
Neuropathological Findings in a Patient with Epilepsy and the Parry-Romberg Syndrome
6
Neuropathological Findings in a Patient with Epilepsy and the Parry–Romberg Syndrome *Javier DeFelipe, ²Tomás Segura, *Jon I. Arellano, *‡Angel Merchán, §Jesús DeFelipe-Oroquieta, \ Pilar Martín, \ Fernando Maestú, ¶Santiago Ramón y Cajal, **Alicia Sánchez, and §Rafael G. Sola *Instituto Cajal, Madrid, ²Department of Neurology, Hospital General de Albacete, ‡Universidad Europea CEES, §Department of Neurosurgery Hospital de la Princesa, \ Facultad de Psicología, Universidad Autónoma, ¶Department of Pathology, Clínica Puerta de Hierro, and **Department of Neurophysiology, Hospital de la Princesa, Madrid, Spain Summary: Purpose: The Parry–Romberg syndrome is an un- usual disorder frequently associated with epilepsy. The origin of this disease, and the cause of epilepsy, are unknown. This study is the first reported case of the Parry–Romberg syndrome, with intractable temporal lobe epilepsy, in which detailed mi- croanatomic analyses have been performed on resected brain tissue obtained after surgical intervention. Methods: Standard histopathologic methods and correlative light and electron microscopy, combined with immunocyto- chemical techniques, were used to study in detail the synaptic microorganization of the resected hippocampal formation. Results: After surgery, the patient was seizure free (follow- up period of 4 years and 7 months). The resected temporal lobe showed a variety of dramatic microanatomic alterations (small groups of ectopic cells, neuronal loss, gliosis, and activated microglial cells) in mesial structures, including the entorhinal cortex, subiculum, and dentate gyrus. At the electron- microscopic level, we found that in the dentate gyrus, the num- ber of synapses in the cell-sparse region adjacent to the ectopic mass of neurons was almost twice that found in the molecular and polymorph cell layers, indicating the intrusion of neuritic processes and synapse formation. In addition, the symmetrical axosomatic synapses characteristically found on granule cells, which are likely derived from g-aminobutyric acid (GABA)er- gic inhibitory basket cells, were not observed. Conclusion: The complete seizure relief after surgery sug- gests that the pacemaker region(s) of seizure activity were within the resected tissue. However, we do not know which of the multiple neuropathologic findings reported here were the primary cause of seizure activity. Nevertheless, the changes found in the dentate gyrus circuitry appear to be among the most important alterations that would lead to epilepsy. Key Words: Epilepsy—Neuronal loss—Gliosis—Hippocampus— Electron microscopy. The Parry-Romberg syndrome (PRS), also known as progressive hemifacial atrophy, is a rare disorder char- acterized by a progressive hemifacial atrophy of the skin and adipose tissue and, in some cases, results in atrophy of muscle, cartilage, and underlying bony structures. The atrophy process commonly appears during the first, or early in the second, decade of life and affects mainly the face, but can extend to the eye, larynx, and brain. The disease was first described by Parry in 1825 (1) and later by Romberg, who detailed the clinical findings in 1846 (2). Romberg named the process “trophoneurosis of the face,” emphasizing the implication of the nervous system in this syndrome. However, there have been only occa- sional reports about neurologic disorders in PRS, mainly those that also had epileptic seizures (3). Some authors have considered PRS a clinical variant of linear sclero- derma and have attributed it to the same autoimmune origin (4), but other explanations have been proposed, such as a regional sympathetic innervation abnormality or an underlying inflammatory process (5). The patho- genic mechanism of the brain lesions is still unknown, because of the scarcity of neuropathological studies con- cerning PRS and because most of these have consisted of gross anatomic observations (6). Even less is known about the relation between PRS and the brain alterations that coexist with the medically intractable epilepsy in some of these patients (3). Here we report the clinical, laboratory, neuroimaging, neuropathologic, and microanatomic features of a patient with PRS in whom medically intractable temporal lobe epilepsy developed; she had relief of seizures after uni- lateral removal of the anterior temporal lobe. Revision accepted June 15, 2001. Address correspondence and reprint requests to Dr. J. DeFelipe at Instituto Cajal (CSIC), Avenida Dr. Arce, 37, 28002-Madrid, Spain. E-mail: [email protected] Epilepsia, 42(9):1198–1203, 2001 Blackwell Science, Inc. © International League Against Epilepsy 1198
