
Compromised emotional competence: Seeds of violence sown early?

J Mol Cell Cardiol 31, 2063–2076 (1999)
Article No. jmcc.1999.1037, available online at http://www.idealibrary.com on
The Absence of Desmin Leads toCardiomyocyte Hypertrophy and CardiacDilation with Compromised SystolicFunctionDerek J. Milner1∗, George E. Taffet2∗, Xuejun Wang3∗, Thuy Pham2,Tetsutaro Tamura3, Craig Hartley2, A. Martin Gerdes3 andYassemi Capetanaki1
1Department of Cell Biology and 2Department of Medicine Section of Cardiovascular Science andGeriatrics, Baylor College of Medicine, Houston, Texas 77030, USA; 3Department of Anatomy,University of South Dakota, School of Medicine, Vermillion, SD 57069, USA
(Received 19 November 1998, accepted in revised form 27 August 1999)
D. J. M, G. E. T, X. W, T. P, T. T, C. H, A. M. G Y. C. TheAbsence of Desmin Leads to Cardiomyocyte Hypertrophy and Cardiac Dilation with Compromised SystolicFunction. Journal of Molecular and Cellular Cardiology (1999) 31, 2063–2076. Desmin is the muscle-specificmember of the intermediate filament family of cytoskeletal proteins, expressed both in striated and smooth muscletissues. In mature striated muscle fibers, the desmin filament lattice surrounds the Z-discs, interconnects themto each other and links the entire contractile apparatus to the sarcolemmal cytoskeleton, cytoplasmic organellesand the nucleus. There have been increasing reports of human cardiomyopathies associated with abnormalaccumulation and aggregation of desmin filaments. Recently identified desmin mutations in humans sufferingfrom skeletal muscle myopathy and cardiomyopathy suggest that these diseases might arise as a consequence ofimpaired function of desmin filaments. Previous generation of desmin null mice in our laboratory demonstratedthat the absence of desmin results in myocyte ultrastructural defects and myocyte cell death leading to fibrosisand calcification of the myocardium. However, the effects that these defects have on cardiac function were notaddressed. To further our understanding of desmin function in vivo, and in order to address the direct involvementof desmin in cardiomyopathy, we investigated the effect of the absence of desmin on myocardial mass, myocytesize and shape, changes in gene expression and cardiac systolic and diastolic function in mice. Morphometriccharacterization of isolated cardiomyocytes demonstrated a 24% increase in cell volume in the desmin null mice,solely due to an increase in transverse section area, suggesting for the first time that mice lacking the intermediatefilament protein desmin develop concentric cardiomyocyte hypertrophy. This type of hypertrophy was accompaniedby induction of embryonic gene expression and later by ventricular dilatation, and compromised systolic function.These results demonstrate that desmin is essential for normal cardiac function, and they suggest that the absenceof an intact desmin filament system, rather than accumulation of the protein, may be responsible for the pathologyseen in some of the desmin associated cardiomyopathies. 1999 Academic Press
K W: Desmin; Intermediate filaments; Cardiomyopathy; Hypertrophy; Dilation; Doppler.
cardiomyopathy (HCM), dilated cardiomyopathyIntroduction(DCM) and restrictive cardiomyopathy, may be in-herited or acquired, but in all forms the underlyingThe main forms of cardiomyopathy, hypertrophic
Please address all correspondence to: Dr Yassemi Capetanaki, Department of Cell Biology, Baylor College of Medicine, Houston, Texas77030. E-mail: [email protected]∗ The first three authors contributed equally to this work.
0022–2828/99/112063+14 $30.00/0 1999 Academic Press

D. J. Milner et al.2064
cause is frequently unknown. Recently, mutations could influence nuclear events (Li et al., 1994;Weitzer et al., 1995). However, gene targeting ap-in several genes, all encoding sarcomeric proteins
such as b-myosin heavy chain, cardiac troponin T, proaches demonstrate that prenatal muscle de-velopment can take place in desmin null mice (Lia-tropomyosin, myosin binding protein-C, myosin
regulatory light chain, myosin essential light chain et al., 1996; Milner et al., 1996).There are increasing reports of desmin-relatedand cardiac troponin I, have been identified as
causes of familial HCM (for review see Keating and skeletal and cardiac myopathies that have as ahallmark abnormal deposits of desmin aggregates.Sanguinetti, 1996; Vikstrom and Leinwand, 1996;
Maron, 1997; Marian and Roberts, 1998; Towbin, These myopathies are characterized by both skeletalmuscle weakness and cardiac abnormalities, in-1998). In contrast, a few studies have linked DCM
to genes encoding enzymes involved in metabolic cluding restrictive cardiomyopathy, cardiomyocytehypertrophy, dilation of the ventricular chambers,pathways, including oxidative phosphorylation and
fatty acid b-oxidation (for review see Keating and conduction blocks, arrhythmias and heart failure.Hypoplasia of the aorta and thinned intestinal wallsSanguinetti, 1996). However, the majority of DCM-
linked genes encode proteins of the membrane cyto- have also been reported (Vajsar et al., 1993; Arizaet al., 1995; Goebel, 1995). However, underlyingskeleton. Such proteins include dystrophin, the gene
responsible for X-linked dilated cardiomyopathy and molecular mechanisms giving rise to these my-opathies are unknown. Recently, mutations in theDuchenne muscular dystrophy (Towbin, 1993,
1995; Bies et al., 1997), a-dystroglycan (Towbin, highly conserved carboxy-terminal end (Goldfarbet al., 1998) or the middle (Munoz-Marmol et al.,1995; Bies et al., 1997), d-sarcoglycan (Nigro et
al., 1997) and metavinculin (Maeda et al., 1997). 1998) of the desmin rod domain were identifiedin patients with generalized myopathy includingIn addition to the membrane cytoskeleton, striated
muscle contains at least two more cytoskeletal cardiomyopathy and heart failure. These regionsare important in IF assembly, so these mutationssystems, the intrasarcomeric cytoskeleton, located
within the myofibrillar lattice and composed of titin are likely to impair filament assembly. In the lattercase, this was shown to be true in vitro. On theand nebulin, and the extrasarcomeric cytoplasmic
filaments composed primarily of desmin inter- other hand, a point mutation at the non-helicalcarboxyl-terminus of desmin was recently linked tomediate filaments (IFs), which are muscle specific,
and lower amounts of synemin and paranemin DCM (Li et al., 1999). The function of this regionremains unclear. Thus, myopathy might developIFs which are not muscle-specific (for reviews see
Lazarides and Capetanaki, 1986; Capetanaki et al., directly due to altered function or loss of functionof desmin filaments. Alternatively, myopathy may1997). Of these cytoskeletal proteins, so far in mice
only desmin has been linked to myopathy (Li et al., develop indirectly due to disturbance of other nor-mal processes by the aggregated filaments. No1996; Milner et al., 1996; Capetanaki and Milner,
1998). human myopathy with complete absence of desminhas so far been reported. To address these issuesDesmin, encoded by a single copy gene (Ca-
petanaki et al., 1984), is expressed in all striated and further investigate the role of desmin in cardiacmuscle structure and function, we have previouslyand smooth muscle tissues (for review see Lazarides,
1982; Lazarides and Capetanaki, 1986). It is one generated desmin null mice through gene targeting(Milner et al., 1996). Mice lacking desmin dem-of the earliest known muscle-specific genes to be
expressed during cardiac and skeletal muscle de- onstrate numerous muscle architectural and ul-trastructural defects. Cardiac muscle is the mostvelopment (Furst et al., 1989; Herrmann et al.,
1989; Schaart et al., 1989). The desmin IF lattice severely affected, with calcified lesions and fibrousscar tissue developing throughout the degeneratingsurrounds the Z-discs, interconnects them to each
other and links the entire contractile apparatus to myocardium. Structural abnormalities include lossof lateral alignment of myofibrils, perturbation ofthe sarcolemmal cytoskeleton, cytoplasmic or-
ganelles and the nucleus (for review see Capetanaki myofibril anchorage to the sarcolemma, abnormalmitochondrial organization, and loss of nuclearand Milner, 1998). This allows the formation of
a continuous cytoskeletal network that could be shape and positioning. Loose cell adhesion andincreased intercellular space are also prominentinvolved in several diverse functions, including,
among others, maintenance of cellular integrity, defects (Capetanaki et al., 1997; Capetanaki andMilner, 1998). However, the effect of these defectsforce transmission and mechanochemical signaling.
