
Atm-deficient mice: an osteoporosis model with defective osteoblast differentiation and increased...
11
Atm-deficient mice: an osteoporosis model with defective osteoblast differentiation and increased osteoclastogenesis Naslin Rasheed 1,{ , Xueying Wang 1,{ , Qing-Tian Niu 2 , James Yeh 2 and Baojie Li 1, * 1 The Institute of Molecular and Cell Biology Proteos, 61 Biopolis Drive, Singapore 138673, Republic of Singapore and 2 Department of Medicine, Winthrop-University Hospital, Mineola, NY 11501, USA Received September 15, 2005; Revised and Accepted April 26, 2006 Atm is a Ser/Thr kinase involved in DNA damage response and is required for genome integrity and stem cell renewal. Here, we report an additional role for Atm in bone remodeling. Atm 2/2 mice showed reduced bone mass, especially at the trabecular bones, accompanied by a decrease in bone formation rate and defective differentiation of osteoblasts, but normal numbers of osteoprogenitor cells and osteoblasts. Atm might affect osteoblast differentiation by modulating the expression of osterix, a lineage-specific transcription factor essential for osteoblast maturation, likely via the bone morphogenetic proteins pathway. Atm 2/2 mice also displayed a marked increase in osteoclastogenesis and bone resorption, although Atm had no cell-autonomous effect on osteoclast differentiation and resorption. Increased osteoclastogenesis could be caused by a substantial reduction in testosterone and estradiol levels in male and female mice, respectively. The steroid hormone deficiency is a result of gonad developmental defects, which led to an increase in serum gonadotrophic hormone, FSH via a feedback regulation. Overall, these results indicate that Atm deficiency leads to osteoporosis mainly as a result of hypogonadism-induced bone resorption together with compromised osteoblast differentiation, and that Atm plays a positive role in regulating expression of osteoblast-specific transcription factor, osterix. INTRODUCTION Ataxia-telangiectasia (A-T) is an incurable progressive neurodegenerative disorder. Ataxia usually occurs at a very early stage and A-T patients are often confined to wheelchairs before the age of 10. Most A-T patients die in their teens or early twenties (1). The disease is caused by mutations in ATM (A-T mutated) and is accompanied by other syndromes including immunodeficiency, infertility, cancer predisposition, radiosensitivity and premature aging (2 – 4). Most of these symptoms are recapitulated in Atm knockout mice (5–7). The ATM gene encodes a Ser/Thr kinase, which is activated by double-stranded DNA breaks (DSBs) (8,9). Its main func- tion is to modify downstream molecules such as p53 and Brca1 to regulate cell cycle progression, apoptosis and DNA repair. As a consequence, A-T cells or murine Atm 2/2 cells exhibit genomic instability including telomere shortening and premature senescence. Impaired DNA damage response and repair are believed to be the underlying cause for cancer predis- position, immunodeficiency and infertility in A-T patients. ATM has been implicated in various other cellular processes as well (10). For instance, ATM was reported to regulate insulin signaling and this may explain the diabetic phenotype of A-T patients (11). ATM has also been implicated in regulat- ing oxidative stress response and A-T cells and Atm-deficient murine cells were found to accumulate reactive oxygen species (ROS) and ROS-induced damage (12–14). This could be attributed to the failure of Atm 2/2 cells to up-regulate anti- oxidant proteins through the transcription factor, Nrf2 (15). An aberration in oxidative stress response was found to affect hematopoietic stem cell renewal in Atm 2/2 mice (16). In addition, Atm deficiency was reported to accelerate aging and stem cell renewal defect of mice with telomere dysfunction (the telomerase RNA component Terc-deficient mice) (17). # 2006 The Author(s) This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/ licenses/by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited. { The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors. *To whom correspondence should be addressed. Tel: þ65 65869679; Fax: þ65 67791117; Email: [email protected] Human Molecular Genetics, 2006, Vol. 15, No. 12 1938–1948 doi:10.1093/hmg/ddl116 Advance Access published on April 27, 2006 by guest on December 21, 2014 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Atm-deficient mice: an osteoporosis model with defective osteoblast differentiation and increased...

Atm-deficient mice: an osteoporosis modelwith defective osteoblast differentiation andincreased osteoclastogenesis
Naslin Rasheed1,{, Xueying Wang1,{, Qing-Tian Niu2, James Yeh2 and Baojie Li1,*
1The Institute of Molecular and Cell Biology Proteos, 61 Biopolis Drive, Singapore 138673, Republic of Singapore
and 2Department of Medicine, Winthrop-University Hospital, Mineola, NY 11501, USA
Received September 15, 2005; Revised and Accepted April 26, 2006
Atm is a Ser/Thr kinase involved in DNA damage response and is required for genome integrity and stem cellrenewal. Here, we report an additional role for Atm in bone remodeling. Atm2/2 mice showed reduced bonemass, especially at the trabecular bones, accompanied by a decrease in bone formation rate and defectivedifferentiation of osteoblasts, but normal numbers of osteoprogenitor cells and osteoblasts. Atm mightaffect osteoblast differentiation by modulating the expression of osterix, a lineage-specific transcriptionfactor essential for osteoblast maturation, likely via the bone morphogenetic proteins pathway. Atm2/2
mice also displayed a marked increase in osteoclastogenesis and bone resorption, although Atm had nocell-autonomous effect on osteoclast differentiation and resorption. Increased osteoclastogenesis couldbe caused by a substantial reduction in testosterone and estradiol levels in male and female mice,respectively. The steroid hormone deficiency is a result of gonad developmental defects, which led to anincrease in serum gonadotrophic hormone, FSH via a feedback regulation. Overall, these results indicatethat Atm deficiency leads to osteoporosis mainly as a result of hypogonadism-induced bone resorptiontogether with compromised osteoblast differentiation, and that Atm plays a positive role in regulatingexpression of osteoblast-specific transcription factor, osterix.
INTRODUCTION
Ataxia-telangiectasia (A-T) is an incurable progressiveneurodegenerative disorder. Ataxia usually occurs at a veryearly stage and A-T patients are often confined to wheelchairsbefore the age of 10. Most A-T patients die in their teens orearly twenties (1). The disease is caused by mutations inATM (A-T mutated) and is accompanied by other syndromesincluding immunodeficiency, infertility, cancer predisposition,radiosensitivity and premature aging (2–4). Most of thesesymptoms are recapitulated in Atm knockout mice (5–7).
The ATM gene encodes a Ser/Thr kinase, which is activatedby double-stranded DNA breaks (DSBs) (8,9). Its main func-tion is to modify downstream molecules such as p53 andBrca1 to regulate cell cycle progression, apoptosis and DNArepair. As a consequence, A-T cells or murine Atm2/2 cellsexhibit genomic instability including telomere shortening and
premature senescence. Impaired DNA damage response andrepair are believed to be the underlying cause for cancer predis-position, immunodeficiency and infertility in A-T patients.ATM has been implicated in various other cellular processesas well (10). For instance, ATM was reported to regulateinsulin signaling and this may explain the diabetic phenotypeof A-T patients (11). ATM has also been implicated in regulat-ing oxidative stress response and A-T cells and Atm-deficientmurine cells were found to accumulate reactive oxygenspecies (ROS) and ROS-induced damage (12–14). This couldbe attributed to the failure of Atm2/2 cells to up-regulate anti-oxidant proteins through the transcription factor, Nrf2 (15). Anaberration in oxidative stress response was found to affecthematopoietic stem cell renewal in Atm2/2 mice (16). Inaddition, Atm deficiency was reported to accelerate aging andstem cell renewal defect of mice with telomere dysfunction(the telomerase RNA component Terc-deficient mice) (17).
# 2006 The Author(s)This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/2.0/uk/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work isproperly cited.
{The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.*To whom correspondence should be addressed. Tel: þ65 65869679; Fax: þ65 67791117; Email: [email protected]
Human Molecular Genetics, 2006, Vol. 15, No. 12 1938–1948doi:10.1093/hmg/ddl116Advance Access published on April 27, 2006
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Yet, the exact molecular mechanism by which ATM deficiencycauses these defects are not well understood.
