
Central Nervous System Anomalies
22
CONGENITAL ANOMALIES 0095-5108/00 $15.00 + .OO CENTRAL NERVOUS SYSTEM ANOMALIES Richard A. Bronsteen, MD, and Christine H. Comstock, MD Historically, central nervous system (CNS) abnormalities are noteworthy because they were the first group of fetal malformations to be detected by prenatal ultrasound. This article reviews many of the CNS abnormalities, and provides information on prognosis that it is hoped will be helpful to both the physician and patient. Because of space limitations, this article is not intended to be a complete review of all CNS anomalies. Reference is made to several recent books dedicated to this subject.", l7 NEURAL TUBE DEFECTS Most physicians providing prenatal care are quite familiar with neural tube defects because of the long-standing and widespread use of maternal serum .screening for these disorders, and the more recent availability of folic acid supplementation for preconception prophylaxis. Neural tube defects are lesions caused by incomplete closure of the embryonic neural tube. This defect occurs early in the organogenesis process and can lead to abnormal mesenchymal tissue formation that normally covers the neuroectoderm. These defects can be classified as open (neural tissue exposed or covered only by a thin membrane) or closed (covered by normal skin). In this country the incidence is 1 per 1000 births.lg The more common forms of neural tube defects are anencephaly and spina bifida. Rarer forms include iniencephaly, encephalocele, and craniora- chisclusis. These are multifactorial diseases, with incidences varying with geo- graphic location; family history; drug exposure (valproic acid); genetic associa- tions (autosomal recessive Meckel-Gruber, Walker-Warburg syndromes); and dietary factors (folate). An affected parent has a 3% to 5% chance of having a chld with spina bifida. Ths risk is similarly increased by a history of a prior From the Division of Fetal Imaging, Department of Obstetrics and Gynecology, William Beaumont Hospital, Royal Oak, Michigan CLINICS IN PERINATOLOGY VOLUME 27 NUMBER 4 DECEMBER 2000 791
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Central Nervous System Anomalies

CONGENITAL ANOMALIES 0095-5108/00 $15.00 + .OO
CENTRAL NERVOUS SYSTEM ANOMALIES
Richard A. Bronsteen, MD, and Christine H. Comstock, MD
Historically, central nervous system (CNS) abnormalities are noteworthy because they were the first group of fetal malformations to be detected by prenatal ultrasound. This article reviews many of the CNS abnormalities, and provides information on prognosis that it is hoped will be helpful to both the physician and patient. Because of space limitations, this article is not intended to be a complete review of all CNS anomalies. Reference is made to several recent books dedicated to this subject.", l7
NEURAL TUBE DEFECTS
Most physicians providing prenatal care are quite familiar with neural tube defects because of the long-standing and widespread use of maternal serum .screening for these disorders, and the more recent availability of folic acid supplementation for preconception prophylaxis. Neural tube defects are lesions caused by incomplete closure of the embryonic neural tube. This defect occurs early in the organogenesis process and can lead to abnormal mesenchymal tissue formation that normally covers the neuroectoderm. These defects can be classified as open (neural tissue exposed or covered only by a thin membrane) or closed (covered by normal skin). In this country the incidence is 1 per 1000 births.lg The more common forms of neural tube defects are anencephaly and spina bifida. Rarer forms include iniencephaly, encephalocele, and craniora- chisclusis. These are multifactorial diseases, with incidences varying with geo- graphic location; family history; drug exposure (valproic acid); genetic associa- tions (autosomal recessive Meckel-Gruber, Walker-Warburg syndromes); and dietary factors (folate). An affected parent has a 3% to 5% chance of having a chld with spina bifida. Ths risk is similarly increased by a history of a prior
From the Division of Fetal Imaging, Department of Obstetrics and Gynecology, William Beaumont Hospital, Royal Oak, Michigan
CLINICS IN PERINATOLOGY
VOLUME 27 NUMBER 4 DECEMBER 2000 791

792 BRONSTEEN & COMSTOCK
affected child and increases to 10% with two prior affected children. These risks can be significantly decreased through the use of preconception folate supplementation.
Anencephaly
Anencephaly is the most common of the open neural tube defects. This significant defect has historic importance in that it was the first fetal anomaly to be diagnosed by ultrasound over 35 years The bony skull above the base of the skull and orbits is missing. The cerebral hemispheres are also absent, although the cerebellum, brainstem, and midbrain are still present. On ultra- sound, h s defect should be detected reliably in the second and third trimesters through the absence of the usual cranial outline, and the frog-eyed appearance of the fetal orbits protruding into the amniotic fluid. When detected early, there may be a significant amount of exposed cerebral tissue. Exencephaly is the presence of exposed brain with absent protective covering of bone and skin, and is felt to be an embryologic precursor to anencephaly.1s, 37 First-trimester diagno- sis can be difficult because of the potential presence of a significant amount of cerebral tissue at that time. Associated defects are noted in one third to one half of these fetuses, and polyhydramnios is common, although it is usually not seen until the third trimester.*O, 29 Loss of cranial tissue tends to be symmetric in anencephaly; asymmetric tissue loss suggests the diagnosis of an amniotic band syndrome. When anencephaly is seen in conjunction with an open upper spinal defect, the condition is called craniorachischisis.
Cephalocele
Cephaloceles are herniations of the meninges and possibly brain tissue, through cranial defects. Of these lesions, 90% are encephaloceles (herniation of brain tissue and meninges) and 10% are meningoceles (herniation of meninges alone). They are more commonly skin covered than open. These lesions are typically found in the midline occipital region, with frontal, parietal, and nasal locations less commonly encountered. When present in the lower midline occipi- tal region, cephaloceles can be difficult to differentiate from cervical meningo- celes. Occasionally tissue can protrude through defects in the base of the skull and be seen in the nasal and orbital areas (sincipital encephaloceles) or from the pharyngeal area. Other neurologic findings, most commonly hydrocephalus, can be seen. Spinal defects are seen in 7% to 15% of these cases.1° Encephaloceles can be seen in conjunction with other anomalies, and in Meckel-Gruber syn- drome (occipital encephalocele with polydactyly and cystic kidneys), an autoso- ma1 recessive condition with a 25% recurrence risk. Prenatal diagnosis can be made with ultrasound, although careful scanning should be done to detect the defect in the cranium. If not seen, then alternative diagnoses of cystic hygromas, cervical spinal defects, teratomas, and hemangiomas should also be considered. Neonatal outcomes depend primarily on whether brain tissue is present within the hernia sac, because a worse prognosis is predicted with the presence of other defects or genetic syndromes, and with larger lesions.

