
Amyloid-like self-assembly of a dodecapeptide sequence from the adenovirus fiber shaft: Perspectives...
7
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Amyloid-like self-assembly of a dodecapeptide sequence from the adenovirus fiber shaft: Perspectives...

This article appeared in a journal published by Elsevier. The attached
copy is furnished to the author for internal non-commercial research
and education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or
licensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of the
article (e.g. in Word or Tex form) to their personal website or
institutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies are
encouraged to visit:
http://www.elsevier.com/copyright

Author's personal copy
Amyloid-like self-assembly of a dodecapeptide sequence from the adenovirus !bershaft: Perspectives from molecular dynamics simulations
Phanourios Tamamis, Georgios Archontis !Department of Physics, University of Cyprus, PO20537, CY1678 Nicosia, Cyprus
a b s t r a c ta r t i c l e i n f o
Article history:Received 10 April 2010Received in revised form 17 May 2010
Keywords:Peptide-based nanostructures;Amyloids;Adenovirus;Molecular dynamics;Replica-exchange;Implicit-solvent
Peptide and protein self-assembly is related to the fundamental problems of protein folding and misfoldingand has potential applications in medicine, materials science and nanotechnology. Sequence repeats fromself-assembling proteins may provide useful elementary building blocks of peptide-based nanostructures.Sequences from the adenovirus !ber shaft self-assemble into amyloid-like !brils outside their native context.In earlier simulations we studied the self-assembly of two shaft sequences, the octapeptide NSGAITIG andthe hexapeptide GAITIG. Based on these simulations, cysteine residues were substituted at the !rst twopositions of the octapeptide, yielding amyloid !brils capable of binding to silver, gold and platinumnanoparticles. Here, we study by implicit-solvent replica-exchange simulations the self-assembly of a longershaft sequence, the dodecapeptide LSFDNSGAITIG. The simulations provide insights on the molecularorganization of the corresponding !bers. Individual molecules tend to adopt hairpin-like conformations inthe observed intermolecular !-sheets, in line with the experimentally determined amyloid !ber diametersand the conformation of the peptide in the adenovirus !ber shaft. By analyzing the arrangement of individualpeptides in the intermolecular sheets, we suggest possible structural models of the corresponding !bers andinterpret their stability by energetic calculations.
© 2010 Elsevier B.V. All rights reserved.
1. Introduction
During the last decade, extensive experimental [1–13] andcomputational studies [11,13–26] have focused on the self-assemblyof proteins and peptides into non-crystalline and insoluble [27]amyloid !bers [1–4], nanostructures of various morphologies [5–7]and systems responsive to external stimuli [6,8]. Sequences occurringnaturally in !brous proteins may constitute elementary buildingblocks of peptide-based nanostructures [9]. Insights on the self-assembly of such sequences may contribute to applications inmedicine, nanotechnology and materials science and to our under-standing of biomolecular folding and association.
The adenovirus type-2 !ber shaft is a 582-residue protein,consisting of a virus-binding N-terminal tail, a central shaft and aglobular, receptor-binding C-terminal domain (reviewed in Papani-kolopoulou et al. [28]). A stable trimeric domain, containing part ofthe shaft and the C-terminal head (residues 319–582) folds into anovel, triple !-spiral fold [29]. Speci!c shaft sequences with 6–41residues self-assemble into amyloid-like !brils [9,30]. In recentwork we studied the aggregation properties of two such sequences,the octapeptide NSGAITIG and the hexapeptide GAITIG [26]. Oursimulations suggested that the N-terminal residues of the octapep-
tide did not participate in the !bers. In line with this observation,cysteine residues were recently substituted at positions 1 and 2; thenewly designed peptides formed amyloid !brils capable of bindingto silver, gold and platinum nanoparticles [31].
The shaft dodecapeptide LSFDNSGAITIG (residues 381–392),hereafter denoted as LSFD, forms also amyloid-like !bers outsidethe native context [9,30]. The same peptide self-assembles at theair–water interface into a monomolecular layer composed by "atantiparallel !-sheets [32–34]. This result suggests that amyloidpeptides may self-assemble on surfaces, in a different organizationthan bulk amyloid !brils adsorbed on surfaces. The crystallineorder in these layers is much greater than the corresponding orderin layers formed at the air–water interface by the Alzheimer's A!peptide [32]. Consequently it has been suggested that the transferof such highly ordered peptide assemblies onto solid surfaces (i.e.LSFDNSGAITIG monolayers on quartz) could be used for nanome-ter-scale surface patterning [32]. Therefore, the study of the LSFDself-assembly properties in bulk and at the air–water interfacepresents signi!cant interest. Here, we undertake a !rst step in thisdirection, by studying the aggregation properties of LSFD in bulkwater with implicit-solvent molecular dynamics (MD) simulations.
