
Aus der Medizinischen Universitätsklinik
155
Aus der Medizinischen Universitätsklinik - Abteilung Innere Medizin I (Schwerpunkt Hämatologie und Onkologie) - der Albert-Ludwigs-Universität Freiburg i. Br. IDENTIFIZIERUNG UND FUNKTIONELLE UNTERSUCHUNG VON ZIELGENEN DES LEUKÄMIE-SPEZIFISCHEN TRANSKRIPTIONSFAKTORS AML1/ETO INAUGURAL-DISSERTATION zur Erlangung des Medizinischen Doktorgrades der Medizinischen Fakultät der Albert-Ludwigs-Universität Freiburg Vorgelegt 2010 von Jesús Duque Afonso geboren in Santa Cruz de Tenerife, Spanien
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Aus der Medizinischen Universitätsklinik

Aus der Medizinischen Universitätsklinik
- Abteilung Innere Medizin I (Schwerpunkt Hämatologie und Onkologie) -
der Albert-Ludwigs-Universität Freiburg i. Br.
IDENTIFIZIERUNG UND FUNKTIONELLE UNTERSUCHUNG
VON ZIELGENEN DES LEUKÄMIE-SPEZIFISCHEN
TRANSKRIPTIONSFAKTORS AML1/ETO
INAUGURAL-DISSERTATION
zur
Erlangung des Medizinischen Doktorgrades
der Medizinischen Fakultät
der Albert-Ludwigs-Universität Freiburg
Vorgelegt 2010
von Jesús Duque Afonso
geboren in Santa Cruz de Tenerife, Spanien

Dekan: Prof. Dr. Dr. h.c. mult. Hubert E. Blum
1. Gutachter: Prof. Dr. Michael Lübbert
2. Gutachter: Fr. Prof. Dr. Charlotte Niemeyer
Jahr der Promotion: 2011

Diese Doktorarbeit wurde von September 2004 bis Juli 2007 unter der Betreuung von Prof.
Dr. M. Lübbert in der Abteilung I der Inneren Medizin der Universitätsklinik Freiburg
angefertigt.
Teile dieser Doktorarbeit wurden in folgende Publikationen aufgenommen:
Duque-Afonso J., Yalcin A. Berg T., Abdelkarim M., Heidenreich O. and Lübbert M.. The HDAC class I specific inhibitor Entinostat (MS-275) effectively relieves epigenetic silencing of the LAT2 gene mediated by AML1/ETO. (Oncogene, Januar 2011, angenommen) Duque-Afonso J., Solari L., Essig A. Berg T., Pahl H. L. and Lübbert M. Regulation of the adaptor molecule LAT2, an in vivo target gene of AML1/ETO, during myeloid differentiation (British Journal of Haematology, Dezember 2010, angenommen) Müller AM., Duque J., Shizuru J.A. and Lübbert M.. Complementing Mutations in Core Binding Factor Leukemias: From Mouse Models to Clinical Therapeutic Implications. Oncogene 2008; 27(44): 5759-73 Claus R., Fliegauf M., Stock M., Duque J., Kolanczyk M. and Lübbert M. Inhibitors of DNA methylation and histone deacetylation independently relieve AML1/ETO-mediated lysozyme repression. J. Leuk. Biol. 2006; 80: 1462-72

Inhaltsverzeichnis
Abkürzungverzeichnis
Abbildungsverzeichnis
Tabellenverzeichnis
1.- Einleitung ......................................................................................................................................................... 1
1.1.- Akute Myeloische Leukämie ................................................................................................................. 1
1.2.- Pathogenese akuter myeloischer Leukämien. ....................................................................................... 2
1.2.1.- Die „Two-hit“-Hypothese............................................................................................................ 2
1.2.2.- Klinische Daten bestätigen die „Two-hit“-Hypothese .............................................................. 3
1.2.3.- Bestätigung der „Two-hit“ -Hypothese in Maus-Modellen...................................................... 3
1.3.- Das chimäre Fusionsprotein AML1/ETO ............................................................................................. 4
1.3.1.- Das AML1/RUNX1-Gen und der CBF-Transkriptionfaktorkomplex.................................... 5
1.3.2.- Das ETO-Gen............................................................................................................................... 6
1.3.3.- Das AML1/ETO-Fusionsgen ...................................................................................................... 7
1.3.4.- AML1- und AML1/ETO-Zielgene ............................................................................................. 8
1.4.- Epigenetische Aktivität von chimärer Fusionsproteinen in AML ...................................................... 9
1.4.1.- Die epigenetische Regulation der Gentranskription................................................................. 9
1.4.1.1.- DNA-Methylierung .............................................................................................................. 10
1.4.1.2- Histonmodifikation ............................................................................................................... 12
1.4.1.3.- Die Kooperation von DNA-Methylierung und Histonmodifikation in der Regulation der
Gentranskription................................................................................................................................ 12
1.4.1.4.- Die pharmakologische Beeinflussbarkeit der epigenetischen Genregulation:
demethylierende Substanzen und HDAC-Inhibitoren.................................................................... 13
1.4.2.- Das PML/RARα-Fusionsprotein .............................................................................................. 14
1.4.2.1.- PML/RARα: ein Modell der Kooperation von DNA-Methyltransferase und
Histondeacetylasen bei der Entstehung der AML........................................................................... 14
1.4.2.2.- RARβ2: Tumorsuppressor-Gen und Zielgen des PML/RARα Fusionsproteins. ........... 15
1.4.3.- Epigenetische Ereignisse in der Pathogenese des AML1/ETO-Proteins: therapeutische
Konsequenzen. ...................................................................................................................................... 15
1.5.- Die U937-9/14/18-Zelllinie .................................................................................................................... 16
1.6.- Methoden zur Analyse der DNA-Methylierung ................................................................................. 18
1.6.1.- Restriction Landmark Genomic Scanning (RLGS)............................................................... 18
1.6.2.- Bisulfit-Sequenzierung .............................................................................................................. 20
1.7.- LAT2-Gen als neues Zielgen von AML1/ETO ................................................................................... 20
1.8.- Myeloische Differenzierung.................................................................................................................. 23
2.- Ziele des Projektes ......................................................................................................................................... 25
3.- Materialien und Methoden ........................................................................................................................... 26
3.1.- Materialien............................................................................................................................................. 26
3.1.1.- Zellen .......................................................................................................................................... 26
3.1.1.1.- Zelllinien ............................................................................................................................... 26

3.1.1.2.- Bakterien............................................................................................................................... 27
3.1.1.3.- Primäre Zellen von gesunden Spendern ............................................................................ 28
3.1.1.4.- Primäre Zellen von Patienten ............................................................................................. 28
3.1.2.- Materialien für Zellkultur ........................................................................................................ 29
3.1.2.1.-Zellkulturmedien, Zusätze und Reagenzien ....................................................................... 29
3.1.2.2.- Sonstige Materialien, Plastikwaren .................................................................................... 29
3.1.3.- Materialien für die Bakterienkultur ........................................................................................ 30
3.1.4.- Materialien für molekularbiologische Arbeiten...................................................................... 30
3.1.4.1.- Chemikalien und Reagenzien.............................................................................................. 30
3.1.4.2.- Standardlösungen und Puffer ............................................................................................. 33
3.1.4.3.- Radiochemikalien................................................................................................................. 35
3.1.4.4.- Enzyme.................................................................................................................................. 35
3.1.4.5.- Nukleinsäuren ...................................................................................................................... 35
3.1.4.6.- Antikörper ............................................................................................................................ 37
3.1.4.7.- Kits ........................................................................................................................................ 38
3.1.4.8.- Sonstige Materialien ............................................................................................................ 39
3.1.5.- Geräte ......................................................................................................................................... 40
3.1.6.- Bioinformatische Software........................................................................................................ 41
3.2.- Methoden ............................................................................................................................................... 43
3.2.1.- Aufarbeitung des primären Materials. .................................................................................... 43
3.2.1.1.- Isolierung der peripheren Blutzellen.................................................................................. 43
3.2.1.2.- Lymphozyten-Isolierung ..................................................................................................... 43
3.2.1.3.- Monozyten-Isolierung.......................................................................................................... 44
3.2.1.4.- Granulozyten-Isolierung ..................................................................................................... 44
3.2.1.5.- Patientenproben ................................................................................................................... 44
3.2.1.6.- Isolierung von CD34+ Zellen .............................................................................................. 45
3.2.1.7.- Isolierung von Alveolar-Makrophagen. ............................................................................. 45
3.2.2. Zellkultur..................................................................................................................................... 45
3.2.2.1.- Kultivierung der Zellen ....................................................................................................... 46
3.2.2.2.- Einfrieren und Auftauen der Zellen................................................................................... 46
3.2.2.3.- Zellzahl- und Viabilitäts-Bestimmung durch Trypan Blau Färbung..............................47
3.2.2.4.- Zellzahl- und Viabilitäts-Bestimmung durch Färbung mit Türklösung......................... 47
3.2.2.5.- Abtrennung toter Zellen und Zelltrimmern mittels Ficollgradientenzentrifugation..... 48
3.2.2.6.- Beurteilung der Morphologie durch Zytospin und Färbung ........................................... 48
3.2.2.6.2- May-Grünwald Giemsa Färbung (Pappenheim Färbung)........................................... 48
3.2.2.6.3.- Benzidin Färbung........................................................................................................... 48
3.2.3.- Behandlung der Zellen .............................................................................................................. 49
3.2.3.1.- Induktion der Expression des AML1/ETO-Proteins in der 9/14/18-Zelllinie................. 49
3.2.3.2.- Myeloische Differenzierung der HL60, U937, NB4 und K562 Zelllinien. ....................... 49
3.2.3.3.- Ex vivo myeloische Differenzierung der CD34+ Zellen. ................................................... 50
3.2.3.4.- Behandlung der Kasumi-1 Zellinie mit epigenetisch aktiven Substanzen. ..................... 50
3.2.4.- Isolierung der Nukleinsäuren ................................................................................................... 51

3.2.4.1.- DNA-Isolierung mittels Phenol-Chloroform Extraktion. ................................................. 51
3.2.4.2.- DNA-Isolierung mittels Qiagen Kit .................................................................................... 52
3.2.4.3.- RNA-Isolierung mittels Guanidiniumthiocyanat. ............................................................. 52
3.2.4.4.- RNA-Isolierung mittels Qiagen Kit .................................................................................... 53
3.2.4.5.- Photometrische Bestimmung der DNA- und RNA-Konzentration.................................. 53
3.2.5.- cDNA Synthese........................................................................................................................... 53
3.2.5.1.- DNAse Verdau...................................................................................................................... 53
3.2.5.2.- Reverse Transkription – Polymerasekettenreaktion (RT-PCR)...................................... 54
3.2.6.- Protein-Isolierung...................................................................................................................... 55
3.2.6.1.- Isolierung von Membranproteinen..................................................................................... 55
3.2.6.2.- Isolierung von nukleären und zytoplasmatischen Proteinen............................................ 55
3.2.6.3.- Proteinquantifizierung mittels Bradford Assay ................................................................ 56
3.2.7.- Sonden-Herstellung für Southern Blot .................................................................................... 56
3.2.7.1.- Entwicklung der Primer...................................................................................................... 56
3.2.7.2.- Amplifikation der Sonde durch die Polymerasekettenreaktion (PCR)........................... 56
3.2.7.3.- Auftrennung von DNA-Fragmenten durch Gelelektrophorese ....................................... 57
3.2.7.4.- Isolierung von DNA-Fragmenten aus Agarose-Gelen ...................................................... 57
3.2.7.5.- Klonierung des PCR Produkts............................................................................................ 57
3.2.7.6.- Transformation von kompetenten Bakterien.................................................................... 58
3.2.7.7.- Herstellung von Bakterien-Stocks ...................................................................................... 58
3.2.7.8.- Mini-Präparation von Plasmid-DNA ................................................................................. 58
3.2.7.9.- Maxi-Präparation von Plasmid-DNA................................................................................. 59
3.2.7.10.- Extraktion der Sonde aus dem Plasmid. .......................................................................... 59
3.2.8.- Sonden-Herstellung für Northern Blot .................................................................................... 59
3.2.9.- Restriktionsverdau der genomischen DNA ............................................................................. 60
3.2.10.- Southern Blot ........................................................................................................................... 60
3.2.10.1.- Vorbereitung und Auftrennung der genomischen DNA................................................. 60
3.2.10.2.- Transfer der DNA nach der Southern Blot Methode ..................................................... 60
3.2.11.- Northern Blot ........................................................................................................................... 61
3.2.11.1.- Vorbereitung und Auftrennung der RNA ....................................................................... 61
3.2.11.2.- Transfer der RNA nach der Northern Blot Methode ..................................................... 61
3.2.12.- Hybridisierung der Membranen von Southern und Northern Blot.................................... 62
3.2.12.1.- Radioaktive Markierung von DNA-Fragmenten ............................................................ 62
3.2.12.2.- Hybridisierung mit radioaktiv markierten DNA-Sonden .............................................. 62
3.2.13.- Western Blot ............................................................................................................................ 63
3.2.13.1.- Vorbereitung der Proben .................................................................................................. 63
3.2.13.2.- Elektrophoretische Auftrennung im Gel ......................................................................... 63
3.2.13.3.- Blotting................................................................................................................................ 64
3.2.13.4.- Protein-Detektion mittels Antikörper. ............................................................................. 65
3.2.14.- Durchflusszytometrie .............................................................................................................. 65
3.2.14.1.- Vorbereitung der Zellen für die Messung von Oberflächen-Antigenen........................ 65
3.2.14.2.- Messung der Oberflächen-Antigene................................................................................. 66

3.2.15.- "Real-time"-RT-PCR.............................................................................................................. 66
3.2.15.1.- Mechanismen...................................................................................................................... 66
3.2.15.2.- Durchführung..................................................................................................................... 67
3.2.15.3.- Relative Quantifizierung ................................................................................................... 68
3.2.16.- Chromatin-Immunopräzipitation (ChIP).............................................................................. 69
3.2.17.- Densitometrie ........................................................................................................................... 70
3.2.18.- Statistische Auswertung .......................................................................................................... 70
4.- Ergebnisse....................................................................................................................................................... 71
4.1 Biologische Effekte der AML1/ETO-Induktion in 9/14/18-Zellen. ..................................................... 71
4.2.- Funktionelle Untersuchung des AML1/ETO-Zielgens LAT2 (WBSCR5/NTAL/LAB).................. 72
4.2.1.- Charakterisierung der Repression des LAT2-Gens in einer AML1/ETO induzierbaren
Zelllinie .................................................................................................................................................. 72
4.2.2.- Untersuchung der LAT2-mRNA- und -Protein-Expression in myeloischen Zelllinien....... 73
4.2.3.- Untersuchung der Protein-Expression des LAT2-Gens in primären AML-Blasten von
Patienten mit unterschiedlichen chromosomalen Aberrationen....................................................... 75
4.2.4.- Untersuchung der Re-Expression des LAT2-Gens mittels siRNAs gegen AML1/ETO in
Kasumi-1-Zellen.................................................................................................................................... 79
4.2.5.- Re-Expression der LAT2-mRNA durch epigenetisch aktive Substanzen in der
AML1/ETO-positiven Kasumi-1-Zelllinie. ......................................................................................... 81
4.2.6.- Histonmodifikation nach der Behandlung mit MS-275 auf dem LAT2-Promotor in
Kasumi-1-Zellen.................................................................................................................................... 83
4.2.7.- Histonmodifikation nach der Induktion von AML1/ETO auf dem LAT2-Promotor in
9/14/18-Zellen. ....................................................................................................................................... 84
4.3.- Identifizierung neuer AML1/ETO-Zielgene mittels RLGS-Methode............................................... 86
4.3.1- RLGS-Ergebnisse ....................................................................................................................... 86
4.3.2.- Klonierung und Annotation der detektierten Sequenzen ...................................................... 88
4.3.3.- Funktionelle Annotationen der Gene....................................................................................... 89
4.3.4.- Southern Blots............................................................................................................................ 90
4.4.- Untersuchung der Expression des LAT2-Gens während induzierter myeloischer Differenzierung
von AML-Zelllinien. ......................................................................................................................................93
4.4.1.- Regulation von LAT2-mRNA und -Protein in der ATRA-induzierten granulozytären
Differenzierung der NB4-Zelllinie. ................................................................................................... 93
4.4.2.- Regulation von LAT2-mRNA und -Protein in der ATRA-induzierten granulozytären
Differenzierung der U937-Zelllinie. .................................................................................................. 95
4.4.3.- Regulation von LAT2-Protein in der ATRA-induzierten granulozytären Differenzierung
der HL60-Zelllinie. ............................................................................................................................. 98
4.4.4.- Regulation von LAT2-mRNA und -Protein in der DMSO-induzierten granulozytären
Differenzierung der HL60-Zelllinie. ............................................................................................... 100
4.4.5.- Regulation von LAT2-mRNA und Protein in der PMA-induzierten monozytären
Differenzierung der HL60-Zelllinie. ............................................................................................... 101
4.4.6.- Regulation von LAT2-Protein in der PMA-induzierten monozytären Differenzierung der
U937-Zelllinie. .................................................................................................................................. 104

4.4.7.- Expression von LAT2-Protein bei der Hemin-induzierten erythrozytären Differenzierung
der K562-Zelllinie............................................................................................................................. 106
4.4.8.- Expression von LAT2-Protein in der PMA-induzierten megakaryozytären
Differenzierung der K562-Zelllinie................................................................................................. 108
4.5.- Expression von LAT2-Protein in humanen primären Zellen.......................................................... 110
4.6.- Regulation von LAT2-Protein in der granulozytären und monozytären-dendritischen
Differenzierung der CD34 + Zellen durch Zytokine. ............................................................................... 112
5.- Diskussion..................................................................................................................................................... 114
Das Fusionsprotein AML1/ETO ................................................................................................................ 114
Epigenetische Regulation vom LAT2-Gen durch AML1/ETO ............................................................... 114
Screening von AML1/ETO Zielgenen auf Promoter- DNA-Methylierungsveränderungen. ................ 118
Regulation vom LAT2-Gen während der myeloischen Differenzierung. ............................................... 119
6 .- Zusammenfassung ...................................................................................................................................... 122
7.- Danksagung.................................................................................................................................................. 123
8.- Bibliographie................................................................................................................................................ 124
9.- Lebenslauf .................................................................................................................................................... 134

Abbildungsverzeichnis
Abbildung 1.- Schematische Darstellung des AML1- und AML1/ETO- Proteins.........................................Seite 6
Abbildung 2.- Chemische Reaktion von Cytosin zu 5´-Methyl-Cytosin......................................................Seite 10
Abbildung 3.- Schematische Darstellung der DNA-Methylierung im normalen und Tumor-Gewebe........Seite 11
Abbildung 4.- Schematische Darstellung der Cytidin-Analoga 5-Azacytidin und 5-Aza-2'deoxycytidin
(Decitabine)....................................................................................................................................................Seite 13
Abbildung 5.- Das AML1/ETO-Protein rekrutiert HDACs und DNMTs....................................................Seite 16
Abbildung 6.- Das Ecdyson-induzierbare System........................................................................................Seite 17
Abbildung 7.- RLGS-Methode......................................................................................................................Seite 19
Abbildung 8.- Bisulfit-Konversion der DNA................................................................................................Seite 20
Abbildung 9.- Transduktionssignalwege des LAT2-Proteins in Mastzellen................................................Seite 22
Abbildung 10.- Schematische Darstellung der Myelopoese.........................................................................Seite 24
Abbildung 11.- Funktionelle Analyse der AML1/ETO-induzierbaren U-937 Zellen..................................Seite 74
Abbildung 12.- Repression von LAT2-mRNA und -Protein nach der ektopen Expression vom AML1/ETO in
der 9/14/18-Zelllinie.......................................................................................................................................Seite 76
Abbildung 13.- LAT2-Expression in verschiedenen Zelllinien....................................................................Seite 77
Abbildung 14.- LAT2-Protein-Expression in Knochenmarksproben von Patienten mit AML von
unterschiedlichen Karyotypen........................................................................................................................Seite 78
Abbildung 15.- LAT2 Protein-Expression in Knochenmarksproben von Patienten mit AML zu Zeitpunkt der
Erst-Diagnose (ED) und der Vollremission (CR)..........................................................................................Seite 81
Abbildung 16.- Re-Expression der LAT2-mRNA nach Knock-down von AML1/ETO mittels siRNAs in der
Kasumi-1-Zelllinie.........................................................................................................................................Seite 83
Abbildung 17.- LAT2-mRNA Re-Expression durch epigenetische aktive Therapien in der AML1/ETO positiven
Kasumi-1-Zellen.............................................................................................................................................Seite 85
Abbildung 18.- Induktion der aktivierenden Histon-Modification durch MS-275 und AML1/ETO -vermittelte
Rekrutierung von HDAC-Aktivität in den 9/14/18-Zellen............................................................................Seite 87
Abbildung 19.- Intensitätsveränderung von Spot 2D24 (Dematin-Gen) in RLGS Gelen nach der Induktion des
AML1/ETO-Proteins in 9/14/18....................................................................................................................Seite 89
Abbildung 20.- RLGS-Ergebnisse................................................................................................................Seite 91
Abbildung 21.- Southern Blot für Dematin-Gen...........................................................................................Seite 93
Abbildung 22.- Southern Blot von PTPN11-Gen.........................................................................................Seite 95
Abbildung 23.- Untersuchung des biologischen Effektes der durch ATRA induzierten granulozytären
Differenzierung der NB4-Zellen....................................................................................................................Seite 97
Abbildung 24.- LAT2-mRNA- und Protein-Expression in der granulozytären Differenzierung der NB4- Zellen
durch ATRA...................................................................................................................................................Seite 98
Abbildung 25.- Untersuchung des biologischen Effektes der durch ATRA induzierten granulozytären
Differenzierung der U937-Zellen...................................................................................................................Seite 99
Abbildung 26.- LAT2-mRNA- und -Protein-Expression in der granulozytären Differenzierung der U937
Zelllinie durch ATRA..................................................................................................................................Seite 100

Abbildung 27.- Untersuchung des biologischen Effektes der durch ATRA induzierten granulozytären
Differenzierung der HL60-Zellen................................................................................................................Seite 101
Abbildung 28.- LAT2-mRNA- und -Protein-Expression in der granulozytären Differenzierung der HL60-
Zelllinie durch ATRA..................................................................................................................................Seite 102
Abbildung 29.- Untersuchung des biologischen Effektes der durch ATRA induzierten granulozytären
Differenzierung der HL60-Zellen................................................................................................................Seite 103
Abbildung 30.- Untersuchung des biologischen Effektes der durch PMA induzierten monozytären-macrophagen
Differenzierung der HL60-Zellen................................................................................................................Seite 105
Abbildung 31.- LAT2-mRNA- und -Protein-Expression in der monozytären-macrophagen Differenzierung der
HL60-Zelllinie durch PMA..........................................................................................................................Seite 106
Abbildung 32.- Untersuchung des biologischen Effektes der durch PMA induzierten monozytären-macrophagen
Differenzierung der U937-Zellen................................................................................................................Seite 108
Abbildung 33.- Untersuchung des biologischen Effektes der durch Hemin induzierten erythrozytären
Differenzierung der K562-Zellen.................................................................................................................Seite 109
Abbildung 34.- LAT2-Protein-Expression während der durch Hemin induzierten erythrozytären Differenzierung
der K562-Zelllinie........................................................................................................................................Seite 110
Abbildung 35.- Untersuchung des biologischen Effektes der durch PMA-induzierten megakaryozytären
Differenzierung der K562-Zellen.................................................................................................................Seite 111
Abbildung 36.- LAT2-Protein-Expression in der megakaryozytären Differenzierung der K562-Zelllinie durch
PMA.............................................................................................................................................................Seite 113
Abbildung 37.- LAT2-Protein-Expression in primären Blutzellen und alveolären Makrophagen.............Seite 114
Abbildung 38.- LAT2-Protein-Expression in der myeloischen Differenzierung von normalen CD34+ Zellen.
......................................................................................................................................................................Seite 115

Tabellenverzeichnis
Tabelle 1.- Zusammenfassung der AML1- und AML1/ETO-Zielgene..........................................................Seite 8
Tabelle 2.- Reagenzien für eine „real-time“-RT-PCR…...............................................................................Seite 68
Tabelle 3.- Temperatur- und Zeit-Bedingungen für eine „real-time“-RT-PCR............................................Seite 68
Tabelle 4.- Klinischen Daten der untersuchten Patienten..............................................................................Seite 79
Tabelle 5.- Ergebnisse der sequenzierten RLGS-Spots................................................................................Seite 92

Abkürzungsverzeichnis
Aberr. Aberrationen
A/E AML1/ETO
AG: Arbeitsgruppe
ALL: Akute lymphatische Leukämie
AIMS: Amplification of inter-methylated sites
AML: Akute myeloische Leukämie
AML1: Akute myeloische Leukämie Gen 1
APL: Akute promyeloische Leukämie
ATRA: All trans-Retinsäure
BCL-2: B cell CLL/lymphoma 2
bp: Basenpaare
BSA: Bovine Serum Albumine
CBF: Core binding factor
CBFB: Core binding factor subunit β
cDNA: Zirkuläre Desoxyribonucleinsäure
C/EBPα: CAATT enhancer binding protein α
Cp: Cross points
CPM: counts pro minute
CSF-R: Colony stimulating factor receptor
CREB: c-AMP response element binding
CTP: Cytosine Triphosphate
dL: Deciliter
DAC: Decitabine
DC: Dendritische Zellen
DDT: Dithiothreitol
DMSO: Dimethylsulfoxid
DNA: Desoxyribonukleinsäure
DNMT: DNA Methyltransferase
Dnmt1: DNA Methyltransferase Typ 1
DSMZ: Deutsche Sammlung von Mikroorganismen und Zellkulturen
EAP: EBER-associated protein
EDTA: Ethylendiamintetraessigsäure
ENU: N-Ethyl-N-Nitrosurea
ETO: Eight-Twenty-One Gen
EVI1: ecotropic viral integration site 1
FAB: French-American-British
FACS: Fluorescence-activated cell sorting
FBS: Fetal Bovine Serum
FcЄR I: Fc epsilon receptor I
FITC: fluorescein isothiocyanate

FLT3: fms-related tyrosine kinase 3
FS: Vorwärtsbeugung
FW: Forward
GAPDH: Glyceraldehyd-3-Phosphate Dehydrogenase
gDNA: Genomische DNA
GEM: glycosphingolipid enriched membrane
G-CSF: Granulocyte colony stimulating factor
GM-CSF: Granulocyte-macrophage colony stimulating factor
GTP: Guanosine Triphosphate
GUSB: β-Glukoronidase
H3: Histon H3
H4: Histon H4
HAT: Histon-Acetyl Transferase
Hb: Hämoglobin
HCl: Salzsäure
HDAC: Histon-Deacetylase
HDAC1: Histon-Deacetylase Typ 1
bHLH: basic Helix-Loop-Helix
HSW Buffer: “High Stringency Wash” Puffer
IL-3: Interleukin 3
IL-4: Interleukin 4
IL-6: Interleukin 6
inv: Inversion
ITD: Internal tandem duplication
JAK2: Janus Kinase 2
kB: Kilobasen
kDa: Kilodalton
KM: Knochenmark
KK: Komplexer Karyotyp
LAB: Linker for activation of B cells
LAT: Linker for activation of T cells
LB Medium: Luria Broth Base Medium
LSW Buffer: Low Stringency Wash Buffer
M: Molar
MBD: Methyl binding protein
M-CSF: Macrophage colony stimulating factor
MDS: Myelodysplastische Syndrom
MES buffer: 3–(N–Morpholino) Ethansulfonsäure-Puffer
MIP-1α: Macrophage inflammatory protein - 1α
MPO: Myeloperoxidase
min: Minuten

MOPS: 3-N-morpholino Propan-sulfon-säure
MSP: Methylierungs-spezifische-PCR
MDR1: multiple drug resistance Gen 1
MTG8: myeloid translocation gene on 8q22
MYH11: Myosin heavy chain 11
ml: Milliliter
µl: Mikroliter
µg: Mikrogramm
µM: Mikromolar
mm: Millimeter
mM: Millimolar
mg/mL: Milligramm pro Milliliter
MTA: Medizinisch-Technischer Assistent
NaCl: Natriumchlorid
NaCitrate: Natriumcitrat
NaF: Natriumfluorid
NaOH: Natriumlauge
Na3VO4: Natrium-Orthovanadat
NCBI: National Center for Biotechnology Information
NEB: New England Biolabs
NN: Normalkaryotyp
nm: Nanometer
ng: Nanogramm
NP40: Nonidet P40 Lösung
NTAL: Non T cell activation linker
OH: Ohio
PB: Peripheres Blut
PBS: Phosphat-gepufferte Kochsalzlösung
PE: Phykoerythrin
PCR: Polymerase-Ketten-Reaktion
PLC-γ1: Phospholipase C gamma 1
PMA: Phorbol 12-Myristat 13-Acetat
pmol: picomol
PML: Promyelocytic leukemia Gene
PMSF: Phenylmethylsulphonylfluorid
Pon A: Ponasteron A
p14arf: p14arf /CDKN2A Gen
PTPN11: Protein tyrosine phosphatase, non-receptor type 11 (Noonan syndrome 1) Gen
RARβ2: Retinoid acid receptor beta 2
RARa: Retinoid acid receptor alpha
RDA: Representational Difference Analysis

RLGS: Restriction Landmark Genomic Scanning
RNA: Ribonukleinsäure
rpm: Revolutionen pro Minute
RT-Reaktion: Reverse Transkriptase Reaktion
RUNX1: Runt related Transkription-faktor 1
RHD: Runt homology domain
RW: Reverse Primer
s.: Siehe
SAHA: Suberoylanilid- hidroxam-säure
sAML: Sekundäre AML
SCF: Stem cell factor
SDS: Natriumdodecylsulfat
sec Sekunden
SS: Seitwärtsstreuung
SSC: Kochsalz/Natriumcitrat-Puffer
Std: Stunden
t: Translokation
tAML: Therapy-related AML
TBS-T: Gepufferte Kochsalzlösung mit Tween
TE Buffer: Tris EDTA Puffer
TSA: Trichostatin A
U/mL: Einheiten pro Milliliter
UV Ultraviolett
usw: Und so weiter
vs: versus
V: Volt
WBSCR5: Williams Beuren Syndrome Critical Region 5 Gen
WHO: World Health Organisation
µL: Mikroliter
µg: Mikrogramm
µg/ µL: Mikrogramm pro Mikroliter
X-Gal: 5-bromo-4-chloro-3-indolzl-b-D-Galactopyranosid
WBC: white blood cells, Leukozyten
Tsd: Tausend

1.- Einleitung Seite - 1 -
1.- Einleitung
1.1.- Akute Myeloische Leukämie
Die akute myeloische Leukämie (AML) ist eine hämatologische Krankheit, die durch
eine erhöhte Zahl von Blasten im Knochenmark und eine Blockade ihrer Reifung
charakterisiert ist. Dies führt zu einer Insuffizienz der Hämatopoese mit oder ohne
Leukozytose. In den Industrieländern liegt die Inzidenz bei rund 2-3 Fällen pro 100000
Einwohner pro Jahr. Diese Zahl steigt aber im Alter auf bis zu 12 Fälle pro 100000 Personen
über 65 Jahre an (Löwenberg et al., 1999).
In der ersten French American British (FAB) Klassifikation waren mehr als 30 %
Blasten im Knochenmark für die Diagnose einer AML notwendig (Bennett et al., 1976). Je
nach morphologischen Charakteristika der AML-Blasten kann man nach der FAB-
Klassifikation 8 verschiedene Subgruppen unterteilen. (Bennett et al., 1976, 1980, 1985,
1991).
Diese Klassifikation wurde von der WHO (World Health Organisation) durch eine
Einteilung aus der Kombination von Morphologie, Immunophänotyp, genetischen und
klinischen Charakteristika ergänzt und revidiert. In der WHO-Klassifikation reichen 20 %
Blasten im Knochenmark für die Diagnose einer AML aus (Harris et al., 1999). Die WHO-
Klassifikation unterscheidet 5 Gruppen: AML mit rekurrenten chromosomalen
Translokationen, AML mit multilineärer Dysplasie, AML und Myelodysplastisches Syndrom
(MDS) nach vorausgegangener Therapie, AML nicht in anderen Gruppen klassifiziert, und
biphänotypische AML (Harris et al., 1999).
In der Ära der Zytogenetik konnte gezeigt werden, dass die verschiedenen
chromosomalen Translokationen eine bessere Korrelation mit der Prognose der AML haben
(Arthur et al., 1989) als die FAB-Klassifikation. Es wurde auch festgestellt, dass bestimmte
Translokationen sehr häufig in manchen Subgruppen der FAB-Klassiffikation vorkommen,
wie z. B. die Translokation t(15;17) in der FAB M3 Subgruppe (de Thé et al., 1990), und die
Inversion inv(16) in der M4eo Subgruppe (Le Beau et al., 1983). Infolgedessen wurde
angenommen, dass die verschiedenen chromosomalen Aberrationen eine Rolle in der
Entstehung der AML und der Charakteristika der Blasten spielen.

1.- Einleitung Seite - 2 -
1.2.- Pathogenese akuter myeloischer Leukämien.
Für den molekularen Pathomechanismus der AML sind chromosomale und genetische
Veränderungen von entscheidender Bedeutung. Dies sind beispielweise:
Chromosomentranslokationen t(8;21), t(15;17), inv(16), Deletionen eines Chromosomenteils
oder eines ganzen Chromosoms (5q-, 7q-, 20q-, 12p-), Trisomien (8, 13, 21), Aktivierung von
Onkogenen (Punkmutationen in N-Ras oder K-Ras Genen, Mutationen und Insertionen in Typ
III-Tyrosinkinaserezeptoren wie FLT3 und c-Kit) und Geninaktivierung durch Promoter-
Hypermethylierung (p15, Oestrogen Gen Rezeptor, E-cadherin) (Löwenberg et al., 1999)
1.2.1.- Die „Two-hit“-Hypothese
In den letzten Jahren hat sich die „Two-hit“ Hypothese für die Entstehung der AML
von Knudson (Gilliland, 2001) sowohl in klinischen Studien als auch in Mausmodellen
bestätigen lassen. Nach dieser Hypothese gibt es zwei verschiedene genetische Ereignisse für
die Entstehung der AML. Zum einen gibt es die der Klasse I-Mutationen, die den Zellen ein
proliferatives Signal und eine Hemmung der Apoptose vermitteln. Beispiele von Klasse I-
Mutationen sind die Punkmutationen in Codon 12, 13 und 61 der Onkogene N-Ras und K-Ras
(Bacher et al., 2006; Bowen et al., 2005; Gari et al., 1999; Valk et al., 2004a); Mutationen,
Deletionen und Insertionen in der juxtamembranären oder in der Tyrosinkinase-Domäne der
Rezeptoren FLT3 (Kottaridis et al., 2001; Kuchenbauer et al., 2005; Schnittger et al., 2002;
Thiede et al., 2002) und c-Kit (Boissel et al., 2006; Paschka et al., 2006; Schessl et al., 2005)
und Mutationen in der Janus Kinase 2 (JAK2) (Lee et al., 2006; Schnittger et al., 2007;
Schnittger et al., 2006). Mutationen der Klasse II verursachen eine Blockade der
Differenzierung in den betroffenen Zellen. Translokationen, in denen Transkriptionsfaktoren
(wie z.B. AML1/ETO, PML/RARα, CBFB/MYH11) involviert sind, und Punktmutation von
Transkriptionsfaktoren der Hämatopoese wie z.B. AML1 (Michaud et al., 2002), CEBPα
(Pabst et al., 2001b) und Pu.1 (Mueller et al., 2002) gehören zu solchen Klasse II Mutationen.
Klasse I-und Klasse II-Mutationen kooperieren in der Pathogenese akuter myeloischer
Leukämien.

1.- Einleitung Seite - 3 -
1.2.2.- Klinische Daten bestätigen die „Two-hit“-Hypothese
Diese Hypothese wurde in den letzten Jahren durch Mutations-Screening in
verschiedenen klinischen AML-Studien bestätigt. Es wurde nachgewiesen, dass einige Klasse
I Mutationen in bestimmten Karyotypen von AML-Patienten häufiger sind. Mutationen von
FLT3-Rezeptor finden sich in 30 % der Patienten mit der Translokation (15;17) (Kuchenbauer
et al., 2005) und mit Normalkaryotyp (NN) (Schnittger et al., 2002; Thiede et al., 2002).
Mutationen des c-Kit-Rezeptors sind in 17-30 % der Patienten mit "Core Binding Factor
Leukämie", das heißt mit Translokation t(8;21) oder Inversion inv(16) beschrieben (Boissel et
al., 2006; Paschka et al., 2006; Schessl et al., 2005). N-Ras Punktmutationen sind in 10-30 %
der Patienten mit inv(16) vorhanden (Bacher et al., 2006).
Die Untersuchung verschiedener Mutationen in Verbindung mit dem Karyotyp in
AML-Patienten hat einen groβen Einfluss auf die Prognose und die klinische Behandlung.
Zum Beispiel haben AML1/ETO-positive Patienten allgemein eine gute Prognose, bei
Koexistenz einer Mutation des c-Kit-Rezeptors zeigt sich eine schlechtere Prognose (Paschka
et al., 2006) mit entsprechenden therapeutischen Konzequenzen wie Therapieintensivierung
(z.B. allogene Stamzelltransplantation in erster kompletter Remission). Zurzeit werden neue
spezifische Medikamente entwickelt, die die Effekte von FLT3- und c-Kit-Mutationen
blockieren oder N-Ras- und K-Ras-Onkogene inhibieren können (Tallman et al., 2005). Diese
Medikamente können bei Vorliegen der Mutationen z. B. in Kombination mit einer
Chemotherapie eingesetzt werden.
1.2.3.- Bestätigung der „Two-hit“ -Hypothese in Maus-Modellen.
Die „Two-hit“ Hypothese für die Entstehung von Leukämien konnte auch in
Mausmodellen bestätigt werden. Transgene knock-in Mäuse für AML1/ETO (Yergeau et al.,
1997) und CBFB/MYH11 (Castilla et al., 1999) verstarben am 14. Tag der embryonalen
Entwicklung an intrakraniellen Blutungen und fehlender Leberhämatopoese. Dieser Phänotyp
ist sehr ähnlich im Vergleich mit AML1- (Okuda et al., 1996) und CBFB- (Wang et al., 1996)
knock-out Mäusen. Das zeigt die dominante Rolle der Fusionsgene AML1/ETO und
CBF/MYH11 über den Transkriptionskomplex CBF („Core Binding Factor“) und die
Blockade der myeloischen Differenzierung, die diese Fusionsgene in AML Blasten
verursachen. Im Weiteren wurden AML1/ETO Knock-in Mäuse mit einem Tetracyclin-
induzierbaren System (Rhoades et al., 2000), mit einer Cre/loxP Rekombination (Higuchi et

1.- Einleitung Seite - 4 -
al., 2002) und unter Kontrolle des myeloischen spezifischen Promoters hMRP8 (Yuan et al.,
2001) generiert. Obwohl die Hämatopoese einer präleukämischen Phase ähnlich war,
entwickelten die Mäuse keine akute myeloische Leukämie. Nur unter zusätzlicher
Behandlung mit dem DNA-alkylierten-Mutagen N-Ethyl-N-nitrosurea (ENU) (Yuan et al.,
2001) oder wenn die Knochenmarkstammzellen mit dem Transgen einer aktivierten Rezeptor
Kinase wie TEL/EVI1 (Grisolano et al., 2003) oder FLT3-ITD (Schessl et al., 2005)
transfiziert wurden, entwickelten die AML1/ETO-Knock-in Mäuse eine AML-ähnliche
Krankheit. Weitere Hinweise dafür zeigten Knock-in Mäuse für PML/RARα in Kombination
mit dem FLT3-ITD Mutant (Kelly et al., 2002) und die Knock-in Mäuse für CBF/MYH11 in
Kombination mit ENU (Castilla et al., 1999) oder retroviraler Transfektion (Castilla et al.,
2004), die jeweils APL bzw AML-ähnlichen Krankheiten entwickelten und daran starben. In
Sp1/PU.1 knock-out Mäusen, deren Proerythroblasten eine Blockade in der Differenzierung
zeigten und eine Erythroleukämie zu einem späteren Zeitpunkt entwickelten, wurden
sekundäre genetische Ereignisse zur Entstehung der akuten Erythroleukämie untersucht. 86 %
der Tumorzellen wiesen eine Mutation des c-Kit-Rezeptors im letzten Stadium der Krankheit
auf (Kosmider et al., 2005).
Um spezifischere Therapien der einzelnen Subtypen der AML zu entwickeln, ist ein
fundamentales Verständnis dieser molekularen Veränderungen in der Leukämogenese von
entscheidender Bedeutung.
1.3.- Das chimäre Fusionsprotein AML1/ETO
Die Translokation t(8;21) führt zur Produktion eines Fusionsproteins, welches aus 177
Aminosäuren des N-terminalen Teils des AML1- (RUNX1) Gens von Chromosom 21,
einschlieβlich der DNA-Bindungs- und der CBF-Interaktionsdomäne, und fast dem gesamten
ETO-Gen von Chromosom 8 besteht (Miyoshi et al., 1991). Diese reziproke chromosomale
Translokation war die erste, die bei AML nachgewiesen werden konnte (Rowley, 1973).
Morphologisch sind die Blasten, die diese Translokation tragen, Myeloperoxidase positiv,
haben häufig mehrere Nukleolen, Vakuolen, Globuli und rötliche Granula. Charakteristisch
sind auch Auerstäbchen im Zytoplasma (Nucifora and Rowley, 1995). Immunophänotypisch
sind die Blasten häufig positiv für die myeloischen Antigene CD13, CD15, die Stammzell-
Antigene CD34 und HLA-DR und den B-Zell Marker CD19 sowie das Antigen CD56. Die
Blasten sind negativ bezüglich der Expression des MDR1-Gens und der Expression des CD33

1.- Einleitung Seite - 5 -
Antigens (Hurwitz et al., 1992; Kita et al., 1992; Orazi et al., 1992). Die Blasten mit der
Translokation t(8;21) gehören gemäß FAB-Klassifikation hauptsächlich zur Subgruppe M2.
1.3.1.- Das AML1/RUNX1-Gen und der CBF-Transkriptionfaktorkomplex
Das physiologische AML1-Gen ist ein Transkriptionsfaktor, der zur Familie der
RUNX-Gene gehört. Die RUNX-Gene haben einen sehr hohen Grad an Sequenzhomologie
zum Drosophila Transkriptionsfaktor "runt" (Gergen and Wieschaus, 1986; Ogawa et al.,
1993) und spielen eine wichtige Rolle in der embryonalen Entwicklung verschiedener
Organsysteme: AML1/RUNX1 in der Hämatopoese (Okuda et al., 1996; Wang et al., 1996),
AML3/RUNX2 in der Osteogenese (Otto et al., 1997) und AML2/RUNX3 in der
Neurogenese (Levanon et al., 2002) und der Entwicklung der gastrischen epithelialen Zellen
(Li et al., 2002).
Das AML1-Protein gehört zu einem aktivierenden Transkriptionskomplex, der als
„Core Binding Factor“ (CBF) bekannt ist. Das AML1/RUNX1-Protein besitzt eine DNA
Bindungstelle in der „Runt Homologous Domäne“ (RHD), welche die Sequenz TGTGGT an
den Promotoren von Zielgenen erkennt (Meyers et al., 1993; Otto et al., 2003). Es hat eine
sehr hohe Homologie zur „runt“ Domäne der transkriptionellen Regulatoren der
Segmentierungsgene von Drosophila (Gergen and Wieschaus, 1986). Es gibt 3
unterschiedliche Splicing-Varianten: AML1a (Meyers et al., 1995), AML1b und AML1c
(Miyoshi et al., 1995), die möglicherweise verschiedene Funktionen in der Regulation der
Genexpression haben könnten.
Die CBFB-Untereinheit besitzt keine DNA-Bindungsdomäne, aber sie interagiert mit
dem AML1/RUNX1-Protein in der „RHD“-Region. Die CBFB-Bindung erhöht die Affinität
des CBF-Komplexes an den DNA-Doppelstrang (Ogawa et al., 1993). Diese Interaktion
zwischen CBFB und AML1/RUNX1 stabilisiert und schützt vor der Degradation durch
Ubiquitinierung (Huang et al., 2001). Der CBF-Komplex reguliert die Transkription von
verschiedenen für die Hämatopoese wichtigen Genen durch Interaktion mit transkriptionellen
Co-Aktivatoren wie p-300 und CREB-binding Protein, die ihrerseits mit Histon-Acetyl-
Transferasen (HAT) interagieren und sie an Genpromotoren rekrutieren (Kitabayashi et al.,
1998). HATs sind Proteine, die die Aminosäuren Lysin und Arginin der Histone H3 und H4
acetylieren und somit die Konformation von Histonen zu einer aktiven Form verändern
können (Santos-Rosa and Caldas, 2005). Dadurch wird Chromatin im Bereich von
Promotoren der AML1-Zielgene in eine zugänglichere Struktur verwandelt, so dass die

1.- Einleitung Seite - 6 -
Transkription dieser Gene stattfinden kann. RUNX1 rekrutiert zusätzlich SUV39
Methyltransferase und kann auch durch Methylierung von Lysin 9 im Histon 3 (H3) die
Expression der Gene beeinflussen (Reed-Inderbitzin, Oncogene 2006).
1.3.2.- Das ETO-Gen
Das ETO/MTG8-Gen befindet sich auf Chromosom 8 Bande q22. Obwohl seine
Expression im Gehirn sehr hoch ist (Erickson et al., 1994) und 2 putative Zinc-Fingermotive
hat (Lo Coco et al., 1997), bleibt seine physiologische Funktion unbekannt. Auf eine Funktion
bei der Entwicklung des Darms wurde in einer Arbeit verwiesen (Calabi et al., 2001). Es
Abbi ldung 1.- Schematische Darstellung des AML1- und AML1/ETO - Proteins. Das AML1-Protein ist auf dem langen Arm des Chromosoms 8 lokalisiert. Die RHD Domäne des AML1 Proteins erkennt im Bereich des Promotors seiner Zielgene die Konsensus-Sequenz TGT/cGGT. Das CBFB-Protein interagiert mit AML1 und steigert die DNA-Affinität des CBF-Komplexes. Auβerdem stabilisiert und schützt sie den CBF-Komplex vor Ubiquitin-Degradation. Durch Interaktion von AML1 mit p300 und CREB-Binding-Protein werden Histon-Acetylasen an Zielgene rekrutiert, und damit wird die Konformation der Histone zu einer aktiveren Form umgewandelt. In der t(8; 21) wird das AML1-Gen mit ETO fusioniert. AML1 behält die RHD-Domäne und somit die DNA-Bindungs-Domäne und seine Interaktion mit CBFB. ETO interagiert mit dem Co-Repressor-Komplex mit N-Cor und m-Sin3a. Dadurch werden Histon-Deacetylasen an AML1-Zielgene rekrutiert und die Konformation der Histone wird in eine inaktivere Form umgewandelt.
Chr. 8
Chr. 21
AML1
CBFßP300
HAT
CCGTGT/cGGTATGTGTCTGACTGCTGAGTATGA
Targetgenes
IL-3
GM-CSF
CSF1-R
C/EBP-α
MPO
T cell receptor subunits
CBFß
CCGTGT/cGGTATGTGTCTGACTGCTGAGTATGA
Targetgenes
IL-3
GM-CSF
CSF1-R
C/EBP-α
MPO
T cell receptor subunits
AML1
ETO
NCor mSin3
HDAC1
t(8;21)
AML1
AML1/
ETO
WBSCR5
WBSCR5

1.- Einleitung Seite - 7 -
konnte gezeigt werden, dass das ETO-Protein über die Zink-Finger-Domäne direkt an die
nukleären Proteine N-Cor und mSin3a binden kann (Lutterbach et al., 1998). Diese Proteine
interagieren ihrerseits gleichzeitig mit den Histone-Deacetylasen (HDAC) 1 (Wang et al.,
1998), HDAC2 and HDAC3 (Amann et al., 2001). Dies bewirkt, dass ETO durch Interaktion
mit DNA-bindenden Transkriptionsfaktoren und durch Rekrutierung eines Co-Repressor
Komplexes die Genexpression regulieren kann (Alcalay et al., 2003; Gelmetti et al., 1998).
1.3.3.- Das AML1/ETO-Fusionsgen
Das AML1/RUNX1-Gen ist in verschiedene chromosomale Translokationen in AML
und ALL involviert. Es wird vermutet, dass die resultierende klinische Entität von der
Funktion der Partner des AML1-Gens abhängig ist (Alcalay et al., 2001). Es finden sich
Fusionen mit EVI-1 (Mitani et al., 1994), MDS-1 und EAP (Nucifora et al., 1993) auf
Chromosom 3 in AML der Erwachsenen und mit TEL auf Chromosom 12 (Golub et al.,
1995) in ALL bei Kindern. Häufigste Kooperationspartner von AML1 ist das ETO-Gen auf
Chromosom 8 in AML der Erwachsenen.
In der Translokation t(8;21) wird der N-terminale Teil des AML1-Gens von
Chromosom 21 mit nahezu dem ganzen ETO-Gen von Chromosom 8 verbunden (Erickson et
al., 1992). Es gibt unterschiedliche Transkript-Varianten, jedoch behält das AML1-Protein
immer die DNA-Bindungsdomäne und die Interaktion mit der CBFB-Untereinheit, die eine
gewisse Stabilität des chimären Fusionsproteins bewirkt und die DNA-Affinität verstärkt
(Ogawa et al., 1993). Der ETO-Teil des Fusionsproteins rekrutiert HDACs durch seine
Partner N-Cor und mSin3a an AML1- bzw. AML1/ETO-Zielgene, deren Expression durch
eine Konformationsänderung der lokalen Histone gehemmt wird (Lutterbach et al., 1998;
Wang et al., 1998).
Es wurde sowohl in einer AML1/ETO-positiven Zelllinie als auch in AML-Blasten
von Patienten gezeigt, dass AML1/ETO auch durch einen anderen epigenetischen
Mechanismus, nämlich die Rekrutierung von DNA-Methyltransferase (DNMTs) an den
Zielpromotor, die Expression des AML1/ETO-Zielgens IL-3 hemmen kann (Liu et al., 2005).
AML1/ETO kann auch durch Protein-Protein Interaktion die Expression von Genen
reprimieren, die nicht durch AML1 reguliert sind (Mao et al., 1999) und kompetitiv die
Funktion von Transkriptionsfaktoren inhibieren (Westendorf et al., 1998). Letztlich wurde
gezeigt, dass AML1/ETO durch die Interaktion mit den E Proteinen (Klasse A der bHLH

1.- Einleitung Seite - 8 -
Proteine) E2A und HEB die Expression der entsprechenden Zielgene reprimieren kann
(Zhang et al., 2004).
1.3.4.- AML1- und AML1/ETO-Zielgene
Der Nachweis und die funktionelle Untersuchung von AML1/ETO Zielgenen hat zu
einem besseren Verständnis des Pathomechanismus dieses Fusionsproteins geführt.
AML1 und AML1/ETO regulieren die Expression verschiedener Gene, die zumeist
eine wichtige Rolle in der Hämatopoese spielen. AML1 aktiviert die Transkription der Gene
durch Rekrutierung seines Ko-Aktivatorkomplexes, AML1/ETO hingegen hindert die Trans-
Aktivierung dieser AML1-Zielgene durch Rekrutierung von HDACs (Gelmetti et al., 1998)
und möglicherweise DNMTs (Liu et al., 2005). Entsprechende Beispiele sind in der folgenden
Tabelle genannt:
Tabelle 1.- Zusammenfassung der AML1- und AML1/ETO-Zielgene
Gen Name Funktion des Proteins Art der Regulation Referenzen
IL-3
Wachstumsfaktor von hämatopoetischen Stammzellen und myeloischen Progenitoren.
AML1/ETO Repression mittels HDAC und DNMT in Zelllinien und myeloischen Blasten.
(Cameron et al., 1994) (Uchida et al., 1997) (Liu et al., 2005)
p14ARF
p14ARF bindet an MDM2 und fördert seine Degradation. Infolgedessen wird p53 stabilisiert und der Zellzyklus wird in G1 und G2/M Phase gestoppt
AML1/ETO bindet an den p14 ARF Promotor und reprimiert seine Expression
(Linggi et al., 2002)
T-Zell Rezeptor δ Funktion in der Entwicklung der T-Zellen
AML1-Bindung ist notwendig für die Aktivierung der Transkription.
(Redondo et al., 1992)
C/EBPα
Notwendig für die Differenzierung der Granulozyten. Aktiviert die Transkription des G-CSF Rezeptors.
C/EBPα Expression ist herunterreguliert in AML1/ETO positiven Blasten und Zelllinien. AML1/ETO blockiert die Selbst-regulation von C/EBPα.
(Pabst et al., 2001a)
M-CSF Rezeptor
Involviert in Differenzierung, Proliferation und Überleben von Monozyten.
C/EBPα und AML1 aktivieren seiner Transkription in vitro.
(Zhang et al., 1994) (Petrovick et al., 1998)
GM-CSF Zytokin, Wachstum und Differenzierung der Granulozyten,
AML1/ETO blockiert die Transaktivierung des GM-CSF Promotors durch AML1
(Frank et al., 1995)

1.- Einleitung Seite - 9 -
Monozyten, Eosinophilen und Erythrozyten wird gefördert.
Myeloperoxidase (MPO)
Microbizides Protein vorhanden in primärer Granula myeloischer Zellen.
Erstes identifiziertes myeloisches Gen; reguliert durch AML1-Bindung im Promotor in vitro.
(Nuchprayoon et al., 1994)
Neutrophile Elastase
Microbizides Protein vorhanden in primären Granula der myeloischen Zellen.
AML1 bindet und aktiviert die Transkription in vitro.
(Nuchprayoon et al., 1994)
Pu.1
Reguliert die Transkription von myeloisch-spezifischen Genen wie G-CSF-R und GM-CSF-R
Die mRNA von PU.1 ist nicht detektierbar in Geweben von AML1 (-/-) und CBFB (-/-) Mäuse.
(Okada et al., 1998) (Vangala et al., 2003)
G-CSF Rezeptor
Spezifischer myeloischer Rezeptor des Wachstumfaktors G-CSF.
Indirekte Hochregulation durch C/EBPα, nach ektopischer AML1/ETO Induktion.
(Shimizu et al., 2000)
BCL-2
Reguliert den programmierten Zelltod und seine Überexpression kann Apoptose verzögern und verhindern.
Hohe Expression in AML1/ETO positiven Zelllinien, Hochregulation nach der Induktion von AML1/ETO in U937 und sequenzspezifische Bindung von AML1 und AML1/ETO.
(Klampfer et al., 1996)
MIP-1α
Proinflammatorisches Zytokin, das auch die Proliferation der hämatopoetischen Stamzellen hemmt.
AML1 bindet an dem Promotor in vitro und AML1/ETO hemmt die Aktivierung des Promoters mittles PMA.
(Bristow and Shore, 2003)
LAT2
Adaptormolekül, das durch den B-Zell Rezeptor, c-Kit Rezeptor und FcєR I aktiviert werden kann.
Herunterregulation der mRNA in AML1/ETO induzierbaren Zellen, AML1/ETO positiven Zelllinien und AML1/ETO positiven Patientenblasten.
(Fliegauf et al., 2004) (Ross et al., 2004) (Valk et al., 2004b)
1.4.- Epigenetische Aktivität von chimärer Fusionsproteinen in AML
1.4.1.- Die epigenetische Regulation der Gentranskription
Die Epigenetik beschäftigt sich mit vererbbaren Modifikationen der Genexpression,
die ohne eigentliche Veränderung der DNA-Sequenz einhergehen. Die wichtigsten
Mechanismen der Epigenetik sind die DNA-Methylierung und die
Chromatinstrukturveränderungen, die vorwiegend auf Histonmodifikationen beruhen.

