Introduction to SN2, E2, SN1, and E1 Mechanisms - Harvard ...

1
1
Title: Activation of NFκB by human papillomavirus 16 E1 limits E1-dependent viral replication 2
through degradation of E1. 3
4
Running title: A possible negative feedback loop between E1 and NFκB 5
6
Tomomi Nakahara, Katsuyuki Tanaka, Shin-ichi Ohno*, Nagayasu Egawa**, Takashi Yugawa, 7
Tohru Kiyono#. 8
Division of Carcinogenesis and Cancer Prevention, National Cancer Center Research Institute, 5-9
1-1 Tsukiji, Chuo-ku, Tokyo 104-0045, Japan. 10
#Address correspondence to Tohru Kiyono: [email protected] 11
*Present address: Department of Reproduction, National Research Institute for Child Health and 12
Development, 2-10-1 Okura, Setagaya-ku, Tokyo, 157-8535, Japan 13
**Present address: Division of Virology, National Institute for Medical Research, The Ridgeway, 14
Mill Hill, London, NW7 1AA, United Kingdom 15
The abstract: 211 words, Importance: 116 words. 16
The text: 12,398 words 17
18
19
JVI Accepted Manuscript Posted Online 25 February 2015J. Virol. doi:10.1128/JVI.00389-15Copyright © 2015, American Society for Microbiology. All Rights Reserved.

2
Abstract 20
NFκB is a family of transcription factors that regulate gene expression involved in many 21
processes such as the inflammatory response and cancer progression. Little is known about 22
associations of NFκB with the HPV life cycle. We have developed a tissue culture system to 23
conditionally induce E1-dependent replication of HPV16 genome in human cervical 24
keratinocytes, and found that expression of HPV16 E1, a viral helicase, results in reduction of 25
IκBα and subsequent activation of NFκB in a manner dependent on helicase activity. Exogenous 26
expression of a degradation-resistant mutant of IκBα, which inhibits the activation of NFκB, 27
enhanced E1-dependent replication of the viral genome. Wortmannin, a broad inhibitor of 28
phosphoinositide 3-kinases (PI3Ks), and to a lesser extent, VE-822, an ATR kinase inhibitor, but 29
not KU55933, an ATM kinase inhibitor, suppressed the activation of NFκB and augmented E1-30
dependent replication of HPV16 genome. Interestingly, the enhancement of E1-dependent 31
replication of the viral genome was associated with increased stability of E1 in the presence of 32
Wortmannin as well as the IκBα mutant. Collectively, we propose that expression of E1 induces 33
NFκB activation at least in part through the ATR-dependent DNA damage response and NFκB in 34
turn limits E1-dependent replication of HPV16 through degradation of E1, so that E1 and NFκB 35
may constitute a negative feedback loop. 36
37

3
Importance 38
A major risk factor in HPV-associated cancers is persistent infection with high-risk HPVs. To 39
eradicate viruses from infected tissue, it is important to understand molecular mechanisms 40
underlying the establishment and maintenance of persistent infection. In this study, we obtained 41
evidence that HPV16 E1, a viral DNA helicase essential for amplification of the viral genomes, 42
induces NFκB activation and that this limits E1-dependent genome replication of HPV16. These 43
results suggest that NFκB mediates a negative feedback loop to regulate HPV replication and this 44
feedback loop could be associated with control of the viral copy numbers. We could thus show 45
for the first time that NFκB activity is involved in the establishment and maintenance of 46
persistent HPV infection. 47
48

4
Introduction 49
Human papillomaviruses (HPVs) are a large family of non-enveloped, small DNA viruses 50
with an approximately 8-kbp double stranded circular genome that infect stratified squamous 51
epithelium in various anatomical sites such as skin, the anogenital tract and the oral cavity. Virus 52
infection can cause hyperproliferative lesions, ranging from verrucas to cancer. To date, at least 53
180 types of HPVs have been cloned from clinical lesions and classified as either cutaneous or 54
mucosal types based on their predilection for different sites of infection. A subset of mucosal 55
HPVs, high risk (HR)-HPVs such as HPV16, is strongly associated with anogenital cancers 56
including nearly 100% of cervical cancers and some proportion of head and neck cancers. 57
Altogether, it is estimated that HR-HPV infections are responsible for more than 5% of all 58
cancers worldwide. A major risk factor for HR-HPV associated carcinogenesis is persistent 59
infection of HR-HPVs. Therefore, it is important to elucidate molecular mechanisms underlying 60
the establishment and maintenance of persistent infection of HR-HPVs in order to allow 61
eradication of the viruses from infected tissue (1) . 62
The life cycle of HPVs is characterized by tight associations with differentiation processes in 63
the stratified epithelium. The production of progeny virions is exclusively restricted to upper 64
differentiated layers of stratified epithelium. The viruses enter nuclei of basal, mitotic cells and 65
establish their genomes as nuclear episomes at 50 - 100 copies per cell following transient initial 66
amplification. In basal cells, viral early genes are expressed at minimal levels and the copy 67
number is maintained constant by limiting replication of the viral genome on average once in S 68
phase, termed maintenance replication. As the infected cells initiate differentiation, levels of the 69
viral early genes are increased and the expression of late viral genes including major and minor 70

5
capsid proteins, L1 and L2, respectively, is induced. The viral genomes are drastically amplified 71
to many thousands by continued replication at a high level, termed productive amplification, and 72
eventually encapsidated into progeny virions (2). Two viral early proteins, E1 and E2 are 73
necessary for HPV genome replication along with the cellular replication machinery. A basic 74
mechanism of E1-dependent replication of papillomavirus genomes is well elucidated in which 75
E1 binds to the replication origin of the viral genome in cooperation with E2, forming a 76
hexameric DNA helicase and unwinding the viral DNA by power of ATP hydrolysis. E1 also 77
recruits host cell replication factors such as DNA polymerase α, topoisomerase I and replication 78
protein A (RPA). However, it is poorly understood how this E1-dependent replication is 79
regulated during the three phases of viral genome replication, initial amplification, maintenance 80
replication and productive amplification in the viral life cycle (3, 4). 81
Recently, several studies have indicated that DNA damage response (DDR) pathways are 82
involved in HPV genome replication. Key regulators of DDR are phosphoinositide 3-kinase like 83
kinases (PI3KKs), ataxia telangiectasia mutated (ATM), ATM and Rad3-related (ATR) and 84
DNA-dependent protein kinase (DNA-PKcs). Upon sensing DNA damage, these PIKKs initiate 85
signaling inducing the cell cycle checkpoints and DNA repair machinery or apoptosis (5-7). 86
Moody and Laimins reported a series of studies demonstrating that activation of ATM is required 87
for productive amplification but not for stable maintenance of HPV31 in CIN612 cells isolated 88
from a cervical intraepithelial neoplasia grade I (CIN) biopsy. HPV31 E7, an oncogene of HR-89
HPV, was shown to activate ATM and its downstream mediator checkpoint kinase 2 (CHK2). (8-90
10). Sakakibara et al. and Fradet-Turcotte et al. reported that E1 proteins of various HPVs, 91
including HPV16 and 31 and Bos taurus papillomavirus 1 (BPV1), also cause activation of ATM 92

6
(11, 12). In addition, it is reported that transient replication of HPV18 genomes in U2OS cells 93
induces activation of ATR mediated DDRs which depend on the expression of E1 and E2 (13). 94
Nuclear factor κ-light-chain-enhancer of activated B cells (NFκB) comprises a family of 95
transcription factors that activate or repress expression of a large number of genes in a myriad of 96
physiological and pathological processes contributing to immune and inflammatory responses, 97
cellular stress responses, differentiation, cell proliferation and apoptosis. Currently, five 98
members of the family are known in mammals, RELA/p65, p50/p105, p52/p100, RELB and c-99
REL that form homo-or hetero-dimers. The activity of NFκB is negatively regulated primarily 100
through its cytoplasmic retention by binding to inhibitors of NFκB (IκBs) including IκBα. Upon 101
stimulation, phosphorylation dependent degradation of IκBs by an ubiquitin proteasome pathway 102
liberates NFκBs into the nucleus (14, 15). Such activation of NFκB is a fundamental immediate 103
early step of immune activation, inducing expression of interferons (IFNs) as well as 104
inflammatory cytokines. Many viruses have been shown to target NFκB pathways to evade 105
innate immune responses. On the other hand, accumulated studies have revealed that activation 106
of NFκB signaling is induced and incorporated in the viral life cycle and is important for 107
pathogenicity of oncogenic viruses such as Epstein Barr virus (EBV), Kaposi sarcoma herpes 108
virus (KSHV) and human adult T leukemia virus -1 (HTLV-1)(16). 109
Increased expression of NFκB has been described for HPV associated lesions such as cervical 110
cancers as well as their precursor lesions, and laryngeal papillomas and cancers (17-22). Several 111
studies have shown that E6 and E7 proteins of HPVs modulate NFκB pathways (23-30). 112
However, the results of these studies have been contradictory as to whether E6 and/or E7 activate 113
or suppress. NFκB p65 (RELA) has been shown to bind to long control region (LCR) of HPV16 114

7
and suppress its promoter, p97, activity (31). Recently, it was also reported that E2 proteins of 115
various HPVs enhance tumor necrosis factor (TNF) α induced activation of NFκB in HaCaT 116
cells (32). NFκB plays an important role in maintaining homeostasis of epithelium and cellular 117
differentiation of keratinocytes (33, 34). Altogether, the available data suggests involvement of 118
NFκB in the pathogenesis and the life cycle of HPVs. In the present study, we investigated the 119
relationship between NFκB activity and replication of HPV16 genomes. As the outcomes and 120
pathways of NFκB signaling are known to vary dependent on the cell type, we developed a tissue 121
culture system to efficiently induce E1-dependent replication of the HPV16 genome in human 122
cervical keratinocytes (HCKs), a natural host of HPV replication. We found that NFκB is 123
activated upon E1 expression, which in turn limits E1-dependent replication of HPV16 genome 124
in mitotic, undifferentiated HCKs. The activation of NFκB was induced through DDR upon E1 125
expression. Implications of the NFκB-mediated suppression of E1-dependent replication for the 126
three phases of viral genome replication in the viral life cycle will be discussed. 127
128

8
Materials and Methods 129
Cells and tissue culture 130
Human cervical keratinocytes immortalized with TERT (HCK1T) were described previously 131
(35). HCK1T-HPV16 cells were generated by co-transfecting 1.25μg of pAd/HPV16/neo with 132
1.25μg of pxCANCre to HCK1T using lipofectamine 2000 (Life Technologies, Grand Island, 133
NY) according to the manufacturer’s protocol. Cells were selected with 150 μg/ml of G418 for 2 134
days starting from 2 days post-transfection and then maintained until G418 resistant colonies 135
became visible. These colonies were isolated and the presence of HPV16 genomes as episomes 136
was confirmed by Southern blotting. Oligo-clonal HCK1T that contained HPV16 genomes as 137
episomes was named HCK1T-HPV16 and used in this study. HCK1T tetON E1, tetON E1+E2 138
and tetON E2 were generated by retrovirus gene transfer and subsequent drug selection as 139
indicated below. HCK1T-HPV16 tetON E1+E2, tetON E1K483A+E2, tetON E1 and tetON E2 140
were generated by lentivirus infection. HCK1T-HPV16 tetON E1+E2 clone A5 and B2 141
introduced with expression of IκBα wild type, the IκBα mutant and the multi cloning site (MCS) 142
control were generated by retrovirus infection and subsequent drug selection. HCK1T-HPV16 143
and its derivatives were maintained in Keratinocyte-Serum Free medium (K-SFM) at 37°C in 5% 144
CO2 incubator. HCK1T and its derivatives not containing an HPV16 genome were maintained in 145
EpiLife medium (Life Technologies, Grand Island, NY). CIN612-9E cells were maintained on 146
feeder cells in E medium as described previously (36). Inhibitors and doxycycline were dissolved 147
in Dexamethasone (DMSO) and stored as high concentration stocks at -20°C until used at the 148
indicated concentrations. The concentrations of the stock solutions were as follows: Doxycycline 149
(Cat. Z1311N, TAKARA BIO Inc. Shiga, Japan) at 1 mg/ml, KU55933 (Cat. 118500, Merk 150

9
Millipore, Billerica, MA, USA), Wortmannin (Cat. 230-02341, WAKO Pure Chemical Industries, 151
Ltd., Osaka, Japan), SB203580 (Cat. 20-2173, Funakoshi Co., Ltd., Tokyo, Japan), VE-822 (Cat. 152
S7102, Selleck Chemicals, Houston, TX, USA) and Nu7026 (Cat. 2828, Tocris Bioscience, 153
Minneapolice, MN, USA) at 10 mM. Human TNFα (Cat. 203-15263, WAKO Pure Chemical 154
Industries, Ltd.) was dissolved and stored in phosphate buffered saline (PBS) at 10 μg/ml. 155
Plasmids 156
pAd/HPV16/neo encoding the full length of HPV16 genome flanked with two loxP sites was 157
constructed by replacing an XbaI-NotI fragment encoding EGFP of the original vector, 158
pAd/HPV16/Cre (37), with an XbaI-NotI fragment coding a neomycin resistant gene of pMSCV-159
neo (TAKARA BIO INC, Shiga, Japan). pxCANCre was obtained from the RIKEN DNA bank 160
(RDB10748, RIKEN Bioresource center, Tsukuba, Japan). Lentivirus vectors, CSII-CMV-161
tetON-ADV and CSII-TRE-Tight-HA16E1, were described previously (38). The codon 162
optimized HPV16 E2 cDNA (GeneScript, Piscataway, NJ) was used to construct CSII-TRE-163
Tight-HA16E2, in which the N-terminal hemaglutinin (HA)-tagged HPV16 E2 was inserted 164
under a tetracycline responsible promoter. CSII-TRE-Tight-HA16E1K483A was generated by site-165
directed mutagenesis (QuickChange Site-Directed Mutagenesis kit, Agilent Technologies, Santa 166
Clara, CA). CSII-TRE-Tight-HA16E1-PB and CSII-TRE-Tight-3xFLAG16E2-PB were 167
generated by inserting an expression cassette of the blastcidin (PB) resistant gene under the PGK 168
promoter into downstream of HA16E1 or 3xFLAG16E2. pQCXIZeo-tetON was generated by 169
inserting rtTA (tetON) from pRevTet-ON (Code 631007, BIO INC, Shiga, Japan) under CMV 170
promoter of pQCXIZeo, in which a puromycin resistant gene downstream of IRES of the 171
retroviral vector pQCXIP (Code.631516, TAKARA BIO INC, Shiga, Japan) was replaced by a 172

10
zeocin resistant gene from pCXbleo (Gene accession No. AB086388, a kind gift from Dr. Akagi, 173
Osaka Bioscience Institute). pQCXIZeo- IkBαWT and IkBαMT (Ser32/36Ala) were generated 174
by inserting cDNAs of IkBα and IkBαMT (Ser32/36Ala) derived from pCMV-IkBα and pCMV-175
IkBαMT (Ser32/36Ala) (Cat. 631923, TAKARA BIO INC, Shiga, Japan) into pQCXIZeo using 176
Gateway recombination (Life Technologies, Grand Island, NY). pCMV-6xHis-Ub expressing 177
histidine-tagged ubiquitin (His-Ub) was a kind gift from Dr. Bohmann (University of Rochester, 178
USA). 179
Retrovirus gene transduction 180
The production and infection of recombinant lentiviruses and retroviruses were accomplished 181
as previously described (39). To generate populations of HCK1T-HPV16 tetON E1, E1K483A 182
and/or E2, HCK1T-HPV16 cells were seeded at one day before and inoculated with CSII-CMV-183
tetON-ADV and one or corresponding combinations of lentiviruses, CSII-TRE-Tight-HA16E1, 184
CSII-TRE-Tight-HA16E1K483A and CSII-TRE-Tight-HA16E2 at multiplicity of infection (MOI) 185
20. For generation of HCK1T-HPV16 tetON E1 and FLAG-E2, a pooled population of HCK1T-186
HPV16 tetON E1 was infected with lentiviruses containing CSII-TRE-Tight-3xFLAG16E2 at 187
MOI 20. To generate HCK1T tetON E1, tetON E1+E2 and tetON E2, HCK1T cells were first 188
infected with pQCXIZeo-tetON at MOI 3 followed by zeocin selection (2 μg/ml). The generated 189
HCK1T-tetON cells were then infected with lentiviruses, CSII-TRE-Tight-HA-HPV16E1-PB 190
and/or CSII-TRE-Tight-3XFLAG-HPV16 E2-PB, at MOI 3 and selected with blastcidin (2 μg 191
/ml). Parental HCK1T-HPV16 and HCK1T-HPV16 tetON E1+E2 clones A5 and B2 were 192
infected with retroviruses, pQCXIZeo-MCS, IkBαWT or IkBαMT, and selected with zeocin (10 193
μg/ml). 194

