
Analysis of gene expression during myc oncogene-induced lymphomagenesis in the bursa of Fabricius

Molecular Cell, Vol. 11, 905–914, April, 2003, Copyright 2003 by Cell Press
Myc-Mediated Proliferation and Lymphomagenesis,but Not Apoptosis, Are Compromised by E2f1 Loss
2002), none have yet been shown to induce p53 directly.One of their common targets is the Arf tumor suppres-sor, whose product (p19Arf in the mouse and p14ARF in
Troy A. Baudino,1 Kirsteen H. Maclean,1
Jennifer Brennan,1 Evan Parganas,1,3 Chunying Yang,1
Aaron Aslanian,4 Jacqueline A. Lees,4
humans) inhibits Mdm2, thereby stabilizing p53 (Sherr,Charles J. Sherr,2,3,* Martine F. Roussel,2,5
2001). This led to the parsimonious idea that Myc (Zindyand John L. Cleveland1,5,*et al., 1998) and E2f1 (Bates et al., 1998) might induce1Department of Biochemistryp53 through the agency of Arf and to the ancillary hy-2 Genetics & Tumor Cell Biologypothesis that Myc may work upstream of E2f1 in so3 Howard Hughes Medical Institutedoing (Leone et al., 2001).St. Jude Children’s Research Hospital
It remains unclear whether Myc directly binds to theMemphis, Tennessee 38105minimal Arf promoter to induce its expression. Yet, in4 Department of Biologyprimary mouse embryo fibroblasts (MEFs), myeloid pro-Center for Cancer Researchgenitors, and in B lymphoid cells in culture or in vivo,Massachusetts Institute of TechnologyMyc is a potent Arf inducer, and its ability to triggerCambridge, Massachusetts 02139p53-dependent apoptosis is compromised in cells that5 Department of Molecular Scienceshave sustained Arf mutations (Zindy et al., 1998; EischenUniversity of Tennesseeet al., 1999; Jacobs et al., 1999; Schmitt et al., 1999). InMemphis, Tennessee 38163E�-Myc transgenic mice, where Myc overexpression istargeted to B lymphocytes by the immunoglobulin heavychain enhancer-promoter (E�), Burkitt-type lymphomasSummaryappear with a mean latency of about 6 months (Adamset al., 1985). In the early stages of disease, Myc drivesMyc and E2f1 promote cell cycle progression, butB cell proliferation, which is counterbalanced by apopto-overexpression of either can trigger p53-dependentsis. However, loss of Arf or p53 function inhibits theapoptosis. Mice expressing an E�-Myc transgene inapoptotic program, canceling this failsafe mechanismB lymphocytes develop lymphomas, the majority ofand enabling Myc to act in an unopposed manner towhich sustain mutations of either the Arf or p53 tumorpromote cell proliferation. Tumors arising in E�-Mycsuppressors. E�-Myc transgenic mice lacking one ortransgenic mice exhibit either Arf or p53 inactivatingboth E2f1 alleles exhibited a slower onset of lymphomamutations, overexpress Mdm2, or may sustain muta-development associated with increased expression oftions affecting other regulators that effectively diminishthe cyclin-dependent kinase inhibitor p27Kip1 and a re-the function of the Arf–Mdm2–p53 pathway (Eischen etduced S phase fraction in precancerous B cells. Inal., 1999, 2001; Jacobs et al., 1999; Schmitt et al., 1999,contrast, Myc-induced apoptosis and the frequency2002; Inoue et al., 2001). As expected, crossing E�-Mycof Arf and p53 mutations in lymphomas were unaf-transgenic mice onto an Arf null background dramati-fected by E2f1 loss. Therefore, Myc does not requirecally enhances lymphoma onset and aggressiveness,E2f1 to induce Arf, p53, or apoptosis in B cells, butresulting in the death of all animals by less than 2 monthsdepends upon E2f1 to accelerate cell cycle progres-of age.sion and downregulate p27Kip1.
Like Myc, E2f1 overexpression can activate Arf (De-Gregori et al., 1997; Bates et al., 1998) and can triggerIntroductionp53-dependent apoptosis (Qin et al., 1994; Wu and Lev-ine, 1994; Shan and Lee, 1994). However, in several
Early observations that transcription factors such asanimal model systems, Arf does not appear to mediate
Myc and E2f1 can induce both cell proliferation and cell the activation of p53 by E2f1 (Russell et al., 2002; Tolbertdeath in primary diploid cells raised an obvious paradox et al., 2002; Tsai et al., 2002a, 2002b). Although com-as to how such apparently conflicting outcomes could plexes of retinoblastoma (Rb) family proteins and thebe elicited by these proteins (Askew et al., 1991; Evan E2fs to which they bind repress the Arf promoter (Row-et al., 1992; Wu and Levine, 1994; Qin et al., 1994; Shan land et al., 2002), Arf seems not to be periodically ex-and Lee, 1994). However, it is now generally thought pressed during the cell cycle like many E2f-responsivethat physiological levels of Myc and E2f signaling are genes. This implies that Arf is insulated from transientnot only well tolerated but are required to properly pro- signals associated with cell cycle-dependent E2f regula-mote cell cycle progression, whereas a surfeit of such tion and might only respond to persistently elevated E2fsignals is countermanded by a p53-dependent apo- activity, such as may be encountered in tumor cellsptotic program that effectively suppresses tumor forma- lacking Rb or p16Ink4a, or in those overexpressing cyclintion (Evan and Vousden, 2001). Although both Myc and D-dependent kinases.different E2fs can enter into higher order chromatin- Studies of an E2f-Arf connection are further compli-associated complexes that activate or repress many cated by the fact that the E2f family includes six mem-cellular genes (Eisenman, 2001; Trimarchi and Lees, bers that interact with two distinct DP proteins to form
heterodimers (Nevins, 2001; Trimarchi and Lees, 2002).E2f1-, E2f2-, and E2f3-containing dimers are specifically*Correspondence: [email protected] (J.L.C.), sherr@stjude.
org (C.J.S.) regulated by Rb, whereas E2f4- and E2f5-DP complexes
Molecular Cell906
can interact with Rb family members p107 and p130.E2f6 lacks the domain required for interaction with Rbfamily members, but can enter into repressor complexeswith other proteins (Trimarchi et al., 2001; Ogawa et al.,2002). E2f1 can act as an oncogene or tumor suppressor,depending on the biologic setting (Yamasaki et al., 1996;Field et al., 1996). Loss of E2f1 in Rb-deficient mouseembryos (Tsai et al., 1998) or in certain tumor types(Pan et al., 1998) inhibits both proliferation and p53-dependent apoptosis. Some studies suggest that E2f1activity selectively triggers apoptosis, whereas E2f2 andE2f3 govern cell cycle progression (DeGregori et al.,1997; Lissy et al., 2000; Leone et al., 2001). In contrast,other results suggest that the Rb-regulated E2fs haveoverlapping functions and that their thresholds dictatewhether cells proliferate or die (Humbert et al., 2000;Ziebold et al., 2001; Wu et al., 2001; Saavedra et al.,2002).