In aqueous solution, LSFD forms amyloid !brils with a diameterof 21 Å, much smaller than the maximum length of the peptides.Thus, it has been suggested that the peptides adopt a hairpinconformation in the amyloid !bers [9] as in the spiral fold [29]. Ourresults indeed support the formation of !-sheets with individual
Journal of Non-Crystalline Solids 357 (2011) 717–722
! Corresponding author. Tel.: +357 22 89 28 22; fax: +357 22 89 28 21.E-mail address: [email protected] (G. Archontis).
0022-3093/$ – see front matter © 2010 Elsevier B.V. All rights reserved.doi:10.1016/j.jnoncrysol.2010.05.083
Contents lists available at ScienceDirect
Journal of Non-Crystalline Solids
j ourna l homepage: www.e lsev ie r.com/ locate / jnoncryso l

Author's personal copy
peptides in hairpin-like conformations. We analyze the structuralproperties of these sheets and identify important stabilizinginteractions. In this way, we obtain insights on the likely organiza-tion of the peptides in their amyloid !bers. We also compare ourresults against experimental information on the LSFD amyloid !brils[9,30] and earlier simulations of an LSFD monomer in aqueoussolution [35].
2. Methods
2.1. System
The simulation system consisted of three monomers with thesequence NH3
+–LSFDNSGAITIG–NH2; terminal groups were chosenfor consistency with the experimental sequences.
2.2. Force !eld
The peptide atomic charges, van der Waals and stereochemicalparameters were taken from the CHARMM19 all-atom force !eld forconsistency with FACTS 19 parametrization [36]. Solvent effects weretaken into account implicitly by the recent (C19) implicit-solventmodel FACTS of Ca"isch [37]; the peptide dielectric constant was setto "=2. A non-polar solvation energy surface termwas included, witha surface-tension coef!cient of 0.025 kcal/mol/Å2. A 7.5-Å cutoff wasused for the non-bonded interactions [37]. The equations of motionwere integrated by the leap-frog algorithm, with a 2.0-fs timestep.Bonds involving hydrogen atoms were constrained to standard valueswith the SHAKE algorithm [38,39]. The temperature was controlled bythe Langevin method; the friction coef!cients were set to 5.0 ps!1 forheavy atoms and 0 ps!1 for hydrogen atoms. The monomers wereplaced in a periodically replicated 110-Å cubic box, modeling a4.2 mg/ml solution. This concentration is within the range ofexperimental conditions in which the LSFD peptides form !bers [9].All simulations were conducted with the program CHARMM, versionc35a2 [39].
2.3. Simulation method
The system was simulated by the replica-exchange method[40–45]. We employed 10 replicas with temperatures 290, 300,310, 320, 331, 342, 353, 364, 375 and 386 K. The obtainedexchange probabilities between adjacent replicas were 27±1%.Replica exchanges were attempted at 10-ps intervals; the totalsimulation length at each temperature was 1.2 #s. We analyzed120,000 snapshots (conformations) extracted at 100-ps intervals,spanning the 1.2 #s replica simulations at 300 K.
2.4. Secondary-structure assignment
The secondary-structure content was assigned by the STRIDEalgorithm [46]. Additional analyses were performed with in-houseFORTRAN programs, available upon request.
2.5. De!nition of conformational states
We classi!ed conformational states based on the existence ofhairpin-like (U-shaped) structures. A peptide was in U-shapedconformation if two of the three distances 3C$–8C$, 4C$–9C$, and5C$–10C$ were smaller than 10 Å and at least one intramolecular!-bridge (linking segments 2–6 and 8–11) and/or a !-turn existed inthe region 4–10. If this condition was not satis!ed, the conformationwas classi!ed as extended (I-shaped). Intermolecular !-sheets wereclassi!ed into seven elementary states, depending on the number(two or three) and shape (hairpin-like, U, or extended, I) ofparticipating peptides: U3, IU2, UI2, I3, U2, UI, and I2.
2.6. Polar parameter and nematic order parameters
We assessed the degree of order in sheets with all three peptides ina U-shape conformation by the polar order-parameter P1 and thenematic order-parameter P2, de!ned in Eq. (1). These parameters are
Scheme 1. Representative examples of three-peptide !-sheets with all peptides inhairpin-like (U-shaped) conformations. The respective values of the polar orderparameters P1 and P1! are included in the !gure. Molecular vectors used in the de!nitionof P1 and P1! are shown by black and red arrows, respectively. The C$ atoms of the !rstand last residues are shown in blue and yellow circles, respectively. The hydrogen-bonding interaction patterns are indicated in parentheses.
