
b-Catenin activation synergizes with Pten loss and Myc overexpression in Notch-independent T-ALL
12
doi:10.1182/blood-2012-12-471904 Prepublished online June 25, 2013; 2013 122: 694-704 Deepika Kaveri, Philippe Kastner, Doulaye Dembélé, Claus Nerlov, Susan Chan and Peggy Kirstetter in Notch-independent T-ALL -Catenin activation synergizes with Pten loss and Myc overexpression β http://bloodjournal.hematologylibrary.org/content/122/5/694.full.html Updated information and services can be found at: (1470 articles) Lymphoid Neoplasia Articles on similar topics can be found in the following Blood collections http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requests Information about reproducing this article in parts or in its entirety may be found online at: http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprints Information about ordering reprints may be found online at: http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtml Information about subscriptions and ASH membership may be found online at: Copyright 2011 by The American Society of Hematology; all rights reserved. Washington DC 20036. by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly only. For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.org From
Transcript of b-Catenin activation synergizes with Pten loss and Myc overexpression in Notch-independent T-ALL

doi:10.1182/blood-2012-12-471904Prepublished online June 25, 2013;2013 122: 694-704
Deepika Kaveri, Philippe Kastner, Doulaye Dembélé, Claus Nerlov, Susan Chan and Peggy Kirstetter in Notch-independent T-ALL-Catenin activation synergizes with Pten loss and Myc overexpressionβ
http://bloodjournal.hematologylibrary.org/content/122/5/694.full.htmlUpdated information and services can be found at:
(1470 articles)Lymphoid Neoplasia �Articles on similar topics can be found in the following Blood collections
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://bloodjournal.hematologylibrary.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://bloodjournal.hematologylibrary.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Washington DC 20036.by the American Society of Hematology, 2021 L St, NW, Suite 900, Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom

Regular Article
LYMPHOID NEOPLASIA
b-Catenin activation synergizes with Pten loss and Myc overexpressionin Notch-independent T-ALLDeepika Kaveri,1 Philippe Kastner,1,2 Doulaye Dembele,1 Claus Nerlov,3,4 Susan Chan,1 and Peggy Kirstetter1
1Institut de Genetique et de Biologie Moleculaire et Cellulaire, Institut National de la Sante et de la Recherche Medicale U964, Centre National de la
Recherche Scientifique UMR 7104, Universite de Strasbourg, Illkirch, France; 2Universite de Strasbourg, Faculte de Medecine, Strasbourg, France;3European Molecular Biology Laboratory Mouse Biology Unit, Monterotondo, Italy; and 4Medical Research Council, Weatherall Institute of Molecular
Medicine, Oxford, United Kingdom
Key Points
• Wnt activation, Pten loss, andMyc translocation synergizeto define a novel subset ofmurine Notch-independentT-ALL.
Wnt signaling is important for T-cell differentiation at the early CD42CD82 stage and is
subsequently downregulated with maturation. To assess the importance of this down-
regulation, we generated a mouse line (R26-bcat) in which high levels of active b-catenin
are maintained throughout T-cell development. Young R26-bcat mice show a differentia-
tion block at the CD41CD81 double-positive (DP) stage. These DP cells exhibit impaired
apoptosis upon irradiation or dexamethasone treatment. All R26-bcat mice develop T-cell
leukemias at 5 to 6 months of age. R26-bcat leukemias remain dependent on b-catenin
function but lack Notch pathway activation. They exhibit recurrent secondary genomic
rearrangements that lead to Myc overexpression and loss of Pten activity. Because b-catenin activation and Myc translocations were
previously found in murine T-cell acute lymphoblastic leukemias (T-ALLs) deficient for Pten, our results suggest that activation of the
canonical Wnt pathway is associated with a subtype of Notch-independent T-ALLs that bear Myc gene rearrangements and Pten
mutations. (Blood. 2013;122(5):694-704)
Introduction
T-cell acute lymphoblastic leukemia (T-ALL) represents around15% of pediatric and 25% of adult acute lymphoblastic leukemiaand results from the proliferation of T lymphoid blasts in the bonemarrow (BM) and other organs. Advances in therapy have led tolong-term survival for 75% to 80% of children but only 30% to 40%of adults. T-ALL is genetically heterogeneous and includes severalmajor subtypes associated with specific chromosomal rearrange-ments and defined by distinct gene expression signatures (mostlyTal-like, Tlx1/3-like, or Hox-like).1-3 In addition, Notch1 gain-of-function mutations affect;65% of T-ALLs across genetic subtypes,suggesting that the majority of T-ALLs are dependent on Notchpathway activation.4 Notch1 mutations, however, appear to be as-sociated with a favorable therapeutic response, while Notch-independent T-ALLs have more pejorative prognoses.5-7 Thus,a better understanding of the molecular pathways associated withNotch-independent T-ALLs is required to define new therapies.
The Wnt pathway is an interesting candidate. This pathway isoncogenic in many types of cancers, including leukemias. Wnt acti-vation is detected in chronic myeloblastic leukemia, acute myelo-blastic leukemia (AML), chronic lymphoid leukemia, and multiplemyeloma and occurs mostly through the epigenetic inactivation (byDNA methylation) of genes encoding Wnt pathway inhibitors.8
The Wnt/b-catenin pathway is activated by the binding of Wntligands to a receptor complex composed of Frizzled receptors (Fzd)and one of the LDL receptor-related proteins, Lrp5/6. As a result,Dishevelled (Dvl) is activated and sequesters glycogen synthase kinase
3b (GSK3b) away from the “destruction complex” composed ofAxin1, adenomatous polysis coli (APC), and GSK3b. This dis-placement abolishes the phosporylation and proteosomal degradationof the b-catenin transcriptional coactivator. b-Catenin then accumu-lates and translocates to the nucleus, where it binds to members of theTCF/LEF family of transcription factors and modulates the expressionof a broad range of target genes.8
Wnt signaling plays a key role in ab T-cell development. In thethymus, early progenitors can be phenotypically divided intodouble-negative (DN) 1 (CD42CD82CD32CD252CD441), DN2(CD42CD82CD32CD251CD441), DN3 (CD42CD82CD32
CD251CD442), and DN4 (CD42CD82CD32CD252CD442) thy-mocytes. Successful rearrangement of the b chain of the T-cellreceptor (TCR) in DN3 cells allows them to differentiate into DN4thymocytes. DN4 cells further mature into double-positive (DP;CD41CD81CD3lo) thymocytes, susceptible to positive and nega-tive selection events that determine their developmental outcomeinto single-positive (SP) CD41 or CD81 T cells.9 LEF1 is expressedhighly in mature SP thymocytes, while TCF1 is abundantly expressedat all stages, where it has been shown to bind enhancers ofT-cell–specific genes.10,11 b-Catenin is expressed in DN1-3 cellsand subsequently downregulated in DN4 and DP cells.12 At thefunctional level, loss of Wnt signaling in TCF12/2 and TCF12/2
LEF12/2 thymocytes blocks T-cell development at the DN3 stageand Wnt pathway inhibitors block thymocyte differentiation at theDN stage in vitro.10,13,14 Wnt signaling may also be important for
Submitted December 6, 2012; accepted May 31, 2013. Prepublished online as
Blood First Edition paper, June 25, 2013; DOI 10.1182/blood-2012-12-
471904.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge
payment. Therefore, and solely to indicate this fact, this article is hereby
marked “advertisement” in accordance with 18 USC section 1734.
© 2013 by The American Society of Hematology
694 BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom

cell survival, because CD4-Cre–mediated deletion of TCF1, ortransgenic expression of catenin b interacting protein 1 (ICAT, aninhibitor of b-catenin/TCF interaction), reduces the survival of DPcells in the thymus, while overexpression of b-catenin enhancesDP cell survival in vitro.15-17 Surprisingly, loss of b-catenin inhematopoietic cells does not affect T-cell differentiation.18,19 How-ever, varying APC levels result in a gradient of developmentaldefects at the DN3 and DN4 stages.20 These data suggest animportant, albeit still unclear, function of the Wnt pathway atmultiple stages of T-cell development.
To investigate the relevance ofWnt downregulation in late DN andDP thymocytes, we generated a mouse line (R26-bcat) expressingphysiological levels of an activated form of b-catenin in thymocytesfrom the DN4 stage. We show that b-catenin activation enhances thesurvival of DP thymocytes and leads to a developmental block of thesecells. Furthermore, R26-bcat mice develop T-cell leukemias that lackNotch activation and are associated with genomic defects leading toMyc overexpression and Pten loss. Our data suggest thatWnt pathwayactivation may delineate a specific subclass of T-ALL.
Materials and methods
See supplemental Materials and methods for further details, available on theBlood Web site.
All mouse procedures were approved by the Institut de Genetique et deBiologie Moleculaire et Cellulaire (IGBMC) ethical committee (Illkirch,France) and were in accordance with national regulations.