Given these ambiguities, we were motivated to testwhether the proapoptotic and/or cell cycle-promotingeffects of Myc overexpression were mediated by E2f1.Using the E�-Myc lymphoma model, our studies revealthat Myc-induced cell cycle progression, but not apo-
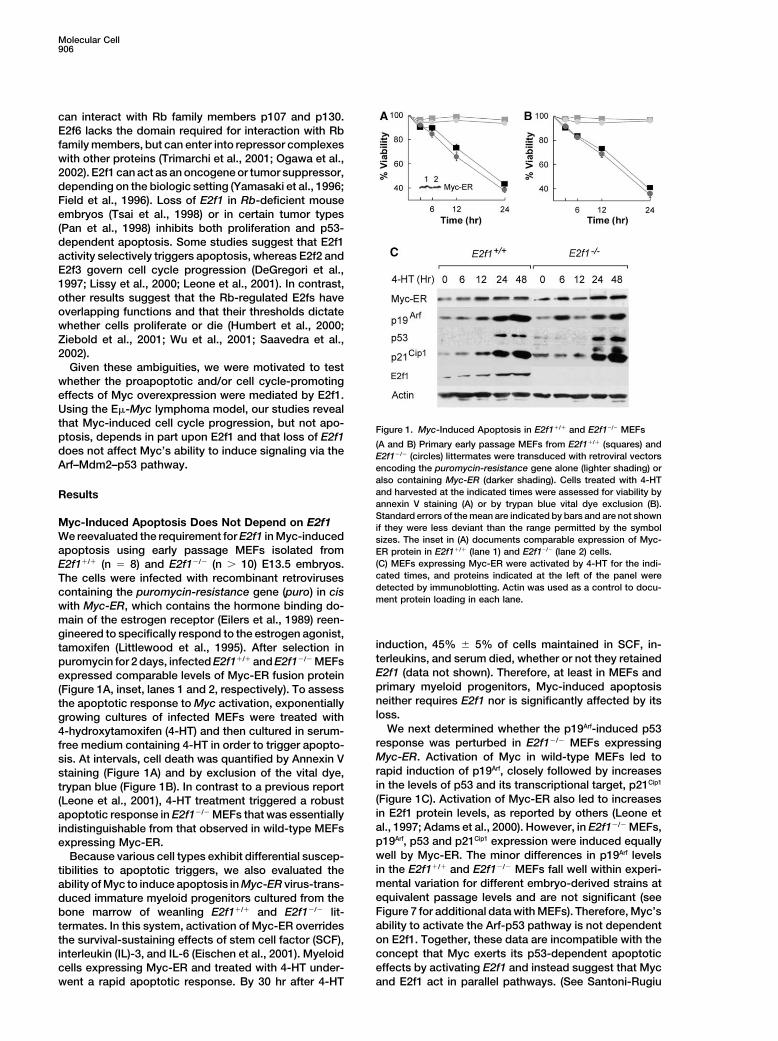
Figure 1. Myc-Induced Apoptosis in E2f1�/� and E2f1�/� MEFsptosis, depends in part upon E2f1 and that loss of E2f1
(A and B) Primary early passage MEFs from E2f1�/� (squares) anddoes not affect Myc’s ability to induce signaling via the E2f1�/� (circles) littermates were transduced with retroviral vectorsArf–Mdm2–p53 pathway. encoding the puromycin-resistance gene alone (lighter shading) or
also containing Myc-ER (darker shading). Cells treated with 4-HTand harvested at the indicated times were assessed for viability byResultsannexin V staining (A) or by trypan blue vital dye exclusion (B).Standard errors of the mean are indicated by bars and are not shown
Myc-Induced Apoptosis Does Not Depend on E2f1 if they were less deviant than the range permitted by the symbolWe reevaluated the requirement for E2f1 in Myc-induced sizes. The inset in (A) documents comparable expression of Myc-apoptosis using early passage MEFs isolated from ER protein in E2f1�/� (lane 1) and E2f1�/� (lane 2) cells.
(C) MEFs expressing Myc-ER were activated by 4-HT for the indi-E2f1�/� (n � 8) and E2f1�/� (n � 10) E13.5 embryos.cated times, and proteins indicated at the left of the panel wereThe cells were infected with recombinant retrovirusesdetected by immunoblotting. Actin was used as a control to docu-containing the puromycin-resistance gene (puro) in cisment protein loading in each lane.
with Myc-ER, which contains the hormone binding do-main of the estrogen receptor (Eilers et al., 1989) reen-gineered to specifically respond to the estrogen agonist,
induction, 45% � 5% of cells maintained in SCF, in-tamoxifen (Littlewood et al., 1995). After selection interleukins, and serum died, whether or not they retainedpuromycin for 2 days, infected E2f1�/� and E2f1�/� MEFsE2f1 (data not shown). Therefore, at least in MEFs andexpressed comparable levels of Myc-ER fusion proteinprimary myeloid progenitors, Myc-induced apoptosis(Figure 1A, inset, lanes 1 and 2, respectively). To assessneither requires E2f1 nor is significantly affected by itsthe apoptotic response to Myc activation, exponentiallyloss.growing cultures of infected MEFs were treated with
We next determined whether the p19Arf-induced p534-hydroxytamoxifen (4-HT) and then cultured in serum-response was perturbed in E2f1�/� MEFs expressingfree medium containing 4-HT in order to trigger apopto-Myc-ER. Activation of Myc in wild-type MEFs led tosis. At intervals, cell death was quantified by Annexin Vrapid induction of p19Arf, closely followed by increasesstaining (Figure 1A) and by exclusion of the vital dye,in the levels of p53 and its transcriptional target, p21Cip1trypan blue (Figure 1B). In contrast to a previous report(Figure 1C). Activation of Myc-ER also led to increases(Leone et al., 2001), 4-HT treatment triggered a robustin E2f1 protein levels, as reported by others (Leone etapoptotic response in E2f1�/� MEFs that was essentiallyal., 1997; Adams et al., 2000). However, in E2f1�/� MEFs,indistinguishable from that observed in wild-type MEFsp19Arf, p53 and p21Cip1 expression were induced equallyexpressing Myc-ER.well by Myc-ER. The minor differences in p19Arf levelsBecause various cell types exhibit differential suscep-in the E2f1�/� and E2f1�/� MEFs fall well within experi-tibilities to apoptotic triggers, we also evaluated themental variation for different embryo-derived strains atability of Myc to induce apoptosis in Myc-ER virus-trans-equivalent passage levels and are not significant (seeduced immature myeloid progenitors cultured from theFigure 7 for additional data with MEFs). Therefore, Myc’sbone marrow of weanling E2f1�/� and E2f1�/� lit-ability to activate the Arf-p53 pathway is not dependenttermates. In this system, activation of Myc-ER overrideson E2f1. Together, these data are incompatible with thethe survival-sustaining effects of stem cell factor (SCF),concept that Myc exerts its p53-dependent apoptoticinterleukin (IL)-3, and IL-6 (Eischen et al., 2001). Myeloideffects by activating E2f1 and instead suggest that Myccells expressing Myc-ER and treated with 4-HT under-
went a rapid apoptotic response. By 30 hr after 4-HT and E2f1 act in parallel pathways. (See Santoni-Rugiu

Myc-Induced Proliferation Depends on E2f1907
Figure 2. Survival of E�-Myc Transgenic Mice of Different E2f1 Ge-notypes
Animals were monitored for 1 year for tumor development. Tics Figure 3. Expression of p53, p19Arf, and E2f1 Proteins in Lympho-above the curves designate surviving animals of the indicated geno- mas Arising in E�-Myc Transgenic Mice of Different E2f1 Genotypestypes that are less than 1 year old or that remained alive after 1 year Proteins, indicated at the left of the three panels, were detected byof observation. Given the number of animals studied, the apparent immunoblotting. Each lane corresponds to a different tumor, withdifferences in rates of tumor incidence between the E2f1�/� and E2f1 genotypes noted at the right. Mutant forms of p53 do not induceE2f1�/� cohorts are not significant, whereas the differences between Mdm2 to catalyze their own destruction and therefore accumulate tothese cohorts and E2f1�/� mice were (p � 0.001). inappropriately high levels. Due to disruption of the p53-Arf feed-
back loop, p19Arf levels are also elevated in tumors that have sus-tained p53 mutations. Hence, increased p19Arf and p53 protein levels
et al. [2000] for similar conclusions drawn from studies generally go hand in hand. However, tumors overexpressing Mdm2also exhibit high levels of p19Arf protein without associated p53with human cells.)expression (e.g., tumors 1 and 4 in the E2f1�/� series).
E2f1 Loss Impairs Myc-Induced LymphomagenesisTo address the requirements for E2f1 in Myc-inducedtumorigenesis, we utilized E�-Myc transgenic mice, expression in all Myc-induced tumors arising on the
heterozygous E2f1�/� background (Figure 3, middlewhich develop a protracted course of lethal B cell lym-phoma akin to that observed in Burkitt’s lymphoma pa- panel). Comparison of tumors from E�-Myc transgenic
E2f1�/� and E2f1�/� mice suggested that, overall, theretients where MYC expression is driven by translocationsinvolving immunoglobulin loci. Both Arf and p53 impair were lower E2f1 protein levels expressed in the hetero-
zygotes (Figure 3, middle versus top panels), althoughMyc-induced lymphomagenesis in this model system(Eischen et al., 1999; Jacobs et al., 1999; Schmitt et al., it is difficult to make a strong case, given the range of
variation in protein expression seen among tumors of1999). If E2f1 were required for Myc to trigger the Arf-p53 apoptotic checkpoint in vivo, then germline deletion any one genotype. At face value, however, the delayed
onset of lymphomas in heterozygotes appears to be dueof E2f1, like that of Arf or p53, should negate apoptosisand accelerate lymphomagenesis in E�-Myc transgenic to E2f1 haploinsufficiency.