718 P. Tamamis, G. Archontis / Journal of Non-Crystalline Solids 357 (2011) 717–722
Author's personal copy
widely used in the structural characterization of liquid crystals[47,48], and have been employed successfully in simulation studiesof peptide aggregation [17,26].
P1=1N
"N
i=1zid; P2=
1N
"N
i=1
32
zid! " 2
!12
!1"
In Eq. (1), N is the number of molecules in the simulation, zi is aunit vector along a suitably de!nedmolecular direction, and d is a unitvector along a preferred direction of alignment, which emerges fromthe properties of the system [49]. In our calculations, we employedtwo different sets of molecular vectors. In the !rst set (P1, P2), zi wasde!ned along the lines connecting the C$ geometric centers ofresidue moieties Ser2-Phe3-Ile11-Gly12 and Ser6-Gly7-Ala8-Ile9(Scheme 1).
In the second set (P1!, P2!), z i#! was de!ned along the lines
connecting the geometric centers of C$ atoms of the residuemoieties 2–5 and 9–12. Residue 1 was excluded because in themajority of 3-peptide U-shaped !-sheets it was disordered. P1 candifferentiate between parallel (i.e. U:U:U; P1=1) or antiparallel/mixed (U:#:U/U:U:#; P1=1/3) arrangements (the symbol “:”denotes intermolecular !-sheet interaction); P1! can differentiatebetween parallel or antiparallel/mixed hydrogen-bonding interac-tions of adjacent strands in the sheets, depending on the P1 value;typical examples are presented in Scheme 1. The parameters P2 andP2! differentiate between perfectly ordered (P2,P2! ~1) and disor-
dered conformations (P2, P2!b##########################81 = 40%N! "
p, for a system of N
peptides; here, N=3 and##########################81 = 40%N! "
p=0.46 [50]). All parameters
were computed with WORDOM [51]. In our calculations, structureswere ordered enough to constitute elementary structural units ofamyloid !bers if P2,P2!N0.5.
Free-energy surfaces were constructed by the two-dimensionalprobability P P1; P#
1$ %
as
G P1; P#1
$ %= !kBT1n P P1; P
#1
$ %& ': !2"
3. Results and discussion
3.1. Earlier experimental and computational studies
The LSFD amyloid !bers have a 21-Å diameter. This is approxi-mately half the length of the fully extended peptide (41.4 Å),re"ecting the fact that the peptides are not extended in the !bers.The !ber X-ray diffraction pattern suggests the existence ofantiparallel !-sheets. Based on this result and the !ber diameter, ithas been suggested that the peptides are folded into hairpin-like (U-shaped) conformations in the amyloid !bers, in a parallel (U:U:U:U…),antiparallel (U:#:U:#…) or mixed orientation (Fig. 9B of ref. [9]).Explicit-water MD studies of an isolated LSFD peptide supported theexistence of hairpin-like conformers [35]. Experimental studies of theLSFD peptide in the air/water interface con!rmed the existence ofantiparallel hydrogen-bonding interactions [32,52].