1.- Einleitung Seite - 10 -
1.4.1.1.- DNA-Methylierung
Bis zu 5 % der Cytosine in der genomischen DNA von Säugerzellen sind methyliert.
Diese Modifikation wird durch DNA Methyltransferasen (DNMTs) mit S-adenosyl-methionin
als Methylgruppendonor katalysiert (Bestor et al., 1988). Bisher sind 3 DNMTs beschrieben.
DNMT1 bewahrt nach der DNA-Replikation das identische DNA Methylierungsmuster in der
Tochterzelle ("maintainance DNMT") (Turker and Bestor, 1997). DNMT3a und DNMT3b
haben die Funktion von „de novo“ Methyltransferasen und spielen eine wichtige Rolle in der
Tumorgenese (Robertson et al., 1999).
Methylcytosine finden sich fast ausschließlich im Kontext von CpG-Dinukleotiden.
Die CpG Dinukleotide sind im gesamten Genom unterrepräsentiert, es finden sich jedoch
„Cluster“ von CpGs in Promotorregionen und regulatorischen Regionen von Genen (Larsen et
al., 1992). Die CpG reichen Regionen werden CpG-Inseln genannt, wenn sie bestimmte
Voraussetzungen erfüllen (Fragment > 200 bp, "observed/expected" Ratio>0.6) (Gardiner-
Garden and Frommer, 1987).
Die Funktionen der DNA-Methylierung sind vielfältig. Es wurde beschrieben, dass sie
während der embryonalen Entwicklung eine wichtige Rolle spielt (Kafri et al., 1992). DNA-
Methylierung ist in genomisches „Imprinting“ involviert, dadurch ist nur ein Allel, mütterlich
oder väterlich, von einem Gen in der Zelle exprimiert (Falls et al., 1999). Sie hat eine
Abbildung 2.- Chemische Reaktion von Cytosin zu 5´-Methyl -Cytosin. Eine Methyl-Gruppe von S-adenyl-Methionin wird in Position hinzugefügt. Das Produkt der Reaktion ist ein 5´-Methyl-Cytosin und S-adenyl-Homocystein. Diese Reaktion wird durch DNA Methyltransferasen katalysiert. (Modifiziert nach DNA Methylation, CRC Press, Manel Esteller).
NH2 NH2
O O
N
N N
N CH3
Cytosine 5-methylCytosine
S-adenyl-methionine
S-adenyl-homocysteine
DNA Methyltransferase

1.- Einleitung Seite - 11 -
wichtige Funtion in der Inaktivierung des X-Chromosoms bei Frauen (Riggs and Pfeifer,
1992) und bei der Alterung von Zellen (Cooney, 1993).
Die Hauptbedeutung von DNA-Methylierung liegt in der Kontrolle der
Gentranskription (Holliday and Pugh, 1975). Die CpG-Methylierung in regulativen Regionen
von Promotoren und 5' lokalisierten Exons korreliert in vielen Genen mit transkriptioneller
Inaktivierung. So konnte gezeigt werden, dass Promotoren und Exon 1 von Genen in einem
aktiven Zustand zumeist hypomethyliert sind. In inaktiven Genen hingegen sind sie
hypermethyliert (Gonzalez-Zulueta et al., 1995).
Es gibt zwei Hauptmechanismen, mit denen DNA-Methylierung die Genexpression
reguliert. Einerseits interferieren die methylierten Cytosine mit der Bindung von
Transkriptionsfaktoren (Kass et al., 1997). Andererseits gibt es Proteine, die preferentiell an
methylierte Sequenzen im Promotorbereich binden. Die Methylierung des Promotors in einer
CpG-Insel führt zur Bindung von Methyl-CpG-bindenden Proteinen (MBDs) und
Transkriptions-Korepressoren wie HDACs, die den Transkriptionsstart blockieren (Jones et
al., 1998) (Nan et al., 1998). Bisher sind nur vier sogenannte Methyl-DNA-bindende Proteine
beschrieben: MDBP-1 (Huang et al., 1984), MDBP2 (Jost and Hofsteenge, 1992), MeCP1
(Meehan et al., 1989) und MeCP2 (Lewis et al., 1992).
Abbildung 3.- Schematische Darstellung der DNA-Methylierung im normalen und Tumor-Gewebe. Im normalen Gewebe sind Promotoren überwiegend demethyliert und die CpG-Dinukleotide der Exons methyliert. In Tumorzellen kommt häufig eine Hypermethylierung des Promotors und eine Hypomethylierung des Exons vor. Die Hypermethylierung des Genpromotors führt in Tumorzellen zu einer Repression der Genexpression (Modifiziert nach Baylin et al., 2005).

1.- Einleitung Seite - 12 -
1.4.1.2- Histonmodifikation
Histone sind die Zellkernproteine, die wichtige Ordnungsprinzipien der DNA-
Verpackung ermöglichen. Acht dieser Proteine (vier Typen) formen einen Nukleosomkern,
um den die DNA verpackt ist. Das humane Genom wird in unterschiedlichem Ausmaβ
kondensiert: Euchromatin besteht aus aktiven Regionen, in denen das Chromatin eine lockere
Packung hat, Heterochromatin besteht dem gegenüber aus inaktiven Regionen, in denen eine
festere Packung der Histone vorliegt (Santos-Rosa and Caldas, 2005).
Der Verpackungsgrad, und damit die Transkription der verschiedenen Regionen der
Chromosomen, wird durch Histonmodifikationen reguliert. Die 3 wichtigsten
posttranslationalen Histonmodifizierungen sind Phosphorylierung der Serin-Reste,
Methylierung der Lysin- und Arginin-Reste und, vor allem, Acetylierung der Lysine. Die
Kombination der verschiedenen Histonmodifikationen mit den verschiedenen Aminosäure-
Resten führt zu einer aktiven oder inaktiven Konformation. Es wurde gezeigt, dass
Methylierung von Lysin 4 (Santos-Rosa et al., 2002), Acetylierung von Lysin 9 und 14 des
Histons H3 und von Lysin 12 des Histons H4 zu einer aktiven Form führt (Strahl and Allis,
2000). Hingegen bewirkte die Methylierung von Arginin 3 des Histons H4 (Lee et al., 2005)
sowie Mono- bis Trimethylierung von Lysin 9 des Histons H3 und von Lysin 20 des Histons
H4 eine inaktive Form (Schotta et al., 2004).
Die am ausführlichsten untersuchte Histonmodifikation ist die
Acetylierung/Deacetylierung. Sie wird von verschiedenen Enzymfamilien vermittelt, von
Histon-Acetyltransferasen (HAT) und den Histon-Deacetylasen (HDAC).
1.4.1.3.- Die Kooperation von DNA-Methylierung und Histonmodifikation in der
Regulation der Gentranskription.
Eine sehr interessante Entdeckung, die die Beziehung zwischen DNA-Methylierung,
Histonmodifikation und transkriptioneller Regulation verdeutlicht, war, dass die Proteine
MeCP2 und MBD2 mit Histondeacetylasen ko-präzipitieren (Jones et al., 1998; Nan et al.,
1998; Ng et al., 1999). Weitere Hinweise auf eine Kooperation zwischen der DNA
Methylierung und der Histonmodifikation wurden durch die Interaktion von DNMT1 und
DNMT3a mit HDAC1 und HDAC2 gezeigt (Fuks et al., 2000; Robertson et al., 2000;
Rountree et al., 2000). Dadurch sind DNA-Methylierung und Histonmodifikationen in der
epigenetischen Regulation der Transkription untrennbar miteinander verbunden.

1.- Einleitung Seite - 13 -
1.4.1.4.- Die pharmakologische Beeinflussbarkeit der epigenetischen Genregulation:
demethylierende Substanzen und HDAC-Inhibitoren.
Während genetische Mutationen in Tumorzellen irreversibel sind, sind epigenetische
Veränderungen pharmakologisch reversibel. Sowohl die DNA-Methylierung als auch die
Histonmodifikationen können mit verschiedenen Substanzen gehemmt werden, die als
Medikamente zur Behandlung von Neoplasien eingesetzt werden können.
Die erste „demethylierende“ Substanz (=DNMTs Inhibitor), die in der Behandlung
von Tumoren benutzt wurde, war 5-Azacytidine (Vidaza®) (Glover and Leyland-Jones, 1987).
Ein Nebenprodukt dieser Substanz, 5-Aza- 2´-deoxycytidin (Dacogen®, Decitabine, DAC),
zeigte einen stärkeren Effekt als demethylierende Substanz. 5-Azacytidin wird nur in 10 % in
die DNA und in 90% in die RNA inkorporiert, während 5-Aza 2'-deoxycytidin hingegen zu
100 % in die DNA eingebaut wird (Creusot et al., 1982). 5-Aza 2'-deoxycytidin wird während
der Replikationsphase in DNA eingebaut und inaktiviert DNMTs durch eine irreversible
kovalente Bindung (Juttermann et al., 1994). 5-Aza 2'-deoxycytidin wird zurzeit in der
Behandlung von AML und MDS erfolgreich eingesetzt (Rüter et al., 2006). Beide DNMT-
Inhibitoren wurden erst kürzlich durch die FDA für die Behandlung von MDS in den
Vereinigten Staaten zugelassen.
Abbildung 4.- Schematische Darstellung der Cytidin-Analoga 5-Azacytidin und 5-Aza 2'deoxycytidin (Decitabine), die in der klinische Behandlung von AML- und MDS- Patienten eingesetzt werden. 5-Azacytidin wird teilweise (10%) in DNA inkorporiert, wohingegen 5-Aza 2'deoxycytidin komplett in DNA eingebaut wird. Sie wirken als Inhibitoren der DNMTs.
NH2
O
R
N
5-Azacytidine
NH2
O
dR
N N
5-Aza 2' -deoxycytidine
N

1.- Einleitung Seite - 14 -
Als HDAC Inhibitoren sind verschiedene Substanzen beschrieben. Trichostatin A
(TSA) hat in vitro einen starken Effekt in der Reexpression von Genen durch Inhibition der
HDACs geziegt (Nervi et al., 2001; Suzuki et al., 2000). Es wurde für andere Substanzen, wie
z. B. MS-275 (Hu et al., 2003; Saito et al., 1999), SAHA (Kelly et al., 2003; Richon et al.,
1996), Depsipeptide (Nakajima et al., 1998) beschrieben, dass sie auch die Aktivität von
HDACs hemmen können. Auβerdem wurde nachgewiesen, dass die antiepileptische Substanz
Valproinsäure einen Effekt in der HDAC-Hemmung hat (Phiel et al., 2001). Deshalb wird
Valproat zurzeit in verschiedenen klinischen Studien in der Behandlung von AML (Bug et
al., 2005) und MDS (Kuendgen et al., 2004) eingesetzt.
Eine sehr wichtige Entdeckung war, dass die partielle Demethylierung von
bestimmten Genen durch Decitabine mit einen HDAC Inhibitor wie TSA einen
synergistischen Effekt in der Re-Aktivierung der Gentranskription hat (Cameron et al., 1999).
Diese Arbeit bildet die Rationale, um demethylierenden Substanzen und HDAC Inhibitoren in
Kombination bei der Behandlung von Tumorerkrankungen einzusetzen.
1.4.2.- Das PML/RARα-Fusionsprotein
1.4.2.1.- PML/RARα: ein Modell der Kooperation von DNA-Methyltransferase und
Histondeacetylasen bei der Entstehung der AML.
Als Beispiel für einen epigenetischen Pathomechanismus der Entstehung einer AML
gilt die Akute Promyelozyten-Leukämie (APL, AML M3) mit der Translokation t(15;17), die
zur Bildung des chimären Fusionsproteins PML/RARα führt. Dieses Fusionsprotein rekrutiert
HDACs und DNMTs an Promotoren von Zielgenen mit der Konsequenz, dass diese nicht
mehr exprimiert werden können (Di Croce et al., 2002). Das bewirkt, dass die myeloischen
Zellen im Promyelozyten-Stadium angehalten werden und sich nicht weiter differenzieren
können. In diesem grundlegenden Artikel wurde das erste Mal gezeigt, dass die beiden
epigenetischen Ereignisse (DNA methylierung udn Histone acetylierung) mit einem chimären
Fusionsprotein bei der Entstehung eines Tumors kooperieren.
Die APL Blasten zeigen eine hohe Sensitivität für All- trans-Retinsäure (ATRA) in
vitro (Chomienne et al., 1990) und in vivo (Castaigne et al., 1990). Die All-trans-Retinsäure
führt zu einer Differenzierung der Blasten in reife Granulozyten, charakterisiert durch
Expression von CD15 und die Anwesenheit von Auerstäbchen. Die Behandlung der APL mit
ATRA führt häufig zu einer Remission der Leukämie. Das PML/RARα Protein wird durch

1.- Einleitung Seite - 15 -
ATRA auf post-translationaler Ebene degradiert. Das Verhältnis PML-RARα/RARα wird
gesenkt und die myeloische Differenzierung der APL Zellen wird wieder ermöglicht (Raelson
et al., 1996). Pharmakologische Konzentrationen von ATRA können auch das PML/RARα
Fusionsprotein von rekrutierten HDACs (Grignani et al., 1998) und DNMTs (Di Croce et al.,
2002) freisetzen und somit kann die Expression der PML/RARα Zielgene wie z. B. RARβ2
stattfinden.
1.4.2.2.- RARβ2: Tumorsuppressor-Gen und Zielgen des PML/RARα Fusionsproteins.
Das „retinoid acid receptor β2“Gen (RARβ2) ist ein klassiches Tumorsuppressor-Gen,
dessen Repression auf mRNA- Ebene im Mammakarzinom (Swisshelm et al., 1994;
Widschwendter et al., 1997), Ovarialkarzinom (Caliaro et al., 1994) und
Plattenepithelkarzinom (Crowe, 1998) bestätigt wurde. Die Hemmung der Synthese von
RARβ2 mRNA kann durch Promotor-Hypermethylierung ausgelöst werden, wie z. B. im
primären Kolonkarzinom (Coté et al., 1998) und Mammakarzinom (Bovenzi et al., 1999)
festgestellt wurde. In Kolonkarzinom-Zelllinien wurde gezeigt, dass RARβ2 durch
Hypermethylierung reprimiert ist und durch 5-Aza-2'deoxycytidin demethyliert und re-
exprimiert werden kann (Coté and Momparler, 1997; Cote et al., 1998).
RARβ2 ist ein Zielgen des leukämie-spezifischen PML/RARα Fusionsproteins (Di
Croce et al., 2002). Sein Promotor wird nach der ektopen Expression von PML/RARα in PR9
Zellen hypermethyliert und nach der Gabe von ATRA und 5-Aza-2'-deoxycytidin in NB4
Zellen und APL Blasten von Patienten demethyliert und re-exprimiert.
1.4.3.- Epigenetische Ereignisse in der Pathogenese des AML1/ETO-Proteins:
therapeutische Konsequenzen.
Da AML1/ETO ein chimäres Fusionsprotein ist, in dem ein Traskriptionsfaktor und
die Rekrutierung der Histon-Deacetylasen impliziert sind, ist ein ähnlicher Pathomechanismus
wie im PML/RARα Fusionsprotein denkbar. AML1/ETO könnte möglicherweise zusätzlich
DNMTs an die Promotoren seiner Zielgene rekrutieren, um ihre Transkription durch DNA-
Methylierung zu hemmen.
Zusammenfassend sind diese Daten Hinweise auf einen epigenetischen
Pathomechanismus der AML1-ETO-positiven AML und bilden eine mögliche Rationale für
den Einsatz demethylierender Substanzen (5-Azacytidine, 5-Aza-2-Deoxycytidine) in

1.- Einleitung Seite - 16 -
Kombination mit HDAC-Inhibitoren in der Therapie der AML, um damit die Expression der
unterdrückten Genen wiederherzustellen.
1.5.- Die U937-9/14/18-Zelllinie
Die Rolle des Fusionsproteins AML1/ETO bei der Leukämogenese und der genaue
Pathomechanismus sind noch nicht ganz geklärt. Die stabile ektope Expression des
Fusionsgens wird von den meisten Zelllinien nicht toleriert und die transienten
Transfektionen und Reporter-Konstrukte geben nicht genügend Information über die „in
vivo“ Funktion dieses Proteins. Um Zielgene des AML1/ETO-Proteins zu entdecken und um
die zellulären Effekte zu verstehen, wurde von Dr. Manfred Fliegauf in der Arbeitsgruppe von
Prof. Lübbert ein induzierbares AML1/ETO-System in der U937-Zelllinie entwickelt
(Fliegauf et al., 2004).
Abbildung 5.- Molekularer Pathomechanismus vom AML1/ETO. Das AML1/ETO-Protein rekrutiert HDACs und DNMTs an seine Zielgene und so wird die Expression durch einen epigenetischen Mechanismus unterdrückt. Nach dem Einsatz der DNA-demethylierenden Substanzen und HDAC-Inhibitoren kann die Expression der AML1/ETO-Zielgene wiederhergestellt werden.
CCGTGT/cGGTATGTGTCTG
Hypomethylating agents :
5-azacytidine,
5-aza 2´-deoxycytidine
HDAC inhibitors: valproic acid, TSA,
MS-275, SAHA
CBFßAML1
ETO
NCor mSin3
HDAC1
CGTCGATCGTCGACGTATAATGCTCTC
DNMT1
MeCP2
MDB2
CCGTGT/cGGTATGTGTCTGACTGCTGAGTATGA
Targetgenes
WBSCR5
IL-3
LZM
LST1
P14
CCGTGT/cGGTATGTGTCTG
Hypomethylating agents :
5-azacytidine,
5-aza 2´-deoxycytidine
HDAC inhibitors: valproic acid, TSA,
MS-275, SAHA
CBFßAML1
ETO
NCor mSin3
HDAC1
CGTCGATCGTCGACGTATAATGCTCTC
DNMT1
MeCP2
MDB2
CCGTGT/cGGTATGTGTCTGACTGCTGAGTATGA
Targetgenes
WBSCR5
IL-3
LZM
LST1
P14

1.- Einleitung Seite - 17 -
Der stabile transfizierte Klon, 9/14/18, ist mit einem Ecdyson-induzierbaren Zwei-
Vektor-Expressionssystem ausgestattet (No et al., 1996). Der erste Vektor (pVgRXR) kodiert
einen konstitutiv exprimierten heterodimeren Rezeptor (VgEcR/RXR), der durch ein
Insektenhormon (Ecdyson) aktiviert werden kann. Der aktivierte Rezeptor kann die
Expression des gewünschten Gens regulieren, welches unter der Kontrolle eines
induzierbaren Promotors auf dem zweiten Vektor (pIND-GenX) liegt. Als induzierende
Substanzen können die Ecdyson-Agonisten Muriston und Ponasteron A benutzt werden.
Bisher wurde kein Effekt dieser Moleküle oder des Hormonrezeptors in Säugerzellen
beschrieben.
Abbildung 6.- Das Ecdyson-induzierbare System besteht aus 2 verschiedenen Vektoren. Ein Vektor trägt das Fusionsgen unter der Kontrolle eines pIND-Promoters. Der zweite Vektor exprimiert einen RXR-Rezeptor und VgEcR (Ecdysone Rezeptor) konstitutiv. Wenn der Ligand (Muristone, Ponasteron) hinzugegeben wird, kommt es zu einer Heterodimerisierung der RXR und VgEcR Rezeptoren. Dadurch wird der pIND-Promoter aktiviert und die Expression des Fusionsgens AML1/ETO induziert.
The Ecdysone-inducible Expressions-System(adapted from Invitrogen, Gronigen, NL)
AML1/ETO
AML1/ETO
AML1/ETO
AML1/ETO

1.- Einleitung Seite - 18 -
1.6.- Methoden zur Analyse der DNA-Methylierung
Die Methoden zur Analyse von DNA-Methylierung können in 2 Gruppen klassifiziert
werden. Zum einen Methoden, die DNA-Methylierung auf globaler genomweiter Ebene
untersuchen. Sie haben den Zweck neue Gene oder chromosomale Regionen zu finden, in
denen Methylierungsveränderungen vorliegen, oder den gesamten Methyl-Cytosinen Gehalt
der DNA zu bestimmen. Zu dieser Gruppen gehören beispielweise: Restriction Landmark
Genomic Scanning (RLGS) (Plass et al., 1996; Smiraglia and Plass, 2002, , 2003), methyl-
Cytosine ChIP (Weber et al., 2005) , MethylScope (Lippman et al., 2004), AIMS (Paz et al.,
2003), capillary electrophoretic analysis of genomic DNA (Stach et al., 2003)
Zum anderen gibt es Methoden, die die DNA Methylierung in bestimmten Genen
untersuchen. Diese haben das Ziel methylierte CpGs in einem bestimmten Gen zu
quantifizieren. Zu dieser Gruppe gehören Southern Blot mit methylsensitiven Enzymen (Bird
and Southern, 1978), Bisulfite DNA Sequenzierung (Frommer et al., 1992), Methyl-Sensitive
PCR (MSP) (Herman et al., 1996), COBRA (Xiong and Laird, 1997) und Methylierung-
Sensitive single nucleotide Primer Extension (MS-SnuPE) (Gonzalgo and Jones, 1997).
RLGS wurde als Screening Methode für DNA-Methylierungsveränderung nach der
Induktion des AML1/ETO-Proteins in dieser Arbeit benutzt, und Southern Blots wurden nach
Restriktion mit einem methylsensitiven Enzym (Not I) als Validierungsexperiment der RLGS-
Daten durchgeführt. Diese Methode wurde deshalb ausgewählt, um neue robuste
Methylierungsveränderungen innerhalb CpG-reicher Regionen der möglichen AML1/ETO-
Zielgene zu identifizieren.
1.6.1.- Restriction Landmark Genomic Scanning (RLGS)
In Kooperation mit der Arbeitsguppe von Prof. Dr. C. Plass (University of Columbus,
Ohio, USA) wurde eine RLGS-Analyse mit den induzierbaren Zellen durchgeführt, um
Methylierungsveränderungen nach der Induktion von AML1-ETO auf einer globalen DNA-
Ebene zu untersuchen. Das Ziel dieser Methode ist die Identifizierung von differentiell
methylierten Sequenzen.
Die RLGS-Methode basiert auf einem sequentiellen Restriktionsverdau, kombiniert
mit zweidimensionaler Gelelektrophorese: zuerst wird intakte genomische DNA mit dem
methylsensitiven Enzym Not I geschnitten. Not I schneidet nur an demethylierten CpGs

1.- Einleitung Seite - 19 -
innerhalb der Restriktionsstellen. Die überlappenden Enden werden mit radioaktiven GTP-
und CTP-Nukleotiden gefüllt. Dann wird eine Gel-Elektrophorese auf einem zwei-
dimensionalen Polyacrylamid-Gel durchgeführt. Nach Auftrennung in der ersten Dimension
wird ein zweiter Restriktionsverdau in situ mit einem nicht-Methylierungs-sensitiven Enzym
durchgeführt. Anschließend erfolgt eine weitere Elektrophorese. Schließlich kann man im
Autoradiogramm die Signale von sogennanten Spots, die DNA-Fragmenten entsprechen,
detektieren und so zwei Proben miteinander vergleichen. Wenn eine Veränderung von einem
Spot gefunden wird, ist eine schnelle Identifikation der Spots durch den Vergleich mit
Master-Profilen, erstellt aus peripheren Blut-Lymphozyten, möglich. Ansonsten ist eine
Identifikation der Spots durch direkte Subklonierung und Sequenzierung möglich.
Abbildung 7.- RLGS Methode. A.- Die genomische DNA wurde mit dem nicht methylierungssensitive Restriktionsenzym EcoR V und dem methylierungssensitive Restriktionsenzym Not I verdaut. Die geschnittene DNA wurde mit radioaktiv markierten Cytosinen und Guanidinen an der Not I Restriktionsstelle aufgefüllt. Eine zwei dimensionale Gelelektrophorese wird durchgeführt, und die verschiedenen DNA-Fragmente werden durch Autoradiographie detektiert. B.- Methylierungsveränderungen werden nach dem Vergleich von 2 Gelen von verschiedenen Proben nachgewiesen. C.- Beispiel einer Autoradiographie von einer zwei dimensionalen Gelelektrophorese nach der RLGS Methode.
NotI
NotI
NotI
NotI
NotI
NotI
unmethylated Allele-specificmethylation
methyated
NotI
NotI
NotI
NotI digest
RLGS1st-D
2nd-D
1st-D
2nd-D
1st-D
2nd-D
endlabeling
NotI
NotI
NotI
NotI
NotI
NotI
unmethylated Allele-specificmethylation
methyated
NotI
NotI
NotI
NotI digest
RLGS1st-D
2nd-D
1st-D
2nd-D
1st-D
2nd-D
endlabeling
Plass Nat. Gen. 1996Smiraglia and Plass Oncogene 2002Smiraglia and Plass Ann. N.Y. Aca. Sci. 2003
23
1

1.- Einleitung Seite - 20 -
1.6.2.- Bisulfit-Sequenzierung
Die Bisulfitbehandlung genomischer DNA (Clark et al., 1994; Frommer et al., 1992)
ermöglicht die Unterscheidung zwischen einzelnen methylierten und unmethylierten
Cytosinen über einem definierten DNA-Abschnitt. Der Schlüsselmechanismus für den
Nachweis methylierter Cytosine ist durch die selektive chemische Reaktion vom Natrium-
Bisulfit mit den unmethylierten Cytosinen festgelegt. Hierbei wird das unmethylierte Cytosin
durch Desaminierung in Uracil umgewandelt und das methylierte Cytosin bleibt unverändert.
1.7.- LAT2-Gen als neues Zielgen von AML1/ETO
Mit Hilfe des AML1/ETO-induzierbaren Systems in U937-Zellen wurden in einer
cDNA-RDA Analyse Gene identifiziert, deren Expression nach der Induktion des
AML1/ETO Proteins reguliert wurden. Ein Kandidat für differentielle Expression war das
Abbildung 8.- Bisulfit -Konversion der DNA. Der entscheidende Schritt, um Methyl-Cytosine mit Bisulfit-Sequenzierung nachzuweisen, ist die Bisulfit- Konversion der DNA. Nach der Sulfonierungs Reaktion wird eine Bisulfit-Gruppe im Cytosin aufgebaut. Bisulfit-Cytosin wird in Bisulfit-Uracil nach der Desaminierung konvertiert. Bisulfit-Uracil wird alkalisch desulfoniert und damit in ein Uracil-Molekül umgewandelt. Diese Reaktion findet nur an unmethylierten Cytosinen statt. (Modifiziert nach DNA Methylation, CRC Press, Manel Esteller).
NH2 NH2
O O
OO
O O
N
N N
NN
N
+HN
+HN
SO3-
SO3-
HSO3-
OH -
H2ONH4+
HSO3-
OH -
Cytosine CytosineSulfonate
Uracil UracilSulfonate
1.-Sulfonation
2.- HydrolyticDeamination
3.- AlkaliDesulfonation

1.- Einleitung Seite - 21 -
LAT2 (WBSCR5/NTAL/LAB) Gen. Die AML1/ETO-vermittelte Repression dieses Gens
wurde in AML1/ETO-induzierbaren Zellen sowie in verschiedenen AML1/ETO-positiven vs
negativen Zelllinien und in primären Blasten von AML-Patienten validiert (Fliegauf et al.,
2004).
Das Williams Beuren Syndrom (WBS) ist eine Krankheit, die durch eine
chromosomale Mikrodeletion der Bande 7q11.23, von ca. 8x106 bp, verursacht wird. 24 Gene
sind in dieser Mikrodeletion beinhaltet und eines davon ist das Williams Beuren Syndrom
Critical Region 5 (WBSCR5) Gen. Die Diagnose wird mittels FISH Analyse mit dem
Kosmid, der Teil des Elastin-Gens enhält, gestellt. Das Elastin-Gen befindet sich in der Mitte
der gemeinsamen Deletion. (Lowery et al., 1995). Kinder mit diesem Syndrom haben
folgenden Phänotyp in unterschiedlicher Ausprägung: dismorphologe Gesichtform,
kardiovaskuläre Abnormalitäten (z. B. supravalvuläre Aortenstenose und periphäre
Pulmonalstenose), ophthalmologische und auditive Fehlfunktionen (z. B. Hyperakusis und
"Visual-Spatial processing" Dysfunktion), sowie eine Hyperkalziämie. Allerdings haben
Kinder mit WBS auch bemerkenswerte neurologische und kognitive Eigenschaften, wie einen
umfangreichen Wortschatz, musikalische Kreativität und ein ausgeprägtes soziales Gespür.
Andererseits fallen bei diesen Kindern aber auch Schwächen im kognitiven Bereich auf wie z.
B. Aufmerksamkeitsstörungen, Ängste und Phobien (Tassabehji, 2003). Bisher wurde nur die
Fehlfunktion des Elastin-Gens in Maus-Modellen mit supravalvulärer Aortenstenose
nachgewiesen (Li et al., 1998). Die Rolle anderer Gene für den Phänotyp des WBS ist
weitestgehends noch ungeklärt.
Das Produkt des WBSCR5-Gens ist ein 28 kDa Protein, inzwischen auch Linker for
Activation of T cells 2 (LAT2), Non T Cell Activation Linker (NTAL) (Brdicka et al., 2002)
oder Linker for Activation of B cells (LAB) (Janssen et al., 2003) genannt. Das LAT2-Protein
gehört zur Familie der Binder- oder Adaptormoleküle. Dies sind Membranproteine, die durch
eine nicht-kovalent Protein-Protein Interaktion die multimolekulare Transduktionsignal-
Komplexe organisieren und regulieren. Die extrazellulären Signale werden durch die
Adaptormoleküle von den Rezeptoren zum Zytoplasma vermittelt. Dadurch werden nicht nur
die extrazellulären Signale von Rezeptoren weitergeleitet, sondern auch laterale Signale, wie
z. B. die Rekrutierung anderer Rezeptoren, und die Modifizierung der Aktivität von
Membranproteinen, wie z. B. Integrine, mittels Adaptormoleküle reguliert (Simeoni et al.,
2004).
Das LAT2-Protein wird nach Cross-linking des B-Zell-Rezeptors in B-Zellen (Janssen
et al., 2003), dem FCєRI (High Affinity Receptor for IgE) und dem c-Kit- Rezeptor in

1.- Einleitung Seite - 22 -
Mastzellen (Tkaczyk et al., 2004) phosphoryliert und dadurch aktiviert. Nach seiner
Aktivierung interagiert das LAT2-Protein mit Grb2, Sos-1, Gab-1 und c-Cbl (Brdicka et al.,
2002) . LAT2 kann die Entwicklung von Thymozyten in „Linker for Activation of T cells“
(LAT) und homologe Proteine von LAB in T Zellen von Knock-out Mäusen wiederherstellen
(Janssen et al., 2003). Diese Daten weisen auf eine partiell redundante Rolle von LAT2
(NTAL/LAB) und seinem Homolog in T Zellen, LAT, hin.
LAT2 reguliert negativ die Degranulation von Mastzellen nach der Aktivierung in
LAT2 (NTAL/LAB) Knock-out Mäusen (Zhu et al., 2004). Diese Funktion von LAT2 wird
durch die Hyperaktivität des LAT-Proteins in LAT2-Knock-out Mäusen verursacht, da LAT
das PLC-γ1 und dadurch den Kalziumfluss regulieren kann und mehr mRNA und
phosphoryliertes LAT-Protein vorhanden ist (Volna et al., 2004). Überraschend war, dass
keine Veränderung in der Funktion und Entwicklung der B-Zellen von LAT2-Knock-out
Abbildung 9.- Transduktionssignalwege des LAT2-Proteins in Mastzellen. Das LAT2-Protein kann durch den c-Kit-Rezeptor und FCεR I phosphoryliert und dadurch aktiviert werden. Nach seiner Aktivierung interagiert es mit zytosolischen Molekülen wie Grb2, Gab-1, Sos-1 und c-CBL. Diese Moleküle sind in der Aktivierung verschiedener Transduktionssignalwege (MAPK-Kinase, PI3-Kinase und Protein-Degradation durch Ubiquitinierung) beteiligt. Modifiziert nach Tkaczyk et al, Chem Immunol. Allerg. 2005

1.- Einleitung Seite - 23 -
Mäusen zu beobachten war. (Wang et al., 2005). Letzlich könnte die Abwesenheit von
LAT2-Protein eine Hyperaktivität der T Zellen verursachen, und damit eine konsequente
Autoimmune Erkrankung in LAT2-Knock-out Mäusen auslösen (Zhu et al., 2006).
Die Funktion dieses Gens und die Bedeutung seiner Repression ist in der Myelopoese
bisher ungeklärt und deshalb sind weitere Untersuchungen notwendig. In dieser Arbeit
werden funktionelle Untersuchungen von LAT2 in der Hämatopoese durchgeführt, um die
Rolle bei der Entstehung von AML abzuklären.
1.8.- Myeloische Differenzierung
Die embryonale Entwicklung des Menschen braucht die regulierte und koordinierte
Expression von verschiedenen Transkriptionsfaktoren, die zugleich Kaskaden der
Genexpression induzieren. Dieser regulierte Prozess hat die Differenzierung der embryonalen
Stammzellen in den verschiedenen Gewebe zufolge. Die Hämatopoese ist wie die embryonale
Entwicklung durch verschiedene Transkriptionfaktoren reguliert, die von den Stammzellen
bis hin zu reifen Zellen führt (Skalnik, 2002).
Während der Differenzierung der Zellen in der myeloischen Reihe werden Gene für
wichtige Funktionen der Zellen in verschiedenen Reifungsstufen ab- und angeschaltet. Zum
Beispiel wird Myeloperoxidase im Übergang von der myeloblastären zur promyelozytären
Phase exprimiert (Borregaard et al., 1995), wobei CD11b in der letzten Stufe der
granulozytären (metamyelozytäre und segmentierte Phase) und monozytären Differenzierung
hochreguliert wird (Rosmarin et al., 1989). Die Expression dieser Gene ist durch spezifische
myeloische Transkriptionfaktoren wie AML1 (Wang and Speck, 1992), CCAAT/enhancer
binding Protein α (C/EBPα) (Landschulz et al., 1989), PU.1 (Chen et al., 1995) reguliert, die
häufig durch Mutationen, chromosomale Aberrationen und epigenetische Disregulation in
ihrer Expression und Funktion bei hämatologischen Erkrankungen verändert wird.
Der molekulare Mechanismus der Genregulation beruht auf der Interaktion von
Transkriptionfaktoren und dem Promotor-Enhancer solcher Gene, wie am Beispiel der
Neutrophilen Elastase gezeigt wurde (Oelgeschlager et al., 1996). Außerdem werden die
Promotoren von Proteinase 3 und MPO während der myeloischen Zelldifferenzierung
(Lübbert et al., 1991b; Lübbert et al., 1999) demethyliert, ihre Chromatinstruktur verändert,
und dadurch wird die Expression dieser Gene induziert.

1.- Einleitung Seite - 24 -
Myeloische Zelllinien wurden aus Patienten mit AML oder CML in der Blastenkrise
etabliert. Sie sind auf verschiedenen Stufen der myeloischen Zelldifferenzierung blockiert und
können durch Substanzen wie DMSO, Phorbolester, ATRA, 1,25-Dihydroxyvitamin D3 in
vitro differenziert werden (Lübbert et al., 1991a). Diese Differenzierungsmodelle der
Myelopoese werden benutzt, um die Expression myeloischer Gene während der
verschiedenen Phasen der Zelldifferenzierung zu untersuchen. Diese Arbeit hatte das Ziel, die
Expression von LAT2-Gen während der myeloischen, erythrozytären und megakaryozytären
Differenzierung von verschiedenen Zelllinien zu untersuchen.
Abbildung 10.- Schematische Darstellung der Myelopoese. Die myeloische Progenitorzellen
können sich in Myeloblasten oder Pronormoblasten differenzieren. Die Pronormoblasten sind Vorläuferzellen der roten Reihe. Myeloblasten können sich weiter in Promyelozyten, Megakaryozyten, unreife Monoblasten, Eosinophile und Basophile differenzieren. In jeder Differenzierungsreihe werden unter der Kontrolle von myeloischen spezifischen Traskriptionsfaktoren wichtige Gene für die Funktion der Zellen exprimiert oder abgeschaltet. Modifiziert nach http://www.userlogic.com/anll/studyaids/maturationchart/maturation_chart.html

1.- Einleitung Seite - 25 -
CD34+ ist der Oberflächemarker für hämatopoetische Stammzellen, die sich in alle Zellen des
myeloischen und lymphatischen Systems differenzieren können (Srour et al., 1991). Die
Kultivierung dieser Zellen erfolgt in serumfreiem Medium mit einem Zytokincocktail (IL-3,
IL-6, SCF und FLT3) (Guo et al., 2006). Mit G-CSF kann die Differenzierung in
Granulozyten (Berliner et al., 1995) und mit GM-CSF und IL-4 in Monozyten/Dendritische
Zellen (Ferlazzo et al., 2000) ex vivo erfolgen. Diese Differenzierungmodelle wurden in der
vorliegenden Arbeit benutzt, um die Expression der Gene während der CD34+
Zelldifferenzierung zu untersuchen.

2.- Ziele des Projektes Seite - 25 -
2.- Ziele des Projektes
Das erste Ziel dieser Doktorarbeit war die Validierung der RLGS-Daten mittels
Southern Blots, um neue Zielgene des AML1/ETO-Proteins zu entdecken. Mit der
Validierung der RLGS-Daten konnte ein neuer epigenetischer Mechanismus des AML1/ETO-
Proteins bestätigt werden, durch den das chimäre Fusionsprotein die Expression seiner
Zielgene beeinflussen kann. Bisher wurde nur ein Artikel in der wissenschaflichen Literatur
veröffenlicht, in dem eine methylierende Aktivität des AML1/ETO-Proteins in einer
bekannten AML1/ETO-Zielgen gezeigt wurde. Diese Arbeit wäre die Erste, in der Zielgene
von AML1/ETO durch Methylierung auf ihren Promotoren mit einem randomisierten
globalen Screening untersucht wurde.
Eine weitere Fragestellung dieser Doktorarbeit war, ob die durch Methylierung von
Zielgenen des AML1/ETO-Proteins, deren Expression nach der Induktion des Fusionsproteins
verändert wird. Die Charakterisierung der ermittelten Gene in Bezug auf das Vorhandensein
von CpG-Inseln und die Bestätigung der Vermittlung des Effekts durch AML1-ETO wären
die nächsten Schritte.
Funktionelle Analysen der validierten Gene als Beitrag zum Verständnis ihrer Rolle
für die Pathogenese der AML waren das dritte Ziel dieser Doktorarbeit. Ein von unserer
Arbeitsgruppe identifiziertes AML1/ETO-Zielgen, LAT2, wurde durch funktionelle Analysen
untersucht, um seine Rolle in der Hämatopoese und Leukemogenese aufzuklären. Da die
Funktion dieses Gens bisher in der Hämatologie nicht untersucht wurde, war die
Untersuchung seiner Funktion und die Methodik dafür offen. Den Mechanismus der
Repression durch AML1/ETO zu untersuchen und diese Repression durch epigenetische
aktive Therapien zu hemmen, war ebenso ein Ziel dieser Arbeit.
Die Entdeckung neuer Zielgene und der Mechanismus der Repression vom
AML1/ETO konnte weiter die Pathogenese der AML aufklären und die Therapie der AML
durch neue zielgerichtete Substanzen verbessern. Dies könnte künftig die Überlebensdauer
und die Lebensqualität von AML-Patienten optimieren.
Momentan wird auch nach molekularen Markern gesucht, die die Wirkung der
zielgerichteten Therapien überwachen. Ein Zielgen, das durch epigenetische aktive Therapien
in vitro re-exprimiert ist, wäre geeignet für das Monitoring der Effektivität dieser Therapien.

3.- Material und Methoden Seite - 26 -
3.- Materialien und Methoden
3.1.- Materialien
3.1.1.- Zellen
3.1.1.1.- Zelllinien
Alle Zelllinien wurden von der Deutschen Sammlung von Mikroorganismen und Zellkulturen
(DSMZ) GmbH, Braunschweig bezogen.
• Kasumi-1
Die Zelllinie Kasumi-1 wurde 1989 aus dem peripheren Blut eines siebenjährigen japanischen
Jungen mit akuter myeloischer Leukämie im zweiten Rezidiv nach
Knochenmarktransplantation gewonnen (Asou et al., 1991). Die Kasumi-1-Zellen wurden
einer akuten myeloblastischen Leukämie, nach der Französich-Amerikanisch-Britischen
Klassifikation FAB-M2, zugeordnet. Die Kasumi-1-Zelllinie weist eine chromosomale
Abnormalität auf, die Translokation t(8;21)(q22;q22). Das resultierende Fusionsgen heißt
AML1/ETO.
• HL-60
HL-60 ist eine Zelllinie, die 1976 aus dem peripheren Blut einer 35-jährigen Patientin mit
akuter myeloischer Leukämie etabliert wurde. Es erfolgte die Zuordnung der Zellen zu einer
akuten myeloblastischen Leukämie in Reifung FAB M2 (Dalton et al., 1988). Die HL60
Zelllinie weist einen komplexen Karyotyp auf.
• U-937
Die Zelllinie U-937 wurde 1974 aus dem Pleura-Exsudat eines 37-jährigen Mannes mit
generalisiertem histiozytären Lymphom gewonnen (Sundstrom and Nilsson, 1976). Der
histiozytäre Ursprung dieser Zelle wurde durch den Nachweis hoher Lysozym-Produktion
und starker Esterase-Aktivität gesichert. Diese Zelllinie weist einen komplexen Karyotyp auf
und hat die t(10;11)(p14;q23), die zur MLL chromosomalen Aberrationen gehört und typisch
für die AML FAB M5 ist.

3.- Material und Methoden Seite - 27 -
• NB-4
NB-4-Zellen wurden aus dem Knochenmark einer 23-jährigen Frau mit akuter
Promyelozyten-Leukämie (FAB M3) im zweiten Rezidiv etabliert (Lanotte et al., 1991).
Diese Zelllinie weist die Translokation t(15;17)(q22;q11-12) auf und trägt somit das
Fusionsgen PML-RARα.
• LCL-GK
LCL-GK-Zelllinie ist eine Epstein-Barr Virus (EBV) immortalisierte lymphoblastische B-
Zelllinie (Kapp et al., 1999).
• Jurkat
Die Jurkat-Zelllinie wurde im Jahr 1976 aus dem peripherischen Blut eines 14-jährigen
Patienten mit einer akuten lymphoblastischen Leukämie (ALL) in erstem Rezidiv isoliert. Die
Zellen gehören zur Kategorie „menschlicher T Zell Leukämie“ Zelllinie (Schneider et al.,
1977).
• 9/14/18
Die 9/14/18-Zelllinie wurde aus der U937-Zelllinie mit einem Ecdyson- induzierbaren
Expressionsvektor, pVgRXR und dem AML1/ETO Konstrukt im pIND-Vektor (Invitrogen)
gemacht (Fliegauf et al., 2004). Diese Zelllinie wurde in der AG Prof. Lübbert entwickelt
und ist eine von 3 Zelllinien, in denen AML1/ETO induzierbar ist. In anderen Zelllinie wird
die AML1/ETO konditionelle Expression durch Zink und Tetrazyklin-off induziert (Alcalay
et al., 2003; Burel et al., 2001).
• K562
Die K562 Zelllinie wurde 1970 aus dem Pleuraerguß einer 53-jährigen Patientin mit
chronischen myeloischen Leukemia (CML) in Blastenkrise isoliert. Die Zellen bilden
Hämoglobin und tragen das Philadelphia-Chromosom mit dem BCR-ABL Fusionsgen
(Lozzio and Lozzio, 1975).
3.1.1.2.- Bakterien
E. Coli OneShot Mach1tm T1RChemically Competent (Invitrogen) mit dem pENTR/D-
TOPO® Cloning Kit wurden benutzt.

3.- Material und Methoden Seite - 28 -
3.1.1.3.- Primäre Zellen von gesunden Spendern
• Periphere mononukleäre Zellen
Peripheren mononukleären Zellen des peripheren Blutes wurden aus Eigenblutspenden durch
Auftrennung im Dichtegradienten mit Ficolllösung gewonnen.
• Granulozyten
Nach Ficollgradienten-Zentrifugation von peripherem Blut wurden die Granulozyten aus der
untersten Phase isoliert, nach osmolarischer Lyse der Erythrozyten.
• Monozyten
Sie wurden aus den peripheren mononukleären Zellen des peripheren Blutes isoliert und von
den Lymphozyten durch ihre eigene Adhärenz in einer Kulturflasche getrennt.
• Lymphozyten
Die Zellen, die von den mononukleären Zellen des peripheren Blutes keine eigene Adhärenz
hatten, wurden als Lymphozyten isoliert.
3.1.1.4.- Primäre Zellen von Patienten
• Blasten
Patienten-Proben wurden aus der Tumorbank der Abteilung I der Inneren Medizin des
Univertätsklinikum Freiburg genommen, nach Genehmigung von Prof. Dr. Finke, Prof. Dr.
Veelken und PD. Dr. Martens. Alle Patienten hatten der Klinik die Erlaubnis für die Nutzung
ihrer Proben für Forschungszwecke unterschrieben. Manche Patientenproben, die in der
Tumorbank nicht vorhanden waren, stammen aus dem Molekulardiagnose-Labor der
Abteilung.
• CD34+ Zellen
Die CD34+ Zellen wurden aus dem Blut von Patienten mit soliden Tumoren isoliert. Diese
Zellen werden als "normale Stammzellen (CD34+ Zellen)" bezeichnet, da sie aus Patienten
ohne hämatologische Krankheit gewonnen wurden.

