
El Kit de herramientas de PARP Section 5 Resources - NYS PTA
Upload
independentCategory
view
0download
0
Zn21-Induced ERK Activation Mediates PARP-1-Dependent Ischemic-Reoxygenation Damage toOligodendrocytes
MARIA DOMERCQ,1,2 SUSANA MATO,1,2 FEDERICO N. SORIA,1,2 M. VICTORIA S�ANCHEZ-G�OMEZ,1,2
ELENA ALBERDI,1,2 AND CARLOS MATUTE1,2*1Departamento de Neurociencias, Centro de Investigaciones Biom�edicas en Red (CIBERNED), Universidad del Pa�ıs Vasco,Leioa, Spain2Achucarro Basque Center for Neuroscience, Parque Tecnol�ogico de Bizkaia, Zamudio, Bizkaia, Spain
KEY WORDSoligodendrocytes; ischemia; ERK1/2 signaling
ABSTRACTMuch of the cell death following episodes of anoxia and is-chemia in the mammalian central nervous system has beenattributed to extracellular accumulation of glutamate andATP, which causes a rise in [Ca21]i, loss of mitochondrialpotential, and cell death. However, restoration of blood flowand reoxygenation are frequently associated with exacerba-tion of tissue injury (the oxygen paradox). Herein wedescribe a novel signaling pathway that is activated duringischemia-like conditions (oxygen and glucose deprivation;OGD) and contributes to ischemia-induced oligodendroglialcell death. OGD induced a retarded and sustained increasein extracellular signal-regulated kinase 1/2 (ERK1/2) phos-phorylation after restoring glucose and O2 (reperfusion-likeconditions). Blocking the ERK1/2 pathway with the MEKinhibitor UO126 largely protected oligodendrocytes againstischemic insults. ERK1/2 activation was blocked by thehigh-affinity Zn21 chelator TPEN, but not by antagonists ofAMPA/kainate or P2X7 receptors that were previouslyshown to be involved in ischemic oligodendroglial celldeath. Using a high-affinity Zn21 probe, we showed that is-chemia induced an intracellular Zn21 rise in oligodendro-cytes, and that incubation with TPEN prevented mitochon-drial depolarization and ROS generation after ischemia.Accordingly, exposure to TPEN and the antioxidant Troloxreduced ischemia-induced oligodendrocyte death. Moreover,UO126 blocked the ischemia-induced increase in poly-[ADP]-ribosylation of proteins, and the poly[ADP]-ribose po-lymerase 1 (PARP-1) inhibitor DPQ significantly inhibitedischemia-induced oligodendroglial cell death—demonstrat-ing that PARP-1 was required downstream in the Zn21-ERK oligodendrocyte cell death pathway. Chelation of cyto-solic Zn21, blocking ERK signaling, and antioxidants maybe beneficial for treating CNS white matter ischemia-reper-fusion injury. Importantly, all the inhibitors of this pathwayprotected oligodendrocytes when applied after the ischemicinsult. VVC 2012 Wiley Periodicals, Inc.
INTRODUCTION
Damage to oligodendrocytes and myelin underlies thefunctional impairment that follows hypoxic-ischemic dis-eases (stroke), cerebral palsy (periventricular leukomala-cia), spinal cord injury, and multiple sclerosis. Gluta-
mate, the main excitatory transmitter of the CNS, con-tributes to oligodendrocyte excitotoxicity and axonaldysfunction after ischemia through its actions on iono-tropic AMPA/kainate (Tekkok et al., 2007) and NMDAreceptors (Karadottir et al., 2005; Micu et al., 2006;Salter and Fern, 2005). In addition to glutamate recep-tors, oligodendrocytes express purinergic receptors andthere is evidence that ATP release and P2X7 receptoractivation contribute to white matter damage in multi-ple sclerosis and ischemia (Domercq et al., 2010; Matuteet al., 2007).
A central event in glutamate and ATP excitotoxicity isCa21 influx upon receptor activation; Ca21 accumulationwithin mitochondria leads to depolarization of this or-ganelle and increased production of radical oxygen spe-cies (ROS) (Domercq et al., 2010; Matute et al., 2006;Orrenius et al., 2003; S�anchez-G�omez et al., 2003). Wepreviously reported that glutamate receptor overactiva-tion in oligodendrocytes induces rapid activation ofERK1/2 signaling and increased Dusp6 phosphataseexpression, and that levels of phosphorylated ERK1 and2 (pERK1/2)—which are controlled by Dusp6 phospha-tase levels—are pivotal in determining cell death or sur-vival (Domercq et al., 2011). ERK activation could beinduced by a cytosolic Zn21 increase (Zhang et al., 2006)since AMPA/kainate receptors are also permeable toZn21 (Sensi et al., 2009). ERK1/2 activation acts as apro-survival signal in oligodendrocytes through itsactions at different steps of the mitochondrial apoptoticpathway and by controlling AMPA receptor permeability(Domercq et al., 2011).
ERK activation is generally associated with cell sur-vival and proliferation; however, depending on the stim-uli and cell types involved, activation of ERK can alsopromote cell death. Some studies suggest that the bal-ance among the intensity and duration of pro-apoptotic
Grant sponsors: Fundaci�on de Investigaci�on M�edica Mutua Madrile~na,CIBERNED, the Spanish Ministry of Education and Science, the Basque Govern-ment, Instituto de Salud Carlos III (to F.N.S.).
*Correspondence to: Carlos Matute, Departamento de Neurociencias, Universi-dad del Pa�ıs Vasco, E-48940 Leioa, Spain. E-mail: [email protected]
Received 1 June 2012; Accepted 19 October 2012
DOI 10.1002/glia.22441
Published online 22 December 2012 in Wiley Online Library (wileyonlinelibrary.com).
GLIA 61:383–393 (2013)
VVC 2012 Wiley Periodicals, Inc.

versus anti-apoptotic signals transmitted by ERK1/2determines whether a cell survives or undergoes celldeath (Mebratu and Tesfaigzi, 2009; Murphy and Blenis,2006). However, the molecular mechanisms that definethe conditions for ERK-mediated cell death remainpoorly understood. Here we examined the functionalrole of ERK1/2 in OGD-induced oligodendroglial dam-age. In contrast to the previously obtained resultsregarding the role of ERK1/2 signaling in oligodendro-glial excitotoxicity (Domercq et al., 2011), here we foundthat ERK1/2 activation contributed significantly toOGD-induced oligodendrocyte death. OGD triggered asustained and delayed activation of ERK1/2; this effectwas not mediated by activation of the AMPA/kainate orP2X7 receptors, but was secondary to Zn21-induced oxi-dative stress and it resulted in PARP-1 activation.Blockage of this signaling pathway protected oligoden-drocytes from ischemia-reperfusion damage.
MATERIALS AND METHODSOxygen and Glucose Deprivation in
Oligodendrocyte Cultures
Primary cultures of oligodendrocytes derived fromoptic nerves of 12-day-old Sprague-Dawley rats wereobtained as previously described (Matute et al., 1997).Cells were maintained at 37�C under 5% CO2 in achemically defined medium (Matute et al., 1997). After 2to 4 days in vitro, cultures were composed of at least98% O4/GalC1 cells. One hour of in vitro ischemia wasachieved by replacing O2 with N2 and external glucose(10 mM) with sucrose, and adding iodoacetate (IAA) toblock glycolysis in an extracellular solution containing130 mM NaCl, 5.4 mM KCl, 1.8 mM CaCl2, 26 mMNaHCO3, 0.8 mM MgCl2, and 1.18 mM NaH2PO4 (pH7.4). Cell death was determined 24 h later using calcein-AM (Invitrogen) as previously described (Matute et al.,2007). Antagonists were present during in vitro ischemiaand during the subsequent 24 h incubation period,except where otherwise indicated. The total calcein fluo-rescence on each coverslip was measured using a Syn-ergy-HT fluorimeter (BioTek Instruments) and datawere expressed as percentage of cell death versus that ofthe respective control with or without antagonists; pres-ence of antagonists did not alter cell survival (data notshown). All experiments (n � 3) were performed at leastin triplicate and plotted as mean 6 SEM.
