
Astrocytes regulate GluR2 expression in motor neurons and their vulnerability to excitotoxicity
Upload
independentCategory
view
4download
0
Neurobiology of Disease xxx (2012) xxx–xxx
YNBDI-02584; No. of pages: 8; 4C: 7
Contents lists available at SciVerse ScienceDirect
Neurobiology of Disease
j ourna l homepage: www.e lsev ie r .com/ locate /ynbd i
P2X7 receptor blockade prevents ATP excitotoxicity in neurons and reduces braindamage after ischemia
Joana Arbeloa a,1, Alberto Pérez-Samartín a,1, Miroslav Gottlieb b, Carlos Matute a,⁎a CIBERNED and Laboratory of Neurobiology, Department of Neurosciences, University of the Basque Country, 48940-Leioa, Spainb Institute of Neurobiology, Slovak Academy of Sciences, 04001-Kosice, Slovak Republic
⁎ Corresponding author. Fax: +34 94 6015055.E-mail address: [email protected] (C. Matute).
1 Both authors contributed equally to this study.Available online on ScienceDirect (www.scienced
0969-9961/$ – see front matter © 2011 Elsevier Inc. Alldoi:10.1016/j.nbd.2011.12.014
Please cite this article as: Arbeloa, J., et al.,ischemia, Neurobiol. Dis. (2012), doi:10.10
a b s t r a c t
a r t i c l e i n f oArticle history:Received 13 July 2011Revised 9 November 2011Accepted 4 December 2011Available online xxxx
Keywords:ExcitotoxicityIschemiaNeuronal damageP2X7
Overactivation of subtype P2X7 receptors can induce excitotoxic neuronal death by calcium (Ca2+) overload.In this study, we characterize the functional properties of P2X7 receptors using electrophysiology and Ca2+
monitoring in primary cortical neuron cultures and in brain slices. Both electrical responses and Ca2+ influxinduced by ATP and benzoyl-ATP were reduced by Brilliant Blue G (BBG) at concentrations which specificallyinhibit P2X7 receptors. In turn, oxygen-glucose deprivation (OGD) caused neuronal death that was reducedwith BBG application. OGD in neuron cultures and brain slices generated an inward current, which wasdelayed and reduced by BBG. To assess the relevance of these in vitro findings, we used middle cerebral arteryocclusion in rats as a model of transient focal cerebral ischemia to study the neuroprotective effect of BBG invivo. Treatment with BBG (twice per day, 30 mg/kg) produced a 60% reduction in the extent of brain damagecompared to treatment with vehicle alone. These results show that P2X7 purinergic receptors mediate tissuedamage after OGD in neurons and following transient brain ischemia. Therefore, these receptors are a rele-vant molecular target for the development of new treatments to attenuate brain damage following stroke.
© 2011 Elsevier Inc. All rights reserved.
Introduction
Cell surface purine/pyrimidine nucleotide receptors, termed P2receptors (Ralevic and Burnstock, 1998), are activated by adenosinetriphosphate (ATP) and subdivided into two major groups: metabo-tropic receptors, P2Y, which are G-protein coupled [P2Y1,2,4,6,11–14], and ionotropic receptors, P2X, which are ligand-gated ion channels [P2X1–7] (Burnstock, 2007). P2X receptors arecation-selective channels with almost equal permeability to Na+
and K+, significant permeability to Ca2+, and, at least concerningP2X7 receptors, permeable to molecules up to 700 Da in size(Surprenant et al., 1996). P2X receptors are expressed throughoutthe central and peripheral nervous systems (Burnstock andKnight, 2004; Gever et al., 2006) and are involved in a widerange of physiologic and pathologic processes (Khakh and North,2006). Specifically, P2X7 receptors in peripheral tissues mediate in-flammation, cell proliferation, and apoptosis (Burnstock, 2007)while in the nervous system they are involved in modulation ofneurotransmitter release, as well as microglial and astroglial activa-tion (Sperlágh et al., 2006).
During and after stressful events and pathological conditionssuch as ischemia, damaged neurons and nonneuronal cells release
irect.com).
rights reserved.
P2X7 receptor blockade prev16/j.nbd.2011.12.014
ATP into the extracellular space (Braun et al., 1998; Juranyi et al.,1999; Melani et al., 2005; for recent reviews, see also Rossi et al.,2007 and Yenari et al., 2010). Previous studies reported conflictingdata concerning the participation of P2X7 receptors in neuronaldeath after excitotoxicity. Deletion of P2X7 receptors did not affectcell death induced by transient cerebral ischemia and P2X antago-nists did not affect ischemic or excitotoxic death (Le Feuvre et al.,2002). However, more recent studies found that traumatic damageto the spinal cord caused the release of ATP and lethal overactivationof P2X7 receptors in neurons (Wang et al., 2004). In addition, block-age of P2X7 receptors is protective in white matter injury (Domercqet al., 2010). In this study we reanalyzed the idea that P2X7 recep-tors mediate ischemic brain damage. Here, we provide evidencethat these receptors are indeed crucial in triggering neuronal deathafter ischemia both in vitro and in vivo.
Material and methods
Animals
All experiments were conducted under the supervision and withthe approval of our internal animal ethics committee (University ofthe Basque Country, UPV/EHU). Animals were handled in accordancewith the European Communities Council Directive. All possible effortswere made to minimize animal suffering and the number of animalsused.
ents ATP excitotoxicity in neurons and reduces brain damage after

2 J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
Cortical neuron culture
Cortical neurons were obtained from the E18 Sprague–Dawley ratembryos according to previously described procedures (Cheung et al.,1998; Larm et al., 1996). Neurons were resuspended in B27 Neuroba-sal medium plus 10% fetal bovine serum (FBS) and then seeded ontopoly-L-ornithine-coated plates or glass coverslips at 1.5×105 cells perwell. The medium was replaced by serum-free, B27-supplementedNeurobasal medium 24 h later. The cultures were essentially free ofastrocytes and microglia and were maintained at 37 °C and 5% CO2.Cultures were used at 8–10 days in vitro (div).
Oxygen and glucose deprivation (OGD) in neuronal cultures
In vitro ischemia (1 h) was achieved by replacing O2 with N2 andexternal glucose (10 mM) with sucrose and adding iodoacetate(IAA; 20, 50, and 100 μM) to block glycolysis in an extracellular solu-tion containing (in mM) NaCl (130), KCl (5.4), CaCl2 (1.8), NaHCO3
(26), MgCl2 (0.8), and NaH2PO4 (1.18) (pH 7.4). Cell death was deter-mined 24 h later using calcein-AM (Invitrogen, Barcelona, Spain) aspreviously described (Matute et al., 2007) using a Synergy-HT fluo-rimeter (BioTek Instruments). The antagonist was applied duringOGD and during the next 24 h incubation period. Data wereexpressed as percentage of cell death versus the respective controlwith or without antagonist, which did not alter cell survival (datanot shown).
Electrophysiology and Ca2+ imaging in cultured cells
Whole-cell voltage-clamp recordings of cortical neurons wereperformed at room temperature using the EPC-7 patch-clamp ampli-fier (HEKA Elektronik, Lambrecht, Germany). Currents were recordedat a holding membrane potential of −70 mV. Extracellular bath solu-tion with a pH of 7.3 contained the following (in mM): NaCl (150),KCl (5), CaCl2 (2.5), MgCl2 (1), HEPES (10), and glucose (10). Divalentcation-free extracellular solutions were obtained by omitting Ca2+
and Mg2+. Patch-clamp pipettes (5–7 MΩ) were filled with internalsolution at a pH of 7.3 containing the following (in mM): potassiumgluconate (140), CaCl2 (1), MgCl2 (2), HEPES (10), EGTA (10), Na-GTP (0.2), and Mg-ATP (2). For electrophysiology monitoring, ische-mia was induced chemically using the glycolytic blocker IAA(1 mM), the oxidative phosphorylation inhibitor antimycin(0.25 μM), and replacing glucose with sucrose.
