
Infusion of Platelet-derived Growth Factor or Basic Fibroblast ...
Upload
independentCategory
view
3download
0
Advanced Glycation End Products Enhance Expression ofPro-apoptotic Genes and Stimulate Fibroblast Apoptosis throughCytoplasmic and Mitochondrial Pathways*
Received for publication, June 7, 2004, and in revised form, November 29, 2004Published, JBC Papers in Press, December 6, 2004, DOI 10.1074/jbc.M406313200
Zoubin Alikhani, Mani Alikhani, Coy M. Boyd, Kiyoko Nagao, Philip C. Trackman,and Dana T. Graves‡
From the Department of Periodontology and Oral Biology, Boston University School of Dental Medicine,Boston, Massachusetts 02118
Both aging and diabetes are characterized by the for-mation of advanced glycation end products (AGEs).Both exhibit other similarities including deficits inwound healing that are associated with higher rates offibroblast apoptosis. In order to investigate a potentialmechanism for enhanced fibroblast apoptosis in diabe-tes and aged individuals, experiments were carried outto determine whether the predominant advanced glyca-tion end product in skin, N-�-(carboxymethyl) lysine(CML)-collagen, could induce fibroblast apoptosis. Invivo experiments established that CML-collagen but notunmodified collagen induced fibroblast apoptosis andthat apoptosis was dependent upon caspase-3, -8, and -9activity. In vitro experiments demonstrated that CML-collagen but not control collagen induced a time- anddose-dependent increase in fibroblast apoptosis. By useof blocking antibodies, apoptosis was shown to be medi-ated through receptor for AGE signaling. AGE-inducedapoptosis was largely dependent on the effectorcaspase, caspase-3, which was activated through bothcytoplasmic (caspase-8-dependent) and mitochondrial(caspase-9) pathways. CML-collagen had a global effectof enhancing mRNA levels of pro-apoptotic genes thatincluded several classes of molecules including ligands,receptors, adaptor molecules, mitochondrial proteins,and others. However, the pattern of expression was notidentical to the pattern of apoptotic genes induced bytumor necrosis factor �.
Advanced glycation end products (AGEs)1 result from non-enzymatic reactions of carbohydrates and oxidized lipids withproteins (1, 2). Lysine side chains condense with aldehyde orketone structures to form a reversible Schiff’s base. This struc-
ture isomerizes to a more reactive ketoamine (Amadori prod-uct), which then may undergo additional rearrangement, de-hydration, condensation, and oxidation reactions. Thesestructures can result in protein cross-links and in a variety ofprotein modifications that collectively are known as advancedglycation end products (3). N-�-(Carboxymethyl) lysine (CML)structure is a prevalent AGE and is generated principally byoxidative cleavage of Amadori intermediates (3).
AGEs form in tissues and accumulate during aging. AGEformation occurs throughout life, and AGEs are found at sig-nificantly higher levels in aged individuals (4, 5). Their forma-tion is considered to be one of the major etiologic factors in thepathophysiology of aging (6–8). AGEs have also been studiedin relation to development of many age-related chronic diseasesincluding cataract formation, neurodegenerative disorderssuch as Alzheimer disease, osteoarthritis and changes observedin myocardial dysfunction (9–14).
AGE formation is enhanced by hyperglycemia and has beenlinked to many of the complications of diabetes. By functionallyblocking AGE activity or by applying AGEs in vivo, it has beenshown that AGEs contribute to several pathologic processes.These include diabetes-associated cataract formation, nephrop-athy, retinopathy, neuropathy, and periodontal disease (15–20). AGEs also impair osseous wound healing (21).
Originally it was thought that most of the effects of AGEswere the result of cross-linking of matrix and other proteins (3).Although cross-linking is physiologically important, glycationcan alter proteins so that they acquire signaling properties,which they do not otherwise possess. Several AGE cell surfacereceptors/binding proteins have been identified. The most thor-oughly studied is receptor for advanced glycation end products(RAGE). RAGE is a multi-ligand member of the immunoglob-ulin superfamily of cell surface receptors that binds manyligands including those of the S100/calgranulin family (22).Other receptors/binding proteins include macrophage scav-enger receptor types I and II, oligosaccharyl transferase-48(AGE-R1), 80K-H phosphoprotein (AGE-R2), and galectin-3(AGE-R3). These are expressed on a wide range of cells includ-ing smooth muscle cells, monocytes, macrophages, endothelialcells, podocytes, astrocytes, microglia, and fibroblasts. Most ofthe biologic activities associated with AGEs have been shown tobe transduced by RAGE, whereas the scavenger receptors arethought to regulate removal of AGEs (23, 24). AGEs can mod-ulate inflammatory events by stimulating production of reac-tive oxygen species, chemotaxis, activation of monocytes/macrophages, and stimulation of interleukin-1 and TNFproduction (25–30).
It is well known that AGEs form in the skin as part of theaging process and as a result of diabetes (31–33). Accumulation
* This work was supported by National Institutes of Health GrantsDE07559, DE11254, AR49920, and DE14066. The costs of publication ofthis article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence should be addressed: Dept. of Periodon-tology and Oral Biology, Boston University School of Dental Medicine,700 Albany St., Boston, MA 02118. Tel.: 617-638-8547; Fax: 617-638-4924; E-mail: [email protected].
1 The abbreviations used are: AGE, advanced glycation end product;RAGE, receptor for advanced glycation end product; CML, N-�-(car-boxymethyl) lysine; TNF, tumor necrosis factor; PBS, phosphate-buff-ered saline; TUNEL, terminal deoxynucleotidyl transferase-mediatednick end labeling; AFC, 7-amino-4-trifluoromethyl coumarin; ELISA,enzyme-linked immunosorbent assay; EMSA, electrophoretic mobilityshift analysis; RT, reverse transcription; NF-�B, nuclear factor �B; Z,benzyloxycarbonyl; fmk, fluoromethyl ketone; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 280, No. 13, Issue of April 1, pp. 12087–12095, 2005© 2005 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org 12087
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from
of AGEs in skin has been related to tissue stiffening and lack ofelasticity during aging (5, 31, 32). However, AGEs in connec-tive tissue can have detrimental effects through cell signaling.For example, RAGE activation in dermal fibroblasts reducescollagen synthesis and matrix production (34). AGEs also con-tribute to impaired diabetic wound healing, in part throughinterfering with formation of an extracellular matrix (35).
One mechanism through which AGEs may affect pathologicprocesses is by enhanced apoptosis as supported by in vitrostudies. AGEs are pro-apoptotic for cultured retinal pericytes,corneal endothelial cells, neuronal cells, and renal mesangialcells (36–38). The mechanisms by which AGEs lead to apo-ptosis are not well understood. There are reports suggestingthat AGEs may enhance apoptosis indirectly through increas-ing oxidative stress or via induced expression of pro-apoptoticcytokines (36, 38, 39). Interestingly, enhanced apoptosis ofthese cells is also associated with diabetic complications suchas retinopathy, neuropathy, nephropathy, and accelerated vas-culopathy (38–40). Enhanced apoptosis has been linked tomany of the detrimental effects of aging, which is also associ-ated with AGE accumulation (41). Because fibroblasts playimportant roles in the maintenance and healing of dermalconnective tissue, the accumulation of AGEs in skin may havea detrimental effect, in part, through promoting fibroblast apo-ptosis. To address this issue, we investigated the apoptoticeffect of AGEs on fibroblasts and the mechanisms throughwhich AGEs induce apoptosis in these cells.
MATERIALS AND METHODS
CML-collagen—CML-collagen was prepared by chemical modifica-tion of acid-soluble bovine skin collagen (Sigma), as described previ-ously (21, 42). Briefly, 50 mg of collagen was dissolved in 25 ml of 1 mM
HCl freshly made in sterile water and incubated at 37 °C with occa-sional mixing. Sterile PBS (25 ml) was added, followed by sodiumcyanoborohydride (1.42 g) and glyoxylic acid (0.715 g). Control collagenwas prepared at the same time, except that no glyoxylic acid was added.