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Neuropathological Findings in a Patient with Epilepsy and the Parry-Romberg Syndrome

Neuropathological Findings in a Patient with Epilepsy and theParry–Romberg Syndrome
*Javier DeFelipe, †Tomás Segura, *Jon I. Arellano, *‡Angel Merchán,§Jesús DeFelipe-Oroquieta,\Pilar Martín, \Fernando Maestú, ¶Santiago Ramón y Cajal,
**Alicia Sánchez, and §Rafael G. Sola
*Instituto Cajal, Madrid, †Department of Neurology, Hospital General de Albacete, ‡Universidad Europea CEES, §Departmentof Neurosurgery Hospital de la Princesa,\Facultad de Psicología, Universidad Autónoma, ¶Department of Pathology,
Clínica Puerta de Hierro, and **Department of Neurophysiology, Hospital de la Princesa, Madrid, Spain
Summary: Purpose:The Parry–Romberg syndrome is an un-usual disorder frequently associated with epilepsy. The originof this disease, and the cause of epilepsy, are unknown. Thisstudy is the first reported case of the Parry–Romberg syndrome,with intractable temporal lobe epilepsy, in which detailed mi-croanatomic analyses have been performed on resected braintissue obtained after surgical intervention.
Methods:Standard histopathologic methods and correlativelight and electron microscopy, combined with immunocyto-chemical techniques, were used to study in detail the synapticmicroorganization of the resected hippocampal formation.
Results:After surgery, the patient was seizure free (follow-up period of 4 years and 7 months). The resected temporal lobeshowed a variety of dramatic microanatomic alterations (smallgroups of ectopic cells, neuronal loss, gliosis, and activatedmicroglial cells) in mesial structures, including the entorhinalcortex, subiculum, and dentate gyrus. At the electron-
microscopic level, we found that in the dentate gyrus, the num-ber of synapses in the cell-sparse region adjacent to the ectopicmass of neurons was almost twice that found in the molecularand polymorph cell layers, indicating the intrusion of neuriticprocesses and synapse formation. In addition, the symmetricalaxosomatic synapses characteristically found on granule cells,which are likely derived fromg-aminobutyric acid (GABA)er-gic inhibitory basket cells, were not observed.
Conclusion:The complete seizure relief after surgery sug-gests that the pacemaker region(s) of seizure activity werewithin the resected tissue. However, we do not know which ofthe multiple neuropathologic findings reported here were theprimary cause of seizure activity. Nevertheless, the changesfound in the dentate gyrus circuitry appear to be among themost important alterations that would lead to epilepsy.KeyWords: Epilepsy—Neuronal loss—Gliosis—Hippocampus—Electron microscopy.
The Parry-Romberg syndrome (PRS), also known asprogressive hemifacial atrophy, is a rare disorder char-acterized by a progressive hemifacial atrophy of the skinand adipose tissue and, in some cases, results in atrophyof muscle, cartilage, and underlying bony structures. Theatrophy process commonly appears during the first, orearly in the second, decade of life and affects mainly theface, but can extend to the eye, larynx, and brain. Thedisease was first described by Parry in 1825 (1) and laterby Romberg, who detailed the clinical findings in 1846(2). Romberg named the process “trophoneurosis of theface,” emphasizing the implication of the nervous systemin this syndrome. However, there have been only occa-sional reports about neurologic disorders in PRS, mainly
those that also had epileptic seizures (3). Some authorshave considered PRS a clinical variant of linear sclero-derma and have attributed it to the same autoimmuneorigin (4), but other explanations have been proposed,such as a regional sympathetic innervation abnormalityor an underlying inflammatory process (5). The patho-genic mechanism of the brain lesions is still unknown,because of the scarcity of neuropathological studies con-cerning PRS and because most of these have consisted ofgross anatomic observations (6). Even less is knownabout the relation between PRS and the brain alterationsthat coexist with the medically intractable epilepsy insome of these patients (3).
