
Androgen-mediated cholesterol metabolism in LNCaP and PC3 cell lines is regulated through two...
14
The Prostate 68:20 ^33 (2008) Androgen-Mediated Cholesterol Metabolism in LNCaP and PC-3 Cell Lines is Regulated Through Two Different Isoforms of Acyl-Coenzyme A:Cholesterol Acyltransferase (ACAT) Jennifer A. Locke, 1 Kishor M. Wasan, 2 * Colleen C. Nelson, 1 Emma S. Guns, 1,2 and Carlos G. Leon 2 * 1 Department of Urologic Sciences,University of British Columbia,The Prostate Centre at Vancouver General Hospital, Vancouver, British Columbia,Canada 2 Division of Pharmaceutics and Biopharmaceutics, Faculty of Pharmaceutical Sciences,University of British Columbia, Vancouver, British Columbia,Canada BACKGROUND. The objective of this work was to determine the effect of an androgen agonist, R1881, on intracellular cholesterol synthesis and esterification in androgen-sensitive (AS) prostate cancer (LNCaP) cells. METHODS. We investigated the activity and expression of cholesterol metabolism enzymes, HMG-CoA-reductase and ACAT in the LNCaP and PC-3 (androgen-independent control) models. RESULTS. Microsomal PC-3 HMG-CoA-reductase activity was increased with R1881 despite having similar cholesterol levels while increased cholesterol levels in microsomes from LNCaPs treated with R1881 (Lþ) were associated with increased HMG-CoA reductase activity. Increased intracellular cholesteryl esters (CE) found in (Lþ) were not associated with an increased ACAT1 activity. There was no effect from androgen treatment on ACAT1 protein expression in theses cells; however, ACAT2 expression was induced upon R1881 treatment. In contrast, we found an increase in the in vitro ACAT1 activity in PC-3 cells treated with androgen (Pþ). Only ACAT1 expression was induced in Pþ. We further assessed the expression of STAT1a, a transcriptional activator that modulates ACAT1 expression. STAT1a expression and phosphorylation were induced in Pþ. To determine the role of the AR on ACAT1 expression and esterification, we treated PC-3 cells overexpressing the androgen receptor with R1881 (PARþ). AR expression was decreased in PARþ cells; ACAT1 protein expression and cholesterol ester levels were also decreased, however, ACAT2 remained unchanged. STAT1a expression was decreased in PARþ. CONCLUSIONS. Overall, these findings support the importance of cholesterol metabolism regulation within prostate cancer cells and unravel a novel role for STAT1a in prostate cancer metabolism. Prostate 68: 20–33, 2008. # 2007 Wiley-Liss, Inc. KEY WORDS: ACAT; androgen receptor; HMG-CoA reductase; LNCAP cells; PC-3 cells; R1881; STAT1a INTRODUCTION Androgens are known to play a central role in malignant prostate cancer growth and therefore hor- mone ablation therapy and/or surgical castration are front line treatments of this disease [1]. Most cancer cells are terminated by these treatments, however, over time, the cancer recurs in a more aggressive and devastating manner termed as hormone refractory Grant sponsor: Terry Fox Foundation/National Cancer Institute of Canada; Grant sponsor: Michael Smith Foundation for Health Research. *Correspondence to: Drs. Kishor M. Wasan and Carlos G. Leon, 2146 East Mall, Vancouver, British Columbia, Canada V6T 1Z3. E-mail: [email protected]; [email protected] Received 12 April 2007; Accepted 11 September 2007 DOI 10.1002/pros.20674 Published online 13 November 2007 in Wiley InterScience (www.interscience.wiley.com). ȣ 2007 Wiley-Liss, Inc.
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Androgen-mediated cholesterol metabolism in LNCaP and PC3 cell lines is regulated through two...

The Prostate 68:20 ^33 (2008)
Androgen-MediatedCholesterolMetabolisminLNCaPandPC-3Cell Lines is RegulatedThrough
TwoDifferent IsoformsofAcyl-CoenzymeA:CholesterolAcyltransferase (ACAT)
Jennifer A. Locke,1 Kishor M. Wasan,2* Colleen C. Nelson,1
Emma S. Guns,1,2 and Carlos G. Leon2*1DepartmentofUrologic Sciences,Universityof British Columbia,The Prostate Centreat VancouverGeneralHospital,
Vancouver, British Columbia,Canada2Divisionof Pharmaceuticsand Biopharmaceutics, Facultyof Pharmaceutical Sciences,Universityof British Columbia,
Vancouver, British Columbia,Canada
BACKGROUND. The objective of this work was to determine the effect of an androgenagonist, R1881, on intracellular cholesterol synthesis and esterification in androgen-sensitive(AS) prostate cancer (LNCaP) cells.METHODS. We investigated the activity and expression of cholesterol metabolism enzymes,HMG-CoA-reductaseandACATin theLNCaPandPC-3 (androgen-independent control)models.RESULTS. Microsomal PC-3 HMG-CoA-reductase activity was increased with R1881 despitehaving similar cholesterol levels while increased cholesterol levels in microsomes from LNCaPstreated with R1881 (Lþ) were associated with increased HMG-CoA reductase activity. Increasedintracellular cholesteryl esters (CE) found in (Lþ) were not associated with an increased ACAT1activity.Therewasnoeffect fromandrogen treatmentonACAT1protein expression in theses cells;however,ACAT2expressionwas induceduponR1881 treatment. In contrast,wefoundan increasein the in vitro ACAT1 activity in PC-3 cells treated with androgen (Pþ). Only ACAT1 expressionwas induced in Pþ.We further assessed the expression of STAT1a, a transcriptional activator thatmodulates ACAT1 expression. STAT1a expression and phosphorylation were induced in Pþ. Todetermine the role of the AR on ACAT1 expression and esterification, we treated PC-3 cellsoverexpressing theandrogen receptorwithR1881 (PARþ).ARexpressionwasdecreased inPARþcells; ACAT1 protein expression and cholesterol ester levels were also decreased, however,ACAT2 remained unchanged. STAT1a expression was decreased in PARþ.CONCLUSIONS. Overall, these findings support the importance of cholesterol metabolismregulation within prostate cancer cells and unravel a novel role for STAT1a in prostate cancermetabolism. Prostate 68: 20–33, 2008. # 2007 Wiley-Liss, Inc.
KEY WORDS: ACAT; androgen receptor; HMG-CoA reductase; LNCAP cells; PC-3cells; R1881; STAT1a
INTRODUCTION
Androgens are known to play a central role inmalignant prostate cancer growth and therefore hor-mone ablation therapy and/or surgical castration arefront line treatments of this disease [1]. Most cancercells are terminated by these treatments, however,over time, the cancer recurs in a more aggressive anddevastating manner termed as hormone refractory
Grant sponsor: Terry Fox Foundation/National Cancer Institute ofCanada; Grant sponsor: Michael Smith Foundation for Health Research.
*Correspondence to: Drs. Kishor M.Wasan and Carlos G. Leon, 2146East Mall, Vancouver, British Columbia, Canada V6T 1Z3.E-mail: [email protected]; [email protected] 12 April 2007; Accepted 11 September 2007DOI 10.1002/pros.20674Published online 13 November 2007 in Wiley InterScience(www.interscience.wiley.com).
� 2007Wiley-Liss, Inc.

disease [2]. The mechanism by which androgensare involved in the progression to this androgen-independent (AI) phenotype remains unclear. We andothers have recently shown that upstream and parallelsurvival pathways of androgen synthesis such ascholesterol synthesis, lipid metabolism and fatty acidsynthesis are implicated in prostate cancer progression[3–6]. Hager et al. initially investigated the role ofcholesterol metabolism in prostate cancer and foundthat during early stage development, large accumu-lations of membrane associated lipids developed [4].Furthermore, Swinnen et al. [7] correlated androgenstimulation in prostate cancer cells with the accumu-lation of neutral lipids (triglyceride and cholesterolesters). Sonn et al. [8] discovered that exogenous fatintake has also been correlated with prostate cancermortality and serum androgen levels. Based on thesestudies, androgens are believed to play a significantrole in lipogenesis involved in the progression to theAI phenotype but the underlying mechanisms of howthis occurs remain to be elucidated. The androgenreceptor (AR) is known to be a central mediator in thisprogression [9,10] as re-ignition of the AR in the AIphenotype has been documented in hormone refrac-tory human tissues [2,11,12]. One contending hypoth-esis to explain the role of AR in the emergence of the AIphenotype is that it becomes hypersensitive to lowcirculating levels of androgens following castration[9,10]. The role of lipogenesis in this AR-sensitiveprogression to AI is uncertain.
