Progress in Mapping Human Epilepsy Genes
12
Epi/ep.siu, 35(Suppl. I):S29-S40. I994 Raven Press, Ltd.. New York 0 International League Against Epilepsy Progress in Mapping Human Epilepsy Genes *?$Antonio V. Delgado-Escueta, *?$Jose M. Serratosa, *?$Amy Liu, §Karen Weissbecker, *$/(Marc0 T. Medina, *?Manyee Gee, *?$Lucy J. Treiman, and *lTRobert S. Sparkes *Cal$ornia Comprehensive Epilepsy Program, tNeurology and Research Services, West Los Angeles VeteransAflairs Medical Center, $Department of Neurology and YDivision of Medical Genetics, Department of Medicine, UCLA School of Medicine. Los Angeles, California; $Biometry and Genetics. Louisiana State University, U.S.A.; and 11 National Autonomous University of Honduras, Tegucigalpa, Honduras Summary: The chromosomal loci for seven epilepsy genes have been identified in chromosomes Iq, 6p, 8q, 16p, 20q, 21q, and 22q. In 1987, the first epilepsy locus was mapped in a common benign idiopathic generalized epilepsy syn- drome, juvenile myoclonic epilepsy (JME). Properdin factor or Bf, human leukocyte antigen (HLA), and DNA markers in the HLA-DQ region were genetically linked to JME and the locus, named EJMI, was assigned to the short arm of chromosome 6. Our latest studies, as well as those by White- house et al., show that not all families with JME have their genetic locus in chromosome 6p, and that childhood absence epilepsy does not map to the same EJMl locus. Recent re- sults, therefore, favor genetic heterogeneity for JME and for the common idiopathic generalized epilepsies. Heterogeneity also exists in benign familial neonatal convulsions, a rare form of idiopathic generalized epilepsy. Two loci are now recognized; one in chromosome 20q (EBNI) and another in chromosome 8q. Heterogeneity also exists for the broad group of debilitating and often fatal progressive myoclonus epilep- sies (PME). The gene locus (EPMI) for both the Baltic and Mediterranean types of PME or Unvemcht-Lundborg dis- ease is the same and is located in the long arm of chromosome 21. Lafora type of PME does not map to the same EPMI locus in chromosome 2 1. PME can be caused by the juvenile type of Gaucher’s disease, which maps to chromosome Iq, by the juvenile type of neuronal ceroid lipofuscinoses (CLN3), which maps to chromosome 16p, and by the ‘.‘cherry-red- spot-myoclonus” syndrome of Guazzi or sialidosis type I, which has been localized to chromosome 10. A point mu- tation in the mitochondria1 tRNALyS coding gene can also cause PME in children and adults (MERIT). Key Words: Benign familial neonatal convulsion-Chromosome 6p- Epilepsy-juvenile myoclonic- I -Gene mapping-Progressive myoclonic epilepsy. Currently, the focus of research on the genetics of the epilepsies is the identification of mutations causing epilepsies; the manner in which these epilepsy muta- tions are perpetuated, and the abnormal properties of the neuron glia system through which the mutations are expressed and result in clinical epilepsy. Such re- search seeks to explain, in molecular terms, the entire series of events by which an epilepsy genotype is con- verted into the clinical seizure phenotype. Five ques- tions can be asked: (a) What types of genetic mutations can lead to epilepsy? (b) How would such mutations be inherited and expressed? (c) Are cellular structures of neuron, synapses, and glia (including blood-brain bamer) self-assembled by means of the “code script” information inherent in the mutated sequences of the Address correspondenceand reprint requests to Dr. A. V. Delgado- Escueta at West Los Angeles DVA Medical Center. Wilshire and Sawtelle Boulevards, Room 3405, Los Angeles, CA 90073, U S A . epilepsy protein? (d) Are there other secondarily af- fected components produced by the mutated code script (so-called “downstream lesions”)? (e) In epilepsy and during seizures, what type of information might be responsible for the development of differences be- tween cells during embryogenesis? Various approaches by different investigations are being used to answer these questions. One approach that has provided some glimmer of hope to answer these questions is mapping of epilepsy genes. Seven separate epilepsy genes have now been chromosomally localized (Table 1). Among the benign idiopathic gen- eralized epilepsies are juvenile myoclonic epilepsy (JME)-one gene locus termed epilepsy-juvenile myo- clonic- 1 or EJM I is in chromosome 6p (Greenberg et al., 1987, 1988; Delgado-Escueta et al., 1989, 1990; Durner et al., 1991; Weissbecker et al., 1991); and be- nign familial neonatal convulsion (BFNC) for which two gene loci have been identified: EBNl in chromo- S29
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Progress in Mapping Human Epilepsy Genes

Epi/ep.siu, 35(Suppl. I):S29-S40. I994 Raven Press, Ltd.. New York 0 International League Against Epilepsy
Progress in Mapping Human Epilepsy Genes
*?$Antonio V. Delgado-Escueta, *?$Jose M. Serratosa, *?$Amy Liu, §Karen Weissbecker, *$/(Marc0 T. Medina, *?Manyee Gee, *?$Lucy J. Treiman, and *lTRobert S. Sparkes
*Cal$ornia Comprehensive Epilepsy Program, tNeurology and Research Services, West Los Angeles Veterans Aflairs Medical Center, $Department of Neurology and YDivision of Medical Genetics, Department of Medicine, UCLA School
of Medicine. Los Angeles, California; $Biometry and Genetics. Louisiana State University, U.S.A.; and 11 National Autonomous University of Honduras, Tegucigalpa, Honduras
Summary: The chromosomal loci for seven epilepsy genes have been identified in chromosomes Iq, 6p, 8q, 16p, 20q, 21q, and 22q. In 1987, the first epilepsy locus was mapped in a common benign idiopathic generalized epilepsy syn- drome, juvenile myoclonic epilepsy (JME). Properdin factor or Bf, human leukocyte antigen (HLA), and DNA markers in the HLA-DQ region were genetically linked to JME and the locus, named EJMI, was assigned to the short arm of chromosome 6. Our latest studies, as well as those by White- house et al., show that not all families with JME have their genetic locus in chromosome 6p, and that childhood absence epilepsy does not map to the same EJMl locus. Recent re- sults, therefore, favor genetic heterogeneity for JME and for the common idiopathic generalized epilepsies. Heterogeneity also exists in benign familial neonatal convulsions, a rare form of idiopathic generalized epilepsy. Two loci are now recognized; one in chromosome 20q (EBNI) and another in
chromosome 8q. Heterogeneity also exists for the broad group of debilitating and often fatal progressive myoclonus epilep- sies (PME). The gene locus (EPMI) for both the Baltic and Mediterranean types of PME or Unvemcht-Lundborg dis- ease is the same and is located in the long arm of chromosome 21. Lafora type of PME does not map to the same EPMI locus in chromosome 2 1. PME can be caused by the juvenile type of Gaucher’s disease, which maps to chromosome Iq, by the juvenile type of neuronal ceroid lipofuscinoses (CLN3), which maps to chromosome 16p, and by the ‘.‘cherry-red- spot-myoclonus” syndrome of Guazzi or sialidosis type I, which has been localized to chromosome 10. A point mu- tation in the mitochondria1 tRNALyS coding gene can also cause PME in children and adults (MERIT). Key Words: Benign familial neonatal convulsion-Chromosome 6p- Epilepsy-juvenile m yoclonic- I -Gene mapping-Progressive myoclonic epilepsy.