All samples were then incubated at 37 °C for 24 h. AGE collagen andcontrol collagen were then exhaustively dialyzed against distilled wa-ter. In dose-response experiments where higher concentrations of CML-collagen were used, AGE and control collagen was dialyzed againstPBS. Both CML-collagen and control collagen were soluble at the con-centrations stored and tested. In total, 3–8% of lysine residues inCML-collagen were converted to CML, as determined by the trinitro-benzenesulfonic acid assay (43). The percentage of modification of col-lagen that we have generated is 10-fold less than the amount used in arecent report to assess CML binding and activation of NF-�B (42) andonly a small amount higher than that reported for the skin of aged ordiabetic individuals (31). CML-collagen was highly reactive on Westernblots with anti-CML monoclonal antibody 6D12 (Wako, Richmond, VA),whereas control collagen was not reactive. The amount of endotoxincontamination was measured by Pyrochrome Limulus Amebocyte Ly-sate assay (Associates of Cape Cod, Inc., Woods Hole, MA) and found tobe low (0.094 � 0.01 ng/mg lipopolysaccharide in control collagen and0.087 � 0.01 ng/mg lipopolysaccharide in CML-collagen).
Animals—CD1 mice were purchased from Charles River Laborato-ries, (Waltham, MA). These are outbred mice that were selected be-cause they do not exhibit strain-associated responses, as has beenreported in some cases where a particular strain of mouse has beenstudied (45, 46). All procedures involving mice were approved by theBoston University Medical Center Institutional Animal Care and UseCommittee. Mice were anesthetized with injection of ketamine (80mg/kg) and xylazine (10 mg/kg) in sterile PBS. CML-collagen or unmod-ified collagen was injected into the loose connective tissue adjacent tocalvarial bone at a point on the midline of the skull located between theears. Injection at this anatomic site can be reproducibly achieved. Foreach data point, there were six mice (n � 6). We undertook preliminaryexperiments to identify a dose for CML-collagen that gave a moderatenumber of apoptotic cells. On this basis, 100 �g of CML-collagen or anequal amount of unmodified collagen was injected. Mice were eutha-nized 24 h after injection. In addition to CML-collagen or unmodifiedcollagen alone, some animals were treated by intraperitoneal injectionof caspase-3, -8, or -9 inhibitor (1 mg/kg) 1 h before CML-collageninjection and locally (25 �g) at the time of CML-collagen injection. Thecaspase-3 inhibitor Z-DEVD-fmk, the caspase-8 inhibitor Z-IETD-fmk,and the caspase-9 inhibitor Z-LEHD-fmk were purchased from R&DSystems (Minneapolis, MN). Control mice received CML-collagen or
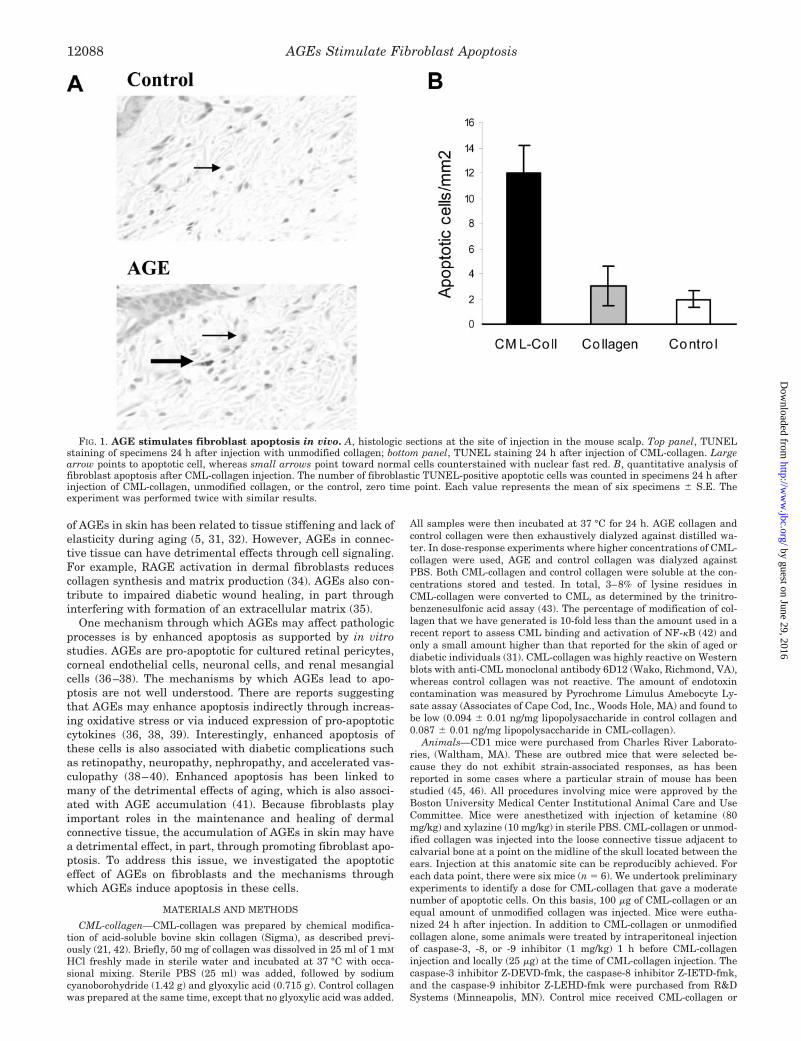
FIG. 1. AGE stimulates fibroblast apoptosis in vivo. A, histologic sections at the site of injection in the mouse scalp. Top panel, TUNELstaining of specimens 24 h after injection with unmodified collagen; bottom panel, TUNEL staining 24 h after injection of CML-collagen. Largearrow points to apoptotic cell, whereas small arrows point toward normal cells counterstained with nuclear fast red. B, quantitative analysis offibroblast apoptosis after CML-collagen injection. The number of fibroblastic TUNEL-positive apoptotic cells was counted in specimens 24 h afterinjection of CML-collagen, unmodified collagen, or the control, zero time point. Each value represents the mean of six specimens � S.E. Theexperiment was performed twice with similar results.
AGEs Stimulate Fibroblast Apoptosis12088
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from

unmodified collagen containing 2% Me2SO (Sigma-Aldrich).Preparation of Histologic Sections—Animals were euthanized by de-
capitation, and their heads were fixed for 72 h in cold 4% paraformal-dehyde and decalcified by incubation with cold Immunocal (Decal Corp.,Congers, NY) for �12 days with solution changed daily. Paraffin-em-bedded sagittal sections were prepared at a thickness of 5–6 �m.
TUNEL Assay and Quantitative Histologic Analysis—Apoptotic cellswere detected by an in situ TUNEL assay by means of a TACS 2TdT-Blue Label kit purchased from Trevigen (Gaithersburg, MD), fol-lowing the manufacturer’s instructions. Sections were counterstainedwith nuclear fast red. The number of fibroblastic apoptotic cells wasdetermined in six specimens per group by counting the number ofTUNEL-positive cells that had the characteristic microscopic appear-ance of fibroblasts. Counts and measurements were confirmed by re-analysis of all the specimens by an independent examiner. The intra-and inter-examiner variations were �10%. Student’s t test was used todetermine significant differences between the experimental and controlgroups at the p � 0.05 level.
Caspase Activity—Caspase-3, -8, and -9 activities from in vitro and invivo experiments were assayed with fluorometric kits purchased fromR&D Systems. Briefly, after sacrifice at the indicated time points,murine scalps were immediately dissected from the calvaria and frozenin liquid nitrogen. Frozen tissues were pulverized, and lysates wereprepared using cell lysis buffer provided by R&D Systems. After cen-trifugation, total protein was quantitated using a BCA protein assay kit(Pierce). Caspase-3 activity was detected by using the specific caspase-3fluorogenic substrate, DEVD peptide conjugated to 7-amino-4-triflu-oromethyl coumarin (AFC). Caspase-8 activity was detected by usingthe specific caspase-8 fluorogenic substrate, IETD-AFC. Caspase-9 ac-tivity was detected by using the specific caspase-9 fluorogenic sub-strate, LEHD-AFC. Measurements were made on a fluorescent micro-plate reader using filters for excitation (400 nm) and detection ofemitted light (505 nm). In some assays, recombinant caspase-3 enzyme(R&D Systems) was used as a positive control. Buffers without celllysate and cell lysate without substrate were used as negative controls.The same caspase-3, -8, and-9 inhibitors were used in both in vivo and
in vitro studies. In addition, the pan-caspase (general caspase) inhibitorZ-VAD-fmk was purchased from R&D Systems for in vitro assays. Foreach group, there were six specimens (n � 6).