Slower tumor development on E2f1�/� and E2f1�/� back-mice. To address this issue, E�-Myc transgenic micewere mated to E2f1�/� animals, and F1 offspring were grounds would not be expected if E2f1 mediated the
Myc-induced Arf response. Therefore, we evaluated theinterbred to obtain E2f1�/�, E2f1�/�, and E2f1�/� E�-Myc transgenic mice. Littermates were monitored for functional status of Arf and p53 in these lymphomas.
The hallmark of p53 mutation is expression of high levelsdevelopment of lymphoma, and tumors were taken forfurther analysis. of dominant-negative p53 protein, which forms tran-
scriptionally inactive tetramers that are unable to acti-On this genetic background, all E2f1�/� E�-Myc trans-genic mice rapidly developed B cell lymphomas and vate Mdm2 and catalyze p53 destruction (Kubbutat et
al., 1997; Haupt et al., 1997). Accumulation of mutantdied within a half year of birth (Figure 2). Rather thanaccelerate disease, loss of E2f1 protected E�-Myc p53 proteins was observed in a significant fraction of
lymphomas arising in animals of all three genotypestransgenic mice from developing lymphomas (p �0.001). E2f1 null animals exhibited a mean latency for (Figure 3). Levels of p19Arf were uniformly elevated in
tumors that accumulated mutant p53, due to loss of Arftumor onset of 24 weeks, and some survived for morethan a year. Surprisingly, E2f1�/� E�-Myc mice also had feedback repression by p53 (Stott et al., 1998; Kamijo
et al., 1998; Robertson and Jones, 1998). As seen ina markedly retarded course of disease that was notsignificantly different from that of E2f1�/� animals (p � past studies (Eischen et al., 1999), p19Arf expression was
increased in some lymphomas that lacked p53 muta-0.26) (Figure 2). Thus, E2f1 did not mimic the roles ofArf and p53 as tumor suppressors. tions (see, for example, E2f1�/� tumors, Figure 3, lanes
1 and 4, and E2f1�/� tumors, Figure 3, lanes 10 and 13);The latter results suggested that E�-Myc does notselect for loss of E2f1 and, in turn, that deletion of only these expressed high levels of Mdm2 (data not shown).
Additionally, Southern blotting analyses revealed thatone functional E2f1 allele was sufficient to protect micefrom disease. In fact, we detected residual E2f1 protein 25%–30% of tumors of all three genotypes displayed

Molecular Cell908
biallelic Arf deletions (data not shown). Other indepen-dently examined cohorts of E�-Myc transgenic micesustained p53 mutations or Arf deletions at overall fre-quencies similar to those seen here (Eischen et al., 1999;Inoue et al., 2001). Therefore, E2f1 loss had no detect-able effect on Myc’s propensity to disrupt the Arf–Mdm2–p53 signaling axis during the course of lympho-magenesis.
Loss of E2f1 Impairs Proliferation of E�-MycTransgenic B CellsPrecancerous transgenic B cells of E�-Myc mice displayincreased rates of proliferation offset by apoptosis(Eischen et al., 1999; Jacobs et al., 1999; Schmitt etal., 1999), which keeps disease in check until Arf-p53signaling is disrupted. We initially assessed the effectsof E2f1 loss on Myc-induced cell death by culturingpre-B cells from the bone marrow of precancerous,weanling E2f1�/�, E2f1�/�, and E2f1�/� E�-Myc trans-genic littermates. The apoptotic indices of E�-Myctransgenic bone marrow-derived B cells cultured onstromal feeder layers and IL-7 were increased relativeto B cells from nontransgenic mice, regardless of E2f1genotype (Figure 4A). Although the mean apoptotic in-dex for B cells derived from E2f1�/� animals was some-what lower than that of E2f1�/� counterparts, these dif-ferences were not statistically significant.
We next performed in situ DNA end-labeling assaysto detect apoptotic cells in lymph nodes from E�-Myctransgenic mice of different E2f1 genotypes early in thecourse of disease and from age-matched nontransgenicmice (Figure 4B). Lymph node sections from youngE2f1�/� E�-Myc transgenic mice displayed significantlyhigher levels of apoptosis than the spontaneous indexobserved in nontransgenic B cells, and E2f1 loss led tono diminution (see Figure 4B legend for quantitation of
Figure 4. Apoptosis of B Lymphocytes from E�-Myc Transgenicapoptotic cells). Note, however, that continuous MycMice of Different E2f1 Genotypes
expression eventually led to mutations in Arf or p53(A) Bone marrow from nontransgenic (black bar) and E�-Myc trans-(Figure 3), which virtually eliminate apoptosis in lympho-genic mice (gray bars) of the indicated E2f1 genotypes was cultured
matous tissues later in the course of disease. in an ex vivo system employing stromal feeder layers and IL-7 thatProgrammed expression of c-Myc in B cells of E�- supports the growth of pre-B cells at the expense of myeloid progen-
itors. The apoptotic index of B220� B cells was estimated by FACSMyc transgenic mice also accelerates their rate of prolif-of cells stained with propidium iodide and by calculating the per-eration, which is further magnified in the absence of Arf-centage of cells with fragmented DNA (sub-2N DNA content).p53 signaling (Eischen et al., 1999). When we compared(B) Apoptosis in lymph nodes was determined using an in situ DNAthe proliferation of pre-B cells cultured ex vivo from theend-labeling procedure specific for apoptotic cells. Representative
bone marrow of precancerous E�-Myc E2f1�/�, E2f1�/�, stained sections are shown for lymph nodes derived from nontrans-and E2f1�/� mice, loss of one or both E2f1 alleles was genic (wild-type) mice and from E�-Myc transgenic mice of the
indicated E2f1 genotypes. The darkly stained cells are apoptotic.accompanied by significant reductions in the S phaseThe numbers of apoptotic cells per mm2 were 4.3 � 1.8 (nontrans-fractions of E�-Myc transgenic B cells (Figure 5A). Togenic control); 19.3 � 2 (E2f1�/�, E�-Myc); 19.7 � 3 (E2f1�/�, E�-document similar effects in vivo, 8-week-old precancer-Myc); and 22.3 � 4 (E2f1�/�, E�-Myc).ous mice were injected intraperitoneally with BrdU, and
bone marrow, spleen, and lymph node cells were har-vested 12 hr later for analysis (Figures 5B–5D, respec-
cells, as E2f1�/� and E2f1�/� B cells from nontransgenictively). E�-Myc transgenic B cells displayed a markedlystrains grew at comparable rates (Figures 5B–5D, darkhigher proliferative index in all of these hematopoieticfilled bars). Therefore, whereas normal B cells compen-compartments when compared to B cells from age-sate for its loss, a critical threshold of E2f1 is requiredmatched nontransgenic mice (Figures 5B–5D, �/� ge-for Myc to provoke the accelerated growth of B cells.notype, light versus dark filled bars). This was particu-
larly evident in bone marrow, where 44% of B cells fromE�-Myc mice were labeled with BrdU (Figure 5B). Again, Loss of E2f1 Blocks c-Myc’s Ability
to Downregulate p27Kip1loss of either one or two E2f1 alleles dramatically re-duced the proliferative index of B220� B cells in E�- Myc accelerates cell cycle entry and proliferation, at
least in part through its ability to promote the degrada-Myc transgenic animals. The effects of E2f1 loss on cellcycle progression were only manifested in transgenic B tion of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1

Myc-Induced Proliferation Depends on E2f1909
Figure 5. S Phase Fraction in B Cells from E�-Myc Transgenic Mice of Different E2f1 Genotypes
(A) Pre-B cells cultured from the bone marrow of E�-Myc transgenic mice were pulsed for 1 hr with BrdU, and cells that had incorporatedBrdU into replicating DNA were enumerated by use of an antibody-dependent fluorescence assay.(B–D) Two mice of each genotype received intraperitoneal injections of BrdU, and cells from bone marrow (B), spleens (C), and lymph nodes(D) of E�-Myc transgenic mice (gray bars) or nontransgenic littermates (black bars) were harvested 12 hr later and assayed for BrdU incorporationinto DNA as in (A). The bars indicate averages from two independent experiments. E2f1 genotypes are indicated below the panels.