CENTRAL NERVOUS SYSTEM ANOMALIES 793
lniencephaly
Iniencephaly is a rare malformation, combining a skull defect in the occipital region with an open spinal defect. These fetuses have a retroflexed head and cervical spine, with the fetal head facing upward. A short spine and other associated defects are common with this lethal
Spina Bifida
Spina bifida is the second most common neural tube defect and encom- passes a wide range of defects. Ths group of disorders can be separated into three classes. In its mildest form, spina bifida occulta, the vertebral arches fail to fuse, but there is a skin covering and no exposed neural tissue. This form is usually asymptomatic, and is often accompanied by alterations of the overlying skin, such as pigmented areas, sinus formation, or tufts of hair. No herniation of membranes or neural tissue is present. Spina bifida cystica includes closed spinal defects with an obvious back mass. This category includes closed myelo- meningoceles and meningoceles, and lipomyelomeningoceles. A meningocele is a protruding defect with the meninges, without neural tissue. When neural tissue is also included within the hernia, the defect is called a myelomeningocele. When a lipomatous growth is noted extending onto the dorsal aspect of the sac, the prefix lip0 is added.
Open spinal defects, or spina bifida aperta, are the clinically most severe of the spinal lesions. An open myelomeningocele includes a defect in the covering skin and bone, and a bulging sac containing neural tissue. These open defects are more common than their closed forms. Symptoms can arise from both the underlying damaged nerve roots and from hydrocephalus (which is present in three quarters of these patients). These lesions are most commonly found involv- ing the lumbar region. Chiari I1 malformations (downward displacement of the cerebellar vermis medulla into the upper aspects of the cervical spinal canal) are commonly seen with myelomeningoceles. The size and level of the spinal defect determine the neonatal prognosis. If untreated, open lesions have a high morbid- ity and mortality from infection and hydrocephalus. Lesions above L3 are associ- ated with complete paraplegia. Lesions below L3 result in variable motor and sensory defects in the lower extremities. Bowel and bladder function can also be compromised.
Ultrasound evaluation for spina bifida includes both spinal and cranial imaging. Spinal findings include widening of the posterolateral spinal ossifica- tion centers, absence of the continuity of the skin over the spine, and a bulging sac past the dorsal skin line. These are illustrated in Figures 1 and 2. Cranial findings suggestive of spina bifida include the presence of ventriculomegaly; small head size; an elongated cerebellum with obliteration of the cisterna magna from a Chiari I1 malformation (banana sign); and scalloping of the frontal skull region (lemon sign). The banana and lemon signs are illustrated in Figure 3. In a review of nine studies evaluating ultrasound findings with open neural tube defects (nearly 300 patients), the lemon and banana signs were the most sensi- tive, both 'being present in 97% of the cases.76 The lemon sign is best obtained superior to the plane used for biparietal diameter (BPD) measurements, at the level of the lateral ventricles. Because of increasing density of the fetal skull with increasing gestational age, the scalloping of the frontal bones usually resolves in the third trimester.74 This is a highly sensitive test for open neural tube defects only in the second trimester; its absence between 14 and 24 weeks

794 BRONSTEEN & COMSTOCK
Figure 1. Coronal view of the spine showing the widening of the lumbar vertebrae because of spina bifida. The T12 vertebra is marked. The site of the lesion at L4-5 is also shown.
makes spina bifida unlikely. Unfortunately, its presence is not diagnostic for spina bifida because, although the specificity for this sign is reasonably high at 99%, the 1% incidence t h s figure implies is quite large for a disease with an incidence of 1 per 1000.74 Posterior fossa changes with open neural tube defects are caused by the Chiari I1 malformation with herniation of the cerebellum into the upper spinal canal. The cerebellum takes on a crescent or banana shape and
Figure 2. Sagittal view of an open myelomeningocele. The arrows point to the herniated neural sac and the disruption in the bony vertebrae.

CENTRAL NERVOUS SYSTEM ANOMALIES 795
Figure 3. Cranial findings in a fetus with an open spinal defect. The arrow points to the crescent or banana-shaped cerebellum from the Chiari I I malformation. The anterior inden- tions of the lemon sign are also seen.
the cistema magna becomes obliterated. This banana sign in the second trimester is both sensitive and specific for open neural tube defects, without the false- positives of the lemon sign.74 In the third trimester, the obliterated cisterna magna may be easier to see, and more helpful than the banana sign. The posterior fossa changes in the presence of a spinal defect are called an Amold- Chiari type I1 malformation. Hydrocephalus develops in up to 90% of these cases because of the previously mentioned Chiari I1 malformation, and conversely one third hydrocephalic fetuses have a spinal defect.51
VENTRICULOMEGALY
Ventriculomegaly is defined as an enlargement of the cerebral ventricular system, most commonly the lateral ventricles. Cerebral spinal fluid (CSF) is predominantly produced within the choroid plexuses of the ventricles and flows through the ventricles, entering the subarachnoid space from the fourth ventricle. From here it flows around the brain or spine to be absorbed into the venous system through arachnoid granulations. Ventricular enlargement typically results from one of three processes:
1. Increased relative fluid pressure within the ventricles in relation to the venous system because of decreased resorption or, rarely, increased pro- duction
2. Abnormal formation of fetal brain tissue resulting in an increase in ventricular size
3. Atrophy of existing brain tissue

796 BRONSTEEN & COMSTOCK
The terms hydrocephalus and ventriculomegaly are often used interchangeably. More appropriately, the term ventriculomegaly is a generic term encompassing all forms of ventricular enlargement. Hydrocephalus is most appropriately used for the first category listed; ventricular enlargement because of increased fluid caused by either increased production or decreased clearance. Increased produc- tion can arise from a choroid plexus papilloma or an aneurysm of the vein of Galen (because of increased venous pressure). More commonly, hydrocephalus caused by decreased clearance of CSF (also called obstructive hydrocephalus) resulting from blockages either within the ventricular system (noncommunicat- ing) or outside of the ventricles at the site of CSF resorption (communicating). The most common cause of noncommunicating hydrocephalus is from aque- ductal stenosis. A case of severe hydrocephalus from aqueductal stenosis is shown in Figure 4. The obstruction typically occurs in the midtrimester as the aqueduct elongates and narrows with brain growth. An x-linked form is known, but most cases are idiopathic in origin. Communicating hydrocephalus is the result of hemorrhage or infection blocking fluid resorption at the arachnoid granulations. In a recent series the frequency of hydrocephalus was listed as 0.81 cases per 1000 live bird1s.6~ As with most other CNS abnormalities, ventricu- lomegaly is seen in association with other anomalies, abnormal karyotypes, and genetic syndromes. In general, the diagnosis of hydrocephalus carries a poor prognosis, and over three quarters of these babies have associated anomalies.
The diagnosis of ventriculomegaly can be made with a combination of qualitative and quantitative assessments. The choroid plexus usually fills the body of the lateral ventricle. In the early second trimester the choroid takes up
Figure 4. Severe hydrocephalus caused by aqueductal stenosis. The thin rim of cerebral tissue at the periphery separates this from hydrancephaly and the presence of a midline septum differentiates this from holoprosencephaly.