3.- Material und Methoden Seite - 29 -
3.1.2.- Materialien für Zellkultur
3.1.2.1.-Zellkulturmedien, Zusätze und Reagenzien
5-Aza-2'-deoxycytidin (DAC, Decitabine) #A3656 Sigma Aldrich
ATRA (all trans Retin-Säure) #A2625 Sigma
Dimethylsulfoxid (DMSO) #D8418 Sigma Aldrich
Phosphate Buffered Saline (D-PBS) Gibco BRL / Invitrogen
Fötales Kälberserum (FCS) Gibco BRL / Invitrogen
Geneticin (G418) #11811-064 Gibco RBL
Hemin #51280 Sigma
Lymphocyte Separation Medium (LSM 1077) PAA Laboratories Gmbh
MS-275 #02112316 Schering
Natriumpyruvat (100 mM) Gibco BRL / Invitrogen
Penicillin-Streptomycin (10000 U/mL) Gibco BRL / Invitrogen
PMA (Phorbol 12-myristate 13-acetate) #P1585 Sigma
Ponasteron A #H101-03 Invitrogen
Suberoylanilid Hydroxam Säure (SAHA)
#270288M005
Alexis Biochemicals
RPMI 1640 mit L-Glutamine Gibco BRL / Invitrogen
Trichostatin A #T8552 Sigma
Trypanblau 0.4% Gibco BRL / Invitrogen
Türks Lösung Merck
Valproic acid #P4543
Zeocin # R325005 Invitrogen
3.1.2.2.- Sonstige Materialien, Plastikwaren
Glaspipetten (2,5,10,25 ml) Corning
Kryoröhrchen (1,8ml) Corning
Mikro-Schraubröhrchen Sarstedt
Neubauer Zählkammer Marienfeld
Reaktionsgefäße ( 1,5 , 2 ml ) Eppendorf, Greiner
Sterile Schraubdeckelröhrchen (15, 50 ml) Greiner

3.- Material und Methoden Seite - 30 -
Sterile Spitzen Molecular Bio Products , Corning
Zellkulturflaschen Nunc
Pipetboy Integra Biosciences
3.1.3.- Materialien für die Bakterienkultur
Ampicillin, Natriumsalz Merck
Bacto-Agar Difco
Bacto-Hefe-Extrakt Difco
Bacto-Trypton Difco
LB (Luria-Bertani)-Medium (1% Bacto-Trypton, 0.1% NaCl, 0.5% Bacto-Hefe-Extrakt, pH
7.0): Zur Herstellung von LB-Medium wurden die einzelnen Komponenten in H2O aufgelöst,
der pH mit NaOH-Lösung eingestellt und das Medium anschließend autoklaviert. Die
Aufbewahrung erfolgte bei +4ºC im Kühlschrank. Antibiotika (50 µg/ml Ampicillin) wurden
stets unmittelbar vor Beimpfung des Mediums zugesetzt.
Alternativ wurde auch folgendes Fertigpulver verwendet:
LB-Medium Gibco BRL / Invitrogen
LB-Agarplatten (15 g Bacto-Agar pro 1000 ml LB-Medium, 150 µg/ml Ampicillin):
Zur Herstellung von LB-Agarplatten wurden 15g Bacto-Agar zu 1000 ml LB-Medium vor
dem Autoklavieren zugesetzt. Nach dem Abkühlen (ca. 37°C) wurden Antibiotika
zugefügt (150 µg/ml Ampicillin), die Platten gegossen und bei 4°C aufbewahrt.
3.1.4.- Materialien für molekularbiologische Arbeiten
3.1.4.1.- Chemikalien und Reagenzien
Agarose Serva
Ammoniumacetat Merck
Aprotinin Sigma-Aldrich

3.- Material und Methoden Seite - 31 -
Aqua ad iniectabilia Braun
Aqua Roti-Phenol Roth
Borsäure Merck
Rind Serumalbumin (BSA) Sigma-Aldrich, Invitrogen
Bromphenolblau Merck
Chloroform Merck
Dimethylsulfoxid (DMSO) Sigma Aldrich
Dithiothreitol (DTT) 0,1 M Invitrogen
ECLPlus Western Blot Detektionsreagenzien Amersham
EDTA Sigma Aldrich
Essigsäure Merck
Ethanol Merck, BDH Prolabo
Ethidiumbromid Qbiogen
Formaldehyd Riedel de Häen
Formamid Merck
GammaBind G Sepharose GE Healthcare
Glyzerin Serva
Glykogen Ambion
Guanidiniumthiocyanat Fluka
HEPES Sigma-Aldrich
Isoamylalkohol Merck
Isopropanol Roth
Kaliumhydrogencarbonat Merck
Lachssperma-DNA Sigma-Aldrich
Leupeptin Sigma-Aldrich
Light Cycler 480 SYBR Green I Master Mix Roche
Magermilchpulver AppliChem
Methanol Merck
3- N-morpholino propanesulfonic acid
(MOPS)
Serva
Natriumacetat, wasserfrei Merck
Natriumchlorid Fluka
Natriumchlorid (5M) Merck
Natriumcitrat Merck

3.- Material und Methoden Seite - 32 -
Natriumdeoxycholat Merck
DiNatriumdihydrogenphosphat Merck
Natriumhydrogenphosphat Merck
Natronlauge 1mol/l Merck
Natronlauge 5mol/l Merck
N-Lauryl-Sarcosine Sigma-Aldrich
Nonidet P40 (NP40) Sigma-Aldrich
NuPAGE Antioxidant Invitrogen
NuPAGE MES SDS Running Buffer (20x) Invitrogen
NuPAGE Sample Loading Buffer (4x) Invitrogen
NuPAGE Sample Reducing Agent (10x) Invitrogen
NuPAGE Transfer Buffer (20x) Invitrogen
PBS Invitrogen
Pepstatin A Sigma-Aldrich
Phenylmethansulfonylfluorid (PMSF) Sigma-Aldrich
Propidium-Iodid Sigma-Aldrich
Protein-G-Plus-Agarose Santa Cruz Biotechnology
Proteinase K Merck
Restriktionspuffer NEB 1-4 New England Biolabs
RNase A Sigma-Aldrich
RNAsin Plus (RNAse Inhibitor) Promega
Salzsäure 1mol/l Merck
Salzsäure 5 mol/l Merck
Dodecylsulfat-Natriumsalz (SDS) Serva
SeeBluePlus2 Gel Marker Invitrogen
Sephadex G50 fine Pharmacia
Sephadex G50 medium Pharmacia
Sukrose Sigma-Aldrich
tri-Natriumcitrat Merck
tri-Natriumcitrat-Dihydrat Merck
Tris-Base Sigma-Aldrich
Tris-Hydrochlorid Sigma-Aldrich
Titriplex III / EDTA Merck
Triton X-100 Sigma-Aldrich

3.- Material und Methoden Seite - 33 -
Tween 20 Sigma-Aldrich,
5-bromo-4-chloro-3-indolyl-b-D-
Galactopyranosid (X-Gal)
Merck
Xylencyanol Sigma-Aldrich
β-Mercaptoethanol Sigma-Aldrich
3.1.4.2.- Standardlösungen und Puffer
Die Herstellung von Puffern und gepufferten Salzlösungen sowie alle Methoden für Arbeiten
mit Nukleinsäuren wurden, soweit nicht anders angegeben, ausgeführt wie in Sambrook,
Fritsch, Maniatis, “Molecular Cloning“ (Cold Spring Harbor Laboratory Press, 1989)
beschrieben. Für alle molekularbiologischen und biochemischen Arbeiten wurde hochreines
Nuklease- und Protease-freies Wasser verwendet (Wasser für Injektionszwecke). Für
Bakterienmedien, Agarosegel-Elektrophorese-Puffer sowie Transfer-Puffer und
Waschlösungen wurde meist Wasser verwendet.
Benzidin Stocklösung 3 % Benzidin Lösung gelöst in 90 % Essigsäure und 10 %
Wasser.
Church Puffer 0.33 M Na2HPO4, 0.16 M NaH2PO4, 7 % SDS,
1 mM EDTA pH 8.0
Chloroform Lösung 24 Volumen Chloroform
1 Volumen Isoamylalkohol
Denaturierungs-Lösung 0.5 M NaOH
1.5 M NaCl
DNA-Lyse Puffer 20 mM Tris-HCl pH 8.0, 100 mM EDTA pH 8.0, 0.4
mg/mL Proteinase K und 0.4 % SDS
DNA-Ladepuffer 0,15% Bromphenolblau
0,15% Xylencyanol
42% Sucrose
in 1xTBE
Erststrangpuffer (5x) für
cDNA Synthese
Invitrogen
High-stringency wash (HSW) 0.1 x SSC

3.- Material und Methoden Seite - 34 -
Puffer 0.1 % SDS
Lösung D 4 M Guanidiumsothiocyanat,
25 mM Natrium Citrate pH 7.0,
0.5 % Natrium-Lauryl-Sarkosyn
Lösung D+ Lösung D
+ 0,1 M β-Mercaptoethanol
(bei 4°C aufbewahrt,
meist frisch angesetzt)
Low-stringency wash (LSW)
Puffer
2 x SSC
0.1 % SDS
Membranprotein-Lysie Puffer 50 mM Tris HCl pH 7.4, 1 % NP-40, 1 % Laurylmaltoside,
150 mM NaCl, 1 mM EDTA, 1mM PMSF, 1mM Na3VO4,
1 mM NaF
MOPS, 20x 400 mM 3-(N-Morpholino) propansulfonsäure
100 mM Natriumacetat
20 mM EDTA
pH 7.0
Phenol-Chloroform Lösung 25 Vol Phenol, 24 Vol Chloroform, 1 Vol Isoamylalkohol
Prehybridisierungslösung ChurchBuffer
+ 100 µg/mL Salmon Sperm DNA
RNA-Ladepuffer 50 % Formamid
17.5 % Formaldehyd 37%ig
1-2 % Ficoll
In 1x MOPS-Puffer + Bromphenolblau
SDS, 20% 20.0 g SDS in ca. 90.0 ml H2O lösen (in 68°C Wasserbad),
mit HCl pH auf 7.0, einstellen auf 100.0 ml H2O.
Sephadex G50 20.0 g Sephadex in 500 ml Flasche mit H2O auffüllen
über Nacht quellen lassen
H2O abgießen
mit H2O auffüllen
ausschlieβend autoklavieren
SSC 10x 1.5 M NaCl,
0.15 M NaCitrate, pH 7.0
TAE (50x) Für 1000ml
3 mal

3.- Material und Methoden Seite - 35 -
242 g Tris-Base, 57.1 ml Essigsäure, 100 ml 0.5 M EDTA
ph 8.0, auf 1000 ml mit Wasser.
TBE (10x) Für 1000ml
890 mM Tris-Borat (108 g Tris-Base + 55 g Borsäure pro
Liter), 25mM EDTA pH 8.0
TBS 40mM Tris, 270 mM NaCl
TBS-T 40mM Tris, 270 mM NaCl, 1 mL Tween 20, pH 7.6
TE-Buffer 10 mM Tris HCl pH 7.4, 1 mM EDTA pH 8.0
Western Blot Blocking-Puffer TBS-T mit 5 % fett-freiem Magermilchpulver
Western Blot Running Buffer 1x NUPAGE® MES SDS Running Buffer
Western Blot Transfer Buffer 1x NUPAGE® Transfer Buffer,
10 % Methanol für eine Membrane
20 % Methanol für zwei Membrane
3.1.4.3.- Radiochemikalien
[α32P]- dCTP (3000 Ci/mmol) Amersham
3.1.4.4.- Enzyme
Superscript II Reverse Transcriptase Invitrogen
HotStarTaq DNA Polymerase Qiagen
Klenow-Polymerase Amersham
EcoR V New England Biolabs
Not I New England Biolabs
EcoR I New England Biolabs
RNase A Qiagen
rDNase 1 (DNA-free) Ambion
Proteinase K Merck
3.1.4.5.- Nukleinsäuren
Lachssperma-DNA Sigma-Aldrich

3.- Material und Methoden Seite - 36 -
Deoxyribonucleotide triphosphate (dNTPs) (dATP,
dCTP, dGTP, dTTP) Fermentas
1 kb Molekulargewichtstandard Fermentas / Invitrogen
100 bp Molekulargewichtstandard Fermentas / Invitrogen
Oligo(dT) Primer Invitrogen
Oligonukleotide als forward und reverse Primer wurden von Herrn Dr. Igloi (Institut für
Biologie III, Universität Freiburg) und von der Firma Operon bezogen,
Sequenzen der Oligonukleotide
für Southern Blot Sonden Annealing Temp.
Dematin forw CTCAGGGAACCCTCTGTGTT
Dematin rev AGCCATTGTTCTCTGGCTTC 55,3 °C
PTPN11 forw GGTTCCCTCCTCGTCCC
PTPN11 rev TCTGATAGGGCTGTGGTCAG 60,7 °C
CDC28 forw TTGGAATCTGGAATATGGGC
CDC28 rev GTTAGTGAGCCAAGACTG 53,9 °C
OTX1 forw GCAACACCTCGTGTATGCAG
OTX1 rev AGTGCAGGCAATGGACATCT 55,0 °C
PELI2 forw CTTCGAAACGTCCTCTACGC
PELI2 rev GCTGGAGTTCGATGGGATTA 56,4 °C
Sequenzen der Oligonukleotide
für Northern Blot Sonden Annealing Temp.
LAT2-1 forw ACAAGAGGCAACACCAGGAG
LAT2-1rev GGACCCCGTAAAGCTCTGTT 55,0 °C
Lysozyme forw GCCAAATGGGAGAGTGGTTA
Lysozyme rev ATCACGGACAACCCTCTTTG 55,0 °C
β-Aktin forw GGACTTCGAGCAAGAGATGG
β-Aktin rev AGCACTGTGTTGGCGTACAG 55,0 °C
GAPDH forw GAAGGTGAAGGTCGGAGTC
GAPDH rev GAAGATGGTGATGGGATTTC 55,0 °C
Sequenzen der Oligonukleotide
für Bisulfit-Sequenzierung Annealing Temp.
RARβ2 forw TTTAAAGTGTGGGTTGGG 54,8 °C

3.- Material und Methoden Seite - 37 -
RARβ2 rev TCCCAAATTCTCCTTCCAAA
Die Myeloperoxiedase (MPO) Northern Blot Sonde wurde aus cDNA hergestellt.
Die Oligonukleotide für die „real-time PCR“ wurden von Operon / Qiagen hergestellt.
Sequenzen der Oligonukleotide
für "real-time"-RT-PCR Annealing Temp.
LAT2-1 forw GAACCGGATTACGTGAATGG
LAT2-1 rev TAGCTCTGGGAAATGGTCCT 60,0 °C
GAPDH forw GAGTCAACGGATTTGGTCGTATT
GAPDH rev GAATTTGCCATGGGTGGAAT 60,0 °C
GUSB forw CGCCCTGCCTATCTGTATTC
GUSB rev TCCCCACAGGGAGTGTGTAG 60,0 °C
AML1/ETO forw CCGAGAACCTCGAAATCGTACT
AML1/ETO rev TCAGCCTAGATTGCGTCTTCAC 60,0 °C
Sequenzen der Oligonukleotide
für "real-time"-DNA-PCR Annealing Temp.
LAT2-1 forw CATTGCCAGAGACTGGTGAA,
LAT2-1 rev TTTCCCCTGACCTGTGTAGG 60,0 °C
p21 forw CAGCTGCATTGGGTAAATCC
p21 rev CACGAAGTGAGCCACAAATC 60,0 °C
3.1.4.6.- Antikörper
Antikörper für Western Blot
Antikörper Firma Verdünnung
Esel anti Ziege-HRP #sc2033 Santa Cruz
Antibodies 1:2000
Kaninchen anti-NTAL, polyklonal #11-485-C100 ExBio 1:1000
Maus anti-NTAL, monoklonal #11-374-C100 ExBio 1:1000
Maus anti-β-Aktin monoclonal # A2228 Sigma Aldrich 1:50000
Ziege anti Kaninchen -HRP- #sc2004 Santa Cruz 1:2000

3.- Material und Methoden Seite - 38 -
Antibodies
Ziege anti Maus -HRP- #sc2005 Sigma Aldrich 1:20000
Ziege anti-ETO, polyklonal #sc9737 Santa Cruz
Antibodies 1:1000
Antikörper für FACS
Maus anti-Human CD41a-PE monoklonal #555467 BD Pharmigen
Maus anti-Human CD61FITC monoklonal #555753 BD Pharmigen
Maus IgG1-FITC #X0927 DakoCytomation
Maus IgG1-PE #0928 DakoCytomation
Maus anti-Human CD11b-PE monoklonal #555388 BD Biosciences
Maus anti-Human CD11c-PE monoklonal #555392 BD Biosciences
Antikörper für ChIP
Rabbit anti-Human AcH3 #06599 Upstate
Rabbit anti-Human AcH4 #06598 Upstate
Rabbit anti-Human 3meH3K4 #06745 Upstate
Rabbit anti-Human AcH3K9 #07352 Upstate
3.1.4.7.- Kits
Bio-Rad Protein Assay Bio Rad
DNAfree Ambion
Nuclear Extract Kit Active Motif
RNAse free DNAse Set Qiagen
RNAeasy Mini Kit Qiagen
DNeasy Tissue Kit Qiagen
Megaprime DNA Labelling Kit Amersham
QIAGEN Plasmid Maxi Kit Qiagen
QIAPrep Spin MiniPrep Kit Qiagen
QIAquick™ Gel Extraction Kit Qiagen
EZ DNA Methylation Kit Zymo Research
TOPO TA Cloning Kit for Sequencing Invitrogen
ECL Plus Western blotting Detektion System Amershams
Nucleospin Exbio
SuperScript™ III Reverse Transcriptase Invitrogen

3.- Material und Methoden Seite - 39 -
3.1.4.8.- Sonstige Materialien
384 Loch-Microtiterplaten Roche
96-Loch-Microtiterplatten Greiner
Aluminium-Folie
Autoklavierklebeband 3M
Blotting Pads NuPAGE / Invitrogen
Kryoboxen mit Deckel 50 mm, Rastereinsatz 10x10 RZ
Erlenmeyerkolben (100, 250, 1000 ml) Schott
Filterpapiere / Blottingpapiere Whatman, Schleicher & Schüll
Flatcap PCR-Tubes (0.5 ml) Peqlab
Glaswolle, silanisiert Serva
Haushaltsfolie, Saran Roth
Hybond N+ Nylonmembran Amersham
Hybond-P Membranen Amersham
Hyperfilm Amersham
Impfösen und -nadeln Nunc
Kanüle 26 G (0.45 mm) B. Braun
Messbecher Kartell
Multiply-Strip mit Deckelkette, dünn (0.2 ml) Sarstedt
NuPAGE 4-12% Bis-Tris Gel Invitrogen
Pasteurpipetten Brand
PCR-Tubes Abgene
PCR-Tubes, dünn (0.2 ml) Peqlab
QPCR Seal Roche
Reaktionsgefäß (0.5, 1.5, 2.0 ml) Eppendorf, Greiner
Reaktionsgefäß safe lock (0.5, 1.5, 2.0 ml) Eppendorf
Röhrchen, konisch (225 ml) Falcon
Röhrchen, Polypropylen (15, 50 ml) Falcon
Schaumstoffplatten, Schwämme Bauhaus, Freiburg
Skalpelle Feather
Thermo-Fast 96 Detektionsplatte Abgene
Thermoprintpapier K65HM Mitsubishi
Wägeschalen (40/40/10, 80/80/20, 140/140/20) Bender& Hobein

3.- Material und Methoden Seite - 40 -
Wägeschalen (40/40/10, 80/80/20, 140/140/20) Bender & Hobein
X-OMAT-AR Röntgenfilme Kodak
3.1.5.- Geräte
ABI Prism 7700 Sequenzierer Applied Biosystems
Auflicht-Mikroskop Leica DMIL Leica
Cyan ADP (Durchflußzytometer) DakoCytomation
Durchflusszytometer FACSCalibur Becton & Dickinson
Entwickler X-Omat M35 Kodak
Gelkammern BioRad
Gelkammern Hans Thoma Laborbedarf,
Freiburg
Gelkammern DNA sub cell Bio-Rad
Gelkammern Wide mini sub cell Bio-Rad
Heizblock TCR100 Roth
Heizblock DB-2D Techne
Heizblock Dri-Block Techne
Heizblock TCR100 Roth
Hybridisation Oven 640 Affymetrix
Inkubator Heraeus 6000 Heraeus
Kühlschrank -80°C Heraeus
Kühlschrank -20°C Liebherr
Kühlschrank 4°C Liebherr
Kühlschrank HFU586STD-U12 -80°C Heraeus
Kühlschrank UF80-450S Colora
Kühlzentrifuge 5417R Eppendorf
Mikroskop Zeiss Axioskop 2 MOT und AxioCam Zeiss
Mikrowelle AEG/Siemens
Netzteile LKB ECPS 3000/150 Pharmacia
Netzteile LKB EPS 500/400 Pharmacia
Optimax X-Ray Film Processor Typon
Photoprinter PGGE (Video Copy Processor) Mitsubishi Electric Corporation

3.- Material und Methoden Seite - 41 -
Schüttelinkubator Incubator shaker series 25 New Brunswick scientific Co.,
Inc.
Spektrometer DU640 Beckman
Spektrometer, GeneQuant Pro Amersham
Sterile Werkbank CaminAir HB2448 Heraeus
Sterile Werkbank Variolab Mobilien W90 Waldner Laboreinrichtungen
T3 Thermocycler Biometra
T-Gradient Thermocycler Biometra
Thermocycler PTC200 MJ Research, Biozym Diagnostik
Thermomagnetrührer Heidolph
Thermomixer Comfort 1,5ml Eppendorf
Tischzentrifuge 5417C Eppendorf
Tischzentrifuge Biofuge 13 Heraeus
Tischzentrifuge Biofuge 15 Heraeus Heraeus
Ultraviolett Transilluminator UVP Herolab
UV Stratalinker 2400 Stratagene
Vacuum Concentrator BA-VC-300 Bachhofer
Vortexer Heidolph
Vortexer Janke und Kunkel, IKA-
Labortechnik
Waage basic BA1103 Sartorius
Waage portable PT120 Sartorius
Waage portable PT6 Sartorius
Wasserbad W26/PC3, Haake
XCell SureLock Mini-Cell NuPage / Invitrogen
Zentrifuge Megafuge 1.0R Heraeus
Zentrifuge Megafuge 3.0R Heraeus
Zentrifuge Varifuge 3.0R Heraeus
3.1.6.- Bioinformatische Software
Chromas Technelysium

3.- Material und Methoden Seite - 42 -
CpGPlot http://www.ebi.ac.uk/emboss/cpgplot/
European Bioinformatics Institute (EBI) (Cambridge, England)
Repeat Masker (http://repeatmasker.genome.washington.edu/)
University of Washington, USA
Primer3 (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi)
Whitehead Institute for Biomedical Research, Massachusetts, USA
NCBI Blast (http://www.ncbi.nlm.nih.gov/BLAST/)
National Center for Biotechnology Information, USA
MethPrimer (www.urogene.org/methprimer/)
Urology Research Center, Veterans Affairs Medical Center and
University of California, San Francisco, USA
Blast/Special/alignment
2 Sequences
(http://www.ncbi.nlm.nih.gov/blast/bl2seq/wblast2.cgi)
National Center for Biotechnology Information, USA
NEB Cutter (http://tools.neb.com/NEBcutter2/index.php)
New England Biolabs (Ipswich, MA, USA)
Universal Probe Library (https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp)
Roche Diagnostics (Basel, Schweiz)
Image J (http://rsb.info.nih.gov/ij/)
National Institutes of Health, USA
Summit v 4.2 (www.dakousa.com)
Dakocytomation, California, USA
FlowJo v. 6.2 (www.flowjo.com)
Tree Star inc, Oregon, USA

3.- Material und Methoden Seite - 43 -
3.2.- Methoden
3.2.1.- Aufarbeitung des primären Materials.
3.2.1.1.- Isolierung der peripheren Blutzellen
Mononukleäre Zellen aus Blut gesunder Spendern wurden mittels
Ficollgradientenzentrifugation isoliert und als Zellpellets für DNA- und Proteinisolierung
eingefroren. Blut von gesunden Spendern wurde in zwei 10 ml EDTA Röhrchen
aufgenommen. Die peripheren mononukleären Zellen wurden so rasch wie möglich isoliert.
Dazu wurden zunächst 20 ml Ficoll Lösung in ein steriles Schraubdeckelröhrchen pipettiert.
10 ml Blut wurden mit 10 ml PBS Puffer verdünnt und vorsichtig auf die Ficolllösung
geschichtet. Nach Zentrifugation für 30 min bei 2300 rpm ohne Bremse reicherten sich die
mononukleären Zellen an der Phasengrenze als feine gelbliche Bande an. Im Zellpellet
befanden sich die Erythrozyten und Granulozyten. Die peripheren mononuklearen Zellen
(MNC) wurden aus der Interphase geerntet, mit PBS gewaschen und anschließend mit Türks-
Lösung gefärbt und gezählt. In Abhängigkeit von der Zellenausbeute wurden 5x106 bis 1x107
Zellen als Zellpellets bei -80°C eingefroren.
3.2.1.2.- Lymphozyten-Isolierung
Die Lymphozyten und Monozyten wurden aus der Interphase nach dem
Ficollgradientenzentrifugation isoliert. Die Zellen, die in der Interphase lagen, wurden in
37ºC warmen RPMI 1640 Medium, Gibco, plus 10 % FCS aufgenommen und in einer 175
cm2 Zellkulturflasche bei 37°C und 5 % CO2 für 1 Stunde inkubiert.
Nach dieser Zeit wurden die nicht adhärenten Zellen durch Beklopfen der Flasche abgelöst
und in einem 50 ml Röhrchen zusammen mit dem Überstand überführtzugegeben. Die
adhärenten Zellen wurden zweimal mit 10 ml PBS gewaschen und diese Flüssigkeit zu den
nicht-adhärenten Zellen in die 50 ml Röhrchen pipettiert.
Die nicht-adhärenten Zellen wurden mit 1800 rpm und 5 min. zentrifugiert und mit 10 ml
PBS gewaschen. Anschließend wurden sie mit Türks-Lösung gefärbt und in einer Neubauer
Zählkammer gezählt und als Lymphozyten definiert.

3.- Material und Methoden Seite - 44 -
3.2.1.3.- Monozyten-Isolierung
Die adhärenten Zellen, die auf dem Boden der Zellkulturflasche haften, wurden mit Trypsin-
EDTA Lösung abgelöst. Dabei wurde der Flaschenboden mit 2 ml Trypsin-EDTA-Lösung
bedeckt und 5-10 min. bei 37°C inkubiert. Durch leichtes Klopfen auf die Unterseite des
Gefäßes wurden die Zellen abgelöst und wurden durch Auswaschen der Flasche mit PBS als
Monozyten geerntet. Die Zellzahl wurde nach Färbung mit Türks-Lösung in einer Neubauer-
Zählkammer bestimmt.
3.2.1.4.- Granulozyten-Isolierung
Die Granulozyten wurden aus der untersten Phase des Fikollgradienten isoliert. Zuerst wurde
das Pellet mit den Granulozyten und den Erythrozyten in 50 ml Lösung (25 ml PBS, 25 ml 3%
Dextran gelöst in 0.9 % NaCl) resuspendiert und 30 bis 60 min. bei 4 ºC sedimentiert. Der
Überstand wurde 10 min. bei 1600 rpm zentrifugiert. Um die noch vorhandenen Erythozyten
zu lysieren, wurde das Pellet für 30 sec. mit 10 ml eiskalten 0.2 % NaCl Lösung und dann für
30 sec. mit 10 ml eiskalter 1.6 % NaCl-Lösung gemischt.
Danach wurden die Zellen 10 min. bei 1600 rpm zentrifugiert, der Überstand verworfen und
das Pellet mit den Granulozyten einmal mit PBS gewaschen. Die Zellzahl wurde mit Türks-
Lösung in einem Neubauer-Zählkammer bestimmt.
3.2.1.5.- Patientenproben
Die Patienten, deren Proben wir auf Proteinexpression hinuntersucht haben, wurden aus den
behandelten AML-Patienten der Universitätsklinik Freiburg ausgewählt. Kriterien, um
Patienten für unseren Projekt einzuschlieβen, waren: 1) AML, diagnostisiert in der
Universitätsklinik Freiburg, 2) schriftliche Erlaubnis für die Verwendung der Proben für
Forschungszwecke unterschrieben, 3) bekannter Karyotyp der Blasten festgestellt und 4)
ausreichend Proben in der Tumorbank für anderen Zwecke. Nach einer schriftlichen
Vorstellung unseres Vorhabens erhielten wir die Genehmigung von der Tumorbank für die
Benutzung dieser Proben.
Die meisten Proben waren aus dem Knochenmark der Patienten nach
Ficollgradientenzentrifugation gewonnen und in einem mit 50 % FCS und 10 % DMSO

3.- Material und Methoden Seite - 45 -
angereicherten RPMI 1640 Medium eingefroren. Die Auftrennung mittels
Fikollgradientenzentrifugation und das Einfrieren der Proben wurden von MTAs der Abteilung
I der Inneren Medizin durchgeführt.
In weiterem wurden die Proben der Patienten in einem Wasserbad bei 37ºC aufgetaut und in
10 ml RPMI Medium, Gibco, zweimal gewaschen, um das DMSO so schnell wie möglich zu
entfernen. Dann wurden die Zellen in 1 mL PBS aufgelöst und nach Trypanblau Färbung
gezählt. Anschließend wurden die Proteine nach der Methode für die Isolierung der
Membranproteine gewonnen (s. u. 3.2.6.).
3.2.1.6.- Isolierung von CD34+ Zellen
CD34+ Zellen sind hämatologische Stammzellen und werden in der Klinik für die
Stammzelltransplantation eingesetzt. Diese Zellen kann man durch Zugabe verschiedener
Zytokine in die Differenzierung zu lymphatischen und myeloischen Zellen treiben. Die CD34+
Vorläuferzellen werden durch G-CSF Gabe mobilisiert und in das Blut ausgeschwemmt. Sie
werden dann mittels einer Leukapherese (Cobspektra) abgesammelt. Aus diesem
Leukapherisatprodukt werden die CD34+ Zellen durch Antikörpermarkierung mit
Magnetbeads und dem sogenannten Clinimacs angereichert. Anhand eines
Durchflußzytometers wird dann die Reinheit der CD34+ Zellen bestimmt.
3.2.1.7.- Isolierung von Alveolar-Makrophagen.
Die Alveolar-Makrophagen stammten aus einer bronchoalveolären Lavage von Patienten, bei
denen zu Diagnose Zwecken eine Bronchoskopie notwendig war. Die Zellen der Patienten
wurden von Fr. Dr. Brassel aus der Abteilung für Pneumologie, des Universitätsklinikums
Freiburg zur Verfügung gestellt. Nach der bronchoalveolären Lavage wurden die Zellen
zweimal in PBS gewaschen, mit der Türk-Färbung in einer Neubauer-Zählkammer gezählt und
als Zellpellet eingefroren.
3.2.2. Zellkultur
Alle Zellen wurden bei 37°C, 5% CO2 und 100 % Luftfeuchtigkeit im Inkubator kultiviert.
Sämtliche Zentrifugationsschritte von Zellen, die weiter in Kultur gehalten wurden, wurden
für 5 min bei 1200 rpm in einer Heraeuszentrifuge durchgeführt. Zellkulturarbeiten wurden

3.- Material und Methoden Seite - 46 -
ausschließlich an einer sterilen Werkbank verrichtet. Die fertig angesetzten Kulturmedien
wurden bei 4 °C aufbewahrt und vor der Benutzung im 37 °C Wasserbad vorgewärmt.
3.2.2.1.- Kultivierung der Zellen
Alle Medienansätze mit RPMI-1640, Gibco, wurden mit 5 ml Penicillin-Streptomycin-
Lösung je 500 ml versetzt (100 U/ml). Die Zellkultur wurde mit einer Zellkonzentration von
ca. 5x105 Zellen/ml gestartet. Mediumwechsel und Aufteilung der Zellen erfolgten alle zwei
bis drei Tage. Die Zellkonzentration wurde im weiteren Kulturverlauf zwischen 0.25 und
1x106 Zellen/ml gehalten. Eine deutliche Menge an Zelldebris war stets während der Kultur
der Kasumi-1 Zelllinie zu beobachten.
Die Zelllinie Kasumi-1 erhielt das Medium RPMI-1640, Gibco, mit 20% FCS. Zusätzlich
wurde gelegentlich noch 5 ml Natriumpyruvat-Lösung zum Medium hinzugefügt. Die
Zelllinien HL60, U937, 9/14/18 und K562 wurden in RPM1-1640 mit 10% FCS und 100
U/mL Penicilin-Streptomycin kultiviert. Die Zellen wurden alle zwei Tage aufgeteilt und mit
frischem Medium versetzt, so dass Zellkonzentrationen zwischen 0.5 und 2x106 Zellen/ml
erreicht wurden. Die Zelllinie NB4 wurde in RPMI-1640, Gibco, mit 10 oder 20 % FCS unter
den gleichen Bedingungen wie HL60 und U937 kultiviert.
Zum Mediumwechsel wurden die Zellen für 5 min bei 1200 rpm in Falcon-Röhrchen
zentrifugiert. Der Überstand mit dem alten Medium wurde verworfen, das Zellpellet in
frischem Medium resuspendiert und auf die Zellkulturflaschen verteilt.
3.2.2.2.- Einfrieren und Auftauen der Zellen
Sowohl das Einfrieren als auch das Auftauen von Zellen erfolgte sehr zügig, um toxische
Wirkungen von DMSO auf die Zellen möglichst gering zu halten.
Zum Auftauen wurden die Kryoröhrchen mit den Zellen kurz im 37°C Wasserbad
geschwenkt. Nach dem Auftauen der Zellsuspension wurde diese direkt in 10 ml 37ºC
warmes Medium pipettiert und für 5 min bei 1200 rpm zentrifugiert, um das DMSO
auszuwaschen. Das Zellpellet wurde dann in frischem Kulturmedium aufgenommen und in
der gewünschten Dichte auf Kulturflaschen aufgeteilt.
Zum Einfrieren wurden die Zellen 5 min bei 1200 zentrifugiert und anschließend in einer
Zelldichte zwischen 0.5 und 1x107 in Einfrierungsmedium (20% FCS, 10% DMSO und 70%
RPMI-1640, Gibco) aufgenommen. Je 1 ml der Zellsuspenison wurde in Kryoröhrchen

3.- Material und Methoden Seite - 47 -
aliquotiert. Das langsame Einfrieren der Zellen wurde durch die Lagerung der Röhrchen bei -
80ºC in einem isolierenden Isopropanol enthaltenden Gefäß gewährleistet. Nach 2-3 Wochen
wurden die Zellen in flüssigem Stickstoff gelagert.
3.2.2.3.- Zellzahl- und Viabilitäts-Bestimmung durch Trypan Blau Färbung
Zellzahl und Viabilität der Zellen wurden mittels Trypanblau-Färbung bestimmt. Dieser
Färbung liegt das Prinzip zu Grunde, dass lebende Zellen den Farbstoff Trypanblau nicht
aufnehmen, während tote Zellen angefärbt werden.
Zur Bestimmung der Zellzahl wurden 50 µl Zellsuspension mit dem gleichen Volumen
0,4%iger Trypanblaulösung vermischt. Die Mischung aus Farbstoff und Zellen wurde in einer
Neubauer-Zählkammer gegeben und jeweils die vier großen Quadranten unter dem
Durchlichtmikroskop ausgezählt. Blau gefärbte Zellen wurden als tot gewertet, ungefärbte
helle Zellen als lebend. Da jeder Quadrant einem Volumen von 0,1 mm3 (10-4ml) entspricht,
wurden die Zellzahl und Viabilität in 1 ml mit folgende Formeln berechnet.
Zellzahl/ml = Anzahl gezählter Zellen / Zahl der ausgezählten Eckquadrate x 2
(Verdünnungsfaktor) x104
Viabilität = Anzahl lebender Zellen / Summe aller Zellen (lebende + tote) x 100 %
3.2.2.4.- Zellzahl- und Viabilitäts-Bestimmung durch Färbung mit Türklösung
Die Anzahl solcher Zellen, die direkt aus dem Blut gewonnen wurden, wurde mit der
Türklösung bestimmt, um Artefakte mit Erythrozyten zu vermeiden. Die Türklösung färbt nur
die Zellen der weiβen Reihe und nicht die Zellen der roten Reihe, allerdings kann damit nicht
die Viabilität der Zellen bestimmt werden.
Die Methode ist analog der Trypanblau Färbung, doch wurden die Zellen in einem Verhältnis
vom 1:100 mit der Türklösung versetzt. Dann wurden die Zellen mit einem Neubauer
Zählkammer gezählt und die Zellzahl wurde mit einem Wert von 100 als Verdünnungsfaktor
errechnet.

3.- Material und Methoden Seite - 48 -
3.2.2.5.- Abtrennung toter Zellen und Zelltrimmern mittels
Ficollgradientenzentrifugation
Zur Abtrennung von toten Zellen und Debris wurde die Ficollgradienten Zentrifugation
durchgeführt. Dazu wurde zunächst in einem 50 ml Falcon Röhrchen 1 Volumen der Ficoll-
Lösung gegeben und diese mit dem entsprechenden Volumen an Zellsuspension vorsichtig
überschichtet. Anschließend wurde für 30 min bei 2300rpm ohne Bremse zentrifugiert.
Während sich die toten Zellen und Debris im Pellet sammeln, reichern sich die lebenden
Zellen in einer weiß-gelben Schicht an der Phasengrenze an. Diese Zellen wurden vorsichtig
mit der Pipette entnommen, zweimal mit PBS gewaschen und dann wieder in Kultur
genommen.
3.2.2.6.- Beurteilung der Morphologie durch Zytospin und Färbung
3.2.2.6.1.- Zytospin
Die Zellen wurden zentrifugiert, mit PBS gewaschen und anschließend in einer Dichte von
1 x 105 Zellen in 200 µl PBS resuspendiert. 100 µl dieser Zellsuspension wurden in die
vorbereiteten Trichter pipettiert und bei 800 rpm 3 min auf die Objektträger zentrifugiert. Die
Objektträger wurden dann an der Luft getrocknet und anschließend mit der Panoptischen
Färbung nach Pappenheim gefärbt.
3.2.2.6.2- May-Grünwald Giemsa Färbung (Pappenheim Färbung)
Hierzu wurden die Zytospinpräparate 3 min mit May-Grünwald-Lösung bedeckt,
anschließend mit Wasser abgespült und weitere 3 min in Wasser stehen gelassen. Im weiteren
wurden die Präparate mit Giemsa-Gebrauchslösung (1:20 in PBS verdünnt) für 20 min gefärbt
und abschließend wieder mit Wasser abgespült. Die Objektträger trockneten dann
schrägstehend an der Luft.
3.2.2.6.3.- Benzidin Färbung
Für die Benzidin Färbung wurde die Benzidin Lösung immer frisch hergestellt. Es wurden 1
Teil Benzidin Stammlösung (3 % Benzidin Lösung gelöst in 90 % Essigsäure und 10 %
Wasser), 1 Teil Wasserstoffperoxid 30 % und 5 Teile bidestiliertes Wasser gemischt.

3.- Material und Methoden Seite - 49 -
2 ml Zellsuspension wurden mit 0.2 ml Benzidin Lösung gemischt, 5 min. bei Raumtemperatur
inkubiert und dann wurden die gefärbten und farblosen Zellen in einer Neubauer Zählkammer
gezählt. Das Photographieren der Zellen erfolgte innerhalb einer Stunde, da die Färbung sehr
unstabil ist.
3.2.3.- Behandlung der Zellen
3.2.3.1.- Induktion der Expression des AML1/ETO-Proteins in der 9/14/18-
Zelllinie
Die 9/14/18-Zelllinie wurde in 600 µg/ml Geneticin und 150 µg/ml Zeocin kultiviert, um die
Präsenz der Vektoren zu gewährleisten. Die AML1/ETO-Expression wurde durch 5 µM in
Ethanol gelöstem Ponasteron A induziert. Zur Kontrolle wurde das gleiche Volumen Ethanol
den nicht-induzierten Zellen hinzugegeben. Bei Induktionsexperimenten mit einer Dauer von
mehr als 48 Stunden wurde alle 48 Stunden ein Mediumwechsel unter Hinzugabe von
Ponasteron A durchgeführt.
3.2.3.2.- Myeloische Differenzierung der HL60, U937, NB4 und K562 Zelllinien.
Die HL60-, U937-, NB4- und K562-Zelllinien wurden für myeloische Differenzierungs-Assays
benutzt. Zellen wurden in einer Dichte von 2.5 x105 Zellen/ml auf ca. 20-40 ml Medium auf
den Kulturflaschen verteilt. An jedem Tag wurde die Zellzahl und die Viabilität mit Trypan-
Blau-Färbung bestimmt, und es wurden Zellen für weitere Expressionsanalyse mittels Western
und Northern Blot geerntet. Am letzten Tag wurden 2x105 Zellen für Morphologieanalysen
mittels May-Grünwald-Giemsa-Färbung und 1x105 Zellen für Antigen-Expressionanalyse
mittels Durchflusszytometrie (FACS) genommen.
Bei der ATRA-Behandlung der HL60-, U937- und NB4-Zellen wurde das ATRA (in Ethanol
aufgelöst) jeden Tag in einer Endkonzentration von 10-6 M zugegeben. Das Medium wurde
jeden Tag gewechselt und die Substanzen neu hinzugegeben. Bei der Hemin-Behandlung der
K562-Zellen wurde auch täglich die Substanz ersetzt und das Medium gewechselt.
Für die PMA-Behandlung (in Wasser aufgelöst) der HL60- und K562-Zellen wurden die
Substanzen nur einmal am ersten Tag zugegeben und das Medium wurde während des
Versuches nicht gewechselt.

3.- Material und Methoden Seite - 50 -
3.2.3.3.- Ex vivo myeloische Differenzierung der CD34+ Zellen.
Nach dem Auftauen der CD34+ Zellen in einem auf 37ºC vorgewärmten Wasserbad wurden
die Zellen zweimal in 10 ml CellGro Medium gewaschen und anschlieβend in diesem Medium
resuspendiert. Die Zellen wurden mit der Trypanblau-Färbung gezählt und in einer
Konzentration von 1x105 Zellen/ml Medium auf 75 cm2 Flaschen verteilt. Als Kulturmedium
wurde das CellGro Medium benutzt, versetzt mit einem Expansion-Cocktail (modifiziert nach
Guo et al. Leukemia 2006) aus den folgenden Zytokinen: SCF (100 ng/ml), FLT3 (100 ng/ml),
IL3 (50 ng/ml) und IL6 (20 ng/ml).
Durch Zugeben von G-CSF (50 ng/ml) differenzieren die Zellen in reife Granulozyten, durch
GM-CSF (50 ng/ml) wird die monozytäre Differenzierung angeregt und mit GM-CSF (50
ng/ml) plus IL-4 (50 ng/ml) erfolgt eine monozytär-dendritische Differenzierung. Als
Kontrolle wurde ein Teil der Zellen nur mit dem Expansion-Cocktail kultiviert. Die Zellen
wurden am Tag 0, 1, 3, 7 und 14 als Zellpellet bei -80ºC geerntet und jeweils 1x105 Zellen
wurden für Antigenanalyse mittels Durchflusszytometrie und die morphologische
Untersuchung mittels May-Grünwald Giemsa Färbung gewonnen.
3.2.3.4.- Behandlung der Kasumi-1 Zellinie mit epigenetisch aktiven Substanzen.
Die Zellen wurden mit 5-Aza-2'-deoxycytidine (Decitabine, DAC) (in PBS aufgelöst)
Konzentrationen zwischen 25 und 200 nM behandelt. Zu Beginn der Behandlung wurden die
Zellen auf eine Konzentration von 2,5x105 Zellen/ml gebracht. DAC wurde in einer
Konzentration von 100 µM in sterilem PBS gelöst. Die Reagenzien wurden unter sterilen
Bedingungen in die Kulturflaschen eingebracht. Aufgrund der Instabilität von DAC musste
die Substanz alle 24 Stunden mit frischem Medium frisch angesetzt werden. Die Behandlung
erfolgte daher in 3 Pulsen jede 24 Stunden. Die Zellen wurden innerhalb dieser Zeit täglich
zentrifugiert, mit frischem Medium versorgt und gezählt. Anschließend wurden die Zellen für
weitere 72 Stunden ohne Mediumwechsel in Kultur gehalten. Die Zellen wurden sowohl am
sechsten Tag, als auch zu unterschiedlichen Zeitpunkten während der Behandlung geerntet.
Als Leerkontrollen wurden jeweils Ansätze mit entsprechenden Volumina an PBS versetzt.
Wenn die Kasumi-1 Zellen mit der Kombination von demethylierenden Substanzen (DAC)
und HDAC Inhibitoren ATRA, TSA (in Ethanol aufgelöst), MS275 (in DMSO aufgelöst),
SAHA (in Methanol aufgelöst) behandelt wurden, haben wir drei Pulse vom DAC in den

3.- Material und Methoden Seite - 51 -
ersten drei Tage gegeben. Nach 72 Stunden wurde der HDAC-Inhibitor mit frischem Medium
zugegeben. Die Zellen wurden anschlieβend am sechsten Tag gezählt und geerntet.
Das Ernten der Zellen erfolgte nach Bestimmung der Zellzahl und Viabilität zu verschiedenen
Zeitpunkten während oder nach der Behandlung. Dazu wurden 0,5 bis 1x107 Zellen in 1,5 ml
Reaktionsgefäße überführt und bei 4°C für 5 min bei 3000 rpm zentrifugiert. Nach Absaugen
des Mediums im Überstand wurden die Zellpellets je nach Weiterverwendung in RNA-Lyse-
Puffer oder als Pellet im -80°C Gefrierschrank gelagert.
3.2.4.- Isolierung der Nukleinsäuren
3.2.4.1.- DNA-Isolierung mittels Phenol-Chloroform Extraktion.
Zelllinien und mononukleäre Zellen aus dem peripheren Blut wurden in DNA Lysis-Buffer
resuspendiert und über Nacht in einem bei 55ºC vorgewarmten Wasserbad lysiert. Während
der DNA Isolierung wurden abgeschnittene Pipettenspitzen zur Vermeidung von Scherung
der DNA-Doppelstränge verwendet. Auf 107 Zellen wurde 1 ml DNA Lysis Puffer gegeben
und die Zellen wurden in einem 15 ml Röhrchen resuspendiert.
An folgendem Tag wurde die DNA aus dem Lysat nach Phenol/Chloroform-Extraktion und
Natrium Acetat-Isopropanol -Fällung gewonnen. Zu einem Volumen des Lysates wurde ein
Volumen von Phenol gegeben. Die Suspension wurde 15 min. in einem horizontalen Schüttler
gemischt und dann für 5 min. bei 2500 rpm zentrifugiert. Die obere wässrige Phase wurde
vorsichtig mit einer angeschnitteten Pipettenspitze abgenommen und in ein frisches 15 ml
Röhrchen überführt. Dazu wurde ein Volumen von Phenol/Chloroform/Isoamylalkohol
(25/24/1) gegeben und es wurde wieder 15 min. im horizontalen Schüttler gemischt und 5
min. bei 2000 rpm zentrifugiert. Die obere wässrige Phase wurde in ein neues 15 ml
Röhrchen überführt. Nach Zugabe von einem Volumen Chloroform/Isoamylalkohol (49/1)
wurde im horizontalen Schüttler gemischt, um die Reste vom Phenol zu entfernen.
Anschlieβlich wurde 5 min. bei 2000 rpm zentrifugiert.
Die DNA wurde mit 0,3 M Natrium Acetat pH 4,8 und 60 % Endvolumen von Isopropanol
bei Raumtemperatur gefällt. Die gefällte DNA wurde in 70 %iger Ethanollösung gewaschen
und 15 min. bei Raumtemperatur trocken gelassen. Anschlieβlich wurde die DNA in TE
Puffer resuspendiert, bei 37 ° in einem Brutschrank gelöst und im Kühlschrank bei +4ºC
aufbewahrt.

3.- Material und Methoden Seite - 52 -
3.2.4.2.- DNA-Isolierung mittels Qiagen Kit
Genomische DNA wurde mit dem DNeasy Tissue Kit von Qiagen entsprechend den
Vorgaben des Herstellers isoliert, wenn kleine Menge DNA für die folgenden Experimente
(PCR, bisulfite Konversion) notwendig waren. Dazu wurden die Proben zuerst mit Proteinase
K lysiert und in Puffer resuspendiert, die eine optimale Bindung der DNA in die Silica-Gel-
Membranen ergeben. Nach einer Zentrifugation ist die DNA an die Säule gebunden, und die
Kontaminanten sind abgetrennt. Danach wurden Enzyme und Reste von Kontaminanten von
der DNA mit den verschiedenen Waschschritten entfernt. Anschlieβend wurde die DNA in
TE Buffer gelöst und bei +4ºC gelagert.
3.2.4.3.- RNA-Isolierung mittels Guanidiniumthiocyanat.
Die RNA-Isolierung wurde nach der Methode von Chomczynski und Sacchi (Anal Biochem
1987) durchgeführt.
Die Zellen wurden direkt nach der Zellkultur in eiskaltem PBS gewaschen und in einem
Alliquot von 1x107 zentrifugiert. Dann wurden sie in 1 ml frischer Lösung D+ resuspendiert,
lysiert und ggf. bis zur weiteren Verarbeitung bei –80°C eingefroren.
Zur Reinigung der RNA wurde zunächst eine Phenol/Chloroform-Extraktion durchgeführt
und anschließend mit Isopropanol gefällt. Die weitere Beschreibung der RNA-Isolierung
bezieht sich auf jeweils 500 µl dieses Isolates in 1.5 ml RNase-freien Reaktionsgefäßen. Nach
Zugabe von 50 µl 2 M Natriumacetat pH 4.0 und erneutem Vortexen wurde das Lysat durch
mehrmaliges Aufziehen in Insulin-Spritzen mit Kanülen (26 G, Durchmesser 0.45 mm) weiter
homogenisiert. Im Weiteren wurden 500 µl H2O-gesättigtes Phenol zugesetzt, wiederum
gevortext und bis zu 15 min auf einem Schüttler gemischt. 100 µl Chloroform/Isoamylalkohol
im Volumenverhältnis 49:1 wurden zugesetzt, intensiv gevortext und die Ansätze für 15 min
auf Eis inkubiert. Zur Trennung der Phasen wurde es für 15 min bei 13000 rpm und 4°C
zentrifugiert, die wässrige Phase abgenommen und in ein frisches Reaktionsgefäß transferiert.
Die RNA wurde anschließend durch Zugabe von 500 µl Isopropanol und Lagerung über
Nacht bei -80°C präzipitiert. Das Präzipitat wurde durch erneute Zentrifugation für 15 min bei
13000 rpm und 4°C pelletiert, und der Überstand wurde verworfen. Das RNA-Pellet wurde
dann einmal in eiskaltem 70%igen Ethanol gewaschen, getrocknet und in einem geeigneten
Volumen RNAse freiem Wasser aufgelöst. Die RNA-Konzentration wurde photometrisch
bestimmt und die Aufbewahrung der RNA erfolgte bei -80°C.

3.- Material und Methoden Seite - 53 -
3.2.4.4.- RNA-Isolierung mittels Qiagen Kit
Die RNA Isolierung wurde mit dem RNeasy Kit von Qiagen durchgeführt. Diese Methode
nutzt die selektiven Nukleinsäure Bindungseigenschaften von Silicagel-Membranen. Alle
Schritte wurden bei Raumtemperatur durchgeführt. Die Zellen wurden dabei zunächst mit dem
stark denaturierenden, Guanidiniumthiocyanate enthaltenden RLT-Puffer lysiert. Durch diese
Behandlung werden RNAsen effizient inaktiviert, sodass die Isolierung von intakter RNA
gewährleistet ist. Anschließend wurde das Zelllysat durch mehrmaliges Passieren einer 20-
Gauge Kanüle (0,9 mm Durchmesser) homogenisiert. Um die optimale Bindungskapazität der
RNA an die Säulen zu erreichen, wurde 1 Volumen 70%iges Ethanol hinzugefügt. Die
Zelllysate wurden auf die RNeasy-Säulen geladen. Nach kurzer Zentrifugation bei 10000 rpm
lag die Gesamt-RNA an die Membran gebunden vor. Durch weitere Waschschritte mit 700 µl
Puffer RW1 und zweimal mit 500 µl des ethanolhaltigen Puffer RPE wurden alle anderen
Bestandteile entfernt. Im letzen Schritt wurde RNA in 40 µl RNAse-freiem Wasser eluiert
(siehe auch RNeasy Mini Protokoll für die Isolierung von Gesamt RNA aus tierischen Zellen).
Die RNA wurde anschließend bei -80°C gelagert
3.2.4.5.- Photometrische Bestimmung der DNA- und RNA-Konzentration
Die Konzentrationsbestimmung von DNA und RNA erfolgte photometrisch unter
Verwendung von Quarzkapillaren mit dem Densitometer GeneQuant Pro. Hierbei wurde die
Eigenschaft der Nukleinsäuren ausgenutzt, Licht bei einer Wellenlänge von 260 nm maximal
zu extingieren. Mittels der optischen Dichte bei 260 nm (OD260) und des Warburgschen
Extinktionskoeffizienten lässt sich die Konzentration berechnen. Proteine hingegen
absorbieren Licht bei 280 nm maximal. Das Verhältnis OD260/OD280 ist ein Maß für die
Proteinverunreinigung, Werte unter 1.7 weisen auf einen zu hohen Proteingehalt hin. Der
Leerwert wurde mit Wasser oder TE Buffer gemessen.
3.2.5.- cDNA Synthese
3.2.5.1.- DNAse Verdau
Bei einer Verwendung der RNA für quantitative RT-PCR und bei der Herstellung von Sonden
für Northern Blots wurde die RNA zusätzlich DNase behandelt, um die Kontamination mit

3.- Material und Methoden Seite - 54 -
genomischer DNA zu minimieren. Der DNase-Verdau erfolgte dabei entweder direkt auf der
RNeasy-Säule mit Hilfe des RNAse-Free DNase Sets von Qiagen oder nach Eluation mit dem
DNA-free Set von Ambion durchgeführt.
Bei der DNAse Behandlung auf der Säule wurde zunächst Lyse, Homogenisation und
Absorption der RNA an die Silicagelmembran wie oben beschrieben durchgeführt. Nach
einem Waschschritt mit 350 µl Puffer RW1 wurde die RNA für 15 min bei Raumtemperatur
mit DNAse I in RDD Puffer inkubiert. Anschließen wurde mit weiteren 350 µl Puffer RW1
gewaschen und der Durchfluss verworfen. Für die folgenden Waschchritte und die Eluation
wurde nach dem RNeasy-Standardprotokoll (siehe oben) verfahren.
Die DNAse-Behandlung mit dem DNA-free Set von Ambion wurde in 50µl Reaktionen
durchgeführt. Dabei wurden 5 µl Volumen 10x DNase I-Puffer und 1µl rDNase I (2U/µl) zu
der gewünschten RNA-Menge dazugegeben und mit RNAse freiem Wasser auf 50µl
Reaktionsvolumen aufgefüllt. Nach einem Inkubationsschritt für 20-30 min bei 37°C wurde
0.1 Volumen DNase-Inactivation-Reagenz dazugegeben und die Lösung für weitere 2 min bei
Raumtemperatur inkubiert. Während dieser Zeit wurde der Ansatz zwei bis dreimal durch
Pipettieren gemischt. Es folgte eine Zentrifugation bei 10000 g für 1.5 min. Der RNA-
enthaltende Überstand wurde in ein frisches Reaktionsgefäß überführt und bei -80°C gelagert.
Das DNAse- und DNAse-Inactivation-Reagenz-enthaltende Pellet wurde verworfen.
3.2.5.2.- Reverse Transkription – Polymerasekettenreaktion (RT-PCR).
Die cDNA-Synthese erfolgte mit RNA-Startkonzentrationen zwischen 250 ng und 2 µg.
Dabei wurde zunächst die RNA mit 250 ng Oligo(dT)12-18 Primern (Invitrogen) gemischt und
Wasser wurde bis auf ein Gesamtvolumen von 10 µl hinzugefügt. Zur Hybridisierung der
Primer an den poly(A)-Schwanz der mRNA wurde der Ansatz für 5 min bei 65°C inkubiert,
anschließend auf Eis plaziert und kurz zentrifugiert. Im nächsten Schritt wurden 2 µl 10 mM
dNTP Mix (Fermentas), 4 µl 5x First Strand Puffer und 2µl 0.1 M DTT (beide Invitrogen)
hinzugefügt. Optional wurden noch 1µl RNAsin (Promega) zu dem Ansatz gegeben. Nach
Inkubation für 2 min bei 25°C wurde 1 µl (200 Einheiten) Superscript II Reverse
Transkriptase hinzugefügt. Nach weiteren Inkubationsschritten für 10 min bei 25°C, 50 min
bei 42°C und 15 min bei 70°C wurde die gewonnene cDNA bei -20°C gelagert. Als
Negativkontrolle zum Ausschluβ einer Kontamination mit genomischer DNA wurde für jeden
Ansatz eine negative RT-Reaktion durchgeführt. Dabei wurden alle Schritte wie oben

3.- Material und Methoden Seite - 55 -
beschrieben durchgeführt, aber anstelle des Enzyms Superscript II wurde 1µl H20 zu dem
Reaktionsansatz hinzugefügt.
3.2.6.- Protein-Isolierung
3.2.6.1.- Isolierung von Membranproteinen
Das LAT2-Protein liegt in der GEM (Glycosphingolipid Enriched Membrane), eine Region
der Zellmembran, die sehr wichtig für die Initiierung der Aktivierung von Immunorezeptor-
Transduktionsignalwege ist (Brdicka et al., 2000). Die GEMs sind kleinen Inseln in der
Zellmembran (<70 nm), reich an Glykosphingolipiden, Sphingomyelin und Cholesterin
(Brown, 1998). Die Hauptcharakteristika sind ihre Insolubilität in Detergenzen wie z.B Triton
X-100 und NP-40 bei 0-4 °C. Sie sind jedoch löslich in N-alkyl-glykosidic Detergenzen wie
z. B. N-Lauryl-Maltoside (Brown and Rose, 1992).
Um Membranproteine zu isolieren, wurden die Zellen zuerst nach der Zellkultur in PBS
gewaschen, dann wurden die Zellen im frischen Membranprotein-Lysis-Puffer resuspendiert,
gevortext und 15 min. bei 4°C in einem orbitalen Schüttler inkubiert. Das Lysat wurde 15
min. bei 13000 rpm und 4°C zentrifugiert und der Überstand wurde in ein neues Gefäß
gegeben. Die Proteinkonzentration wurde mit dem Biorad Bradford-Assay (s.u.) bestimmt,
danach wurden die Proteine aliquotiert und bei -80°C aufbewahrt.
3.2.6.2.- Isolierung von nukleären und zytoplasmatischen Proteinen
Die Isolierung von kompletten Zelllysaten erfolgte mit dem Nuclear Extract Kit (Active
Motif, Rixensart, Belgien) und nach den Herstellersempfehlungen durchgeführt. Dabei
wurden 3x106 Zellen in 100 µl des vollständigen Lyse-Puffers (bestehend aus 89 µl Lyse
Puffer AM1, 10 µl 10mM DDT und und 1 µl Proteaseinhibitorcocktail) resuspendiert und für
10s gevortext. Nach einer 10 minütigen Inkubationsphase auf Eis wurden die Lysate für
weitere 30s gevortext und anschließend für 20 min bei 13 000 rpm und 4°C zentrifugiert. Der
Überstand mit dem Zelllextrakt und den darin enthaltenden Proteinen wurde abgenommen
und das Pellet verworfen. Die Proteinlysate wurden alliquotiert und bei -80°C gelagert.