Lipid metabolism involves a multitude of enzymesand complex pathways within each cell, ultimatelyleading to the uptake, storage, synthesis, transportand excretion of important survival molecules suchas cholesterol. Cholesterol, a molecular precursor ofandrogen, fatty acid and lipid synthesis, has beenintensively characterized in terms of its cellularmetabolism within other model systems. Exogenouscholesterol is brought into the cell through the SR-BIand LDL receptors, synthesis of cholesterol within thecell occurs via the HMG-CoA reductase enzyme,transport of cholesterol within the cell occurs via theACAT enzymes, storage of cholesterol is conducted inthe formof cholesteryl esters and excretion from the celloccurs through ABCA1, ABCG5, and ABCG8 [13,14].
Cholesterol metabolism is hypothesized to beregulated by a group of transcription factors knownas the sterol regulatory element-binding proteins(SREBPs) which are in turn regulated by androgens[13]. This occurs through a negative feedback loop ofcholesterol synthesis within the cell [15]. SREBP’sremain in the ER with scaffolding proteins, SREBPcleavage activating protein, SCAP and retention pro-tein, Insig [15]. Low cholesterol levels trigger thecleavage of Insig allowing SCAP-SREBP to travel to
the Golgi. In the Golgi, SREBP is cleaved by twoproteases called S1P and S2P, and it is then transcrip-tionally activated [14]. Recently, Brusselmans et al. [16]showed that androgen treatment channeled inter-mediates of the mevalonate/isoprenoid pathwaytowards cholesterol synthesis. RNA interference ofsqualene synthase, key enzyme in determining theswitch toward sterol biosynthesis, affected the choles-terol content in lipid rafts and also attenuated cellproliferation.
Swinnen et al. [17] proposed that the conformationalchange that leads to the cleavage of this complex relieson the androgen regulation of the escort protein, SCAP.Upon synthetic androgen (R1881) stimulation of cells,Heemers et al. verified an increase in expressionof SCAP and SREBPs within the nucleus [17]. Thisincrease in expression did not occur in androgen-unresponsive cells and thus this process is believed tobe androgen dependent in nature [7].
There is significant evidence suggesting a role ofandrogens in cholesterol metabolism via the SREBPs[3][17], however the role of androgens in these andother cholesterol metabolizing processes have yet to beinvestigated specifically inprostate cancer progression.These processes are central to cell survival andproliferation [5] and thus regulation of these metabo-lism enzymes may be important in the transition fromandrogen-dependence to androgen-independence. Inthis paper, we explore the effect of synthetic androgen,R1881, on cholesterol synthesis and esterificationenzymes, HMG-CoA reductase and Acyl-CoenzymeA:cholesterol acyltransferase (ACAT) in an androgen-sensitive LNCaP cell model as well as an androgen-insensitive PC-3 cell model. These models encompassboth early stage androgen-dependence and late stageandrogen-independence and thus are useful in vitromodels to investigate this transformation.
HMG-CoA reductase is an important enzyme inthe biosynthesis of cholesterol as it catalyzes theconversion of HMG-CoA to mevalonate, a precursorof cholesterol [13,18]. This is believed to be the rate-limiting step in cholesterol metabolism and is impor-tant in the maintenance of membrane fluidity andsteroid hormone synthesis [19]. There are two classesof this enzyme, the first found in eukaryotes andthe second found in eubacteria and archaea [19,20]. Thehuman form consists of three domains: catalytic, linkerand anchor whereas the second form consists of onlythe catalytic domain [21].
Regulation of HMG-CoA reductase takes placeduring transcription, translation and post-translationprocesses [14]. Transcriptionally, this enzyme is underthe control of the SREBP factors. Upon cleavage ofthe SREBP-SCAP-Insig complex and transport to thenucleus via Golgi, this process is thought to lead to the
The Prostate DOI 10.1002/pros
Androgen-MediatedCholesterolMetabolism 21

activation of the lipogenic gene, HMG-CoA reductase[14]. Degradation of HMG-CoA reductase occurs inthe transmembrane region when cholesterol levels arehigh. When HMG-CoA reductase binds with Insig,SCAP dissociates leading to its rapid degradationthrough a ubiquitin-proteasome mechanism. Catalyticactivity of HMG-CoA reductase appears to be undercontrol of the post-translational modification, phos-phorylation [13,21]. The complex role of androgensin the regulation of HMG-CoA reductase activity inprostate cancer has not been extensively investigatedand is therefore addressed in this work.
ACAT is an integral membrane protein localizedin the endoplasmic reticulum and it is important incatalyzing the transfer of cholesterol to cholesterolesters [2,22]. These cholesterol esters are further storedas lipid droplets within the cell. Two ACAT genes arefound in mammals, ACAT1 and ACAT2. ACAT1controls the cholesterol ester formation in the brain,adrenal glands, macrophages and kidneys whileACAT2 controls the cholesterol ester formation in theliver and intestine [14]. These two genes share highstructural homology in their transmembrane domainsand phosphorylation sites. Regulation of these twogenes is quite diverse in their localization andexpression mechanisms [2]. ACAT1 expression isregulated by transcriptional factors such as STAT1aand Sp1 [23] while ACAT2 is regulated by Cdx2 andHNF1a [24]. STAT1a induction by interferon gammahas been shown to induceACAT1 expression in humanmonocytes and thus this may be a possible activationpathway for this isoform [23]. Further, ACAT2 doesnot contain sterol responsive elements in its promoterregion and therefore is not hypothesized to beregulated by SREBPs. It is poorly understood how thisenzyme is activated and regulated within the cell.Cholesterol has been shown in vitro to initiate ACATaction but the mechanism behind this process isunknown [13]. Furthermore, ACAT expression hasbeen affected in many diseases such as humanatherosclerosis, hypercholesterolemia and possiblyAlzheimer’s [25,26]. The mechanism of ACAT actionwithin the cell is thus an important concept to addressin order to develop new treatments against thesediseases and others such as prostate cancer.
Our results uncovered in the study of HMG-CoAreductase and ACAT expressions and activities inLNCaP and PC-3 cell models of prostate cancer furthersuggest that an in depth understanding of the roleof cholesterol metabolism enzymes as well as theirtranscriptional cascades involved in prostate cancerprogressionmay lead to the development of novel drugtargets. This will inevitably improve treatment optionsfor patients thereby increasing their overall qualityof life.
MATERIALSANDMETHODS
Cell Culture andAndrogenTreatment
PC-3 and PC-3 AR2 cells were cultured in DMEMwith L-glutamine, penicillin streptomycin (PS) andeither 10% full bovine serum (FBS) or charcoal strippedserum (CSS; Hyclone, Logan, UT). LNCaP cells werecultured in RPMI-1640 (without phenol red) withL-glutamine without sodium bicarbonate and with PSand either 10% FBS or CSS. PC-3 and LNCaP cells werepurchased from the American Type Culture Collection(Rockville, MD) and were used within passages 10–25and 40–47, respectively. PC-3 AR2 cells were kindlydonated by Dr. TJ Brown (Division of ReproductiveScience, Toronto Hospital Research Institute, Ontario,Canada). When the cells reached 40% confluency, themedia was changed from full (FBS) to stripped (CSS)serum.After 2 days, cells were treatedwith 1 nMR1881(Dupont, Boston,MA) synthetic androgen.After 4 daysof treatment, cells were pelleted and frozen at �808C.
MTSViabilityAssay
In a 96-well plate, PC-3 or LNCaP cellswere culturedin 100 ml media per well. Each well was treated with20 ml of MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy-methoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium](Promega, Madison, WI) and left to incubate at 378C inthe dark for 30 min. The plate was then read at 490 nmon the spectrometer. The viability of the cells wasdetermined based on comparing absorbance measure-ments of samples and a control of untreated cells.
Prostate Specif|c Antigen (PSA)Assay
Media was obtained during each treatment stepof experiment: media alone, before splitting into T175flasks, before change to stripped media, before treat-ment and before pelleting.Media fractions of 50 ml fromeach step were analyzed using a PSA ELISA kit fromClinPro (Union City, CA). First the fractions wereincubated in zero buffer (ClinPro Catalogue # TM-107)for 1 hr, then enzyme conjugate for another hour, TMB(3,3,5,5-Tetramethylbenzidine) for 20 min and finallystop solution to terminate the reaction. The absorbancewas read at 450 nm. PSA expression was evaluated interms of absorbance and calculated using a 6-pointstandard curve.
