streptomycin (50 �g/mL.igma, Stockholm, Sweden), epidermal growth factor (EGF; 20 ng/L, R&D Systems, Stockholm, Sweden) and basic fibroblast growth
actor (bFGF; 20 ng/mL, R&D Systems, Stockholm, Sweden). Theedium was supplemented with mitogens by the addition of 3 ml of
roliferation medium at 2 and 5 days in vitro. Secondary neuro- 7pheres (P1) and later passages were generated by mechanicalissociation of primary neurospheres (P0). Individual flasks represent
ndividual animals.
eurosphere differentiation
fter 7 days in vitro, the neurospheres were plated on poly-L-ysine/Laminin (15/0.01 mg/mL, Sigma, Stockholm, Sweden)oated glass coverslips. We induced the differentiation by with-rawal of EGF and bFGF. The differentiation medium washanged after 3 days. At day 5, we fixed the cultures using 4%araformaldehyde in PBS.
mmunohistochemistry
or immunohistochemical analysis, animals were deeply anesthe-ized with pentobarbital and transcardially perfused with saline for
min, followed by 4% paraformaldehyde in PBS for 10 min.Brains were post-fixed in the same fixative overnight at 4 °C,
ryopreserved in 30% sucrose in PBS and subsequently, 30 �mhick coronal sections were cut using a microtome (Zeiss) andere preserved in an antifreeze solution. For immunohistochem-
stry, we pre-incubated the brain sections/cultures for 1 h with alocking solution (5% goat serum, 0.25% Triton X-100), followedy an overnight incubation of the appropriate primary antibody atoom temperature. The following primary antibodies were used:ouse anti-�-III-tubulin (1:1000), mouse anti-2=,3=-cyclic nucleotide=-phosphodiesterase(oligodendrocyte specific marker) (CNPase;:300, monoclonal, Sigma, Stockholm, Sweden), rabbit anti-glialbrillary acidic protein (astrocyte specific marker) (GFAP; 1:1000,olyclonal, Dako, Stockholm, Sweden) OX-42/CD11b (1:200, Sero-ec, Oxford, UK) and Iba1 (1:500, rabbit, Wako Chemicals, Osaka,apan). The corresponding secondary antibodies were incubated for
h (Alexa-488 goat anti-mouse and Alexa-595 goat anti-rabbit;:200; Molecular Probes, Stockholm, Sweden) followed by incuba-ion with DAPI (1:1000, Sigma, Stockholm, Sweden) for 15 min andinsing before mounting on glass slides with PVA-DABco. We per-ormed the analysis with a fluorescence microscope (Leica BX60)sing FITC/CY3 filters at 40� magnification.
eurosphere quantification and differentiationnalysis
sing a 4� objective, we counted all the neurospheres (�40 �m iniameter) in the visual field in five different areas of the T25 cultureask (each corner and the center). The number of neurospheres inndividual flasks was estimated by taking the sum of all neurospheresounted in the five fields. We estimated the mean diameter of neu-ospheres by randomly measuring 10 neurospheres in each cultureask using a 20� objective and an eyepiece grid scale. The numberf neurons (�-III-tubulin), astrocytes (GFAP) and oligodendrocytesCNPase) was assessed for each experimental condition (individualnimals) by selecting five neurospheres of similar size (�100 �m iniameter) (Roybon et al., 2005), and analyzing only the cells thatigrated out from the neurospheres and attached to the Poly-L-
ysine/Laminin-coated surface. The numbers of neurons, astrocytes,nd oligodendrocytes were counted as a percentage of approxi-ately 150 DAPI positive cells per field (monitored in five fields forach sphere). Only cells with a clear immunoreactivity/morphologyere included in the analysis.
onditioned medium and flasks with microglial cells
ultures of SVZ-derived NSPCs from a lesioned brain gave rise tomonolayer of microglial cells, possibly originating from the SVZ
r the adjacent injured striatal tissue. We studied the effect ofoluble factors (secreted from microglial cells) on neurosphereeneration by using conditioned medium (centrifuged at 450 g, for
min, at 4 °C) from the different experimental groups (Ctrl, MCAO
aapanaTcad
S
FstfWfcwdms
Da
TgdiigtaeAd1hbmn
If
WcaoWennScsma2
TpNutiSZ
Tp
WNnw(psfhwnwTia
Cb
WwcmeflQpwmSiSsfstcnrme
Mse
Ti
T. Deierborg et al. / Neuroscience 171 (2010) 1386–13961388
nd QA) obtained 7 days after neurosphere generation. Dissoci-ted cells from neurospheres (P4) derived from naive brains werelated at a density of 200,000 cells/ml. We evaluated the numbernd the size of the neurospheres after 7 days in culture. The totalumber of cells was counted following mechanical dissociation ofll the neurospheres from each culture flask in a Bürker chamber.o determine whether the microglia were proliferating, microglialultures were incubated (at 5–7 days in vitro) with the thymidinenalogue 5-bromodeoxyuridine (BrdU; 50 mM, Chemicon) for 2ays and the number of proliferating cells was counted.
tatistics
or statistical comparison of the number and the size of neuro-pheres from control, MCAO and QA lesions, the data were logransformed and analysed using analysis of variance (ANOVA)ollowed by Fischer’s post hoc test. The non-parametric Mann–
hitney U-test was applied for the time and differentiation studiesollowed with Bonferroni correction to compensate for multipleomparisons between experimental groups. We used ANOVAith Scheffe’s post hoc test to analyze groups treated with con-itioned medium. Experiments were performed in duplicates orore. Data are expressed as mean�SEM and P�0.05 was con-
idered as significantly different.