(Vlach et al., 1996; Bouchard et al., 1999; Muller et al., tured B cells of E2f1�/� and E2f1�/� E�-Myc transgenicmice (Figure 6B). The effects of E2f1 loss on expression1997; Perez-Roger et al., 1999; O’Hagan et al., 2000).
Loss of Kip1 accelerates Myc-induced lymphomagene- of p27Kip1 were only evident in the context of Myc overex-pression, as levels of p27Kip1 were comparable in B cellssis, without overtly affecting the Myc apoptotic program
(Martins and Berns, 2002). We therefore assessed from nontransgenic E2f1�/�, E2f1�/�, and E2f1�/� mice(data not shown).whether increased levels of p27Kip1 accompanied the
reduced proliferation of E2f1�/� and E2f1�/� E�-Myc To determine whether the accumulation of p27Kip1 inE2f1-deficient cells reflected a defect in its Myc-depen-transgenic B cells. Indeed, levels of p27Kip1 were mark-
edly elevated in E2f1-deficient, E�-Myc transgenic B dent degradation, we compared p27Kip1 levels in E2f1�/�
and E2f1�/� MEFs expressing Myc-ER after treatmentcells propagated ex vivo relative to those in B cellsexplanted from E2f1�/� E�-Myc transgenic animals (Fig- with 4-HT. As expected, there was a marked decrease
in p27Kip1 levels in E2f1�/� MEFs following Myc activation;ure 6A). We next sorted B220� B cells from spleensand lymph nodes of precancerous E�-Myc transgenic by contrast, this response was disabled in their E2f1�/�
counterparts (Figure 6C), despite the fact that signalingmice and analyzed p27Kip1 levels by immunoblotting.Again, p27Kip1 accumulated to higher levels in the uncul- to the Arf-p53 pathway was intact (Figure 1 and Figure
7 below).We next transduced E2f1�/� and E2f1�/� MEFs with
viruses encoding either Myc-ER, E2f1-ER, or both (Fig-ure 7). The retroviral vector expressing E2f1-ER con-tained puro, whereas that encoding Myc-ER expressedgreen fluorescent protein (GFP). Doubly infected cellswere selected for 2 days in medium containing puromy-cin and tested for GFP expression (�95% positive). Be-cause both Myc and E2f1 are highly proapoptotic, cellsinduced with 4-HT were maintained in 10% fetal calfserum to support their viability. As expected, p19Arf (Fig-ure 7) and p53 (data not shown) were induced uponactivation of either Myc-ER or E2f1-ER by 4-HT in bothE2f1�/� and E2f1�/� MEF strains, confirming that bothexpression vectors were functional. In cells containingE2f1, p27Kip1 turnover was accelerated upon Myc-ER but
Figure 6. Myc and E2f1 Jointly Regulate p27Kip1 Levels not E2f1-ER activation (Figure 7A). In contrast, Myc-ER(A) The relative levels of p27Kip1 protein in cultured precancerous had no such effect in MEFs lacking E2f1 (Figure 7B).E�-Myc transgenic pre-B cells of the indicated E2f1 genotypes are Importantly, reexpression of E2f1 in E2f1�/� MEFs re-illustrated.
stored Myc’s ability to downregulate p27Kip1 (Figure 7B,(B) B220� B cells harvested from the spleens and lymph nodeslanes labeled “Both”). Therefore, E2f1 is required forof E�-Myc transgenic mice of the indicated E2f1 genotypes were
analyzed directly without intervening cell culture for expression of Myc to downregulate p27Kip1 and to promote cell cyclep27Kip1 protein. progression.(C) E2f1�/� and E2f1�/� MEFs (genotypes indicated at the top) trans-duced with Myc-ER were treated with 4-HT for the indicated times(hr). Cells were lysed, and p27Kip1 levels were determined by immu- Discussionnoblotting. Note that the higher levels of p27Kip1 observed in E2f1 nullMEFs were not degraded in response to Myc activation. Activation of
Myc-Induced Apoptosis Does Not Require E2f1Arf was confirmed in 4-HT treated cells of both genotypes (data notMyc triggers an apoptotic program that must be by-shown, but see Figure 7 for representative data). Actin was used
as a control for protein loading in each experiment. passed for it to exhibit its full oncogenic potential (Evan

Molecular Cell910
ori et al., 1997; Bates et al., 1998) in which E2f1-inducedapoptosis is p53 dependent (Qin et al., 1994; Wu andLevine, 1994; Shan and Lee, 1994; Lomazzi et al., 2002).However, several studies have challenged the idea thatE2f1 requires p19Arf to activate the p53-dependent apo-ptotic program. For example, the rate of onset of epithe-lial tumors of the choroid plexus induced by a truncatedT antigen incapable of interacting with p53 dependson deregulated E2f1 activity (Pan et al., 1998). Yet, theappearance of such tumors is accelerated on a p53 null,but not an Arf null, background (Tolbert et al., 2002). Ina transgenic setting in which either Myc or E2f1 expres-sion is driven by a keratin-5 promoter, Arf loss dimin-ished apoptosis in Myc-induced skin tumors but in-creased apoptosis in E2f1-induced tumors (Russell etal., 2002). In each setting, E2f1 induced p53, whetheror not Arf was disabled.