Currently, the focus of research on the genetics of the epilepsies is the identification of mutations causing epilepsies; the manner in which these epilepsy muta- tions are perpetuated, and the abnormal properties of the neuron glia system through which the mutations are expressed and result in clinical epilepsy. Such re- search seeks to explain, in molecular terms, the entire series of events by which an epilepsy genotype is con- verted into the clinical seizure phenotype. Five ques- tions can be asked: (a) What types of genetic mutations can lead to epilepsy? (b) How would such mutations be inherited and expressed? (c) Are cellular structures of neuron, synapses, and glia (including blood-brain bamer) self-assembled by means of the “code script” information inherent in the mutated sequences of the
Address correspondence and reprint requests to Dr. A. V. Delgado- Escueta at West Los Angeles DVA Medical Center. Wilshire and Sawtelle Boulevards, Room 3405, Los Angeles, CA 90073, USA.
epilepsy protein? (d) Are there other secondarily af- fected components produced by the mutated code script (so-called “downstream lesions”)? (e) In epilepsy and during seizures, what type of information might be responsible for the development of differences be- tween cells during embryogenesis?
Various approaches by different investigations are being used to answer these questions. One approach that has provided some glimmer of hope to answer these questions is mapping of epilepsy genes. Seven separate epilepsy genes have now been chromosomally localized (Table 1). Among the benign idiopathic gen- eralized epilepsies are juvenile myoclonic epilepsy (JME)-one gene locus termed epilepsy-juvenile myo- clonic- 1 or EJM I is in chromosome 6p (Greenberg et al., 1987, 1988; Delgado-Escueta et al., 1989, 1990; Durner et al., 1991; Weissbecker et al., 1991); and be- nign familial neonatal convulsion (BFNC) for which two gene loci have been identified: EBNl in chromo-
S29

S30 A. V. DELGADO-ESCUETA ET AL.
TABLE 1. Epilepsy genes
Epilepsies Chromosome Authors/yrs
Idiopathic generalized epilepsies Juvenile myoclonic epilepsy (EJM I )
Juvenile myoclonic epilepsy (possible EJM2) Benign familial neonatal convulsions (EBN I ) Benign familial neonatal convulsions (possible EBN2) Childhood absence epilepsy (possible ECAI)
Unverricht-Lundborg type (EPM 1) (Baltic) Unverricht-Lundborg type (EPM I ) (Mediterranean) Juvenile type of neuronal ceroid lipofuscinoses
Juvenile Gaucher's disease "Cherry-red-spot-myoclonus" syndrome or sialidosis
Myoclonus epilepsy and ragged-red fibers
Progressive myoclonus epilepsies
(CLN3)
type I
Greenberg et al., 1987. 198863 Delgado-Escueta et al., 1989. 1990: Durner et al.. 1991: Weissbecker et al., 1991
Liu et al., 1992, 1993: Whitehouse et al., 1993 Leppert et al.. 1989: Malafosse et al.. 1992 Lewis et al.. 1993: Ryan, 1990. 1993 Serratosa et al.. 1993
Lehesjoki et al., I99 I . I992 Malafosse et al.. 1 9 9 2 ~ Gardiner et al.. 1990
Barneveld et al.. 1983 Mueller et al., 1985
Shoffner et al.. 1990
some 20q (Leppert et al., 1989; Malafosse et al., 1992~) and EBN2 in chromosome 8q (Ryan et al., 1990; Ryan 1993; Lewis et al., 1993). Among the debilitating and often fatal progressive myoclonus epilepsies (PME) are Baltic-Mediterranean or Unvemcht-Lundborg (EPMI) mapped to chromosome 21q (Lehesjoki et al., 199 1, 1992: Malafosse et al., 19926); the juvenile type of Gaucher's disease mapped to chromosome 1 q (Bar- neveld et al., 1983): juvenile ceroid lipofucsinoses (CLN-3) in chromosome 16p (Gardiner et al., 1990): and the "cherry-red-spot-myoclonus" syndrome of Guazzi or sialidosis type I mapped to chromosome 10 (Mueller et al., 1985).
IDIOPATHIC GENERALIZED EPILEPSIES
The most common forms of idiopathic generalized epilepsies are JME, childhood absence epilepsy (CAE), and epilepsy with grand ma1 seizures on awakening (EGMA). JME is estimated to account for -10% of all epilepsies: 4.3% according to Janz (1985), 1 1.4% according to Wolf and Goosses (1986), and 3-4% ac- cording to Asconape and Penry ( 1984) and Loiseau et al. ( 1990). Early epidemiologic studies calculated that
23% of all epilepsies involve patients who present ex- clusively with generalized tonic-clonic seizures (GTCS) (Janz, 1969), whereas absence with or without grand ma1 appear in 6.5% ofall epileptic patients (Janz, 1969). Other clinical studies have given childhood absence epilepsy a prevalence of 7.5% of all epilepsy patients (Loiseau et al., 1990). Epilepsy with grand ma1 seizures on awakening, with or without other types of seizures, accounts for -30% of epilepsies, according to Janz ( 1985). However, EGMA, which has tonic-clonic grand ma1 as the only type of seizure, affected only 1.2% of the patients in a recent series by Loiseau et al. (1990). Table 2 shows the percentage of the common forms of idiopathic generalized epilepsies found in four different series.
JME JME (Janz, 1969, 1985; Asconape and Penry, 1984;
Delgado-Escueta and Enrile-Bascal, 1984) (Herpin- Janz syndrome) is a specific and common form of nonprogressive idiopathic generalized epilepsy char- acterized by adolescent onset (mean age of onset is 14.2 years; range 8-30 years) myoclonias, tonic-clonic or clonic tonic-clonic convulsions and absence sei-
TABLE 2. Percentage of common idiopathic generalized epilepsies
Author/yr (data base) JME CAE JAE EGMA Total ICE (%) (%I
- Janz 1969, 1985 (6,500 patients) 4.3 7.8 3 Tsuboi and Christian, 1973 (466 patients) 7. I 15" 15" 31.5 Wolf and Goosses, I986 ( 1.044 patients) 11.4 8.9 I 7.5 25.4 Loiseau et al., 1990 (986 patients) 3.3 7.5 2.4 1.2
- 53 29
JME, juvenile myoclonic epilepsy; CAE, childhood absence epilepsy: JAE, juvenile absence epilepsy: EGMA, epilepsy with grand ma1 on awakening; ICE, idiopathic generalized epilepsies.
Includes all absence except those of Lennox-Gastaut syndrome and psychomotor seizures.
Epilepsia. Vol. 35. Suppl. I. 1994

MAPPING HUMAN EPILEPSY GENES S3 1
zures. As mentioned earlier, JME accounts for -3- 1 1% of all epilepsies and is the most common cause of idiopathic GTCS in adolescents and young adults. Myoclonic and tonic-clonic seizures occur mainly just after awakening and are triggered by lack of sleep, fa- tigue, and alcohol. Ictal electroencephalograms (EEGs) show high-amplitude multispikes followed by slow waves during myoclonias and interictal EEGs manifest 3.5- to 6-Hz diffuse multispike wave complexes. EGMA is an overdiagnosed syndrome. In many patients, myoclonias are recognized only after repeated interview or documentation of seizures on closed-circuit televi- sion and EEG (CCTV-EEG) recordings. A large pro- portion of patients referred to our clinic with a diagnosis of EGMA had myoclonic jerks or absence seizures pre- ceding or independent of tonic-clonic seizures on CCTV-EEG. Many were not aware of and denied the jerks or absence seizures in the initial interview. Myo- clonic jerks are the only seizure type in 5% of JME patients. The prevalence of myoclonic seizures as the only expression of the JME syndrome in the general population is difficult to ascertain, because the majority of these patients may interpret these jerks as clumsiness, nervousness, or other complaints (Table 3). The prev- alence of JME is probably greater than is generally estimated.
Although considered a single clinical syndrome, sev- eral lines of evidence suggest genotypic heterogeneity in JME: (a) there is phenotypic variability in probands, (b) clinical and EEG affectedness of family members in pedigrees of JME suggest different family sub- types, (c) segregation analysis, and (d) genetic linkage mapping.
