
A preliminary neuropathological study of Japanese encephalitis in humans and a mouse model
Upload
independentCategory
view
3download
0
Abstract We report neuropathological, biochemical andmolecular studies on two patients with childhood ataxiawith diffuse central nervous system hypomyelination(CACH) syndrome, a leukodystrophy recently defined ac-cording to clinical and radiological criteria. Both had se-vere cavitating orthochromatic leukodystrophy without at-rophy, predominating in hemispheric white matter, whereasU-fibers, internal capsule, corpus callosum, anterior com-missure and cerebellar white matter were relatively spared.The severity of white matter lesions contrasted with therarity of myelin breakdown products and astroglial andmicroglial reactions. In the white matter, there was an in-crease in a homogeneous cell population with the mor-phological features of oligodendrocytes, in many in-stances presenting an abundant cytoplasm like myelina-tion glia. These cells were negative for glial fibrillaryacidic protein and antibodies PGM1 and MIB1. Somewere positive for myelin basic protein, proteolipid protein(PLP), and myelin oligodendrocyte glycoprotein, but the
majority were positive for human 2′-3′ cyclic nucleotide3′ phosphodiesterase and all were positive for carbonicanhydrase II, confirming that they are oligodendrocytes.Myelin protein and lipid content were reduced. The PLPgene, analyzed in one case, was not mutated or dupli-cated. The increased number of oligodendrocytes withoutmitotic activity suggests an intrinsic oligodendroglial de-fect or an abnormal interaction with axons or other glialcells. This neuropathological study supports the notionthat CACH syndrome constitutes a specific entity.
Key words Leukodystrophy · Oligodendrocyte · Myelin · Orthochromatic · CACH syndrome
Introduction
A large proportion of childhood leukodystrophies, most ofwhich are genetically inherited, have no known etiology[15]. Phenotypic classification of these myelin disordersis essential for linkage analysis in affected families. Anew leukoencephalopathy termed childhood ataxia withdiffuse central nervous system hypomyelination (CACH)syndrome or leukoencephalopathy with vanishing whitematter was recently identified on clinical and neuroradio-logical grounds [13, 33, 34, 38]. This disorder only affectsmyelin of the central nervous system (CNS) and has anautosomal recessive mode of inheritance, but the geneticdefect is not known. Previous neuropathological reportsof two brain biopsies and one autopsy showed extensivecavitating leukoencephalopathy but with no significanthistological findings [34, 38]. Here, we report neu-ropathological data of two new patients with CACH syn-drome and a biochemical and molecular study of one ofthem. The cavitating white matter lesions which charac-terize this orthochromatic leukodystrophy are associated,in the less affected white matter areas, with an unusuallyincreased oligodendrocyte density.
D. Rodriguez · A. Gelot · B. della Gaspera · O. Robain ·G. Ponsot · L. L. Sarliève · S. Ghandour · A. Pompidou · A. Dautigny · P. Aubourg · D. Pham-Dinh
Increased density of oligodendrocytes in childhood ataxia with diffuse central hypomyelination (CACH) syndrome: neuropathological and biochemical study of two cases
Acta Neuropathol (1999) 97 :469–480 © Springer-Verlag 1999
Received: 23 February 1998 / Revised: 12 August 1998, 20 October 1998 / Accepted: 21 October 1998
REGULAR PAPER
D. Rodriguez (Y) · G. Ponsot · P. AubourgService de Neuropédiatrie, Hôpital Saint Vincent de Paul, AP-HP, 74–82 avenue Denfert-Rochereau, F-75674 Paris Cedex 14, Francee-mail: [email protected], Fax: +33-1-40-48-83-56
D. Rodriguez · P. AubourgPathologie métabolique et hormonale du développement, INSERM U342, Hôpital Saint Vincent de Paul, Paris, France
A. Gelot · A. PompidouService d’Histo-Embryologie, Cytogénétique, Anatomopathologie, Hôpital Saint Vincent de Paul, AP-HP, Paris, France
B. della Gaspera · A. Dautigny · D. Pham-DinhLaboratoire de Neurogénétique Moléculaire, CNRS URA 1488, Université de Paris VI, Paris, France
O. RobainINSERM U29, Hôpital Port-Royal, Paris, France
L. L. Sarliève · S. GhandourLaboratoire de Neurobiologie Moléculaire des Interactions cellulaires, CNRS UPR 416, Université Louis Pasteur, Strasbourg, France

Materials and methods
Clinical data
Patient 1
This male patient had a normal family history with healthy, non-consanguineous parents and an unaffected 14-year-old sister. Helearned to walk at 15months but speech was mildly delayed. Thefirst disabilities in walking were noted after a minor head traumawhen he was 7.5years old. He was mildly macrocephalic (headcircumference at +2SD, like his father). Neurological examinationshowed cerebellar ataxia and diplegia. Computed tomography scanand magnetic resonance imaging (MRI) showed diffuse abnormal-ities of the cerebral white matter. Hemispheric white matter signalintensity was low on T1-weighted images, and partially low andsimilar to the CSF signal on proton density images. On T2-weightedimages, signal intensity of hemispheric and cerebellar white matterwas homogeneously increased with two particularly intense spotsin the pontine tegmentum. U-fibers were spared. Visual- and somato-sensory-evoked potentials showed a decrease in central conduc-tion. Brain stem auditory potentials, motor and sensory peripheralnerve conduction and electromyogram were normal. Laboratorytests were negative for all peroxysomal, lysosomal and mitochon-drial leukodystrophies. N-Acetylaspartic acid, oligosaccharides,amino acids, organic acids and sialic acids were normal in urine.The karyotype was also normal. The disease was characterized byprogressive deterioration with optic atrophy, seizures, vomitingand bulbar symptoms. At age 10, the patient became comatose after a minor head trauma and died 2months later.