In vitro studies, using both C2C12 cells as well as on the function of the heart were not assessed.In order to address the direct involvement ofembryonic stem cells, have suggested that desmin is
important in muscle differentiation and its absence desmin in cardiomyopathy and normal cardiac

Desmin Cytoskeleton in Cardiomyopathy 2065
function, we investigated the effect of the absence et al., 1988) and a-skeletal actin. Equivalent loadingwas assessed using a probe for 18S RNA.of desmin on myocardial mass, myocyte shape and
cardiac systolic and diastolic function. We reportfor the first time that mice lacking the intermediatefilament protein desmin develop cardiac hyper- Exercise testing and histologytrophy with ventricular dilatation, compromisedsystolic function and heart failure later in life. Mice were exercised using a swimming protocol
similar to that used by Geisterfer-Lowrance et al.(1996). Mice were exercised twice a day, beginningat 10 min per session on the first day. The durationMaterials and Methodsof swimming was increased by 10 min per day toa final duration of 90 min per session, which wasGeneration of desmin null micemaintained for the duration of the experiment. Thetotal duration of each experiment was 8 weeks. UpMice lacking desmin were generated by gene tar-to 50% of the desmin null mice died before the endgeting via homologous recombination as previouslyof this protocol. For histological examination ofdescribed (Milner et al., 1996). Targeted AB2.2 EScardiac muscle, hearts were removed after sacrifice,cell lines were microinjected into C57Bl/6 blast-washed briefly in phosphate-buffered saline andocysts and subsequently transferred into pseudo-fixed for 12–36 h in 10% neutral buffered formalin.pregnant females. Chimeras were bred to C57Bl/6The specimens were processed, embedded and sec-females and germline transmission of the ES celltioned using standard histological methods, andgenome was obtained as verified by the productiontissue sections were stained with hematoxylin andof agouti offspring. Heterozygotes were crossed toeosin.produce mice homozygous for the disrupted allele
(Des−/−). Northern and western blot analysis onRNA and protein prepared from mouse leg muscleverified the absence of desmin message and protein Wall thickness measurementsin mice homozygous for the disrupted allele (Milneret al., 1996). For assessment of cardiac wall thickness, the heart
was excised and rinsed in cold cardioplegic solution,the aorta was cannulated with a 22-gauge bluntneedle and the left atrial appendage with a PE-50Isolation of RNA and Northern analysiscatheter pushed across the mitral valve into the LVand secured in place. Hearts were fixed for 10 minMice were killed by cervical dislocation and heartsby aortic perfusion of 10% zinc-buffered formalinwere excised, trimmed and weighed, and heartwith the LV drain held vertically 16 cm above theweight/body weight calculations were tabulated.base of the heart to maintain constant LV pressureAll blood was removed from the chambers beforeat>16 cm H2O. After the cannulas were removed,hearts were weighed. For analysis of heart weight/fixation by immersion continued for 2 h, after whichbody weight ratios, a Student’s t-test was used tothe hearts were dehydrated, cleared, and embeddeddetermine the presence of intergroup differences,in paraffin. The entire heart was cross-sectionedand statistical significance was set at P<0.05. Datainto 5 lm sections from base to apex. Sections werewere expressed as mean±.. The atria were thenstained with hematoxylin and eosin or Massoncut away, and the ventricles were snap frozen intrichrome stain. Average wall thickness was cal-liquid nitrogen. Frozen ventricles were stored atculated from measurements made on digitized im-−70°C until RNA isolation was performed. Totalages of the 5-lm sections.ventricular RNA was isolated using the Totally RNA
isolation kit from Ambion (Austin, TX). Three micewere used for each RNA preparation. Individualventricles of respective genotypes and ages were Isolation of ventricular myocytespooled for RNA isolation. Northern blotting wasperformed as previously described using standard Ventricular myocytes were enzymatically isolated
with a modified protocol described previously (Li ettechniques (Capetanaki et al., 1983). Blots weresequentially probed using cDNA fragments specific al., 1996; Wolska and Solaro, 1996). Ap-
proximately 7-month-old desmin-null and age-for ANF (Seidman et al., 1984), phospholamban(Ganim et al., 1992), SERCA 2A (Gunteski-Hamblin matched wild-type male mice were heparinized

D. J. Milner et al.2066
(5000 U/kg, i.p.) 30 min before induction of an- Doppler studiesesthesia (50 mg/kg of pentobarbital sodium, i.p.).Hearts were excised, trimmed, weighed, and placed Mice were anesthetized by an intraperitoneal
injection with a rodent anesthesia mixture (ace-in a petri dish filled with Solution A which iscomposed of Jolkik’s Minimum Essential Medium promazine 1.4 mg/ml, xylazine 8.6 mg/ml, ke-
tamine 42.8 mg/ml) at a dose of 0.5 ll/g bodyEagle (pH 7.2) (Sigma, St Louis, MO) supplementedwith (in m) 34.5 NaHCO3, 1 MgSO4, 20 BDM (2, weight and were taped to a temperature-controlled
laminated plastic board with copper electrodes3-butanedione monoxide; Sigma), and 0.2 EGTA(ethylene glycol bis (B-amino-ethyl ether)-N,N′-te- placed such that all four limb leads could be used
for electrocardiographic monitoring. Body fur attraacetic acid). A retrograde aortic cannulation wasdone under a dissecting microscope within 6 min. the left lower sternal border was clipped lightly
and the skin in that area was wetted with warmThe heart was mounted in a Langendorff perfusionapparatus and perfused first with Solution A for water to improve sound transmission. A 10-MHz
pulsed Doppler probe was positioned at the xiphoid5 min and then with Solution B for 8 min. SolutionB is similar to Solution A, but contains 150 U/ml applying only minimal pressure. The pulsed Dop-
pler range gate depth was set at 4–7 mm deepof type II collagenase (Worthington Biochem. Co.,Lakewood, NJ) with the replacement of EGTA with to obtain optimal signals from the LV inflow and
outflow tracks substernally. An ECG timing signal0.1% BSA. The digested heart was put back intoSolution A and the myocardium of both ventricles was superimposed on the Doppler display using
an R-wave trigger. The result is a dashed verticalincluding septum minced. The dissociated vent-ricular myocytes were filtered through a 250 lm line on the display (Fig. 4).
