
MGMT expression predicts PARP-mediated resistance to temozolomide
33
1 MGMT expression predicts PARP-mediated resistance to temozolomide Oihane Erice 1,2* , Michael P. Smith 1* , Rachel White 3 , Ibai Goicoechea 1,4 , Jorge Barriuso 1 , Chris Jones 5 , Geoffrey P. Margison 6 , Juan Carlos Acosta 3 , Claudia Wellbrock 1,8 and Imanol Arozarena 1,7,8 1 Manchester Cancer Research Centre, The University of Manchester, Michael Smith Building, Oxford Road, Manchester, M13 9PT, UK 2 Current address: Division of Hepatology and Gastroenterology, Biodonostia Research Institute, Calle Doctor Begiristain, San Sebastian, 20014, Spain 3 Edinburgh Cancer Research UK Centre and MRC Institute of Genetics and Molecular Medicine, Western General Hospital. University of Edinburgh, Crewe Road South, Edinburgh, EH4 2XR, UK 4 Current address: Oncology area, Biodonostia Research Institute, Calle Doctor Begiristain, San Sebastian, 20014, Spain 5 Divisions of Molecular Pathology and Cancer Therapeutics, Institute of Cancer Research, 15 Cotswold Road, Sutton, SM2 5NG, UK 6 Centre for Occupational and Environmental Health, The University of Manchester, Stopford Building, Oxford Road, Manchester, M13 9PL, UK 7 Current address: School of Applied Sciences. University of Huddersfield. HD1 3DH *These authors contributed equally to this work 8 Corresponding authors: Imanol Arozarena and Claudia Wellbrock. Email: [email protected], Tel: +44-148-4472722 Email: [email protected], Tel: +44-161-2755189 on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810
Transcript of MGMT expression predicts PARP-mediated resistance to temozolomide

1
MGMT expression predicts PARP-mediated resistance to temozolomide
Oihane Erice1,2*, Michael P. Smith1*, Rachel White3, Ibai Goicoechea1,4, Jorge
Barriuso1, Chris Jones5, Geoffrey P. Margison6, Juan Carlos Acosta3, Claudia
Wellbrock1,8 and Imanol Arozarena1,7,8
1 Manchester Cancer Research Centre, The University of Manchester, Michael
Smith Building, Oxford Road, Manchester, M13 9PT, UK
2Current address: Division of Hepatology and Gastroenterology, Biodonostia
Research Institute, Calle Doctor Begiristain, San Sebastian, 20014, Spain
3Edinburgh Cancer Research UK Centre and MRC Institute of Genetics and
Molecular Medicine, Western General Hospital. University of Edinburgh, Crewe
Road South, Edinburgh, EH4 2XR, UK
4Current address: Oncology area, Biodonostia Research Institute, Calle Doctor
Begiristain, San Sebastian, 20014, Spain
5Divisions of Molecular Pathology and Cancer Therapeutics, Institute of Cancer
Research, 15 Cotswold Road, Sutton, SM2 5NG, UK
6Centre for Occupational and Environmental Health, The University of
Manchester, Stopford Building, Oxford Road, Manchester, M13 9PL, UK
7Current address: School of Applied Sciences. University of Huddersfield. HD1
3DH
*These authors contributed equally to this work
8Corresponding authors: Imanol Arozarena and Claudia Wellbrock.
Email: [email protected], Tel: +44-148-4472722
Email: [email protected], Tel: +44-161-2755189
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

2
Running title: MGMT mediated response to temozolomide and PARP inhibitors
Key words: melanoma, temozolomide, DNA damage, repair, MGMT, PARP
List of abbreviations: TMZ: temozolomide; MGMT: methylguanine-DNA-
methyl-transferase; OLAP: olaparib; ABT: ABT-888 or veliparib; MMS:
methylmethanesulfonate; CRC: colorectal cancer; MM: malignant melanoma;
DDR: DNA damage response
Financial information:
C. Wellbrock is a recipient of a program grant by Cancer Research UK
(C11591/A10202). J. Barriuso is funded through a fellowship by Marie Curie
(FP7-PEOPLE-2012-IEF-329702). J. C. Acosta holds fellowships by Cancer
Research UK (C47559/A16243) and Medical Research Council (R42576 MRC -
Chancellors Fellowship).
Person to whom reprints should be sent: Imanol Arozarena, School of Applied
Sciences. University of Huddersfield. HD1 3DH
Conflict of Interest Statement: The authors declare no conflict of interest.
Word count: 5020 words
Figures: 6 main figures plus 5 supplementary figures.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

3
ABSTRACT
Melanoma and other solid cancers are frequently resistant to chemotherapies
based on DNA alkylating agents such as dacarbazine and temozolomide (TMZ).
As a consequence, clinical responses are generally poor. Such resistance is partly
due to the ability of cancer cells to use a variety of DNA repair enzymes to
maintain cell viability. Particularly, the expression of MGMT has been linked to
TMZ resistance, but co-targeting MGMT has proven difficult due to dose limiting
toxicities. Here we show that the MGMT mediated resistance of cancer cells is
profoundly dependent on the DNA repair enzyme PARP. Both in vitro and in vivo
we observe that MGMT-positive cancer cells strongly respond to the combination
of TMZ and PARP inhibitors, while MGMT-deficient cells do not. In melanoma
cells TMZ induced an anti-proliferative senescent response, which was greatly
enhanced by PARP-inhibitors in MGMT-positive cells. In summary, we provide
compelling evidence to suggest that the stratification of cancer patients upon the
MGMT status would enhance the success of combination treatments using TMZ
and PARP inhibitors.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

4
INTRODUCTION
Melanoma is a form of skin cancer notoriously resistant to current therapies.
Before the arrival of targeted therapies against the BRAF and MEK components
of the hyper-activated MAPK pathway, alkylating agent-based chemotherapy was
the first line treatment for decades. In around 40% of all cases, patients qualify
for BRAF and MEK inhibitors and targeted therapies provide a good clinical
response. However, after 2-8 months, the vast majority of patients relapse (1-3).
For these patients their hopes lie in chemotherapy or immunotherapy. The only
FDA approved chemotherapeutic agent is the intravenously administered
prodrug dacarbazine, which is metabolically converted in the liver to MTIC, a
toxic monofunctional DNA alkylating agent (4, 5). An alkylating agent related to
dacarbazine but with excellent oral bioavailability is temozolomide (TMZ), which
can cross the blood brain barrier and therefore represents an alternative to
dacarbazine for melanoma with brain metastases (6-8). TMZ is an attractive
chemotherapy agent for patients with unresectable metastatic melanoma.
However single-agent escalated dose TMZ compared to single-agent dacarbazine
treatment did not improve overall survival or progression-free survival (9, 10),
and overall response rates for both drugs were 15-20% (5). TMZ is FDA
approved for anaplastic astrocytoma and glioblastoma, but again with poor
overall responses of around 26.5% (11). This suggests that in order to improve
the efficacy of TMZ, we need to enhance our understanding about the mode of
action of TMZ as a DNA damaging and cytotoxic drug.
TMZ methylates DNA bases predominantly at oxygen in position 6 and
nitrogen at position 7 in guanine and nitrogen at position 3 in adenine (O6-metG,
N7-metG and, N3-metA respectively). O6-metG is the most toxic and mutagenic
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