Here we report the clinical, laboratory, neuroimaging,neuropathologic, and microanatomic features of a patientwith PRS in whom medically intractable temporal lobeepilepsy developed; she had relief of seizures after uni-lateral removal of the anterior temporal lobe.
Revision accepted June 15, 2001.Address correspondence and reprint requests to Dr. J. DeFelipe at
Instituto Cajal (CSIC), Avenida Dr. Arce, 37, 28002-Madrid, Spain.E-mail: [email protected]
Epilepsia,42(9):1198–1203, 2001Blackwell Science, Inc.© International League Against Epilepsy
1198

MATERIALS AND METHODS
Case reportA 30-year-old woman, with normal milestones and
without past familial history of neurologic disease, beganto develop progressive left facial hemiatrophy at age 18years. PRS was diagnosed, and the patient underwentcosmetic surgical treatment with reconstruction of theaffected area. At age 23 years, epileptic seizures started,which consisted principally of brief speech-arrest epi-sodes, fixed expression, and automatic activity for a du-ration of 1 to several minutes (partial complex seizures).Generalized tonic–clonic seizures never occurred. At age28 years, she was diagnosed with a left Fuch’s hetero-chromic iridocyclitis. During the following 2 years, theepileptic disease continued to progress until seizurecontrol could not be achieved with medical treatment(polytherapy). The patient was referred to our epilepsysurgery unit (Hospital de la Princesa) for surgical man-agement. At admission, the neurologic examination wasnormal. The patient showed a facial hemiatrophy (Fig.1A) and heterochromia of the irides. All standard bio-logic tests were normal, including antinuclear antibodiesand rheumatoid factor. Cerebrospinal fluid (CSF) pro-teins were slightly increased (59 mg/100 ml) withoutintrathecal production of immunoglobulins. A magneticresonance imaging (MRI) study demonstrated a largeabnormal cortical area with blurring of the sulci andsubcortical hyperintensity on T2-weighted sequences,that involved the left parietal, occipital, and temporallobes (Fig. 1C). The99mTc-HMPAO single-photon emis-sion computed tomography (SPECT) study of the brain
showed an extensive left parietooccipital hypoperfusionarea (Fig. 1B).
Video-EEG monitoring was recorded from bilateralforamen ovale electrodes. This study showed frequentunilateral multiple spikes and slow-wave discharges onthe left ovale electrode, without clinically detectablechanges in the patient. Four times during the monitoring,the right ovale electrode showed paroxysmal activity(sharp waves between 100 and 200 ms), always after theleft discharges. In these situations, the scalp EEGshowed generalized ictal EEG changes, and the patienthad a partial complex seizure.
In 1996 the patient underwent surgery, and a left tem-poral craniotomy was performed. Surgical procedureconsisted of a tailored anterior temporal cortical resec-tion including the amygdala and the anterior portion ofthe hippocampus and adjacent cortex (leaving intact thesuperior temporal gyrus). After surgery, the patient re-mained seizure free (follow-up period of 4 years,7 months).
Furthermore, pre- and postsurgical psychopathologicaland neuropsychological tests were done after a signedconsent was obtained from the patient. Before surgery,the patient showed a mood disorder with manic featurescontributing to greater vulnerability to affective andanxiety disorders. Intellectual performance was “lownormal” (FIQ 4 80) and showed short- and long-termmemory disorders for verbal material. After surgery, thepersonality and psychological features were improved,with the exception of a decrease in the control of emo-tions and an increase in emotional interference duringcognition. A postsurgical improvement in verbal IQ was
FIG. 1. A: The Parry–Romberg syndromepatient with facial hemiatrophy on the leftside (arrow). B: Axial (upper part) and co-ronal (lower part) 99mTc-HMPAO single-photon emission computed tomography(SPECT) acquisitions showing an exten-sive left parietooccipital hypoperfusionarea. C: Axial (upper part) and coronal(lower part) T2-weighted acquisitions show-ing a large abnormal cortical area with at-rophy and blurring of the sulci that involvedthe left parietal, occipital, and temporallobes. D: Higher magnification of C, show-ing atrophy of the left parahippocampal gy-rus (right arrow). Left arrow indicates thenormal appearance of the parahippocam-pal gyrus in the right side.