Measurements of Reactive Oxygen Species andMitochondrial Potential
To assay the ROS levels, cells were loaded with 10 lMchloromethyl 20,70-dichlorodihydrofluorescein diacetate(CM-H2DCFDA; Invitrogen) at 37�C for 20 min at differ-ent times after OGD. Cells were then washed twice withHBSS without phenol red to eliminate excess dye. Fluo-rescence was measured using a Synergy-HT fluorimeterwith the excitation and emission wavelengths suggested
by the supplier for CM-H2DCFDA. Values were plottedas the percentage of DCFDA versus control.
The changes in mitochondrial membrane potentialwere monitored by reduction of 5,50,6,60-tetrachloro-1,10,3,30-tetraethylbenzimidazolcarbocyanine iodide dye(JC-1; Invitrogen), according to the manufacturer’sinstructions. At different times after OGD, cells wereloaded with 3 lM JC-1, incubated 37�C for 15 min, andthen washed twice with HBSS without phenol red toeliminate excess dye. In the cytosol, the monomeric formof this dye fluoresces green (emission read at 527 nmwhen excited at 485 nm) whereas within the mitochon-drial matrix, highly concentrated JC-1 forms aggregatesthat fluoresce red (emission at 590 nm when excited at485 nm). Both JC-1 monomers and aggregates were de-tectable using a Synergy-HT fluorimeter (Bio-TekInstruments). Changes in the membrane potential werecalculated as the red/green ratio in each condition, andresults were normalized to the control. All experiments(n � 3) were performed at least in triplicate and plottedas mean 6 SEM.
Western Blot
Total protein was extracted from oligodendrocyte cul-tures by scraping the cells in SDS/sample buffer. Sam-ples were boiled for 5 min and subjected to SDS/PAGEin 12.5% polyacrylamide gels. After electroblotting onnitrocellulose membranes, proteins were visualizedusing primary antibodies to phosphoERK1/2 (1:500; CellSignaling), total ERK (1:500; Cell Signaling), and poly(ADP-ribose) (PAR; 1:1,000; Alexis), followed by second-ary peroxidase-coupled goat anti-rabbit antibodies(1:5,000; Sigma). After washing, the blots were devel-oped using an enhanced chemiluminescence detectionkit according to the manufacturer’s instructions (SuperSignal ULTRA, Pierce). Images were acquired with aChemiDoc MP system (BioRad) and quantified usingScion Image Software.
Zn21 and Ca21 Imaging
For [Ca21] and [Zn21] recording, cells were loaded for5 to 10 min at 37�C with the cell-permeant acetoxy-methyl dye derivatives of fura-2M (5 lM; MolecularProbes, Inc., Eugene, OR) and FluoZin-3 (1 lM) in cul-ture medium. Cells were subsequently washed for atleast 10 min at room temperature in HBSS containing137 mM NaCl, 20 mM HEPES, 10 mM glucose, and 2mM CaCl2 (pH 7.4). Experiments were carried out in acoverslip chamber that was continuously perfused at 1mL/min with a buffer containing HBSS with 20 mMHEPES, 2 mM CaCl2, and 10 mM glucose. The perfusionchamber was mounted on the stage of a Zeiss invertedepifluorescence microscope (Axiovert 35), equipped witha 150-W xenon lamp Polychrome IV (TILL PhotonicsGMBH) and a Plan Neofluar 403 oil-immersion objec-tive (Zeiss, Germany). Fura-2 was excited alternately at
384 DOMERCQ ET AL.
GLIA
340 and 380 nM, and FluoZin-3 at 490 nM. Emissionwas monitored at 520 nM. [Ca21]i measurements wereexpressed as the ratio of F340/F380, whereas FluoZin-3fluorescence was normalized to starting values andexpressed as F/F0. Images were acquired every 10 to 20 sfor Ca21 and Zn21 with a high-resolution digital black/white CCD camera (ORCA, Hamamatsu Photonics Ib�er-ica, Barcelona, Spain) using the AquaCosmos softwareprogram (Hamamatsu Photonics Ib�erica), and back-ground fluorescence was subtracted at the beginning ofeach experiment. Data were analyzed with Excel andPrism software. Ischemia was induced chemically withthe glycolytic blocker iodoacetate (50 lM) and the oxida-tive phosphorylation inhibitor antimycin (2.5 lM) in amedium lacking glucose because recordings were per-formed in an open chamber where O2 could diffuse tothe cells (K�arad�ottir et al., 2005).
RESULTSSustained ERK1/2 Phosphorylation InducesOligodendroglial Cell Death After Ischemia
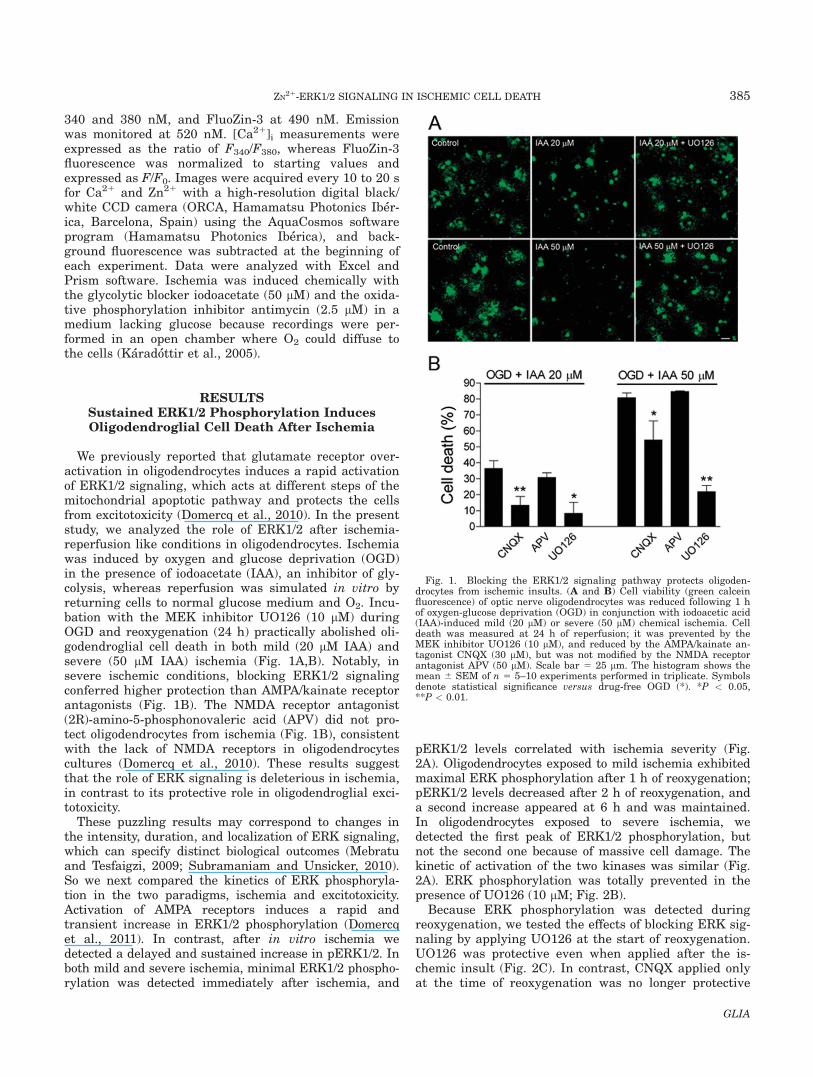
We previously reported that glutamate receptor over-activation in oligodendrocytes induces a rapid activationof ERK1/2 signaling, which acts at different steps of themitochondrial apoptotic pathway and protects the cellsfrom excitotoxicity (Domercq et al., 2010). In the presentstudy, we analyzed the role of ERK1/2 after ischemia-reperfusion like conditions in oligodendrocytes. Ischemiawas induced by oxygen and glucose deprivation (OGD)in the presence of iodoacetate (IAA), an inhibitor of gly-colysis, whereas reperfusion was simulated in vitro byreturning cells to normal glucose medium and O2. Incu-bation with the MEK inhibitor UO126 (10 lM) duringOGD and reoxygenation (24 h) practically abolished oli-godendroglial cell death in both mild (20 lM IAA) andsevere (50 lM IAA) ischemia (Fig. 1A,B). Notably, insevere ischemic conditions, blocking ERK1/2 signalingconferred higher protection than AMPA/kainate receptorantagonists (Fig. 1B). The NMDA receptor antagonist(2R)-amino-5-phosphonovaleric acid (APV) did not pro-tect oligodendrocytes from ischemia (Fig. 1B), consistentwith the lack of NMDA receptors in oligodendrocytescultures (Domercq et al., 2010). These results suggestthat the role of ERK signaling is deleterious in ischemia,in contrast to its protective role in oligodendroglial exci-totoxicity.