5
DNA modification produced by TMZ and is quickly repaired by the enzyme 06-
methyl-guanine DNA methyl transferase (MGMT). MGMT incorporates the O6-
methyl adduct into a cysteine and the enzyme is then degraded by proteolysis
(4). On the other hand N3-metA and N7-metG repair are mediated by the base
excision repair (BER) machinery in a process that involves Poly-(ADP-ribose)-
polymerase proteins (PARP), which ADP-ribosylate DNA and proteins. When
acting on DNA, PARP proteins create a docking site for the incorporation of other
components of the BER machinery, which complete the repair process (4).
The most evident mechanism of resistance to both DTIC and TMZ is MGMT
activity, and MGMT down-regulation sensitises cancer cells to TMZ (12).
Moreover, in elderly glioblastoma patients MGMT status predicts clinical
response to TMZ (13) Unfortunately, therapies combining MGMT inhibitors with
TMZ in melanoma or colorectal cancer patients have so far failed to improve
outcome due to exacerbated treatment-related haematological toxicity (14-16).
More recently several small molecule inhibitors have been developed to target
other DNA repair mechanisms, with the aim to use them as synthetically lethal
single agents or in combination therapies with chemotherapeutic agents such as
TMZ. Notably, current clinical trials are testing PARP inhibitors as monotherapy
in BRCA-deficient breast cancer patients or in combination with DNA alkylating
agents (17, 18). Combination therapies of DTIC or TMZ and PARP inhibitors are
better tolerated than those with MGMT inhibitors, but myelosuppression and
liver toxicity still represent clinical concerns (19, 20), and overall survival rates
were not improved compared to patients treated with alkylating alone (21-23).
Therefore further research is warranted to identify markers that help stratify
patients for TMZ based therapies to improve clinical outcome. Our study
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

6
suggests that MGMT mediated resistance to TMZ in melanoma and other cancer
types require PARP and that MGMT expression correlates with improved
response to combination of TMZ with PARP inhibitors.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

7
MATERIAL AND METHODS
Cell culture
Melanoma cell lines were provided by Prof. Richard Marais and Dr. Adam
Hurlstone and have been described recently (24). Colorectal carcinoma cells
were obtained from Prof. Stephen Taylor, originally from ATCC. Glioblastoma cell
lines have been described recently (25). Cell stocks were expanded and vials
kept in liquid nitrogen. New aliquots were thawed every 5-7 weeks. Melanoma
cell lines were authenticated in house by short tandem repeat profiling. Cells
were cultured in Dulbecco's Modified Eagle's Medium or in RPMI-164 medium
(SIGMA) as previously indicated, supplemented with 0.5% penicillin and
streptomycin (SIGMA) and 10% bovine calf serum (PAA, Yeovil, UK). Cells were
grown at 37ºC in a 5%CO2 environment. WM98.1-MGMT expressing cells were
established by transfection of a pcDNA3 vector encoding the cDNA for human
MGMT. MGMT expressing cells were selected using G418 as pcDNA3 expresses
the Neomycin resistance gene.
Reagents
Temozolomide and methylmethanesulfonate were from SIGMA. Olaparib,
veliparib (ABT-888), selumetinib (AZD-6244) and lomeguatrib were from
Selleck Chemicals, Newmarket, UK.
Proliferation assays
Cells were plated in 96-wel plates and treated with serial increasing
concentrations (range: 0.15M to 1mM) of drugs as indicated. After five days
cells were fixed and stained with toluidine blue, the concentration necessary to
inhibit cell growth to a 50% (GI50) was calculated as previously described (24).
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

8
Colony formation assays
Cells were plated in 12-well plates and treated, from the top-left well, with DMSO,
5, 25, 75, 250 or 1000M TMZ for 24 hours (Supplementary Figure 1). Then cells
were washed and fresh, drug-free medium was replaced. Cells were left to form
colonies for 10-15 days before being fixed and stained with a solution of 4%
Formaldehyde (Fisher Scientific) and 0.5% Crystal violet (SIGMA) in PBS.
Remaining dye was solubilised in PBS, 1% Sodium Dodecyl Sulphate (SDS, Fisher
Scientific).
Xenograft Assay
Animal studies described within were approved by The University of Manchester
Ethical Review Board and performed according to UK Home Office regulations.
Briefly GFP-expressing human melanoma cells were injected into the pericardial
cavity of 48 hr post-fertilization zebrafish embryos. Embryos were treated at 1
day post-injection with DMSO (SIGMA), TMZ, olaparib or the combination of both.
On day 1 and 5 pictures were taken and relative growth quantified as previously
described (26).
Western blotting
Cell lysates were prepared using RIPA buffer and analysed as described (27).
Primary antibodies used were: MGMT (Thermo Fisher Scientific), phospho-
Thre68-CHK-2 and Ser345-CHK-1, MLH-1, MSH-2, MSH-6 and cleaved caspase 3
(Cell Signalling), ERK2 and beta-tubulin (Santa Cruz Biotechnologies).
MGMT activity assays
Briefly, increasing amounts of PARP or MGMT inhibitors were incubated with
recombinant MGMT (~6 fmoles) for 1h at room temperature, then substrate
DNA was added and incubation continued for 1h. After incubation, assay samples
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

9
were processed as described previously in (28) and radioactivity quantified by
scintillation counting.
FACS analysis
100000 cells were treated as indicated for 3 days, fixed in 80% ice-cold ethanol
in PBS. Cells were then washed in PBS and incubated in a solution containing PBS,
RNase A and Propidium Iodide (SIGMA) at 37ºC for 1 hour. The analysis was
performed using FACS-Calibur (Becton Dickinson).
Senescence-associated β-galactosidade (SA-β-gal) assay
After 5 days of treatment cells were fixed in 0.5% glutaraldehyde and stained
overnight with X-gal solution: 0.12mM K3Fe(CN)6, 0.12mM K4Fe(CN)6, 1mM
MgCl2 and 1mg/ml 5-Bromo-4-cloro-3-indolyl--D-galactopiranoside in PBS pH
6.0. Cell nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI). Bright
field and UV images were taken using an inverted fluorescence microscope. The
percentage of SA-β-Gal-positive cells is determined upon counting of at least 100
cells.
High Content Analysis of the senescent response.
For the High Content Analysis, cells where plated in 96-well plates and stained
by immunofluorescence. Two fluorescence images corresponding to DAPI (to
identify individual cells) and gene-primary antibody/AlexaFluor488-secondary
antibody were acquired and analysed as previously described (29). Each well
was scored a percentage positive of reference channel to the total cells.
Antibodies used were: anti--H2AX (Millipore), anti-BrdU (Invitrogen), anti-
p21Cip1 (Sigma), anti-p53 (Santa Cruz) and anti-phospho-(Ser/Thr)Q (Cell
Signaling).
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