LipidAnalysis
Cells were lysed in 400 ml of modified RIPA buffer(50 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40plus protease inhibitors) for 30 min. Three hundredmicroliters of this was extracted with 4ml chloroform/
The Prostate DOI 10.1002/pros
22 Locke et al.

methanol (2:1) and 1.2 ml 0.9% NaCl at 378C for 1 hr.Samples were centrifuged at 2,000 rpm for 1 min inorder to separate phases. The top phase was discardedand the bottom phase containing the lipids was drieddown under nitrogen gas flow. Samples were recon-stituted in 100 ml isopropanol for total and freecholesterol quantification. Ten microliters of samplein triplicate were analyzed for total cholesterol content(492 nm) (Stanbio, Boerne, TX) as well as for freecholesterol content (650 nm) (Wako, Richmond, VI).Cholesteryl ester concentration was determined bysubtracting the free cholesterol from the total chole-sterol.
Cells incubatedwith [1(2)-14C]-AceticAcid (55.0mCi/mmol in 20 ml, Amersham Biosciences, Baie d’Urfe,Quebec, Canada), a precursor of cholesterol, for 96 hrwere lysed and extracted following method above.Remaining lipid residue were reconstituted in 75 mlchloroform and sealed with ParafilmTM. Samples wereblotted onto a silical gel TLC plate and developedin 80 ml of hexane/ethyl acetate (9:1, v/v). Afterthe samples had separated in the TLC chamber for90 min. they were visualized using iodine crystals. Thecholesterol and cholesteryl oleate bandswere analyzedusing a scintillation counter.
Lipid content was corrected by protein concentra-tion which was measured using a BIO-RAD proteindetermination kit (Hercules, CA) as per manufacturerinstructions. Absorbencies were read at 590 nm on thespectrometer and compared to a BSA standard curvein order to determine total protein concentration ineach sample.
Microsome Isolation
Cell pellets were resuspended in isolation buffer(50 mM Tris pH 7.8, 1 mM EDTA, 1 mM fresh PMSFplus protease inhibitors) (Sigma, St. Louis, MO),homogenized by incubating on ice for 30 min and bydisrupting them by stroking using a microcentrifugetube homogenizer. Then lysates were centrifuged inpolycarbonate centrifuge bottles at 16,000g for 15min at48C. The supernatant of this was centrifuged at 40,000gfor 60min at 48C. The pellet from this was resuspendedin isolation buffer and centrifuged at 40,000g foranother 45 min at 48C. This pellet was resuspended inBufferA (50mMTris pH7.8, 1mMEDTA, 1mMPMSF)containing protease inhibitors (Sigma). Total proteincontent in this mixture was quantified and this amountwas used in the following activity analyses.
HMG-CoAReductaseActivityAssay
Fifty micrograms of sample was placed in 27 mlhomogenization buffer (30 mM EDTA, 250 mM NaCl,
1 mM DTT, 50 mM potassium phosphate pH 7.4) andwater up to a total volume of 125 ml. This mixture wasincubated for 5 min in a 378C water bath. Treatmentwith 50 ml Cofactor-substrate solution containingradioactive substrate for 3 min was conducted andstopped using 25 ml 12 N HCl. An internal standard3H-Mevalonic acid was then added in a 20 ml volume.This samplewas incubated for 30min in the 378Cwaterbath. After incubation, 50 ml of each samplewas blottedon a silica gel thin layer chromatography (TLC) sheet7 ml at a time. Cholesterol was developed for 60 min in50ml of (1:1 v/v) benzene-acetoneandvisualizedusingiodine crystals. Bands were cut from the TLC sheet andanalyzed for 3H and 14C activity using a scintillationcounter. Enzyme activities are expressed as picomolesof mevalonic acid synthesized per min per mg protein(pmoles/min per mg).
ACATActivityAssay
Fifty micrograms of sample was mixed with 6.7 mlof 50 mg/ml BSA and ACAT buffer (0.1 M Tris HCl,0.25 M sucrose, and 1 mM EDTA, pH 7.5) to a finalvolume of 200 ml. This mixture was incubated for 5minin the 378C water bath. After incubation, the samplewas treated with 2.5 ml (56.0 mCi/mmol in 200 ml)14C-oleyl coenzyme A for 3 min. This reaction wasstopped using 1 ml of chloroform/methanol (1:1 v/v).After the reaction was stopped 2 ml more of chloro-form/methanol (2:1), 20 ml 3H cholesterol, 1 ml 0.9%NaCl, 50 ml of 1 mM cold cholesteryl oleate and 10 mlcold cholesterol were added to the mixture. This wasincubated for 60 min in the 378C water bath. Sampleswere centrifuged at 1,000g for 3 min for phaseseparation and then the top layer was aspirated away.The nitrogen blower was then used to dry down thesamples. Residues were treated and analyzed by thesame method outlined in the radioactive cholesteroland cholesteryl ester quantification experiment above.ACAT activities are expressed as picomoles of choles-teryl oleate synthesized per min per mg protein. OurACAT assay uses oleyl coenzyme A as substrate whichis preferentially used by ACAT1 [27].
Western Blot Analysis
Cell lysates or microsomes were separated by SDS–PAGE, electroblotted into nitrocellulose membraneand probed with different antibodies: androgen recep-tor (AR 441) 1:300 dilution (Santa Cruz Biotechnology,Santa Cruz, CA), HMG-CoA reductase, phosphotyro-sine STAT1a (Upstate Biotechnology, Lake Placid,NY),LDLr (Fitzgerald Industries, Concord, MA), ACAT1,STAT1a and actin (Santa Cruz Biotechnology) andACAT2 (kindly provided by Dr L. Rudel, Wake Forest
The Prostate DOI 10.1002/pros
Androgen-MediatedCholesterolMetabolism 23

University, Winston-Salem, NC). Actin was used as aprotein loading control. Protocols for HMG-CoAreductase, ACAT2 and actin immunoblotting arepreviously described [19]. ACAT1 antibody was usedin a 1:300 dilution in blocking buffer (5% milk in TBS-Tween-20 at 0.1%). STAT1a and phosphotyrosineSTAT1a immunoblotting were performed as describedelsewhere [28].
Statistical Analysis
Normalized protein expressions were comparedbetween treatment groups by an unpaired t-test(INSTAT; GraphPad). Critical differences were asses-sed by Tukey post-hoc tests. A difference was consi-dered significant if the probability of chance explainingthe results was reduced to less than 5% (P< 0.05). If theequal variance test failed, then a Mann–Whitney RankSum Test was performed. Using this test, a differencewas considered significant if the probability of chanceexplaining the results was reduced to less than 0.1%(P< 0.001). All data are expressed as mean� standarddeviation.
RESULTS
Protein Expression in Response toAndrogenAgonistTreatment (R1881) in PC-3 and LNCaPCells
The expression of the androgen receptor (AR) wasassessed by immunoblotting with a specific antibodyin control and R1881-treated PC-3 (n¼ 7) and LNCaP(n¼ 3) cells. We found an increase in AR expression inboth LNCaP and PC-3 cells treated with R1881 (17- and4-fold compared to untreated cells respectively)(Fig. 1A).
At a functional level the response of the prostate cellsto R1881 was assessed by measuring PSA (prostatespecific antigen) expression. LNCaP cells (n¼ 4)treated with R1881 showed a marked increase in PSAexpression (P< 0.001), while PSA expression wasunchanged in PC-3 cells (n¼ 4) (Fig. 1B). Furthermore,LNCaP cells (n¼ 5), but not PC-3 cells (n¼ 5), are moreviable when stimulated with R1881 as compared to notreatment (P¼ 0.002) by MTS assay (Fig. 1C).
Additionally we assessed the expression of the LDLreceptor in PC-3 and LNCaP cells (Fig. 1D). The LDLreceptor was highly expressed in PC-3 cells, though itsexpression did not change significantly upon R1881treatment (n¼ 7). LNCaP cells presented a low expres-sion of the LDLr (data not shown).
The HMGCoA reductase protein levels were higherin PC-3 cells (Fig. 1E) compared to LNCaP cells (datanot shown), however there was no significant changein expression in untreated and R1881-treated cells ineither cell line.
LipidAnalysis: CholesterolandCholesteryl EstersLevels in PC-3 and LNCaPCells
The levels of free cholesterol and cholesteryl esterswere similar in PC-3 cells (n¼ 3) treated with R1881compared to the controls (Fig. 2A). LNCaP cells (n¼ 3)treated with R1881 showed a decrease in free choles-terol (P< 0.001) and an increase in cholesteryl esters(P< 0.001) (Fig. 2B). In order to reproduce these results,an in vitro radiolabeling assay coupled with TLCseparation and radiometric detection was utilized.Following this method, PC-3 (n¼ 4) cholesterol andcholesteryl ester levels remained unchanged followingR1881 treatment (Fig. 2C), while both LNCaP (n¼ 4)total cholesterol (P< 0.001) and cholesteryl ester(P< 0.001) levels increased with R1881 treatment(Fig. 2D).