Results
Wnt pathway regulation is required during T-cell development
We previously described the R26-NC allele, which is generated by theknock in of sequences encoding a Myc-tagged stable form ofb-catenin (S33Y) preceded by a floxed stop cassette into the Rosa26locus.21 This mutation allows the production of a constitutively activeform of b-catenin in cells expressing the Cre recombinase. To activateb-catenin in T cells, we combined the homozygous alleles of R26-NCwith the CD4-Cre transgene (tg).22 R26NC/NCCD4-CreTg mice werenamed R26-bcat mice, and R26NC/NCCD4-Cre2 mice were used ascontrols. R26-bcat thymocytes strongly expressed the b-cat-S33Yprotein (supplemental Figure 1A). As expected, this expression wasdetected in R26-bcat thymocytes from the DN3-DN4 stage on, whenendogenous b-catenin levels were downregulated in control thymo-cytes (supplemental Figure 1B). Importantly, b-catenin levels re-mained steady at all stages tested and were similar to that ofphysiological b-catenin in DN2 cells (supplemental Figure 1B).
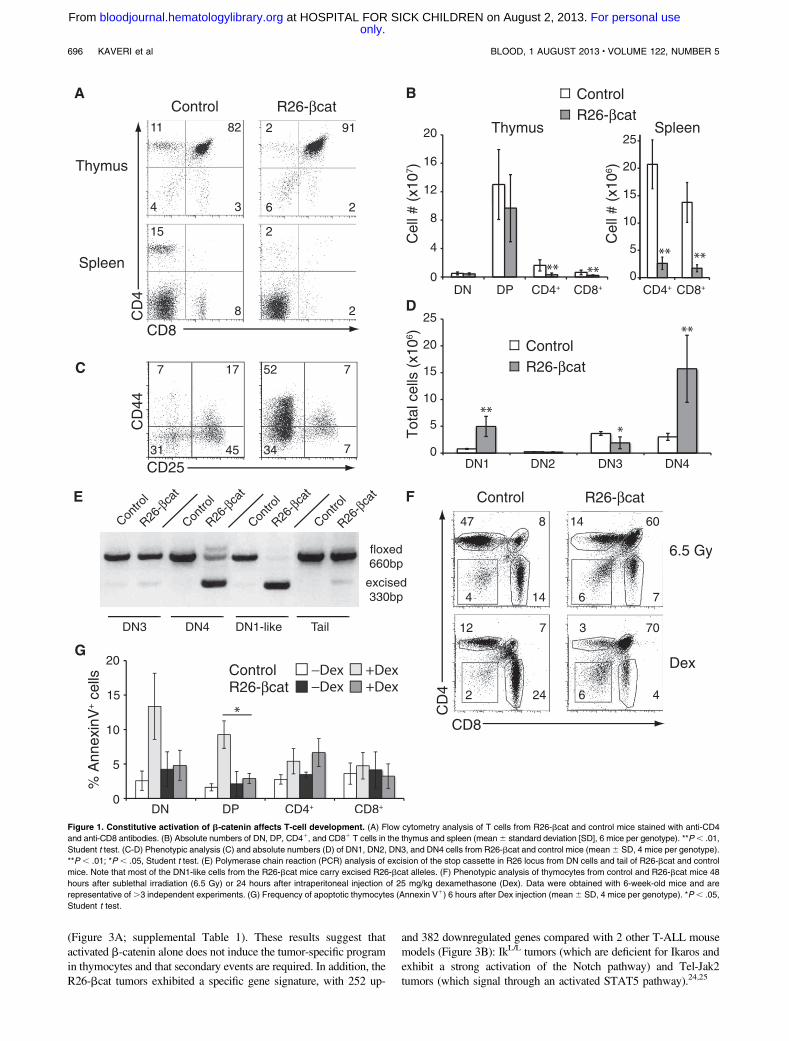
To examine the effects of sustained Wnt signaling in T-cell dif-ferentiation, we analyzed the thymus of young R26-bcat mice (6-week-old). Flow cytometry analysis showed that R26-bcat micehave reduced numbers of SP thymocytes (5-fold less CD41 and3-fold less CD81 cells; Figure 1A-B) and normal number of totalthymocytes (supplemental Figure 1C). These data suggest a block atthe DP stage, which was reflected in the spleen with low numbers ofmature T cells (Figure 1A-B). Intriguingly, R26-bcat mice showeda 6-fold increase in DN1 and DN4 cells and a decrease in DN3 cells(Figure 1C-D). Since the activity of the Cre recombinase starts at theDN3-DN4 stage (Figure 1E), the accumulation of cells with a DN1phenotype was unlikely due to the expression of b-cat-S33Y in theDN1 population. We hypothesized that CD44, a known Wnt target
gene, was induced in DN4 cells as a consequence of activatedb-catenin expression.23 Indeed, the DN1-like cells lacked c-kit1
cells, showed complete excision of the floxed cassette, and expressedhigh levels of intracellular b-catenin (Figure 1E; supplementalFigure 1B,D). Unlike DN2 and DN3 cells, the DN1-like cells ex-hibited increased proliferation (supplemental Figure 1E). Becausea similar DN1-like population has also been observed in micewith reduced APC function, we concluded that the expressionof b-catenin in DN3-DN4 thymocytes leads to the accumulationof DN4 cells ectopically expressing CD44.20 Interestingly,R26-bcat DN4 cells incorporated less BrdU than control DN4cells, suggesting a differentiation block rather than increased pro-liferation in this population.
Since Wnt signaling has been implicated in the regulation ofthymocyte survival, we tested the apoptotic response of R26-bcatthymocytes to sublethal irradiation (6.5 Gy) or dexamethasone treat-ment. Strikingly, R26-bcat DP thymocytes were resistant to treat-ment (Figure 1F). Further, the percentage of Annexin-V1 DP cellsdid not increase in R26-bcat mice after dexamethasone treatment,indicating that R26-bcat DP thymocytes were not undergoingapoptosis (Figure 1G). Thus, the constitutive expression of b-cateninin thymocytes increases the survival of DP cells.
b-Catenin activation leads to T-cell leukemia development
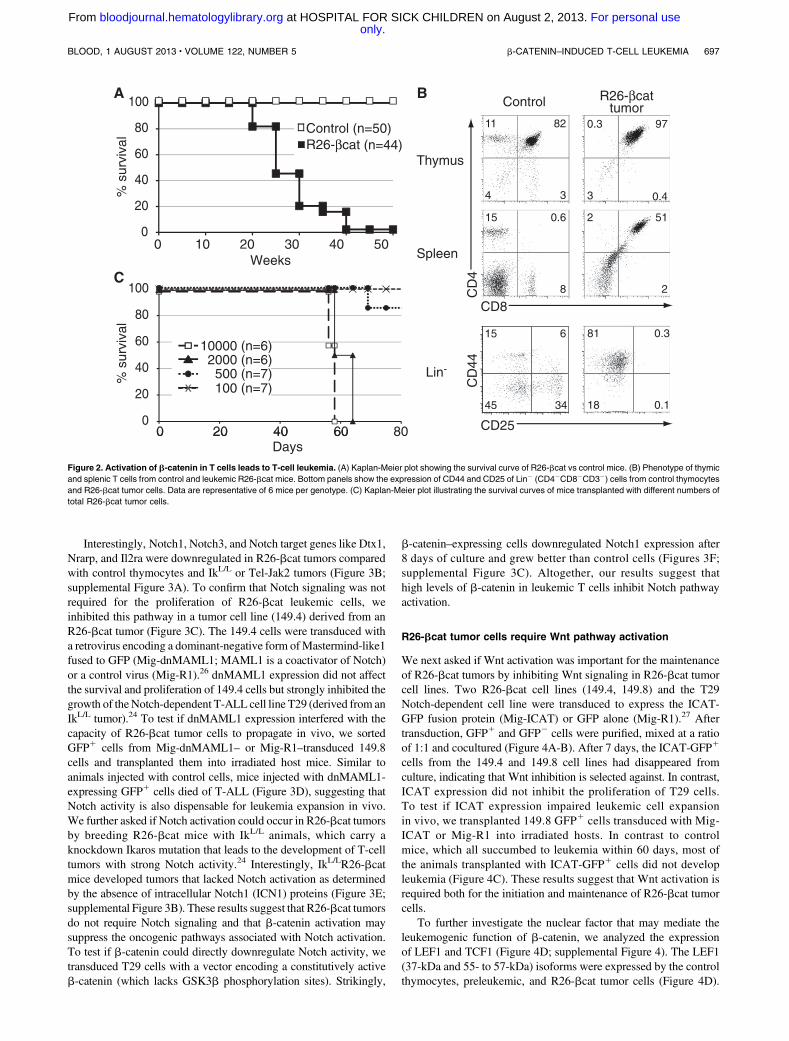
R26-bcat mice died of T-cell leukemia between 20 and 40 weeksof age (Figure 2A). Mice that were moribund around 24 weeks ofage died of the primary tumor in the thymus, while mice that fellsick around 30 weeks of age showed metastasis in peripheral organs(Figure 2B, data not shown). The tumor cells mainly exhibited a DPphenotype with high levels of CD3 and TCRb (Figure 2B; sup-plemental Figure 2A). Db2-Jb2 rearrangement analysis showed thatthe majority of R26-bcat tumors were mono- or oligoclonal (sup-plemental Figure 2B). Thus, b-catenin activation in the thymus leadsto the development of T-cell tumors.