10
Statistical analysis
Unless indicated otherwise, data are from assays performed in triplicate, with
error bars to represent standard deviations or errors from the mean. Statistics
used were: predominately Student t-test and One-way ANOVA with Dunnett's
Multiple Comparison Test performed using GraphPad Prism version 4.00 for Mac
OS, GraphPad Software, www.graphpad.com.
RESULTS
PARP inhibition specifically sensitises MGMT proficient cells to TMZ
To assess the ability of PARP inhibitors (PARPi) to sensitise melanoma cells to
TMZ we treated a panel of 12 melanoma cell lines with increasing concentrations
of TMZ in the absence or presence of the PARP inhibitor olaparib. We used
olaparib at a concentration of 0.25M, which did not affect cell proliferation
alone (Supplementary Figure 2A), but is reported to synergise with alkylating
agents to inhibit growth (30, 31). Olaparib significantly sensitised melanoma
cells to a five-day treatment with TMZ as seen by the GI50 values for TMZ
(Figure 1A and Supplementary Figure 1B). However, the cell lines segregated
into two defined groups: one with high GI50 values for TMZ, which showed
sensitisation to olaparib, and one with no significant difference (Figure 1A). We
therefore tested for differences in DNA repair regulators and found that all cell
lines belonging to the "high-GI50" group expressed enhanced levels of MGMT;
only one MGMT-expressing cell line segregated in the "low-GI50" group (Figure
1B). Expression data was complemented with methylation-specific PCR, where
MGMT promoter methylation was detectable in all MGMT-ve cells (Figure 1B).
When we grouped the cell lines upon MGMT status (MGMT+ve and MGMT-ve),
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

11
this revealed that PARP inhibition only sensitized to TMZ in MGMT+ve
melanoma cells (Figure 1C). Indeed, the Potentiation Factor 50 (PF50, ratio of
the GI50 of TMZ in mono-therapy compared to the GI50 of TMZ in the presence
of PARP inhibitor) of olaparib was significantly higher in MGMT+ve cells than in
MGMT-ve cells where the PF50 was close to 1, indicating no potentiation (Figure
1D, Supplementary Figure 2B). Similar results were obtained from colony
formation assays where cells are tested for they capacity to repair DNA damage
induced by TMZ during just 24 hours. These assays confirmed that only MGMT
expressing cells responded to the combination of TMZ and olaparib (Figure 1E
and Supplementary Figure 2C).
Lastly we analysed whether the observed differential sensitivity to TMZ in the
presence of PARP inhibitors occurred also in vivo. To do so we used a human
melanoma xenograft assay in which we injected GFP-expressing A375
(MGMT+ve) or WM266-4 (MGMT-ve) cells into the pericardial cavity of zebrafish
embryos. This approach allows fast and efficient assessment of drug combination
treatments of tumour cells in an in vivo setting, which can be recapitulated in
other pre-clinical models (26, 32). Embryos were treated with DMSO or TMZ in
the presence or absence of olaparib and after 4 days xenograft volume was
assessed (Figure 1F). Again, A375 (MGMT+ve) xenografts responded to the
combination of olaparib and TMZ (Figure 1G), but did not affect xenograft
growth in WM266-4 (MGMT-ve) derived xenografts (Figure 1H). Together, our
data suggest that in human melanoma cells MGMT expression correlated with
synergistic action of TMZ and PARP inhibitors.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

12
MGMT is active in TMZ resistant cells
To establish whether MGMT expression correlated with its activity we used the
MGMT inhibitor lomeguatrib. We treated cell lines for 5 days with TMZ in the
absence or presence of 20M lomeguatrib (LOM), which only sensitised MGMT
expressing melanoma cells to TMZ. Accordingly the average PF50 for MGMT+ve
cells was significantly higher than in MGMT-ve cells (Figure 2A-C). As expected
the GI50 for MGMT+ve cells was significantly higher than for MGMT-ve cells.
Colony formation assays provided very similar results (Figure 2D-F). These data
confirmed that MGMT expression and activity are correlated in our panel of
melanoma cell lines, demonstrating that MGMT activity provides resistance to
TMZ and correlates with TMZ and PARP inhibitor synergy. Importantly, every
melanoma cell line expressed detectable levels of the Mismatch Repair (MMR)
proteins MSH-6, MSH-2 and MLH-1 (Supplementary Figure 3). This result,
together with the responses to TMZ and olaparib or lomeguatrib previously
observed, strongly suggested that all our melanoma cells are MMR proficient and
the observed differences are not related to MMR deficiency.
The MGMT-dependent response to TMZ-PARPi treatment is also seen in
other cancer types
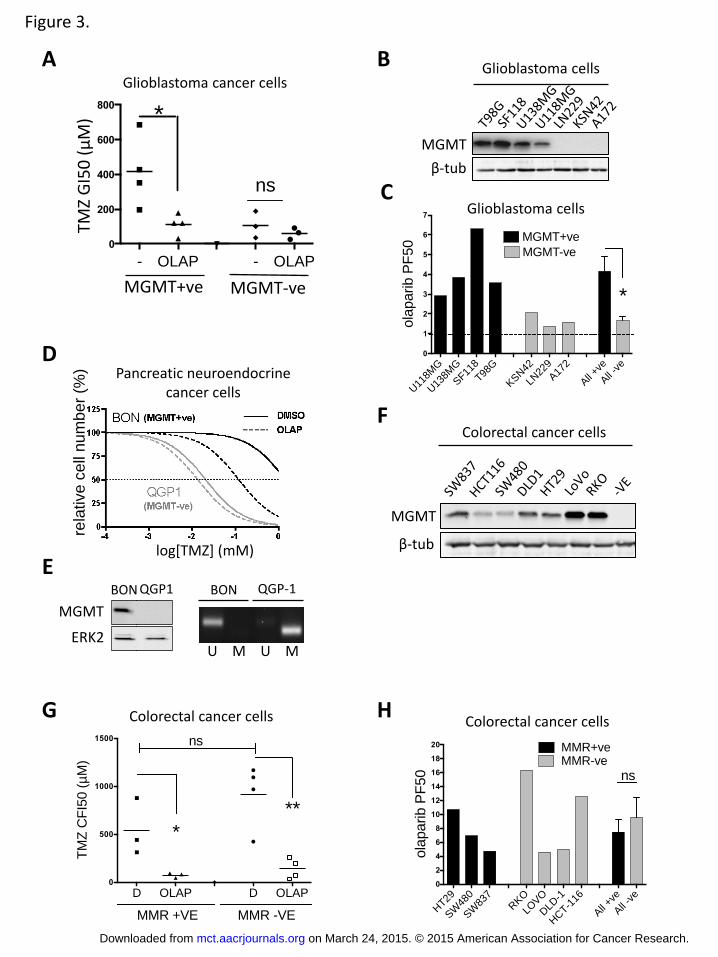
To determine whether the impact of MGMT expression on the PARPi response is
specific for melanoma cells or cancer type-independent, we assessed the
response to TMZ and olaparib in cells from three cancer types where TMZ is
used as a therapeutic option: Glioblastoma, Pancreatic Neuroendocrine tumours
(NETs) and colorectal carcinoma (11, 33, 34). We treated seven glioblastoma cell
lines (four MGMT+ve and three MGMT-ve cell lines) known to be MMR+ve ((25))
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