3.- Material und Methoden Seite - 56 -
3.2.6.3.- Proteinquantifizierung mittels Bradford Assay
Die Konzentrationsbestimmung erfolgte in 96-well Microtiterplatten mit Flachboden und
dafür wurde der Bradford Protein Assay (BioRad) entsprechend den Herstellerempfehlungen
eingesetzt. Es handelt sich hierbei um einen einfachen kolorimetrischen Assay zur Messung
der totalen Protein-Konzentration, (Bradford, 1976):
1 µl des Proteinlysats wurden mit 200 µl PBS und 50 µl Phosphorsäure und Methanol haltiges
Bradford-Reagenz gemischt und die Absorption der Wellenlänge wurde bei 595 nm im
Spektometer gemessen. Alle Proteinlysate wurden als Triplikate gemessen. Die Ermittlung
der Proteinkonzentrationen erfolgte mit Rinderserumalbumin (BSA) als Standard.
3.2.7.- Sonden-Herstellung für Southern Blot
3.2.7.1.- Entwicklung der Primer
Die Sequenzen, die wir vom RLGS Versuch bekommen haben, wurden zuerst mit dem
Program Repeat Masker der Universität von Washington
(http://repeatmasker.genome.washington.edu/) analysiert, um repetitive Elemente der DNA in
unserer Sonde zu vermeiden. Danach wurden die Primer mit dem Program Primer3
(http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi) in den Bereichen, in denen es
keine repetitiven Elemente gab, nach optimalen PCR Bedingungen (Oligonucleotides 20 bp
lang, Annealing Temperature 60 °C, GC< 60 %, usw) entwickelt. Die Primer wurden mit
Hilfe von NCBI Blast (http://www.ncbi.nlm.nih.gov/BLAST/) auf ausreichend Spezifität für
die uns interessierenden Sequenzen untersucht. Die Oligonukleotide für "forward" und
"reverse" Primer wurden von Herrn Dr. Igloi (Fakultät der Biologie, Universität Freiburg)
oder von Operon (Qiagen) hergestellt.
3.2.7.2.- Amplifizierung der Sonde durch die Polymerasekettenreaktion (PCR)
Mit den Primern haben wir zuerst eine Gradienten-PCR mit folgenden Bedingungen
durchgeführt: 100 ng gDNA, 5-10 pmol FW and RW Primer, 10 µl Eppendorf PCR Master
Mix und Wasser auf 25 µl angesetzt. Dann haben wir mit optimaler Annealing-Temperatur
eine Standard-PCR mit einem gröβeren Volumen (50µl) durchgeführt. Die PCR-Produkte
wurden in einem Agarose-Gel aufgetrennt, die jeweils gewünschte richtige Banden wurde

3.- Material und Methoden Seite - 57 -
ausgeschnitten und schließlich wurde mit Gel-Extraktion (Gel Extraktion Kit, Qiagen) das
PCR Produkt isoliert.
3.2.7.3.- Auftrennung von DNA-Fragmenten durch Gelelektrophorese
Je nach Länge der aufzutrennenden DNA-Fragmente wurde Agarose in Konzentrationen von
0.7% bis 2.0% in 1x TAE-Puffer oder 1xTBE Puffer durch Aufkochen vollständig aufgelöst.
Nach Abkühlen auf ca. 50°C wurde das Gel in einen mit entsprechendem Aussparungskamm
vorbereiteten Gelträger gegossen. Wenn das Gel im +4ºC Raum erstarrten war wurde, wurden
die PCR Produkte mit 6x DNA-Ladepuffer gemischt und vorsichtig in die Taschen des
erstarrten Gels pipettiert. In einer mit 1x TAE oder 1x TBE als Laufpuffer gefüllten
Horizontalkammer wurden die Proben bei Spannungen zwischen 50 und 130 V im
elektrischen Feld aufgetrennt. Zusätzlich zu den Proben wurden jeweils 5 µl Längenstandards
(100 bp, 1 kb) als Maßstab für die Länge der aufgetrennten Fragmente geladen. Nach der
Elektrophorese wurde das Gel 10-15 min. in 0,025 % Ethidium Bromide gefärbt.
Anschließend wurde das Gel unter Ultraviolett-Transillumination (254 nm) fotografiert.
3.2.7.4.- Isolierung von DNA-Fragmenten aus Agarose-Gelen
Die entsprechende Bande mit der richtigen Größe wurde aus dem Gel unter UV-Licht mit
einem Skalpell geschnitten und in einem sterilen 1.5 ml Reaktionsgefäß bei -20 ºC gelagert.
Das PCR Produkt wurde mittels des Qiagen „QIAquick Gel Extraction Kit“ entsprechend den
Vorgaben des Herstellers extrahiert, aufgereinigt, und in einem geeigneten Volumen H2O
eluiert.
3.2.7.5.- Klonierung des PCR Produkts
Das PCR Produkt wurde mit dem TOPO-TA-Cloning-Kit (Invitrogen) subkloniert. Zuerst
wurden 4 µl vom PCR Produkt mit 1 µl Salt Lösung (1,2 M NaCl, 0,06 M MgCl2, Invitrogen)
und 1 µl Vektor (pCR®2.1-TOPO®, Invitrogen) in einem Reaktionsgefäβ gemischt. Nach 5
min. Inkubationszeit bei Raumtemperatur wurde die Reaktionslösung auf Eis abgekühlt.

3.- Material und Methoden Seite - 58 -
3.2.7.6.- Transformation von kompetenten Bakterien.
Kompetente Bakterien (E. coli OneShot Mach1tm T1RChemically Competent, Invitrogen)
wurden auf Eis aufgetaut. Die Ligationslösung wurde zu den Bakterien gegeben. Nach einer
Inkubationszeit von 30 min. auf Eis wurden die Bakterien 30 sec. auf 42 ºC in der Hitzeblock
erwärmt und anschließend kurz auf Eis gestellt. Dann wurden sie in 250 µl S.O.C Medium
(Invitrogen) resuspendiert und in einen 15 ml Röhrchen für Bakterienkultur überführt. Die
Bakterien wurden 1 Stunde bei 37 ºC in einem Schüttler inkubiert und in der Folge wurden
zwischen 50 und 200 µl von den Bakterien zur Selektion auf LB Platten, versetzt mit 150
µg/ml Ampicilin (Sigma) und 40 µl X-Gal (20 mg/ml, Sigma), ausgestrichen. Anschlieβend
wurden die Bakterien bei 37 ºC über Nacht inkubiert.
Am nächsten Tag wurden die LB-Platten 4 Stunden bei +4ºC gelagert, um die weiβe
Kolonien, die das PCR Produkt im Plasmid enthalten, besser zu unterscheiden. Die blauen
Kolonien waren die Bakterien, den Vektor, aber kein PCR Produkt erhielten. Die weissen
Kolonien wurden mit einem Zahnstocher gepickt, in 4 ml LB Medium versetzt mit 150 µg/ml
Ampicilin in einen Röhrchen geimpft und über Nacht bei 37ºC in einem Schüttler wachsen
gelassen.
Am nächsten Tag wurden die Bakterien in ein steriles Reaktionsgefäβ überführt und 5 min.
bei 10000 rpm zentrifugiert. Das Bakterien-Pellet konnte für Bakterien-Stocks oder für
Plasmid-Extraktion benutzt werden.
3.2.7.7.- Herstellung von Bakterien-Stocks
Zur Aufbewahrung nach Transformation mit rekombinanten Plasmiden wurden Aliquots der
Kultur einzelner Bakterien-Klone in 1.5 ml Reaktionsgefäße überführt, mit Glycerin in einer
Endkonzentration von 20% versetzt, in flüssigem Stickstoff schockgefroren und anschließend
bei -80°C gelagert.
3.2.7.8.- Mini-Präparation von Plasmid-DNA
Nach der Isolierung der Bakterien konnten wir mit dem Mini Prep Kit (Qiagen) die Plasmide
extrahieren. Das Bakterien-Pellet wurde in 250 µl Puffer P1 (50 mM Tris/HCl pH 8.0, 10 mM
EDTA, 100 µg/ml RNase A) resuspendiert und der Ansatz in ein 1.5 ml Reaktionsgefäß

3.- Material und Methoden Seite - 59 -
übertragen. Die Isolierung der Plasmide erfolgte anschließend nach Angaben des Herstellers
und Verwendung der mitgelieferten Puffer über die Bindung an eine Säule. Die so
gewonnenen Plasmide wurden anschließend in H2O aufgenommen.
3.2.7.9.- Maxi-Präparation von Plasmid-DNA
Die Isolierung größerer Mengen Plasmid-DNA bis zu 100 mg erfolgte mit dem „QIAGEN
Plasmid Maxi Kit“ von Qiagen. 100 bis 500 ml LB-Medium wurden mit 2 bis 5 ml einer
Starter-Kultur beimpft und über Nacht für ca. 12 bis 16 h bei 37°C und 300 rpm im Schüttler
inkubiert. Die Zellen wurden dann bei 3000 rpm und 4°C pelletiert und das Pellet in 10 ml
Puffer P1 resuspendiert. Die anschließende Plasmid-Präparation erfolgte nach Angaben des
Herstellers unter Verwendung der „QIAGEN-tips 500“ und der entsprechenden Puffer. Die so
gewonnenen Plasmide wurden anschließend in H2O aufgenommen und die Konzentration
photometrisch ermittelt.
3.2.7.10.- Extraktion der Sonde aus dem Plasmid.
Zwischen 5-10 µg Plasmide wurden 2-3 Stunden mit 2 µl (40 Einheiten) vom
Restriktionenzym EcoR I (New England Biolabs) und NEB Puffer EcoR I (New England
Biolabs) verdaut, immer wenn das Insert keine EcoR I-Schnittstelle hatte. Die Produkte des
Restriktionsverdaus wurde in einem 1,5 %igen Agarose Gel getrennt, und schließlich wurde
die gewünschte Bande ausgeschnitten und mit dem Gel Extraktion Kit (Qiagen) isoliert.
3.2.8.- Sonden-Herstellung für Northern Blot
Die Entwicklung der Primer wurde wie so oben (3.2.7.1) für Sonden Herstellung für Southern
Blot durchgeführt. Als Template für die PCR wurde cDNA einer Zelllinie benutzt, die das uns
interessierende Gen exprimiert. Die RNA wurde von der Zelllinie mit dem RNeasy Kit von
Qiagen isoliert (sieh oben 3.2.2.4) und davon cDNA synthetisiert (sieh oben 3.2.5). Die
weitere Herstellung der Sonde wurde mit der oben beschriebenen Prozedur für die Southern
Blot Sonde durchgeführt.

3.- Material und Methoden Seite - 60 -
3.2.9.- Restriktionsverdau der genomischen DNA
10 µg genomische DNA von jeder Zelllinie und von den mononukleären Zellen aus dem Blut
wurden über Nacht verdaut: es wurden 60 Einheiten EcoR V (nicht methylsensitive Enzyme)
für den einfach Verdau und zusätzlich 30 Einheiten Not I (methylsensitive Enzyme) für den
Doppelverdau, BSA in einer Endkonzentration von 100 µg/ml und der NEB 3
Reaktionsbuffer in einfacher Konzentration benutzt. Die Ansätze wurden je nach der
verwendeten Restriktionsendonuklease nach den Angaben der Hersteller inkubiert.
3.2.10.- Southern Blot
3.2.10.1.- Vorbereitung und Auftrennung der genomischen DNA
Die genomische DNA wurde zuerst durch Gelelektrophorese aufgetrennt und danach wurde
die DNA auf eine Membran nach der Southern Blot Methode (Southern, 1975) übergetragen.
Die durch Restriktionenzyme verdaute gDNA wurde mit abgeschnittenen Spitzepipetten auf
ein 0.7 %iges Agarose Gel in 1xTAE Puffer aufgetragen. Die Gelelektrophorese wurde über
Nacht bei 20-30 V durchgeführt. Zur Ladekontrolle wurde das Gel mit Ethidiumbromid
(Sigma) gefärbt und fotographisch dokumentiert.
3.2.10.2.- Transfer der DNA nach der Southern Blot Methode
Die DNA im Gel wurde dann durch Behandlung mit 0.25 M HCl für mindestens 20 min oder
bis zum Umschlag der Bromophenol-Farbe im DNA-Ladepuffer depuriniert, anschließend in
Denaturierungs-Lösung (0.5 M NaOH, 1.5 M NaCl) für eine Stunde (zweimal 30 min.)
denaturiert.
Der Transfer erfolgte über Nacht auf eine positiv geladene Nylon-Membran (Hybond N+,
Amersham), als Transferpuffer wurde Denaturierungs-Lösung verwendet. Vor dem Aufbau
der Blot-Apparatur wurde die Membran zunächst kurz in H2O und anschließend in
Denaturierungs-Lösung äquilibriert. Der Aufbau der Blots erfolgte prinzipiell nach dem
Prinzip des Kapillar-Transfer-Systems, wie in Sambrook et al. (1989) beschrieben. Als
Modifikation wurde jedoch statt des vorgegeben Docht-Prinzips ein Schaumstoff-Schwamm
als Basis verwendet (Fourney et al., 1988), durch den ein gleichmäßiger Transferpuffer-Strom
über die gesamte Gelfläche gewährleistet wurde.

3.- Material und Methoden Seite - 61 -
Nach dem Transfer wurde die Membran zur Entfernung von Gelresten vorsichtig kurz in 2x
SSC gewaschen und anschließend an der Luft getrocknet. Eine Immobilisierung der DNA auf
der Nylon-Membran erfolgte durch zweimalige Bestrahlung mit einem UV-Crosslinker (UV
Stratalinker 2400, Stratagene; Einstellung: Auto crosslink entspricht 30 sec). Vor der
Hybridisierung wurde die Membran erneut mit 2x SSC benetzt.
3.2.11.- Northern Blot
3.2.11.1.- Vorbereitung und Auftrennung der RNA
Vorbereitend wurden Gelkammern, Träger und Kämme, die zur elektrophoretischen
Auftrennung von RNA dienen sollten, zur Denaturierung von RNasen mindestens 12 Stunden
in 2 M NaOH eingelegt. Agarose wurde in RNAse freien H2O durch Kochen aufgelöst, nach
dem Abkühlen auf ca. 60°C mit 10x MOPS-Puffer und 37%igem Formaldehyd versetzt
(Endkonzentration: 1% Agarose, 1x MOPS-Puffer, 0.66 M Formaldehyd) und in den
entsprechenden Gelträger gegossen.
15 µg RNA wurden in 10 µl H2O gelöst, mit 30 µl RNA Laddebuffer (75 % Formamide, 1x
MOPS Buffer, 10 % Formaldehyde, 10 % Glycerol und Bromophenol Blau) gemischt und 15
min. bei 65 °C denaturiert. Danach wurde die RNA 5 min. auf Eis gestellt und durch
Elektrophorese in einem 1 %igen Agarose-Gel unter denaturierenden Bedingungen (1x
MOPS und 0,66 M Formaldehyde) für 3-4 Stunden bei 70 V aufgetrennt. Als
Elektrophoresepuffer wurde 1x MOPS benutzt. Qualität und Quantität der RNA wurden durch
Ethidiumbromid-Färbung der ribosomalen RNA (rRNA) der 28S- und 18S-Banden
abgeschätzt.
3.2.11.2.- Transfer der RNA nach der Northern Blot Methode
Vor dem Transfer der RNA vom Gel auf eine Nylon-Membran (Hybond N+, Amersham)
wurde das Gel für 30 min in 0.05 M NaOH/1x SSC inkubiert und anschließend zweimal für
20 min in 10x SSC gewaschen. Der prinzipielle Aufbau der Blot-Vorrichtung erfolgte wie in
Sambrook et al. (1989). Vergleichbar der Southern-Blot-Technik wurde das Verfahren wie
oben beschrieben durch Verwendung eines Schaumstoff-Schwammes als Basis modifiziert
(Fourney et al., 1988). Der Transfer erfolgte über Nacht in 10x SSC als Transferpuffer. Nach

3.- Material und Methoden Seite - 62 -
dem Transfer wurde die Membran zur Entfernung von Gelresten kurz in 2x SSC geschwenkt
und anschließend an der Luft getrocknet.
Die Effizienz und Qualität des Transfers wurde durch Kontrolle des Gels und der Membran
unter UV-Licht überprüft. Durch zweimaliges Bestrahlung mit einem UV-Crosslinker (UV
Stratalinker 2400, Stratagene; Einstellung: Auto crosslink entspricht 30 sec) wurde die RNA
auf der Nylon-Membran immobilisiert. Vor der Hybridisierung wurde die Membran erneut
mit 2x SSC benetzt.
3.2.12.- Hybridisierung der Membranen von Southern und Northern Blot
3.2.12.1.- Radioaktive Markierung von DNA-Fragmenten
Die Sonde wurde mit dem Kit „Megaprime DNA Labelling System“ (Amersham)
entsprechend den Herstellerempfehlungen markiert. Hierzu wurden 5-25 ng Sonde in 5 µl
angesetzt, mit 5 µl Random Primer gemischt und 5 min. bei 100 °C denaturiert. Dann wurden
21 µl Wasser, 10 µl Labelling Buffer, 2 µl Klenow Enzyme und 5 µl α-32P-dCTP (radioaktiv,
3000 Ci/mmol von Amersham) im Arbeitsraum für radioaktive Tätigkeit zugegeben und für 30
min bei 37°C inkubiert. Die inkorporierte Radioaktivität wird anschließend durch einen
Sephadex-Filter (Amersham) mit Hilfe einer Zentrifuge (5 min., 2500 rpm) von restlichen
Radioisotopen getrennt. Schließlich wurde die Sonde bei 100°C denaturiert, auf Eis abgekühlt
und die Aktivität der markierten Sonde mit einem β-Counter (Ercicount 2000, Scotlab)
gemessen.
3.2.12.2.- Hybridisierung mit radioaktiv markierten DNA-Sonden
Zuerst wurden die Northern- und Southern-Blot-Membranen für 15 min. in 2x SSC Buffer
gewaschen. Die Membranen wurden mit der Nukleinsäure tragenden Seite nach innen gerollt
in eine Hybridisierungsröhre (Glaszylinder mit abdichtendem Schraubdeckel) eingebracht.
Dann wurden sie 3-4 Stunden bei 65°C, mit 10 mL vorgewärmten Church Buffer (0.33 M
Na2HPO4, 0.16 M NaH2PO4, 7 % SDS, 1 mM EDTA pH 8.0) und 100 µl Salmon Sperm
DNA (10 mg/mL) in einem Hybridisierungsofen vorinkubiert. Danach wurde die radioaktiv
markierte Sonde (wie oben beschrieben) zu den Membranen pipettiert und anschlieβend
wurde die Hybridisierung über Nacht bei 60-65 ºC durchgeführt.

3.- Material und Methoden Seite - 63 -
Am nächsten Tag wurde die Membran zweimal 10 min. bei 42°C in LSW Buffer (1xSSC, 0.1
SDS) und zweimal 20 min. bei 65°C in HSW Buffer (0.2 SSC, 0.1 SDS) gewaschen. Dann
wurde die Radioaktivität auf der Membran mit einem β- Counter (LB122 Berthold, UMO)
gemessen. Wenn die Radioaktivität einer Membran unter 40 CPM war, wurde die Membran
mit einer Plastikfolie in eine Filmkassette eingepackt und ein Film auf die Membran im
Dunkelraum gelegt. Die Kassette wurde dazu 3-4 Tage, in Abhängigkeit von der
Radioaktivität, bei -80°C aufbewahrt. Dann wurde der Film entwickelt und, je nach Stärke des
Signals ein weiterer Film auf die Membrane für eine längere oder kürzere Dauer der
Exposition gelegt.
3.2.13.- Western Blot
Der Western Blot dient der Detektion bestimmter Proteine. Die Proteine werden zunächst
elektrophoretisch nach ihrer Größe aufgetrennt und auf eine Nitrozellulosemembran
transferiert. Anschließend erfolgt die Inkubation mit einem antigenspezifischen
Primärantikörper, der das gesuchte Protein bindet. Durch Zugabe eines Enzyms gekoppelten
sekundären Antikörpers, der an den Fc-Teil des spezifischen Antikörpers bindet erfolgt die
Substrat Reaktion.
Die Versuchsdurchführung erfolgte mit dem NuPAGE® Elektrophoresis System (Invitrogen)
und der XCell SureLockTM Mini-Cell (Invitrogen) entsprechend des Standardprotokolls
3.2.13.1.- Vorbereitung der Proben
30 µg Protein wurde mit 4 µl NuPAGE® LDS Puffer 4x (Invitrogen) pro Probe gemischt und
auf 15 µl mit Wasser verdünnt. Dann wurden sie 10 min. bei 70 °C denaturiert und 5 min. auf
Eis gestellt. Wenn der Western Blot unter reduzierenden Bediengungen durchgeführt wurde,
wie z. B. bei der Detektion mit dem anti-ETO-Antikörper, wurde 1.5 µl NuPAGE® Reducing
Agent (Invitrogen) hinzugefügt
3.2.13.2.- Elektrophoretische Auftrennung im Gel
Zur Elektrophorese wurde das NuPAGE® Elektrophorese System (Invitrogen) entsprechend
den Herstellerempfehlungen verwendet. Hierzu wurden ein NuPAGE® 4-12 % Bis Tris Gel
und der 1x NUPAGE® MES SDS Buffer (Invitrogen) benutzt. Das Gel wurde aus der

3.- Material und Methoden Seite - 64 -
Plastikfolie geschnitten und mit destilliertem Wasser gewaschen. Nach vorsichtiger
Herausnahme des Kamms wurden die Taschen mit Running Puffer gespült. Anschließend
wurden die XCell SureLockTM Mini-Cell Kammer zusammengebaut und die Gele eingesetzt.
Es wurden 500 µl NuPAGE® Antioxidant (Invitrogen) als Laufpuffer eingesetzt, wenn
reduzierenden Bedingungen für die Proteindetektion notwendig waren. Als Laufkontrolle
wurde 5 µl des gefärbten Proteinmarkers SeePlus Blue 2 (Invitrogen) eingesetzt und als
Entwicklungskontrolle wurde gelegentlich ein fluoreszierender Proteinmarker (Invitrogen)
benutzt. Die Gelelektrophorese wurde 1.5 Stunden bei 150 V und bei Raumtemperatur
durchgeführt.
3.2.13.3.- Blotting
Für das Blotten wurde zunächst der 1x NuPAGE® Transfer Buffer (Invitrogen) mit 10 %
Methanol (J.T. Baker, Deventer-Holland) vorbereitet. Bei dem gleichzeitigen Blotting von 2
Gelen wurden 200 ml Methanol (20% Gesamtvolumen) hinzugefügt, um einen effizienten
Transfer beider Gele zu gewährleisten. Mit Hilfe des zugehörigen Gel-Messers wurden die
Gele aus der Plastikhülle herausgelöst und dem Blotten zugeführt.
Auch der Transfer erfolgte in dem X-Cell IITM Blot Modul (Invitrogen) entsprechend den
Herstellerempfehlungen. Zum Tranfer wurde zunächst eine Hybond-P Membran (Amersham)
für 30 sec. in Methanol aktiviert, 5 min. in destilliertem Wasser gespült und 5 min. im
Transfer Buffer äquilibriert. Die zugehörigen Blotting Pads wurden solange in Transfer Puffer
gelegt, bis sie vollständig mit Puffer gefüllt und keine Luftblasen mehr vorhanden waren. Das
auf Gelgröße geschnittene Filterpapier wurde ebenfalls im Transfer Puffer aufgefeuchtet.
Anschließend wurden die Blotting Pads, Filterpapiere, Gel(e) und Membranen(en) nach
Herstelleremphelung in der Transferkammer zusammengebaut.
Die Proteine wurden für 1.5 Stunden bei 30 V und auf Eis mit Hilfe der Transfer Buffer (1x
NUPAGE® Transfer Buffer, 10 % Methanol) auf die Membran geblottet.
Nach dem Blotten wurde mit einer Ponceau-Rot-Färbung kontrolliert, ob die Proteine
gleichmäßig auf die Membrane transferiert waren. Dann wurde die Membrane zuerst kurz in
Wasser und danach in TBS-T (40mM Tris, 270 mM NaCl, 1 ml Tween 20, pH 7.6)
gewaschen.

3.- Material und Methoden Seite - 65 -
3.2.13.4.- Protein-Detektion mittels Antikörper.
Die Membran wurde eine Stunde im Blocking-Puffer (TBS-T mit 5 % fett-freiem
Magermilchpulver) inkubiert. Danach wurde die Membran kurz mit TBS-T gewaschen und
mit dem ersten Antikörper in einer vorher bestimmten Konzentration (s. 3.1.4.6.) inkubiert.
Das Immunoblotting erfolgte über Nacht bei 4°C auf einem horizontalen Schüttler.
Am nächsten Tag wurde die Membran 4 x 5 min. bei Raumtemperatur in TBS-T in einem
horizontalen Schüttler gewaschen. Danach wurde der Zweit-Antikörper in vorher bestimmter
Konzentration zugegeben (s. 3.1.4.6.). Die Membran wurde mit dem Zweit-Antikörper für
eine Stunde bei Raumtemperatur im Schüttler inkubiert, danach wurde die Membran wieder 4
x 5 min. mit TBS-T gewaschen.
Als Detektionsmethode wurde das ECL Plus Western blotting Detektions System (Amersham
Biosciences) verwendet. Lösungen A und B wurden in einem Verhältnis vom 40:1 gemischt,
auf die Membran pipettiert und es wurde 5 min. bei Raumtemperatur inkubiert. Dann wurde
die Membran auf Zellstoff kurz getrocknet und mit der Proteinseite nach unten in eine
Plastikfolie gepackt. Schließlich wurde sie in einer Filmkassette mit Autoklavierband fixiert
und HyperFilm (Amersham) für verschiedene Zeiten (1, 5, 30 min.) exponiert und dann in
einem Entwicklungsgerät (Optimax X-Ray Film Procesor, Typon) entwickelt.
3.2.14.- Durchflusszytometrie
Die Durchflusszytometrie ist eine Methode, bei der verschiedene Parameter einzelner Zellen
gleichzeitig bestimmt werden. Die Zellen werden durch einen Fluss geführt, in dem die Form,
Granularität, Grösse, usw, mit Hilfe eines Lasers gemessen werden können.
Wenn die Zellen vor der Durchflusszytometrie mit einem Fluoreszenz-markierten Antikörper
inkubiert werden, kann man auch den Anteil der Antikörper tragenden Zellen messen. Damit
werden oberflächen Zellmarker gemessen und ganzen Zellpopulationen charakterisiert. In
unserem myeloischen Differenzierung-Assays gewinnen wir so Information über den Anteil
der Zellen, die differenziert sind.
3.2.14.1.- Vorbereitung der Zellen für die Messung von Oberflächen-Antigenen
Die Zellen wurden nach der Zellkultur mit PBS gewaschen, gezählt und in Portionen von
1x105 Zellen auf FACS Röhrchen aufgeteilt. Dann wurden die Zellen 5 min. bei 1200 rpm

3.- Material und Methoden Seite - 66 -
zentrifugiert und der Überstand verworfen. 5 µl Antikörper, gebunden an Fluorezenz-
Farbstoffe (PE und FITC), wurde zu den Zellen gegeben. Die Inkubation mit dem Antikörper
erfolgte 20 min. bei +4ºC im Dunkel. Nach dieser Zeit wurden die Zellen mit PBS
gewaschen, zentrifugiert und in 1 mL PBS resuspendiert kurz vor der Messung. In einem
FACS Gerät (Cyan ADP, Dakocytomation) wurden die Zellen auf Fluorezenz-Intensität
analysiert. Die Werte wurden mit der Software Summit v 4.2 ausgewertet und ggf mit dem
FlowJo Software in Graphiken dargestellt.
3.2.14.2.- Messung der Oberflächen-Antigene
Zuerst wurde das Gerät nach der FS (Vorwärtsbeugung) und SS (Seitwärtsstreuung)
Eigenschaften der Zellen kalibriert. Zellpopulationen, die eine regelmäßige FS und SS
aufwiesen, wurden mit Hilfe eines elektronischen Fensters für weitere Messungen
selektioniert. In einem weiteren elektronischen Fenster wurde die Zahl der Zellen gemessen,
die eine bestimmte Fluorezenzintensität trugen und wurden in einem Histogramm
representiert. Um die reproduzierbare Messung zu erzielen, wurden die Verstärkung der
Lichtwelle so ausgewählt, dass ungefärbten Zellen innerhalb der log 100 und log 101-Stufe
lagen. Es wurden pro Ansatz 10000 Zellen gemessen. Die Fluorezenzintensität, ab der die
Zellen als positiv gewertet wurden, wurde so gewählt, dass ein Prozent der mit einem Isotyp-
Kontrolle-Antikörper inkubierten Zellen positiv waren. Anschlieβend wurden von dieser
selektionierten Zellpopulation wenigstens 10000 Zellen für die uns interessierenden Antigene
gemessen.
3.2.15.- "Real-time"-RT-PCR
3.2.15.1.- Mechanismen
Das Prinzip der quantitativen PCR beruht auf der herkömmlichen Polymerasekettenreaktion
und bietet zusätzlich die Möglichkeit der Quantifizierung durch Fluoreszenzmessungen
mittels eines Lasers während der PCR-Zyklen. Bei verschiedenen Fluorezenzssystemen ist
das Prinzip gleich: die Fluorezenzintensität wird am Ende jedes Zyklus gemessen und diese
Daten werden während des PCR-Laufes mit der Zyklenzahl für weitere Auswertungen in
einer Graphik repräsentiert. Da die Fluorezenzintensität während der Exponentialphase der
PCR direkt mit der Menge des PCR Produktes korreliert, ist eine Quantifizierung des
synthetisierten Produkts und der Startkonzentration möglich.

3.- Material und Methoden Seite - 67 -
Es wurde der SYBR I GREEN Farbstoff in dieser Arbeit benutzt, weil die Etablierung nur
eine klassische PCR erfordert und das Verfahren sich als ausreichend sensitiv und spezifisch
für unseren Zweck erwies. Wenn dieser Farbstoff zwischen 2 DNA Ketten interkaliert, wird
er fluoreszierend (bei 530 nm). Um spezifische von unspezifischen Produkten und von
Primer-Dimeren zu unterscheiden, wurde jeweils am Ende des PCR Laufes eine
Schmelzkurve bestimmt. Proben, bei denen die Schmelzkurve mehrere Produkte zeigte,
wurden von der Auswertung ausgeschloßen.
Die Primersequenzenzen wurden mit Hilfe der Universal Probe Library Software der Roche
Webseite (https://www.roche-applied-science.com/sis/rtpcr/upl/index.jsp) und der Software
Primer3 entwickelt (http://frodo.wi.mit.edu/cgi-bin/ primer3/ primer3_www.cgi). Für jedes
Primerpaar wurde die PCR Reaktion solange optimiert, bis sie eine Effizienz von mehr als 1,9
ohne unspezifische Produkte erreicht hatten.
3.2.15.2.- Durchführung
Die quantitative Polymerasekettenreaktion aus cDNA-Templates wurde im Light Cycler 480
(Roche diagnostics) durchgeführt. Der Light Cycler 480 arbeitet mit 384-Well-PCR-
Mikrotiterplatten. Zur relativen Quantifizierung wurde das SYBR I GREEN® (Roche)
Fluorezenzstoff verwendet, der sich im Light Cycler 480 SYBR I GREEN Master Mix
befindet. Die Reaktionen wurden in 10 µl Ansätzen in einer 384-Well Mikrotiterplatte
(Roche) durchgeführt. Die gewonnene cDNA aus der RT-Reaktion (s.o. 3.2.5.2.) wurde in
einer Endkonzentration von 5-50 ng verwendet.
Für die relative Quantifizierung wurden Verdünnungsreihen von cDNA aus U937 (1:10,
1:100, 1:1000, 1:10000, 1:100000) hergestellt. Alle Proben und Standards wurden als
Duplikate gemessen und der Mittelwert für weitere Rechnungen benutzt. Als
Negativkontrollen wurden sowohl Ansätze ohne Template zum Ausschluβ eine
Kontamination des Reaktionsansatzes, als auch minus RT-Reaktionen (ohne Reverse
Transkriptase) zum Ausschluß einer Amplifikation von genomischer DNA verwendet. Nach
Zugabe aller Reaktionskomponenten wurden die Platten mit selbstklebenden optischen Folien
(Roche) verschlossen.

3.- Material und Methoden Seite - 68 -
Tabelle 2.- Reagenzien für eine "real-time"-RT- PCR
PCR-Reaktionskomponenten Endkonzentration
SyBrGreen QPCR Master Mix Roche 1x
Primer FW (100 pmol/µl)
Primer RW (100 pmol/µl)
0,5 µM
0,5 µM
Template: cDNA 5-50 ng (aus RNA Menge)
H2O auf 10µl
Die Reaktionen wurden im Light Cycler 480 von Roche Diagnostics mit 45 Zyklen unter den
in der Tabelle aufgeführten Bedingungen durchgeführt.
Tabelle 3.- Temperatur- und Zeit-Bedingungen für eine "real-time" PCR
Schritt Temperatur Zeit Ramp Rate Zyklen Art der
Akquisition
1. Enzymaktivierung 95°C 10 min 4.8 °C/sec 1
2. Denaturierung 95°C 15 sec 4.8 °C/sec
3. Annealing 60°C 15 sec 2.5 °C/sec
4. Extension 72 °C 20 sec 4.8 °C/sec
45
einfach
5. Meltingkurve
5.1.
5.2.
95 °C
60 °C
10 sec
10 sec
4.8 °C/sec
2.5 °C/sec
durchgehend
6. Cooling 37 °C 1 min 2.5 °C/sec
3.2.15.3.- Relative Quantifizierung
In einer PCR Reaktion wird, die Zyklusnummer, bei der die Fluorezenzintensität über den
Hintergrundfluorezenz in den einzelnen Reaktionen detektiert werden kann, als "Cross point"
(Cp) bezeichnet. Diese Cp Werte ist für jede einzelne Probe charakteristisch und wird als das

3.- Material und Methoden Seite - 69 -
"Maximun der Zweiten Ableitung" der Intensitätskurve berechnet. Die Cp Werte dienen als
Grundlage für die weitere Quantifizierung.
Zur Berechnung der unterschiedlichen Expressionslevel wurde die Menge des untersuchten
Gens nach Behandlung der Zellen mit der Menge des Zielgens in der unbehandelten Kontrolle
verglichen. Als interne Kontrolle wurde die Menge von einem Haushaltsgen in allen Proben
mitbestimmt. Die Bedingungen, die an solche Haushaltsgene gestellt werden, sind eine
konstante Expression, die nicht durch die Behandlung der Zellen beeinträchtigt wird und ein
Expressionslevel ähnlich dem des uns interessierenden Gens.
Zur relativen Quantifizierung und zur Qualitätskontrolle unserer PCR wurde die
Standardkurvenmethode gewählt. Zur Generierung der Standardkurve wurde eine
Verdünnungsreihe (1:10, 1:100, 1:1000, 1:10000, 1:100000) von cDNA der U937 Zellen
gewählt. Da eine lineare, umgekehrt proportionale Beziehung zwischen dem Logarithmus der
eingesetzten cDNA-Menge und dem Cp-Wert besteht, kann aus den festgelegten
Ausgangsmengen der Standardproben eine Standardkurve generiert werden. Der Logarithmus
der Ausgangsmenge der Proben wird gegen den Cp aufgetragen.
Mit Hilfe einer Exponentialfunktion kann die Ausgangsmenge in den Proben berechnet
werden. Eine solche Standardkurve und die daraus errechneten Ausgangsmengen der
unbekannten Proben werden sowohl für das Zielgen, als auch für das Haushaltsgen generiert.
Da jeder PCR Ansatz als Duplikat durchgeführt wurde, wurde anschließend der Mittelwerte
der Kopienanzahl jeder Probe bestimmt. Die relative Expression wurde so berechnet, dass die
Expression des uns interessierenden Gens durch die Expression des Haushaltgens geteilt
wurde.
Im nächsten Schritt der Normalisierung werden die errechneten Kopienzahlen des Zielgens
durch die Kopienzahl des Haushaltsgens geteilt. Um die Änderung der Expression in
Zahlenwerte fassen zu können, wurde die Expression des normalisierten Zielgens in Bezug zu
einem Referenzwert gesetzt. Als Referenzwert diente die Expression in unbehandelten oder
uninduzierten Zellen.
3.2.16.- Chromatin-Immunopräzipitation (ChIP)
Das Protokoll wurde bereits beschrieben (Metzger et al., 2005). Die Zellen wurden für 10
min. mit 1 % Formaldehyd bei 37 °C vernetzt ("cross-linked") und zweimal mit PBS
gewaschen. Zellpellets wurden für 10 min. mit SDS Lyse-Buffer (1 % SDS, 10 mM EDTA,
50 mM Tris-HCl) auf Eis lysiert und schlieβlich wurde die DNA in Fragmenten von zwischen

3.- Material und Methoden Seite - 70 -
200 bp-1 kb Gröβe mit Ultraschall beschallt (4 Pulse für 30 sec., Amplitude 20 %, 1 sec
on/0.3 sec. Off). Nach dem Pre-clearing für 2 Stunden mit Protein A-Sepharose wurde 5 %
des Volumens entnommen und als Input bezeichnet. Das Lysat wurde dann in ChIP-Dilution
Puffer (1 % Triton X-100, 2 nM EDTA, 20 nM Tris/HCl pH 8.1, 150 nM NaCl) verdünnt und
über Nacht mit 5 µg des primären Antikörpers inkubiert. Am folgenden Tag wurden die
Antikörper-Protein-DNA Komplexe mit Protein A-Sepharose präzipitiert und das Pellet
wurde mit ChIP-Dilution-Puffer, TSE I-Puffer (0.1 % SDS, 1 % Triton X-100, 2 mM EDTA,
20 mM Tris/HCl, 150 mM NaCl), TSE II-Puffer (0.1 % SDS, 1 % Triton X-100, 2 mM
EDTA, 20 mM Tris-HCl, 500 mM NaCl), LiCl-Puffer (0.25 M LiCl, 1 % NP40, 1 % Na-
Deoxycholat, 1 mM EDTA, 10 mM Tris-HCl) und zweimal mit TE-Puffer (10 mM Tris-HCl,
1 mM EDTA) gewaschen. Die Desorption erfolgte zweimal nach der Inkubation bei
Raumtemperatur mit einem frisch zubereiteten 1 % SDS, 0.1 M NaHCO3 Puffer und die
Formaldehyd-Vernetzung wurde für 4-6 Stunden bei 65 °C rückgängig gemacht. Die gelöste
DNA wurde mit dem „PCR-Purification Kit“ (Qiagen) isoliert. Die isolierte DNA wurde
mittels einer „real time“-DNA-PCR quantifiziert und mit dem Input verglichen.
3.2.17.- Densitometrie
Die Densitometrie wurde mit dem software ImageJ (http://rsb.info.nih.gov/ij/) durchgeführt.
Image J ist ein freies Software Programm, das von der "National Institutes of Health" der
Vereinigten Staaten entwickelt wurde. Die Fläche und die Intensität der interessierenden Bände
wurden gemessen und diese Werte wurde gegen das Produkt von der Fläche und der Intensität
von einem Haushaltsgen relativiert. Diese Werte wurden dann gegen die Positivkontrolle oder
unbehandelte Zellen normalisiert.
3.2.18.- Statistische Auswertung
Der Mittelwert von mindestens drei unabhängigen Experimenten oder Klonen und die
Standardabweichung dieser Werte wurden auf statistische Signifikanz mit dem ANOVA t-
Test geprüft. Die klinischen Daten der Patienten wurden mit dem Fisher´s exact Test auf
statistische Signifikanz geprüft.

4.- Ergebnisse Seite - 71 -
4.- Ergebnisse
4.1 Biologische Effekte der AML1/ETO-Induktion in 9/14/18-Zellen.
Das AML1/ETO-Protein wurde mit 5 µM Ponasteron A in 9/14/18-Zellen induziert.
Ponasteron A und frisches Medium wurden alle 48 Stunden zu den Zellen gegeben. Viabilität
und Zellwachstum wurden am ersten und dann jeden zweiten Tag nach der Induktion von
AML1/ETO durch Trypan Blau Färbung bestimmt. Dadurch wurde festgestellt, dass die
Induktion des AML1/ETO-Proteins eine Zellwachstum-Inhibition (Zellkonzentration von
16,3 +/- 4,2 Zellen x 105 /ml in den nicht-induzierten Zellen vs. 9,9 +/- 2,9 Zellen x 105 /ml in
den induzierten Zellen, Wachstum-Inhibition von 39,3% am 4. Tag) (Abb.11a) und
Apoptose/Nekrose der Zellen (Viabilität von 89,2 +/- 5,2 % in den nicht-induzierten Zellen
vs. 72,4 +/- 11,6 % in den induzierten Zellen am 4. Tag) verursacht (s. Abbildung 11b).
Eine solche Zellwachstums-Inhibition und Apoptose-Induktion nach der ektopen
AML1/ETO-Expression in 9/14/18-Zellen wurden schon in den Arbeiten von (Berg et al.,
2007; Fliegauf et al., 2004; Lu et al., 2006)) beschrieben. Die Unterschiede im Zellwachstum
und in der Viabilität zwischen den induzierten und nicht induzierten Zellen waren umso
größer, je länger die Induktion des AML1/ETO-Proteins dauerte.
Abbildu ng 11.- Funktionelle Analyse der AML1/ETO -induzierbaren U-937-Zellen. A und B.- Zellwachstum und Viabilität wurden durch Trypan-Blau-Färbung bestimmt und die AML1/ETO-induzierten Zellen wurden mit den nicht-induzierten Zellen verglichen.
9/14/18 +/- Pon A
0
5
10
15
20
25
0 24 48 72 96 120
Time (hrs)
Cel
l con
cent
ratin
(x10
*5/m
l)
9/14/18 + Pon A 9/14/18 - Pon A
9/14/18 +/- Pon A
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72 96 120
Time (hrs)
Via
blity
9/14/18 + Pon A 9/14/18 - Pon A
A.- B.-9/14/18 +/- Pon A
0
5
10
15
20
25
0 24 48 72 96 120
Time (hrs)
Cel
l con
cent
ratin
(x10
*5/m
l)
9/14/18 + Pon A 9/14/18 - Pon A
9/14/18 +/- Pon A
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72 96 120
Time (hrs)
Via
blity
9/14/18 + Pon A 9/14/18 - Pon A
A.- B.-

4.- Ergebnisse Seite - 72 -
4.2.- Funktionelle Untersuchung des AML1/ETO-Zielgens LAT2
(WBSCR5/NTAL/LAB).
Funktionelle Untersuchungen wurden mit Hilfe eines bereits identifizierten
AML1/ETO-Zielgens der AG von Prof. Lübbert durchgeführt. Mit Hilfe der AML1/ETO-
induzierbaren Zellen wurden verschiedene Gene in einer cDNA-RDA-Analyse identifiziert,
deren Expression durch das AML1/ETO-Protein reguliert wurde. Die AML1/ETO-vermittelte
Repression des LAT2-Gens wurde mittels Northern Blots in verschiedenen AML1/ETO-
positiven vs. -negativen Zelllinien und in primären AML-Blasten validiert (Fliegauf et al.,
2004).
4.2.1.- Charakterisierung der Repression des LAT2-Gens in einer AML1/ETO
induzierbaren Zelllinie
Im ersten Schritt der funktionellen Untersuchungen wurde die Repression der LAT2-
mRNA nach der Induktion des AML1/ETO-Proteins in 9/14/18-Zellen mittels Northern Blot
bestätigt. Die relative Expression der LAT2-mRNA in 9/14/18-Zellen 48 Stunden nach der
AML1/ETO-Induktion war 73,8 +/- 0,4 % % der Expression der nicht-behandelten 9/14/18-
Zellen. (Abb. 12a). Ebenso konnten die Daten von Dr. M. Fliegauf, die Repression von der
LAT2- mRNA durch AML1/ETO, auf der Protein-Ebene bestätigt werden (Daten nicht
gezeigt).
Eine weitere Charakterisierung der AML1/ETO-Repression der LAT2-mRNA wurde
zu sehr frühen Zeitpunkten mittels „real-Time“-RT-PCR durchgeführt. Eine Repression von
LAT2 mRNA (0,76 +/- 0,13-fach im Vergleich zur Grundexpression in der 9/14/18-Zellen)
wurde schon nach 4 Stunden der Induktion des AML1/ETO Proteins nachgewiesen (Abb
12b). Die Expression von AML1/ETO war 2 Stunden 9,5-fach höher und 4 Stunden nach der
Induktion mit Ponasteron A 15,5-fach höher als die Grundexpression in 9/14/18-Zellen (Abb.
12c).

