
Apolipoprotein E affects the central nervous system response to injury and the development of...
24
Apolipoprotein E Affects the Central Nervous System Response to Injury and the Development of Cerebral Edema John R. Lynch, MD, 1,2 Jose A. Pineda, MD, 1,3 Duncan Morgan, BS, 2 Lin Zhang, MD, 4 David S. Warner, MD, 1,4 Helen Benveniste, MD, PhD, 4 and Daniel T. Laskowitz, MD 1,2 Apolipoprotein E has been implicated in modifying neu- rological outcome after traumatic brain injury, although the mechanisms by which this occurs remain poorly de- fined. To investigate the role of endogenous apolipopro- tein E following acute brain injury, noninvasive magnetic resonance imaging was performed on anesthetized mice following closed head injury. Effacement of the lateral ventricle was used as a radiographic surrogate for cerebral edema. At 24 hours following injury, apolipoprotein E-deficient animals had a greater degree of cerebral edema as compared to matched controls. In addition, the brains of apolipoprotein E-deficient animals had a signif- icantly greater upregulation of tissue necrosis factor messenger ribonucleic acid as compared to controls as early as 1-hr post injury. Thus, modulation of the endog- enous central nervous system inflammatory response may be one mechanism by which apolipoprotein E affects out- come following acute brain injury. Ann Neurol 2002;51:113–117 There are three common human isoforms of apoli- poprotein E (apoE), designated apoE2, apoE3, and apoE4. 1 Numerous clinical reports have demonstrated that apoE modifies neurological recovery following closed head injury in an isoform-specific manner. 2–5 To model the effect of endogenous apoE on acute brain trauma, C57BL/6 mice and apoE-deficient mice matched for age, sex, and genetic background were subjected to closed head injury using a stereotactically guided pneumatic compression device to exert an acute acceleration/deceleration injury. This injury causes ce- rebral edema, which can increase intracranial hyperten- sion and lead to herniation of brain tissue. The latter represents a significant source of neurological morbid- ity in the clinical setting following closed head injury. In order to obtain additional information regarding the effect of cerebral edema in producing anatomic distor- tions in situ, we utilized magnetic resonance imaging (MRI) as a noninvasive assessment measurement of edema. Astrocytic and microglial activation with the re- sultant secretion of inflammatory mediators is believed to promote breakdown of the blood brain barrier and subsequent development of cerebral edema. In particu- lar, TNF has been suggested to play an important role in this regard. 7 This is of particular interest given recent in vitro and in vivo data suggesting that one role of apoE in the injured central nervous system may be to downregulate glial activation and the endogenous inflammatory response. 8 –12,15 We measured the differ- ential upregulation of TNF messenger RNA (mRNA) after closed head injury in apoE-deficient mice com- pared to wild-type controls. Materials and Methods Animals All experiments were performed with the approval of the Duke University Animal Care and Use Committee. Briefly, 8 –16- week-old male C57-BL/6 mice and apoE-deficient mice matched for gender and age that had been previously back bred for 10 generations to C57BL/6 background were obtained com- mercially from Jackson Laboratories (Bar Harbor, ME). Head Injury Model with Controlled Pneumatic Impact Device Mice were anesthetized with 4.3% isoflurane in oxygen at a FiO2 of 50% in an anesthesia induction box for 90 seconds. The trachea was intubated and the lungs were mechanically ventilated and anesthetized with 1.4% isoflurane in 50% O 2 and 50% N 2 . Body temperature was maintained at 37°C us- ing surface heating/cooling. Each mouse was positioned in a stereotactic device. The top of the skull was exposed to iden- tify anatomical landmarks. Animals were subjected to con- trolled skull impact with a pneumatic impactor (Air-Power, High Point, NC) using a 2.0mm steel tip impounder at a controlled velocity (6.0 0.2m/sec) and vertical displace- ment (3.0mm). The animals were allowed to recover spon- taneous ventilation prior to extubation. Following recovery, mice were allowed free access to food and water. Assessment of Functional Effects of apoE Following Head Injury Neurological severity score: Motor function was assessed at 1 and 24 hours following injury using a modified neuroseverity score that included beam walking on 3cm, 2cm, and 1cm beams, ability to exit from circle (30cm in diameter), pres- ence of seeking behavior, hemiplegia, ability to walk straight, and presence of a startle reflex. This generated a score from 0 (normal) to 10 (moribund). This scale is weighted toward From the 1 Multidisciplinary Neuroprotection Laboratory, and De- partments of 2 Medicine (Neurology), 3 Pediatrics, and 4 Anesthesiol- ogy, Duke University Medical Center, Durham, NC. Received Apr 10, 2001, and in revised form Aug 15, 2001. Ac- cepted for publication Sep 5, 2001. Published online Dec 3, 2001 Address correspondence to Dr Laskowitz, Department of Medicine (Neurology), Box 2900, Duke University Medical Center, Durham, NC 27710. E-mail: [email protected] BRIEF COMMUNICATIONS © 2001 Wiley-Liss, Inc. 113 DOI 10.1002/ana.10098
-
Upload
independent -
Category
Documents
-
view
5 -
download
0
Transcript of Apolipoprotein E affects the central nervous system response to injury and the development of...

Apolipoprotein E Affectsthe Central Nervous SystemResponse to Injury andthe Development ofCerebral EdemaJohn R. Lynch, MD,1,2 Jose A. Pineda, MD,1,3
Duncan Morgan, BS,2 Lin Zhang, MD,4
David S. Warner, MD,1,4 Helen Benveniste, MD, PhD,4
and Daniel T. Laskowitz, MD1,2
Apolipoprotein E has been implicated in modifying neu-rological outcome after traumatic brain injury, althoughthe mechanisms by which this occurs remain poorly de-fined. To investigate the role of endogenous apolipopro-tein E following acute brain injury, noninvasive magneticresonance imaging was performed on anesthetized micefollowing closed head injury. Effacement of the lateralventricle was used as a radiographic surrogate for cerebraledema. At 24 hours following injury, apolipoproteinE-deficient animals had a greater degree of cerebraledema as compared to matched controls. In addition, thebrains of apolipoprotein E-deficient animals had a signif-icantly greater upregulation of tissue necrosis factor �messenger ribonucleic acid as compared to controls asearly as 1-hr post injury. Thus, modulation of the endog-enous central nervous system inflammatory response maybe one mechanism by which apolipoprotein E affects out-come following acute brain injury.
Ann Neurol 2002;51:113–117
There are three common human isoforms of apoli-poprotein E (apoE), designated apoE2, apoE3, andapoE4.1 Numerous clinical reports have demonstratedthat apoE modifies neurological recovery followingclosed head injury in an isoform-specific manner.2–5
To model the effect of endogenous apoE on acutebrain trauma, C57BL/6 mice and apoE-deficient micematched for age, sex, and genetic background weresubjected to closed head injury using a stereotacticallyguided pneumatic compression device to exert an acuteacceleration/deceleration injury. This injury causes ce-
rebral edema, which can increase intracranial hyperten-sion and lead to herniation of brain tissue. The latterrepresents a significant source of neurological morbid-ity in the clinical setting following closed head injury.In order to obtain additional information regarding theeffect of cerebral edema in producing anatomic distor-tions in situ, we utilized magnetic resonance imaging(MRI) as a noninvasive assessment measurement ofedema. Astrocytic and microglial activation with the re-sultant secretion of inflammatory mediators is believedto promote breakdown of the blood brain barrier andsubsequent development of cerebral edema. In particu-lar, TNF� has been suggested to play an importantrole in this regard.7 This is of particular interest givenrecent in vitro and in vivo data suggesting that one roleof apoE in the injured central nervous system may beto downregulate glial activation and the endogenousinflammatory response.8–12,15 We measured the differ-ential upregulation of TNF� messenger RNA (mRNA)after closed head injury in apoE-deficient mice com-pared to wild-type controls.
Materials and MethodsAnimalsAll experiments were performed with the approval of the DukeUniversity Animal Care and Use Committee. Briefly, 8–16-week-old male C57-BL/6 mice and apoE-deficient micematched for gender and age that had been previously back bredfor 10 generations to C57BL/6 background were obtained com-mercially from Jackson Laboratories (Bar Harbor, ME).
Head Injury Model with Controlled PneumaticImpact DeviceMice were anesthetized with 4.3% isoflurane in oxygen at aFiO2 of 50% in an anesthesia induction box for 90 seconds.The trachea was intubated and the lungs were mechanicallyventilated and anesthetized with 1.4% isoflurane in 50% O2
and 50% N2. Body temperature was maintained at 37°C us-ing surface heating/cooling. Each mouse was positioned in astereotactic device. The top of the skull was exposed to iden-tify anatomical landmarks. Animals were subjected to con-trolled skull impact with a pneumatic impactor (Air-Power,High Point, NC) using a 2.0mm steel tip impounder at acontrolled velocity (6.0 � 0.2m/sec) and vertical displace-ment (3.0mm). The animals were allowed to recover spon-taneous ventilation prior to extubation. Following recovery,mice were allowed free access to food and water.
Assessment of Functional Effects of apoE FollowingHead InjuryNeurological severity score: Motor function was assessed at 1and 24 hours following injury using a modified neuroseverityscore that included beam walking on 3cm, 2cm, and 1cmbeams, ability to exit from circle (30cm in diameter), pres-ence of seeking behavior, hemiplegia, ability to walk straight,and presence of a startle reflex. This generated a score from 0(normal) to 10 (moribund). This scale is weighted toward
From the 1Multidisciplinary Neuroprotection Laboratory, and De-partments of 2Medicine (Neurology), 3Pediatrics, and 4Anesthesiol-ogy, Duke University Medical Center, Durham, NC.
Received Apr 10, 2001, and in revised form Aug 15, 2001. Ac-cepted for publication Sep 5, 2001.
Published online Dec 3, 2001
Address correspondence to Dr Laskowitz, Department of Medicine(Neurology), Box 2900, Duke University Medical Center, Durham,NC 27710. E-mail: [email protected]
BRIEF COMMUNICATIONS
© 2001 Wiley-Liss, Inc. 113DOI 10.1002/ana.10098

motor function, and minor modifications of this scale havebeen described previously.6
Magnetic Resonance ImagingAnesthetized, spontaneously breathing mice were scanned us-ing a 7.1, 15cm bore superconducting Oxford magnet withshielded gradient coils controlled by a Signa console (GeneralElectric Medical Systems, Milwaukee, WI). We used a three-dimensional diffusion-weighted spin-echo pulse sequencewith the following acquisition parameters: (a) 2.4-hour scantime; (b) 20ms echo time; (c) 600ms repetition time; (d)diffusion gradients of 35 Gauss/cm applied in the slice di-rection; and (e) b value of 965mm2/sec. All images were ac-quired at a spatial resolution of 58�m � 58�m �469�m � 1.6 � 10�3mm3. The raw data were recon-structed by Fourier transform and displayed as magnitudeimages.
Ventricular volumes were measured on the three-dimensional diffusion data sets by manually outlining regionsof interest in each hemisphere using National Institutes ofHealth Image 1.68.
Wet-to-Dry Method of Assessing BrainWater ContentBrains were removed from mice 24 hours after injury, dis-sected along the midline saggital plane, and immediatelyweighed. As previously described,17 to determine water con-tent, the brains were then placed in a desiccating oven at105°C for 48 hours and then reweighed.
Measurement of Cytokine mRNAAt 1 and 24 hours following injury, cohort animals were per-fused with normal saline and decapitated. The left and righthemispheres were separated and placed in a 3cc volume ofTrizol. Prompt homogenization was performed by rapidlypassing the tissue through an 18g and 22g needle respectivelyuntil virtually no tissue debris was visible (mRNA was quan-tified by RNAse protection assay [RPA]). RNA isolation wasperformed using Trizol reagent manufactured by Gibco-BRL(Carlesbad, CA) and their recommended protocol was fol-lowed. RPA results were resolved using a 5% acrylamide gel,which was subsequently dried and placed on a phosphorim-aging screen for overnight exposure. Numerical data werecollected and analyzed using the Molecular DynamicsStorm� phosphorimaging system and the ImageQuant�(Sunnyvale, CA) software package provided. To normalizefor any potential differences in loading conditions, mRNAwas expressed as ratio of gene of interest to the housekeepinggene GAPDH.
Statistical AnalysisAll results were analyzed using Student’s two-tailed t test.Values are expressed as mean � standard error of the mean.
ResultsPrior to injury, there were no significant differences be-tween C57BL/6 (n � 7) and apoE-deficient (n � 7)mice with respect to weight (32 � 1gm vs 31 � 1gm;p � 0.3) or functional neurological status as deter-
mined by a blinded observer on a 10-point neurologi-cal scale.6
There was no asymmetry between left and right ven-tricular volumes in the two noninjured control groups.However, at 24 hours following injury, we found thatboth apoE-deficient and C57BLJ/6 mice had signifi-cant effacement of the lateral ventricle ipsilateral to theclosed head injury side compared to the right, nonin-jured hemisphere (Fig 1). The apoE-deficient mice hadan average decrease in left lateral ventricular volume of30%, as compared to 15% in C57BL6/J mice (p �0.01). Figure 2 shows diffusion images at the level ofdorsal hippocampus from 3 apoE-deficient and 3C57BLJ/6 mice. All apoE-deficient mice show efface-ment of the left lateral ventricle ipsilateral to the injurysite and a subtle midline shift. In contrast, the onlyone of the C57BLJ/6 mice shows clear ventricle efface-ment (see Fig 2, top). At 6-week follow-up, there wasno longer any evidence of ventricular asymmetry in ei-ther group. Ventricular volumes were symmetric andsimilar to controls, suggesting a complete resolution ofcerebral edema.
To demonstrate that radiographic evidence of ven-tricular effacement is a valid surrogate for cerebraledema, we directly measured water content in the in-jured and control hemispheres. Using the standardwet-to-dry method, we found significantly increasedwater content in the injured hemisphere of the apoE-deficient animals as compared to the wild-type controls(80.12 � 1.22mg vs 78.35 � 1.02mg in apoE-deficient and wild-type controls, respectively; n � 12,p � 0.038. Data expressed as mean � standard errorof the mean). However, there were no significant dif-ferences in the control nonlesioned hemisphere ofapoE-deficient and wild-type animals.
To determine whether animals received a uniforminjury, a blinded observer using a 10-point neurologi-cal scale assessed neurological function after controlledhead injury, in which 0 represents a normal exam, andincreasing scores represent progressive motor deficit.6
At 1 hour following injury, there were no significantdifferences in neurological function between groups(median � 5 � 1 vs 5 � 2 in wild-type versus apoE-deficient animals, respectively; p � 0.5). Animals werereassessed at 24 hours and again there was no differ-ence in motor function between groups (3 � 1 vs 3 �1 in wild-type vs apoE-deficient animals, respectively;p � 0.9), although neurological function had im-proved from baseline in both groups.
To determine whether TNF� was downregulated asa function of endogenous apoE in our model of con-trolled head injury, cohort animals were sacrificed andperfusion fixed at different time points following in-jury. Messenger RNA was extracted from brain ho-mogenates, and TNF� mRNA levels were quantifiedin the injured hemisphere by RNAse protection assay
114 Annals of Neurology Vol 51 No 1 January 2002

Fig 1. Eight representative diffusion magnetic resonance images from a three-dimensional data set obtained 24 hours after closedhead injury in a C57BLJ/6 mouse. The injury site is clearly seen as a high signal intensity area in the most superior part of thecortex (slices 1 and 2). More ventrally the effacement of the left lateral ventricle is clearly seen. We have demonstrated how measur-ing left and right lateral ventricle volume indirectly assessed the amount of edema. The apoE-deficient mice had an average decreasein left lateral ventricular volume of 30%, as compared to 15% in C57BL6/J mice (p � 0.01). Absolute ventricular volumes were2.83 � 0.10mm3 at baseline. After injury the volumes were 2.38 � 0.17mm3 in the wild-type mice and 2.09 � 0.27mm3 inthe apoE-deficient mice. Seven animals were used per group. Data expressed as mean � standard error of the mean.