RESULTS
istinct microgliosis in the striatum of rats at onend four weeks following brain injury
o develop an ex vivo system to study the effect of micro-lial cells on NSPCs, we activated microglial cells by in-ucing experimental stroke (tMCAO) and excitotoxic brain
njury in rats. We first examined striatal microgliosis follow-ng brain injury using immunohistochemistry for the micro-lial marker CD11b at 1 and 4 weeks post-injury. At bothime points, the microglial hyperplasia appeared identicalt the cellular level with hypertrophic microglial cells thatxhibited characteristic bushy cellular ramifications (Fig. 1,1–D2). At 1 week post-injury, the microgliosis was evenlyistributed in the injured brain parenchyma (Fig. 1A1,C1), whereas, after 4 weeks, the microglial distributionad changed to a more dense cell accumulation at theorder of the injury (Fig. 1A1, D1), resembling the glial scaricrogliosis (Arranz et al., 2010) known to hamper neuro-al plasticity (Rolls et al., 2009).
ncreased number and size of neurospheres derivedrom brain lesions
e next examined whether brain injury induces dynamichanges in the proliferation of NSPCs located in the SVZ. Wessessed the effects of excitotoxic lesion and ischemic stroken the proliferation of NSPCs grown as neurospheres in vitro.e isolated the SVZ 7 days after injury from rats with isch-
mic brain injury (n�5), with a striatal QA-lesion (n�5) andaive animals (n�5) and compared the number and size ofeurospheres generated over a period of 7 days in culture.trikingly, we observed that both types of brain injury in-reased the number and the size of neurospheres. Neuro-pheres derived from ischemic and QA lesions were bothore numerous (by 69% and 102%, respectively; Fig. 2A)nd larger in diameter (by 23%, and 40%, respectively; Fig.
B) than the neurospheres derived from naive animals. uhese results confirm that different brain injuries increaseroliferation of NSPCs derived from the SVZ and imply thatSPCs grown as neurospheres are a valid model that can besed to study neurogenesis following brain injury. Moreover,
hese in vitro data are consistent with previous reports show-ng that brain lesions promote proliferation of NSPCs in theVZ in vivo (Arvidsson et al., 2002; Tattersfield et al., 2004;hang et al., 2004a).
ime-dependent increase in neurosphereroliferation
e next examined the temporal effect of a brain lesion onSPC proliferation. We assessed the number and the size ofeurospheres generated from SVZ isolated 1, 2, 3 and 4eeks after brain injury, for example after a striatal QA-lesion
n�5 animals for all time points). Following a 7-day cultureeriod, we found a 4-fold increase in the number of neuro-pheres generated from NSPCs isolated 1 and 2 weeksollowing a QA-lesion (Fig. 3C). Notably, the neurospheresarvested a week after lesion were 30% larger comparedith control (Fig. 3B, C). However, the number and the size ofeurospheres isolated 3 and 4 weeks following a QA-lesionere comparable with control neurospheres (Fig. 3A–C).hese data are in agreement with earlier observations show-
ng that maximal cell proliferation in the SVZ occurs 7 daysfter brain injury (Zhang et al., 2004a).
ultures of neurospheres derived from lesionedrains generate a monolayer of microglial cells
e next investigated whether the proliferation of NSPCsas influenced by the presence of microglial cells in ourultures. Immunocytochemistry for the specific microglialarkers Iba1 and CD11b consistently revealed the pres-nce of a microglial cell layer, on the bottom of the cultureask, in neurosphere cultures derived from ischemic orA-lesioned brains (Fig. 4A, C, D). These cells activelyroliferated, as shown by the incorporation of BrdU, whichas added to the medium (Fig. 4D). The microglia wereost likely derived from the medial striatum adjacent to theVZ (Duan et al., 1998). Microglial cells were rarely found
n cultures derived from sham-lesioned brains (Fig. 4B).imilarly, we rarely detected microglial cells when neuro-phere cultures were prepared from SVZ tissue isolatedrom 3 and 4 weeks following brain injuries (data nothown). The results strongly suggest an intimate link be-ween the appearance of a monolayer of microglial cell inultures derived from injured brains and the increase ineurosphere proliferation. In analogy with our results, aecent study described that microglia proliferate and accu-ulate in the rat SVZ during the first 2 weeks followingxperimental stroke (Thored et al., 2009).
icroglial cells, derived from SVZ of injured brains,ecrete soluble factors with a robust proliferativeffect
o confirm whether the microglia present in our culturesncreases neurosphere formation and proliferation we
sed complementary paradigms; microglia-conditioned
m(
nSb
pov(f
Fmlmat n of this a
T. Deierborg et al. / Neuroscience 171 (2010) 1386–1396 1389
edium, and the co-culture of microglia and neurospheresFig. 5A).
We first plated single cell suspensions of dissociatedeurospheres derived from NSPCs generated from theVZ of naive brains, grown in the presence of EGF and
ig. 1. Microgliosis in the striatum at 1 and 4 wk following brainicroglial/macrophage marker CD11b demonstrates general and homo
ateral ventricle (left) at 1 wk following a striatal lesion (A, C). Four waking up a microglial barrier that delineates the striatal injury (B, D).t 1 and 4 wk post-injury. A1 and A2 have the same magnification aso color in this figure legend, the reader is referred to the Web versio
FGF, according to conventional protocols. We then ex- f
osed these single cell cultures to conditioned mediumbtained from cultures containing microglial cells har-ested from ischemic (n�4) and QA-lesioned brainsn�4). As a control (n�4), we used conditioned mediumrom cultures generated from naive brains, in which only a
uced by excitotoxic lesions (A, B) or ischemic stroke (C, D). They distributed microglial activation in the injured striatum adjacent to theain injury, the microglial cells show a scar-like accumulation of cellssimilarity in the morphology of the activated bushy-shaped microgliaD1 and B2, C2 D2, respectively. For interpretation of the referencesrticle.
injury indgenousl
k after brNote theB1, C1,
ew or no microglial cells were present. We found a sub-

ssdnbnnogu
mgorbmttoc
ipwdg
Fb2c(casRs
F(edlaM
T. Deierborg et al. / Neuroscience 171 (2010) 1386–13961390
tantial increase in the proliferation of “naïve neuro-pheres” (generated from naive NSPCs) when grown for 7ays in conditioned medium derived from microglia origi-ating from ischemic (9-fold) and QA-lesioned (11-fold)rains (Fig. 5B). The increase in proliferation was evidentot only in the number (Fig. 5B1) and the size (Fig. 5B2) ofeurospheres, but also when we counted the total numberf cells in the neurospheres (Fig. 5B3). These data sug-est that microglial cells release soluble factors that stim-late the proliferation of NSPCs.