Clinical variability in probands and eight .family subtypes suggest heterogeneity in JME
Variable features between probands were the first signs suggesting possible heterogeneity. We studied 1 I5 patients with JME. All 1 I5 patients have myoclonias,
TABLE 3. Common symploms or cornplaints used by juvenile myoclonic epilepsy patienls
to explain myoclonic jerks
Nervousness of hands or legs Ataxia (impaired gait) Tics (nervous habits) Slurred speech (jerks of mouth and tongue muscles) Unilateral jerks as focal seizures (especially during antiepileptic
Frequent sighs and deep breathing Early-morning nervousness (need the first morning hours to get
Difficulty eating, chewing, and swallowing
drug treatment)
steady)
Modifed from Lee et al. (1987).
whereas 95% had GTCS and 5% had myoclonias only. One-third of patients also manifest absence of child- hood or juvenile onset. These patients were considered to have “classical JME.” In addition to these common features of JME, we observed sudden drops in six pa- tients and only rare myoclonias in another eight pa- tients. These latter patients were classified by us as hav- ing “non-classical JME.”
We also observed different clinical features among families of patients with classic JME. We observed eight possible subtypes of families based on the affectedness of the family members. Most commonly, classical JME is present in affected nonproband family members (47 of I I5 pedigrees). In 25 families, affected family mem- bers only have GTCS. In another 2 I families, absences ofjuvenile or childhood or early childhood onset were found in affected family members some of whom pre- sented with GTCS. In six pedigrees, clinically asymp- tomatic siblings had abnormal EEGs. Specific diffuse 3.5- to 6-Hz multispike wave complexes were present in these asymptomatic siblings of six pedigrees. Non- specific epileptiform patterns (diffuse irregular 3-7 Hz spike and sharp wave formations) werc present in non- proband members of another two families. Based on the combinations of affectedness in family members, we divided families into eight subgroups (Table 4).
Segregation analysis favors autosomal dominant transmission with incomplete penetrance and sporadic cues or heterogeneity in JME
Of I 15 families, we completed validation of clinical and EEG traits in 249 nonproband family members of 52 families originally collected from ‘our epilepsy clinics. Of these 52 families, 48 were used for segre- gation analyses. In the other four families, the proband was an only child, and therefore the pedigree provided no information for segregation analysis. Families were ascertained through a single patient with JME. These JME patients had been referred for diagnosis and treat- ment. Table 5 summarizes the data base and the af- fectedness of parents and siblings in these 48 families.
Because every sibship had been ascertained through only one proband, we initially estimated the proportion of affected siblings or segregation ratio @2 for each mating group by using the proband method described by Weinberg in I9 12 to correct for single ascertain- ment. We are presently performing complex analysis using the SAGE computer program package. The as- certainment method was unambiguously single selec- tion.
We examined which mode of transmission could best explain most, if not all, of the information we obtained from the nuclear families of our 48 sibships (Delgado-
Epilepsiu, L’ol. 35. Suppl. I . 1994

S32 A. V. DELGADO-ESCUETA ET AL.
TABLE 4. Siih1jpr.s of I15 JhifE,furnilie.s“: bawd on ulli’c.tc’clness of’pohund und.fumi1.v member.$
No. of enrolled Subtype no. Description of family subtypes fam i I ies
(A) Proband with “classical JME” I
Ila
Ilb
111 IV
(B) Proband with “nonclassical JME” V
VI
VII
V l l l
Total
Proband and affected family members have JME: MS + GTCS + absence, adolescent onset in all affected family members
Proband has JME and asymptomatic sibling has EEG 3.5- to 6-Hz multispike wave complexes
Proband has JME and asymptomatic sibling(s) has EEG nonspecific epileptiform patterns
Proband has JME but affected family members have GTCS Proband has JME but affected family members have CAE (6) or ECAE
( I ) or JAE (15) with or without GTCS
Proband has CAE with GTCS; during adolescence rare MS appear,
Proband has JAE, GTCS. but rare MS at adolescence; CAE or GTCS in
Proband has GTCS and rare adolescent onset MS; CAE or GTCS in
Proband has GTCS. MS. and adolescent-onset drop attacks; CAE or
CAE or GTCS is present in affected family members
family members
family members
GTCS in family members
47
6
2
25 21
3
2
3
6
I I5
MS, myoclonic seizures; JME, juvenile myoclonic epilepsy; CAE, childhood absence epilepsy: JAE. juvenile absence epilepsy; ECAE. early childhood absence epilepsy; GTCS, generalized tonic-clonic seizures. Nonspecific EEG patterns are irregular 3-Hz and 4- to 7-Hz spike and slow wave formations, 4- to 7-Hz sharp and slow wave formations, and rhythmic 6- to 12-Hz paroxysms mixed with spikes.
Only 52 families, so far, have had EEGs in all nuclear members. The EEGs of 78 families are now being completed.
TABLE 5. Dufu buse und busis ,j>r clussifving uf2ctedncw during segregulion anu1wi.s
No. of families Clinically affected EEG Family classification
21 8
Proband only Proband and sibling
6 Proband only
2 [ ] * I Proband only
4
9
Proband and one parent
Proband, one parent and sibling
Proband only
Proband only
Proband and sibling
37 Normal by normal matings
Normal EEGs on all members, except proband Proband and one sibling have 3.5- to 6-Hz
Proband and one sibling have 3.5- to 6-Hz
Proband and one sibling have nonspecific
multispike wave complexes
multispike wave complexes
epileptiform EEG proxysms
16 Normal by affected matings
Normal EEGs on all members except proband
Proband and sibling have 3.5- to 6-Hz multispike wave complexes
One parent has nonspecific epileptiform EEG paroxysms
One parent and sibling have nonspecific epileptiform EEG paroxysms
One parent has nonspecific epileptiform EEG paroxysms
Simplex by clinical and EEG Multiplex clinically and by
specific EEG pattern Multiplex by specific EEG
paroxysms only Multiplex by nonspecific
EEG paroxysms only
Multigenerational clinically
Multigenerational and multiplex clinically and by specific EEG paroxysms
Multigenerational by nonspecific EEG paroxysms
Multigenerational and multiplex by nonspecific EEG paroxysms
Multigenerational by nonspecific EEG paroxysms and multiplex by clinical
Total: 48 of 52 families were used for segregation analysis; 4 families were not used because there were no offspring aside from the proband. “One of these two pedigrees is represented under both mating types (they are the same pedigree).
These families would be classified as simplex and normal by normal matings when nonspecific epileptiform EEG paroxysms are excluded. These three families would be classified as multiplex clinically under normal by normal mating when nonspecific epileptiform EEG paroxysms
in parents are excluded and were originally counted under the eight-pedigree multiplex by clinical and specific EEG paroxysms.
Epilepsia. Vol. 35, Sirppl. 1. I994

MAPPING HUMAN EPILEPSY GENES s33
Escueta et al., unpublished data). Every sibship was ascertained by only one proband and the progeny from all mating types were analyzed statistically. Three def- initions of the affected status of parents and siblings were used. In the first, most broad analysis, all family members with a history of clinical generalized seizures or an abnormal EEG (specific or nonspecific) were classified as affected. In the second analysis, individuals with nonspecific EEGs were classified as not affected whereas individuals with specific spike-wave abnor- malities were classified as affected along with individ- uals with seizures. In the final analysis, only individuals with a history of seizures were considered affected (fe- brile convulsions not included).
Initial estimation for segregation ratio or p’ of all pedigrees across all mating types, when all individuals with abnormal EEGs were classified as affected, was 0.28 (SE = +0.04), but the segregation ratio dropped considerably to f i = 0.15 when we excluded all par- oxysmal forms of EEGs, specific and nonspecific. This ratio is similar to that obtained by Obeid and Pana- yiotopoulos ( 1988) and Panayiotopoulos and Obeid (1989) in Saudi Arabian families who did not have their EEGs examined. Matings in which both parents are unaffected yielded an estimate of the segregation ratio of f i of 0.20 lower than the expected 0.25 in re- cessive inheritance. Some of many possible reasons for a segregation ratio less than expected under Mendelian inheritance include (a) a mixture of sporadic or non- genetic cases with “genetic” cases; (b) phenotypes de- fined incorrectly as when we exclude the EEG JME trait in asymptomatic individuals; and (c) heterogeneity (Delgado-Escueta et al., unpublished data).