Cell Culture—Primary human adult dermal fibroblasts were pur-chased from Cambrex (Walkersville, MD). Cells were propagated andmaintained in Dulbecco’s modified Eagle’s medium (Cambrex) supple-mented with 10% fetal bovine serum, gentamicin (100 �g/ml), andamphotericin B (100 ng/ml) at 37 °C in a humidified atmosphere of 5%CO2. Experiments with CML-collagen were performed in culture me-dium supplemented with 0.5% fetal bovine serum. Assays were per-formed when the cultures reached 75–85% confluence. In most exper-iments, 200 �g/ml CML-collagen or unmodified control collage wasused, which is equivalent to 2 �M. Apoptosis of fibroblasts was deter-mined by an ELISA technique measuring histone-associated DNA frag-ments (Roche Applied Science), following the manufacturer’s instruc-tions. In these studies, 20,000 fibroblasts/cm2 were treated with CML-collagen (200 �g/ml) or unmodified collagen (200 �g/ml) for 24 h.Apoptosis was determined by ELISA, and the cell numbers were as-sessed in corresponding wells to normalize apoptosis measurements.
For caspase activity measurements, the same technique was used asdescribed above for in vivo studies. In some cases, cells were treatedwith caspase-3, caspase-8, caspase-9, caspase-8 � caspase-9, or pan-caspase inhibitors (50 �M) at the time of CML-collagen stimulation. Thedose of each inhibitor was selected based on our previous results (47).
FIG. 2. AGE stimulates fibroblast apoptosis in vitro. A, primaryhuman adult dermal fibroblasts were incubated with CML-collagen orunmodified collagen (200 �g/ml) for 1, 3, 6, and 24 h. Apoptosis wasdetermined by ELISA. The experiment was performed twice with sim-ilar results. B, primary human adult dermal fibroblasts were incubatedwith CML-collagen or unmodified collagen (0–800 �g/ml) for 24 h, andapoptosis was measured by ELISA. Each value represents the mean offive replicates � S.E. The experiment was performed three times withsimilar results.
FIG. 3. CML-collagen-induced apoptosis is mediated throughRAGE. Primary human adult dermal fibroblasts were incubated withCML-collagen (200 �g/ml) in the presence or absence of anti-RAGEpolyclonal antibody specific for the extracellular domain of RAGE (10�g/�l) or non-immune serum (10 �g/�l) for 24 h. In some cases, cellswere incubated with antiserum or non-immune serum alone. The ex-tent of apoptosis was determined by ELISA. Each value represents themean of five replicates � S.E. The experiment was performed threetimes with similar results.
FIG. 4. Caspase activity is stimulated by CML-collagen in fi-broblasts in vitro. Caspase-3,-8, and -9 activities were measured byfluorometric assays in lysates from cultured primary human adultdermal fibroblasts incubated with CML-collagen or unmodified collagen(200 �g/ml) for 24 h. Each value represents the mean of five replicates �S.E. The experiment was performed four times with similar results.
AGEs Stimulate Fibroblast Apoptosis 12089
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from

Control cells were incubated in assay medium supplemented with ve-hicle alone (2% Me2SO) in the culture media.
Inhibition of RAGE—Antibodies to the extracellular domain ofRAGE have been shown to inhibit binding of CML modified proteins toRAGE and have been used to assess the impact of RAGE activation ofcellular events (42, 48). To study the role of RAGE, fibroblast cultureswere incubated with CML-collagen or unmodified collagen (200 �g/ml)in the presence or absence of anti-RAGE polyclonal antibodies specificfor the extracellular domain of RAGE (10 �g/�l) or non-immune serum(10 �g/�l) for 24 h (Biocompare, San Francisco, CA). As controls, cellswere incubated with 10 �g/ml RAGE antiserum or non-immune serumwithout AGE stimulation. The extent of apoptosis was determined byELISA. Statistical difference between samples was determined by one-way analysis of variance followed by Tukey’s multiple comparison tests.
EMSA—Fibroblast cultures were incubated in assay medium for 1 hwith 200 �g/ml CML-collagen or unmodified collagen in the presence orabsence of a specific NF-�B inhibitor, SN50 (100 �g/ml) (Biomol, Plym-outh Meeting, PA). Nuclear proteins were extracted using protein ex-traction kit (Pierce) following the manufacturer’s instructions. Concen-trations of nuclear proteins were measured by using BCA protein assaykit (Pierce). Interaction between NF-�B in the protein extract and DNAprobe was investigated using EMSA kit from Panomics (Redwood City,CA) following the manufacturer’s instruction.
Microarray—Fibroblast cell cultures were exposed to CML-collagen(200 �g/ml) or TNF-� (20 ng/ml) for 6 h. TNF-� was purchased fromR&D Systems. Control cells were exposed to either unmodified collagen(200 �g/ml) or vehicle alone (PBS). RNA was extracted using theRNeasy Mini Kit (Qiagen, Valencia, CA). Human Apoptosis GEArray Qseries kit that includes 96 key genes involved in apoptosis was pur-chased from SuperArray, Inc. (Bethesda, MD). Total RNA was used asthe template for 32P-labeled cDNA probe synthesis following the man-ufacturer’s instructions. After overnight hybridization with labeledprobe, the GEArray membranes were exposed using a PhosphorImager.The resulting digital image was converted to a raw data file usingScanalyze software (www.microarray.org/software.html). GEArray An-
alyzer software (SuperArray, Inc.) was used for data analysis. Themicroarray was performed twice with similar results, and the meanvalues of the two experiments are presented for each gene. The microar-ray for TNF-� stimulation was performed simultaneously with the AGEstimulation array.
RT-PCR—Five �g of total RNA was used to produce cDNA using aFirst Strand cDNA Synthesis Kit (SuperArray, Inc.). PCR was per-formed on the synthesized cDNA using the SingleGene PCR Kit pur-chased from SuperArray, Inc. following the manufacturer’s protocol.Specific primers were used for each gene, and GAPDH was used asinternal control. This kit has the advantage that GAPDH and the gene ofinterest are amplified in the same tube and for the same number of cycles.Based on a pilot study, 24 and 28 cycles were chosen to compare the geneexpression. After electrophoresis on a 2% agarose gel containing 0.5 �g/mlethidium bromide, bands were visualized on a UV-box. The density ofeach band was measured and compared using Quantity One software(Bio-Rad). The optical density of each band was normalized by the valueof the GAPDH in the same lane. Two separate RT-PCRs were performedwith similar results. Student’s t test was used to determine significantdifferences between the experimental and control groups.
RESULTS
In Vivo Induction of Apoptosis by CML-collagen—To studythe effect of AGEs on fibroblast apoptosis in vivo, 100 �g ofCML-collagen or control collagen was injected in the scalp ofCD1 mice. Histologic sections were examined for fibroblastapoptosis by their characteristic appearance in the TUNELassay (Fig. 1). Fibroblast apoptosis could be detected in theCML-collagen-treated group. In comparison, there was virtu-ally no induction of fibroblast apoptosis in mice treated withunmodified collagen compared with the zero time point (Fig.1A). Quantitative analysis indicated that CML-collagen stim-ulated a 4-fold increase in apoptotic cells compared with un-
FIG. 5. Caspase inhibitors preventAGE-stimulated fibroblast apoptosisand caspase-3 activity in vitro. Pri-mary human adult dermal fibroblastswere incubated with unmodified or CML-collagen (200 �g/ml) with or withoutcaspase inhibitors (50 �M) for 24 h as de-scribed under “Materials and Methods.”A, the extent of apoptosis was determinedby ELISA and performed twice with sim-ilar results. Each value represents themean of five replicates � S.E. B,caspase-3 activity was measured using afluorometric assay in lysates from cell cul-tures obtained after 24 h of stimulationwith CML-collagen (200 �g/ml) with orwithout caspase-8 or -9 inhibitors as de-scribed under “Materials and Methods.”The experiment was performed threetimes with similar results. Each valuerepresents the mean of five replicates �S.E.
AGEs Stimulate Fibroblast Apoptosis12090
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from
modified collagen (Fig. 1B) (p � 0.05). When expressed as thepercentage of TUNEL-positive fibroblasts, the levels were0.85 � 0.05 in the CML-collagen-inoculated group and 0.21 �0.01 (p � 0.05) for control collagen-treated group.