Doppler outputs were captured on a personalmesh into centrifuge tubes, which had been filledwith 2.5% glutaraldehyde. Isolated myocyte pre- computer and the images were analysed off-line
using the NIH Image program. For each studyparations with less than 70% rod-shaped cells wereexcluded from this study. All the procedures and 3–6 beats were analysed. Data were quantified in
a Lotus 1-2-3 spreadsheet. The systolic parametersanimal care were in accordance of institutionalguidelines. shown in Table 3 were utilized to evaluate the
aortic outflow patterns. (See Fig. 4 for typicalpulsed Doppler output.) Additionally, the aorticvelocity time integral (also termed stroke distance)is defined as the area under the flow velocitycurve. The aortic velocity time integral is theMorphometry of isolated myocytesequivalent of stroke volume except in thesestudies the aortic valve area is unknown. AverageThe cell volume was determined from>5000 myo-
cytes per sample with a Z2 COULTER particle count acceleration is calculated by dividing peak aorticflow velocity by the acceleration time (time toand size analyzer (Coulter Co., Miami, FL; 33). The
Coulter Analyzer determines the size of cells by peak flow velocity). Average velocity is the aorticvelocity time integral multiplied by the heartmeasuring the change in electric resistance across
a small aperture resulting from the displacement rate.The diastolic parameters shown in Table 3 wereof electrolyte when the cells move through the
aperture. All data are expressed as mean±.. utilized: peak early (E) flow velocity, peak atrial (A)flow velocity, E/A ratio of maximum velocities ofThe cell length and profile area of myocytes was
measured with a Jandel Video Analysis System early and atrial flow (see Fig. 4 for typical trans-mitral pulsed Doppler output). The E deceleration(Jandel Scientific, San Rafael, CA). The cell length
is defined as the longest distance between the two rate is calculated by dividing peak E velocity by theE deceleration time (Nishimura et al., 1989a,b).ends of a myocyte. The cross sectional area (CSA),
and major (A) and minor (B) diameters of the Normalized filling rate is calculated by dividing thepeak E flow velocity by the E velocity time integraltransverse section of myocytes (assuming elliptical
cross sectional shape for ventricular myocytes) were (Bowman et al., 1988). Doppler shifts in frequencyin Khz were multiplied by 7.5 to calculate velocitycalculated with the following formulae:in cm/s at 10 MHz (Taffet et al., 1998). An unpairedStudent’s t-test was used to determine the presenceof intergroup differences. For all studies, P<0.05CSA=cell volume/cell length;
A diameter=profile area/cell length; was used to determine statistical significance. Dataare expressed as mean±...B diameter=4×CSA/(p×A diameter).

Desmin Cytoskeleton in Cardiomyopathy 2067
this relationship was maintained in older animals.ResultsWhen wall thickness was measured mor-phometrically in sections from hearts fixed by per-Cardiomyocyte hypertrophy and chamber dilation in thefusion, no statistically significant increase wasabsence of desminobserved (Table 2). It seems, however, that there issome age dependent increase in both wild type andDesmin null animals were shown to have numerousnull mice (Table 2).muscular abnormalities, with cardiac muscle dem-
onstrating the most severe defects (Milner et al.,1996). As early as 1 week after birth, calcified
Morphometric characterization of desmin-null isolatedlesions and areas of fibrous replacement of muscleventricular myocytes reveals concentric cellulartissue can be seen throughout the myocardiumhypertrophy(Milner et al., 1996). While these lesions can occur
anywhere in the myocardium, they appear to beThe average cell volume of ventricular myocytesmost prominent in the right ventricle and towardisolated from 7-month-old desmin-null mice wasthe inner and outer surfaces of the myocardium.24% greater than that from wild-type controls,In addition to demonstrating these lesions, desminwhile the measured cell length and profile areanull hearts are frequently enlarged when comparedremained unchanged (see Table 3). Therefore theto hearts of normal mice (Fig. 1). Desmin null miceincrease in myocyte size was solely due to anmay exhibit a small degree of premature lethality,increase in the area of transverse section, as is seenas 6% of individuals die suddenly without warningin cells from hearts displaying concentric hyper-before reaching 1 year of age. Additionally, desmintrophy. Assuming elliptic shape for the cross sectionnull mice also demonstrate impaired exercise cap-of ventricular myocytes, the major diameters wereacity. In three independent swimming trials, 50%identical between the wild type and desmin-nullof desmin null animals were unable to completegroups, but the minor diameter increased aboutthe exercise regimen and died during swimming.30% in desmin-null cardiomyocytes.Usually, upon gross inspection, the enlargement
of desmin null hearts is not very profound [Fig.1(a)]. However, in the hearts of mutant animals
The absence of desmin results in changes in cardiacthat have died either suddenly in the absence ofgene expression associated with pressure overloadapplied stress, or as a result of stress caused byhypertrophyexercise, the enlargement may be quite severe [Fig.