MicrosomalHMG-CoAReductaseActivity andExpression in PC-3 and LNCaPCells
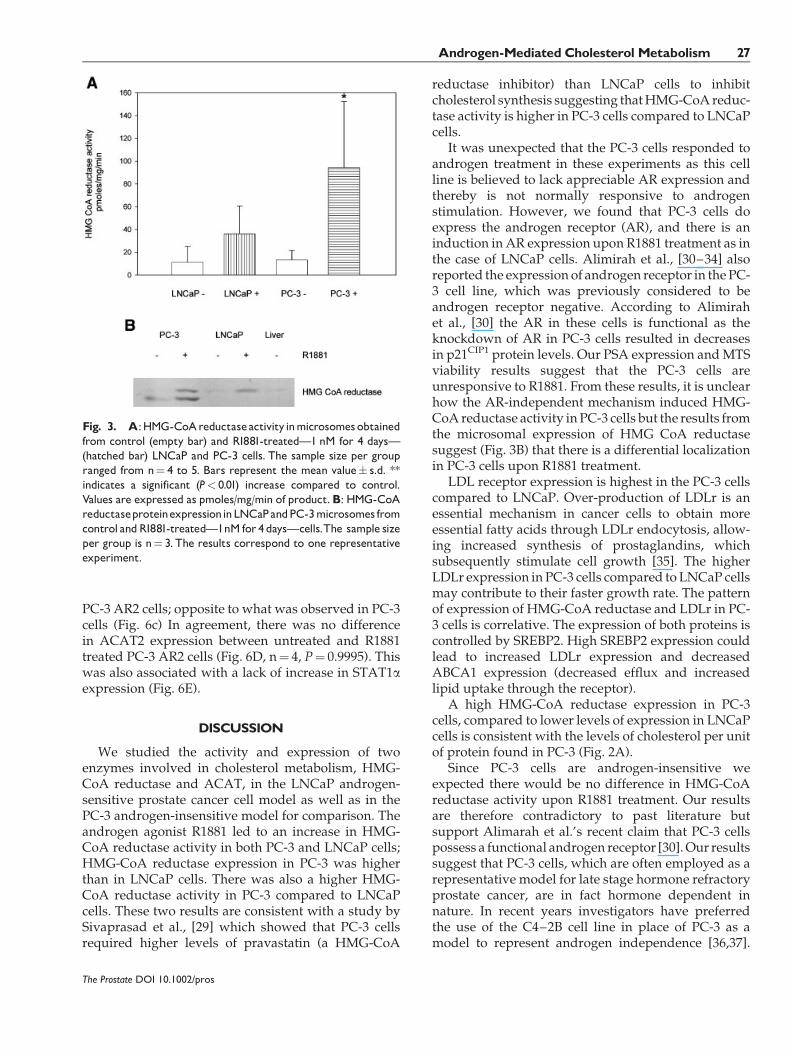
Results shown in Figure 3A indicate a significantlyhigher HMG-CoA reductase activity associated withthe R1881-treated PC-3 cells compared to the untreatedPC-3 cells (n¼ 4, P¼ 0.0329).
Similarly LNCaP cells treated with R1881 (Fig. 3A)showed a higher HMG-CoA reductase activity com-pared to untreated cells, albeit not significantly differ-ent (n¼ 4, P¼ 0.1266). HMG-CoA reductase activity inPC-3 cells treatedwithR1881washigher than anyof theother groups.
There was an increase in HMG-CoA reductaseprotein expression in microsomes in PC-3 treated withR1881 compared to untreated cells (Fig. 3B). LNCaPmicrosomes showed a lower expression of HMG-CoAreductase compared to PC-3 cells (Fig. 3B).
ACAT1Activity and Expression (ACAT1andACAT2WithActin as Control) in PC-3 and LNCaPCells
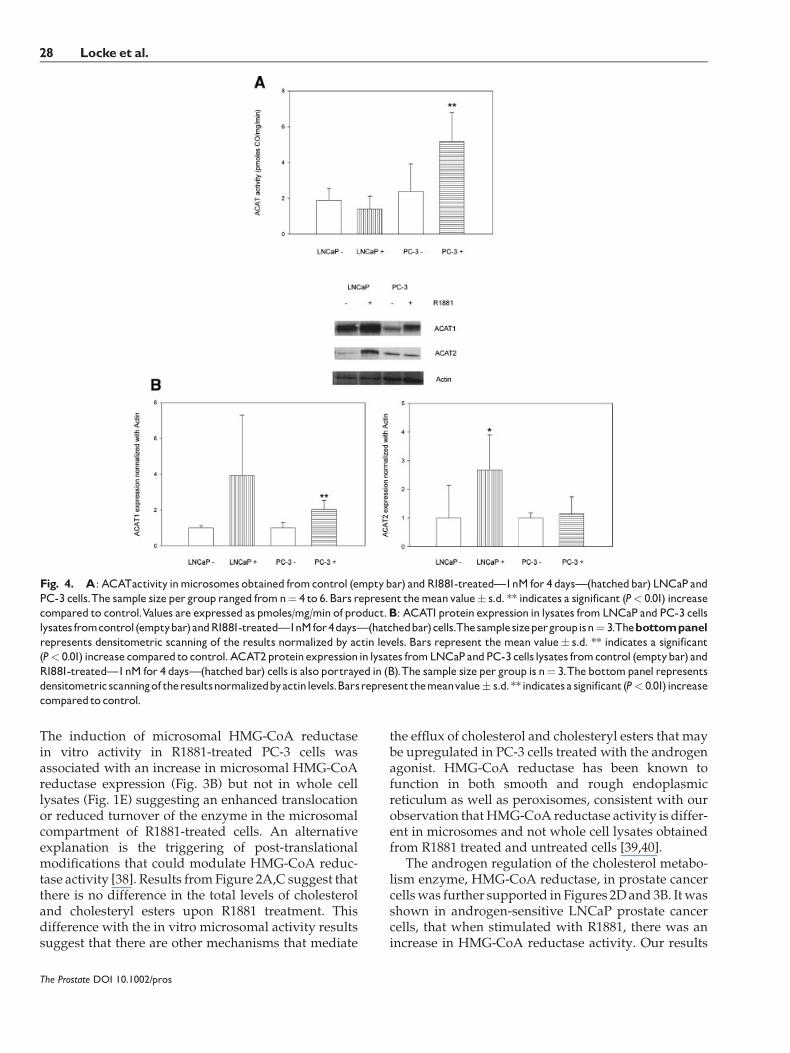
In Figure 4A there is a higher in vitroACAT1 activityassociated with the PC-3 (n¼ 4) treated with R1881than the untreated cells (P¼ 0.045). LNCaP cells (n¼ 6)treatedwith R1881 exhibited a reducedACAT1 activity(Fig. 4A) which was not significantly different tocontrol cells (P¼ 0.180).
To investigate the molecular mechanisms account-ing for the increased in vitro ACAT activity found inthe PC-3 cells treated with R1881, we analyzed theexpression of the two known ACAT isoforms, ACAT1and ACAT2. ACAT1 expression was induced in PC-3cells (n¼ 5) treated with R1881 (P¼ 0.0023) (Fig. 4B),which is consistent with the increased ACAT1 activitythat we detected previously. ACAT2 expression wasnot significantly affected in PC-3 cells. In the LNCaPcells, we found an increase in ACAT2 expression
The Prostate DOI 10.1002/pros
24 Locke et al.

The Prostate DOI 10.1002/pros
Fig. 1. A: Androgen receptor protein expression in LNCaP and PC-3 cells lysates from control (empty bar) and R1881-treated�1nM for4 days�(hatched bar) cells. The sample size per group ranged from n¼ 3 to 7. The bottom panel represents densitometric scanning ofthe results normalizedby actin levels.Bars represent themeanvalue� s.d. ** indicates a significant (P< 0.01) increase compared to control.B: PSAproteinexpression in LNCaP andPC-3 cells lysates fromcontrol (emptybar) andR1881-treated�1nMfor 4days�(hatchedbar) cells.The sample sizeper group ranged fromn¼ 4 to 5 (P< 0.001).Bars represent themeanvalue� s.d. ** indicates a significant (P< 0.01) increasecompared to control.C: MTSviability absorbencies in LNCaP and PC-3 cells lysates from control (empty bar) and R1881-treated�1nM for4 days�(solidbar) cells.The sample sizeper groupwas n¼ 5.Bars represent themeanvalue� s.d. ** indicates a significant (P< 0.01) increasecompared to control.D: LDL receptor protein expression in lysates from PC-3 cells from control (empty bar) and R1881-treated�1nM for4 days�(hatchedbar) cells.The sample sizeper group is n¼ 7.Thebottompanelrepresents densitometric scanningof theresults normalizedby actin levels. Bars represent the mean value� s.d.E: HMG CoA reductase protein expression in PC-3 lysates from control (empty bar)andR1881-treated�1nMfor 4days�(hatchedbar) cells.The sample sizepergroup isn¼ 9 and11.Thebottompanelrepresents densitometricscanningof theresultsnormalizedbyactinlevels.Barsrepresentthemeanvalue� s.d.