Our studies indicate that E2f1 loss does not diminishMyc-induced apoptosis in settings in which this Mycfunction is clearly Arf-dependent. Conditional activationof Myc-ER-induced apoptosis in cultured primary MEFswas unaffected by loss of E2f1. Similar results wereobtained using primary E2f1 null myeloid cells trans-duced with Myc-ER, as well as with cultured E�-MycFigure 7. E2f1 Is Required for Myc-Induced Downregulation oftransgenic B cells harvested from weanling, precancer-p27Kip1
ous E2f1 null animals. We cannot preclude the possibilityMEFs from E2f1�/� (A) and E2f1�/� (B) mice were transduced withthat E2f1 might contribute to Myc-induced apoptosis inMyc-ER or E2f1-ER retroviral vectors containing either GFP or theother cell types. Nonetheless, these results contradictpuro gene, respectively. Cells infected with Myc-ER or both con-
structs together (Both) were selected for 48 hr in puromycin; cells those of a previous study, in which the ability of Myc totransduced with E2f1-ER or both vectors were stained for GFP to induce apoptosis was markedly and specifically re-confirm protein expression (�95% GFP positive). After selection, duced in E2f1 null primary MEFs, and where its abilitycells were treated with 4-HT in complete serum-containing medium
to trigger S phase entry appeared to depend insteadfor the indicated times (hr), and proteins in cell lysates, indicatedupon the activities of E2f2 and E2f3 (Leone et al., 2001).at the left of the panel, were detected by immunoblotting usingWe cannot easily reconcile these differences. Bothactin as a control for protein loading. E2f1-ER did not trigger p27Kip1
downregulation, whereas Myc-ER stimulated this process (A). Acti- groups used 129 X C57BL/6 E2f1 null mice backcrossedvation of Myc-ER in E2f1 null cells did not lead to p27Kip1 downregula- onto a C57BL/6 genetic background. Whereas Leonetion, but this was restored in the presence of both activated Myc- and coworkers used high multiplicity adenovirus infec-ER and E2f1-ER (B). tion or transient transfection to overexpress Myc, we
employed retroviral transduction of a conditional Myc-ER allele activated by tamoxifen. In both cases, apopto-
and Vousden, 2001). Overexpression of Myc kills primary sis was triggered by serum deprivation. Although notMEFs, myeloid progenitors, and B lymphocytes in cul- observed at the protein level (Leone et al., 2001), it isture, a process exacerbated by removal of survival fac- conceivable that other E2fs or Rb family members,tors from the culture medium. In these cell types, Myc whose extent of recruitment could differ in indepen-induces the expression of p19Arf, which, by inactivating dently derived embryos, might developmentally com-Mdm2, helps to enhance p53-dependent apoptosis pensate for disruption of E2f1 in the germline.(Zindy et al., 1998). In turn, cells cultured from Arf null Whatever the explanation, our studies in animals sup-or p53 null mice are resistant to Myc’s apoptotic effects port the results that we obtained with cultured cells.and exhibit a clear survival advantage when compared Lymphomagenesis induced by an E�-Myc transgeneto cells that maintain a functional Arf–Mdm2–p53 signal- was inhibited on an E2f1 null background, although apo-ing pathway. B cells harvested directly from lymphoid ptosis in precancerous B cells from lymph nodes,organs of E�-Myc transgenic mice exhibit an increased spleen, and bone marrow was seemingly unaffected.S phase fraction, but like cultured primary cells, their The fact that E�-Myc-induced lymphomas arising inproliferative expansion is initially counterbalanced by E2f1 null mice sustained the same frequencies of ArfMyc-induced apoptosis (Eischen et al., 1999; Jacobs et loss and p53 mutations as those observed in E2f1�/�
al., 1999; Schmitt et al., 1999, 2002). Selection for loss animals provides the single most compelling argumentof Arf or p53 in vivo similarly compromises Myc’s ability that Myc-induced signaling through the Arf-p53 axis wasto kill B cells, converting Myc into a pure promoter of not influenced by E2f1 loss in premaligant B cells. Inproliferation and thereby accelerating lymphomagenesis. striking contrast to the retardation in lymphoma onset
How Myc triggers Arf expression remains unclear, but following loss of E2f1, disruption of the Arf transcrip-some studies have implicated E2f1 as a downstream tional activator, Dmp1, significantly accelerates E�-mediator of Myc’s effects. Myc can induce E2f1 (Leone Myc-induced lymphomagenesis (Inoue et al., 2001).et al., 1997; Adams et al., 2000), and in turn, E2f1 overex- Loss of even a single allele of Dmp1 removes the Myc-
dependent selective pressure driving Arf or p53 muta-pression activates Arf and p53 in cultured cells (DeGreg-

Myc-Induced Proliferation Depends on E2f1911
tions, the aggregate frequency of which is reduced by vated in E�-Myc transgenic B cells lacking E2f1 versusthose that retained the gene. Myc’s ability to provoke�80% when compared to those in E�-Myc-transgenic
animals, whether E2f1-deficient or not. Lymphoma de- accelerated proliferation has been linked to its capacityto induce the proteasomal degradation of p27Kip1 medi-velopment in E�-Myc transgenic mice is unaffected on
an Rb�/� background (Schmitt et al., 1999) or in a “pure” ated by the SCFSkp2 E3 ubiquitin protein ligase (Bartekand Lukas, 2001; Slingerland and Pagano, 2002). The SInk4a-deficient strain that retains Arf function (Krimpen-
fort et al., 2001), providing further evidence that disrup- phase promoting effects of both Myc and the E2fs canaffect this process in several ways. First, various E2fstion of the Rb pathway and deregulation of E2fs do not
mimic Myc’s effects. Together, these results reinforce directly activate cyclin E (Dyson 1998; Nevins 2001),resulting in an increase in cyclin E-Cdk2 complexes,the view that E2f1 function is dispensable for Myc-
induced apoptosis in B cells and are incompatible with p27Kip1 phosphorylation on Thr187, and its phosphoryla-tion-dependent recognition by the SCF as cells transita model in which Myc’s apoptotic effects are mediated
through E2f1. G1 into S phase (Vlach et al., 1997; Sheaff et al., 1997).Second, Myc induces cyclin D2 and Cdk4 (Bouchard etUsing transgenic mice in which a keratin-5 promoter
drives Myc expression in epithelium, Rounbehler et al. al., 1999; Hermeking et al., 2000), enhancing the forma-tion of a complex that sequesters p27Kip1 and lowering(2002) observed that Myc-induced, p53-dependent apo-
ptosis in papillomas was increased in animals lacking its ability to block Cdk2 activity (Sherr and Roberts,1999). Third, E2fs activate Skp2 synthesis (Marti et al.,E2f1. Moreover, Myc overexpression cooperated with
E2f1 loss to accelerate, rather than retard, tumor devel- 1999), while Myc increases the expression of Cullin-1(O’Hagan et al., 2000), each a component of the SCFopment. Although, as in B cells, E2f1 is not required for
Myc to induce apoptosis in mouse keratinocytes, E2f1 complex. Fourth, in an E2f-independent manner (San-toni-Rugiu et al., 2000), Myc activates Cdc25a, whichplays a tumor suppressive role in these cells. Such re-
sults underscore the complexities of Myc-E2F interac- encodes a phosphatase that promotes Cdk2 activity(Galaktionov et al., 1996; Vigo et al., 1999). Fifth, Myctions in different animal tissues and are consistent with
earlier reports that E2f1 exhibits both oncogenic and may enter into a transcriptional complex that repressesthe Kip1 promoter (Yang et al., 2001). Finally, anothertumor suppressive properties (Yamasaki et al., 1996;
Field et al., 1996). less well-characterized G1 phase-specific pathway cantrigger p27Kip1 degradation even in the absence of itsphosphorylation on Thr187 (Malek et al., 2001). AlthoughE2f1 Is Required for Myc to Provoke ProliferationE2f1 loss inhibited Myc’s ability to trigger degradationand to Downregulate p27Kip1
of p27Kip1, E2f1 overexpression alone failed to downregu-Myc accelerates the proliferation of primary cells ex vivolate the protein, consistent with the idea that Myc andand augments the S phase fraction of B cells in situ inE2f1 collaborate in promoting this process. Importantly,E�-Myc transgenic mice. Cells that overexpress Myca critical threshold of E2f1 seems to be required forand lack Arf or p53 function not only resist apoptosisp27Kip1 downregulation by Myc, as this was also inhibitedbut proliferate much faster than those that retain thesein E2f1�/� cells. However, we do not know whether thegenes (Eischen et al., 1999; Jacobs et al., 1999; Schmittincreased levels of p27Kip1 in E2f1 null cells are the causeet al., 1999). In MEFs, Arf overexpression per se doesor consequence of their extended cell generation time.not trigger apoptosis but instead leads to p53-depen-
Loss of Kip1 accelerates lymphoma development indent cell cycle arrest (Kamijo et al., 1997). This impliesE�-Myc transgenic mice, without affecting Myc-inducedthat Myc provides collateral signals that reprogram suchapoptosis (Martins and Berns, 2002). In contrast, ascells to die (Juin et al., 1999; Eischen et al., 2001).shown here, E2f1 loss compromises Myc-driven prolifer-Like Myc, E2fs can positively and negatively regulateation and lymphomagenesis. Thus, E2f1-induced prolif-many periodically expressed genes whose functions areeration is rate limiting for lymphomagenesis in E�-Myckey to DNA metabolism and replication (Nevins, 2001;transgenic mice, and acceleration of cell cycle progres-Trimarchi and Lees, 2002). Although it has been sug-sion, as well as the dismantling of the apoptotic pro-gested that E2f1 and E2f3 play differential roles in regu-gram, contributes to the rate of tumor onset.lating apoptosis and S phase entry, respectively (De-
Gregori et al., 1997; Lissy et al., 2000; Leone et al., 2001),in-depth analyses of E2f1�/�, E2f3�/�, and E2f1�/�/ E2f1 Haploinsufficiency Affects
Myc-Induced TumorigenesisE2f3�/� double null mice have instead revealed thatthese two E2fs can play overlapping roles in both these Loss of only one E2f1 allele impaired Myc-induced prolif-
eration and lymphomagenesis almost to the same de-processes (Yamasaki et al., 1998; Humbert et al., 2000;Ziebold et al., 2001; Saavedra et al., 2002). Loss of either gree as total loss of E2f1. In all tumors arising in the
E2f1�/� background, the wild-type E2f1 allele was re-E2f1 or E2f3 might decrease the rate of Myc-inducedtumorigenesis by affecting cell cycle progression. In nor- tained and expressed. Therefore, a critical threshold of
E2f1 must be achieved to support Myc-induced prolifer-mal B cells, E2f1 disruption had no overt effect on thefraction of cells in S phase, implying that other E2fs can ation, and a reduced dosage of transcriptionally active
E2fs may lead to haploinsufficient effects.compensate. However, its loss impaired the proliferationof E�-Myc transgenic B cells, both ex vivo and in vivo, Absence of E2f3, the most abundant activating E2f
species in MEFs, impairs expression of E2f-responsiveand retarded lymphoma development. Hence, the abilityof sustained Myc signaling to promote B cell division genes and thereby retards cell proliferation (Humbert et
al., 2000). The proliferative rates of E2f3�/� MEFs aredepends upon E2f1.Levels of the Cdk inhibitor p27Kip1 were markedly ele- intermediate, indicating that E2f3 acts in a dose-depen-

Molecular Cell912
Immunoblottingdent manner and that 2-fold changes in its level canExtracts from MEFs, primary pre-B cells, or lymphomas from E�-affect the phenotype. While loss of E2f1 or E2f2 aloneMyc transgenic mice, were prepared as previously described (Kam-has little effect on the proliferation of MEFs, the com-ijo et al., 1998). Protein (100 �g per lane) was electrophoretically
bined absence of E2fs 1, 2, and 3 completely blocks separated in 12% polyacrylamide gels containing sodium dodecylproliferation, underscoring their cooperative effects sulfate, transferred to membranes (Protran, Schleicher & Schuell,
Dassel, Germany), and blotted with antibodies specific for p19Arf(Humbert et al., 2000; Wu et al., 2001). Importantly, the(Abcam, Cambridge UK), p53 (Ab7, Calbiochem, San Diego, CA),E�-Myc model shows that loss of E2f1, while not overtlyp27Kip1 (K25020, BD Transduction Labs, San Diego, CA), p21Cip1 (F5,affecting proliferation of normal B cells, cannot be com-Santa Cruz Inc., Santa Cruz, CA), c-Myc (N-262, Santa Cruz), andpensated sufficiently by other E2fs to support the in-E2f1 (BD Transduction Labs, San Diego, CA).