For Ca2+ recording, cells were loaded with fura-2 AM (5 μM; Invi-trogen, Barcelona, Spain) in culture medium for 30 min at 37 °C. Ex-periments were carried out in a chamber perfused with a buffercontaining (in mM) NaCl (150), KCl (5), CaCl2 (2.5), MgCl2 (1),HEPES (10), and glucose (10) at 1 ml/min. Cells were visualizedwith a high-resolution digital B/W CCD camera (ORCA; HamamatsuPhotonics Iberica, Barcelona, Spain). Image acquisition (acquisitionrate 1/300 ms) and data analysis were carried out using the AquaCos-mos software program (Hamamatsu Photonics Iberica). [Ca2+]i wasestimated by the 340/380 ratio method, using a Kd value of 224 nM.
Electrophysiology in brain slices
Brain slice recordings were carried out using 12–13 day old Spra-gue Dawley rats. Animals were anesthetized with isofluorane. Thebrain was rapidly dissected and tissue block was cut in horizontalslices (300 μm thick) on a vibratome (Pelco 100, Pelco, Clovis, CA,USA) in ice cold artificial cerebrospinal fluid containing (in mM):NaCl (126), NaHCO3 (24), NaH2PO4 (1), KCl (2.5), CaCl2 (2.5), MgCl2(2), and D-glucose (10) (bubbled with 95% O2/5% CO2) at pH 7.4.Slices were allowed to recover at least 1 h and were then transferredto a chamber with continuous flow (1.5 ml/min). Cells were visual-ized using upright microscopy (Leica DMLFSA, Germany). Whole
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
cell voltage-clamp recordings of cortical neurons were performedusing a Multiclamp 700B amplifier (Molecular Devices, U.S.A.). Pi-pettes (4–7 MΩ) were filled with the same internal solution usedabove. To simulate ischemia, glucose was replaced by 7 mM sucroseand 95% O2/5% CO2 was replaced with 95% N2/5% CO2.
Transient middle cerebral artery occlusion
Transient focal ischemia was induced by middle cerebral arteryocclusion (MCAO) in male Wistar rats (270–300 g) using the intra-luminal filament technique (Longa et al., 1989). Rats were anesthe-tized with 4% halothane in an anesthetic chamber and maintainedduring surgery with 1.5% halothane using a rodent mask. Body tem-perature was maintained at 37 °C with a heat pad. MCAO was carriedout for 90 min by inserting a 4-0 nylon monofilament via the right ex-ternal carotid artery into the internal carotid artery to block the originof the middle cerebral artery (MCA). The P2X7 receptor antagonistBBG (30 mg/kg body weight per day), which crosses the blood–brain-barrier (Peng et al., 2009), was administrated intraperitoneallyevery 12 h beginning 30 min after the onset of ischemia. Animalswere euthanized 3 days after ischemia, the brain was removed andinfarct volume and number of degenerated neurons were calculatedas described below. Sham-operated controls were surgically treatedas the ischemic group, but the middle cerebral artery was notoccluded.
Neurological examination
Neurological deficit was assessed in each animal on a numericalscale of 0–4 at the end of ischemic insult and 60 min after MCAOand, later, at intervals of 24 h. The scoring system based onBederson et al. (1986) was used: 0, no detectable deficits; 1, forelimbflexion and torso turning to the contralateral side when lifted by tail;2, same behavior as grade 1 and decreased resistance to lateral push;3, same behavior as grade 2 with unilateral circling; 4, no spontane-ous walking and a depressed level of consciousness. Rats with a neu-rological deficit lower than 2 were excluded from the study.
Determination of brain infarct and histological analysis
Analysis of cerebral ischemic damage was carried out using 2,3,5-triphenyl tetrazolium chloride (TTC), FluoroJade C staining, and NeuNimmunohistochemistry. TTC stains dehydrogenases and its absenceallows quantification of the infarct area, whereas FluoroJade C is amarker for degenerating neurons (Schmued et al., 2005).
The animals were euthanized after reperfusion, under chloral hy-drate anesthesia followed by decapitation. The brains were rapidlydissected out and the forebrains cut into seven coronal sections,2 mm thick, using a rat brain matrix (Activational Systems, MI,USA). Analysis of cerebral ischemic damage was carried out using2,3,5-triphenyltetrazolium chloride (TTC, Sigma). The sections werestained by incubating them in a 1% solution at 37 °C for 15 min andfixed in 10% buffered formalin. Anterior and posterior sides of all sec-tions were scanned using a high-resolution scanner (Hewlett PackardScanjet). The non-ischemic hemisphere, ischemic hemisphere, andinfarct area of each brain section was measured in a blinded manner,using Image J software (National Institutes of Health, Bethesda, Mary-land, USA). The average infarct area (mm2) in each section was calcu-lated by the following formula: (infarct area on the anterior surface+infarct area on the posterior surface)/2.
The corrected infarct area in each slice was calculated to compen-sate for brain edema (Callaway et al., 2000). Corrected infarct vol-umes (mm3) were calculated by multiplying the corrected area bythe slice thickness and summing the volume.
Coronal sections after TTC staining were cryostat cut at 10 μm, sec-tions mounted onto gelatinized microscope slides, and stored at
ents ATP excitotoxicity in neurons and reduces brain damage after

3J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
−20 °C until staining. Some sections were air dried and stained with0.0001% Fluoro Jade C for 20 min, washed, and coverslipped with DPX(Schmued et al., 2005). The slides were examined using Zeiss Axoplanmicroscope and images acquired using a digital camera. Fluoro-Jade Cpositive cells were counted.
For immunohistochemistry, sections were incubated with primaryantibodies (mouse monoclonal anti-NeuN (1:200; Millipore, Madrid,Spain) overnight at 4 °C and subsequently with fluorescent secondaryAlexa 594 goat anti-mouse (1:200; Molecular Probes) antibodies for2 h at room temperature, and then coverslipped for image analysis.Microphotographs were taken from the ipsilateral and contralateralsides of the cerebral cortex with a 20× objective and NeuN positivecells were counted.
Statistical analysis
All data are reported as mean±SEM. p values are from Student'st-tests and were considered significant when less than 0.05. Formore than two groups, ANOVA and Bonferroni post hoc test weredone. The electrophysiological dose–response experiment curveswere obtained by non linear dose–response sigmoidal regression.The effect of BBG on the neurological score was examined by Kruskal–Wallis analysis and the difference between the two groups wasanalyzed with Mann–Whitney U test. All data were analyzed usingGraphPad Prism v. 4 software.