Estimates (j = 0.40) fit the expectation of a fully penetrant autosomal dominant inheritance in matings between affected and unaffected. Thirteen normal by affected matings based on clinical affectedness only provided further proof for an autosomal dominant in- heritance with reduced penetrance in these families (j = 0.42). Although an autosomal recessive trait could not be rejected in normal by affected matings, we con- sidered this unlikely because one affected homozygous parent would have to be mamed to an unaffected het- erozygous parent. Such would be possible in the pres- ence of a high gene frequency for JME, which is not true or in the presence of consanguinity, which was absent in the patients of our data set. Thus, our pre- liminary segregation analysis suggests several possibil- ities. The first possibility is that the majority of JME families follow an incompletely penetrant autosomal dominant mode of inheritance with or without an ad- mixture of sporadic cases (nonhereditary or dominant new mutations). The second possibility is that genetic heterogeneity exists within our population.
Genetic linkage mapping supports genotypic heterogeneity in JME
At the Human Gene Mapping 9 Workshop in 1987 (Greenberg et al., 1987) we reported that JME may be linked to the Bf-HLA loci in chromosome 6p. Using clinical and EEG characteristics in the identification of affected family members, we reported a maximum lod score of 3.05 at recombination fraction of 0.10 un- der a recessive model of inheritance, assuming a pen- etrance of 0.6 using HLA and Bf (properdin factor) as markers. We obtained these results from 11 nuclear families that were informative for Bf or HLA (see Table 6 for data base and affectedness assignments). Evidence of possible linkage was strengthened when Greenberg et al. ( 1988) increased the original sample of families to 18 that were informative for Bf and 4 that were informative for HLA. The maximum lod score ob- tained for this analysis was 3.78 (0, = Of = 0.01), as- suming autosomal dominant inheritance and 90% penetrance. Assuming autosomal recessive inheritance with complete penetrance, the maximum lod score was 3.05 (0, = Of = 0.01). This suggested that a JME locus was present in the 6p2 1.3 area (Greenberg et al., 19886; Delgado-Escueta et al., 1989, 1990).
Since then, two studies by Weissbecker et al. (1 99 1) and Durner et al. ( 199 1) confirmed this linkage. Weiss- becker et al. (1 99 1) used HLA serologic markers and clinical data obtained from Janz’ Berlin clinic and Durner et al. ( 199 1 ) used DNA markers in the HLA- DQ region in these same families. There were 143 fam- ily members of 25 Berlin pedigrees. In the studies of Durner et al. ( 199 l), lod scores were 3.65 when asymp- tomatic family members with abnormal EEGs were considered affected.
The observed different clinical manifestations among probands with JME and the eight subtypes of JME families reviewed in Table 4 raise several questions. Because other forms of idiopathic generalized epilepsies are observed in family members of patients with JME, as in subphenotypes 111, IV, and VII, does it mean that other generalized epilepsies, such as CAE and grand mal, have their genetic locus in chromosome 6p iden- tical to the genetic locus of JME? Is there heterogeneity within the primary generalized epilepsies? Is there het- erogeneity within JME? Our present results suggest that there is heterogeneity within the common primary generalized epilepsies and within JME. Our latest stud- ies using chromosome 6p reference DNA markers show that there are families with JME that do not map to the HLA region of chromosome 6p (Liu et al., 1992). Furthermore, in families of children with CAE with or without GTCS, we can exclude linkage to the same EJMl locus (Serratosa et al., 1993). In the last year, Serratosa et al. ( 1993) have studied 127 family members
Epilepsia. Vol. 35, Suppl. I . 1994

s34 A. V. DELGADO-ESCUETA ET AL.
TABLE 6. Data base and basis ,for classifying aflectedness during genetic linkage analysis in Los Angeles families
Original I I Subsequent 33 families families"
(Greenberg (Greenberg et al.. et al., Clinical 1987) 1988) affectedness EEG affectedness Family classification
4
I h
2
0
Normal by normal matings
14 Proband only Normal EEGs on all nonproband members
7 Proband and one sibling by 3.5- to 6-Hz multispike wave complexes
2 Proband only Proband and one sibling by 3.5- to 6-Hz multispike wave complexes
3 Proband only Proband and one sibling by nonspecific epileptiform EEG paroxysms
Proband and sibling
Normal by affected matings
parent and multispike wave complexes sibling
I 3 Proband, one Proband and sibling by 3.5- to 6-Hz
3 (2<) 4 Proband only One parent has nonspecific epileptiform EEG paroxysms
Simplex by clinical and EEG
Multiplex by clinical and specific EEG pattern
Multiplex by specific EEG paroxysms only
Multiplex by nonspecific EEG paroxysms only
Multigenerational and multiplex by clinical and specific EEG paroxysms
Multigenerational by nonspecific EEG paroxysms
"These included the original I I families. Multiplex by clinical but other siblings are asymptomatic with epileptiform EEG patterns. Siblings also have epileptiform nonspecific patterns.
of 12 probands with CAE. Results favor a locus for CAE outside the chromosome 6p locus for EJMl and outside the EPMl locus in chromosome 2 I .
From 1991 to 1992, we screened 12 JME families with DQa and eight other chromosome 6p reference markers above (D6105, D6S89, D6S109, F13A1) and below (D6S29, GLO 1, TCTE I , D6Z 1) HLA. JME was assumed to be an autosomal dominant trait with pen- etrance of 70%. The disease allele was given a frequency of 0.001 to account for its morbid risk in the popula- tion. Lod scores were consistently negative when pair- wise analyses between the 9 loci located in chromosome 6p and JME were carried out. In light of earlier studies, which suggested genetic linkage of JME to HLA, these results of Liu et al. (1992) suggest locus heterogeneity, and the possibility of a second locus for JME outside chromosome 6p which must now be sought. Thus, the pathogenesis of JME in the Berlin families analyzed by Weissbecker et al. (1991), and Durner et al. (1991) and Los Angeles pedigrees studied by Greenberg et al. (1987, 1988) and by Liu et al. (1992), may involve mutations at different chromosomal sites. There may be more than one locus which leads to the same EEG and clinical phenotypes of JME. We are therefore in- vestigating the role of heterogeneity in JME, linkage of JME to chromosome 6p markers, and linkage to polymerase chain reaction (PCR)/microsatellites in other areas of the human genome.
The EJMl locus and JME family subtypes I , Ira, and IIb
Because of strong exclusionary evidence presented by Whitehouse et al. ( 1993) and our own JME families who do not show linkage to chromosome 6p markers (Liu et al., 1992), we reviewed the family subtypes of the original 1 1 JME families reported in 19'87 to be informative for Bf or HLA. We are presently correlating the results of these HLA serology with chromosome 6p DNA markers. In studies of linkage analysis that have been completed, we are also comparing the subgroups of JME families that show positive lod scores, as in the original 1987 study, and those that show negative lod scores, as reported by Liu et al. (1992). It is important to note that in these early studies on genetic linkage mapping of 22 infor.mative Los An- geles JME families, completed and reported in 1987 and 1988, we rejected probands with CAE only, pure GTCS only, myoclonic absence only, and photogenic epilepsies. Of 1 1 families informative for Bf or HLA, two JME families with unaffected parents were mul- tiplex because of specific 3.5- to 6-Hz multispike wave paroxysms and two other JME families were multiplex on account of nonspecific epileptiform patterns. Two other JME families were multiplex on account of a sibling affected with JME (Table 6). In this early study, no siblings or parents had CAE. Three families were multigenerational by virtue of a parent with nonspecific
Epilep.siu. Lid. 3.5. Siippl. 1. I994

MAPPING HUMAN EPILEPSY GENES s3.5
epileptiform paroxysms. Thus, in 6 of 1 1 informative families, affectedness of a family member was deter- mined by the EEG. An early concern was whether ge- netic linkage was to the EEG trait or to both the EEG and clinical traits of JME (Greenberg et al., 1987, 1988b).