Next, we asked whether the factors secreted by theicroglial cells could efficiently induce the formation androwth of neurospheres from naive NSPCs in the absencef exogenously added growth factors. We grew naive neu-ospheres in the presence of microglial cells derived fromrains that had sustained lesions. In this set of experi-ents, we first studied the proliferative effect induced by
he microglial cells using basic medium, without the addi-ion of EGF and bFGF. In absence of EGF and bFGF, webserved a 5-fold increase in the number of neurospheres
ig. 2. Neurospheres cultured from brains lesioned by an ischemicMCAO) or intrastriatal excitotoxic injury with QA increase the prolif-ration of neurospheres. The number of neurospheres (A) and theiameter of the neurospheres (B) are increased following a brain
esion. The injury was induced 7 d before the dissection and dissoci-tion of SVZ cells. Evaluation was done after 7 d of culturing.ean�SEM (n�5), * denotes P�0.05.
ompared with cultures grown without microglia, suggest-c*
ng that microglial cells produce mitogenic factors thatartially compensate for the lack of EGF and bFGF. Next,e grew the naive NSPCs in new fresh proliferation me-ium containing EGF and bFGF in the presence of micro-lia and observed an additional 58% increase in the pro-
ig. 3. The proliferation of neurospheres cultured from lesionedrains is increased only if they are isolated and cultured during the firstwk following the lesion. (A) The number of neurospheres is in-
reased if the SVZ is dissected out within 2 wk following a QA lesion.B) The size of neurospheres, measured as the diameter, was in-reased only if the SVZ was cultured 7 d after a QA lesion. The valuesre expressed as a percentage of the corresponding controls (neuro-pheres cultured from naive rats and cultured at the same time). (C–F)epresentative pictures showing the difference in the number and theize of neurospheres taken out 1 and 4 wk following a brain lesion and
ultured for 1 wk. Mean�SEM (n�5). M–W U-test, * denotes P�0.05,* denotes P�0.01.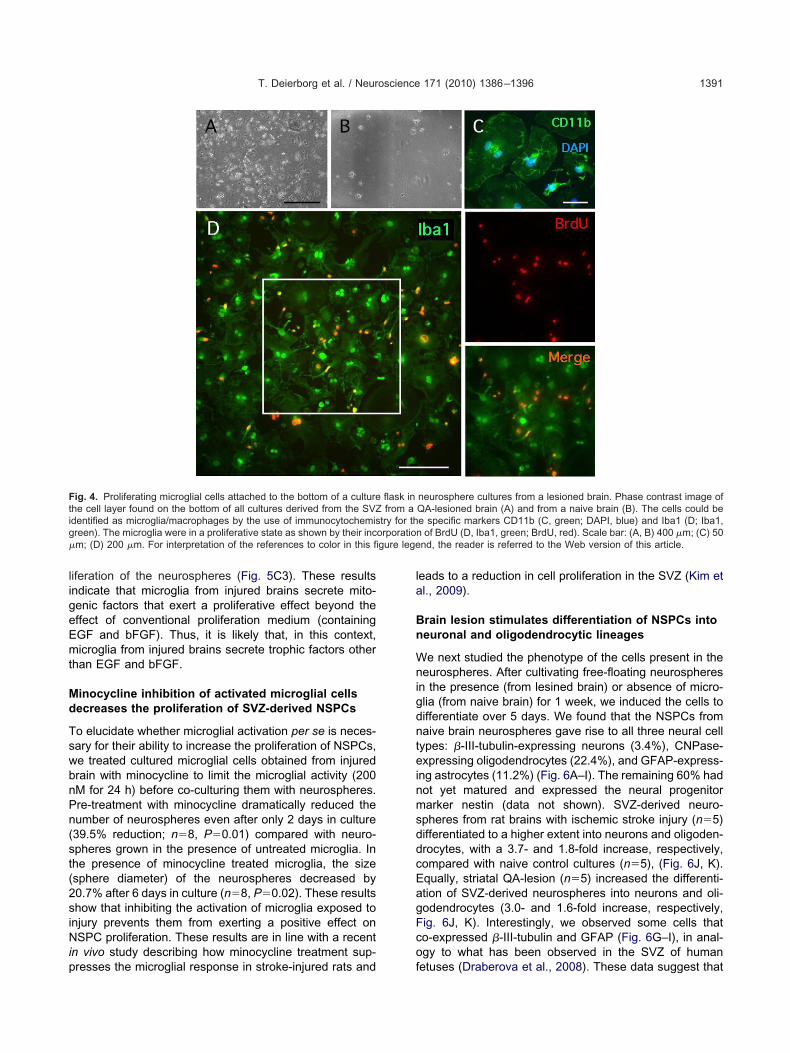
ligeEmt
Md
TswbnPn(st(2siNip
la
Bn
WnigdnteinmsddcEagFco
Ftig orporation� gure lege
T. Deierborg et al. / Neuroscience 171 (2010) 1386–1396 1391
iferation of the neurospheres (Fig. 5C3). These resultsndicate that microglia from injured brains secrete mito-enic factors that exert a proliferative effect beyond theffect of conventional proliferation medium (containingGF and bFGF). Thus, it is likely that, in this context,icroglia from injured brains secrete trophic factors other
han EGF and bFGF.