1(b)]. Comparison of histological sections throughTo better define the cardiac phenotype of the desmin-hearts from those shown in Figure 1(a) revealsdeficient mice, we carried out a detailed analysis ofslight dilation of both the right and left ventricularhypertrophy-associated gene expression patterns.chambers [Fig. 2(a)]. In this case no severe changeBoth types of hypertrophy, pressure overload (PO)in the thickness of the myocardium is observed.and volume overload (VO), display distinct molecularHistological inspection also frequently reveals areasphenotypes (Calderone et al., 1995). For example,of disorganized and ragged appearing myocytes thatatrial natriuretic factor (ANF) expression is inducedare commonly seen in hypertrophic myocardiumin the ventricles of hypertrophic myocardium in both(Milner et al., 1996). When more severely enlargedPO and VO, while a-skeletal actin expression is in-hearts are examined, particularly after suddenduced only in PO hypertrophy (Calderone et al.,death, much more prominent dilation of the cham-1995). Expression of the sarcoplasmic reticulumbers can be observed, with moderate or severeCa2+ ATPase 2A (SERCA 2A) is decreased in POthinning of the myocardial wall [Fig. 2(b)]. Prom-hypertrophy, but is unchanged in VO hypertrophy.inent enlargement and dilation may be observed inPhospholamban expression is reduced in both forms.older desmin null animals as well.To investigate the behavior of these hypertrophicTo determine if cardiac hypertrophy is occurringmarkers in the myocardium of desmin null mice,in desmin null animals, hearts were excised andnorthern hybridization was carried out using RNAweighed, and heart weight/body weight ratiosisolated from the ventricles of normal and desmin(HW/BW) were calculated and compared (Table 1).null mice at 1 month and 6 months of age. At 1At 1 week of age, no significant increase in HW/month, both ANF and a-skeletal actin are clearlyBW of desmin null animals compared to those ofinduced at high levels in desmin null ventricles whenwild type controls was observed. However, by 1compared to the basal level of expression in wild typemonth of age a significant increase (>20%) of HW/
BW in desmin null animals was observed, and ventricular muscle (Fig. 3). Similar results with these

D. J. Milner et al.2068
Figure 1 Severe enlargment of the heart in desmin deficient mice. (a) Gross comparison of hearts from 5-month-oldwild-type (+/+) and desmin null mice (−/−) demonstrating mild enlargement and the presence of calcified lesionsin the desmin null heart (arrows). (b) Gross comparison of hearts from exercised wild-type (+/+) and desmin null(−/−) mice. Massive enlargement of the desmin null heart is observed. The desmin null animal died in the 6th weekof an 8-week exercise protocol.
transcripts are seen at 6 months of age as well. Age-dependent loss of cardiac performance in theabsence of desminSERCA 2A RNA shows a slight decrease in 1-month-
old desmin null myocardium when compared to wildLeft ventricular systolic function (Table 4) was ex-type, but shows a much more dramatic decrease at
6 months. Phospholamban RNA levels appear un- amined in young wild-type and desmin null mice bymonitoring left ventricular outflow. Typical aorticchanged at 1 month of age, but are obviously de-
creased in desmin null myocardium by 6 months of Doppler interogation is shown in Figure 4. Therewas no decrement in function produced by thisage (Fig. 3). Thus, the expression pattern of these
markers indicates that desmin null myocardium dis- genetic manipulation on peak outflow velocity andaverage flow velocity in the young mice. Averageplays the molecular phenotype seen in pressure over-
load hypertrophy, in agreement with the acceleration, a relatively afterload independentmeasure of function, was almost identical in bothmorphometric analysis.

Desmin Cytoskeleton in Cardiomyopathy 2069
Figure 2 Severe dilation of the chambers of desmin null hearts. (a) Histological sections of the wild-type (+/+) anddesmin null (−/−) hearts seen in Figure 1(a). Dilation of the chambers of desmin null hearts is observed, with littlechange in thickness of the ventricular walls. (b) Hearts from Figure 1(b) cut longitudinally to reveal ventricularchambers. There is severe dilation of the chambers in the desmin null (−/−) heart and thinning of the ventricularwalls, especially in the right ventricle.
Table 1 Comparison of heart weight/body weight ratios for desmin nulland control mice
Genotype 1 week 1 month 3 months 6 months
des +/+ 5.78±0.55 4.52±0.31 4.35±0.31 4.59±0.48des −/− 6.64±0.66 5.56±0.30∗∗ 5.32±0.42∗ 5.79±0.37∗∗
Compared to wild-type group: ∗ P<0.01; ∗∗ P<0.005, Student’s t-test.
young groups. None of the systolic intervals, pre- months the peak aortic flow velocity decreased 20%in the desmin null in contrast to increasing 10–15%ejection time, ejection time, acceleration time (time
to peak flow), were different between the young in the wild type mice. Average velocity and averageacceleration also followed the same pattern of beinggroups.
In contrast, the systolic function (peak outflow decreased with middle age in desmin null mice.Importantly there were no differences in heart ratevelocity, acceleration) in the 13–14-month-old de-
smin null mice was significantly less than the age- under anesthesia during the studies.At the heart rates studied, left ventricular fillingmatched controls (P<0.05). From aged 3 to 13

D. J. Milner et al.2070
Table 2 Cardiac wall thickness measurements
Young (6 months) Old (12 months)
Wild type Desmin −/− Wild type Desmin −/−
Free wall thickness 1.64±0.22 1.67±0.26∗ 1.88±0.19 1.81±0.22∗(mm)Septum thickness 1.55±0.16 1.40±0.15∗ 1.98±0.17 1.69±0.11∗(mm)
∗ P<0.05.
Table 3 Morphometric data of myocytes isolated from desmin null and wild-type mice
Groups n Cell vol. (lm3) Length (lm) CSA (lm2) A diameter (lm) B diameter (lm)
Wild type 8 34.63±3.79 142.6±3.5 244.0±26.8 29.7±2.8 10.3±1.0Desmin−/− 6 42.81±4.20∗ 139.7±10.0 309.0±47.8∗ 29.9±1.7 13.4±1.6∗
Compared to wild-type group: ∗ P<0.01, Student’s t-test.A diameter=profile area/cell length; B diameter=4×CSA/(p×A diameter).
is biphasic, an early (E) wave dependent upon 1998; Li et al., 1999). Furthermore, these resultssuggest that at least part of the pathology seen incardiac relaxation and an atrial (A) wave due to
atrial systole (Table 5). Typical transmitral Doppler humans could result as a consequence of the ab-sence of an intact, functional desmin filament net-patterns are shown in Figure 4. The ratio of E/A
peak velocities and areas revealed enhanced dia- work within myocytes.The results of our morphometric studies onstolic function in the young desmin null mouse.