Because of early suggestions of possible genetic link- age ofJME subgroups I, IIa, and IIb with Bf-HLA (Liu et al., 1992), and our more recent results showing ex- clusion of linkage between CAE and chromosome 6p markers (Serratosa et al., 1993), we have tightened up our rejection criteria, emphasizing rejection of any probands with absence only, or GTCS only, or drop seizures as part of JME and the importance of sepa- rating out JME families whose family members have early childhood, childhood, or juvenile absence. In other words, we are now concentrating recruitment on the JME subgroups I (families with JME in all affected members), IIa, and IIb (families with asymptomatic siblings who have epileptiform EEG paroxysms), listed in Table I . This was the original group studied in 1987 with the exception of one patient who had adolescent drop attacks as part of the JME syndrome. We are also investigating the possible contribution of racial origins to heterogeneity of JME.
BFNC heterogeneity is also present-EBN1 locus in chromosome 20 and EBN2 in chromosome 8
BFNC is included in the rare syndromes of benign neonatal convulsions that show a favorable neurolog- ical outcome. Psychomotor development is normal. In 80% of cases, unprovoked partial or generalized clonic seizures, sometimes with apneic spells, start on the 2nd and 3rd days of life. In contrast to the syndrome of benign infantile neonatal convulsions, BFNC is a fa- milial disorder (Whitehouse et al., 1993) with auto- soma1 dominant transmission: Seizures usually stop by the age of 6 months without any neurologic sequelae. The subsequent development of childhood or adult epilepsy has been documented in - 10-14% ofpatients.
Leppert et al. (1989) studied four generations of a single family-1 9 members of which were affected with BFNC-and linked their epilepsy to genetic markers CMM6 and RMR6 on chromosome 20. CMM6 and RMR6 belong to a series of markers known as a variable number of tandem repeats, which use the 9-base-pair core sequence GNNGTGGG.
The linkage analysis of this four-generation pedigree, including 50 members and 19 affected individuals (Leppert et al., 1989), indicated strong evidence that a genetic locus of BFNC is located on the long arm of chromosome 20. In six French pedigrees, Malafosse et al. (1 9 9 2 ~ ) confirmed close linkage of BFNC to the same DNA markers CMM6 and RMR6. Pooling both
samples, a maximum lod score of 5.77 (8 = 0.064) for CMM6 and 5.04 (0 = 0) for RMR6 was obtained. The results from a homogeneity test between these two sets of data did not reveal any significant difference.
The EBNl gene is highly penetrant, clearly segregates as a single autosomal dominant gene, and has clear, unique phenotypic characters that apparently help simplify the diagnosis. Although it is a rare form of epilepsy syndrome, BFNC was considered an ideal syndrome for genetic analysis because of its clear elec- troclinical manifestations and apparent homogeneity. However, the presence of genetic heterogeneity and the existence of two distinct genetic loci for BFNC in two large families of BFNC was demonstrated in recent studies by Ryan et al. (1991), Ryan (1993), and Lewis et al. ( 1993). The maximum lod score of 1.66 obtained for family 2 in the study by Ryan et al. ( 199 1) suggested but did not prove that the BFNC locus in this kindred was linked to the marker loci CMM6 (D20S19). Given previous reports by Leppert et al. ( 1989) and Malafosse et al. (1992~) of linkage of BFNC to these markers, it appears quite likely that the disease gene in family 2 of Ryan et al. (199 1 ) maps to chromosome 20q. In contrast, the odds were >20,000: 1 in family 1 against linkage to CMM6 (D20S19) at 10% recombination or less. In April 1993, Ryan (1993) and subsequently Lewis et al. ( 1993) reported further on the same family 1 with BFNC and demonstrated that the locus was not in chromosome 20q but in chromosome 8q.
PROGRESSIVE MYOCLONUS EPILEPSIES
The PMEs are a heterogeneous group of debilitating, often fatal epileptic encephalopathies characterized by segmental arrhythmic myoclonus, massive myoclonias, GTCS or clonic seizures, with or without absence, de- mentia, and other progressive neurologic manifesta- tions, especially cerebellar (Lafora, 19 1 1 ; Lafora and Glueck, 191 1; Evans, 1962; Guazzi et al., 1968; Ko- skiniemi et al., 1974a,b; Fukuhara et al., 1980; Barn- veld et al., 1983; Mueller et al., 1985; Berkovic et al., 1986, 1989; Gardiner et al., 1990; Durner et al., 1991). Because a biochemical pathology ‘can usually be dem- onstrated, the PME are considered forms of symptom- atic generalized epilepsies (Commission, 1989).
Unverricht-Lundborg or Baltic-Mediteranean PME: PMEl locus
The Baltic type of debilitating but not fatal form of PME was first described by Unvemcht (1891, 1895) in Estonia and by Lundborg (1903) in Sweden. The characteristic clinical features of a slow progressive na- ture and stimulus sensitivity of myoclonus start be- tween age 6 and 18 years. Proprioceptive stimuli, such as tendon tapping, or passive joint movements, and
Epilepsia. Vol. 35. Suppl. 1. 1994

S36 A. V. DELGADO-ESCUETA ET AL.
auditory and light stimuli are common precipitants of myoclonias. Generalized clonic and tonic-clonic sei- zures, as well as myoclonic seizures mostly appear on awakening and should be differentiated from JME. Resistance to antiepileptic drug (AED) treatment, slowly progressive but mild ataxia, and mild intellectual deterioration and dementia, which develop late in the course of the disease, differentiate this disorder from JME. The progression of this autosomal-recessive dis- order may vary considerably between and within fam- ilies. After a period of -5 years, the patient is usually incapacitated. The gene for Unvemcht-Lundborg type was recently assigned to chromosome 2 1q22.3 by link- age analysis in 12 Finnish families with 68 members, 26 of whom were affected. The maximum multipoint lod score was 10.08 (Lehesjoki, 199 1, 1992). Multipoint linkage analysis determined a location of the disease gene at 6.0 centimorgans distal to locus BCEZ and 0.8 centimorgans proximal to locus D2 1 S 154. Malafosse et al. (1992~) showed that the Mediterranean type of progressive myoclonus epilepsy is most probably due to the same mutation as that of the Baltic PME (Ma- lafosse et al., 19926). Families with the Ramsay Hunt syndrome are now being examined with the same chromosomal markers to determine whether that syndrome is caused by the same mutation in chro- mosome 2 1.
Lafora’s disease This fatal autosomal recessive form of PME usually
starts with epileptic seizures during early adolescence, although symptoms may start as late as age 18 years. Patients die 5-10 years after the first symptoms. The first manifestations are usually GTCS, absence seizures, or drop attacks, followed soon by small, irregular, and asymmetric myoclonic jerks. As the disease progresses, myoclonus becomes almost constant. Trains of photic- sensitive, high-voltage, usually bilaterally synchronous, spike-wave, and polyspike-wave complexes interrupt a disorganized slow EEG background. Dementia, apraxia, and visual loss leads to a vegetative state (La- fora, 191 1; Lafora and Glueck, 191 1; Roger et al., 1992). Periodic acid-Schiff-positive cytoplasmic in- clusion bodies originally described by Lafora contain polyglycosans and are found in the brain, striated mus- cle, liver, and skin (Lafora, 191 1; Lafora and Glueck, 19 1 1 ). A skin biopsy is the diagnostic procedure of choice. The gene locus for Lafora’s disease has not yet been mapped. Exclusion of linkage to the chromosome 21q22.3 region where EPMl locus lies has been re- ported in three Italian families (Lehesjoki et al., 1992).