In Vitro Induction of Apoptosis by CML-collagen—In vitrostudies were carried out to measure the direct effect of AGEs onfibroblast apoptosis under well-defined conditions. Apoptosis ofhuman adult primary skin fibroblasts was measured by therelative amount of histone-associated DNA fragments in thecytoplasm as determined by ELISA. A time course experimentdetermined that AGE-induced apoptosis was not detected be-fore 6 h (Fig. 2A). At the 6 h time point, a 1.7-fold increase wasnoted, whereas at 24 h, a 3-fold increase in apoptosis wasdetected. The increase at 6 and 24 h was statistically signifi-cant (p � 0.05). CML-collagen induced a dose-dependent in-crease in fibroblast apoptosis, whereas control collagen did not(Fig. 2B). At 200 �g/ml, CML-collagen compared with unmod-ified collagen induced a 3-fold increase in apoptosis, which wasstatistically significant (p � 0.05). Saturating levels werereached at 400 �g/ml CML-collagen with a 5-fold increase inapoptosis (p � 0.05). In subsequent studies, a concentration of200 �g/ml CML-collagen or control collagen and a time point of24 h were used unless stated otherwise.
Role of RAGE in Induction of Apoptosis by CML-collagen—To determine the significance of RAGE activation in the apop-totic response to AGEs, human adult dermal fibroblasts wereincubated with CML-collagen (200 �g/ml) in the presence orabsence of anti-RAGE polyclonal antibodies specific for theextracellular domain of RAGE (10 �g/ml) or non-immune se-rum (10 �g/ml) for 24 h (Fig. 3). CML-collagen stimulated asignificant increase in fibroblast apoptosis (p � 0.05). However,anti-RAGE antibody was able to completely block the apoptoticeffects of AGE so that there was no difference between fibro-blasts incubated with control collagen and cells incubated withCML-collagen plus RAGE antibody. Matched pre-immune se-rum had no effect on CML-collagen-induced apoptosis, demon-strating specificity of the antibody. As additional controls, cellsincubated with RAGE antibody or pre-immune serum in theabsence of CML-collagen had no direct effect on apoptosis. Theresults obtained are consistent with functional studies in whichRAGE antibody inhibited activation of cells by CML-albumin(data not shown).
In Vitro Effects of CML-collagen on Activation of Caspases-3,-8, and -9—To further investigate mechanisms of AGE-inducedfibroblast apoptosis, the activation of initiator and executionercaspases was measured in adult human fibroblasts after stim-ulation by CML-collagen or unmodified collagen (Fig. 4). CML-collagen stimulated a 5-fold increase in caspase-3, a 4.3-foldincrease in caspase-8, and a 3.2-fold increase in caspase-9activity compared with cells treated with unmodified collagenalone. Each was statistically significant (p � 0.05).
Effects of Different Caspase Inhibitors on Apoptosis inVitro—To investigate the effect of caspase-3, caspase-8, andcaspase-9 activity on AGE-induced apoptosis in human dermalfibroblasts, cells were incubated with a pan-caspase andcaspase-3, -8, or -9 inhibitors at the time of CML-collagenstimulation (Fig. 5A). The concentration of inhibitors used was50 �M, based on previous studies that demonstrated that thisconcentration inhibits virtually all activity in a highly specificmanner (49–52). The pan-caspase inhibitor reduced apoptosiscompared with CML-collagen alone by 92%. Caspase-3 inhibi-tor reduced the apoptotic effect of CML-collagen by 85%, a levelsimilar to that of the pan-caspase inhibitor. Caspase-8 inhibi-tor alone and caspase-9 inhibitor alone significantly reducedapoptosis by 65% and 32%, respectively (p � 0.05). However,when caspase-8 and -9 inhibitors were combined, the result
was similar to that of the caspase-3 inhibitor (p � 0.05). Takentogether, these results suggest that AGEs function primarilythrough caspase-3, whose induction can be accounted for byactivation from both caspases-8 and-9. The latter is demon-strated by (p � 0.05), establishing that caspase-3 activity issignificantly reduced in the presence of caspase-8 and -9 inhib-itors (p � 0.05) (Fig. 5B).
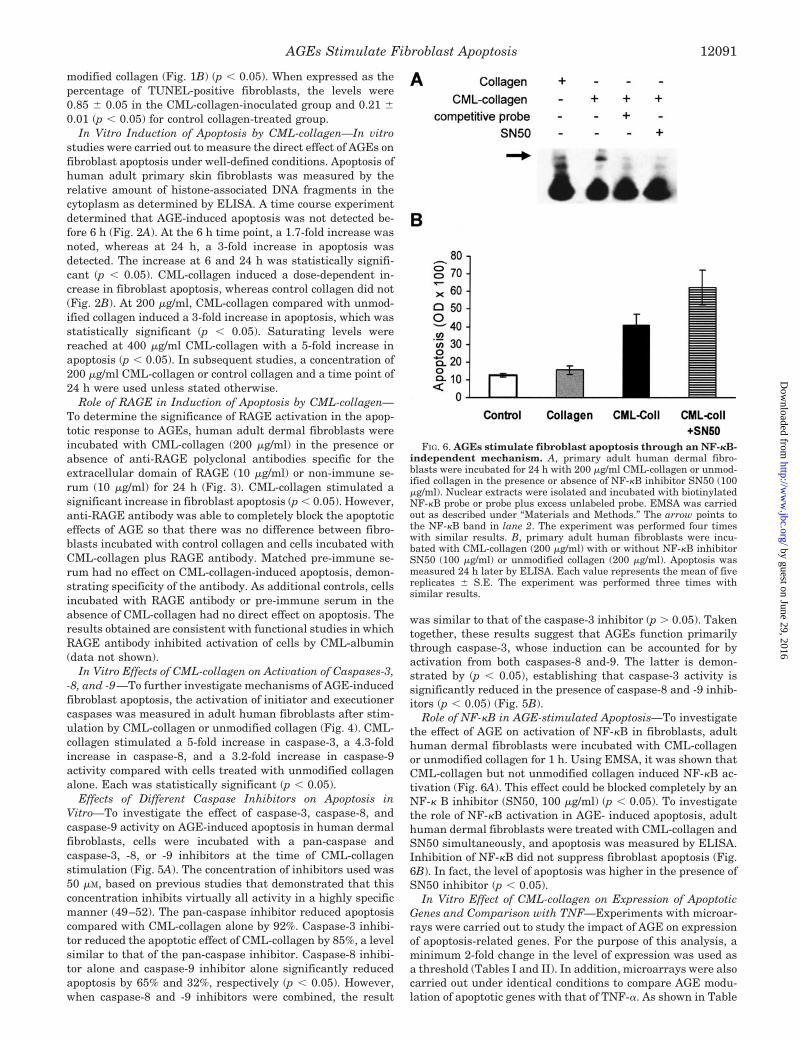
Role of NF-�B in AGE-stimulated Apoptosis—To investigatethe effect of AGE on activation of NF-�B in fibroblasts, adulthuman dermal fibroblasts were incubated with CML-collagenor unmodified collagen for 1 h. Using EMSA, it was shown thatCML-collagen but not unmodified collagen induced NF-�B ac-tivation (Fig. 6A). This effect could be blocked completely by anNF-� B inhibitor (SN50, 100 �g/ml) (p � 0.05). To investigatethe role of NF-�B activation in AGE- induced apoptosis, adulthuman dermal fibroblasts were treated with CML-collagen andSN50 simultaneously, and apoptosis was measured by ELISA.Inhibition of NF-�B did not suppress fibroblast apoptosis (Fig.6B). In fact, the level of apoptosis was higher in the presence ofSN50 inhibitor (p � 0.05).
In Vitro Effect of CML-collagen on Expression of ApoptoticGenes and Comparison with TNF—Experiments with microar-rays were carried out to study the impact of AGE on expressionof apoptosis-related genes. For the purpose of this analysis, aminimum 2-fold change in the level of expression was used asa threshold (Tables I and II). In addition, microarrays were alsocarried out under identical conditions to compare AGE modu-lation of apoptotic genes with that of TNF-�. As shown in Table
FIG. 6. AGEs stimulate fibroblast apoptosis through an NF-�B-independent mechanism. A, primary adult human dermal fibro-blasts were incubated for 24 h with 200 �g/ml CML-collagen or unmod-ified collagen in the presence or absence of NF-�B inhibitor SN50 (100�g/ml). Nuclear extracts were isolated and incubated with biotinylatedNF-�B probe or probe plus excess unlabeled probe. EMSA was carriedout as described under “Materials and Methods.” The arrow points tothe NF-�B band in lane 2. The experiment was performed four timeswith similar results. B, primary adult human fibroblasts were incu-bated with CML-collagen (200 �g/ml) with or without NF-�B inhibitorSN50 (100 �g/ml) or unmodified collagen (200 �g/ml). Apoptosis wasmeasured 24 h later by ELISA. Each value represents the mean of fivereplicates � S.E. The experiment was performed three times withsimilar results.