We assessed the ability of R26-bcat tumor cells to reinitiate thetumor by transplanting 104 R26-bcat tumor cells (CD45.21) intohost mice. The recipients were followed and sacrificed when leukemiccells appeared in the blood. The BM, thymus, and spleen contained70% to 90% CD45.21 cells with a DP phenotype similar to that ofthe primary tumor (supplemental Figure 2C), showing the malignantpotential of the R26-bcat tumors. To determine if the R26-bcat tumorswere heterogeneous and if the self-renewal potential was restricted toa subset of cells, serial dilution of R26-bcat tumors cells were trans-planted in host mice. All mice transplanted with at least 23 103 cellsdeveloped leukemia while only 1 in 7 mice transplanted with 53 102
cells developed the disease (Figure 2C). Similar results were obtainedusing purified DP tumor cells (data not shown). These results indicatethat the R26-bcat tumors are heterogeneous and that a minority ofcells (1/1187; Poisson statistics) can reinitiate the leukemia.
R26-bcat tumor cells are Notch-independent
To identify the molecular pathways involved in tumor develop-ment, we profiled the global gene expression of transformed andpretransformed DP cells using Affymetrix microarrays (GeneExpression Omnibus accession number GSE48203). DP thymo-cytes from 6-week-old mice (with normal numbers of thymocytesand a polyclonal TCR repertoire) were used to define the pre-transformed transcriptome. These analyses showed that the tran-scriptome of transformed cells diverged significantly from that ofcontrol cells, with . 1800 deregulated genes (Figure 3A). In con-trast, only 154 genes were deregulated in the pre-transformed cells
BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5 b-CATENIN–INDUCED T-CELL LEUKEMIA 695
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
(Figure 3A; supplemental Table 1). These results suggest thatactivated b-catenin alone does not induce the tumor-specific programin thymocytes and that secondary events are required. In addition, theR26-bcat tumors exhibited a specific gene signature, with 252 up-
and 382 downregulated genes compared with 2 other T-ALL mousemodels (Figure 3B): IkL/L tumors (which are deficient for Ikaros andexhibit a strong activation of the Notch pathway) and Tel-Jak2tumors (which signal through an activated STAT5 pathway).24,25
Figure 1. Constitutive activation of b-catenin affects T-cell development. (A) Flow cytometry analysis of T cells from R26-bcat and control mice stained with anti-CD4
and anti-CD8 antibodies. (B) Absolute numbers of DN, DP, CD41, and CD81 T cells in the thymus and spleen (mean6 standard deviation [SD], 6 mice per genotype). **P, .01,
Student t test. (C-D) Phenotypic analysis (C) and absolute numbers (D) of DN1, DN2, DN3, and DN4 cells from R26-bcat and control mice (mean6 SD, 4 mice per genotype).
**P , .01; *P , .05, Student t test. (E) Polymerase chain reaction (PCR) analysis of excision of the stop cassette in R26 locus from DN cells and tail of R26-bcat and control
mice. Note that most of the DN1-like cells from the R26-bcat mice carry excised R26-bcat alleles. (F) Phenotypic analysis of thymocytes from control and R26-bcat mice 48
hours after sublethal irradiation (6.5 Gy) or 24 hours after intraperitoneal injection of 25 mg/kg dexamethasone (Dex). Data were obtained with 6-week-old mice and are
representative of.3 independent experiments. (G) Frequency of apoptotic thymocytes (Annexin V1) 6 hours after Dex injection (mean6 SD, 4 mice per genotype). *P, .05,
Student t test.
696 KAVERI et al BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
Interestingly, Notch1, Notch3, and Notch target genes like Dtx1,Nrarp, and Il2ra were downregulated in R26-bcat tumors comparedwith control thymocytes and IkL/L or Tel-Jak2 tumors (Figure 3B;supplemental Figure 3A). To confirm that Notch signaling was notrequired for the proliferation of R26-bcat leukemic cells, weinhibited this pathway in a tumor cell line (149.4) derived from anR26-bcat tumor (Figure 3C). The 149.4 cells were transduced witha retrovirus encoding a dominant-negative form ofMastermind-like1fused to GFP (Mig-dnMAML1; MAML1 is a coactivator of Notch)or a control virus (Mig-R1).26 dnMAML1 expression did not affectthe survival and proliferation of 149.4 cells but strongly inhibited thegrowth of theNotch-dependent T-ALL cell line T29 (derived from anIkL/L tumor).24 To test if dnMAML1 expression interfered with thecapacity of R26-bcat tumor cells to propagate in vivo, we sortedGFP1 cells from Mig-dnMAML1– or Mig-R1–transduced 149.8cells and transplanted them into irradiated host mice. Similar toanimals injected with control cells, mice injected with dnMAML1-expressing GFP1 cells died of T-ALL (Figure 3D), suggesting thatNotch activity is also dispensable for leukemia expansion in vivo.We further asked if Notch activation could occur in R26-bcat tumorsby breeding R26-bcat mice with IkL/L animals, which carry aknockdown Ikaros mutation that leads to the development of T-celltumors with strong Notch activity.24 Interestingly, IkL/LR26-bcatmice developed tumors that lacked Notch activation as determinedby the absence of intracellular Notch1 (ICN1) proteins (Figure 3E;supplemental Figure 3B). These results suggest that R26-bcat tumorsdo not require Notch signaling and that b-catenin activation maysuppress the oncogenic pathways associated with Notch activation.To test if b-catenin could directly downregulate Notch activity, wetransduced T29 cells with a vector encoding a constitutively activeb-catenin (which lacks GSK3b phosphorylation sites). Strikingly,
b-catenin–expressing cells downregulated Notch1 expression after8 days of culture and grew better than control cells (Figures 3F;supplemental Figure 3C). Altogether, our results suggest thathigh levels of b-catenin in leukemic T cells inhibit Notch pathwayactivation.
R26-bcat tumor cells require Wnt pathway activation
We next asked if Wnt activation was important for the maintenanceof R26-bcat tumors by inhibiting Wnt signaling in R26-bcat tumorcell lines. Two R26-bcat cell lines (149.4, 149.8) and the T29Notch-dependent cell line were transduced to express the ICAT-GFP fusion protein (Mig-ICAT) or GFP alone (Mig-R1).27 Aftertransduction, GFP1 and GFP2 cells were purified, mixed at a ratioof 1:1 and cocultured (Figure 4A-B). After 7 days, the ICAT-GFP1
cells from the 149.4 and 149.8 cell lines had disappeared fromculture, indicating that Wnt inhibition is selected against. In contrast,ICAT expression did not inhibit the proliferation of T29 cells.To test if ICAT expression impaired leukemic cell expansionin vivo, we transplanted 149.8 GFP1 cells transduced with Mig-ICAT or Mig-R1 into irradiated hosts. In contrast to controlmice, which all succumbed to leukemia within 60 days, most ofthe animals transplanted with ICAT-GFP1 cells did not developleukemia (Figure 4C). These results suggest that Wnt activation isrequired both for the initiation and maintenance of R26-bcat tumorcells.
To further investigate the nuclear factor that may mediate theleukemogenic function of b-catenin, we analyzed the expressionof LEF1 and TCF1 (Figure 4D; supplemental Figure 4). The LEF1(37-kDa and 55- to 57-kDa) isoforms were expressed by the controlthymocytes, preleukemic, and R26-bcat tumor cells (Figure 4D).
Figure 2. Activation of b-catenin in T cells leads to T-cell leukemia. (A) Kaplan-Meier plot showing the survival curve of R26-bcat vs control mice. (B) Phenotype of thymic
and splenic T cells from control and leukemic R26-bcat mice. Bottom panels show the expression of CD44 and CD25 of Lin2 (CD42CD82CD32) cells from control thymocytes
and R26-bcat tumor cells. Data are representative of 6 mice per genotype. (C) Kaplan-Meier plot illustrating the survival curves of mice transplanted with different numbers of
total R26-bcat tumor cells.
BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5 b-CATENIN–INDUCED T-CELL LEUKEMIA 697
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
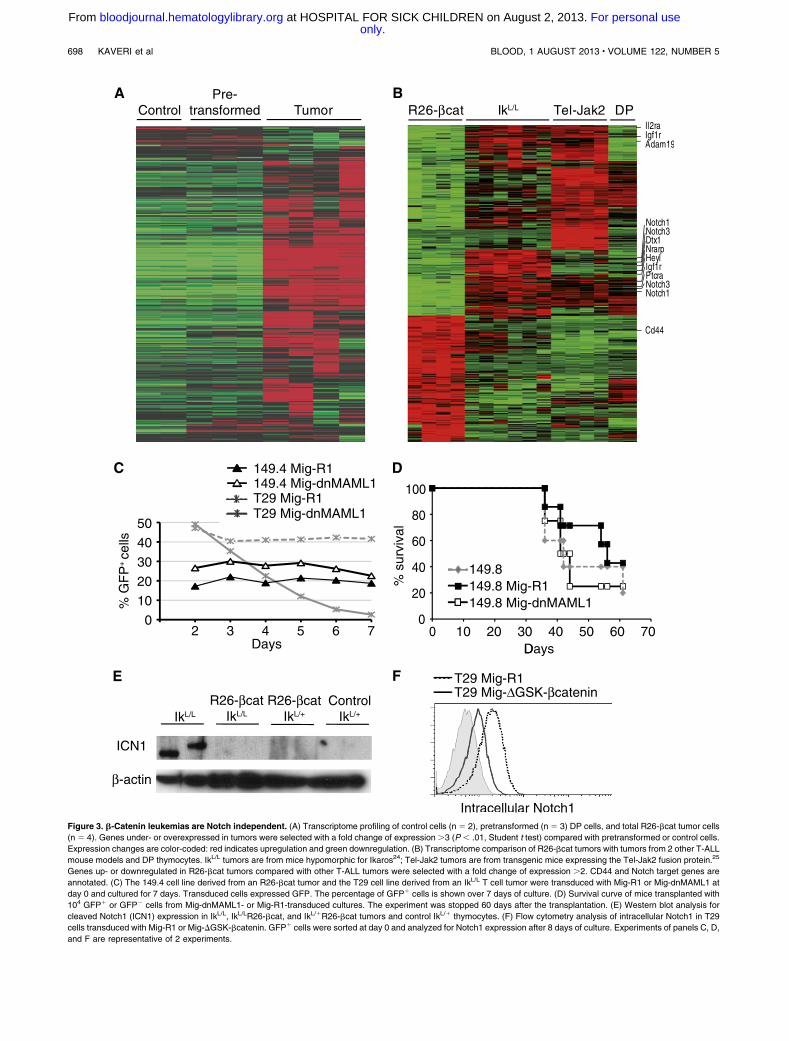
Figure 3. b-Catenin leukemias are Notch independent. (A) Transcriptome profiling of control cells (n 5 2), pretransformed (n 5 3) DP cells, and total R26-bcat tumor cells
(n 5 4). Genes under- or overexpressed in tumors were selected with a fold change of expression.3 (P , .01, Student t test) compared with pretransformed or control cells.
Expression changes are color-coded: red indicates upregulation and green downregulation. (B) Transcriptome comparison of R26-bcat tumors with tumors from 2 other T-ALL
mouse models and DP thymocytes. IkL/L tumors are from mice hypomorphic for Ikaros24; Tel-Jak2 tumors are from transgenic mice expressing the Tel-Jak2 fusion protein.25
Genes up- or downregulated in R26-bcat tumors compared with other T-ALL tumors were selected with a fold change of expression .2. CD44 and Notch target genes are
annotated. (C) The 149.4 cell line derived from an R26-bcat tumor and the T29 cell line derived from an IkL/L T cell tumor were transduced with Mig-R1 or Mig-dnMAML1 at
day 0 and cultured for 7 days. Transduced cells expressed GFP. The percentage of GFP1 cells is shown over 7 days of culture. (D) Survival curve of mice transplanted with
104 GFP1 or GFP2 cells from Mig-dnMAML1- or Mig-R1-transduced cultures. The experiment was stopped 60 days after the transplantation. (E) Western blot analysis for
cleaved Notch1 (ICN1) expression in IkL/L, IkL/LR26-bcat, and IkL/1R26-bcat tumors and control IkL/1 thymocytes. (F) Flow cytometry analysis of intracellular Notch1 in T29
cells transduced with Mig-R1 or Mig-DGSK-bcatenin. GFP1 cells were sorted at day 0 and analyzed for Notch1 expression after 8 days of culture. Experiments of panels C, D,
and F are representative of 2 experiments.
698 KAVERI et al BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
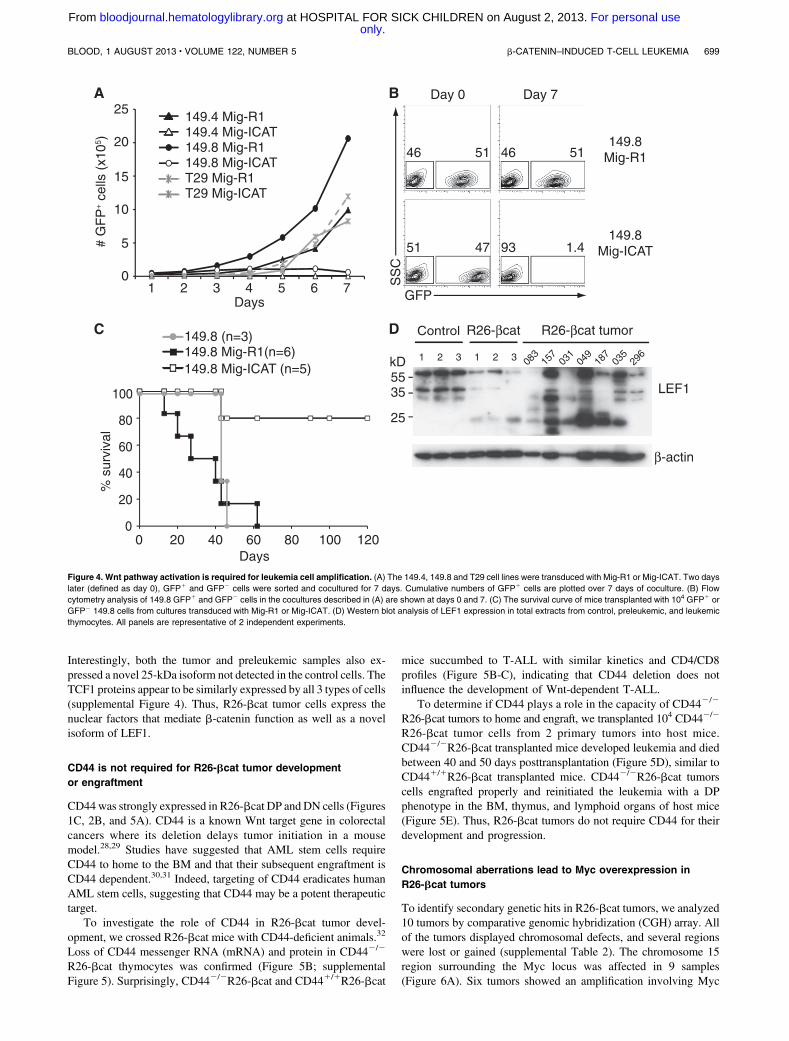
Interestingly, both the tumor and preleukemic samples also ex-pressed a novel 25-kDa isoform not detected in the control cells. TheTCF1 proteins appear to be similarly expressed by all 3 types of cells(supplemental Figure 4). Thus, R26-bcat tumor cells express thenuclear factors that mediate b-catenin function as well as a novelisoform of LEF1.
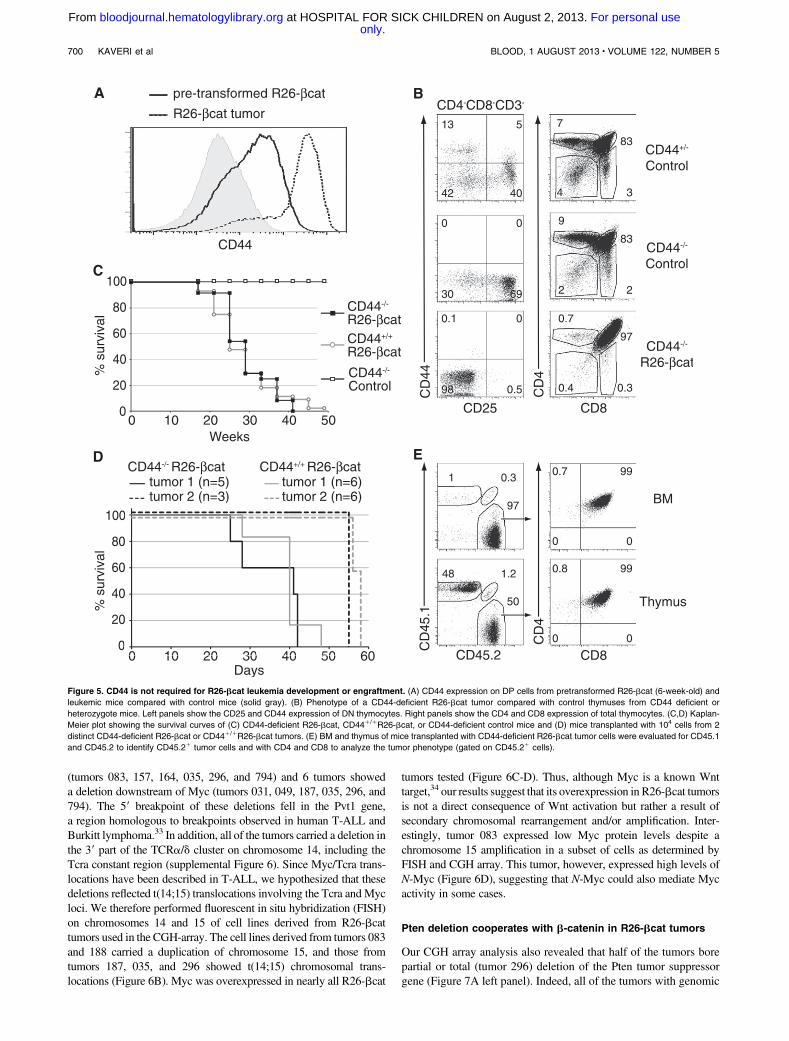
CD44 is not required for R26-bcat tumor development
or engraftment
CD44was strongly expressed in R26-bcat DP andDN cells (Figures1C, 2B, and 5A). CD44 is a known Wnt target gene in colorectalcancers where its deletion delays tumor initiation in a mousemodel.28,29 Studies have suggested that AML stem cells requireCD44 to home to the BM and that their subsequent engraftment isCD44 dependent.30,31 Indeed, targeting of CD44 eradicates humanAML stem cells, suggesting that CD44 may be a potent therapeutictarget.