13
and observed that MGMT expression correlated with the response to TMZ
combined with olaparib (Figure 3A-C). Similarly, whereas the NET cell line BON,
which expresses high levels of MGMT responded to olaparib (and lomeguatrib)
co-treatment, QGP-1 NET cells, which lack MGMT, were unaffected by PARP
inhibition (Figure 3D-E). Again both NET cell lines where found to be MMR+ve
(Supplementary Figure 4A). Furthermore, a panel of seven MGMT expressing
colorectal cancer cell lines of known MMR status (35) responded to olaparib co-
treatment. (Figure 3F-G). Importantly, when grouped by MMR status the PF50
for olaparib was similar for both groups (Figure 3H). (Details of GI50s and PF50s
for each cell line can be found in Supplemental Figure 4B).
MGMT over-expression confers PARP dependent resistance to TMZ
Our data suggest that MGMT expression provides resistance to TMZ in a PARP
dependent manner. To further confirm these findings, we ectopically expressed
MGMT in MGMT-ve melanoma cells (WM98-1) (Fig. 4A). As expected, MGMT
over-expression conferred resistance to TMZ but lomeguatrib sensitised WM98-
1-MGMT clones to TMZ induced growth inhibition (Fig. 4B). Crucially, the MGMT-
produced resistance could be overcome by treatment with olaparib (Fig.4C).
Similar results were obtained using veliparib (Supplemental figure 5A-B).
Furthermore, we observed that when MGMT activity is inhibited, PARP
inhibitors no longer sensitised to TMZ (Figure 4E-F). We show that PARP is
required for the MGMT mediated resistance to TMZ. As mentioned above TMZ
produces N3- and N7-methyl adducts, which are repaired by PARP mediated
BER. To assess the requirement of PARP in this repair, we used the alkylating
agent Methylmethanesulfonate (MMS), which exclusively produces N3- and N7-
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

14
methyl adducts (36). Both olaparib and veliparib significantly sensitised WM98-
1 and WM98-1-MGMT cells to MMS (Figure 4G and Supplemental Figure 5C).
Similar results were obtained in a panel of melanoma cells confirming that PARP
is required for efficient BER in melanoma cells, irrespective of their MGMT status
(Figure 4H and Supplemental Figure 5D). Finally, to rule out the possibility that
PARP inhibitors non-specifically interfere with MGMT activity, we performed in
vitro MGMT activity assays and confirmed that, unlike lomeguatrib, even high
concentrations of olaparib or veliparib failed to inhibit MGMT (Supplementary
Figure 5E).
PARP inhibition potentiates TMZ induced cell cycle arrest in MGMT
proficient cells
To further understand the action of TMZ and PARP inhibition in the context of
the MGMT expression status, we studied the induction of DNA single and double
strand breaks measuring histone H2AX phosphorylation (H2AX) after a 16h
treatment. PARP inhibition only synergised with TMZ to induce DNA damage in
MGMT+ve cells (Figure 5A, B). Analysis of CHK-1 and 2 phosphorylation, (Thr68
and Ser345 respectively), at 24h of treatment revealed that in MGMT-ve
WM266-4 cells TMZ activated CHK1/2 at lower concentrations than in MGMT+ve
A375 cells (Figure 5C-D). While PARP inhibition did not enhance the activation
of CHK2 by TMZ in WM266-4 cells, in A375 cells TMZ-induced CHK-2
phosphorylation was potentiated by olaparib. On the other hand CHK-1
phosphorylation did not correlate consistently with olaparib co-treatment.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

15
We next analysed the cell cycle profile of A375 and WM266-4 cells after 72 hours
of treatment. In WM266-4 cells sub-optimal concentrations of TMZ (5M, unable
to affect cell cycle progression) failed to cooperate with olaparib to promote a
cell cycle arrest while 20M TMZ (GI50= 73.26M), produced a marked G2/M
arrest (Figure 5E). In contrast, in A375 cells (GI50= 439.65M) combining
olaparib with a sub-optimal concentration of TMZ (50M) produced a dramatic
arrest in the G2/M phase of the cell cycle (Figure 5E). Strikingly we were not able
to detect the appearance of a sub-G1 phase, which is associated with cell death
(37). In agreement with the absence of a sub-G1 population, we did not observe
any caspase3 cleavage in response to TMZ in melanoma cells, whereas this was
detectable in colorectal cancer cells (Figure 5F). Moreover, co-treatment of A375
cells with concentrations of TMZ and OLAP that potently affect cell proliferation
failed to induce caspase3 cleavage (Figure 5F, lower panel). On the other hand
we observed that melanoma cells treated with TMZ displayed a very flat
appearance with a marked increase in cell size/surface (Figure 5G). Because
these are common traits of a senescent response, we decided to gain more
insight into the role of senescence in the response of melanoma cells to TMZ (38).
Characterisation of the senescent response induced by the combination of
TMZ and PARP inhibition
A 5day treatment with growth inhibitory concentrations of TMZ induced
Senescence Associated -galactosidase (SA--gal) staining in MGMT-ve WM98-1
as well as MGMT+ve A375 and WM98-1-MGMT cells (Figure 6, A to C),
confirming previous observations suggesting that TMZ induces senescence
rather than apoptosis in melanoma cells (39). However, in MGMT+ve cells
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

16
treated with concentrations of TMZ that induced only a weak SA--gal activity,
PARP inhibition strongly enhanced the senescent response (Figure 6B-C). High
Content Analysis of A375 cells, used to assess the expression of several known
markers and mediators of senescence (29), confirmed that the combination of
TMZ and PARPi induced a potent DNA damage response (DDR, maintained over
5 days of treatment), as indicated by ATM and ATR phosphorylation (ST/Q) and
a H2AX signal (Figure 6D). The activation of the DDR was accompanied by a
significant increase in p53 levels and the cell cycle inhibitor p21 (Figures 6E).
Finally, as expected from a long-term cell cycle arrest we observed a dramatic
reduction in the number of cells undergoing DNA synthesis, as revealed by BrdU
incorporation assay (Figure 6F). In summary, when PARP is inhibited in MGMT
expressing melanoma cells TMZ induced DNA damage triggers a senescence
response that correlates with p53-mediated up-regulation of p21.
DISCUSSION
Because melanomas are notoriously resistant to chemotherapy, we assessed the
potential of combining TMZ and PARP inhibition in melanoma cells. Strikingly,
we observed that PARP, required for Base Excision Repair, plays a role in MGMT
mediated resistance to TMZ. In MGMT-ve cells PARP function appears to be
dispensable for the repair of TMZ induced DNA damage, because olaparib does
not enhance the TMZ induced effects on growth. On the other hand, in MGMT+ve
cells the MGMT mediated resistance to TMZ induced DNA damage is dependent
on PARP, as evidenced by the synergy observed between PARP inhibitors and
TMZ to inhibit proliferation.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

17
We suggest that, upon TMZ treatment, a threshold of N3-, N7-methylation needs
to be reached before PARP-dependent DNA repair becomes relevant to the anti-
proliferative effect of TMZ. In the absence of MGMT, 06-meG is not repaired (4),
and the immediate toxicity of 06-meG prevents this threshold to be reached.
Alternatively, tolerance to 06-meG due to deficient MMR might allow to reach this
threshold, which would explain previous results, obtained in MMR-ve/MGMT-ve
leukemia cells (40).
So far the combination of MGMT inhibitors with dacarbazine or TMZ has failed in
the clinic, but our data suggest that the MGMT status could be a predictive
marker for response to therapies combining temozolomide and PARP inhibitors.
To date, early phase clinical trials using temozolomide and veliparib in CNS and
prostate cancer patients have shown modest activity (21, 41). On the other hand
one Phase II trial in melanoma patients showed an increase in progression-free
survival over historical records (23). Although trials are in progress it is clear
that there is still a need for biomarkers predicting response to specific therapies
in order to increase clinical response. Indeed the upcoming clinical trial in
glioblastoma combining TMZ and veliparib in patients with MGMT promoter
methylation (A071102, NCT02152982, www.clinicaltrials.gov) will hopefully
shed more light on the role of MGMT as a marker.
Because we observe a role for MGMT in the TMZ+PARPi mediated growth
inhibition in cell lines from four different cancer types our data suggest a
common mechanism of response to TMZ. Indeed Palma et al. 2009 observed that
a first cycle of TMZ and veliparib was most effective in MGMT+ve tumours from
different origin. Initially, MGMT-ve tumours did not benefit from PARP inhibitor
co-treatment while a 2nd cycle demonstrated efficacy in tumours that underwent
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