(RPA). We found that as early as 1 hour following in-jury, apoE-deficient mice had significantly greaterTNF� mRNA levels compared to control mice (ratioTNF�/GAPDH 0.0018 vs 0.0034; p � 0.006; Fig 3).
DiscussionOur observations suggest an increase in hemisphericedema in apoE-deficient animals as compared to wild-type controls at 24 hours following closed head injury.This is consistent with a prior report demonstrating thathistological damage and long-term behavioral sequelaewere more severe in apoE-deficient mice after head in-
jury.16 The two groups of mice in this study receivedthe same initial insult using the pneumatic compressiondevice, but the apoE-deficient mice suffered greater in-juries compared to C57BLJ/6 as evaluated by MRI. Onepotential mechanism by which this may occur is thatapoE downregulates glial activation and subsequent se-cretion of inflammatory cytokines such as TNF�. Thisfunction of apoE is consistent with prior in vitro and invivo data.8–12,15 We did observe a more robust increasein the levels of TNF� mRNA in the brains of apoE-deficient animals, which more than likely reflects an el-evation of TNF� in the brain after closed head injury.
Fig 2. Diffusion images at the level of the dorsal hippocampus acquired 24 hours after closed head injury from 3 apoE-deficientand 3 C57BLJ/6 mice are shown. Left lateral effacement and a subtle midline shift (as indirectly indicated by the red line) can beappreciated in all 3 apoE-deficient mice. In contrast, only 1 (no. 9) of the 3 C57BLJ/6 mice shown demonstrates such changes.
116 Annals of Neurology Vol 51 No 1 January 2002

Although the role of TNF� in the injured centralnervous system has not been fully elucidated, it is up-regulated by neurons and astrocytes following humantraumatic brain injury and appears to play an integralrole in the breakdown of the blood brain barrier andsubsequent development of cerebral edema in hu-mans.13 In addition, inhibition of TNF� in a rodentmodel of closed head injury resulted in decrease ofblood brain barrier breakdown and the subsequent de-velopment of cerebral edema.14 In this study we dem-onstrate that apoE-deficient mice exhibit enhancementof TNF� mRNA synthesis following closed head in-jury relative to wild-type controls.
These results are consistent with prior observationsthat apoE suppresses microglial and astrocytic activa-tion in vitro,8,9,11,12,15 and the inflammatory cyto-kines, including TNF�, are more robustly upregulatedafter injury in apoE-deficient mice in vivo.10 This ex-cess production of TNF� might promote more severeblood brain barrier breakdown and thus more extensiveedema.
Our results suggest that the presence of endogenousapoE might be protective in closed head injury bymodulating glial activation, TNF� production andthus decrease the amount of cerebral edema in theinjured brain. The results of this study may serve asa framework for understanding clinically relevantisoform-specific differences in neurological outcome
following acute brain injury, as well as designing noveltherapeutic strategies in this clinical setting.
This work was supported by the National Institutes of Health(1K08NS01949, T32GM08600, R01NS37235), a Novartis PilotGrant, and two Paul Beeson Physician Faculty Awards to D. Las-kowitz and H. Benveniste. Imaging was performed at Center for invivo Microscopy supported by funds from the National Institutes ofHealth Research Grants NCRR P41 RR05959 (Principle Investiga-tor Dr. G. Allan Johnson).
References1. Weisgraber KH. Apolipoprotein E: structure-function relation-
ships. Adv Protein Chem 1994;45:249–302.2. Sorbi S, Nacmias N, Piacentini S, et al. ApoE as a prognostic
factor for post-traumatic coma. Nature Med 1995;1:852.3. Teasdale, GM, Nicoll, JA, Murray, G, et al. Association of apo-
lipoprotein E polymorphism with outcome after head injury.Lancet 1997;350:1069–1071.
4. Friedman, G, Froom, P, Sazbon, L, et al. ApolipoproteinE-epsilon 4 genotype predicts a poor outcome in survivors oftraumatic brain injury. Neurology 1999;52:244–248.
5. Jordan, BD, Relkin NR, Raydin LD, et al. Apolipoprotein Eepsilon-4 associated with chronic traumatic brain injury in box-ing. JAMA 1997;278:136–140.
6. Chen Y, Constantini S, Trembovler V, et al. An experimentalmodel of closed head injury in mice: pathophysiology, histopa-thology, and cognitive deficits. J Neurotrauma 1996;13:557–568.
7. Ramilo O, Saez-Llorens X, Mertsola J, et al. Tumor necrosisfactor alpha/cachectin and interleukin 1 beta initiate meningealinflammation. J Exp Med 1990;172:497–507.
8. Laskowitz D, Goel S, Bennett ER, et al. Apolipoprotein E sup-presses glial cell secretion of TNF�. J Neuroimmunology 1997;76:70–74.
9. Laskowitz DT, Thekdi A., Thekdi S, et al. Downregulation ofglial activation by apolipoprotein E and apoE-mimetic peptides.Exp Neurol 2001;167:74–85.
10. Lynch JR, Morgan D, Mance J, et al. Apolipoprotein E mod-ulates glial activation and the endogenous central nervous sys-tem response. J Neuroimmunology 2001;114:107–113.
11. Barger SW, Harmon AD. Microglial activation by Alzheimeramyloid precursor protein and modulation by apolipoprotein ENature 1997;388:878–881.
12. Hu J, Ladu MJ, Van Eldik LJ. Apolipoprotein E attenuatesbeta-amyloid-induced astrocyte activation. J Neurochem 1998;71:1626–1634.
13. Ott L, McClain CJ, Gillespie M, Toung B. Cytokines and met-abolic dysfunction after severe head injury. J Neurotrauma1994;11:447–472.
14. Shohami E, Bass R, Wallach D, et al. Inhibition of tumor ne-crosis factor alpha (TNFalpha) activity in rat brain is associatedwith cerebroprotection after closed head injury. J Cereb BloodFlow Metab 1996;16:378–384.
15. Laskowitz DT, Matthew WD, Bennett ER, et al. Endogenousapolipoprotein E suppresses LPS-stimulated microglial nitric-oxide production. Neuroreport 1998;9:615–618.
16. Chen Y, Lomnitski L, Michaelson DM, Shohami E. Motor andcognitive deficits in apolipoprotein-E deficient mice after closedhead injury. Neuroscience 1997;80:1255–1262.
17. Pineda JA, Aono M, Sheng H, et al. Extracellular superoxidedismutase overexpression improves behavioral outcome fromclosed head injury in the mouse. J Neurotrauma 2001;18:625–634.
Fig 3. Baseline brain TNF� messenger RNA levels were notsignificantly different in apoE-deficient vs wild-type mice. At 1hour following closed head injury, TNF� messenger RNA hadgreater expression in the brains of apoE-deficient mice as com-pared to matched wild-type controls (**p � 0.01). To controlfor loading conditions, quantification of genes of interest wererepresented as a ratio to the housekeeping gene GAPDH. Sixanimals were used per time point. Data represented asmean � standard error of the mean.
Lynch et al: Apolipoprotein E Affects the Central Nervous System 117

Novel HeteroplasmicmtDNA Mutation in aFamily with HeterogeneousClinical PresentationsP. Corona, MSc,1 E. Lamantea, MSc,1 M. Greco, PhD,1
F. Carrara, BSc,1 A. Agostino, MSc,1 D. Guidetti, MD,2
M. T. Dotti, MD,3 C. Mariotti, MD,1
and M. Zeviani, MD, PhD1
The protean manifestations of a novel maternally inher-ited point mutation of the mitochondrial genome are re-ported. The proband showed isolated, spastic paraparesis.A brother, who had suffered from a multisystem progres-sive disorder, ultimately died of cardiomyopathy. An-other brother is healthy. The proband’s mother showedtruncal ataxia, dysarthria, severe hearing loss, mental re-gression, ptosis, ophthalmoparesis, distal cyclones, anddiabetes mellitus. A muscle biopsy performed in the pro-band failed to show the morphological abnormalities typ-ical of mitochondrial disorders; the activities of respira-tory chain complexes were normal. However, complex Iand IV activities were low in the muscle homogenate ofthe affected mother and brother. Sequence analysis ofmtDNA showed a heteroplasmic mutation of the tRNAIle
gene (G4284A). The mutation load was approximately55%, 80%, and 90% in the muscle mtDNA of the pro-band, his mother, and his affected brother, respectively.Mutation was undetected in the healthy brother, as wellas in 100 control samples. Several cybrid clones contain-ing homoplasmic mutant mtDNA from the probandshowed significant reductions of complex IV activity andmaximum oxygen consumption rate, compared with ho-moplasmic wild-type clones derived from the same sub-ject.
Ann Neurol 2002;51:118–122
Mutations of mitochondrial DNA (mtDNA) are re-sponsible for a wide spectrum of syndromes, oftencharacterized by a combination of symptoms that affectmuscle, brain, and peripheral nerves, and occasionallythe heart.1,2 In several cases, syndromes are rather well
defined. In other cases, overlap presentations or the un-usual combination of symptoms, slow progression, andqualitative and quantitative variations in differentmembers of the same family can complicate the diag-nosis.2,3 Additional difficulty can be attributable to theabsence or scarcity of diagnostic clues that are consid-ered typical of mitochondrial encephalomyopathies,such as morphological abnormalities in the muscle bi-opsy.4 The family reported exemplifies these diagnosticproblems.
Case ReportThe family tree is reported in Figure 1. The proband,subject III-3, is a 35-year-old man with an 8-year his-tory of progressive spastic paraparesis. He was reportedto have bilateral genu valgum and what was character-ized as a “clumsy walk” since childhood. A brain mag-netic resonance image (MRI) performed when he was31 years old disclosed the presence of a poroencephaliccavitation in the medial part of the left frontal lobe,associated with moderate atrophy of the anterior seg-ment of the corpus callosum. The lesion was attributedto an old, possibly perinatal, infarction in the territoryof the left callosomarginal artery. Nevertheless, the pa-tient’s psychomotor development was reported to benormal. He obtained a master’s degree in life sciencesand, in spite of walking problems, practiced severalsports during adolescence. The clinical examination,carried out when he was 35 years of age, disclosed thepresence of a “pure” spastic paraparesis with bilateralankle clonus and extensor plantar reflexes. No otherneurological or systemic abnormalities were found atphysical examination. An electrocardiogram (EKG) andechocardiogram were both normal. Electromyographyshowed a myopathic pattern; the latencies of visual and
From the 1Division of Biochemistry and Genetics, National Neuro-logical Institute C. Besta, Milan; 2Division of Neurology, PublicHealth Hospital Santa Maria Nuova, Reggio Emilia; 3Neurometa-bolic Unit, Institute of Neurological Sciences, University of Siena,Siena; Italy.
Received Jul 24, 2001, and in revised form Sep 7, 2001. Acceptedfor publication Sep 10, 2001.
Published online Dec 28, 2001
Address correspondence to Dr Zeviani, Divisione di Biochimica eGenetica, Istituto Nazionale Neurologico Carlo Besta, via Celoria11, 20133 Milano, Italy. E-mail: [email protected].
Fig 1. Family tree. The proband is indicated by an arrow.Black symbols indicate the three affected subjects reported inthis study. Asterisk indicates subjects reported to be affected bya neurologic disorder which could not be investigated further(see text). The presence of hearing loss or diabetes mellitus inother members of the pedigree is also indicated.
118 © 2001 Wiley-Liss, Inc.DOI 10.1002/ana.10059