11
Cell collection 195
The cultural supernatant was harvested and “detached cells” were collected by centrifugation at 196
2,000 x g for 5 minutes at 4˚C. The cells staying attached were washed with PBS twice and 197
removed from a tissue culture plate by incubation with trypsin. The “attached cells” were then 198
incubated with a soybean trypsin inhibitor (0.25 mg/ml in PBS, Life Technologies, Grand Island, 199
NY) and collected by centrifugation at 2,000 x g for 5 minutes at 4˚C. When necessary, 200
“attached cells” were counted using a Coulter Counter (Beckman Coulter, Inc. Brea, CA, USA). 201
The “attached cells” and “detached cells” were combined and the mixtures were washed with 202
PBS twice. The combined cells were lysed with a buffer specific to each assay. 203
Southern blotting and quantitative real-time PCR (qPCR) 204
Total genomic DNA for Southern blotting was isolated after several passages and at least one 205
freezing and thawing from transfection as described previously (38). For qPCR, total genomic 206
DNA was isolated using a Wizard SV Genomic Purification System following the 207
manufacturer’s protocol (Cat. A2361, Promega, Madison, WI, USA). For Southern blotting, 10 208
μg of total genomic DNA was digested either with BamHI or XhoI overnight and 209
electrophoresed through a 0.7% agarose gel. The gel was alkali transferred onto a nylon 210
membrane using a TurboBlotter (Cat. 10416336, GE healthcare, Fairfield, Connecticut, USA). 211
The probe used for Southern blotting was randomly labeled with biotin by using a NEBlot 212
Phototope kit (New England BioLabs. Inc., Ipswich, MA, USA). The linearized plasmid 213
encoding the full length of HPV16 was used as a template. Hybridization and chemiluminescent 214
detection was performed as described previously. The LAS3000 charge-coupled device (CCD) 215
imaging system (Fujifilm Co. Ltd., Japan) was employed for detection. Quantification of images 216

12
was conducted using software NIH image J and Multi Gauge (Fujifilm, Co. Ltd.). Realtime PCR 217
to quantify the viral genome copy number was performed as described previously. In brief, 10-218
100 ng of total genomic DNA was mixed with a mastermix of KAPA SYBR FAST qPCR kits 219
(Kapa Biosystems, Woburn, MA) and 300 nM of each primer and then subjected to a real time 220
PCR reaction using StepOnePlus (Life Technologies, Grand Island, NY). Serial dilutions of 221
linearized HPV16 genome from pUC-HPV16 plasmid DNA by BamHI digestion were used as 222
standards to measure the amount of HPV16 DNA. All real-time PCRs were run in triplicate and 223
at least three independent experiments were performed. The copy number of HPV16 per cell was 224
calculated based on the assumption that total human genomic DNA is 6.6 pg/diploid cell. P-225
values were determined by Sudent’s t- test. Two sets of primer pairs were used for qPCR to 226
confirm that the viral copy numbers estimated by each pair were consistent. The pairs of primers 227
used were as follows: E6 pairs (HPV16-97F, 5’-GAACTGCAATGTTTCAGGACCC-3’and 228
HPV16-174R, 5’-TGTATAGTTGTTTGCAGCTCTGTGC-3) and L2 pairs (HPV16-4481F, 5’-229
ACAGATACACTTGCTCCTGTAAGACC-3’ and HPV16-4665R, 5’- 230
GCAGGTGTGGTATCAGTTGAAGTAGT-3’). 231
RNA extraction and reverse transcription (RT)-qPCR 232
Total RNA was isolated using an RNeasy Plus Mini kit (Cat. 74136, Qiagen, Venlo, 233
Netherlands) following the manufacturer’s instructions. 1 μg of total RNA was subjected to 10 μl 234
RT reaction using a PrimeScript RT reagent kit (Cat. RR037A, TAKARA BIO INC, Shiga, 235
Japan) and 1 μl of the RT products was used for real time PCR reaction to measure mRNAs of 236
the interest. In the case of β-actin mRNA, 10 time-diluted RT products were used due to 237
abundance of the mRNA. The PCR mixtures were prepared using KAPA SYBR FAST qPCR 238

13
kits and the PCR run was achieved with StepOnePlus. The relative mRNA levels of the interest 239
were calculated by a delta-delta CT method using β-actin mRNA as an internal control. All PCRs 240
were run in triplicate. All experiments were repeated at least three times. P-values were 241
determined by Student’s t- test. The following primers were used. β-actin: Forward 5’-242
ACCAACTGGGACGACATGGAGAAA-3’ and Reverse 5’-243
TAGCACAGCCTGGATAGCAACGTA-3’, TNFα: Forward, 5’- 244
CGAGTGACAAGCCTGTAGC-3’ and Reverse, 5’-GGTGTGGGTGAGGAGCACAT-3’, IL-6: 245
Forward, 5’-AAATTCGGTACATCCTCGACGGCA-3’ and Reverse, 5’-246
AGTGCCTCTTTGCTGCTTTCACAC-3’, IL-8: Forward, 5’- 247
AGCCTTCCTGATTTCTGCAGCTCT-3’, and Reverse, 5’-248
AATTTCTGTGTTGGCGCAGTGTGG-3’, E1^E4: Forward, 5’-249
GCTGATCCTGCAAGCAACGAAGTATC-3’, and Reverse, 5’-250
GGATTGGAGCACTGTCCACTGAG-3’, HA-E1: Forward, 5’-251
CCTTATGACGTGCCAGATTACGC-3’, and Reverse, 5’-252
GTCATTTTCGTTCTCATCGTCTGAGATG-3’. These primers were designed based on the 253
published data (40). 254
Western blotting 255
Cells were collected as described above and lysed in lysis buffer (50 mM Tris-HCl, 250 mM 256
NaCl, 5 mM EDTA, 1% NP-40, 20% glycerol, 0.1% SDS, 1% Deoxycholate) containing a 257
proteinase inhibitor cocktail (Nacalai tesque, Kyoto, Japan) and phosphatase inhibitors (500 μM 258
sodium orthovanadate, 100 mM sodium fluoride, 10 mM sodium pyrophosphate) and then 259
subjected to brief sonication in an ice-cold water bath. A protein concentration of cell lysates was 260

14
measured by using a DC Protein Assay kit (BIO-RAD, Hercules, CA, USA) after centrifugation 261
at 20,000g for 15 minutes. When necessary, an insoluble fraction was lysed in an SDS sample 262
buffer (50mM Tris-HCl, 2%SDS and 10% glycerol). The equivalent protein amounts of cell 263
lysates were subjected to SDS-polyaclylamide gel electrophoresis (SDS-PAGE) and Western 264
blot analysis was conducted as described previously (38). The primary antibodies used in this 265
study were follows. Mouse monoclonal antibodies: HA (16B12; Covance, Princeton, NJ), FLAG 266
(M2; Sigma-Aldrich, St.Louis, MO, USA ), Vinculin (hVIN-1; Sigma-Aldrich), pATM(S1981) 267
(10H11E12; EMD Millipore, Billerica, MA, USA), CHK2 (clone 7; EMD Millipore), ATM 268
(2C1: Santa Cruz Biotechnology, Santa Cruz, CA, USA), CHK1 (G-4; Santa Cruz 269
Biotechnology), IκBα (L35A5; Cell Signaling Technology, Danvers, MA, USA), His tag (Cat. 270
70796; EMD Millipore), Histone H3 (Ab10799, Abcam, Chambridge, UK). Rabbit monoclonal 271
antibodies: pCHK1(S345) (133D3), RELA (D14E12) and rabbit polyclonal antibodies: Caspase-272
3 (Cat. 9662), PARP-1 (Cat. 9542), pCHK2(T68) (Cat. 2661), α-Tubulin (Cat.2144), 273
p38MAPK(T180/Y182) (Cat. 9211) and p38MAPK (Cat. 9212), all purchased from Cell 274
Signaling Technology. Rabbit polyclonal antibodies: NBS1 (Cat. NB100-143, Novus 275
Biologicals, Littleton, CO, USA), GFP (A6455, Life Technologies, Grand Island, NY), pDNA-276
PKcs(S2056) (Ab18192, Abcam) and DNA-PKcs (Ab70250, Abcam). A goat polyclonal MCM2 277
antibody was also purchased from Santa Cruz Biotechnology (sc-9839). Secondary antibodies 278
used were horseradish peroxidase (HRP) -conjugated anti-mouse, anti-rabbit (Jackson 279
ImmunoResearch Laboratories, West Grove, PA) and anti-goat antibodies (sc2020, Santa Cruz 280
Biotechnology). 281
Indirect immunofluorescence analysis 282

15
The cells were first rinsed with PBS twice and fixed in 4% paraformaldehyde (4% PFA) in PBS 283
for 15 minutes at room temperature. The fixed cells were incubated with 0.01% of Triton-X in 284
PBS for 10 minutes after rinsing twice with PBS, and then incubated with the blocking buffer 285
(2% Bovine Serum Albumin, BSA, in PBS) for 30 minutes at room temperature. Incubation with 286
the primary antibody diluted in the blocking buffer was done overnight at 4˚C and the samples 287
were then washed with PBS three times each for 5 minutes. Incubation with secondary 288
antibodies was done for 30 minutes at room temperature and the samples were then sealed with a 289
mounting medium containing 4', 6-diamidino-2-phenylindole (DAPI) (Prolong Gold, Life 290
technologies, Grand Island, NY) to counterstain nucleus. Fluorescence images were examined 291
using a Leica FW4000 microscope (Leica Microsystems, Wetzlar, Germany). 292
Ubiquitin conjugation assays 293
293FT cells were seeded at 1 x 106 cells in 6-cm tissue culture dishes on the day before 294
transfection. 3.2 μg of pCMV-6xHis-Ub, 0.8 μg of CSII-CMV-HA16E1 or CSII-CMV-MCS, 1 295
μg of pCMV-IκBαMT or an empty vector and 0.2 μg of pEGFP-C3 (TAKARA BIO INC., Shiga, 296
Japan) were mixed and incubated with 18 μl of polyethylenimine (PEI) max solution (1 mg/ml) 297
in 800 μl of serum-free medium for 15 minutes at room temperature. The DNA-PEI max solution 298
was then added to the cells followed by further incubation. At 18 hours post-transfection, the 299
cells were removed by trypsin and divided into three 6-cm tissue culture dishes and incubated 300
with DMSO, 10 μM of Wortmannin or 20 ng/ml of TNFα from 20 hours post-transfection. Cells 301
transfected with pCMV-IκBαMT were also divided and incubated with a vehicle or TNFα. At 48 302
hours post-transfection, the cells were harvested according to a published protocol (41). Briefly, 303
the cells were lysed with the lysis buffer [2% SDS, 150 mM NaCl, 10 mM Tris-HCl pH8.0, 304

16
proteinase inhibitors, phosphatase inhibitors and 2 mM N-ethylmaleimide (NEM)] and 305
immediately boiled for 10 minutes. The cell lysates were subjected to sonication, then diluted in 306
the dilution buffer (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% Triton-X100, 2 307
mM NEM) and rotated at 4˚C for 30 minutes. The equivalent protein amounts of the lysates were 308
incubated with anti-HA tag mAb-magnetic beads (Cat. M132-9; Medical and Biological 309
Laboratories Co., Ltd. Nayoga, Japan) overnight with rotation at 4˚C. The immunoprecipitates 310
were collected using MagneSphere Magnetic Separation Stands (Promega, Madison, WI, USA) 311
and extensively washed. The immunoprecipitates were suspended in 2x Laemmli sample buffer 312
and subjected to Western blotting. 313
Cell cycle analysis 314
The HCK1T-HPV16 tetON E1+E2 clone B2 and A5 were collected as described above and 315
fixed in chilled 70% EtOH for at least 1 hour at -20˚C. The fixed cells were then rinsed with PBS 316
twice, resuspended in PBS containing 0.05% Triton X and 0.5mg/ml of RNase A (Nacalai tesque, 317
Kyoto, Japan) and incubated for 30 minutes at 37˚C. Propidium iodide (PI) was added to a final 318
concentration of 0.05 μg/ml and the cells were subjected to flow cytometry using a cell analyzer 319
EC800 (SONY, Tokyo, Japan). 320
321

17
Results 322
Conditional expression of E1 and E2 induces amplification of the HPV16 genome as 323
episomes in human cervical keratinocytes (HCKs). 324
In order to study E1-dependent replication of HPV16 genome in human cervical keratinocytes, 325
we first established immortal human cervical keratinocytes stably maintaining HPV16 genomes 326
as episomes as described in the Materials and Methods. We confirmed that HCK1T-HPV16 cells 327
were able to maintain HPV16 genomes as episomes at a constant copy number (approximately 328
30 copies/cell) for at least 3 months and several freeze and thaw cycles (Fig.1A, lanes 4, 6, 8 and 329
10). They were co-infected with lentivirus vectors expressing the reverse tetracyclin-regulated 330
transactivator (rtTA; tetON) together with those expressing HA-tagged HPV16 E1 (HA16E1), a 331
helicase defective mutant of E1 (HA16E1K484A), or HA-tagged HPV16 E2 under the control of 332
tetracyclin-responsive promoter or combinations of HA16E2 and HA16E1 or HA16E1K484A. The 333
same numbers of those cells were seeded the day before and then incubated with either vehicle or 334
1 μg per ml of doxycycline (DOX) for 24 hours. The total genomic DNA was isolated and the 335
copy number of HPV16 genomes was measured by Southern blotting and quantitative real time 336
PCR (qPCR) (Fig 1A and B). Expression levels of HA16-E1 and -E2 were also analyzed by 337
Western blotting (Fig 1F). Episomal copies of HPV16 genomes increased by approximately 100 338
fold in HCK1T-HPV16 cells designed to express both E1 and E2 (tetON E1+E2) upon 339
incubation with DOX (Fig.1A, lanes 4, 5, 8 and 9 and Fig.1B), whereas no increase was 340
detectable in those designed to express E1K483A and E2 (tetON E1K483A+E2) with the DOX 341
treatment (Fig.1A, lanes 6, 7, 10 and 11 and Fig.1B) even though the level of E1K483A appeared 342
higher than that of the wild type E1 (Fig.1F, lanes 2 and 4). The copy number of HPV16 343