In an effort to study the function of Atm in bone develop-ment, we analyzed bone remodeling in Atm knockout miceand their wild-type littermates. Rapid bone growth in sizeand mineral deposition takes place during puberty and slowsdown thereafter. Bone is then constantly remodeled with oldbones being replaced by newly formed bones after pubertyuntil pre-menopause (18,19). In both, the growth spurtduring puberty and the remodeling, steroid hormones playimportant roles. Bone formation is a function of osteoblaststhat are derived from bone marrow mesenchymal stem cells(MSCs). Osteoblast differentiation from MSCs is a multiple-step process that requires the expression of lineage-specifictranscription factors such as Runx2 and Osterix (Osx), alongwith other more generally expressed transcription factorslike Atf4 and Dlx5 (20,21). Results obtained from knockoutmice studies indicate that both Runx2 and Osx are essentialfor osteoblast maturation and bone calcification and that Osxacts downstream of Runx2. Moreover, ectopic expression ofeither Runx2 or Osx is sufficient to induce the expression ofosteoblast-specific proteins (22). Osteoblast differentiation/maturation is promoted by growth factors and cytokines,among which bone morphogenetic proteins (BMPs) and Wntare well studied. For example, BMP2 and BMP4 are able todrastically induce the expression of Osx and promote osteo-blast differentiation (23). On the other hand, bone resorptionis a function of osteoclast that shares the same precursor asmacrophages and is derived from bone marrow hematopoieticstem cells (HSCs). Furthermore, there exists a functional inter-action between osteoblasts/osteoprecursors and osteoclasts, asthe former express cytokines such as RANKL, M-CSF andOPG to regulate differentiation of the latter, which expressthe cognate receptors for these cytokines (24,25). A coordi-nated action of both osteoblasts and osteoclasts is critical inmaintaining optimal bone density and bone mass.
Bone disorders occur when the balance between boneformation and bone resorption is disrupted. Osteoporosis isone of the most common aging-associated bone disordersand is characterized by reduced bone mass and deteriorationof bone microstructures, leaving one at an increased risk ofbone fractures (26). There are mainly two types: post-menopausal osteoporosis, affecting 40% of women andcaused by hyperactivity of osteoclasts due to estrogen short-age; and aging-associated osteoporosis, affecting both menand women and caused by a decline in bone formation (lossof 20–30% of cancellous bones and 5–10% of corticalbones) (18,20,27). Estrogen deficiency is known to inducebone resorption. This can be mediated by osteoblasts as estro-gen deficiency was found to induce expression of M-CSF andRANKL, which stimulate osteoclastogenesis. Estrogendeficiency is also believed to stimulate bone formation dueto the coupling of bone resorption and bone formation, atleast at the early stage of estrogen deficiency (18,19). Butbone resorption outpaces bone formation in units (BMUs),leading to a net loss of bone and resulting in osteoporosis.We report here that Atm2/2 mice show osteoporotic pheno-types, accompanied by decreased bone formation andincreased bone resorption. Bone formation defect is likelycaused by defective osteoblast differentiation, rather than a
shortage of osteoprogenitor cells. The differentiation defectis associated with a downregulation of Osx but not Runx2 orAtf4, suggesting that reduction in osterix might mediate thedefective differentiation. Furthermore, Atm deficiency com-promised the induction of Osx by BMP2, which is synthesizedand secreted by osteoblasts and plays critical roles in osteo-blast differentiation and bone formation. The increased boneresorption is very likely caused by hypogonadism-inducedosteoclastogenesis. Our data also suggest that Atm deficiencyresults in hypogonadism due to defective gonad developmentitself rather than a shortage of gonadotrophins. Therefore,Atm-deficient mice represent a model for osteoporosis withsteroid shortage and decreased bone formation.
RESULTS
Atm2/2 mice showed reduced bone mass
To determine whether Atm deficiency affected bone remodel-ing, mice deficient for Atm and their control littermates(4-months-old) were analyzed for bone mass by dual X-rayabsorptiometry and bone histomorphometry. The mutantmice showed a slight reduction in bone mineral density anda 60% reduction in the trabecular bone volume (Table 1).The number of trabecular bones was decreased by 40% andthe separation of trabecular bones was markedly increased(Table 1). Therefore, the mutant mice exhibit an osteoporoticphenotype at the cancellous bones rather than the corticalbones. These results suggest that the balance between boneformation and bone resorption during bone remodeling wasdisrupted in Atm2/2 mice. We also found that both maleand female Atm2/2 mice showed osteoporotic phenotypes(data not shown). Histomorphometry analysis of 6-week-oldmice revealed a 25% reduction in bone volume in theabsence of Atm (12.6+ 1.7% for 2/2 versus 16.6+ 2.5%for þ/þ), a 20% reduction in the number of trabecularbones (4.98+ 1.2 for 2/2 versus 6.18+ 0.8 for þ/þ, #/mm). We decided to focus on 4-month-old Atm2/2 mice asthey showed more severe osteoporotic phenotypes and are inthe phase of bone remodeling, whereas 6-week-old mice areat the stage of pubertal growth spurt that is under thecontrol of growth hormone and insulin-like growth factor 1(IGF-1), which decline after puberty.
Decreased bone formation in Atm2/2 mice
One cause of bone loss is defective bone formation by osteo-blasts. To measure the bone formation rates in Atm2/2 andcontrol mice, calcein was injected twice at an interval of 7days into both mutant and wild-type mice, which were sacri-ficed 2 days after the second injection. Histomorphometricanalysis of the femurs revealed decreased bone formationrates in Atm2/2 mice at the trabecular bones, but not muchdifference was seen at the periosteal or endosteal surface ofcortical bones (Table 1 and data not shown). However, nosignificant change was observed in osteoblast surface andthe number of osteoblasts in Atm2/2 mice, suggesting thatthe decrease in bone formation rate could be a result ofcompromised osteoblast differentiation/maturation, ratherthan the decline in the number of osteoblasts (Table 1), and
Human Molecular Genetics, 2006, Vol. 15, No. 12 1939
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

that reduced bone formation might contribute to theosteoporotic phenotypes of Atm2/2 mice.
Defective differentiation/maturation ofAtm2/2 osteoblasts
To test whether decreased bone formation was caused by acell-autonomous effect of Atm on osteoblasts, we studiedthe differentiation potential of osteoblasts in the presence orabsence of Atm. Primary calvarial osteoblasts were isolatedand their differentiation was monitored by the specificmarkers, ALP and mineralization. As shown in Figure 1 Aand B, histochemical staining of ALP and quantitative assayof ALP revealed a decrease in ALP activities in Atm2/2 osteo-blast cultures. We found a similar decrease in mineralizationbone nodules in Atm2/2 osteoblast cultures (Fig. 1C). RT–PCR assays also showed a reduction of procollagen a1 (datanot shown). These results indicate that Atm has acell-autonomous effect on osteoblast differentiation, consistentwith the reduced bone formation seen in Atm2/2 mice.
Atm deficiency did not alter the number ofbone marrow osteoprogenitor cells
To test whether Atm has a role in osteoblast stem cell renewal,we compared the numbers of osteoprogenitor cells in the bonemarrow of Atm2/2 and control mice. One difference weobserved is that the colonies from Atm2/2 mice were signifi-cantly smaller in size than those from the control mice, indi-cating that Atm2/2 osteoprogenitor cells might proliferate ata reduced rate in vitro. We also observed a compromisedALP staining in Atm2/2 cultures. However, careful countingof these colony forming units (upto day 10) did not reveal asignificant difference in the number of ALP positive colonyformation units, an indication of the number of osteopro-genitor cells (Fig. 1D and E). More importantly, even in8-month-old mice, Atm2/2 mice showed a normal numberof osteoprogenitor cells, similar to that of 4-month-old mice,indicating that aging (upto 8 months) has no significant
effect on the number of osteoprogenitor cells in mice (datanot shown). These results clearly show that Atm did notaffect the stem cell population of osteoblast lineage duringbone remodeling.