brainstem-evoked potentials were moderately delayed.Blood lactate and pyruvate levels were both normal atrest and after standard exercise. A muscle biopsy wasmorphologically normal, as were the activities of themitochondrial respiratory chain (RC) complexes inboth muscle homogenate (Table) and cultured fibro-blasts (not shown). An older brother of the proband,subject III-2, had died at 27 years of age of heart fail-ure as a result of rapidly progressive dilating cardiomy-opathy. This patient had suffered from petit mal epi-lepsy during infancy and had a single episode ofgeneralized tonic–clonic seizures when he was 17; myo-clonic jerks of the distal segments of upper limbs, eye-lids, and facial muscles developed from age 16 on. Ad-ditional neurological symptoms ensued during thesubsequent years, including severe truncal ataxia, mod-erate dysarthria and dysmetria, bilateral sensorineuralhearing loss, markedly slowed saccades, and change inmood and personality, which evolved into overt mentaldeterioration. Bilateral macular degeneration was notedwhen he was 23 years old, and an episode of transitoryhemiparesis on the right side was reported at age 26years. Computed tomography (CT) scan and MRI ofthe brain displayed marked cortical and subcortical at-rophy. The electroencephalogram showed the presenceof diffuse slow waves with no paroxysms. Visual andbrainstem-evoked potentials were reduced with severelydelayed latencies; nerve conduction velocity was alsomoderately reduced. Additional symptoms included di-abetes mellitus type 2 since age 18 years, the presenceof a single lipoma on the back of the neck, and a hy-pogonadal appearance. Endocrine tests suggested a hy-pogonadotropic hypogonadism attributable to hypo-thalamic dysfunction. At age 25, rapidly progressiveheart failure developed due to hypertrophic-dilatingcardiomyopathy, leading to death at age 27 years. AnEKG performed at age 25 showed the presence of acomplete left bundle branch block and ischemic abnor-malities. The echocardiogram showed a reduced ejec-tion fraction with hypomotility of a dilated and hyper-trophic left ventricle. Blood lactate at rest was 3.5mg% (normal values �2 mg%). A muscle biopsy failed
to show ragged-red fibers. Histochemical analysis wasnot performed. However, biochemical assays, carriedout years later in our laboratory, disclosed a dramaticdecrease in complex IV (COX) activity and a less pro-found decrease of complex I and complex V activities(Table).
The mother of the proband, subject II-3, is a 64-year-old woman with a 10-year history of profound bi-lateral hearing loss, type 2 diabetes mellitus, and acomplex neurological syndrome, including truncalataxia, mild dysarthria, myoclonic jerks of the handsand forearms, proximal muscle weakness, severe oph-thalmoparesis, and mild mental deterioration. Bloodlactate at rest was normal (0.9 mg%). Both the EKGand the echocardiogram showed no abnormalities.MRI of the brain disclosed cortical and subcortical dif-fuse atrophy. A muscle biopsy showed that the homog-enate disclosed a combined reduction of the activitiesof complex I and complex IV (Table).
A younger brother of the proband (subject III-4) is a33-year-old, apparently healthy, man. Other membersof the maternal lineage of this family were reported tobe affected. In particular, subject I-2, the proband’smaternal grandmother, had adult diabetes mellitus andprofound hearing loss. The latter was the major com-plaint of the older maternal aunt of the proband (II-1),whose only daughter (III-1) was reported to be affectedby muscular dystrophy. A maternal uncle (III-5) haddied at 8 years as a result of “encephalopathy.”
MethodsMorphological and Biochemical AnalysesMorphological examination of skeletal muscle and biochem-ical assays of the individual RC and citrate synthase (CS) onmuscle homogenate and digitonin-treated fibroblasts werecarried out as described.5 The maximum oxygen consump-tion rate was measured in intact cells by polarography6 in thepresence of 25 �M dinitrophenol (DNP).
Analysis of mtDNASouthern blot analysis of linearized mtDNA, single-strandconformation polymorphism analysis of the 22 mitochon-
Table. Respiratory Chain Activities in Muscle Homogenate
Pt II-3 Pt III-2 Pt III-3 Control Range
NADH:CoQ1 reductase/CS (complex I) 9.5 10.9 21.4 17–33Succinate dehydrogenase/CS (SDH) 16.6 15.7 17.8 10–20Succinate:CoQ1 reductase/CS (complex II) 27.7 23.3 34.3 18–35DBH2:cyt. c reductase/CS (complex III) 91 124 188 70–150Cytochrome c oxidase/CS (complex IV) 65 5.5 202 80–180ATPase/CS (complex V) 97 82 165 100–200Citrate synthase (CS)a 148 104 120 80–210
aExpressed as nanomoles/min/mg.
CoQ1 � coenzyme Q1; DBH2 � decyl-ubiquinol; cyt. c � cytochrome c.
Corona et al: Novel Mutation in tRNAIle mtDNA Gene 119
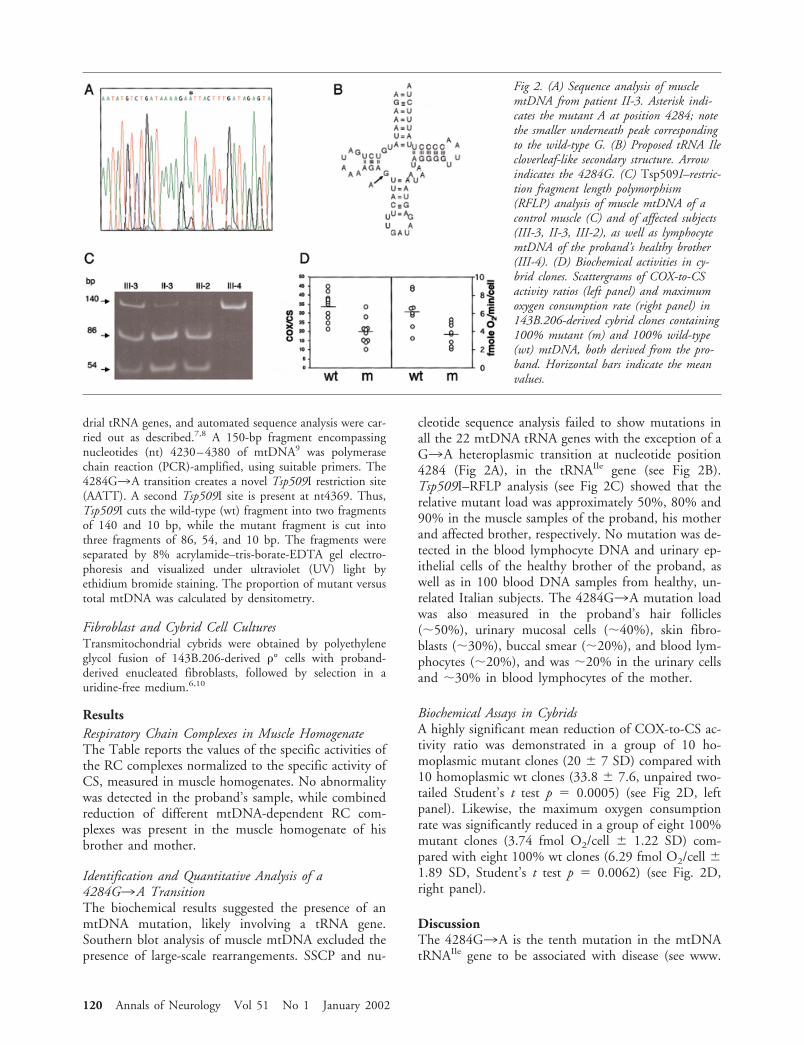
drial tRNA genes, and automated sequence analysis were car-ried out as described.7,8 A 150-bp fragment encompassingnucleotides (nt) 4230–4380 of mtDNA9 was polymerasechain reaction (PCR)-amplified, using suitable primers. The4284G3A transition creates a novel Tsp509I restriction site(AATT). A second Tsp509I site is present at nt4369. Thus,Tsp509I cuts the wild-type (wt) fragment into two fragmentsof 140 and 10 bp, while the mutant fragment is cut intothree fragments of 86, 54, and 10 bp. The fragments wereseparated by 8% acrylamide–tris-borate-EDTA gel electro-phoresis and visualized under ultraviolet (UV) light byethidium bromide staining. The proportion of mutant versustotal mtDNA was calculated by densitometry.
Fibroblast and Cybrid Cell CulturesTransmitochondrial cybrids were obtained by polyethyleneglycol fusion of 143B.206-derived �° cells with proband-derived enucleated fibroblasts, followed by selection in auridine-free medium.6,10
ResultsRespiratory Chain Complexes in Muscle HomogenateThe Table reports the values of the specific activities ofthe RC complexes normalized to the specific activity ofCS, measured in muscle homogenates. No abnormalitywas detected in the proband’s sample, while combinedreduction of different mtDNA-dependent RC com-plexes was present in the muscle homogenate of hisbrother and mother.
Identification and Quantitative Analysis of a4284G3A TransitionThe biochemical results suggested the presence of anmtDNA mutation, likely involving a tRNA gene.Southern blot analysis of muscle mtDNA excluded thepresence of large-scale rearrangements. SSCP and nu-
cleotide sequence analysis failed to show mutations inall the 22 mtDNA tRNA genes with the exception of aG3A heteroplasmic transition at nucleotide position4284 (Fig 2A), in the tRNAIle gene (see Fig 2B).Tsp509I–RFLP analysis (see Fig 2C) showed that therelative mutant load was approximately 50%, 80% and90% in the muscle samples of the proband, his motherand affected brother, respectively. No mutation was de-tected in the blood lymphocyte DNA and urinary ep-ithelial cells of the healthy brother of the proband, aswell as in 100 blood DNA samples from healthy, un-related Italian subjects. The 4284G3A mutation loadwas also measured in the proband’s hair follicles(50%), urinary mucosal cells (40%), skin fibro-blasts (30%), buccal smear (20%), and blood lym-phocytes (20%), and was 20% in the urinary cellsand 30% in blood lymphocytes of the mother.
Biochemical Assays in CybridsA highly significant mean reduction of COX-to-CS ac-tivity ratio was demonstrated in a group of 10 ho-moplasmic mutant clones (20 � 7 SD) compared with10 homoplasmic wt clones (33.8 � 7.6, unpaired two-tailed Student’s t test p � 0.0005) (see Fig 2D, leftpanel). Likewise, the maximum oxygen consumptionrate was significantly reduced in a group of eight 100%mutant clones (3.74 fmol O2/cell � 1.22 SD) com-pared with eight 100% wt clones (6.29 fmol O2/cell �1.89 SD, Student’s t test p � 0.0062) (see Fig. 2D,right panel).
DiscussionThe 4284G3A is the tenth mutation in the mtDNAtRNAIle gene to be associated with disease (see www.
Fig 2. (A) Sequence analysis of musclemtDNA from patient II-3. Asterisk indi-cates the mutant A at position 4284; notethe smaller underneath peak correspondingto the wild-type G. (B) Proposed tRNA Ilecloverleaf-like secondary structure. Arrowindicates the 4284G. (C) Tsp509I–restric-tion fragment length polymorphism(RFLP) analysis of muscle mtDNA of acontrol muscle (C) and of affected subjects(III-3, II-3, III-2), as well as lymphocytemtDNA of the proband’s healthy brother(III-4). (D) Biochemical activities in cy-brid clones. Scattergrams of COX-to-CSactivity ratios (left panel) and maximumoxygen consumption rate (right panel) in143B.206-derived cybrid clones containing100% mutant (m) and 100% wild-type(wt) mtDNA, both derived from the pro-band. Horizontal bars indicate the meanvalues.
120 Annals of Neurology Vol 51 No 1 January 2002

gen.emory.edu/mitomap.html). Four mutations havebeen reported in patients affected by chronic progres-sive external ophthalmoplegia,11–14 while five addi-tional mutations were found in individuals affected bycardiomyopathy of variable severity.15–19 Although thepathogenetic mechanisms of some of these mutationshave not been characterized completely, the tRNAIle
gene can well be considered a major mutational hot-spot in mtDNA disorders. The 4284G3A transitionaffects the boundary between the DHU and the anti-codon stems of the putative tRNAIle cloverleaf, a re-gion of uncertain functional significance, and involvesa poorly conserved nucleotide. Thus, its functionalconsequences cannot be deduced by an obvious delete-rious effect on the structure or function of tRNAIle orby evolutionistic considerations. However, several linesof evidence support the pathogenicity of the4284G3A transition:
1. It was heteroplasmic and, as often observed inpathogenic mutations of mtDNA, the mutantload was higher in a postmitotic tissue (ie, mus-cle), than in rapid turnover tissues.
2. The degree of heteroplasmy in muscle was con-cordant with the clinical severity, which was rel-atively low in the proband, who was affected byisolated spastic paraparesis, higher in his mother,affected by a late-onset multisystem neurologicaldisorder, and even higher in the proband’sbrother, who suffered from juvenile-onset rapidlyprogressive encephalocardiomyopathy.
3. The mutation segregated with the disease, whichwas undetectable in the blood lymphocytes of acohort of 100 control subjects, and in the bloodand urinary epithelium of a normal family mem-ber (subject III-4), although the presence of themutation cannot be excluded in other tissues ofthis subject.
4. The mutation load was correlated with impair-ment of respiratory chain activities in muscle ho-mogenates. In addition, statistically significantdifferences in the activities of COX and maxi-mum oxygen consumption rate were obtained intwo populations of cybrid clones carrying ho-moplasmic mutant and homoplasmic wt mito-chondrial genomes, respectively. As frequentlyobserved in mitochondrial disorders, the thresh-old at which the mutant load leads to an overtbiochemical impairment appears to be higherthan the threshold at which clinical symptomsare produced, as the RC activities were withinthe normal range in the muscle sample from aclinically affected patient, ie, the proband. In ad-dition, some of the 100% mutant cybrid clonesdisplayed biochemical activities within the nor-mal range, suggesting a relatively mild deleterious
effect of the mutation on the RC biochemistry,as measured by our standard assays.
5. The complex clinical and laboratory features inthe mother and affected brother of the probandwere indeed suggestive of a mitochondrial cause.
The onset of rapidly progressive heart failure in sub-ject III-2 confirms the frequent association betweenmutations of tRNAIle gene and cardiomyopathy. Pa-tient III-3, the proband, was affected by isolated spasticparaparesis. In this patient, morphological examinationof the muscle biopsy was completely uninformative.
The possibility of a mitochondrial cause was consid-ered only for the presence of a suggestive family his-tory. A causative link between the poroencephalic le-sion shown by MRI and the mtDNA mutation foundin this patient remains an interesting, but unproved,hypothesis. Although pyramidal signs, including spasticparaparesis, have been reported in mtDNA-related dis-ease, they are usually accompanied by other neurolog-ical and extraneurological symptoms. We cannot ex-clude that additional clinical features will present in thefuture in our patient, but it is tempting to speculatethat his monosymptomatic, relatively benign, clinicalcourse can be correlated with the lower mutation loadfound in this subject, compared with the mutationload detected in his more severely affected relatives.
This work was supported by Fondazione Telethon-Italy (1180) (toM.Z.), by Ricerca Finalizzata Ministero Sanita. (ICS 030.3/RF98.37), and by Fondazione Pierfranco e Luisa Mariani.
We are indebted to Ms B Geehan for revising the manuscript. Wethank Dr Thomas Klopstock for critical discussion and Dr MarinaMora for technical advice.
References1. Hirano M, Davidson M, DiMauro S. Mitochondria and the
heart. Curr Opin Cardiol 2001;16:201–210.2. Smeitink J, van den Heuvel L, DiMauro S. The genetics and
pathology of oxidative phosphorylation. Nature Rev Genet2001;2:342–352.
3. DiMauro S, Andreu AL. Mutations in mtDNA: are we scrapingthe bottom of the barrel? Brain Pathol 2000;10:431–441.
4. Zeviani M, Tiranti V, Piantadosi C. Mitochondrial disorders.Medicine 1998;77:59–72.
5. Tiranti V, Munaro M, Sandona D, et al. Nuclear DNA originof cytochrome c oxidase deficiency in Leigh’s syndrome: geneticevidence based on patient’s derived rho° transformants. HumMol Genet 1995;4:2017–2023.
6. Mariotti C, Tiranti V, Carrara F, et al. Defective respiratorycapacity and mitochondrial protein synthesis in transformantcybrids harboring the tRNA leu (UUR) mutation associatedwith maternally inherited myopathy and cardiomyopathy.J Clin Invest 1994;93:1102–1107.
7. Zeviani M, Moraes CT, DiMauro S, et al. Deletions of mito-chondrial DNA in Kearns-Sayre syndrome. Neurology 1988;38:1339–1346.
Corona et al: Novel Mutation in tRNAIle mtDNA Gene 121