Androgen-MediatedCholesterolMetabolism 25

(Fig. 4B) (P< 0.05) but not ACAT1. The lack ofinduction of ACAT1 expression in LNCaP cells treatedwith the agonists, explains why we did not observe anincreased microsomal ACAT1 activity.
Mechanismof Induction of ACAT1Expressionin PC-3 CellsTreatedWith R1881
We investigated the expression of STAT1a, anACAT1 transcriptional factor, in PC-3 and LNCaPcells. The untreated PC-3 showed higher levels ofSTAT1a expression than the LNCaP cells (not shown).When the cellswere treatedwithR1881, thePC-3 (n¼ 3)showed a significant induction in STAT1a (P¼ 0.0082;Fig. 5A). To determine if the changes in STAT1aexpression were correlated with changes in activation,we used an anti-phosphotyrosine STAT1a antibody.We found a significant increase in STAT1a phosphor-ylation in R1881-treated PC-3 cells (n¼ 3, P¼ 0.0314)(Fig. 5B). As a negative control, we measured theprotein levels of HMG CoA reductase in these cells. As
expected HMG CoA reductase expression was notaffected (data not shown).
Role of theARinACAT1and, ACAT2Protein Expression andCholesteryl
Esterif|cation in PC-3 Cells
PC-3AR2 cells showed a reduction inAR expressionlevels upon R1881 treatment (Fig. 6A, P¼ 0.0073). ARfunction was previously documented for PC-3 AR2cells and thus was not investigated here [28].
In order to examine the effect of the changes in ARexpression in the PC-3 AR2 cells, we determined thelevels of cholesterol and cholesteryl esters in these cells.PC-3 AR2 cells (n¼ 4) treated with R1881 showeda decrease in both total cholesterol (P¼ 0.025) andcholesteryl esters (P< 0.001) upon R1881 treatment(Fig. 6B).
We then investigated the protein expression ofACAT1 and ACAT2 in PC-3 AR2 cells to account forthis reduction in cholesteryl ester formation. UponR1881 treatment, ACAT1 expression was decreased in
The Prostate DOI 10.1002/pros
Fig. 2. A,B:Freecholesterol, totalcholesterol, andcholesterylesterconcentrationsincontrol (emptybar)andR1881-treated(hatchedbar)PC-3(A)andLNCaP(B)cells.Barsrepresentthemeanvalue� s.d.of threeindependentexperimentseachanalyzedbytriplicatemeasurements.**indicatesa significant(P< 0.001)increasecomparedtocontrol.Cholesterolandcholesterylesterlevelsmeasuredinmicrogramsandnormal-izedto totalproteincontent.C,D:Totalcholesterolandcholesterylesterlevelsincontrol(emptybar)andR1881-treated(hatchedbar)PC-3(C)and LNCaP (D) cells.Bars represent themean14C counts� s.d. of four experiments each analyzedby triplicatemeasurements. ** indicates asignificant (P< 0.05) increase compared to control.Cholesterol and cholesteryl ester levelsmeasured in 14C counts and normalized to totalproteincontent.
26 Locke et al.
PC-3 AR2 cells; opposite to what was observed in PC-3cells (Fig. 6c) In agreement, there was no differencein ACAT2 expression between untreated and R1881treated PC-3 AR2 cells (Fig. 6D, n¼ 4, P¼ 0.9995). Thiswas also associated with a lack of increase in STAT1aexpression (Fig. 6E).
DISCUSSION
We studied the activity and expression of twoenzymes involved in cholesterol metabolism, HMG-CoA reductase and ACAT, in the LNCaP androgen-sensitive prostate cancer cell model as well as in thePC-3 androgen-insensitive model for comparison. Theandrogen agonist R1881 led to an increase in HMG-CoA reductase activity in both PC-3 and LNCaP cells;HMG-CoA reductase expression in PC-3 was higherthan in LNCaP cells. There was also a higher HMG-CoA reductase activity in PC-3 compared to LNCaPcells. These two results are consistent with a study bySivaprasad et al., [29] which showed that PC-3 cellsrequired higher levels of pravastatin (a HMG-CoA
reductase inhibitor) than LNCaP cells to inhibitcholesterol synthesis suggesting thatHMG-CoAreduc-tase activity is higher in PC-3 cells compared to LNCaPcells.
It was unexpected that the PC-3 cells responded toandrogen treatment in these experiments as this cellline is believed to lack appreciable AR expression andthereby is not normally responsive to androgenstimulation. However, we found that PC-3 cells doexpress the androgen receptor (AR), and there is aninduction inAR expression upon R1881 treatment as inthe case of LNCaP cells. Alimirah et al., [30–34] alsoreported the expression of androgen receptor in the PC-3 cell line, which was previously considered to beandrogen receptor negative. According to Alimirahet al., [30] the AR in these cells is functional as theknockdown of AR in PC-3 cells resulted in decreasesin p21CIP1 protein levels. Our PSA expression andMTSviability results suggest that the PC-3 cells areunresponsive to R1881. From these results, it is unclearhow the AR-independent mechanism induced HMG-CoA reductase activity inPC-3 cells but the results fromthe microsomal expression of HMG CoA reductasesuggest (Fig. 3B) that there is a differential localizationin PC-3 cells upon R1881 treatment.
LDL receptor expression is highest in the PC-3 cellscompared to LNCaP. Over-production of LDLr is anessential mechanism in cancer cells to obtain moreessential fatty acids through LDLr endocytosis, allow-ing increased synthesis of prostaglandins, whichsubsequently stimulate cell growth [35]. The higherLDLr expression in PC-3 cells compared to LNCaP cellsmay contribute to their faster growth rate. The patternof expression of HMG-CoA reductase and LDLr in PC-3 cells is correlative. The expression of both proteins iscontrolled by SREBP2. High SREBP2 expression couldlead to increased LDLr expression and decreasedABCA1 expression (decreased efflux and increasedlipid uptake through the receptor).
A high HMG-CoA reductase expression in PC-3cells, compared to lower levels of expression in LNCaPcells is consistent with the levels of cholesterol per unitof protein found in PC-3 (Fig. 2A).
Since PC-3 cells are androgen-insensitive weexpected there would be no difference in HMG-CoAreductase activity upon R1881 treatment. Our resultsare therefore contradictory to past literature butsupport Alimarah et al.’s recent claim that PC-3 cellspossess a functional androgen receptor [30].Our resultssuggest that PC-3 cells, which are often employed as arepresentative model for late stage hormone refractoryprostate cancer, are in fact hormone dependent innature. In recent years investigators have preferredthe use of the C4–2B cell line in place of PC-3 as amodel to represent androgen independence [36,37].
The Prostate DOI 10.1002/pros
Fig. 3. A:HMG-CoAreductaseactivityinmicrosomesobtainedfrom control (empty bar) and R1881-treated�1 nM for 4 days�(hatched bar) LNCaP and PC-3 cells. The sample size per groupranged from n¼ 4 to 5. Bars represent the mean value� s.d. **indicates a significant (P< 0.01) increase compared to control.Values are expressed as pmoles/mg/min of product.B: HMG-CoAreductaseproteinexpressioninLNCaPandPC-3microsomes fromcontrol andR1881-treated�1nM for 4days�cells.The sample sizeper group is n¼ 3. The results correspond to one representativeexperiment.