4.- Ergebnisse Seite - 73 -
4.2.2.- Untersuchung der LAT2-mRNA- und -Protein-Expression in myeloischen
Zelllinien.
Im Weiterem wurde die Expression von LAT2 auf mRNA- und Protein- Ebene in
verschiedenen myeloischen Zelllinien untersucht. Die B-Zelllinie LCL-GK und die T-
Zelllinie Jurkat wurden als Positiv- bzw. Negativkontrollen benutzt (Brdicka et al., 2002). In
Northern- sowie in Western Blots war die LAT2-Expression von Kasumi-1-Zellen
(AML1/ETO positiv) so niedrig wie in der Negativkontrolle (Jurkat-Zellen, T-Zelllinie).
Weitere myeloische Zelllinien exprimierten LAT2-mRNA und -Protein unterschiedlich:
Abbildung 12.- Repression von LAT2-mRNA und -Protein nach der ektopen Expression von AML1/ETO in der 9/14/18-Zelllinie. A.- Durch Northern Blot wurde die Repression der LAT2-mRNA 48 Stunden nach der Induktion des AML1/ETO-Proteins bestätigt. Die Hybridisierung mit einer AML1/ETO-Sonde zeigte, dass die mit Ponasteron A induzierten Zellen das AML1/ETO-Transkript exprimieren. B.- und C.- Die Kinetik der Repression der LAT2-mRNA durch AML1/ETO wurde durch "real time"-RT-PCR untersucht. Die Repression der LAT2-mRNA wurde schon 4 Stunden nach der Induktion mit Ponasteron A nachgewiesen. Die Expression der AML1/ETO-mRNA war zu diesem Zeitpunkt 15,5 Fach höher als die AML1/ETO-Expression der 9/14/18-Zellen.
C.-
B.-A.-
EtBr
Ponasterone A + -Time (hours) 0 48 48
9/14/18U937
AML1/ETO
LAT2
GAPDH 0
0,2
0,4
0,6
0,8
1
1,2
1,4
0 2 4 6 8 10
Time (hrs)
Rel
ativ
e LA
T2/
GA
PD
H e
xpre
ssio
n
0
4
8
12
16
20
0 2 4 6 8 10
Time (hrs)
Rel
ativ
e A
ML1
-ET
O/G
AP
DH
exp
ress
ion
C.-
B.-A.-
EtBr
Ponasterone A + -Time (hours) 0 48 48
9/14/18U937
AML1/ETO
LAT2
GAPDH
EtBr
Ponasterone A + -Time (hours) 0 48 48
9/14/18U937
AML1/ETO
LAT2
GAPDH 0
0,2
0,4
0,6
0,8
1
1,2
1,4
0 2 4 6 8 10
Time (hrs)
Rel
ativ
e LA
T2/
GA
PD
H e
xpre
ssio
n
0
4
8
12
16
20
0 2 4 6 8 10
Time (hrs)
Rel
ativ
e A
ML1
-ET
O/G
AP
DH
exp
ress
ion

4.- Ergebnisse Seite - 74 -
Die relative Expression der LAT2/GAPDH-mRNA in 9/14/18-, oder HL60- Zelllinien
war so stark wie in der LCL-GK-Zelllinie (B-Zelllinie). Die U937-Zelllinie hatte eine 2-fach
stärkere und die NB4-Zelllinie eine 3-fache stärkere relative LAT2/ ß -Aktin -Expression als
die LCL-GK Zelllinie (Abb. 13a). Die Membranen der Northern Blots wurden mit einer ß-
Aktin-Sonde als Ladekontrolle hybridisiert
Diese Daten konnten durch Western Blots bestätigt werden. Die relative LAT2/β-Aktin-
Expression war 1,5-fach höher in 9/14/18-, 3-fach in HL60-, 2-fach in NB4- und U937-Zellen
als in der LCL-GK Zelllinie. Die relative LAT2/β-Aktin Expression von Jurkat- und Kasumi-
1-Zellen war unter 10 % der Expression in LCL-GK-Zellen (Abb. 13b).
Abbildung 13.- LAT2 -Expression in verschiedenen Zelllinien. A.- LAT2-mRNA-Expression wurde mittels Northern Blot in verschiedenen myeloischen Zelllinien analysiert. Die LCL-GK ist eine B-Zelllinie, die als Positivkontrolle galt, und Jurkat ist eine T-Zelllinie, die als Negativkontrolle galt. Die Membranen der Northern Blots wurden mit einer ß-Aktin-Sonde als Ladekontrolle hybridisiert. B.- LAT2-Protein-Expression wurde mittels Western Blot in verschiedenen myeloischen Zelllinien analysiert. ß-Aktin wurde als Ladekontrolle benutzt und die relative LAT2/ß-Aktin Expression wurde zwischen den verschiedenen Zelllinien verglichen.
A.-
B.-
LAT2
EtBr
LCL-GK Jurkat 9/14/18 HL60 NB4 U937 Kas- 1
β-ACTIN
LAT2
ß-Actin
LCL-GK Jurkat 9/14/18 HL60 NB4 U937 Kas- 1
A.-
B.-
LAT2
EtBr
LCL-GK Jurkat 9/14/18 HL60 NB4 U937 Kas- 1
β-ACTIN
LAT2
ß-Actin
LCL-GK Jurkat 9/14/18 HL60 NB4 U937 Kas- 1

4.- Ergebnisse Seite - 75 -
4.2.3.- Untersuchung der Protein-Expression des LAT2-Gens in primären AML-Blasten
von Patienten mit unterschiedlichen chromosomalen Aberrationen.
Knochenmarksproben von 43 AML Patienten der Universitätklinik Freiburg wurden
auf Expression des LAT2-Proteins mittels Western Blots untersucht. Die
Knochenmarksproben der Patienten wurden so ausgewählt, dass eine representative Zahl von
AML Patienten mit verschiedenen Karyotypen untersucht wurde.
Abbildung 14.- LAT2-Protein-Expression in Knochenmarksproben von Patienten mit AML unterschiedlicher Karyotypen. Die mononukleären Zellen von Knochenmarksproben der AML-Patienten wurden mittels Fikollgradient getrennt. Diese Zellen wurden in Einfriermedium eingefroren und in der Tumorbank aufbewahrt. Nach dem Auftauen dieser Zellen wurde Protein isoliert. Die Expression des LAT2-Proteins wurde mittels Western Blot analysiert. Kein Patienten (6/6) mit der t(8; 21), mit Aberrationen auf Chromosom 7 (3/3) und mit der t(15; 17) (4/4) exprimierten das LAT2 auf der Proteinebene.
inv(16) inv(16)** inv(16)* (8;21) Complex Complex inv(16) -7 U937-7q
LAT2
ß-Actin
LAT2
ß-Actin
11q23 (8;21) (8;21) (8;21) NN NN inv(16) Complex U937
LAT2
ß-Actin
NN NN NN inv(16) +10 NN NN NN
inv(16) inv(16)** inv(16)* (8;21) Complex Complex inv(16) -7 U937-7q
LAT2
ß-Actin
LAT2
ß-Actin
11q23 (8;21) (8;21) (8;21) NN NN inv(16) Complex U937
LAT2
ß-Actin
NN NN NN inv(16) +10 NN NN NN
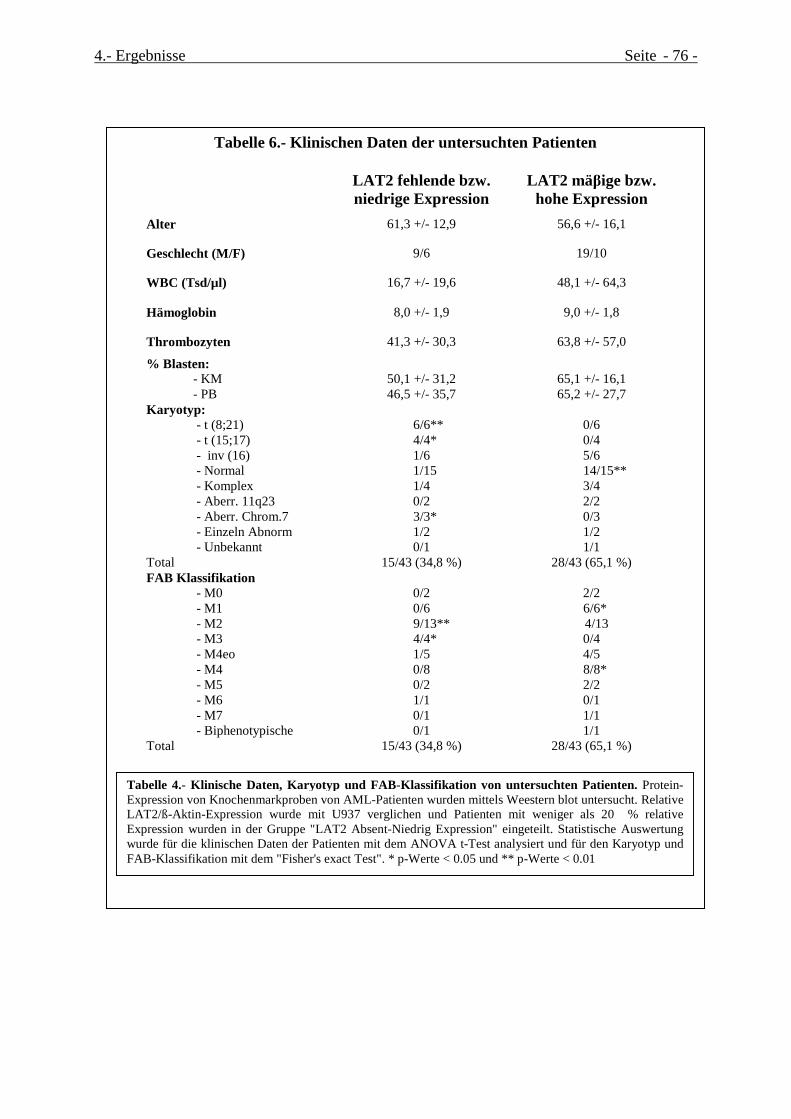
4.- Ergebnisse Seite - 76 -
Tabelle 6.- Klinischen Daten der untersuchten Patienten
LAT2 fehlende bzw. niedrige Expression
LAT2 mäβige bzw. hohe Expression
Alter 61,3 +/- 12,9 56,6 +/- 16,1
Geschlecht (M/F) 9/6 19/10
WBC (Tsd/µl) 16,7 +/- 19,6 48,1 +/- 64,3
Hämoglobin 8,0 +/- 1,9 9,0 +/- 1,8
Thrombozyten 41,3 +/- 30,3 63,8 +/- 57,0
% Blasten: - KM - PB
50,1 +/- 31,2 46,5 +/- 35,7
65,1 +/- 16,1 65,2 +/- 27,7
Karyotyp: - t (8;21) - t (15;17) - inv (16) - Normal - Komplex - Aberr. 11q23 - Aberr. Chrom.7 - Einzeln Abnorm - Unbekannt Total
6/6** 4/4* 1/6
1/15 1/4 0/2
3/3* 1/2 0/1
15/43 (34,8 %)
0/6 0/4 5/6
14/15** 3/4 2/2 0/3 1/2 1/1
28/43 (65,1 %) FAB Klassifikation - M0 - M1 - M2 - M3 - M4eo - M4 - M5 - M6 - M7 - Biphenotypische Total
0/2 0/6
9/13** 4/4* 1/5 0/8 0/2 1/1 0/1 0/1
15/43 (34,8 %)
2/2
6/6* 4/13
0/4 4/5
8/8* 2/2 0/1 1/1 1/1
28/43 (65,1 %) Tabelle 4.- Klinische Daten, Karyotyp und FAB-Klassifikation von untersuchten Patienten. Protein-
Expression von Knochenmarkproben von AML-Patienten wurden mittels Weestern blot untersucht. Relative LAT2/ß-Aktin-Expression wurde mit U937 verglichen und Patienten mit weniger als 20 % relative Expression wurden in der Gruppe "LAT2 Absent-Niedrig Expression" eingeteilt. Statistische Auswertung wurde für die klinischen Daten der Patienten mit dem ANOVA t-Test analysiert und für den Karyotyp und FAB-Klassifikation mit dem "Fisher's exact Test". * p-Werte < 0.05 und ** p-Werte < 0.01

4.- Ergebnisse Seite - 77 -
Das relative LAT2/ß-Aktin-Protein-Verhältnis wurde durch Densitometrie ermittelt.
Diese Werte wurden mit dem relativen LAT2/ß-Aktin-Protein-Verhältnis in den U937-Zellen
verglichen. Patienten wurden der Gruppe der LAT2-absent-niedrig-Expressoren zugerechnet,
wenn die relative LAT2/ß-Aktin-Expression unter 20 % der U937-Zelllinie lag.
Es gab keinen signifikanten Unterschied zwischen den klinischen Daten der AML-
Patienten (Tab. 6). Die Patienten mit niedriger LAT2-Expression waren 61,3 +/- 12,9 Jahre
alt und die Patienten mit normaler Expression waren 56,6 +/- 16,1 Jahre alt. Das Verhältnis
Männer/Frauen war bei den Patienten mit niedriger Expression 1,5 zu 1 und in den Patienten
mit einer normalen LAT2-Expression 2 zu 1. Einen deutlichen Unterschied gab es zwischen
den Mittelwerten der Zahl der peripheren Leukozyten, 16,7 +/- 19,6 Tsd/µl bei den Patienten
mit niedriger LAT2-Expression vs 48,1 +/- 64,3 Tsd/µl bei den Patienten mit der normalen
Expression, allerdings nicht statistische signifikant. Die Hämoglobin-Werte waren fast gleich,
8,0 +/- 1,9 Hb/dL bei den Patienten mit niedriger LAT2-Expression und 9,0 +/- 1,8 Hb/dL bei
den Patienten mit normaler Expression. Die Zahl der Thrombozyten waren 41,3 +/- 30,3
Tsd/µl bei den Patienten mit niedriger LAT2 Expression und 63,8 +/- 57,0 Tsd/µl bei den
Patienten mit normaler Expression.
Der Blastenanteil im Knochenmark der Patienten mit niedriger LAT2-Expression war
50,1 +/- 31,2 %, während im peripheren Blut dieser Patienten der Blastenanteil 46,5 +/- 35,7
% war. Bei den Patienten mit normaler LAT2 Expression war der Blastenanteil 65,1 +/- 16,1
% im Knochenmark und 65,2 +/- 27,7 % im peripheren Blut. Keine statistische Signifikanz
wurde bei den klinischen Daten der Patienten mit dem ANOVA t-test gefunden.
Nach der Untersuchung von Knochenmarksproben von 43 AML-Patienten wurden 15
Patienten gefunden, die das LAT2 Protein nicht exprimierten. In den Knochenmarksproben
aller Patienten mit der t(8;21) fand sich keine Expression des LAT2-Proteins.
Interessanterweise war die Expression des LAT2-Proteins bei 4 Patienten mit t(15; 17) sowie
bei 3 Patienten mit einer Deletion des langen Armes vom Chromosom 7 abwesend.
Andererseits wurde das LAT2-Protein bei 27 von 30 Patienten mit anderen Karyotypen,
darunter Normalkaryotyp (NN), inv(16), Aberrationen in 11q23, komplexen Karyotypen und
einzeln Chromosomen-Abnormalitäten detektiert (Abb. 14).
Im Bezug auf die FAB-Klassifikation konnte festgestellt werden, dass 9 von 13
Patienten der FAB M2 Subgruppe, keine Expression des LAT2-Proteins aufwiesen
(überwiegend AML1/ETO-Positiv). Bei 4 FAB M3 Patienten (alle positiv für t(15; 17)) und 3
Patienten mit Aberrationen von Chromosom 7 wurde die LAT2-Expression mittels Western

4.- Ergebnisse Seite - 78 -
blot nicht detektiert. Dagegen war LAT2-Protein in allen Patienten der FAB M1 und FAB M4
Subgruppe nachweisbar.
Zwei Knochenmarksproben von Patienten mit inv (16) aus der dritten Kontrolle einer
Kompletter-Remission wurden untersucht, um zu bestätigen, dass die Expression des LAT2-
Proteins in den Patientenproben nur aus den leukämischen Zellen stammt.
Mittels Western Blot konnten wir nachweisen, dass die Expression des LAT2-
Proteins in den Knochenmarksproben der Patienten im Kompletter-Remission nicht
nachweisbar und nur in den Proben der gleichen Patienten bei der Erst-Diagnose der AML
detektierbar war (Abb. 15).
Abbildung 15.- LAT2-Protein-Expression in Knochenmarksproben von Patienten mit AML zu Zeitpunkt der Erst-Diagnose(ED) und der Vollremission (CR). Die Expression des LAT2-Proteins wurde in Knochenmarksproben von Patienten bei der Diagnose und in kompletter Remission untersucht. Die Expression des LAT2-Proteins wurde nur im pathologischen Knochenmark nachgewiesen. Die Patienten, deren Knochenmark in CR untersucht wurden, sind in Abb. 14 mit * und ** gekennzeichnet.
LAT2
ß-Actin
ED CR ED CR
Pat.: 1 2
LAT2
ß-Actin
ED CR ED CR
Pat.: 1 2

4.- Ergebnisse Seite - 79 -
4.2.4.- Untersuchung der Re-Expression des LAT2-Gens mittels siRNAs gegen
AML1/ETO in Kasumi-1-Zellen.
In Kooperation mit PD. Dr. Heidenreich (Universität Tübingen) wurde die Expression
des LAT2-Gens in einem AML1/ETO-Knock-Down-System mittels siRNAs in der Kasumi-
1-Zelllinie untersucht. Dieses System wurde bereits publiziert (Heidenreich, Blood 2003;
Martinez BMC Cancer 2004; Dunne Oncogene 2006). Die Kasumi-1-Zellen wurden mit
einem Plasmid ohne Oligonukleotid (Mock), mit einem Oligonukleotid gegen AML1/ETO-
mRNA (siAGF1) und mit einem nicht verwandten Oligonukleotid (siAGF6) elektroporiert.
Nach 16 Stunden wurde ein Teil der Zellen geerntet. Ein weiterer Teil wurde am 4. Tag nach
der ersten Elektroporation ein zweites Mal elektroporiert. 16 Stunden nach der zweiten
Elektroporation wurden die Zellen dann geerntet. Das Kultivieren der Zellen und die
Elektroporation der siRNAs wurden im Labor von PD. Dr. Heidenreich durchgeführt; die
RNA-Isolierung, Northern Blots und „real-Time“-RT-PCR, sowie Protein-Isolierung und
Western Blots wurden in Freiburg durchgeführt.
Mit Northern Blots konnte nachgewiesen werden, dass die LAT2-mRNA nach der
ersten und zweiten Elektroporation (jeweils 30 % und 50 % höhere Expression als die
Expression von den Mock-transfizierten Kasumi-1-Zellen) schwach re-exprimiert wurde
(Daten nicht gezeigt), allerdings in deutliche Grenze der Methode. Eine weitere, quantitative
Nachweis Methode war notwendig. In diesem Versuch wurde auch nachgewiesen, dass die
Lysozym-mRNA nach der Herunterregulation der AML1/ETO mRNA sehr stark re-
exprimiert wurde (s. Abb. 16a). Diese Daten wurde im Verlauf dieser Doktorarbeit publiziert
(Claus et al., 2006). Die Membranen von Northern Blots wurden mit einer ß-Aktin-Sonde als
Ladekontrolle hybridisiert.
Die Expression der LAT2-mRNA wurde mittels „real-Time“-RT-PCR untersucht, um
die Daten des Northern Blots zu bestätigen. Durch Quantifizierung mit „real-Time“-RT-PCR
wurde eine Hochregulation auf das 4,5 +/- 0,8-fache nach der ersten Elektroporation und auf
das 9,6 +/- 3,8-fach nach der zweiten Elektroporation im Vergleich zur Expression der
„Mock“-transfizierten Kasumi-1 Zellen ermittelt (Abb. 16c). Die Herunterregulation der
AML1/ETO-mRNA wurde ebenfalls mittels „real-Time“-RT-PCR in diesem Versuch
quantifiziert. Nach der ersten Elektroporation wurde die Expression von AML1/ETO um 51,8
+/- 25,9 % und nach der zweiten Elektroporation um 39,7 +/- 14,1 % reduziert im Vergleich
zur Expression der "Mock"-transfizierten Kasumi-1-Zellen (Abb. 16d). Diese Daten konnten
auch mittels Western Blots auf Protein-Ebene bestätigt werden. (s. Abb. 16b)

4.- Ergebnisse Seite - 80 -
Abbildung 16.- Re-Expression der LAT2-mRNA nach Knock-down von AML1/ETO mittels siRNAs in der Kasumi-1-Zelllinie. A.- Northern Blot von Kasumi-1-Zellen: „Mock“ elektroporierte Zellen als Kontrolle, das siAGF1-Oligonukleotid war gegen AML1/ETO und das siAGF6 Oligonukleotid war als nicht verwandtes Oligonukleotid hergestellt wurden. Die Membran wurde mit einer AML1/ETO-Sonde als Kontrolle der Herrunterregulation der AML1/ETO-mRNA, LYSOZYM als Kontrolle für einen AML1/ETO-Zielgen und ß-AKTIN als Ladekontrolle hybridisiert. B.- Die Repression von AML1/ETO durch siRNAs konnte mittels Western Blots in Kasumi-1 Zellen gezeigt werden. C.- und D.- Die Re-expression des LAT2-Gens durch den AML1/ETO-Knock-Down wurde mittels „real-Time-PCR“ bestätigt. Die Expression von LAT2- und AML1/ETO-mRNA wurde mit der GAPDH-Expression verglichen und mit der Expression der "Mock" transfizierten Zellen normalisiert.
D.-
C.-A.-
AML1/ETO
EtBr
LYSOZYME
Kasumi-11° Electroporation 2°Electroporation
Mock siAGF1 siAGF6 Mock siAGF1 siAGF6
ß-Actin0
3
6
9
12
15
18
I. Mock I. siAGF1 I. siAGF6 II. Mock II. siAGF1 II. siAG F6R
elat
ive
LAT
2/G
AP
DH
exp
ress
ion
0
0,3
0,6
0,9
1,2
1,5
I. Mock I. siAGF1 I. siAGF6 II. Mock II. siAGF1 II. siAG F6AM
L1-E
TO
/GA
PD
H r
ela
tive
exp
ress
ion
B.-
AML1/ETO(85 kDa)β-Actin(42 kDa)
Kasumi-1
1° Electroporation
Mock siAGF1 siAGF6
D.-
C.-A.-
AML1/ETO
EtBr
LYSOZYME
Kasumi-11° Electroporation 2°Electroporation
Mock siAGF1 siAGF6 Mock siAGF1 siAGF6
ß-Actin0
3
6
9
12
15
18
I. Mock I. siAGF1 I. siAGF6 II. Mock II. siAGF1 II. siAG F6R
elat
ive
LAT
2/G
AP
DH
exp
ress
ion
0
0,3
0,6
0,9
1,2
1,5
I. Mock I. siAGF1 I. siAGF6 II. Mock II. siAGF1 II. siAG F6AM
L1-E
TO
/GA
PD
H r
ela
tive
exp
ress
ion
B.-
AML1/ETO(85 kDa)β-Actin(42 kDa)
Kasumi-1
1° Electroporation
Mock siAGF1 siAGF6

4.- Ergebnisse Seite - 81 -
4.2.5.- Re-Expression der LAT2-mRNA durch epigenetisch aktive Substanzen in der
AML1/ETO-positiven Kasumi-1-Zelllinie.
AML1/ETO rekrutiert an seine Zielgene HDACs der Klasse I, HDAC1-3 (Amann, MCB
2001) sowie DNMT1 (Liu, Cancer Res. 2005). Dadurch kann AML1/ETO durch
epigenetische Veränderungen die Expression verschiedener Genen beeinflussen (Fliegauf,
Oncogene 2004; Alcalay, JCI 2003).
Um einen solchen epigenetischen Einfluß auf die LAT2-Expression zu untersuchen,
wurde die Kasumi-1-Zelllinie mit verschiedenen, epigenetisch aktiven Therapien
(demethylierende Substanzen wie 5-aza-2'-deoxycytidine/Decitabine/DAC oder mit HDAC-
Inhibitoren wie MS-275, TSA und SAHA) behandelt. Die Änderung der LAT2-mRNA
Expression wurde durch „real time“-RT-PCR quantifiziert.
Die Zellen wurden 3 Tage mit verschiedenen Konzentrationen von HDAC-Inhibitoren
oder mit ATRA behandelt. Für weitere Experimenten wurden equitoxische Konzentrationen
der Substanzen so ausgewählt, dass die Viabilität der Zellen über 80 % blieb und eine
maximale Zellwachstum-Inhibition erreicht wurde. Die LAT2-mRNA wurde nach der
cDNA-Traskription mittels „real time“-RT-PCR quantifiziert und mit der Expression von ß-
Glukuronidase (GUSB) relativiert.
Die Expression der LAT2-mRNA wurde 3 Tagen nach der Behandlung mit 1 µM MS-
275 deutlich re-exprimiert (4,3 +/- 1,3-fach höher als die Grundexpression in Kasumi-1) (
Abb. 17a). Die Substanz ATRA (1µM) und die HDAC-Inhibitoren TSA (0,1-0,2 µM) und
SAHA (0,5-1 µM) hatten nach 3 Tage Behandlung kaum einen Effekt auf die Expression der
LAT2 mRNA (Daten nicht gezeigt).
Kasumi-1 Zellen wurden mit MS-275 behandelt. Alle 2 Stunden wurde Aliquots
geerntet. Daraus wurde die RNA isoliert und mit einer Reverse Transkriptase in cDNA
synthetisiert. Zeit-abhängig wurde mit MS-275 in einer Konzentration vom 0,1 µM die LAT2
mRNA re-exprimiert. Durch „real time“-RT-PCR konnte eine maximale Re-Expression (9,4
+/- 2,6 fach höher als die Grundexpression) nach 8 Stunden nachgewiesen werden und die
Expression der LAT2-mRNA blieb nach 24 Stunden nocht etwa 3 fach-höher als die
Grundexpression in den nicht-behandelten Kasumi-1-Zellen (Abb. 17b).
Die AML1/ETO bedingte Hemmung der LAT2-Expression konnte dosisabhängig
auch durch Decitabine (DAC) aufgehoben werden (Abb. 17c). Nach 3 Gaben von DAC alle
24 Stunden wurde LAT2-mRNA am 6. Tag durch „real-Time“-RT- PCR gemessen. Die
relativen LAT2/GUSB Expression der behandelten Kasumi-1-Zellen war 1,4 +/- 0,3-fach mit

4.- Ergebnisse Seite - 82 -
25 nM DAC; 1,6 +/- 0,6-fach mit 50 nM DAC; 1,6-+/- 0,1-fach höher mit 100 nM DAC und
2,9 +/- 0,2-fach höher mit 200 nM DAC als die relative LAT2/GUSB-mRNA Expression in
den nicht-behandelten Kasumi-1-Zellen.
In der Arbeit von (Cameron et al., 1999) wurde gezeigt, dass die HDAC Inhibitoren
und die demethylierenden Substanzen einen synergistischen Effekt auf die Re-expression der
unterdrückten Gene haben können. Daher wurden die HDAC-Inhibitoren und Decitabine in
Kombination benutzt, um einen synergistischen Effekt in der Re-expression der LAT2-
mRNA in der Kasumi-1-Zelllinie zu untersuchen. Es wurden niedrigere Konzentrationen als
in Einzelgaben benutzt, um die Toxizität der Substanzen zu minimieren. Die Behandlung
erfolgte mit 3 Gaben von DAC alle 24 Stunden und anschließend wurde am 3. Tag der HDAC
Inhibitor für weitere 3 Tage eingesetzt.
Abbildung 17.- LAT2-mRNA Re-expression durch epigenetisch aktive Therapien in AML1/ETO-positiven Kasumi-1 Zellen. A.- Kasumi-1-Zellen wurden für 3 Tagen mit verschiedenen HDAC-Inhibitoren behandelt und die LAT2-mRNA Expression wurde am Tag 3 mittels „real-time“-RT-PCR quantifiziert und gegen die β-Glukuronidase normalisiert. B.- Mit dem HDAC-Inhibitor MS-275 in einer Konentration von 1 µM konnte die Expression der LAT2-mRNA in einer Zeit-abhängigen Art re-exprimiert werden. C.- Die Kasumi-1-Zellen wurden mit 3 Gaben alle 24 Stunden mit der demethylierenden Substanz Decitabine (DAC) behandelt. Am 6. Tag wurde die RNA der Zellen isoliert. Nach der Transkription in cDNA mit einer Reverse Transkriptase wurde die LAT2-mRNA mittels „real-time“-RT-PCR quantifiziert. Mit Decitabine konnte die Expression der LAT2-mRNA auch in einer Konzentrationsabhängigen Art in den Kasumi-1-Zellen induziert werden. D.- Die Kasumi-1-Zellen wurden mit 3 Gaben von DAC alle 24 Stunden behandelt. Am 3. Tag wurde MS-275 gegeben. Die Zellen wurden am 6. Tag geerntet und die LAT2-mRNA Expression durch „real time“-RT-PCR gemessen. Decitabine und MS-275 in Kombination konnten die Expression der LAT2-mRNA in einer synergistischen Form in Kasumi-1-Zellen induzieren. Die Säulen repräsentieren den Mittelwert und die Pfeile die Standardabweichung von 3 unabhängigen Experimenten
Kasumi-1 +/- MS275
0
3
6
9
12
15
0 2 4 6 8 10 12 24
Time (hrs)
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
Kasumi-1 +/- single epigenetically active substances
0
1
2
3
4
5
6
untreatedcells
1 µM ATRA 1 µM S275 0,1 µM TSA 1 µM SAHA
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
Kasumi 1+/- DAC
0
1
2
3
4
5
0 DAC 25 nM DAC 50 nM DAC 100 nM DAC 200 nM DAC
Rel
ativ
e L
AT
2/G
US
B E
xpre
ssio
n
Kasumi-1 +/- combined epigenetically active substances
0
2
4
6
8
10
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
50 nM50 nM25 nM25 nM00DAC
100 nM0100 nM0100 nM0MS-275
A. B.
C. D.
Kasumi-1 +/- MS275
0
3
6
9
12
15
0 2 4 6 8 10 12 24
Time (hrs)
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
Kasumi-1 +/- single epigenetically active substances
0
1
2
3
4
5
6
untreatedcells
1 µM ATRA 1 µM S275 0,1 µM TSA 1 µM SAHA
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
Kasumi 1+/- DAC
0
1
2
3
4
5
0 DAC 25 nM DAC 50 nM DAC 100 nM DAC 200 nM DAC
Rel
ativ
e L
AT
2/G
US
B E
xpre
ssio
n
Kasumi-1 +/- combined epigenetically active substances
0
2
4
6
8
10
Rel
ativ
e LA
T2/
GU
SB
Exp
ress
ion
50 nM50 nM25 nM25 nM00DAC
100 nM0100 nM0100 nM0MS-275
A. B.
C. D.

4.- Ergebnisse Seite - 83 -
MS-275 in einer Konzentration von 100 nM hatte nur einen leichten Effekt auf die Re-
expression von LAT2-mRNA (1,4 +/- 0,5-fach höher). Die LAT2-mRNA konnte auch in
einzelnen Therapien mit 25 nM DAC allein (1,5 +/- 0,2-fach höher) und mit 50 nM DAC (2,9
+/- 1,5-fach höher) reexprimiert werden. Eine Kombination von 100 nM MS-275 mit 25 nM
DAC zeigte eine LAT2-mRNA Re-expression von 3,2 +/- 1,2-fach verglichen mit den nicht-
behandelten Zellen; 100nM MS275 plus 50 nM DAC zeigte eine 5,8 +/- 1,4 fach Induktion
verglichen mit den nicht-behandelten Kasumi-1-Zellen (s.Abbildung 17d). Durch diesen
Versuch wurde ein synergistischer Effekt von einer demethylierenden Substanz mit einem
HDAC-Inhibitor auf die LAT2-mRNA-Expression in den Kasumi-1-Zellen festgestellt.
4.2.6.- Histonmodifikation nach der Behandlung mit MS-275 auf dem LAT2-Promotor
in Kasumi-1-Zellen.
Die nächste Fragestellung war, ob der HDAC-Inhibitor MS-275 einen direkten Effekt
auf die aktivierenden Histon-Markierungen des LAT2-Promotors hat. Hierzu wurden die
Histon-Acetylierung von H3 (AcH3) und H4 (AcH4), die Histon-Acetylierung von Lysin 9
von Histon H3 (AcH3K9) und die Tri-methylierung-Histon H3 Lysin 4 (3meH3K4), die nach
der Acetylierung von Histon H3 durch die Histon-Methyltransferase MLL4 nach Behandlung
mit HDAC-Inhibitoren katalysiert wird untersucht (Nightingale et al., 2007). Nach der
Chromatin Immunopräzipitation (ChIP) wurde eine „real time“-DNA-PCR durchgeführt, um
eine direkte Quantifizierung der isolierten DNA durchzuführen. Außerdem wurde der
Promotor von p21WAF untersucht, weil bekannt ist, dass p21WAF-mRNA durch Acetylierung
der Histone seines Promotors nach der Behandlung mit HDAC-Inhibitoren induziert wird
(Richon et al., 2000). Nach 24 Stunden Behandlung mit 1µM MS-275 wurden AcH3,
AcH3K9 und 3meH3K4 auf dem LAT2-Promotor in der Kasumi-1 Zellen stark induziert,
nicht aber AcH4 (Abb. 18a). Eine vergleichbare Induktion dieser Histon-Markierungen wurde
auf dem p21WAF-Promotor gefunden (Abb. 18b). Diese Ergebnisse sprechen dafür, dass der
Promotor von LAT2 durch HDAC-Inhibitoren in den Kasumi-1-Zellen moduliert werden
kann, und dass die mögliche AML1/ETO-vermittelte Rekrutierung von HDACs auf den
Promotor pharmakologisch gehemmt werden kann.

4.- Ergebnisse Seite - 84 -
4.2.7.- Histonmodifikation nach der Induktion von AML1/ETO auf dem LAT2-
Promotor in 9/14/18-Zellen.
AML1/ETO rekrutiert HDACs zu den Promotoren seiner Zielgene (Amann et al.,
2001; Gelmetti et al., 1998). Dadurch werden die Histone zu einer geschlossenen
Konformation um den Promotor angeornet und die Transkription der AML1/ETO-Zielgene
wird gehemmt. Deshalb wurde untersucht, ob AML1/ETO HDACs auf den LAT2-Promotor
rekrutiert und welche Histon-Markierungen dadurch beeinflusst werden. Auf dem LAT2-
Promotor der U937-Zellen wurden alle untersuchten Histon-Markierungen (AcH3, AcH4,
AcH3K9 und 3meH3K4) nach der ChIP-Methode angereichert detektiert. Im Gegensatz
hierzu waren sie jedoch nahezu nicht nachweisbar in den Kasumi-1-Zellen (Abb. 18c).
A.-
B.-
C.-
D.-
ChIP for p21 promoter
0
0,2
0,4
0,6
0,8
1
1,2
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to in
pu
t
Kas-1 untreated
Kas-1 + MS275
ChIP for LAT2 promoter
0
0,2
0,4
0,6
0,8
1
1,2
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to In
put
Kas-1 untreated
Kas-1 + MS275
Time course 9/14/18 for AcH3
0
0,5
1
1,5
2
2,5
0 4 12 24 36 48
Time (hrs) after adding PonA
Rel
ativ
e va
lues
to in
put
ChIP for LAT2 promoter
0
0,5
1
1,5
2
2,5
3
3,5
4
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to In
put
U937 Kasumi-1
A.-
B.-
C.-
D.-
ChIP for p21 promoter
0
0,2
0,4
0,6
0,8
1
1,2
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to in
pu
t
Kas-1 untreated
Kas-1 + MS275
ChIP for LAT2 promoter
0
0,2
0,4
0,6
0,8
1
1,2
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to In
put
Kas-1 untreated
Kas-1 + MS275
Time course 9/14/18 for AcH3
0
0,5
1
1,5
2
2,5
0 4 12 24 36 48
Time (hrs) after adding PonA
Rel
ativ
e va
lues
to in
put
ChIP for LAT2 promoter
0
0,5
1
1,5
2
2,5
3
3,5
4
Input AcH3 AcH4 AcH3K9 3meH3K4 NIgG
Rel
ativ
e va
lues
to In
put
U937 Kasumi-1
Abbildung 18.- Induktion der aktivierenden Histon-Moifikationen durch MS-275 und
AML1/ETO-vermittelte Rekrutierung von HDAC-Aktivitä t auf dem LAT2-Promotor. A. Kasumi-1-Zellen wurden für 24 Stunden mit 1 µM MS-275 behandelt und dann wurden mehrere Histon-Markierungen nach Chromation-Immunopräzipitation (ChIP) auf dem LAT2-Promotor mittels "real time"-RT-PCR untersucht. AcH3, AcH3K9 und 3meH3K4 wurden nach MS-275-Behandlung in den Kasumi-1-Zellen angereichert nachgewiesen. B. Vergleichbare Aktivierung der Histon-Markierungen wurden auf dem Promotor von p21waf gefunden. C. Aktivierende Histon-Markierungen der U937-Zellen und der Kasumi-1-Zellen wurden verglichen. Abb. 18D. Die Acetylierung von Histon H3 wurde nach der konditionalen Expression von AML1/ETO in 9/14/18-Zellen untersucht. Eine maximale Depletion dieser Histon-Markierung wurde nach 24 Stunden nachgewiesen, allerdings war sie transient. Die Säulen repräsentieren den Mittelwert und die Pfeile die Standardabweichung von 3 unabhängigen Experimenten.

4.- Ergebnisse Seite - 85 -
Darüberhinaus die Acetylierung von H3 und H4 nach der konditionalen Expression von
AML1/ETO in 9/14/18-Zellen untersucht. In einem „Time-Course“ wurde nach 4 Stunden
schon eine Verminderung und nach 24 Stunden eine maximale Depletion der Acetylierung
von H3 nachgewiesen, allerdings war diese, wie die Repression von LAT2-Expression nach
der Induktion von AML1/ETO in der 9/14/18-Zellen, transient (Abb 18d). Zusammenfassend
deuten diese Ergebnisse darauf hin, dass AML1/ETO HDAC-Aktivität auf den LAT2-
Promotor rekrutiert, die durch HDAC-Inhibitoren wie MS-275 aufgehoben werden kann.

4.- Ergebnisse Seite - 86 -
4.3.- Identifizierung neuer AML1/ETO-Zielgene mittels RLGS-Methode
4.3.1- RLGS-Ergebnisse
In den funktionell charakterisierten 9/14/18 System wurde durch die Arbeitsgruppe von Prof.
Plass (Columbus, OH, USA) eine RLGS-Analyse der beschriebenen Methodik entsprechend
durchgeführt (s. 1.6.1). Die Induktion des AML1/ETO-Proteins wurde mittels Western Blot in
Freiburg kontrolliert. Dort wurden die DNA-Isolierung, die RLGS und die Auswertung der
Gele durchgeführt. Schließlich wurde die Validierung der RLGS-Ergebnisse mit einem
unabhängigen Experiment mittels Southern Blot in Freiburg durchgeführt.
Nach Durchführung der RLGS-Analyse übermittelte Prof. Plass eine Liste mit Spots,
die sich als verändert zeigten. Jeder Spot auf dieser Liste entspricht einem DNA-Fragment
flankiert von einer Not I und EcoR V Schnittstelle, welches entweder detektierbar oder
abwesend war, wenn induzierte Zellen mit nicht-induzierten Zellen verglichen wurden.
Manche dieser Spots verfügen über eine definierte Adresse im oben beschriebenen
Masterprofil. Damit ist die Identifizierung der Spots einfacher und schneller machbar, denn
manche dieser Spots waren für die Master Profile bereits sequenziert worden.
Abbildung 19.- Intensitätsveränderung von Spot 2D24 (Dematin-Gen) in RLGS-Gelen nach der Induktion des AML1/ETO-Proteins in 9/14/18. Die genomische DNA der 9/14/18-Zellen wurde mit dem methylierungssensitiven Enzym Not I und mit dem nicht-methylierungssensitiven Enzym EcoR V verdaut. Die Sticky-Endungen wurden mit Radioaktiven „C“s und „G“s Nukleotiden gefüllt. Die radioaktiv markierte DNA wurde auf einem bidimensionalen Polyacrylamid Gel aufgetragen und elektrophoretisch getrennt. Mit Hilfe eines radioaktiv sensitiven Filmes wurden die Spots (DNA-Fragmenten) detektiert. Die Abbildung zeigt ein Intensitätsveränderung des Spot 2D24 nach der Induktion von AML1/ETO. Dieser Spot konnte weiter mit Southern Blots in einem unabhängigen Versuch validiert werden.
9/14/18 -96 h Pon A
RLGS fragment 2D24 EPB49 (Dematin)9/14/18 +96 h Pon A

4.- Ergebnisse Seite - 87 -
In den durchgeführten Experimenten wurden die nicht-induzierten Zellen des Klons
9/14/18 mit induzierten 9/14/18-Zellen zu zwei unterschiedlichen Zeitpunkten (48 und 96
Stunden) nach der Induktion verglichen. Es wurden verschiedene Spotveränderungen
zwischen den zwei Proben gefunden, die möglicherweise einem Methylierungs- oder
Demethylierungsereignis entsprechen könnten. Diese Spots wurden subkloniert und
sequenziert oder in der Datenbank des Master Profile gesucht. Ziel der durchgeführten
Experimente ist es, diese Sequenzen mit Southern Blots zu validieren und gegebenenfalls
weiter zu untersuchen.
Interessanterweise gab es insgesamt 35 neu aufgetretene Spots (8 von ihnen waren in
den Master-Profilen vorhanden) und 8 verschwundene Spots (7 von ihnen waren in den
Master-Profilen vorhanden), als ein Vergleich der induzierten mit den nicht-induzierten
Zellen durchgeführt wurde (Abb. 20). Der Spot 2D24 (Dematin) wurde als verschwundener
Spot 48 und 96 Stunden nach der Induktion gewertet (Abb. 19). Die verschwundenen Spots
entsprechen einem Methylierungsereignis in einer Not I Restriktionsstelle (GCGGCCGC) in
Abbildung 20.- RLGS-Ergebnisse. A.- Die „Spots“ von RLGS wurden als möglichen Methylierugs- und Demethylierungs-Ereignisse bezeichnet, wenn ein Unterschied in der Intensität des Spots nach dem Vergleich mit den RLGS-Gelen von den induzierten und nicht-induzierten Zellen zum gleichen Zeitpunkt gefunden wurde. Die „Spots“-Bezeichnung sind ihre Adresse in dem Master Profile. B.- Die Sequenzen der „Spots“ wurden mit Hilfe der „BLAST“ Software der PubMed Webseite zu einem bestimmten Gen adressiert.
6:2C2, 2C66, 2D24, 2D174D2, 2E43
5:2D10, 2D20, 2G93, 3E2, 4G87
96 +/96 -
1:2D24
3:1F07, 1G26, 2G51
48 +/48 -
Verschwundene Spots und ihreAdresse in der “Master profile”.
Neu aufgetretene Spots in den induzierten Zellen und ihre Adresse in der “Master Profile”.
Sample Paar (Stundennach der induktionvom AML1/ETO mitPonasterone A)
6:2C2, 2C66, 2D24, 2D174D2, 2E43
5:2D10, 2D20, 2G93, 3E2, 4G87
96 +/96 -
1:2D24
3:1F07, 1G26, 2G51
48 +/48 -
Verschwundene Spots und ihreAdresse in der “Master profile”.
Neu aufgetretene Spots in den induzierten Zellen und ihre Adresse in der “Master Profile”.
Sample Paar (Stundennach der induktionvom AML1/ETO mitPonasterone A)
2D20: OTX1 gene (Homeobox family gene)2D24: Dematin (Protein membrane band 4.9 of erythrocytes)2D10:2C66: PELI2 (linker for Toll/IL-1 signalling pathway)
1F07: CDC28 Protein kinase 2 gene 2G93: Homo sapiens protein tyrosine phosphatase, non-receptor type 11 (Noonan syndrome 1) (PTPN11)2D17: ion transporter protein (NRITP)
Klonierte Sequenzen und ihre Genname:
MöglicheDemethylierung-
Erreignisse
MöglicheMethylierung-
ErreignisseB.-
A.-

4.- Ergebnisse Seite - 88 -
AML1/ETO-induzierten Zellen. Ob das Neuauftreten eines Spots ein
Demethylierungsereignis signalisiert, weiß man deshalb nicht, weil bisher nur
Methylierungsereignisse in Tumoren untersucht worden sind.
4.3.2.- Klonierung und Annotation der detektierten Sequenzen
Nach der Lokalisierung der Spots erfolgte die Identifizierung entweder durch
Sequenzierung oder durch den Vergleich mit dem Master Profile. Die Gene wurden mit der
NCBI Genbank (BLAST Software, www.ncbi.nlm.nhi.gov) verifiziert. Bisher wurden 7 Spots
sequenziert: 6 davon sind Fragmente, die zu unterschiedlichen Genen gehören.
Interessanterweise spielen 3 dieser Gene eine Rolle in der Hämatopoese (OTX1, Protein band
4.9/Dematin und Noonan Syndrom Gen/PTPN11).
OTX1 ist ein Gen, das zur HOMEOBOX Familie gehört und von dem überraschend
eine Rolle in der Hämatopoese der Mäuse gefunden wurde (Levantini et al., 2003). KO-
Mäuse für OTX1 hatten Anämie und eine niedrige Expression der erythrozytären
Transkriptionfaktoren SCL und GATA-1.
Das Dematin-Gen (Proteinband 4.9 der Erythrozyten) kodiert ein Protein, das eine
strukturelle Funktion in der Erythrozytenmembrane hat und als Bindeglied zwischen dem
Spectrin-Aktin des Zytoskeletts und der Zellmembran dient (Lutchman et al., 2002). EKLF-1,
ein essentieller Transkriptorfaktor für primitive und definitive Erythropoese, bindet an den
Dematin-Promotor und reguliert dessen Expression (Hodge et al., 2006).
Das Noonan-Syndrom-Gen 1 (PTPN11) gehört zu der Protein-Tyrosin-Phosphatase-
Familie. Heterozygote (+/-) Mäuse für dieses Gen haben ein Phänotyp ähnlich dem von
Menschen, die am Noonan Syndrom erkrankt sind, mit Aortenstenose und Reurgitation,
kraniofazialen Abnormalitäten und myeloproliferativen Erkrankungen. Angeborene
Mutationen des PTPN11-Gens werden in mehr als 50 % der Patienten mit diesem Syndrom
beschrieben (Tartaglia et al., 2001). Somatische Mutationen im PTPN11-Gen sind in 34 % der
Kinder mit JMML (Juveniler Myelomonozytärer Leukämie) zu finden einer
myeloproliferativen Erkrankung des Kinderalters, zu einem kleinen Anteil bei Kindern mit
AML und MDS (Tartaglia et al., 2003) und zu 6 % in B-ALL-Patienten (Yamamoto et al.,
2006)..
Weitere durch RLGS-Analyse gefundene Gene haben eine Funktion in der Regulation
des Zellzyklus (CDC28 Proteinkinase 2). Ein weiteres (PELI2) hat wiederum eine Funktion in
der Kaskade der Signaltransduktion von Toll/IL-1. Möglicherweise erlaubt also der Einsatz
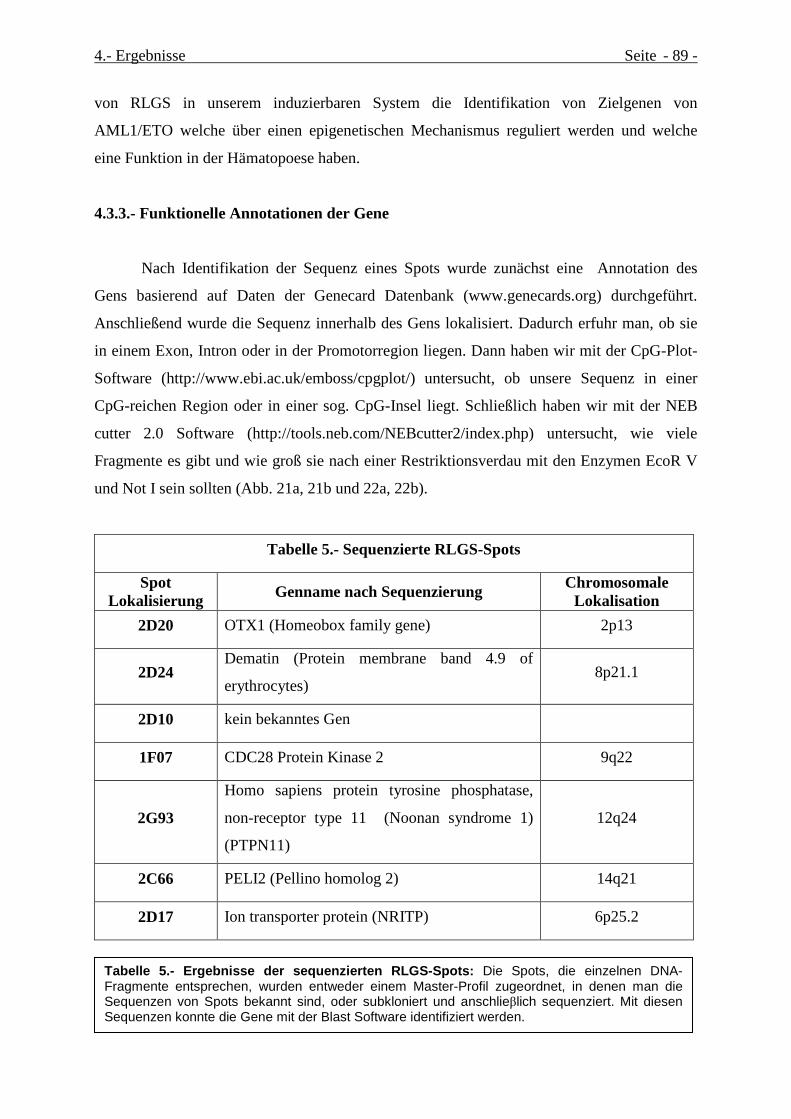
4.- Ergebnisse Seite - 89 -
von RLGS in unserem induzierbaren System die Identifikation von Zielgenen von
AML1/ETO welche über einen epigenetischen Mechanismus reguliert werden und welche
eine Funktion in der Hämatopoese haben.
4.3.3.- Funktionelle Annotationen der Gene
Nach Identifikation der Sequenz eines Spots wurde zunächst eine Annotation des
Gens basierend auf Daten der Genecard Datenbank (www.genecards.org) durchgeführt.
Anschließend wurde die Sequenz innerhalb des Gens lokalisiert. Dadurch erfuhr man, ob sie
in einem Exon, Intron oder in der Promotorregion liegen. Dann haben wir mit der CpG-Plot-
Software (http://www.ebi.ac.uk/emboss/cpgplot/) untersucht, ob unsere Sequenz in einer
CpG-reichen Region oder in einer sog. CpG-Insel liegt. Schließlich haben wir mit der NEB
cutter 2.0 Software (http://tools.neb.com/NEBcutter2/index.php) untersucht, wie viele
Fragmente es gibt und wie groß sie nach einer Restriktionsverdau mit den Enzymen EcoR V
und Not I sein sollten (Abb. 21a, 21b und 22a, 22b).
Tabelle 5.- Sequenzierte RLGS-Spots
Spot Lokalisierung Genname nach Sequenzierung
Chromosomale Lokalisation
2D20 OTX1 (Homeobox family gene) 2p13
2D24 Dematin (Protein membrane band 4.9 of
erythrocytes) 8p21.1
2D10 kein bekanntes Gen
1F07 CDC28 Protein Kinase 2 9q22
2G93
Homo sapiens protein tyrosine phosphatase,
non-receptor type 11 (Noonan syndrome 1)
(PTPN11)
12q24
2C66 PELI2 (Pellino homolog 2) 14q21
2D17 Ion transporter protein (NRITP) 6p25.2
Tabelle 5. - Ergebnisse der sequenzierten RLGS -Spots : Die Spots, die einzelnen DNA-Fragmente entsprechen, wurden entweder einem Master-Profil zugeordnet, in denen man die Sequenzen von Spots bekannt sind, oder subkloniert und anschlieβlich sequenziert. Mit diesen Sequenzen konnte die Gene mit der Blast Software identifiziert werden.

4.- Ergebnisse Seite - 90 -
Eine Validierung dieser Ergebnisse wurde von Sequenzen, die für Gene identifiziert
wurden, mittels Southern Blot ausgestrebt. Zuerst mussten Zellen neu induziert werden. Dann
wurde die DNA isoliert und mittels Southern Blot Methode auf eine Nylon-Membran
transferiert. Parallel wurden Primerpaare in Regionen der RLGS-Sequenzen ohne repetitive
Elemente gesucht. Mit diesen Primerpaar wurde DNA mittels PCR amplifiziert und in einem
Vektor kloniert. Plasmide mit Inserts wurden in gröβer Menge hergestellt. Schließlich wurden
die Vektoren mit Restriktionsenzymen geschnitten und die Inserts als radioaktiv-markierte
Hybridizierungssonde benutzt.
4.3.4.- Southern Blots
Abbildung 21.- Southern Blot zur Methylierungsanalyse des Dematin-Gens. A.- Das Gen wurden mit der Software CpG plot nach CpG-Inseln auf seinem Promotor analysiert. B.- Untersuchung der EcoR V und Not I Schnittstelle im untersuchtem Gen und Lokalisierung der DNA-Sonde. Dann wurden die Gröβe der Fragmente von Southern Blot je nach Methylierungsstatus der Not I Schnittstelle berechnet. C.- Southern Blot für das Dematin-Gen. Die Membran wurde mit einer radioaktiv-markierten DNA-Sonde hybridisiert. In diesem Southern Blot konnte nachgewiesen werden, dass die CpG-Dinukleotide innerhalb der Not I Schnittstelle des Dematin-Gens in periphären Blutmononukleären Zellen (PMN) demethyliert ist (Bande bei 1.5 kb), in Kasumi-1 methyliert ist (Bande bei >20 kb), in nicht-induzierten Zellen partiell methyliert ist (Bande bei >20 kb und bei 1.5 kb) und in den induzierten Zellen komplett methyliert ist (Bande bei 1,5 kb).
M 1x 2x 1x 2x 1x 2x 1x 2x
PWL Kas-1 9/14/18 9/14/18 - 48 Std + 48 Std
M = Marker
- = Not treated with Ponasterone A
+ = Treated with Ponasterone A
1x = Digested with ECORV
2x = Digested with ECORV & NOT I
Spot 2D24: Dematin
(Membranprotein Band 4.9 der Erythrozyten)
= Probe
CpG Demethylated
CpG Partial methylated
CpG Methylated
1.5 kb
17 kb
1.5 kB band
17 kB band
1.5 and 17 kB bands
Expected bandsPossible methylationstatus of Not I site
A.-
B.-
C.-

4.- Ergebnisse Seite - 91 -
Southern Blots wurden durchgeführt, um die Daten der RLGS-Analysen zu validieren
und um die Reproduzierbarkeit der Versuche festzustellen.
Als Negativkontrolle wurden Lymphozyten aus peripherem Blut benutzt, weil bekannt
ist, dass ihre DNA überwiegend demethyliert ist. Als interne Kontrolle wurden unsere
Versuche mit der U937-Zelllinie durchgeführt, um festzustellen, wie der Methylierungsstatus
jeweiligen der Not I Schnittstelle ist. Letztlich wurde die 9/14/18-Zelllinie mit und ohne
Induktion von AML1-ETO verwendet. Die Zellen wurden zu 2 verschiedenen Zeitpunkten
(48 und 96 Stunden nach der Induktion des AML1/ETO-Proteins) geerntet. Für 5 Sequenzen
aus der primären RLGS-Analyse wurden Southern Blots zur Validierung durchgeführt (2D20,
2G93, 2D24, 1F07, 2C66). Diese entsprechen folgenden Genen: OTX1, PTPN11/Noonan
Syndrom Gen, Dematin/Proteinband 4.9 der Erythrozyten, CDC28 Proteinkinase 2 und
PELI2.
a) Im Dematin-Gen (Spot 2D24), von dem das Protein Band 4.9 aus der Membran von
Erythrozyten kodiert wird, haben wir eine Methylierungsveränderung in den AML1/ETO
induzierten Zellen festgestellt. In mononukleärem Blutzellen ist dieses CpG-Dinukleotid
demethyliert. In der Kasumi-1-Zelllinie ist es komplett methyliert, in U937- und in den nicht-
induzierten 9/14/18-Zellen ist es partiell methyliert, aber in den induzierten 9/14/18-Zellen ist
es zu beiden Zeitpunkten (48 und 96 Std) komplett methyliert (Abb. 21c).
b) Im PTPN11 Gen oder Noonan Syndrom Gen (Spot 2G93) zeigte sich, dass die Not
I-Schnittstelle in Blutlymphozyten und Kasumi-1 Zellen demethyliert ist. Trotzdem wurde ein
Demethylierungsereignis von partiell methyliert (10:1) in den nicht- induzierten 9/14/18-
Zellen zu partiell methyliert (2:1) in den induzierten 9/14/18 Zellen nach Quantifizierung des
Signals der Autoradiogramme nachgewiesen. Das bedeutet, dass unter AML1/ETO Einfluss
diese Not I-Schnittstelle teilweise demethyliert wird. Dieses Ergebnis entspricht dem
Ergebnis der RLGS-Analyse (s. Abbildung 22c).
c) Die Not I-Schnittstelle im OTX1-Gen (Spot 2D20) ist in Blutlymphozyten
demethyliert, partiell methyliert in Kasumi-1-Zellen und komplett methyliert sowohl in U937-
Zellen als auch in nicht-induzierten bzw induzierten 9/14/18-Zellen. Nach der Induktion des
AML1/ETO-Proteins gab es keine Methylierungsveränderung, die durch Southern Blots
nachgewiesen werden konnten (Daten nicht gezeigt).
d) Für die Gene PELI2 (Spot 2C66) und CDC28 Proteinkinase 2 (Spot 1F07) konnten
wir ebenfalls keine Unterschiede zwischen den nicht-induzierten und induzierten 9/14/18-
Zellen nachweisen (Daten nicht gezeigt).