To investigate the role of CD44 in R26-bcat tumor devel-opment, we crossed R26-bcat mice with CD44-deficient animals.32
Loss of CD44 messenger RNA (mRNA) and protein in CD442/2
R26-bcat thymocytes was confirmed (Figure 5B; supplementalFigure 5). Surprisingly, CD442/2R26-bcat and CD441/1R26-bcat
mice succumbed to T-ALL with similar kinetics and CD4/CD8profiles (Figure 5B-C), indicating that CD44 deletion does notinfluence the development of Wnt-dependent T-ALL.
To determine if CD44 plays a role in the capacity of CD442/2
R26-bcat tumors to home and engraft, we transplanted 104 CD442/2
R26-bcat tumor cells from 2 primary tumors into host mice.CD442/2R26-bcat transplanted mice developed leukemia and diedbetween 40 and 50 days posttransplantation (Figure 5D), similar toCD441/1R26-bcat transplanted mice. CD442/2R26-bcat tumorscells engrafted properly and reinitiated the leukemia with a DPphenotype in the BM, thymus, and lymphoid organs of host mice(Figure 5E). Thus, R26-bcat tumors do not require CD44 for theirdevelopment and progression.
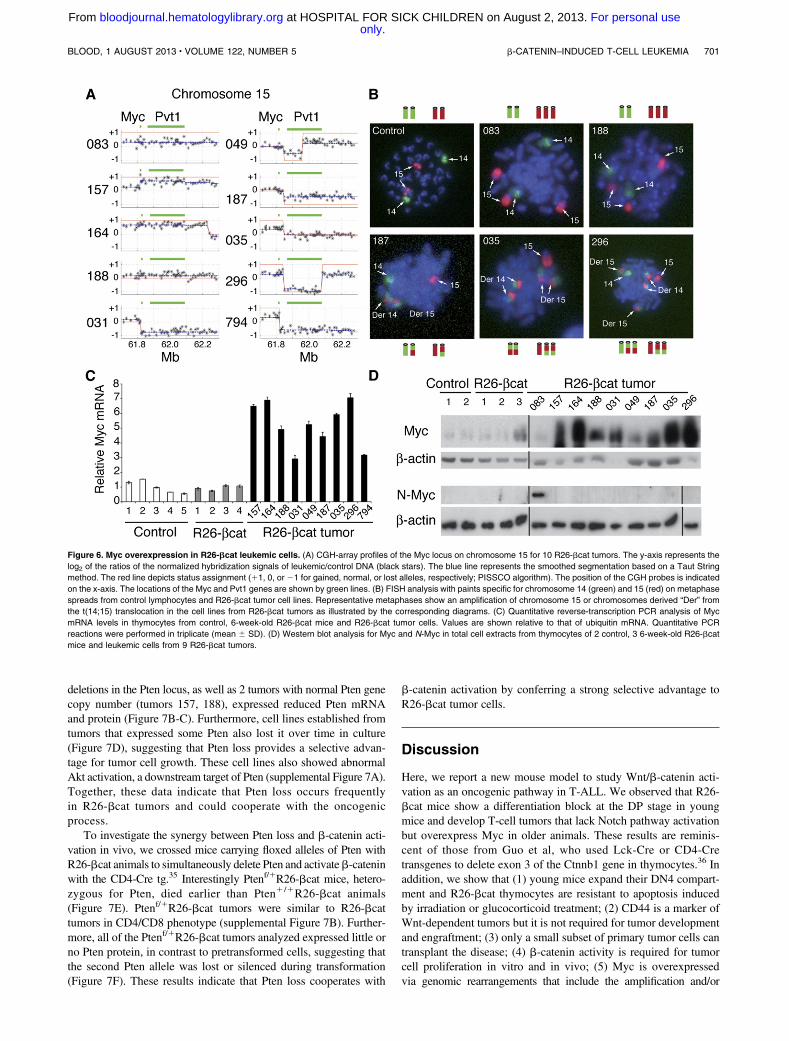
Chromosomal aberrations lead to Myc overexpression in
R26-bcat tumors
To identify secondary genetic hits in R26-bcat tumors, we analyzed10 tumors by comparative genomic hybridization (CGH) array. Allof the tumors displayed chromosomal defects, and several regionswere lost or gained (supplemental Table 2). The chromosome 15region surrounding the Myc locus was affected in 9 samples(Figure 6A). Six tumors showed an amplification involving Myc
Figure 4. Wnt pathway activation is required for leukemia cell amplification. (A) The 149.4, 149.8 and T29 cell lines were transduced with Mig-R1 or Mig-ICAT. Two days
later (defined as day 0), GFP1 and GFP2 cells were sorted and cocultured for 7 days. Cumulative numbers of GFP1 cells are plotted over 7 days of coculture. (B) Flow
cytometry analysis of 149.8 GFP1 and GFP2 cells in the cocultures described in (A) are shown at days 0 and 7. (C) The survival curve of mice transplanted with 104 GFP1 or
GFP2 149.8 cells from cultures transduced with Mig-R1 or Mig-ICAT. (D) Western blot analysis of LEF1 expression in total extracts from control, preleukemic, and leukemic
thymocytes. All panels are representative of 2 independent experiments.
BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5 b-CATENIN–INDUCED T-CELL LEUKEMIA 699
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
(tumors 083, 157, 164, 035, 296, and 794) and 6 tumors showeda deletion downstream of Myc (tumors 031, 049, 187, 035, 296, and794). The 59 breakpoint of these deletions fell in the Pvt1 gene,a region homologous to breakpoints observed in human T-ALL andBurkitt lymphoma.33 In addition, all of the tumors carried a deletion inthe 39 part of the TCRa/d cluster on chromosome 14, including theTcra constant region (supplemental Figure 6). Since Myc/Tcra trans-locations have been described in T-ALL, we hypothesized that thesedeletions reflected t(14;15) translocations involving the Tcra and Mycloci. We therefore performed fluorescent in situ hybridization (FISH)on chromosomes 14 and 15 of cell lines derived from R26-bcattumors used in the CGH-array. The cell lines derived from tumors 083and 188 carried a duplication of chromosome 15, and those fromtumors 187, 035, and 296 showed t(14;15) chromosomal trans-locations (Figure 6B). Myc was overexpressed in nearly all R26-bcat
tumors tested (Figure 6C-D). Thus, although Myc is a known Wnttarget,34 our results suggest that its overexpression in R26-bcat tumorsis not a direct consequence of Wnt activation but rather a result ofsecondary chromosomal rearrangement and/or amplification. Inter-estingly, tumor 083 expressed low Myc protein levels despite achromosome 15 amplification in a subset of cells as determined byFISH and CGH array. This tumor, however, expressed high levels ofN-Myc (Figure 6D), suggesting that N-Myc could also mediate Mycactivity in some cases.
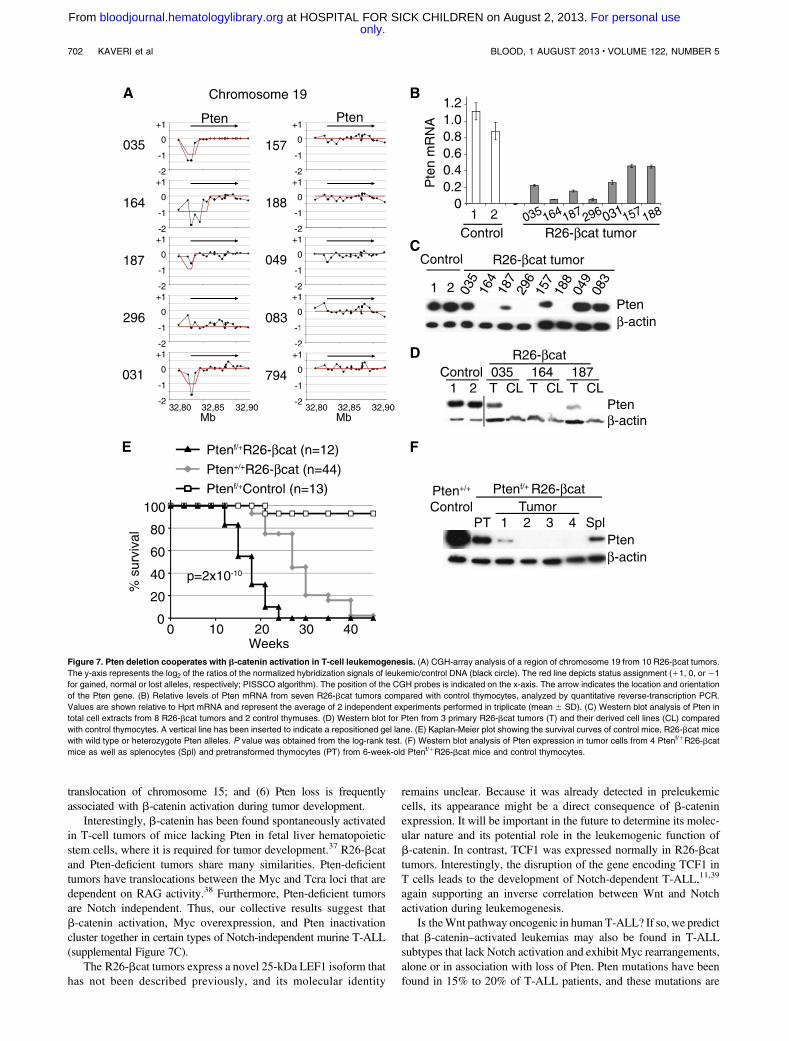
Pten deletion cooperates with b-catenin in R26-bcat tumors
Our CGH array analysis also revealed that half of the tumors borepartial or total (tumor 296) deletion of the Pten tumor suppressorgene (Figure 7A left panel). Indeed, all of the tumors with genomic
Figure 5. CD44 is not required for R26-bcat leukemia development or engraftment. (A) CD44 expression on DP cells from pretransformed R26-bcat (6-week-old) and
leukemic mice compared with control mice (solid gray). (B) Phenotype of a CD44-deficient R26-bcat tumor compared with control thymuses from CD44 deficient or
heterozygote mice. Left panels show the CD25 and CD44 expression of DN thymocytes. Right panels show the CD4 and CD8 expression of total thymocytes. (C,D) Kaplan-
Meier plot showing the survival curves of (C) CD44-deficient R26-bcat, CD441/1R26-bcat, or CD44-deficient control mice and (D) mice transplanted with 104 cells from 2
distinct CD44-deficient R26-bcat or CD441/1R26-bcat tumors. (E) BM and thymus of mice transplanted with CD44-deficient R26-bcat tumor cells were evaluated for CD45.1
and CD45.2 to identify CD45.21 tumor cells and with CD4 and CD8 to analyze the tumor phenotype (gated on CD45.21 cells).