Atm deficiency down-regulated the expression of osterix
Calvarial osteoblasts deficient for Atm showed a compromiseddifferentiation, manifested by a decrease in ALP expressionand a decease in bone nodule formation in vitro (Fig. 1). Tounderstand the molecular mechanisms behind the differen-tiation defect observed in Atm2/2 osteoblasts, we tested theexpression of the transcription factors that are critical forosteoblast differentiation, as well as other osteo-markers.The same number of Atm2/2 and control osteoblasts wereplated and cultured in differentiation medium. At day 1, 4and 7, a plate was harvested and total RNA was isolated.These RNA samples were used to perform semi-quantitativeRT–PCR assays. It was found that Atm2/2 osteoblastsexpressed the same levels of Runx2 and Atf4 as wild-typeosteoblasts. Yet, the mutant osteoblasts showed significantlyreduced levels of osterix (Fig. 2A). Western blot analysis con-firmed reduced expression of Osx at the protein level inAtm2/2 osteoblasts (Fig. 2B). As Osx is essential for osteo-blast differentiation and bone calcification and that the levelsof Osx determine the expression of osteoblast-specificmarkers, we believe that the downregulation of Osx mightmediate the effects of Atm deficiency on osteoblastdifferentiation.
These results suggest that Atm might participate in a signal-ing pathway that controls Osx expression. One of the beststudied pathways that regulate Osx expression is theBMP-Smad1/5/8 pathway, although it is likely that Osx is
Table 1. Histomorphometric parameters analyzed in wild-type and Atm2/2
mice
Atmþ/þ Atm2/2
Bone mineral density 0.06+ 0.01 0.05+ 0.01�
Trabecular number (#/mm) 3.19+ 1.11 1.90+ 1.03�
Trabecular separation (mcm) 451.89+ 229.47 884.0+ 259.31�
Trabecular area (%) 13.65+ 3.78 5.40+ 1.89�
Inter-label width (mcm) 9.04+ 1.44 7.58+ 1.20�
Bone formation rate/bone Pm(mcm/day)
63.71+ 16.42 44.13+ 8.43�
Peri-osteal ILW (mcm) 6.41+ 1.61 5.48+ 0.55Peri-osteal-MAR (mcm/day) 0.92+ 0.23 0.78+ 0.08Peri-osteal-BFR (mcm/day) 43.07+ 21.97 36.73+ 11.87Osteoblast surface/bone
surface (%)25.05+ 10.28 23.30+ 7.99
Osteoblast number/bonesurface (#/mcm)
23.35+ 10.34 22.19+ 7.25
�P, 0.05 (n ¼ 8).Figure 1. Atm2/2 calvarial osteoblasts showed defective osteoblast differen-tiation. (A) Calvarial osteoblasts were cultured for different periods of timeand then stained for ALP. (B) ALP quantitation assay which was normalizedto the total protein levels. (C) Von Kossa staining for mineralization in weeks3 and 4. (D) ALP-CFU staining. Bone marrow cells were extracted as permaterials and methods and the same number of cells was plated. Afterbeing cultured for different periods of time, the plates were stained forALP. (E) ALP-CFU results from four Atm2/2 mice versus control mice.�P , 0.05.
1940 Human Molecular Genetics, 2006, Vol. 15, No. 12
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

not a direct target gene of Smad1/5/8 (28). BMP2-inducedup-regulation of Osx was also significantly compromised inAtm2/2 osteoblasts (Fig. 2C), suggesting an important rolein the BMP-induced Osx upregulation and a possible linkbetween the BMP-Smads pathway and the Atm signalingpathway. The data also point to an important role for Osx insensing various stimuli and in controlling osteoblast differen-tiation (29,30).
Increased bone resorption in Atm2/2 mice
We have shown that Atm2/2 mice exhibited an osteoporoticphenotype, accompanied by decreased bone formation andosteoblast differentiation. Yet, the disparity between thereduction in bone mass at cancellous bones and the extent ofthe reduction in bone formation rate suggests that bone resorp-tion might also be altered in Atm2/2 mice at the phaseof remodeling. Further histomorphometric analysis of4-month-old mice revealed a significant increase in the boneresorption surface and in the number of osteoclasts in Atm2/2
mice (Fig. 3A and B). Moreover, Atm2/2 mice showed an
increase in excretion of urine deoxypyridinoline (DPD) cross-links, a marker for in vivo bone resorption (Fig. 3C). However,no obvious increase in bone resorption and osteoclastogenesiswas observed in 6-week-old Atm2/2 mice (data not shown).These data indicate that Atm2/2 mice have increased boneresorption in addition to decreased bone formation duringbone remodeling but not during pubertal growth spurt.
Atm did not affect osteoclast differentiation andresorption in vitro
Increased bone resorption and osteoclastogenesis could becaused by a cell-autonomous role for Atm in osteoclastlineage. Alternatively, Atm might indirectly affect osteoclasto-genesis, e.g. via hormonal regulation. To distinguish the twopossibilities, we compared osteoclast differentiation inAtm2/2 and control bone marrow monocytes. The numberof TRAP positive osteoclasts are similar for both Atm2/2
and control monocyte cultures (Fig. 4A and B), indicatingthat Atm does not directly affect osteoclast differentiation.Pit formation assay on dentine discs revealed no significantdifference in osteoclast resorption activities between Atm2/2
and control osteoclasts (Fig. 4C). These results indicate thatAtm may not have a cell-autonomous effect on osteoclastdifferentiation or resorption.
Figure 2. Atm2/2 calvarial osteoblasts showed reduced expression of osterixand compromised BMP2-induced Smad1/5/8 activation. (A) RT–PCR assaysfor the transcription factors that are involved in osteoblast differentiation.Atm2/2 calvarial osteoblasts showed reduced levels of Osx, but normallevels of Runx2 and Atf4. Atm2/2 and control calvarial osteoblasts wereplated for differentiation as described in Figure 1A. At different periods oftime, a plate was collected for total RNA isolation. RT–PCR assays werecarried out to determine the mRNA levels of above mentioned transcriptionfactors or cytokines. The Osx level of wild-type control culture at day 1was set at 1. (B) Western blot analysis of Osx protein levels in Atm2/2 andcontrol osteoblasts. Cells were cultured as described in (A) and were collectedfor western blot analysis. The Osx protein level of Atm2/2 was set at 1(n ¼ 3). (C) Atm deficiency compromised BMP2-induced Osx up-regulation.Quantitation data from semi-quantitative RT–PCR assays.
Figure 3. Increased bone resorption in Atm2/2 mice. In vivo histomorphome-try indicated that Atm2/2 showed increased osteoclast surface per bonesurface area (A) and increased number of osteoclast per bone surface area(B) when compared with control mice. (C) Atm2/2 mice showed increasedlevels of urine DPD, an in vivo marker for bone resorption. �P, 0.05.
Human Molecular Genetics, 2006, Vol. 15, No. 12 1941
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Osteoblasts can synthesize and secrete cytokines to regulateosteoclast differentiation from HSC in the bone marrow.Among the best studied cytokines are RANKL and M-CSF,both required for proliferation and differentiation of osteo-clast, and OPG, a decoy receptor for RANKL that inhibitsosteoclast differentiation (24,25). We then compared theexpression of these cytokines in Atm2/2 and control osteo-blasts using semi-quantitative RT–PCR assays. No significantdifference was observed for all three cytokines at mRNA levels(Fig. 4C), suggesting that Atm deficiency might not affect thepotential of osteoblasts in supporting osteoclastogenesis.