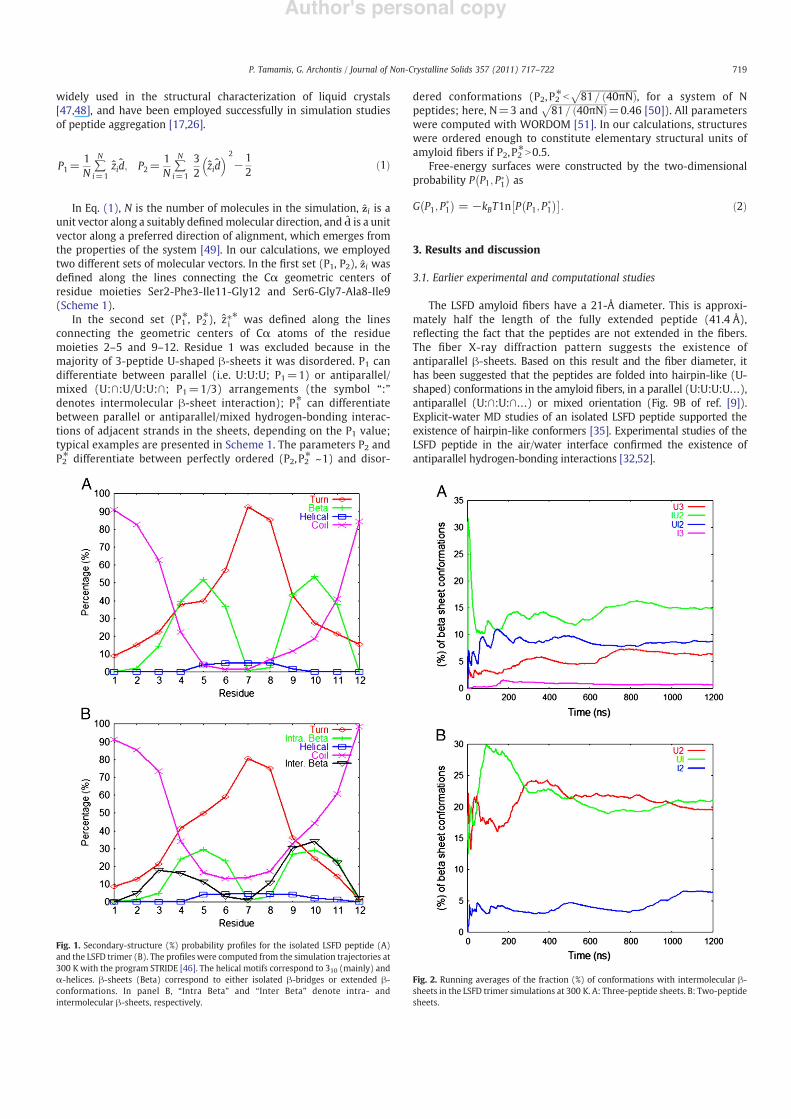
Fig. 1. Secondary-structure (%) probability pro!les for the isolated LSFD peptide (A)and the LSFD trimer (B). The pro!les were computed from the simulation trajectories at300 K with the program STRIDE [46]. The helical motifs correspond to 310 (mainly) and$-helices. !-sheets (Beta) correspond to either isolated !-bridges or extended !-conformations. In panel B, “Intra Beta” and “Inter Beta” denote intra- andintermolecular !-sheets, respectively.
Fig. 2. Running averages of the fraction (%) of conformations with intermolecular !-sheets in the LSFD trimer simulations at 300 K. A: Three-peptide sheets. B: Two-peptidesheets.
719P. Tamamis, G. Archontis / Journal of Non-Crystalline Solids 357 (2011) 717–722
Author's personal copy
3.2. Accuracy of the FACTS19 implicit-solvent model
To check the accuracy of the implicit-solvent model, we comparedsimulations of a single LSFDNSGAITIG peptide in implicit-(FACTS19)with explicit-solventMD results of the same system [35]. The implicit-solvent simulations employed the replica-exchange methodology,with eight optimized temperatures spanning the range 283–432 K.The total simulation length at each temperature was 300 ns. Theexplicit-solvent simulations were conducted at 350 K, to acceleratethe hairpin formation.
The secondary-structure content of the monomer in the implicit-solvent simulations (at 300 K) is presented in Fig. 1A. The agreementwith the earlier explicit-solvent results of ref. [35] is excellent, if weadd together the turn and bend populations in the latter (both areclassi!ed as turns in STRIDE [46]). The main conformer predicted byour simulations is a hairpin, with disordered terminal ends, segments4–6 and 9–11 engaged in !-sheet interactions and segment 7–8 in apredominantly type-II" !-turn. A similar hairpin was the mainconformer in the explicit-solvent simulations [35]. The correspondingsequence (residues 381–392) adopts a slightly different hairpinconformation in the adenovirus shaft, with segments 2–4 (382–384)
and 8–10 (388–390) in sheet interactions and 5–7 (385–387) in areverse turn [29]. This difference may be caused by tertiary contactswith the surrounding protein [35].
3.3. Conformational analysis of the LSFD trimer
The secondary-structure population in the trimer simulations isshown in Fig. 1B. We observe the frequent formation of intermolec-ular !-sheets, in accordance with the amyloidogenic properties of thepeptides [9]. The propensity for intramolecular !-sheets is reducedwith respect to the monomer simulations due to the competitionbetween intra- and intermolecular structures. Residues 9–11 partic-ipate simultaneously in intra- and intermolecular !-sheets. This isconsistent with the high intermolecular !