PARRY-ROMBERG SYNDROME 1199
Epilepsia, Vol. 42, No. 9, 2001

observed (FIQ4 90), as there was in declarativememory.
Tissue preparationSampled tissues submitted for neuropathologic analy-
sis consisted of pieces of 2 × 3 × 2 cm ofneocortex fromthe anterolateral middle and inferior temporal gyri, and apiece of 2 × 2 × 1.5 cm ofmesial temporal structures,which included the anterior portion of the hippocampalformation (hippocampus, subiculum, and entorhinal cor-tex). They were fixed in 4% paraformaldehyde in phos-phate buffer. H&E and thionin stains were used forbasic histopathologic studies. Immunocytochemistrywas performed according to the indirect peroxidase–antiperoxidase method of Sternberger with the followingantibodies: anti–glial fibrillary acidic protein (GFAP;rabbit polyclonal antibody diluted 1:50; and mousemonoclonal antibody, dilution 1:200) (Sigma, St. Louis,MO, U.S.A.), and for the histocompatibility human leu-cocyte associate-DR (HLA-DR) antigen identified bymonoclonal antibody LN-3 (dilution 1:12; ICN Biomedi-cals Inc., Costa Mesa, CA., U.S.A.). Hippocampal sec-tions were osmicated in 1% osmium tetroxide and thenflat-embedded in Araldite resin. The dentate gyrus andsamples specifically excised from a microarray of het-erotopic neurons (see later) was resectioned at 60–70 nmand analyzed by using a Jeol 1200EX electron micro-scope. Volumetric density of synapses of neuropil in thedentate gyrus was tabulated according to the disectormethod of Sterio.