Steroid hormone shortage in Atm2/2 mice
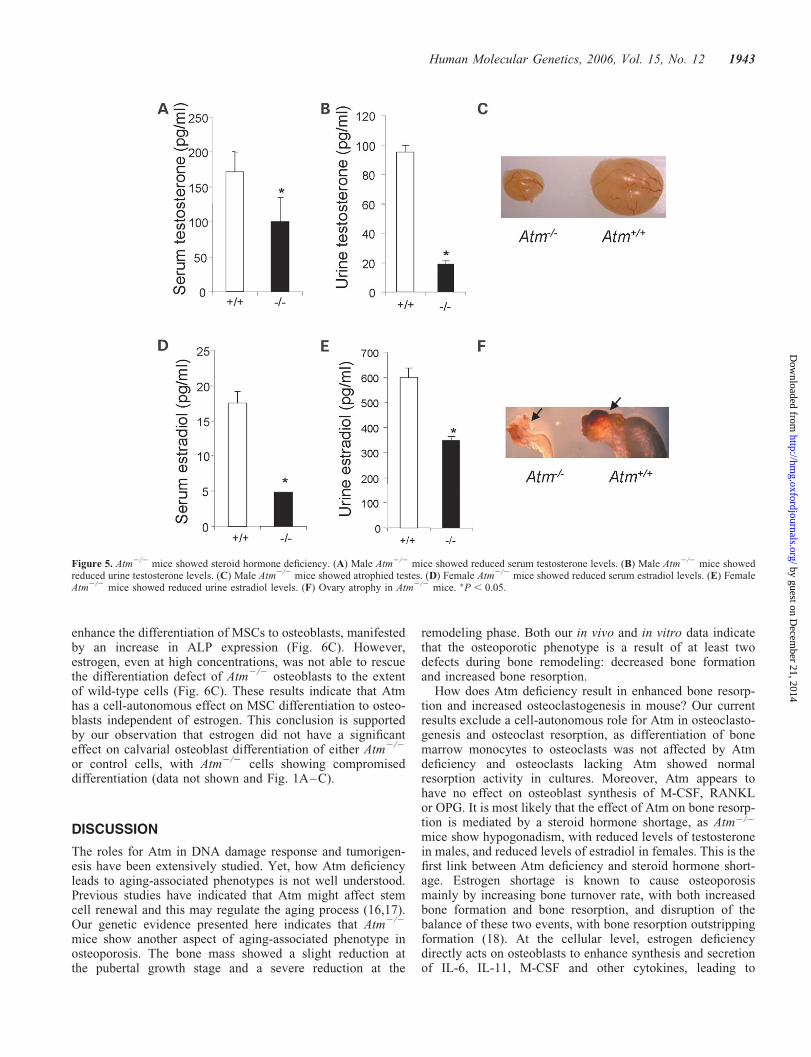
We have shown that Atm2/2 mice display an osteoporoticphenotype and exhibit increased bone resorption. Yet, Atmdeficiency did not affect either osteoclast differentiationfrom bone marrow monocytes or osteoclast resorption activity.These results suggest that Atm deficiency augments boneresorption and osteoclastogenesis without a cell-autonomouseffect on osteoclasts. One of the best studied risk factors forosteoporosis is deficiency of steroid hormones, which arecrucial for proper bone remodeling by inhibiting osteoclasto-genesis. For example, post-menopausal women and ovari-ectomized mice show increased osteoclastogenesis andosteoporosis (31,32). Steroid hormone shortage is known topromote osteoclastogenesis, osteoclast activity and survival.One mechanism is through controlling the production ofcytokines such as M-CSF and RANKL by osteoblasts. Itwas previously reported that Atm2/2 mice are infertile dueto defective gametogenesis (7,33). To test whether production
of steroid hormones is altered in Atm2/2 mice, we measuredserum and urine levels of testosterone and estrogen in maleand female mice, respectively. Male Atm2/2 mice had mark-edly reduced levels of testosterone and significantly atrophiedtestes when compared with the control mice (Fig. 5A–C). Amarked decrease in estradiol levels and atrophied ovarieswere also observed in female Atm2/2 mice (Fig. 5D–F).Because of the crucial roles for testosterone and estradiol inosteoclastogenesis and bone remodeling, the osteoporotic phe-notypes of Atm2/2 mice might be at least partially caused byincreased osteoclastogenesis resulting from hypogonadism.
Hypogonadism can be a result of shortage of gonadotrophichormones that are secreted by pituitary, or due to developmen-tal defect of gonads. Previous studies indicated that Atm2/2
mice are infertile due to defects in gametes maturation asAtm is required for proper DNA recombination and meiosis(6,33,34). To understand what causes hypogonadism inAtm2/2 mice, we analyzed the serum levels of FSH and LH,two of the most important gonadotrophic hormones. It wasfound that Atm2/2 mice showed a significant increase ofserum FSH in both male and female (Fig. 6A and B),suggesting that the hypogonadism is unlikely to be causedby a defect in the hypothalamus–pituitary axis. Instead, therise in FSH reflects a feedback regulation on hypothalamus–pituitary–gonadotrophin release by steroid hormonesdeficiency. This feedback regulation has been observed inpost-menopausal women, gonadectomized mice, as well asin mice with targeted estrogen receptors [reviewed in(35,36)]. On the other hand, LH levels were not significantlyaltered based on two assays (data not shown). Similar caseshave been reported in which FSH was selectively elevatedby gonadectomy (37,38). We cannot exclude the possibilitythat the assays were not sensitive enough for low levels ofLH in mouse serum. Nevertheless, these results overallsuggest that hypogonadism of Atm2/2 mice is most likelydue to an autonomous effect of Atm deficiency on develop-ment of ovary and testis, rather than a defect in gonadotrophinsynthesis and secretion. Moreover, the pituitary appearedmorphologically normal in Atm2/2 mice (data not shown),in contrast to the severely hypotrophied gonads (Fig. 5).This is supported by the observation that even 6-week-oldmice showed upregulation of FSH and that Atm2/2 micedisplay spermatocyte degeneration as early as postnatal day8 and oocyte degeneration in the later stage of embryogenesis.Degeneration leads to extensive apoptotic gamete precursorsand a structural alteration of ovary and testis (33).
Atm-regulated osteoblast differentiationindependent of estrogen
It has been previously reported that estrogen as well as andro-gen can directly affect the osteoblast differentiation, althoughinconsistent results were obtained (18,19). It is believed thatthe variable results are due to difference in the expression ofestrogen receptor isoforms, stages of differentiation and celllines used (19). To test whether the differentiation defect ofAtm2/2 osteoblasts involves steroid hormone shortage, bonemarrow MSCs from Atm2/2 and control mice were treatedwith increasing amounts of estrogen and their differentiationwas compared. It was found that estrogen could indeed
Figure 4. Atm showed no cell-autonomous effect on osteoclast differentiationand resorption. (A) Osteoclast differentiation assays. Bone marrow monocyteswere isolated and plated into 96-well-plates in the presence of 50 ng/mlRANKL and 30 ng/ml M-CSF. Representative images of TRAP staining areshown. (B) Quantitation data from (A) showed no significant difference inthe osteoclast differentiation potential of wild-type and Atm2/2mice (3–4and 7–8-months-old). (C) Pit formation on the dentine discs indicates thatosteoclast resorption activity is not affected by Atm deficiency. (D) RT–PCR assays for the cytokines that are synthesized by osteoblast to regulateosteoclastogenesis.
1942 Human Molecular Genetics, 2006, Vol. 15, No. 12
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from
enhance the differentiation of MSCs to osteoblasts, manifestedby an increase in ALP expression (Fig. 6C). However,estrogen, even at high concentrations, was not able to rescuethe differentiation defect of Atm2/2 osteoblasts to the extentof wild-type cells (Fig. 6C). These results indicate that Atmhas a cell-autonomous effect on MSC differentiation to osteo-blasts independent of estrogen. This conclusion is supportedby our observation that estrogen did not have a significanteffect on calvarial osteoblast differentiation of either Atm2/2
or control cells, with Atm2/2 cells showing compromiseddifferentiation (data not shown and Fig. 1A–C).
DISCUSSION
The roles for Atm in DNA damage response and tumorigen-esis have been extensively studied. Yet, how Atm deficiencyleads to aging-associated phenotypes is not well understood.Previous studies have indicated that Atm might affect stemcell renewal and this may regulate the aging process (16,17).Our genetic evidence presented here indicates that Atm2/2
mice show another aspect of aging-associated phenotype inosteoporosis. The bone mass showed a slight reduction atthe pubertal growth stage and a severe reduction at the
remodeling phase. Both our in vivo and in vitro data indicatethat the osteoporotic phenotype is a result of at least twodefects during bone remodeling: decreased bone formationand increased bone resorption.
How does Atm deficiency result in enhanced bone resorp-tion and increased osteoclastogenesis in mouse? Our currentresults exclude a cell-autonomous role for Atm in osteoclasto-genesis and osteoclast resorption, as differentiation of bonemarrow monocytes to osteoclasts was not affected by Atmdeficiency and osteoclasts lacking Atm showed normalresorption activity in cultures. Moreover, Atm appears tohave no effect on osteoblast synthesis of M-CSF, RANKLor OPG. It is most likely that the effect of Atm on bone resorp-tion is mediated by a steroid hormone shortage, as Atm2/2
mice show hypogonadism, with reduced levels of testosteronein males, and reduced levels of estradiol in females. This is thefirst link between Atm deficiency and steroid hormone short-age. Estrogen shortage is known to cause osteoporosismainly by increasing bone turnover rate, with both increasedbone formation and bone resorption, and disruption of thebalance of these two events, with bone resorption outstrippingformation (18). At the cellular level, estrogen deficiencydirectly acts on osteoblasts to enhance synthesis and secretionof IL-6, IL-11, M-CSF and other cytokines, leading to
Figure 5. Atm2/2 mice showed steroid hormone deficiency. (A) Male Atm2/2 mice showed reduced serum testosterone levels. (B) Male Atm2/2 mice showedreduced urine testosterone levels. (C) Male Atm2/2 mice showed atrophied testes. (D) Female Atm2/2 mice showed reduced serum estradiol levels. (E) FemaleAtm2/2 mice showed reduced urine estradiol levels. (F) Ovary atrophy in Atm2/2 mice. �P , 0.05.