Androgen-Mediated CholesterolMetabolism 27
The induction of microsomal HMG-CoA reductasein vitro activity in R1881-treated PC-3 cells wasassociated with an increase in microsomal HMG-CoAreductase expression (Fig. 3B) but not in whole celllysates (Fig. 1E) suggesting an enhanced translocationor reduced turnover of the enzyme in the microsomalcompartment of R1881-treated cells. An alternativeexplanation is the triggering of post-translationalmodifications that could modulate HMG-CoA reduc-tase activity [38]. Results from Figure 2A,C suggest thatthere is no difference in the total levels of cholesteroland cholesteryl esters upon R1881 treatment. Thisdifference with the in vitro microsomal activity resultssuggest that there are other mechanisms that mediate
the efflux of cholesterol and cholesteryl esters that maybe upregulated in PC-3 cells treated with the androgenagonist. HMG-CoA reductase has been known tofunction in both smooth and rough endoplasmicreticulum as well as peroxisomes, consistent with ourobservation thatHMG-CoA reductase activity is differ-ent in microsomes and not whole cell lysates obtainedfrom R1881 treated and untreated cells [39,40].
The androgen regulation of the cholesterol metabo-lism enzyme, HMG-CoA reductase, in prostate cancercellswas further supported in Figures 2Dand 3B. Itwasshown in androgen-sensitive LNCaP prostate cancercells, that when stimulated with R1881, there was anincrease in HMG-CoA reductase activity. Our results
The Prostate DOI 10.1002/pros
Fig. 4. A: ACATactivity inmicrosomes obtained fromcontrol (emptybar) andR1881-treated�1nM for 4 days�(hatchedbar) LNCaP andPC-3 cells.The sample sizeper group ranged fromn¼ 4 to 6.Bars represent themeanvalue� s.d. ** indicates a significant (P< 0.01) increasecompared to control.Values are expressed as pmoles/mg/min of product.B: ACAT1protein expression in lysates fromLNCaP and PC-3 cellslysates fromcontrol(emptybar)andR1881-treated�1nMfor4days�(hatchedbar)cells.Thesamplesizepergroupisn¼ 3.Thebottompanelrepresents densitometric scanning of the results normalized by actin levels. Bars represent the mean value� s.d. ** indicates a significant(P< 0.01) increase compared to control.ACAT2proteinexpressionin lysates fromLNCaP andPC-3cells lysates fromcontrol (emptybar) andR1881-treated�1nM for 4 days�(hatchedbar) cells is also portrayed in (B).The sample size per group is n¼ 3.Thebottompanel representsdensitometric scanningof theresultsnormalizedbyactinlevels.Barsrepresentthemeanvalue� s.d.**indicatesa significant(P< 0.01) increasecomparedto control.
28 Locke et al.

are in agreement with the work published by Brussel-mans et al. [16] with the addition that we provide amolecular mechanism for the changes in enzymaticactivities in the cholesterol metabolism pathway.
Microsomal in vitro ACAT1 activity is increasedupon androgen stimulation in PC-3 cells (but not inLNCaP, Fig. 4A). When the AR is overexpressed as inPC-3 AR2 cells, and the cells are treated with R1881 theAR expression is decreased, there is a reduction inACAT1 protein expression and activity as shown by
immunoblot and the radioactive acetate assays. Thissuggests that the AR and STAT1a may play a role inACAT1 expression and activity.
Induction of cholesteryl ester levels by R1881 havebeen reported by Carpten et al., [41] in LNCaP cells,indicating that either one or both of the ACATisoform(s) is (are) expressed in these cells OR there isan induction in cholesteryl ester import (or a reductionof cholesteryl ester secretion) upon R1881 treatmentAND/OR there is a decrease in cholesteryl hydrolaseactivity. ACAT1 expression has been shown to beinduced by fatty acids [27,42] which have been shownto be upregulated in prostate cells [6]. An induction infatty acids has been shown to stimulate low densitylipoprotein receptor expression [43,44], which is inagreement with the high LDLr expression observed inPC-3 cells. Moreover in this case ACAT activity isenhanced, as in the case of PC-3 cells.
In Figure 4A it appears that the ACAT activityin treated and untreated LNCaP cells is similar. Anexplanation is that ACAT2 the enzyme induced inLNCaP cells utilizes preferentially linolenoyl CoA assubstrate [27] and not oleyl CoA as in the case ofACAT1. This would explain why we could only detectin vitro ACAT activity in PC-3where ACAT1may playa more important role than ACAT2 which shows nochange in expression in that model.We speculated thatif androgen and the androgen receptor played a role incholesterol esterification in PC-3 cells, overexpressingthe receptor would affect the cholesterol ester levels.In fact when we compare the results from Figures 2Cand 6B we found an increase in cholesterol esterifica-tion in the untreated AR overexpressing cells. There isalso an increase in ACAT1 expression due to the ARoverexpression (not shown). When the AR expressionis decreased by the R1881 treatment in the PC-3 AR2
The Prostate DOI 10.1002/pros
Fig. 5. A: STAT1aexpressioninLNCaPandPC-3cells.Cellswereeither untreatedor incubatedwith1nMR1881for 4days.Cellswerethen washed and lysed with modified RIPA buffer on ice andthe lysates were cleared by centrifugation. Lysates were separatedby SDS^PAGE, transferred to a nitrocellulose and probedwith ananti-STAT1a antibody. The levels of STAT1a expression in LNCaPwere lower thaninPC-3 cells (not shown).Thebottompanel rep-resents densitometric scanning of the results normalized by actinlevels.Bars represent themeanvalue� s.d. * indicates a significant(P< 0.05) increase compared to control (n¼ 3).B: STAT1atyrosinephosphorylation in PC-3 cells.Cells were either untreated or incu-batedwith1nMR1881for 4 days.Cells were thenwashed and lysedwithmodifiedRIPAbufferoniceandthelysateswereclearedbycen-trifugation.Lysateswere separatedby SDS^PAGE, transferred to anitrocellulose and probed with anti-phosphotyrosine STAT1aand actin antibodies. The bottom panel represents densitometricscanning of the results normalized by actin levels. Bars representthe mean value� s.d. * indicates a significant (P< 0.05) increasecomparedto control (n¼ 3).
Androgen-MediatedCholesterolMetabolism 29

The Prostate DOI 10.1002/pros
Fig. 6. A: ARproteinexpressioninPC-3andPC-3AR2cells.Cellswereeitheruntreatedor incubatedwith1nMR1881for4days.CellswerethenwashedandlysedwithmodifiedRIPAbufferonice and thelysateswere clearedbycentrifugation.Lysateswere separatedbySDS^PAGE,transferred to a nitrocellulose andprobedwith an anti-AR antibody.The levels of AR expression in R1881-treated PC-3 were comparable tountreatedPC-3AR2cells.PC-3AR2R1881treatedcells showedareductioninARexpressionlevels.Thebottompanelrepresentsdensitomet-ric scanningof theresultsnormalizedbyactinlevels.Barsrepresentthemeanvalue� s.d. * indicatesa significant (P< 0.05)decreasecomparedto control (n¼ 4).B:Total cholesterol andcholesteryl ester levels incontrol (emptybar) andR1881-treated (hatchedbar) PC-3AR2cells.Barsrepresentthemean14Ccounts� s.d.of fourexperimentseachanalyzedby triplicatemeasurements.**indicatesa significant(P< 0.05)increasecompared to control.Cholesterol and cholesteryl ester levelsmeasuredin14C counts andnormalized to total protein content.C^E: ACAT1,ACAT2, andSTAT1aexpressioninPC-3andPC-3AR2cells.Cellswereeitheruntreatedornotasdescribedabove (A).Thelevels ofARexpres-sion inR1881-treatedPC-3were comparable tountreatedPC-3AR2cells.Thebottompanelrepresents densitometric scanningof theresultsnormalizedbyactinlevels.Barsrepresentthemeanvalue� s.d. * indicates a significant (P< 0.05)decreasecomparedto control (n¼ 4).
30 Locke et al.

cells (Fig. 6A), there is also a decrease in cholesterylester levels (Fig. 6B) further suggesting an associationbetween AR and cholesteryl esterification. From thesetwo pieces of evidence we state that androgen affectscholesterol esterification in PC-3 cells.
ACAT1 expression has been shown to be induced byinterferon gamma (IFN-g) [23]. There is evidence of acrosstalk between androgen and interferon signaling[45]. There are several reports on cells treated withandrogens, including R1881 which show that IFN-g-dependent genes are induced by the androgen treat-ment [46,47]. The interferon-activated ribonucleaseRNAse L has been identified as a candidate for thehereditary prostate cancer activated allele HPC1 [41].Recently, Bettoun et al. [45] described the physicalinteraction of the androgen receptor and RNAse L.They proposed a model were the activation of ARsignaling or downregulation of IFN-g signalingthrough mutation of RNAse L contribute to cellsurvival and suppression of apoptosis, accounting forthe effect of RNAse mutation on prostate cancerprogression. The androgen receptor expression hasbeen shown to be induced by interferons [48]. How-ever, the IFN-g pathway is not functional in LNCaP, asJAK1 is not expressed in these cells [49]. Anotherexample of the deficiency of JAK-STAT signaling isprovided by the work from Kominsky et al., [50]. Theyfound that IFN-g did not induce the tyrosine phos-phorylation of STAT1a in LNCaP cells and that at afunctional level interferon gamma treatment inducedgrowth arrest in PC-3 but not in LNCaP cells [50].