inocycline inhibition of activated microglial cellsecreases the proliferation of SVZ-derived NSPCs
o elucidate whether microglial activation per se is neces-ary for their ability to increase the proliferation of NSPCs,e treated cultured microglial cells obtained from injuredrain with minocycline to limit the microglial activity (200M for 24 h) before co-culturing them with neurospheres.re-treatment with minocycline dramatically reduced theumber of neurospheres even after only 2 days in culture39.5% reduction; n�8, P�0.01) compared with neuro-pheres grown in the presence of untreated microglia. Inhe presence of minocycline treated microglia, the sizesphere diameter) of the neurospheres decreased by0.7% after 6 days in culture (n�8, P�0.02). These resultshow that inhibiting the activation of microglia exposed tonjury prevents them from exerting a positive effect onSPC proliferation. These results are in line with a recent
n vivo study describing how minocycline treatment sup-
ig. 4. Proliferating microglial cells attached to the bottom of a culturehe cell layer found on the bottom of all cultures derived from the SVZdentified as microglia/macrophages by the use of immunocytochemisreen). The microglia were in a proliferative state as shown by their incm; (D) 200 �m. For interpretation of the references to color in this fi
resses the microglial response in stroke-injured rats and f
eads to a reduction in cell proliferation in the SVZ (Kim etl., 2009).
rain lesion stimulates differentiation of NSPCs intoeuronal and oligodendrocytic lineages
e next studied the phenotype of the cells present in theeurospheres. After cultivating free-floating neurospheres
n the presence (from lesined brain) or absence of micro-lia (from naive brain) for 1 week, we induced the cells toifferentiate over 5 days. We found that the NSPCs fromaive brain neurospheres gave rise to all three neural cellypes: �-III-tubulin-expressing neurons (3.4%), CNPase-xpressing oligodendrocytes (22.4%), and GFAP-express-
ng astrocytes (11.2%) (Fig. 6A–I). The remaining 60% hadot yet matured and expressed the neural progenitorarker nestin (data not shown). SVZ-derived neuro-
pheres from rat brains with ischemic stroke injury (n�5)ifferentiated to a higher extent into neurons and oligoden-rocytes, with a 3.7- and 1.8-fold increase, respectively,ompared with naive control cultures (n�5), (Fig. 6J, K).qually, striatal QA-lesion (n�5) increased the differenti-tion of SVZ-derived neurospheres into neurons and oli-odendrocytes (3.0- and 1.6-fold increase, respectively,ig. 6J, K). Interestingly, we observed some cells thato-expressed �-III-tubulin and GFAP (Fig. 6G–I), in anal-gy to what has been observed in the SVZ of human
neurosphere cultures from a lesioned brain. Phase contrast image ofQA-lesioned brain (A) and from a naive brain (B). The cells could bee specific markers CD11b (C, green; DAPI, blue) and Iba1 (D; Iba1,
of BrdU (D, Iba1, green; BrdU, red). Scale bar: (A, B) 400 �m; (C) 50nd, the reader is referred to the Web version of this article.
flask infrom a
try for th
etuses (Draberova et al., 2008). These data suggest that

bN
fl
FEibiin(MP
T. Deierborg et al. / Neuroscience 171 (2010) 1386–13961392
rain injury could increase the cellular differentiation of
ig. 5. Conditioned medium from flasks containing microglial cells, haxperimental outline. To obtain microglial cultures, SVZ and medial st
schemic and QA-lesioned brains 7 d after the lesion was induced. Afottom of the culture flask and all neurospheres were discarded from
n respective medium or flasks. Following 7 d of culturing, neurospncreased in number (B1), size (B2) and total number of cells/ml (B3) caive brains and without the presence of microglial cells. (C) CulturingCtrl) or 7 d old flasks from QA-lesioned brain with microglial cellsean�SEM, (n�4). ANOVA and Scheffe’s post hoc, (B1–3). Student�0.001.
SPCs into neurons and oligodendrocytes. Possibly, in- r
ammatory molecules and microglia could have an activerom a lesioned brain, have a proliferative effect on neurospheres. (A)re dissected out and dissociated from naive animals (Ctrl), and from
f culturing, a confluent layer of microglial cells could be found on thee medium. Dissociated neurospheres from naive brains were seededculture flasks containing conditioned medium from lesioned brainswith neurospheres maintained in control medium that is culture from
iated naive neurospheres with new proliferation medium in new flasksottom further increased the neurosphere proliferation (cells/ml, C3).C3) was applied. * denotes P�0.05; ** denotes P�0.01; *** denotes
rvested friatum weter 7 d othe culturheres inomparedof dissocon the b’s t-test, (
ole in this differentiation (Taupin, 2008).