Patient 2
This male patient had healthy unrelated parents and two healthyyounger siblings. He walked at 14months, but motor stagnationwas noted at 16months. Neurological examination showed spasticdiplegia, cerebellar ataxia, but normal speech at the age of 2.5years. Brain MRI showed abnormal hemispheric, cerebellar andbrain stem white matter, as in patient 1 (Fig.1A). Visual-, somato-sensory- and brain stem auditory-evoked potential latenciesshowed delayed responses. Sensory and motor peripheral nerveconduction was normal. Biochemical tests for identified leukodys-trophies were negative as in patient 1. The patient deterioratedslowly. He no longer walked at age 4, developed seizures andswallowing difficulties at age 5 and died at age 6 during a feverwith recurrent apnea. Autopsy was unremarkable.
Neuropathological analysis
The brains were removed immediately after death and quicklyfixed or frozen in cooled isopentane. Several fragments were fixedin 4% paraformaldehyde-1% glutaraldehyde in phosphate buffer,post-fixed in 4% osmium tetroxide, embedded in epoxy and cut onan ultramicrotome in semithin and thin sections for ultrastructuralanalysis (Philips CM10). The remaining fragments were fixed informalin, embedded in paraffin or frozen after immersion in a 10%sucrose solution, and cut into 8-µm sections. Paraffin sections werestained with hematoxylin and eosin (H&E), cresyl-violet (CV),Luxol-fast-blue (LFB), periodic acid-Schiff (PAS) or silver im-pregnated by the Bodian technique. Frozen fixed sections wereused for lipid revelation (Oil-Red-O, ORO) and immunostaining.
Specific CNS cell types were identified using the followingprimary antibodies. Oligodendrocytes and myelin were stainedwith a monoclonal mouse anti-human myelin basic protein (MBP)IgG2 (Chemicon, Temecula, Calif.) diluted 1:50, a mouse mono-clonal antibody against human 2′-3′ cyclic nucleotide 3′ phospho-diesterase (CNP; Chemicon) diluted 1:25, and a rabbit polyclonalanti-bovine myelin oligodendrocyte glycoprotein (MOG) serum[3]. On frozen tissues, an antibody against a C-terminal fragment
common to proteolipid protein (PLP) and DM20 [28] and an anti-rat carbonic anhydrase (CA) II antibody diluted 1:200 [11] wereused. Astrocytes were stained with mouse anti-human glial fibril-lary acid protein (GFAP; Dakopatts, Denmark) diluted 1:25. Mi-croglia-macrophages were stained with a monoclonal mouse anti-human CD68 IgG3, PGM1 (Dakopatts) diluted 1:100 [16]. Mitoticactivity was detected with a mouse monoclonal antibody againstnuclear antigen 2KI67, MIB1 (Immunotech, France) diluted 1:20.A 13-week-old fetus was used as a positive control. The Vectastainavidin-biotin-peroxidase system (Vector laboratories) was used forproduct detection. Some slides were double immunostained toidentify the mitotically active (anti-MIB1/anti-GFAP, anti-MIB1/anti-PGM1, anti-MIB1/anti-CNPase) and hypercellularity (anti-GFAP/anti-PGM1) cell types, revealed with peroxidase and alka-line phosphatase.
Cell counts were performed on matched brain specimens frompatients and a control who died at the age of 10years from Lesh-Nyhan disease. Using an ocular net micrometer with 100 squares,total cell number, with the exception of vascular cells, was countedby the same person, in comparable areas of white matter, on paraf-fin-embedded slides stained with CV [36, 39]. Labeled cells werealso counted after GFAP, PGM1, CNP, CAII, and MIB1 im-munostaining. Five counts were made for each of the values re-ported in this study. Results are expressed as the mean for eachsample.
Biochemical and molecular analyses
Postmortem cerebral and cerebellar white matter from patient 1and from the Lesh-Nyhan patient were used to prepare total ho-mogenates or to isolate myelin. Myelin was purified according toNorton and Poduslo [26]. Total lipids and myelin lipids were ex-tracted [8] from the cerebellum and measured by weighing thedried lipid extract. Polar lipids, galactolipids and phospholipidswere separated by high performance thin-layer chromatography(HPTLC) and quantified as described [14]. UDP-galactose:ce-ramide-galactosyltransferase (EC 2.4.1.45, CGalT) was assessedin brain homogenates [24]. One unit corresponds to 1nmol ofgalactose transferred per hour. Total cerebellar homogenates wereused for SDS-PAGE and immunoblotting [17]. PLP immunoblot-ting was performed as described [28]. Total RNA extracted fromcerebral white matter was reverse-transcribed as previously de-scribed [29] according to standard procedures [32]. The coding se-quence of the PLP cDNA was subsequently amplified using spe-cific primers (the sequence of PLP-specific primers can be ob-tained upon request), and sequenced using the T7 sequencing kit(Pharmacia Biotech). Southern blots were hybridized with a 32P-la-beled human PLP cDNA probe. For quantitative analysis, PLP signals were compared to signals obtained by hybridization withthe XV2C probe, an internal marker of the X chromosome.