This enhancement was no longer present after 13 isolated ventricular myocytes demonstrate an in-crease in cardiomyocyte cell volume in desminmonths of age. On the other hand, the atrial filling
fraction was lowest in the young desmin null mouse, null myocytes when compared to contols. Thisincrease is due to an increased cross-sectionalbut there were no differences between the groups
when assessed at 13 months of age. area, which is caused by an increase in minordiameter, or myocyte thickness. The myocyte sizeand shape changes resemble the concentric formof cardiomyocyte hypertrophy often caused byDiscussionincreased systolic wall stress, such as pressureoverload. Likewise, the expression patterns ofOur previous work (Milner et al., 1996) and the
work of an independent group (Li et al., 1996; the hypertrophic markers ANF, a-skeletal actin,phospholamban and SERCA 2A observed by north-Thornell et al., 1997) has demonstrated that the
absence of desmin in the mouse results in a patho- ern analysis of ventricular RNA from desmin nullanimals also resemble those seen in the concentriclogical entity consistent with cardiomyopathy, char-
acterized by degeneration of the myocardium and form of cardiac hypertrophy. Downregulation ofphospholamban and SERCA 2A is also believedultrastructural abnormalities. The aim of the pres-
ent work was to investigate the consequences of the to accompany the development of heart failure.One explanation for the changes in cell size andcardiomyocyte structural abnormalities on cardiac
performance. We demonstrate that mice lacking shape of ventricular myocytes in desmin-null micecould be that lack of desmin filaments shifts thedesmin develop cardiomyocyte hypertrophy which
later leads to chamber dilatation with compromised share of tension normally undertaken by desminfilaments to myofibrils. Thus, cardiac myocytessystolic function and heart failure. This supports
the prediction that mutations in the desmin gene may need more myofibrils to handle even normalmechanical stress when there is a lack of desminwill give rise to cardiac dysfunction in humans.
Indeed, there are now human mutations in the filaments, and thus respond to this need byhypertrophic growth. Another possible explanationdesmin gene associated with cardiomyopathy that
were reported during the preparation of this manu- is that degenerating myocytes release growthfactors that stimulate hypertrophic growth of thescript (Goldfarb et al., 1998; Munoz-Marmol et al.,

Desmin Cytoskeleton in Cardiomyopathy 2071
in desmin null myocardium, however the com-pensatory mechanisms appear to be successful inearly life.
While desmin null hearts are hypertrophic asjudged by heart weight/body weight measurementsand increased myocyte volume and CSA, as wellas induction of embryonic gene expression, grossexamination of desmin null hearts from non-ex-ercised animals generally does not show a largeincrease in heart size, and no increase in ventricularwall thickness. This can be explained by myocyteloss due to cell death and compensatory hyper-trophy of remaining myocytes. Histological analysisof desmin null myocardium reveals the presence ofcalcified lesions and areas of fibrous replacementthat result from myocyte death (Milner et al., 1996).Myocyte number can be estimated by dividing thetotal volume of myocytes (calculated from heartweight, myocyte volume percent and specificgravity) by mean myocyte volume (Campbelland Gerdes, 1988). Compared to wild-type mice(5.105±>0.629 mg/mm3, n=6), the heart weight/myocyte volume ratio is less in desmin-null mice(3.886±0.664 mg/mm3, n=8). Myocyte volumepercent is likely to be reduced based on histologicalreports (Milner et al., 1996). Consequently, heartsfrom desmin-null mice may have a reduction in
Figure 3 Expression of hypertrophic markers in the myocyte number. The relative preservation of sys-heart of desmin null mice. Northern analysis of cardiac tolic function until late in life is, in part, evidencegene expression in the ventricles of wild-type (+/+) and of the efficacy of this compensatory hypertrophy.desmin null (−/−) mice at 1 month and 6 months of
Currently, we have a very limited understandingage demonstrating induction of atrial natriuretic factorof how the physical stimulus of mechanical load is(ANF) and a-skeletal actin (a-Sk act) and decrease in
phospholamban (PLB) and sarcoplasmic reticulum cal- converted into intracellular signals that alter genecium pump isoform 2A (SERCA 2A); (18S) 18S RNA. expression and eventually lead to hypertrophy
(MacLellan et al., 1997; Sadoshima and Izumo,1997). In addition to secretion of a number ofgrowth factors, increase of Ca2+ levels (Sadoshimaconcentric form, such as bFGF, TGFb, endothelin-and Izumo, 1997) and induction of cardiac gene1 and angiotensin II. Currently, we do not have
any data on the status of growth factor expression expression by the calcineurin activated NF-AT3/
Table 4 Doppler outflow measurements
3 months 13–14 months
WT Des−/− WT Des−/−n=5 n=7 n=7 n=9
Peak aortic velocity (cm/s) 79.8±4.4 89.5±2.9 91.8±5.8 73.1±4.6∗Velocity time integral (cm) 3.8±0.2 4.7±0.6 5.2±0.3 4.3±0.2∗Ejection time (ms) 76±3 77±3 88±3 86±2Acceleration time (ms) 16±1 18±2 20±1 23±2Average acceleration (cm/ 5195±361 5028±267 4725±296 3647±541∗
s2)Stroke integral (cm/min) 1285±82 1327±97 1541±115 1206±114Heart rate (/min) 264±20 262±17 283±6 266±22
∗ P<0.05.
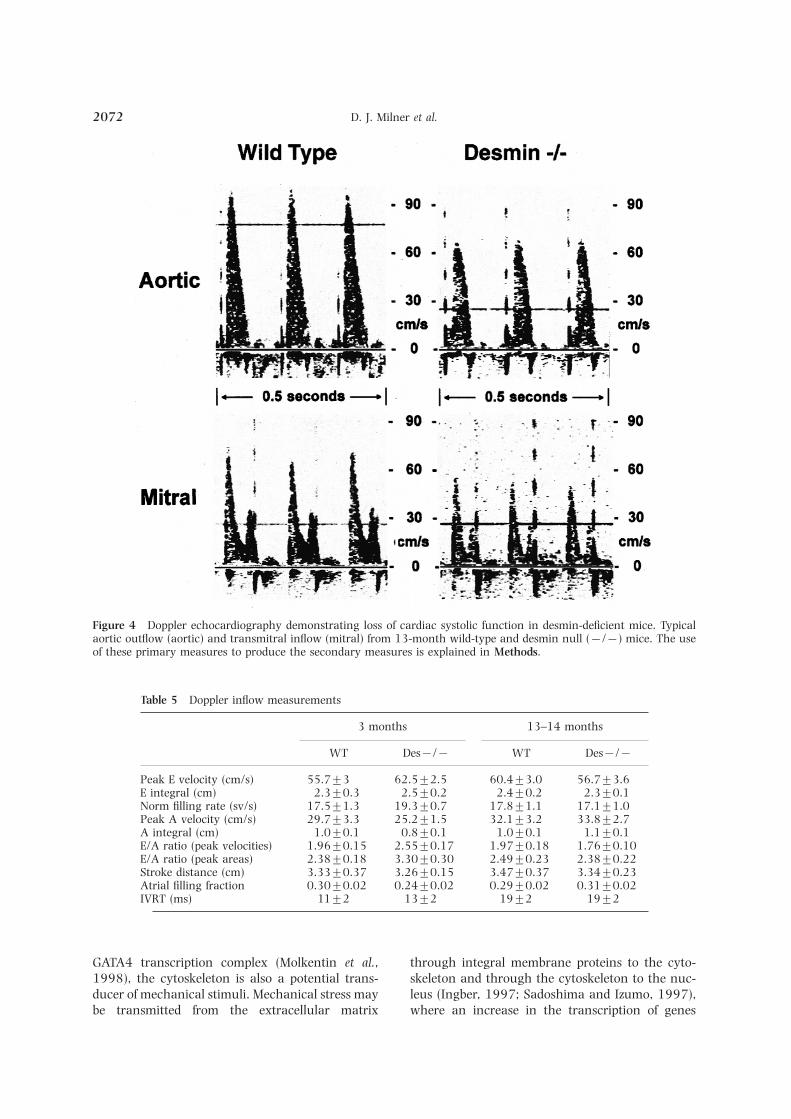
D. J. Milner et al.2072
Figure 4 Doppler echocardiography demonstrating loss of cardiac systolic function in desmin-deficient mice. Typicalaortic outflow (aortic) and transmitral inflow (mitral) from 13-month wild-type and desmin null (−/−) mice. The useof these primary measures to produce the secondary measures is explained in Methods.