Results
Cortical neurons express functional P2X7 receptors
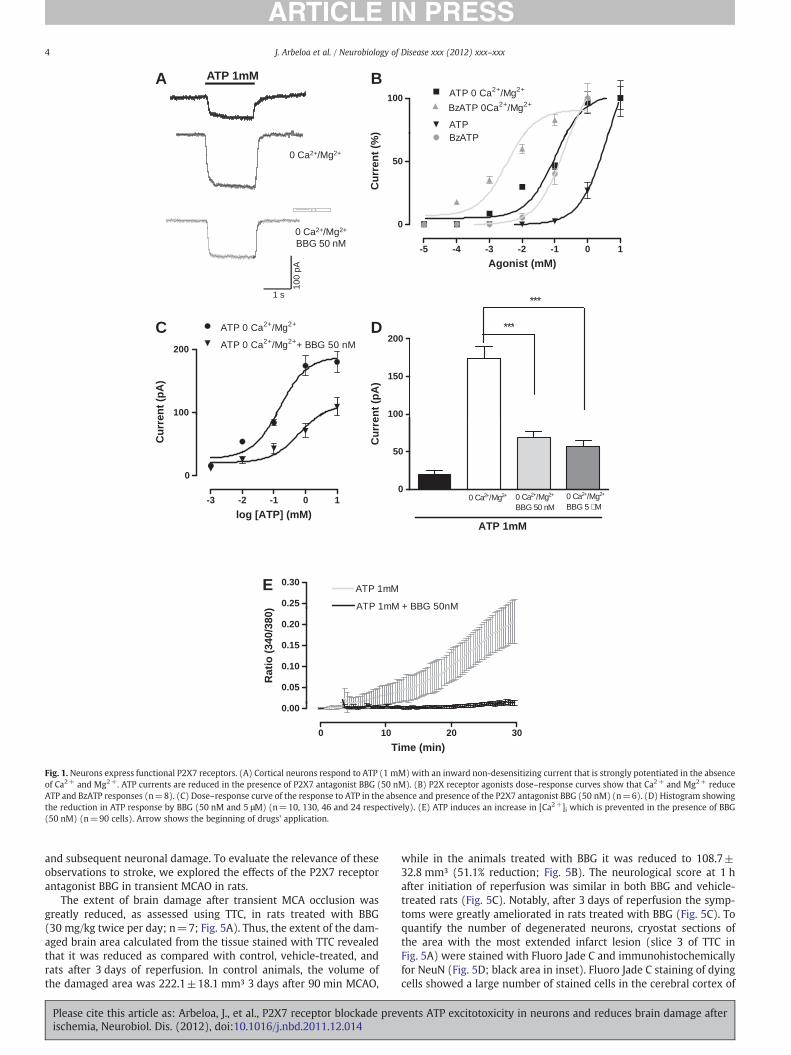
We initially studied the properties of P2X7 receptors in primarycortical neuron cultures using electrophysiological and Ca2+ moni-toring techniques. In normal extracellular Ringer bath solution, ATP(1 mM) induced a non-desensitizing inward current (20.13±4.75pA, n=10; Figs. 1A, D). This current was strongly potentiated(173.68±16.04 pA, n=130; Figs. 1A, D) when the extracellular solu-tion was free of divalent cations Ca2+ and Mg2+. Although the re-sponses had the same kinetics the EC50 changed from 4.2 mM to153.4 μM in the presence or absence of Ca2+ and Mg2+, respectively(Fig. 1B). The ATP analog BzATP (100 μM) also induced an inwardcurrent (110.8±7.6 pA, n=25) and the dose–response analysis indi-cated that BzATP was an order of magnitude more potent than ATP, aspecific feature of the P2X7 receptors (Moatassim and Dubyak, 1992).Its EC50 value in Ca2+- and Mg2+- free medium was 9.7 μM (Fig. 1B).
BBG is a potent P2X7 antagonist and at 50 nM is selective for theP2X7 subtype (Jiang et al., 2000; Anderson and Nedergaard, 2006).BBG at 50 nM substantially reduced ATP (1 mM) responses (68.2±8.3 pA, n=40; Figs. 1A, C, and D). A further slight reduction ofthe responses to ATP (1 mM) was observed when BBG was appliedat 5 μM (56.3±7.8 pA, n=40; Fig. 1D), which supports the ideathat other BBG sensitive P2X receptors are involved in ATP responsesin cortical neurons in culture, but only marginally (Jiang et al., 2000).A similar blockade of the ATP (1 mM) responses was observed withthe more recently developed P2X7 antagonist A438079 (100 μM;65.3±5.38% reduction).
To further characterize the functional properties of P2X7 recep-tors, we next monitored the concentration of intracellular Ca2+ incultured neurons after application of ATP. Application of ATP 1 mMcaused a sustained increase in cytosolic Ca2+ that was completelyblocked by BBG (50 nM) (Fig. 1E).
Together, this pharmacological profile of responses to ATP indi-cates that P2X7 receptors are the major P2X receptor in cortical neu-rons, and that other P2X receptors contribute only marginally toelectrophysiologic and Ca2+ responses to ATP in these cells.
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
P2X7 antagonist prevents neurons from death after OGD
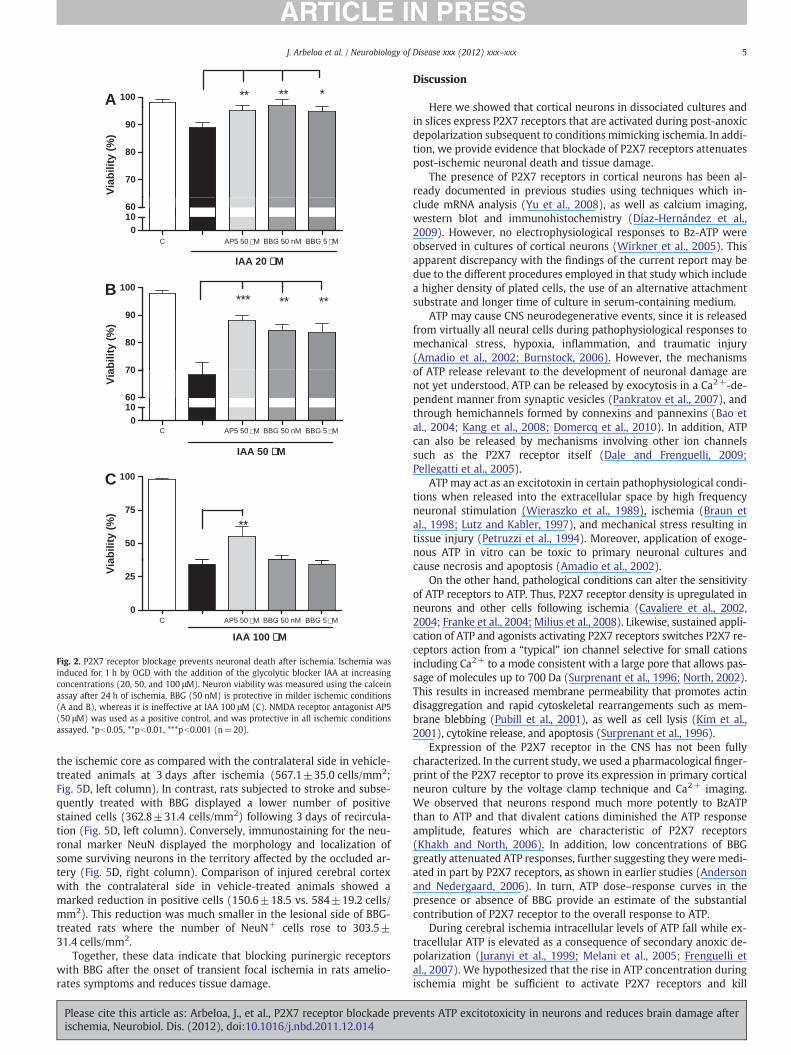
To examine the role of P2X receptors in cell death after ischemia,we mimicked ischemic conditions by depriving neurons of oxygenand glucose in the presence of various concentrations of the glycolyticinhibitor IAA. Under these conditions, neuronal death was dependenton IAA concentration (20 to 100 μM) (Fig. 2). The effects of ischemiawere greatly reduced by BBG at 50 nM and 5 μM when the ischemicconditions were less severe (IAA at 20 or 50 μM), and the protectiveeffect of BBG was comparable to that of the NMDA blocker AP5(50 μM; Fig. 2). However, and in contrast to AP5, BBG was not effec-tive in attenuating ischemic neuronal death at more stringent condi-tions (IAA at 100 μM; Fig. 2). As the protective effect on neuronaldeath observed with lower and higher concentrations of BBG weresimilar, these findings indicate that P2X7 receptors mediate ischemicdamage to neurons.