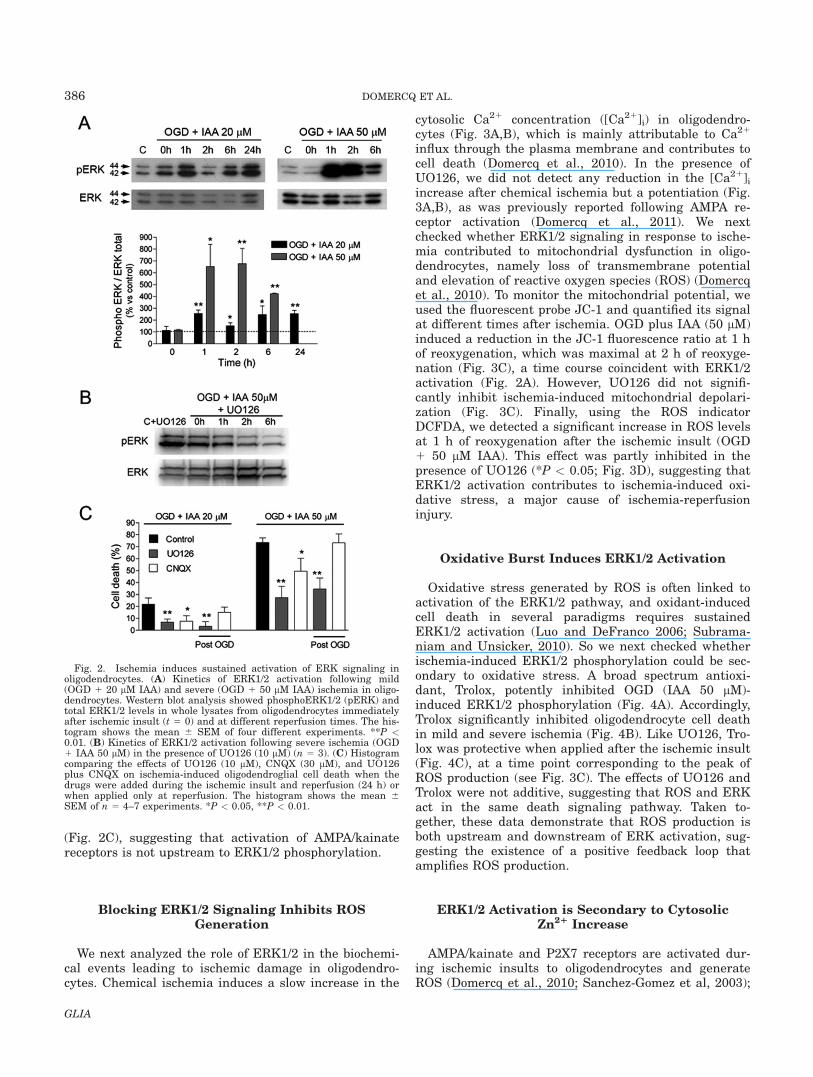
These puzzling results may correspond to changes inthe intensity, duration, and localization of ERK signaling,which can specify distinct biological outcomes (Mebratuand Tesfaigzi, 2009; Subramaniam and Unsicker, 2010).So we next compared the kinetics of ERK phosphoryla-tion in the two paradigms, ischemia and excitotoxicity.Activation of AMPA receptors induces a rapid andtransient increase in ERK1/2 phosphorylation (Domercqet al., 2011). In contrast, after in vitro ischemia wedetected a delayed and sustained increase in pERK1/2. Inboth mild and severe ischemia, minimal ERK1/2 phospho-rylation was detected immediately after ischemia, and
pERK1/2 levels correlated with ischemia severity (Fig.2A). Oligodendrocytes exposed to mild ischemia exhibitedmaximal ERK phosphorylation after 1 h of reoxygenation;pERK1/2 levels decreased after 2 h of reoxygenation, anda second increase appeared at 6 h and was maintained.In oligodendrocytes exposed to severe ischemia, wedetected the first peak of ERK1/2 phosphorylation, butnot the second one because of massive cell damage. Thekinetic of activation of the two kinases was similar (Fig.2A). ERK phosphorylation was totally prevented in thepresence of UO126 (10 lM; Fig. 2B).
Because ERK phosphorylation was detected duringreoxygenation, we tested the effects of blocking ERK sig-naling by applying UO126 at the start of reoxygenation.UO126 was protective even when applied after the is-chemic insult (Fig. 2C). In contrast, CNQX applied onlyat the time of reoxygenation was no longer protective
Fig. 1. Blocking the ERK1/2 signaling pathway protects oligoden-drocytes from ischemic insults. (A and B) Cell viability (green calceinfluorescence) of optic nerve oligodendrocytes was reduced following 1 hof oxygen-glucose deprivation (OGD) in conjunction with iodoacetic acid(IAA)-induced mild (20 lM) or severe (50 lM) chemical ischemia. Celldeath was measured at 24 h of reperfusion; it was prevented by theMEK inhibitor UO126 (10 lM), and reduced by the AMPA/kainate an-tagonist CNQX (30 lM), but was not modified by the NMDA receptorantagonist APV (50 lM). Scale bar 5 25 lm. The histogram shows themean 6 SEM of n 5 5–10 experiments performed in triplicate. Symbolsdenote statistical significance versus drug-free OGD (*). *P < 0.05,**P < 0.01.
385ZN21-ERK1/2 SIGNALING IN ISCHEMIC CELL DEATH
GLIA
(Fig. 2C), suggesting that activation of AMPA/kainatereceptors is not upstream to ERK1/2 phosphorylation.