Table 5 Doppler inflow measurements
3 months 13–14 months
WT Des−/− WT Des−/−
Peak E velocity (cm/s) 55.7±3 62.5±2.5 60.4±3.0 56.7±3.6E integral (cm) 2.3±0.3 2.5±0.2 2.4±0.2 2.3±0.1Norm filling rate (sv/s) 17.5±1.3 19.3±0.7 17.8±1.1 17.1±1.0Peak A velocity (cm/s) 29.7±3.3 25.2±1.5 32.1±3.2 33.8±2.7A integral (cm) 1.0±0.1 0.8±0.1 1.0±0.1 1.1±0.1E/A ratio (peak velocities) 1.96±0.15 2.55±0.17 1.97±0.18 1.76±0.10E/A ratio (peak areas) 2.38±0.18 3.30±0.30 2.49±0.23 2.38±0.22Stroke distance (cm) 3.33±0.37 3.26±0.15 3.47±0.37 3.34±0.23Atrial filling fraction 0.30±0.02 0.24±0.02 0.29±0.02 0.31±0.02IVRT (ms) 11±2 13±2 19±2 19±2
GATA4 transcription complex (Molkentin et al., through integral membrane proteins to the cyto-skeleton and through the cytoskeleton to the nuc-1998), the cytoskeleton is also a potential trans-
ducer of mechanical stimuli. Mechanical stress may leus (Ingber, 1997; Sadoshima and Izumo, 1997),where an increase in the transcription of genesbe transmitted from the extracellular matrix

Desmin Cytoskeleton in Cardiomyopathy 2073
coding for metabolic and contractile proteins takes function (linkage of the sarcomere to the sar-colemma, force transduction from the sarcomere toplace. The desmin cytoskeletal network is an at-
tractive potential transducer of mechanical stress, the extracellular matrix, mechanochemical sig-naling, etc). Similarly, knockout of the muscle LIMas it links the contractile apparatus to the sar-
colemma, the nucleus and other organelles (Lockard protein (MLP), a protein involved in linking thecardiomyocyte actin cytoskeleton to the contractileand Bloom, 1993; Bloom et al., 1996; for review,
see Capetanaki and Milner, 1998). Indeed, in vitro apparatus at the Z-disc, also leads to disruption ofcardiomyocyte architecture and DCM in micestudies have shown that the absence of desmin can
influence nuclear events (Li et al., 1994; Weitzer et (Arber et al., 1997). That genetic alterations ofdesmin and MLP can also lead to a DCM-like pheno-al., 1995; Capetanaki et al., 1997). While it is
obvious that desmin is not absolutely required for type in mice provides support for a general schemewhere genetic alterations in sarcomeric proteins,hypertrophy to take place, as desmin null hearts
display hypertrophy, desmin may play an important responsible for force generation, will lead to HCM,while those of cytoskeletal proteins, mainly involvedrole in the maintenance of the response to hy-
pertrophic stimuli, which seems to be compromised in force transmission, to DCM. This is supported,though indirectly, by the recent identification ofin its absence. This could explain the dominance
of the dilated appearance vs the hypertrophic ap- mutations in the cardiac actin gene of humanswith DCM (Olson et al., 1998). Although this is apearance, and eventual heart failure in desmin
null animals after exercise overload. Furthermore sarcomeric protein, the fact that these mutationsare in the domains of actin that attach to the Z-it suggests that the hypertrophy to dilatation switch
in many human cases might be indeed due to discs and intercalated discs also suggests that forcetransmission and normal signaling through thecompromised growth response when any part of
the cytoskeleton is affected. It will be interesting to cytoskeleton must be compromised, leading to celldeath, dilation and heart failure similar to what issee if desmin null hearts can mount the proper
hypertrophic resonse to pressure and volume over- seen in the most severely affected desmin nullhearts. Further support is also provided by studiesload stimuli in vivo, and if desmin null car-
diomyocytes can display proper hypertrophic where overexpression of the actin-capping proteintropomodulin leads to dilation (Sussman et al.,growth in response to mechanical stretch or treat-
ment with hypertrophic growth factors in vitro. 1998a,b).The systolic function studies demonstrated thatGross and histological observation of desmin null
hearts from animals which have died suddenly, the peak aortic flow velocity and average aorticacceleration are depressed in mid-age desmin nulleither in the absence of applied stress or after
exercise, demonstrate chamber dilation and thin- mice. This may reflect the decrease in total myocytemass and the limitations of the hypertrophic re-ning of the myocardial wall. Our Doppler meas-
urements also demonstrate an age-dependent sponse. One must consider that the animals thatsurvive to mid-age and tolerate the anesthesia fordecrease in systolic function in desmin null animals;
however, the extent of this decline is comparatively the Doppler study may be the least affected by thegenetic manipulation. Although peak aortic flowmild. Systolic dysfunction is a characteristic of DCM
(Towbin, 1998, 1993). These observations suggest velocity and average aortic acceleration are clearlydependent upon systolic function of the ventricle,that the cardiomyopathy seen in desmin null mice
resembles DCM. This is consistent with the recent they are also modified by cardiac afterload; that is,a ventricle with compromised function can ejectidentification of a missense mutation at the carboxy
terminus of desmin in humans suffering from idio- blood into a low resistance, high compliance aorta.Similarly, they may be modified by anestheticpathic DCM (Li et al., 1999). As abnormalities of
dystrophin have been shown to lead to DCM in agents. The desmin null mouse aorta, characterizedby low pulse wave velocity, a marker for vesselhumans, it is not surprising to note that desmin
and dystrophin proteins have similar intracellular wall stiffness (Taffet and Capetanaki, unpublishedresults), may be just such a compliant vessel. Thedistribution, especially in cardiac muscle. Both de-
smin and dystrophin are found at the sarcolemma, preserved peak aortic flow velocity seen in theyoung desmin null mouse is perhaps a reflection ofwith desmin primarily present at the costamere
(Lazarides and Capetanaki, 1986). Likewise, a frac- the reduced afterload rather than an increase incontractile function.tion of cardiac dystrophin can be localized to the