700 KAVERI et al BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
deletions in the Pten locus, as well as 2 tumors with normal Pten genecopy number (tumors 157, 188), expressed reduced Pten mRNAand protein (Figure 7B-C). Furthermore, cell lines established fromtumors that expressed some Pten also lost it over time in culture(Figure 7D), suggesting that Pten loss provides a selective advan-tage for tumor cell growth. These cell lines also showed abnormalAkt activation, a downstream target of Pten (supplemental Figure 7A).Together, these data indicate that Pten loss occurs frequentlyin R26-bcat tumors and could cooperate with the oncogenicprocess.
To investigate the synergy between Pten loss and b-catenin acti-vation in vivo, we crossed mice carrying floxed alleles of Pten withR26-bcat animals to simultaneously delete Pten and activateb-cateninwith the CD4-Cre tg.35 Interestingly Ptenf/1R26-bcat mice, hetero-zygous for Pten, died earlier than Pten1/1R26-bcat animals(Figure 7E). Ptenf/1R26-bcat tumors were similar to R26-bcattumors in CD4/CD8 phenotype (supplemental Figure 7B). Further-more, all of the Ptenf/1R26-bcat tumors analyzed expressed little orno Pten protein, in contrast to pretransformed cells, suggesting thatthe second Pten allele was lost or silenced during transformation(Figure 7F). These results indicate that Pten loss cooperates with
b-catenin activation by conferring a strong selective advantage toR26-bcat tumor cells.
Discussion
Here, we report a new mouse model to study Wnt/b-catenin acti-vation as an oncogenic pathway in T-ALL. We observed that R26-bcat mice show a differentiation block at the DP stage in youngmice and develop T-cell tumors that lack Notch pathway activationbut overexpress Myc in older animals. These results are reminis-cent of those from Guo et al, who used Lck-Cre or CD4-Cretransgenes to delete exon 3 of the Ctnnb1 gene in thymocytes.36 Inaddition, we show that (1) young mice expand their DN4 compart-ment and R26-bcat thymocytes are resistant to apoptosis inducedby irradiation or glucocorticoid treatment; (2) CD44 is a marker ofWnt-dependent tumors but it is not required for tumor developmentand engraftment; (3) only a small subset of primary tumor cells cantransplant the disease; (4) b-catenin activity is required for tumorcell proliferation in vitro and in vivo; (5) Myc is overexpressedvia genomic rearrangements that include the amplification and/or
Figure 6. Myc overexpression in R26-bcat leukemic cells. (A) CGH-array profiles of the Myc locus on chromosome 15 for 10 R26-bcat tumors. The y-axis represents the
log2 of the ratios of the normalized hybridization signals of leukemic/control DNA (black stars). The blue line represents the smoothed segmentation based on a Taut String
method. The red line depicts status assignment (11, 0, or 21 for gained, normal, or lost alleles, respectively; PISSCO algorithm). The position of the CGH probes is indicated
on the x-axis. The locations of the Myc and Pvt1 genes are shown by green lines. (B) FISH analysis with paints specific for chromosome 14 (green) and 15 (red) on metaphase
spreads from control lymphocytes and R26-bcat tumor cell lines. Representative metaphases show an amplification of chromosome 15 or chromosomes derived “Der” from
the t(14;15) translocation in the cell lines from R26-bcat tumors as illustrated by the corresponding diagrams. (C) Quantitative reverse-transcription PCR analysis of Myc
mRNA levels in thymocytes from control, 6-week-old R26-bcat mice and R26-bcat tumor cells. Values are shown relative to that of ubiquitin mRNA. Quantitative PCR
reactions were performed in triplicate (mean 6 SD). (D) Western blot analysis for Myc and N-Myc in total cell extracts from thymocytes of 2 control, 3 6-week-old R26-bcat
mice and leukemic cells from 9 R26-bcat tumors.
BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5 b-CATENIN–INDUCED T-CELL LEUKEMIA 701
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom
translocation of chromosome 15; and (6) Pten loss is frequentlyassociated with b-catenin activation during tumor development.
Interestingly, b-catenin has been found spontaneously activatedin T-cell tumors of mice lacking Pten in fetal liver hematopoieticstem cells, where it is required for tumor development.37 R26-bcatand Pten-deficient tumors share many similarities. Pten-deficienttumors have translocations between the Myc and Tcra loci that aredependent on RAG activity.38 Furthermore, Pten-deficient tumorsare Notch independent. Thus, our collective results suggest thatb-catenin activation, Myc overexpression, and Pten inactivationcluster together in certain types of Notch-independent murine T-ALL(supplemental Figure 7C).
The R26-bcat tumors express a novel 25-kDa LEF1 isoform thathas not been described previously, and its molecular identity
remains unclear. Because it was already detected in preleukemiccells, its appearance might be a direct consequence of b-cateninexpression. It will be important in the future to determine its molec-ular nature and its potential role in the leukemogenic function ofb-catenin. In contrast, TCF1 was expressed normally in R26-bcattumors. Interestingly, the disruption of the gene encoding TCF1 inT cells leads to the development of Notch-dependent T-ALL,11,39
again supporting an inverse correlation between Wnt and Notchactivation during leukemogenesis.
Is theWnt pathway oncogenic in human T-ALL? If so, we predictthat b-catenin–activated leukemias may also be found in T-ALLsubtypes that lack Notch activation and exhibit Myc rearrangements,alone or in association with loss of Pten. Pten mutations have beenfound in 15% to 20% of T-ALL patients, and these mutations are
Figure 7. Pten deletion cooperates with b-catenin activation in T-cell leukemogenesis. (A) CGH-array analysis of a region of chromosome 19 from 10 R26-bcat tumors.
The y-axis represents the log2 of the ratios of the normalized hybridization signals of leukemic/control DNA (black circle). The red line depicts status assignment (11, 0, or 21
for gained, normal or lost alleles, respectively; PISSCO algorithm). The position of the CGH probes is indicated on the x-axis. The arrow indicates the location and orientation
of the Pten gene. (B) Relative levels of Pten mRNA from seven R26-bcat tumors compared with control thymocytes, analyzed by quantitative reverse-transcription PCR.
Values are shown relative to Hprt mRNA and represent the average of 2 independent experiments performed in triplicate (mean 6 SD). (C) Western blot analysis of Pten in
total cell extracts from 8 R26-bcat tumors and 2 control thymuses. (D) Western blot for Pten from 3 primary R26-bcat tumors (T) and their derived cell lines (CL) compared
with control thymocytes. A vertical line has been inserted to indicate a repositioned gel lane. (E) Kaplan-Meier plot showing the survival curves of control mice, R26-bcat mice
with wild type or heterozygote Pten alleles. P value was obtained from the log-rank test. (F) Western blot analysis of Pten expression in tumor cells from 4 Ptenf/1R26-bcat
mice as well as splenocytes (Spl) and pretransformed thymocytes (PT) from 6-week-old Ptenf/1R26-bcat mice and control thymocytes.