OGD induces an inward current in neuron cultures which is modulatedby BBG
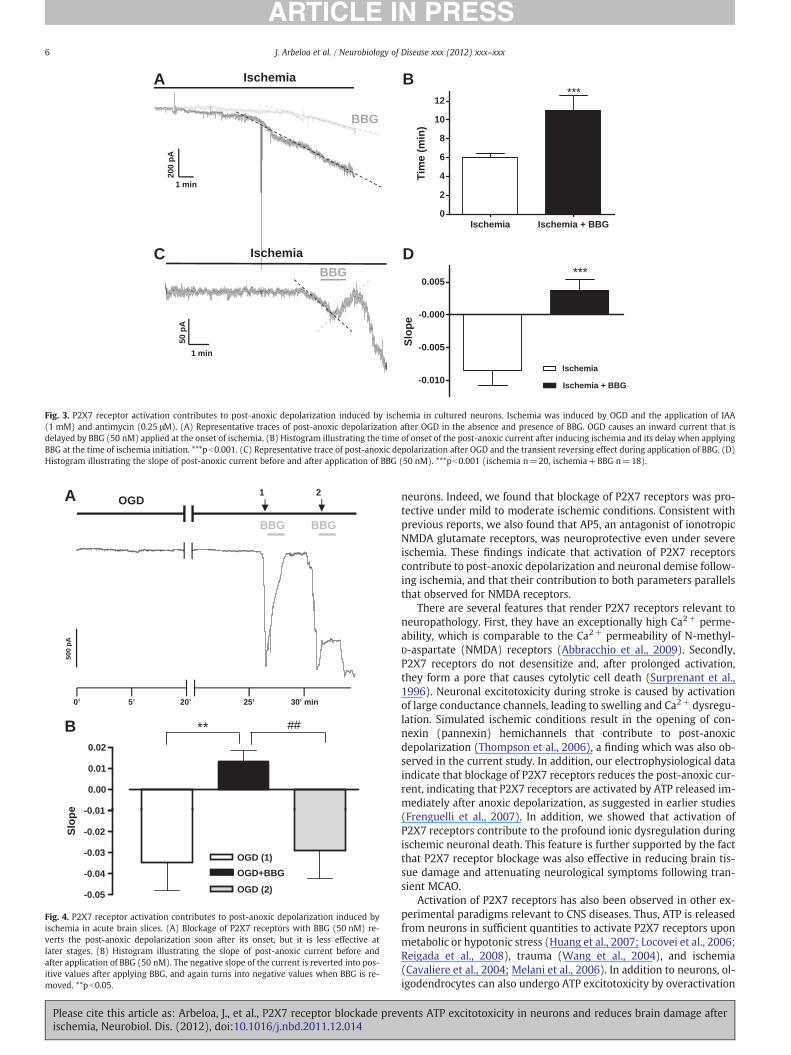
We next studied whether P2X7 receptor activation contributes topost-anoxic depolarization after the onset of ischemia OGD. Ischemicconditions were simulated by oxygen and glucose deprivation plusthe addition of IAA 1 mM and antimycin 0.25 μM to the cultures.These ischemic conditions activated an inward current in neuronswithin 5.65±0.5 min (Fig. 3A). Ischemia-induced currents had a pur-inergic receptor-mediated component as their onset was delayed to11±1.5 min in the presence of BBG (Figs. 3A and B) and their slopewas significantly lower (m=−0.0085±0.0018 in control ischemiaversus m=−0.0046±0.0009 in ischemia during exposure to BBG).
We then tested the effect of BBG after the onset of post-anoxiccurrent. To that end, we measured the current slopes before andafter applying BBG; results were m=−0.0085±0.0022 andm=0.0037±0.0017, respectively (Figs. 3C and D). This change inthe current slope from negative to positive indicates that P2X7 recep-tors opened during ischemia are blocked by BBG. In addition, we test-ed the pannexin 1 hemichannel blocker probenecid (1 mM) andfound that its application after the onset of the post-anoxic currenthad a similar effect than BBG (slope m=0.0029±0,001085, pb0.01as compared to ischemia control).
These findings suggest that during ischemia, P2X7 receptors areactivated and that their activation contributes to post-anoxic depolar-ization in neurons, and that the pannexin 1 hemichannel opening alsomakes a substantial contribution.
Blockade of P2X7 receptors suppresses the post-anoxic ischemic currentin neurons in brain slices
We next examined if activation of P2X7 receptors during anoxicdepolarization also occurs in neurons in brain slices following acuteischemia, a more integral preparation than neuronal cultures. As incultures of cortical neurons, ischemia activated an inward currentthat in slices showed a latency of 18.1±2.3 min (n=14; Fig. 4A). Ad-dition of BBG (50 nM) to the perfusion solution after the onset of thepost-anoxic depolarization effectively reversed the inward current(Figs. 4A, B). Thus, the slope of the current changed from −0.035±0.013 to 0.013±0.051 (Figs. 4A, B). Basal membrane current also re-covered substantially (61%±9.5). However, the efficacy of BBGdecayed with the progression of the ischemic insult (Figs. 4A, B).
Blockade of P2X7 receptor reduces brain damage after MCAO
The findings above indicate that activation of P2X7 receptors con-tributes to the onset of the post-anoxic depolarization current leadingto neuronal demise in neuronal cultures and in acute brain slices, andthat blockage of P2X7 receptors attenuates the post-ischemic current
ents ATP excitotoxicity in neurons and reduces brain damage after
BA ATP 1mM
100 ATP 0 Ca2+/Mg2+
BzATP 0Ca2+/Mg2+
ATP
0 Ca2+/Mg2+
0 C 2 /M 20
50
BzATP
Cu
rren
t (%
)
p A
C D
1 s10
0 pA
0 Ca2+/Mg2+
BBG 50 nM -5 -4 -3 -2 -1 0 1
Agonist (mM)
***
***
ATP 0 Ca2+/Mg2++ BBG 50 nM
ATP 0 Ca2+/Mg2+
100
150
200***
-3 -2 -1 0 1
Cu
rren
t (p
A)
0
50
0 Ca2+/Mg2+ 0 Ca2+/Mg2+
BBG 5 µM0 Ca2+/Mg2+
BBG 50 nM
Cu
rren
t (p
A)
E0.25
0.30ATP 1mM
ATP 1mM + BBG 50nM
log [ATP] (mM)ATP 1mM
0.05
0.10
0.15
0.20
ATP 1mM + BBG 50nM
Rat
io (
340/
380)
0 10 20 30
0.00
Time (min)
100
200
0
Fig. 1. Neurons express functional P2X7 receptors. (A) Cortical neurons respond to ATP (1 mM) with an inward non-desensitizing current that is strongly potentiated in the absenceof Ca2+ and Mg2+. ATP currents are reduced in the presence of P2X7 antagonist BBG (50 nM). (B) P2X receptor agonists dose–response curves show that Ca2+ and Mg2+ reduceATP and BzATP responses (n=8). (C) Dose–response curve of the response to ATP in the absence and presence of the P2X7 antagonist BBG (50 nM) (n=6). (D) Histogram showingthe reduction in ATP response by BBG (50 nM and 5 μM) (n=10, 130, 46 and 24 respectively). (E) ATP induces an increase in [Ca2+]i which is prevented in the presence of BBG(50 nM) (n=90 cells). Arrow shows the beginning of drugs' application.