CENTRAL NERVOUS SYSTEM ANOMALIES 797
a significant portion of the cerebral hemispheres. As the gestation continues, both the lateral ventricle and the choroid decrease in size. A lateral ventricular body with shrunken-appearing choroid plexus is suggestive of a pathologic situation. This dangling choroid is usually noted with its free posterior portion pointing to the floor under the influence of gravity. Although unilateral dilation of the lateral ventricles has been reported, this is usually a bilateral process. Reverberation artifacts usually limit visualization of the lateral ventricle closest to the transducer.
Quantitatively, various components of the ventricular system can be mea- sured and used to define ventriculomegaly. Currently, many use the width of the atria of the lateral ventricles as the point of reference.8 A measurement above 10 mm is abnormal. This measurement is easy to obtain, reproducible, and is gestational age independent with its size relatively constant through the last two trimesters of pregnancy. When multiple measurements are taken, the smallest measurement is probably the best one. Errors in measurement, such as tangential measurements, and poor visualization of sonolucent cerebral tissue give falsely elevated values. False-normal readings are not typical. The measurements are taken from a transaxial plane just superior to the plane used for the BPD measurement. Calipers should not include the ventricular wall, but should be placed perpendicular to them and not the cranial midline. The point of measure- ment is not crucual because points of measurement just anterior or posterior to the atria (at the distal body of the lateral ventricle, the atrium, or the proximal occipital lobe) all give similar results.36 A 3-mm separation from the medial ventricular wall to the medial margin of the choroid can also used to evaluate ventriculomegaly.38, 43
Determinants of prognosis for fetuses with hydrocephalus include the pres- ence of associated genetic or other structural abnormalities, and the degree of ventricular dilation. In a recent review, including nearly 700 cases of hydrocepha- lus, isolated cases constituted only 20% of the cases.54 Chromosomal problems were present in 8%, CNS anomalies in 47%, and non-CNS anomalies in 36%. Spina bifida is seem in approximately one fourth’ of the cases. As a group, the prognosis for fetuses with ventriculomegaly is not favorable. Even with a fetus diagnosed with isolated ventriculomegaly, there is a 30% mortality rate, and approximately 60% of the survivors are normal.3l Where isolated ventriculomeg- aly is diagnosed in utero, there is only a 40% chance of producing a normal child. Obviously, fetuses with ventriculomegaly and associated anomalies have a worse prognosis.
Recently, the appropriateness of the cutoff of 10 mm as diagnostic of ventri- culomegaly has come into question. Despite the poor prognostic outcomes expected, many fetuses listed as abnormal because of mild atrial enlargement above 10 mm turn out to be normal on neonatal assessment. More normal neonates than expected, given the previously mentioned prognosis, were seen following the diagnosis of ventriculomegaly using this diagnostic point. Is the statistical cutoff of 10 mm appropriate as a pathologic cutoff, especially in boy fetuses where normal values exceed those for girls?56 The diagnosis of ventriculomegaly leads to increased patient anxiety, and possible expensive invasive testing. This issue has been addressed nicely in a recent editorial, and with recent outcome data.”
The atrial measurement was popularized by Cardoza et a1,8 who chose an atrial width of 10 mm as the cutoff between normal and enlarged. This 10-mm figure was four standard deviations above the mean in his group. Others have also supported the 10-mm cutoff for separating normal from abnormal, although
26, 57, 65

798 BRONSTEEN & COMSTOCK
Table 1. DISTRIBUTION OF LATERAL VENTRICULAR ATRIAL MEASUREMENTS IN NORMAL POPULATIONS
Author Mean Width Standard Deviation
Cardoza et als 7.6 Pilu et a15* 6.9 Heiserman et a136 6.5 Alagappan et a13 6.6 Farrell et alZ4 5.4 Pate1 et a156 6.1 Achiron et all 6.6
0.6 1.3 1.3 1.4 1.2 1.3 1.2
most of these studies found h s cutoff to be 2.5 to three standard deviations above the means (Table 1).
A recent review of isolated mild ventriculomegaly presented 31 cases and reviewed the 1iteratu1-e.~~ Looking at the data presented in that review, and adding three other recent articles not included in the review, the overall outcome is shown in Table 2.2 22, 65 Overall, over 250 fetuses were evaluated. Most articles used a definition of an atrial diameter of 10 to 15 mm to define this group, although some references used a lower cutoff or included cases with unilateral dilation. Where possible the results are stratified in Table 2 by those with an atrial diameter equal to or above 12 mm as opposed to those less than 12 mm. From these data it can be seen that even mild ventriculomegaly is a pathologic entity. Even in its mildest form (10 to 12 mm dilation), a 3.7% rate of abnormal karyotypes, and 7.7% developmental delay rate were encountered. Certainly, mild isolated ventriculomegaly has a much better prognosis than more dismal outcome seen with more severe forms of the disease, with nearly 80% of the fetuses found to be normal on follow-up.
Fetuses with normal atrial measurements ,(below 10 mm), but increased distance between the medial ventricular wall and medial border of the choroid (above 3 mm), have also been shown to be at increased risk of an abnormal outcome?* Evaluating 74 such fetuses, 20% were classified as abnormal, with a wide range of abnormalities from isolated polydactyly to severe defects. Cases with isolated increased medial ventricle wall-choroid distances did better (15.5% abnormal) than those with associated defects (37.5% abnormal).
These data support the current ongoing practice of using 10 mm as the cutoff for ventriculomegaly. This group, statistically defined as abnormal, has been confirmed by outcome data also to be pathologically abnormal. These data support the concept that there is a spectrum of disease, starting from isolated mild ventricular enlargement where mortality and abnormal karyotype are 3% to 4% each and normal outcome is seen in 80% of the cases, to isolated hydro- cephalus where approximately 60% are normal, to all patients with ventriculo-
Table 2. OUTCOME OF FETUSES WITH MILD ISOLATED VENTRICULOMEGALY
Overall % %Atria = 10-12 % Atria >12-15
Abnormal karyotype 4.3 3.7 4.3 Perinatal deaths 3.4 Developmental delays 12.1 7.7 16.1

CENTRAL NERVOUS SYSTEM ANOMALIES 799
megaly where mortality rates can reach 85% and associated defects are com- m01-1.~~
AGENESIS OF THE CORPUS CALLOSUM
The corpus callosum is the largest of three bundles of fibers (commissures) that connect the two cerebral hemispheres. It is located in the middle of the cerebral hemispheres, and forms the roof of the lateral and third ventricles, and the floor of the interhemispheric fissure. Embryologically, the corpus callosum begins forming at its rostra1 end at 12 weeks, with caudal extension over the next several months. Once formed, by 18 to 20 weeks, the thin structure thckens as the second half of pregnancy and first year of life progresses.
Abnormal formation of the corpus callosum can be either partial or complete agenesis. In the former, it is typically the caudal end that does not develop. In the complete form, agenesis of the corpus callosum (ACC), thick bundles of fibers (bundles of Probst) are seen running parallel to the interhemispheric fissure along the superomedial wall of the lateral ventricles. The incidence of ACC in the general population has been estimated at 0.7% based on a pneumoencephalogram
ACC can be found in both syndromic and nonsyndromic forms. The non- syndromic forms of ACC are the most common. Absence of this structure is a characteristic finding in four genetic syndromes (Aicardi’s, Anderman’s, acrocal- losal, and Shapiro’s syndromes); can variably be present in over 20 others (including Dandy-Walker and Arnold-Chiari 11); and can be seen in trisomies, typically for chromosomes 8, 13, and 18. There have been reports of inherited cases of ACC with autosomal recessive, sex-linked recessive, and autosomal dominant transmissions reported.
Both functional and structural defects are commonly reported in association with ACC. Autopsy data show the association of CNS abnormalities in 85% of the cases, and other anomalies in 62%.55 Pediatric data show ACC associated with mental retardation in 73%, seizures in 42%, and ocular abnormalities in 42%.40 The pessimistic view given by those data on the outcome of cases with ACC is probably not appropriate for prenatal estimates and counseling because these data are probably biased toward a negative outcome, because of patient collection from selected abnormal populations Pediatric data also suggest this probable bias, and because children with earlier diagnosis of ACC do poorer than those with a diagnosis at a later age.41
Studies reporting the outcome of prenatally detected ACC do not share the biases of pediatric studies based on already abnormal patients and provide a better foundation on which to base counseling for pregnant patients. Unfortu- nately, data here are limited by the small size of cases with prenatal detection of ACC where follow-up is available. Results of a recent literature review and two subsequent series are shown in Table 3.4, 33, 75 In approximately 40% of the cases the absent corpus callosum was an isolated finding. Here the outcome was good with 81% normal on follow-up. When other anomalies are seen, only 18% were normal on follow-up with a significant increase in mental-motor delay and mortality.
The ultrasound diagnosis of ACC was first reported a decade and a half ago.16 Prenatal detection is limited by several factors. Although completely formed by 20 weeks, the corpus callosum is still thin and not well visualized in the axial plane, the most common one used in routine obstetric fetal head scanning. Coronal and sagittal views are better suited to visualize this structure,