470
Fig.1A–F Telencephalic white matter. A Flair magnetic reso-nance image of patient 2 at age 5years 9months. Hemisphericwhite matter shows low signal intensity, similar to CSF, and iscrossed by a meshwork of tissue, whereas signal intensity in the in-ternal and external capsules is high. B Coronal section of the fixedbrain of patient 2: white matter is diffusely cavitated, and appearsflattened. There is no ventricular enlargement or thinning of thecorpus callosum. C Closer observation reveals the radial cobwebpattern of remaining fiber bundles (case 2). D Macroscopic frontalsection after MBP immunostaining showing widespread cavitationand preservation of subcortical myelin (case 2). E Frontal sectionrevealing diffuse myelin depletion and relative preservation ofperivascular myelin (case 1). F Sparse myelinated bundles in sub-cortical areas (top left). They become progressively disorganizedwhen going deeper, except in perivascular areas (case 2) (MBPmyelin basic protein). E, F Luxol-fast blue (LFB). E × 2, F × 10
E

471

Results
Neuropathology
Brain weight was 1639g in case1 and 1277g in case2,compared to normal (1400g). The external aspect wasnormal with no cerebral or cerebellar atrophy. The neu-ropathological abnormalities were identical in both pa-tients, but more severe in the second case. On sectioning,white matter was affected throughout but to various de-grees (Table1). No atrophy of the cerebrum or corpus cal-losum was noted. The ventricles were not enlarged. Allgray matter structures appeared normal. The most severelesions were observed in the telencephalic white matterthat was grayish, gelatinous and flattened with cavitationrestricted to central white matter areas in case1, but morediffuse in case2 where only a thin subcortical ribbon ofwhite matter was present (Fig.1B, C). In these cavitatedareas, the white matter resembled a cobweb with sparseand thin myelinated bundles extending from the ventricu-lar wall to the cortical ribbon (Fig.1C). Myelin was betterpreserved in U-fibers, internal and external capsules, cor-pus callosum, anterior commissure and fornix (Fig.1B,D). Lesions were less severe in infratentorial structures inboth cases (Fig.2A, B, E). Cerebellar white matter wasdiscolored, particularly in central areas like the nucleusdentatus hilus which appeared grayish (Fig.2E). In thebrain stem (Fig.2A, B), pyramidal and cerebellar tractswere discolored. In both cases the tractus tegmentalis cen-tralis and cerebellar tract decussation in the pons were re-placed by two para-median bright white dots (open arrowsin Fig.2A, B). The remaining tracts appeared normal.
Histological analysis confirmed the preservation ofgray structures (neocortex, basal ganglia and brain stem
nuclei), except the cerebellar cortex where Purkinje cellswere partially lost. White matter was diffusely involvedbut to various degrees (Table1). In discolored white mat-ter areas (i.e. subcortical cerebral and cerebellar whitematter, internal and external capsules, cerebral peduncles,pyramidal and cerebellar tracts) myelin fibers were dis-persed by vacuoles and their density was decreased (Fig.1E). In addition, mild astrocytosis was noted. Deeper intocerebellar white matter, these focal swellings of the myelinsheath became more numerous, giving rise to a spongi-form, and finally to a pre-cavitated network. Simultane-ously, reactive astrocytes were detected and ramified mi-croglia became phagocytic macrophages. In cavitated ar-eas of the telencephalum (Fig.1D–F), lace-like tissue wasnearly devoid of myelin, axons and cells. The rare re-maining myelin sheaths, stained with CNP, MBP, PLP andMOG, were mostly located in the perivascular space.They displayed numerous vacuoles that surrounded axons(see Fig.4I). Most of the axons were naked and wereloosely threaded among sparse disseminated reactive as-trocytes, phagocytic macrophages and oligodendrocytes(see Fig.4H). In cavitated areas, large astrocytes with anincreased GFAP expression were observed, contrastingwith the mild fibrillary astrogliosis in subcortical areas(Fig.3C, D). The cytoplasm of macrophages was or-thochromatic, rarely PAS positive and became su-danophilic, filled with myelin debris, close to cavitatedzones (Figs. 3E, F; 4D, F). In the pons, the superior cere-bellar peduncles and the tractus tegmentalis centralis weretotally devoid of myelin (Fig.2C). Axons were present,but naked. These areas contained a homogeneous layer ofmacrophages filled with ORO-positive PAS-negative ma-terial (Fig.2D).