WmibdsetsM
o(fisp2emtm
FdDiar Web vers
T. Deierborg et al. / Neuroscience 171 (2010) 1386–1396 1393
DISCUSSIONe have developed an ex vivo model to study activatedicroglia-induced proliferation of NSPCs following brain
njury in vivo. We found that microglia activated because ofrain injury influenced NSPC proliferation. Specifically, weemonstrated an increase in cell proliferation in neuro-pheres generated from NSPCs isolated from the ipsilat-ral SVZ tissue following both ischemic stroke and excito-
oxin-induced lesion. Enhanced neurogenesis has beenuggested to occur in stroke patients (Jin et al., 2006;
ig. 6. Differentiation towards neurons and oligodendrocytes was enifferentiated into all neural lineages; neurons (�-III-tubulin: A, G, C aand F). Neurospheres obtained from ischemic or QA lesioned brain
ncreased differentiation into neurons (�-III-tubulin, J). The differentstrocytic differentiation was unaffected (L). Mean�SEM (n�5). M–Weferences to color in this figure legend, the reader is referred to the
acas et al., 2006). The notion that enhanced proliferation r
f NSPCs following stroke promotes functional recoveryNygren et al., 2006; Thored et al., 2006) makes thesendings potentially clinically relevant. The ambiguous re-ponse of microglia (pro-/anti-inflammatory) to injury de-ends on the types of stimulus (Hanisch and Kettenmann,007) and receptor activation (Ribes et al., 2010). Theffects of microglia often differ between in vivo and in vitroodels (Zietlow et al., 1999; Eskes et al., 2003). It is,
herefore, important to create in vitro models that closelyimic the activation pattern of microglia in vivo and can
fter a brain lesion. Neurospheres from naive control brains could berocytes (GFAP: B, E, H, C, F, and I) and oligodendrocytes (CNPase:tured for 7 d and subsequently differentiated for 5 d have a markedly
oligodendrocytes (CNPase) was also increased (K), whereas theith Bonferroni correction, * denotes P�0.05. For interpretation of theion of this article.
hanced and I), asts and culiation into
U-test w
eplicate their effects on other cells in vivo. We believe that

ohd
BS
Mraeapeppbta2
eNibldepea2tMonwe2eclahsf
Lp
Eairt(ai2ae
ogmi(cmpmti
Mp
Wmmga(pvt2uegntchu
Tc
HtpbeswcH
wanitidtttsi2
T. Deierborg et al. / Neuroscience 171 (2010) 1386–13961394
ur ex vivo assay is a valuable tool which can help to clarifyow activated microglia affect the proliferation and theifferentiation of NSPCs.
rain injury increases cell proliferation ofVZ-derived NSPCs
icroglia are activated following ischemic brain injury (Mo-ioka et al., 1993) and excitotoxic brain lesions (Topper etl., 1993). We observed an increase in neurosphere prolif-ration using tissue harvested 1–2 weeks following ischemicnd excitotoxic brain lesions. This increase in neurosphereroliferation was induced by injury-activated microglia. Thesearly time points following brain injury correspond to theeriod when microglia accumulate in the SVZ following ex-erimental stroke (Thored et al., 2009). Moreover, the num-er of MHC-II positive microglia peaks in the injured stria-um within the first 2 weeks (Thored et al., 2009) and theyre suggested to regulate neurogenesis (Butovsky et al.,006; Ziv et al., 2006).
Experimental stroke and intrastriatial injection of thexcitotoxin QA are known to increase the proliferation ofSPCs in the SVZ, with a maximal effect 1 week after the
njury (Tattersfield et al., 2004; Zhang et al., 2004b; Deier-org et al., 2009). We confirmed this increase in the pro-
iferation of neurospheres by dissecting out the SVZ andissociating the cells 1 week after either ischemic stroke orxcitotoxic striatal lesion. Earlier studies suggest that cellroliferation in the SVZ is sustained up to 2 weeks afterxcitotoxin-induced brain injury (Deierborg et al., 2009)nd thereafter decreases to baseline levels (Zhang et al.,004b). In this respect, our ex vivo model robustly mimicshe dynamic changes in cell proliferation observed in vivo.oreover, the enhanced (3–4-fold) neuronal differentiationf NSPCs that we observed among cells derived fromeurospheres harvested from injured brains is consistentith previous observations showing increased neurogen-sis in vivo following these brain lesions (Arvidsson et al.,002; Tattersfield et al., 2004; Zhang et al., 2004b). Inter-stingly, this stroke-induced neurogenesis in the SVZ isoncomitant with trophic microglial cells expressing insu-in-like growth factor-1 in the vicinity of the SVZ (Thored etl., 2006). In addition, these neurospheres generatedigher numbers of oligodendrocytes in line with previoustudies reporting an increased density of oligodendrocytesollowing focal ischemia in the rat (Gregersen et al., 2001).
esion-induced microglial activation promotes NSPCroliferation
arly literature often suggested that microglia might neg-tively influence plasticity (including neurogenesis) follow-
ng brain injury (Monje et al., 2003; Liu et al., 2007). Recenteports convey a more complex role of microglia, stressinghat they sometimes promote brain repair following injurysee Introduction). Thus, the classical view of microglia, asuniform cell type that exacerbates neuronal damage and
nhibits neurogenesis, is under question (Schwartz et al.,006). When we treated microglia derived from injureddult brain with minocycline, we observed reduced prolif-
ration of NSPCs derived from the SVZ, indicating that, in cur model, activated microglia actually enhance stem/pro-enitor cell proliferation. In a similar way, inhibition oficroglial activation with minocyline in vivo following stroke
n rats reduces the cell proliferation in the neurogenic SVZKim et al., 2009), in stark contrast to the effect of mino-ycline on hippocampal neurogenesis. Possibly, activatedicroglia in the SVZ/striatum secrete other factors as com-ared with those present in the dentate gyrus. Indeed,icroglia in the SVZ show a greater response to injury, in
erms of activation and proliferation, compared to microglian non-neurogenic regions (Goings et al., 2006).