18
re-expression of MGMT (42). On the other hand cancer type specific responses
cannot be ruled out as it has been suggested that only MGMT-ve leukemia and
glioblastoma multiforme cells would respond to TMZ combined with the PARP
inhibitor veliparib (40, 43, 44). It is nevertheless important to consider that
variables such as differences in PARPi action, bioavailability or the application of
particular treatment regimes need to be taken into account to correlate in vitro
and in vivo efficacy of TMZ and PARP inhibitor combinations as well as clinical
efficacy (31, 42-45). Indeed, in order to synergise with TMZ, both drugs work
through catalytic PARP inhibition and trapping PARP-DNA complexes. Although
olaparib has been shown to be more effective to trap PARP-DNA complexes than
veliparib, we show evidence that the role of PARP in MGMT induced resistance
can be overcome by both inhibitors (31). This could be explained by the different
concentrations at which olaparib and veliparib were used (0.25M and 5M,
respectively).
Our data provide compelling evidence of MGMT as a potential marker for
TMZ+PARPi based therapies. If confirmed clinically, this has the potential for
improving clinical responses and reducing toxicity (41).
Importantly, MGMT status assessment remains a challenge and although MGMT
promoter methylation has already entered clinical guidelines as predictive
biomarker in elderly patients with glioblastoma it has not been fully validated
across cancer types (13, 46). We observed that some MGMT expressing
melanoma cell lines and, notably, also some MGMT expressing gliobastoma cell
lines are in fact positive for promoter methylation (Fig. 1B and (25)), suggesting
that a positive signal for promoter methylation does not always correlate with
MGMT expression. Thus, the MGMT promoter status alone might not be
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

19
sufficient as marker in a clinical setting. As already described for glioblastoma
and NET tumours we propose that this issue could be overcome by combining
promoter methylation with immunohistochemistry (47, 48).
Notably, we observed that TMZ and olaparib failed to induce apoptosis in
melanoma cells, but rather induced senescence, which is most probably
regulated through the p53/p21axis (39, 49). Whether treatment with mono-
alkylating agents inhibits mechanisms of cell death, or if the apoptotic machinery
is turned-off as cells get arrested in the G2/M phase of the cell cycle warrants
further investigation.
In summary, our data provide strong evidence that, specifically in cells
expressing MGMT, the combination of TMZ and PARP inhibitors might be of
benefit and improve responses.
REFERENCES
1. Belden S, Flaherty KT. MEK and RAF inhibitors for BRAF-mutated cancers. Expert Rev Mol Med. 2012;14:e17. 2. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507-16. 3. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694-703. 4. Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L. Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res. 2007;56:275-87. 5. Mouawad R, Sebert M, Michels J, Bloch J, Spano JP, Khayat D. Treatment for metastatic malignant melanoma: old drugs and new strategies. Crit Rev Oncol Hematol. 2010;74:27-39. 6. Koekkoek JA, Wiggenraad RG, Zwinkels H, Oosterkamp HM, Taphoorn MJ. Survival over 6 years in a patient with brain metastases from melanoma treated with temozolomide. BMJ Case Rep. 2012;2012.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

20
7. Siena S, Crino L, Danova M, Del Prete S, Cascinu S, Salvagni S, et al. Dose-dense temozolomide regimen for the treatment of brain metastases from melanoma, breast cancer, or lung cancer not amenable to surgery or radiosurgery: a multicenter phase II study. Ann Oncol. 2010;21:655-61. 8. Zhu W, Zhou L, Qian JQ, Qiu TZ, Shu YQ, Liu P. Temozolomide for treatment of brain metastases: A review of 21 clinical trials. World J Clin Oncol. 2014;5:19-27. 9. Middleton MR, Grob JJ, Aaronson N, Fierlbeck G, Tilgen W, Seiter S, et al. Randomized phase III study of temozolomide versus dacarbazine in the treatment of patients with advanced metastatic malignant melanoma. J Clin Oncol. 2000;18:158-66. 10. Patel PM, Suciu S, Mortier L, Kruit WH, Robert C, Schadendorf D, et al. Extended schedule, escalated dose temozolomide versus dacarbazine in stage IV melanoma: final results of a randomised phase III study (EORTC 18032). Eur J Cancer. 2011;47:1476-83. 11. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987-96. 12. Verbeek B, Southgate TD, Gilham DE, Margison GP. O6-Methylguanine-DNA methyltransferase inactivation and chemotherapy. Br Med Bull. 2008;85:17-33. 13. Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA-08 randomised, phase 3 trial. Lancet Oncol. 2012;13:707-15. 14. Khan OA, Ranson M, Michael M, Olver I, Levitt NC, Mortimer P, et al. A phase II trial of lomeguatrib and temozolomide in metastatic colorectal cancer. Br J Cancer. 2008;98:1614-8. 15. Ranson M, Hersey P, Thompson D, Beith J, McArthur GA, Haydon A, et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J Clin Oncol. 2007;25:2540-5. 16. Ranson M, Middleton MR, Bridgewater J, Lee SM, Dawson M, Jowle D, et al. Lomeguatrib, a potent inhibitor of O6-alkylguanine-DNA-alkyltransferase: phase I safety, pharmacodynamic, and pharmacokinetic trial and evaluation in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2006;12:1577-84. 17. Basu B, Yap TA, Molife LR, de Bono JS. Targeting the DNA damage response in oncology: past, present and future perspectives. Curr Opin Oncol. 2012;24:316-24. 18. Lee JM, Ledermann JA, Kohn EC. PARP Inhibitors for BRCA1/2 mutation-associated and BRCA-like malignancies. Ann Oncol. 2014;25:32-40. 19. Bedikian AY, Papadopoulos NE, Kim KB, Hwu WJ, Homsi J, Glass MR, et al. A phase IB trial of intravenous INO-1001 plus oral temozolomide in subjects with unresectable stage-III or IV melanoma. Cancer Invest. 2009;27:756-63. 20. Plummer R, Jones C, Middleton M, Wilson R, Evans J, Olsen A, et al. Phase I study of the poly(ADP-ribose) polymerase inhibitor, AG014699, in combination with temozolomide in patients with advanced solid tumors. Clin Cancer Res. 2008;14:7917-23.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