Neuronal ceroid-lipofuscinoses PME can be caused by three types of neuronal ceroid
lipofuscinoses: late infantile (Bielschowsky-Jansky
disease), juvenile (Spielmeyer-Vogt-Sjogren’s disease), and adult (Kufs disease) (Zeman et al., 1970; Roger et al., 1992). The infantile type (Santavuori’s disease or CLN-I), which has been mapped to the short arm of chromosome 1 (Jarvela et al., 1991), is not consid- ered a PME syndrome (Zeman et al., 1970; Berkovic et al., 1989). The late infantile form, on the other hand, does cause a fatal form of PME, is an autosomal re- cessive disorder, starts in early childhood between 2.5 and 4 years of age and is characterized by myoclonic, GTCS, atonic, and atypical absence seizures. It is usu- ally mistaken for Lennox-Gastaut syndrome. Myo- clonus is initially segmental, asymmetric, and spon- taneous but becomes almost continuous and sensitive to any stimulus after a few months. Intellectual loss, blindness, spasticity, and ataxia lead to a decorticate state and death between 6 and 10 years of age. The slow type of irregular spikes and polyspike-wave dis- charges interrupt the slow, disorganized EEG back- ground. Photic stimulation at frequencies <3 Hz can induce posterior high-amplitude, polyphasic spikes, corresponding to giant visual evoked potentials (Roger et al., 1992).
The juvenile form of ceroid lipofuscinosis (Batten diseuse or Spielmeyer- Vogt disease or CLN3) also causes a fatal form of PME. It starts in early childhood or childhood (4-10 years) with loss of central vision that progresses to blindness. Absence seizures and GTCS appear 2 years later with ataxia, dysarthria, and mental deterioration. Myoclonias are aggravated by passive movements. Paroxysmal bursts in a slow, dis- organized EEG background are exacerbated with sleep. Photic stimulation has no effects clinically or on the EEG. Death occurs in the late teens or early twenties. Vacuolated lymphocytes in peripheral blood and char- acteristic fingerprint profiles and curvilinear inclusions in ultrastructural study of skin biopsy or lymphocytes confirm the diagnosis. Juvenile ceroid-lipofuscinosis has been mapped by Gardiner et al. (1 990) to chro- mosome 16p using linkage analysis. The locus symbol for the disease is CLN3.
Gaucher’s disease Of all forms of Gaucher’s disease or cerebroside lip-
idoses, only type 3 or the juvenile type presents as PME in childhood or early adulthood. Severe intention my- oclonus, abnormal saccadic horizontal eye movements, and supranuclear gaze paralysis are the first symptoms. Cerebellar deficits, generalized and partial seizures, and some degree of dementia are common. Hepatospleno- megaly is usually present. Death usually occurs 10 years from the diagnosis. The EEG shows rapid 6- to 10-Hz diffuse multispike and rhythmic sharp waves, most prominent posteriorly and 6- to 10-Hz photic stimu-
Epilepsia, Vol. 35, Suppl. I . 1994

MAPPING HUMAN EPILEPSY GENES s3 7
lation induces a photoparoxysmal or photomyoclonic response. This autosomal recessive disease is caused by a deficiency of glucocerebrosidase (Tsuji et al., 1987). Neurotoxic glucocerebrosides accumulate due to a homozygous mutation in nucleotide 1448 of the glucocerebrosidase gene. The gene for glucocerebro- sidase has been mapped to the q2 1 -q3 1 region of chro- mosome I (Barnveld et al., 1983).
Sialidosis type I The cherry-red-spot myoclonzis syndrome (Engel et
al., 1977; Roger et al., 1992) or sialidosis type I, first described by Guazzi et al. ( 1968), and usually associated with an isolated neuroaminidase deficiency, causes a PME syndrome in childhood and adolescence appear- ing between the ages of 8 and 15 years. Macular cheny- red spots are seen in the fundus with visual failure. These are combined with easily controlled seizures and painful “burning hands and feet.” There is usually no dementia. Patients exhibit massive, bilaterally syn- chronous, and symmetric, stimulus-sensitive myo- clonic jerks as well as independent sporadic, stimulus- insensitive facial myoclonus. Active and passive movements, the thought of movements, touch, but not light or sound precipitate massive myoclonias. Low- voltage fast activity with 10- to 20-Hz vertex-positive spikes in the EEG fire synchronously with 10- to 20- Hz electromyogram discharges during massive myo- clonus. The partial facial myoclonias have no EEG correlate (Evans, 1962). An inherited primary defi- ciency of neuroaminidase was first demonstrated in the severe (type 11) form of sialidosis. A similar defi- ciency was observed in sialidosis type I. Sialidosis type I1 has abnormal somatic features, including coarse fa- cies and dysostosis multiplex. There are rare reports that cherry-red spots and myoclonus can also be ob- served in older children with type I1 sialidosis. Sialidosis is caused by a mutation in a structural gene encoded in chromosome 10pter-q23 (Mueller et al., 1985).
Mitochondria] disease can also cause the syndrome of PME: myoclonus epilepsy and ragged-red fibers (MERRF)
A tRNALyS mutation in mitochondrial DNA can cause a childhood or adult form of PME as reported by Fukuhara et al. (1980). Cerebellar ataxia is present with action myoclonus epilepsy, GTCS, and ragged- red fiber myopathy. Deafness, dementia, dysarthria, short stature, optic atrophy, neuropathy, lactic acidosis, hypoventilation, and migraine may also be present. Inheritance is consistent with mitochondrial (maternal) transmission. Muscle biopsy reveals subsarcolemmal aggregates of mitochondria, the so-called ragged-red fibers (Fukuhara et al., 1980; DiMauro et al., 1985; Berkovic et al., 1986, 1989). Some patients present oc-
casional, spontaneous, bilateral massive myoclonias associated with generalized spike-wave discharges. Photic stimulation also induces 2- to 5-Hz diffuse spike- wave epileptiform activity of posterior predominance, or even a photomyoclonic response. Giant visual evoked potentials are recorded in all cases (Roger et al., 1992). Several investigators (Shoffner, 1990) have demonstrated, in a number of patients with MERRF syndrome, the presence of a disease-related A-to-G substitution at nucleotide 8344 in the mitochondrial DNA (mtDNA), which affects the pseudouridine loop of the mitochondrial tRNALyS. A causal relationship between this genetic mutation and MERRF disease is present and the A-to-G mutation in the mitochondrial tRNALyS gene is disease-specific for MERRF. Investi- gators have also observed that the rate of mitochondrial protein synthesis proceeded normally, but the sodium dodecyl sulfate-pdyacrylamide gel electrophoresis technique analysis of mitochondrial translation prod- ucts showed a reduction in the higher-molecular- weight products and an overproduction of smaller pro- teins.
REVISED STRATEGY FOR MAPPING EPILEPSY GENES
Results of the MERRF studies have widespread im- plications for the heterogeneous generalized epilepsies. Further studies on mitochondrial genetics are indi- cated, given the higher incidence of seizures in relatives of female than of male probands in Doose’s myoclonic astatic epilepsy of early childhood epilepsies (Doose and Baier, I987), in the Heidelberg families with JME (Tsuboi and Christian, 1973) and in photogenic epi- lepsies (Wolf and Goosses, 1986), particularly when there is only female-to-male or female-to-female trans- mission. Presently, there is no explanation for why risks are higher for the siblings and offspring of female com- pared with male patients (Ottman et al., 1988). Aside from the possibility of a mitochondrial disorder, ge- nomic imprinting is another possible explanation (Hall, 1990).
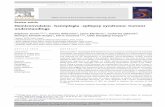
As a consequence of the above developments, es- pecially the difficult problem that heterogeneity poses as has been observed in tuberous sclerosis and Alzhei- mer’s disease (Smith et al., 1990; Karlinsky et al., 1992; Northrup et al., 1992; Schellenberg et al., 1992), we have modified our strategy in our new studies for lo- cating the genetic loci of JME, CAE, early childhood familial myoclonic epilepsy, and EGMA. Figure 1 il- lustrates the screening of families first with chromo- some 6p markers, chromosomes 8q, 16p, 1 q, 20q, 2 lq, and 1Oq markers (where loci for EBNZ, CLN-3, ju- venile Gaucher’s, EBNl, EPMI, and sialidosis type I are located, respectively) and then chromosomal sites
Epilep.siu. Ibl. 35, Siippl. I . 1994

S38
FIG. 1. Strategy for mapping common idiopathic generalized epilepsies. JME, juvenile myoclonic epilepsy; CAE, childhood absence epilepsy; EGMA, epilepsy with grand ma1 on awakening; ECFME, early childhood familial myoclonic epilepsy; EMJl , epilepsy juvenile myoclonus gene locus; TNF, tumor necrosis factor; PME, progressive myoclonic epilepsy; chr, chromosome.
homologous to mouse spike wave loci, namely, chro- mosomes lq, 2q, 1 1 p, 12q, 15q, 16q. If all these studies do not favor genetic linkage, we proceed to screen the rest of the human genome with PCR/microsatellites (Weber and May, 1989; Weber, 1990; Weissenbach et al., 1992).