creased rates of cell division driven by oncogene acti-vation. Acknowledgments
We thank Lili Yamasaki for her generous gift of E2f1 null mice,Experimental ProceduresKristian Helin for providing the E2f1-ER retroviral vector, and Chris-tine Eischen for initiating the E�-Myc � E2f1 null breeding. We thankInterbreeding of Mice and Tumor SurveillanceRose Mathew, Elsie White, Sara Norton, and Mehmet Kocak forE2f1 null mice (129/svj � C57BL/6, backcrossed to C57BL/6) (Yama-expert assistance and our Animal Resources Center for help withsaki et al., 1996) were interbred with a C57BL/6 strain of E�-Mycmice. This work was supported by NIH grants CA76379 andtransgenic mice (Adams et al., 1985; Sidman et al., 1988). Fl offspringDK44158 (J.L.C.), CA71907 (M.F.R.), CA42063 (J.A.L.), Cancer Cen-were intercrossed to obtain E2f1�/�, E2f1�/�, and E2f1�/� transgenicter Core Grant CA21765, and by the American Lebanese Syrianlittermates. Animals were observed daily for signs of morbidity andAssociated Charities (ALSAC) of St. Jude Children’s Research Hos-tumor development. Tumors that arose were harvested after sacri-pital. T.A.B. was supported by NRSA grant 1F32 DK10154. C.J.S.fice of animals, snap-frozen in liquid nitrogen, and processed foris an Investigator of the Howard Hughes Medical Institute.analysis of DNA and protein.
Received: December 20, 2002Cell Culture Revised: February 4, 2003MEFs from day 13.5 embryos were isolated according to Todaro Accepted: February 14, 2003and Green (1963) as described previously (Kamijo et al., 1997). Cells Published: April 24, 2003from single embryos were plated into two T25 flasks and incubatedat 37C in a 9% CO2 humidified chamber. Plating after disaggrega- Referencestion of embryos was considered passage 1, and the first replating3 days later as passage 2. Cells at passages 3–5 were infected with Adams, J.M., Harris, A.W., Pinkert, C.A., Corcoran, L.M., Alexander,retroviruses containing puro alone or puro in cis with Myc-ER or W.S., Cory, S., Palmiter, R.D., and Brinster, R.L. (1985). The c-mycE2f1-ER and selected in puromycin for 48 hr before analysis. Cells oncogene driven by immunoglobulin enhancers induces lymphoidinfected with a Myc-ER retrovirus expressing GFP instead of puro malignancy in transgenic mice. Nature 318, 533–538.were monitored by fluorescence-activated flow cytometry (FACS).
Adams, M.R., Sears, R., Nuckolls, F., Leone, G., and Nevins, J.R.For double infection with Myc-ER (GFP) and E2f1-ER (puro) vectors,(2000). Complex transcriptional regulatory mechanisms control ex-cells selected in puromycin were monitored for GFP expressionpression of the E2F3 locus. Mol. Cell. Biol. 20, 3633–3639.(�95% positive) before protein analysis.Askew, D.S., Ashmun, R.A., Simmons, B.C., and Cleveland, J.L.Primary bone marrow-derived pre-B cell cultures were generated(1991). Constitutive c-myc expression in an IL3-dependent myeloidfrom 8-week-old E2f1�/�, E2f1�/�, and E2f1�/� E�-Myc transgeniccell line suppresses cell cycle arrest and accelerates apoptosis.mice as described (Borzillo and Sherr, 1989; Eischen et al., 1999).Oncogene 6, 1915–1922.After 2 weeks in culture during which myeloid progenitors died, cells
were immunophenotyped and confirmed to be �90% pre-B cells Bartek, J., and Lukas, J. (2001). p27 destruction: Cks1 pulls the(i.e., B220�, CD43�, IgM�, and negative for T cell- or myeloid/ trigger. Nat. Cell Biol. 3, E95–E98.macrophage-specific markers).
Bates, S., Phillips, A.C., Clarke, P.A., Stott, F., Peters, G., Ludwig,R.L., and Vousden, K.H. (1998). p14ARF links the tumour suppressorsRB and p53. Nature 395, 124–125.Analysis of BrdU-Labeled Cells
Rates of proliferation of B220� B cells were measured using a Flow Borzillo, G.V., and Sherr, C.J. (1989). Early pre-B-cell transformationKit as described by the manufacturer (BD Biosciences Pharmingen, induced by the v-fms oncogene in long-term mouse bone marrowSan Diego, CA). B cells growing in culture were labeled with 10 �M cultures. Mol. Cell. Biol. 9, 3973–3981.of BrdU for 1 hr. In vivo labeling was performed via intraperitoneal Bouchard, C., Thieke, K., Maier, A., Saffrich, R., Hanley-Hyde, J.,injection of 100 �l of 10 mg/ml BrdU in sterile PBS. Animals were Ansorge, W., Reed, S., Sicinski, P., Bartek, J., and Eilers, M. (1999).sacrificed 12 hr post injection, and bone marrow, spleen, and lymph Direct induction of cyclin D2 by Myc contributes to cell cycle pro-nodes were harvested. Cells were resuspended, incubated with anti- gression and sequestration of p27. EMBO J. 18, 5321–5333.body against B220 (BD Biosciences Pharmingen, San Diego, CA),
DeGregori, J., Leone, G., Miron, A., Jakoi, L., and Nevins, J. (1997).washed, and collected by centrifugation. Cells were further pro-Distinct roles for E2F proteins in cell growth control and apoptosis.cessed and stained with FITC anti-BrdU antibody as described byProc. Natl. Acad. Sci. USA 94, 7245–7250.the same manufacturer. Washed cells were resuspended inDyson, N. (1998). The regulation of E2F by pRB-family proteins.7-amino-actinomycin D and analyzed by dual parameter FACS.Genes Dev. 12, 2245–2262.