800 BRONSTEEN & COMSTOCK
Table 3. NEONATAL OUTCOME OF PRENATALLY DETECTED ACC
Normal on Mental- Follow-up Motor Genetic NN
Total Assessment Delay Trisomy Syndrome IUFD Death No. Y O % % Y O Yo Yo
_ _ Isolated 42 81 11 3 5 With other
anomalies 57 18 36 6 6 15 18
IUFD = In utero fetal demise; NN = neonatal. *Number of cases excluding voluntary terminations of pregnancy where follow-up was given.
but are not part of most routine scans. The presence of indirect findings must be used to increase suspicion for ACC. These findings are caused by changes produced in the surrounding brain tissue by the absence of the corpus callosum, and its replacement by the longitudinally running bundles of Probst. These indirect ultrasound findings are listed next:
1. Absent cavum septi pellucidi 2. Elevated third ventricle; interhemispheric cyst seen between the bodies
of the lateral ventricles 3. Abnormal lateral ventricles
Thinned or slitlike frontal horns that do not approach the midline; on coronal scan have a concave medial border and more vertical orienta- tion than normal
Lateral displacement, axis is more parallel to the midline Dilated occipital horns (colpocephaly)
Figure 5 also illustrates some of these findings. Unfortunately, even these indirect signs may only be subtle at the time of routine second-trimester scan- ning, and many cases are not detected until the third trimester4, 33
HOLOPROSENCEPHALY
Holoprosencephaly is a condition resulting from absent or incomplete di- verticulation and cleavage of the embryonic forebrain (prosencephalon) into the cerebral hemispheres and lateral ventricles. This disorder presents with a spec- trum of changes of the brain and face. Depending on the degree of abnormalities present, this condition can be classified in order of increasing severity as lobar, semilobar, and alobar. The cause is heterogeneous. Most cases seen are sporadic, but karyotypic abnormalities can be seen, most typically with chromosomes 13 and 18. Familial forms have been found with both autosomal dominant and autosomal recessive inheritance. More common in abortuses, where it may reach a frequency of 0.4% of all conceptuses, the occurrence of holoprosencephaly is rare later in pregnancy, reported to occur between 1 in 2500 and in 16,000 live birthsz1, 45
In alobar holoprosencephaly, the most severe of the three variants, a single ventricular cavity is present, with no formation of separate lateral ventricles. The thalami are fused and both the corpus callosum and falx are absent. No interhemispheric fissure is present. Optic tracts and olfactory bulbs are absent. Midfacial defects are common including the following:

CENTRAL NERVOUS SYSTEM ANOMALIES 801
Figure 5. Axial view of a fetus with an absent corpus callosum. The plane shown is above that used for a biparietal diameter measurement. Note the presence of an interhemispheric cyst (vertical arrow), which is the elevated third ventricle. The frontal horns of the lateral (horizontal arrow) are compressed and pushed laterally by the bundles of Probst. This gives a railroad track appearance to the frontal horns. The occipital lobe is not dilated in this case.
Cyclopia: a single orbit with a single or partially divided eye, and a proboscis
Ethmocephaly: hypotelorism with a high midline proboscus Cebocephaly: hypotelorism with a single nasal orbit Cleft lip and palate: usually seen with hypotelorism Less severe midfacial defects including hypotelorism, absent nasal bones, iris
coloboma, single central incisor, and an absent frenulum
Infants with this form of holoprosencephaly are profoundly affected, and do not survive more than a year.
In semilobar prosencephaly, some cleavage of the cranial tissue posteriorly is present. Partial formation of the occipital horns is noted, and the posterior portions of the falx and interhemispheric fissure are seen. The thalami may only be partially fused. As with the alobar form, anteriorly the ventricles are fused, and the corpus callosum, optic tracts, and olfactory bulbs are missing. Mental retardation is usually present, and most do not survive infancy. Facial defects as mentioned previously can be seen here, although the severer forms (cyclopia and ethmocephaly) are usually seen in association with the alobar form.
In the mildest form, lobar holoprosencephaly, anterior cleavage is also present, and the degree of anatomic distortion is much less than the other forms. Frontal horns can now be identified, although fusion between the right and left halves in the midline is still present. The corpus callosum is absent. The roof of the anterior end of the frontal horns takes on a squared or flat appearance. The interhemispheric fissure and falx may be present anteriorly, although not fully developed. This appearance is similar to that seen with septo-optic dysplasia (this latter condition is diagnosed after birth by the absence of optic tracts.
above the eye

802 BRONSTEEN & COMSTOCK
Unfortunately, although gross brain formation is more advanced, severe mental retardation can still be present.
Holoprosencephaly is most commonly seen as sporadic occurrence. Its oc- currence can also be the result of teratogenic exposure (retinoic acid alcohol); maternal diabetes; as a part of a genetic syndrome (Smith-Lemli-Opitz); or with chromosomal abnormalities including autosomal dominant and recessive modes of inheritance. Approximately 55% of these cases have been reported with abnormal karyotypes, usually trisomy 13.59
The genetic aspect of this disorder has attracted considerable interest recently.47* 62 Recurrent chromosomal anomalies in patients with holoprosenceph- aly implicate several regions in the chromosomal arrangement as being involved with the disordered tissue formation noted. One of these loci, 7q36, the "sonic hedgehog" gene, is responsible for production of a protein important in neural differentiation. This loci is involved in the familial forms of holoprosencephaly. Interestingly, cholesterol levels can modify the function of the sonic hedgehog protein, also leading to holoprosencephaly. This is the proposed mechanism behind the association between the Smith-Lemli-Opitz syndrome (with its inher- ited defects in cholesterol synthesis) and holoprosencephaly. Microforms of fa- milial holoprosencephaly are also reported. Here, mild craniofacial defects can be present with normal intracranial imaging. Such findings include microcephaly, hypotelorism, single central maxillary incisor, absent frenulum, midface hypo- plasia, and iris c010boma.~~ These individuals are at increased risk of having offspring with holoprosencephaly.
Recurrence risks vary. A 1% recurrence rate is predicted if a chromosomal abnormality is present; otherwise a 6% recurrence rate is seen, unless the defect is part of a genetic syndrome, or autosomal dominant pattern.I4
CHOROID PLEXUS CYSTS
It has been over a decade and a half since the antenatal diagnosis of choroid plexus cysts (CPCs) was first reported.13 The finding of a CPC now a relatively common one on prenatal scans. A recent review of 33 studies found these cysts to be present in nearly 1% of fetuses scanned.60 By itself the CPC is typically a benign entity, usually regressing as the pregnancy progresses. Their presence has been shown, however, to be associated with an increased risk of an abnormal karyotype, specifically trisomy 18. Although at one time felt to be associated with Down syndrome, CPCs are no longer felt to be a marker for this ane~ploidy.~, 32
Trisomy 18 (Edwards' syndrome) is the second most common chromosomal defect after Down syndrome. The presence of trisomy 18 has significant implica- tions for the ongoing pregnancy. %s is essentially a lethal diagnosis because survivors are rare. Approximately two thirds of these fetuses diagnosed in the midtrimester do not survive to te~m.4~ Those that go into the third trimester may manifest growth disorders, and can be delivered under urgent and emergency conditions for nonreassuring fetal status. Of those liveborn, most die within the first several months of life. Only 10% of liveborns live to their first birthday.
The significance of a prenatal finding of isolated CPCs is currently unsettled in terms of the advisability of obtaining a genetic consult or genetic amniocente- sis to evaluate for trisomy 18. Most trisomy 18 fetuses (86%) have abnormalities seen on prenatal ~l t rasounds.~~ These abnormalities can range from severe de-