Human Molecular Genetics, 2006, Vol. 15, No. 12 1943
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

increased osteoclastogenesis (19). Estrogen is also believed todirectly affect osteoblast differentiation although no consensushas been reached. We found that estrogen had no significanteffect on calvarial osteoblast differentiation, but showed astimulatory effect on differentiation of bone marrow MSCsinto osteoblasts. This is consistent with previous studiesusing primary bone marrow MSCs (39). Yet Atm2/2 andcontrol cultures displayed a similar response to estrogen,suggesting that Atm plays no role in estrogen-dependentpathways. On the basis of the well-established link betweenhypogonadism and the development of osteoporosis (18,31),we believe that osteoporotic phenotypes of Atm2/2 mice canbe attributed to hypogonadism, and that female Atm2/2
mice can be used as an osteoporosis model for aged post-menopausal women, who suffer from both estrogen deficiencyand impaired bone formation.
Our results also suggest that hypogonadism of Atm2/2 miceis of peripheral origin rather than central origin. If hypogonad-ism is a result of defects at hypothalamus–pituitary, gonado-trophic hormones would be low. Instead, we observed anelevated FSH levels even in young mice. This is an indicationof a feedback regulation of gonadotrophins by steroid hor-mones. Our conclusion is further supported by the observationthat Atm2/2 mice display gamete degeneration/apoptosis inembryogenesis or very early postnatal development (33,35).This study may also provide a clue to how Atm deficiencymight promote the aging process. Aging is accompanied bya marked decline of steroid and other hormones, which arebelieved to possess anti-aging activities (40,41). Further
studies will be necessary to test whether some of the otherphenotypic features observed in Atm2/2 mice could beattributed to hypogonadism.
Atm2/2 mice showed reduced bone formation, with thenumber of osteoblasts per bone surface not altered in thebone remodeling phase. Neither is the number of osteopro-genitor cells altered in the bone marrow, suggesting thatstem cell renewal for osteoblast lineage was not significantlyaffected in Atm2/2 mice. These observations also suggestthat compromised bone formation is unlikely to be causedby the depletion of osteoprogenitor cells or a lack of osteo-blasts. This is further supported by our observation that thebone marrow of mice upto 8 months did not lose the abilityof osteoblastogenesis.
However, calvarial osteoblast differentiation to matureosteocytes is defective in Atm2/2 calvarial osteoblast cultures,suggesting that reduced bone formation in Atm2/2 mice mightbe, at least in part, contributable to osteoblast differentiationdefect. Although steroid hormone shortage has been reportedto affect osteoblast differentiation, our results indicate thatAtm has a cell-autonomous effect on osteoblast differentiation,independent of estrogen. First, calvarial osteoblasts, which arenot responsive to steroid hormones, showed compromiseddifferentiation in the absence of Atm. Secondly, althoughdifferentiation of bone marrow MSC into osteoblast can bepromoted by estrogen, Atm2/2 and control wild-type osteo-blasts displayed similar response to estrogen even at veryhigh concentrations. In other words, estrogen could not over-come the differentiation defect of Atm2/2 osteoblasts.Thirdly, estrogen deficiency is generally accompanied byincreased bone formation in addition to increased bone resorp-tion [(42) and reviewed in (18)]. We observed the opposite,reduced bone formation. This underscores the role for Atmin osteoblast differentiation and function. These resultsclearly indicate that Atm has a cell-autonomous effect onosteoblast differentiation and on bone formation in vivo.This conclusion is further supported by the findings thatdeficiency of c-Abl or p53, both interacting proteins anddownstream targets of Atm (43,44), leads to altered osteoblastdifferentiation and bone formation as well (29,45), withoutdirectly affecting osteoclast differentiation or function.
What are the molecular mechanisms by which Atmdeficiency causes defects in osteoblast differentiation andbone formation? From the available data, we believe thatdown-regulation of Osx might at least partially mediate theeffect. This is because Osx appears to act at the center ofosteoblast differentiation. In Osx-deficient mice, no matureosteoblasts were formed and no bone calcification wasobserved in vivo; up-regulation of Osx, by ectopic expressionor by BMP induction, leads to elevated expression ofosteo-specific markers (22). Osx has been shown to mediatethe effect of p53 deficiency and Cox2 inhibition on osteoblastdifferentiation (29,30). Moreover, c-Abl deficiency leads todefective osteoblast differentiation, associated with reducedexpression of Osx (29). Up to now, how expression of Osxis regulated is not well understood. Osx expression can beinduced by BMP2/4 (23), or repressed by TNF1 and p53(29,46). We found that Atm deficiency diminished the BMPinduced up-regulation of Osx, suggesting that Atm mightparticipate in a step(s) from BMP2-triggered signaling
Figure 6. Upregulation of FSH in Atm2/2 mice. (A) Serum levels of FSH inmale mice. (B) Serum levels of FSH in female mice. (C) Estrogen could notrescue the differentiation defect of bone morrow osteoprogenitor cells ofAtm2/2 mice. Bone marrow cells were isolated from 2–3-month-old miceand plated. Different amounts of estrogen were added to the culture. After 7days, the plates were stained for ALP.
1944 Human Molecular Genetics, 2006, Vol. 15, No. 12
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

pathway to the activation of Osx transcription. But how Atmregulates the transcription of Osx or the BMP signalingpathway awaits further investigation. Atm can be localizedin both the cytoplasm and the nucleus and exerts differentialfunctions (11,47). For example, Atm was reported to regulatethe expression of IGFR in the nucleus in response to DNAdamage and this may contribute to cell growth and survival(48,49). Atm can also phosphorylate eIF-4E-binding protein1 in the cytoplasm and regulate insulin signaling (20). Thebone phenotypes and Atm2/2 osteoblasts might provide a suit-able system to study the crosstalk between BMP signalingpathway and Atm.
Our previous and current studies suggest that both Atm andp53 are able to regulate the expression of Osx and osteoblastdifferentiation (29). It is well documented that in response toDNA damage, especially DSBs, Atm acts upstream of p53and is required for p53 phosphorylation at Ser15, leading tostabilization and/or activation of p53 (9). Here, p53 channelsthe Atm’s effect in DNA damage response. However, thesetwo proteins appear not to use the same pathway to regulateOsx. First, Atm deficiency leads to a downregulation of Osx,whereas p53 deficiency leads to an upregulation. Secondly,p53 was found to directly suppress Osx promoter activity,whereas Ser15 phosphorylation, a major site of Atm, doesnot significantly affect the suppressive activity of p53 in thesame settings (29). Thirdly, there is no evidence to support arelationship between DNA damage and osteoblast differen-tiation. Thus, Atm and p53 appear to regulate Osx expressionin different ways. This is similar to the observation that Atm2/2
MEFs undergo premature senescence, whereas p532/2 MEFscan escape senescence. It is also in line with the concept thatAtm and p53 functionally interact in a more complex mannerin cell proliferation and radiosensitivity (50,51). Their preciserelationship in regulation of Osx expression awaits furtherinvestigation on mice deficient for both Atm and p53 andprimary osteoblasts derived from these mice.
In summary, we defined a novel role for Atm in bone remo-deling. Atm deficiency leads to an osteoporotic phenotype inmouse due to reduced bone formation and increased boneresorption. Atm controls bone formation mainly by regulatingosteoblast differentiation via the transcription factor Osx. Atmcontrols bone resorption through its effect on gonadogenesisand synthesis/secretion of steroid hormones. These resultsindicate that Atm2/2 mice is an osteoporosis animal modelsimilar to that affecting post-menopausal women. Moreover,our results also support an involvement of Atm, a Ser/Thrkinase, in regulating the expression of an osteoblast-specifictranscription factor, osterix.