Blocking ERK1/2 Signaling Inhibits ROSGeneration
We next analyzed the role of ERK1/2 in the biochemi-cal events leading to ischemic damage in oligodendro-cytes. Chemical ischemia induces a slow increase in the
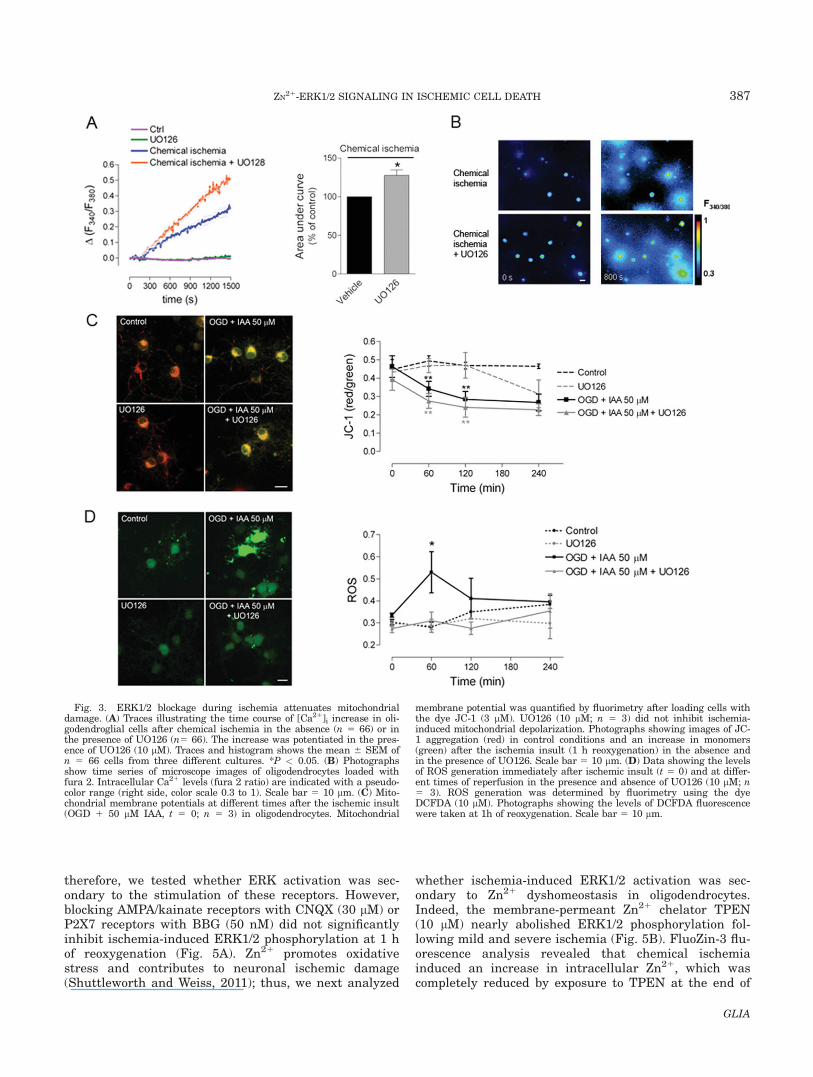
cytosolic Ca21 concentration ([Ca21]i) in oligodendro-cytes (Fig. 3A,B), which is mainly attributable to Ca21
influx through the plasma membrane and contributes tocell death (Domercq et al., 2010). In the presence ofUO126, we did not detect any reduction in the [Ca21]iincrease after chemical ischemia but a potentiation (Fig.3A,B), as was previously reported following AMPA re-ceptor activation (Domercq et al., 2011). We nextchecked whether ERK1/2 signaling in response to ische-mia contributed to mitochondrial dysfunction in oligo-dendrocytes, namely loss of transmembrane potentialand elevation of reactive oxygen species (ROS) (Domercqet al., 2010). To monitor the mitochondrial potential, weused the fluorescent probe JC-1 and quantified its signalat different times after ischemia. OGD plus IAA (50 lM)induced a reduction in the JC-1 fluorescence ratio at 1 hof reoxygenation, which was maximal at 2 h of reoxyge-nation (Fig. 3C), a time course coincident with ERK1/2activation (Fig. 2A). However, UO126 did not signifi-cantly inhibit ischemia-induced mitochondrial depolari-zation (Fig. 3C). Finally, using the ROS indicatorDCFDA, we detected a significant increase in ROS levelsat 1 h of reoxygenation after the ischemic insult (OGD1 50 lM IAA). This effect was partly inhibited in thepresence of UO126 (*P < 0.05; Fig. 3D), suggesting thatERK1/2 activation contributes to ischemia-induced oxi-dative stress, a major cause of ischemia-reperfusioninjury.
Oxidative Burst Induces ERK1/2 Activation
Oxidative stress generated by ROS is often linked toactivation of the ERK1/2 pathway, and oxidant-inducedcell death in several paradigms requires sustainedERK1/2 activation (Luo and DeFranco 2006; Subrama-niam and Unsicker, 2010). So we next checked whetherischemia-induced ERK1/2 phosphorylation could be sec-ondary to oxidative stress. A broad spectrum antioxi-dant, Trolox, potently inhibited OGD (IAA 50 lM)-induced ERK1/2 phosphorylation (Fig. 4A). Accordingly,Trolox significantly inhibited oligodendrocyte cell deathin mild and severe ischemia (Fig. 4B). Like UO126, Tro-lox was protective when applied after the ischemic insult(Fig. 4C), at a time point corresponding to the peak ofROS production (see Fig. 3C). The effects of UO126 andTrolox were not additive, suggesting that ROS and ERKact in the same death signaling pathway. Taken to-gether, these data demonstrate that ROS production isboth upstream and downstream of ERK activation, sug-gesting the existence of a positive feedback loop thatamplifies ROS production.
ERK1/2 Activation is Secondary to CytosolicZn21 Increase
AMPA/kainate and P2X7 receptors are activated dur-ing ischemic insults to oligodendrocytes and generateROS (Domercq et al., 2010; Sanchez-Gomez et al, 2003);
Fig. 2. Ischemia induces sustained activation of ERK signaling inoligodendrocytes. (A) Kinetics of ERK1/2 activation following mild(OGD 1 20 lM IAA) and severe (OGD 1 50 lM IAA) ischemia in oligo-dendrocytes. Western blot analysis showed phosphoERK1/2 (pERK) andtotal ERK1/2 levels in whole lysates from oligodendrocytes immediatelyafter ischemic insult (t 5 0) and at different reperfusion times. The his-togram shows the mean 6 SEM of four different experiments. **P <0.01. (B) Kinetics of ERK1/2 activation following severe ischemia (OGD1 IAA 50 lM) in the presence of UO126 (10 lM) (n 5 3). (C) Histogramcomparing the effects of UO126 (10 lM), CNQX (30 lM), and UO126plus CNQX on ischemia-induced oligodendroglial cell death when thedrugs were added during the ischemic insult and reperfusion (24 h) orwhen applied only at reperfusion. The histogram shows the mean 6SEM of n 5 4–7 experiments. *P < 0.05, **P < 0.01.
386 DOMERCQ ET AL.
GLIA
therefore, we tested whether ERK activation was sec-ondary to the stimulation of these receptors. However,blocking AMPA/kainate receptors with CNQX (30 lM) orP2X7 receptors with BBG (50 nM) did not significantlyinhibit ischemia-induced ERK1/2 phosphorylation at 1 hof reoxygenation (Fig. 5A). Zn21 promotes oxidativestress and contributes to neuronal ischemic damage(Shuttleworth and Weiss, 2011); thus, we next analyzed
whether ischemia-induced ERK1/2 activation was sec-ondary to Zn21 dyshomeostasis in oligodendrocytes.Indeed, the membrane-permeant Zn21 chelator TPEN(10 lM) nearly abolished ERK1/2 phosphorylation fol-lowing mild and severe ischemia (Fig. 5B). FluoZin-3 flu-orescence analysis revealed that chemical ischemiainduced an increase in intracellular Zn21, which wascompletely reduced by exposure to TPEN at the end of
Fig. 3. ERK1/2 blockage during ischemia attenuates mitochondrialdamage. (A) Traces illustrating the time course of [Ca21]i increase in oli-godendroglial cells after chemical ischemia in the absence (n 5 66) or inthe presence of UO126 (n5 66). The increase was potentiated in the pres-ence of UO126 (10 lM). Traces and histogram shows the mean 6 SEM ofn 5 66 cells from three different cultures. *P < 0.05. (B) Photographsshow time series of microscope images of oligodendrocytes loaded withfura 2. Intracellular Ca21 levels (fura 2 ratio) are indicated with a pseudo-color range (right side, color scale 0.3 to 1). Scale bar 5 10 lm. (C) Mito-chondrial membrane potentials at different times after the ischemic insult(OGD 1 50 lM IAA, t 5 0; n 5 3) in oligodendrocytes. Mitochondrial
membrane potential was quantified by fluorimetry after loading cells withthe dye JC-1 (3 lM). UO126 (10 lM; n 5 3) did not inhibit ischemia-induced mitochondrial depolarization. Photographs showing images of JC-1 aggregation (red) in control conditions and an increase in monomers(green) after the ischemia insult (1 h reoxygenation) in the absence andin the presence of UO126. Scale bar 5 10 lm. (D) Data showing the levelsof ROS generation immediately after ischemic insult (t 5 0) and at differ-ent times of reperfusion in the presence and absence of UO126 (10 lM; n5 3). ROS generation was determined by fluorimetry using the dyeDCFDA (10 lM). Photographs showing the levels of DCFDA fluorescencewere taken at 1h of reoxygenation. Scale bar 5 10 lm.