CENTRAL NERVOUS SYSTEM ANOMALIES 803
fects to subtle findings. In a recent review of CPCs and aneuploidy, 54% of trisomy 18 babies were found to have CPCS.~O In most cases of trisomy 18, the CPCs were seen along with other abnormalities, but overall approximately 4% of trisomy 18 have isolated CPCS.~ This is compared with the normal popula- tion, where CPCs are present nearly 1% of the time, and in most of these cases the cysts are an isolated finding.60 Of concern with isolated CPCs leading to genetic evaluation is the relatively frequent finding of these cysts in the normal population (approximately YO) and the small contribution to this total from trisomy 18 fetuses (1 per 3000 livebirths; less than 5% are predicted to have isolated CPCs). Because of the potential for a large number of false-positives with isolated CPCs, genetic evaluation in these cases could lead to excessive testing with resulting cost- and procedure-related loss of normal fetuses com- pared with the small number of abnormal fetuses identified. Despite these concerns, and predicted low trisomy 18 detection rate, a 1% aneuploidy rate, mostly caused by trisomy 18, was seen in a review of over 3200 reported cases of isolated CPCS.~O
Other factors along with the presence of CPCs can be used to assist in determining the genetic risk assessment. Most CPCs (87%) are an isolated find- ing.6O The presence of associated anomalies along with the CPCs resulted in a 38% aneuploidy rate. Size, persistence, and number of cysts was not a good discriminating factor. Increased maternal age, as with Down syndrome, increases the risk for trisomy 18.25, 68 In reviewing nine prospective studies, the presence of isolated CPCs resulted in a likelihood ratio of 9 for trisomy 18, over the baseline risk.34 By combining this increased risk from isolated cysts, with prior work on risk because of maternal and gestational age, a risk table has been constructed to calculate trisomy 18 risk given maternal age and gestational age for fetuses with isolated CPCS.'~ The chance of having a fetus at 16 to 18 weeks with a finding of isolated CPCs was at least 1 in 200 for women who are 32 years or older. Alternatively, Snidjers et aP8 calculated that isolated CPCs gave a likelihood ratio of only 1.5 for trisomy 18, and recommended no correction of the baseline maternal age-related risk when these cysts are an isolated finding.68
Maternal serum triple screen testing can detect 60% of trisomy 18 fetuses. Although not tested yet on a large scale, in series with isolated CPCs, an abnormal maternal serum screen has been shown to be predictive of trisomy 18 in limited studies." 70 Although further evaluation is needed, one would predict that maternal serum screening should be helpful in further evaluating trisomy 18 risk when isolated CPCs are present.
The final words on isolated CPCs and the need for genetic testing have yet to be written, and the issue is far from settled. With isolated CPCs, we are faced with a group with a small but potentially increased risk of trisomy 18. Uncertainties arise from the relative infrequency of both trisomy 18 and isolated CPCs in these fetuses, leading to difficulties in the statistical interruption of the data. Also of concern is the ability of different ultrasound units to detect subtle limb defects, which may be the only other anomaly in trisomy 18 fetuses. Physicians need to take care of their patients in a way that minimizes needless anxiety, cost, and pregnancy loss, yet leaves them with an option for appropriate prenatal diagnosis. A careful scan of the fetus should be done when CPCs are seen. In cases where CPCs are seen in association with other anomalies, with abnormal maternal serum screening, or with other genetic risk factors (such as advanced maternal age), genetic testing should be considered. There is currently not uniform agreement on the need for genetic testing when isolated CPCs are seen in an otherwise low-risk patient.
804 BRONSTEEN & COMSTOCK
ARTERIOVENOUS MALFORMATIONS
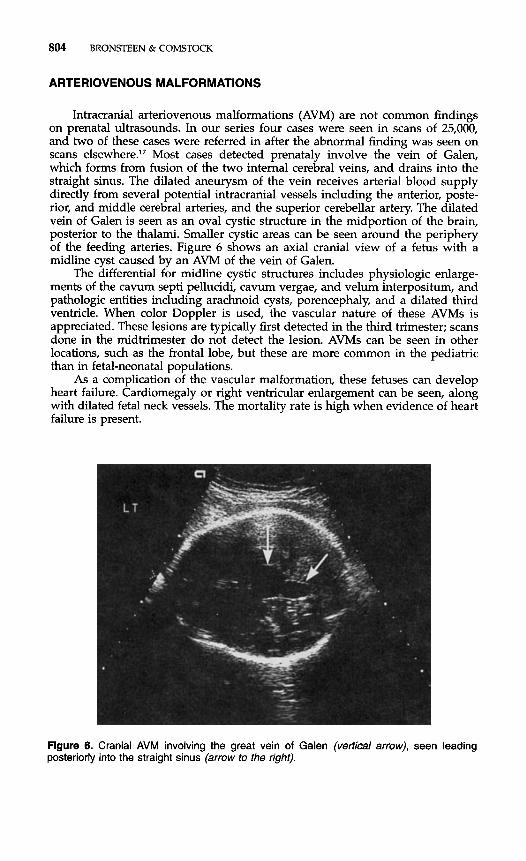
Intracranial arteriovenous malformations (AVM) are not common findings on prenatal ultrasounds. In our series four cases were seen in scans of 25,000, and two of these cases were referred in after the abnormal finding was seen on scans e1~ewhere.l~ Most cases detected prenataly involve the vein of Galen, which forms from fusion of the two internal cerebral veins, and drains into the straight sinus. The dilated aneurysm of the vein receives arterial blood supply directly from several potential intracranial vessels including the anterior, poste- rior, and middle cerebral arteries, and the superior cerebellar artery. The dilated vein of Galen is seen as an oval cystic structure in the midportion of the brain, posterior to the thalami. Smaller cystic areas can be seen around the periphery of the feeding arteries. Figure 6 shows an axial cranial view of a fetus with a midline cyst caused by an AVM of the vein of Galen.
The differential for midline cystic structures includes physiologic enlarge- ments of the cavum septi pellucidi, cavum vergae, and velum interpositurn, and pathologic entities including arachnoid cysts, porencephaly, and a dilated third ventricle. When color Doppler is used, the vascular nature of these AVMs is appreciated. These lesions are typically first detected in the third trimester; scans done in the midtrimester do not detect the lesion. AVMs can be seen in other locations, such as the frontal lobe, but these are more common in the pediatric than in fetal-neonatal populations.
As a complication of the vascular malformation, these fetuses can develop heart failure. Cardiomegaly or right ventricular enlargement can be seen, along with dilated fetal neck vessels. The mortality rate is high when evidence of heart failure is present.
Figure 6. Cranial AVM involving the great vein of Galen (vertical arrow), seen leading posteriorly into the straight sinus (arrow to the right).