8. Tiranti V, Carrara F, Confalonieri P, et al. A novel mutation(8342G3A) in the mitochondrial tRNA(Lys) gene associatedwith progressive external ophthalmoplegia and myoclonus.Neuromusc Disord 1999;9:66–71.
9. Anderson S, Bankier AT, Barrell BG, et al. Sequence and or-ganization of the human mitochondrial genome. Nature 1981;290:457–465.
10. Tiranti V, Munaro M, Sandona D, et al. Nuclear DNA originof cytochrome c oxidase deficiency in Leigh’s syndrome: geneticevidence based on patient’s derived rho0 transformants. HumMol Genet 1995;4:2017–2023.
11. Chinnery PF, Johnson MA, Taylor RW, et al. A novel mitochon-drial tRNA isoleucine gene mutation causing chronic progressiveexternal ophthalmoplegia. Neurology 1997;49:1166–1168.
12. Silvestri G, Servidei S, Rana M, et al. A novel mitochondrialDNA point mutation in the tRNA(Ile) gene is associated withprogressive external ophtalmoplegia. Biochem Biophys ResCommun 1996;220:623–627.
13. Taylor RW, Chinnery PF, Bates MJ, et al. A novel mitochon-drial DNA point mutation in the tRNA(Ile) gene: studies in apatient presenting with chronic progressive external ophthalmo-plegia and multiple sclerosis. Biochem Biophys Res Commun1998;243:47–51.
14. Franceschina L, Salani S, Bordoni A, et al. A novel mitochon-drial tRNA(Ile) point mutation in chronic progressive externalophthalmoplegia. J Neurol 1998;245:755–758.
15. Taniike M, Fukushima H, Yanagihara I, et al. MitochondrialtRNA Ile mutation in fatal cardiomyopathy. Biochem BiophysRes Commun 1992;186:47–53.
16. Merante F, Myint T, Tein I, et al. An additional mitochondrialtRNA(Ile) point mutation (A-to-G at nucleotide 4295) causinghypertrophic cardiomyopathy. Hum Mutat 1996;8:216–222.
17. Casali C, Santorelli FM, D’Amati G, et al. A novel mtDNApoint mutation in maternally inherited cardiomyopathy. Bio-chem Biophys Res Commun 1995;213:588–593.
18. Tanaka M, Ino H, Ohno K, et al. Mitochondrial mutation infatal infantile cardiomyopathy. Lancet 1990;336:1452.
19. Santorelli FM, Mak SC, Vazquez-Acevedo M, et al. A novelmitochondrial DNA point mutation associated with mitochon-drial encephalocardiomyopathy. Biochem Biophys Res Com-mun 1995;216:835–840.
Increase in Hand MuscleStrength of Stroke Patientsafter SomatosensoryStimulationAdriana B. Conforto, MD, Alain Kaelin-Lang, MD,and Leonardo G. Cohen, MD
It has been proposed that somatosensory input in theform of peripheral nerve stimulation can influence func-tional measures of motor performance. We studied theeffects of median nerve stimulation on pinch musclestrength (a function mediated predominantly by mediannerve innervated muscles) in the affected hand of chronicstroke patients. A 2-hour period of median nerve stimu-lation elicited an increase in pinch strength that outlastedthe stimulation period. The improvement in musclestrength correlated with stimulus intensity and was iden-tified in the absence of motor training. These results sug-gest that somatosensory stimulation may be a promisingadjuvant to rehabilitation of the motor deficits in strokepatients.
Ann Neurol 2002;51:122–125
Somatosensory input can modulate reorganization insensorimotor cortical and subcortical structures.1–4 Ithas been suggested that somatosensory input in theform of peripheral nerve stimulation (PNS) could con-tribute to improved motor function after stroke.5 Here,we studied the effects of median nerve stimulation(MNS) on pinch strength in the affected hand ofchronic stroke patients in a randomized, crossover de-sign.
Patients and MethodsEight patients (7 men, 1 woman; mean age 65 years; range38–81 years) with hemiparesis caused by ischemic strokeparticipated in this study. The protocol was approved by theNINDS Investigational Review Board, and all subjects gavewritten informed consent. Lesions identified on computer-ized tomography or magnetic resonance imaging were lo-cated in the left internal capsule (3), left pons (1), right in-
From the Human Cortical Physiology Section, NINDS, NationalInstitutes of Health, Bethesda, MD.
Received Jun 29, 2001, and in revised form Sep 13. Accepted forpublication Sep 14, 2001.
Published online Dec 3, 2001
Address correspondence to Dr Cohen and Dr Kaelin-Lang, HumanCortical Physiology Section, National Institutes of Health, Building10, Room 5N23, 10 Center Drive, MSC 1430, Bethesda, MD20892-1430. E-mail: [email protected]
122 © 2001 Wiley-Liss, Inc.DOI 10.1002/ana.10070

ternal capsule and basal ganglia (2), right frontoparietalcortex, basal ganglia and internal capsule (1), and right pons(1). The average time after stroke was 5 years and 6 months(range 14 months to 7 years).
Previously, we performed a pilot study and found that a2-hour period of median nerve stimulation resulted in anaverage increase of 2.55 � 0.9 Newtons (N) (mean � stan-dard error [SE]) in pinch strength in 5 chronic stroke pa-tients (unpublished observations). Based on these preliminaryresults, we planned the present study in a randomized, pro-spective, crossover design.
Each patient participated in two different sessions sepa-rated by at least 24 hours: 2-hour MNS and 2-hour controlstimulation (CS). Patients were comfortably seated and wereinstructed to remain at rest. Background EMG activity re-corded from surface electrodes was continuously monitoredduring the experiments. In both sessions, silver-silver surfacechloride electrodes (diameter 10mm) were optimally placedto stimulate the median nerve at the wrist in the affectedarm. Trains of electrical stimulation were delivered at 1Hz(Grass stimulator S8800 with stimulus isolation unit [SIU],Grass Instrument Division, Astro-Med Inc., West Warwick,RI). Each train consisted of five single pulses of 1ms dura-tion delivered at 10Hz.5
These stimulus parameters are thought to preferentially
activate large cutaneous and proprioceptive sensory fibers.6
In the MNS session, stimulus intensity was gradually in-creased until the patients reported strong paresthesias in themedian nerve territory in the absence of pain (Fig 1). Aver-age stimulus intensity in the MNS session was 47 � 8.3%above the minimal stimulus intensity required to elicit par-esthesias (mean � SE; range 22–83%). This intensity ofstimulation usually elicited compound muscle action poten-tials smaller than 100�V from the abductor pollicis brevismuscle in the absence of visible finger movements. In the CSsession, stimulus intensity was kept immediately below thatrequired to elicit paresthesias. None of the patients reportedparesthesias in the absence of electrical stimulation. In theneurological exam, Patient 1 had slightly decreased joint po-sition sense, and Patient 8 had tactile hypoesthesia in theaffected hand. Joint position sense, vibration, tactile, andpain sensation were normal in the affected hand of the other6 patients.
Maximal key pinch strength7 of the affected hand wasmeasured according to a protocol that exhibits good validityand test-retest reliability7,8 (Jamar dynamometer, SammonsPreston, Inc., Bolingbrook, IL). Patients held the arm of thedynamometer between the lateral aspect of the middle pha-lanx of the index finger and the thumb pad and were in-structed to squeeze as hard as they could. Muscle strengthmeasurements of five consecutive trials were averaged. Oneinvestigator recorded and analyzed muscle strength measure-ments blind to the intervention type while another investi-gator administered the interventions.
The order of the sessions was randomized. In the first ses-sion, 5 patients received CS and 3 patients received MNS.Two-tailed, paired t tests were used to compare musclestrength measurements before and after interventions (datanormally distributed according to Shapiro-Wilk test, signifi-cance level set to 0.05). Patient 8 was excluded from thismain comparison because of poor compliance during the CSsession. Linear regression analysis was used to investigate therelation between changes in muscle strength and stimulus in-tensity in the MNS sessions (n � 8).
ResultsMuscle strength measurements recorded before the CSand MNS sessions were comparable: 54.40 � 10.63N
Fig 1. Representation of hand areas where patients reportedparesthesias during the median nerve stimulation session.
Fig 2. Absolute changes in muscle strength(Newtons) in the control stimulation ses-sion and the median nerve stimulationsession (n � 7). Error bars represent stan-dard errors of the mean.
Conforto et al: Somatosensory Stimulation of Stroke Patients 123

(mean � SE; range 22.56–103.59N) and 49.72 �8.09N (mean � SE; range 24.53–91.43N), respec-tively (p � 0.22), indicating similar baseline condi-tions in both sessions. Stimulation resulted in an in-crease in pinch strength of 2.41 � 0.74N (p � 0.017)in the MNS session and in a nonsignificant decrease of1.07 � 2.4N (p � 0.67) in the CS session (Fig 2). Inan “intent to treat analysis,” in the control interventionthere were no significant changes in strength with apaired t test (p � 0.62), whereas in the median nervestimulation session there was a significant increase of2.57N (p � 0.006).
Following the MNS session, 2 patients spontaneouslyreported that they could “write better” and “hold ob-jects and play cards more accurately”; this perceptionlasted for approximately 24 hours. No patients re-ported any changes after the CS session. The increasedpinch muscle strength identified in the MNS sessioncorrelated well with the intensity of MNS (r � 0.729,p � 0.04, Fig 3).
DiscussionThe main result of this study is that a 2-hour period ofmedian nerve stimulation increased pinch musclestrength in the affected hand of stroke patients. Thiseffect was clearly identifiable in 6 of 8 individuals andoutlasted the stimulation period. The magnitude of theimprovement in pinch strength was similar in this ran-domized crossover study and in the open-label pilot ex-periments that we had previously performed. Addition-ally, increase in muscle strength was proportional tothe intensity of sensory stimulation, suggesting a causalrelationship. These results are consistent with emerging
evidence that somatosensory input can modify motorfunction.9,10
Prior experiments demonstrated motor improvementin the hand of stroke patients after a combination ofmotor training and manipulation of sensory input(Muellbacher and colleagues, unpublished observa-tions) or electrical muscular stimulation over a periodof several weeks or months.11,12
Hand motor deficits play an important role in strokedisability.13 In some patients, rehabilitative strategiesthat are based on motor practice can be difficult orimpossible to implement because of severe muscleweakness. Our findings in a small sample of stroke pa-tients indicate that a single session of predominantlysensory nerve stimulation can improve pinch musclestrength in the absence of practice. This type of inter-vention may be a promising adjuvant to enhance neu-rorehabilitative strategies when hand weakness makesmotor training difficult or impossible.
Supported by the National Institutes of Neurological Diseases andStroke Intramural Program.
We thank Mark Hallett and Carolyn Wu for helpful discussion andDevera G. Schoenberg for skillful editing.
References1. Asanuma H. Functional role of sensory inputs to the motor
cortex. Prog Neurobiol 1981;16:241–262.2. Kaas JH. Plasticity of sensory and motor maps in adult mam-
mals. Annu Rev Neurosci 1991;14:137–167.3. Recanzone GH, Allard TT, Jenkins WM, Merzenich MM. Re
ceptive-field changes induced by peripheral nerve stimulation inSI of adult cats. J Neurophysiol 1990;63:1213–1225.
Fig 3. Relation between stimulus intensity(intensity of median nerve stimulation[MNS] relative to the threshold to elicitparesthesias in each individual) andchanges in pinch muscle strength (pinchmuscle strength after median nerve stimu-lation relative to prestimulation levels) inthe MNS session (n � 8).
124 Annals of Neurology Vol 51 No 1 January 2002