STAT1a expression is induced in PC-3 cells treatedwith R1881. The lack of induction of STAT1a expres-sion inLNCaPcellsmaybe related to the fact that Jak1 isnot functional in these cells [49]. Interestingly cells thatoverexpressed the AR showed a higher expression ofSTAT1a, however, upon R1881 treatment there was adrastic decrease in STAT1a expression (Fig. 6). Theseresults would suggest that there is an associationbetweenARand STAT1a expression. Protein inhibitorsof STATs (PIAS) have been shown to be involved inandrogen receptor signaling [51].Activation of STAT1amay be a novel finding that explains activation ofinterferon inducible genes in prostate cells treatedwithR1881 as shown by microarray studies [3,46]. Onanother avenue, the STAT and JAKs pathways havealready shown to be involved in PC-3 cell growththrough IL-6 and NFkB protein stimulation [47].STAT1a also plays a role in the development ofdocetaxel resistance in prostate cancer tumor cells[52]. Another gene regulated by the androgen receptorin LNCaP cells is the ATP-binding cassette transporterA1 (ABCA1). Interestingly ABCA1 expression is sup-pressed by androgen treatment [4]. ABCA1 functionsas a cholesterol exporter and its suppressionmay serve
as an additional mechanism to regulate cholesterolaccumulation in prostate cancer cells [53].
We propose the following model to account for ourfindings in the PC-3 cells. Upon androgen treatment,STAT1a is activated either by recruitment to theandrogen receptor (AR) and/or by phosphorylationof a tyrosine kinase that is associated to the AR andactivated upon ligand binding. Activated STAT1a thenis recruited to the nucleus and binds to the GASsequences in the promoter of genes such as ACAT1 todrive gene expression. Both AR and STAT1a arerequired for the induction of ACAT1 expression uponR1881 treatment of PC-3 cells.
In this study, the activity of cholesterol metabolismenzymes, HMG-CoA reductase and ACAT wereinvestigated in androgen stimulated prostate cancercells (PC-3 and LNCaP). It was shown that HMG-CoAreductase activity is apparently regulated by androgendependent and independent pathways and thus thismay be a useful target in developing a treatmentfor prostate cancer. This enzyme’s activity profilesmay also contribute to deciphering the mechanismof change from androgen dependent to androgen-independent prostate cancer. ACAT is also androgenregulated in nature but through a different mechanismthan that of HMG-CoA reductase involving STAT1a.Increased knowledge of these two enzymes and tran-scriptional factor’s involvement in prostate cancerprogression will allow for better optimization oftherapeutic targeting, improving the treatment optionsforprostate cancer patients andultimately leading to anoverall increase in their quality of life.
We have investigated the effects of androgens oncholesterol metabolizing enzymes, HMG-CoA reduc-tase andACAT using two cell models representative ofandrogen-sensitive (LNCaP) and androgen-insensitive(PC-3) prostate cancer. In the future it would beinteresting to investigate the effect of androgens onthese enzymes using our well established in vivoLNCaP xenograft model that follows androgen-dependent to androgen-independent progression byPSA marker [3]. This consistent model may help todevelop better understanding of the molecular mecha-nisms underlying prostate cancer evolution.
ACKNOWLEDGMENTS
The authors wish to thank Dr C. Warburton forhelpful discussion of this manuscript and Dr. T. Brownfor the kind donation of PC-3 AR2 cells. Funding forthis project was provided by a Terry Fox Foundation/National Cancer Institute of Canada Group Grant toC.Nelson,E.GunsandK.Wasan. J. Lockewas fundedonthis project by theMichael Smith Foundation forHealthResearch and VGH and UBC Hospital Foundation.
The Prostate DOI 10.1002/pros
Androgen-MediatedCholesterolMetabolism 31

REFERENCES
1. Buttyan R, Ghafar MA, Shabsigh A. The effects of androgendeprivation on the prostate gland: Cell death mediated byvascular regression. Curr Opin Urol 2000;10:415–420.
2. Burnstein KL. Regulation of androgen receptor levels: Implica-tions for prostate cancer progression and therapy. J Cell Biochem2005;95:657–669.
3. Ettinger SL, Sobel R, Whitmore TG, Akbari M, Bradley DR,Gleave ME, Nelson CC. Dysregulation of sterol responseelement-binding proteins and downstream effectors in prostatecancer during progression to androgen independence. CancerRes 2004;64:2212–2221.
4. HagerMH, SolomonKR, FreemanMR. The role of cholesterol inprostate cancer. Curr Opin Clin Nutr Metab Care 2006;9:379–385.
5. Swinnen JV, Heemers H, Van de ST, de SE, Brusselmans K,Heyns W, Verhoeven G. Androgens, lipogenesis and prostatecancer. J Steroid BiochemMol Biol 2004;92:273–279.
6. Swinnen JV,VanderhoydoncF, ElgamalAA,EelenM,VercaerenI, JoniauS,VanPH,Baert L,GoossensK,HeynsW,VerhoevenG.Selective activation of the fatty acid synthesis pathway in humanprostate cancer. Int J Cancer 2000;88:176–179.
7. Swinnen JV, Van Veldhoven PP, Esquenet M, Heyns W,Verhoeven G. Androgens markedly stimulate the accumulationof neutral lipids in the human prostatic adenocarcinoma cell lineLNCaP. Endocrinology 1996;137:4468–4474.
8. Sonn GA, Aronson W, Litwin MS. Impact of diet on prostatecancer: A review. Prostate Cancer Prostatic Dis 2005;8:304–310.
9. Feldman Brian J, Feldman D. The development of androgen-independent prostate cancer. Nat Rev Cancer 2001;1:34–45.
10. So A, Gleave M, Hurtado-Col A, Nelson C. Mechanisms of thedevelopment of androgen independence in prostate cancer.World J Urol 2005;23:1–9.
11. Suzuki H, Ueda T, Ichikawa T, Ito H. Androgen receptorinvolvement in the progression of prostate cancer. Endocr RelatCancer 2003;10:209–216.
12. Titus Mark A, Schell Michael J, Lih Fred B, Tomer Kenneth B,Mohler James L. Testosterone and dihydrotestosterone tissuelevels in recurrent prostate cancer. Hum Cancer Biol 2005;11:4653–4657.
13. Friesen JA, Rodwell VW. The 3-hydroxy-3-methylglutarylcoenzyme-A (HMG-CoA) reductases. Genome Biol 2004;5:248.
14. Chang TY, Chang CC, Ohgami N, Yamauchi Y. Cholesterolsensing, trafficking, and esterification. Annu Rev Cell Dev Biol2006;22:129–157.
15. Soccio RE, Breslow JL. Intracellular cholesterol transport.Arterioscler Thromb Vasc Biol 2004;24:1150–1160.
16. BrusselmansK, TimmermansL,Vande SandeT,VanVeldhovenPP, Guan G, Shechter I, Claessens F, Verhoeven G, Swinnen JV .Squalene synthase, a determinant of raft-associated cholesterolandmodulator of cancer cell proliferation. J Biol Chem 2007;282:18777–18785.
17. Swinnen JV, Verhoeven G. Androgens and the control of lipidmetabolism in human prostate cancer cells. J Steroid BiochemMol Biol 1998;65:191–198.
18. Robichon C, Dugail I. De novo cholesterol synthesis at thecrossroads of adaptive response to extracellular stress throughSREBP. Biochimie 2007;89:260–264.
19. Panda T, Devi VA. Regulation and degradation of HMGCo-Areductase. Appl Microbiol Biotechnol 2004;66:143–152.
20. Istvan ES. Bacterial and mammalian HMG-CoA reductases:Related enzymes with distinct architectures. Curr Opin StructBiol 2001;11:746–751.
21. Istvan ES, Deisenhofer J. The structure of the catalytic portionof human HMG-CoA reductase. Biochim Biophys Acta 2000;1529:9–18.
22. Leon C, Hill JS, Wasan KM. Potential role of acyl-coenzymeA:Cholesterol transferase (ACAT) inhibitors as hypolipidemicand antiatherosclerosis drugs. Pharm Res 2005; 1578–1588.