387ZN21-ERK1/2 SIGNALING IN ISCHEMIC CELL DEATH
GLIA

the experiment (Fig. 5C,D). The presence of TPEN dur-ing the ischemic insult consistently prevented theincrease in cytosolic Zn21 levels in response to ischemia(Fig. 5E).
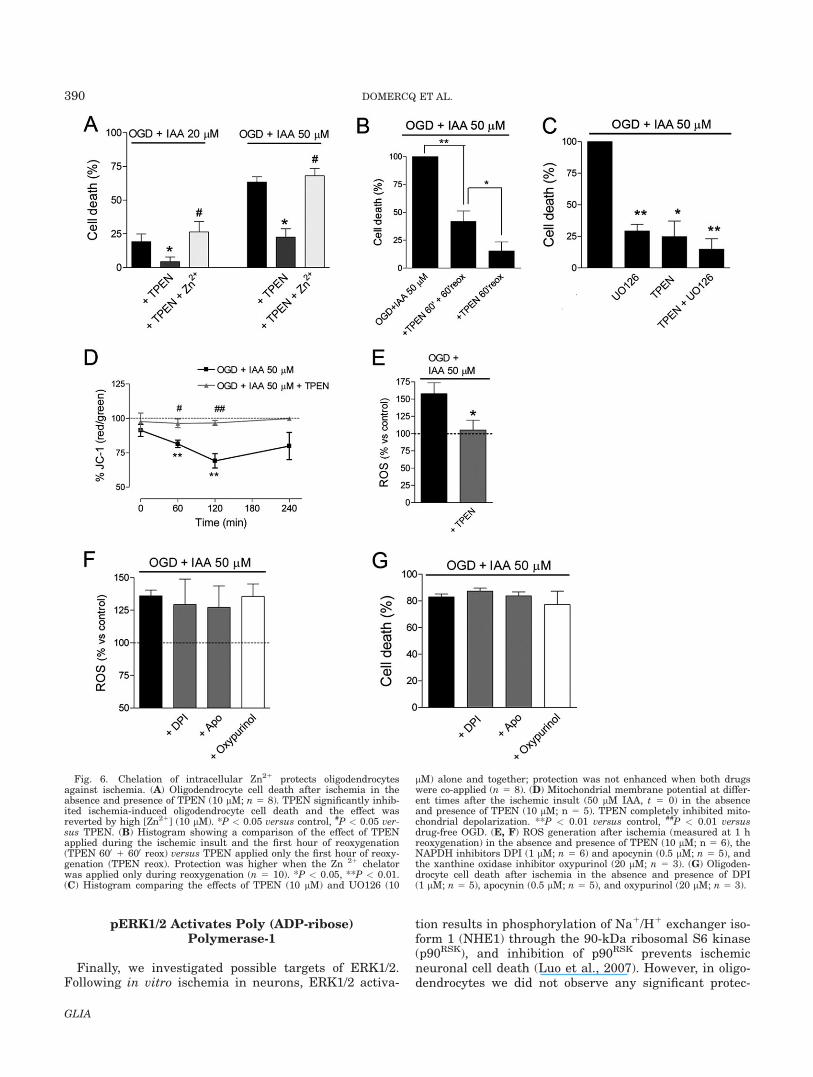
Altered Zn21 homeostasis clearly contributes to ische-mic damage in oligodendrocytes since TPEN signifi-cantly inhibited ischemic cell death and this effect wasreversed by extracellularly applying Zn21 (Fig. 6A).TPEN was toxic to cells when applied during 24 h (datanot shown); therefore, in these experiments we appliedTPEN during the ischemic insult (for 1 h) and the firsthour after reoxygenation (Fig. 6A). Interestingly, weobserved that application of TPEN for only 1 h at thestart of reoxygenation gave even higher protection (Fig.6B). The protective effects of TPEN and UO126 were notpotentiated by co-application of both drugs (Fig. 6C),suggesting that ERK and Zn21 act in the same pathway.
Mitochondrial Zn21 uptake provides clearance of cyto-solic Zn21 in neurons undergoing excitotoxicity or ischemicinsults, and leads to a loss of mitochondrial membranepotential (DWm), as well as to increased generation of reac-tive ROS and eventually cell death (Medvedeva et al.,2009; Sensi et al., 2003). In oligodendrocytes, chelation of
Zn21 with TPEN totally inhibited mitochondrial depolari-zation after ischemia (Fig. 6D). Next, we analyzed oxidativestress in the presence of TPEN. Oxidative burst after ische-mia appeared to be secondary to the alteration of Zn21 ho-meostasis, since ROS generation was completely inhibitedin the presence of TPEN (Fig. 6E). These data suggest thatalteration of Zn21 homeostasis after ischemia-reperfusionprecedes mitochondrial failure and ROS release. In neu-rons, three distinct mechanisms contribute to ROS produc-tion and oxidative stress after ischemia and reperfusion:mitochondria generate an initial burst of ROS, xanthine ox-idase (XO) generates a second phase of ROS, and reoxyge-nation induces an additional increase in ROS generationthat is mediated mainly through NADPH oxidase (NOX;Abramov et al., 2007). However, in oligodendrocytes, ROSgeneration after ischemia was not significantly inhibited bythe NOX inhibitors DPI and apocynin, or by the xanthineoxidase inhibitor oxypurinol (Fig. 6F). Accordingly, NOX orXO inhibition did not prevent ischemia-induced oligoden-drocyte cell death (Fig. 6G). These data suggest Zn21 home-ostasis alteration and mitochondrial depolarization as themain sources of ROS generation contributing to ischemia-induced cell death in oligodendrocytes.
Fig. 4. ROS induces ERK phosphorylation and cell death after is-chemia. (A) Western blot analysis of the effect of the antioxidant Trolox(100 lM) on ERK1/2 phosphorylation after ischemia. ERK phosphoryla-tion was analyzed at 1 h of reperfusion. The histogram shows the mean6 SEM of four different experiments with similar results. (B) Oligoden-drocyte cell death after ischemia in the absence and presence of Trolox(100 lM; n 5 8) and UO126 (10 lM; n 5 8). (C) Histogram showing
protection by Trolox (100 lM) and UO126 (10 lM) applied alone or to-gether against ischemia-induced oligodendroglial cell death when thedrugs were added during the ischemic insult and reperfusion (24 h) orwhen applied only at reperfusion. The histogram shows the mean 6SEM of n 5 3–5 experiments. Symbols denote statistical significanceversus control (#) or versus drug-free OGD (*). #, *P < 0.05, **P < 0.01.