-sheet propensity of thecorresponding moiety (ITI) in the NSGAITIG and GAITIG simulations[26]. Residues in segment 3–6 participate (sometimes simultaneous-ly) in intra- and intermolecular !-sheets. Overall, the C-terminalmoiety has a higher propensity for intermolecular !-sheets, presum-ably due to the higher hydrophobic character of the residuescomprising the former; as we show below, the sheet conformationsare stabilized by non-polar interactions. The extreme residues (Leu1
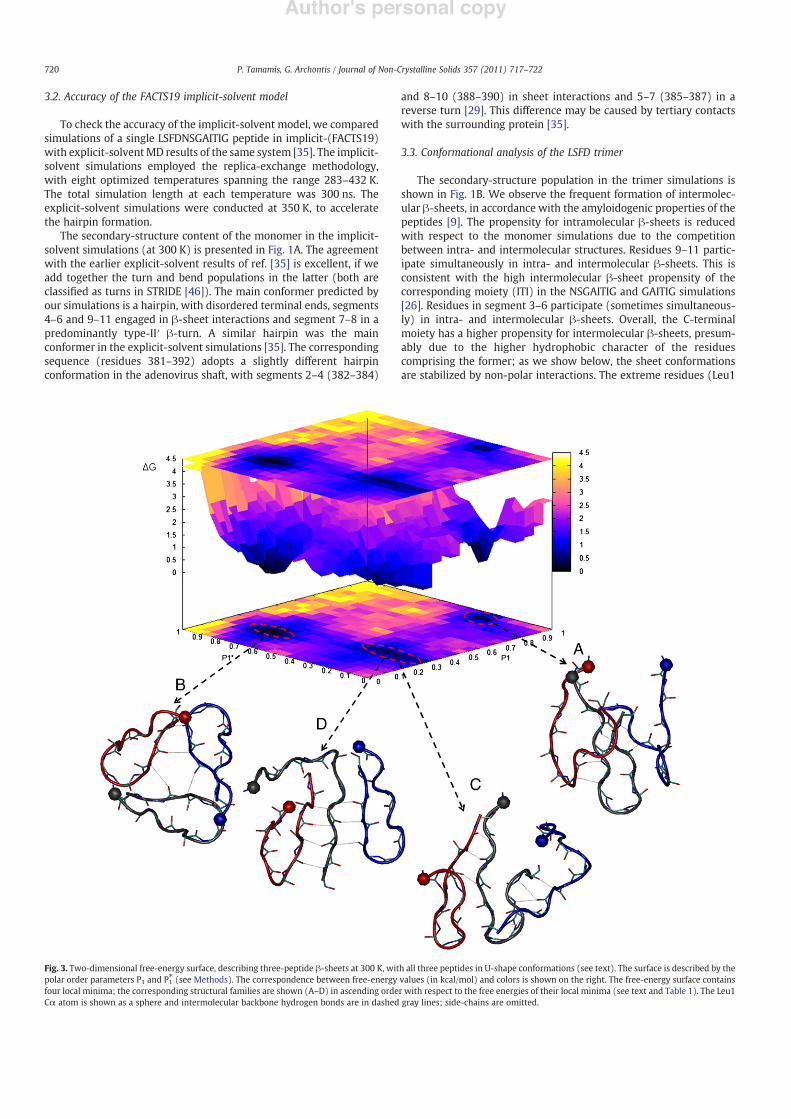
Fig. 3. Two-dimensional free-energy surface, describing three-peptide !-sheets at 300 K, with all three peptides in U-shape conformations (see text). The surface is described by thepolar order parameters P1 and P1! (see Methods). The correspondence between free-energy values (in kcal/mol) and colors is shown on the right. The free-energy surface containsfour local minima; the corresponding structural families are shown (A–D) in ascending order with respect to the free energies of their local minima (see text and Table 1). The Leu1C$ atom is shown as a sphere and intermolecular backbone hydrogen bonds are in dashed gray lines; side-chains are omitted.
720 P. Tamamis, G. Archontis / Journal of Non-Crystalline Solids 357 (2011) 717–722
Author's personal copy
and Gly12) do not participate in either intra- or intermolecular !-sheets.
The intermolecular !-sheets of our trimer simulations can beclassi!ed into seven elementary states, depending on the number (2–3) and shape (hairpin-like, U, or extended, I) of participating peptides:U3, IU2, UI2, I3, U2, UI, and I2. The running averages of the fraction (%) ofconformations corresponding to these states are shown in Fig. 2.Overall, states with U-shaped peptides are more favoured, due to theinherent intramolecular tendency of the sequence to fold into ahairpin. In particular, state U3 is much more dominant than state I3,suggesting that the peptides are folded in hairpin conformations inthe amyloid nanostructures.
3.4. Free-energy surface of state U3
In this sectionwe focus on state U3, which is likely to correspond toan elementary structural unit of the !ber. Additional insights on thestructural diversity of this state can be obtained by the polar (P1 andP1!) and nematic (P2 and P2!) order parameters, de!ned in Methods.7597 (6.33 %) of the analyzed snapshots at 300 K corresponded tostate U3. We treated the !rst 1000 conformations (correspondingapproximately to the !rst 200 ns of the simulation) as part of theequilibration phase. Using the remaining 6597 conformations, wecomputed the two-dimensional free energy G P1; P#
1$ %
of parameters P1and P1! shown in Fig. 3. Representative conformations correspondingto the free-energyminima are also shown in Fig. 3 and their respectiveenergetic properties are summarized in Table 1.