Tissue sections of normal temporal neocortex and me-sial temporal lobe structures, which were processed asdescribed earlier, were used as controls. This tissue wasobtained at autopsy from normal individuals (a 23-year-old man and a 45-year-old woman) dead in traffic acci-dents, and at biopsy from normal tissue removed duringsurgery for intractable epilepsy to gain access to mesialstructures (from two women, 24 and 28 years old).
RESULTS
Light microscopyNeuropathologic assessment of the resected cortical
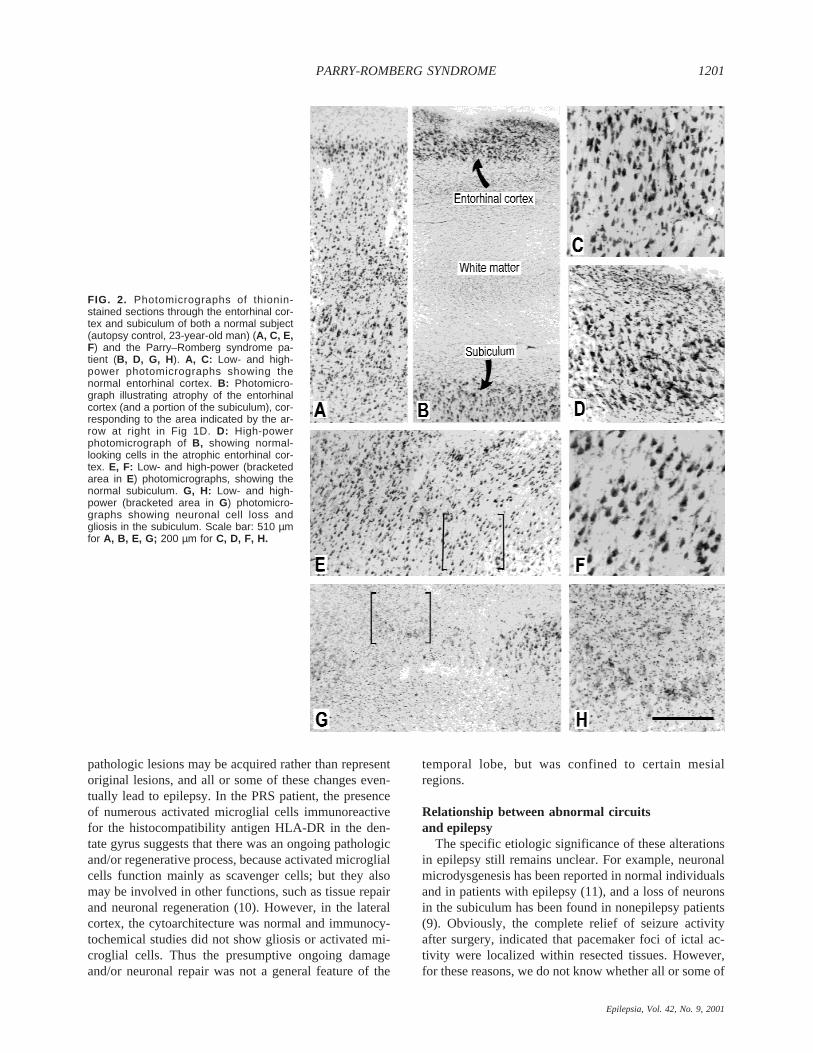
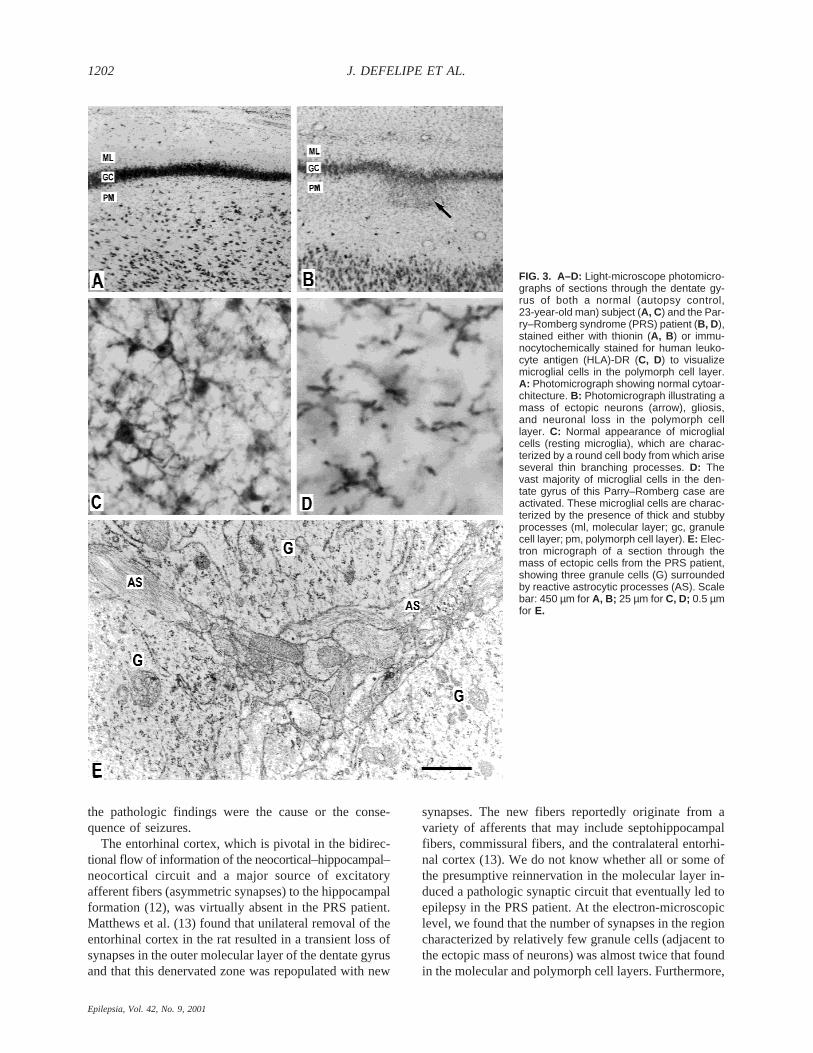
tissue revealed no significant alterations in the lateralneocortex, whereas a variety of pathologic alterationswere found in the mesial temporal structures. These al-terations included (a) a dramatic loss of neurons in theentorhinal cortex, which had only a single layer with afew rows of neurons (Fig. 2A–D) and was reduced toa small segment of a few hundred micrometers long(Fig. 1D); (b) neuronal loss and gliosis in the subiculum(Fig. 2E–H) and in the polymorphic cell layer of thedentate gyrus (Fig. 3A, B); (c) dispersion of the granulecell layer and the presence of small groups of ectopicgranule cells in some regions of the dentate gyrus (Fig.3A, B); (d) presence of numerous hypertrophic astro-
cytes immunoreactives for GFAP, and activated microg-lia strongly immunolabeled by HLA-DR were found inthe dentate gyrus (Fig. 3C, D).