21
21. Hussain M, Carducci MA, Slovin S, Cetnar J, Qian J, McKeegan EM, et al. Targeting DNA repair with combination veliparib (ABT-888) and temozolomide in patients with metastatic castration-resistant prostate cancer. Invest New Drugs. 2014. 22. Khan OA, Gore M, Lorigan P, Stone J, Greystoke A, Burke W, et al. A phase I study of the safety and tolerability of olaparib (AZD2281, KU0059436) and dacarbazine in patients with advanced solid tumours. Br J Cancer. 2011;104:750-5. 23. Plummer R, Lorigan P, Steven N, Scott L, Middleton MR, Wilson RH, et al. A phase II study of the potent PARP inhibitor, Rucaparib (PF-01367338, AG014699), with temozolomide in patients with metastatic melanoma demonstrating evidence of chemopotentiation. Cancer Chemother Pharmacol. 2013;71:1191-9. 24. Arozarena I, Goicoechea I, Erice O, Ferguson J, Margison GP, Wellbrock C. Differential chemosensitivity to antifolate drugs between RAS and BRAF melanoma cells. Mol Cancer. 2014;13:154. 25. Gaspar N, Marshall L, Perryman L, Bax DA, Little SE, Viana-Pereira M, et al. MGMT-independent temozolomide resistance in pediatric glioblastoma cells associated with a PI3-kinase-mediated HOX/stem cell gene signature. Cancer Res. 2010;70:9243-52. 26. Smith MP, Ferguson J, Arozarena I, Hayward R, Marais R, Chapman A, et al. Effect of SMURF2 targeting on susceptibility to MEK inhibitors in melanoma. J Natl Cancer Inst. 2013;105:33-46. 27. Arozarena I, Bischof H, Gilby D, Belloni B, Dummer R, Wellbrock C. In melanoma, beta-catenin is a suppressor of invasion. Oncogene. 2011;30:4531-43. 28. Watson AJ, Sabharwal A, Thorncroft M, McGown G, Kerr R, Bojanic S, et al. Tumor O(6)-methylguanine-DNA methyltransferase inactivation by oral lomeguatrib. Clin Cancer Res. 2010;16:743-9. 29. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978-90. 30. Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13:433-43. 31. Murai J, Zhang Y, Morris J, Ji J, Takeda S, Doroshow JH, et al. Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J Pharmacol Exp Ther. 2014;349:408-16. 32. Chapman A, Fernandez Del Ama L, Ferguson J, Kamarashev J, Wellbrock C, Hurlstone A. Heterogeneous tumor subpopulations cooperate to drive invasion. Cell Rep. 2014;8:688-95. 33. Fine RL, Gulati AP, Krantz BA, Moss RA, Schreibman S, Tsushima DA, et al. Capecitabine and temozolomide (CAPTEM) for metastatic, well-differentiated neuroendocrine cancers: The Pancreas Center at Columbia University experience. Cancer Chemother Pharmacol. 2013;71:663-70. 34. Hochhauser D, Glynne-Jones R, Potter V, Gravalos C, Doyle TJ, Pathiraja K, et al. A phase II study of temozolomide in patients with advanced aerodigestive tract and colorectal cancers and methylation of the O6-methylguanine-DNA methyltransferase promoter. Mol Cancer Ther. 2013;12:809-18.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

22
35. Lengauer C, Kinzler KW, Vogelstein B. DNA methylation and genetic instability in colorectal cancer cells. Proc Natl Acad Sci U S A. 1997;94:2545-50. 36. Pachkowski BF, Tano K, Afonin V, Elder RH, Takeda S, Watanabe M, et al. Cells deficient in PARP-1 show an accelerated accumulation of DNA single strand breaks, but not AP sites, over the PARP-1-proficient cells exposed to MMS. Mutat Res. 2009;671:93-9. 37. Naumann SC, Roos WP, Jost E, Belohlavek C, Lennerz V, Schmidt CW, et al. Temozolomide- and fotemustine-induced apoptosis in human malignant melanoma cells: response related to MGMT, MMR, DSBs, and p53. Br J Cancer. 2009;100:322-33. 38. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463-79. 39. Mhaidat NM, Zhang XD, Allen J, Avery-Kiejda KA, Scott RJ, Hersey P. Temozolomide induces senescence but not apoptosis in human melanoma cells. Br J Cancer. 2007;97:1225-33. 40. Horton TM, Jenkins G, Pati D, Zhang L, Dolan ME, Ribes-Zamora A, et al. Poly(ADP-ribose) polymerase inhibitor ABT-888 potentiates the cytotoxic activity of temozolomide in leukemia cells: influence of mismatch repair status and O6-methylguanine-DNA methyltransferase activity. Mol Cancer Ther. 2009;8:2232-42. 41. Su JM, Thompson P, Adesina A, Li XN, Kilburn L, Onar-Thomas A, et al. A phase I trial of veliparib (ABT-888) and temozolomide in children with recurrent CNS tumors: a Pediatric Brain Tumor Consortium reportdagger. Neuro Oncol. 2014. 42. Palma JP, Wang YC, Rodriguez LE, Montgomery D, Ellis PA, Bukofzer G, et al. ABT-888 confers broad in vivo activity in combination with temozolomide in diverse tumors. Clin Cancer Res. 2009;15:7277-90. 43. Clarke MJ, Mulligan EA, Grogan PT, Mladek AC, Carlson BL, Schroeder MA, et al. Effective sensitization of temozolomide by ABT-888 is lost with development of temozolomide resistance in glioblastoma xenograft lines. Mol Cancer Ther. 2009;8:407-14. 44. Gupta SK, Mladek AC, Carlson BL, Boakye-Agyeman F, Bakken KK, Kizilbash SH, et al. Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin Cancer Res. 2014;20:3730-41. 45. Cen L, Carlson BL, Pokorny JL, Mladek AC, Grogan PT, Schroeder MA, et al. Efficacy of protracted temozolomide dosing is limited in MGMT unmethylated GBM xenograft models. Neuro Oncol. 2013;15:735-46. 46. Weller M, van den Bent M, Hopkins K, Tonn JC, Stupp R, Falini A, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15:e395-403. 47. Kulke MH, Hornick JL, Frauenhoffer C, Hooshmand S, Ryan DP, Enzinger PC, et al. O6-methylguanine DNA methyltransferase deficiency and response to temozolomide-based therapy in patients with neuroendocrine tumors. Clin Cancer Res. 2009;15:338-45. 48. Lalezari S, Chou AP, Tran A, Solis OE, Khanlou N, Chen W, et al. Combined analysis of O6-methylguanine-DNA methyltransferase protein expression and promoter methylation provides optimized prognostication of glioblastoma outcome. Neuro Oncol. 2013;15:370-81.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

23
49. Mirzayans R, Andrais B, Scott A, Murray D. New insights into p53 signaling and cancer cell response to DNA damage: implications for cancer therapy. J Biomed Biotechnol. 2012;2012:170325. Figure Legends
Figure 1. PARP inhibition synergises with TMZ only in MGMT+ve melanoma
cells
A, Melanoma cell lines were treated with TMZ with or without 0.25M olaparib
(OLAP). The graph shows the effect of PARP inhibition on the average GI50 for
TMZ. Each point corresponds to an individual cell line. B. Top, Western-blot for
MGMT expression in melanoma cell lines. ERK2 was used as a loading control.
Bottom, Methylation-specific PCR for un-methylated (U) and methylated (M)
promoter sequences. C-D, Cell lines were grouped upon their MGMT status, GI50
results from A re-analysed and Potentiation Factor 50 calculated. E, Potentiation
Factor results from colony formation assays on MGMT+ve and –ve cells treated
as indicated. Student’s t test was used to compares the effect of each treatment.
*p<0.05, **p<0.01. n.s: not significant. F, Pictures from A375 and WM266-4 cells
injected in zebra fish embryos and treated as indicated for 5 days. G-H,
Quantification of the in vivo effect of PARP inhibition on TMZ (500M) treated
xenografts. One-way Anova was used to compare the effect of each treatment
with DMSO (D) treated animals. ***p<0.001.
Figure 2. Characterization of melanoma cell lines upon MGMT status
A, Representative examples of dose-response curves for MGMT+ve and MGMT-
ve cells treated with TMZ with or without lomeguatrib (LOM). B-C, Graphs show
the average GI50 for TMZ in the presence or absence of lomeguatrib (B) and the
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