MATERIALS AND METHODS
Mice and cell cultures
Atm2/2 mice (129S6/SvEvTac-Atmtm1Awb, Jackson Labora-tories) were crossed to C57BL/6 six times. Primary osteoblastswere isolated from newborn pups as previously described (45).Briefly, calvaria from new-born pups were excised andwashed in PBS. They were then digested in MEM containing0.1% collagenase type I and dispase for 10 min at 378C andthe supernatant is discarded. The calvarias were digested
four more times with fresh enzymatic solutions for 15 mineach at 378C and the supernatants were pooled, which werethen spinned down to collect the cells. The cells were culturedin a-MEM with 15% FCS for 1 week until confluency andthen used for experiments.
In vitro osteoblast differentiation assay andCFU assay of bone marrow cells
Calvarial osteoblasts (passage 2) were used for differentiationassays as described (45). In order to limit the potential effectof cell growth on differentiation, 1.5 � 105 cells were platedinto each well of 12-well plate to ensure that the cells wouldbe confluent the following day. They were cultured in differ-entiation medium (aMEM medium containing 50 mg/mlascorbic acid and 10 mM b-glycerol-phosphate) and werere-fed every 3 days. The relative ALP activity is defined asmillimoles of p-nitrophenol phosphate hydrolyzed perminute per milligram of total protein (units). To assaymineral deposition, cells were cultured for 21 or 28 days.The plates were then stained for nodules with Von Kossamethod. To determine the number of osteoprogenitor cells inthe bone marrow, total bone marrow cells were flushed outfrom femurs of Atm2/2 mice and their control littermates atdifferent ages (1.5, 4 and 8 months). A total of 5 � 106 cellswere plated per well of six-well plates and cultured ina-MEM with 15% FCS. After progressive periods of time inculture, the plates were stained for ALP. Colonies with morethan 20 cells were counted.
Osteoclastogenesis and bone resorption assays
For osteoclast differentiation, bone marrow of 3–4-month-oldmice were flushed, the monocyte fraction was isolated by cen-trifugation on a Ficoll plus lymphocyte separation mediumgradient (ICN Biomedicals), washed and seeded at 7.5 � 104
cells/well of 96-well plates and cultured for 7 days in differen-tiation medium (a-MEM containing 10% FCS (Invitrogen),30 ng/ml M-CSF (R&D Systems), and 50 ng/ml solublerecombinant RANKL (Sigma–Aldrich, St Louis, MO,USA). TRAP staining was carried out using an acid phospha-tase kit (Sigma–Aldrich). Osteoclast resorption function wasassessed by a pit formation assay on dentine slices (Osteosite).Monocytes were cultured for 2–3 days in the presence of30 ng/ml M-CSF and 50 ng/ml soluble recombinant RANKL,counted and plated onto dentine slices that were pre-incubatedwith serum for 2 h. After 7 days, the dentine slices were soni-cated in 0.5 M ammonium hydroxide and then stained withGill haematoxylin or Toluidine blue for 2 min, washed withwater and photographed under a light microscope. Theresorbed areas were measured using a densitometry systemand were normalized to the number of osteoclasts in thewell. Urine levels of DPD cross-links were determined usingcommercial kits (Quidel Corporation) and were normalizedto urine creatinine following the manufacturer’s protocols.
Western blot analysis
Cells were lysed in TNEN buffer (50 mM Tris, 150 mM NaCl,5 mM EDTA, 0.5% NP-40, 0.1% Triton X-100) supplemented
Human Molecular Genetics, 2006, Vol. 15, No. 12 1945
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

with 1 mM NaF, Na2VO3, 1 mM PMSF, 1 mg/ml of aprotonin,leupeptin and pepstatin. Protein concentration was determinedusing the Bio-Rad assay. Proteins were resolved by SDS–PAGE and transferred to polyvinylidene difluoride membranes(PVDF, Millipore). Anti-osterix was generated by injectingrabbits with a synthesized peptide (Biogenes). Anti-actin anti-bodies are obtained from Sigma.
RNA isolation and RT–PCR assay
Total mRNA was isolated from osteoblasts growing on 60 mmdishes using TRIzol reagent (GIBCO), subjected to DNasetreatment (Ambion) and quantitated. A total of 5 mg mRNAwas reverse-transcribed into cDNA using AMV (Roche Diag-nostic) reverse transcriptase. The total reaction was used in thePCR to assay for the presence of osterix, Runx2, or actin withthe following primers: Osterix: forward, 50-TGA GGA AGAAGC CCA TTC AC; reverse, 50-ACT TCT TCT CCC GGGTGT G. Runx2: forward, 50-TGG CAG CAC GCT ATTAAA TC; reverse, 50-TCT GCC GCT AGA ATT CAA AA.b-actin: forward, 50-AGA TGT GGA TCA GCA AGC AG;reverse, 50-GCG CAA GTT AGG TTT TGT CA. Atf4:forward, 50-TTC CAC TCC AGA GCA TTC CT; reverse,50-CAG GTG GGT CAT AAG GTT TG. RankL: forward,50-CAG AAG ACA GCA CTC ACT GC; reverse, 50-GAACCC GAT GGG ATG C. Opg: forward, 50-CTG CCT GGGAAG AAG ATC AG; reverse, 50-TTG TGA AGC TGTGCA GGA AC. M-Csf: forward, 50-CTG GAA GGA GGATCA GCA AG; reverse, 50-ATG TCT GAG GGT CTCGAT GG. Procollagen a1: forward, 50-GCC TTG GAGGAA ACT TTG CTT; reverse, 50-GCT TCC CCA TCATCT CCA TTC. PCR was carried out for 30 cycles of dena-turation (948C/30 s), annealing (578C/30 s), extension (728C/1 min) and one cycle of final extension (728C/10 min),which was just enough to detect the PCR products of osterixand Runx2.
The detection and quantification of target mRNA were per-formed with semi-quantitative RT–PCR. The amplificationfor each mRNA was performed in the linear range for RT–PCR by optimizing the template concentration and limitingthe amplification cycles to below 30 to ensure exponentialamplification. RT–PCR results (negative images of gels)were scanned with a Molecular Dynamics scanning densi-tometer. The relative levels of mRNA or protein of interestwere then determined by measuring the intensity of the corre-sponding bands. All values were averages of cell cultures iso-lated from at least three Atm2/2 mice and their controllittermates and were normalized to the constitutive expressionof the housekeeping genes.
Preparation of bone specimens
All mice were labeled with 15 mg/kg of calcein sub-cutaneously (Sigma Chemical Co.) twice in an interval of 7days before sacrifice. The right femur of each animal was dis-sected free of soft tissue and used for measurement of femoralbone density by a dual energy X-ray absorptometer. The righttibia was dissected and cut into three equal parts. The rightproximal tibia and tibial shaft were fixed in 70% ethanol sol-ution for 2 days, and immersed in Villanueva Osteochrome
Bone Stain (Polysciences Inc., Warrington, PA, USA) for5 days. The specimens were dehydrated by sequentialchanges of ascending concentrations of ethanol (70, 95 and100%) and xylene and then embedded in methyl methacrylate(Eastman Organic Chemicals, Rochester, NY, USA). Frontalsections of the proximal tibia were cut at 5 mm using a micro-tome (Leica RM2155, Germany) and cross-sections of thetibial shaft proximal to the tibiofibular junction were cut at40 mm using a diamond wire Histo-Saw machine (DelawareDiamond Knives Inc., DE, USA). All sections were cover-slipped with Eukitt (Calibrated Instruments Inc., Hawthorne,NY, USA) for static and dynamic histomorphometric analysis.
Bone densitometry and histomorphometric analysis
Right femoral bone mineral content (BMC) and bone mineraldensity (BMD) were determined utilizing a HologicQDR-1000W dual-energy X-ray absorptiometer. Themachine was adapted for an ultra-high-resolution mode withline spacing of 0.0254 cm, resolution of 0.0127 cm and a col-limator of 0.9 cm diameter. The bones were placed in a Petridish. To simulate soft tissue density surrounding the bones, tapwater was poured around the bones to achieve a depth of 1 cm.Results are given for BMC and for area; area BMD is calcu-lated as BMC/area. In addition to results for total femur, thedistal and mid-region of the femur were analyzed as sub-regions. Coefficients of variation for these measurements inour laboratory are 0.8, 1.0 and 0.6%, respectively.