4 J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
and subsequent neuronal damage. To evaluate the relevance of theseobservations to stroke, we explored the effects of the P2X7 receptorantagonist BBG in transient MCAO in rats.
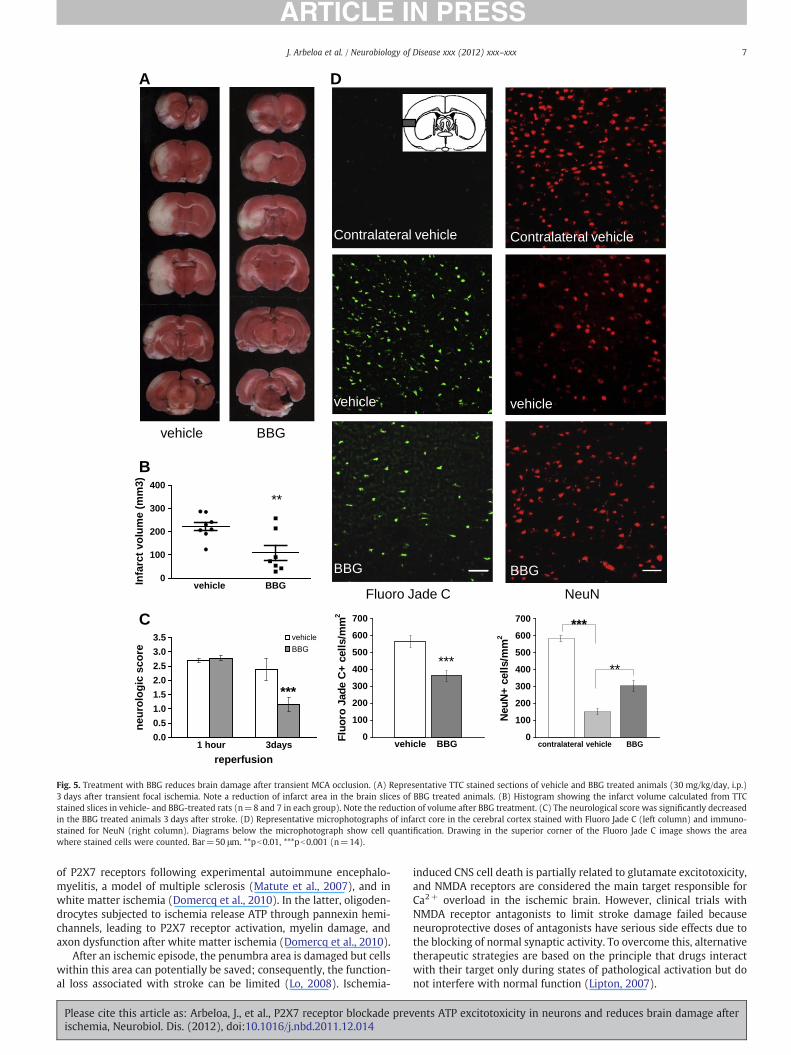
The extent of brain damage after transient MCA occlusion wasgreatly reduced, as assessed using TTC, in rats treated with BBG(30 mg/kg twice per day; n=7; Fig. 5A). Thus, the extent of the dam-aged brain area calculated from the tissue stained with TTC revealedthat it was reduced as compared with control, vehicle-treated, andrats after 3 days of reperfusion. In control animals, the volume ofthe damaged area was 222.1±18.1 mm³ 3 days after 90 min MCAO,
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
while in the animals treated with BBG it was reduced to 108.7±32.8 mm³ (51.1% reduction; Fig. 5B). The neurological score at 1 hafter initiation of reperfusion was similar in both BBG and vehicle-treated rats (Fig. 5C). Notably, after 3 days of reperfusion the symp-toms were greatly ameliorated in rats treated with BBG (Fig. 5C). Toquantify the number of degenerated neurons, cryostat sections ofthe area with the most extended infarct lesion (slice 3 of TTC inFig. 5A) were stained with Fluoro Jade C and immunohistochemicallyfor NeuN (Fig. 5D; black area in inset). Fluoro Jade C staining of dyingcells showed a large number of stained cells in the cerebral cortex of
ents ATP excitotoxicity in neurons and reduces brain damage after
A ** ** *100
70
80
90
B
C AP5 50 µM BBG 50 nM BBG 5 µM
01060
*** ** **
70
80
90
100
C AP5 50 µM BBG 50 nM BBG 5 µM0
10
IAA 50 µM
IAA 20 µM
IAA 100 µM
60
C
50
75
100
**
C AP5 50 µM BBG 50 nM BBG 5 µM0
25
Via
bili
ty (
%)
Via
bili
ty (
%)
Via
bili
ty (
%)
Fig. 2. P2X7 receptor blockage prevents neuronal death after ischemia. Ischemia wasinduced for 1 h by OGD with the addition of the glycolytic blocker IAA at increasingconcentrations (20, 50, and 100 μM). Neuron viability was measured using the calceinassay after 24 h of ischemia. BBG (50 nM) is protective in milder ischemic conditions(A and B), whereas it is ineffective at IAA 100 μM (C). NMDA receptor antagonist AP5(50 μM) was used as a positive control, and was protective in all ischemic conditionsassayed. *pb0.05, **pb0.01, ***pb0.001 (n=20).
5J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
the ischemic core as compared with the contralateral side in vehicle-treated animals at 3 days after ischemia (567.1±35.0 cells/mm2;Fig. 5D, left column). In contrast, rats subjected to stroke and subse-quently treated with BBG displayed a lower number of positivestained cells (362.8±31.4 cells/mm2) following 3 days of recircula-tion (Fig. 5D, left column). Conversely, immunostaining for the neu-ronal marker NeuN displayed the morphology and localization ofsome surviving neurons in the territory affected by the occluded ar-tery (Fig. 5D, right column). Comparison of injured cerebral cortexwith the contralateral side in vehicle-treated animals showed amarked reduction in positive cells (150.6±18.5 vs. 584±19.2 cells/mm2). This reduction was much smaller in the lesional side of BBG-treated rats where the number of NeuN+ cells rose to 303.5±31.4 cells/mm2.
Together, these data indicate that blocking purinergic receptorswith BBG after the onset of transient focal ischemia in rats amelio-rates symptoms and reduces tissue damage.
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
Discussion
Here we showed that cortical neurons in dissociated cultures andin slices express P2X7 receptors that are activated during post-anoxicdepolarization subsequent to conditions mimicking ischemia. In addi-tion, we provide evidence that blockade of P2X7 receptors attenuatespost-ischemic neuronal death and tissue damage.
The presence of P2X7 receptors in cortical neurons has been al-ready documented in previous studies using techniques which in-clude mRNA analysis (Yu et al., 2008), as well as calcium imaging,western blot and immunohistochemistry (Díaz-Hernández et al.,2009). However, no electrophysiological responses to Bz-ATP wereobserved in cultures of cortical neurons (Wirkner et al., 2005). Thisapparent discrepancy with the findings of the current report may bedue to the different procedures employed in that study which includea higher density of plated cells, the use of an alternative attachmentsubstrate and longer time of culture in serum-containing medium.
ATP may cause CNS neurodegenerative events, since it is releasedfrom virtually all neural cells during pathophysiological responses tomechanical stress, hypoxia, inflammation, and traumatic injury(Amadio et al., 2002; Burnstock, 2006). However, the mechanismsof ATP release relevant to the development of neuronal damage arenot yet understood. ATP can be released by exocytosis in a Ca2+-de-pendent manner from synaptic vesicles (Pankratov et al., 2007), andthrough hemichannels formed by connexins and pannexins (Bao etal., 2004; Kang et al., 2008; Domercq et al., 2010). In addition, ATPcan also be released by mechanisms involving other ion channelssuch as the P2X7 receptor itself (Dale and Frenguelli, 2009;Pellegatti et al., 2005).