Eilers, M., Picard, D., Yamamoto, K., and Bishop, J.M. (1989). Chi-Apoptosis Measurements maeras between the MYC oncoprotein and sterioid receptors causeThe viability of Myc-ER-infected MEF strains (E2f1�/� and E2f1�/�) hormone-dependent transformation cells. Nature 340, 66–68.was measured by trypan blue vital dye exclusion and by use of
Eischen, C.M., Weber, J.D., Roussel, M.F., Sherr, C.J., and Cleve-an Annexin-V-FLUOS Staining Kit (Roche Diagnostics, Mannheim,land, J.L. (1999). Disruption of the ARF-Mdm2-p53 tumor suppressorGermany) at specific intervals after the addition of 2 �M 4-HT (Sigma,pathway in Myc-induced lymphomagenesis. Genes Dev. 13, 2658–St. Louis, MO). Apoptosis was measured in precancerous lymph2669.nodes using 7 �m cryosections stained with the ApopTag In Situ
Oligo Ligation Kit (ISOL, Intergen Company, Purchase, NY) as de- Eischen, C.M., Woo, D., Roussel, M.F., and Cleveland, J.L. (2001).Apoptosis triggered by Myc-induced suppression of Bcl-X(L) orscribed by the manufacturer.

Myc-Induced Proliferation Depends on E2f1913
Bcl-2 is bypassed during lymphomagenesis. Mol. Cell. Biol. 21, sequential proteolytic pathways that regulate p27Kip1 in G1 and Sphase. Nature 413, 323–327.5063–5070.
Eisenman, R.N. (2001). Deconstructing myc. Genes Dev. 15, 2023– Marti, A., Wirbelauer, C., Scheffner, M., and Krek, W. (1999). Interac-tion between ubiquitin-protein ligase SCFSKP2 and E2F–1 underlies2030.the regulation of E2F-1 degradation. Nat. Cell Biol. 1, 14–19.Evan, G.I., and Vousden, K.H. (2001). Proliferation, cell cycle and
apoptosis in cancer. Nature 411, 342–348. Martins, C.P., and Berns, A. (2002). Loss of p27(Kip1) but notp21(Cip1) decreases survival and synergizes with MYC in murineEvan, G.I., Wyllie, A.H., Gilbert, C.S., Littlewood, T.D., Land, H.,lymphomagenesis. EMBO J. 21, 3739–3748.Brooks, M., Waters, C.M., Penn, L.Z., and Hancock, D.C. (1992).
Induction of apoptosis in fibroblasts by c-myc protein. Cell 69, Muller, D., Bouchard, C., Rudolph, B., Steiner, P., Stuckmann, I.,Saffrich, R., Ansorge, W., Huttner, W., and Eilers, M. (1997). Cdk2-119–128.dependent phosphorylation of p27 facilitates its Myc-induced re-Field, S.J., Tsai, F.-Y., Kuo, F., Zubiaga, A.M., Kaelin, W.G., Living-lease from cyclin E/cdk2 complexes. Oncogene 15, 2561–2576.ston, D.M., Orkin, S.H., and Greenberg, M.E. (1996). E2F-1 functions
in mice to promote apoptosis and suppress proliferation. Cell 85, Nevins, J.R. (2001). The Rb/E2F pathway and cancer. Hum. Mol.Genet. 10, 699–703.549–561.
Galaktionov, K., Chen, X., and Beach, D. (1996). Cdc25 cell-cycle Ogawa, H., Ishiguro, K., Gaubatz, S., Livingston, D.M., and Nakatani,Y. (2002). A complex with chromatin modifiers that occupies E2F-phosphatase is a target of c-myc. Nature 382, 511–517.and Myc-responsive genes in G0 cells. Science 296, 1132–1136.Haupt, Y., Maya, R., Kazaz, A., and Oren, M. (1997). Mdm2 promotes
the rapid degradation of p53. Nature 387, 296–299. O’Hagan, R.C., Ohh, M., David, G., de Alboran, I.M., Alt, F.W., Kaelin,W.G., Jr., and DePinho, R.A. (2000). Myc-enchanced expressionHermeking, H., Rago, C., Schuhmacher, M., Li, Q., Barrett, J.F.,of Cul1 promotes ubiquitin-dependent proteolysis and cell cycleObaya, A.J., O’Connell, B.C., Mateyak, M.K., Tam, W., Kohlhuber,progression. Genes Dev. 14, 2185–2191.F., et al. (2000). Identification of CDK4 as a target of c-MYC. Proc.
Natl. Acad. Sci. USA 97, 2229–2234. Pan, H., Yin, C., Dyson, J.J., Harlow, E., Yamasaki, L., and Van Dyke,T. (1998). Key roles for E2F1 in signaling p53-dependent apoptosisHumbert, P.O., Verona, R., Trimarchi, J.M., Rogers, C., Dandapani,and cell division. Mol. Cell 2, 283–292.S., and Lees, J.A. (2000). E2f3 is critical for normal cellular prolifera-
tion. Genes Dev. 14, 690–703. Perez-Roger, I., Kim, S.H., Griffiths, B., Sewing, A., and Land, H.(1999). Cyclins D1 and D2 mediate myc-induced proliferation viaInoue, K., Zindy, F., Randle, D.H., Rehg, J.E., and Sherr, C.J. (2001).sequestration of p27kip1 and p21Cip1. EMBO J. 18, 5310–5320.Dmp1 is haplo-insufficient for tumor suppression and modifies the
frequencies of Arf and p53 mutations in Myc-induced lymphomas. Qin, X.Q., Livingston, D.M., Kaelin, W.G., Jr., and Adams, P.D. (1994).Deregulated transcription factor E2F-1 expression leads to S-phaseGenes Dev. 15, 2934–2939.entry and p53-mediated apoptosis. Proc. Natl. Acad. Sci. USA 91,Jacobs, J.J.L., Scheijen, B., Vonchen, J.-W., Kieboom, K., Berns,10918–10922.A., and van Lohuizen, M. (1999). Bmi-1 collaborates with c-Myc in
tumorigenesis by inhibiting c-Myc induced apoptosis via INK4a/ Robertson, K.D., and Jones, P.A. (1998). The human ARF cell cycleregulatory gene promoter is a CpG island which can be silenced byARF. Genes Dev. 13, 2678–2690.DNA methylation and down-regulated by wild-type p53. Mol. Cell.Juin, P., Hueber, A.-O., Littlewood, T., and Evan, G. (1999). c-MycBiol. 18, 6457–6473.induced sensitization to apoptosis is mediated through cytochrome
c release. Genes Dev. 13, 1367–1381. Rounbehler, R.J., Rogers, P.M., Conti, C.J., and Johnson, D.G.(2002). Inactivation of E2f1 enhances tumorigenesis in a Myc trans-Kamijo, T., Zindy, F., Roussel, M.F., Quelle, D.E., Downing, J.R.,genic model. Cancer Res. 11, 3276–3281.Ashmun, R.A., Grosveld, G., and Sherr, C.J. (1997). Tumor suppres-
sion at the mouse INK4a locus mediated by the alternative reading Rowland, B.D., Denissov, S.G., Douma, S., Stunnenberg, H.G., Ber-nards, R., and Pepper, D.S. (2002). E2F transcriptional repressorframe product p19ARF. Cell 91, 649–659.complexes are critical downstream targets of p19ARF/p53-inducedKamijo, T., Weber, J.D., Zambetti, G., Zindy, F., Roussel, M.F., andproliferative arrest. Cancer Cell 2, 55–65.Sherr, C.J. (1998). Functional and physical interactions of the ARF
tumor suppressor with p53 and Mdm2. Proc. Natl. Acad. Sci. USA Russell, J.L., Powers, J.T., Rounbehler, R.J., Rogers, P.M., Conti,C.J., and Johnson, D.G. (2002). ARF differentially modulates apopto-95, 8292–8297.sis induced by E2F1 and Myc. Mol. Cell. Biol. 22, 1360–1368.Krimpenfort, P., Quon, K.C., Mooi, W.J., Loonstra, A., and Berns, A.