icroglia activated by brain injury secrete factorsromoting NSPC proliferation
e are currently investigating which microglia-derivedolecules stimulate the proliferation of NSPCs and theirechanism(s) of action. Earlier work suggests that micro-lia produce multiple pro- and anti-inflammatory cytokines,s well as growth factors, with diverse cellular effectsHanisch, 2002). For example, microglia can produce tro-hic factors and exert a positive effect on: neurogenesis inivo (Butovsky et al., 2006); oligodendrocytic differentia-ion (Nicholas et al., 2001) and astrogliogenesis (Zhu et al.,008). Several mitogens are also known to positively reg-late the proliferation of NSPCs in the SVZ, for examplerythropoietin (Wang et al., 2004), vascular endothelialrowth factor (VEGF; Jin et al., 2002) and endocannabi-oids (Carrier et al., 2004; Aguado et al., 2005). We foundhat microglia-conditioned medium had additive effects onell proliferation in the neurospheres in the presence ofigh concentrations of EGF/bFGF, suggesting that thenknown factor acts through a different pathway.
he complexity of microglial activation and itsontribution to NSPC proliferation
ypoxia, which clearly occurs in experimental stroke, ac-ivates microglia (Abraham et al., 2001). In our study, thero-neurogenic effect of microglia activated by excitotoxicrain damage was equally strong to that seen after isch-mic hypoxia. Therefore, the microglial activation we ob-erved might instead be a direct response to cell deathhere factors, such as nucleotides, released from dyingells could trigger activation (Nimmerjahn et al., 2005;aynes et al., 2006).
One week after striatal lesion, we observed microgliaith a bushy and hypertrophic morphology. They remainedt 4 weeks after the lesion, when we found the microglia too longer be pro-neurogenic. Thus, the morphology of
ndividual microglia does not necessarily correlate withheir pathophysiological state (Perry et al., 2007). Interest-ngly, between 1 and 4 weeks post-injury the anatomicalistribution of activated microglia changed. Thus, initially,hey were present in the whole striatum and at 4 weekshey concentrated around a scar-like region medially, closeo the SVZ. The cells present initially are likely to representupportive microglia reported to occur shortly after a brain
njury (Neumann et al., 2006; Lalancette-Hebert et al.,007), whereas those at 4 weeks may have a more phago-
ytic role (Rolls et al., 2009). Studies on the effects of
agrvA(cN
WsbapfnalsmNrc
ARmpKAFaa
A
A
A
A
B
B
C
C
C
C
C
D
D
D
D
E
E
G
G
H
H
H
J
J
K
T. Deierborg et al. / Neuroscience 171 (2010) 1386–1396 1395
ctivated microglia from a transgenic model of neurode-enerative disease further exemplify the complexity of theiroles. For instance, culture medium conditioned by acti-ated microglia harvested from a presenilin 1 transgeniclzheimer’s mouse model impairs NSPC proliferation
Choi et al., 2008). In conclusion, the nature of the mole-ules secreted by activated microglia and their effects onSPCs depend strictly on the type of activating stimulus.
CONCLUSION
e have generated an ex vivo model to study the re-ponse of NSPCs to microglial cells obtained from injuredrains. Our data imply that microglial cells present incutely damaged SVZ/striatum release factors that initiallyromote proliferation. We also show that NSPCs derivedrom an injured brain differentiate to a higher extent intoeurons and oligodendrocytes. These results suggest thatmicroglial change might underlie the increased cell pro-
iferation observed in the SVZ and the striatum followingtroke. Our results also support the notion that anti-inflam-atory treatment might not necessarily be beneficial forSPC proliferation following injury to the striatum, but
ather might inhibit the proliferative effect of activated mi-roglia in this brain region.
cknowledgments—This work was supported by the Swedishesearch Council (including NeuroFortis, Strong Research Environ-ent on Neurodegeneration and Brain Repair), EU 6th frameworkrogram PROMEMORIA, Crafoord Foundation, Gyllenstiernskarapperup foundation, Royal Physiographic Society, Swedish Strokessociation, Bergvall Foundation, Kock Foundation, Lars Hiertaoundation, Tore Nilsson Foundation and Segerfalk Foundation. Welso thank C. Sjölund and B. Haraldsson for providing technicalssistance and Edina Silajdzic for proof reading.
REFERENCES
braham H, Losonczy A, Czeh G, Lazar G (2001) Rapid activation ofmicroglial cells by hypoxia, kainic acid, and potassium ions in slicepreparations of the rat hippocampus. Brain Res 906:115–126.
guado T, Monory K, Palazuelos J, Stella N, Cravatt B, Lutz B,Marsicano G, Kokaia Z, Guzman M, Galve-Roperh I (2005) Theendocannabinoid system drives neural progenitor proliferation.FASEB J 19:1704–1706.
rranz AM, Gottlieb M, Perez-Cerda F, Matute C (2010) Increasedexpression of glutamate transporters in subcortical white matterafter transient focal cerebral ischemia. Neurobiol Dis 37:156–165.
rvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronalreplacement from endogenous precursors in the adult brain afterstroke. Nat Med 8:963–970.
urns TC, Verfaillie CM, Low WC (2009) Stem cells for ischemic braininjury: a critical review. J Comp Neurol 515:125–144.
utovsky O, Ziv Y, Schwartz A, Landa G, Talpalar AE, Pluchino S,Martino G, Schwartz M (2006) Microglia activated by IL-4 or IFN-gamma differentially induce neurogenesis and oligodendrogenesisfrom adult stem/progenitor cells. Mol Cell Neurosci 31:149–160.
acci E, Ajmone-Cat MA, Anelli T, Biagioni S, Minghetti L (2008) Invitro neuronal and glial differentiation from embryonic or adultneural precursor cells are differently affected by chronic or acuteactivation of microglia. Glia 56:412–425.
arlen M, Meletis K, Goritz C, Darsalia V, Evergren E, Tanigaki K,Amendola M, Barnabe-Heider F, Yeung MS, Naldini L, Honjo T,
Kokaia Z, Shupliakov O, Cassidy RM, Lindvall O, Frisen J (2009)Forebrain ependymal cells are notch-dependent and generateneuroblasts and astrocytes after stroke. Nat Neurosci 12:259–267.