CENTRAL NERVOUS SYSTEM ANOMALIES 805
Figure 7. lntracranial teratoma involving the inferior cerebral hemisphere is shown between the ultrasound markers.
INTRACRANIAL TUMORS
Intracranial tumors are rare findings during the antenatal period. The most common tumor encountered is a teratoma. This tumor is usually supratentorial in location, and has a complex appearance with both solid and cystic areas. If found early, the tumor can be localized to one hemisphere. As the tumor grows, a mass effect can be seen in the remaining tissue, and eventually the entire cranium can be taken up by the abnormal tissue. Complications that can arise include craniomegaly, making delivery difficult; heart failure; and hydrops. Perinatal mortality for intracranial tumors is high.66 Figure 7 shows a fetus with an intracranial teratoma. The neuroepithelial tumors (glioblastomas and astrocytomas) are more typically seen after delivery than during the prenatal pe- riod.
HYDRANENCEPHALY
Another rare abnormality occurring in less than 1 per 10,000 live births, this lesion is marked by absent cerebral cortex, with an intact skull and meninges. It has been hypothesized to occur from or arise from the destruction of normally forming brain tissue during the pregnancy. Bilateral occlusion of the internal carotid arteries has been proposed as an etiologic mechanism. Unlike other cranial defects discussed here, hydranencephaly does not carry an increased risk of other structural defects or an association with an abnormal karyotype. Few of these newborns survive for more than several months, and those who do survive are usually severely retarded. On ultrasound there is absent cerebral tissue, although the falx, posterior fossa, midbrain, and basal ganglia are still present. This entity can be differentiated from severe hydrocephalus and holo-

806 BRONSTEEN & COMSTOCK
prosencephaly by the absence of any cortical tissue. Also, the presence of a falx is usually not seen with holoprosencephaly.
PORENCEPHALY
As with the previously described hydrancephaly, porencephaly involves the destruction of previously formed brain tissue. Hemorrhage, trauma, or infarction within the brain leads to tissue necrosis, with the resulting formation of an intraparenchymal cyst. The remaining cyst may be isolated within the brain tissue, or more commonly empty into either the ventricles or subarachnoid space. Unlike arachnoid or interhemispheric cysts, porencephalic cysts do not produce a mass effect. The ipsilateral ventricle usually enlarges to make up for the loss of brain tissue. The presence of porencephaly is not felt to be associated with other structural defects.
SCHIZENCEPHALY
Schizencephaly presents typically as bilateral clefts within the cerebral pa- renchyma, such that the ventricles widely communicate with the subarachnoid space. Unilateral cases can be seen. The presence of these clefts in the distribution of the middle cerebral artery suggest a possible vascular cause. As with por- encephaly, this diagnosis is not thought to be associated with chromosomal or other structural defects.
POSTERIOR FOSSA ABNORMALITIES
The posterior fossa is bordered by the cranial vault, the tentorium, and the brainstem, and can be evaluated on axial views by angling the posterior portion of the transducer caudally from the plane used to obtain a BPD. Structures to be evaluated include the cerebellum and the cisterna magna.
Enlarged Cisterna Magna
The cisterna magna is typically measured from the posterior aspect of the vermis to the internal surface of the occipital bone. This measurement has been shown to be gestational age independent, averaging 5 mm with a standard deviation of 3 mm.43
When measurements of the cisterna magna above 10 mm are obtained there is an increased risk for several underlying pathologic entities. An enlarged cisterna magna, as shown in Figure 8, can be seen with Dandy-Walker malforma- tions and variants, trisomy 18, arachnoid cysts, and a small cerebellum. The latter finding can be seen with intrauterine growth retardation and trisomy 18. The finding of an enlarged cisterna magna should prompt a careful survey of the fetal anatomy. Also, care should be taken in measuring the cisterna magna. The transducer should be rotated slightly posteriorly (15 to 30 degrees) from the axial plane used to get the BPD measurement. Too steep an angle can falsely increase this measurement.
If after an evaluation of an enlarged cisterna magna the chromosomes are normal, and no other defects are seen, the diagnosis of a normal variant can be

CENTRAL NERVOUS SYSTEM ANOMALIES 807
Figure 8. Enlarged cisterna magna shown by the arrow.
entertained. Communicating hydrocephalus is a potential cause to be considered, although the most common causes for this condition (infection or hemorrhage) are unlikely in the fetus.
Dandy-Walker Syndrome
The Dandy-Walker syndrome is a group of disorders characterized by a complete or partial hypoplasia of the cerebellar vermis and has a frequency of 1 in 25,000 to 35,000 live births.39 In its severer form, the Dandy-Walker malforma- tion (DWM), the cerebellar hemispheres are thinned, separated, and pushed to the sides of the posterior fossa, as shown in Figure 9. The posterior fossa is occupied by a cystic-appearing structure, which communicates with the fourth ventricle. This posterior fossa cyst is the enlarged fourth ventricle. The DWM is probably the result of abnormal formation of the roof of the fourth ventricle rather than because of an obstruction of CSF flow out of the fourth ventricle foramina.27 Hydrocephalus is commonly seen in association with DWM; ACC and encephaloceles can also be If an enlarged occipital lobe of the lateral ventricle is seen with a Dandy-Walker cyst, the possibilities of hydrocephalus and colpocephaly from ACC should be considered.
As with other CNS anomalies discussed in this article, the DWM can be an isolated finding; seen in association with other extra-CNS anomalies; with abnor- mal karyotypes and genetic syndromes (Aicardi's, Walker-Warburg); or because of environmental factors (TORCH infections or alcohol and coumadin exposure). The prognosis is related to the presence of these associated factors. From a collection of larger pediatric series, DWM is an isolated finding approximately

808 BRONSTEEN & COMSTOCK
Figure 9. Dandy-Walker malformation. No cerebellar verrnis is present; the fourth ventricle (arrowhead) is continuous with the enlarged cisterna (posterior fossa cyst); and the cerebel- lar hemispheres (arrow) are pushed anteriolaterally.
40% of the time (range 14% to 57%); cerebellar dysfunction is not typical; and a low intelligence quotient seen in 40% to 70% of the cases.34, 35, 39, 53, 64, Series with prenatally diagnosed cases show a worse prognosis because apparently the more severely affected cases do not survive for inclusion in the pediatric series. In antenatally diagnosed cases, mortality rates are high, with nearly two thirds of the cases dying either in utero or in the newborn period, and chromosomal abnormalities are seen in one third to one half of the cases.5o, 63
Dandy-Walker Variant
In the Dandy-Walker variant (DWV), the cerebellar vermis is present, al- though the inferior portion is hypoplastic. Findings here can be quite subtle. On the typical axial view, the cerebellum, fourth ventricle, and cisterna magna can appear normal. On angling the ultrasound transducer more posteriorly, the sonoluscent subarachnoid space seen between the inferior portions of the cere- bellar hemispheres is enlarged because of the absent portion of the inferior vermis. This finding is more qualitative than quantitative. A U-shaped keyhole configuration of the subarachnoid space extending from the cisterna magna through to the fourth ventricle is typical for the DWV. This is to be distinguished from the normal V-shaped appearance in this area.15 This diagnosis is difficult to make with certainty before 18 weeks of gestation because the cerebellar vermis may not physiologically close until this gestational age.6 Care should be taken to avoid excessive posterior rotation of the transducer, which can give a false impression of a DWV: the spine and thick nuchal tissue should not be visualized with the cerebellum. As with the severer DWM, the diagnosis of a DWV should prompt consideration for other structural defects and an abnormal