ATP may act as an excitotoxin in certain pathophysiological condi-tions when released into the extracellular space by high frequencyneuronal stimulation (Wieraszko et al., 1989), ischemia (Braun etal., 1998; Lutz and Kabler, 1997), and mechanical stress resulting intissue injury (Petruzzi et al., 1994). Moreover, application of exoge-nous ATP in vitro can be toxic to primary neuronal cultures andcause necrosis and apoptosis (Amadio et al., 2002).
On the other hand, pathological conditions can alter the sensitivityof ATP receptors to ATP. Thus, P2X7 receptor density is upregulated inneurons and other cells following ischemia (Cavaliere et al., 2002,2004; Franke et al., 2004; Milius et al., 2008). Likewise, sustained appli-cation of ATP and agonists activating P2X7 receptors switches P2X7 re-ceptors action from a “typical” ion channel selective for small cationsincluding Ca2+ to a mode consistent with a large pore that allows pas-sage of molecules up to 700 Da (Surprenant et al., 1996; North, 2002).This results in increased membrane permeability that promotes actindisaggregation and rapid cytoskeletal rearrangements such as mem-brane blebbing (Pubill et al., 2001), as well as cell lysis (Kim et al.,2001), cytokine release, and apoptosis (Surprenant et al., 1996).
Expression of the P2X7 receptor in the CNS has not been fullycharacterized. In the current study, we used a pharmacological finger-print of the P2X7 receptor to prove its expression in primary corticalneuron culture by the voltage clamp technique and Ca2+ imaging.We observed that neurons respond much more potently to BzATPthan to ATP and that divalent cations diminished the ATP responseamplitude, features which are characteristic of P2X7 receptors(Khakh and North, 2006). In addition, low concentrations of BBGgreatly attenuated ATP responses, further suggesting they were medi-ated in part by P2X7 receptors, as shown in earlier studies (Andersonand Nedergaard, 2006). In turn, ATP dose–response curves in thepresence or absence of BBG provide an estimate of the substantialcontribution of P2X7 receptor to the overall response to ATP.
During cerebral ischemia intracellular levels of ATP fall while ex-tracellular ATP is elevated as a consequence of secondary anoxic de-polarization (Juranyi et al., 1999; Melani et al., 2005; Frenguelli etal., 2007). We hypothesized that the rise in ATP concentration duringischemia might be sufficient to activate P2X7 receptors and kill
ents ATP excitotoxicity in neurons and reduces brain damage after
D
50 p
A
1 min
BBGC
200
pA
1 min
Ischemia
BBG
A B
Ischemia Ischemia + BBG0
2
4
6
8
10
12***
Tim
e (m
in)
-0.010
-0.005
-0.000
0.005
Ischemia
Ischemia + BBG
***
Slo
pe
Ischemia
Fig. 3. P2X7 receptor activation contributes to post-anoxic depolarization induced by ischemia in cultured neurons. Ischemia was induced by OGD and the application of IAA(1 mM) and antimycin (0.25 μM). (A) Representative traces of post-anoxic depolarization after OGD in the absence and presence of BBG. OGD causes an inward current that isdelayed by BBG (50 nM) applied at the onset of ischemia. (B) Histogram illustrating the time of onset of the post-anoxic current after inducing ischemia and its delay when applyingBBG at the time of ischemia initiation. ***pb0.001. (C) Representative trace of post-anoxic depolarization after OGD and the transient reversing effect during application of BBG. (D)Histogram illustrating the slope of post-anoxic current before and after application of BBG (50 nM). ***pb0.001 (ischemia n=20, ischemia+BBG n=18).
A
B
-0.01
0.00
0.01
0.02
** ##
Slo
pe
OGD
5’ 20’0’ 25’ 30’ min
BBG BBG
1 2
500
pA
-0.05
-0.04
-0.03
-0.02
OGD (1)
OGD+BBG
OGD (2)
Fig. 4. P2X7 receptor activation contributes to post-anoxic depolarization induced byischemia in acute brain slices. (A) Blockage of P2X7 receptors with BBG (50 nM) re-verts the post-anoxic depolarization soon after its onset, but it is less effective atlater stages. (B) Histogram illustrating the slope of post-anoxic current before andafter application of BBG (50 nM). The negative slope of the current is reverted into pos-itive values after applying BBG, and again turns into negative values when BBG is re-moved. **pb0.05.
6 J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
neurons. Indeed, we found that blockage of P2X7 receptors was pro-tective under mild to moderate ischemic conditions. Consistent withprevious reports, we also found that AP5, an antagonist of ionotropicNMDA glutamate receptors, was neuroprotective even under severeischemia. These findings indicate that activation of P2X7 receptorscontribute to post-anoxic depolarization and neuronal demise follow-ing ischemia, and that their contribution to both parameters parallelsthat observed for NMDA receptors.
There are several features that render P2X7 receptors relevant toneuropathology. First, they have an exceptionally high Ca2+ perme-ability, which is comparable to the Ca2+ permeability of N-methyl-D-aspartate (NMDA) receptors (Abbracchio et al., 2009). Secondly,P2X7 receptors do not desensitize and, after prolonged activation,they form a pore that causes cytolytic cell death (Surprenant et al.,1996). Neuronal excitotoxicity during stroke is caused by activationof large conductance channels, leading to swelling and Ca2+ dysregu-lation. Simulated ischemic conditions result in the opening of con-nexin (pannexin) hemichannels that contribute to post-anoxicdepolarization (Thompson et al., 2006), a finding which was also ob-served in the current study. In addition, our electrophysiological dataindicate that blockage of P2X7 receptors reduces the post-anoxic cur-rent, indicating that P2X7 receptors are activated by ATP released im-mediately after anoxic depolarization, as suggested in earlier studies(Frenguelli et al., 2007). In addition, we showed that activation ofP2X7 receptors contribute to the profound ionic dysregulation duringischemic neuronal death. This feature is further supported by the factthat P2X7 receptor blockage was also effective in reducing brain tis-sue damage and attenuating neurological symptoms following tran-sient MCAO.
Activation of P2X7 receptors has also been observed in other ex-perimental paradigms relevant to CNS diseases. Thus, ATP is releasedfrom neurons in sufficient quantities to activate P2X7 receptors uponmetabolic or hypotonic stress (Huang et al., 2007; Locovei et al., 2006;Reigada et al., 2008), trauma (Wang et al., 2004), and ischemia(Cavaliere et al., 2004; Melani et al., 2006). In addition to neurons, ol-igodendrocytes can also undergo ATP excitotoxicity by overactivation
ents ATP excitotoxicity in neurons and reduces brain damage after
BBG
Contralateral vehicle
vehicle
vehicle BBG
A D
B
0.0
0.5
1.0
1.5
2.0
2.5
3.0
3.5
neu
rolo
gic
sco
re
vehicle
BBG
reperfusion1 hour 3days
C
Fluoro Jade C NeuN
***
0
100
200
300
400
500
600
700
Neu
N+
cells
/mm
2
contralateral vehicle BBG
***
**
BBG
Contralateral vehicle
vehicle
0
100
200
300
400
vehicle BBGInfa
rct
volu
me
(mm
3)
**
0
100
200
300
400
500
600
700
Fluo
ro J
ade
C+
cells
/mm
2
vehicle BBG
***
Fig. 5. Treatment with BBG reduces brain damage after transient MCA occlusion. (A) Representative TTC stained sections of vehicle and BBG treated animals (30 mg/kg/day, i.p.)3 days after transient focal ischemia. Note a reduction of infarct area in the brain slices of BBG treated animals. (B) Histogram showing the infarct volume calculated from TTCstained slices in vehicle- and BBG-treated rats (n=8 and 7 in each group). Note the reduction of volume after BBG treatment. (C) The neurological score was significantly decreasedin the BBG treated animals 3 days after stroke. (D) Representative microphotographs of infarct core in the cerebral cortex stained with Fluoro Jade C (left column) and immuno-stained for NeuN (right column). Diagrams below the microphotograph show cell quantification. Drawing in the superior corner of the Fluoro Jade C image shows the areawhere stained cells were counted. Bar=50 μm. **pb0.01, ***pb0.001 (n=14).