18
genomes increased slightly (by approximately 3 fold) in cells designed to express E1 alone 344
(tetON E1) upon DOX treatment, while no increase was detected in those designed to express E2 345
alone (tetON E2). The basal copy number of HPV16 genomes was comparable among those 346
cells in the absence of DOX. These results clearly indicated that exogenous E2 expression 347
enhanced E1-dependent replication of the HPV16 genome though we failed to detect HA-tagged 348
E2 by Western blotting. The slight increase in the copy number of HPV16 genomes in the tetON 349
E1 cells could depend on E2 derived from episomal HPV16 genomes. The expression level of E1 350
from episomal genomes appeared to be too low to support detectable replication even when 351
excess amounts of E2 were provided exogenously. In the tetON E1+E2 cells, the copy number of 352
the viral genomes increased in a time dependent manner (Fig.1C); increase was detected as early 353
as at 4 hours and continued up to 24 hours after DOX addition whereas no further increase was 354
detected thereafter. The maximal level of E1 as well as the copy number of HPV16 genomes 355
were observed at the highest concentration of DOX tested (1 μg/mL) (data not shown). Therefore, 356
E1-dependent replication of the HPV16 genome was evaluated at 24 hours after 1 μg per ml of 357
DOX addition in subsequent analyses unless otherwise indicated. As the estimated copy number 358
of the viral genomes by qPCR was well correlated with that obtained by Southern blotting, the 359
copy number of HPV16 genomes was measured by qPCR in the subsequent experiments. 360
The intracellular localization of HA-tagged E1 and E2 was examined by immunofluorescence 361
analysis (IFA) using an anti-HA tag antibody at 24 hours after addition of vehicle or DOX 362
(Fig.1D). In the presence of DOX, HA positive signals were observed in roughly 40% of cells, 363
indicating heterogeneity in expression levels of E1 and E2 in the population. In a fraction of the 364
tetON E1+E2 cells, positive signals with anti-HA antibody were found accumulated to nuclear 365
foci (Fig.1D, E1+E2 DOX+). In contrast, no obvious accumulation of HA positive signals to 366

19
nuclear foci was observed in the tetON E1K483A+E2 cells. Instead, strong HA-positive signals 367
were diffusely distributed throughout nuclei and weaker signals were also detected in the 368
cytoplasm. In the tetON E1 cells, a diffuse pattern of HA-positive signals was detected in nuclei 369
and weaker signals were also detected in cytoplasm in a majority of the positive cells. On the 370
other hand, homogeneous HA positive signals were found mostly confined to nucleus and no 371
obvious accumulation to foci was detected in the tetON E2 cells. Our results coincided with the 372
previous reports that E1 and E2 co-localize to nuclear foci when co-expressed and a helicase 373
defective mutant of E1 does not form nuclear foci with E2 (11, 42). Although we could not 374
distinguish E1 and E2 due to the same tag, taking into account the previous report and the fact 375
that the HA positive nuclear foci were detectable only in cells in which expression of HA-E1 and 376
HA-E2 was expected, it is likely that those nuclear foci represent co-localization of E1 and E2. 377
The nuclear foci formation required helicase activity of E1 and well corresponded with the copy 378
number increase of HPV16 genomes, reinforcing the notion that E1 and E2 form nuclear foci to 379
support viral genome replication. No signals were detected in any of those cells when incubated 380
with a vehicle control, suggesting basal expression levels of exogenous E1 and E2 are very low 381
without DOX treatment (Fig. 1D and F). 382
Sakakibara et al. and Fradet-Turcotte et al. reported that E1 expression suppresses cell 383
proliferation (11, 12). Consistent with their results, we noted that the tetON E1+E2 cells and the 384
tetON E1 cells but not the tetON E1K483A+E2 cells and the tetON E2 cells appeared to be not 385
proliferating very well and some of those cells were detached from bottom of tissue culture 386
plates by 24 hours when incubated with DOX. We counted the cells which stayed attached to the 387
culture plates and found that the number of the tetON E1+E2 cells and the tetON E1 cells grown 388
in the presence of DOX for 24 hours was significantly less than those grown in the presence of a 389

20
vehicle control (Fig.1E, data not shown for the tetON E1 cells). On the other hand, no such 390
difference in the cell numbers was observed in the tetON E1K483A+E2 or the tetON E2 cells 391
(Fig.1E, data not shown for E2 alone). To investigate whether the apparent inhibition of cell 392
proliferation was caused by induction of apoptosis, markers of apoptosis such as caspase-3 and 393
poly (ADP ribose) polymerase-1 (PARP-1) were analyzed by Western blotting. The cells 394
detached from a tissue culture plate as well as the cells remaining attached were collected and 395
equivalent protein amounts of cell lysates were subjected to SDS-PAGE and subsequent Western 396
blot analysis (Fig.1F). The cleaved forms of PARP-1 and caspase-3 were detected in the cells 397
expressing E1 and E2 or E1 alone but not in the cells expressing E1K483A and E2 or E2 alone, 398
suggesting that E1 expression induces apoptosis depending on its helicase activity. 399
HPV16 E1 induces activation of NFκB in human cervical keratinocytes containing 400
episomal HPV16 genomes. 401
To explore associations of NFκB with viral genome replication, we first studied the status of 402
NFκB p65 (RELA) and IκBα in HCK1T-HPV16 cells after induction of E1, E1K483A and/or E2 403
expression. We found that the steady state level of IκBα was reduced upon induction of E1 and 404
E2 or E1 alone but not E1K483A and E2 or E2 alone (Fig.2A, lanes 2 and 8 compared to lanes 1 405
and 7, respectively). On the other hand, the levels of RELA remained comparable. As expected, 406
nuclear translocation of RELA was detected in the cells containing HA positive nuclear foci 407
upon induction of E1 and E2 expression, while RELA was localized to cytoplasm in a majority 408
of the cells incubated with a vehicle control (Fig.2B). These results indicated that NFκB is 409
activated upon induction of E1 and E2 expression. Then transcriptional activation of several 410
NFκB target genes was examined (Fig.2C). Total RNA was isolated following incubation of 411

21
DOX or vehicle and subjected to reverse transcription (RT)-qPCR. All the NFκB target genes 412
examined were up-regulated upon induction of E1 and E2 expression. The mRNA levels of 413
TNF-α, interleukin (IL) 6 and IL8 were increased by 2.5 +/- 0.11, 4.9+/- 1.17 and 14.2 +/- 3.46 414
fold, respectively, whereas no significant increase of these mRNA levels was detected upon 415
induction of E1K483A and E2 expression. Taken together, these results indicate that the expression 416
of E1 and E2 induces activation of NFκB most likely through promoting degradation of IκBα. 417
Viral early transcripts containing spliced E1^E4 ORF were slightly reduced in the tetON E1+E2 418
cells (0.88+/- 0.12) even though the viral copy number increased (Fig.1A). It is possible that 419
exogenous E2 expression suppressed the viral early promoter, p97. However, in the 420
E1K483A+E2 cells, similar reduction of the viral early transcripts was observed (0.73+/-0.12) 421
when neither NFκB activation nor viral genome replication was induced. Therefore, no increase 422
of viral early transcripts despite the increased viral genome copies was likely associated with 423
suppression of p97 by NFκB activation in the tetON E1+E2 cells (31). 424
HPV16 E1 induces activation of NFκB in human cervical keratinocytes in the absence of 425
an episomal HPV genome. 426
We next examined whether E1 can reduce the IκBα level in the absence of an episomal HPV16 427
genome. Parental HCK1T cells were used to establish new cell lines expressing HA-tagged E1 428
and/or 3xFLAG-tagged E2 of HPV16 upon DOX administration. The steady state level of IκBα 429
was reduced in the parental HCK1T cells upon expression of E1 and E2 as well as E1 alone but 430
not E2 alone (Fig.3A, lanes 4 and 6 compared to lanes 3 and 5, respectively), indicating that 431
replication of the viral genomes is not necessary for NFκB activation, E1 expression per se rather 432
causing the activation. Reduction of the IκBα level appeared more prominent in the cells 433

22
expressing only E1 compared to E1 and E2, most likely due to higher expression of E1 (Fig.3A, 434
lanes 4 and 6). By switching the tag from HA to 3xFLAG, we were able to detect E2 by Western 435
blotting. Consistent with reduction of the IκBα level, NFκB target genes were up-regulated upon 436
expression of E1 and E2 as well as E1 alone in the absence of an HPV16 genome (Fig.3B). We 437
noted that endogenous mRNA levels of NFκB target genes were lower in parental HCK1T cells 438
than in HCK1T-HPV16. In addition, endogenous levels of the NFκB target genes were also 439
higher in CIN612-9E cells harboring episomal HPV31 genomes, compared to HCK1T, 440
suggesting constitutive activation of NFκB in the presence of episomal HPV genomes and this 441
was not specific to HCK1T-HPV16 (Fig.3C). Similar to HCK1T-HPV16, cell proliferation of 442
HCK1T was also inhibited and cleaved forms of caspase-3 and PARP-1 were detected upon 443
expression of E1 alone or E1 and E2, but not E2 alone (data not shown). These results are 444
consistent with previous findings that E1-mediated growth suppression does not require E2 or 445
viral genome replication (12, 13). 446
Inhibition of NFκB activation enhances E1-dependent replication of HPV16 genomes. 447
As induction of NFκB activation by E1 has never been described, we were interested in whether 448
this activation of NFκB affects E1-dependent replication of the viral genome. To this end, we 449
examined effects of IκBα overexpression, which prevents activation of NFκB, on E1-dependent 450
replication. As shown in Fig.1, the tetON E1+E2 cells proved to be a heterogeneous cell 451
population in which roughly 40% of the cells express HA-tagged proteins upon DOX treatment. 452
We therefore tried to obtain a clonal cell population expressing E1 and E2 more homogenously 453
upon induction. A total of 50 clones were isolated by limiting dilution and examined for the copy 454
number of HPV16 genomes and the levels of E1 and E2 expression in the presence or absence of 455

23
DOX. There were variations in the basal copy number of HPV16 genomes and the induction 456
levels of E1 among clones; some clones contained only a few copies of HPV16 genomes while 457
others contained 1,000 copies (data not shown). In most clones, the induction levels of E1 were 458
correlated with the increase in HPV16 copy numbers and levels of E1 and IκBα were inversely 459
correlated (Fig 4A) as expected from the results of the pooled population (Fig.2A). In clones A5 460
and B2, we were able to detect a band at a size corresponding to HA16E2 (Fig.4A, HA long 461
exposure), which could not be detected in the parental pooled population upon DOX treatment 462
(Fig.1F). We chose clones A5 and B2 for further studies as these showed the sharpest increase of 463
the viral genomes after DOX addition. The basal copy number of HPV16 genomes in clones A5 464
and B2 were the average of the clones. Wild type IκBα (IκBαWT), an IκBαS32A/S36A mutant 465
(IκBαMT), which is resistant to phosphorylation and subsequent degradation, or an empty vector 466
(MCS) as a control were transduced to clones A5 and B2 by retroviral gene transfer. 467
Accumulation of exogenous IκBαWT as well as IκBαMT regardless of E1 and E2 expression 468
was confirmed while reduction of endogenous IκBα level upon induction of E1 and E2 469
expression was evident in the control cells (Fig.4B, shown for clone B2). TNFα treatment which 470
activates NFκB via degradation of IκBα to parental HCK1T-HPV16 was also included as a 471
positive control (Fig.4B, lanes 1 and 2). The NFκB target genes, TNF-α, IL6 and IL8, were 472
extremely up-regulated following incubation with DOX in the control cells (Fig 4C). In clone B2, 473
the mRNA levels of TNFα, IL6 and IL8 were increased by 65+/-7.6, 88 +/-24.6 and 289 +/-42 474
fold, respectively, upon DOX treatment. The higher levels of these mRNAs compared to those 475
detected in the parental heterogeneous population (Fig.2C) was most likely due to homogeneity 476
of the cells expressing high levels of E1 (Fig.4A left, HA bottom panel). Consistent with the 477
IκBα protein level, transcriptional up-regulation of the NFκB target genes upon induction of E1 478

24
and E2 expression was strongly inhibited although not completely abrogated in the cells 479
expressing IκBαWT as well as IκBαMT. These data verified that over-expression of IκBαWT as 480
well as IκBαMT indeed prevented NFκB activation induced by E1 expression. Of note, basal 481
levels of the NFκB target genes in the absence of DOX were significantly lower in the cells 482
expressing IκBαWT as well as IκBαMT compared to the control cells (MCS), suggesting that 483
over-expression of IκBα also suppresses constitutive activation of NFκB seen in the presence of 484
HPV16 genomes (Fig.3C). We then analyzed replication of the HPV16 genome and cell 485
proliferation upon E1 and E2 expression in the presence of IκBα over-expression (Fig. 4D and E). 486
Interestingly, the copy number of HPV16 genomes following DOX incubation in the cells 487
expressing IκBαWT as well as IκBαMT was significantly higher than that in the control cells, 488
suggesting that the activation of NFκB suppresses E1-dependent replication of the HPV16 489
genome (Fig.4D). The basal copy number of HPV16 genomes in the cells expressing IκBαWT as 490
well as IκBαMT was slightly higher than that in the control cells. Although the difference was 491
not statistically significant, it is possible that the basal activity of NFκB also influences the 492
control of HPV16 copy numbers in the absence of exogenous E1 as discussed later. We noted 493
that the protein level of HA-E1 in the cells expressing IκBαMT appeared to be higher than that in 494
MCS control cells. The elevated level of HA-E1 might have contributed to augmented 495
replication of the HPV16 genome in cells expressing IκBαMT. In contrast, the inhibitory action 496
of E1 on cell proliferation was not affected by over-expression of IκBαWT or IκBαMT (Fig. 4E), 497
suggesting that activation of NFκB is not involved in E1-induced growth suppression or 498
apoptosis. The levels of viral transcripts containing spliced E1^E4 ORF were approximately 1.5 499
and 1.7 fold higher in the cells expressing IκBαWT and IκBαMT, respectively, than that in the 500
control cells in the absence of DOX, implying that the basal activity of NFκB might suppress 501

25
p97 to some extent. The viral transcripts were increased by 2 fold in the control cells and by 5.6 502
fold in the cells with IκBαWT or IκBαMT upon DOX treatment. We still considered these results 503
that activation of NFκB suppresses p97 and that over-expression of IκBαWT or IκBαMT 504
substantially alleviated the NFκB-mediated suppression (Fig.3D). Overall, these data suggest 505
that activation of NFκB limits E1-dependent replication of the HPV16 genome while it is not 506
involved in suppressive effects of E1 on cell proliferation. We repeated the same experiments 507
using clone A5 and obtained essentially the same results as with clone B2 (data not shown). 508
E1 induces NFκB activation by eliciting DNA damage response (DDR) pathways. 509
NFκB can be activated by a variety of upstream signals including DDR. Upon generation of 510
double stranded breaks (DSBs), ATM has been shown to activate NFκB through phosphorylation 511
dependent degradation of IκBα (43). Because the wild type but not the helicase defective mutant 512
of HPV16 E1 has been shown to activate ATM, we initially hypothesized that E1 induces NFκB 513
activation by eliciting DDR, most likely via activation of ATM. To test this hypothesis, 514
activation of DDR was analyzed in heterogeneous cell populations of tetON E1+E2, tetON 515
E1K483A+E2, tetON E1 or tetON E2 or several clones of the tetON E1+E2 cells by Western 516
blotting. Checkpoint kinase 1 and 2 (CHK1 and CHK2) are known phosphorylation targets of 517
ATR and ATM that mediate signal transduction downstream of ATR and ATM, respectively. 518
Antibodies specific to phosphorylated ATM, pATM(S1981) and CHK2, pCHK2(T68), or CHK1, 519
pCHK1(S345), were used as markers for ATM or ATR activation, respectively. NBS1 was also 520
analyzed as it has been shown to be phosphorylated by ATM (5, 44). Upon DOX incubation, 521
phosphorylation of ATM and CHK1 was increased in the cell populations of tetON E1+E2 and 522
tetON E1 though basal levels of the phosphorylated CHK1 appeared also higher in these cells 523