702 KAVERI et al BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom

predominant in cases that lack Notch activation or have weak Notch1mutant alleles.7,40 Currently, no mutations that directly activate theWnt pathway, like those found in colorectal cancers, have beenidentified in human T-ALL. However, Wnt is activated in somehematopoietic malignancies (eg, AML, chronic myeloblastic leuke-mia, and multiple myeloma) through the hypermethylation of genesthat encode Wnt signaling inhibitors like the Sfrp and Dkk families,suggesting that Wnt can be activated by indirect means.8 Indeed,Wntactivation and b-catenin stabilization is common in breast cancer, butdirect mutations affecting this pathway are rare.41 In a recent reviewarticle, Weerkamp et al disclosed that high levels of intracellularb-catenin can be detected in a subset of T-ALL, but this findingawaits formal publishing.42 If true, then b-catenin activation mightplay a role in the immature ETP-ALL subtype, which has a lowincidence of Notch1 mutations, or the TAL/LMO subtype, whichfrequently exhibits Pten mutations.7,43,44 We attempted to identifyhuman T-ALL cases homologous to the R26-bcat leukemias by min-ing existing T-ALL transcriptomic datasets for the R26-bcat–specificsignature. However, these attempts were unsuccessful.3,44,45 Furtherresearch is therefore needed to clarify the involvement of b-cateninactivation in human T-ALL.
Myc (and more rarely N-Myc) overexpression appears to be adriving force in b-catenin–dependent leukemogenesis. Interestingly,Myc is also critical for Notch-dependent T-ALL, where it has beenshown to be a direct target of the Notch pathway, and does not appearto require chromosomal rearrangements for its upregulation.46-49
Although Myc is a target of the Wnt pathway, its expression is notinduced in pretransformed R26-bcat thymocytes. Rather, Myc in-duction in R26-bcat tumor cells is due to rearrangements involvingthe Myc locus and probably Pten inactivation, as Pten has beenshown to inhibit Myc protein accumulation in human T-ALL cells.40
Pten negatively regulates Akt, which in turn phosphorylates GSK3bto inhibit its activity. As GSK3b is required for Myc degradation,loss of Pten favors Myc overexpression.50 Indeed, we observed aninverse correlation between Myc and Pten in 2 R26-bcat tumors(tumors 049 and 083; see Figures 6 and 7). Nevertheless, our analysisof Pten expression as well as Akt andMyc phosphorylation in a panelof R26-bcat cell lines did not reveal a good correlation with Ptenstatus (supplemental Figure 7A), suggesting that Myc accumulationmay not be due to Pten loss in some tumors.
Why is the Notch pathway silent in R26-bcat tumors? Onepossibility is that R26-bcat tumors develop from DP thymocytes,a stage at which Notch signaling is normally diminished.51 Anotheris that Wnt and Notch signaling are mutually antagonistic inT cells. The latter is supported by the phenotype of IkL/LR26-bcattumors. Although IkL/L tumors always show strong Notch acti-vation,24 IkL/LR26-bcat tumors do not, suggesting that Wnt maysuppress Notch activation. Further, intracellular Notch1 is reducedin T29 cells expressing active b-catenin, although this occurredafter several days of expression, suggesting a complex cascade ofregulatory events. Antagonism between Wnt and Notch exists inother biological systems. In Drosophila, Wnt activation inhibitsNotch signaling during wing morphogenesis, due to competition forDv1 by both pathways.52 Conversely, in colon cancer, Notchactivation inhibits Wnt signaling through a mechanism not fullyunderstood.53 The R26-bcat model may therefore provide a tool toinvestigate how Wnt activation suppresses the Notch pathway.
The downstream effectors of Wnt activation in leukemogenesisremain elusive. CD44, a Wnt target gene, is overexpressed in R26-bcat tumors and was an attractive candidate to test in our model.However, CD44 deletion does not change the kinetics of R26-bcat
tumor development or its engraftment properties during trans-plantation. This was surprising given that CD44 was shown tocontribute to colon cancer aggressivity and CD44 plays a role indefining the leukemic stem cell activity in certain cancers.29,30 Incontrast, Wnt activation has been reported to negatively regulateapoptosis, and this appears to be the case for pretransformed R26-bcat DP thymocytes that are resistant to radiation- or glucocorticoid-induced apoptosis. Our results are in agreement with previousreports that TCF1 or b-catenin signaling is required for DP cellsurvival.15,17,54 Increased survival may therefore promote tumorinitiation by favoring the accumulation of secondary mutations.
Finally, we found that relatively high numbers of R26-bcattumor cells (1/1187) are required to initiate a new tumor in hostmice, suggesting that only a minor subset of cells have leukemia-initiating cell activity. Interestingly, Pten-deficient leukemias alsocontain a minor population of cKitmedCD31Lin2 cells with LICactivity that requires b-catenin activation.37 cKitmedCD31Lin2
cells are not present in R26-bcat tumors, and we have not beenable to identify a phenotypically consistent leukemia-initiating cellpopulation.
In summary, we show that the canonicalWnt pathway is oncogenicin T cells and that Wnt activation, Pten loss, and Myc translocationsynergize to define a novel subset of murine Notch-independentT-ALL. These results have implications for future studies in humans.
Acknowledgments
The authors thank T. Mak and A. Suzuki for the floxed Pten mice,J. Meijerink for sharing clinical status of human T-ALL samples,J. Ghysdael for transcriptomic data from Tel-Jak2 tumors, J. Asterfor the Mig-dnMAML1 vector, A. Leutz for the Mig-DGSK-bcatenin vector, W. Pear for the Mig-R1 vector system, G. Nolanfor the Eco-Phoenix cells, P. Marchal for technical assistance, theIGBMC transcriptome and sequencing Platform, and S. Falconeand M. Gendron for animal husbandry.
This work was supported by grants from the Fondation ARC,the Fondation de France, La Ligue Contre le Cancer (equipelabellisee), Institut National du Cancer, and institute funding fromInstitut National de la Sante et de la Recherche Medicale, CentreNational de laRecherche Scientifique, and theUniversite de Strasbourg.D.K. received predoctoral fellowships from the Institut de Genetiqueet de Biologie Moleculaire et Cellulaire International PhD programand from the Fondation pour la Recherche Medicale. P. Kirstetterreceived a postdoctoral fellowship from the Fondation ARC.
Authorship
Contribution: D.K., P. Kastner, S.C., and P. Kirstetter designed andperformed experiments, analyzed data, and wrote the paper; D.D.analyzed data; and N.C. contributed to scientific discussions.
Conflict-of-interest disclosure: The authors declare no compet-ing financial interests.
Correspondence: Peggy Kirstetter, IGBMC, 1 Rue Laurent Fries,67404 Illkirch Cedex, France; e-mail: [email protected]; and SusanChan, IGBMC, 1 rue Laurent Fries, 67404 Illkirch Cedex, France;e-mail: [email protected].
BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5 b-CATENIN–INDUCED T-CELL LEUKEMIA 703
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom

References
1. Ferrando AA, Neuberg DS, Staunton J, et al.Gene expression signatures define noveloncogenic pathways in T cell acute lymphoblasticleukemia. Cancer Cell. 2002;1(1):75-87.
2. Bergeron J, Clappier E, Radford I, et al.Prognostic and oncogenic relevance of TLX1/HOX11 expression level in T-ALLs. Blood. 2007;110(7):2324-2330.
3. Homminga I, Pieters R, Langerak AW, et al.Integrated transcript and genome analyses revealNKX2-1 and MEF2C as potential oncogenes inT cell acute lymphoblastic leukemia. Cancer Cell.2011;19(4):484-497.
4. Weng AP, Ferrando AA, Lee W, et al. Activatingmutations of NOTCH1 in human T cell acutelymphoblastic leukemia. Science. 2004;306(5694):269-271.
5. Paganin M, Ferrando A. Molecular pathogenesisand targeted therapies for NOTCH1-inducedT-cell acute lymphoblastic leukemia. Blood Rev.2011;25(2):83-90.
6. Callens C, Baleydier F, Lengline E, et al. Clinicalimpact of NOTCH1 and/or FBXW7 mutations,FLASH deletion, and TCR status in pediatricT-cell lymphoblastic lymphoma. J Clin Oncol.2012;30(16):1966-1973.
7. Zuurbier L, Petricoin EF III, Vuerhard MJ, et al.The significance of PTEN and AKT aberrationsin pediatric T-cell acute lymphoblastic leukemia.Haematologica. 2012;97(9):1405-1413.
8. Seke Etet PF, Vecchio L, Bogne Kamga P,Nchiwan Nukenine E, Krampera M, NwaboKamdje AH. Normal hematopoiesis andhematologic malignancies: role of canonical Wntsignaling pathway and stromal microenvironment.Biochim Biophys Acta. 2013;1835(1):1-10.
9. Ellmeier W, Sawada S, Littman DR. Theregulation of CD4 and CD8 coreceptor geneexpression during T cell development. Annu RevImmunol. 1999;17:523-554.
10. Okamura RM, Sigvardsson M, Galceran J,Verbeek S, Clevers H, Grosschedl R. Redundantregulation of T cell differentiation and TCRalphagene expression by the transcription factorsLEF-1 and TCF-1. Immunity. 1998;8(1):11-20.
11. Tiemessen MM, Baert MR, Schonewille T, et al.The nuclear effector of Wnt-signaling, Tcf1,functions as a T-cell-specific tumor suppressor fordevelopment of lymphomas. PLoS Biol. 2012;10(11):e1001430.