7J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
of P2X7 receptors following experimental autoimmune encephalo-myelitis, a model of multiple sclerosis (Matute et al., 2007), and inwhite matter ischemia (Domercq et al., 2010). In the latter, oligoden-drocytes subjected to ischemia release ATP through pannexin hemi-channels, leading to P2X7 receptor activation, myelin damage, andaxon dysfunction after white matter ischemia (Domercq et al., 2010).
After an ischemic episode, the penumbra area is damaged but cellswithin this area can potentially be saved; consequently, the function-al loss associated with stroke can be limited (Lo, 2008). Ischemia-
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
induced CNS cell death is partially related to glutamate excitotoxicity,and NMDA receptors are considered the main target responsible forCa2+ overload in the ischemic brain. However, clinical trials withNMDA receptor antagonists to limit stroke damage failed becauseneuroprotective doses of antagonists have serious side effects due tothe blocking of normal synaptic activity. To overcome this, alternativetherapeutic strategies are based on the principle that drugs interactwith their target only during states of pathological activation but donot interfere with normal function (Lipton, 2007).
ents ATP excitotoxicity in neurons and reduces brain damage after

8 J. Arbeloa et al. / Neurobiology of Disease xxx (2012) xxx–xxx
Conclusions
Results of the current study suggest that ATP excitotoxicity viaP2X7 receptors is a relevant mechanism mediating post-ischemicdamage to the brain. Thus, selective activation of the P2X7 receptorduring brain damage makes it an ideal candidate for therapeutic in-tervention to attenuate the consequences of stroke.
Acknowledgments
This work has been supported by CIBERNED, Gobierno Vasco, Uni-versidad del País Vasco and SK-VEGA 2/0146/09. J.A. was the recipientof a fellowship from Gobierno Vasco.
References
Abbracchio, M.P., Burnstock, G., Verkhratsky, A., Ziermann, H., 2009. Purinergic signal-ling in the nervous system: an overview. Trends Neurosci. 32, 19–29.
Amadio, S., D'Ambrosi, N., Cavaliere, F., Murra, B., Sancesario, G., Bernardi, G., Burnstock, G.,Volonte, C., 2002. P2 receptormodulation and cytotoxic function in cultured CNS neu-rons. Neuropharmacology 42, 489–501.
Anderson, C.M., Nedergaard, M., 2006. Emerging challenges of assigning P2X7 receptorfunction and immunoreactivity in neurons. Trends Neurosci. 29, 257–262.
Bao, L., Locovei, S., Dahl, G., 2004. Pannexin membrane channels are mechanosensitiveconduits for ATP. FEBS Lett. 572, 65–68.
Bederson, J.B., Pitts, L.H., Tsuji, M., Nishimura, M.C., Davis, R.L., Bartkowski, H., 1986. Ratmiddle cerebral artery occlusion: evaluation of the model and development of aneurologic examination. Stroke 17, 472–476.
Braun, N., Zhu, Y., Krieglstein, J., Culmsee, C., Zimmermann, H., 1998. Upregulation ofthe enzyme chain hydrolyzing extracellular ATP after transient forebrain ischemiain the rat. J. Neurosci. 18, 4891–4900.
Burnstock, G., 2006. Historical review ATP as a neurotransmitter. Trends Pharmacol.Sci. 27, 166–176.
Burnstock, G., 2007. Physiology and pathophysiology of purinergic neurotransmission.Physiol. Rev. 87, 659–797.
Burnstock, G., Knight, G.E., 2004. Cellular distribution and functions of P2 receptor sub-types in different systems. Int. Rev. Cytol. 240, 31–304.
Callaway, J.K., Knight, M.J., Watkins, D.J., Beart, P.M., Jarrott, B., Delaney, P.M., 2000. Anovel, rapid, computerised method for quantitation of neuronal damage in a ratmodel of stroke. J. Neurosci. Meth. 102, 53–60.
Cavaliere, F., Sancesario, G., Bernardi, G., Volonte, C., 2002. Extracellular ATP and nervegrowth factor intensify hypoglycemia-induced cell death in primary neurons: roleof P2 and NGFRp75 receptors. J. Neurochem. 83, 1129–1138.
Cavaliere, F., Amadio, S., Sancesario, G., Bernardi, G., Volonté, C., 2004. Synaptic P2X7and oxygen/glucose deprivation in organotypic hippocampal cultures. J. Cereb.Blood Flow Metab. 24, 392–398.
Cheung, N.S., Pascoe, C.J., Giardina, S.F., John, C.A., Beart, P.M., 1998. Micromolar L-glu-tamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurones. Neuropharmacology37, 1419–1429.
Dale, N., Frenguelli, B.G., 2009. Release of adenosine and ATP during ischemia and ep-ilepsy. Curr. Neuropharmacol. 7, 160–179.
Díaz-Hernández, M., Díez-Zaera, M., Sánchez-Nogueiro, J., Gómez-Villafuertes, R.,Canals, J.M., Alberch, J., Miras-Portugal, M.T., Lucas, J.J., 2009. Altered P2X7-receptor level and function in mouse models of Huntington's disease and thera-peutic efficacy of antagonist administration. FASEB J. 23, 1893–1906.
Domercq, M., Perez-Samartin, A., Aparicio, D., Alberdi, E., Pampliega, O., Matute, C., 2010.P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 58, 730–740.
Franke, H., Gunther, A., Grosche, J., Schmidt, R., Rossner, S., Reinhardt, R., Faber-Zuschratter, H., Schneider, D., Illes, P., 2004. P2X7 receptor expression after ischemiain the cerebral cortex of rats. J. Neuropath. Exp. Neur. 63, 686–699.
Frenguelli, B.G., Wigmore, G., Llaudet, E., Dale, N., 2007. Temporal and mechanistic dis-sociation of ATP, adenosine release during ischaemia in the mammalian hippocam-pus. J. Neurochem. 101, 1400–1413.
Gever, J.R., Cockayne, D.A., Dillon, M.P., Burnstock, G., Ford, A.P., 2006. Pharmacology ofP2X channels. Pflugers Arch. 452, 513–537.
Huang, Y.J., Maruyama, Y., Dvoryanchikov, G., Pereira, E., Chaudhari, N., Roper, S.D.,2007. The role of pannexin 1 hemichannels in ATP release and cell–cell communi-cation in mouse taste buds. Proc. Natl. Acad. Sci. U. S. A. 104, 6436–6441.
Jiang, L.H., Mackenzie, A.B., North, R.A., Surprenant, A., 2000. Brilliant blue G selectivelyblocks ATP-gated rat P2X(7) receptors. Mol. Pharmacol. 58, 82–88.
Juranyi, Z., Sperlágh, B., Vizi, E.S., 1999. Involvement of P2 purinoceptors and the nitricoxide pathway in [3H]purine outflow evoked by short-term hypoxia and hypogly-cemia in rat hippocampal slices. Brain Res. 823, 183–190.
Kang, J., Kang, N., Lovatt, D., Torres, A., Zhao, Z., Lin, J., Nedergaard, M., 2008. Connexin43 hemichannels are permeable to ATP. J. Neurosci. 28, 4702–4711.