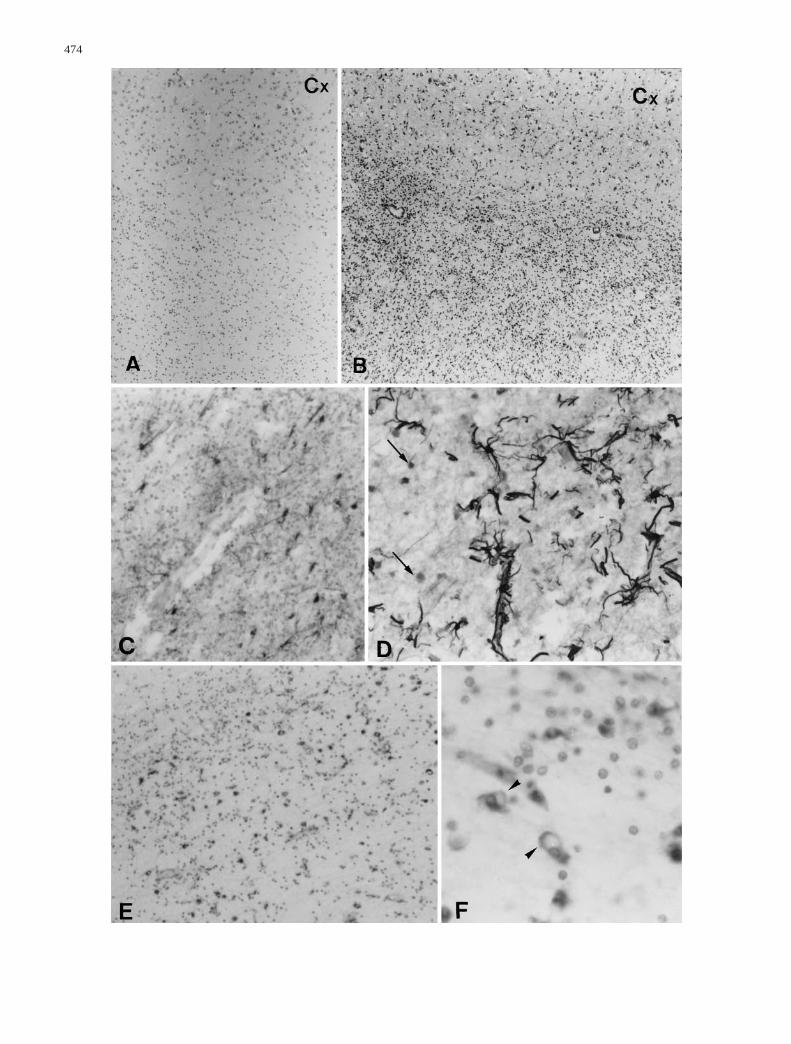
Cell numbers were increased (Fig.3A, B; Table2) by200–300% in telencephalic subcortical white matter and
472
Table 1 Distribution and morphological features of white matter lesions (WM white matter, + present, ++abundant, +++ very abundant,– absent)
Cerebral structures Macroscopic study Microscopic studyMyelin aspect
Myelin Axon Hyper- Macrophages fully loss loss cellu- filled by sudanophilic
larity myelin debris
Cerebral central WM Cavitary (persistent +++ + – ++perivascular myelin)
U-fibers, internal capsule Slightly discolored ++ – ++ –Corpus callosum, anterior comissure, fornix, Slightly discolored ++ – – –
external capsule, cerebral pedunclesCerebellar WM Slightly discolored ++ – + +Nucleus dentatus hilus Grayish aspect ++ – – –Superior cerebellar peduncle Discolored ++ – – –Superior cerebellar peduncle at decussation level Bright dots +++ – – +++Tractus tegmentalis centralis Bright dots +++ – – +++Pyramidal tract Slightly discolored + – + –Olivary nucleus (hilus) Discolored + – – –Inferior cerebellar peduncle Discolored ++ – – –Medial cerebellar peduncle, lemniscus medialis, Normal – – – –
cranial nerve

473
Fig.2A–F Brain stem andcerebellar lesions. A Horizon-tal section through pons andcerebellar peduncles showingpreserved myelin in the medianpeduncle (arrows) and discol-ored myelin in superior and in-ferior cerebellar peduncles (ar-rowheads). In the dorsal para-median pons, the superior cere-bellar peduncle decussation(open arrow), is unusuallybright (case 1). B Horizontalsection at the ponto-bulbarjunction revealing a bright spotcorresponding to the tractustegmentalis centralis (open ar-row) (case 1). C, D Horizontalsections through the ponsstained by LFB, and immunos-tained with anti-PGM1-anti-body, respectively, show thatthe selective loss of myelin intractus tegmentalis centralisand superior cerebellar pedun-cle decussation is correlatedwith macrophage invasion(open arrow). These lesionsare specific to the brain stemand to both fiber tracts (case2). E In the cerebellum, myelinis better preserved, except innucleus dentatus hilus whichappears gray. F Macroscopicsection of the cerebellum re-vealing diffuse white matterdiscoloration with sparing ofsubcortical myelin (LFB) (case 2)
474

by 40–80% in cerebellar white matter, structures in whichmyelin was decreased. In both patients, hypercellularitywas present in remaining telencephalic white matter butalso in cerebellar white matter which was not cavitated.After double immunostaining with anti-GFAP and anti-PGM1, only 10–20% of white matter cells were labeled(in equal proportion for both markers) (Fig.4B), as in thecontrol case (not shown). The remaining unlabelled cellswere morphologically homogeneous with small, dark,round nuclei similar to those of oligodendrocytes, and insome cases an abundant and elongated cytoplasm (Fig.4A, E). Some of these cells were immunolabeled withanti-MBP, -PLP, -MOG (Fig.4C). Nearly 80–90% ex-pressed CNP within well-developed cytoplasmic expan-sions (Fig.4E). All cells with oligodendrocytic morphol-ogy expressed CAII (Fig.4G). Rare MIB1-positive cellswere mostly located in deep cerebral white matter, but notin less affected myelinated areas (not shown). MIB1-pos-itive cells were identified as perivascular macrophagesand astrocytes by double immunostaining. CNP-positivecells were MIB1 negative (not shown). Macrophages andother glial cells contained no pigment and did not autoflu-oresce.
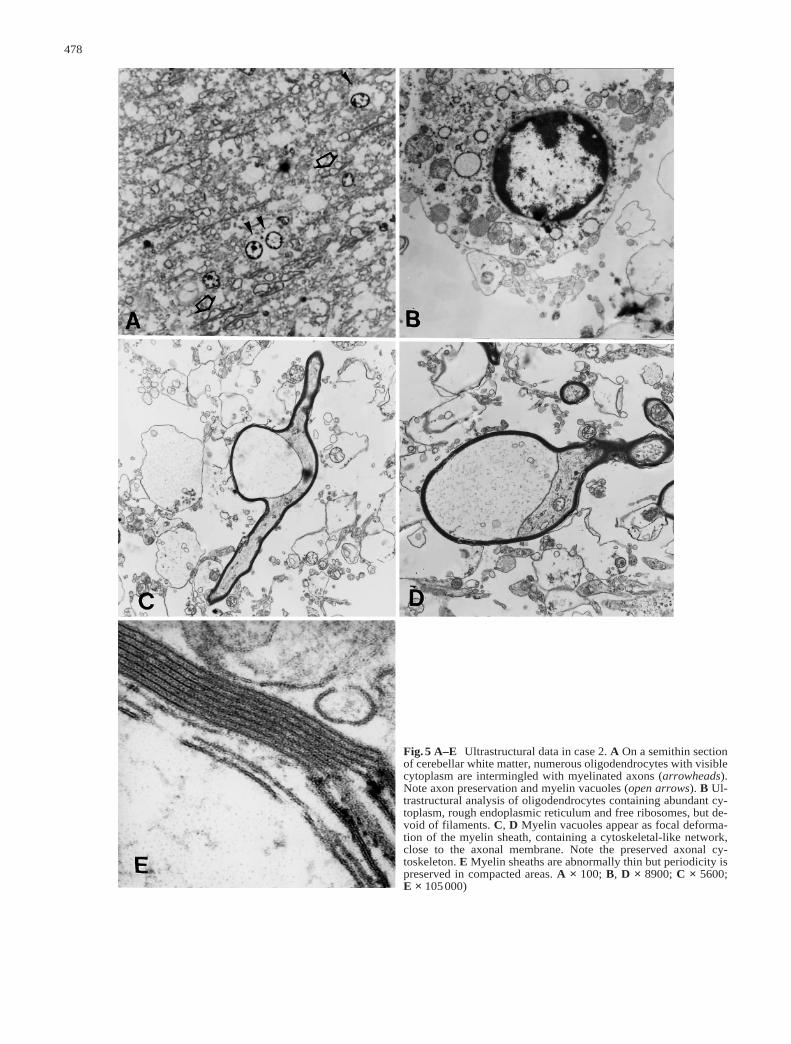
In semithin sections of cerebellar white matter, numer-ous oligodendrocytes with visible cytoplasm were inter-mingled with myelinated axons. In this area, axons werepreserved and myelin vacuoles were present (Fig.5A). Ul-trastructurally, oligodendrocytes were identified by chro-matin lumps, rough endoplasmic reticulum and lack of in-termediate filaments (Fig.5B). In accordance with lightmicroscopic observations, they were abundant in subcor-tical cerebral and cerebellar white matter, and had, for themost part, well-developed cytoplasm containing numer-ous organelles. Myelin sheaths appeared too thin for thediameter of the axons, but periodicity was normal in com-pacted myelin (Fig.5E). Previously observed vacuolescorresponded to focal areas of uncompacted myelin closeto the axonal membrane (Fig.5C, D). Underlying axonshad normal cytoplasm and a well-organized cytoskeleton.Astrocyte cytoplasm contained dense gliofilaments. Thecytoplasmic extensions appeared markedly thick (notshown). In macrophages, membranous debris and lipiddroplets could be identified. No lipofuscin deposits wereseen.