Histomorphometric parameters of cancellous and corticalbones in the proximal tibia and tibial shaft were measuredwith a digitizing morphometry system, which consistsof an epifluorescent microscope (Olympus, BH-2), a colorVideo-Camera and a digitizing pad (Numonics 2206)coupled to an IBM computer and a morphometry program‘OsteoMetrics’ (OsteoMetrics Inc., Atlanta, GA, USA).Measured parameters in cortical bone included total tissuearea, periosteal perimeter, marrow area, endosteal perimeter,periosteal and endosteal single and double-labeled perimeters,inter-labeled widths and intra-cortical resorption area. Theywere then used to calculate percent cortical bone area [(totaltissue area 2 marrow area2 intra cortical resorption area)/total tissue area � 100%], percent intra-cortical porosis [(intra-cortical resorption area/cortical area) � 100%], periosteal andendosteal bone formation rate (BFR) [(double-labeledperimetersþ single-labeled perimeters/2) � inter-labeledwidths/interval time/periosteal perimeters] according to thestandard nomenclature (52).
Measured parameters of cancellous bone included totaltissue area, trabecular bone area and perimeter, single anddouble-labeled perimeters and inter-labeled widths. Theywere then used to calculate percent cancellous bone volume(trabecular bone area/total tissue area � 100%) andcancellous BFR [(double-labeled perimetersþ single-labeledperimeters/2) � inter-labeled widths/interval time/trabecularperimeters]. The region of bone measured in all groups is1–4 mM from the growth plate in the proximal tibia. Allmeasurements and calculations were referenced to thestandard nomenclature (52).
1946 Human Molecular Genetics, 2006, Vol. 15, No. 12
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

Measurement of steroid and gonadotrophic hormones
The plasma and urine levels of testosterone and estradiol weredetermined by ELISA following the manufacturers’ protocol(Cayman Chemicals). Samples were collected from 1.5–4-month-old Atm2/2 and their control littermates. Serumlevels of FSH and LH were determined by NationalHormone and Peptide Program (Torrance, CA, USA) andthe levels of LH was also assessed with ELISA kits fromBioserv Diagnostics.
Statistical analysis
Each experiment was repeated with three or more mutant andcontrol mice. Statistical analysis was performed using anunpaired t-test (STATISTICA). Significant association wasdefined when P , 0.05 (�) was compared with control.
ACKNOWLEDGEMENTS
We thank Drs Ben Li, Qiang Yu, Wai Fook Leong, SimonCool and Tej K. Pandita for helpful discussions, Hang InIan, Deyu Cai and Goh Choon Hong for technical support.This work was supported by the Career Enhancement Awardof American Society of Bone and Mineral Research (toB. Li) and the Agency for Science, Technology and Researchof the Republic of Singapore. N. Rasheed, X. Wang and B. Liare adjunct members of the Department of Medicine ofNational University of Singapore. Funding to pay OpenAccess publication charges for this article was provided bythe Career Enhancement Award from ASBMR.
Conflict of Interest statement. The authors declare that theyhave no conflict of interest.
REFERENCES
1. Sedgewick, R. and Boder, E. (1991) In Vinken, P., Bruyn, G. andKlawans, H. (eds), Handbook of Clinical Neurology. Elsevier, NY,USA, pp. 347–423.
2. Rotman, G. and Shiloh, Y. (1998) ATM: from gene to function.Hum. Mol. Genet., 7, 1555–1563.
3. McKinnon, P.J. (2004) ATM and ataxia telangiectasia. EMBO Rep., 5,772–776.
4. Lavin, M.F. and Shiloh, Y. (1997) The genetic defect inataxia-telangiectasia. Annu. Rev. Immunol., 15, 177–202.
5. Savitsky, K., Bar-Shira, A., Gilad, S., Rotman, G., Ziv, Y., Vanagaite, L.,Tagle, D.A., Smith, S., Uziel, T., Sfez, S. et al. (1995) A single ataxiatelangiectasia gene with a product similar to PI-3 kinase. Science, 268,1749–1753.
6. Xu, Y., Ashley, T., Brainerd, E.E., Bronson, R.T., Meyn, M.S. andBaltimore, D. (1996) Targeted disruption of ATM leads to growthretardation, chromosomal fragmentation during meiosis, immune defects,and thymic lymphoma. Genes Dev., 10, 2411–2422.
7. Elson, A., Wang, Y., Daugherty, C.J., Morton, C.C., Zhou, F.,Campos-Torres, J. and Leder, P. (1996) Pleiotropic defects inataxia-telangiectasia protein-deficient mice. Proc. Natl Acad. Sci. USA,93, 13084–13089.
8. Abraham, R.T. (2001) Cell cycle checkpoint signaling through theATM and ATR kinases. Genes Dev., 15, 2177–2196.
9. Kastan, M.B., Lim, D.S., Kim, S.T., Xu, B. and Canman, C. (2000)Multiple signaling pathways involving ATM. Cold Spring Harb. Symp.Quant. Biol., 65, 521–526.
10. Kastan, M.B. and Lim, D.S. (2000) The many substrates and functions ofATM. Nat. Rev. Mol. Cell Biol., 1, 179–186.
11. Yang, D.Q. and Kastan, M.B. (2000) Participation of ATM in insulinsignalling through phosphorylation of eIF-4E-binding protein 1. Nat. CellBiol., 2, 893–898.
12. Schubert, R., Erker, L., Barlow, C., Yakushiji, H., Larson, D., Russo, A.,Mitchell, J.B. and Wynshaw-Boris, A. (2004) Cancer chemoprevention bythe antioxidant tempol in Atm-deficient mice. Hum. Mol. Genet., 13,1793–1802.
13. Barlow, C., Dennery, P.A., Shigenaga, M.K., Smith, M.A., Morrow, J.D.,Roberts, L.J., Wynshaw-Boris, A. and Levine, R.L. (1999) Loss of theataxia-telangiectasia gene product causes oxidative damage in targetorgans. Proc. Natl Acad. Sci. USA, 96, 9915–9919.
14. Ziv, S., Brenner, O., Amariglio, N., Smorodinsky, N.I., Galron, R.,Carrion, D.V., Zhang, W., Sharma, G.G., Pandita, R.K., Agarwal, M. et al.(2005) Impaired genomic stability and increased oxidative stressexacerbate different features of Ataxia-telangiectasia. Hum. Mol. Genet.,14, 2929–2943.
15. Li, B., Wang, X., Rasheed, N., Hu, Y., Boast, S., Ishii, T., Nakayama, K.,Nakayama, K.I. and Goff, S.P. (2004) Distinct roles of c-Abl and Atm inoxidative stress response are mediated by protein kinase C delta. GenesDev., 18, 1824–1837.
16. Ito, K., Hirao, A., Arai, F., Matsuoka, S., Takubo, K., Hamaguchi, I.,Nomiyama, K., Hosokawa, K., Sakurada, K., Nakagata, N. et al. (2004)Regulation of oxidative stress by ATM is required for self-renewal ofhaematopoietic stem cells. Nature, 431, 997–1002.
17. Wong, K.K., Maser, R.S., Bachoo, R.M., Menon, J., Carrasco, D.R.,Gu, Y., Alt, F.W. and DePinho, R.A. (2003) Telomere dysfunction andAtm deficiency compromises organ homeostasis and accelerates ageing.Nature, 421, 643–648.
18. Riggs, B.L., Khosla, S. and Melton, L.J. III, (2002) Sex steroids and theconstruction and conservation of the adult skeleton. Endocr. Rev., 23,279–302.
19. Syed, F. and Khosla, S. (2005) Mechanisms of sex steroid effects on bone.Biochem. Biophys. Res. Commun., 328, 688–696.
20. Yang, X. and Karsenty, G. (2002) Transcription factors in bone:developmental and pathological aspects. Trends Mol. Med., 8, 340–345.
21. Kobayashi, T. and Kronenberg, H. (2005) Minireview: transcriptionalregulation in development of bone. Endocrinology, 146, 1012–1017.
22. Nakashima, K., Zhou, X., Kunkel, G., Zhang, Z., Deng, J.M., Behringer,R.R. and de Crombrugghe, B. (2002) The novel zinc finger-containingtranscription factor osterix is required for osteoblast differentiation andbone formation. Cell, 108, 17–29.