26
(Fig.5A, left panel). A slower migrating pattern of NBS1 and CHK2 upon DOX incubation was 524
detected, indicating that this was attributable to ATM-mediated phosphorylation of these proteins. 525
In clones of the tetON E1+E2 cells, the phosphorylation of ATM, CHK1 and CHK2 was also 526
increased following incubation with DOX and a slower migrating pattern of NBS1 and CHK2 527
was detected. On the other hand, none of those indications for DDR activation could be detected 528
in the cell populations of tetON E1K483A+E2 and tetON E2, as expected. These results 529
indicated that ATM and ATR pathways are both activated upon induction of E1 and the 530
activation of DDR depends on its helicase activity. We then examined reduction of the IκBα 531
level in the presence of DDR inhibitors such as Wortmannin and KU55933 upon E1 and E2 532
expression. Wortmannin is a broad inhibitor of PI3Ks shown to inhibit ATM, ATR and DNA-533
PKcs while KU55933 is a specific inhibitor of ATM. Clone B2 was pre-incubated with indicated 534
inhibitors at 2 hour prior to addition of DOX and the cell lysates were collected at the indicated 535
time points in the presence or absence of the inhibitors. Activation of DDR and reduction of the 536
IκBα level were analyzed by Western blotting (Fig.5B). A high level of E1 expression was 537
detected at 8 hours following DOX addition. Concomitantly, a slower migrating pattern of NBS1 538
and CHK2 and the phosphorylated form of CHK1 at S345 and CHK2 at T68 were detected and 539
they continued to accumulate up to 24 hours in DMSO treated control cells. Reduction of the 540
IκBα level was also evident as early as 8 hours following DOX addition and continued thereafter 541
(Fig.5B, lanes 1 to 4). In the presence of Wortmannin, a slower migrating pattern of NBS1 was 542
less prominent though present, and appearance of the phosphorylated CHK1 and CHK2 delayed; 543
they were first detected at 16 hours following incubation with DOX as compared to 8 hours in 544
DMSO treated controls. The time dependent accumulation of the phosphorylated CHK1 and 545
CHK2 was reduced although not completely abrogated in the presence of Wortmannin, 546

27
indicating that ATM as well as ATR mediated signal transduction was suppressed albeit not 547
completely. Interestingly, the IκBα level following DOX addition was maintained in the 548
presence of Wortmannin, suggesting that activation of DDR signals results in reduction of the 549
IκBα level (Fig.5B, lanes 5 to 8). In the presence of KU55933, a slower migrating pattern of 550
NBS1 was almost completely absent while the phosphorylation of CHK1 was increased at a level 551
and with kinetics similar to that seen in DMSO treated cells. The phosphorylated form and 552
slower migrating pattern of CHK2 were reduced though not completely abrogated in the 553
presence of KU55933, indicating that ATM but not ATR mediated signals were suppressed as 554
expected (Fig.5B, lanes 9 to 12). The inhibitory effects of KU55933 on reduction of the IκBα 555
level following induction of E1 and E2 expression was much weaker than that of Wortmannin. In 556
the presence of KU55933, reduction of the IκBα level was not observed at 8 hours but became 557
evident at 24 hours following DOX incubation, suggesting that ATM signals partially contribute 558
to reduction of the IκBα level upon E1 and E2 expression. Furthermore, activation of the NFκB 559
target genes was strongly suppressed in the presence of Wortmannin upon induction of E1 and 560
E2 expression, corresponding with the level of IκBα (Fig.5C, left panel). Similar to the 561
overexpression of IκBα, the basal levels of the NFκB target genes in the absence of DOX were 562
significantly lower in the presence of Wortmannin than that in a control. The activation of NFκB 563
target genes was also suppressed in the presence of KU55933. The difference in those mRNA 564
levels between the presence and absence of KU55933 was statistically significant. However, the 565
suppression by KU55933 treatment was not as potent as by Wortmannin treatment, consistent 566
with the extent of the IκBα reduction (Fig.5C, right panel). The levels of the NFκB target genes 567
in the absence of DOX were comparable in the presence and absence of KU55933. The levels of 568
viral early transcripts containing E1^E4 spliced ORF were comparable between DOX treated and 569

28
untreated samples regardless of the presence of the inhibitors. These results suggested that the 570
activation of DDR upon E1 expression results in activation of NFκB, most likely by promoting 571
IκBα degradation. We also examined the effects of Wortmannin and KU55933 on E1-dependent 572
replication of the HPV16 genome (Fig.5D). The viral copy number following DOX incubation 573
was significantly higher in the presence of Wortmannin than that in a DMSO control. On the 574
other hand, they were only slightly higher in the presence of KU55933 following DOX 575
incubation, without statistical significance. The copy number of HPV16 genomes without DOX 576
incubation was comparable among the cells treated with inhibitors and a DMSO control. These 577
results suggested that activation of DDR results in suppression of E1-dependent replication of 578
the viral genome and this suppression is primarily mediated by DDR pathways other than the 579
ATM mediated pathways. Furthermore, when the effects of Wortmannin and KU55933 were 580
investigated, reduction of cell numbers after incubation with DOX was observed regardless of 581
the presence of inhibitors (Fig.5E). Given the fact that Wortmannin as well as KU55933 582
treatment itself modestly inhibited cell proliferation in the absence of DOX, it was not clear 583
whether activation of DDR is involved in inhibitory action of E1 on cell proliferation. Similar 584
results were obtained upon treatment of those inhibitors to parental cell populations of tetON 585
E1+E2 and some of other clones (data not shown). Importantly, the effects of Wortmannin and 586
KU55933 on E1-dependent replication of the viral genome were well correlated with those on 587
reduction of the IκBα level and proved collinear with overexpression of IκBα (Fig.4). 588
Collectively, these results suggest that activation of DDR signals limits E1-dependent replication 589
of HPV16 genome through activation of NFκB. The subtle effects of KU55933 on E1-dependent 590
replication and suppression of cell growth were consistent with previous reports (12, 13). 591

29
We noted that the protein level of E1 was highest at 8 hours following DOX incubation and 592
then gradually decreased, despite the continued presence of DOX (Fig.5B, lanes 1 to 4). In 593
contrast, such decrease in the E1 protein level was completely blocked and accumulation during 594
the observation period was apparent up to 24 hours with Wortmannin treatment (Fig.5B, lanes 5 595
to 8). The decrease in the E1 level was modestly alleviated by KU55933 treatment (Fig.5B, lanes 596
9 to 12). The results were reproducible in several independent experiments. These observations 597
led us to hypothesize that the DDR-dependent activation of NFκB disturbs the protein stability of 598
E1. To eliminate the possibility that Wortmannin somehow enhanced transcription of E1 from a 599
tetracycline inducible promoter, the mRNA level of exogenous HA-E1 was analyzed in the 600
presence or absence of Wortmannin (Fig.5F). The mRNA level of HA-E1 at 24 hours following 601
DOX induction was comparable between the cells treated with Wortmannin and DMSO, 602
indicating that the higher level of E1 protein in the presence of the inhibitor was not due to 603
enhanced transcription. We also considered the possibility that lysis of E1 is partial under our 604
lysis conditions and that treatment of Wortmannin may alter solubility of E1. To address whether 605
our lysis method is responsible for the seemingly increased level of E1 in the presence of 606
Wortmannin, we lysed an insoluble fraction in an SDS sample buffer after extraction of a soluble 607
fraction and the levels of E1 were analyzed in those fractions from equal numbers of cells. In 608
addition, the cells were directly lysed in an SDS sample buffer for comparison (Fig.5G). 609
Minichromosome maintenance protein (MCM) 2, histone H3 and vinculin were also detected to 610
indicate solubility of nuclear, chromatin and cytoplasmic proteins, respectively, under these 611
conditions. Under our lysis conditions, E1 was detected mostly in the soluble fraction (Fig.5G, 612
lanes 1, 3 and 5) and very little E1 remained in the insoluble fraction (Fig.5G, lanes 2, 4 and 6). 613
MCM2 and vinculin were detected mostly in the soluble fraction while histone H3 was detected 614

30
in both. All proteins were well extracted after direct lysis in an SDS sample buffer as expected 615
(Fig.5G, lanes 7 to 9). The level of E1 was higher in the presence of Wortmannin compared to 616
the vehicle treated control regardless of the lysis methods tested, demonstrating that the 617
increased level of E1 was not due to increased solubility (Fig.5G, lanes 3, 5, 8 and 9). 618
ATR, but not DNA-PKcs and p38 mitogen-activated protein kinase (p38MAPK), is 619
involved in E1-dependent activation of NFκB 620
It is reported that p38MAPK activated upon UV exposure induces NFκB activation through 621
casein kinase II (CKII)- and p38MAPK-dependent phosphorylation of IκBα at its C-terminal 622
proline, glutamic acid, serine and threonine-rich (PEST) domain (45, 46). UV induced activation 623
of p38MAPK is thought to be independent of ATM. Because our results suggested that E1 624
induces reduction of the IκBα level by eliciting DDR and this reduction is not primarily mediated 625
by kinase activity of ATM, we questioned whether p38MAPK might be involved. Activation of 626
p38MAPK was investigated using an antibody specific to phosphorylated p38MAPK at T180 627
and Y182, an indicator of p38MAPK activation. Phosphorylation of p38MAPK was increased 628
following incubation with DOX in the pooled cell populations of tetON E1+E2 and tetON E1 629
(Fig.6A) and in clones of tetON E1+E2 cells (Fig.6B, lanes 1 and 2), while it remained 630
unaffected in the cell populations of tetON E1K483A+E2 and tetON E2. These results suggested 631
that E1 induces activation of p38MAPK in a manner dependent on its helicase activity, akin to 632
the activation of DDR. To investigate if the E1 induced activation of p38MAPK is mediated by 633
DDR, the effects of Wortmannin and KU55933 on p38MAPK was examined in clone B2 634
(Fig.6B). In the presence of KU55933, the phosphorylation of p38MAPK was increased to a 635
comparable level as in the presence of the DMSO control upon incubation with DOX, indicating 636

31
that the E1-induced activation of p38MAPK is independent of ATM. Phosphorylation of 637
p38MAPK was increased during 2 hours of pre-incubation with Wortmannin. Although further 638
accumulation of the phosphorylated form was not observed after incubation with DOX, it was 639
not clear whether the E1-induced activation of DDR other than ATM is involved in the 640
activation of p38MAPK. We also examined activation of DNA-PKcs upon E1 and E2 expression 641
as phosphorylation of DNA-PKcs at Serine 2056 (S2056) has been shown to occur in response to 642
DNA damage (47). Phosphorylation of DNA-PKcs was indeed increased upon E1 and E2 643
expression and this was abrogated in the presence of Wortmannin (Fig.6B). The treatment of 644
KU55933 showed little to no effect on the phosphorylation of DNA-PKcs, suggesting that the 645
activation of DNA-PKcs upon E1 and E2 expression is independent of ATM. To explore which 646
one of these molecules mediates the E1 dependent activation of NFκB, the levels of E1 and IκBα 647
at 24 hours after addition of DOX were investigated in the presence of VE-822, Nu7026 and 648
SB203580, specific inhibitors of ATR, DNA-PKcs and p38MAPK, respectively (Fig.6C). In 649
comparison with a vehicle treated control, increased levels of E1 were detected in the presence of 650
Wortmannin and VE-822, among those inhibitors, implying that ATR activity is involved in 651
regulation of the protein stability of E1 (Fig.6C, bottom panel for quantification). On the other 652
hand, apparent inhibition of IκBα reduction was only seen in the presence of Wortmannin. To 653
further analyze the effects of VE-822 on the levels of IκBα and E1, a time course experiment 654
was carried out with HCK1T-HPV16 tetON E1+E2 clone B2 as described in Fig.5. In the 655
presence of VE-822, the phosphorylation of CHK1 was almost completely abrogated, verifying 656
that VE-822 indeed inhibits ATR activity. Importantly, progressive decrease of E1 as well as 657
IκBα levels following DOX addition was prevented, at least till 16 hours, then became evident at 658
24 hours after DOX addition in the presence of VE-822, suggesting that ATR is involved in the 659

32
E1-dependent activation of NFκB and subsequent destabilization of E1. We then examined E1-660
dependent replication of the HPV16 genome and found it was enhanced in the presence of VE-661
822, compared to the control. The basal copy number of HPV16 genomes was also slightly 662
increased in the absence of DOX (Fig.6E). On the other hand, inhibition of cell proliferation was 663
still seen in the presence of VE-822 upon expression of E1 and E2 (Fig.6F). Because the 664
treatment of VE-822 itself modestly suppressed cell proliferation in the absence of DOX, it was 665
not clear whether activation of ATR mediates inhibition of cell proliferation by E1 expression. 666
We found no difference in the levels of E1 and IκBα examined by time course experiments and 667
in E1-dependent replication of the viral genomes in the presence of Nu7026 or SB203580 668
compared to the control (data not shown). The effect of VE-822 in inhibiting reduction of the 669
IκBα level was more potent than that of KU55933 but not Wortmannin, suggesting that ATR and 670
ATM play major and minor roles, respectively, in the E1-dependent activation of NFκB. Since 671
the expression level of E2 could also affect viral genome replication, we analyzed the stability of 672
E2 in the presence of Wortmannin and VE-822 (Fig.6G). To this end, we generated a 673
heterogeneous cell population of HCK1T-HPV16 with tetON inducible expression of HA-E1 674
and 3xFLAG-tagged E2 by lentivirus gene transfer, as HA-tagged E2 was very difficult to detect 675
in our hands. HCK1T-HPV16 tetON HA-E1 and FLAG-E2 induced replication of the HPV16 676
genome as well as original tetON E1+E2 cells did with DOX incubation (data knot shown). A 677
time course experiment revealed that the expression level of FLAG-E2 was overall constant for 678
at least 24 hours and not affected by the presence of Wortmannin or VE-822, indicating that 679
DDR and/or NFκB induced-destabilization is specific to E1. All results could be replicated with 680
clone A5 (data not shown). 681

33
Activation of NFκB mediates DDR induced-destabilization of E1 and promotes 682
polyubiquitination of E1. 683
To address whether the activation of NFκB indeed mediates DDR induced destabilization of 684
E1, a time course experiment was performed with HCK1T-HPV16 tetON E1+E2 clone B2 685
transduced with IκBαWT, IκBαMT or MCS. Following DOX incubation, phosphorylation of 686
CHK1 was increased and a slower migrating pattern of NBS1 appeared, with kinetics and levels 687
comparable among the cells transduced with MCS, IκBαWT and IκBαMT (Fig.7A). These 688
results indicated that over-expression of IκBα does not disrupt the activation of ATM or ATR 689
mediated DDRs. As expected, progressive decrease of the E1 level was evident in the MCS 690
control cells while this was alleviated in the cells expressing IκBαMT, though accumulation of 691
E1 protein was not so evident as that in Wortmannin-treated cells. In the cells expressing 692
IκBαWT, progressive decrease of the E1 level was also observed, but the level remained 693
consistently higher than that in the MCS control cells. The mRNA levels of HA-E1 were 694
comparable between the MCS control and cells expressing IκBαWT, while it was modestly 695
decreased in cells expressing IκBαMT (Fig.7B), indicating that the protein level of E1 in 696
IκBαMT expressing cells was increased independently of transcriptional up-regulation. 697
Furthermore, we examined the effects of a proteasome inhibitor, Epoximicin, on the protein 698
levels of HA-E1 and IκBα (Fig.7C). Because treatment of Epoximicin caused significant damage 699
to cells regardless of DOX incubation, cell lysates of HCK1T-HPV16 tetON E1+E2 clone B2 700
were isolated in the presence or absence of Epoximicin at 12 hours after incubation with vehicle 701
or DOX, the earliest possible time at which destabilization of E1 could be detected. As expected, 702
the protein level of E1 was elevated in the presence of Epoximicin compared to the control 703
(Fig.7C, lanes 2 and 4). Proteasome inhibitors have been shown to inhibit degradation of IκBα 704