Biochemical and molecular studies
During dissection, the cerebellum of the affected braincontained more water than the control. Total protein andlipid contents in the cerebellum were reduced by about 14and 40%, respectively, in case 1 compared to the control(not shown). Despite severe loss of myelin in the cerebel-lum (around 50%), the specific activity of CGalT was, onaverage, about 30% higher than in the control cerebellum(not shown). Loss of polar lipids in the whole cerebellumand myelin (Table 3) was correlated with the loss ofmyelin. Since an increased number of immature-appear-ing oligodendrocytes are present in rumpshaker mice [7],myelin mutants with a defective PLP [35], we searchedfor a similar PLP mutation in case 1. Western blot analy-sis showed that the amount of PLP was decreased, but itsmolecular weight was normal (not shown). Southern blotanalysis showed a normal restriction pattern of the PLPgene, excluding the presence of a major gene rearrange-ment. The intensity of enzyme-restricted DNA fragmentswas identical to the signal observed with an internal X-chromosome-specific probe control (not shown), indicat-ing that the PLP gene was not duplicated in patient 1. Thesequence of the coding region of the PLP gene was nor-mal.
Discussion
Our two patients presented clinical and MRI featurescompatible with the diagnosis of the CACH syndrome[13, 33, 34, 38]. The neuropathological hallmarks of thisnew leukodystrophy were unusual. In both cases, cavitat-ing orthochromatic leukodystrophy without atrophy wasobserved. The intensity of white matter lesions contrastedmarkedly with the paucity of myelin breakdown productsand the relatively mild astroglial and microglial reactions,except within the pons. In this region, numerous macro-phages contained myelin debris. Surprisingly, in less-af-fected white matter areas, oligodendrocytes were abnor-mally numerous but no significant mitotic activity was de-tected. There were no neuronal lesions, apart from a lossof Purkinje cells. Axons were relatively well preservedexcept in cavitated areas. Upon ultrastructural analysis,myelin sheaths appeared thinner than usual. Compactionwas normal, but was disrupted in places by vacuoles. Bio-chemical analysis showed that myelin components werequalitatively normal but several lipids and proteins werereduced. These neuropathological features correlate withthe clinical course of the disease. Lesions in the telen-cephalic white matter displayed features of old lesions, assuggested by the severity of myelin lesions, the paucity ofmyelin debris and the mild astrocytic and microglial reac-tivity. These features could be related to the progressiveevolution of the disease. Lesions of the pons, however,appeared to be acute and recent, with disappearance ofmyelin correlated to presence of numerous macrophagescontaining myelin debris. Bulbar signs appeared late inthe disease, preceding death. The loss of Purkinje cells
475
Fig.3A–F White matter cells. A, B Cell density in cerebral sub-cortical white matter in the control and in case 1, respectively. CMild fibrillary astrogliosis in hypercellular areas (case 1). D Samemagnification as C showing the increase in GFAP-expression andthe large size of astrocytes in cavitated areas (case 2). Note the en-hanced staining in the perivascular area and the presence of ORO-positive, GFAP-negative cells (arrows). E PGM1-positive mi-croglial cells progressively (top left, subcortical area) becomephagocytes close to cavitated areas (bottom right, central area)(case 2). F PGM1-positive cells are distinct from oligodendroglialcells with small, dark, round nuclei. Some are active phagocytes(arrowheads) (case 2) (Cx cortex, ORO Oil Red O, GFAP glial fib-rillary acidic protein). A, B Cresyl violet staining; C, D GFAP im-munostaining; E, F PGM1 immunostaining. A, B × 6.3; C–E × 10;F × 40
F

476

probably resulted from seizures late in the disease. Thecavitated lesions observed on MRI, proton density andflair images were quite similar to those observed macro-scopically (Fig.1A, B).