12. Weerkamp F, Baert MR, Naber BA, et al. Wntsignaling in the thymus is regulated by differentialexpression of intracellular signaling molecules.Proc Natl Acad Sci USA. 2006;103(9):3322-3326.
13. Schilham MW, Wilson A, Moerer P, Benaissa-Trouw BJ, Cumano A, Clevers HC. Criticalinvolvement of Tcf-1 in expansion of thymocytes.J Immunol. 1998;161(8):3984-3991.
14. Staal FJ, Meeldijk J, Moerer P, et al. Wnt signalingis required for thymocyte development andactivates Tcf-1 mediated transcription. Eur JImmunol. 2001;31(1):285-293.
15. Ioannidis V, Beermann F, Clevers H, Held W.The beta-catenin—TCF-1 pathway ensuresCD4(1)CD8(1) thymocyte survival. Nat Immunol.2001;2(8):691-697.
16. Xie H, Huang Z, Sadim MS, Sun Z. Stabilizedbeta-catenin extends thymocyte survival byup-regulating Bcl-xL. J Immunol. 2005;175(12):7981-7988.
17. Hossain MZ, Yu Q, Xu M, Sen JM. ICATexpression disrupts beta-catenin-TCF interactionsand impairs survival of thymocytes and activatedmature T cells. Int Immunol. 2008;20(7):925-935.
18. Koch U, Wilson A, Cobas M, Kemler R,Macdonald HR, Radtke F. Simultaneous loss of
beta- and gamma-catenin does not perturbhematopoiesis or lymphopoiesis. Blood. 2008;111(1):160-164.
19. Jeannet G, Scheller M, Scarpellino L, et al. Long-term, multilineage hematopoiesis occurs in thecombined absence of beta-catenin and gamma-catenin. Blood. 2008;111(1):142-149.
20. Luis TC, Naber BA, Roozen PP, et al. Canonicalwnt signaling regulates hematopoiesis ina dosage-dependent fashion. Cell Stem Cell.2011;9(4):345-356.
21. Kirstetter P, Anderson K, Porse BT, Jacobsen SE,Nerlov C. Activation of the canonical Wnt pathwayleads to loss of hematopoietic stem cellrepopulation and multilineage differentiationblock. Nat Immunol. 2006;7(10):1048-1056.
22. Lee PP, Fitzpatrick DR, Beard C, et al. A criticalrole for Dnmt1 and DNA methylation in T celldevelopment, function, and survival. Immunity.2001;15(5):763-774.
23. Wielenga VJ, Smits R, Korinek V, et al.Expression of CD44 in Apc and Tcf mutant miceimplies regulation by the WNT pathway. Am JPathol. 1999;154(2):515-523.
24. Dumortier A, Jeannet R, Kirstetter P, et al. Notchactivation is an early and critical event duringT-Cell leukemogenesis in Ikaros-deficient mice.Mol Cell Biol. 2006;26(1):209-220.
25. dos Santos NR, Rickman DS, de Reynies A, et al.Pre-TCR expression cooperates with TEL-JAK2to transform immature thymocytes and induceT-cell leukemia. Blood. 2007;109(9):3972-3981.
26. Maillard I, Koch U, Dumortier A, et al. Canonicalnotch signaling is dispensable for the maintenanceof adult hematopoietic stem cells. Cell Stem Cell.2008;2(4):356-366.
27. Tago K, Nakamura T, Nishita M, et al. Inhibitionof Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Genes Dev. 2000;14(14):1741-1749.
28. Marhaba R, Klingbeil P, Nuebel T, Nazarenko I,Buechler MW, Zoeller M. CD44 and EpCAM:cancer-initiating cell markers. Curr Mol Med.2008;8(8):784-804.
29. Zeilstra J, Joosten SP, Dokter M, Verwiel E,Spaargaren M, Pals ST. Deletion of the WNTtarget and cancer stem cell marker CD44 in Apc(Min/1) mice attenuates intestinal tumorigenesis.Cancer Res. 2008;68(10):3655-3661.
30. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE.Targeting of CD44 eradicates human acutemyeloid leukemic stem cells. Nat Med. 2006;12(10):1167-1174.
31. Siapati EK, Papadaki M, Kozaou Z, et al.Proliferation and bone marrow engraftment ofAML blasts is dependent on b-catenin signalling.Br J Haematol. 2011;152(2):164-174.
32. Protin U, Schweighoffer T, Jochum W, Hilberg F.CD44-deficient mice develop normally withchanges in subpopulations and recirculationof lymphocyte subsets. J Immunol. 1999;163(9):4917-4923.
33. Rack KA, Delabesse E, Radford-Weiss I, et al.Simultaneous detection of MYC, BVR1, and PVT1translocations in lymphoid malignancies byfluorescence in situ hybridization. GenesChromosomes Cancer. 1998;23(3):220-226.
34. He TC, Sparks AB, Rago C, et al. Identification ofc-MYC as a target of the APC pathway. Science.1998;281(5382):1509-1512.
35. Suzuki A, Yamaguchi MT, Ohteki T, et al. T cell-specific loss of Pten leads to defects in centraland peripheral tolerance. Immunity. 2001;14(5):523-534.
36. Guo Z, Dose M, Kovalovsky D, et al. Beta-cateninstabilization stalls the transition from double-positive to single-positive stage and predisposesthymocytes to malignant transformation. Blood.2007;109(12):5463-5472.
37. Guo W, Lasky JL, Chang CJ, et al. Multi-geneticevents collaboratively contribute to Pten-nullleukaemia stem-cell formation. Nature. 2008;453(7194):529-533.
38. Guo W, Schubbert S, Chen JY, et al. Suppressionof leukemia development caused by PTEN loss.Proc Natl Acad Sci USA. 2011;108(4):1409-1414.
39. Yu S, Zhou X, Steinke FC, et al. The TCF-1 andLEF-1 transcription factors have cooperative andopposing roles in T cell development andmalignancy. Immunity. 2012;37(5):813-826.
40. Bonnet M, Loosveld M, Montpellier B, et al.Posttranscriptional deregulation of MYC via PTENconstitutes a major alternative pathway of MYCactivation in T-cell acute lymphoblastic leukemia.Blood. 2011;117(24):6650-6659.
41. Bafico A, Liu G, Goldin L, Harris V, Aaronson SA.An autocrine mechanism for constitutive Wntpathway activation in human cancer cells. CancerCell. 2004;6(5):497-506.
42. Weerkamp F, van Dongen JJ, Staal FJ. Notch andWnt signaling in T-lymphocyte development andacute lymphoblastic leukemia. Leukemia. 2006;20(7):1197-1205.
43. Van Vlierberghe P, Ferrando A. The molecularbasis of T cell acute lymphoblastic leukemia.J Clin Invest. 2012;122(10):3398-3406.
44. Zhang J, Ding L, Holmfeldt L, et al. The geneticbasis of early T-cell precursor acute lymphoblasticleukaemia. Nature. 2012;481(7380):157-163.
45. Soulier J, Clappier E, Cayuela JM, et al. HOXAgenes are included in genetic and biologicnetworks defining human acute T-cell leukemia(T-ALL). Blood. 2005;106(1):274-286.
46. Weng AP, Millholland JM, Yashiro-Ohtani Y, et al.c-Myc is an important direct target of Notch1 inT-cell acute lymphoblastic leukemia/lymphoma.Genes Dev. 2006;20(15):2096-2109.
47. Sharma VM, Draheim KM, Kelliher MA. TheNotch1/c-Myc pathway in T cell leukemia. CellCycle. 2007;6(8):927-930.
48. Li X, Gounari F, Protopopov A, Khazaie K, vonBoehmer H. Oncogenesis of T-ALL andnonmalignant consequences of overexpressingintracellular NOTCH1. J Exp Med. 2008;205(12):2851-2861.
49. Palomero T, Ferrando A. Therapeutic targeting ofNOTCH1 signaling in T-cell acute lymphoblasticleukemia. Clin Lymphoma Myeloma. 2009;9(Suppl 3):S205-S210.
50. Hann SR. Role of post-translational modificationsin regulating c-Myc proteolysis, transcriptionalactivity and biological function. Semin CancerBiol. 2006;16(4):288-302.
51. Hasserjian RP, Aster JC, Davi F, Weinberg DS,Sklar J. Modulated expression of notch1 duringthymocyte development. Blood. 1996;88(3):970-976.
52. Axelrod JD, Matsuno K, Artavanis-Tsakonas S,Perrimon N. Interaction between Winglessand Notch signaling pathways mediated bydishevelled. Science. 1996;271(5257):1826-1832.
53. Kim HA, Koo BK, Cho JH, et al. Notch1counteracts WNT/b-catenin signaling throughchromatin modification in colorectal cancer. J ClinInvest. 2012;122(9):3248-3259.
54. Chao DT, Korsmeyer SJ. BCL-XL-regulatedapoptosis in T cell development. Int Immunol.1997;9(9):1375-1384.
704 KAVERI et al BLOOD, 1 AUGUST 2013 x VOLUME 122, NUMBER 5
only.For personal use at HOSPITAL FOR SICK CHILDREN on August 2, 2013. bloodjournal.hematologylibrary.orgFrom




