AGEs Stimulate Fibroblast Apoptosis 12091
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from
I, CML-collagen compared with unmodified collagen induced a2–5-fold increase in the expression of six members of the TNFligand superfamily (LT-�, TNF-�, OX40L, TNFSF9, APRIL,and CD30L). The same six ligands had enhanced mRNA levelsstimulated by TNF. Another six TNF superfamily ligands
(CD27L, CD40L, Lt�, TRAIL, TNFSF12, and FasL) were in-duced more than 2-fold by TNF-� stimulation, but not by AGE.From the TNF receptor family of the genes, Fas, CD27, LT�R,CD40, and TNFRSF9 mRNA levels increased 2–7-fold in re-sponse to both AGE and TNF-�. However, other members ofthis family exhibited increased expression by TNF-� but notAGE stimulation (TRAIL-R3, DR3, TNFRSF14, TNFR1,TNFR2, and OX40). Of the mitochondrial proteins that regu-late apoptosis, AGE and TNF both increased Bax, Blf-1, andNip-3, which are pro-apoptotic members of the Bcl-2 family,and MCL-1, which is an anti-apoptotic member of the Bcl-2family. In addition, TNF-� enhanced the pro-apoptotic proteinsBlk, Bak, and Bad, whereas AGE had no significant effect onexpression of these genes. The caspase family showed signifi-cant increases in expression after both stimuli. Initiatorcaspases-2, -8, and -9 and effector caspases -3, -6, and -7showed a 2–3-fold increase by both AGE and TNF. From theadaptor family, TRAF2, CASPER, TRAF1, RIP, I-TRAF, andFADD increased between 2- and 8-fold in response to bothstimuli. The genes of six adaptor molecules (Flash, Apaf1,DAP-kinase, myD88, CRADD, and TRAF6) increased morethan 2-fold in response to TNF but not to AGE. P53, a cell cycle
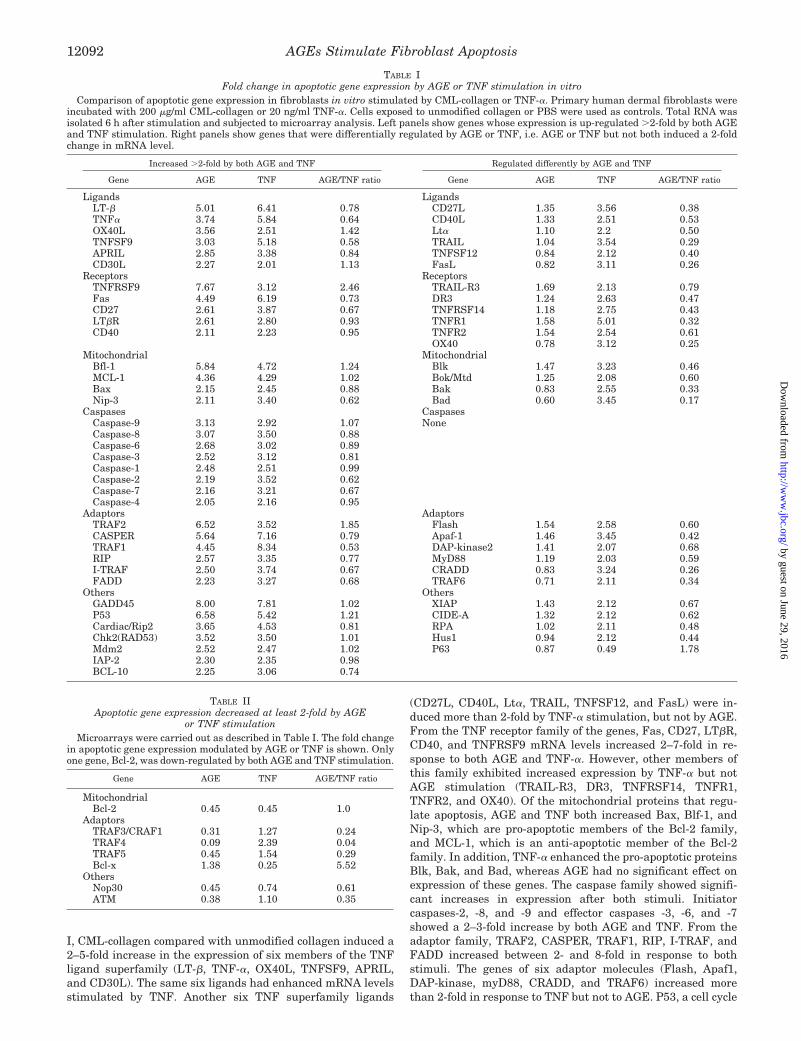
TABLE IFold change in apoptotic gene expression by AGE or TNF stimulation in vitro
Comparison of apoptotic gene expression in fibroblasts in vitro stimulated by CML-collagen or TNF-�. Primary human dermal fibroblasts wereincubated with 200 �g/ml CML-collagen or 20 ng/ml TNF-�. Cells exposed to unmodified collagen or PBS were used as controls. Total RNA wasisolated 6 h after stimulation and subjected to microarray analysis. Left panels show genes whose expression is up-regulated �2-fold by both AGEand TNF stimulation. Right panels show genes that were differentially regulated by AGE or TNF, i.e. AGE or TNF but not both induced a 2-foldchange in mRNA level.
Increased �2-fold by both AGE and TNF Regulated differently by AGE and TNF
Gene AGE TNF AGE/TNF ratio Gene AGE TNF AGE/TNF ratio
Ligands LigandsLT-� 5.01 6.41 0.78 CD27L 1.35 3.56 0.38TNF� 3.74 5.84 0.64 CD40L 1.33 2.51 0.53OX40L 3.56 2.51 1.42 Lt� 1.10 2.2 0.50TNFSF9 3.03 5.18 0.58 TRAIL 1.04 3.54 0.29APRIL 2.85 3.38 0.84 TNFSF12 0.84 2.12 0.40CD30L 2.27 2.01 1.13 FasL 0.82 3.11 0.26
Receptors ReceptorsTNFRSF9 7.67 3.12 2.46 TRAIL-R3 1.69 2.13 0.79Fas 4.49 6.19 0.73 DR3 1.24 2.63 0.47CD27 2.61 3.87 0.67 TNFRSF14 1.18 2.75 0.43LT�R 2.61 2.80 0.93 TNFR1 1.58 5.01 0.32CD40 2.11 2.23 0.95 TNFR2 1.54 2.54 0.61
OX40 0.78 3.12 0.25Mitochondrial Mitochondrial
Bfl-1 5.84 4.72 1.24 Blk 1.47 3.23 0.46MCL-1 4.36 4.29 1.02 Bok/Mtd 1.25 2.08 0.60Bax 2.15 2.45 0.88 Bak 0.83 2.55 0.33Nip-3 2.11 3.40 0.62 Bad 0.60 3.45 0.17
Caspases CaspasesCaspase-9 3.13 2.92 1.07 NoneCaspase-8 3.07 3.50 0.88Caspase-6 2.68 3.02 0.89Caspase-3 2.52 3.12 0.81Caspase-1 2.48 2.51 0.99Caspase-2 2.19 3.52 0.62Caspase-7 2.16 3.21 0.67Caspase-4 2.05 2.16 0.95
Adaptors AdaptorsTRAF2 6.52 3.52 1.85 Flash 1.54 2.58 0.60CASPER 5.64 7.16 0.79 Apaf-1 1.46 3.45 0.42TRAF1 4.45 8.34 0.53 DAP-kinase2 1.41 2.07 0.68RIP 2.57 3.35 0.77 MyD88 1.19 2.03 0.59I-TRAF 2.50 3.74 0.67 CRADD 0.83 3.24 0.26FADD 2.23 3.27 0.68 TRAF6 0.71 2.11 0.34
Others OthersGADD45 8.00 7.81 1.02 XIAP 1.43 2.12 0.67P53 6.58 5.42 1.21 CIDE-A 1.32 2.12 0.62Cardiac/Rip2 3.65 4.53 0.81 RPA 1.02 2.11 0.48Chk2(RAD53) 3.52 3.50 1.01 Hus1 0.94 2.12 0.44Mdm2 2.52 2.47 1.02 P63 0.87 0.49 1.78IAP-2 2.30 2.35 0.98BCL-10 2.25 3.06 0.74
TABLE IIApoptotic gene expression decreased at least 2-fold by AGE
or TNF stimulationMicroarrays were carried out as described in Table I. The fold change
in apoptotic gene expression modulated by AGE or TNF is shown. Onlyone gene, Bcl-2, was down-regulated by both AGE and TNF stimulation.