The parameter P1 differentiates between parallel (i.e. U:U:U;P1=1) or antiparallel/mixed (U:#:U/U:U:#; P1=1/3) arrangementsof the individual peptides (see Methods and Scheme 1). P1! candifferentiate between parallel or antiparallel/mixed hydrogen-bond-ing interactions of adjacent strands in the sheets, depending on thevalues of P1. The free-energy surface contains three free-energy basins(Fig. 3). The !rst basin contains the global free-energy minimum. Themost representative conformation is shown in Fig. 3A. The peptidesform parallel U-shapes, with mixed (parallel/antiparallel) intermo-lecular sheets. Thehigh order of these conformations is re"ected by thelarge average P2 and P2! values. A C$-coordinate cluster analysis showsthat the 3–6 segment of the central peptide (black tube in Fig. 3A,WEBversion) forms an antiparallel !-sheet with segment 8–11 of one edgepeptide (red tube, WEB version); segment 9–11 of the central peptideforms a parallel !-sheet with moiety 8–10 of the second edge peptide(blue tube,WEB version). Conformations in the!rst basin are probablyelementary structural units of the LSFD amyloid !bers.
The second basin contains the second free-energy minimum.Conformations in this basin are not well ordered and are not likely toconstitute elementary structural units of the amyloid !bers. A typicalconformation is shown in Fig. 3B; the peptides form a triangular shapeand the N-terminal residue moieties 1–6 do not participate inintermolecular !-sheets.
The third basin contains two distinct conformations, correspondingto the last two free-energyminima. In the thirdminimum, theU-shapedpeptides are arranged in a mixed style (e.g. U:#:#), in line with the P1value. The hydrogen-bonding interactions among adjacent strands ondifferent peptides are antiparallel, in line with P1! and P1 values. Themost representative conformation of this minimum is shown in Fig. 3C.Segment 10–11 of the central peptide (black tube, WEB version) formsan antiparallel !-sheet with segment 9–10 of one edge peptide (bluetube, WEB version); sector 5–6 of the central peptide forms anantiparallel !-sheet with sector 10–11 of the other edge peptide (redtube, WEB version). Conformations in the third minimum can beconsidered as elementary structural units of the LSFD amyloid !bers.
In the fourth minimum, the U-shapes are antiparallel and areengaged in antiparallel sheets (P1 ~1/3 and P1! ~1/3). The mostrepresentative conformation of this basin (according to C$ clusteringanalysis) is shown in Fig. 3D. The 9–11 moiety of the central peptide
(black tube, WEB version) forms simultaneously an antiparallel !-sheet with 9–11 sectors of the edge peptides (blue and red tubes,WEBversion). The signi!cantly low value of P2! and the fact that N-terminalresidue moiety 1–6 does not participate in intermolecular !-sheetssuggest that these conformations are less likely to be elementary unitsof the amyloid !bers.
The corresponding average total energies for the !rst four minimaare !868, !867, !859 and !857 kcal/mol respectively (Table 1).Structures with high (low) free energies are consistently correlatedwith high (low) energies. The increased stability of the conformationsin the !rst two minima is due to non-polar interactions, presumablydue to their optimum packing.
4. Conclusions
In the present work we have investigated the conformational andaggregation properties of the amyloidogenic dodecapeptide NH3–LSFDNSGAITIG–NH2 in bulk water by implicit-solvent replica-ex-change MD simulations. Our monomer simulations suggest theformation of a hairpin (U-shaped) conformer, with main-chainmoieties 4–6 and 9–11 in an antiparallel !-sheet and the moiety 7–8 in the center of a type-II" !-turn, in line with previous MD explicit-solvent simulations of the same monomer [35].
The trimer simulations are consistent with the existence of !-turnsand/or !-strands in the LSFD aggregates at the early stages for the!bril formation, as depicted by FTIR spectroscopy [9]. We observe thefrequent appearance of intermolecular !-sheets, in accordance withthe peptide amyloidogenic propensity [9]. The peptides adoptfrequently hairpin-like (U-shaped) conformation in these sheets,with moieties 9–11 and (to a lesser extent) 3–5 engaged inintermolecular sheet interactions. The U-shaped conformations areconsistent with the !ber diameter and the meridional re"ections ofthe LSFD amyloid !ber diffraction patterns [9]. Analysis of !-sheetsinvolving all three peptides in a U conformation suggests that theparallel (U:U:U) arrangement is more likely compared to theantiparallel or mixed (U:#:U/U:U:#), due to improved non-polarinteractions.
Our simulations provide insights on the self-assembly of the LSFDpeptide in bulk water. In a future study, we plan to investigate theself-assembly of the same peptide at the air–water interface.Experimental studies suggest that at that environment, LSFD isorganized into monolayers consisting of antiparallel !-sheets [9].This implies that amyloid peptides may self-assemble at surfaces intoa different structure than in bulk solution. Furthermore, such ordered
Table 1Properties of the LSFD trimer free-energy surface constructed by the two-dimensionalprobability of the order parameters P1 and P1! at 300 K (Fig. 3).