34
and subsequent activation of NFκB (48). Although the treatment with Epoximicin somehow 705
reduced the steady state level of IκBα in the absence of DOX, further reduction of IκBα upon E1 706
and E2 expression was abrogated in the presence of Epoximicin. These results could be 707
replicated with clone A5 (data not shown). While the findings were compatible with our 708
hypothesis that activation of DDR-NFκB may induce proteasomal degradation of E1, 709
involvement of proteasomes in destabilization of E1 needed to be further clarified. Therefore, we 710
carried out an ubiquitin conjugation assay using 293FT cells to directly analyze the 711
polyubiquitination of E1. A lentivirus vector expressing HA-E1 under control of the CMV 712
promoter, CSII-CMV-HA16E1, and an expression vector of 6xHis tagged ubiquitin (pCMV-713
6xHis-Ub) were co-transfected into 293FT cells with an expression vector of IκBαMT or an 714
empty vector and cell lysates were collected at 48 hours thereafter. The HA-E1 was 715
immunoprecipitated using magnetic beads conjugated with anti-HA antibodies and subsequently 716
analyzed by Western blotting. The effect of Wortmannin was investigated by incubation from 20 717
hours post-transfection till harvesting. The GFP expression vector was also included in all 718
transfections to monitor the efficiency of the transfections. We found that E1 expression did not 719
conspicuously reduce the IκBα level in 293FT cells (Fig.7D, lanes 7 and 8). To rule out the 720
possibility that co-expression of His-tagged ubiquitin hinders the E1-induced reduction of IκBα, 721
293FT cells were also transfected without pCMV-6xHis-Ub. Regardless of co-expression of His-722
tagged ubiquitin, reduction of the IκBα level was barely detected and effects of Wortmannin on 723
levels of E1 and IκBα were inconspicuous (Fig.7D, lanes 1, 2, 3 and 7, 8, 12). To potentiate 724
NFκB activation in 293FT cells expressing E1, TNFα was added at 20 hours-post transfection. 725
As expected, this TNFα treatment reduced the IκBα level and, to lesser extent, that of E1, 726
compared to the vehicle treated control (Fig.7D, compare lane 2 with lane 4 and lane 8 with lane 727

35
10). In cells co-transfected with an IκBαMT expression vector, the level of E1 appeared lower, 728
probably due to the lower efficiency of the transfection as indicated by GFP expression (Fig.7D, 729
lanes 5, 6, 11 and 12). Nonetheless, protein ladders were detected in the immunoprecpitates at a 730
size above 100 kDa, bigger than the E1 (around 70kDa), indicating polyubiquitination of E1 731
(Fig.7D right). Strikingly, TNFα treatment strongly enhanced polyubiquitination of E1 and this 732
was abolished with the expression of IκBαMT (Fig.7D, lanes 16 and 18). Consistent with the 733
results of Western blotting, effects of Wortmannin on polyubiquitination of E1 were barely 734
detected. These results suggest that activation of NFκB pathway promotes ubiquitin-dependent 735
proteasomal degradation of E1, though the precise mechanisms remained unclear. 736
E1 and E2 expression results in accumulation of cells in S and G2 phases. 737
Several studies have shown that expression of E1 and E2 results in accumulation of cells in S 738
and G2 phases (11-13). To investigate a temporal relationship between the levels of E1 and the 739
cell cycle progression in our system, the cell cycle profile was analyzed at the indicated time 740
points following DOX addition in clones B2 (Fig.8). Accumulation of cells in S and G2/M 741
phases was already evident at 8 hours and increased by 16 hours following induction of E1 and 742
E2 expression. The percentage of the cells in S phase was consistently higher (32% in S and 23% 743
in G2 at 8 hours) than that in G2 phase until 16 hours after DOX addition (46% in S and 26% in 744
G2 phase at 16 hours), suggesting that expression of E1 and E2 delayed S phase progression, 745
consistent with previous reports (12, 13). A proportion of the cells progressed to G2 phase by 24 746
hours. The percentage of the cells in G2 phase (40%) exceeded that of the cells in S phase (33%) 747
at 24 hours and remained consistently higher up to 40 hours with incubation of DOX. Cells 748
containing less than 2N DNA (sub-G1) appeared at 24 hours, coinciding with the detection of 749

36
cleaved forms of PARP-1 and caspase-3 (Fig.1F). These results were replicated with clone A5 750
(data not shown). Taken together with the observation that the level of E1 was highest between 8 751
and 16 hours and progressively reduced by 24 hours (Fig.4, 5 and 6), these results suggest that 752
E1 and E2 expression initially induces cell cycle retardation in S phase, which is attenuated as 753
the level of E1 reduces. 754
NFκB activity regulates copy number of the HPV genome during maintenance replication. 755
As mentioned above, endogenous levels of the NFκB target genes were consistently higher in 756
human keratinocytes stably maintaining episomal HPV genomes than in HCK1T cells (Fig.3C 757
and D). We also noted a slight increase of the HPV16 genome copy number in HCK1T-HPV16 758
tetON E1+E2 cells introduced with IκBαMT compared to MCS in the absence of DOX (Fig.4D). 759
Since our results suggested that NFκB activation limits E1-dependent replication, we postulated 760
that NFκB activity could be involved in maintenance of the HPV genome in proliferating 761
keratinocytes. Therefore, IκBαMT was introduced into parental HCK1T-HPV16 by retrovirus 762
gene transfer as described in Fig.4 and the copy number of HPV16 genomes was measured at 5 763
days after drug selection was completed (Fig.9A, black columns). As expected, the copy number 764
was increased by 30% with statistical significance in the cells expressing IκBαMT compared to 765
the MCS control. Conversely, the copy number of HPV16 genomes was decreased by 766
approximately 50% with incubation of TNFα for 5 days. The copy number of the HPV31 767
genome in CIN612-9E cells was also decreased by TNFα in a dose dependent manner (Fig.9B, 768
black columns). While expression of IκBαMT did not affect cell proliferation, TNFα treatment 769
modestly suppressed cell proliferation in both cell lines (Fig.9A and B, white columns). These 770

37
results imply that constitutive activation of NFκB in the presence of episomal HPV is involved in 771
regulation of maintenance replication of the viral genome. 772
773

38
Discussion 774
Papillomaviruses undergo three phases of genome replication, the initial amplification, 775
maintenance replication and productive amplification (38). While the underlying mechanisms for 776
the differentiation-dependent productive amplification are becoming gradually clear, virtually 777
nothing is known about the molecular mechanisms for transition from the initial amplification to 778
maintenance replication within the basal compartment. Our results showed that NFκB is 779
activated when E1-dependent replication of the HPV16 genome is induced in undifferentiated, 780
basal-like human cervical keratinocytes and inhibition of this NFκB activation enhances E1-781
dependent replication of the HPV16 genome. Thus, our results suggest that E1 and NFκB may 782
form a negative feedback loop to regulate E1-dependent replication of the HPV16 genome 783
(Fig.9C). The stability of E1 was augmented when activation of NFκB was abrogated in the 784
presence of DDR inhibitors or IκBαMT and activation of NFκB by TNFα facilitated 785
polyubiquitination of E1, suggesting that DDR-NFκB signaling may promote degradation of E1. 786
Furthermore, constitutive activation of NFκB was evident in human keratinocytes stably 787
maintaining episomal HPV genomes and inhibition or potentiation of the NFκB activity 788
respectively resulted in increase or decrease of the viral copy numbers (Fig.9A and B). Based on 789
these results, we propose a possible model featuring involvement of an E1-NFκB feedback loop 790
in the establishment and maintenance of persistent HPV infection. This feedback loop could be 791
further ensured by transcriptional inhibition of the viral early promoter, which drives the 792
expression of early genes including E1, by active NFκB. To our knowledge, this is the first 793
evidence implicating NFκB activity in regulation of papillomavirus genome replication. 794
Activation of NFκB by E1 through DDR 795

39
Our results showed that NFκB is activated as a consequence of E1-induced DDR, primarily 796
via the ATR mediated pathway (Fig.6). These results were rather surprising to us since the best 797
studied pathway of NFκB activation upon DNA damage is ATM-mediated induction of IκBα 798
degradation. ATM has been shown to activate the IκB kinase (IKK) complex by directly 799
phosphorylating its regulatory subunit, the NFκB essential modulator (NEMO), and this 800
activation of IKK leads to phosphorylation-dependent degradation of IκBα (49). Interestingly, it 801
was recently shown that degradation of NEMO is enhanced in human keratinocytes containing 802
episomal HPV16 genomes, compared to primary human keratinocytes (50). Therefore, activation 803
of NFκB by the ATM-mediated pathway through NEMO could be inherently limited in HCK1T-804
HPV16 cells. We failed to detect phosphorylation of the IKK complex upon E1 and E2 805
expression and found that an inhibitor of IKKs, IKK inhibitor VII, had no effect on E1-806
dependent replication (T.Nakahara and T.Kiyono, unpublished data). These results are consistent 807
with a modest effect of KU55933 on reduction of the IκBα level, as the ATM mediated 808
activation of NFκB was shown to be dependent of IKKs. The absence of IKK phosphorylation 809
also implies that ATR mediated reduction of IκBα might be independent of IKKs. Similar to our 810
results, it was reported that degradation of IκBα was induced by doxorubicin, a DNA damaging 811
agent, in mouse embryonic fibroblasts (MEFs) devoid of IKKs and this was independent of 812
phosphorylation at S32 and S36 as well as the PEST domain of IκBα. LY294002, a PI3K 813
inhibitor, partially blocked the degradation of IκBα (51). Whereas the level of IκBα was 814
maintained throughout a 24 hours observation period in the presence of Wortmannin, it was 815
decreased after 16 hours following DOX addition even though phosphorylation of CHK1 was 816
more efficiently suppressed throughout in the presence of VE-822 (Fig.6D). The greater effect of 817
Wortmannin in inhibiting E1-induced reduction of IκBα could be due to its pleiotropic effects, 818

40
suppressing both ATM and ATR pathways. Alternatively, it is possible that other PI3Ks are 819
involved. Since no data are available directly implicating ATR in NFκB activation in the 820
literature thus far, further studies are necessary to elucidate the mechanism(s) for ATR mediated 821
activation of NFκB. We also found that E1 induces activation of p38MAPK in a manner 822
dependent on its helicase activity but independent of DDR (Fig.6). Although it is beyond a scope 823
of our current study, the significance of p38MAPK activation in the HPV life cycle warrants 824
future investigation. 825
Inhibition of E1-dependent replication of the viral genome by NFκB. 826
The time course experiments in the present study revealed that the expression level of E1 827
progressively decreased and this was abrogated in the presence of Wortmannin, VE-822 and 828
IκBαMT expression, suggesting that activation of NFκB causes destabilization of E1 (Fig.5, 6 829
and 7). Since the results with a proteasome inhibitor, Epoxicimin, did not provide a direct answer 830
to whether NFκB dependent-reduction of E1 is mediated by an ubiquitin-proteasome pathway, 831
we carried out an ubiquitin conjugation assay in 293FT cells and found polyubiquitination of E1 832
to be greatly enhanced by TNFα treatment (Fig.7D). Thus, it is likely that NFκB promotes 833
ubiquitin-dependent proteasomal degradation of E1. On the other hand, E1 expression scarcely 834
induced reduction of IκBα in 293FT cells. We observed increased phosphorylation of ATM and 835
CHK1 in 293FT cells expressing E1 (data not shown). The lack of conspicuous effect of E1 836
expression on the IκBα level in 293FT cells might be due to the presence of adenovirus E1A, 837
since it has been shown to suppress IκBα degradation (52), or to a cell type difference 838
downstream of DDR activation. It was previously shown that E1 of BPV-1 is targeted by 839
anaphase-promoting complex (APC) for degradation (53, 54). Although the question of whether 840

41
E1 proteins of HPVs, including HPV16, are also targeted by APC has not been resolved, it is 841
possible that the reduction of HPV16 E1 seen in this study was mediated by APC as HPV16 E1 842
contains three putative D-boxes (RxxL) and one KEN box, known recognition motifs for APC. 843
APC is a multicomponent ubiquitin ligase well known to regulate the cell cycle. Several recent 844
studies have indicated that APC is activated upon DNA damage in ATM-dependent as well as 845
ATM-independent manners (55-57). Although it is not clear whether NFκB was involved in the 846
DDR induced APC activation in these studies, it is reasonable to speculate that E1-dependent 847
activation of DDR induces activation of APC, possibly through NFκB. The fact that Wortmannin 848
treatment completely blocked the reduction of E1 while over-expression of IκBαMT blocked 849
reduction of E1 somewhat less efficiently suggests that NFκB-dependent as well as -independent 850
mechanisms for E1 degradation may exist downstream of DDR (Fig.5, 6 and 7). In the BPV1 851
study, polyubiquitination of BPV1 E1 was enhanced in the presence of BPV1 E2 and a plasmid 852
containing the replication origin of BPV-1 in vivo. We observed that a majority of the cells 853
accumulated in S phase by 16 hours and a proportion of these cells progressed to G2 phase by 24 854
hours following DOX treatment, indicating that the degradation of E1 in our experiments most 855
likely occurs during S phase in the presence of E2 and the viral genome (Fig.8). These results are 856
consistent with the finding that polyubiqutination of BPV-1 E1 is facilitated in the S phase 857
extract of 293 cells in vitro. Therefore, it seems likely that APC-mediated degradation of BPV-1 858
E1 in vivo also occurs in S phase and APC is activated by E1-induced DDR. However, our 859
results do not rule out the possibility that other E3 ubiquitin ligases are involved in DDR and/or 860
NFκB-dependent degradation of E1. Whereas several studies have highlighted a significance of 861
ATM activation for productive amplification, contribution of ATR activation to the HPV life 862
cycle has not been indicated. Our results also suggest that ATR may negatively regulate viral 863

42
genome replication through NFκB activation. Previously, King et al. reported that activation of 864
DDR by etoposide had no effect on E1-dependent replication in a transient replication assay 865
using 293T cells (58). Their results are in agreement with our finding of a minor effect of ATM 866
since etoposide primarily induces DSBs by inhibiting topoisomerase II (59). However, they 867
showed elevated phosphorylation of CHK1 by etopside treatment suggesting that ATR was also 868
activated. The limited effect of ATR activation on E1-dependent replication in their study could 869
be because 293T cells were used. The effect of E1 expression on IκBα reduction in 293FT cells 870
was likewise minor in our hands. On the other hand, Edwards et al. recently showed that 871
knockdown of ATR by siRNA and treatment with a CHK1 inhibitor for 2 days resulted in 872
reduction of the viral genome in W12 cells, isolated from a CIN1 biopsy containing HPV16, 873
contrary to our results (60). Since we incubated VE-822 at most for 26 hours, the longer duration 874
and/or differences in approaches to inhibition of the ATR-CHK1 pathway might have affected 875
the results through an NFkB-independent pathway. 876
It was shown recently that DNA damage induces simultaneous activation of NFκB and IRF7 877
and that IFN α and λ, but not ß, were subsequently produced in HeLa cells (61). Several studies 878
have demonstrated that chronic IFNß treatment reduces episomal copies of papillomavirus 879
genome in W12 cells for HPV16, in CIN612 cells for HPV31 and in mouse C127 cells 880
containing BPV-1(62-65). Conversely, it was found that IFN inducible genes were down-881
regulated in cells containing an episomal HPV genome and that loss of viral episomes is 882
associated with endogenous activation of type I IFN-induced antiviral genes (30, 66). The gene 883
product of IFN-induced protein with tetratricopeptide repeats 1 (IFIT1, a.k.a. ISG56) was 884
demonstrated to inhibit E1-dependent replication of viral DNA by direct binding to HPV18 E1 in 885
transient replication assays (67, 68). Our results do not exclude the possibility that E1-dependent 886