The general histological features of leukodystrophy[30], i.e. exclusive white matter involvement, absence ofinflammatory signs, relative axon sparing and thinness ofmyelin sheaths, were observed in our patients. However,reduced brain weight, ventricle enlargement, fibrillary as-trogliosis and loss of oligodendrocytes, all usual in leuko-dystrophies [30], were missing in our patients. The ab-sence of marked astrogliosis, in spite of severe myelinloss, resulting in cavitated lesions which appeared filledby CSF, was unusual. The most prominent histologicalfeature was the increased density of a homogenous cellpopulation with the morphological features of oligoden-drocytes, both in subcortical cerebral white matter sur-rounding cavitated areas and in the less affected regionssuch as cerebellar white matter. These cells were alsoGFAP and PGM1 negative, and CAII positive, confirmingtheir oligodendroglial origin [11]. Some of these oligo-dendrocytes had unusually abundant cytoplasm. Bio-chemical analysis showed that CGalT activity was in-creased. This enzyme is only expressed in oligoden-droglial cells, and its expression is developmentally regu-lated [24]. The increased activity of this enzyme con-trasted with the severe loss of myelin lipids and proteins,and might reflect the immaturity or the increased densityof the oligodendroglial cells.
Oligodendroglial preservation in leukodystrophies hasonly been reported in the early stages of Canavan disease[9], and in some adult cases of pigmentary orthochromaticleukodystrophy [12, 27, 37], where brown pigment accu-mulated in macrophages and glial cells that were autoflu-orescent, orthochromatic and PAS-positive. Neither ofthese diagnoses corresponded to our patients. Orthochro-matic leukodystrophies with cavitation and an increaseddensity of oligodendrocytes were previously reported intwo adults ([10] and case 2 of [39]) and in children ([6]and J-J. Hauw, personal communication), before CACHsyndrome was identified. Neuropathological findings andthe clinical course of childhood cases suggest that theymay have been cases of CACH syndrome. The adult casesraise the possibility that the onset of CACH syndromemay extend into adult life. A primary oligodendrocyte de-fect was evoked in all cases [6, 10, 39]. An increasednumber of cells was previously reported in open-brainbiopsy specimens from two patients with CACH syn-drome [34], but was ascribed to proliferation of astrocytesand microglia. The variability of cell composition in dif-ferent brain areas may have led to an underestimation ofsome glial cell populations, particularly when studieswere restricted to brain biopsy samples. This is the firstreport of increased oligodendrocyte density as a hallmarkof CACH syndrome. The cause of the increase in oligo-dendrocyte density, is a matter of speculation. Oligoden-droglial proliferation due to remyelination, as in multiplesclerosis [4, 22, 25, 31], can be excluded. Hypercellularityin our cases is not correlated with the severity of myelindefect, and no mitotic activity was detected in oligoden-drocytes. An abnormal maturation is suggested by thepresence of oligodendrocytes with abundant, elongatedand organelle-rich cytoplasm, features that are normalduring myelination but which disappear as myelinationprogresses [21]. An increased number of oligodendro-cytes in the myelin mutant rumpshaker mouse has beenattributed to abnormal maturation of oligodendrocytes [7,18, 23], caused by a point mutation in the PLP gene [35].Molecular analysis has, however, excluded a PLP muta-tion in patient 1. Finally, the apoptotic process, that nor-mally reduces the number of oligodendrocytes by 50%during the development [1], could have been defective inthese patients.
Ultrastructural, immunocytochemical and biochemicalstudies showed that the composition and catabolism ofmyelin was normal in our patients. However, the myelinsheaths were too thin and, although compaction was nor-mal, it was disrupted in places by vacuoles. The signifi-cance of the latter is controversial in postmortem tissue,but they have also been described in brain biopsy speci-mens [34]. Whether myelin was initially normal in thesepatients is uncertain. It is clear, however, that demyelina-tion has occurred [38]. It is particularly obvious at thelevel of the pons. The greater severity of the telencephaliclesions in patient 2 compare to patient 1 suggests a pro-gressive loss of myelin, giving rise to cavitated lesions bya secondary axonal loss. Oligodendrocyte maturation, latesteps of myelination, or myelin maintenance may also be
477
Table 2 Cell counts in cases 1 and 2, and in an age-matched con-trol. The results are the mean (SD) of five cell counts in each area
Case 1 Case 2 Control
Hemispheric white matter 1611.3 1495 482.2 (subcortical frontal areas) (52.2) (49.5) (83.4)
Cerebellar white matter 501.7 636.0 360.7(87.1) (18.3) (22.0)
Fig. 4A–I Characteristic features of oligodendrocytes and myelin.A Case 1. Cells in subcortical white matter have small, dark, roundnuclei. Most of them have a large cytoplasm (arrowheads), H&Estaining. B Case 2. Cerebellar white matter double labeled withanti-PGM1 (brown) and anti-GFAP (red). Most of the cells appearunlabeled. C Case 1. The cytoplasm of some oligodendrocytescontains MOG immunoreactivity (arrow). D–F Case 1. Doubleimmunolabeling: CNP (pink) and PGM1 (brown). D Oligodendro-cytes (arrowheads) and macrophages (arrow) are easily identifi-able. Some macrophages (double arrowhead) have an heteroge-neous double-labeled cytoplasm (arrows). E Some of the oligo-dendrocytes have an hypertrophied, homogeneous CNP-positivecytoplasm. F Some of macrophages have an heterogeneousPGM1- and CNP-positive cytoplasm, due to the presence ofmyelin debris. G Case 1. All oligodendrocytes have CAII-positivecytoplasm. H Case 2. Lace-like central network containing mostlynaked axons (arrowheads). The sparse remaining myelinated fiberscontain vacuoles. Note the paucity of cells, LFB-Bodian staining.I Case 2. LFB-Bodian staining shows that vacuoles are in myelin,whereas underlying axons seem preserved (MOG myelin oligo-dendrocyte glycoprotein, CNP cyclic nucleotide phosphodi-esterase, CAII carbonic anhydrase II). A, B, D, H × 40; C, G × 60;E, F, I × 100
F
478
Fig.5 A–E Ultrastructural data in case 2. A On a semithin sectionof cerebellar white matter, numerous oligodendrocytes with visiblecytoplasm are intermingled with myelinated axons (arrowheads).Note axon preservation and myelin vacuoles (open arrows). B Ul-trastructural analysis of oligodendrocytes containing abundant cy-toplasm, rough endoplasmic reticulum and free ribosomes, but de-void of filaments. C, D Myelin vacuoles appear as focal deforma-tion of the myelin sheath, containing a cytoskeletal-like network,close to the axonal membrane. Note the preserved axonal cy-toskeleton. E Myelin sheaths are abnormally thin but periodicity ispreserved in compacted areas. A × 100; B, D × 8900; C × 5600; E × 105000)

involved. Whether these abnormalities result from a de-fect intrinsic to oligodendrocytes, abnormal interactionswith axons or glial cells, or environmental factors re-mained to be elucidated [1, 2, 5, 19, 20]. In conclusion,the neuropathological data reported here support the no-tion that CACH syndrome constitutes a specific entity.Complementary investigations at both morphological,biochemical and molecular level are required for a bettercomprehension of the physiopathogenesis of this syn-drome.