4. Fox K, Glazewski S, Schulze S. Plasticity and stability of so-matosensory maps in thalamus and cortex. Curr Opin Neuro-biol 2000;10:494–497.
5. Ridding MC, Brouwer B, Miles TS, et al. Changes in muscleresponses to stimulation of the motor cortex induced by periph-eral nerve stimulation in human subjects. Exp Brain Res 2000;131:135–143.
6. Panizza M, Nilsson J, Roth BJ, et al. Relevance of stimulusduration for activation of motor and sensory fibers: implicationsfor the study of H-reflexes and magnetic stimulation. Electro-encephalogr Clin Neurophysiol 1992;85:22–29.
7. Mathiowetz V, Kashman N, Volland G, et al. Grip and pinchstrength: normative data for adults. Arch Phys Med Rehabil1985;66:69–74.
8. Mathiowetz V, Weber K, Volland G, Kashman N. Reliabilityand validity of grip and pinch strength evaluations. J HandSurg [Am] 1984;9:222–226.
9. Fraser C, Power M, Hobday D, et al. Driving plasticity in adulthuman motor cortex improves functional performance after ce-rebral injury [abstract]. In: Proceedings of the 15th Interna-tional Congress of Clinical Neurophysiology, May 2001; Bue-nos Aires, Argentina.
10. Struppler A, Jakob C, Muller-Barna P, et al. Eine neue Meth-ode zur Fruhrehabilitation zentralbedingter Lahmungen vonArm und Hand mittels Magnetstimulation. Z EEG EMG1996;27:151–157.
11. Cauraugh J, Light K, Kim S, et al. Chronic motor dysfunctionafter stroke: recovering wrist and finger extension byelectromyography-triggered neuromuscular stimulation. Stroke2000;31:1360–1364.
12. Dimitrijevic MM, Stokic DS, Wawro AW, Wun CC. Modifi-cation of motor control of wrist extension by mesh-glove elec-trical afferent stimulation in stroke patients. Arch Phys MedRehabil 1996;77:252–258.
13. Whitall J, McCombe Waller S, Silver KH, Macko RF. Repet-itive bilateral arm training with rhythmic auditory cueing im-proves motor function in chronic hemiparetic stroke. Stroke2000;31:2390–2395.
Selective HippocampalNeuron Loss in Dementiawith Lewy BodiesAntony J. Harding, PhD, Bronwyn Lakay, BSc(Hons),and Glenda M. Halliday, PhD
Hippocampal volume and neuron number were measuredusing stereological techniques in pathologically con-firmed dementia with Lewy bodies (n � 8), Parkinson’sdisease only (n � 4), and controls (n � 9). We, andothers, have previously shown considerable cell loss inthe CA1 and subiculum subregions in Alzheimer’s dis-ease. In contrast, these regions were spared in dementiawith Lewy bodies where a selective loss of lower presub-iculum pyramidal neurons was found. These findingssuggest a selective loss of frontally projecting hippocam-pal neurons in dementia with Lewy bodies versus thoseprojecting to temporal lobe regions in Alzheimer’s dis-ease.
Ann Neurol 2002;51:125–128
The hippocampus is a complex structure with multipleanatomical and functional compartments. Degenera-tion of the CA1 and subiculum subregions underliesthe dementia found in Alzheimer’s disease (AD),1–3
with gross atrophy of the hippocampus a marker ofthis disease.4–8 At end stage, similar hippocampal at-rophy is found in dementia with Lewy bodies (DLB),9
but the CA1 and subiculum are spared.2,10 The hip-pocampal atrophy in DLB correlates with atrophy andLewy body formation in the frontal lobes, as well aswith the severity of Lewy neurite formation in theCA2/3 subregions of the hippocampus.9 This suggeststhat the subregions of the hippocampus affected inDLB differ significantly from those affected in AD.The present study quantifies the density and numberof neurons in the different hippocampal subdivisions(CA1, CA2, CA3, CA4, dentate gyrus, subiculum, andpresubiculum) comparing cases with DLB to age- andsex-matched controls, nondemented cases of Parkin-son’s disease (PD) only, and previous data publishedon cases with AD.1–3
From the Prince of Wales Medical Research Institute and Universityof New South Wales, Sydney, Australia.
Received Jun 8, 2001, and in revised form Sep 20. Accepted forpublication Sep 20, 2001.
Published online Dec 3, 2001
Address correspondence to Dr Harding, Prince of Wales MedicalResearch Institute, Barker Street, Randwick, Sydney, NSW 2031,Australia. E-mail: [email protected]
© 2001 Wiley-Liss, Inc. 125DOI 10.1002/ana.10071
Materials and MethodsCases were selected using standardized clinicopathologicalprocedures from our regional brain donor program (ap-proved by Human Ethics Committees) as previously de-tailed.9 Cases were excluded if they had significant neuropa-thology (eg, cerebral infarction, head injury, or hepaticencephalopathy) other than those related to Parkinson’s dis-ease (PD) or AD. Four cases without dementia had PD only,8 cases had DLB,11,12 and 9 age- and sex-matched controlswere selected. Neocortical neurofibrillary tangles were absentin all cases and all PD only and 2 DLB cases had no neuriticplaque. Three DLB cases reached Consortium to Establish aRegistry for Alzheimer’s Disease (CERAD) criteria for prob-able AD and the remaining three reached CERAD criteriafor definite AD. There were no significant differences in ageor postmortem delay between the groups (Table).
Brains were fixed in 15% buffered formalin for 2 weeks,the anteroposterior dimensions of each hemisphere recorded,and the cerebrum embedded in agar, prior to being cut into3mm thick coronal slices. All tissue blocks containing theright hippocampus were dissected from the coronal brainslices, cryoprotected and 50�m-thick sections cut from themore posterior end of each hippocampal block using a freez-ing microtome. Sections were mounted onto glass slides,stained with cresyl violet, and coverslipped using DPXmountant.
All tissue analyses were performed blinded to classifica-tion. Quantitation was performed using well-establishedpublished methods.3,9,13 Briefly, the boundaries of the hip-pocampal subdivisions were delineated (dentate gyrus, CA1,
CA2-3, CA4, subiculum, and presubiculum) according tothe criteria of Duvernoy14 and Amaral and Insausti.15 Thecross-sectional areas of the hippocampal subdivisions in eachsection were determined by tracing their boundaries using amicrofiche reader at 19 to 45� magnification, and applyingpoint counting techniques. The volume of each hippocampalsubdivision was determined by multiplying the sum of thecross-sectional areas by the distance between the sections us-ing Cavalieri’s principle. The optical disector technique, us-ing the full section thickness (50�m) as the disector height,was used to estimate neuronal number for each hippocampalregion, as previously described in detail.3,13 Between cases,the number of sections quantified varied from 9 to 12, andthe number of disector frames per subregion varied from 14to 136. The number of nucleolated neurons within the in-clusion boundaries of the frame varied between 93 and 276for the dentate gyrus; 97 and 627 for the CA1; 84 and 293for the CA2-3; 102 and 243 for the CA4; 87 and 287 forthe subiculum; and 93 and 218 for the presubiculum. Re-peated measures of the neuronal number in multiple sectionsfrom multiple cases always gave similar results, even betweendifferent investigators. Neuron density (coefficient of errorrange 0.035–0.054) was determined from the total numberof neurons within the sample volume. Regional neuronalnumber was estimated by multiplying the density and vol-ume.
Statistical analysis was performed on Statview 5.0 program(Abacus Concepts, Berkeley, CA). Group differences wereanalyzed using analysis of variance (ANOVA) with post-hocFisher’s least significant difference test. Means � standard
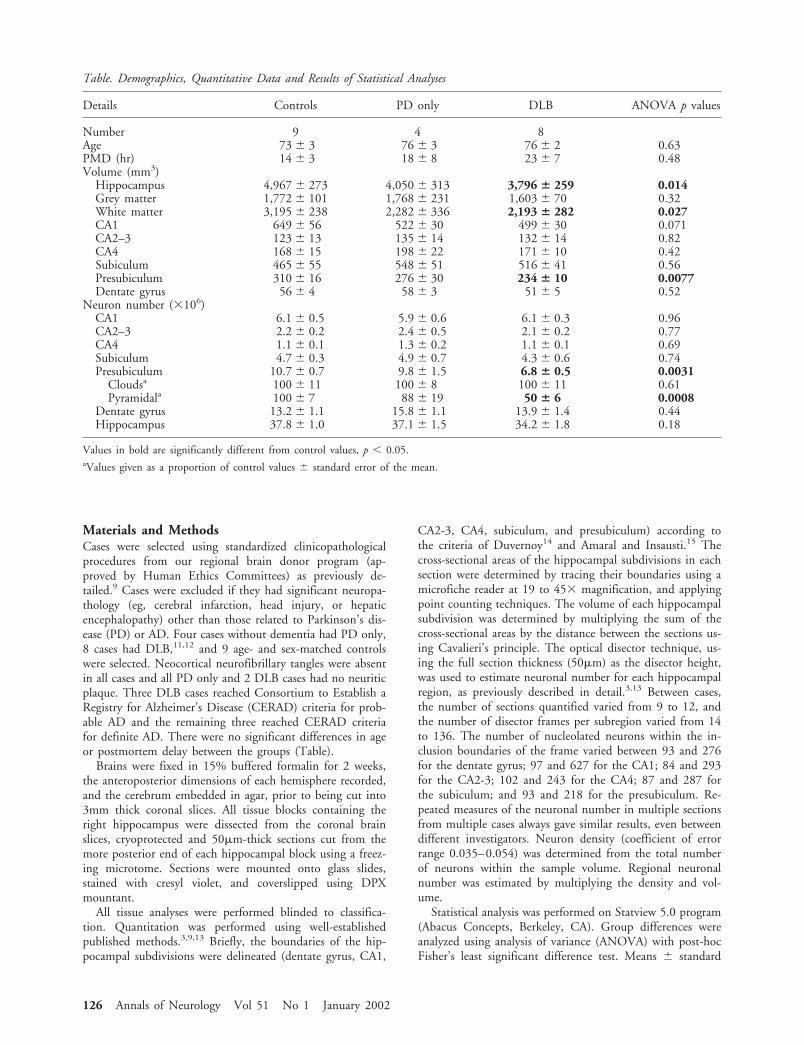
Table. Demographics, Quantitative Data and Results of Statistical Analyses
Details Controls PD only DLB ANOVA p values
Number 9 4 8Age 73 � 3 76 � 3 76 � 2 0.63PMD (hr) 14 � 3 18 � 8 23 � 7 0.48Volume (mm3)
Hippocampus 4,967 � 273 4,050 � 313 3,796 � 259 0.014Grey matter 1,772 � 101 1,768 � 231 1,603 � 70 0.32White matter 3,195 � 238 2,282 � 336 2,193 � 282 0.027CA1 649 � 56 522 � 30 499 � 30 0.071CA2–3 123 � 13 135 � 14 132 � 14 0.82CA4 168 � 15 198 � 22 171 � 10 0.42Subiculum 465 � 55 548 � 51 516 � 41 0.56Presubiculum 310 � 16 276 � 30 234 � 10 0.0077Dentate gyrus 56 � 4 58 � 3 51 � 5 0.52
Neuron number (�106)CA1 6.1 � 0.5 5.9 � 0.6 6.1 � 0.3 0.96CA2–3 2.2 � 0.2 2.4 � 0.5 2.1 � 0.2 0.77CA4 1.1 � 0.1 1.3 � 0.2 1.1 � 0.1 0.69Subiculum 4.7 � 0.3 4.9 � 0.7 4.3 � 0.6 0.74Presubiculum 10.7 � 0.7 9.8 � 1.5 6.8 � 0.5 0.0031
Cloudsa 100 � 11 100 � 8 100 � 11 0.61Pyramidala 100 � 7 88 � 19 50 � 6 0.0008
Dentate gyrus 13.2 � 1.1 15.8 � 1.1 13.9 � 1.4 0.44Hippocampus 37.8 � 1.0 37.1 � 1.5 34.2 � 1.8 0.18
Values in bold are significantly different from control values, p � 0.05.aValues given as a proportion of control values � standard error of the mean.
126 Annals of Neurology Vol 51 No 1 January 2002
errors are given for all variables and a p value less than 0.05accepted as statistically significant.
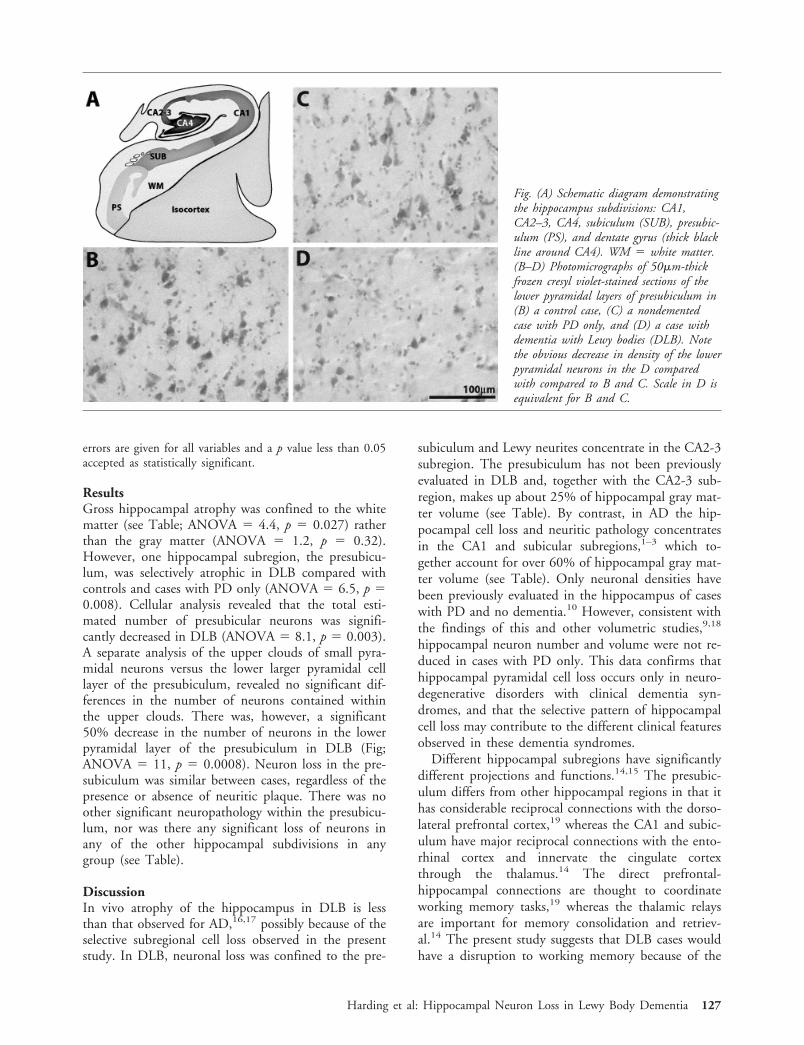
ResultsGross hippocampal atrophy was confined to the whitematter (see Table; ANOVA � 4.4, p � 0.027) ratherthan the gray matter (ANOVA � 1.2, p � 0.32).However, one hippocampal subregion, the presubicu-lum, was selectively atrophic in DLB compared withcontrols and cases with PD only (ANOVA � 6.5, p �0.008). Cellular analysis revealed that the total esti-mated number of presubicular neurons was signifi-cantly decreased in DLB (ANOVA � 8.1, p � 0.003).A separate analysis of the upper clouds of small pyra-midal neurons versus the lower larger pyramidal celllayer of the presubiculum, revealed no significant dif-ferences in the number of neurons contained withinthe upper clouds. There was, however, a significant50% decrease in the number of neurons in the lowerpyramidal layer of the presubiculum in DLB (Fig;ANOVA � 11, p � 0.0008). Neuron loss in the pre-subiculum was similar between cases, regardless of thepresence or absence of neuritic plaque. There was noother significant neuropathology within the presubicu-lum, nor was there any significant loss of neurons inany of the other hippocampal subdivisions in anygroup (see Table).
DiscussionIn vivo atrophy of the hippocampus in DLB is lessthan that observed for AD,16,17 possibly because of theselective subregional cell loss observed in the presentstudy. In DLB, neuronal loss was confined to the pre-
subiculum and Lewy neurites concentrate in the CA2-3subregion. The presubiculum has not been previouslyevaluated in DLB and, together with the CA2-3 sub-region, makes up about 25% of hippocampal gray mat-ter volume (see Table). By contrast, in AD the hip-pocampal cell loss and neuritic pathology concentratesin the CA1 and subicular subregions,1–3 which to-gether account for over 60% of hippocampal gray mat-ter volume (see Table). Only neuronal densities havebeen previously evaluated in the hippocampus of caseswith PD and no dementia.10 However, consistent withthe findings of this and other volumetric studies,9,18
hippocampal neuron number and volume were not re-duced in cases with PD only. This data confirms thathippocampal pyramidal cell loss occurs only in neuro-degenerative disorders with clinical dementia syn-dromes, and that the selective pattern of hippocampalcell loss may contribute to the different clinical featuresobserved in these dementia syndromes.
Different hippocampal subregions have significantlydifferent projections and functions.14,15 The presubic-ulum differs from other hippocampal regions in that ithas considerable reciprocal connections with the dorso-lateral prefrontal cortex,19 whereas the CA1 and subic-ulum have major reciprocal connections with the ento-rhinal cortex and innervate the cingulate cortexthrough the thalamus.14 The direct prefrontal-hippocampal connections are thought to coordinateworking memory tasks,19 whereas the thalamic relaysare important for memory consolidation and retriev-al.14 The present study suggests that DLB cases wouldhave a disruption to working memory because of the
Fig. (A) Schematic diagram demonstratingthe hippocampus subdivisions: CA1,CA2–3, CA4, subiculum (SUB), presubic-ulum (PS), and dentate gyrus (thick blackline around CA4). WM � white matter.(B–D) Photomicrographs of 50�m-thickfrozen cresyl violet-stained sections of thelower pyramidal layers of presubiculum in(B) a control case, (C) a nondementedcase with PD only, and (D) a case withdementia with Lewy bodies (DLB). Notethe obvious decrease in density of the lowerpyramidal neurons in the D comparedwith compared to B and C. Scale in D isequivalent for B and C.
Harding et al: Hippocampal Neuron Loss in Lewy Body Dementia 127