43
activation of NFκB results in up-regulation of type I IFNs and these subsequently inhibit the 887
activity of E1 with regard to viral genome replication. This might explain our finding that 888
expression of IκBαMT enhanced E1-dependent replication of the viral genome very well while it 889
attenuated the degradation of E1 less efficiently than did Wortmannin treatment. 890
A possible function of the NFκB-mediated feedback loop in the viral life cycle 891
In our previous study, we demonstrated that E1 of HPV16 is required for initial and productive 892
amplification while it is dispensable for maintenance replication. Interestingly, Hoffmann et al. 893
reported that HPV16 genomes in W12 cells are maintained by replicating in a once-per-S-phase 894
fashion while HPV31 genomes in CIN612-9E cells are maintained by a random-choice 895
mechanism and introduction of E1 expression from an exogenous promoter in W12 cells 896
switched HPV16 genome replication to a random-choice mechanism. The authors concluded that 897
there are two modes of genome replication, at least for HPV16 during the maintenance phase, 898
and choice of modes is associated with the expression level of E1 (69). In agreement with 899
previous studies (11, 12), our results also showed that excess expression of E1 could be 900
detrimental to cell proliferation in basal-like cervical keratinocytes (Fig.1). Thus, regulation of 901
the E1 protein level appeared to be an essential element in the viral life cycle for establishing 902
viral persistence and determining the mode of viral genome replication. Results of our present 903
study suggest that NFκB activity is involved in regulation of the E1 level and that NFκB 904
activation is induced by E1 itself, constituting a negative feedback loop in proliferating 905
keratinocytes. We propose that E1-dependent initial amplification following virus infection 906
induces NFκB activation and this in turn terminates the initial amplification by inducing 907
degradation of E1, mediating a transition to maintenance replication. Although the expression 908

44
level of exogenous E1 in our experiments could be much higher than that during initial 909
amplification, a lower level of E1 might be sufficient to induce NFκB activation. Karim et al. 910
reported that increase in NFκB activity was detected within 24 hours after infection with HPV16 911
virions isolated from raft cultures (50). Although their interpretation was that NFκB is activated 912
as part of an innate immune response, their results did not exclude the possibility that E1-913
dependent initial amplification contributes to activation of NFκB. Indeed, it is plausible that 914
innate immune responses to virus infection potentiate E1-induced activation of NFκB during the 915
initial amplification. In cervical keratinocytes stably maintaining episomal HPV genomes, 916
constitutive activation of NFκB was detected and manipulation of NFκB activity resulted in 917
increase or decrease of the viral copy numbers (Fig.3 and Fig.9). Although we cannot provide 918
direct evidence that the endogenous E1 level was changed by such manipulation, given that an 919
antibody sufficiently sensitive to detect endogenous E1 has yet to be developed, the results imply 920
that endogenous E1, if present, may induce constitutive activation of NFκB in the presence of 921
episomal HPV genomes and the NFκB activity regulates viral copy numbers through limiting or 922
completely abolishing E1. Thus, we speculate that NFκB activity may play a key role in 923
determining the mode of maintenance replication. Since several groups have shown that E1s of 924
various papillomaviruses induce activation of DDR (11-13), we believe that a negative feedback 925
loop between E1 and NFκB might constitute a common mechanism to regulate E1 levels in the 926
basal compartment of the infected tissue. Interestingly, it has been reported that NFκB is up-927
regulated in basal cells of a fraction of CIN1 when compared with normal tissues (22). The 928
reported percentage NFκB positivity in low grade cervical lesions has varied from study to study; 929
some authors have indicated up-regulation of NFκB is detected only in cervical cancer while 930
others indicated that it is already present in the basal compartment in 30% of CIN1 (17, 19). The 931

45
varied patterns of NFκB activation in the basal compartment of CIN1 may reflect the presence or 932
absence of E1 during the stable maintenance phase. It is also possible that expression of other 933
viral proteins such as E6 contributes to constitutive activation of NFκB in the presence of HPV 934
genomes, as E6 of HPV16 has been shown to induce NFκB by at least two mechanisms (28, 70). 935
Recently, Gunasekharan and Laimins reported that a cellular microRNA, miR-145, contains a 936
seed sequence matching E1 and E2 ORFs of HPV31 and that its over-expression resulted in 937
decrease of viral copy numbers in CIN612-9E under undifferentiating as well as differentiating 938
culture conditions. The level of miR-145 was found to be high in a monolayer culture but 939
suppressed upon differentiation, suggested that miR-145 suppresses E1 and/or E2 levels in basal 940
cells and that the suppression is removed upon differentiation. Interestingly, they found two 941
RELA binding sites in the promoter of miR-145. Although experimental data that NFκB 942
upregulates miR-145 has yet been provided, effects of TNFα on viral genomes could also have 943
been mediated by up-regulation of miR-145 in the case of CIN612-9E. Together with our results, 944
these studies clearly underscore an importance of NFκB activity for viral persistence of HPVs. 945
Currently, we are not sure whether NFκB activation and/or its inhibition of E1-dependent 946
replication occur during productive amplification. As NFκB activity and components of NFκB 947
pathways are intricately associated with differentiation processes in keratinocytes, the outcomes 948
as well as the pathways of NFκB activation could be different in differentiating cells. 949
Interestingly, Satsuka et al. reported that TNFα treatment slightly reduces the copy number of 950
HPV18 replicons in monolayer cultures of keratinocytes whereas it enhanced productive 951
amplification in raft cultures (71). On the other hand, it is also conceivable that NFκB activation 952
is prevented by increased levels of E6 and E7, as these may suppress NFκB activity by various 953
mechanisms (24, 30). 954

46
NFκB is a major transcription factor that up-regulates inflammatory cytokines including TNFα, 955
IL6 and IL8 which in turn activate NFκB itself. A number of studies have provided compelling 956
evidence that chronic inflammation increases risk of cancer development, including that of 957
cervical neoplasms. The ability of NFκB to suppress viral genome replication may facilitate loss 958
of episomal HPV and emergence of viral genome integration, potentiating cancer progression 959
when ectopically activated in basal cells. Clearly, uncovering NFκB functions in the viral life 960
cycle is important to elucidate molecular mechanisms underlying viral persistence and 961
pathogenesis of HPV infection. 962
963

47
Acknowledgements 964
We would like to thank Keiko Ishiyama for her technical assistance. This work was supported 965
in part by Grants-in-Aid for Young Scientists (B) (23790517 and 25860349) to T.N and a Grant-966
in-Aid for Cancer Research from the Ministry of Health, Labor and Welfare of Japan to T.Y. and 967
T.K. 968
969

48
970
971
References 972 973
1. Stanley MA. 2012. Epithelial cell responses to infection with human papillomavirus. 974 Clin Microbiol Rev 25:215-222. 975
2. Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. 2012. 976 The biology and life-cycle of human papillomaviruses. Vaccine 30:F55-F70. 977
3. Kadaja M, Silla T, Ustav E, Ustav M. 2009. Papillomavirus DNA replication - from 978 initiation to genomic instability. Virology 384:360-368. 979
4. Bergvall M, Melendy T, Archambault J. 2013. The E1 proteins. Virology 445:35-56. 980 5. Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. 2003. ATM, ATR and DNA-PK: 981
initiators of the cellular genotoxic stress responses. Carcinogenesis 24:1571-1580. 982 6. Shiloh Y, Ziv Y. 2013. The ATM protein kinase: regulating the cellular response to 983
genotoxic stress, and more. Nature Reviews Molecular Cell Biology 14:197-210. 984 7. Lovejoy CA, Cortez D. 2009. Common mechanisms of PIKK regulation. DNA repair 985
8:1004-1008. 986 8. Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA 987
damage pathway for viral genome amplification upon differentiation. PLoS pathogens 988 5:e1000605. 989
9. Gillespie KA, Mehta KP, Laimins LA, Moody CA. 2012. Human papillomaviruses 990 recruit cellular DNA repair and homologous recombination factors to viral replication 991 centers. Journal of virology 86:9520-9526. 992
10. Moody CA, Fradet-Turcotte A, Archambault J, Laimins LA. 2007. Human 993 papillomaviruses activate caspases upon epithelial differentiation to induce viral genome 994 amplification. Proc Natl Acad Sci U S A 104:19541-19546. 995
11. Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a 996 cellular DNA damage response in viral replication foci. Journal of virology 85:8981-997 8995. 998
12. Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, 999 Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-1000 phase progression and triggers an ATM-dependent DNA damage response. J Virol 1001 85:8996-9012. 1002
13. Reinson T, Toots M, Kadaja M, Pipitch R, Allik M, Ustav E, Ustav M. 2013. 1003 Engagement of the ATR-dependent DNA damage response at the human papillomavirus 1004 18 replication centers during the initial amplification. Journal of virology 87:951-964. 1005
14. Hayden MS, Ghosh S. 2012. NF-κB, the first quarter-century: remarkable progress and 1006 outstanding questions. Genes & development 26:203-234. 1007
15. Perkins ND. 2012. The diverse and complex roles of NF-κB subunits in cancer. Nature 1008 Reviews Cancer 12:121-132. 1009
16. Sun S-C, Cesarman E. 2011. NF-κB as a Target for Oncogenic Viruses. NF-kB in 1010 Health and Disease:197-244. 1011

49
17. Nair A, Venkatraman M, Maliekal TT, Nair B, Karunagaran D. 2003. NF-κB is 1012 constitutively activated in high-grade squamous intraepithelial lesions and squamous cell 1013 carcinomas of the human uterine cervix. Oncogene 22:50-58. 1014
18. Ramdass B, Maliekal TT, Lakshmi S, Rehman M, Rema P, Nair P, Mukherjee G, 1015 Reddy B, Krishna S, Radhakrishna Pillai M. 2007. Coexpression of Notch1 and NF-1016 κB signaling pathway components in human cervical cancer progression. Gynecologic 1017 oncology 104:352-361. 1018
19. Li J, Jia H, Xie L, Wang X, He H, Lin Y, Hu L. 2009. Association of constitutive 1019 nuclear factor-kappaB activation with aggressive aspects and poor prognosis in cervical 1020 cancer. International journal of gynecological cancer : official journal of the International 1021 Gynecological Cancer Society 19:1421-1426. 1022
20. Vancurova I, Wu R, Miskolci V, Sun S. 2002. Increased p50/p50 NF-κB activation in 1023 human papillomavirus type 6-or type 11-induced laryngeal papilloma tissue. Journal of 1024 virology 76:1533-1536. 1025
21. Du J, Chen GG, Vlantis AC, Xu H, Tsang RK, van Hasselt AC. 2003. The nuclear 1026 localization of NFκB and p53 is positively correlated with HPV16 E7 level in laryngeal 1027 squamous cell carcinoma. Journal of Histochemistry & Cytochemistry 51:533-539. 1028
22. Wu Z, Peng X, Li J, Zhang Y, Hu L. 2013. Constitutive Activation of Nuclear Factor 1029 κB Contributes to Cystic Fibrosis Transmembrane Conductance Regulator Expression 1030 and Promotes Human Cervical Cancer Progression and Poor Prognosis. International 1031 Journal of Gynecological Cancer 23:906-915. 1032
23. Havard L, Delvenne P, Frare P, Boniver J, Giannini S. 2002. Differential production 1033 of cytokines and activation of NF-κB in HPV-transformed keratinocytes. Virology 1034 298:271-285. 1035
24. Huang S-M, McCance D. 2002. Down regulation of the interleukin-8 promoter by 1036 human papillomavirus type 16 E6 and E7 through effects on CREB binding protein/p300 1037 and P/CAF. Journal of virology 76:8710-8721. 1038
25. Spitkovsky D, Hehner SP, Hofmann TG, Moller A, Schmitz ML. 2002. The human 1039 papillomavirus oncoprotein E7 attenuates NF-kappa B activation by targeting the Ikappa 1040 B kinase complex. The Journal of biological chemistry 277:25576-25582. 1041
26. Boccardo E, Noya F, Broker TR, Chow LT, Villa LL. 2004. HPV-18 confers 1042 resistance to TNF-α in organotypic cultures of human keratinocytes. Virology 328:233-1043 243. 1044
27. Yuan H, Fu F, Zhuo J, Wang W, Nishitani J, An DS, Chen IS, Liu X. 2005. Human 1045 papillomavirus type 16 E6 and E7 oncoproteins upregulate c-IAP2 gene expression and 1046 confer resistance to apoptosis. Oncogene 24:5069-5078. 1047
28. James MA, Lee JH, Klingelhutz AJ. 2006. Human papillomavirus type 16 E6 activates 1048 NF-κB, induces cIAP-2 expression, and protects against apoptosis in a PDZ binding 1049 motif-dependent manner. Journal of virology 80:5301-5307. 1050
29. Xu M, Katzenellenbogen RA, Grandori C, Galloway DA. 2010. NFX1 plays a role in 1051 human papillomavirus type 16 E6 activation of NFκB activity. Journal of virology 1052 84:11461-11469. 1053
30. Vandermark ER, Deluca KA, Gardner CR, Marker DF, Schreiner CN, Strickland 1054 DA, Wilton KM, Mondal S, Woodworth CD. 2012. Human papillomavirus type 16 E6 1055

50
and E 7 proteins alter NF-kB in cultured cervical epithelial cells and inhibition of NF-kB 1056 promotes cell growth and immortalization. Virology 425:53-60. 1057
31. Fontaine V, van der Meijden E, de Graaf J, ter Schegget J, Struyk L. 2000. A 1058 functional NF-κB binding site in the human papillomavirus type 16 long control region. 1059 Virology 272:40-49. 1060
32. Boulabiar M, Boubaker S, Favre M, Demeret C. 2011. Keratinocyte sensitization to 1061 tumour necrosis factor-induced nuclear factor kappa B activation by the E2 regulatory 1062 protein of human papillomaviruses. Journal of General Virology 92:2422-2427. 1063
33. Pasparakis M. 2012. Role of NF-kappaB in epithelial biology. Immunol Rev 246:346-1064 358. 1065
34. Sur I, Ulvmar M, Toftgard R. 2008. The two-faced NF-kappaB in the skin. 1066 International reviews of immunology 27:205-223. 1067
35. Narisawa-Saito M, Handa K, Yugawa T, Ohno S, Fujita M, Kiyono T. 2007. HPV16 1068 E6-mediated stabilization of ErbB2 in neoplastic transformation of human cervical 1069 keratinocytes. Oncogene 26:2988-2996. 1070
36. Hummel M, Hudson JB, Laimins LA. 1992. Differentiation-induced and constitutive 1071 transcription of human papillomavirus type 31b in cell lines containing viral episomes. 1072 Journal of virology 66:6070-6080. 1073
37. Lee JH, Yi SMP, Anderson ME, Berger KL, Welsh MJ, Klingelhutz AJ, Ozbun MA. 1074 2004. Propagation of infectious human papillomavirus type 16 by using an adenovirus 1075 and Cre/LoxP mechanism. Proc Natl Acad Sci U S A 101:2094-2099. 1076
38. Egawa N, Nakahara T, Ohno S-i, Narisawa-Saito M, Yugawa T, Fujita M, Yamato 1077 K, Natori Y, Kiyono T. 2012. The E1 protein of human papillomavirus type 16 is 1078 dispensable for maintenance replication of the viral genome. Journal of virology 1079 86:3276-3283. 1080
39. Yugawa T, Handa K, Narisawa-Saito M, Ohno S-i, Fujita M, Kiyono T. 2007. 1081 Regulation of Notch1 gene expression by p53 in epithelial cells. Molecular and cellular 1082 biology 27:3732-3742. 1083
40. Conway MJ, Alam S, Ryndock EJ, Cruz L, Christensen ND, Roden RB, Meyers C. 1084 2009. Tissue-spanning redox gradient-dependent assembly of native human 1085 papillomavirus type 16 virions. Journal of virology 83:10515-10526. 1086
41. Choo YS, Zhang Z. 2009. Detection of protein ubiquitination. Journal of visualized 1087 experiments: JoVE. 1088
42. Swindle CS, Zou N, Van Tine BA, Shaw GM, Engler JA, Chow LT. 1999. Human 1089 papillomavirus DNA replication compartments in a transient DNA replication system. 1090 Journal of virology 73:1001-1009. 1091
43. Miyamoto S. 2011. Nuclear initiated NF-kappaB signaling: NEMO and ATM take center 1092 stage. Cell research 21:116-130. 1093
44. Difilippantonio S, Nussenzweig A. 2007. The NBS1-ATM connection revisited. Cell 1094 Cycle 6:2366-2370. 1095
45. Tsuchiya Y, Asano T, Nakayama K, Kato Jr T, Karin M, Kamata H. 2010. Nuclear 1096 IKKβ is an adaptor protein for IκBα ubiquitination and degradation in UV-induced NF-1097 κB activation. Molecular cell 39:570-582. 1098
46. Kato Jr T, Delhase M, Hoffmann A, Karin M. 2003. CK2 is a C-terminal IκB kinase 1099 responsible for NF-κB activation during the UV response. Molecular cell 12:829-839. 1100