23. Celil, A.B. and Campbell, P.G. (2005) BMP-2 and insulin-like growthfactor-I mediate Osterix (Osx) expression in human mesenchymal stemcells via the MAPK and protein kinase D signaling pathways. J. Biol.Chem., 280, 31353–31359.
24. Khosla, S. (2001) Minireview: the OPG/RANKL/RANK system.Endocrinology, 142, 5050–5055.
25. Boyle, W.J., Simonet, W.S. and Lacey, D.L. (2003) Osteoclastdifferentiation and activation. Nature, 423, 337–342.
26. Manolagas, S.C. and Jilka, R.L. (1995) Bone marrow, cytokines, and boneremodeling. Emerging insights into the pathophysiology of osteoporosis.N. Engl. J. Med., 332, 305–311.
27. Harada, S. and Rodan, G.A. (2003) Control of osteoblast function andregulation of bone mass. Nature, 423, 349–355.
28. Celil, A.B., Hollinger, J.O. and Campbell, P.G. (2005) Osx transcriptionalregulation is mediated by additional pathways to BMP2/Smad signaling.J. Cell Biochem., 95, 518–528.
29. Wang, X., Kua, H.Y., Hu, Y., Guo, K., Zeng, Q., Wu, Q., Ng, H.H.,Karsenty, G., de Crombrugghe, B., Yeh, J. and Li, B. (2006) p53 functionsas a negative regulator of osteoblastogenesis, osteoblast-dependentosteoclastogenesis, and bone remodeling. J. Cell Biol., 172, 115–125.
30. Zhang, X., Schwarz, E.M., Young, D.A., Puzas, J.E., Rosier, R.N. andO’Keefe, R.J. (2002) Cyclooxygenase-2 regulates mesenchymal celldifferentiation into the osteoblast lineage and is critically involved in bonerepair. J. Clin. Invest., 109, 1405–1415.
31. Moggs, J.G., Deavall, D.G. and Orphanides, G. (2003) Sex steroids,ANGELS and osteoporosis. Bioessays, 25, 195–199.
32. Compston, J.E. (2001) Sex steroids and bone. Physiol. Rev., 81, 419–447.33. Barlow, C., Liyanage, M., Moens, P.B., Tarsounas, M., Nagashima, K.,
Brown, K., Rottinghaus, S., Jackson, S.P., Tagle, D., Ried, T. et al. (1998)Atm deficiency results in severe meiotic disruption as early as leptonemaof prophase I. Development, 125, 4007–4017.
Human Molecular Genetics, 2006, Vol. 15, No. 12 1947
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from

34. Barlow, C., Hirotsune, S., Paylor, R., Liyanage, M., Eckhaus, M.,Collins, F., Shiloh, Y., Crawley, J.N., Ried, T., Tagle, D. et al. (1996)Atm-deficient mice: a paradigm of ataxia telangiectasia. Cell, 86,159–171.
35. Rulli, S.B. and Huhtaniemi, I. (2005) What have gonadotrophinoverexpressing transgenic mice taught us about gonadal function?Reproduction, 130, 283–291.
36. Huirne, J.A. and Lambalk, C.B. (2001) Gonadotropin-releasing-hormone-receptor antagonists. Lancet, 358, 1793–1803.
37. Ramaswamy, S., Plant, T.M. and Marshall, G.R. (2000) Pulsatilestimulation with recombinant single chain human luteinizing hormoneelicits precocious sertoli cell proliferation in the juvenile male rhesusmonkey (Macaca mulatta). Biol. Reprod., 63, 82–88.
38. Plant, T.M. and Marshall, G.R. (2001) The functional significance of FSHin spermatogenesis and the control of its secretion in male primates.Endocr. Rev., 22, 764–786.
39. Dang, Z.C., van Bezooijen, R.L., Karperien, M., Papapoulos, S.E. andLowik, C.W. (2002) Exposure of KS483 cells to estrogen enhancesosteogenesis and inhibits adipogenesis. J. Bone Miner. Res., 17, 394–405.
40. Allolio, B. and Arlt, W. (2002) DHEA treatment: myth or reality?Trends Endocrinol. Metab., 13, 288–294.
41. Nawata, H., Yanase, T., Goto, K., Okabe, T. and Ashida, K. (2002)Mechanism of action of anti-aging DHEA-S and the replacement ofDHEA-S. Mech. Ageing Dev., 123, 1101–1106.
42. Usui, M., Yoshida, Y., Tsuji, K., Oikawa, K., Miyazono, K., Ishikawa, I.,Yamamoto, T., Nifuji, A. and Noda, M. (2004) Tob deficiencysuperenhances osteoblastic activity after ovariectomy to block estrogendeficiency-induced osteoporosis. Proc. Natl Acad. Sci. USA, 101,6653–6658.
43. Shafman, T., Khanna, K.K., Kedar, P., Spring, K., Kozlov, S., Yen, T.,Hobson, K., Gatei, M., Zhang, N., Watters, D. et al. (1997) Interactionbetween ATM protein and c-Abl in response to DNA damage. Nature,387, 520–523.
44. Baskaran, R., Wood, L.D., Whitaker, L.L., Canman, C.E., Morgan, S.E.,Xu, Y., Barlow, C., Baltimore, D., Wynshaw-Boris, A., Kastan, M.B.et al. (1997) Ataxia telangiectasia mutant protein activates c-Abl tyrosinekinase in response to ionizing radiation. Nature, 387, 516–519.
45. Li, B., Boast, S., de los, S.K., Schieren, I., Quiroz, M., Teitelbaum, S.L.,Tondravi, M.M. and Goff, S.P. (2000) Mice deficient in Abl areosteoporotic and have defects in osteoblast maturation. Nat. Genet., 24,304–308.
46. Lu, X., Gilbert, L., He, X., Rubin, J. and Nanes, M.S. (2006)Transcriptional regulation of the Osterix (Osx, Sp7) promoter bytumor necrosis factor identifies disparate effects of mitogen-activatedprotein kinase and NFkappaB pathways. J. Biol. Chem., 281,6297–6306.
47. Barlow, C., Ribaut-Barassin, C., Zwingman, T.A., Pope, A.J., Brown,K.D., Owens, J.W., Larson, D., Harrington, E.A., Haeberle, A.M.,Mariani, J. et al. (2000) ATM is a cytoplasmic protein in mouse brainrequired to prevent lysosomal accumulation. Proc. Natl Acad. Sci. USA,97, 871–876.
48. Macaulay, V.M., Salisbury, A.J., Bohula, E.A., Playford, M.P.,Smorodinsky, N.I. and Shiloh, Y. (2001) Downregulation of the type 1insulin-like growth factor receptor in mouse melanoma cells is associatedwith enhanced radiosensitivity and impaired activation of Atm kinase.Oncogene, 20, 4029–4040.
49. Peretz, S., Jensen, R., Baserga, R. and Glazer, P.M. (2001)ATM-dependent expression of the insulin-like growth factor-I receptor ina pathway regulating radiation response. Proc. Natl Acad. Sci. USA, 98,1676–1681.
50. Westphal, C.H., Schmaltz, C., Rowan, S., Elson, A., Fisher, D.E. andLeder, P. (1997) Genetic interactions between atm and p53 influencecellular proliferation and irradiation-induced cell cycle checkpoints.Cancer Res., 57, 1664–1667.
51. Westphal, C.H., Rowan, S., Schmaltz, C., Elson, A., Fisher, D.E. andLeder, P. (1997) atm and p53 cooperate in apoptosis and suppression oftumorigenesis, but not in resistance to acute radiation toxicity. Nat.Genet., 16, 397–401.
52. Parfitt, A.M., Drezner, M.K., Glorieux, F.H., Kanis, J.A., Malluche, H.,Meunier, P.J., Ott, S.M. and Recker, R.R. (1987) Bone histomorphometry:standardization of nomenclature, symbols, and units. Report of theASBMR Histomorphometry Nomenclature Committee. J. Bone Miner.
Res., 2, 595–610.
1948 Human Molecular Genetics, 2006, Vol. 15, No. 12
by guest on Decem
ber 21, 2014http://hm
g.oxfordjournals.org/D
ownloaded from




