considerable pyramidal cell loss in the direct hip-pocampal output to the dorsolateral prefrontal cortex.Our previous work showing strong correlations be-tween Lewy body formation in the frontal lobe andfrontal and hippocampal atrophy in DLB9 also supportthis thesis, and further suggest that frontal afferentconnections with the hippocampus may also be dis-rupted by Lewy body formation. It would appear thatthe direct connections between the frontal lobe andhippocampus are significantly affected in DLB. Loss ofthis pathway would significantly affect memory circuitsin a different manner to the loss of the temporal lobeconnections found in AD.20
We acknowledge support from Parkinson’s New South Wales, Aus-tralian Brain Foundation, National Health and Medical ResearchCouncil, Clive and Vera Ramaciotti Foundation, and the Ian PotterFoundation.
We thank Heidi Cartwright for the figure work and Dr. JasmineHenderson for her comments on the manuscript.
References1. West MJ, Coleman PD, Flood DG, Troncoso JC. Differences
in the pattern of hippocampal neuronal loss in normal ageingand Alzheimer’s disease. Lancet 1994;344:769–772.
2. Lippa CF, Smith TW, Swearer JM. Alzheimer’s disease andLewy body disease: a comparative clinicopathological study.Ann Neurol 1994;35:81–88.
3. Harding AJ, Wong A, Svoboda M, et al. Chronic alcohol con-sumption does not cause hippocampal neuron loss in humans.Hippocampus 1997;7:78–87.
4. Laakso M, Partanen K, Riekkinen P, et al. Hippocampal vol-umes in Alzheimer’s disease, Parkinson’s disease with and with-out dementia, and in vascular dementia: an MRI study. Neu-rology 1996;46:679–681.
5. Nagy Z, Jobst K, Esini M, et al. Hippocampal pathology re-flects memory deficit and brain imaging measurements in Alz-heimer’s disease: clinicopathologic correlations using three setsof pathologic diagnostic criteria. Dementia 1996;7:76–81.
6. O’Brien JT, Paling S, Barber R, et al. Progressive brain atrophyon serial MRI in dementia with Lewy bodies, AD, and vasculardementia. Neurology 2001;56:1386–1388.
7. Bobinski M, Wegiel J, Wisniewski HM, et al. Atrophy of hip-pocampal formation subdivisions with stage and duration ofAlzheimer’s disease. Dementia 1995;6:205–210.
8. Lehtovirta M, Laakso MP, Soininen H, et al. Volumes of hip-pocampus, amygdala and frontal lobe in Alzheimer patientswith different apolipoprotein E genotypes. Neuroscience 1995;67:65–72.
9. Cordato NJ, Halliday GM, Harding AJ, et al. Regional brainatrophy in progressive supranuclear palsy and Lewy body dis-ease. Ann Neurol 2000;47:718–728.
10. Churchyard A, Lees AJ. The relationship between dementia anddirect involvement of the hippocampus and amygdala in Par-kinson’s disease. Neurology 1997;49:1570–1576.
11. Harding A, Halliday G. Simplified neuropathological diagnosisof dementia with Lewy bodies. Neuropathol Appl Neurobiol1998;24:195–201.
12. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelinesfor the clinical and pathologic diagnosis of dementia with Lewybodies (DLB): report of the consortium on DLB internationalworkshop. Neurology 1996;47:1113–1124.
13. Harding AJ, Halliday GM, Kril JJ. Variation in hippocampalneuron number with age and brain volume. Cereb Cortex1998; 8:710–718.
14. Duvernoy HM. The human hippocampus. Functional anat-omy, vascularization and serial sections with MRI. Berlin:Springer-Verlag, 1998.
15. Amaral D, Insausti R. Hippocampal formation. In: Paxinos G,ed. The human nervous system. Melbourne: Academic, 1990:712–756.
16. Barber R, Ballard C, McKeith IG, et al. MRI volumetric studyof dementia with Lewy bodies: a comparison with AD and vas-cular dementia. Neurology 2000;54:1304–1309.
17. Barber R, McKeith IG, Ballard C, et al. A comparison of me-dial and lateral temporal lobe atrophy in dementia with Lewybodies and Alzheimer’s disease: magnetic resonance imagingvolumetric study. Dement Geriatr Cogn Disord 2001;12:198–205.
18. Double KL, Halliday GM, McRitchie DA, et al. Regional brainatrophy in idiopathic Parkinson’s disease and diffuse Lewy bodydisease. Dementia 1996;7:304–313.
19. Goldman-Rakic P, Selemon L, Schwartz M. Dual pathwaysconnecting the dorsolateral prefrontal cortex with the hip-pocampal formation and parahippocampal cortex in the rhesusmonkey. Neuroscience 1984;12:719–743.
20. Braak H, Braak E, Yilmazer D, et al. Pattern of brain destruc-tion in Parkinson’s and Alzheimer’s disease. J Neural Trans1996;103:455–490.
128 Annals of Neurology Vol 51 No 1 January 2002

A Novel, Blood-BasedDiagnostic Assay for LimbGirdle Muscular Dystrophy2B and Miyoshi MyopathyMengfatt Ho, DPhil,1 Eduard Gallardo, PhD,2
Diane McKenna-Yasek, RN, BSN,1
Noemi De Luna, PhD,2 Isabel Illa, MD,2
and Robert H. Brown, Jr., MD, DPhil1
Limb girdle muscular dystrophy 2B and Miyoshi myop-athy were recently found to be allelic disorders arisingfrom defects in the dysferlin gene. We have developed anew diagnostic assay for limb girdle muscular dystrophy2B and Miyoshi myopathy, which screens for dysferlinexpression in blood using a commercially availablemonoclonal antibody. Unlike current methods that re-quire muscle biopsy for immunodiagnosis, the newmethod is simple and entails a significantly less invasiveprocedure for tissue sampling. Moreover, it overcomessome of the problems associated with the handling andstorage of muscle specimens. In our analysis of 12 pa-tients with limb girdle muscular dystrophy 2B or Miyo-shi myopathy, the findings obtained using the new assayare fully consistent with the results from muscle immu-nodiagnosis.
Ann Neurol 2002;51:129–133
Limb girdle muscular dystrophy type 2B (LGMD 2B)and Miyoshi myopathy (MM) have been considered dis-tinct clinical entities because different muscle groups areinvolved at onset. LGMD 2B begins with predomi-nantly proximal muscle weakness, whereas initial weak-ness in MM patients invariably affects the distal muscu-lature. However, both disorders are characterized byautosomal recessive inheritance, adult onset, and markedelevations of the muscle enzyme creatine kinase.1
LGMD 2B and MM were recently shown to arisefrom defects in a novel gene that encodes dysferlin.2,3
Remarkably, the same mutation in the dysferlin gene
can cause different clinical presentation, even amongmembers of the same family.4–6 In addition, an ante-rior distal myopathy was recently linked to dysferlinmutation.7 Thus, there is considerable clinical hetero-geneity in dysferlinopathy. The dysferlin gene is large,comprising 55 exons that span a genomic region of150kb.8 It encodes the 237kDa dysferlin protein,composed of 2,080 amino acids. The function of dys-ferlin is unknown, but its homology to a Caenorhabitiselegans protein, fer-1, suggests that it might function incalcium-mediated membrane fusion or trafficking.9
This hypothesis is supported by recent findings thatdysferlin is membrane associated.10–12
An accurate diagnosis of dysferlinopathy requires acombination of clinical evaluation, protein studies (im-munoblot or immunohistochemical analysis), or directgene analysis. Although the latter approach provides themost definitive diagnosis, it is costly, time-consuming,and labor intensive because of the large size of the dys-ferlin gene. Moreover, defects in the dysferlin gene arepredominantly single nucleotide changes with no evi-dence of recurrent mutations, gross rearrangements, ormutational hotspots to aid detection.3,8,10 For these rea-sons, DNA-based diagnosis is difficult as an initialscreening strategy to distinguish dysferlinopathies fromthe other forms of muscular dystrophy.
By contrast, screening for defective protein expressionby immunoblot analysis has proved to be a reliable andrapid means for differential diagnosis in muscular dys-trophies.13 However, current methods require muscle bi-opsy samples. To avoid this painful and invasive proce-dure, we sought to develop a new protein-baseddiagnosis using nonmuscle tissues and a less invasivesampling technique. We have previously shown that dys-ferlin is expressed in multiple tissues,3,11 we therefore in-vestigated its expression in peripheral blood cells. In thepresent study, we report that dysferlin is expressed spe-cifically in monocytes and we describe a novel blood-based diagnostic assay for LGMD 2B and MM.
Patients and MethodsIsolation of Peripheral Blood Mononuclear Cells andImmunoblot AnalysisPeripheral blood mononuclear cells (PBMC) were isolatedfrom whole blood of patients and healthy controls by Ficoll-Hypaque gradient centrifugation according to the manufac-turer’s instructions (Amersham, Buckinghamshire, UK).PBMC were washed twice in phosphate-buffered saline(PBS) and lysed in 10 volumes of protein extraction buffer,M-PER (Pierce, Rockford, IL). Approximately 20�g of pro-tein was separated on a 4 to 15% gradient sodium dodecylsulfate-polyacrylamide gel electrophoresis gel. Immunoblot-ting was performed according to standard methods, usingprimary anti-dysferlin monoclonal antibodies (NCL-Hamletfrom Novacastra, UK) at 1:300 dilution. Immunoreactivebands were detected with the enhanced chemiluminescencesystem (Amersham).
From the 1Day Laboratory for Neuromuscular Research, Massachu-setts General Hospital, Harvard Medical School, Charlestown, Mas-sachusetts; 2Department of Neurology, Neuromuscular DiseasesSection, Hospital Santa Creu i St Pau, Universitat Autonoma deBarcelona, Barcelona, Spain
Received Aug 16, 2001, and in revised form Aug 16, 2001. Ac-cepted for publication Aug 16, 2001.
Published online Dec 28, 2001
Address correspondence to Dr Brown, Day Laboratory for Neuro-muscular Research, Massachusetts General Hospital, Harvard Med-ical School, Bldg. 114, 16th Street, Rm 3125, Charlestown, MA02129.
© 2001 Wiley-Liss, Inc. 129DOI 10.1002/ana.10080

Separation of PBMC into CD14� and CD14� cellpopulationsPBMC (approximately 107 cells) were mixed with 20�l ofCD14-coated microbeads (Milteny Biotec, Germany) and in-cubated at 6 to 12°C for 30 minutes. Unbound microbeadswere removed by washing cells in excess PBS buffer followedby centrifugation at 300g for 10 minutes. The cell pellet wasresuspended in PBS buffer to a concentration of 2 � 108
cells/ml before separation on a MACS apparatus according tomanufacturer’s instructions (Milteny Biotec, Germany).
ImmunocytochemistryPBMC were spun onto microscopic slides using a Cytospin(Shandon, UK) centrifuge. Acetone-fixed preparations werepreincubated in PBS buffer containing 0.5% bovine serumalbumin and 5% normal goat serum. The sections were in-
Fig 1. Dysferlin expression in peripheral blood mononuclear cells (PBMC). (A) Northern blot analysis of dysferlin gene expression inperipheral blood cells from a healthy individual, with a probe corresponding to nucleotides 5364–5732 of the dysferlin cDNA. A7.5kb band corresponding to the dysferlin transcript is detected in peripheral blood cells and skeletal muscle. Additional transcriptsof 4.5kb and 2.0kb are also present in peripheral blood. (B) Western blot analysis of dysferlin expression in multiple tissues, usingthe NCL-Hamlet monoclonal antibody. A prominent 230kDa protein is detected in PBMC, but not in white blood cells (WBC),of two unrelated, healthy individuals. The positive controls include skeletal muscle and brain tissues from mouse and human. (C toE) Immunocytochemical analysis of dysferlin expression in CD14� cells (C) and CD14� cells (D). (E) Western blot analysis of dys-ferlin expression in CD14� cells (lane 1) and CD14� cells (lane 2). Immunoblotting of the 42 kDa actin antigen served as apositive control in both preparations (lanes 1, 2, bottom panel).
130 Annals of Neurology Vol 51 No 1 January 2002