4.- Ergebnisse Seite - 92 -
Nach diesen Ergebnissen wurde erneut eine RLGS-Analyse von den induzierten und
nicht-induzierten 9/14/18-Zellen durchgeführt. Diesmal wurden die Zellen zu einem früheren
Zeitpunkt (24 Stunden) und zu einem späteren Zeitpunkt (144 Std), sowie zu 48 Std. und 96
Std. als Wiederholung untersucht. Überraschend war, dass neue De- und
Methylierungsveränderungen zu allen Zeitpunkten gefunden wurden und die im ersten
Versuch aufgetretenen und verschwundenen Spots nicht mehr beteiligt waren. Auβerdem
wurde eine neue Methylierungssignatur mit späterer Passage der 9/14/18-Zellen gefunden.
Wegen der Instabilität des Epigenoms der 9/14/18-Zellen und der geringen Sensivität
der benutzten Methode wurde zu diesem Zeitpunkt beschlossen, dieses Projekt nicht weiter zu
verfolgen. Neuere Methoden sind notwendig, um globale Methylierungsveränderungen nach
der Induktion des AML1/ETO-Proteins in den 9/14/18-Zellen nachzuweisen. Geeignete
Methoden könnten die „ChIP on CHIP“ Methode mit einem Antikörper gegen Methyl-
Cytosin und die MethScope-Methode der Firma Oriongenomics sein.
Abbildung 22.- Southern Blot zur Methylierungsanalyse des PTPN11-Gens. A.- Analyse der CpG-Inseln des PTPN11-Gen auf dem Promotor und Gen-Sequenz. B.- Untersuchung der Fragmentgröße nach Restriktion mit den EcoR V- und Not I-Enzymen und Lokalisierung der DNA-Sonde für dieses Gen. C.- Southern Blot für das PTPN11-Gen. PBL galt als Negativkontrolle und die Kasumi-1-Zelllinie galt als Positivkontrolle für AML1/ETO.
1x 2x 1x 2x M 1x 2x 1x 2x M 1x 2x 1x 2x
PWL Kas-1 9/14/18 9/14/18 9/14/18 9/14/18- 48 Std + 48 Std - 96 Std + 96 std
M = Marker
- = Not treated with Ponasterone A
+ = Treated with Ponasterone A
1x = Digested with ECORV
2x = Digested with ECORV & NOT I
Spot 2G93: PTPN11-Gene (Noonan Syndrom 1)
= Probe
CpG Demethylated
CpG Partial methylated
CpG Methylated
3.5 kb
>20 kb
3.5 kB band
>20 kB band
3.5 and >20 kB bands
Expected bandsPossible methylationstatus of Not I site
A.-
B.-
C.-

4.- Ergebnisse Seite - 93 -
4.4.- Untersuchung der Expression des LAT2-Gens während induzierter myeloischer
Differenzierung von AML-Zelllinien.
Dr. M. Fliegauf schloss seinen Artikel mit folgenden Wörter ab: „...aber die Funktion dieses
Gens, und die Bedeutung seiner Repression durch AML1/ETO ist in der Myelopoese bisher
ungeklärt und so sind weitere Untersuchungen notwendig“.
Die AML1- und AML1/ETO- Zielgene spielen eine wichtige Rolle in der
Hämatopoese, aber die Funktion in der Hämatopoese des LAT2-Gens, einschließlich die
Bedeutung seiner Repression durch AML1/ETO, ist bisher nicht untersucht.
Daher wurde die Expression dieses Gens sowohl auf RNA- und Protein-Ebene in
Differenzierungsversuchen mit Zelllinien, auf Protein-Ebene in Zytokinen-induzierten
Differenzierungen von normalen CD34+ Zellen, und in primären myeloischen Zellen
untersucht.
4.4.1.- Regulation von LAT2-mRNA und -Protein in der ATRA-induzierten
granulozytären Differenzierung der NB4-Zelllinie.
Die NB4-Zelllinie trägt die t (15; 17) (Lanotte et al., 1991), die das chimäre
Fusionprotein PML/RARα kodiert (Goddard et al., 1991). Die Zellen sind sensitiv für ATRA
und nach ATRA-Gabe differenzieren sie zu Granulozyten (Lanotte et al., 1991). ATRA wurde
in einer Konzentration von 1 µM in 3 Gaben alle 24 Stunden zugesetzt. Das Medium wurde
mit jeder Gabe von ATRA gewechselt.
Zellwachstum und Viabilität wurden täglich mit Trypan-Blau-Färbung bestimmt. Das
Zellwachstum war am 3. Tag in den behandelten Zellen deutlich niedriger (17,5 +/- 4,4 x 105
Zellen/ml in den unbehandelten vs 8,7 +/- 1,5 x 105 Zellen/ml in den behandelten Zellen,
Wachstum Inhibition vom 50,3 %). Es konnte am 3. Tag keine signifikante Veränderung der
Viabilität beobachtet werden (92,7 +/- 0,6 % in den unbehandelten vs 91,3 +/- 1,8 % in den
behandelten Zellen) (Abb. 26a-b). Die Zellen wurden am 3. Tag auf das CD11b Antigen
mittels FACS Analyse untersucht. Die unbehandelten NB4-Zellen waren zu 60,2 +/- 21,2 %
positiv auf CD11b aber die durch ATRA-behandelten NB4-Zellen waren zu 95,7 +/- 4,2 %
positiv auf CD11b (Abb. 26c). Am 3. Tag wurden auch 1x105 Zellen nach Zytospin auf einem
Objekträger „fixiert“. Nach einer May-Grünwald-Giemsa-Färbung wurden die Zellen auf ihre
Morphologie unter dem Mikroskop untersucht (Abb. 26d). Die unbehandelten Zellen hatten
die typische Morphologie der Promyelozyten. Sie waren größer als andere myeloische

4.- Ergebnisse Seite - 94 -
Zelllinien, die in den Differenzierungsversuchen verwendet wurden. Das Zytoplasma war
basophil. Des Weiteren war ebenfalls reichlich dicke Azurgranula zu sehen. Der Kern hatte
einen großen Teil des Zytoplasmas besetzt und lag exzentrisch. Die ATRA-behandelten
Zellen waren kleiner als die nicht-differenzierten Zellen. Das Zytoplasma war rosa-violett
gefärbt; Granula waren kaum zu sehen. Der Kern war kleiner, kondensiert und meistens in 2-3
Lobulen segmentiert.
Die Zellen wurden 0, 6, 12, 24, 48 und 72 Stunden nach Induktion der Differenzierung
geerntet. Die mRNA- und Protein-Expression wurden mittels Northern und Western Blot
untersucht. (Abb. 27a-b.). 6 und 12 Stunden nach Induktion der Differenzierung war LAT2-
mRNA und –Protein verstärkt sichtbar, danach war die Expression niedriger als in den
unbehandelten Zellen. Nach 72 Stunden hat die Herunterregulation bis zu 30 % der
Expression der unbehandelten NB4-Zelllinie auf der mRNA- und Protein-Ebene erreicht. Die
Northern-Blot-Membran wurde mit einer Myeloperoxidase (MPO)-Sonde als
Abbildung 23.- Untersuchung des biologischen Effektes der durch ATRA-induzierten granulozytären Differenzierung der NB4-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. ATRA hat einen hemmenden Effekt auf das Zellwachstum der NB4-Zellen ohne groβen Einfluss auf die Viabilität der Zellen. C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf CD11b-Antigen untersucht. Die NB4-Zellen wurden durch ATRA differenziert, wie die Positivität der Zellen auf CD11b zeigte. D.- Die Differenzierung der Zellen wurden am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt. Die durch ATRA-behandelten NB4-Zellen hatten nach 3 Tagen eine ähnliche Morphologie wie die Granulozyten.
NB4 cells + ATRANB4 cells - ATRA
0 %
2 0 %
4 0 %
6 0 %
8 0 %
1 0 0 %
0 2 4 4 8 7 2T im e (h o u rs )
Via
bilit
y
N B 4 + A T R A N B 4 - A T R A
0
5
1 0
1 5
2 0
2 5
0 2 4 4 8 7 2T im e (h o u rs )
Cel
l con
cent
ratio
n (1
0 *5
/mL)
N B 4 + A T R A N B 4 - A T R A NB4 cells
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
without ATRA With ATRA
pos
tive
cells
to C
D11
bPE
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 95 -
Differenzierungskontrolle und mit einer GAPDH-Sonde als Ladekontrolle hybridisiert. β-
Aktin wurde als Ladekontrolle im Western Blot benutzt.
4.4.2.- Regulation von LAT2-mRNA und -Protein in der ATRA-induzierten
granulozytären Differenzierung der U937-Zelllinie.
Die monoblastäre U937-Zelllinie (Sundstrom and Nilsson, 1976) ist eine ATRA sensitive
Zelllinie (Olsson et al., 1982), die sich mit dieser Substanz in Granulozyten-ähnliche Zellen
differenziert. Für die ATRA-induzierte granulozytäre Differenzierung der U937-Zelllinie
wurde ATRA in einer Konzentration von 1 µM in 3 Gaben alle 24 Stunden angesetzt. Das
Medium wurde mit jeder Gabe von ATRA gewechselt.
Abbildung 24.- LAT2-mRNA- und -Protein-Expression in der granulozytären Differenzierung der NB4-Zellen durch ATRA. A.- Die LAT2-mRNA-Expression wurde zu verschiedenen Zeitpunkten mittels Northern Blot untersucht. MPO galt als Differenzierungsmarker und GAPDH wurde als Ladekontrolle benutzt. B.- Die LAT2-Protein-Expression wurde mittels Western Blot untersucht. LCL-GK ist eine B Zelllinie, die als Positivkontrolle für die Expression von LAT2-Protein galt. Eine Herunterregulation der LAT2-mRNA und –Protein wurde nachgewiesen.
A.-
B.-
ATRA (10-6 M) - + + + + +
Time (hrs) 0 6 12 24 48 72
NB4
LAT2
GAPDH
EtBr
MPO
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
NB4
LAT2
β-Actin
LCL-GK
A.-
B.-
ATRA (10-6 M) - + + + + +
Time (hrs) 0 6 12 24 48 72
NB4
LAT2
GAPDH
EtBr
MPO
ATRA (10-6 M) - + + + + +
Time (hrs) 0 6 12 24 48 72
NB4
LAT2
GAPDH
EtBr
MPO
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
NB4
LAT2
β-Actin
LCL-GKATRA (10-6 M) - + + + + + -
Time (hrs) 0 6 12 24 48 72 72
NB4
LAT2
β-Actin
LCL-GK

4.- Ergebnisse Seite - 96 -
Zellwachstum und Viabilität wurden täglich mit Trypan-Blau-Färbung bestimmt. Das
Zellwachstum war am 3. Tag deutlich niedriger (21,1 +/- 7,7 x 105 Zellen/ml in den
unbehandelten vs. 12,6 +/- 4,3 x 105 Zellen/ml in den behandelten Zellen, Wachstums-
Inhibition vom 43,5 %). Dennoch wurde am 3. Tag keine Veränderung der Viabilität
beobachtet (94,2 +/- 1,0 % in den unbehandelten vs. 94,0 +/- 1,8 % in den behandelten Zellen)
(Abb. 28a-b). Die Inhibition des Zellwachstums war nicht so stark wie bei der ATRA-
induzierten granulozytären Differenzierung der NB4-Zelllinie. Die unbehandelten U937-
Zellen waren zu 30,7 +/- 5,6 % positiv auf CD11b allerdings waren die mit ATRA-
behandelten U937-Zellen zu 94,2 +/- 5,6 % positiv auf CD11b mittels FACS Analyse (Abb.
28c). Die unbehandelten Zellen hatten nach der May-Grünwald-Giemsa-Färbung eine
typische monoblastäre Morphologie. Das Zytoplasma war teilweise basophil (rosa-blau) mit
kaum sichtbaren Granula. Große Vakuolen waren als Zeichen ihrer phagozytären Aktivität im
Abbildung 25.- Untersuchung des biologischen Effektes der durch ATRA induzierten granulozytären Differenzierung der U937-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. ATRA hat einen hemmenden Effekt auf das Zellwachstum der U937-Zellen ohne groβen Einfluss auf die Viabilität. Dieser Effekt ist nicht so stark wie bei den NB4-Zellen. C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf CD11b untersucht. Die U937-Zellen wurden durch ATRA differenziert, wie die Positivität der Zellen auf CD11b zeigte. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt. Die durch ATRA-behandelten U937-Zellen hatten nach 3 Tagen eine ähnliche Morphologie wie die Granulozyten.
U937 cells + ATRAU937 cells - ATRA
0
5
10
15
20
25
30
0 24 48 72
Time (hours)
Cel
l con
cent
ratio
n (1
0 *5
/mL
)
U937 + ATRA U937 - ATRA
0%
20%
40%
60%
80%
100%
0 24 48 72Time (hours)
Via
bili
ty
U937 + ATRA U937 - ATRA
U937 cells
0 ,0%
20,0%
40,0%
60,0%
80,0%
100,0%
without ATRA W ith ATRA
post
ive
cells
to C
D11
bPE
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 97 -
Zytoplasm vorhanden. Der Kern war unregelmäßig und mit Faltenbildungen. Die durch
ATRA-differenzierten Zellen waren kleiner als die unreifen Zellen. Das Zytoplasma war
acidophil (rosa) und unregelmäßig. Der Kern war gelappt und das Chromatin kondensiert
(Abb. 28d).
Die Zellen wurden 0, 6, 12, 24, 48 und 72 Stunden nach Induktion der Differenzierung
geerntet und die mRNA- und Protein-Expression wurden mittels Northern und Western Blot
untersucht (Abb. 29a-b.). Die Expression von LAT2-mRNA und -Protein wurden während
des gesamten Versuchs herunterreguliert. Nach 72 Stunden hat diese Herunterregulation 60 %
(auf der mRNA-Ebene) und 70 % (auf der Protein-Ebene) der Grund-Expression der U937-
Zelllinie erreicht. Die Northern Blot-Membrane wurden mit einer Lysozym-Sonde als
Differenzierungskontrolle und mit einer GAPDH-Sonde als Ladekontrolle hybridisiert. β-
Aktin wurde als Ladekontrolle im Western Blot benutzt.
Abbildung 26.- LAT2-mRNA- und -Protein-Expression in der granulozytären Differenzierung der U937-Zelllinie durch ATRA. A.- Die LAT2-mRNA Expression wurde zu verschiedenen Zeitpunkten mittels Northern Blot untersucht. Lysozym galt als Differenzierungsmarker und GAPDH wurde als Ladekontrolle benutzt. B.- Die LAT2-Protein-Expression wurde mittels Western Blot untersucht. LCL-GK ist eine B Zelllinie, die als Positivkontrolle für die Expression von LAT2-Protein galt. Die Expression der LAT2-mRNA und Protein wurde nach der ATRA-Behandlung der U937-Zellen herunterreguliert. Nach 6 Stunden wurde weniger Proteine im diesen Western Blot geladen.
A.-
B.-
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
GAPDH
EtBr
LYSOZYM
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
β-Actin
LCL-GK
A.-
B.-
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
GAPDH
EtBr
LYSOZYM
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
GAPDH
EtBr
LYSOZYM
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
β-Actin
LCL-GKATRA (10-6 M) - + + + + + -
Time (hrs) 0 6 12 24 48 72 72
U937
LAT2
β-Actin
LCL-GK
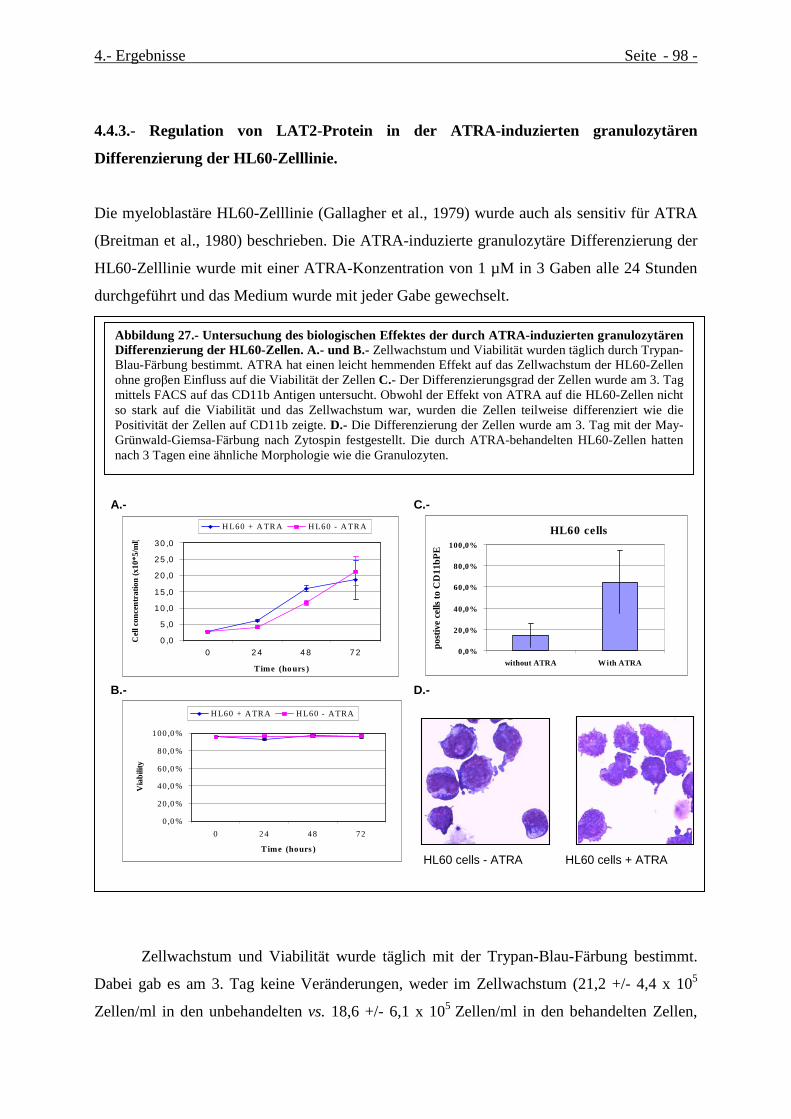
4.- Ergebnisse Seite - 98 -
4.4.3.- Regulation von LAT2-Protein in der ATRA-induzierten granulozytären
Differenzierung der HL60-Zelllinie.
Die myeloblastäre HL60-Zelllinie (Gallagher et al., 1979) wurde auch als sensitiv für ATRA
(Breitman et al., 1980) beschrieben. Die ATRA-induzierte granulozytäre Differenzierung der
HL60-Zelllinie wurde mit einer ATRA-Konzentration von 1 µM in 3 Gaben alle 24 Stunden
durchgeführt und das Medium wurde mit jeder Gabe gewechselt.
Zellwachstum und Viabilität wurde täglich mit der Trypan-Blau-Färbung bestimmt.
Dabei gab es am 3. Tag keine Veränderungen, weder im Zellwachstum (21,2 +/- 4,4 x 105
Zellen/ml in den unbehandelten vs. 18,6 +/- 6,1 x 105 Zellen/ml in den behandelten Zellen,
Abbildung 27.- Untersuchung des biologischen Effektes der durch ATRA-induzierten granulozytären Differenzierung der HL60-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. ATRA hat einen leicht hemmenden Effekt auf das Zellwachstum der HL60-Zellen ohne groβen Einfluss auf die Viabilität der Zellen C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf das CD11b Antigen untersucht. Obwohl der Effekt von ATRA auf die HL60-Zellen nicht so stark auf die Viabilität und das Zellwachstum war, wurden die Zellen teilweise differenziert wie die Positivität der Zellen auf CD11b zeigte. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt. Die durch ATRA-behandelten HL60-Zellen hatten nach 3 Tagen eine ähnliche Morphologie wie die Granulozyten.
HL60 cells + ATRAHL60 cells - ATRA
0 ,0
5 ,0
10 ,0
15 ,0
20 ,0
25 ,0
30 ,0
0 2 4 4 8 7 2
Time (hours )
Cel
l con
cent
ratio
n (x
10*5
/ml)
H L60 + A TRA H L60 - A TRA
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72
Time (hours )
Via
bilit
y
H L60 + A TRA HL60 - ATRA
HL60 cells
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
without ATRA With ATRA
post
ive
cells
to C
D11
bPE
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 99 -
Zellwachstum-Inhibition von 12,3 %) noch in der Viabilität (96,2 +/- 2,2 % in den
unbehandelten vs. 96,1 +/- 1,6 % in den behandelten Zellen) (Abb. 30a-b).
Im Gegenteil, die unbehandelten HL60-Zellen waren zu 14,7 +/- 11,7 % positiv auf
CD11b und die durch ATRA-behandelten HL60-Zellen waren zu 65,0 +/- 29,5 % positiv auf
das CD11b-Antigen mittels FACS-Analyse (Abb. 30c). Diese Daten haben gezeigt, dass die
HL60-Zellen durch ATRA in Granulozyten-ähnliche Zellen differenziert wurden. Die
Behandlung mit ATRA hat das Zellwachstum, Differenzierungsgrad und Viabilität dieser
Zelllinie nicht so stark beeinflusst wie in NB4- oder U937-Zellen.
Die unbehandelten HL60-Zellen hatten eine typische myeloblastäre Morphologie mit
einem sehr basophilen Zytoplasma. Der Kern war oval und die Chromatinstruktur
feinretikuliert. Die durch ATRA-differenzierten Zellen hatten eine reifere Morphologie mit
einem kondensierten, segmentierten Kern, teilweise basophilem Zytoplasma (rosa-blau) und
einen zentralen azidophilen Nukleolus. (Abb. 30d)
Die LAT2-mRNA- und -Protein-Expression wurden 0, 6, 12, 24, 48 und 72 Stunden
nach der Induktion der Differenzierung mittels Northern (Daten nicht gezeigt) und Western
Blot untersucht (Abb. 31). Die Expression des LAT2-Proteins wurde während des Versuches
transient herunterreguliert. Nach 24 Stunden hat die Herunterregulation das Maximum mit
55,7 % der Grundexpression der HL60-Zelllinie auf der Protein-Ebene erreicht. Nach 48 und
72 Stunden hatte die Expression des LAT2-Proteins aber wieder ca. 80 % der Expression auf
Tag 0 erreicht. β-Aktin wurde als Ladekontrolle im Western Blot benutzt. Im Northern Blot
wurde keine Herunterregulation festgestellt, da die Expression der LAT2-mRNA in der
HL60-Zelllinie insgesamt sehr niedrig war.
Abbildung 28.- LAT2-mRNA- und -Protein-Expression in der granulozytären Differenzierung der HL60-Zelllinie durch ATRA. A.- Die LAT2-Protein-Expression wurde zu verschiedenen Zeitpunkten mittels Western Blot untersucht. Eine transiente Herunterregulation wurde auf der Protein-Ebene nachgewiesen. Das Maximumniveau der LAT2-Protein-Repression wurde 24 Stunden nach der ATRA-Behandlung erreicht.
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL60
LAT2
β-Actin
ATRA (10-6 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL60
LAT2
β-Actin

4.- Ergebnisse Seite - 100 -
4.4.4.- Regulation von LAT2-mRNA und -Protein in der DMSO-induzierten
granulozytären Differenzierung der HL60-Zelllinie.
Die HL60-Zelllinie kann nach der Dimethylsulfoxide (DMSO) Behandlung sowohl in
Granulozyten-ähnliche-Zellen (Collins et al., 1978, , 1979) und auch in Monozyten-
Makrophagen-ähnliche-Zellen (Vorbrodt et al., 1979)- kontrolliert mit saurer Phosphatase-;
(Rhyner and Taetle, 1986) differenzieren.
Die DMSO-induzierte granulozytäre Differenzierung der HL60-Zelllinie wurde mit
einer Konzentration von 1,3 % in 3 Gaben alle 24 Stunden durchgeführt und das Medium
wurde mit jeder Gabe gewechselt.
Abbildung 29.- Untersuchung des biologischen Effektes der durch DMSO-induzierten granulozytären Differenzierung der HL60-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. DMSO hat einen leicht hemmenden Effekt auf das Zellwachstum der HL60-Zellen ohne groβen Einfluss auf die Viabilität der Zellen C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf das CD11b Antigen untersucht. Obwohl der Effekt von DMSO auf die HL60-Zellen nicht so stark auf die Viabilität und das Zellwachstum war, wurden die Zellen teilweise differenziert wie die Positivität der Zellen auf CD11b zeigte. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt. Die durch DMSO-behandelten HL60-Zellen hatten nach 3 Tagen eine ähnliche Morphologie wie die Granulozyten.
HL60 cells + DMSOHL60 cells - DMSO
0,0
5,0
10,0
15,0
20,0
25,0
0 24 48 72
Time (hours)
Cel
l con
cent
ratio
n (1
0 *5
/mL
)
HL60 + DMSO HL60-DMSO
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72
Time (hours)
Via
bilit
y
HL60 + DMSO HL60 - DMSO
HL60 cells
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
without DMSO with DMSO
post
ive
cells
to m
yello
id
mar
kers
CD11bPE CD11cPE
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 101 -
Zellwachstum und Viabilität wurde täglich mit Trypan-Blau-Färbung bestimmt. Es
gab schon am 2. Tag zwischen den DMSO-behandelten und den unbehandelten Zellen einen
Unterschied in der Zellwachstumskurve. Dieser Unterschied war am 3. Tag am gröβten (22,9
+/- 1,0 x 105 Zellen/ml in den unbehandelten vs 12,0 +/- 4,7 x 105 Zellen/ml in den
behandelten Zellen, Wachstums-Inhibition von 47,6 %). Dabei wurde keine Veränderung in
der Viabilität der Zellen festgestellt (93,4 +/- 1,5 % in den unbehandelten vs 92,0 +/- 0,6 % in
den behandelten Zellen) (Abb. 29a und 29b).
Die unbehandelten HL60 Zellen waren zu 14,7 +/- 11,7 % positiv auf CD11b und die
DMSO-behandelten Zellen zu 80,6 +/- 25,5 % positiv auf das CD11b-Antigen. Das reife
monozytäre Antigen CD11c wurde am 3. Tag durch FACS gemessen, um die monozytäre
Differenzierung der Zellen festzustellen. Die unbehandelten HL60-Zellen waren zu 6,8 +/- 4,8
% positiv auf CD11c und die DMSO-behandelten Zellen waren zu 31,5 +/-19,9 % positiv auf
das CD11c Antigen. Dies lässt zum Schluss zu, dass DMSO eine gemischt granulozytäre und
monozytäre Differenzierung der HL60-Zelllinie induziert (Abb. 29c).
Nach der May-Grünwald-Giemsa Färbung hatten die unbehandelten Zellen eine
typische myeloblastäre Morphologie (s. o. 4.5.4). Die DMSO-differenzierten HL60-Zellen
waren kleiner als die unbehandelten Zellen und hatten einen kondensierten und segmentierten
Kern (bis 3-4 Lobuli). Das Zytoplasma war basophil (hell blau) und sehr unregelmäßig mit
Segmentierungen (Abb. 29d).
Die LAT2-mRNA- und -Protein-Expression wurden 0, 6, 12, 24, 48 und 72 Stunden
nach der Induktion der Differenzierung mittels Northern und Western Blot untersucht. Die
Ergebnisse dieser Experimente waren inkonklusiv und deshalb werden sie nicht gezeigt.
4.4.5.- Regulation von LAT2-mRNA und Protein in der PMA-induzierten monozytären
Differenzierung der HL60-Zelllinie.
Die HL60-Zellen können in Monozyten differenzieren, wenn sie mit dem Phorbol-Ester PMA
behandelt werden (Huberman and Callaham, 1979; Rovera et al., 1979). Diese
Differenzierung von HL60-Zellen wurde benutzt, um die verschiedenen Schritte der
monozytären bzw. makrophagen Differenzierung zu untersuchen.
Die PMA-induzierte monozytäre Differenzierung der HL60-Zelllinie wurde mit einer
einmalige Gabe von 100 nM PMA durchgeführt und das Medium wurde 3 Tage nicht
gewechselt.

4.- Ergebnisse Seite - 102 -
Zellwachstum und Viabilität wurden täglich mit Trypan-Blau-Färbung bestimmt.
Schon am 2. Tag gab es einen Unterschied in der Zellwachstumskurve zwischen den PMA-
behandelten und unbehandelten Zellen. Dieser Unterschied war am 3.Tag am größten (21,7
+/- 2,3 x 105 Zellen/ml in den unbehandelten vs 4,4 +/- 0,8 x 105 Zellen/ml in den behandelten
Zellen, Zellwachstum-Inhibition von 79,7 %). Ein leichter Einfluss (94,2 +/- 1,7 % in den
unbehandelten vs 82,1 +/- 8,0 % in den behandelten Zellen) wurde am 3. Tag durch PMA auf
die Viabilität der Zellen festgestellt (Abb. 30a und 30b.). Die Viabilität war jedoch nie unter
75 %. Die unbehandelten HL60-Zellen waren zu 14,7 +/- 11,7 % positiv auf CD11b und die
behandelten HL60-Zellen waren zu 82,5 +/- 11,6 % positiv auf CD11b. Das reife monozytäre
Antigen CD11c wurde am 3. Tag mittels FACS gemessen, um die monozytäre-macrophage
Differenzierung der Zellen zu bestimmen. Die unbehandelten HL60-Zellen waren zu 6,8 +/-
4,8 % positiv auf CD11c und die PMA-behandelten Zellen waren zu 90,8 +/- 1,7 % positiv
Abbildung 30.- Untersuchung des biologischen Effektes der durch PMA-induzierten monozytären Differenzierung der HL60-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. Die PMA hat einen stark hemmenden Effekt auf das Zellwachstum der HL60-Zellen und hat einen geringen Einfluss auf die Viabilität der Zellen. C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf das CD11b Antigen und auf das CD11c Antigen untersucht. Die HL60-Zellen wurden durch PMA nur in Monozyten-Makrophagen differenziert, weil die CD11b und CD11c Antigene gleich stark positiv waren. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt. Die durch PMA-behandelten HL60-Zellen hatten nach 3 Tagen eine ähnliche Morphologie wie die Makrophagen.
HL60 cells + PMAHL60 cells - PMA
0
5
10
15
20
25
30
0 24 48 72Time (hours)
Cel
l con
cent
ratio
n (c
ells
x 1
0*5/
ml)
Hl60+ PMA HL60 - PMA
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72
Time (hours)
Via
bilit
y
Hl60 + PMA HL60 - PMA
HL60 ce lls
0,0 %
2 0,0 %
4 0,0 %
6 0,0 %
8 0,0 %
10 0,0 %
without PMA with PMA
posi
tive
cells
to m
yello
id
mar
kers
CD11bPE CD11cPE
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 103 -
auf CD11c. PMA induzierte überwiegend eine monozytäre-macrophage Differenzierung der
HL60-Zellen (Abb. 30c).
Nach der May-Grünwald-Giemsa-Färbung hatten die unbehandelten Zellen eine
typisch myeloblastäre Morphologie wie in 4.5.4 beschieben wurde (s.o.). Die durch PMA
differenzierten Zellen hatten eine Macrophagen-ähnliche Morphologie. Die Zellen waren
größer als die unbehandelten Zellen und adhärent an die Kulturflasche. Das Zytoplasma war
sehr azidophil (rosa und blaue Flecken) und die Anwesenheit von Vakuolen zeigte
Phagozytose. Der Kern war oval und gelappt (Abb. 30d)
Die LAT2-mRNA und -Protein Expression wurden 0, 6, 12, 24, 48 und 72 Stunden
nach Induktion der Differenzierung mittels Northern und Western Blot untersucht (Abb. 31a-
b). Die Expression des LAT2-Gens wurde sowohl auf der mRNA- und als auch auf der
Protein-Ebene hochreguliert. Diese Hochregulation hatte 24 Stunden nach der Behandlung
mit PMA das Maximum mit einem 3,8-fachen der Grundexpression auf mRNA-Ebene
Abbildung 31.- LAT2-mRNA- und -Protein-Expression in der monozytären Differenzierung der HL60-Zelllinie durch PMA. A.- Die LAT2-mRNA-Expression wurde zu verschiedenen Zeitpunkten mittels Northern Blot untersucht. MPO wurde als myeloischer Differenzierungsmarker benutzt und das Ethidiumbromid-Bild als Ladekontrolle. B.- Die LAT2-Protein-Expression wurde mittels Western Blot untersucht. LCL-GK ist eine B Zelllinie, die als Positivkontrolle für die Expression des LAT2-Proteins galt. Die Inkubation der Membran mit einem β-Aktin-Antikörper wurde als Ladekontrolle benutzt. Das LAT2-Gen wurde durch die PMA-Behandlung der HL60-Zellen auf der mRNA- und auf der Protein-Ebene sehr stark hochreguliert.
A.-
B.-
PMA (10-7 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL-60
LAT2
MPO
EtBr
PMA (10-7M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL60
LAT2
β-Actin
LCL-GK
A.-
B.-
PMA (10-7 M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL-60
LAT2
MPO
EtBr
PMA (10-7M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL60
LAT2
β-Actin
LCL-GKPMA (10-7M) - + + + + + -Time (hrs) 0 6 12 24 48 72 72
HL60
LAT2
β-Actin
LCL-GK

4.- Ergebnisse Seite - 104 -
erreicht. Diese Daten wurden mit den absoluten Werten der Densitometrie berechnet, da auch
die Expression von GAPDH- und β-Aktin-mRNA zum Teil reguliert war. Andererseits war
die Hochregulation des LAT2-Proteins 72 Stunden nach der Behandlung 4-fach höher als die
Grundexpression in HL60-Zellen. Die Membran des Northern Blots wurde mit einer MPO-
Sonde als Differenzierungskontrolle und Ethidiumbromid wurde als Ladekontrolle benutzt. β-
Aktin wurde als Ladekontrolle im Western Blot benutzt.
4.4.6.- Regulation von LAT2-Protein in der PMA-induzierten monozytären
Differenzierung der U937-Zelllinie.
Die U937-Zellen werden durch PMA-Behandlung in Monozyten-Makrophagen differenziert
(Dellagi and Brouet, 1984; Welgus et al., 1986). Die PMA-induzierte monozytäre
Differenzierung der U937-Zelllinie wurde mit einer Konzentration von 100 nM in 3 Gaben
alle 24 Stunden durchgeführt und das Medium wurde mit jeder Gabe gewechselt.
Zellwachstum und Viabilität wurden täglich mit der Trypan-Blau-Färbung bestimmt.
Am 3. Tag wurde ein Unterschied zwischen den PMA-behandelten und unbehandelten Zellen
in der Zellwachstumskurve (14,2 +/- 2,1 x 105 Zellen/ml in den unbehandelten vs 8,2 +/- 2,6 x
105 Zellen/ml in den behandelten Zellen, Zellwachstum-Inhibition von 42,2 %) beobachtet. In
der Viabilität der Zellen war am 3. Tag keine Veränderung zu beobachten (95,8 +/- 2,1 % in
den unbehandelten vs 94,7 +/- 1,5 % in den behandelten Zellen) (Abb. 32a-b).
Die unbehandelten U937-Zellen waren zu 28,1 +/- 4,5 % positiv und die durch PMA-
behandelten U937-Zellen waren zu 82,5 +/- 13,5 % positiv auf CD11b. Das CD11c-Antigen
wurde am 3. Tag durch FACS gemessen, um die monozytäre-macrophage Differenzierung
der Zellen zu bestimmen. Die unbehandelten U937-Zellen waren zu 8,2 +/- 0,9 % positiv und
die mit PMA-behandelten Zellen waren zu 81,7 +/- 14,2 % positiv auf CD11c-Antigen. PMA
induzierte überwiegend eine monozytäre Differenzierung der U937-Zelllinie (Abb. 32c).

4.- Ergebnisse Seite - 105 -
Nach der May-Grünwald-Giemsa-Färbung hatten die unbehandelten Zellen eine
monoblastäre Morphologie wie in 4.5.2 beschrieben wurde (s.o.). Die durch PMA
differenzierten U937-Zellen hatten eine Monozyten-ähnliche Morphologie mit bilobulären
gelappten Kernen und teilweise basophilen Zytoplasma (rosa-blau). Die Zellen hatten auch
kleine Vakuolen durch Phagozytose im Zytoplasma und waren leicht adhärent an die
Kulturflasche (Abb. 32d).
Die mRNA- und Protein-Expression wurden 0, 6, 12, 24, 48 und 72 Stunden nach
Induktion der Differenzierung mittels Northern und Western Blot (Daten nicht gezeigt)
untersucht. Die Expression von LAT2 war transient hochreguliert auf der mRNA-Ebene.
Nach 24 Stunden hatte diese Hochregulation das Maximum mit einem 2,4-fachen der
Expression der nicht-behandelten U937-Zellen auf der mRNA-Ebene erreicht. Die GAPDH-
mRNA war am 3. Tag auch leicht reguliert. Die Membran des Northern Blots wurde mit einer
Abbildung 32.- Untersuchung des biologischen Effektes der durch PMA-induzierten monozytären Differenzierung der U937-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. Die PMA hat einen stark hemmenden Effekt auf das Zellwachstum der U937-Zellen und hat einen geringen Einfluss auf die Viabilität der Zellen. C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels FACS auf das CD11b Antigen und auf das CD11c Antigen untersucht. Die U937-Zellen wurden durch PMA nur in Monozyten-Makrophagen differenziert, weil die CD11b und CD11c Antigene gleich stark positiv waren. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin festgestellt.
U937 cells + PMAU937 cells - PMA
0
4
8
12
16
20
0 24 48 72
Time (hours)
Cel
l con
cen
tratio
n (c
ells
x10*
5/m
l)
U937 + PMA U937 - PMA
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72
Time (hours)
Via
bili
ty
U937 + PMA U937-PMA
U937 cells
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
without PMA with PMA
post
ive
cells
to m
yello
id
mar
kers
CD11bPE CD11cPE
A.-
B.-
C.-
D.-
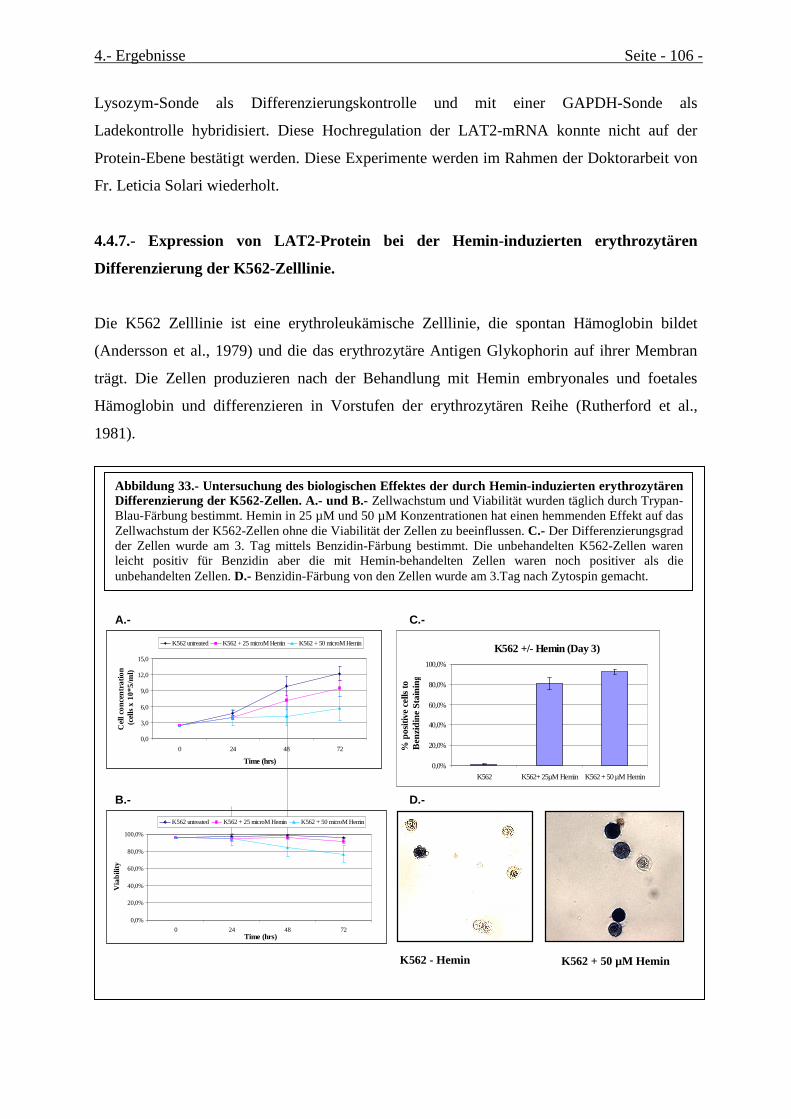
4.- Ergebnisse Seite - 106 -
Lysozym-Sonde als Differenzierungskontrolle und mit einer GAPDH-Sonde als
Ladekontrolle hybridisiert. Diese Hochregulation der LAT2-mRNA konnte nicht auf der
Protein-Ebene bestätigt werden. Diese Experimente werden im Rahmen der Doktorarbeit von
Fr. Leticia Solari wiederholt.
4.4.7.- Expression von LAT2-Protein bei der Hemin-induzierten erythrozytären
Differenzierung der K562-Zelllinie.
Die K562 Zelllinie ist eine erythroleukämische Zelllinie, die spontan Hämoglobin bildet
(Andersson et al., 1979) und die das erythrozytäre Antigen Glykophorin auf ihrer Membran
trägt. Die Zellen produzieren nach der Behandlung mit Hemin embryonales und foetales
Hämoglobin und differenzieren in Vorstufen der erythrozytären Reihe (Rutherford et al.,
1981).
Abbildung 33.- Untersuchung des biologischen Effektes der durch Hemin-induzierten erythrozytären Differenzierung der K562-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. Hemin in 25 µM und 50 µM Konzentrationen hat einen hemmenden Effekt auf das Zellwachstum der K562-Zellen ohne die Viabilität der Zellen zu beeinflussen. C.- Der Differenzierungsgrad der Zellen wurde am 3. Tag mittels Benzidin-Färbung bestimmt. Die unbehandelten K562-Zellen waren leicht positiv für Benzidin aber die mit Hemin-behandelten Zellen waren noch positiver als die unbehandelten Zellen. D.- Benzidin-Färbung von den Zellen wurde am 3.Tag nach Zytospin gemacht.
K562 - Hemin K562 + 50 µM Hemin
K562 +/- Hemin (Day 3)
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
K562 K562+ 25µM Hemin K562 + 50 µM Hemin
% p
ositi
ve c
ells
to B
enzi
dine
Sta
inin
g
0,0
3,0
6,0
9,0
12,0
15,0
0 24 48 72
Time (hrs)
Cel
l co
ncen
tra
tion
(ce
lls x
10
*5/m
l)
K562 untreated K562 + 25 microM Hemin K562 + 50 microM Hemin
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72Time (hrs)
Via
bili
ty
K562 untreated K562 + 25 microM Hemin K562 + 50 microM Hemin
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 107 -
Die Hemin-induzierte erythrozytäre Differenzierung der K562-Zelllinie wurde mit
einer Hemin-Konzentration von 25 und 50 µM in 3 Gaben alle 24 Stunden durchgeführt und
das Medium wurde mit jeder Gabe gewechselt.
Das Zellwachstum und die Viabilität wurden täglich mit der Trypan-Blau-Färbung
bestimmt. Am 3. Tag gab es schon einen Unterschied zwischen den Hemin-behandelten und
den unbehandelten Zellen in der Zellwachstumskurve zu beobachten (12,2 +/- 1,4 x 105
Zellen/ml in den unbehandelten vs 9,5 +/- 1,6 x 105 Zellen/ml in den mit 25 µM Hemin
behandelten, Zellwachstum-Inhibition von 22,1 %; bzw 5,7 +/- 2,3 x 105 Zellen/ml in den mit
50 µM Hemin behandelten Zellen, Zellwachstum-Inhibition von 53,3 %) (Abb. 33a). In der
Viabilität der unbehandelten und mit 25 µM Hemin behandelten Zellen wurde am 3. Tag
keine Veränderung festgestellt (95,6 +/- 0,7 % in den unbehandelten vs 92,0 +/- 2,8 % in den
behandelten K562-Zellen). Einen geringen Unterschied in der Viabilität gab es am 3. Tag
zwischen den unbehandelten Zellen und den mit 50 µM Hemin-behandelten Zellen (76,7 +/-
9,9 %) (Abb. 33b).
Um die Differenzierung zu bestätigen, wurden die Zellen nach Zytospin mit Benzidin
gefärbt. Bei der Benzidin-Färbung wird Hämoglobin durch Wasserstoffperoxid oxidiert und
durch Benzidin kommt es zu einer Blaufärbung. Die Zellen wurden auf Benzidin-Positivität
in einer Neubauer-Zählkammer unter dem Mikroskop gezählt. Die unbehandelten K562-
Zellen waren am 3. Tag zu 1,4 +/- 0,8 % positiv, die durch 25 µM Hemin behandelten K562-
Zellen waren zu 81,0 +/- 5,8 % positiv, und die durch 50 µM Hemin behandelten K562-
Zellen waren zu 92,4 +/- 2,0 % positiv auf Benzidin (Abb. 33c-d). Mit diesen Daten konnte
bestätigt werden, dass Hemin in einer Konzentration von 25 und 50 µM die K562-Zellen
Hämoglobin-Bildung induziert.
Abbildung 34.- LAT2-Protein-Expression während der durch Hemin induzierten erythrozytären Differenzierung der K562 Zelllinie. A.- Die LAT2 Protein-Expression wurde alle 24 Std. und nach 25 µM und 50 µM Hemin mittels Western Blot untersucht. Die U937-Zelllinie wurde als Positivkontrolle für die Expression des LAT2-Proteins benutzt. Die K562-Zelllinie hatte keine Grundexpression des LAT2-Proteins und dieses wird auch nicht während der erythrozytären Differenzierung der K562-Zellen hochreguliert
A.- B.-
LAT2
ß-Actin
Hemin (µM) 0 25 50
K562 (day 3)
U937
LAT2
ß-Actin
Time (hours) 0 24 48 72
Hemin 50 µM - + + +
K562A.- B.-
LAT2
ß-Actin
Hemin (µM) 0 25 50
K562 (day 3)
U937
LAT2
ß-Actin
Time (hours) 0 24 48 72
Hemin 50 µM - + + +
K562
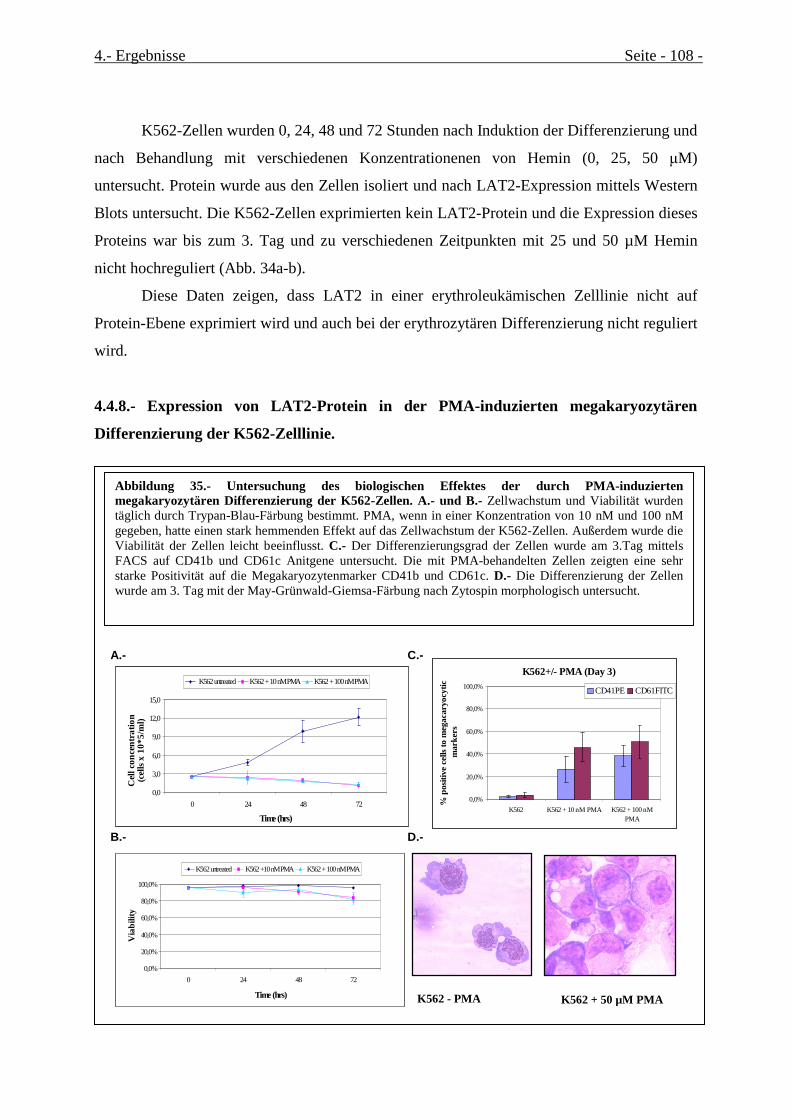
4.- Ergebnisse Seite - 108 -
K562-Zellen wurden 0, 24, 48 und 72 Stunden nach Induktion der Differenzierung und
nach Behandlung mit verschiedenen Konzentrationenen von Hemin (0, 25, 50 µM)
untersucht. Protein wurde aus den Zellen isoliert und nach LAT2-Expression mittels Western
Blots untersucht. Die K562-Zellen exprimierten kein LAT2-Protein und die Expression dieses
Proteins war bis zum 3. Tag und zu verschiedenen Zeitpunkten mit 25 und 50 µM Hemin
nicht hochreguliert (Abb. 34a-b).
Diese Daten zeigen, dass LAT2 in einer erythroleukämischen Zelllinie nicht auf
Protein-Ebene exprimiert wird und auch bei der erythrozytären Differenzierung nicht reguliert
wird.
4.4.8.- Expression von LAT2-Protein in der PMA-induzierten megakaryozytären
Differenzierung der K562-Zelllinie.
Abbildung 35.- Untersuchung des biologischen Effektes der durch PMA-induzierten megakaryozytären Differenzierung der K562-Zellen. A.- und B.- Zellwachstum und Viabilität wurden täglich durch Trypan-Blau-Färbung bestimmt. PMA, wenn in einer Konzentration von 10 nM und 100 nM gegeben, hatte einen stark hemmenden Effekt auf das Zellwachstum der K562-Zellen. Außerdem wurde die Viabilität der Zellen leicht beeinflusst. C.- Der Differenzierungsgrad der Zellen wurde am 3.Tag mittels FACS auf CD41b und CD61c Anitgene untersucht. Die mit PMA-behandelten Zellen zeigten eine sehr starke Positivität auf die Megakaryozytenmarker CD41b und CD61c. D.- Die Differenzierung der Zellen wurde am 3. Tag mit der May-Grünwald-Giemsa-Färbung nach Zytospin morphologisch untersucht.
K562 - PMA K562 + 50 µM PMA
K562+/- PMA (Day 3)
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
K562 K562 + 10 nM PMA K562 + 100 nMPMA
% p
ositi
ve c
ells
to m
egac
aryo
cytic
m
arke
rs
CD41PE CD61FITC
0,0
3,0
6,0
9,0
12,0
15,0
0 24 48 72
Time (hrs)
Cel
l con
cen
tratio
n (c
ells
x 1
0*5
/ml)
K562 untreated K562 + 10 nM PMA K562 + 100 nM PMA
0,0%
20,0%
40,0%
60,0%
80,0%
100,0%
0 24 48 72
Time (hrs)
Via
bili
ty
K562 untreated K562 +10 nM PMA K562 + 100 nM PMA
A.-
B.-
C.-
D.-

4.- Ergebnisse Seite - 109 -
Die K562-Zelllinie kann in die megakaryozytäre Reihe differenzieren, wenn sie mit
Phorbol-Ester (PMA) kultiviert wird (Tetteroo et al., 1984) und exprimiert nach der PMA-
Behandlung die megakaryozytären Antigene CD41b (Integrin alpha IIb) und CD61c (Integrin
beta III)
Das Zellwachstum und die Viabilität wurden täglich mit der Trypan-Blau-Färbung
bestimmt. Am 3. Tag gab es einen Unterschied zwischen den mit PMA-behandelten und
unbehandelten Zellen in der Zellwachstumskurve (12,2 +/- 1,4 x 105 Zellen/ml in den
unbehandelten Zellen vs 1,1 +/- 0,5 x 105 Zellen/ml in den mit 10 nM PMA-behandelten
Zellen, Zellwachstum-Inhibition von 91,0 %; vs 1,3 +/- 0,4 x 105 Zellen/ml in den mit 100 nM
PMA-behandelten Zellen, Zellwachstum Inhibition von 89,3 %) (Abb. 35a). Die Viabilität
der unbehandelten und der mit PMA-behandelten Zellen unterschied sich am 3. Tag
geringfügig (95,6 +/- 0,7 % in den unbehandelten vs 84,2 +/- 5,8 % in den mit 10 nM PMA-
behandelten Zellen vs 82,0 +/- 5,9 % in den mit 100 nM PMA-behandelten Zellen) (Abb.
35b).
Um die durch PMA-induzierte megakaryozytäre Differenzierung der K562-Zellen zu
quantifizieren, wurden die Zellen am 3. Tag auf die Megakaryozytärmarker CD41b und
CD61c mittels FACS untersucht. Die unbehandelten K562-Zellen waren zu 2,0 +/- 1,2 %
positiv, die mit 10 nM PMA-behandelten K562-Zellen zu 26,2 +/- 11,3 % positiv und die mit
100 nM PMA-behandelten K562-Zellen zu 37,9 +/- 9,6 % positiv für CD41b. Die
unbehandelten K562-Zellen waren zu 3,2 +/- 2,0 % positiv, die mit 10 nM PMA-behandelten
K562-Zellen zu 45,5 +/- 12,8 % positiv und die mit 100 nM PMA-behandelten K562 Zellen
zu 50,8 +/- 14,4 % positiv für CD61c. Mit diesen Daten konnte bestätigt werden, dass die
K562-Zellen sich durch PMA-Behandlung zu Megakaryozyten differenzieren lassen (Abb.
35c).
Nach der May-Grünwald-Giemsa-Färbung hatten die unbehandelten K562 Zellen eine
typische erythroleukämische Morphologie mit einem unregelmäßigen Zytoplasma, das
überwiegend basophil war. Der Kern war unregelmäßig und hatte eine lockere
Chromatinstruktur. Die mit PMA-differenzierten K562-Zellen hatten zum Teil eine
charakteristische Polyploidie. Das Zytoplasma war überwiegend azidophil und teilweise
waren Vakuolen vorhanden (Abb. 35d).
Die Expression des LAT2-Proteins wurde am 3. Tag nach der Behandlung mit 10 nM
und 100 nM PMA Konzentration und jeden Tag nach der Behandlung mit 100 nM PMA
mittels Western Blot untersucht (Abb. 36a-b).