Gene AGE TNF AGE/TNF ratio
MitochondrialBcl-2 0.45 0.45 1.0
AdaptorsTRAF3/CRAF1 0.31 1.27 0.24TRAF4 0.09 2.39 0.04TRAF5 0.45 1.54 0.29Bcl-x 1.38 0.25 5.52
OthersNop30 0.45 0.74 0.61ATM 0.38 1.10 0.35
AGEs Stimulate Fibroblast Apoptosis12092
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from

regulator that is strongly pro-apoptotic, increased almost 6-foldin both groups.
In Table II, molecules down-regulated more than 2-fold byAGE and/or TNF stimulation are shown. Expression of theanti-apoptotic gene Bcl-2 was reduced more than 2-fold by bothAGE and TNF. The pro-apoptotic adaptor molecule TRAF4mRNA levels decreased almost 10-fold in response to AGEwhile increasing 2-fold after TNF stimulation. Adaptor mole-cules TRAF3 and TRAF5 decreased in response to AGE but didnot change more than 2-fold after TNF-� stimulation. ThemRNA level of the anti-apoptotic gene Bcl-x was diminished4-fold by TNF but not by AGE. Two other pro-apoptotic genes,Nop30 and ATM, were down-regulated by AGE but not by TNF.A list of other apoptotic genes whose expression was not en-hanced or diminished more than 2-fold is shown in Table III.
To confirm the results of the microarray, RT-PCR usingspecific primers for five selected genes of interest was exam-ined (Fig. 7). Amplification was performed over 24 and 28
cycles to establish that differences obtained were not anartifact of a particular number of amplifications. Densitomet-ric analysis of the resulting bands indicates that the RT-PCRresults confirmed the data obtained for the microarray verywell (Table IV). In every case, the RT-PCR results exhibitedthe same 2-fold change or lack of 2-fold change in cells stim-ulated by CML-collagen or TNF-� compared with the respec-tive control.
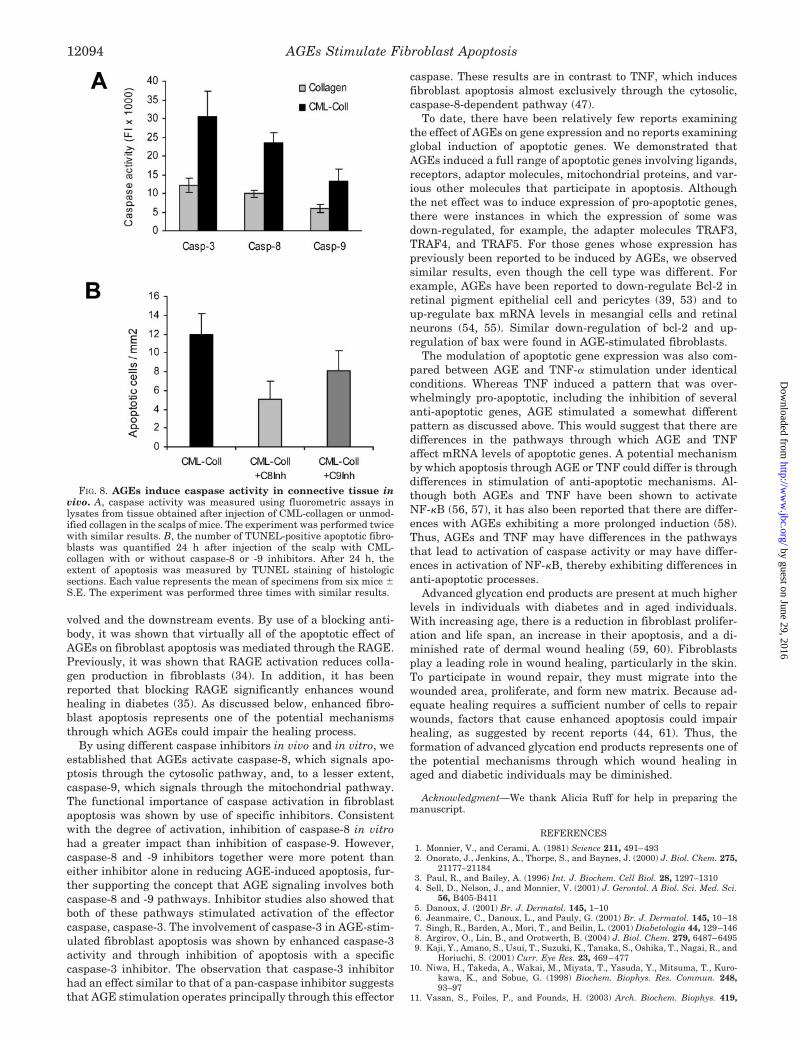
In Vivo Effects of CML-collagen on Activation of DifferentCaspases—To determine whether AGEs also induce caspaseactivity in vivo, experiments were performed in which CML-collagen and unmodified collagen were injected into the scalp,and the activation of caspases was measured (Fig. 8A). CML-collagen (100 �g) increased caspase-3 activity 2.5-fold, in-creased caspase-8 activity 2.4-fold, and increased caspase-9activity 2.2-fold compared with unmodified collagen (p � 0.05).
To study the functional role of caspase-8 and -9 in AGE-induced apoptosis, mice received injection with CML-collagenwith or without specific caspase inhibitors (Fig. 8B). Caspase-8inhibitor decreased AGE-induced fibroblast apoptosis in vivo77%, whereas caspase-9 inhibitor decreased it by 43% (p �0.05). These results agree well with in vitro data.
DISCUSSION
To study the apoptotic effect of AGE stimulation on fibro-blasts and also to investigate mechanisms through whichAGEs induce apoptosis in these cells, CML-collagen was in-jected subcutaneously in the scalps of mice. The direct effects ofAGE stimulation on fibroblasts were examined in culture. Theresults establish that AGEs significantly induce apoptosis infibroblasts in vivo and in vitro and that it is largely dependentupon caspase-8 and -9 activation of caspase-3. To our knowl-edge, these studies are the first to characterize the caspasepathways through which AGEs induce apoptosis in vitro and invivo. Moreover, AGE stimulation has a predominant effect ofenhancing expression of pro-apoptotic genes.
In order to identify the mechanisms through which AGE-induced apoptosis was signaled, we identified the receptor in-
FIG. 7. Comparison of AGE- and TNF-stimulated expression ofapoptotic genes in vitro as measured by RT-PCR. Expression ofselected apoptotic genes was measured by RT-PCR of total RNA ex-tracted from fibroblast cultures stimulated with CML-collagen or un-modified collagen (200 �g/ml) for 6 h. Other cells were stimulated withTNF (20 ng/ml) or PBS alone, and the extracted RNA was used for thesame purpose. RT-PCR was carried out using primer pairs from Super-Array in which an internal standard, GAPDH, is run for the samenumber of cycles as the gene of interest. The amplicon of each gene isshown after 24 and 28 cycles of PCR. The experiment was performedtwice with similar results.
TABLE IIIApoptotic gene expression not increased or decreased �2-fold by both AGE and TNF stimulation
Microarrays were carried out as described in Table I. Genes that did not exhibit a �2-fold change in expression by both AGE and TNF stimulationare shown. Each value presented is the fold induction compared to unmodified protein (for AGE) or buffer alone (for TNF) and represents the meanof two separate microarrays, which gave virtually identical results.