Z-disc, where desmin is prominently localized (Meng Diastolic function studies show no difference inpeak E filling between the groups, but left vent-et al., 1996). Thus it is possible that desmin and
dystrophin may have at least partially overlapping ricular wall thickness, stiffness or regional hetero-

D. J. Milner et al.2074
geneity in function among the left ventricular seg- Acknowledgementsments can also alter peak E height (Nishinura etal., 1989a,b). The functional findings noted above We thank Dr Mark Entman, Dr Robert Roberts,
Dr Michael Schneider and Dr Jeffrey Towbin forare consistent with the lack of modification of LVwall thickness at the earlier time points. Alterations valuable discussions and comments. This work was
supported by NIH grant AR 39617-09, and AHAin active cardiac relaxation impact early diastolicparameters most directly (Nishimura et al., 1989a, Texas affiliate MDA grant to YC and by AG13251
to GET.b; Sys and Brutsaert, 1995). The peak E is in partdependent on sarcoplasmic reticulum functions; forexample, thyroxine treated mice have augmentedsarcoplasmic reticulum calcium uptake and a sig- Referencesnificant increase in peak E velocity (Taffet et al.,1998). Other factors, including modification of dia- A S, H JJ, R J J, H M, S G, Bstolic filling pressures, may have influenced diastolic J, P JC, C KR, C P, 1997. MLP-
deficient mice exhibit a disruption of cardiac cy-filling at the 13-month stage.toarchitectural organization, dilated cardiomyopathy,This is the first demonstration of cardiac hyper-and heart failure. Cell 88: 393–403.trophy and dilation with compromised systolic
A A, C J, F-F MT, L MD,function due to removal of an intermediate M JL, G O, F-V A, N-filament protein. There are several reported my- -P JJ, 1995. Desmin myopathy: a mul-
tisystem disorder involving skeletal, cardiac, andopathies and cardiomyopathies with abnormalsmooth muscle. Hum Pathol 26: 1032–1037.accumulations of granular and filamentous ag-
B RD, M M, R SL, H E, Bgregates of desmin (for review see Goebel, 1995).T, Y JB, C KP, 1997. A 5′ dystrophin
There is no case, however, where desmin is duplication mutation causes membrane deficiency ofcompletely absent. Similarly, there is no evidence, a-dystroglycan in a family with X-linked cardio-
myopathy. J Mol Cell Cardiol 29: 3175–3188.so far, demonstrating that these abnormalities inB S, L VG, B M, 1996. Intermediatedesmin distribution have a causal effect on the
filament-mediated stretch-induced changes in chro-disease. The defects developed in all three musclematin: a hypothesis for growth initiation in cardiac
types of the desmin null mice are reminiscent of myocytes. J Mol Cell Cardiol 28: 2123–2127.those reported in some familial desminopathies B LK, L FA, J CC, M J, W FJT,
Z BL, 1988. Peak filling rate normalized to mitral(Vajsar et al., 1993; Ariza et al., 1995; Goebel etstroke volume: a new Doppler echocardiographic fillingal., 1995). These include dilation of the ventricularindex validated by radionuclide angiographic tech-chambers, hypoplasia of the aorta, thinned in-niques. J Am Coll Cardiol 12: 937–943.
testinal walls and distended and fragmented Z- C A, T N, I NJ J, T CM,lines (Ariza et al., 1995). The data obtained from C WS, 1995. Pressure- and volume-induced left
ventricular hypertrophies are associated with distinctdesmin null mice support the idea that themyocyte phenotypes and differential induction of pep-observed abnormalities in desmin distribution intide growth factor mRNAs. Circulation 92: 2385–several reported myopathies might have a causal2390.
effect. This hypothesis is strongly favored by C SE, G AM, 1988. Regional changes inrecently identified mutations in the desmin gene myocyte size during reversal of thyroid-induced cardiac
hypertrophy. J Mol Cell Cardiol 20: 379–387.in members of families suffering from desmin-C Y, M DJ, 1998. Desmin cytoskeletonrelated generalized myopathy (Goldfarb et al.
im muscle integrity and function. Subcell Biochem 31:1998; Munoz-Marmol et al., 1998) and DCM (Li463–495.
et al., 1999), respectively. Furthermore, they C Y, N J, F CN, L E, 1983.suggest that the myopathy might be the con- Tissue-specific expression of two mRNA species tran-
scribed from a single vimentin gene. Cell 35: 411–420.sequence of the absence of functional desminC Y, N J, L E, 1984. Charac-filaments, as in a null background, rather than
terization and regulation in the expression of a genearising from toxic effects of aggregates. Con-encoding for the intermediate filament protein desmin.
sequently, it could be suggested that the desmin Proc Natl Acad Sci USA 8: 6909–6912.null mouse could provide a useful animal model C Y, M DJ, W G, 1997. Desmin in
muscle formation and maintenance: knockouts andfor those human cardiomyopathies with age-consequences. Cell Struc Func 22: 103–116.dependent and exercise overload-dependent trans-
F̈ DO, O M, W K, 1989. Myogenesis inition from compensatory cardiac hypertrophy tothe mouse embryo: differential onset of expression of
non-compensated state, despite the fact that it is myogenic proteins and the involvement of titin innot a complete replica of human disease in which myofibril assembly. J Cell Biol 109: 517–527.
G JR, L W, P S, G I, K HW, Fdesmin is present but not functional.

Desmin Cytoskeleton in Cardiomyopathy 2075
DG, K V, N JC, D T, K cardial growth factors. In: Marks AR, Taubman MBEG, 1992. Mouse phospholamban gene expression dur- (eds). Molecular Biology of Cardiovascular Diseases. Newing development in vivo and in vitro. Circ Res 71: York: Marcel Dekker, Inc., 327–377.1021–1030. M M, H E, L B, V S, B RD, 1997.
G-L AAT, C M, C DA, I- Dilated cardiomyopathy associated with deficiency of JS, S FJ, S CE, S JG, 1996. A the cytoskeletal protein metavinculin. Circulation 95:mouse model of familial hypertrophic cardiomyopathy. 17–20.Science 272: 731–734. M AJ, R R, 1998. Molecular genetic basis
G AM, 1997. A reliable, efficient, and com- of hypertrophic cardiomyopathy: genetic markers forprehensive approach to assess myocyte remodeling in sudden cardiac death. J Cardiovasc Electrophys 9: 88–96.cardiac hypertrophy and failure. J Cardiac Failure 3: M BJ, 1997. Hypertrophic cardiomyopathy. Lancet63–68. 350: 127–133.