388 DOMERCQ ET AL.
GLIA
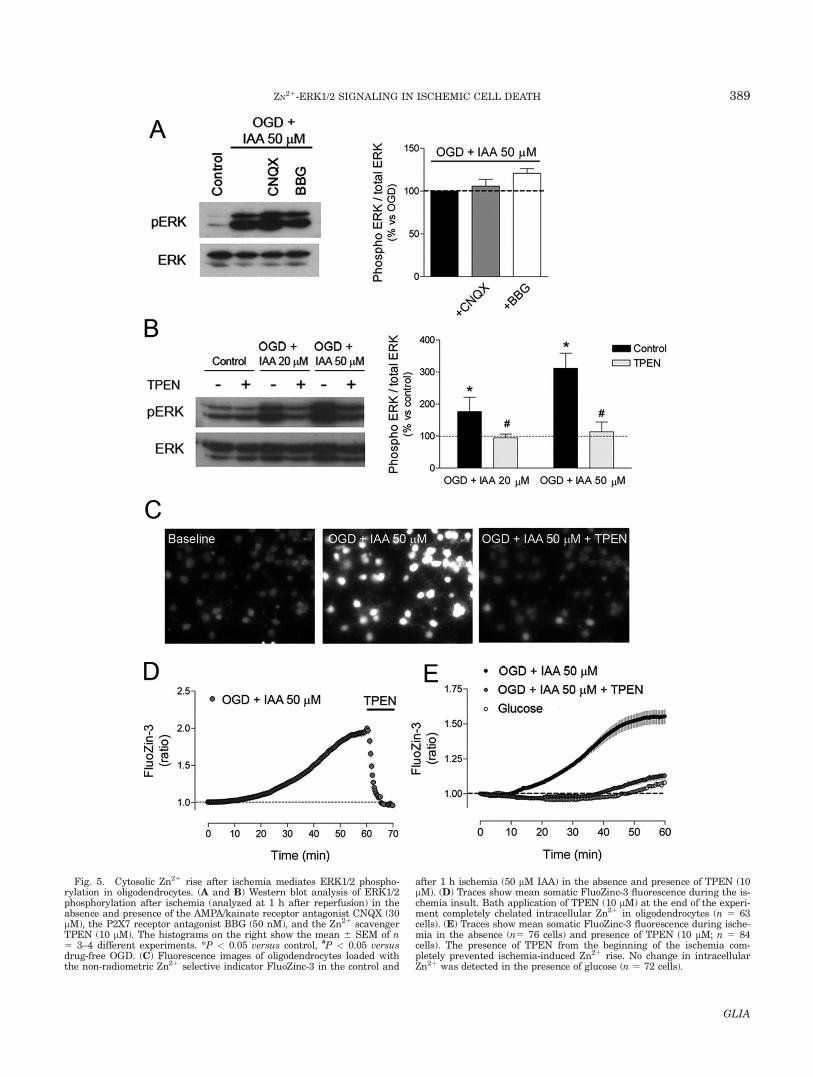
Fig. 5. Cytosolic Zn21 rise after ischemia mediates ERK1/2 phospho-rylation in oligodendrocytes. (A and B) Western blot analysis of ERK1/2phosphorylation after ischemia (analyzed at 1 h after reperfusion) in theabsence and presence of the AMPA/kainate receptor antagonist CNQX (30lM), the P2X7 receptor antagonist BBG (50 nM), and the Zn21 scavengerTPEN (10 lM). The histograms on the right show the mean 6 SEM of n5 3–4 different experiments. *P < 0.05 versus control, #P < 0.05 versusdrug-free OGD. (C) Fluorescence images of oligodendrocytes loaded withthe non-radiometric Zn21 selective indicator FluoZinc-3 in the control and
after 1 h ischemia (50 lM IAA) in the absence and presence of TPEN (10lM). (D) Traces show mean somatic FluoZinc-3 fluorescence during the is-chemia insult. Bath application of TPEN (10 lM) at the end of the experi-ment completely chelated intracellular Zn21 in oligodendrocytes (n 5 63cells). (E) Traces show mean somatic FluoZinc-3 fluorescence during ische-mia in the absence (n5 76 cells) and presence of TPEN (10 lM; n 5 84cells). The presence of TPEN from the beginning of the ischemia com-pletely prevented ischemia-induced Zn21 rise. No change in intracellularZn21 was detected in the presence of glucose (n 5 72 cells).
389ZN21-ERK1/2 SIGNALING IN ISCHEMIC CELL DEATH
GLIA
pERK1/2 Activates Poly (ADP-ribose)Polymerase-1
Finally, we investigated possible targets of ERK1/2.Following in vitro ischemia in neurons, ERK1/2 activa-
tion results in phosphorylation of Na1/H1 exchanger iso-form 1 (NHE1) through the 90-kDa ribosomal S6 kinase(p90RSK), and inhibition of p90RSK prevents ischemicneuronal cell death (Luo et al., 2007). However, in oligo-dendrocytes we did not observe any significant protec-
Fig. 6. Chelation of intracellular Zn21 protects oligodendrocytesagainst ischemia. (A) Oligodendrocyte cell death after ischemia in theabsence and presence of TPEN (10 lM; n 5 8). TPEN significantly inhib-ited ischemia-induced oligodendrocyte cell death and the effect wasreverted by high [Zn21] (10 lM). *P < 0.05 versus control, #P < 0.05 ver-sus TPEN. (B) Histogram showing a comparison of the effect of TPENapplied during the ischemic insult and the first hour of reoxygenation(TPEN 600 1 600 reox) versus TPEN applied only the first hour of reoxy-genation (TPEN reox). Protection was higher when the Zn 21 chelatorwas applied only during reoxygenation (n 5 10). *P < 0.05, **P < 0.01.(C) Histogram comparing the effects of TPEN (10 lM) and UO126 (10
lM) alone and together; protection was not enhanced when both drugswere co-applied (n 5 8). (D) Mitochondrial membrane potential at differ-ent times after the ischemic insult (50 lM IAA, t 5 0) in the absenceand presence of TPEN (10 lM; n 5 5). TPEN completely inhibited mito-chondrial depolarization. **P < 0.01 versus control, ##P < 0.01 versusdrug-free OGD. (E, F) ROS generation after ischemia (measured at 1 hreoxygenation) in the absence and presence of TPEN (10 lM; n 5 6), theNAPDH inhibitors DPI (1 lM; n 5 6) and apocynin (0.5 lM; n 5 5), andthe xanthine oxidase inhibitor oxypurinol (20 lM; n 5 3). (G) Oligoden-drocyte cell death after ischemia in the absence and presence of DPI(1 lM; n 5 5), apocynin (0.5 lM; n 5 5), and oxypurinol (20 lM; n 5 3).
390 DOMERCQ ET AL.
GLIA

tion in the presence of the p90RSK inhibitor SL0101 (50lM; Fig. 7A), excluding a role of NHE1 in ischemia-induced oligodendrocyte death. Poly (ADP-ribose) poly-merase-1 (PARP-1) is another ERK1/2 target involved incell death. Phosphorylated ERK2 can directly activatePARP-1 in the absence of DNA damage (Cohen-Armonet al., 2007) and PARP-1 is involved in caspase-inde-pendent cell death because it consumes NAD1, contrib-uting to further energy depletion under oxidative stressor energy starvation (Wang et al., 2009). In oligodendro-cytes, blocking PARP-1 activity with 3,4-dihydro-5-[4-(1-piperidinyl)butoxyl]-1(2H)-isoquinolinone (DPQ; 30 lM)significantly reduced ischemia-induced cell death (Fig.7A). We also found that ischemia-induced cell death wasindependent of caspase activation, since the broad spec-trum caspase inhibitor zVAD (50 lM) did not affect celldeath (Fig. 7A). Finally, we studied PARP-1 activityusing antibodies to poly [ADP]-ribosylated proteins.Western blot analysis revealed an increase in PAR poly-
mers after ischemia in oligodendroglial homogenatesextracted 2 h after reperfusion (Fig. 7B). Moreover, thiseffect was significantly inhibited by UO126 and TPEN(Fig. 7B), demonstrating that PARP-1 activation wasinduced by Zn21-ERK1/2 signaling pathway.
DISCUSSION
A more complete understanding of the cellular patho-physiology of stroke requires greater attention to whitematter—which is severely damaged by ischemia, produc-ing clinical deficits. Disruption of central conductionpathways may cause loss of motor and sensory function,neurobehavioral syndrome, and cognitive impairment(Goldberg and Ransom, 2003). Here we present twonovel findings: (i) ischemia induces an intracellularZn21 rise in oligodendrocytes, which leads to mitochon-drial depolarization, ROS production, delayed ERK1/2activation, and activation of PARP-1, finally resulting incell death (Fig. 8); and (ii) inhibitors of this pathwayapplied at the onset of reperfusion are protective andtherefore may have therapeutic potential to prevent is-chemia white matter damage.