cubated with NCL-Hamlet (Novacastra, UK), followed byhorseradish peroxidase-labeled secondary antibodies (JacksonImmunoresearch, PA) and visualized by diaminobenzidine(Vector Laboratories, Burlingame, CA).
ResultsDetection of Dysferlin Expression in Peripheral BloodMononuclear CellsAs a first step to develop a nonmuscle diagnostic assayfor dysferlinopathies, we examined dysferlin gene ex-pression in blood cells of healthy individuals by north-ern blot analysis. A weak but distinct band of 7.5kb,corresponding to the dysferlin transcript, was detectedin total RNA isolated from blood (Fig 1A). Smallertranscripts of 4kb and 2.0kb were also detected inblood cells, suggesting the presence of tissue-specificspliced variants.
To determine whether the dysferlin protein is ex-pressed, a Western blot of total white blood cells(WBC), peripheral blood mononuclear cells (PBMC),brain, and skeletal muscle was screened with an anti-dysferlin monoclonal antibody (NCL-Hamlet). Despitethe lower levels of dysferlin gene expression in bloodcompared with muscle, a prominent 230kDa bandcorresponding to the dysferlin protein was detected inPBMC, skeletal muscle, and brain but was absent intotal WBC (Fig 1B). As PBMC consist primarily oflymphocytes and monocytes, this result suggests thatdysferlin is expressed in these cell types.
Immunocytochemical Analysis of Dysferlin Expressionin PBMCTo define further the cell type that expresses dysferlin,we separated PBMC into CD14� and CD14� cells,using magnetic beads coated with CD14 antibodies.This approach distinguishes the two main cell types inPBMC, because CD14 is a specific marker for mono-cytes. As shown in Figure 1C and D, immunocyto-chemical analysis showed dysferlin staining only inCD14� cells, but not in CD14� cells. This result wasconfirmed by Western blot analysis (see Fig 1E). Takentogether, these findings indicate that dysferlin is ex-pressed primarily in monocytes. As monocytes consti-tute only 3 to 7% of total WBC, it is therefore notsurprising that we were unable to detect dysferlin ex-pression in total WBC (see Fig 1A).
Diagnostic Applications of Dysferlin Expressionin PBMCTo evaluate the accuracy of a blood-based diagnosticassay for dysferlinopathies, we compared dysferlin ex-pression in PBMC and skeletal muscle in 12 patientswith LGMD 2B or MM. In each case, dysferlin defi-ciency in skeletal muscle was confirmed by Western
blotting. Conversely, immunoblot analyses of PBMCshowed dysferlin reactivity in controls (all 6 tested) butwas absent in all 12 patients tested (Fig 2, Table), in-dicating there is an excellent correlation between dys-ferlin expression in PBMC and skeletal muscle.
We also investigated the effect of delayed processingof blood samples on the stability of the dysferlin pro-tein. Samples that were processed within 36 hoursshowed no evidence of dysferlin degradation while mi-nor degradation can be seen in samples left for 48hours (see Fig 2). A lower band of 55kDa was de-tected in control samples that were left for �24 hoursbut not in 6-hour-old samples (see Fig 2), suggestingthat this is a partially degraded dysferlin product, as itis present only in control, but not in patient, samples.Thus, only the full-length dysferlin protein should beused as a diagnostic indicator for dysferlin deficiency.
DiscussionWe report the novel finding that dysferlin is expressedin monocytes. More importantly, we showed that dys-ferlin expression in monocytes correlates with its ex-pression in skeletal muscle (see Table). These findingshave led us to develop a new protein-based diagnosticassay for LGMD 2B and MM patients, without theneed for a muscle biopsy.
The new blood-based diagnostic assay offers severaladvantages over current methods of muscle immunodi-agnosis. This simple and rapid technique does not re-quire specialized equipment or training. In addition, itovercomes some of the inherent problems associatedwith the handling and storage of muscle specimens.
Fig 2. Diagnostic screening of dysferlin expression in peripheralblood mononuclear cells (PBMC) by Western blot analysis.Lanes 1, 2, 3, 7, 8 contain PBMC from normal healthy con-trols; lanes 4, 5, 6 (RB 2329, RB 3533, and RB 2079, re-spectively) are from Miyoshi myopathy (MM) patients (see Ta-ble 1 for details of patient genotypes). The effect of delayedprocessing of blood samples on the stability of the dysferlinprotein can be seen by comparing samples that were left for 48hours (lane 1), 36 hours (lane 2), 24 hours (lanes 3, 7), and6 hours (lane 8) before being processed for immunoblot analy-sis.
Ho et al: Limb Girdle Muscular Dystrophy 2B and Miyoshi Myopathy 131

The most significant improvement offered by the newassay is the considerably less invasive tissue samplingmethod compared to muscle biopsy. Thus, it shouldbecome easier to obtain samples from patients to con-firm diagnosis or to guide further testing. Moreover,collection of patient materials will no longer requirethe operating theater, which will be a significant cost-savings benefit.
In addition to diagnostic applications, dysferlin ex-pression in monocytes provides a new paradigm tostudy the biological function of this protein in a non-muscle cell type that is readily accessible and amenableto in-vitro functional assays. Also, for experimentaltherapies intended to increase the levels of dysferlin, wecould now track efficacy by monitoring dysferlin levelsin blood, rather than skeletal muscle. LGMD 2B is notuncommon, accounting for 1 to 25% of limb girdledystrophies.14,15 Unlike MM patients, LGMD 2B pa-tients are not readily diagnosed because there is con-siderable genetic heterogeneity in LGMD, and preciseDNA diagnostic studies are costly and difficult. Thus,the new assay will be particularly useful as a rapidscreen to identify the dysferlin-deficient subgroupwithin LGMDs. More complex DNA analysis can thenbe performed selectively in this subgroup, saving bothtime and costs.
To date, all reports of dysferlin deficiency are asso-ciated with defects in the dysferlin gene.8,10,11 How-ever, it is unknown whether other forms of musculardystrophies could cause a secondary reduction in dys-ferlin levels. Therefore, the new assay will be a usefultool to address this question. Identification of patientswith reduced dysferlin expression but without defectsin the dysferlin gene could lead to the discovery of pro-teins that normally associate with dysferlin and helpelucidate the pathophysiological pathway involved indysferlinopathies.
The authors are very grateful to all the patients who have partici-pated in this study. We thank Drs K. Bejaoui and B. Hosler for thecritical reading of this manuscript.
This work is supported in part by the Muscular Dystrophy Associ-ation (USA), the C. B. Day Investment Company, and Fondo deInvestigacıon Sanitaria (FIS 99/019-01; FIS 01/0979).
References1. Bushby KM. Making sense of the limb-girdle muscular dystro-
phies. Brain 1999;122:1403–1420.2. Bashir R, Britton S, Strachan T, et al. A gene related to Cae-
norhabditis elegans spermatogenesis factor fer-1 is mutated inlimb-girdle muscular dystrophy type 2B. Nat Genet 1998;20:37–42.
3. Liu J, Aoki M, Illa I, et al. Dysferlin, a novel skeletal musclegene, is mutated in Miyoshi myopathy and limb girdle muscu-lar dystrophy. Nat Genet 1998;20:31–36.
4. Illarioshkin SN, Ivanova-Smolenskaya IA, Greenberg CR, etal. Identical dysferlin mutation in limb-girdle muscular dys-trophy type 2B and distal myopathy. Neurology 2000;55:1931–1933.
5. Weiler T, Bashir R, Anderson LV, et al. Identical mutation inpatients with limb girdle muscular dystrophy type 2B or Miyo-shi myopathy suggests a role for modifier gene(s). Hum MolGenet 1999;8:871–877.
6. Weiler T, Greenberg CR, Nylen E, et al. Limb-girdle musculardystrophy and Miyoshi myopathy in an aboriginal Canadiankindred map to LGMD2B and segregate with the same haplo-type. Am J Hum Genet 1996;59:872–878.
7. Illa I, Serrano-Munuera C, Gallardo E, et al. Distal anteriorcompartment myopathy: a dysferlin mutation causing a newmuscular dystrophy phenotype. Ann Neurol 2001;49:130 –134.
8. Aoki M, Liu J, Richard I, et al. Genomic organization of thedysferlin gene and novel mutations in Miyoshi myopathy. Neu-rology 2001;57:271–278.
9. Achanzar W.E, Ward S. A nematode gene required for spermvesicle fusion. J Cell Sci 1997;110:1073–1081.
10. Anderson LV, Davison K, Moss JA, et al. Dysferlin is a plasmamembrane protein and is expressed early in human develop-ment. Hum Mol Genet 1999;8:855–861.
Table. Evaluation of Dysferlin Deficiency by Immunodiagnosis in Peripheral Blood Mononuclear Cells and Skeletal Muscle
PatientClinical
Diagnosis MutationDysferlin Expression
in PBMCDysferlin Expressionin Skeletal Muscle
ImmunocytochemicalAnalysis of PBMC
RB 2329 MM E1883X, 6319 � 1G (�) (�) (�)RB 1859 MM Deletion AG, at 6071 (�) (�) n.d.RB 2079 DACM Deletion G at 5966 (�) (�) (�)II 1101 MM n.d. (�) (�) (�)II 1102 MM n.d. (�) (�) (�)II 1103 MM n.d. (�) (�) (�)II 1104 MM n.d. (�) (�) (�)II 1303 LGMD 2B n.d. (�) (�) (�)II 1304 LGMD 2B n.d. (�) (�) (�)II 1305 LGMD 2B n.d. (�) (�) (�)II 1306 LGMD 2B n.d. (�) (�) (�)RB 3533 MM n.d. (�) (�) n.d.
For positive controls, all 6 PBMC samples obtained from healthy individuals showed positive dysferlin reactivity by Western blot analysis.MM � Miyoshi myopathy; LGMD 2B � limb girdle muscular dystrophy; DACM � distal anterior compartment myopathy (ref 7); X � stopcodon; n.d. � not determined; PBMC � peripheral blood mononuclear cells; (�) � absence of dysferlin staining.
132 Annals of Neurology Vol 51 No 1 January 2002