51
47. Chen BP, Uematsu N, Kobayashi J, Lerenthal Y, Krempler A, Yajima H, Lobrich 1101 M, Shiloh Y, Chen DJ. 2007. Ataxia telangiectasia mutated (ATM) is essential for 1102 DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. The 1103 Journal of biological chemistry 282:6582-6587. 1104
48. Traenckner EB, Pahl HL, Henkel T, Schmidt KN, Wilk S, Baeuerle PA. 1995. 1105 Phosphorylation of human I kappa B-alpha on serines 32 and 36 controls I kappa B-alpha 1106 proteolysis and NF-kappa B activation in response to diverse stimuli. The EMBO journal 1107 14:2876-2883. 1108
49. Shih VF, Tsui R, Caldwell A, Hoffmann A. 2011. A single NFkappaB system for both 1109 canonical and non-canonical signaling. Cell research 21:86-102. 1110
50. Karim R, Tummers B, Meyers C, Biryukov JL, Alam S, Backendorf C, Jha V, 1111 Offringa R, van Ommen G-JB, Melief CJ. 2013. Human Papillomavirus (HPV) 1112 Upregulates the Cellular Deubiquitinase UCHL1 to Suppress the Keratinocyte's Innate 1113 Immune Response. PLoS pathogens 9:e1003384. 1114
51. Tergaonkar V, Bottero V, Ikawa M, Li Q, Verma IM. 2003. IkappaB kinase-1115 independent IkappaBalpha degradation pathway: functional NF-kappaB activity and 1116 implications for cancer therapy. Molecular and cellular biology 23:8070-8083. 1117
52. Shao R, Tsai EM, Wei K, von Lindern R, Chen Y-H, Makino K, Hung M-C. 2001. 1118 E1A inhibition of radiation-induced NF-κB activity through suppression of IKK activity 1119 and IκB degradation, independent of Akt activation. Cancer research 61:7413-7416. 1120
53. Mechali F, Hsu C-Y, Castro A, Lorca T, Bonne-Andrea C. 2004. Bovine 1121 papillomavirus replicative helicase E1 is a target of the ubiquitin ligase APC. Journal of 1122 virology 78:2615-2619. 1123
54. Malcles M-H, Cueille N, Mechali F, Coux O, Bonne-Andrea C. 2002. Regulation of 1124 bovine papillomavirus replicative helicase e1 by the ubiquitin-proteasome pathway. 1125 Journal of virology 76:11350-11358. 1126
55. Sudo T, Ota Y, Kotani S, Nakao M, Takami Y, Takeda S, Saya H. 2001. Activation 1127 of Cdh1‐dependent APC is required for G1 cell cycle arrest and DNA damage‐1128 induced G2 checkpoint in vertebrate cells. The EMBO journal 20:6499-6508. 1129
56. Takahashi A, Imai Y, Yamakoshi K, Kuninaka S, Ohtani N, Yoshimoto S, Hori S, 1130 Tachibana M, Anderton E, Takeuchi T, Shinkai Y, Peters G, Saya H, Hara E. 2012. 1131 DNA damage signaling triggers degradation of histone methyltransferases through 1132 APC/C(Cdh1) in senescent cells. Molecular cell 45:123-131. 1133
57. Wiebusch L, Hagemeier C. 2010. p53- and p21-dependent premature APC/C-Cdh1 1134 activation in G2 is part of the long-term response to genotoxic stress. Oncogene 29:3477-1135 3489. 1136
58. King LE, Fisk JC, Dornan ES, Donaldson MM, Melendy T, Morgan IM. 2010. 1137 Human papillomavirus E1 and E2 mediated DNA replication is not arrested by DNA 1138 damage signalling. Virology 406:95-102. 1139
59. Heisig P. 2009. Type II topoisomerases--inhibitors, repair mechanisms and mutations. 1140 Mutagenesis 24:465-469. 1141
60. Edwards TG, Helmus MJ, Koeller K, Bashkin JK, Fisher C. 2013. Human 1142 papillomavirus episome stability is reduced by aphidicolin and controlled by DNA 1143 damage response pathways. Journal of virology 87:3979-3989. 1144

52
61. Brzostek-Racine S, Gordon C, Van Scoy S, Reich NC. 2011. The DNA damage 1145 response induces IFN. Journal of immunology 187:5336-5345. 1146
62. Pett MR, Herdman MT, Palmer RD, Yeo GS, Shivji MK, Stanley MA, Coleman N. 1147 2006. Selection of cervical keratinocytes containing integrated HPV16 associates with 1148 episome loss and an endogenous antiviral response. Proceedings of the National 1149 Academy of Sciences of the United States of America 103:3822-3827. 1150
63. Turek LP, Byrne JC, Lowy DR, Dvoretzky I, Friedman RM, Howley PM. 1982. 1151 Interferon induces morphologic reversion with elimination of extrachromosomal viral 1152 genomes in bovine papillomavirus-transformed mouse cells. Proc. Natl. Acad. Sci. U. S. 1153 A. 79:7914-7918. 1154
64. Chang YE, Laimins LA. 2000. Microarray analysis identifies interferon-inducible genes 1155 and Stat-1 as major transcriptional targets of human papillomavirus type 31. J Virol 1156 74:4174-4182. 1157
65. Chang YE, Pena L, Sen GC, Park JK, Laimins LA. 2002. Long-term effect of 1158 interferon on keratinocytes that maintain human papillomavirus type 31. J Virol 76:8864-1159 8874. 1160
66. Nees M, Geoghegan JM, Hyman T, Frank S, Miller L, Woodworth CD. 2001. 1161 Papillomavirus type 16 oncogenes downregulate expression of interferon-responsive 1162 genes and upregulate proliferation-associated and NF-κB-responsive genes in cervical 1163 keratinocytes. Journal of virology 75:4283-4296. 1164
67. Saikia P, Fensterl V, Sen GC. 2010. The inhibitory action of P56 on select functions of 1165 E1 mediates interferon's effect on human papillomavirus DNA replication. Journal of 1166 virology 84:13036-13039. 1167
68. Terenzi F, Saikia P, Sen GC. 2008. Interferon‐inducible protein, P56, inhibits HPV 1168 DNA replication by binding to the viral protein E1. The EMBO journal 27:3311-3321. 1169
69. Hoffmann R, Hirt B, Bechtold V, Beard P, Raj K. 2006. Different modes of human 1170 papillomavirus DNA replication during maintenance. Journal of virology 80:4431-4439. 1171
70. An J, Mo D, Liu H, Veena MS, Srivatsan ES, Massoumi R, Rettig MB. 2008. 1172 Inactivation of the CYLD deubiquitinase by HPV E6 mediates hypoxia-induced NF-1173 kappaB activation. Cancer cell 14:394-407. 1174
71. Satsuka A, Yoshida S, Kajitani N, Nakamura H, Sakai H. 2010. Novel human 1175 papillomavirus type 18 replicon and its application in screening the antiviral effects of 1176 cytokines. Cancer science 101:536-542. 1177
1178 1179
1180

53
Figure legends 1181
Figure.1 Conditional induction of E1-dependent replication in immortalized human 1182
cervical karatinocytes containing HPV16 genome (HCK1T-HPV16). (A) A representative 1183
image of Southern blotting for HPV16 genomes in 10 μg of total genomic DNA isolated from 1184
cells with the indicated gene expression at 24 hours after DOX (1μg/ml) addition. The serially 1185
diluted, linearized full length of HPV16 genomes were included as copy number standards (lanes 1186
1 to 3). The DNAs with BamHI digestion which linearizes the HPV16 genome, and Xho I 1187
digestion which does not cut the HPV16 genome, are shown in lanes 4 to 7 and lanes 8 to 11, 1188
respectively. Arrowheads indicate HPV16 DNAs which correspond to Form I, a supercoiled 1189
DNA, and Form II, a relaxed circular DNA. (B) qPCR results for the copy number of HPV16 1190
genomes with the indicated gene expression at 24 hours and (C) at the indicated time points 1191
following DOX incubation. (D) Indirect immunoflorescence analysis (IFA) for HA-E1 and E2. 1192
As a negative control, HCK1T-HPV16 with tetON E1+E2 incubated with a vehicle control is 1193
shown. (E) The number of cells with the indicated gene expression that stayed attached to tissue 1194
culture plates is shown. (F) Western blot analysis for the levels of E1 and the markers of 1195
apoptosis, PARP-1 and caspase-3, in HCK1T-HPV16 cells with the indicated gene expression 1196
following incubation with DOX (lanes 2, 4, 6 and 8) or vehicle as a control (lanes 1, 3, 5 and 7). 1197
Arrowheads indicate cleaved forms of PARP-1 and caspase-3. α-tubulin was detected as a 1198
loading control. The averages of results from at least three independent experiments are shown. 1199
P-values were evaluated by Student’s t-test. 1200
Figure 2. Activation of NFκB upon induction of E1 and/or E2 expression in HCK1T-1201
HPV16 cells. (A) The status of NFκBp65 (RELA) and its inhibitor, IκBα, was analyzed by 1202

54
Western blotting for HCK1T-HPV16 cells with the indicated gene expression at 24 hours after 1203
DOX addition. α-tubulin was included as a loading control. (B) Localization of RELA and HA- 1204
E1 and/or E2 was examined in HCK1T-HPV16 tetON E1+E2 by indirect IFA. A rabbit 1205
polyclonal antibody to RELA and a mouse monoclonal antibody to HA were simultaneously 1206
incubated and secondary antibodies, Alexa 488 conjugated anti-rabbit IgG and Alexa 596 1207
conjugated anti-mouse IgG, respectively, were used for detection. Nucleus was counterstained by 1208
DAPI. (C) Relative mRNA levels of indicated NFκB target genes and viral transcripts containing 1209
E1^E4 spliced ORF were measured by RT-qPCR. The mRNAs of the indicated genes were 1210
normalized to ß-actin mRNA. Fold increase of the indicated mRNAs in DOX treated samples to 1211
vehicle treated samples are shown. The averages of at least three independent experiments are 1212
shown. 1213
Figure 3. Activation of NFκB upon induction of E1 and/or E2 expression in HCK1T in the 1214
absence of an episomal HPV16 genome. (A) The level of IκBα and expression of E1 and E2 1215
were analyzed in parental HCK1T that do not contain an HPV16 genome by Western blotting. 1216
HCK1T-tetON cells were included as a negative control. Cell lysates were prepared at 24 hours 1217
following incubation with DOX (lanes 2, 4, 6 and 8) or a vehicle (lanes 1, 3, 5 and 7). Vinculin 1218
was detected as a loading control. (B) qRT-PCR analysis for mRNA levels of the NFκB target 1219
genes in HCK1T-tetON E1+E2 or E1 alone. The mRNAs of the indicated genes were normalized 1220
to ß-actin mRNA. Fold increase of the indicated mRNAs in DOX treated samples to vehicle 1221
treated samples are shown. The averages of at least three independent experiments are shown. 1222
(C) RT-PCR analysis for mRNA levels of the NFκB target genes in HCK1T, HCK1T-HPV16 1223
and CIN612-9E cells. As a negative control, an RNA isolated from HCK1T not subjected to RT 1224
reaction was used as a template for PCR reactions (lane 1). 1225

55
Figure 4. Effects of over-expression of IκBα on E1-dependent replication of HPV16 and cell 1226
proliferation of subclones of HCK1T-HPV16 cells with tetON E1+E2. (A) Shown are results 1227
of Western blot analysis (left) for E1 and E2 levels and the steady state levels of IκBα in 1228
representative clones and the results of qPCR for HPV16 genomes (right) following incubation 1229
with DOX or vehicle for 24 hours. Images with short and long exposure with the anti-HA 1230
antibody were included to indicate expression of HA-E2. α-tubulin is shown as a loading control 1231
for the top three panels and vinculin for the HA panel comparing clones to the parental 1232
population. (B) Western blot analysis to verify the over-expression of IκBα in clone B2 1233
transduced with retroviruses carrying an empty vector designated as MCS (lanes 3 and 4) or the 1234
vector expressing IκBαS32/36A mutant (IκBαMT) (lanes 5 and 6) or wild type IκBα (IκBαWT) 1235
(lanes 7 and 8). Parental HCK1T-HPV16 treated with TNFα for 15 minutes was included as a 1236
positive control for degradation of IκBα (lanes 1 and 2). Vinculin was detected as a loading 1237
control. (D) RT-qPCR analysis of NFκB target genes and viral early transcripts containing 1238
spliced E1^E4 ORF. Shown are levels of the indicated mRNAs relative to MCS control without 1239
DOX treatment. Two asterisks indicate a p-value < 0.01 for the comparison of the mRNAs in 1240
MCS with those in IκBαWT- or IκBαMT- expressing cells in the absence of DOX. Differences 1241
for all of the mRNA levels in between MCS and IκBαWT- or IκBαMT- expressing cells with 1242
DOX treatment were statistically significant (P<0.01). Highlighting statistical significance was 1243
omitted for DOX treated samples to avoid making graphs too busy. (D) The copy number of 1244
HPV16 genomes was measured by qPCR with indicated gene expression. An asterisk indicates a 1245
p-value <0.05. (E) The number of cells staying attached to tissue culture plates is shown. An 1246
asterisk indicates a p-value < 0.05. The averages of at least three independent experiments are 1247
shown. 1248

56
Figure 5. Effects of inhibitors of DDR on activation of NFκB, E1-dependent replication of 1249
HPV16 genomes and cell proliferation. (A) Western blot analysis for activation of ATM and 1250
ATR mediated DDR pathways. The left panel (lanes 1 to 8) shows the results of original cell 1251
populations of HCK1T-HPV16 with the indicated gene expression. The right panel (lanes 9 to 1252
18) shows the results for representative clones of HCK1T-HPV16 tetON E1+E2 cells. Vinculin 1253
or α-tubulin indicates a loading control in each panel. White arrowheads indicate a slower 1254
migration pattern of NBS1 and CHK2. (B) A time course experiment for DDR activation and the 1255
level of IκBα in the presence of DMSO as a control, 10 μM of Wortmannin or 2.5 μM of 1256
KU55933. (C) Transcriptional activation of NFκB target genes and the viral early transcripts was 1257
examined by RT-qPCR in the presence or absence of the inhibitors incubated with or without 1258
DOX for 24 hours. An asterisk indicates a p-value < 0.05 for the comparison of the mRNAs in 1259
between DMSO treated and Wortmannin treated cells in the absence of DOX. The mRNAs of 1260
NFκB target genes but not the viral early transcripts in Wortmannin or KU55933 treated cells 1261
compared to those in DMSO treated cells in the presence of DOX were significantly reduced 1262
(P<0.05). Highlighting p-values less than 0.05 was omitted for DOX treated samples. (D) The 1263
copy number of HPV16 genomes was measured by qPCR at 24 hours after DOX addition in the 1264
presence of the indicated inhibitors. (E) The number of cells staying attached to tissue culture 1265
plates in the presence or absence of inhibitors was counted. An asterisk indicates a p-value < 1266
0.05. (F) Relative mRNA levels of HA-E1 were analyzed by RT-qPCR in the presence or 1267
absence of Wortmannin following incubation with DOX or vehicle for 24 hours. (G) Western 1268
blot analysis for solubility of E1 in the presence of Wortmannin or vehicle. S and I indicate 1269
soluble (lanes 1, 3 and 5) and insoluble (lanes 2, 4 and 6) fractions, respectively. The averages or 1270
representative images of at least three independent experiments are shown. 1271