Acknowledgments We would like to thank H. Juguelin and J.-L.Nussbaum for their participation in biochemical studies, Dr. A.Tourbah (CHNodes XV-XX, Paris) for the MRI study, A. Cheva-lier and J. Denni for technical assistance, G. Matteo for preparingthe illustrations, and Dr. M. Ruberg for critical review of themanuscript. This work was supported by grants from the CaisseNationale d’Assurance Maladie – Hôpitaux de Paris (CANAM-HP) to D.R.; the Association Française contre les Myopathies(AFM) to A.G.; the Centre National de la Recherche Scientifique(CNRS), the European Leucodystrophies Association (ELA), theAssociation de Recherche contre la Sclérose en Plaques (ARSEP)and the European Economic Community (Biomed II) to A.D. andD.P.-D., and a fellowship of the European Economic Community(Biomed II) to B.D.G.
References
1.Barres BA, Raff MC (1994) Control of oligodendrocyte num-ber in the developing rat optic nerve. Neuron 12: 935–942
2.Barres BA, Lazar MA, Raff MC (1994) A novel role for thy-roid hormone, glucocorticoids and retinoic acid in timingoligodendrocyte development. Development 120: 1097–1108
3.Birling MC, Roussel G, Nussbaum F, Nussbaum JL (1993)Biochemical and immunohistochemical studies with specificpolyclonal antibodies directed against bovine myelin/oligoden-drocyte glycoprotein. Neurochem Res 18: 937–945
4.Brück W, Schmied M, Suchanek G, Brück Y, Breitschopf H,Poser S, Piddlesden S, Lassmann H (1994) Oligodendrocytesin the early course of multiple sclerosis. Ann Neurol 35: 65–73
5.Canoll D, Mussacchio JM, Hardy R, Reynolds R, MarchionniMA (1996) GGF/Neuregulin is a neuronal signal that promotesthe proliferation and survival and inhibits the differentiation ofoligodendrocytes progenitors. Neuron 17: 229–243
6.Deisenhammer E, Jellinger K (1976) Hehlenbindende Neu-tralfett-leukodystrophie. Mit Schubverlauf. Neuropediatrie 7:111–121
7.Fanarraga ML, Sommer IU, Griffiths IR, Montague P, GroomeNP, Nave KA, Schneider A, Brophy PJ, Kennedy PGE (1993)Oligodendrocyte development and differentiation in the rump-shaker mutation. Glia 9: 146–156
8.Folch J, Lees MB, Sloane-Stanley GH (1957) A simple methodfor the isolation and purification of total lipids from animal tis-sues. J Biol Chem 226: 497–509
9.Friede LR (1989) Developmental neuropathology, 2nd edn.Springer, Berlin Heidelberg New York
10.Gautier JC, Gray F, Awada A, Escourolle R (1984) Leucodys-trophie orthochromatique cavitaire de l’adulte: proliferation etinclusions oligodendrogliales. Rev Neurol (Paris) 140: 493–501
11.Ghandour MS, Langley OK, Vincendon G, Gombos G, FilippiD, Limozin N, Dalmasso D, Laurent G (1980) Immunochemi-cal and immunohistochemical study of carbonic anhydrase II inadult rat cerebellum: a marker for oligodendrocytes. Neuro-science 5: 559–571
12.Gray F, Destée A, Bourre JM, Gherardi R, Krivosic I, Warot P,Poirier J (1987) Pigmentary type of orthochromatic leukodys-trophy (OLD). A new case with ultrastructural and biochemicalstudy. J Neuropathol Exp Neurol 46: 585–596
13.Hanefeld F, Holzbach U, Kruse B, Wilichowski E, Christen H-J, Frahm J (1993) Diffuse white matter disease in three chil-dren: an encephalopathy with unique features on magnetic res-onance imaging and proton magnetic resonance spectroscopy.Neuropediatrics 24: 244–248
14.Heape A, Juguelin H, Fabre M, Boiron F, Garbay B, FournierM, Bonnet J, Cassagne C (1986) Correlation between the mor-phology and the lipid and protein compositions in the periph-eral nervous system of individual 8-day-old normal and trem-bler mice. Dev Brain Res 25: 173–180
15.Heim P, Claussen M, Hoffmann B, Conzelmann E, Gartner J,Harzer K, Hunneman DH, Kohler W, Kurlemann G, Kohl-schutter A (1997) Leukodystrophy incidence in Germany. AmJ Med Genet 71: 475–478
16.Hulette CM, Downey BT, Burger PC (1992) Macrophagemarkers in diagnostic neuropathology. Am J Surg Pathol 92:493–499
17.Laemmli UK (1970) Cleavage of structural proteins during theassembly of the head bacteriophage T4. Nature 227: 680–685
18.Lunn KF, Fanarraga ML, Ducan ID (1995) Myelin mutants:new models and new observations. Microsc Res Tech 32:183–203
19.McKinnon RD, Dubois-Dalcq M (1996) Cytokines and growthfactors in the development and regeneration of oligodendro-cytes. In: Ransohoff RM, Benveniste EN (eds) Cytokines andthe CNS. CRC Press, Boca Raton, pp 85–113