Min &Ga P1/P1!
rangeb,cP1/P1!
averagevaluesb
P2/P2!
averagevaluesb
Non-bonded energies (kcal/mol)d
Polar Non-polar Totale
1 0.00 0.87–1.00/0.22–0.35
0.93/0.28 0.80/0.73 !791.7(5.7)
!76.5(9.7)
!868.2(10.1)
2 0.08 0.15–0.30/0.65–0.85
0.22/0.73 0.31/0.32 !794.5(5.0)
!73.1(7.3)
!867.6(8.0)
3 0.37 0.25–0.35/0.00–0.35
0.30/0.18 0.51/0.59 !793.1(5.4)
!66.3(8.9)
!859.8(9.9)
4 0.37 0.25–0.35/0.00–0.35
0.31/0.16 0.86/0.36 !795.8(5.3)
!61.2(9.5)
!857.0(10.4)
a Free energies relative to the global minimum.b The polar (P1, P1!) and nematic (P2, P2!) order parameters are de!ned in the
Methods section.c Ranges of values over conformations in the corresponding free-energy minima
are listed.d Average values and standard deviations (in parentheses) over conformations in the
corresponding free-energy minima are listed.e Sum of polar and non-polar energies.
721P. Tamamis, G. Archontis / Journal of Non-Crystalline Solids 357 (2011) 717–722

Author's personal copy
!-sheet interfacial structures could serve as scaffolds for the growth ofminerals [33].
Acknowledgements
All simulations were conducted in Biophysics clusters at theUniversity of Cyprus. PhT was supported by a grant from the A.G.Leventis Foundation. We thank an anonymous referee for helpfulcomments.
References
[1] F. Chiti, C.M. Dobson, Ann. Rev. Biochem. 75 (2006) 333.[2] F. Bemporad, G. Galloni, S. Campioni, G. Plakoutsi, N. Taddei, F. Chiti, Acc. Chem.
Res. 39 (2006) 620.[3] R. Nelson, D. Eisenberg, Adv. Prot. Chem. 73 (2006) 235.[4] R.S. Harrison, P.C. Sharpe, Y. Singh, D.P. Fairlie, Rev. Physiol. Biochem. Pharmacol.
159 (2007) 1.[5] E. Gazit, Chem. Soc. Rev. 36 (2007) 1263.[6] X.J. Zhao, S.G. Zhang, Macrom. Biosci. 7 (2007) 13.[7] G. Colombo, P. Soto, E. Gazit, Trends Biotech. 25 (2007) 211.[8] K. Chockalingam, M. Blenner, S. Banta, Protein Eng. Des. Sel. 20 (2007) 155.[9] K. Papanikolopoulou, G. Schoehn, V. Forge, V.T. Forsyth, C. Riekel, J.F. Hernandez,
R.W.H. Ruigrok, A. Mitraki, J. Biol. Chem. 280 (2005) 2481.[10] M.R. Sawaya, S. Sambashivan, R. Nelson, M.I. Ivanova, S.A. Sievers, M.I. Apostol, M.
J. Thompson, M. Balbirnie, J.J.W. Wiltzius, H.T. McFarlane, A.O. Madsen, C. Riekel,D. Eisenberg, Nature 447 (2007) 453.
[11] T.P. Knowles, A.W. Fitzpatrick, S. Meehan, H.R. Mott, M. Vendruscolo, C.M. Dobson,M.E. Welland, Science 318 (2007) 1900.
[12] P. Mesquida, C.K. Riener, C.E. MacPhee, R.A. McKendry, J. Mat. Sci. Mat. Med. 18(2007) 1325.
[13] D. Zanuy, Y. Porat, E. Gazit, R. Nussinov, Structure 12 (2004) 439.[14] N. Mousseau, P. Derreumaux, Acc. Chem. Res. 38 (2005) 885.[15] B.Y. Ma, R. Nussinov, Curr. Op. Chem. Biol. 10 (2006) 445.[16] C.K. Hall, V.A. Wagoner, Meth. Enzymol. 412 (2007) 338.[17] M. Cecchini, F. Rao, M. Seeber, A. Ca"isch, J. Chem. Phys. 121 (2004) 10748.[18] E. Paci, J. Gsponer, X. Salvatella, M. Vendruscolo, J. Mol. Biol. 340 (2004) 555.[19] R. Pellarin, E. Guarnera, A. Ca"isch, J. Mol. Biol. 374 (2007) 917.[20] P.H. Nguyen, M.S. Li, G. Stock, J.E. Straub, D. Thirumalai, Proc. Natl Acad. Sci. USA
104 (2007) 111.[21] R.D. Hills, C.L. III Brooks, J. Mol. Biol. 368 (2007) 894.[22] B. Tarus, J.E. Straub, D. Thirumalai, J. Mol. Biol. 379 (2008) 815.[23] W. Song, G. Wei, N. Mousseau, P. Derreumaux, J. Phys. Chem. B 112 (2008) 4410.