The role of Zn21 in ischemia has been previouslydescribed in neurons (Sensi et al., 2009; Shuttleworthand Weiss, 2011). After ischemia or prolonged seizures,Zn21 accumulates in hippocampal pyramidal neuronsand its release from stores already present within neu-rons; Zn21 chelation protects hippocampal CA1 neuronsfrom forebrain ischemia (Sensi et al., 2009). In oligoden-drocytes, previous studies have described increase intra-
Fig. 7. Role of PARP-1 in ischemia-induced oligodendrocyte celldeath. (A) Oligodendrocyte cell death after ischemia in the absence andpresence of the 90-kDa ribosomal S6 kinase inhibitor SL0101 (50 lM;n 5 7), the pan-caspase inhibitor zVAD (50 lM; n 5 7), and the poly-(ADP-ribose) polymerase-1 enzyme (PARP-1) inhibitor DPQ (30 lM;n 5 9). (B) Representative western blot probed for poly[ADP]-ribosyla-tion in homogenates from oligodendrocytes after ischemia (2 h reoxyge-nation) in the absence and presence of UO126 (10 lM) and TPEN(10 lM). Histogram shows densitometry of three independent experi-ments. *P < 0.05.
Fig. 8. Ischemia-induced oligodendroglial cell death pathway. Ische-mia induces a cytoplasmic [Zn21]i rise, which depolarizes mitochondriaand generates ROS. Zinc induces sustained activation of ERK1/2, whichis secondary to ROS generation or, alternatively, secondary to ERK1/2phosphatase (MKP) inhibition. Phosphorylated ERK1/2 further enhan-ces ROS generation and activates PARP-1, leading to caspase-independ-ent oligodendrocyte cell death. In contrast to this cell death signalingpathway, the transient activation of ERK1/2 secondary to AMPA recep-tor stimulation induces a pro-survival response.
391ZN21-ERK1/2 SIGNALING IN ISCHEMIC CELL DEATH
GLIA

cellular Zn21 in response to the potent oxidant peroxyni-trite (Li et al., 2011; Zhang et al., 2006, 2007). Here wedemonstrated that ischemia induced an intracellularZn21 rise that induced oligodendroglial cell damage; theZn21 chelator TPEN—a molecule that shows a very lowaffinity for Ca21 but is extremely avid for Zn21—com-pletely reversed the intracellular Zn21 rise and pre-vented oligodendrocyte cell death. Further experimentsare required to determine the origin of the Zn21 increasein oligodendrocytes after ischemia. Our findingsexcluded the possibility of Zn21 entry through AMPA/kainate receptors, because antagonists of these receptorsdid not block ischemia-induced ERK activation. Ischemicacidosis is a potent trigger of Zn21 mobilization frommethallothionein, which could cause Zn21 accumulationin the cytoplasm. Oligodendrocytes express the methal-lothionein-III isoform (Miyazaki et al., 2002) and releaseZn21 from this intracellular pool in response to oxidativestressors (Zhang et al., 2006). Mobilization of intracellu-lar Zn21 from Zn21 buffering proteins could also be par-ticularly important during reperfusion injury (Lin et al.,2011; Shuttleworth and Weiss, 2011).
Our data suggest that the main source of ROS gener-ation in oligodendrocytes after ischemia is Zn21-inducedmitochondrial depolarization. Other sources of ROSgeneration in ischemic neurons, like xanthine oxidaseand NAPDH oxidase (Abramov et al., 2007), have noclear role in ROS generation in oligodendrocytes. Zn21
rise could trigger ERK1/2 activation through directinhibition of ERK1/2 phosphatase MKP3, which isindependent of oxidative stress (Ho et al., 2008). How-ever, the ERK1/2 activation in oligodendrocytes is alsoblocked by the ROS scavenger Trolox, suggesting thatERK activation is secondary to the oxidative burstgenerated by Zn21. Moreover, although AMPA/kainatereceptors in oligodendrocytes induce ERK1/2 phospho-rylation (Domercq et al., 2010) and are permeable toZn21 (Sensi et al., 2009), ERK1/2 activation atreoxygenation was independent of glutamate and P2X7receptor activation and was instead mediated throughthe Zn21-ROS pathway.
The MAPK kinase signaling pathway regulates cellu-lar activities including gene expression, mitosis, embryo-genesis, cell differentiation, movement, metabolism, sur-vival, and cell death (Mebratu and Tesfaigzi, 2009). Pre-vious observations link the duration of ERK activationto cell fate decisions in rat pheochromocytoma PC-12cells; transient ERK activation by epidermal growth fac-tor (EGF) induced cell proliferation, whereas sustainedERK activation by nerve growth factor (NGF) induceddifferentiation (Marshall, 1995). Similarly, the kineticsand duration of ERK1/2 activation may direct ERK1/2toward downstream targets that will either promote orlimit neuronal survival (de Bernardo et al., 2004; Levin-thal and Defranco, 2005; Luo and DeFranco, 2006; Sub-ramaniam and Unsicker, 2010). Accordingly, weobserved a dual role of ERK signaling in oligodendro-cytes depending on the type of stimulus. Whereas arapid and transient ERK activation following AMPA re-ceptor stimulation prevents oligodendroglial cell death
(Domercq et al., 2011), herein we showed that delayedand sustained ERK activation secondary to ischemia-reperfusion mediates oligodendroglial cell death. How-ever, we cannot exclude a transient activation of ERKduring the ischemic insult. Indeed, in the presence ofUO126 we observed a higher increase in intracellularcalcium during chemical ischemia, which could be due toERK modulation of the Ca21 permeability of AMPAreceptors (Domercq et al., 2011). However, this ERKactivation did not appear to contribute significantly tocell death. In contrast, activation of ERK at reoxygena-tion was sustained and contributed significantly to oligo-dendrocyte cell death.
Downstream to ERK1/2 activation, the Zn21-ROS-ERKcell death pathway induced PARP-1 activation, whichwas involved in ischemia-induced oligodendrocyte celldeath. ERK1/2, particularly ERK2, activates PARP-1through direct interaction with the enzyme (Cohen-Armon, 2007) and there is mounting evidence that PARP-1 plays an essential role in excitotoxic and ischemic neu-ronal cell death (Eliasson et al., 1997; Wang et al., 2009).However, our study describes for the first time an associ-ation of late activation of ERK1/2 on reoxygenation withPARP activity in ischemic oligodendrocytes. PARP-1 acti-vation in oligodendrocytes has been described in responseto stress (Zhang and Rosenberg, 2004) and to severeAMPA/kainate receptor stimulation (Sanch�ez-G�omez etal., 2003), and is involved in oligodendrocyte depletionand demyelination in the cuprizone model of multiplesclerosis (Veto et al., 2010). Cell death mediated throughPARP-1 is independent of caspase and occurs via therelease of apoptosis-inducing factor (AIF) from the mito-chondria (Yu et al., 2002). Ischemia-induced oligodendro-cyte cell death is consistently independent of caspases.
Despite increasing knowledge of the pathologicalmechanisms of ischemia, limited advances have beenmade to counter the deleterious effects of cerebral ische-mia and the only available treatment is thrombolysiswith tissue plasminogen activator (tPA). Most therapeu-tic approaches have focused on protecting neurons fromthe main pathogenic mechanisms causing ischemicinjury, such as excitotoxicity. However, the pathologicalevents during the ischemic insult do not have muchtherapeutic value due to its narrow temporal window;understanding the mechanism of damage after the is-chemic insult could contribute to developing more usefultherapies. In summary, the present study shows that is-chemia-reperfusion induced intracellular Zn21 accumu-lation that activates the ROS-ERK1/2-PARP-1 signallingpathway. Inhibitors of this pathway are potentiallypromising for the treatment of white matter ischemia.
REFERENCES
Abramov AY, Scorziello A, Duchen MR. 2007. Three distinct mecha-nisms generate oxygen free radicals in neurons and contribute to celldeath during anoxia and reoxygenation. J Neurosci 27:1129–1138.
Cohen-Armon M. 2007. PARP-1 activation in the ERK signaling path-way. Trends Pharmacol Sci 28:556–560.
392 DOMERCQ ET AL.
GLIA

Cohen-Armon M, Visochek L, Rozensal D, Kalal A, Geistrikh I, KleinR, Bendetz-Nezer S, Yao Z, Seger R. 2007. DNA-independent PARP-1activation by phosphorylated ERK2 increases Elk1 activity: A link tohistone acetylation. Mol Cell 25:297–308.
de Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J, MenaMA. 2004. Role of extracellular signal-regulated protein kinase inneuronal cell death induced by glutathione depletion in neuron/gliamesencephalic cultures. J Neurochem 91:667–682.