Electron microscopyResults of correlative light and ultrastructural studies
of the focus of heterotopic neurons adjacent to the den-tate gyrus showed granule cells with normal appearance,containing the usual complement of organelles. How-ever, reactive astrocytic processes were frequently foundattached to the granule cell somata (Fig. 3E), and axo-somatic synapses were rarely observed. Quantitative ul-trastructural studies was performed on three neuropilregions, which we labeled regions 1, 2, and 3. Region 1corresponded to a portion of the molecular layer; region2, to an area showing very few granule cells; and region3, to a portion of the polymorph cell layer. In all threeregions, a large number of reactive astrocytic processesand numerous, relatively large dendritic profiles cuttransversally (1.5- to 2.5-mm diameter) were found. Thesynaptic densities were for region 1, 4.88 × 108; region 2,8.30 × 108; and region 3, 5.15 × 108 synapses/mm3 (dataobtained from 10 disectors through neuropil areas of 35mm2 from each region examined). Thus within region 2,synaptic densities were nearly twice those of upper andlower dentate gyrus layers. Furthermore, these synapseswere mostly of the asymmetric type, whereas symmetricsynapses were observed only occasionally. As asymmet-ric synapses are considered to be excitatory, and sym-metric synapses, inhibitory, it is likely that this increasewas due mainly to a sprouting of excitatory axons (7).
DISCUSSION
Some of the cytoarchitectonic changes observed inthis case, or similar alterations, have been associatedwith intractable temporal lobe epilepsy. For example,ectopic neurons and granule cell dispersion in the dentategyrus (8) and neuronal loss in the polymorph cell layer ofthe dentate gyrus and subiculum (9) have all been ob-served in temporal lobe epilepsy patients. However,other alterations, such as marked neuronal loss in theentorhinal cortex, are not typically found in these pa-tients (but see ref. 7). Furthermore, we do not knowwhether the multiple pathologic tissues found in the pres-ent study were congenital, acquired, or induced by PRS.
Unfortunately, no neuroimaging studies were per-formed before the onset of epilepsy. Therefore, the rela-tion between the development of brain abnormalities,epilepsy, and hemifacial atrophy could not be estab-lished. Epilepsy in the present PRS patient was initiatedlate, at age 23 years, 5 years after the beginning of pro-gressive development of left-facial hemiatrophy. Thissuggests the possibility that the neuropathologic findingswere part of an ongoing process, that led to reorganiza-tion of the previously undamaged temporal lobe; that is,
J. DEFELIPE ET AL.1200
Epilepsia, Vol. 42, No. 9, 2001
pathologic lesions may be acquired rather than representoriginal lesions, and all or some of these changes even-tually lead to epilepsy. In the PRS patient, the presenceof numerous activated microglial cells immunoreactivefor the histocompatibility antigen HLA-DR in the den-tate gyrus suggests that there was an ongoing pathologicand/or regenerative process, because activated microglialcells function mainly as scavenger cells; but they alsomay be involved in other functions, such as tissue repairand neuronal regeneration (10). However, in the lateralcortex, the cytoarchitecture was normal and immunocy-tochemical studies did not show gliosis or activated mi-croglial cells. Thus the presumptive ongoing damageand/or neuronal repair was not a general feature of the
temporal lobe, but was confined to certain mesialregions.