TNF ligand family TNF receptor family Bcl-2 family Caspases Adaptor Other
TNFSF14 TRAIL-R, Trail receptor(DR5), TRAIL-R4,and CD30
Biml, HRK, Bcl-w,and Bik
Caspase-10, caspase-5,caspase-14, andcaspase-13
Bar and Trip CIDE-B, DFF40, DFFA, Chk1,IAP-1, Nod/CARD4, Apollon/Bruce, survivin, and NAIP/BIRC1
TABLE IVApoptotic gene expression modulated by CML-collagen or TNF-� in
fibroblasts in vitro, as measured by RT-PCRPrimary human dermal fibroblasts were incubated with 200 �g/ml
CML-collagen or modified collagen or 20 ng/ml TNF-� or vehicle alone(sterile PBS). Total RNA was isolated 6 h after stimulation and sub-jected to RT-PCR as described under “Materials and Methods.” Eachlane represents the gene of interest and an internal standard, GAPDH,amplified for 24 or 28 cycles. Each value represents the mean foldincrease of each experimental group compared with the respectivecontrol obtained in two separate experiments.
GeneCML-collagen/control TNF/control
24 cycles 28 cycles 24 cycles 28 cycles
P53 3.43 3.72 3.15 3.33TNF SF 9 2.8 3.06 2.8 2.7Bax 2.23 2.02 2.24 2.13TRAIL 1.5 1.01 2.58 2.74Bcl-2 0.43 0.49 0.42 0.44
AGEs Stimulate Fibroblast Apoptosis 12093
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from
volved and the downstream events. By use of a blocking anti-body, it was shown that virtually all of the apoptotic effect ofAGEs on fibroblast apoptosis was mediated through the RAGE.Previously, it was shown that RAGE activation reduces colla-gen production in fibroblasts (34). In addition, it has beenreported that blocking RAGE significantly enhances woundhealing in diabetes (35). As discussed below, enhanced fibro-blast apoptosis represents one of the potential mechanismsthrough which AGEs could impair the healing process.
By using different caspase inhibitors in vivo and in vitro, weestablished that AGEs activate caspase-8, which signals apo-ptosis through the cytosolic pathway, and, to a lesser extent,caspase-9, which signals through the mitochondrial pathway.The functional importance of caspase activation in fibroblastapoptosis was shown by use of specific inhibitors. Consistentwith the degree of activation, inhibition of caspase-8 in vitrohad a greater impact than inhibition of caspase-9. However,caspase-8 and -9 inhibitors together were more potent thaneither inhibitor alone in reducing AGE-induced apoptosis, fur-ther supporting the concept that AGE signaling involves bothcaspase-8 and -9 pathways. Inhibitor studies also showed thatboth of these pathways stimulated activation of the effectorcaspase, caspase-3. The involvement of caspase-3 in AGE-stim-ulated fibroblast apoptosis was shown by enhanced caspase-3activity and through inhibition of apoptosis with a specificcaspase-3 inhibitor. The observation that caspase-3 inhibitorhad an effect similar to that of a pan-caspase inhibitor suggeststhat AGE stimulation operates principally through this effector
caspase. These results are in contrast to TNF, which inducesfibroblast apoptosis almost exclusively through the cytosolic,caspase-8-dependent pathway (47).
To date, there have been relatively few reports examiningthe effect of AGEs on gene expression and no reports examiningglobal induction of apoptotic genes. We demonstrated thatAGEs induced a full range of apoptotic genes involving ligands,receptors, adaptor molecules, mitochondrial proteins, and var-ious other molecules that participate in apoptosis. Althoughthe net effect was to induce expression of pro-apoptotic genes,there were instances in which the expression of some wasdown-regulated, for example, the adapter molecules TRAF3,TRAF4, and TRAF5. For those genes whose expression haspreviously been reported to be induced by AGEs, we observedsimilar results, even though the cell type was different. Forexample, AGEs have been reported to down-regulate Bcl-2 inretinal pigment epithelial cell and pericytes (39, 53) and toup-regulate bax mRNA levels in mesangial cells and retinalneurons (54, 55). Similar down-regulation of bcl-2 and up-regulation of bax were found in AGE-stimulated fibroblasts.
The modulation of apoptotic gene expression was also com-pared between AGE and TNF-� stimulation under identicalconditions. Whereas TNF induced a pattern that was over-whelmingly pro-apoptotic, including the inhibition of severalanti-apoptotic genes, AGE stimulated a somewhat differentpattern as discussed above. This would suggest that there aredifferences in the pathways through which AGE and TNFaffect mRNA levels of apoptotic genes. A potential mechanismby which apoptosis through AGE or TNF could differ is throughdifferences in stimulation of anti-apoptotic mechanisms. Al-though both AGEs and TNF have been shown to activateNF-�B (56, 57), it has also been reported that there are differ-ences with AGEs exhibiting a more prolonged induction (58).Thus, AGEs and TNF may have differences in the pathwaysthat lead to activation of caspase activity or may have differ-ences in activation of NF-�B, thereby exhibiting differences inanti-apoptotic processes.
Advanced glycation end products are present at much higherlevels in individuals with diabetes and in aged individuals.With increasing age, there is a reduction in fibroblast prolifer-ation and life span, an increase in their apoptosis, and a di-minished rate of dermal wound healing (59, 60). Fibroblastsplay a leading role in wound healing, particularly in the skin.To participate in wound repair, they must migrate into thewounded area, proliferate, and form new matrix. Because ad-equate healing requires a sufficient number of cells to repairwounds, factors that cause enhanced apoptosis could impairhealing, as suggested by recent reports (44, 61). Thus, theformation of advanced glycation end products represents one ofthe potential mechanisms through which wound healing inaged and diabetic individuals may be diminished.
Acknowledgment—We thank Alicia Ruff for help in preparing themanuscript.
REFERENCES
1. Monnier, V., and Cerami, A. (1981) Science 211, 491–4932. Onorato, J., Jenkins, A., Thorpe, S., and Baynes, J. (2000) J. Biol. Chem. 275,
21177–211843. Paul, R., and Bailey, A. (1996) Int. J. Biochem. Cell Biol. 28, 1297–13104. Sell, D., Nelson, J., and Monnier, V. (2001) J. Gerontol. A Biol. Sci. Med. Sci.
56, B405-B4115. Danoux, J. (2001) Br. J. Dermatol. 145, 1–106. Jeanmaire, C., Danoux, L., and Pauly, G. (2001) Br. J. Dermatol. 145, 10–187. Singh, R., Barden, A., Mori, T., and Beilin, L. (2001) Diabetologia 44, 129–1468. Argirov, O., Lin, B., and Orotwerth, B. (2004) J. Biol. Chem. 279, 6487–64959. Kaji, Y., Amano, S., Usui, T., Suzuki, K., Tanaka, S., Oshika, T., Nagai, R., and
Horiuchi, S. (2001) Curr. Eye Res. 23, 469–47710. Niwa, H., Takeda, A., Wakai, M., Miyata, T., Yasuda, Y., Mitsuma, T., Kuro-
kawa, K., and Sobue, G. (1998) Biochem. Biophys. Res. Commun. 248,93–97
11. Vasan, S., Foiles, P., and Founds, H. (2003) Arch. Biochem. Biophys. 419,
FIG. 8. AGEs induce caspase activity in connective tissue invivo. A, caspase activity was measured using fluorometric assays inlysates from tissue obtained after injection of CML-collagen or unmod-ified collagen in the scalps of mice. The experiment was performed twicewith similar results. B, the number of TUNEL-positive apoptotic fibro-blasts was quantified 24 h after injection of the scalp with CML-collagen with or without caspase-8 or -9 inhibitors. After 24 h, theextent of apoptosis was measured by TUNEL staining of histologicsections. Each value represents the mean of specimens from six mice �S.E. The experiment was performed three times with similar results.