4.- Ergebnisse Seite - 110 -
Die K562-Zelllinie hat keine oder eine niedrige Expression des LAT2-Proteins und die
Expression wurde weder mit Hemin (25 µM und 50 µM) (Abb. 34) in der erythrozytären
Differenzierung noch mit PMA (10 nM und 100 nM) (Abb. 36) in der megakaryozytären
Differenzierung der K562-Zellen gesteigert. Diese Daten wurden bei der Untersuchung des
gleichen Versuchs mit Hilfe von Affymetryx-Microarrays auf mRNA-Ebene bestätigt
(Ergebnisse der Doktorarbeit von cand. med. Lena Kasten, in Zusammenarbeit mit Dr.
Dietmar Pfeiffer der Abteilung I der Inneren Medizin, Uniklinikum Freiburg).
Die K562-Zelllinie hat nach der DSMZ (www.dsmz.de) den folgenden Karyotyp:
"human hypotriploid karyotype without sharp mode -61-68<3n>XX, -X, -3, +7, -13, -18,
+3mar, del(9)(p11/13), der(14)t(14;?)(p11;?), der(17)t(17;?)(p11/13;?),
der(?18)t(15;?18)(q21;?q12), del(X)(p22)". Bisher wurde in den K562-Zellen keine Deletion
des langen Arms von Chromosom 7 und damit des LAT2-Gens, 7q11.23, beschrieben.
Deshalb ist es anzunehmen, dass das LAT2-Protein (Western Blot Ergebnisse) und -
mRNA (Affymetrix Chips) in der erythroleukämischen Zelllinie K562 nicht exprimiert wird
und keine Regulation des LAT2-Gens in der erythrozytären und in der megakaryozytären
Differenzierung diese Zelllinie stattfindet.
Abbildung 36.- LAT2-Protein-Expression in der megakaryozytären Differenzierung der K562-Zelllinie durch PMA. A.- Die LAT2-Protein-Expression wurde alle 24 Stunden nach der Behandlung mit einer Konzentration von 10 nM und 100 nM PMA mittels Western Blot untersucht. Die U937-Zelllinie wurde als Positivkontrolle für die Expression von LAT2-Protein benutzt. Die K562-Zelllinie hatte keine Grundexpression des LAT2-Proteins und wird auch nicht während der mit PMA-induzierten megakaryozytären Differenzierung der K562 Zellen hochreguliert
A.- B.-
LAT2
ß-Actin
PMA (nM) 0 10 100
K562 (day 3)
U937
LAT2
ß-Actin
Time (hours) 0 24 48 72
PMA 100 nM - + + +
K562
U937
A.- B.-
LAT2
ß-Actin
PMA (nM) 0 10 100
K562 (day 3)
U937
LAT2
ß-Actin
PMA (nM) 0 10 100
K562 (day 3)
U937
LAT2
ß-Actin
Time (hours) 0 24 48 72
PMA 100 nM - + + +
K562
U937

4.- Ergebnisse Seite - 111 -
4.5.- Expression von LAT2-Protein in humanen primären Zellen.
Um die Daten unserer Myeloiddifferenzierungsversuche in primären Zellen zu
validieren, wurden verschiedene primären Blutzellen (Granulozyten, Lymphozyten und
Monozyten) und Alveolar-Makrophagen (als reife Form von Monozyten) isoliert und die
LAT2-Protein Expression wurde mittels Western Blot untersucht.
In Granulozyten und Blutlymphozyten war die Expression des LAT2-Proteins
abwesend und in Monozyten war die Expression des LAT2-Proteins hoch (Abb. 37). Die
Expression des LAT2-Proteins in den alveolären Makrophagen war stärker als in den
Monozyten des gleichen Spenders. Die stärkere Expression von LAT2-Protein in den
alveolären Makrophagen zusammen mit den Daten der monozytären-makrophagen-
Differenzierung von HL60-Zellen und monozytären Differenzierung von U937-Zellen, lässt
eine Funktion vom LAT2 in diesen Zellen auf die Differenzierungsfähigkeit der Monozyten
vermuten.
Abbildung 37.- LAT2-Protein-Expression in primären Blutzellen und alveolären Makrophagen. Nach der Isolation von Protein wurde die LAT2-Protein-Expression mittels Western Blot untersucht. Die U937-Zelllinie wurde als Positivkontrolle für die Expression von LAT2-Protein benutzt. Die Monozyten exprimierten das LAT2-Protein. Die alveolären Makrophagen hatten eine stärkere Expression als die Monozyten. Die Blutlymphozyten und die Granulozyten zeigten keine Expression des LAT2-Proteins.
Lympho. Mono. Gran. U937Mono. BAL
LAT2
ß-Actin
Patient Gesunde Spender
Lympho. Mono. Gran. U937Mono. BAL
LAT2
ß-Actin
Patient Gesunde Spender

4.- Ergebnisse Seite - 112 -
4.6.- Regulation von LAT2-Protein in der granulozytären und monozytären-
dendritischen Differenzierung der CD34 + Zellen durch Zytokine.
Nach dem Auftauen wurden die CD34+ Zellen in serumfreiem Medium kultiviert, das
mit den Zytokinen FLT3-Ligand, SCF, IL-3 und IL-6 versetzt wurde.
Die granulozytäre Differenzierung der CD34 +-Zellen wurde mit 50 Einheiten/mL G-CSF
durchgeführt und die monozytär-dendritische Differenzierung wurde mit 50 Einheiten/ml
GM-CSF + 50 Einheiten/ml IL-4 induziert. Die Zellen wurden zu mehreren Zeitpunkten
mittels FACS auf bestimmte Differenzierungsantigene untersucht. Zytospins wurden nach
May-Grünwald-Giemsa gefärbt, um morphologische Veränderung der Zellen nach der
monozytären bzw. granuloytären Differenzierung nachzuweisen. Es wurde Protein für
Western Blots isoliert und die Expression des LAT2-Proteins untersucht.
Die Expression vom LAT2-Protein war in beiden Induktionen am 3. und 7. Tag
transient hochreguliert und am 14. Tag herunterreguliert (s. Abbildung 38). Die Expression
Abbildung 38.- LAT2-Protein-Expression in der myeloischen Differenzierung von normalen CD34+ Zellen. A. Die isolierten normalen CD34+ wurden kultiviert und mit den Zytokinen G-CSF und GM-CSF + IL-4 jeweils granulozytär oder monozytär-DC differenziert. U937 wurde als Positivkontrolle für die Expression vom LAT2-Protein benutzt. Eine transient Hochregulation des LAT2-Proteins wurde am 3. und 7. Tag in beiden Differenzierungsmodellen nachgewiesen. Am Tag 14 war die Expression des LAT2-Proteins in den G-CSF behandelten Zellen niedriger und in den GM-CSF+ IL-4 behandelten Zellen stärker ausgeprägt. B. Cytospins wurden zu verschiedenen Zeitpunkten gemacht und mit May-Grünwald-Giemsa gefärbt, um morphologische Veränderungen der Zellen nachzuweisen
A
Day 0 3 7 14 3 7 14
CD34+ cells
LAT2
ß-Actin
Cytokine + G-CSF + GMCSFCocktail and IL4
U937Day 0 3 7 14 3 7 14
CD34+ cells
LAT2
ß-Actin
Cytokine + G-CSF + GMCSFCocktail and IL4
U937
B
Day 0 Day 7 + G-CSF Day 14 + G-CSF Day 7 + GM-CSFand IL4
Day 14 + GM-CSFand IL4

4.- Ergebnisse Seite - 113 -
des LAT2-Proteins war am 14. Tag in den durch G-CSF differenzierten Zellen niedriger als
in den Zellen, die durch GM-CSF und IL-4 differenziert wurden. Die Zellen, die ohne G-CSF,
GM-CSF und IL-4 behandelt wurden, hatten am Tag 14 eine höhere Expression als die mit G-
CSF behandelten Zellen und niedriger als die mit GM-CSF und IL-4 behandelten Zellen
(Daten nicht gezeigt). Die Hochregulation vom LAT2-Protein in der ex vivo Differenzierung
der CD34+ Zellen korreliert und bestätigt unsere Daten der monozytären bzw. granulozytären
Differenzierung der NB4-, HL60- und U937-Zelllinien.

5.- Diskussion Seite - 114 -
5.- Diskussion
Das Fusionsprotein AML1/ETO
Der molekulare Pathomechanismus des chimären Fusionsproteins AML1/ETO beruht auf der
Rekrutierung von HDACs an die Promotoren seiner Zielgene (Amann et al., 2001; Gelmetti et
al., 1998). Diese modifizieren die Acetylierung der Lysin- und Arginin-Reste von Histonen,
wodurch des Chromatins in eine geschlossene Struktur umgewandelt wird. So wird die
Transkription der AML1/ETO-Zielgene gehemmt, die überwiegend eine wichtige Rolle
während der Hämatopoese spielen und für verschiedene Schritte der Reifung
hämatopoetischer Zellen notwendig sind. Während der Durchführung dieser Doktorarbeit
wurde ein Artikel veröffenlicht, in dem ein Komplex aus AML1/ETO mit DNMT1 auf dem
Promotor von IL-3-Gen in AML1/ETO-positiven Zelllinien und primären AML-Blasten
identifiziert wurde (Liu et al., 2005).
Epigenetische Regulation vom LAT2-Gen durch AML1/ETO
Das LAT2- (NTAL/LAB/WBSCR5) Gen wurde als ein Zielgen in der AG.von Prof. Lübbert
in AML1/ETO-induzierbaren Zellen identifiziert (Fliegauf et al., 2004). Anderen Autoren
haben dieses Ergebnis nach Untersuchung mit Affymetrix Arrays des Transkriptoms der
Blasten von einer groβen Patientenzahl bestätigt (Fliegauf et al., 2004; Ross et al., 2004; Valk
et al., 2004b). In dieser Arbeit wurden funktionelle Untersuchungen an LAT2 durchgeführt,
um seine Rolle während der Myelopoese und während der Entstehung der AML aufzuklären.
Zuerst wurde die Repression von LAT2 durch AML1/ETO auf Protein-Ebene in 9/14/18-
Zellen, AML1/ETO-positiven Zelllinien und in Knochenmarksproben von AML-Patienten
untersucht. In Anwesenheit von AML1/ETO ist die LAT2-Expression sowohl in vitro
(AML1/ETO-induzierbare Zellen und Kasumi-1) als auch in vivo (Blasten von AML-
Patienten) stark reprimiert. Zusätzlich ist die LAT2-Protein-Expression in
Knochenmarksproben von Patienten mit Aberrationen von Chromosom 7 (n=3) und in
Patientenproben mit t(15;17) (n=4) nicht exprimiert. Das könnte auf eine Funktion von LAT2
als Tumorsuppressor-Gen in der Entstehung der AML hindeuten. Da das LAT2-Gen nur in
einer Kopie bei den Patienten mit Aberrationen von Chromosom 7 (chomosomale
Lokalisation 7q11.23) vorliegt, könnte dieses durch epigenetische Depression oder nonsense
Mutationen nicht exprimiert werden und dadurch die Entstehung einer AML begünstigen. Um
diese Fragestellung zu beantworten, sollen Mikrodeletionen im Bereich des LAT2-Gens
mittels SNPs-Arrays in den KM-Proben von diesen Patienten untersucht werden. LAT2 war
jedoch in 14 von 15 KM-Proben von Patienten mit Normalkaryotyp vorhanden. Das zeigte,

5.- Diskussion Seite - 115 -
dass LAT2-Repression mit verschiedenen chromosomalen und genetischen Aberrationen
korreliert.
Nach der FAB-Klassifikation haben AML-Blasten mit einer myeloblastären (FAB M2) und
promyelozytären Morphologie (FAB M3) keine oder niedrige LAT2-Expression und die
Patienten mit einer frühe myeloblastäre (FAB M1) und myelomonozytären Morphologie
(FAB M4) eine normale oder hohe LAT2-Expression. Das bestätigt wiederum die Daten der
myeloischen Differenzierungsversuche, bei denen die LAT2-Expression während der
granulozytären Differenzierung herunterreguliert aber während der monozytären
Differenzierung hochreguliert wird.
Die LAT2-Expression wurde in KM-Patientenproben bei der Diagnose und bei der Komplett-
Remissionskontrollen von 2 Patienten mit inv(16) untersucht. Mittels Western Blot wurde das
LAT2-Protein nur in den Knochenmarksproben bei der Diagnose detektiert. Dies zeigt, dass
LAT2-Expression nur in einem pathologischen KM zu finden ist und die LAT2-Repression
sehr wahrscheinlich in Patienten mit t(8;21), t(15;17) und Aberrationen von Choromosom 7
aus den AML-Blasten entsteht. Außerdem wurde in den klinischen Daten der Patienten kein
Unterschied festgestellt.
In Kollaboration mit PD. Dr. O. Heidenreich (Universität von Tübingen), der ein Knock-
Down-System von AML1/ETO mittels siRNAs in der Kasumi-1-Zelllinie entwickelt hat
(Heidenreich et al., 2003), wurde die Expression von LAT2-mRNA untersucht. Nach der
ersten und zweiten Elektroporation mit den siRNAs gegen AML1/ETO wurde eine 4,5- und
9,5-fache Hochregulation von LAT2-mRNA mit "real-time" RT-PCR nachgewiesen. Dieses
Ergebnis deutet an, dass LAT2 auch in diesem System ein transkriptionelles AML1/ETO-
Zielgen ist und dass LAT2-mRNA nach dem Knock-Down von AML1/ETO in Kasumi-1-
Zellen re-exprimiert werden kann. Mit diesem AML1/ETO Knock-down System wurden
andere transkriptionelle AML1/ETO-Zielgene nachgewiesen, die Funktionen bei der
myeloischen Differenzierung haben, die ein antiproliferatives Potential haben oder die mit
einer schlechteren Prognosen korreliert sind (Dunne et al., 2006). Interessanterweise wurde
bei 3 dieser Gene (BPI, IGFBP7 und SLA) die Bindung vom AML1/ETO am Promotor mit
Chromatin Immunopräzipitation (ChIP) Assays bestätigt. Es wäre sinvoll zu untersuchen, ob
die Bindung von AML1/ETO am LAT2-Promotor in den 9/14/18-Zellen (AML1/ETO-
induzierbaren Zellen) und Kasumi-1-Zellen mit ChIP Assay detektiert werden kann.
Die nächste Fragestellung war, ob der AML1/ETO-Effekt auf die Expression von LAT2
pharmakologisch gehemmt werden kann. Da AML1/ETO HDACs und DNMTs zu den
Promotoren der Zielgene rekrutiert, wurde untersucht, ob LAT2-mRNA nach der Behandlung

5.- Diskussion Seite - 116 -
mit HDAC- und DNMT-Inhibitoren in Kasumi-1 Zellen re-exprimiert wird. Unterschiedliche
Effekte wurden bei der Expression von LAT2 nach der Behandlung mit verschiedenen
HDAC-Inhibitoren beobachtet. Das Benzamid-Derivat MS-275 (SDX-275®) hatte den
stärksten Effekt auf die Re-expression der LAT2-mRNA bei equitoxischen Konzentrationen
der Substanzen. Equitoxische Konzentrationen wurden in dieser Arbeit definiert, als die
Konzentration, bei der ein Effekt auf Histon-Acetylierung mit Western Blot detektiert wurde
und die Zellen über 80% Viablilität hatten. Dem unterschiedlichen Effekt von HDAC-
Inhibitoren könnte zugrunde liegen, dass jeder HDAC-Inhibitor nur bestimmte HDACs oder
eine HDAC-Klasse bindet und inhibiert. MS-275 blockiert HDAC-Klasse I (HDAC1-3) mit
einer ähnlichen IC50 Konzentration wie TSA, kann aber im Gegenteil zu TSA HDAC 8 nicht
inhibieren (Hu et al., 2003). Andere Proteine (wie Transkriptionsfaktoren) können außerdem
durch HDAC-Inhibitoren abgezielt werden und ihre Funktion bzw. Expression beeinflusst
werden (Lin et al., 2006)
In einem grundlegenden Artikel wurde gezeigt, dass HDAC-Inhibitoren (z. B. TSA) in
Kombination mit DNMTs-Inhibitoren (z. B. Decitabine) einen synergistischen Effekt auf die
Re-expression reprimierter Gene in Tumorzellen haben (Cameron et al., 1999). In dieser
Arbeit wurde der DNMT-Inhibitor Decitabine in Kombination mit dem HDAC-Inhibitor MS-
275 benutzt, um einen synergistischen Effekt auf die Re-expression von LAT2 in Kasumi-1-
Zellen zu erzielen. MS-275 wurde für diese Fragestellung ausgewählt, weil er der effektivste
HDAC-Inhibitor auf die Re-expression von LAT2 in dem vorigen Versuchen war. MS-275
(100 nM) in Kombination mit einer 3-tägigen Decitabine Behandlung (25 oder 50 nM) hatte
einen synergistischen Effekt auf die LAT2-mRNA Re-expression. Dieses Ergebnis zeigte,
dass eine Kombination von HDAC-Inhibitoren und DNMT-Inhibitoren bei der Behandlung
von AML-Patienten eingesetzt werden könnte, bei denen die AML durch eine aberrante
Rekrutierung von HDACs und/oder DNMTs auf Zielgene verursacht wird. Es wurde in dieser
Arbeit gezeigt, dass Tumorsuppressor-Gene (wie z. B. MLH1, TIMP-3, p15 und p16), deren
Promotoren hypermethyliert sind, in der Kombination mit einem HDAC-Inhibitor und einem
DNMT-Inhibitor in Tumorzellen re-exprimiert werden können (Cameron et al., 1999). Nach
einer Expression-Analyse mit Affymetrix Arrays wurden mehrere Gene gefunden, deren
Expression durch die Kombination von DNMT- und HDAC-Inhibitor synergistisch in
Kolonkarzinom-Zelllinien re-exprimiert werden konnten. Allerdings wurde der Promotor von
der Hälfte der Gene als hypermethyliert gefunden (Suzuki et al., 2002). Es wurde
veröffentlicht, dass nur 50 % der Gene, deren Expression durch Decitabine induziert wurde,
eine putative CpG-Insel hatten. Von diesen Genen wurde nur Demethylierung am Promotor

5.- Diskussion Seite - 117 -
des Myeloperoxidase (MPO) Gens nach Decitabine-Behandlung nachgewiesen (Schmelz et
al., 2005). Nach Anfertigung dieser Doktorarbeit wurde der Methylierungsstatus des LAT2-
Promotors von unserer Arbeitsgruppe mittels Bisulfit-Sequenzierung in Kasumi-1-Zellen
untersucht. Dabei wurde herausgefunden, dass über 95 % der CpG-Dinukleotide nicht-
methyliert waren. Zusammenfassend deuten diese Daten darauf hin, dass Decitabine die
Expression von Genen, unter diesen das LAT2-Gen, durch einen anderen Mechanismus als
DNA-Methylieurung beeinflusst.
Es wurde untersucht, ob die durch AML1/ETO-vermittelte Repression von LAT2 mit
epigenetisch aktiven Substanzen aufgehoben werden kann und ob der LAT2-Promotor
epigenetisch beeinflussbar ist. Dafür wurden Chromatin-Immunopräzipition (ChIP) mit
Antikörpern gegen die aktivierenden Histon-Markierung Acetyl-Histon-H3 (AcH3), Acetyl-
Histon-H4 (AcH4), Acetyl-Histon-H3-Lysin-9 (AcH3K9) und Tri-Methyl-Histon-H3-Lysin-4
(3meH3K4) durchgeführt. Anschließend wurde die isolierte-DNA mit einer "real-time"-DNA-
PCR quantifiziert. 3meH3K4 spielt eine sehr wichtige Rolle als Bindeglied zwischen Histon-
Acetylierung und -Methylierung, da die Histon-Methyltransferase MLL4 nach der
Acetylierung von Histon-H3 durch HDAC-Inhibitoren diesen spezifischen Lysin-Rest von
Histone-H3 methyliert (Nightingale et al., 2007). Wir haben nachgewiesen, dass AcH3,
AcH3K9 und 3meH3K4, aber nicht AcH4, nach der Behandlung mit MS-275 auf dem LAT2-
Promotor in Kasumi-1-Zellen induziert wurden. Da p21WAF-mRNA durch HDAC-Inhibitoren
hochreguliert wurde (Richon et al., 2000) und als Positivkontrolle dieser Art von Behandlung
von verschiedenen Autoren benutzt wird, haben wir mittels ChIP die gleichen aktivierenden
Histon-Markierungen auf diesem Promotor untersucht. Ähnliche Ergebnisse wurden
nachgewiesen. Acetylierung von Histon H4 wurde in diesen Versuchen nicht induziert,
obwohl die Gesamt-Acetylierung von Histon H4 mittels Western Blot als hochreguliert
gefunden wurde (Ergebnisse nicht gezeigt). Es könnte sein, dass die Acetylierung von
Histon-H4 für die Expression von LAT2 und p21WAF nicht notwendig ist, der Antikörper für
Histon H4 nicht für ChIP, jedoch für Western Blot geeignet ist (obwohl Unterschiede
zwischen den Kasumi-1 und U937 Zellen in AcH4 gefunden wurden) oder dass der HDAC-
Inhibitor MS-275 die Acetylierung von Histon H4 auf diesen Promotoren nicht induziert.
Eine weitere Fragestellung war, ob AML1/ETO die untersuchten aktivierenden Histon-
Markierungen beeinflussen kann. Dafür wurden die Histon-Markierungen des LAT2-
Promotors von U937- und Kasumi-1-Zellen mittels ChIP verglichen. Deutliche Unterschiede
wurden in allen untersuchten Histon-Markierungen (auch in der Acetylierung von Histon H4)
gefunden. Um die Fragestellung zu beantworten, ob diese Unterschiede von AML1/ETO

5.- Diskussion Seite - 118 -
abhängig sind, wurde während eines "Time-Course"-Versuchs nach der Induktion von
AML1/ETO die Acetylierung von Histon H3 und Histon H4 mittels ChIP in den 9/14/18-
Zellen untersucht. Es konnte nachgewiesen werden, dass die Acetylierung von Histon H3
nach 24 Stunden, nicht aber die von Histon H4 (Ergebnisse nicht gezeigt) maximal
herunterreguliert wurde. Allerdings ist dieser Effekt, wie auch die Repression von LAT2 auf
mRNA- und Protein-Ebene, transient. Diese Daten deuten darauf hin, dass AML1/ETO die
Acetylierung vom LAT2-Promotor möglicherweise durch die Rekrutierung von HDACs
beeinflussen kann. Daraus könnte man schließen, dass der LAT2-Promotor durch HDAC-
Inhibitoren epigenetisch modulierbar ist und dass die AML1/ETO-vermittelten
Veränderungen auf den Promotoren seiner Zielgene pharmakologisch aufgehoben werden.
Substanzen mit HDAC-Inhibitorische Aktivität könnten somit möglicherweise in der
Behandlung von AML1/ETO-positiven Patienten eingesetzt werden.
Ein direkter Effekt von AML1/ETO auf dem LAT2-Promotor kann nur durch ChIP von
AML1/ETO untersucht werden, aber diese Experimente wurden wegen fehlender Spezifität
der kommerziellen verfügbaren Antikörper auf dem chimären Fusionsprotein nicht
durchgeführt (Anti-AML1- sowie Anti-ETO-Antikörper erkennen jeweils das endogene,
wildtyp-AML1 bzw. -ETO-Proteine). Es gibt verschiedene Methoden, um AML1/ETO
spezifisch zu immunpräzipitieren. Eine davon ist die Doppel-ChIP-Methode, in der
sequenziell ein ChIP mit einem primären Antikörper und, nach Spaltung der Antikörper mit
DTT-Lösung, einem zweiten ChIP mit einem zweiten Antikörper durchgeführt wird.
Screening von AML1/ETO Zielgenen auf Promoter- DNA-
Methylierungsveränderungen.
In dieser Arbeit wurde untersucht, ob AML1/ETO, als epigenetischer Modifikator, zusätzlich
DNMTs an die Promotoren seiner Zielgene rekrutiert. Ein Ziel dieser Arbeit war
AML1/ETO-Zielgene auf DNA-Methylierungs-Ebene zu identifizieren.
Dafür wurde eine globale Analyse (RLGS-Methode) benutzt, um Veränderungen der DNA-
Methylierung genomweit nach der konditionellen Expression des AML1/ETO-Fusionsgens in
den 9/14/18-Zellen zu untersuchen. Diese Untersuchung ist nach unserer Kenntnis die erste,
die den Einfluss von AML1/ETO mit einer genomweiten Analyse auf die DNA-Methylierung
untersucht hat. Es konnte nachgewiesen werden, dass die konditionale Expression von
AML1/ETO die DNA-Methylierung verschiedener Sequenzen bzw. Gene, beeinflusst.
Allerdings waren diese Methylierungsveränderungen unter unseren experimentellen
Bedingungen nicht robust. Mögliche Gründe dafür waren, dass a) die 9/14/18-Zellen ein

5.- Diskussion Seite - 119 -
nicht-stabiles Epigenom gezeigt haben, da die Zellen einen unterschiedlichen
Methylierungsmuster nach späteren Passagen hatten; b) subtile Methylierungsänderungen als
Reaktion auf einen einzelnen Transkriptionfaktor durch die RLGS-Methode möglicherweise
nicht detektierbar sind, deshalb sind wohl sensitivere Methoden für DNA-Methylierung bei
weiteren Untersuchungen notwendig; c) die konditionale ektope Expression des AML1/ETO-
Proteins in der 9/14/18-Zelllinie einen randomisierten Effekt auf DNA-Methylierung haben
könnte. Da die Auflösung der RLGS-Methode bei etwa 2000 Sequenzen begrenzt ist, könnten
Methylierungsveränderungen nach der Expression von AML1/ETO auch auβerhalb dieser
Grenze stattfinden. Trotz der genannten Schwierigkeiten, konnten wir mit einer sehr
spezifischen Methode (Southern Blot nach Restriktionsverdau mit methylsensitiven
Enzymen) in zwei unabhängigen AML1/ETO-Induktionen der 9/14/18-Zellen eine
Methylierungsveränderung innerhalb der CpG-Insel des Dematin-Promotors identifizieren.
Dematin ist ein Membran-Protein der Erythrozyten, das als Bindeglied zwischen dem
Aktinzytoskeletton und der Membran funktioniert. Damit konnte gezeigt werden, dass dieses
mögliche AML1/ETO-Zielgen eine Funktion in hämatopoetischen Zellen hat (in diesem Fall,
bei den Erythrozyten) und dass die DNA-Methylierung möglicherweise eine Rolle in der
Pathogenese des AML1/ETO-Fusionsproteins spielen könnte. Wir vermuteten allerdings, dass
Dematin keine Funktion in Leukämie-Zellen oder während der Leukemogenese hat, da seine
Expression und Funktion bisher nur bei Erythrozyten beschrieben wurde.
Regulation vom LAT2-Gen während der myeloischen Differenzierung.
Die AML1- und AML1/ETO-Zielgene haben überwiegend eine Funktion in der
Hämatopoese. Der Transkriptionsfaktor AML1 schaltet während der verschiedenen
Reifungsstufen der hämatopoetischen Zellen die Transkription der Zielgene ein- oder aus,
damit diese ihre Funktion während der Differenzierung der Zellen und in den verschiedenen
Vorstufen der hämatopoetischen Zellen kontrolliert ausüben können. Da die Funktion von
LAT2 noch nicht in der Hämatopoese untersucht wurde, wurden in dieser Arbeit
Expressionsuntersuchungen durchgeführt. Es wurde die LAT2-Expression während der
myeloische Differenzierung von Zelllinien und in normalen CD34+-Zellen, in verschiedenen
primären Zellen und während der megakaryozytären und erythrozytären Differenzierung von
K562-Zellen untersucht. LAT2-Expression wurde während der ATRA-induzierten
granulozytären Differenzierung von NB4-, U937- und HL60-Zellen herunterreguliert..
Auβerdem wurde LAT2 während der PMA-induzierten monozytären Differenzierung der
HL60 Zellen hochreguliert nachgewiesen. In dem Differenzierungs-Modell von normalen

5.- Diskussion Seite - 120 -
CD34+ Zellen wurde LAT2-Protein während der granulozytären Differenzierung mittels G-
CSF herunterreguliert und während der monozytären/DC Differenzierung mittels GM-CSF
plus IL-4 hochreguliert im Vergleich zu den unbehanelten CD34+-Zellen. Eine starke
transiente Hochregulation von LAT2 wurde am Tag 7 in beiden Differenzierungen (mittels G-
CSF und GM-CSF/IL4) gefunden. In primären Zellen war das LAT2-Protein in Granulozyten
abwesend, aber sehr stark in Monozyten exprimiert. Alveolar-Makrophagen exprimierten
LAT2-Protein sogar stärker als Monozyten des gleichen Patienten.
Im Gegensatz dazu wurde das LAT2-Protein in der erythroleukämischen Zelllinie K562 nicht
exprimiert und während der mit PMA-induzierten megakaryozytären und der mit Hemin-
induzierten erythrozytären Differenzierung der Zellen nicht inuiert. Deshalb nehmen wir an,
dass LAT2 keine Rolle in der Erythropoese oder Megakaryopoese spielt. Diese Daten wurden
indirekt bestätigt, in Untersuchungen der Expression von mehreren Transmembran-Adaptor-
Molekülen, darunter auch LAT2 Protein, in verschiedenen Geweben von Patienten mit
hämatologischen Erkrankungen (Tedoldi et al., 2006). Die Expression von LAT2 wurde
sowohl in der erythrozytären Reihe als auch in der megakaryozytären Reihe mit
Immunohistochemie nicht detektiert.
Zusammenfassend deuten diese Ergebnisse daraufhin, dass LAT2 eine Funktion während der
monozytären Differenzierung oder in Monozyten, nicht aber während der granulozytären
Differenzierung oder in Granulozyten hat. Sehr wahrscheinlich spielt LAT2 auch keine Rolle
während der Megakaryopoese und Erythropoese, da seine Expression während der
induzierten K562-Differenzierung mit PMA und Hemin nicht reguliert wurde.
Untersuchungen in Monozyten/Makrophagen von LAT2-Knock-out-Mäusen sind notwendig,
um Defekte der Differenzierung oder Funktion dieser Zellpopulation festzustellen. Zhu und
Mitarbeiter (Zhu et al., 2006) sind zu der gleichen Schlussfolgerung gekommen, da die
Autoimmune Krankheit der LAT2-Knock-Out-Mäuse nicht vollständig durch einen Defekt
der T-Zell-Funktion aufgelöst werden könnte. Sie haben darauf hingewiesen, dass weitere
Untersuchungen in Zellpopulationen mit höherer LAT2-Expression, wie z. B. in
Makrophagen, notwendig sind, um den Phänotyp dieser Mäuse weiter aufzuklären. LAT2-
Protein kann durch verschiedene Rezeptoren phosphoryliert und aktiviert werden, wie z. B.
FCεR I und c-Kit in Mastzellen (Tkaczyk et al., 2004), B-Zell-Rezeptor in B-Zellen (Janssen
et al., 2003) und T-Zell-Rezeptor (Zhu et al., 2006) in T-Zellen. Wir vermuten, dass LAT2
während der monozytären Differenzierung durch einen myeloischen Rezeptor, wie z.B. M-
CSF-Rezeptor, aktiviert werden könnte. Ko-Immunopräzipitation von Proteinen und
Phosphorylierung-Assays wären für diese Fragestellung geeignet. Falls LAT2 eine Rolle

5.- Diskussion Seite - 121 -
während der monozytären Differenzierung spielen würde, könnte der Differenzierungs-Block
bei der Monozytären-Reifung von AML1/ETO positiven Zellen (Okuda et al., 1998) teilweise
durch die fehlende LAT2-Expression erklärt werden.

6.- Zusammenfassung Seite - 122 -
6 .- Zusammenfassung
Die Translokation t(8;21) ist eine der häufigsten chromosomalen Aberrationen bei der
akuten myeloischen Leukämie (AML) des Erwachsenen. Sie führt zur Produktion des
chimären Transkriptionsfaktors AML1/ETO, in dem der Transkriptionsfaktor AML1
(RUNX1) auf Chromosom 21 und das ETO-Gen auf Chromosom 8 fusioniert werden. Die
Arbeitsgruppe von Prof. Lübbert hat ein Ecdyson-induzierbares Expressions-System des
Fusionsproteins AML1/ETO in der U937-Zelllinie entwickelt. Mit diesem können auch
Zielgene dieses Proteins untersucht werden, um ein besseres Verständnis dieser Form der
AML zu erhalten. Wir haben funktionelle Untersuchungen an einem neuen identifizierten
AML1/ETO Zielgen, LAT2 (WBSCR5/NTAL/LAB), durchgeführt. LAT2-Expression wurde
mit Northern und Western Blot in AML1/ETO-positiven und -negativen Zelllinien untersucht.
In Blasten von AML-Patienten mit AML1/ETO war das LAT2-Protein mittels Western Blot
nicht detektierbar. Damit konnten wir nachweisen, dass LAT2 in AML1/ETO-positiven
Zelllinien und AML-Blasten reprimiert ist. Die LAT2-mRNA-Expression wurde nach dem
Knock-Down vom AML1/ETO mittels siRNAs in den Kasumi-1 Zellen untersucht. In diesem
System wurde eine Hochregulation der LAT2-mRNA-Expression nach Knock-Down von
AML1/ETO festgestellt, was als starke Evidenz für eine direkte Repression zu werten ist.
AML1/ETO-vermittelte-Repression von LAT2 wurde mittels unterschiedlicher Histon-
Deacetylasen (HDAC)-Inhibitoren und dem DNA-Methyltransferasen (DNMT)-Inhibitor
Decitabine antagonisiert. Der HDAC-Inhibitor MS275 induziert die LAT2-mRNA am
stärksten bei equitoxischen Konzentrationen und hat einen synergistischen Effekt auf die
Expression von LAT2 in Kombination mit Decitabine in Kasumi-1-Zellen. Die Re-expression
von LAT2 durch MS-275 korrelierte außerdem mit der Induktion der Histon-Modifikationen
AcH3, AcH3K9 und 3meH3K4 (mittels ChIP-Assay auf dem LAT2-Promotor in den Kasumi-
1-Zellen gezeigt). Die LAT2-Expression wurde während der myeloischen Differenzierung
untersucht. Während LAT2 während der granulozytären Differenzierung von myeloischen
Zelllinien und normalen CD34+ Vorläufer-Zellen herrunterreguliert ist, ist es in der
monozytären Differenzierung hochreguliert. Zusammenfassend lassen diese Daten eine
Funktion von LAT2 bei der monozytären Differenzierung vermuten. Die Repression von
LAT2 in Leukämien mit AML1/ETO-Expression ist mit einem Reifungsblock assoziert, der
durch epigenetische Therapie teilweise aufgehoben werden kann.

7.-Danksagung Seite - 123 -
7.- Danksagung
Diese Arbeit wurde am Universitätsklinikum Freiburg in der Abteilung Innere
Medizin I (Schwerpunkt Hämatologie/Onkologie), Ärztlicher Direktor Prof. Dr. Roland
Mertelsmann, in der Arbeitsgruppe von Prof. Dr. Lübbert angefertigt.
Besonders bedanken möchte ich mich bei meinem Betreuer, Herrn Prof. Dr. Michael
Lübbert, der mir die ausgezeichnete Chance anbot, meine Doktorarbeit am
Universitätsklinikum Freiburg in seiner Arbeitsgruppe durchzuführen. Durch die Hilfe von
Herrn Prof. Dr. Lübbert wurde ich in die Methodik und in das selbständige Arbeiten in der
Forschung eingeführt.
Ebenso möchte ich mich bei Herrn Prof. Dr. Roland Mertelsmann bedanken, der mich
als Doktorand in seiner Abteilung aufgenommen und kontinuierliches Interesse und
Unterstützung für meine Arbeit gezeigt hat.
Frau Prof. Dr. C. Niemeyer danke ich herzlich für das Übernehmen des Zweit-
gutachtens dieser Doktorarbeit.
Mein Dank richtet sich auch an Prof. Dr. Florian Otto, PD Dr. Ralph Wäsch und Dr.
Michael Schwabe für zahlreiche Diskussionen im Bereich der Molekularbiologie und
Hämatologie. In diesem Zusammenhang möchte ich mich bei Prof. Christoph Plass und Dr.
Björn Hackanson für ihre Hilfsbereitschaft und Kooperation mit der RLGS-Analyse und
Auswertung der Daten bedanken. PD Dr. Olaf Heidenreich danke ich für die Bereitstellung
von den Proben des AML1/ETO Knock-Down-Systems mittels siRNAs in den Kasumi-1
Zellen. Dr. Rainer Claus, Dr. Björn Hackanson, Dr. Tobias Berg, Dr. Claudia Müller-Schmah
und Dr. Bernhard Kleine danke ich für die sprachlichen Korrekturen am Manuskript.
Allen Mitarbeitern von Laboratorien Nothnagel und der Abteilung I der Inneren
Medizin danke ich für die angenehme Arbeitsatmosphäre und die Hilfe, die sie mir während
der Durchführung meiner Doktorarbeit geleistet haben. Insbesonders danke ich den
Mitarbeitern meiner Arbeitsgruppe, die mich während dieser Zeit innerhalb und außerhalb des
Labors unterstützt haben.
Meiner Freundin Corina Waldkircher, meiner Familie und meinen Freunden möchte
ich ganz herzlich danken, weil diese Arbeit ohne ihre Unterstützung nicht realisierbar
gewesen wäre.
Für die finanzielle Unterstützung danke ich dem Deutschen Verein zur Förderung der
Leukämie- und Tumorforschung im Jahr 2004-2005 und dem DAAD und der Fundación La
Caixa im Jahr 2005-2007.

8.- Bibliographie Seite - 124 -
8.- Bibliographie Alcalay M, Meani N, Gelmetti V, Fantozzi A, Fagioli M, Orleth A, Riganelli D, Sebastiani C, Cappelli E,
Casciari C, Sciurpi MT, Mariano AR, Minardi SP, Luzi L, Muller H, Di Fiore PP, Frosina G, Pelicci PG. 2003. Acute myeloid leukemia fusion proteins deregulate genes involved in stem cell maintenance and DNA repair. J Clin Invest 112:1751-1761.
Alcalay M, Orleth A, Sebastiani C, Meani N, Chiaradonna F, Casciari C, Sciurpi MT, Gelmetti V, Riganelli D, Minucci S, Fagioli M, Pelicci PG. 2001. Common themes in the pathogenesis of acute myeloid leukemia. Oncogene 20:5680-5694.
Amann JM, Nip J, Strom DK, Lutterbach B, Harada H, Lenny N, Downing JR, Meyers S, Hiebert SW. 2001. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol 21:6470-6483.
Andersson LC, Jokinen M, Gahmberg CG. 1979. Induction of erythroid differentiation in the human leukaemia cell line K562. Nature 278:364-365.
Arthur DC, Berger R, Golomb HM, Swansbury GJ, Reeves BR, Alimena G, Van Den Berghe H, Bloomfield CD, de la Chapelle A, Dewald GW, et al. 1989. The clinical significance of karyotype in acute myelogenous leukemia. Cancer Genet Cytogenet 40:203-216.
Asou H, Tashiro S, Hamamoto K, Otsuji A, Kita K, Kamada N. 1991. Establishment of a human acute myeloid leukemia cell line (Kasumi-1) with 8;21 chromosome translocation. Blood 77:2031-2036.
Bacher U, Haferlach T, Schoch C, Kern W, Schnittger S. 2006. Implications of NRAS mutations in AML: a study of 2502 patients. Blood 107:3847-3853.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. 1976. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol 33:451-458.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. 1980. A variant form of hypergranular promyelocytic leukaemia (M3). Br J Haematol 44:169-170.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. 1985. Criteria for the diagnosis of acute leukemia of megakaryocyte lineage (M7). A report of the French-American-British Cooperative Group. Ann Intern Med 103:460-462.
Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. 1991. Proposal for the recognition of minimally differentiated acute myeloid leukaemia (AML-MO). Br J Haematol 78:325-329.
Berg T, Guo Y, Abdelkarim M, Fliegauf M, Lübbert M. 2007. Reversal of p15/INK4b hypermethylation in AML1/ETO-positive and -negative myeloid leukemia cell lines. Leuk Res 31:497-506.
Berliner N, Hsing A, Graubert T, Sigurdsson F, Zain M, Bruno E, Hoffman R. 1995. Granulocyte colony-stimulating factor induction of normal human bone marrow progenitors results in neutrophil-specific gene expression. Blood 85:799-803.
Bestor T, Laudano A, Mattaliano R, Ingram V. 1988. Cloning and sequencing of a cDNA encoding DNA methyltransferase of mouse cells. The carboxyl-terminal domain of the mammalian enzymes is related to bacterial restriction methyltransferases. J Mol Biol 203:971-983.
Bird AP, Southern EM. 1978. Use of restriction enzymes to study eukaryotic DNA methylation: I. The methylation pattern in ribosomal DNA from Xenopus laevis. J Mol Biol 118:27-47.
Boissel N, Leroy H, Brethon B, Philippe N, de Botton S, Auvrignon A, Raffoux E, Leblanc T, Thomas X, Hermine O, Quesnel B, Baruchel A, Leverger G, Dombret H, Preudhomme C. 2006. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 20:965-970.
Borregaard N, Sehested M, Nielsen BS, Sengelov H, Kjeldsen L. 1995. Biosynthesis of granule proteins in normal human bone marrow cells. Gelatinase is a marker of terminal neutrophil differentiation. Blood 85:812-817.
Bovenzi V, Le NL, Cote S, Sinnett D, Momparler LF, Momparler RL. 1999. DNA methylation of retinoic acid receptor beta in breast cancer and possible therapeutic role of 5-aza-2'-deoxycytidine. Anticancer Drugs 10:471-476.
Bowen DT, Frew ME, Hills R, Gale RE, Wheatley K, Groves MJ, Langabeer SE, Kottaridis PD, Moorman AV, Burnett AK, Linch DC. 2005. RAS mutation in acute myeloid leukemia is associated with distinct cytogenetic subgroups but does not influence outcome in patients younger than 60 years. Blood 106:2113-2119.
Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248-254.
Brdicka T, Imrich M, Angelisova P, Brdickova N, Horvath O, Spicka J, Hilgert I, Luskova P, Draber P, Novak P, Engels N, Wienands J, Simeoni L, Osterreicher J, Aguado E, Malissen M, Schraven B, Horejsi V. 2002. Non-T cell activation linker (NTAL): a transmembrane adaptor protein involved in immunoreceptor signaling. J Exp Med 196:1617-1626.

8.- Bibliographie Seite - 125 -
Brdicka T, Pavlistova D, Leo A, Bruyns E, Korinek V, Angelisova P, Scherer J, Shevchenko A, Hilgert I, Cerny J, Drbal K, Kuramitsu Y, Kornacker B, Horejsi V, Schraven B. 2000. Phosphoprotein associated with glycosphingolipid-enriched microdomains (PAG), a novel ubiquitously expressed transmembrane adaptor protein, binds the protein tyrosine kinase csk and is involved in regulation of T cell activation. J Exp Med 191:1591-1604.
Breitman TR, Selonick SE, Collins SJ. 1980. Induction of differentiation of the human promyelocytic leukemia cell line (HL-60) by retinoic acid. Proc Natl Acad Sci U S A 77:2936-2940.
Bristow CA, Shore P. 2003. Transcriptional regulation of the human MIP-1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res 31:2735-2744.
Brown DA, Rose JK. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 68:533-544.
Brown RE. 1998. Sphingolipid organization in biomembranes: what physical studies of model membranes reveal. J Cell Sci 111 ( Pt 1):1-9.
Bug G, Ritter M, Wassmann B, Schoch C, Heinzel T, Schwarz K, Romanski A, Kramer OH, Kampfmann M, Hoelzer D, Neubauer A, Ruthardt M, Ottmann OG. 2005. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer 104:2717-2725.
Burel SA, Harakawa N, Zhou L, Pabst T, Tenen DG, Zhang DE. 2001. Dichotomy of AML1-ETO functions: growth arrest versus block of differentiation. Mol Cell Biol 21:5577-5590.
Calabi F, Pannell R, Pavloska G. 2001. Gene targeting reveals a crucial role for MTG8 in the gut. Mol Cell Biol 21:5658-5666.
Caliaro MJ, Marmouget C, Guichard S, Mazars P, Valette A, Moisand A, Bugat R, Jozan S. 1994. Response of four human ovarian carcinoma cell lines to all-trans retinoic acid: relationship with induction of differentiation and retinoic acid receptor expression. Int J Cancer 56:743-748.
Cameron EE, Bachman KE, Myohanen S, Herman JG, Baylin SB. 1999. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nat Genet 21:103-107.
Cameron S, Taylor DS, TePas EC, Speck NA, Mathey-Prevot B. 1994. Identification of a critical regulatory site in the human interleukin-3 promoter by in vivo footprinting. Blood 83:2851-2859.
Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, Degos L. 1990. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood 76:1704-1709.
Castilla LH, Garrett L, Adya N, Orlic D, Dutra A, Anderson S, Owens J, Eckhaus M, Bodine D, Liu PP. 1999. The fusion gene Cbfb-MYH11 blocks myeloid differentiation and predisposes mice to acute myelomonocytic leukaemia. Nat Genet 23:144-146.
Castilla LH, Perrat P, Martinez NJ, Landrette SF, Keys R, Oikemus S, Flanegan J, Heilman S, Garrett L, Dutra A, Anderson S, Pihan GA, Wolff L, Liu PP. 2004. Identification of genes that synergize with Cbfb-MYH11 in the pathogenesis of acute myeloid leukemia. Proc Natl Acad Sci U S A 101:4924-4929.
Chen HM, Zhang P, Voso MT, Hohaus S, Gonzalez DA, Glass CK, Zhang DE, Tenen DG. 1995. Neutrophils and monocytes express high levels of PU.1 (Spi-1) but not Spi-B. Blood 85:2918-2928.
Chomienne C, Ballerini P, Balitrand N, Daniel MT, Fenaux P, Castaigne S, Degos L. 1990. All-trans retinoic acid in acute promyelocytic leukemias. II. In vitro studies: structure-function relationship. Blood 76:1710-1717.
Clark SJ, Harrison J, Paul CL, Frommer M. 1994. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 22:2990-2997.
Claus R, Fliegauf M, Stock M, Duque JA, Kolanczyk M, Lübbert M. 2006. Inhibitors of DNA methylation and histone deacetylation independently relieve AML1/ETO-mediated lysozyme repression. J Leukoc Biol 80:1462-1472.
Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. 1978. Terminal differentiation of human promyelocytic leukemia cells induced by dimethyl sulfoxide and other polar compounds. Proc Natl Acad Sci U S A 75:2458-2462.
Collins SJ, Ruscetti FW, Gallagher RE, Gallo RC. 1979. Normal functional characteristics of cultured human promyelocytic leukemia cells (HL-60) after induction of differentiation by dimethylsulfoxide. J Exp Med 149:969-974.
Cooney CA. 1993. Are somatic cells inherently deficient in methylation metabolism? A proposed mechanism for DNA methylation loss, senescence and aging. Growth Dev Aging 57:261-273.
Coté S, Momparler RL. 1997. Activation of the retinoic acid receptor beta gene by 5-aza-2'-deoxycytidine in human DLD-1 colon carcinoma cells. Anticancer Drugs 8:56-61.
Coté S, Sinnett D, Momparler RL. 1998. Demethylation by 5-aza-2'-deoxycytidine of specific 5-methylcytosine sites in the promoter region of the retinoic acid receptor beta gene in human colon carcinoma cells. Anticancer Drugs 9:743-750.
Creusot F, Acs G, Christman JK. 1982. Inhibition of DNA methyltransferase and induction of Friend erythroleukemia cell differentiation by 5-azacytidine and 5-aza-2'-deoxycytidine. J Biol Chem 257:2041-2048.

8.- Bibliographie Seite - 126 -
Crowe DL. 1998. Retinoic acid receptor beta induces terminal differentiation of squamous cell carcinoma lines in the absence of cyclin-dependent kinase inhibitor expression. Cancer Res 58:142-148.
de Thé H, Chomienne C, Lanotte M, Degos L, Dejean A. 1990. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor alpha gene to a novel transcribed locus. Nature 347:558-561.
Dellagi K, Brouet JC. 1984. Alteration of vimentin intermediate filaments expression during differentiation of HL60 and U937 human leukemic cell lines. Leuk Res 8:611-616.
Di Croce L, Raker VA, Corsaro M, Fazi F, Fanelli M, Faretta M, Fuks F, Lo Coco F, Kouzarides T, Nervi C, Minucci S, Pelicci PG. 2002. Methyltransferase recruitment and DNA hypermethylation of target promoters by an oncogenic transcription factor. Science 295:1079-1082.
Dunne J, Cullmann C, Ritter M, Soria NM, Drescher B, Debernardi S, Skoulakis S, Hartmann O, Krause M, Krauter J, Neubauer A, Young BD, Heidenreich O. 2006. siRNA-mediated AML1/MTG8 depletion affects differentiation and proliferation-associated gene expression in t(8;21)-positive cell lines and primary AML blasts. Oncogene 25:6067-6078.
Erickson P, Gao J, Chang KS, Look T, Whisenant E, Raimondi S, Lasher R, Trujillo J, Rowley J, Drabkin H. 1992. Identification of breakpoints in t(8;21) acute myelogenous leukemia and isolation of a fusion transcript, AML1/ETO, with similarity to Drosophila segmentation gene, runt. Blood 80:1825-1831.
Erickson PF, Robinson M, Owens G, Drabkin HA. 1994. The ETO portion of acute myeloid leukemia t(8;21) fusion transcript encodes a highly evolutionarily conserved, putative transcription factor. Cancer Res 54:1782-1786.
Falls JG, Pulford DJ, Wylie AA, Jirtle RL. 1999. Genomic imprinting: implications for human disease. Am J Pathol 154:635-647.
Ferlazzo G, Klein J, Paliard X, Wei WZ, Galy A. 2000. Dendritic cells generated from CD34+ progenitor cells with flt3 ligand, c-kit ligand, GM-CSF, IL-4, and TNF-alpha are functional antigen-presenting cells resembling mature monocyte-derived dendritic cells. J Immunother 23:48-58.
Fliegauf M, Stock M, Berg T, Lübbert M. 2004. Williams-Beuren syndrome critical region-5/non-T-cell activation linker: a novel target gene of AML1/ETO. Oncogene 23:9070-9081.
Fourney RM, Dietrich KD, Aubin RA, Paterson MC. 1988. HindIII polymorphism in the human c-sis proto-oncogene. Nucleic Acids Res 16:8197.
Frank R, Zhang J, Uchida H, Meyers S, Hiebert SW, Nimer SD. 1995. The AML1/ETO fusion protein blocks transactivation of the GM-CSF promoter by AML1B. Oncogene 11:2667-2674.
Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A 89:1827-1831.
Fuks F, Burgers WA, Brehm A, Hughes-Davies L, Kouzarides T. 2000. DNA methyltransferase Dnmt1 associates with histone deacetylase activity. Nat Genet 24:88-91.
Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. 1979. Characterization of the continuous, differentiating myeloid cell line (HL-60) from a patient with acute promyelocytic leukemia. Blood 54:713-733.
Gardiner-Garden M, Frommer M. 1987. CpG islands in vertebrate genomes. J Mol Biol 196:261-282. Gari M, Goodeve A, Wilson G, Winship P, Langabeer S, Linch D, Vandenberghe E, Peake I, Reilly J. 1999. c-
kit proto-oncogene exon 8 in-frame deletion plus insertion mutations in acute myeloid leukaemia. Br J Haematol 105:894-900.
Gelmetti V, Zhang J, Fanelli M, Minucci S, Pelicci PG, Lazar MA. 1998. Aberrant recruitment of the nuclear receptor corepressor-histone deacetylase complex by the acute myeloid leukemia fusion partner ETO. Mol Cell Biol 18:7185-7191.
Gergen JP, Wieschaus E. 1986. Dosage requirements for runt in the segmentation of Drosophila embryos. Cell 45:289-299.
Gilliland DG. 2001. Hematologic malignancies. Curr Opin Hematol 8:189-191. Glover AB, Leyland-Jones B. 1987. Biochemistry of azacitidine: a review. Cancer Treat Rep 71:959-964. Goddard AD, Borrow J, Freemont PS, Solomon E. 1991. Characterization of a zinc finger gene disrupted by the
t(15;17) in acute promyelocytic leukemia. Science 254:1371-1374. Golub TR, Barker GF, Bohlander SK, Hiebert SW, Ward DC, Bray-Ward P, Morgan E, Raimondi SC, Rowley
JD, Gilliland DG. 1995. Fusion of the TEL gene on 12p13 to the AML1 gene on 21q22 in acute lymphoblastic leukemia. Proc Natl Acad Sci U S A 92:4917-4921.
Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen T, Beart RW, Van Tornout JM, Jones PA. 1995. Methylation of the 5' CpG island of the p16/CDKN2 tumor suppressor gene in normal and transformed human tissues correlates with gene silencing. Cancer Res 55:4531-4535.
Gonzalgo ML, Jones PA. 1997. Rapid quantitation of methylation differences at specific sites using methylation-sensitive single nucleotide primer extension (Ms-SNuPE). Nucleic Acids Res 25:2529-2531.