WHY MAP EPILEPSY GENES?
Once the mutated code scripts in the common gen- eralized epilepsies, rare benign epilepsies, and fatal or debilitating PME syndromes are identified, we can be- gin to investigate the mechanisms-on a molecular, cellular, and systemic level-by which such inheritance leads to a specific epilepsy syndrome. This will lead to an understanding of how cells are assembled in the face of such abnormal code scripts, and how devel- opment is affected during embryogenesis and the im- mediate postnatal period.
These developments will enable us to finally unravel the code for genotypic heterogeneity within the mon- ogenic generalized epilepsies and the PME as well as to catalog which of the multifactorial symptomatic partial epilepsies have an epilepsy susceptibility gene. A family history of seizures is more common among individuals with posttraumatic epilepsy (7%) than it is among persons with head injuries but no epilepsy (2%) (Evans, 1962; Caveness, 1963; Caveness et al., 1979). A history of seizures in first-degree relatives is present
in 60% of patients with posttraumatic epilepsy but in only 30% of head-injured, seizure-free persons. Seizures (2% of near relatives) and EEG abnormalities (23% of relatives) are also more common in epileptic persons with infantile hemiplegias than in nonepileptic persons with infantile hemiplegias (Rimoin and Metrakos, 1961).
Improving the classification of epilepsy genotypes will undoubtedly improve calculations of sibling risk for seizures and abnormal EEG patterns. This should, in turn, improve the accuracy of risk assessments and facilitate genetic counseling, providing an accurate and balanced picture of a possible family burden.
Identifying the gene defect in the common or rare benign monogenic epilepsies and the fatal PME will brighten prospects for gene therapy (see McNamara, 1993). Somatic or germ cell gene replacement will have a decided advantage over present AED treatment by requiring fewer treatment deliveries, if not one curative treatment delivery. The duration of gene action will depend on the target cells. Delivery of genes by mature somatic cells is limited by the life span of the somatic cell, whereas delivery through progenitor stem cells can have a particularly long-lived effect. Should untoward effects occur, a “suicide gene” can be used to kill the gene delivery construct.
Acknowledgmenk This work was supported by National Institutes of Health grant NS21908.

MAPPING HUMAN EPILEPSY GENES s39
REFERENCES
Asconape J, Penry JK. Some clinical and EEG aspects of benign juvenile myoclonic epilepsy. Epilepsia 1984:25: 108-14.
Barnveld RA, Keijzer W. Tegelders FPW, et al. Assignment of the gene for human B-glucocerebrosidase to the region of q2 I-q3 I of chromosome I using monoclonal antibodies. Human Genet 1983:64:227-3 I .
Berkovic SF, Andermann F. Carpenter S, Wolfe LS. Progressive my- oclonus epilepsies: specific causes and diagnosis. N Engl J Med
Berkovic SF. Carpenter S, Evans A, et al. Myoclonus epilepsy and ragged-red fibres (MERRF) 1. A clinical. pathological, biochem- ical, magnetic resonance spectrographic and positron emission tomographic study. Brain 1989;112: 1231-60.
Caveness WF. Onset and cessation of fits following craniocerebral trauma. J Neirroswg 1963:10:570-82.
Caveness WF. Meirowsky AM, Rish BL et al. The nature of post- traumatic epilepsy. J Neiirusirrg 1979;50:545-53.
Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia I989:30:389-99.
Delgado-Escueta AV, Enrile-Bacsal FE. Juvenile myoclonic epilepsy of Janz. Neiiru1ug.v I984:34:285-94.
Delgado-Escueta AV, Greenberg DA, Treiman L, et al. Mapping the gene for juvenile myoclonic epilepsy. Epilepsia 1989;30(suppl4):
Delgado-Escueta AV, Greenberg D. Weissbecker K, et al. Gene map- ping in the idiopathic generalized epilepsies: juvenile myoclonic epilepsy, childhood absence epilepsy, epilepsy with grand ma1 seizures, and early childhood myoclonic epilepsy. Epilepsia
DiMauro S, Bonilla E, Zeviani M, Nakagawa M, De Vivo DC. Mi- tochondrial myopathies. Ann Neiirol 1985:17:52 1-38.
Doose H, Baier WK. Genetic factors in epilepsies with primarily generalized minor seizures. Neiiropediairics 1987: 18(suppl I ): 1 - 64.
Durner M. Sander T, Greenberg DA, Johnson K. Beck-Mannagetta G. Janz D. Localization of idiopathic generalized epilepsy on chromosome 6p in families of juvenile myoclonic epilepsy pa- tients. Neiirologv 1991:41:165 1-5.
Engel J. Rapin I, Giblin D. Electrophysiological studies in two patients with cherry-red-spot-myoclonus syndrome. Epilepsiu 1977: I8:73- 87.
1986:3 15:296-305.
S8- 18.
1990:31(~~ppl 3):S19-29.
Evans JH. Post-traumatic epilepsy. Newolug). 1962; I2:665-74. Fukuhara N, Tokiguchi S, Shirakawa S, Tsubaki T. Myoclonus epi-
lepsy associated with ragged-red fibers (mitochondria1 abnor- malities): disease entity or syndrome? Light and electron- microscopic studies of two cases and review of the literature. J NeiiroI Sci 1980:47: I 17-33.
Gardiner M, Sandford A. Deadman M, et al. Batten disease (Spielmeyer-Vogt disease. juvenile onset neuronal ceroid lipo- fuscinosis) gene (CLN3) maps to human chromosome 16. Ge- nomics 1990:8:387-90.
Greenberg DA, Delgado-Escueta AV, Widelitz H, et al. A locus in- volved in the expression of juvenile myoclonic epilepsy and of an associated EEG trait may be linked to HLA and Bf. Cyrogenei Cell Genet 1987:46:623.
Greenberg DA. Delgado-Escueta AV. Widelitz H, et al. Juvenile myoclonic epilepsy (JME) may be linked to the Bfand HLA loci in human chromosome 6. Am J Med Genei 1988:31:185-92.
Guazzi GC, Ghetti B, Bertolino A. Fiore C. Vecchio M, Striano S. Epilepsia mioclonica giovanile con macchia rossociliega al fondo dell'occhio. I. Studio genetic0 e clinico. Foliu Neitropsychiui 1968;l 1:737-58.
Hall JG. Genomic imprinting: review and relevance to human dis- eases. Am J Hum Gene/ 1990:46:857-73.
Janz D. Die Epilepsien. Stuttgart: Thieme. 1969. Janz D. Epilepsy with impulsive petit ma1 (juvenile myoclonic epi-
lepsy). Acia Neirml Scund 1985:72:449-59. Jarvela D, Schleutker J, Haataja L. et al. Infantile neuronal ceroid
lipofuscinosis (CLN I ) maps to the short arm of chromosome I . Genomics I99 1 :9: 170-3.
Karlinsky H, Vaula G, Haines JL, et al. Molecular and prospective phenotypic characterization ofa pedigree with familial Alzheimer's disease and a missense mutation in codon 7 I7 of the B-amyloid precursor protein gene. Neurology 1992;42: 1445-53.
Koskiniemi M, Donner M, Majuri H, Haltia M, Norio R. Progressive myoclonus epilepsy: a clinical and histopathological study. Acla Neiirol Scand 1974~;50:307-32.