Domercq M, Alberdi E, S�anchez-G�omez MV, Ariz U, P�erez-Samart�ın A,Matute C. 2011. Dual-specific phosphatase-6 (Dusp6) and ERK medi-ate AMPA receptor-induced oligodendrocyte death. J Biol Chem286:11825–11836.
Domercq M, Perez-Samartin A, Aparicio D, Alberdi E, Pampliega O,Matute C. 2010. P2X7 receptors mediate ischemic damage to oligo-dendrocytes. Glia 58:730–740.
Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J,Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. 1997.Poly(ADP-ribose) polymerase gene disruption renders mice resistantto cerebral ischemia. Nat Med 3:1089–1095.
Goldberg MP, Ransom BR. 2003. New light on white matter. Stroke34:330–332.
Ho Y, Samarasinghe R, Knoch ME, Lewis M, Aizenman E, DeFrancoDB. 2008. Selective inhibition of mitogen-activated protein kinasephosphatases by zinc accounts for extracellular signal-regulated ki-nase 1/2-dependent oxidative neuronal cell death. Mol Pharmacol74:1141–1151.
K�arad�ottir R, Cavelier P, Bergersen LH, Attwell D. 2005. NMDA recep-tors are expressed in oligodendrocytes and activated in ischaemia.Nature 438:1162–1166.
Levinthal DJ, Defranco DB. 2005. Reversible oxidation of ERK-directedprotein phosphatases drives oxidative toxicity in neurons. J BiolChem 280:5875–5883.
Li S, Lin W, Tchantchou F, Lai R, Wen J, Zhang Y. 2011. Protein kinaseC mediates peroxynitrite toxicity to oligodendrocytes. Mol Cell Neuro-sci 48:62–71.
Lin CL, Tseng HC, Chen RF, Chen WP, Su MJ, Fang KM, Wu ML.2011. Intracellular zinc release-activated ERK-dependent GSK-3b-p53 and Noxa-Mcl-1 signaling are both involved in cardiac ischemic-reperfusion injury. Cell Death Differ 18:1651–1663.
Luo J, Kintner DB, Shull GE, Sun D. 2007. ERK1/2-p90RSK-mediatedphosphorylation of Na1/H1 exchanger isoform 1. A role in ischemicneuronal death. J Biol Chem 282:28274–28284.
Luo Y, DeFranco DB. 2006. Opposing roles for ERK1/2 in neuronal oxi-dative toxicity: Distinct mechanisms of ERK1/2 action at early versuslate phases of oxidative stress. J Biol Chem 281:16436–16442.
Marshall CJ. 1995. Specificity of receptor tyrosine kinase signaling:Transient versus sustained extracellular signal-regulated kinase acti-vation. Cell 80:179–185.
Matute C, Domercq M, S�anchez-G�omez MV. 2006. Glutamate-mediatedglial injury: Mechanisms and clinical importance. Glia 53:212–224.
Matute C, S�anchez-G�omez MV, Mart�ınez-Mill�an L, Miledi R. 1997. Glu-tamate receptor-mediated toxicity in optic nerve oligodendrocytes.Proc Natl Acad Sci USA 94:8830–8835.
Matute C, Torre I, P�erez-Cerd�a F, P�erez-Samart�ın A, Alberdi E, Etxe-barria E, Arranz AM, Ravid R, Rodr�ıguez-Antig€uedad A, S�anchez-G�omez M, Domercq M. 2007. P2X(7) receptor blockade prevents ATPexcitotoxicity in oligodendrocytes and ameliorates experimental auto-immune encephalomyelitis. J Neurosci 27:9525–9533.
Mebratu Y, Tesfaigzi Y. 2009. How ERK1/2 activation controls cell pro-liferation and cell death: Is subcellular localization the answer? CellCycle 8:1168–1175.
Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. 2009. IntracellularZn21 accumulation contributes to synaptic failure, mitochondrialdepolarization, and cell death in an acute slice oxygen-glucose depri-vation model of ischemia. J Neurosci 29:1105–1114.
Micu I, Jiang Q, Coderre E, Ridsdale A, Zhang L, Woulfe J, Yin X,Trapp BD, McRory JE, Rehak R, Zamponi GW, Wang W, Stys PK.2006. NMDA receptors mediate calcium accumulation in myelinduring chemical ischaemia. Nature 439:988–992.
Miyazaki I, Asanuma M, Higashi Y, Sogawa CA, Tanaka K, Ogawa N.2002. Age-related changes in expression of metallothionein-III in ratbrain. Neurosci Res 43:323–333.
Murphy LO, Blenis J. 2006. MAPK signal specificity: The right place atthe right time. Trends Biochem Sci 31:268–275.
Orrenius S, Zhivotovsky B, Nicotera P. 2003. Regulation of cell death:The calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565.
Salter MG, Fern R. 2005. NMDA receptors are expressed in developingoligodendrocyte processes and mediate injury. Nature 438:1167–1171.
S�anchez-G�omez MV, Alberdi E, Ibarretxe G, Torre I, Matute C. 2003.Caspase-dependent and caspase-independent oligodendrocyte deathmediated by AMPA and kainate receptors. J Neurosci 23:9519–9528.
Sensi SL, Paoletti P, Bush AI, Sekler I. 2009. Zinc in the physiologyand pathology of the CNS. Nat Rev Neurosci 10:780–791.
Sensi SL, Ton-That D, Sullivan PG, Jonas EA, Gee KR, Kaczmarek LK,Weiss JH. 2003. Modulation of mitochondrial function by endogenousZn21 pools. Proc Natl Acad Sci USA 100:6157–6162.
Shuttleworth CW, Weiss JH. 2011. Zinc: New clues to diverse roles inbrain ischemia. Trends Pharmacol Sci 32:480–486.
Subramaniam S, Unsicker K. 2010. ERK and cell death: ERK1/2 inneuronal death. FEBS J 277:22–29.
Tekk€ok SB, Ye Z, Ransom BR. 2007. Excitotoxic mechanisms of ische-mic injury in myelinated white matter. J Cereb Blood Flow Metab27:1540–1552.
Veto S, Acs P, Bauer J, Lassmann H, Berente Z, Setalo G, Borgulya G,Sumegi B, Komoly S, Gallyas F Jr, Illes Z. 2010. Inhibiting poly-(ADP-ribose) polymerase: A potential therapy against oligodendrocytedeath. Brain 133:822–834.
Wang Y, Dawson VL, Dawson TM. 2009. Poly(ADP-ribose) signals to mi-tochondrial AIF: A key event in parthanatos. Exp Neurol 218:193–202.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poi-rier GG, Dawson TM, Dawson VL. 2002. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing fac-tor. Science 297:259–263.
Zhang Y, Aizenman E, DeFranco DB, Rosenberg PA. 2007. Intracellularzinc release, 12-lipoxygenase activation and MAPK dependent neuro-nal and oligodendroglial death. Mol Med 13:350–355.
Zhang Y, Rosenberg PA. 2004. Caspase-1 and poly (ADP-ribose)polymerase inhibitors may protect against peroxynitrite-inducedneurotoxicity independent of their enzyme inhibitor activity. Eur JNeurosci 20:1727–1736.
Zhang Y, Wang H, Li J, Dong L, Xu P, Chen W, Neve RL, Volpe JJ,Rosenberg PA. 2006. Intracellular zinc release and ERK phosphoryla-tion are required upstream of 12-lipoxygenase activation in peroxyni-trite toxicity to mature rat oligodendrocytes. J Biol Chem 281:9460–9470.
393ZN21-ERK1/2 SIGNALING IN ISCHEMIC CELL DEATH
GLIA
Copyright © 2022 FDOKUMEN
















![Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography](https://static.fdokumen.com/doc/165x107/6335c3a302a8c1a4ec01e906/synthesis-18f-radiolabeling-and-evaluation-of-poly-adp-ribose-polymerase-1.jpg)