[24] G. Bellesia, J.E. Shea, J. Chem. Phys. 130 (2009) 145103.[25] P. Tamamis, L. Adler-Abramovich, M. Reches, K. Marshall, P. Sikorski, L. Serpell, E.
Gazit, G. Archontis, Biophys. J. 96 (2009) 5020.[26] P. Tamamis, E. Kasotakis, A. Mitraki, G. Archontis, J. Phys. Chem. B 113 (47) (2009)
15639.[27] L.K. Thompson, PNAS 100 (2) (2003) 383.[28] K. Papanikolopoulou, S. Teixeria, H. Belrhali, V.T. Forsyth, A. Mitraki, M.J. van Raaij,
J. Mol. Biol. 342 (2004) 219.[29] M.J. van Raaj, A. Mitraki, G. Lavigne, S. Cusack, Nature 401 (1999) 935.[30] M. Luckey, J.-F. Hernandez, G. Arlaud, V.T. Forsyth, R.W.H. Ruigrok, A. Mitraki, FEBS
Lett. 468 (2000) 23.[31] E. Kasotakis, E. Mossou, L. Adler-Abramovich, E.P. Mitchell, V.T. Forsyth, E. Gazit, A.
Mitraki, Biopolymers 92 (2009) 164.[32] M. Lepère, A.H. Muenter, C. Chevallard, P. Guenoun, G. Brezesinski, Colloids Surf. A
Physicochem. Eng. Aspects 303 (2007) 73.[33] M. Lepère, C. Chevallard, G. Brezesinski, M. Goldmann, P. Guenoun, Angew. Chem.
Int. Ed. 48 (2009) 5005.[34] G. Thakur, M. Micic, R.M. Leblanc, Colloids Surf. B Biointerfaces 74 (2009) 436.[35] V. Knecht, J. Phys. Chem. B 112 (31) (2008) 9476.[36] E. Neria, S. Fischer, M. Karplus, J. Chem. Phys. 105 (1996) 1902.[37] U. Haberthür, A. Ca"isch, J. Comput. Chem. 29 (2008) 701.[38] J.P. Ryckaert, G. Ciccotti, H.J.C. Berendsen, J. Comput. Phys. 23 (1997) 327.[39] B.R. Brooks, C.L. Brooks III, A.D. Mackerell Jr., L. Nilsson, R.J. Petrella, B. Roux, Y.
Won, G. Archontis, C. Bartels, S. Boresch, A. Ca"isch, L. Caves, Q. Cui, A.R. Dinner, M.Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V.Ovchinnikov, E. Paci, R.W. Pastor, C.B. Post, J.Z. Pu, M. Schaefer, B. Tidor, R.M.Venable, H.L. Woodcock, X. Wu, W. Yang, D.M. York, M. Karplus, J. Comput. Chem.30 (2009) 1545.
[40] R. Swendsen, J. Wang, Phys. Rev. Lett. 57 (1986) 2607.[41] U. Hansmann, Chem. Phys. Lett. 281 (1997) 140.[42] Y. Sugita, Y. Okamoto, Chem. Phys. Lett. 314 (1999) 141.[43] H. Nymeyer, S. Gnanakaran, A.E. Garcia, Meth. Enzymol. 30 (2004) 119.[44] K. Hukushima, K. Nemoto, J. Phys. Soc. Jpn 65 (1996) 1604.[45] K.Y. Sanbonmatsu, A.E. Garcia, Proteins 46 (2002) 225.[46] D. Frishman, P. Argos, Proteins 23 (1995) 566.[47] S. Chandrasekhar, Liquid Crystals, Cambridge University Press, Cambridge,
England, 1992.[48] P.G. de Gennes, J. Prost, The Physics of Liquid Crystals, 2nd ed.Oxford University
Press, Oxford, 1993.[49] M.P. Allen, D.J. Tildesley, Computer Simulation of Liquids, Oxford Science, Oxford,
UK, 1987.[50] T.P. Doerr, D. Heman, H. Mathur, P.L. Taylor, Europhys. Lett. 59 (2002) 398.[51] M. Seeber, M. Cecchini, F. Rao, G. Settanni, A. Ca"isch, Bioinformatics 23 (2007)
2625.[52] M. Lepère, C. Chevallard, J.F. Hernandez, A. Mitraki, P. Guenoun, Langmuir 23
(2007) 8150.
722 P. Tamamis, G. Archontis / Journal of Non-Crystalline Solids 357 (2011) 717–722




