(2001). Loss of p16Ink4a confers susceptibility to metastatic melanoma Saavedra, H.I., Wu, L., de Bruin, A., Timmers, C., Rosol, T.J.,Weinstein, M., Robinson, M.L., and Leone, G. (2002). Specificity ofin mice. Nature 413, 83–86.E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss ofKubbutat, M.H., Jones, S.N., and Vousden, K.H. (1997). RegulationRb. Cell Growth Differ. 13, 215–225.of p53 stability by Mdm2. Nature 387, 299–303.Santoni-Rugiu, E., Falck, J., Mailand, N., Bartek, J., and Lukas, J.Leone, G., DeGregori, J., Sears, R., Jakoi, L., and Nevins, J.R. (1997).(2000). Involvement of Myc activity in a G(1)/S-promoting mecha-Myc and Ras collaborate in inducing accumulation of active cyclinnism parallel to the pRb/E2F pathway. Mol. Cell. Biol. 20, 3497–3509.E/Cdk2 and E2F. Nature 387, 422–426.Schmitt, C.A., McCurrach, M.E., De Stanchina, E., and Lowe, S.W.Leone, G., Sears, R., Huang, E., Rempel, R., Nuckolls, F., Park, C.-H.,(1999). INK4a/ARF mutations accelerate lymphomagenesis and pro-Giangrande, P., Wu, L., Saavedra, H.I., Field, S.J., et al. (2001). Mycmote chemoresistance by disabling p53. Genes Dev. 13, 2670–2677.requires distinct E2F activities to induce S phase and apoptosis.
Mol. Cell 8, 105–113. Schmitt, C.A., Fridman, J.S., Yang, M., Lee, S., Baranov, E., Hoffman,R.M., and Lowe, S.W. (2002). A senescence program controlled byLissy, N.A., Davis, P.K., Irwin, M., Kaelin, W.G., and Dowdy, S.F.p53 and p16INK4a contributes to the outcome of cancer therapy. Cell(2000). A common E2F-1 and p73 pathway mediates cell death109, 335–346.induced by TCR activation. Nature 407, 642–645.Shan, B., and Lee, W.H. (1994). Deregulated expression of E2F-1Littlewood, T.D., Hancock, D.C., Danielian, P.S., Parker, M.G., andinduces S-phase entry and leads to apoptosis. Mol. Cell. Biol. 14,Evan, G.I. (1995). A modified estrogen receptor ligand-binding do-8166–8173.main as an improved switch for the regulation of heterologous pro-
teins. Nucleic Acids Res. 23, 1686–1690. Sheaff, R., Groudine, M., Gordon, M., Roberts, J., and Clurman, B.(1997). Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 11,Lomazzi, M., Moroni, M.C., Jensen, M.R., Frittoli, E., and Helin, K.1464–1478.(2002). Suppression of the p53- or pRB-mediated G1 checkpoint is
required for E2F-induced S-phase entry. Nat. Genet. 31, 190–194. Sherr, C.J. (2001). The INK4a/ARF network in tumour suppression.Nat. Rev. Mol. Cell Biol. 2, 731–737.Malek, N.P., Sundberg, H., McGrew, S., Nakayama, K., Kyriakides,
T.R., and Roberts, J.M. (2001). A mouse knock-in model exposes Sherr, C., and Roberts, J.M. (1999). CDK inhibitors: positive and

Molecular Cell914
negative regulators of G1-phase progression. Genes Dev. 13, 1501–1512.
Sidman, C.L., Marshall, J.D., and Harris, A.W. (1988). Genetic studieson E�-myc transgenic mice. Curr. Top. Microbiol. Immunol. 141,94–99.
Slingerland, J., and Pagano, M. (2002). Regulation of the cdk inhibitorp27 and its deregulation in cancer. J. Cell. Physiol. 183, 10–17.
Stott, F.J., Bates, S., James, M.C., McConnell, B.B., Starborg, M.,Brookes, S., Palmero, I., Ryan, K., Hara, E., Vousden, K.H., andPeters, G. (1998). The alternative product from the human CDKN2Alocus, p14ARF, participates in a regulatory feedback loop with p53and MDM2. EMBO J. 17, 5001–5014.
Todaro, G.J., and Green, H. (1963). Quantitative studies of the growthof mouse embryo cells in culture and their development into estab-lished lines. J. Cell Biol. 17, 299–313.
Tolbert, D., Lu, X., Yin, C., Tantama, M., and Van Dyke, T. (2002).p19ARF is dispensable for oncogenic stress-induced p53-mediatedapoptosis and tumor suppression in vivo. Mol. Cell. Biol. 22,370–377.
Trimarchi, J.M., and Lees, J.A. (2002). Sibling rivalry in the E2F family.Nat. Rev. Mol. Cell Biol. 3, 11–20.
Trimarchi, J.M., Fairchild, B., Wen, J., and Lees, J.A. (2001). TheE2F6 transcription factor is a component of the mammalian Bmi1-containing polycomb complex. Proc. Natl. Acad. Sci. USA 98, 1519–1524.
Tsai, K.Y., Hu, Y., Macleod, K.F., Crowley, D., Yamasaki, L., andJacks, T. (1998). Mutation of E2f-1 suppresses apoptosis and inap-propriate S phase entry and extends survival of Rb-deficient mouseembryos. Mol. Cell 2, 293–304.
Tsai, K.Y., MacPherson, D., Rubinson, D.A., Crowley, D., and Jacks,T. (2002a). ARF is not required for apoptosis in Rb mutant mouseembryos. Curr. Biol. 12, 159–163.
Tsai, K.Y., MacPherson, D., Rubinson, D.A., Nikitin, A.Y., Bronson,R., Mercer, K.L., Crowley, D., and Jacks, T. (2002b). ARF mutationaccelerates pituitary tumor development in Rb�/�mice. Proc. Natl.Acad. Sci. USA 99, 16865–16870.
Vigo, E., Muller, H., Prosperini, E., Hateboer, G., Cartwright, P., Mo-roni, M.C., and Helin, K. (1999). CDC25A phosphatase is a target ofE2F and is required for efficient E2F-induced S phase. Mol. Cell.Biol. 19, 6379–6395.
Vlach, J., Hennecke, S., Alevizopoulos, K., Conti, D., and Amati, B.(1996). Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1
is abrogated by c-Myc. EMBO J. 15, 6595–6604.
Vlach, J., Hennecke, S., and Amati, B. (1997). Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitorp27Kip1. EMBO J. 16, 5334–5344.
Wu, X., and Levine, A.J. (1994). p53 and E2F-1 cooperate to mediateapoptosis. Proc. Natl. Acad. Sci. USA 91, 3602–3606.
Wu, L., Timmers, C., Maiti, B., Saavedra, H.I., Sang, L., Chong, G.T.,Nuckolls, F., Giangrande, P., Wright, F.A., Field, S.J., et al. (2001).The E2F1-3 transcription factors are essential for cellular prolifera-tion. Nature 414, 457–462.
Yamasaki, L., Jacks, T., Bronson, R., Goillot, E., Harlow, E., andDyson, N.J. (1996). Tumor induction and tissue atrophy in micelacking E2F-1. Cell 85, 537–548.
Yamasaki, L., Bronson, R., Williams, B.O., Dyson, N.J., Harlow, E.,and Jacks, T. (1998). Loss of E2F-1 reduces tumorigenesis andextends lifespan of Rb1(�/�) mice. Nat. Genet. 18, 360–364.
Yang, W., Shen, J., Wu, M., Arsura, M., FitzGerald, M., Suldan, Z.,Kim, D.W., Hofmann, C.S., Pianetti, S., Romieu-Mourez, R., et al.(2001). Repression of transcription of the p27(Kip1) cyclin-depen-dent kinase inhibitor gene by c-Myc. Oncogene 20, 1688–1702.
Ziebold, U., Reza, T., Caron, A., and Lees, J.A. (2001). E2F3 contrib-utes both to the inappropriate proliferation and to the apoptosisarising in Rb mutant embryos. Genes Dev. 15, 386–391.
Zindy, F., Eischen, C.M., Randle, D.H., Kamijo, T., Cleveland, J.L.,Sherr, C.J., and Roussel, M.F. (1998). Myc signaling via the ARFtumor suppressor regulates p53-dependent apoptosis and immor-talization. Genes Dev. 12, 2424–2433.
Copyright © 2022 FDOKUMEN




