8.- Bibliographie Seite - 127 -
Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG. 1998. Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia. Nature 391:815-818.
Grisolano JL, O'Neal J, Cain J, Tomasson MH. 2003. An activated receptor tyrosine kinase, TEL/PDGFbetaR, cooperates with AML1/ETO to induce acute myeloid leukemia in mice. Proc Natl Acad Sci U S A 100:9506-9511.
Guo Y, Engelhardt M, Wider D, Abdelkarim M, Lübbert M. 2006. Effects of 5-aza-2'-deoxycytidine on proliferation, differentiation and p15/INK4b regulation of human hematopoietic progenitor cells. Leukemia 20:115-121.
Harris NL, Jaffe ES, Diebold J, Flandrin G, Muller-Hermelink HK, Vardiman J, Lister TA, Bloomfield CD. 1999. World Health Organization classification of neoplastic diseases of the hematopoietic and lymphoid tissues: report of the Clinical Advisory Committee meeting-Airlie House, Virginia, November 1997. J Clin Oncol 17:3835-3849.
Heidenreich O, Krauter J, Riehle H, Hadwiger P, John M, Heil G, Vornlocher HP, Nordheim A. 2003. AML1/MTG8 oncogene suppression by small interfering RNAs supports myeloid differentiation of t(8;21)-positive leukemic cells. Blood 101:3157-3163.
Herman JG, Graff JR, Myohanen S, Nelkin BD, Baylin SB. 1996. Methylation-specific PCR: a novel PCR assay for methylation status of CpG islands. Proc Natl Acad Sci U S A 93:9821-9826.
Higuchi M, O'Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR. 2002. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell 1:63-74.
Hodge D, Coghill E, Keys J, Maguire T, Hartmann B, McDowall A, Weiss M, Grimmond S, Perkins A. 2006. A global role for EKLF in definitive and primitive erythropoiesis. Blood 107:3359-3370.
Holliday R, Pugh JE. 1975. DNA modification mechanisms and gene activity during development. Science 187:226-232.
Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. 2003. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther 307:720-728.
Huang G, Shigesada K, Ito K, Wee HJ, Yokomizo T, Ito Y. 2001. Dimerization with PEBP2beta protects RUNX1/AML1 from ubiquitin-proteasome-mediated degradation. Embo J 20:723-733.
Huang LH, Wang R, Gama-Sosa MA, Shenoy S, Ehrlich M. 1984. A protein from human placental nuclei binds preferentially to 5-methylcytosine-rich DNA. Nature 308:293-295.
Huberman E, Callaham MF. 1979. Induction of terminal differentiation in human promyelocytic leukemia cells by tumor-promoting agents. Proc Natl Acad Sci U S A 76:1293-1297.
Hurwitz CA, Raimondi SC, Head D, Krance R, Mirro J, Jr., Kalwinsky DK, Ayers GD, Behm FG. 1992. Distinctive immunophenotypic features of t(8;21)(q22;q22) acute myeloblastic leukemia in children. Blood 80:3182-3188.
Janssen E, Zhu M, Zhang W, Koonpaew S, Zhang W. 2003. LAB: a new membrane-associated adaptor molecule in B cell activation. Nat Immunol 4:117-123.
Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. 1998. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet 19:187-191.
Jost JP, Hofsteenge J. 1992. The repressor MDBP-2 is a member of the histone H1 family that binds preferentially in vitro and in vivo to methylated nonspecific DNA sequences. Proc Natl Acad Sci U S A 89:9499-9503.
Juttermann R, Li E, Jaenisch R. 1994. Toxicity of 5-aza-2'-deoxycytidine to mammalian cells is mediated primarily by covalent trapping of DNA methyltransferase rather than DNA demethylation. Proc Natl Acad Sci U S A 91:11797-11801.
Kafri T, Ariel M, Brandeis M, Shemer R, Urven L, McCarrey J, Cedar H, Razin A. 1992. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev 6:705-714.
Kapp U, Yeh WC, Patterson B, Elia AJ, Kagi D, Ho A, Hessel A, Tipsword M, Williams A, Mirtsos C, Itie A, Moyle M, Mak TW. 1999. Interleukin 13 is secreted by and stimulates the growth of Hodgkin and Reed-Sternberg cells. J Exp Med 189:1939-1946.
Kass SU, Landsberger N, Wolffe AP. 1997. DNA methylation directs a time-dependent repression of transcription initiation. Curr Biol 7:157-165.
Kelly LM, Kutok JL, Williams IR, Boulton CL, Amaral SM, Curley DP, Ley TJ, Gilliland DG. 2002. PML/RARalpha and FLT3-ITD induce an APL-like disease in a mouse model. Proc Natl Acad Sci U S A 99:8283-8288.
Kelly WK, Richon VM, O'Connor O, Curley T, MacGregor-Curtelli B, Tong W, Klang M, Schwartz L, Richardson S, Rosa E, Drobnjak M, Cordon-Cordo C, Chiao JH, Rifkind R, Marks PA, Scher H. 2003. Phase I clinical trial of histone deacetylase inhibitor: suberoylanilide hydroxamic acid administered intravenously. Clin Cancer Res 9:3578-3588.

8.- Bibliographie Seite - 128 -
Kita K, Nakase K, Miwa H, Masuya M, Nishii K, Morita N, Takakura N, Otsuji A, Shirakawa S, Ueda T, et al. 1992. Phenotypical characteristics of acute myelocytic leukemia associated with the t(8;21)(q22;q22) chromosomal abnormality: frequent expression of immature B-cell antigen CD19 together with stem cell antigen CD34. Blood 80:470-477.
Kitabayashi I, Yokoyama A, Shimizu K, Ohki M. 1998. Interaction and functional cooperation of the leukemia-associated factors AML1 and p300 in myeloid cell differentiation. Embo J 17:2994-3004.
Klampfer L, Zhang J, Zelenetz AO, Uchida H, Nimer SD. 1996. The AML1/ETO fusion protein activates transcription of BCL-2. Proc Natl Acad Sci U S A 93:14059-14064.
Kosmider O, Denis N, Lacout C, Vainchenker W, Dubreuil P, Moreau-Gachelin F. 2005. Kit-activating mutations cooperate with Spi-1/PU.1 overexpression to promote tumorigenic progression during erythroleukemia in mice. Cancer Cell 8:467-478.
Kottaridis PD, Gale RE, Frew ME, Harrison G, Langabeer SE, Belton AA, Walker H, Wheatley K, Bowen DT, Burnett AK, Goldstone AH, Linch DC. 2001. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood 98:1752-1759.
Kuchenbauer F, Schoch C, Kern W, Hiddemann W, Haferlach T, Schnittger S. 2005. Impact of FLT3 mutations and promyelocytic leukaemia-breakpoint on clinical characteristics and prognosis in acute promyelocytic leukaemia. Br J Haematol 130:196-202.
Kuendgen A, Strupp C, Aivado M, Bernhardt A, Hildebrandt B, Haas R, Germing U, Gattermann N. 2004. Treatment of myelodysplastic syndromes with valproic acid alone or in combination with all-trans retinoic acid. Blood 104:1266-1269.
Landschulz WH, Johnson PF, McKnight SL. 1989. The DNA binding domain of the rat liver nuclear protein C/EBP is bipartite. Science 243:1681-1688.
Lanotte M, Martin-Thouvenin V, Najman S, Balerini P, Valensi F, Berger R. 1991. NB4, a maturation inducible cell line with t(15;17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 77:1080-1086.
Larsen F, Gundersen G, Lopez R, Prydz H. 1992. CpG islands as gene markers in the human genome. Genomics 13:1095-1107.
Le Beau MM, Larson RA, Bitter MA, Vardiman JW, Golomb HM, Rowley JD. 1983. Association of an inversion of chromosome 16 with abnormal marrow eosinophils in acute myelomonocytic leukemia. A unique cytogenetic-clinicopathological association. N Engl J Med 309:630-636.
Lee DY, Teyssier C, Strahl BD, Stallcup MR. 2005. Role of protein methylation in regulation of transcription. Endocr Rev 26:147-170.
Lee JW, Kim YG, Soung YH, Han KJ, Kim SY, Rhim HS, Min WS, Nam SW, Park WS, Lee JY, Yoo NJ, Lee SH. 2006. The JAK2 V617F mutation in de novo acute myelogenous leukemias. Oncogene 25:1434-1436.
Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, Kremer E, Otto F, Brenner O, Lev-Tov A, Groner Y. 2002. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. Embo J 21:3454-3463.
Levantini E, Giorgetti A, Cerisoli F, Traggiai E, Guidi A, Martin R, Acampora D, Aplan PD, Keller G, Simeone A, Iscove NN, Hoang T, Magli MC. 2003. Unsuspected role of the brain morphogenetic gene Otx1 in hematopoiesis. Proc Natl Acad Sci U S A 100:10299-10303.
Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, Bird A. 1992. Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell 69:905-914.
Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. 1998. Elastin is an essential determinant of arterial morphogenesis. Nature 393:276-280.
Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima T, Bae SC, Ito Y. 2002. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell 109:113-124.
Lin HY, Chen CS, Lin SP, Weng JR, Chen CS. 2006. Targeting histone deacetylase in cancer therapy. Med Res Rev 26:397-413.
Linggi B, Müller-Tidow C, van de Locht L, Hu M, Nip J, Serve H, Berdel WE, van der Reijden B, Quelle DE, Rowley JD, Cleveland J, Jansen JH, Pandolfi PP, Hiebert SW. 2002. The t(8;21) fusion protein, AML1 ETO, specifically represses the transcription of the p14(ARF) tumor suppressor in acute myeloid leukemia. Nat Med 8:743-750.
Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, Carrington JC, Doerge RW, Colot V, Martienssen R. 2004. Role of transposable elements in heterochromatin and epigenetic control. Nature 430:471-476.

8.- Bibliographie Seite - 129 -
Liu S, Shen T, Huynh L, Klisovic MI, Rush LJ, Ford JL, Yu J, Becknell B, Li Y, Liu C, Vukosavljevic T, Whitman SP, Chang KS, Byrd JC, Perrotti D, Plass C, Marcucci G. 2005. Interplay of RUNX1/MTG8 and DNA methyltransferase 1 in acute myeloid leukemia. Cancer Res 65:1277-1284.
Lo Coco F, Pisegna S, Diverio D. 1997. The AML1 gene: a transcription factor involved in the pathogenesis of myeloid and lymphoid leukemias. Haematologica 82:364-370.
Löwenberg B, Downing JR, Burnett A. 1999. Acute myeloid leukemia. N Engl J Med 341:1051-1062. Lowery MC, Morris CA, Ewart A, Brothman LJ, Zhu XL, Leonard CO, Carey JC, Keating M, Brothman AR.
1995. Strong correlation of elastin deletions, detected by FISH, with Williams syndrome: evaluation of 235 patients. Am J Hum Genet 57:49-53.
Lozzio CB, Lozzio BB. 1975. Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45:321-334.
Lu Y, Xu YB, Yuan TT, Song MG, Lübbert M, Fliegauf M, Chen GQ. 2006. Inducible expression of AML1-ETO fusion protein endows leukemic cells with susceptibility to extrinsic and intrinsic apoptosis. Leukemia 20:987-993.
Lübbert M, Herrmann F, Koeffler HP. 1991a. Expression and regulation of myeloid-specific genes in normal and leukemic myeloid cells. Blood 77:909-924.
Lübbert M, Miller CW, Koeffler HP. 1991b. Changes of DNA methylation and chromatin structure in the human myeloperoxidase gene during myeloid differentiation. Blood 78:345-356.
Lübbert M, Tobler A, Daskalakis M. 1999. Cytosine demethylation of the proteinase-3/myeloblastin primary granule protease gene during phagocyte development. Leukemia 13:1420-1427.
Lütchman M, Kim AC, Cheng L, Whitehead IP, Oh SS, Hanspal M, Boukharov AA, Hanada T, Chishti AH. 2002. Dematin interacts with the Ras-guanine nucleotide exchange factor Ras-GRF2 and modulates mitogen-activated protein kinase pathways. Eur J Biochem 269:638-649.
Lutterbach B, Westendorf JJ, Linggi B, Patten A, Moniwa M, Davie JR, Huynh KD, Bardwell VJ, Lavinsky RM, Rosenfeld MG, Glass C, Seto E, Hiebert SW. 1998. ETO, a target of t(8;21) in acute leukemia, interacts with the N-CoR and mSin3 corepressors. Mol Cell Biol 18:7176-7184.
Mao S, Frank RC, Zhang J, Miyazaki Y, Nimer SD. 1999. Functional and physical interactions between AML1 proteins and an ETS protein, MEF: implications for the pathogenesis of t(8;21)-positive leukemias. Mol Cell Biol 19:3635-3644.
Meehan RR, Lewis JD, McKay S, Kleiner EL, Bird AP. 1989. Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell 58:499-507.
Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Gunther T, Buettner R, Schule R. 2005. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437:436-439.
Meyers S, Downing JR, Hiebert SW. 1993. Identification of AML-1 and the (8;21) translocation protein (AML-1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol 13:6336-6345.
Meyers S, Lenny N, Hiebert SW. 1995. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Mol Cell Biol 15:1974-1982.
Michaud J, Wu F, Osato M, Cottles GM, Yanagida M, Asou N, Shigesada K, Ito Y, Benson KF, Raskind WH, Rossier C, Antonarakis SE, Israels S, McNicol A, Weiss H, Horwitz M, Scott HS. 2002. In vitro analyses of known and novel RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood 99:1364-1372.
Mitani K, Ogawa S, Tanaka T, Miyoshi H, Kurokawa M, Mano H, Yazaki Y, Ohki M, Hirai H. 1994. Generation of the AML1-EVI-1 fusion gene in the t(3;21)(q26;q22) causes blastic crisis in chronic myelocytic leukemia. Embo J 13:504-510.
Miyoshi H, Ohira M, Shimizu K, Mitani K, Hirai H, Imai T, Yokoyama K, Soeda E, Ohki M. 1995. Alternative splicing and genomic structure of the AML1 gene involved in acute myeloid leukemia. Nucleic Acids Res 23:2762-2769.
Miyoshi H, Shimizu K, Kozu T, Maseki N, Kaneko Y, Ohki M. 1991. t(8;21) breakpoints on chromosome 21 in acute myeloid leukemia are clustered within a limited region of a single gene, AML1. Proc Natl Acad Sci U S A 88:10431-10434.
Mueller BU, Pabst T, Osato M, Asou N, Johansen LM, Minden MD, Behre G, Hiddemann W, Ito Y, Tenen DG. 2002. Heterozygous PU.1 mutations are associated with acute myeloid leukemia. Blood 100:998-1007.
Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. 1998. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res 241:126-133.
Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN, Bird A. 1998. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393:386-389.

8.- Bibliographie Seite - 130 -
Nervi C, Borello U, Fazi F, Buffa V, Pelicci PG, Cossu G. 2001. Inhibition of histone deacetylase activity by trichostatin A modulates gene expression during mouse embryogenesis without apparent toxicity. Cancer Res 61:1247-1249.
Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D, Bird A. 1999. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet 23:58-61.
Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM. 2007. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem 282:4408-4416.
No D, Yao TP, Evans RM. 1996. Ecdysone-inducible gene expression in mammalian cells and transgenic mice. Proc Natl Acad Sci U S A 93:3346-3351.
Nuchprayoon I, Meyers S, Scott LM, Suzow J, Hiebert S, Friedman AD. 1994. PEBP2/CBF, the murine homolog of the human myeloid AML1 and PEBP2 beta/CBF beta proto-oncoproteins, regulates the murine myeloperoxidase and neutrophil elastase genes in immature myeloid cells. Mol Cell Biol 14:5558-5568.
Nucifora G, Begy CR, Erickson P, Drabkin HA, Rowley JD. 1993. The 3;21 translocation in myelodysplasia results in a fusion transcript between the AML1 gene and the gene for EAP, a highly conserved protein associated with the Epstein-Barr virus small RNA EBER 1. Proc Natl Acad Sci U S A 90:7784-7788.
Nucifora G, Rowley JD. 1995. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood 86:1-14.
Oelgeschlager M, Nuchprayoon I, Luscher B, Friedman AD. 1996. C/EBP, c-Myb, and PU.1 cooperate to regulate the neutrophil elastase promoter. Mol Cell Biol 16:4717-4725.
Ogawa E, Maruyama M, Kagoshima H, Inuzuka M, Lu J, Satake M, Shigesada K, Ito Y. 1993. PEBP2/PEA2 represents a family of transcription factors homologous to the products of the Drosophila runt gene and the human AML1 gene. Proc Natl Acad Sci U S A 90:6859-6863.
Okada H, Watanabe T, Niki M, Takano H, Chiba N, Yanai N, Tani K, Hibino H, Asano S, Mucenski ML, Ito Y, Noda T, Satake M. 1998. AML1(-/-) embryos do not express certain hematopoiesis-related gene transcripts including those of the PU.1 gene. Oncogene 17:2287-2293.
Okuda T, Cai Z, Yang S, Lenny N, Lyu CJ, van Deursen JM, Harada H, Downing JR. 1998. Expression of a knocked-in AML1-ETO leukemia gene inhibits the establishment of normal definitive hematopoiesis and directly generates dysplastic hematopoietic progenitors. Blood 91:3134-3143.
Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR. 1996. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84:321-330.
Olsson IL, Breitman TR, Gallo RC. 1982. Priming of human myeloid leukemic cell lines HL-60 and U-937 with retinoic acid for differentiation effects of cyclic adenosine 3':5'-monophosphate-inducing agents and a T-lymphocyte-derived differentiation factor. Cancer Res 42:3928-3933.
Orazi A, Kotylo P, Neiman RS. 1992. Acute leukemia with t(8;21) can express T-lineage-associated markers. Blood 80:1855-1856.
Otto F, Lübbert M, Stock M. 2003. Upstream and downstream targets of RUNX proteins. J Cell Biochem 89:9-18.
Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. 1997. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell 89:765-771.
Pabst T, Mueller BU, Harakawa N, Schoch C, Haferlach T, Behre G, Hiddemann W, Zhang DE, Tenen DG. 2001a. AML1-ETO downregulates the granulocytic differentiation factor C/EBPalpha in t(8;21) myeloid leukemia. Nat Med 7:444-451.
Pabst T, Mueller BU, Zhang P, Radomska HS, Narravula S, Schnittger S, Behre G, Hiddemann W, Tenen DG. 2001b. Dominant-negative mutations of CEBPA, encoding CCAAT/enhancer binding protein-alpha (C/EBPalpha), in acute myeloid leukemia. Nat Genet 27:263-270.
Paschka P, Marcucci G, Ruppert AS, Mrozek K, Chen H, Kittles RA, Vukosavljevic T, Perrotti D, Vardiman JW, Carroll AJ, Kolitz JE, Larson RA, Bloomfield CD. 2006. Adverse prognostic significance of KIT mutations in adult acute myeloid leukemia with inv(16) and t(8;21): a Cancer and Leukemia Group B Study. J Clin Oncol 24:3904-3911.
Paz MF, Wei S, Cigudosa JC, Rodriguez-Perales S, Peinado MA, Huang TH, Esteller M. 2003. Genetic unmasking of epigenetically silenced tumor suppressor genes in colon cancer cells deficient in DNA methyltransferases. Hum Mol Genet 12:2209-2219.
Petrovick MS, Hiebert SW, Friedman AD, Hetherington CJ, Tenen DG, Zhang DE. 1998. Multiple functional domains of AML1: PU.1 and C/EBPalpha synergize with different regions of AML1. Mol Cell Biol 18:3915-3925.

8.- Bibliographie Seite - 131 -
Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. 2001. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276:36734-36741.
Plass C, Shibata H, Kalcheva I, Mullins L, Kotelevtseva N, Mullins J, Kato R, Sasaki H, Hirotsune S, Okazaki Y, Held WA, Hayashizaki Y, Chapman VM. 1996. Identification of Grf1 on mouse chromosome 9 as an imprinted gene by RLGS-M. Nat Genet 14:106-109.
Raelson JV, Nervi C, Rosenauer A, Benedetti L, Monczak Y, Pearson M, Pelicci PG, Miller WH, Jr. 1996. The PML/RAR alpha oncoprotein is a direct molecular target of retinoic acid in acute promyelocytic leukemia cells. Blood 88:2826-2832.
Redondo JM, Pfohl JL, Hernandez-Munain C, Wang S, Speck NA, Krangel MS. 1992. Indistinguishable nuclear factor binding to functional core sites of the T-cell receptor delta and murine leukemia virus enhancers. Mol Cell Biol 12:4817-4823.
Rhoades KL, Hetherington CJ, Harakawa N, Yergeau DA, Zhou L, Liu LQ, Little MT, Tenen DG, Zhang DE. 2000. Analysis of the role of AML1-ETO in leukemogenesis, using an inducible transgenic mouse model. Blood 96:2108-2115.
Rhyner K, Taetle R. 1986. Induction of colony-stimulating factor response in myeloid leukaemia cell lines. Br J Haematol 64:601-612.
Richon VM, Sandhoff TW, Rifkind RA, Marks PA. 2000. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. PNAS 97:10014-10019.
Richon VM, Webb Y, Merger R, Sheppard T, Jursic B, Ngo L, Civoli F, Breslow R, Rifkind RA, Marks PA. 1996. Second generation hybrid polar compounds are potent inducers of transformed cell differentiation. Proc Natl Acad Sci U S A 93:5705-5708.
Riggs AD, Pfeifer GP. 1992. X-chromosome inactivation and cell memory. Trends Genet 8:169-174. Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. 2000. DNMT1 forms a complex with
Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat Genet 25:338-342.
Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA, Jones PA. 1999. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res 27:2291-2298.
Rosmarin AG, Weil SC, Rosner GL, Griffin JD, Arnaout MA, Tenen DG. 1989. Differential expression of CD11b/CD18 (Mo1) and myeloperoxidase genes during myeloid differentiation. Blood 73:131-136.
Ross ME, Mahfouz R, Onciu M, Liu HC, Zhou X, Song G, Shurtleff SA, Pounds S, Cheng C, Ma J, Ribeiro RC, Rubnitz JE, Girtman K, Williams WK, Raimondi SC, Liang DC, Shih LY, Pui CH, Downing JR. 2004. Gene expression profiling of pediatric acute myelogenous leukemia. Blood 104:3679-3687.
Rountree MR, Bachman KE, Baylin SB. 2000. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet 25:269-277.
Rovera G, Santoli D, Damsky C. 1979. Human promyelocytic leukemia cells in culture differentiate into macrophage-like cells when treated with a phorbol diester. Proc Natl Acad Sci U S A 76:2779-2783.
Rowley JD. 1973. Chromosomal patterns in myelocytic leukemia. N Engl J Med 289:220-221. Rüter B, Wijermans PW, Lübbert M. 2006. Superiority of prolonged low-dose azanucleoside administration?
Results of 5-aza-2'-deoxycytidine retreatment in high-risk myelodysplasia patients. Cancer 106:1744-1750.
Rutherford T, Clegg JB, Higgs DR, Jones RW, Thompson J, Weatherall DJ. 1981. Embryonic erythroid differentiation in the human leukemic cell line K562. Proc Natl Acad Sci U S A 78:348-352.
Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O. 1999. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci U S A 96:4592-4597.
Santos-Rosa H, Caldas C. 2005. Chromatin modifier enzymes, the histone code and cancer. Eur J Cancer 41:2381-2402.
Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T. 2002. Active genes are tri-methylated at K4 of histone H3. Nature 419:407-411.
Schessl C, Rawat VP, Cusan M, Deshpande A, Kohl TM, Rosten PM, Spiekermann K, Humphries RK, Schnittger S, Kern W, Hiddemann W, Quintanilla-Martinez L, Bohlander SK, Feuring-Buske M, Buske C. 2005. The AML1-ETO fusion gene and the FLT3 length mutation collaborate in inducing acute leukemia in mice. J Clin Invest 115:2159-2168.
Schmelz K, Sattler N, Wagner M, Lubbert M, Dorken B, Tamm I. 2005. Induction of gene expression by 5-Aza-2'-deoxycytidine in acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS) but not epithelial cells by DNA-methylation-dependent and -independent mechanisms. Leukemia 19:103-111.
Schneider U, Schwenk HU, Bornkamm G. 1977. Characterization of EBV-genome negative "null" and "T" cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer 19:621-626.

8.- Bibliographie Seite - 132 -
Schnittger S, Bacher U, Kern W, Haferlach C, Haferlach T. 2007. JAK2 seems to be a typical cooperating mutation in therapy-related t(8;21)/ AML1-ETO-positive AML. Leukemia 21:183-184.
Schnittger S, Bacher U, Kern W, Schroder M, Haferlach T, Schoch C. 2006. Report on two novel nucleotide exchanges in the JAK2 pseudokinase domain: D620E and E627E. Leukemia 20:2195-2197.
Schnittger S, Schoch C, Dugas M, Kern W, Staib P, Wuchter C, Loffler H, Sauerland CM, Serve H, Buchner T, Haferlach T, Hiddemann W. 2002. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood 100:59-66.
Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. 2004. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18:1251-1262.
Shimizu K, Kitabayashi I, Kamada N, Abe T, Maseki N, Suzukawa K, Ohki M. 2000. AML1-MTG8 leukemic protein induces the expression of granulocyte colony-stimulating factor (G-CSF) receptor through the up-regulation of CCAAT/enhancer binding protein epsilon. Blood 96:288-296.
Simeoni L, Kliche S, Lindquist J, Schraven B. 2004. Adaptors and linkers in T and B cells. Curr Opin Immunol 16:304-313.
Skalnik DG. 2002. Transcriptional mechanisms regulating myeloid-specific genes. Gene 284:1-21. Smiraglia DJ, Plass C. 2002. The study of aberrant methylation in cancer via restriction landmark genomic
scanning. Oncogene 21:5414-5426. Smiraglia DJ, Plass C. 2003. The development of CpG island methylation biomarkers using restriction landmark
genomic scanning. Ann N Y Acad Sci 983:110-119. Southern EM. 1975. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J
Mol Biol 98:503-517. Srour EF, Brandt JE, Briddell RA, Leemhuis T, van Besien K, Hoffman R. 1991. Human CD34+ HLA-DR-
bone marrow cells contain progenitor cells capable of self-renewal, multilineage differentiation, and long-term in vitro hematopoiesis. Blood Cells 17:287-295.
Stach D, Schmitz OJ, Stilgenbauer S, Benner A, Dohner H, Wiessler M, Lyko F. 2003. Capillary electrophoretic analysis of genomic DNA methylation levels. Nucleic Acids Res 31:E2.
Strahl BD, Allis CD. 2000. The language of covalent histone modifications. Nature 403:41-45. Sundstrom C, Nilsson K. 1976. Establishment and characterization of a human histiocytic lymphoma cell line
(U-937). Int J Cancer 17:565-577. Suzuki H, Gabrielson E, Chen W, Anbazhagan R, van Engeland M, Weijenberg MP, Herman JG, Baylin SB.
2002. A genomic screen for genes upregulated by demethylation and histone deacetylase inhibition in human colorectal cancer. Nat Genet 31:141-149.
Suzuki T, Yokozaki H, Kuniyasu H, Hayashi K, Naka K, Ono S, Ishikawa T, Tahara E, Yasui W. 2000. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int J Cancer 88:992-997.
Swisshelm K, Ryan K, Lee X, Tsou HC, Peacocke M, Sager R. 1994. Down-regulation of retinoic acid receptor beta in mammary carcinoma cell lines and its up-regulation in senescing normal mammary epithelial cells. Cell Growth Differ 5:133-141.
Tallman MS, Gilliland DG, Rowe JM. 2005. Drug therapy for acute myeloid leukemia. Blood 106:1154-1163. Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, Ion A,
Jeffery S, Kalidas K, Patton MA, Kucherlapati RS, Gelb BD. 2001. Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat Genet 29:465-468.
Tartaglia M, Niemeyer CM, Fragale A, Song X, Buechner J, Jung A, Hahlen K, Hasle H, Licht JD, Gelb BD. 2003. Somatic mutations in PTPN11 in juvenile myelomonocytic leukemia, myelodysplastic syndromes and acute myeloid leukemia. Nat Genet 34:148-150.
Tassabehji M. 2003. Williams-Beuren syndrome: a challenge for genotype-phenotype correlations. Hum Mol Genet 12 Spec No 2:R229-237.
Tedoldi S, Paterson JC, Hansmann ML, Natkunam Y, Rudiger T, Angelisova P, Du MQ, Roberton H, Roncador G, Sanchez L, Pozzobon M, Masir N, Barry R, Pileri S, Mason DY, Marafioti T, Horejsi V. 2006. Transmembrane adaptor molecules: a new category of lymphoid-cell markers. Blood 107:213-221.
Tetteroo PA, Massaro F, Mulder A, Schreuder-van Gelder R, von dem Borne AE. 1984. Megakaryoblastic differentiation of proerythroblastic K562 cell-line cells. Leuk Res 8:197-206.
Thiede C, Steudel C, Mohr B, Schaich M, Schakel U, Platzbecker U, Wermke M, Bornhauser M, Ritter M, Neubauer A, Ehninger G, Illmer T. 2002. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood 99:4326-4335.
Tkaczyk C, Horejsi V, Iwaki S, Draber P, Samelson LE, Satterthwaite AB, Nahm DH, Metcalfe DD, Gilfillan AM. 2004. NTAL phosphorylation is a pivotal link between the signaling cascades leading to human mast cell degranulation following Kit activation and Fc epsilon RI aggregation. Blood 104:207-214.

8.- Bibliographie Seite - 133 -
Turker MS, Bestor TH. 1997. Formation of methylation patterns in the mammalian genome. Mutat Res 386:119-130.
Uchida H, Zhang J, Nimer SD. 1997. AML1A and AML1B can transactivate the human IL-3 promoter. J Immunol 158:2251-2258.
Valk PJ, Bowen DT, Frew ME, Goodeve AC, Lowenberg B, Reilly JT. 2004a. Second hit mutations in the RTK/RAS signaling pathway in acute myeloid leukemia with inv(16). Haematologica 89:106.
Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, Beverloo HB, Moorhouse MJ, van der Spek PJ, Lowenberg B, Delwel R. 2004b. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 350:1617-1628.
Vangala RK, Heiss-Neumann MS, Rangatia JS, Singh SM, Schoch C, Tenen DG, Hiddemann W, Behre G. 2003. The myeloid master regulator transcription factor PU.1 is inactivated by AML1-ETO in t(8;21) myeloid leukemia. Blood 101:270-277.
Volna P, Lebduska P, Draberova L, Simova S, Heneberg P, Boubelik M, Bugajev V, Malissen B, Wilson BS, Horejsi V, Malissen M, Draber P. 2004. Negative regulation of mast cell signaling and function by the adaptor LAB/NTAL. J Exp Med 200:1001-1013.
Vorbrodt A, Meo P, Rovera G. 1979. Regulation of acid phosphatase activity in human promyelocytic leukemic cells induced to differentiate in culture. J Cell Biol 83:300-307.
Wang J, Hoshino T, Redner RL, Kajigaya S, Liu JM. 1998. ETO, fusion partner in t(8;21) acute myeloid leukemia, represses transcription by interaction with the human N-CoR/mSin3/HDAC1 complex. Proc Natl Acad Sci U S A 95:10860-10865.
Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, Bushweller JH, Bories JC, Alt FW, Ryan G, Liu PP, Wynshaw-Boris A, Binder M, Marin-Padilla M, Sharpe AH, Speck NA. 1996. The CBFbeta subunit is essential for CBFalpha2 (AML1) function in vivo. Cell 87:697-708.
Wang SW, Speck NA. 1992. Purification of core-binding factor, a protein that binds the conserved core site in murine leukemia virus enhancers. Mol Cell Biol 12:89-102.
Wang Y, Horvath O, Hamm-Baarke A, Richelme M, Gregoire C, Guinamard R, Horejsi V, Angelisova P, Spicka J, Schraven B, Malissen B, Malissen M. 2005. Single and combined deletions of the NTAL/LAB and LAT adaptors minimally affect B-cell development and function. Mol Cell Biol 25:4455-4465.
Weber M, Davies JJ, Wittig D, Oakeley EJ, Haase M, Lam WL, Schubeler D. 2005. Chromosome-wide and promoter-specific analyses identify sites of differential DNA methylation in normal and transformed human cells. Nat Genet 37:853-862.
Welgus HG, Connolly NL, Senior RM. 1986. 12-o-Tetradecanoyl-phorbol-13-acetate-differentiated U937 cells express a macrophage-like profile of neutral proteinases. High levels of secreted collagenase and collagenase inhibitor accompany low levels of intracellular elastase and cathepsin G. J Clin Invest 77:1675-1681.
Westendorf JJ, Yamamoto CM, Lenny N, Downing JR, Selsted ME, Hiebert SW. 1998. The t(8;21) fusion product, AML-1-ETO, associates with C/EBP-alpha, inhibits C/EBP-alpha-dependent transcription, and blocks granulocytic differentiation. Mol Cell Biol 18:322-333.
Widschwendter M, Berger J, Daxenbichler G, Muller-Holzner E, Widschwendter A, Mayr A, Marth C, Zeimet AG. 1997. Loss of retinoic acid receptor beta expression in breast cancer and morphologically normal adjacent tissue but not in the normal breast tissue distant from the cancer. Cancer Res 57:4158-4161.
Xiong Z, Laird PW. 1997. COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res 25:2532-2534.
Yamamoto T, Isomura M, Xu Y, Liang J, Yagasaki H, Kamachi Y, Kudo K, Kiyoi H, Naoe T, Kojma S. 2006. PTPN11, RAS and FLT3 mutations in childhood acute lymphoblastic leukemia. Leuk Res 30:1085-1089.
Yergeau DA, Hetherington CJ, Wang Q, Zhang P, Sharpe AH, Binder M, Marin-Padilla M, Tenen DG, Speck NA, Zhang DE. 1997. Embryonic lethality and impairment of haematopoiesis in mice heterozygous for an AML1-ETO fusion gene. Nat Genet 15:303-306.
Yuan Y, Zhou L, Miyamoto T, Iwasaki H, Harakawa N, Hetherington CJ, Burel SA, Lagasse E, Weissman IL, Akashi K, Zhang DE. 2001. AML1-ETO expression is directly involved in the development of acute myeloid leukemia in the presence of additional mutations. Proc Natl Acad Sci U S A 98:10398-10403.
Zhang DE, Fujioka K, Hetherington CJ, Shapiro LH, Chen HM, Look AT, Tenen DG. 1994. Identification of a region which directs the monocytic activity of the colony-stimulating factor 1 (macrophage colony-stimulating factor) receptor promoter and binds PEBP2/CBF (AML1). Mol Cell Biol 14:8085-8095.
Zhang J, Kalkum M, Yamamura S, Chait BT, Roeder RG. 2004. E protein silencing by the leukemogenic AML1-ETO fusion protein. Science 305:1286-1289.
Zhu M, Koonpaew S, Liu Y, Shen S, Denning T, Dzhagalov I, Rhee I, Zhang W. 2006. Negative regulation of T cell activation and autoimmunity by the transmembrane adaptor protein LAB. Immunity 25:757-768.
Zhu M, Liu Y, Koonpaew S, Granillo O, Zhang W. 2004. Positive and negative regulation of FcepsilonRI-mediated signaling by the adaptor protein LAB/NTAL. J Exp Med 200:991-1000.

9.- Lebenslauf Seite - 134 -
9.- Lebenslauf
GRUNDDATEN
Vorname Atilio Jesús
Name Duque Afonso
Geburtsort Santa Cruz de Tenerife
Alter 29 Jahre
Nationalität Spanisch
E-mail [email protected]
AUSBILDUNGSDATEN
1984-1986 Colegio Echeyde (Tenerife, Spanien)
1986-1988 Colegio Echeyde (Tenerife, Spanien)
1988-1994 Colegio Miguel Pintor (Tenerife, Spanien)
1994-1998 I.B. Anaga (Tenerife, Spanien)
1998 Abitur
1998-2004 Studium, Hauptfach Medizin, Universidad de La Laguna (Tenerife, Spanien)
Juli 2004 Approbation in Medizin (Spanien)
2004- Promotion zur Erlangung des medizinischen Doktorgrades an der Universität
Freiburg, Deutschland
2004-2005 Forschungsstipendium des Deutschen Vereins zur Förderung der Leukämie
und Tumorforschung
2005-2007 Forschungsstipendium von „La Caixa“-DAAD
März 2007 Aprobation in Medizin (Deutschland)
2007-
Facharztausbildung in der Abteilung I der Inneren Medizin
(Hämatologie/Onkologie) an der Uniklinik Freiburg, Deutschland.

9.- Lebenslauf Seite - 135 -
ZUSÄTZLICHE MEDIZINISCHE AUSBILDUNG
Februar 2005 Ärztliche Fortbildung über „Fortschritte in der Hämatologie“
April 2005 Symposium „Hämatologische Neoplasien bei älteren Patienten“
Januar 2006 III. Post-ASH Symposium des Tumorzentrums Freiburg.
Februar 2006 „3. Annual Symposium of the European LeukemiaNet“ und „7. Annual
Symposium of the German Competence Network“ in Heidelberg
Februar 2006 International Symposium „Acute Leukemias XI: Prognostic Factors and
Treatment Strategies“ in München.
März 2006 Symposium „Das Multiple Myelom 2006: Auf dem Weg zur Heilung?“
April 2006 Symposium „Grundlagen und Perspektiven in der Behandlung Thorakaler
Tumoren“
Mai 2006 Ärztliche Fortbildung „Hämatologische Neoplasien“
September 2006 Ärztliche Fortbildung „CML - Neue Aspekte für Diagnostik und Therapie“
Oktober 2006 Spanischer National-Kongress „XLVII Reunión Nacional de la AEHH“ und
„XXII Congreso Nacional de la SETH“ in Granada (Spanien)
November 2006 Ärztliche Fortbildung „Prostatakarzinom“ in Freiburg.
Januar 2007 „IV. Post ASH Symposium des Tumorzentrums Freiburg“
ZUSÄTZLICHE WISSENSCHAFTLICHE AUSBILDUNG
Oktober 2004 V. Freiburg Symposium “Molekulare Hämatologie“ in Freiburg
März 2005 XIV. Workshop der Abteilung I der Inneren Medizin in Lenzkirch.
Oktober 2005 Strahlenschutzunterweisung der Uniklinik Freiburg.
März 2006 XV. Workshop der Abteilung I der Inneren Medizin in Lenzkirch
Juni 2006 Veranstaltung „New developments in Immunotherapy of Malignant Lymphoma“
in Freiburg.
Februar 2007 EHA Meeting "The role of epigenetics in hematological malignancies" in
Cannes, Frankreich.
März 2007 XVI. Workshop der Abteilung I der Inneren Medizin in Lenzkirch

9.- Lebenslauf Seite - 136 -
ZUSÄTZLICHE QUALIFIKATIONEN
Muttersprache Spanisch
Fremdsprachen Englisch Oberstufe von der Escuela Oficial de Idiomas, im Jahr 2000
Deutsch Oberstufe von der Escuela Oficial de Idiomas, im Jahr 2004
Französisch Mittelstufe von der Escuela Oficial de Idiomas, im Jahr 2004
EDV Kenntnisse
Grundlegende PC Kenntnisse insbesondere Erfahrung mit Windows XP, MS Word 2003, Excel 2003, Power Point 2003 und Internet Explorer,
AUSLANDSERFAHRUNG
Jahr 1999 2 Wochen freiwillige Tätigkeit in Tánger in einem Behindertenheim der
Franziskaner des Weissen Kreuzes (Marokko).
Jahr 2001-2002 7. und 8. Semester mit dem Erasmus Programm an der Universität Freiburg.
Jahr 2003 3- wöchiges Praktikum in der Abteilung I der Inneren Medizin in Freiburg.
Jahr 2004 12. Semester an der Universität Louis Pasteur in Straßburg (Frankreich).
Jahr 2004-2007 Forschungsprojekt in der AG Prof.Lübbert (Abteilung I der Inneren Medizin,
Uniklinik Freiburg) im Rahmen meiner Doktorarbeit.
Jahr 2007- Facharztausbildung in der Abteilung I der Inneren Medizin
(Hämatologie/Onkologie) der Uniklinik Freiburg
LABORERFAHRUNG
Jahr 2000 Projekt im molekulardiagnostischen Labor des "Hospital Universitario de
Canarias" auf Tenerife (Spanien)
2004- Forschungsprojekt im Rahmen der Promotion in der Abteilung I der Inneren
Medizin des Universitätsklinikums Freiburg.

9.- Lebenslauf Seite - 137 -
PUBLIKATIONSLISTE
Originalarbeiten :
Duque-Afonso J., Yalcin A. Berg T., Abdelkarim M., Heidenreich O., Lübbert M. The HDAC class I specific inhibitor Entinostat (MS-275) effectively relieves epigenetic silencing of the LAT2 gene mediated by AML1/ETO. (Oncogene, Januar 2011, angenommen)
Duque-Afonso J., Solari L., Essig A., Berg T., Pahl HL, Lübbert M. Regulation of the adaptor molecule LAT2, an in vivo target gene of AML1/ETO, during myeloid differentiation (British Journal of Haematology, Dezember 2010, angenommen)
Almstedt M.*, Blagitko-Dorfs N.*, Duque-Afonso J.*, Karbach J.*, Pfeifer D., Jäger E., Lübbert M. The DNA demethylating agent 5-aza-2’-deoxycytidine induces expression of immunogenic Cancer/testis antigens in myeloid leukemia cells. Leukemia Research 2010; 34(7): 899-905. * These authors contributed equally.
Hauswald S., Duque-Afonso J., Wagner MM., Schertl FM., Lübbert M., Peschel C., Keller U., and Licht T., Histone Deacetylase Inhibitors Induce Pleiotropic Anticancer Drug Resistance In Acute Myeloid Leukemia Cells by Modulation of ABC-Transporter Genes. Clin. Cancer Res. 2009; 15(11): 3705-3715.
Becker H., Pfeifer D., Afonso JD., Nimer SD., Veelken H., Schwabe M., Lübbert M. Two cell lines of t(8;21) acute myeloid leukemia with activating c-KIT exon 17 mutation: models for the “second hit” hypothesis. Leukemia 2008; 22(9):1792-4.
Claus R., Fliegauf M., Stock M., Duque J., Kolanczyc M., Lübbert M. Inhibitors of DNA methylation and histone deacetylation independently relieve AML1/ETO-mediated lysozyme repression. Jounal of Leukocyte Biology, 2006, 80 (6): 1462-1472.
Übersichtsartikel
Lübbert M., Duque-Afonso J., Kuendgen A. Epigenetic therapy of myelodysplastic syndromes: an update. Educational book, EHA Meeting 2010.
Müller AM., Duque J., Shizuru JA., Lübbert M. Complementing mutations in Core Binding Factor (CBF) leukemias: mechanisms und therapeutical implications. Oncogene 2008; 27(44):5759-73
Duque J., Lübbert M., Kirschbaum M. Pharmacological modifications of epigenetics: DNA methyltransferase and HDAC inhibitors. Buchkapitel, Innovative Leukemia and Lymphoma Therapy, Informa Healthcare USA, Inc., Erscheinung 04/08
Hackanson B., Becker H., Berg T., Binder M., Dierks C., Duque-Afonso J., Lairmore MD., Schafer HS., Schnitzler M., Zeiser R., Martens U., Mertelsmann R. and Lübbert M. XXIII International Association for Comparative Research on Leukemia and Related Diseases Symposium: from Molecular Pathogenesis to Targeted Therapy in Leukemia and Solid Tumors. Cancer Res 2008;68 5512-5518.
Abstracts Duque J, Carmona N., Rivero Y, Barrios Y, Salido E. Analyse des Polymorphismus im IRS-1 Gen bei Diabetes Mellitus Typ 2 bei den Einwohnern vom Norden von Teneriffa. XIV. Kongress der Medizinstudenten von der Universität La Laguna, Tenerife. Abstraktsprogramm Duque J, Berg T., Lübbert M., The AML1/ETO target gene WBSCR5 (NTAL/LAB) is regulated during myeloid differentiation. “Annals of Hematology“ Vol 85, Supplement 1, 2006, Abstrakt P-7.

9.- Lebenslauf Seite - 138 -
Duque J., Berg T., Lübbert M., The AML1/ETO target gene WBSCR5 (NTAL/LAB) is regulated during myeloid differentiation. “Haematologica“ Vol 91., 2006, Abstrakt C-096
Claus R, Fliegauf M, Stock M, Duque J, Kolanczyc M, Lübbert M. AML1/ETO mediated lyzozyme repression is indepently relieved by inhibitors of DNA methylation and histone deacetylation. Blood 2006. Nov 2006; 108: Abstract #4310
Duque J, Berg T, Heidenreich, Lübbert M The AML1/ETO-mediated repression of the WBSCR5 (NTAL/LAB/LAT2) gene is relieved in Kasumi-1 myeloid leukemia cells. EHA/ESH Workshop „The role of epigenetics in hematological malignancies“, Mandelieu (Frankreich), Februar 2007
Duque J., Hackanson B., Berg T., Plass C., Lübbert M. Does conditional AML1/ETO expression alter DNA methylation patterns of Hematopoiesis associated genes? A Global analysis by Restriction Landmark Genomic Scanning (RLGS). EHA/ESH Workshop „The role of epigenetics in hematological malignancies“, Mandelieu (Frankreich), Februar 2007
Kleine B., Berg T, Duque J, Lübbert M Epigenetic modifications at the P15/INK4B 5´ Region in AML1/ETO positive myeloid leukemias. EHA/ESH Workshop „The role of epigenetics in hematological malignancies“, Mandelieu (Frankreich), Februar 2007
Duque J, Berg T, Heidenreich, Lübbert M The AML1/ETO target gene WBSCR5 (NTAL/LAB/LAT2) is regulated during myeloid differentiation and derepressed by both HDAC inhibitiors and DNMT inhibitors in AML1/ETO positive leukemia cells DGHO 2007, Basel (Schweiz), Oktober 2007
Duque-Afonso J, Berg T, Heidenreich O, Lübbert M. Epìgenetic repression of the adaptor molecule LAT2 by the leukemic fusion protein AML1/ETO. Blood 2007. Dec 2007; 108: Abstract #987 Travel Award
Duque-Afonso J, Solari L, Lübbert M. The AML1/ETO target LAT2 membrane adaptor molecule is regulated during normal monocytic differentiation. Blood 2007. Dec 2007; 108: Abstract #2401 Travel Award
Duque-Afonso J., Heidenreich O., Lübbert M. Epigenetic regulation of the adaptor molecule LAT2 by the leukemic fusion protein AML1/ETO. DGHO Oktober 2008, Wien (Österreich), Vortrag
Duque-Afonso J., Essig A., Berg T., Heidenreich O., Pahl HL., Lübbert M. The adaptor molecule LAT2 is epigenetically regulated by the leukemic fusion protein AML1/ETO. 3. CCRTP Stanford University September 2009, San Francisco (USA)
Duque-Afonso J., Prasse A., Wäsch R., Bertz H., Finke J., Marks R. Low incidence of pulmonary Graft-versus-Host disease in older patients after reduced toxicity conditioning with fludarabin, carmustine and melphalan prior to hematopoietic stem cell transplantation. EBMT Meeting, März 2010, Vienna (Austria)
Poster Präsentationen
März 2005 XIV. Workshops der Abteilung I der Inneren Medizin (Freiburg)
Februar 2006 „International Symposium Acute Leukemias XI“ in München
März 2006 XV. Workshops der Abteilung I der Inneren Medizin Freiburg.
Februar 2007 EHA/ESH Workshop „The Role of Epigenetics in hematological malignancies“, Mandelieu (Frankreich)
März 2007 XVI. Workshops der Abteilung I der Inneren Medizin Freiburg. Best Poster Award
Oktober 2007 DGHO Jahrestagung, Basel (Schweiz)

9.- Lebenslauf Seite - 139 -
Dezember 2007 49.ASH Meeting, Atlanta (USA) Travel Award
September 2009 3. CCRTP Stanford University September 2009, San Francisco (USA)
März 2010 EBMT Meeting, Vienna (Austria)
Dezember 2010 52. ASH Meeting, Orlando (USA)
Vorträge Mai 2000 XI. Kongress der Medizinstudenten von der Universität La Laguna, Tenerife (Spanien).
Dezember 2004 Vorstellung des Promotionsthemas im „Work in Progress“ Seminar.
März 2005 XIV. Workshops der Abteilung I der Inneren Medizin (Freiburg).
Dezember 2005 Vortrag im „Work in Progress“ Seminar.
März 2006 XV. Workshops der Abteilung I der Inneren Medizin (Freiburg).
Oktober 2006
Vortrag mit dem Titel „Regulation of the AML1/ETO target gene WBSCR5 during myelopoiesis“ am „XLVII Reunión Nacional de la AEHH“ und „ XXII Congreso Nacional de la SETH“ in Granada (Spanien).
März 2007 XVI. Workshops der Abteilung I der Inneren Medizin (Freiburg).
März 2007 Vortrag in „Combined target therapy“ Ph.D.-Studentenworkshop, Freiburg
April 2007 Vortrag im „Work in Progress“ Seminar, Freiburg.
Mai 2007 Vortrag mit dem Titel “Epigenetic regulation of the AML1/ETO gene WBSCR5 and its role during the myeolopoiesis” am 14th Annual Meeting of the European Cancer Center, Strassburg (Frankreich)
Februar 2008 XVII. Workshops der Abteilung I der Inneren Medizin (Freiburg).
Juli 2008 Vortrag in Real Time PCR Workshop, unterstützt von Roche Diagnostics, Freiburg.
Oktober 2008 Vortrag mit dem Titel “Epigenetic regulation of the adaptor molecule LAT2 by the leukemic fusion protein AML1/ETO” am DGHO Kongress , Wien (Österreich)
März 2009 XVIII. Workshops der Abteilung I der Inneren Medizin (Freiburg).
Mai 2009 Vortrag mit dem Titel “The adaptor molecule LAT2 is epigenetically repressed by the leukemic fusion protein AML1/ETO” am 16th Annual Meeting of the European Cancer Center, Freiburg (Deutschland)
Februar 2010 XIX. Workshops der Abteilung I der Inneren Medizin (Freiburg).
September 2010 1.Comprehensive Cancer Center Research Programm in Medical Oncology (Hinterzarten)
Freiburg im Breisgau, 03. Februar 2011 Jesús Duque Afonso




