Khakh, B.S., North, R.A., 2006. P2X receptors as cell surface ATP sensors in health anddisease. Nature 442, 527–532.
Please cite this article as: Arbeloa, J., et al., P2X7 receptor blockade previschemia, Neurobiol. Dis. (2012), doi:10.1016/j.nbd.2011.12.014
Kim, M., Jiang, L.H., Wilson, H.L., North, R.A., Surprenant, A., 2001. Proteomic andfunctional evidence for a P2X7 receptor signalling complex. EMBO J. 20,6347–6358.
Larm, J.A., Cheung, N.S., Beart, P.M., 1996. (S)-5-Fluorowillardiine-mediated neurotox-icity in cultured murine cortical neurones occurs via AMPA and kainate receptors.Eur. J. Pharmacol. 314, 249–254.
Le Feuvre, R., Brough, D., Rothwell, N., 2002. Extracellular ATP and P2X7 receptors inneurodegeneration. Eur. J. Pharmacol. 447, 261–269.
Lipton, S.A., 2007. Pathologically activated therapeutics for neuroprotection. Nat. Rev.Neurosci. 8, 803–808.
Lo, E.H., 2008. A new penumbra: transitioning from injury into repair after stroke. Nat.Med. 14, 497–500.
Locovei, S., Bao, L., Dahl, G., 2006. Pannexin 1 in erythrocytes: function without a gap.Proc. Natl. Acad. Sci. U. S. A. 103, 7655–7659.
Longa, E.Z., Weinstein, P.R., Carlson, S., Cummins, R., 1989. Reversible middle cerebralartery occlusion without craniectomy in rats. Stroke 20, 84–91.
Lutz, P.L., Kabler, S., 1997. Release of adenosine and ATP in the brain of the freshwaterturtle (Trachemys scripta) during long-term anoxia. Brain Res. 769, 281–286.
Matute, C., Torre, I., Pérez-Cerdá, F., Pérez-Samartín, A., Alberdi, E., Etxebarria, E.,Arranz, A.M., Ravid, R., Rodriguez-Antigüedad, A., Sánchez-Gómez, M.V., Domercq,M., 2007. P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytesand ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 27,9525–9533.
Melani, A., Turchi, D., Vannucchi, M.G., Cipriani, S., Gianfriddo, M., Pedata, F., 2005. ATPextracellular concentrations are increased in the rat striatum during in vivo ische-mia. Neurochem. Int. 47, 442–448.
Melani, A., Amadio, S., Gianfriddo, M., Vannucchi, M.G., Volontè, C., Bernardi, G., Pedata,F., Sancesario, G., 2006. P2X7 receptor modulation on microglial cells and reduc-tion of brain infarct caused by middle cerebral artery occlusion in rat. J. Cereb.Blood Flow Metab. 26, 974–982.
Milius, D., Sperlagh, B., Illes, P., 2008. Up-regulation of P2X7 receptor-immunoreactivity by in vitro ischemia on the plasma membrane of cultures ratcortical neurons. Neurosci. Lett. 446, 45–50.
Moatassim, C., Dubyak, G.R., 1992. A novel pathway for the activation of phospholipaseD by P2z purinergic receptors in BAC1.2F5 macrophages. J. Biol. Chem. 267,23664–23673.
North, R.A., 2002. Molecular physiology of P2X receptors. Physiol. Rev. 82, 1013–1067.Pankratov, Y., Lalo, U., Verkhratsky, A., North, R.A., 2007. Quantal release of ATP in
mouse cortex. J. Gen. Physiol. 129, 257–265.Pellegatti, P., Falzoni, S., Pinton, P., Rizzuto, R., Di Virgilio, F., 2005. A novel recombinant
plasma membrane-targeted luciferase reveals a new pathway for ATP secretion.Mol. Biol. Cell 16, 3659–3665.
Peng, W., Cotrina, M.L., Han, X., Yu, H., Bekar, L., Blum, L., Takano, T., Tian, G.F., Goldman,S.A., Nedergaard, M., 2009. Systemic administration of an antagonist of the ATP-sensitive receptor P2X7 improves recovery after spinal cord injury. Proc. Natl.Acad. Sci. U. S. A. 106 12489-1243.
Petruzzi, E., Orlando, C., Pinzani, P., Sestini, R., Del Rosso, A., Dini, G., Tanganelli, E.,Buggiani, A., Pazzagli, M., 1994. Adenosine triphosphate release by osmoticshock and hemoglobin A1C in diabetic subjects erythrocytes. Metabolism 43,435–440.
Pubill, D., Dayanithi, G., Siatka, C., Andres, M., Dufour, M.N., Guillon, G., Mendre, C.,2001. ATP induces intracellular calcium increases and actin cytoskeleton disaggre-gation via P2X receptors. Cell Calcium 29, 299–309.
Ralevic, V., Burnstock, G., 1998. Receptors for purines and pirimidines. Pharmacol. Rev.50, 413–492.
Reigada, D., Lu, W., Zhang, M., Mitchell, C.H., 2008. Elevated pressure triggers a physi-ological release of ATP from the retina: possible role for pannexin hemichannels.Neuroscience 157, 396–404.
Rossi, D.J., Brady, J.D., Mohr, C., 2007. Astrocyte metabolism and signaling during brainischemia. Nat. Neurosci. 10, 1377–1386.
Schmued, L.C., Stowers, C.C., Scallet, A.C., Xu, L., 2005. Fluoro-Jade C results in ultrahigh resolution and contrast labeling of degenerating neurons. Brain Res. 1035,24–31.
Sperlágh, B., Vizi, S., Wirkner, K., Illes, P., 2006. P2X7 receptors in the nervous system.Prog. Neurobiol. 78, 327–346.
Surprenant, A., Rassendren, F., Kawashima, E., North, R.A., Buell, G., 1996. The cytolyticP2Z receptor for extracellular ATP identified as a P2X receptor (P2X7). Science 272,735–738.
Thompson, R.J., Zhou, N., MacVicar, B.A., 2006. Ischemia opens neuronal gap junctionhemichannels. Science 312, 924–927.
Wang, X., Arcuino, G., Takano, T., Lin, J., Peng,W.G.,Wan, P., Li, P., Xu, Q., Liu, Q.S., Goldman,S.A., Nedergaard, M., 2004. P2X7 receptor inhibition improves recovery after spinalcord injury. Nat. Med. 10, 821–827.
Wieraszko, A., Goldsmith, G., Seyfried, T.N., 1989. Stimulation dependent release ofadenosine triphosphate from hippocampal slices. Brain Res. 485, 244–250.
Wirkner, K., Köfalvi, A., Fischer, W., Günther, A., Franke, H., Gröger-Arndt, H., Nörenberg,W., Madarász, E., Vizi, E.S., Schneider, D., Sperlágh, B., Illes, P., 2005. Supersensitivityof P2X receptors in cerebrocortical cell cultures after in vitro ischemia. J. Neurochem.95, 1421–1437.
Yenari, M.A., Kauppinen, T.M., Swanson, R.A., 2010. Microglial activation in stroke:therapeutic targets. Neurotherapeutics 7, 378–391.
Yu, Y., Ugawa, S., Ueda, T., Ishida, Y., Inoue, K., Kyaw Nyunt, A., Umemura, A., Mase, M.,Yamada, K., Shimada, S., 2008. Cellular localization of P2X7 receptor mRNA in therat brain. Brain Res. 1194, 45–55.
ents ATP excitotoxicity in neurons and reduces brain damage after
Copyright © 2022 FDOKUMEN




