Relationship between abnormal circuitsand epilepsy
The specific etiologic significance of these alterationsin epilepsy still remains unclear. For example, neuronalmicrodysgenesis has been reported in normal individualsand in patients with epilepsy (11), and a loss of neuronsin the subiculum has been found in nonepilepsy patients(9). Obviously, the complete relief of seizure activityafter surgery, indicated that pacemaker foci of ictal ac-tivity were localized within resected tissues. However,for these reasons, we do not know whether all or some of
FIG. 2. Photomicrographs of thionin-stained sections through the entorhinal cor-tex and subiculum of both a normal subject(autopsy control, 23-year-old man) (A, C, E,F) and the Parry–Romberg syndrome pa-tient (B, D, G, H). A, C: Low- and high-power photomicrographs showing thenormal entorhinal cortex. B: Photomicro-graph illustrating atrophy of the entorhinalcortex (and a portion of the subiculum), cor-responding to the area indicated by the ar-row at right in Fig 1D. D: High-powerphotomicrograph of B, showing normal-looking cells in the atrophic entorhinal cor-tex. E, F: Low- and high-power (bracketedarea in E) photomicrographs, showing thenormal subiculum. G, H: Low- and high-power (bracketed area in G) photomicro-graphs showing neuronal cell loss andgliosis in the subiculum. Scale bar: 510 µmfor A, B, E, G; 200 µm for C, D, F, H.
PARRY-ROMBERG SYNDROME 1201
Epilepsia, Vol. 42, No. 9, 2001
the pathologic findings were the cause or the conse-quence of seizures.
The entorhinal cortex, which is pivotal in the bidirec-tional flow of information of the neocortical–hippocampal–neocortical circuit and a major source of excitatoryafferent fibers (asymmetric synapses) to the hippocampalformation (12), was virtually absent in the PRS patient.Matthews et al. (13) found that unilateral removal of theentorhinal cortex in the rat resulted in a transient loss ofsynapses in the outer molecular layer of the dentate gyrusand that this denervated zone was repopulated with new
synapses. The new fibers reportedly originate from avariety of afferents that may include septohippocampalfibers, commissural fibers, and the contralateral entorhi-nal cortex (13). We do not know whether all or some ofthe presumptive reinnervation in the molecular layer in-duced a pathologic synaptic circuit that eventually led toepilepsy in the PRS patient. At the electron-microscopiclevel, we found that the number of synapses in the regioncharacterized by relatively few granule cells (adjacent tothe ectopic mass of neurons) was almost twice that foundin the molecular and polymorph cell layers. Furthermore,
FIG. 3. A–D: Light-microscope photomicro-graphs of sections through the dentate gy-rus of both a normal (autopsy control,23-year-old man) subject (A, C) and the Par-ry–Romberg syndrome (PRS) patient (B, D),stained either with thionin (A, B ) or immu-nocytochemically stained for human leuko-cyte antigen (HLA)-DR (C, D) to visualizemicroglial cells in the polymorph cell layer.A: Photomicrograph showing normal cytoar-chitecture. B: Photomicrograph illustrating amass of ectopic neurons (arrow), gliosis,and neuronal loss in the polymorph celllayer. C: Normal appearance of microglialcells (resting microglia), which are charac-terized by a round cell body from which ariseseveral thin branching processes. D: Thevast majority of microglial cells in the den-tate gyrus of this Parry–Romberg case areactivated. These microglial cells are charac-terized by the presence of thick and stubbyprocesses (ml, molecular layer; gc, granulecell layer; pm, polymorph cell layer). E: Elec-tron micrograph of a section through themass of ectopic cells from the PRS patient,showing three granule cells (G) surroundedby reactive astrocytic processes (AS). Scalebar: 450 µm for A, B; 25 µm for C, D; 0.5 µmfor E.