23. Yang JB, DuanZJ, YaoW, LeeO, Yang L, YangXY, SunX, ChangCC, Chang TY, Li BL. Synergistic transcriptional activation ofhuman acyl-coenzyme A: Cholesterol acyltransterase-1 gene byinterferon-gamma and all-trans-retinoic acid THP-1 cells. J BiolChem 2001;276:20989–20998.
24. SongBL,WangCH, YaoXM, YangL, ZhangWJ,WangZZ, ZhaoXN, Yang JB, Qi W, Yang XY, Inoue K, Lin ZX, Zhang HZ,Kodama T, Chang CC, Liu YK, Chang TY, Li BL. Human acyl-CoA:Cholesterol acyltransferase 2 gene expression in intestinalcaco-2 cells and in hepatocellular carcinoma. Biochem J 2006;394:617–626.
25. Andela VB, Gordon AH, Zotalis G, Rosier RN, Goater JJ, LewisGD, Schwarz EM, Puzas JE, O’Keefe RJ. NFkappaB: A pivotaltranscription factor in prostate cancer metastasis to bone.Clin Orthop Relat Res 2003;(415 Suppl):S75–S85.
26. Brown MS, Goldstein JL. The SREBP pathway: Regulation ofcholesterol metabolism by proteolysis of a membrane-boundtranscription factor. Cell 1997;89:331–340.
27. Seo T, Oelkers PM, Giattina MR, Worgall TS, Sturley SL,Deckelbaum RJ. Differential modulation of ACAT1 and ACAT2transcription and activity by long chain free fatty acids incultured cells. Biochemistry 2001;40:4756–4762.
28. Leon C, Nandan D, Lopez M, Moeenrezakhanlou A, Reiner NE.Annexin V associates with the IFN-gamma receptor andregulates IFN-gamma signaling. J Immunol 2006;176:5934–5942.
29. Sivaprasad U, Abbas T, Dutta A. Differential efficacy of3-hydroxy-3-methylglutaryl CoA reductase inhibitors on thecell cycle of prostate cancer cells. Mol Cancer Ther 2006;5:2310–2316.
30. Alimirah F, Chen J, Basrawala Z, XinH, ChoubeyD.DU-145 andPC-3 human prostate cancer cell lines express androgenreceptor: Implications for the androgen receptor functions andregulation. FEBS Lett 2006;580:2294–2300.
31. Buchanan G, Craft PS, Yang M, Cheong A, Prescott J, Jia L,Coetzee GA, Tilley WD. PC-3 cells with enhanced androgenreceptor signaling: A model for clonal selection in prostatecancer. Prostate 2004;60:352–366.
32. Culig Z,KlockerH, Eberle J, Kaspar F,HobischA,CronauerMV,Bartsch G. DNA sequence of the androgen receptor in prostatictumor cell lines and tissue specimens assessed by means of thepolymerase chain reaction. Prostate 1993;22:11–22.
33. Jarrard DF, Kinoshita H, Shi Y, Sandefur C, Hoff D, Meisner LF,Chang C, Herman JG, Isaacs WB, Nassif N. Methylation of theandrogen receptor promoter CpG island is associated with lossof androgen receptor expression in prostate cancer cells. CancerRes 1998;58:5310–5314.
34. Edelstein RA, Carr MC, Caesar R, Young M, Atala A, FreemanMR. Detection of human androgen receptor mRNA expressionabnormalities by competitive PCR. DNA Cell Biol 1994;13:265–273.
35. Chen Y, Hughes-Fulford M. Human prostate cancer cells lackfeedback regulation of low-density lipoprotein receptor and itsregulator, SRE BP2. Int J Cancer 2000;91:41–45.
The Prostate DOI 10.1002/pros
32 Locke et al.

36. Jia L, Coetzee GA. Androgen receptor-dependent PSA expres-sion in androgen-independent prostate cancer cells does notinvolve androgen receptor occupancy of the PSA locus. CancerRes 2005;65:8003–8008.
37. Westendorf JJ, Hoeppner L. Down-regulation of androgenreceptor by 3,30-diindolylmethane contributes to inhibition ofcell proliferation and induction of apoptosis in both hormone-sensitive LNCaP and insensitive C4-2B prostate cancer cells.Urol Oncol 2007;25:180–181.
38. Omkumar RV, Darnay BG, Rodwell VW. Modulation of syrianhamster 3-hydroxy-3-methylglutaryl-CoA reductase activity byphosphorylation. role of serine 871. J Biol Chem 1994;269:6810–6814.
39. Kovacs WJ, Faust PL, Keller GA, Krisans SK. Purification ofbrain peroxisomes and localization of 3-hydroxy-3-methylglu-taryl coenzyme A reductase. Eur J Biochem 2001;268:4850–4859.
40. Reinhart MP, Billheimer JT, Faust JR, Gaylor JL. Subcellularlocalization of the enzymes of cholesterol biosynthesis andmetabolism in rat liver. J Biol Chem 1987;262:9649–9655.
41. Carpten J, Nupponen N, Isaacs S, Sood R, Robbins C, Xu J,FaruqueM,MosesT, EwingC,GillandersE,HuP,BujnovszkyP,Makalowska I, Baffoe-Bonnie A, Faith D, Smith J, Stephan D,Wiley K, Brownstein M, Gildea D, Kelly B, Jenkins R, HostetterG, Matikainen M, Schleutker J, Klinger K, Connors T, Xiang Y,Wang Z, De MA, Papadopoulos N, Kallioniemi OP, Burk R,Meyers D, GronbergH,Meltzer P, Silverman R, Bailey-Wilson J,Walsh P, Isaacs W, Trent J. Germline mutations in theribonuclease L gene in families showing linkage with H PC1.Nat Genet 2002;30:181–184.
42. Castoria G, LombardiM, BaroneMV, Bilancio A, Di DM, De FA,Varricchio L, Bottero D, Nanayakkara M, Migliaccio A,Auricchio F. Rapid signalling pathway activation by androgensin epithelial and stromal cells. Steroids 2004;69:517–522.
43. Liu Y. Fatty acid oxidation is a dominant bioenergetic pathwayin prostate cancer. Prostate Cancer Prostatic Dis 2006;9:230–234.
44. Rumsey SC, Galeano NF, Lipschitz B, Deckelbaum RJ. Oleateand other long chain fatty acids stimulate low density lip-oprotein receptor activity by enhancing acyl coenzyme A:Cho-lesterol acyltransferase activity and altering intracellularregulatory cholesterol pools in cultured cells. J Biol Chem1995;270:10008–10016.
45. BettounDJ, ScafonasA, RutledgeSJ,HodorP, ChenO,GamboneC, Vogel R, Elwee-Witmer S, Bai C, Freedman L, Schmidt A.Interaction between the androgen receptor and RNase Lmediates a cross-talk between the interferon and androgensignaling pathways. J Biol Chem 2005;280:38898–38901.
46. Clark DT, Soory M. The metabolism of cholesterol and certainhormonal steroids by treponema denticola. Steroids 2006;71:352–363.
47. Culig Z, Steiner H, Bartsch G, Hobisch A. Interleukin-6regulation of prostate cancer cell growth. J Cell Biochem 2005;95:497–505.
48. Basrawala Z, Alimirah F, Xin H, Mohideen N, Campbell SC,Flanigan RC, Choubey D. Androgen receptor levels areincreased by interferons in human prostate stromal andepithelial cells. Oncogene 2006;25:2812–2817.
49. DunnGP, SheehanKC,Old LJ, Schreiber RD. IFNunresponsive-ness in LNCaP cells due to the lack of JAK1 gene expression.Cancer Res 2005;65:3447–3453.
50. Kominsky SL, Hobeika AC, Lake FA, Torres BA, Johnson HM.Down-regulation of neu/HER-2 by interferon-gamma in pros-tate cancer cells. Cancer Res 2000;60:3904–3908.
51. Nishida T, TerashimaM, Fukami K. PIASy-mediated repressionof the ets-1 is independent of its sumoylation. Biochem BiophysRes Commun 2006;345:1536–1546.
52. Patterson SG,Wei S, Chen X, SallmanDA, Gilvary DL, Zhong B,Pow-Sang J, Yeatman T, Djeu JY. Novel role of Stat1 in thedevelopment of docetaxel resistance in prostate tumor cells.Oncogene 2006;25:6113–6122.
53. Oram JF, Heinecke JW. ATP-binding cassette transporter A1: Acell cholesterol exporter that protects against cardiovasculardisease. Physiol Rev 2005;85:1343–1372.
The Prostate DOI 10.1002/pros
Androgen-Mediated CholesterolMetabolism 33




