24
Potentitation Factor 50 for each cell line (C). D, Colony formation assay on four
MGMT+ve and –ve cells treated treated with TMZ +/- LOM. E, Potentiation Factor
50 from D. F, Representative examples of colony formation assays using A375
(MGMT+ve) and WM9 (MGMT-ve) cells. D: DMSO.
Figure 3. TMZ-PARPi synergy in other cancer types
A-B, Effect of TMZ with or without olaparib of seven glioblastoma cell lines
grouped upon their MGMT status as determined by western-blot (B). C,
Potentiation Factor 50 of olaparib for each cell line. D, Dose-response assay on
neuroendocrine pancreatic BON (MGMT+ve) and QGP-1 (MGMT-ve) cell treated
with serial increasing concentrations of TMZ in the absence (full lines) or
presence (dotted lines) of olaparib. E, MGMT expression in BON and QGP-1 cells
assessed by western-blot and promoter methylation. F, MGMT levels on seven
colorectal cancer cell lines. G, Colony formation assay on 7 colorectal cancer cell
lines grouped upon MMR status and treated as indicated. H, Olaparib
Potentiation Factor 50 for each cell line used in G. Student’s t test was used for
statistical comparisons. * p<0.05, **p<0.001.
Figure 4. MGMT re-expression restores PARPi-TMZ synergy.
A, Western-blot for MGMT expression in lysates from WM98-1 melanoma cell
clones expressing either an empty vector or MGMT. B, Graphs show the average
GI50 for 3 empty vector and 4 MGMT-expressing clones treated with TMZ with
or without lomeguatrib or olaparib. C, Potentiation Factor 50 of olaparib on cells
treated in B. D, Colony formation assay on WM98-1-MGMT cells treated with
TMZ with or without olaparib in the presence or absence of constitutive
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

25
inhibition of MGMT. E, Representative colony formation assay from C. F,
Potentiation factor for olaparib on cells treated as in D. G-H, Average GI50 for
methylmethane-sulfonate (MMS) in the presence or absence of olaparib in
WM98-1 cells expressing MGMT or empty vector (G) or in a panel of MGMT+ve
and –ve melanoma cells lines (H).
Figure 5. Melanoma cell response to TMZ and PARP inhibition
A-B, Quantification of H2AX expression in WM266-4 and A375 cells treated as
determined by immunofluorescence. C-D, Western-blot of phospho Thr68-CHK1
(ppCHK1), phospho Ser345-CHK2 (ppCHK2), of WM266-4 and A375 cells treated
as indicated. E, Effects on the cell cycle: Distribution in G1, S and G2/M phases of
the cell cycle of WM266-4 and A375 cells treated for 3 days with TMZ with or
without olaparib. F, upper panel: Western-blot of cleaved caspase 3 of RKO
colorectal cells (CRC) or A375 melanoma cells (MM) treated with 500M TMZ for
3 days. Lower panel, cleaved caspase 3 expression in A375 cells treated as
indicated for 3 days. AZD: selumetinib (AZD-6244: 2M). G, Barr and whisker
graph showing the increase in cell size upon treatment of MGMT-ve (WM98-1)
and +ve (A375 and WM98-1-MGMT) cells with TMZ for 5 days as determined
using ImageJ. F-G, Induction of beta-galactosidase activity upon treatment of
WM98-1 (F) or A375 (G) cells with TMZ with or without olaparib for 5 days.
Figure 6. Characterisation of TMZ/PARPi induced senescence.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

26
A-B-C. Induction of Senescence-associated -galactosidase activity upon
treatment of WM98.1 (A), WM98-1-MGMT (B) or A375 (C) cells as indicated for
5 days. D. Quantification of the DNA damage response using antibodies against
the consensus sequence phosphorylated by ATM and ATR (S/TQ) and H2AX. E,
TMZ and olaparib cooperate to significantly increase p53 and p21 protein levels.
F, TMZ and olaparib synergise to block DNA synthesis as determined by
quantification of BrDU incorporation. One-way Anova was used to compare the
effect of the double treatment with DMSO or TMZ alone.
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

Figure 1.
A B
D
E F
C
MGMT
β-tub
TMZ
GI5
0 (
μM
)
G
DO4 MM415 501mel A375P WM1361 D10
UMUMUMUMUMUM
CJM WM9 WM266-4WM98.1Mel-JusoWM852
UMUMUMUMUMUM
D04
MM
415
501m
el
A37
5
WM
1361
D10
CJM
WM
9
WM
266.
4
WM
98.1
Mel-J
uso
WM
852
All +v
e
All -v
e0
1
2
3
4
4.5
8.0
11.5
ola
parib P
F50
MGMT+ve
MGMT-ve
**
D04
MM
415
501m
el
A37
5
WM
1361
D10
CJM
WM
9
WM
266.
4
WM
98.1
Mel-J
uso
WM
852
All +ve
All -v
e0
1
2
3
44.0
6.5
9.0
ola
parib P
F50
MGMT+ve
MGMT-ve
**
TMZ
GI5
0 (
μM
)
D OLAP D OLAP
0
250
500
750
1000
16002000
**ns
MGMT+ve MGMT-ve
TM
Z G
I50 (m
M)
**
H A375P xenografts (MGMT+ve)
D OLAP T500 T500+ OLAP
0
1
2
3
4
5
6
*
**
Rela
tive g
row
th to d
ay 0
WM266-4 xenografts (MGMT-ve)
D OLAP T500 T500+ OLAP
0
1
2
3
4
5
6
***
n.s.
Rela
tive g
row
th to d
ay 0
DMSO OLAP TMZ TMZ+ OLAP
WM
26
64
A
37
5
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