J. DEFELIPE ET AL.1202
Epilepsia, Vol. 42, No. 9, 2001

these synapses were mostly of the asymmetric type,which are excitatory in function (7). This suggests theintrusion of neuritic processes and excitatory synapseformation. In addition, axosomatic symmetric synapsesnormally found on granule cells, which are likely derivedfrom g-aminobutyric acid (GABA)ergic inhibitory bas-ket cells (14), were not observed, suggesting that basketcells also were affected. In conclusion, the increaseddensity of synapses adjacent to the ectopic mass of neu-rons, together with the loss of axosomatic putatively in-hibitory terminals, may have led to hyperexcitatorysynaptic circuits and seizure activity. However, there area number of other hypotheses that explain hyperexcit-ability in epileptic tissue (15), and the multiple patholo-gies found in the present study preclude us fromestablishing an obvious etiology for seizures in this pa-tient.
Further neuropsychological data and methods areavailable from http://www.cajal.csic.es/romberg/neuropsychology.htm. The complete methods for pro-cessing of tissue and details on control tissue areavailable from http://www.cajal.csic.es/romberg/methods.htm
Acknowledgment: We are very grateful to the patient forher collaboration and authorization to use the data. We alsothank Guy Elston for critically reading the manuscript, Azu-cena Ortiz for technical assistance, and Steven Jones for edi-torial assistance. This work was supported by FIS grant 98/0933, DGCYT grant PM99-0105, Comunidad de Madrid grants0.8.5/0023/1999 and 08.5/0036/2000, and a Basque Countrygrant to J.I.A.
REFERENCES1. Parry CH.Collections from unpublished papers.London: Unter-
wood, 1825.
2. Romberg MH. Trophoneurosen. In:Klinische ErgebnisseBerlin:Forstner, 1846:75–81.
3. Wolf SD, Verity MA. Neurological complications of progressivefacial hemiatrophy. J Neurol Neurosurg Psychiatry1974;37:997–1004.
4. García de la Torre I, Castello SJ, Esgleyes RT, et al. Autoantibod-ies in Parry-Romberg syndrome: a serologic study of 14 patients.J Rheumatol1995;22:73–7.
5. Terstegge K, Kunath B, Felber S, et al.of brain involvement inprogressive facial hemiatrophy (Romberg disease): reconsiderationof a syndrome. Am J Neuroradiol1994;15:145–50.
6. Dupont S, Catala M, Hasboun D, et al. Progressive facial hemiat-rophy and epilepsy: a common underlying dysgenetic mechanism.Neurology1997;48:1013–8.
7. Houser CR. Neuronal loss and synaptic reorganization in temporallobe epilepsy. In: Delgado-Escueta AV, Wilson WA, Olsen RW,et al. eds.Jasper’s basic mechanisms of the epilepsies: advancesin Neurology. Philadelphia: Lippincott Williams and Wilkins,1999:79:743–61.
8. Houser CR. Granule cell dispersion in the dentate gyrus of humanswith temporal lobe epilepsy.Brain Res1990;535:195–204.
9. Babb TL, Brown WJ. Pathological findings in epilepsy. In: EngelJ Jr, ed. Surgical treatment of the epilepsies.New York: RavenPress, 1987:511–40.
10. Kreutzberg GW. Microglia: a sensor for pathological events in theCNS.Trends Neurosci 1996;19:312–8.
11. Kaufmann WE, Galaburda AM. Cerebrocortical microdysgenesisin neurologically normal patients.Neurology1989;39:238–44.
12. Witter MP. Organization of the entorhinal-hippocampal system:a review of current anatomical data. Hippocampus1993;3:33–44.
13. Matthews DA, Cotman C, Lynch G. An electron microscopic studyof lesion-induced synaptogenesis in the dentate gyrus of the adultrat, II: reappearance of morphologically normal synaptic contacts.Brain Res1976b;115:23–41.
14. Ribak CE, Seress L. Five types of basket cells in the hippocampaldentate gyrus: a combined Golgi and electron microscope study.J Neurocytol1983;12:577–97.
15. Bernard C, Cossart R., Hirsch JC, te al. What is GABAergic in-hibition? How is it modified in epilepsy? Epilepsia2000;41 (suppl6):S90–5.
PARRY-ROMBERG SYNDROME 1203
Epilepsia, Vol. 42, No. 9, 2001




