arrier EJ, Kearn CS, Barkmeier AJ, Breese NM, Yang W, Nithipa-tikom K, Pfister SL, Campbell WB, Hillard CJ (2004) Cultured ratmicroglial cells synthesize the endocannabinoid 2-arachidonylglyc-erol, which increases proliferation via a CB2 receptor-dependentmechanism. Mol Pharmacol 65:999–1007.
hoi SH, Veeraraghavalu K, Lazarov O, Marler S, Ransohoff RM,Ramirez JM, Sisodia SS (2008) Non-cell-autonomous effects ofpresenilin 1 variants on enrichment-mediated hippocampal pro-genitor cell proliferation and differentiation. Neuron 59:568–580.
urtis MA, Kam M, Nannmark U, Anderson MF, Axell MZ, Wikkelso C,Holtas S, van Roon-Mom WM, Bjork-Eriksson T, Nordborg C,Frisen J, Dragunow M, Faull RL, Eriksson PS (2007) Humanneuroblasts migrate to the olfactory bulb via a lateral ventricularextension. Science 315:1243–1249.
eierborg T, Soulet D, Roybon L, Hall V, Brundin P (2008) Emergingrestorative treatments for Parkinson’s disease. Prog Neurobiol85:407–432.
eierborg T, Staflin K, Pesic J, Roybon L, Brundin P, Lundberg C(2009) Absence of striatal newborn neurons with mature pheno-type following defined striatal and cortical excitotoxic brain injuries.Exp Neurol 219:363–367.
raberova E, Del Valle L, Gordon J, Markova V, Smejkalova B,Bertrand L, de Chadarevian JP, Agamanolis DP, Legido A, KhaliliK, Draber P, Katsetos CD (2008) Class III beta-tubulin is constitu-tively coexpressed with glial fibrillary acidic protein and nestin inmidgestational human fetal astrocytes: implications for phenotypicidentity. J Neuropathol Exp Neurol 67:341–354.
uan WM, Widner H, Cameron RM, Brundin P (1998) Quinolinicacid-induced inflammation in the striatum does not impair thesurvival of neural allografts in the rat. Eur J Neurosci 10:2595–2606.
riksson PS, Perfilieva E, Bjork-Eriksson T, Alborn AM, Nordborg C,Peterson DA, Gage FH (1998) Neurogenesis in the adult humanhippocampus. Nat Med 4:1313–1317.
skes C, Juillerat-Jeanneret L, Leuba G, Honegger P, Monnet-Ts-chudi F (2003) Involvement of microglia-neuron interactions in thetumor necrosis factor-alpha release, microglial activation, and neu-rodegeneration induced by trimethyltin. J Neurosci Res 71:583–590.
oings GE, Kozlowski DA, Szele FG (2006) Differential activation ofmicroglia in neurogenic versus non-neurogenic regions of the fore-brain. Glia 54:329–342.
regersen R, Christensen T, Lehrmann E, Diemer NH, Finsen B(2001) Focal cerebral ischemia induces increased myelin basicprotein and growth-associated protein-43 gene transcription inperi-infarct areas in the rat brain. Exp Brain Res 138:384–392.
anisch UK (2002) Microglia as a source and target of cytokines. Glia40:140–155.
anisch UK, Kettenmann H (2007) Microglia: active sensor and ver-satile effector cells in the normal and pathologic brain. Nat Neuro-sci 10:1387–1394.
aynes SE, Hollopeter G, Yang G, Kurpius D, Dailey ME, Gan WB,Julius D (2006) The P2Y12 receptor regulates microglial activationby extracellular nucleotides. Nat Neurosci 9:1512–1519.
in K, Wang X, Xie L, Mao XO, Zhu W, Wang Y, Shen J, Mao Y,Banwait S, Greenberg DA (2006) Evidence for stroke-inducedneurogenesis in the human brain. Proc Natl Acad Sci U S A103:13198–13202.
in K, Zhu Y, Sun Y, Mao XO, Xie L, Greenberg DA (2002) Vascularendothelial growth factor (VEGF) stimulates neurogenesis in vitroand in vivo. Proc Natl Acad Sci U S A 99:11946–11950.
im BJ, Kim MJ, Park JM, Lee SH, Kim YJ, Ryu S, Kim YH, Yoon BW(2009) Reduced neurogenesis after suppressed inflammation byminocycline in transient cerebral ischemia in rat. J Neurol Sci
279:70–75.
L
L
M
M
M
M
N
N
N
N
N
P
R
R
R
R
R
S
S
T
T
T
T
T
W
Z
Z
Z
Z
Z
T. Deierborg et al. / Neuroscience 171 (2010) 1386–13961396
alancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J (2007)Selective ablation of proliferating microglial cells exacerbates isch-emic injury in the brain. J Neurosci 27:2596–2605.
iu Z, Fan Y, Won SJ, Neumann M, Hu D, Zhou L, Weinstein PR, LiuJ (2007) Chronic treatment with minocycline preserves adult newneurons and reduces functional impairment after focal cerebralischemia. Stroke 38:146–152.
acas J, Nern C, Plate KH, Momma S (2006) Increased generation ofneuronal progenitors after ischemic injury in the aged adult humanforebrain. J Neurosci 26:13114–13119.
emezawa H, Minamisawa H, Smith ML, Siesjo BK (1992) Ischemicpenumbra in a model of reversible middle cerebral artery occlusionin the rat. Exp Brain Res 89:67–78.
onje ML, Toda H, Palmer TD (2003) Inflammatory blockade restoresadult hippocampal neurogenesis. Science 302:1760–1765.
orioka T, Kalehua AN, Streit WJ (1993) Characterization of microglialreaction after middle cerebral artery occlusion in rat brain. J CompNeurol 327:123–132.