CENTRAL NERVOUS SYSTEM ANOMALIES 809
kary~type."~ The prognosis with DWV has not been as well studied as the severer DWM, but from two series with prenatal diagnosis of DWV, the morbidity and mortality appear to be ~ignificant.~, 23 Combining data from these two series shows a mortality rate of 44%, an abnormal karyotype in 45%, and other structural defects in 69%. When compared with the outcome for DWM, the mortality rate and associated defects were similar, and an abnormal karyotype was seen more often with the variant form (53% versus 32y0).~ Associated defects and abnormal karyotype appear to play a significant role in prognosis because follow-up outcome was reported as normal in 75% of the isolated cases of DWV, as opposed to a normal outcome in only 20% of the nonisolated cases.23
Arachnoid Cysts
Arachnoid cysts in the posterior fossa can mimic a DWM. A less common abnormality, h s cyst usually displaces the cerebellum as a single unit rather than separating the hemispheres in the midline. Cysts from the quadrigeminal cistern (arising from the arachnoid tissue) can be present in the posterior fossa. These cysts lie in between the cerebellum and the third ventricle.
Cerebellar Abnormalities
Cerebellar size should be measured when an enlarged cisterna magna is noted and compared with existing nomograms for appropriate cerebellar size.=* 46 Although a small cerebellum can be a normal variant with no adverse implications, one needs to be aware of the possibility of a pathologic cause. Although at one time proposed to be a growth-independent measure of fetal age, the cerebellum has been found to be decreased in size in some intrauterine growth retarded fetuses." 61 As previously mentioned, the cerebellum can be abnormal (absent or banana shaped) with open spinal defects. Cerebellar hypo- plasia has an association with chromosomal abnormalities.
Although they are comprised of a diverse group of anomalies, a common theme runs through the various CNS malformations discussed previously. They are often seen in association with other defects, both within and outside of the CNS. They are associated with abnormal karyotypes (trisomies 8, 13, and 18 are typical) and are also part of various genetic syndromes. The eventual prognosis for the affected baby depends on the primary abnormality, and any associated anomalies, both structural and genetic.
ACKNOWLEDGMENTS
The authors thank Celia Feld for her assistance in the preparation of this manuscript.
References
1. Achiron R, Schimmel M, Achiron A, et al: Fetal mild idiopathic lateral ventriculomeg- aly: Is there a correlation with fetal trisomy? Ultrasound Obstet Gynecol 3539, 1993
2. Achiron R, Yagel S, Rotstein Z, et al: Cerebral lateral ventricular asymmetry: Is this a normal ultrasonographic finding in the fetal brain? Obstet Gynecol 89:233, 1997
3. Alagappan R, Browning I'D, Laorr A, et al: Distal lateral ventricular atrium: Reevalua- tion of normal range. Radiology 193:405, 1994

810 BRONSTEEN & COMSTOCK
4. Bennett GL, Bromley B, Benacerraf BR Agenesis of the corpus callosum: Prenatal detection usually is not possible before 22 weeks of gestation. Radiology 199:447, 1996
5. Bromley B, Lieberman E, Benacerraf BR Choroid plexus cysts: Not associated with Down syndrome. Ultrasound Obstet Gynecol 8232, 1996
6. Bromley B, Nadel AS, Pauker S, et al: Closure of the cerebellar vermis: Evaluation with second trimester US. Radiology 193:761, 1994
7. Brown T, Kliewer MA, Hertzberg BS, et a1 A role for maternal serum screening in detecting chromosomal abnormalities in fetuses with isolated choroid plexus cysts: A prospective multicentre study. Prenat Diagn 19:405, 1999
8. Cardoza JD, Goldstein RB, Filly RA: Exclusion of fetal ventriculomegaly with a single measurement: The width of the lateral ventricular atrium. AJR Am J Roentgenol 151:767, 1988
9. Chang MC, Russell SA, Callen PW, et al: Sonographic detection of inferior vermian agenesis in Dandy-Walker malformations: Prognostic implications. Radiology 193:765, 1994
10. Chervenak FA, Isaacson G, Mahoney MJ, et al: Diagnosis and management of fetal cephalocele. Obstet Gynecol 64:86, 1984
11. Chervenak FA, Kurjak A, Comstock CH (eds): Ultrasound and the Fetal Brain. Cam- forth, England, Parthenon Press, 1995
12. Choong S, Meagher S Management of choroid plexus cysts in the mid-trimester fetus. Aust N Z J Obstet Gynaecol 39:1, 1999
13. Chudleigh P, Pearce JM, Campbell S The prenatal diagnosis of transient cysts of the fetal choroid plexus. Prenat Diagn 4:135, 1984
14. Cohen MM: An update on the holoprosencephalic disorders. J Pediatr 101:865, 1982 15. Comstock CH, Boa1 DB: Enlarged fetal cistema magna: Appearance and sigruficance.
Obstet Gynecol 66:25S, 1985 16. Comstock CH, Culp D, Gonzalez J, et al: Agenesis of the corpus callosum in the Fetus:
Its evolution and significance. J Ultrasound Med 4:613, 1985 17. Comstock CH, Kirk J S Arteriovenous malformations. Locations and evolution in the
fetal brain. J Ultrasound Med 10:361, 1991 18. Cox GG, Rosenthall SJ, Holsapple JW: Exencephaly: Sonographic findings and radio-
logic-pathologic correlation. Radiology 155:755, 1985 19. Cragan JD, Roberts HE, Edmonds LD, et al: Surveillance for anencephaly and spina
bifida and the impact of prenatal diagnosis-United States, 198.51994. MMWR CDC Surveil1 Summ 44(SS-4):1, 1995
20. David TJ, Nixon A Congenital malformations associated with anencephaly and in- iencephaly. J Med Genet 13:263, 1976
21. DeMyer W Holoprosencephaly. In Vinken PJ, Bruyn GW (eds): Handbook of Clinical Neurology, vol30. Amsterdam, Elsevier, 1977, p 431
22. Den Hollander NS, Vinkesteijn A, Schmitz-Van Splunder P, et al: Prenatally diagnosed fetal ventriculomegaly: Prognosis and outcome. Prenat Diagn 18:557, 1998
23. Estroff JA, Scott MR, Benacerraf BR Dandy-Walker variant: Prenatal sonographic features and clinical outcome. Radiology 185:755, 1992
24. Farrell TA, Hertzberg BS, Kliewer MA, et al: Fetal lateral ventricles: Reassessment of normal values for atrial diameter at US. Radiology 193:409, 1994
25. Ferguson-Smith MA, Yates JRW Maternal age specific rates for chromosome aberra- tions and factors influencing them: Report of a collaborative European study on 52,965 amniocenteses. Prenat Diagn 45, 1984
26. Filly RA, Goldstein RB: The fetal ventricular atrium: Fourth down and 10 mm to go. Radiology 193:315, 1994
27. Gardner E, ORahilly R, Prolo D: The Dandy-Walker and Arnold-Chiari malformations: Clinical, developmental and teratological considerations. Arch Neurol 32393, 1975
28. Goldstein I, Reece EA, Pilu G, et al: Cerebellar measurements with ultrasonography in the evaluation of fetal growth and development. Am J Obstet Gynecol1561065,1987
29. Goldstein RB, Filly RA: Prenatal diagnosis of anencephaly: Spectrum of sonographic appearances and distinction from the amniotic band syndrome. AJR Am J Roentgenol 151:547, 1988