AGEs Stimulate Fibroblast Apoptosis12094
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from

89–9612. Liu, J., Masurekar, M., Vatner, D., Jyothirmayi, G., Regan, T., Vatner, S.,
Meggs, L., and Malhotra, A. (2003) Am. J. Physiol. Heart Circ. Physiol. 285,H2587–H2591
13. Saudek, D., and Kay, J. (2003) Curr. Rheumatol. Rep. 5, 33–4014. DeGroot, J., Verzijl, N., Jacobs, K., Budde, M., Bank, R., Bijlsma, J., TeKip-
pele, J., and Lafeber, F. (2001) Osteoarthritis Cartilage 9, 720–72615. Braak, H., and Braak, E. (1995) Neurobiol. Aging 16, 271–27816. Bownlee, M. (1995) Annu. Rev. Med. 46, 223–23417. Monnier, V. (1989) Prog. Clin. Biol. Res. 304, 1–2218. Palinski, W., Koschinsky, T., Butler, S., Miller, E., Vlassara, H., Cerami, A.,
and Witztum, J. (1995) Arterioscler. Thromb. Vasc. Biol. 15, 571–58219. Lalla, E., Lamster, I., Feit, M., Huang, L., Spessot, A., Qu, W., Kislinger, T.,
Lu, Y., Stern, D., and Schmidt, A. (2000) J. Clin. Investig. 105, 1117–112420. Vlassara, H., and Palace, M. (2002) J. Intern. Med. 251, 87–10121. Santana, R., Xu, L., Chase, H., Amar, S., Graves, D., and Trackman, P. (2003)
Diabetes 52, 1502–151022. Hofmann, M., Drury, S., Fu, C., Qu, W., Taguchi, A., Lu, Y., Avila, C., Kam-
bham, N., Bierhaus, A., Nawroth, P., Neurath, M., Slattery, T., Beach, D.,Mcclary, J., Nagashima, M., Morser, J., Stern, D., and Schmidt, A. (1999)Cell 97, 889–901
23. Stern, D., Yan, S., Yan, S., and Schmidt, A. (2002) Ageing Res. Rev. 1, 1–1524. Bucciarelli, L., Wendt, T., Rong, L., Lalla, E., Hofmann, M., Goova, M., Tagu-
chi, A., Yan, S., Yan, S., Stern, D., and Schmidt, A. (2002) Cell Mol. Life Sci.59, 1117–1128
25. Guzdek, A., and Stalinska, K. (1992) Folia Histochem. Cytobiol. 30, 167–16926. Schmidt, A., Yan, S., Brett, J., Mora, R., Nowygrod, R., and Stern, D. (1993)
J. Clin. Investig. 91, 2155–216827. Vlassara, H., Brownlee, M., Manogue, K., Dinarello, C., and Pasagian, A.
(1988) Science 240, 1546–154828. Naitoh, T., Kitahara, M., and Tsuruzoe, N. (2001) Cell. Signal. 13, 331–33429. Munch, G., Thome, J., Foley, P., Schinzel, R., and Riederer, P. (1997) Brain
Res. Rev. 23, 134–14330. Wendt, T., Tanji, N., Guo, J., Hudson, B. I., Bierhaus, A., Ramasamy, R.,
Arnold, B., Nawroth, P. P., Yan, S. F., D’Agati, V., and Schmidt, A. M.(2003) J. Am. Soc. Nephrol. 14, 1383–1395
31. Dyer, D., Dunn, J., Thorpe, S., Bailie, K., Lyons, T., McCance, D., and Baynes,J. (1993) J. Clin. Investig. 91, 2463–2469
32. Bucala, R., and Cerami, A. (1992) Adv. Pharmacol. 23, 1–433. Schnider, S., and Kohn, R. (1981) J. Clin. Investig. 67, 1630–163534. Owen, W., Hou, F., Stuart, R., Kay, J., Boyce, B., Chertow, G., and Schmidt, A.
(1998) Kidney Int. 53, 1365–137335. Goova, M. T., Li, J., Kislinger, T., Qu, W., Lu, Y., Bucciarelli, L. G., Nowygrod,
S., Wolf, B. M., Caliste, X., Yan, S. F., Stern, D. M., and Schmidt, A. M.(2001) Am. J. Pathol. 159, 513–525
36. Kasper, M., Roehlecke, C., Witt, M., Fehrenbach, H., Hofer, A., Miyata, T.,Weigert, C., Funk, R., and Schleicher, E. (2000) Am. J. Respir. Cell Mol.Biol. 23, 485–491
37. Denis, U., Lecomte, M., Paget, C., Ruggiero, D., Wiernperger, N., and Lagarde,M. (2002) Free Radic. Biol. Med. 33, 236–247
38. Kaji, Y., Amano, S., Usui, T., Osbika, T., Yamahiro, K., Isbida, S., Suzuki, K.,Tanaka, S., Adamis, A., Nagai, R., and Horiuchi, S. (2003) Investig. Oph-thalmol. 44, 521–528
39. Yamagishi, S., Inagaki, Y., Amano, S., Okamoto, T., Takeuchi, M., and Makita,Z. (2002) Biochem. Biophys. Res. Commun. 296, 877–882
40. Huang, D., Wang, J., Kivisakk, P., Rollins, B., and Ransohoff, R. (2001) J. Exp.Med. 193, 713–726
41. Higami, Y., and Shimokawa, I. (2000) Cell Tissue Res. 301, 125–13242. Kislinger, T., Fu, C., Huber, B., Qu, W., Taguchi, A., Du Yan, S., Hofmann, M.,
Yan, S., Pischesrieder, M., Stern, D., and Schmidt, A. (1999) J. Biol. Chem.274, 31740–31749
43. Habeeb, A. (1966) Anal. Biochem. 14, 328–33644. Liu, R., Desta, T., He, H., and Graves, D. (2004) Endocrinology 145, 2997–300345. Belyavskyi, M., Levy, G., and Leibowitz, J. (1998) Adv. Exp. Med. Biol. 440,
619–62546. Shi, Z., Wakil, A., and Rockey, D. (1997) Proc. Natl. Acad. Sci. U. S. A. 94,
10663–1066847. Alikhani, M., Alikhani, Z., He, H., Liu, R., Popek, I., and Graves, D. (2003)
J. Biol. Chem. 278, 52901–5290848. Oldfield, M., Bach, L., Forbes, J., Nikolic-Paterson, D., McRober, A., Thallas,
V., Atkins, R., Osicka, T., Jerums, G., and Cooper, M. (2001) J. Clin.Investig. 108, 1853–1863
49. Alikhani, M., Alikhani, Z., and Graves, D. T. (2004) J. Dent. Res. 83, 671–67650. Agarwal, C., Singh, R., and Agarwal, R. (2002) Carcinogenesis 23, 1869–187651. Vu, C., Bortner, C., and Cidlowski, J. (2001) J. Biol. Chem. 276, 37602–3761152. Wang, X., Mori, T., Sumii, T., and Lo, E. (2002) Stroke 33, 1882–188853. Honda, S., Farboud, B., Hjelmeland, L., and Handa, J. (2001) Investig. Oph-
thalmol. Vis. Sci. 42, 2419–242554. Yamagishi, S., Inagaki, Y., Okamoto, T., Amano, S., Koga, K., Takeuchi, M.,
and Makita, Z. (2002) J. Biol. Chem. 277, 20309–2031555. Reber, F., Geffarth, R., Kasper, M., Reichenbach, A., Schleicher, E., Sienger,
A., and Funk, R. (2003) Graefes Arch. Clin. Exp. Ophthalmol. 241, 213–22556. Yeh, C., Sturgis, L., Haidacher, J., Zhang, X., Sherwood, S., Bjercke, R.,
Juhasz, O., Crow, M., Tilton, R., and Denner, L. (2001) Diabetes 50,1495–1504
57. Anisowicz, A., Messineo, M., Lee, S., and Sager, R. (1991) J. Immunol. 147,520–527
58. Bierhaus, A., Schiekofer, S., Schwaniger, M., Andrassy, M., Humpert, P.,Chen, J., Hong, M., Luther, T., Henle, T., Koting, I., Morcos, M., Hofmann,M., Tritschler, H., Weigle, B., Kasper, M., Smith, M., Perry, G., Schmidt, A.,Stern, D., Haring, H., Schleicher, E., and Nawroth, P. (2001) Diabetes 50,2792–2808
59. Bruce, S., and Deamond, S. (1991) Exp. Gerontol. 26, 17–2760. Cook, J., and Dzubow, L. (1997) Arch. Dematol. 133, 1273–127761. Darby, I., Bisucci, T., Hewitson, T., and MacLellan, D. (1997) Int. J. Biochem.
Cell Biol. 29, 191–200
AGEs Stimulate Fibroblast Apoptosis 12095
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from

Dana T. GravesZoubin Alikhani, Mani Alikhani, Coy M. Boyd, Kiyoko Nagao, Philip C. Trackman andStimulate Fibroblast Apoptosis through Cytoplasmic and Mitochondrial PathwaysAdvanced Glycation End Products Enhance Expression of Pro-apoptotic Genes and
doi: 10.1074/jbc.M406313200 originally published online December 6, 20042005, 280:12087-12095.J. Biol. Chem.
10.1074/jbc.M406313200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/280/13/12087.full.html#ref-list-1
This article cites 61 references, 23 of which can be accessed free at
by guest on June 29, 2016http://w
ww
.jbc.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