Figure 2.
A B
C D
A375 (MGMT+ve)
WM9 (MGMT-ve)
rela
tive c
ell
num
ber
(%)
log[TMZ] (mM)
E F
TMZ
GI5
0 (
μM
)
D LOM D LOM
0
250
500
750
10001000
1500
2000
MGMT+ve MGMT-ve
** **
TM
Z G
I50 (m
M)
ns
D04
MM
415
501m
el
A37
5
WM
1361
D10
CJM
WM
9
WM
266.
4
WM
98.1
Mel-J
uso
WM
852
All +v
e
All -v
e0
1
2
3
4
4.5
8.0
11.5
ola
parib P
F50
MGMT+ve
MGMT-ve
**
TMZ
GI5
0 (
μM
)
D LOM D LOM0
25
50
250
500
750
MGMT+ve MGMT-ve
** **
IC50 (m
M)
ns
D10
WM
1361
501m
el
A37
5
WM
98.1
WM
266.
4CJM
Mel Jus
o
All +v
e
All -v
e0
1
2
3
410
20
30
40
50
ola
parib P
F50
MGMT+ve
MGMT-ve
**
A375 (MGMT+ve)
WM9 (MGMT-ve)
D
LO
M
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810
U11
8MG
U13
8MG
SF11
8
T98G
KSN42
LN22
9
A17
2
All +v
e
All -v
e0
1
2
3
4
5
6
7
ola
parib P
F50
MGMT+ve
MGMT-ve
*
- OLAP - OLAP
0
200
400
600
800
TM
Z G
I50 (m
M) *
ns
Figure 3.
MGMT
β-tub
A B
D
Glioblastoma cancer cells
F
TMZ
GI5
0 (
μM
)
MGMT+ve MGMT-ve
Pancreatic neuroendocrine cancer cells
rela
tive c
ell
num
ber
(%)
log[TMZ] (mM)
G H
C
Glioblastoma cells
Glioblastoma cells
MGMT
ERK2
BON-1CQGP
MGMT
ERK2
BON QGP1
MGMT
ERK2
BON-1CQGP
U M U M
BON QGP-1
E
MGMT
β-tub
Colorectal cancer cells
Colorectal cancer cells Colorectal cancer cells
D OLAP D OLAP0
500
1000
1500
TM
Z C
FI5
0 (
mM
)
MMR +VE MMR -VE
*
ns
**
HT29
SW
480
SW
837
RKO
LOVO
DLD
-1
HCT-1
16
All +v
e
All -v
e0
2
4
6
8
10
12
14
16
18
20
ola
parib P
F50
MMR+veMMR-ve
ns
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

Figure 4.
A B
C D
E F
G
MGMT
ERK2
MGMT+ve clones
1 2 3 4
WM98.1
D OLAP D OLAP0
50
100
150
MM
S G
I50 (
mM
)
MGMT+ve MGMT-ve
*** ***
D LOM0
2
4
6
8
10
ola
parib P
F50
**
H
TMZ
GI5
0 (
μM
)
DLO
M
OLA
P DLO
M
OLA
P
0
200
400
600
800
**ns
***
vector MGMT
TM
Z C
FI5
0 (
mM
)
WM98.1
vector
1
vector
2
vector
3
MGM
T.1
MGM
T.2
MGM
T.3
MGM
T.4
All ve
ctor
All M
GM
T0
2
4
6
8
10
12
14
ola
parib P
F50
MGMT-veMGMT+ve
**
WM98.1
TMZ
GI5
0 (
μM
)
WM98.1-MGMT
Melanoma cells
WM98.1-MGMT
D
OLA
P D
OLA
P D
OLA
P
0
50
100
150
200
MM
S C
FI5
0 (
mM
)
vector MGMT c1 MGMT c2
*** *** ***
WM98.1
MM
S G
I50
(μ
M)
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810
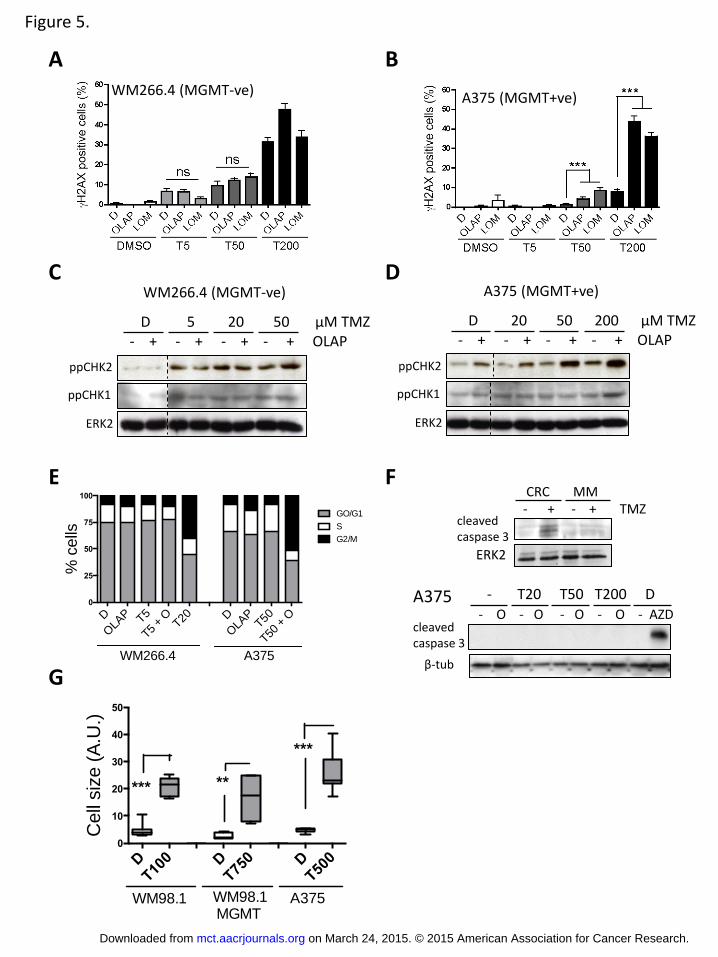
Figure 5.
A B
C WM266.4 (MGMT-ve) A375 (MGMT+ve)
E F
G
D
- + - + TMZ CRC MM
cleaved caspase 3
ERK2
ppCHK2
ppCHK1
ERK2
- + - + - + - + OLAP D 20 50 200 μM TMZ
- + - + - + - + OLAP
ppCHK2
ppCHK1
ERK2
D 5 20 50 μM TMZ
WM266.4 (MGMT-ve) A375 (MGMT+ve)
cleaved caspase 3
A375 - O - O - O - O - AZD
- T20 T50 T200 D
β-tub
DT100 D
T750 D
T500
0
10
20
30
40
50
Cell
siz
e (
A.U
.)
WM98.1 A375WM98.1 MGMT
*** **
***
D
OLA
P T5
T5 +
OT20 D
OLA
PT50
T50 +
O
0
25
50
75
100
GO/G1
S
G2/M
WM266.4 A375
% c
ells
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

Figure 6.
A B
C D
E F
A375 (MGMT+ve)
WM98.1-vector (MGMT-ve)
WM98.1- clon 3 (MGMT+ve)
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810

Published OnlineFirst March 16, 2015.Mol Cancer Ther Oihane Erice, Michael P Smith, Rachel White, et al. temozolomideMGMT expression predicts PARP-mediated resistance to
Updated version
10.1158/1535-7163.MCT-14-0810doi:
Access the most recent version of this article at:
Material
Supplementary
http://mct.aacrjournals.org/content/suppl/2015/03/13/1535-7163.MCT-14-0810.DC1.html
Access the most recent supplemental material at:
Manuscript
Authorbeen edited. Author manuscripts have been peer reviewed and accepted for publication but have not yet
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
on March 24, 2015. © 2015 American Association for Cancer Research. mct.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on March 16, 2015; DOI: 10.1158/1535-7163.MCT-14-0810




![Synthesis, [18F] radiolabeling, and evaluation of poly (ADP-ribose) polymerase-1 (PARP-1) inhibitors for in vivo imaging of PARP-1 using positron emission tomography](https://static.fdokumen.com/doc/165x107/6335c3a302a8c1a4ec01e906/synthesis-18f-radiolabeling-and-evaluation-of-poly-adp-ribose-polymerase-1.jpg)