G HH, 1995. Desmin-related neuromuscular dis- M H, L JJ, F J, H P, T BS, 1996.orders. Muscle Nerve 18: 1306–1320. The association of cardiac dystrophin with myofibrils/
G LG, P KY, C L, G S, Z-disc regions in cardiac muscle suggests a novel roleL HS, V O, N JW, S-M C, in the contractile apparatus. J Biol Chem 271: 12364–S K, D MC, 1998. Missense mutations 12371.in desmin associated with familial cardiac and skeletal M DJ, W G, T D, B A, Cmyopathy. Nat Genet 19: 402–403. Y, 1996. Disruption of muscle architecture and myo-
G-H AM, G J, S GE, 1988. A novel cardial degeneration in mice lacking desmin. J Cell BiolCa2+ pump expressed in brain, kidney, and stomach is 134: 1255–1270.encoded by an alternative transcript of the slow-twitch M JD, L JR, A CL, M B, Rmuscle sarcoplasmic reticulum Ca-ATPase gene. Iden- J, R J, G SR, O EN, 1998. A calcineurin-tification of cDNAs encoding Ca2+ and other cation- dependent transcriptional pathway for cardiac hyper-transporting ATPases using an oligonucleotide probe trophy. Cell 93: 215–228.derived from the ATP-binding site. J Biol Chem 263: M-M AM, S G, I M, C15032–15040. PA, Y Y, R X, V E, M JL, C J, F-
H H, F B, F WW, 1989. Expression -F MT, N-P JJ, A A,of intermediate filament proteins during development F E, 1998. A dysfunctional desmin mutation in aof Xenopus laevis, II. Identification and molecular patient with severe generalized myopathy. Proc Natlcharacterization of desmin. Development 105: 299– Acad Sci USA 95: 11312–11317.307. N V, O Y, B A, P G, M Y,
I DE, 1997. Tensegrity: the architectural basis of P L, N G, V C, A C,cellular mechanotransduction. Ann Rev Physiol 59: M AM, 1997. Identification of the syrian ham-575–599. ster cardiomyopthy gene. J Hum Mol Genet 6: 601–607.
K MT, S MC, 1996. Molecular genetic N RA, H PR, H LK, T AJ,insights into cardiovascular disease. Science 272: 681– 1989a. Assessment of diastolic function of the heart:685. background and current applications of Doppler echo-
L E, 1982. Intermediate filaments: a chemically cardiography. Part I. Physiologic and pathophysiologicheterogeneous, developmentally regulated class of pro- features. Mayo Clin Proc 64: 71–81.teins. Ann Rev Biochem 51: 219–250. N RA, H PR, H LK, T AJ,
L E, C Y, 1986. The striated muscle 1989b. Assessment of diastolic function of the heart:cytoskeleton: expression and assembly in development. background and current applications of Doppler echo-In Molecular Biology of Muscle Development, 749–772. cardiography. Part II. Clinical studies. Mayo Clin Proc
L D, T T, G O, B PE, Q M, 64: 181–204.Z WA, H R, B LL, M DL, R O TM, M VV, T NS, T YS, KR, 1999. A desmin mutation responsible for idiopathic MT, 1998. Actin mutations in dilated cardiomyopathy,dilated cardiomyopathy. Circulation 100: 461–464. a heritable form of the heart failure. Science 280:
L F, W X, C JM, G AM, 1996. Rapid 750–752.transition of cardiac myocytes from hyperplasia to S J, I S, 1997. The cellular and molecularhypertrophy during postnatal development. J Mol Cell response of cardiac myocytes to mechanical stress. AnnCardiol 28: 1737–1746. Rev Physiol 59: 551–571.
L H, C SK, M DJ, M MI, K IR, S G, V C, L W, R F,C Y, 1994. Inhibition of desmin expression 1989. Desmin and titin expression in early post-im-blocks myoblast fusion and interferes with the my- plantation mouse embryos. Development 107: 581–ogenic regulators MyoD and myogenin. J Cell Biol 124: 616.827–841. S CE, B KD, K KA, S JA, S
L Z, C-G E, P-R M, M JG, 1984. Nucleotide sequences of the human andM, P S, P D, B C, 1996. Cardio- mouse atrial natriuretic factor genes. Science 226:
1206–1209.vascular lesions and skeletal myopathy in mice lackingdesmin. Dev Biol 175: 362–366. S MA, W S, C N, K R, H
TE, P R, W SA, K TR, 1998a. MyofibrilL VG, B S, 1993. Trans-cellular desmin-laminB intermediate filament network in cardiac myocytes. J degeneration caused by tropomodulin overexpression
leads to dilated cardiomyopathy in juvenile mice. J ClinMol Cell Cardiol 25: 303–309.ML WR, H J, S MD, 1997. Myo- Invest 101: 51–61.

D. J. Milner et al.2076
S MA, B S, U C-S, D MP, P velopmental Mechanics of Heart Disease. New York: Fu-tura Publishing Co Inc, 121–132.B, S D, T L, K L, 1998b. Altered
expression of tropomodulin in cardiomyocytes disrupts T JA, 1998. The role of cytoskeletal proteins incardiomyopathies. Curr Opin Cell Biol 10: 131–139.the sarcomeric structure of myofibrils. Circ Res 82:
94–103. V J, B LE, F RM, M EG, 1993.Familial desminopathy: myopathy with accumulationS SU, B DL, 1995. Diagnostic significance
of impaired LV systolic relaxation in heart failure. of desmin-type intermediate filaments. J Neurol Neuro-surg Psych 56: 644–648.Circulation 92: 3377–3380.
T GE, H CJ, W X, P T, M HL, V KL, L LA, 1996. Contractile proteinmutations and heart disease. Curr Opin Cell Biol 8:E ML, 1998. Noninvasive indexes of cardiac
systolic and diastolic function in hyperthyroid and 97–105.W G, M DJ, K J-U, B A, Csenescent mouse. Am J Physiol 271: M68–M72.
T L-E, C L, L Z, M M, P Y, 1995. Cytoskeletal control of myogenesis: a desminnull mutation blocks the myogenic pathway duringD, 1997. Null mutation in the desmin gene gives rise
to a cardiomyopathy. J Mol Cell Cardiol 29: 2107–2124. embryonic stem cell differentiation. Dev Biol 17: 422–439.T JA, 1993. Molecular genetic aspects of cardio-
myopathy. Biochem Med Metabol Biol 49: 285–320. W BM, S RJ, 1996. Method for isolation ofadult mouse cardiac myocytes for study of contractionT JA, 1995. Biochemical and molecular charac-
terization of X-linked dilated cardiomyopathy (XLCM). and microfluorometry. Am J Physiol 271: H1250–H1255.In: Clark EB, Markwald RR, Takao A (eds). De-
Copyright © 2022 FDOKUMEN




