20.McMorris FA, McKinnon RD (1996) Regulation of oligoden-drocyte development and CNS myelination by growth factors:prospects for therapy of demyelinating disease. Brain Pathol 6:313–329
479
Table 3 Polar lipid composi-tion in whole cerebellum andin myelin from the cerebellumof case 1 and the age-matchedcontrol. The results are themean (SD) of three separateexperiments, expressed as the% of total polar lipids. Othervalues represent the average oftwo separate experiments(HFA long chain hydroxy fattyacid, NFA long chain nonhy-droxy fatty acid)
Whole cerebellum Myelin
Case 1 Control Case 1 Control
Sphingomyelin 5.9 11.8 6.8 (0.3) 8.6 (2.1)Phosphatidylcholine 22.0 19.7 11 (0.3) 10.8 (1.2)Phosphatidylserine + inositol 19.0 10.5 12.2 (1.3) 9.6 (1.2)Phosphatidylethanolamine + plasmalogen 38.0 35.7 41.7 (6.9) 46.3 (2.6)HFA-sulfatides 0.8 1.7 1.3 (0.1) 0.9 (0.1)NFA-sulfatides 0.8 1.7 0.6 (0.1) 0.9 (0.1)HFA-cerebrosides 12.2 18.0 21.6 (4.4) 17.3 (2.6)NHFA-cerebrosides 1.0 1.0 5.1 (1.0) 5.7 (1.0)Total phospholipids 79.0 65.9 64.9 66.7Total sphingolipids 20.7 34.2 35.4 33.4

21.Mori S, Leblond P (1970) Electron microscopic identificationof three classes of oligodendrocytes and a preliminary study oftheir proliferative activity in the corpus callosum of young rats.J Comp Neurol 139: 1–30
22.Morris CS, Esiri MM, Sprinkle TJ, Gregson N (1994) Oligo-dendrocyte reactions and cell proliferation markers in humandemyelinating diseases. Neuropathol Appl Neurobiol 20: 272–281
23.Nave KA (1994) Neurological mouse mutants and the genes ofmyelin. J Neurosci Res 38: 607–612
24.Neskovic NM, Rebel G, Harth S, Mandel P (1981) Biosynthe-sis of galactocerebrosides and glucocerebrosides in glial celllines. J Neurochem 37: 1363–1370
25.Norton WT (1996) Do oligodendrocytes divide? NeurochemRes 21: 495–503
26.Norton WT, Poduslo SE (1973) Myelination in rat brain:method of myelin isolation. J Neurochem 21: 749–757
27.Okeda R, Matsuo T, Kawahara Y, Eishi Y, Tamai Y, TanakaM, Kamaki M, Yamadera H (1989) Adult pigment type (Pfeif-fer) of sudanophilic leukodystrophy: pathological and morpho-metrical studies on two autopsy cases of siblings. Acta Neu-ropathol 78: 533–542
28.Pham-Dinh D, Birling M-C, Roussel G, Dautigny A, Nuss-baum J-L (1991) Proteolipid DM-20 predominates over PLP inperipheral nervous system. NeuroReport 2: 89–92
29.Pham-Dinh D, Allinquant B, Ruberg M, Gaspera B della,Dautigny A (1994) Cloning and expression of the humanmyelin/oligodendrocyte glycoprotein cDNA. J Neurochem 63:2353–2356
30.Powers JM (1996) A neuropathological overview of the neu-rodystrophies and neurolipidoses. Handb Clin Neurol 22: 1–32
31.Raine CS, Wu E (1993) Multiple sclerosis: remyelination inacute lesions. J Neuropathol Exp Neurol 52: 199–204
32.Sambrook J, Fritsch EF, Maniatis T (eds) (1989) Molecularcloning: a laboratory manual. Cold Spring Harbor Laboratorypress, Cold Spring Harbor
33.Schiffmann R, Trapp BD, Moller JR, Kaye EM, Parker CC,Brady RO, Barton NW (1992) Childhood ataxia with diffusecentral nervous system hypomyelination. Ann Neurol 32: 484
34.Schiffmann R, Moller JR, Trapp BD, Shih HH-L, Farrer RG,Katz DA, Alger JR, Parker CC, Hauer PE, Kaneski CR, HeissJD, Heiss EM, Kaye EM, Quarles RH, Brady RO, Barton NW(1994) Childhood ataxia with diffuse central nervous systemhypomyelination. Ann Neurol 35: 331–340
35.Schneider A, Montague P, Griffiths I, Fanarraga M, KennedyP, Brophy P, Nave KA (1992) Uncoupling of hypomyelinationand glial cell death by a mutation in the proteolipid proteingene. Nature 358: 758–761
36.Schombach J, Hu KH, Friede RL (1968) Cellular and chemicalchanges during myelination: histologic, autoradiographic, his-tochemical and biochemical data on myelination in the pyrami-dal tract and corpus callosum of rat. J Comp Neurol 134: 21–38
37.Van Bogaert L, Nyssen R (1936) Le type tardif de la leucodys-trophie progressive familiale. Rev Neurol (Paris) 65: 21–45
38.Van der Knaap MS, Barth PG, Gabreëls FJM, Franzoni E,Begeer JH, Stroink H, Rotteveel JJ, Walk J (1997) A newleukoencephalopathy with vanishing white matter. Neurology48: 845–855
39.Watanabe I, Muller J (1967) Cavitating “diffuse sclerosis”. J Neuropathol Exp Neurol 26: 437–455
480
Copyright © 2022 FDOKUMEN




