akao N, Brundin P (1997) Effects of alpha-phenyl-tert-butyl nitroneon neuronal survival and motor function following intrastriatal in-jections of quinolinate or 3-nitropropionic acid. Neuroscience76:749–761.
eumann J, Gunzer M, Gutzeit HO, Ullrich O, Reymann KG, Dinkel K(2006) Microglia provide neuroprotection after ischemia. FASEB J20:714–716.
icholas RS, Wing MG, Compston A (2001) Nonactivated microgliapromote oligodendrocyte precursor survival and maturationthrough the transcription factor NF-kappaB. Eur J Neurosci 13:959–967.
immerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cellsare highly dynamic surveillants of brain parenchyma in vivo. Sci-ence 308:1314–1318.
ygren J, Wieloch T, Pesic J, Brundin P, Deierborg T (2006) Enrichedenvironment attenuates cell genesis in subventricular zone afterfocal ischemia in mice and decreases migration of newborn cells tothe striatum. Stroke 37:2824–2829.
erry VH, Cunningham C, Holmes C (2007) Systemic infections andinflammation affect chronic neurodegeneration. Nat Rev Immunol7:161–167.
ibes S, Ebert S, Regen T, Czesnik D, Scheffel J, Zeug A, BunkowskiS, Eiffert H, Hanisch UK, Hammerschmidt S, Nau R (2010) Fi-bronectin stimulates Escherichia coli phagocytosis by microglialcells. Glia 58:367–376.
ickhag M, Deierborg T, Patel S, Ruscher K, Wieloch T (2008) Apo-lipoprotein D is elevated in oligodendrocytes in the peri-infarctregion after experimental stroke: influence of enriched environ-ment. J Cereb Blood Flow Metab 28:551–562.
olls A, Shechter R, Schwartz M (2009) The bright side of the glial scarin CNS repair. Nat Rev Neurosci 10:235–241.
oybon L, Brundin P, Li JY (2005) Stromal cell-derived inducingactivity does not promote dopaminergic differentiation, but en-hances differentiation and proliferation of neural stem cell-derived
astrocytes. Exp Neurol 196:373–380.oybon L, Deierborg T, Brundin P, Li JY (2009) Involvement of Ngn2,Tbr and NeuroD proteins during postnatal olfactory bulb neurogen-esis. Eur J Neurosci 29:232–243.
chwartz M, Butovsky O, Bruck W, Hanisch UK (2006) Microglialphenotype: is the commitment reversible? Trends Neurosci 29:68–74.
vendsen CN, ter Borg MG, Armstrong RJ, Rosser AE, Chandran S,Ostenfeld T, Caldwell MA (1998) A new method for the rapid andlong term growth of human neural precursor cells. J NeurosciMethods 85:141–152.
attersfield AS, Croon RJ, Liu YW, Kells AP, Faull RL, Connor B(2004) Neurogenesis in the striatum of the quinolinic acid lesionmodel of Huntington’s disease. Neuroscience 127:319–332.
aupin P (2008) Adult neurogenesis, neuroinflammation and thera-peutic potential of adult neural stem cells. Int J Med Sci 5:127–132.
hored P, Arvidsson A, Cacci E, Ahlenius H, Kallur T, Darsalia V,Ekdahl CT, Kokaia Z, Lindvall O (2006) Persistent production ofneurons from adult brain stem cells during recovery after stroke.Stem Cells 24:739–747.
hored P, Heldmann U, Gomes-Leal W, Gisler R, Darsalia V, TaneeraJ, Nygren JM, Jacobsen SE, Ekdahl CT, Kokaia Z, Lindvall O(2009) Long-term accumulation of microglia with proneurogenicphenotype concomitant with persistent neurogenesis in adult sub-ventricular zone after stroke. Glia 57:835–849.
opper R, Gehrmann J, Schwarz M, Block F, Noth J, Kreutzberg GW(1993) Remote microglial activation in the quinolinic acid model ofHuntington’s disease. Exp Neurol 123:271–283.
ang L, Zhang Z, Wang Y, Zhang R, Chopp M (2004) Treatment ofstroke with erythropoietin enhances neurogenesis and angiogen-esis and improves neurological function in rats. Stroke 35:1732–1737.
hang R, Zhang Z, Wang L, Wang Y, Gousev A, Zhang L, Ho KL,Morshead C, Chopp M (2004a) Activated neural stem cells con-tribute to stroke-induced neurogenesis and neuroblast migrationtoward the infarct boundary in adult rats. J Cereb Blood FlowMetab 24:441–448.
hang R, Zhang Z, Zhang C, Zhang L, Robin A, Wang Y, Lu M, ChoppM (2004b) Stroke transiently increases subventricular zone celldivision from asymmetric to symmetric and increases neuronaldifferentiation in the adult rat. J Neurosci 24:5810–5815.
hu P, Hata R, Cao F, Gu F, Hanakawa Y, Hashimoto K, Sakanaka M(2008) Ramified microglial cells promote astrogliogenesis andmaintenance of neural stem cells through activation of Stat3 func-tion. FASEB J 22:3866–3877.
ietlow R, Dunnett SB, Fawcett JW (1999) The effect of microglia onembryonic dopaminergic neuronal survival in vitro: diffusible sig-nals from neurons and glia change microglia from neurotoxic toneuroprotective. Eur J Neurosci 11:1657–1667.
iv Y, Avidan H, Pluchino S, Martino G, Schwartz M (2006) Synergybetween immune cells and adult neural stem/progenitor cells pro-motes functional recovery from spinal cord injury. Proc Natl Acad
Sci U S A 103:13174–13179.(Accepted 22 September 2010)(Available online 29 September 2010)




