11. Matsuda C, Aoki M, Hayashi YK, et al. Dysferlin is a surfacemembrane-associated protein that is absent in Miyoshi myop-athy. Neurology 1999;53:1119–1122.
12. Selcen D, Stilling G, Engel AG. The earliest pathologic alter-ations in dysferlinopathy. Neurology 2001;56:1472–1481.
13. Anderson LV, Davison K. Multiplex Western blotting systemfor the analysis of muscular dystrophy proteins. Am J Pathol1999;154:1017–1022.
14. Passos-Bueno MR, Vainzof M, Moreira ES, et al. Seven auto-somal recessive limb-girdle muscular dystrophies in the Brazil-ian population: from LGMD2A to LGMD2G. Am J MedGenet 1999;82:392–398.
15. Fanin M, Pegoraro E, Matsuda-Asada C, et al. Calpain-3 anddysferlin protein screening in patients with limb-girdle dystro-phy and myopathy. Neurology 2001;56:660–665.
Association Studies ofMultiple Candidate Genesfor Parkinson’s Diseaseusing SingleNucleotide PolymorphismsYoshio Momose, MD,1,2 Miho Murata, MD, PhD,2
Kazuhiro Kobayashi, PhD,1 Masaji Tachikawa, MSc,1
Yuko Nakabayashi, BSc,1 Ichiro Kanazawa, MD, PhD,2
and Tatsushi Toda MD, PhD1
We studied 20 single nucleotide polymorphisms in 18candidate genes for association with Parkinson’s disease.We found that homozygosity for the V66M polymor-phism of the brain-derived neurotrophic factor (BDNF)gene occurs more frequently in patients with Parkinson’sdisease than in unaffected controls (�2 � 5.46) and con-firmed an association with the S18Y polymorphism ofthe UCH-L1 gene. Our results provide genetic evidencesupporting a role for BDNF in the pathogenesis of Par-kinson’s disease.
Ann Neurol 2002;51:133–136
Single nucleotide polymorphisms (SNPs) are single-base differences in DNA sequences that can be ob-
served between individuals in the population. As such,they represent high-potential polymorphic markers use-ful in the study of susceptibility genes for multifactorialdiseases such as Parkinson’s disease (PD) or those re-lated to drug responsiveness.1
PD, one of the most common human neurodegen-erative diseases, is characterized by resting tremor, cog-wheel rigidity, bradykinesia, and impaired postural re-flexes. This disease affects 1 to 2% of persons olderthan 65 years of age. Recent studies have indicated thepresence of genetic factors to the pathogenesis of PD.For example (1) there are large families with a historyof PD, in which the existence of heredity is suggested;(2) approximately 10% of patients with PD have apositive family history2; (3) a recent large-scale surveyin Iceland showed that the risk ratio for PD was 6.7for siblings, 3.2 for offspring, and 2.7 for nephews andnieces of patients with PD3; and (4) a twin study using[18F]-dopa PET showed that the concordance rate forPD, including subclinical cases, is approximately threetimes higher in monozygotic twins (55%) than in dizy-gotic twins (18%).4 Causal genes for Mendelian-inherited PD have been reported, including �-synuclein(4q21–23, autosomal dominant [AD]),5 parkin(6q25.2–27, autosomal recessive [AR]),6 UCH-L1(4p14, AD),7 PARK3 (2p13, AD),8 PARK4 (4p15,AD),9 PARK6 (1p35–36, AR),10 PARK7 (1p36, AR).11
Thus far, 84 association studies on 14 genes have ex-amined relationships between PD and polymor-phisms12; however, SNPs that influence PD as stronglyas APOE-ε4 influences Alzheimer’s disease have notbeen identified.
To identify susceptibility genes for PD and obtaininformation helpful for establishing tailor-made medi-cines, we initiated large-scale association studies usingSNPs in various candidate genes and further surveyedthe correlation between SNPs and susceptibility to var-ious side effects.
Patients and MethodsA total of 232 patients (114 men and 118 women) with PDand 249 normal controls (127 men and 122 women) wereexamined in this study. The mean age of onset was 54.1 �10.5 (mean � SD) years [range:24–80]; 18 patients showedearly onset of PD (�40 years of age), and 13 patients had apositive family history of PD. Patients who carried parkinmutations were excluded. All patients and controls were ofJapanese ancestry. Patients were evaluated by certified neu-rologists specializing in PD.
The diagnosis of idiopathic PD was based on the presenceof two or more of the cardinal features of PD (tremor, ri-gidity, bradykinesia, and postural instability) and a lack ofother signs or clinical history that might indicate alternativediagnoses or causes. We were careful to exclude secondaryparkinsonism. Informed consent was obtained from each pa-tient, and approval for the study was obtained from the Uni-versity Ethical Committees.
From the 1Division of Functional Genomics, Department of Post-Genomics and Diseases, Osaka University Graduate School of Med-icine, Osaka, Japan; 2Department of Neurology, Graduate School ofMedicine, University of Tokyo, Tokyo, Japan.
Received Jun 18, 2001, and in revised form Sep 24, 2001. Acceptedfor publication Sep 24, 2001.
Published online Dec 28, 2001
Address correspondence to: Dr Toda, Division of FunctionalGenomics, Department of Post-Genomics and Diseases, Osaka Uni-versity Graduate School of Medicine, 2-2-B9 Yamadaoka, Suita,Osaka 565-0871, Japan. E-mail: [email protected]
© 2001 Wiley-Liss, Inc. 133DOI 10.1002/ana.10079
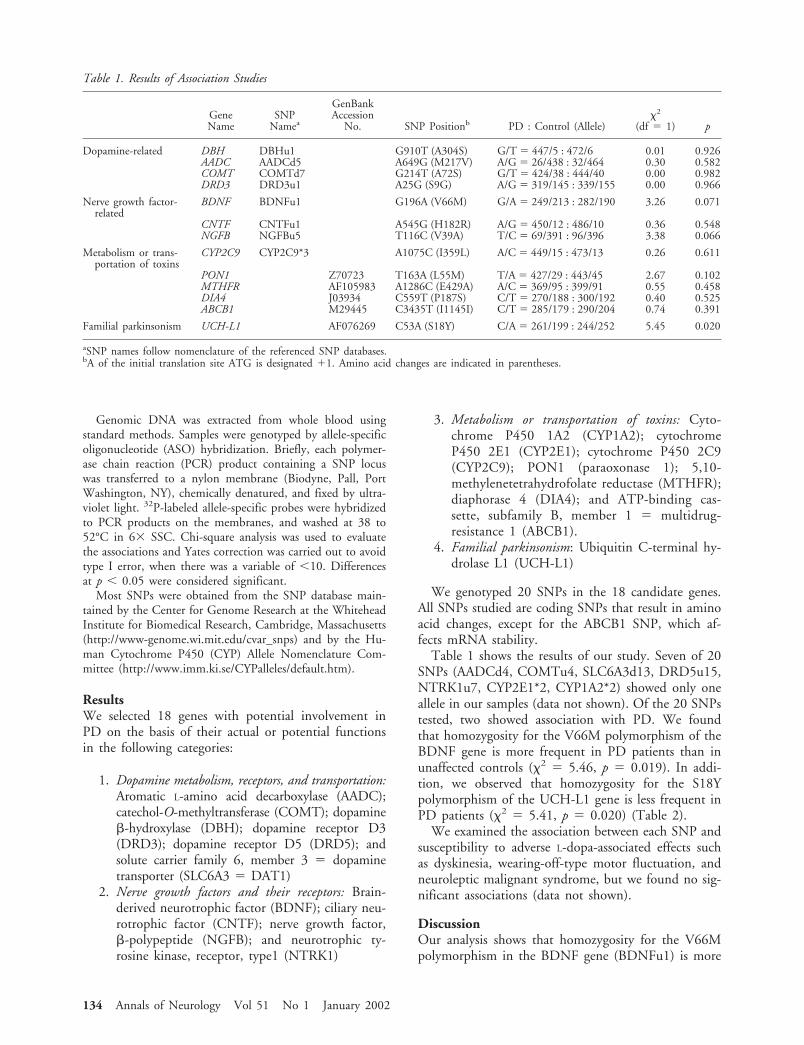
Genomic DNA was extracted from whole blood usingstandard methods. Samples were genotyped by allele-specificoligonucleotide (ASO) hybridization. Briefly, each polymer-ase chain reaction (PCR) product containing a SNP locuswas transferred to a nylon membrane (Biodyne, Pall, PortWashington, NY), chemically denatured, and fixed by ultra-violet light. 32P-labeled allele-specific probes were hybridizedto PCR products on the membranes, and washed at 38 to52°C in 6� SSC. Chi-square analysis was used to evaluatethe associations and Yates correction was carried out to avoidtype I error, when there was a variable of �10. Differencesat p � 0.05 were considered significant.
Most SNPs were obtained from the SNP database main-tained by the Center for Genome Research at the WhiteheadInstitute for Biomedical Research, Cambridge, Massachusetts(http://www-genome.wi.mit.edu/cvar_snps) and by the Hu-man Cytochrome P450 (CYP) Allele Nomenclature Com-mittee (http://www.imm.ki.se/CYPalleles/default.htm).
ResultsWe selected 18 genes with potential involvement inPD on the basis of their actual or potential functionsin the following categories:
1. Dopamine metabolism, receptors, and transportation:Aromatic L-amino acid decarboxylase (AADC);catechol-O-methyltransferase (COMT); dopamine�-hydroxylase (DBH); dopamine receptor D3(DRD3); dopamine receptor D5 (DRD5); andsolute carrier family 6, member 3 � dopaminetransporter (SLC6A3 � DAT1)
2. Nerve growth factors and their receptors: Brain-derived neurotrophic factor (BDNF); ciliary neu-rotrophic factor (CNTF); nerve growth factor,�-polypeptide (NGFB); and neurotrophic ty-rosine kinase, receptor, type1 (NTRK1)
3. Metabolism or transportation of toxins: Cyto-chrome P450 1A2 (CYP1A2); cytochromeP450 2E1 (CYP2E1); cytochrome P450 2C9(CYP2C9); PON1 (paraoxonase 1); 5,10-methylenetetrahydrofolate reductase (MTHFR);diaphorase 4 (DIA4); and ATP-binding cas-sette, subfamily B, member 1 � multidrug-resistance 1 (ABCB1).
4. Familial parkinsonism: Ubiquitin C-terminal hy-drolase L1 (UCH-L1)
We genotyped 20 SNPs in the 18 candidate genes.All SNPs studied are coding SNPs that result in aminoacid changes, except for the ABCB1 SNP, which af-fects mRNA stability.
Table 1 shows the results of our study. Seven of 20SNPs (AADCd4, COMTu4, SLC6A3d13, DRD5u15,NTRK1u7, CYP2E1*2, CYP1A2*2) showed only oneallele in our samples (data not shown). Of the 20 SNPstested, two showed association with PD. We foundthat homozygosity for the V66M polymorphism of theBDNF gene is more frequent in PD patients than inunaffected controls ( 2 � 5.46, p � 0.019). In addi-tion, we observed that homozygosity for the S18Ypolymorphism of the UCH-L1 gene is less frequent inPD patients ( 2 � 5.41, p � 0.020) (Table 2).
We examined the association between each SNP andsusceptibility to adverse L-dopa-associated effects suchas dyskinesia, wearing-off-type motor fluctuation, andneuroleptic malignant syndrome, but we found no sig-nificant associations (data not shown).
DiscussionOur analysis shows that homozygosity for the V66Mpolymorphism in the BDNF gene (BDNFu1) is more
Table 1. Results of Association Studies
GeneName
SNPNamea
GenBankAccession
No. SNP Positionb PD : Control (Allele) 2
(df � 1) p
Dopamine-related DBH DBHu1 G910T (A304S) G/T � 447/5 : 472/6 0.01 0.926AADC AADCd5 A649G (M217V) A/G � 26/438 : 32/464 0.30 0.582COMT COMTd7 G214T (A72S) G/T � 424/38 : 444/40 0.00 0.982DRD3 DRD3u1 A25G (S9G) A/G � 319/145 : 339/155 0.00 0.966
Nerve growth factor-related
BDNF BDNFu1 G196A (V66M) G/A � 249/213 : 282/190 3.26 0.071
CNTF CNTFu1 A545G (H182R) A/G � 450/12 : 486/10 0.36 0.548NGFB NGFBu5 T116C (V39A) T/C � 69/391 : 96/396 3.38 0.066
Metabolism or trans-portation of toxins
CYP2C9 CYP2C9*3 A1075C (I359L) A/C � 449/15 : 473/13 0.26 0.611
PON1 Z70723 T163A (L55M) T/A � 427/29 : 443/45 2.67 0.102MTHFR AF105983 A1286C (E429A) A/C � 369/95 : 399/91 0.55 0.458DIA4 J03934 C559T (P187S) C/T � 270/188 : 300/192 0.40 0.525ABCB1 M29445 C3435T (I1145I) C/T � 285/179 : 290/204 0.74 0.391
Familial parkinsonism UCH-L1 AF076269 C53A (S18Y) C/A � 261/199 : 244/252 5.45 0.020
aSNP names follow nomenclature of the referenced SNP databases.bA of the initial translation site ATG is designated �1. Amino acid changes are indicated in parentheses.
134 Annals of Neurology Vol 51 No 1 January 2002

frequent in PD patients. The neurotrophin BDNF aidsthe survival, growth, and maintenance of neurons inthe central and peripheral nervous systems. It protectsdopaminergic neurons from stresses such as deprivationof serum or MPTP-induced damage.13 Studies haveshown that BDNF concentrations are reduced in thenigrostriatal region of the brain in PD as comparedwith controls.14
The V66M polymorphism lies within the proBDNFsequence, which is cleaved posttranslationally. Al-though how this polymorphism might influence sus-ceptibility to PD remains unclear, some possible sce-narios have been suggested. First, the polymorphismmay affect production of mature, biologically activeBDNF by altering proBDNF processing. Lower levelsof mature BDNF may reduce protection of dopaminer-gic neurons from environmental toxins such as pesti-cides. Second, Mowla and colleagues15 recently re-ported that some proBDNF is released extracellularlyand is biologically active, as demonstrated by its abilityto mediate receptor activation. Thus, the V66M poly-morphism has the potential to affect the activity of ex-tracellular proBDNF activity as well. Finally, otherSNPs that associate with a more powerful influence onthe onset of PD might exist in linkage disequilibriumwith BDNFu1.
UCH-L1 represents 1 to 2% of total soluble brainprotein and is thought to hydrolyze polymeric ubiq-uitin and ubiquitin conjugates to produce monomericubiquitin. Immunoreactivity for this protein is foundin Lewy bodies. A missense mutation in the UCH-L1gene has been reported in relation to PD.7 In that pa-per, Leroy and colleagues showed that this mutation,I93M, causes a partial loss of catalytic thiol proteaseactivity of this enzyme, which may lead to aberrationsin the proteolytic pathway and aggregation of proteins.Some association studies demonstrated that carriers ofthe S18Y polymorphism have a significantly lower riskof PD.16–18 Our results are in accordance with these
studies. The amino acid sequence of UCH-L1 is highlyconserved among rat, mouse, and humans, with ap-proximately 95% sequence identity. Isoleucine (I) atposition 93 is conserved in all three species, and mu-tation of this residue to methionine (M) is causative infamilial PD in a German family. The amino acid atposition 18 is serine (S) or tyrosine (Y) in humans andalanine (A) in rat and mouse. The amino acid changeat this position may exert a milder functional influencethan the change at position 93. Another study has sug-gested that the S18Y polymorphism in UCH-L1 doesnot influence the risk of the development of PD in theAustralian population.19 Further investigation isneeded to clarify the potential role of this polymor-phism in PD. Another coding SNP of UCH-L1,M124L, was recognized in a French family with PD.20
Although it did not co-segregate and was considerednot to be pathogenic in this family, it would be ofinterest to know whether this SNP has an associationwith sporadic PD. Considering the significance of theubiquitin-protease system in neurodegenerative dis-eases, the role of UCH-L1 is of great interest.
Recent research has established that SNPs influencethe variability in patients’ responses to drugs. Althoughwe found no significant results in our current stratifiedanalyses, we consider it important to establish tailor-made treatments by utilizing SNPs for patients withPD. We are increasing the number of patients andSNPs in our studies to test for association.
In summary, we report the first genetic associationbetween BDNFu1 and the onset of PD. This findingprovides the evidence supporting a role for BDNF inthe pathogenesis of PD and information that mayprove useful for future gene therapy approaches usingneurotrophic factors.
This work was supported by research grants from the Ministry ofHealth, Labor, and Welfare (H12-Genome-025) and the Ministry
Table 2. Frequency of the BDNF Gene Polymorphism (G196A, V66M) and UCH-L1 Gene Polymorphism (C53A, S18Y)
Allele Genotype
A G Total AA AG or GG Total
BDNFPD 213 249 462 48 183 231Control 190 282 472 30 206 236
( 2 � 3.26, df � 1, p � 0.071) ( 2 � 5.46, df � 1, p � 0.019)
UCH-L1PD 199 261 460 40 190 230Control 252 244 496 65 183 248
( 2 � 5.45, df � 1, p � 0.020) ( 2 � 5.41, df � 1, p � 0.020)
PD � Parkinson’s disease.
Momose et al: Association Studies for PD by SNPs 135

of Education, Culture, Sports, Science, and Technology (12204035)in Japan.
References1. Gray IC, Campbell DA, Spurr NK. Single nucleotide polymor-
phisms as tools in human genetics. Hum Mol Genet 2000;9:2403–2408.
2. Elbaz A, Grigoletto F, Baldereschi M, et al. Familial aggrega-tion of Parkinson’s disease. Neurology 1999;52:1876–1882.
3. Sveinbjornsdottir S, Hicks AA, Jonsson T, et al. Familial aggre-gation of Parkinson’s disease in Iceland. N Engl J Med 2000;343:1765–1770.
4. Piccini P, Burn DJ, Ceravolo R, et al. The role of inheritancein sporadic Parkinson’s disease: evidence from a longitudinalstudy of dopaminergic function in twins. Ann Neurol 1999;45:577–582.
5. Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation inthe �-synuclein gene identified in families with Parkinson’s dis-ease. Science 1997;276:2045–2047.
6. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkingene cause autosomal recessive juvenile parkinsonism. Nature1998;392:605–608.
7. Leroy E, Boyer R, Auburger G, et al. The ubiquitin pathway inParkinson’s disease. Nature 1998;395:451–452.
8. Gasser T, Muller-Myhsok B, Wszolek ZK, et al. A susceptibil-ity locus for Parkinson’s disease maps to chromosome 2p13.Nat Genet 1998;18:262–265.
9. Farrer M, Gwinn-Hardy K, Muenter M, et al. A chromosome4p haplotype segregating with Parkinson’s disease and posturaltremor. Hum Mol Genet 1999;8:81–85.
10. Valente EM, Bentivoglio AR, Dixon PH, et al. Localization ofa novel locus for autosomal recessive early-onset Parkinsonism,PARK6, on human chromosome 1p35–p36. Am J Hum Genet2001;68:895–900.
11. van Duijn CM, Dekker MCJ, Bonifati V, et al. PARK7, a novellocus for autosomal recessive early-onset parkinsonism, on chro-mosome 1p36. Am J Hum Genet 2001;69:629–634.
12. Tan EK, Khajavi M, Thornby JI, et al. Variability and validityof polymorphism association studies in Parkinson’s disease.Neurology 2000;55:533–538.
13. Hyman C, Hofer M, Barde YA, et al. BDNF is a neurotrophicfactor for dopaminergic neurons of the substantia nigra. Nature1991;350:230–232.
14. Mogi M, Togari A, Kondo T, et al. Brain-derived growth factorand nerve growth factor concentrations are decreased in thesubstantia nigra in Parkinson’s disease. Neurosci Lett 1999;270:45–48.
15. Mowla SJ, Farhadi HF, Pareek S, et al. Biosynthesis and post-translational processing of the precursor to brain-derived neu-rotrophic factor. J Biol Chem 2001;276:12660–12666.
16. Maraganore DM, Farrer MJ, Hardy JA, et al. Case-controlstudy of the ubiquitin carboxy-terminal hydrolase L1 gene inParkinson’s disease. Neurology 1999;53:1858–1860.
17. Wintermeyer P, Kruger R, Kuhn W, et al. Mutation analysisand association studies of the UCHL1 gene in German Parkin-son’s disease patients. Neuroreport 2000;11:2079–2082.
18. Zhang J, Hattori N, Leroy E, et al. Association between a poly-morphism of ubiquitin carboxy-terminal hydrolase L1 (UCH-L1) gene and sporadic Parkinson’s disease. Parkinsonism RelatDisord 2000;6:195–197.
19. Mellick GD, Silburn PA. The ubiquitin carboxy-terminalhydrolase-L1 gene S18Y polymorphism does not confer protec-tion against idiopathic Parkinson’s disease. Neurosci Lett 2000;293:127–130.
20. Farrer M, Destee A, Becquet E, et al. Linkage exclusion inFrench families with probable Parkinson’s disease. Mov Disord2000;15:1075–1083.
136 Annals of Neurology Vol 51 No 1 January 2002




