CENTRAL NERVOUS SYSTEM ANOMALIES 811
30. Grogono JL: Children with agenesis of the corpus callosum. Dev Med Child Neurol 10:613, 1968
31. Gupta JK, Bryce FC, Lilford RJ: Management of apparently isolated fetal ventriculo- megaly. Obstet Gynecol Surv 49:716, 1994
32. Gupta JK, Cave M, Lilford RJ, et al: Clinical significance of fetal choroid plexus cysts. Lancet 346:724, 1995
33. Gupta JK, Lilford RJ: Assessment and management of fetal agenesis of the corpus callosum. Prenat Diagn 15:301, 1995
34. Gupta JK, Thomton JG, Lilford RJ: Management of fetal choroid plexus cysts. Br J Obstet Gynecol 104:881, 1997
35. Hart MN, Malaumd N, Ellis WG: The Dandy-Walker syndrome: A clinicopathological study based on 28 cases. Neurology 22:771, 1972
36. Heiseman J, Filly RA, Goldstein RB: The effect of measurement errors on the sono- graphic evaluation of ventriculomegaly. J Ultrasound Med 10:121, 1991
37. Hendricks SK, Cyr DR, Nyberg DA, et al: Exencephaly-clinical and ultrasonic correla- tion to anencephaly. Obstet Gynecol 72:898, 1988
38. Hertzberg BS, Lile R, Foosaner DE, et al: Choroid plexus-ventricular wall separation in fetuses with normal-sized cerebral ventricles at sonography: Postnatal outcome. AJR Am J Roentgen01 63:405, 1994
39. Hirsch JF, Pierre-Kahn A, Reiner D, et al: The Dandy-Walker malformation. A review of 40 cases. J Neurosurg 61:515, 1984
40. Jeret JS, Serur D, Wisniewski, et al: Clinicopathological findings associated with agene- sis of the corpus callosum. Brain Dev 9:255, 1987
41. Lacey DJ: Agenesis of the corpus callosum. Clinical features in 40 children. Am J Dis Child. 139:953, 1985
42. Lee W, Barton S, Comstock C, et al: Transverse cerebellar diameter: A useful predictor of gestational age for fetuses with asymmetric growth retardation. Am J Obstet Gynecol 165:1044, 1991
43. Mahony BS, Callen PW, Filly RA, et al: The fetal cistema magna. Radiology 153:773, 1984
44. Mahony BS, Nyberg DA, Hirsch JH, et al: Mild idiopathic lateral cerebral ventricular dilatation in utero: Sonographic evaluation. Radiolow 169:715, 1989
45,
46,
47.
48.
49.
50.
51.
52.
53.
54.
55.
56.
57.
Matsunaga E, Shiota Y: fiofoprosencephaly in hum: embryos: Epidemiological stud- ies of 150 cases. Teratology 16:261, 1977 McLeary RD, Kuhns LR, Barr M: Ultrasonography of the fetal cerebellum. Radiology 151:439, 1984 Ming JE, Muenke M: Holoprosencephaly: From Homer to hedgehog. Clin Genet 53:155, 1998 Murray JC, Johnson JA, Bird TD: Dandy-Walker malformation: Etiologic heterogeneity and empiric recurrence risks. Clin Genet 28:272, 1985 Nicolaides KH, Rodeck CH, Gosden C M Rapid karyotyping in non-lethal fetal malfor- mations. Lancet k283, 1986 Nyberg DA, Cyr DR, Mack LA, et al: The Dandy-Walker malformation: Prenatal sonographic diagnosis and its clinical significance. J Ultrasound Med 765, 1988 Nyberg DA, Mack LA, Hirsch J, et al: Abnormalities of fetal cranial contour in sonographic detection of spina bifida: Evaluation of the “lemon” sign. Radiology 167387, 1988 Nyberg DA, Pretorius DH: Cerebral Malformations. St. Louis, MO, Mosby Yearbook, 1990 Osenbach RK, Menezes A H Diagnosis and management of the Dandy-Walker malfor- mation: 30 years of experience. Pediatr Neurosurg 18:179, 1991 Paidas MJ, Cohen A: Disorders of the central nervous system. Semin Perinatol 18:266, 1994 Parrish M, Roessmann U, Lehvinsohn MW Agenesis of the corpus callosum. A study on the frequency of associated malformations. Ann Neurol 6:349, 1979 Pate1 MD, Goldstein RB, Tung S, et al: Fetal cerebral ventricular atrium: Difference in size according to sex. Radiology 194:713, 1995 Pilu G, Falco P, Gabrielli, et al: The clinical significance of fetal isolated cerebral

812 BRONSTEEN & COMSTOCK
borderline ventriculomegaly: Report of 31 cases and review of the literature. Ultra- sound Obstet Gynecol 14:320, 1999
58. Pilu G, Reece A, Goldstein I, et al: Sonographic evaluation of the normal developmental anatomy of the fetal cerebral ventricles: 11. The atria. Obstet Gynecol 73:250, 1989
59. Peebles DM: Holoprosencephaly. Prenat Diagn 18:477, 1998 60. Peleg D, Yankowitz J: Choroid plexus cysts and aneuploidy. J Med Genet 35:554, 1998 61. Reece EA, Goldstein I, Pilu G, et al: Fetal cerebellar growth unaffected by intrauterine
growth retardation: A new parameter for prenatal diagnosis. Am J Obstet Gynecol 157:632, 1987
62. Roessler E, Muenke M: Holoprosencephaly: A paradigm for the complex genetics of brain development. J Inherit Metab Dis 21:481, 1998
63. Russ PD, Pretorius DH, Johnson MJ: Dandy-Walker syndrome: A review of fifteen cases evaluated by prenatal sonography. Am J Obstet Gynecol 161:401, 1989
64. Sawaya R, McLaurin RL: Dandy-Walker syndrome: Clinical analysis of 23 cases. J Neurosurg 55:89, 1981
65. Senat MV, Bernard JP, Schwarzler P, et al: Prenatal diagnosis and follow-up of 14 cases of unilateral ventriculomegaly. Ultrasound Obstet Gynecol 14:327, 1999
66. Sherer DM, Onyeije CI: Prenatal ultrasonographic diagnosis of fetal intracranial tu- mors: A review. Am J Perinatol 5:319, 1998
67. Shields LE, Carpenter LA, Smith KM, et al: Ultrasonographic diagnosis of trisomy 18: Is it practical in the early second trimester? J Ultrasound Med 17327, 1998
68. Snijders RJM, Shawa L, Nicolaides KH: Fetal choroid plexus cysts and trisomy 18: Assessment of risk based on ultrasound findings and maternal age. Prenat Diagn 141119, 1994
69. Stoll C, Alembik Y, Dott B, et al: An epidemiologic study of environmental and genetic factors in congenital hydrocephalus. Eur J Epidemiol 8:797, 1992
70. Sullivan A, Giudice T, Vavelidis F, et al: Choroid plexus cysts: Is biochemical testing a valuable adjunct to targeted ultrasonography? Am J Obstet Gynecol 181:260, 1999
71. Sunden B: Acta Obstet Gynecol Scand 43:6, 1964 72. Tal Y, Freigang B, Dunn HG, et al: Dandy-Walker syndrome: Analysis of 21 cases. Dev
Med Child Neurol 22189, 1980 73. Timor-Tritsch IE, Monteagudo A, Cohen HL (eds): Ultrasonography of the Prenatal
and Neonatal Brain. Stamford, CT, Appleton & Lange, 1996 74. Van den Hof MC, Nicolaides KH, Campbell J, et al: Evaluation of the lemon and
banana signs in one hundred thirty fetuses with open spina bifida. Am J Obstet Gynecol 162:322, 1990
75. Vergani E Ghidini A, Strobelt N, et al: Prognostic indicators in the prenatal diagnosis of agenesis of corpus callosum. Am J Obstet Gynecol 170:753, 1994
76. Watson WJ, Chescheir NC, Katz VL, et al: The role of ultrasound in evaluation of patients with elevated maternal serum alpha-fetoprotein: A review. Obstet Gynecol 78:123, 1991
Address reprint requests to Richard A. Bronsteen, MD Division of Fetal Imaging
William Beaumont Hospital 3601 West 13 Mile Road
Royal Oak, MI 48073




