Koskiniemi M, Toivakka E, Donner M. Progressive myoclonus epi- lepsy: electroencephalographic findings. Acla Neurol Scund
Lafora GR. Uber das Vorkommen amplifier Korpechen im innern der Ganglienzellen; zugleich Ein Beitrag zum Studium der amy- loiden Substanz in Nervensystem. Virchow Arch [A] 191 1:205:
Lafora GR, Glueck B. Beitrag zur Histopathologie der myoklonischen Epilepsie. Z Gesamte Neurol P.s.vchiatr I9 I I :6: I - 14.
Lee AG, Delgado-Escueta AV, Maldonado H. Swartz BE, Walsh GO. Closed circuit television videotaping and electroencephalog- raphy biotelemetry in primary generalized epilepsies. In: Gumnit RJ, ed. Intensive neurodiagnosiic monitoring. New York: Raven Press, I987:27-68. (Advances in neurology: Vol 46).
Lehesjoki A-E, Koskiniemi M, Pandolfo M, et al. Linkage studies in progressive myoclonus epilepsy: Unvenicht-Lundborg and La- fora's diseases. Neurology 1992:42: 1545-50.
Lehesjoki A-E. Koskiniemi M, Sistonen P, et al. Localization of a gene for progressive myoclonus epilepsy to chromosome 2 lq22. Proc Nail Acad Sci USA 1991:88:3696-9.
Leppert M, Anderson VE. Quattlebaum T, et al. Benign familial neonatal convulsions linked to genetic markers on chromosome 20. Nailire 1989:337:647-8.
Lewis TB, Leach RJ. Ward K, OConnell P, Ryan SG. Genetic het- erogeneity in benign neonatal convulsions: identification of a new locus in chromosome 8q, Am J Hum Genet 1993;53:670-5.
Liu A. Delgado-Escueta AV, Weissbecker K, et al. Juvenile myoclonic epilepsy and reference markers of chromosome 6p [Abstract]. Epilepsia I992:33(suppl 3):73.
Loiseau J. Loiseau P, Guyot M, Duche B, Dartigues JF. Aublet B. Survey of seizure disorders in the French Southwest, I: incidence of epileptic syndromes. Epilepsia 1990:3 1:39 1-6.
Lundborg H. Die progressive mjw1onu.c epilepsie (Unverrichi 's mvoklonie). Uppsala: Almquist & Wiskell, 1903:j -207.
Malafosse A, Leboyer M, Dulac L, et al. Confirmation of linkage of benign familial neonatal convulsions to D20S20. !firm Gene1
Malafosse A, Lehesjoki A. Genton P. et al. Identical genetic locus for Baltic and Mediterranean myoclonus. Lancet 1992h; 339: 1080- I .
McNamara JO. Identification of genetic defect of an epilepsy: strat- egies for therapeutic advances. Epilepsiu I994:35(suppl 1 ):S5 1- 7 (this issue).
Mueller OT, Henry WM, Haley LL. Byers MG. Eddy RL. Show TB. Sialidosis and galactosialidosis: chromosomal assignment of two genes associated with neuroaminidase deficiency syndrome. Proc NallAcadSci USA 1985:83:1817-19. ,
Northrup H. Kwiatkiowski DJ, Roach ES, et al. Evidence for genetic heterogeneity in tuberous sclerosis: one locus on chromosome 9 and at least one locus elsewhere. A m J Hum Gmei 1992:5 1 :709- 20.
Obeid T, Panayitopoulos CP. Juvenile myoclonic epilepsy: a study in Saudi Arabia. Epilepsia 1988:29:280-2.
Ottman R. Annegers JF, Hauser WA, Kurland LT. Higher risk of seizures in offspring of mothers than of fathers with epilepsy. Am J Hum Genei 1988:43:257-64.
Panayiotopoulos CP, Obeid T. Juvenile myoclonic epilepsy: an au- tosomal recessive disease. Ann Neurol 1989:25:440-3.
Rimoin DL, Metrakos JK. La genetique des manifestations convul- sives chez des familles d'hemiplegiques. In: Cauverier J, et al., eds. Second Iniernntional Conference of Human Gen1.tic.s. Inter- naiional Congress Series no. 32. London: Excerpta Medica. I96 I : abstract 195.
1974b;50:333-59.
295-303.
1992~:89:54-8.
Epilcp.siu. l i d 35. Siippl. I . 1994

S40 A. V. DELGADO-ESCUETA ET AL.
Roger J, Genton P. Bureau M. Dravet C. Progressive myoclonus epilepsies in childhood and adolescence. In: Roger J. Bureau M, Dravet C. Dreifuss FE, Perret A. Wolf P, eds. Epileptic sjwdromes in infancy. childhood and udole.mvce. London: John Libbey, 1992: 13-23.
Ryan S. Benign Familial Neonatal Convulsion: evidence for chro- mosome 8q locus. Presented at the Alsace International Workshop on Idiopathic Generalized Epilepsies, April 1993.
Ryan S, Wiznitzer M, Hollman C, et al. Benign familial neonatal convulsions: evidence for clinical and genetic heterogeneity. Ann Neiirol I99 1 :29:469-73.
Schellenberg GD, Bird TD, Wijsman EM, et al. Genetic linkage ev- idence for a familial Alzheimer’s disease locus on chromosome 14. Science 1992;258:668-7 1 .
Serratosa JM. Delgado-Escueta AV. Pascual-Castroviejo I, et al. Childhood absence epilepsy: exclusion of genetic linkage to chro- mosome 6p markers. Epilepsia 1993:34(suppl 2):S149.
Shoffner JM. Lott MT. Lezza AMS, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochon- drial DNA tRNALy’ mutation. Cell 1990;61:931-7.
Smith M. Smalley S, Cantor R, et al. Mapping of a gene determining tuberous sclerosis to human chromosome I Iq 14- I lq23. Genomics I990;6: 105- 14.
Tsuboi T, Christian W. On the genetics of primary generalized epilepsy with sporadic myoclonias of impulsive petit mal: a clinical and electroencephalographic study of 399 probands. H i m Gene/ 1973;19:155-82.
Tsuji S. Choudary PV, Martin BM. et al. A mutation in the human
glucocerebrosidase gene in neuronopathic Gaucher’s disease. N Engl J Med 1987;316:570-5.
Unverricht H. Die Mvoclonic.. Vienna: Franz Deuticke. I89 I . Unverricht H. Uber familiare Myoklonie. D/sch Z Nervenheilk
Weber JL. Informativeness of human (dG-dT)n polymorphisms. Genotnits 1990;7:524-30.
Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet 1989;44:388-96.
Weinberg W. Methode und Fehlerquellen der Untersuchung auf Mendelschen Zahlen bein Menschen. Anch Rass Ges Biol1912;9:
Weissbecker K. Durner M, Janz D. et al. Confirmation of linkage between a juvenile myoclonic epilepsy-locus and the HLA-region of chromosome 6. Am J Med Gene/ 1991;38:32-6.
Weissenbach J, Gyapay G, Dib C, et al. A second-generation linkage map of the human genome. Nature 1992:539:794-801.
Whitehouse W, Rees M, Curtis D, et al. Linkage analysis of idiopathic generalized epilepsy (ICE) and marker loci on chromosome 6p in families of patients with juvenile myoclonic epilepsy: no evi- dence for an epilepsy locus in the HLA region. Am J H i m Gene!
Wolf P. Goosses R. Relation of photosensitivity to epileptic syn- dromes. J Neiirol Neiirosurg P.r.vchiatrj1 1986;49: 1386-9 I .
Zeman W, Donahue S. Dyken P, Green J. The neuronal ceroid- lipofuscinoses (Batten-Vogt syndrome). In: Vinken PJ, Bruyn GW. eds. Handbook qfclinical neiirologj: vol. 10. Amsterdam: North- Holland, I970:588-679.
I895;7:32-67.
165-74.
199355~652-62.
Epilepsiu. Vol. 35. Suppl. 1. 1994