57
Figure 6. ATR, but not DNA-PKcs and p38MAPK, is involved in the E1-dependent 1272
reduction of IκBα. (A) Activation of p38MAPK was examined in HCK1T-HPV16 populations 1273
with the indicated gene expression. (B) Effects of Wortmannin and KU55933 on activation of 1274
p38MAPK and DNA-PKcs in clone B2. Cells lysate were prepared at 0 and 24 hours after DOX 1275
addition following pre-incubation with the inhibitors for 2 hours. Vinculin was detected as a 1276
loading control. (C) Western blot analysis for the levels of IκBα and E1 at 24 hours after 1277
incubation with DOX in the presence of indicated inhibitors. A bottom graph shows intensities of 1278
E1 with inhibitors relative to that with a vehicle control. The intensities of E1 were normalized to 1279
that of vinculin. Concentrations of inhibitors used were 10 μM for Wortmannin, 0.5 nM for VE-1280
822 (ATR inhibitor), 20 μM for Nu7026 (DNA-PKcs inhibitor), 2.5 μM for KU55933 and 10μM 1281
for SB203580 (p38MAPK inhibitor). (D) Western blot analysis for a time course experiment in 1282
the presence of Wortmannin (lanes 5 to 8), VE-822 (lanes 9 to 12) or a vehicle control (lanes 1 to 1283
4). (D) The copy number of HPV16 genomes was measured by qPCR and (E) the number of 1284
cells staying attached was examined after incubation with DOX or vehicle in the presence of 1285
DMSO or VE-822 for 24 hours. Astarisks indicate p<0.05. (G) Western blot analysis for the 1286
stability of E2 in the presence of indicated inhibitors as described in Fig.5. The averages or 1287
representative images of at least three independent experiments are shown. 1288
Figure 7. Activation of NFκB mediates destabilization of E1 and promotes 1289
polyubiquitination. (A) Time course experiments for clone B2 transduced with MCS (lanes 1 to 1290
4), IκBαWT (lanes 9 to 12) or IκBαMT (lanes 5 to 8) following incubation with DOX. A white 1291
arrowhead indicates a slower migrating pattern of NBS1. Vinculin was detected as a loading 1292
control. (B) RT-qPCR analysis for HA-E1 in the indicated cells upon DOX incubation. (C) 1293
Western blot analysis for the levels of E1 and IκBα in clone B2 incubated with vehicle (lanes 1 1294

58
and 2) or 10nM of Epoximicin (lanes 3 and 4) for 12 hours. (D) An ubiquitin conjugation assay 1295
was performed as described in the Materials and Methods. The left panel (lanes 8 to 13) shows 1296
results of Western blot analysis for input cell lysates subjected to immunoprecipitation (IP). The 1297
right panel (lanes 14 to 19) shows results of IP-WB to detect His-Ub conjugation to HA-E1. The 1298
cell lysates from 293FT cells transfected without a His-Ub expression vector (lanes 1 to 6) were 1299
also included. Wor and TNF indicate treatment with 10 μM of Wortmannin and 20 ng/ml of 1300
TNFα, respectively, from 20 hours post-transfection. IkB indicates co-transfection with an 1301
expression vector of IκBαMT. 1302
Figure 8. E1 and E2 expression induces accumulation of cells in S and G2 phases. 1303
Clones B2 were collected at indicated time points after a vehicle (above) or DOX (bottom) 1304
addition and cell cycle profiles were analyzed by flow cytometry. Representative profiles are 1305
shown. The graphs indicate the percentages of cells in the cell cycle phases. 1306
Figure 9. The effect of constitutive activation of NFκB in maintenance replication of HPV 1307
genome. (A) The copy number of HPV16 genomes in HCK1T-HPV16 transduced with 1308
retrovirus carrying an empty (MCS) or IκBαMT expressing vector was measured at 5 days after 1309
drug selection was completed. (B) The copy number of HPV16 or HPV31 was measured at 5 1310
days after incubation of TNFα at indicated concentrations (black columns). The cell proliferation 1311
was also compared (White columns). (C) A schematic presentation for a negative feedback loop 1312
between E1 and NFκB. E1 induces activation of NFκB most likely through degradation of IκBα 1313
as a consequence of DDR activation. NFκB activation in turn limits E1 dependent replication 1314
and disrupts the stability of E1 through facilitating proteasomal degradation. 1315

E1K483A+E2 E2+ ++
HP
V16
gen
ome
copy
/cel
l
B
0.1
2.5
2.7
2.9
3.1
3.3Vehicle
+Dox(24h)
E1+E2 E1K483A+E2
E1E2
p<0.05
p<0.05
(x103)
0.1
1
HCK1T-HPV16 tetON A
8Kbp
100
10 1 + + + +- - - -
E1+
E2
E1K
483A
+E2
E1+
E2
E1K
483A
+E2
Cop
y nu
mbe
rst
anda
rds
2 3 4 5 6 7 8 9 10 11
DOX (24h)
Form Ι
Form ΙΙ
Figure 1
E
+Dox(24h) Vehicle
E1+E2 E1K483A+E2
0.5
1.0
1.5
2.0
2.5
Cel
l Num
ber
(x10
5 )
p<0.05
C
HP
V16
gen
ome
copy
/cel
l
200 4 8 12 16 24
0.5
1
2
3
1.5
2.5
3.5HCK1T-HPV16 tetON E1+E2(x103)
24 48 72
Vehi
cle hr +DOXhr +DOX
F
1α-tubulin
E1
+
E1+
E2
E1K
483A
+E2
E2
- + - + - + -
HA(E1)
PARP-1
Caspase-3
DOX(24h)
2 3 4 5 6 7 8
Cleaved
Cleaved
αHA
DAPI
Merge
E1+E2 E1+-DOX(24h)
D
Figure.1 Conditional induction of E1-dependent replication in immortalized human cervical karatinocytes containing HPV16 genome (HCK1T-HPV16). (A) A representative image of Southern blotting for HPV16 genomes in 10 μg of total genomic DNA isolated from cells with the indicated gene expression at 24 hours after DOX (1μg/ml) addition. The serially diluted, linearized full length of HPV16 genomes were included as copy number standards (lanes 1 to 3). The DNAs with BamHI digestion which linearizes the HPV16 genome, and Xho I digestion which does not cut the HPV16 genome, are shown in lanes 4 to 7 and lanes 8 to 11, respectively. Arrowheads indicate HPV16 DNAs which correspond to Form I, a supercoiled DNA, and Form II, a relaxed circular DNA. (B) qPCR results for the copy number of HPV16 genomes with the indicated gene expression at 24 hours and (C) at the indicated time points following DOX incubation. (D) Indirect immunoflorescence analysis (IFA) for HA-E1 and E2. As a negative control, HCK1T-HPV16 with tetON E1+E2 incubated with a vehicle control is shown. (E) The number of cells with the indicated gene expression that stayed attached to tissue culture plates is shown. (F) Western blot analysis for the levels of E1 and the markers of apoptosis, PARP-1 and caspase-3, in HCK1T-HPV16 cells with the indicated gene expression following incubation with DOX (lanes 2, 4, 6 and 8) or vehicle as a control (lanes 1, 3, 5 and 7). Arrowheads indicate cleaved forms of PARP-1 and caspase-3. α-tubulin was detected as a loading control. The averages of results from at least three independent experiments are shown. P-values were evaluated by Student’ s t-test.
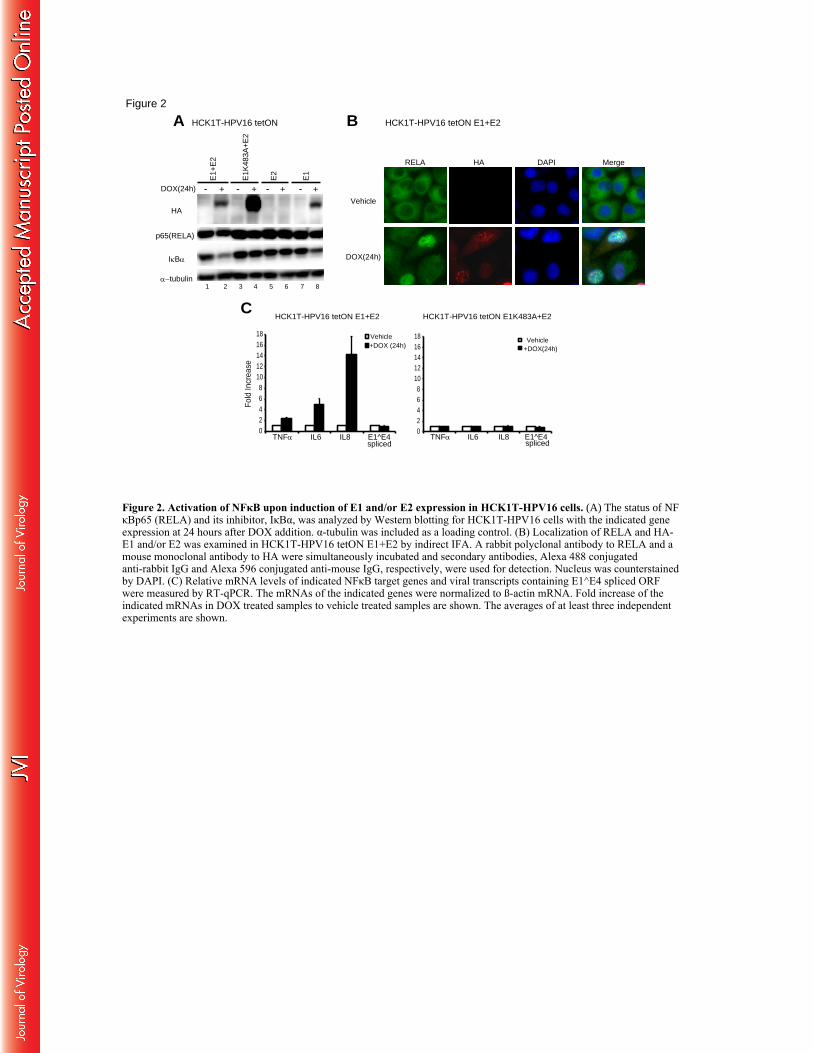
HCK1T-HPV16 tetON E1+E2
RELA HA DAPI Merge
Vehicle
DOX(24h)
02468
1012141618
TNFα IL6 IL8 E1^E4spliced
Vehicle+DOX (24h)
Fold
Incr
ease
HCK1T-HPV16 tetON E1+E2
02468
1012141618
TNFα IL6 IL8 E1^E4spliced
Vehicle+DOX(24h)
HCK1T-HPV16 tetON E1K483A+E2
B
C
A HCK1T-HPV16 tetON
HAE
1+E
2
E1K
483A
+E2
- + - +DOX(24h)
IκBα
α−tubulin
p65(RELA)
1 2 3 4
- + - +
E2
E1
5 6 7 8
Figure 2
Figure 2. Activation of NFκB upon induction of E1 and/or E2 expression in HCK1T-HPV16 cells. (A) The status of NFκBp65 (RELA) and its inhibitor, IκBα, was analyzed by Western blotting for HCK1T-HPV16 cells with the indicated gene expression at 24 hours after DOX addition. α-tubulin was included as a loading control. (B) Localization of RELA and HA- E1 and/or E2 was examined in HCK1T-HPV16 tetON E1+E2 by indirect IFA. A rabbit polyclonal antibody to RELA and a mouse monoclonal antibody to HA were simultaneously incubated and secondary antibodies, Alexa 488 conjugated anti-rabbit IgG and Alexa 596 conjugated anti-mouse IgG, respectively, were used for detection. Nucleus was counterstained by DAPI. (C) Relative mRNA levels of indicated NFκB target genes and viral transcripts containing E1^E4 spliced ORF were measured by RT-qPCR. The mRNAs of the indicated genes were normalized to ß-actin mRNA. Fold increase of the indicated mRNAs in DOX treated samples to vehicle treated samples are shown. The averages of at least three independent experiments are shown.

A HCK1T tetON
1 2 3 4 5 6 7 8
- + - + - +
IκBα
Vinculin
HA(E1)
E1+
E2
E2
E1
DOX(24h)
FLAG(E2)
p65(RELA)
- +
Vehicle +DOX (24h)
10
60
160
80
100
120
140
180
200
220
Fold
Incr
ease
TNFα IL6 IL8 TNFα IL6 IL8
E1+E2 E1
B HCK1T tetON
C
HC
K1T
HC
K1T
-HP
V16
TNFαIL6IL8
β-Actin
CIN
612-
9E
no R
T
1 2 3 4
Figure 3
Figure 3. Activation of NFκB upon induction of E1 and/or E2 expression in HCK1T in the absence of an episomal HPV16 genome. (A) The level of IκBα and expression of E1 and E2 were analyzed in parental HCK1T that do not contain an HPV16 genome by Western blotting. HCK1T-tetON cells were included as a negative control. Cell lysates were prepared at 24 hours following incubation with DOX (lanes 2, 4, 6 and 8) or a vehicle (lanes 1, 3, 5 and 7). Vinculin was detected as a loading control. (B) qRT-PCR analysis for mRNA levels of the NFκB target genes in HCK1T-tetON E1+E2 or E1 alone. The mRNAs of the indicated genes were normalized to ß-actin mRNA. Fold increase of the indicated mRNAs in DOX treated samples to vehicle treated samples are shown. The averages of at least three independent experiments are shown. (C) RT-PCR analysis for mRNA levels of the NFκB target genes in HCK1T, HCK1T-HPV16 and CIN612-9E cells. As a negative control, an RNA isolated from HCK1T not subjected to RT reaction was used as a template for PCR reactions (lane 1).

Figure 4
Figure 4. Effects of over-expression of IκBα on E1-dependent replication of HPV16 and cell proliferation of subclones of HCK1T-HPV16 cells with tetON E1+E2. (A) Shown are results of Western blot analysis (left) for E1 and E2 levels and the steady state levels of IκBα in representative clones and the results of qPCR for HPV16 genomes (right) following incubation with DOX or vehicle for 24 hours. Images with short and long exposure with the anti-HA antibody were included to indicate expression of HA-E2. α-tubulin is shown as a loading control for the top three panels and vinculin for the HA panel comparing clones to the parental population. (B) Western blot analysis to verify the over-expression of IκBα in clone B2 transduced with retroviruses carrying an empty vector designated as MCS (lanes 3 and 4) or the vector expressing IκBαS32/36A mutant (IκBαMT) (lanes 5 and 6) or wild type IκBα (IκBαWT) (lanes 7 and 8). Parental HCK1T-HPV16 treated with TNFα for 15 minutes was included as a positive control for degradation of IκBα (lanes 1 and 2). Vinculin was detected as a loading control. (D) RT-qPCR analysis of NFκB target genes and viral early transcripts containing spliced E1^E4 ORF. Shown are levels of the indicated mRNAs relative to MCS control without DOX treatment. Two asterisks indicate a p-value < 0.01 for the comparison of the mRNAs in MCS with those in IκBαWT- or IκBαMT- expressing cells in the absence of DOX. Differences for all of the mRNA levels in between MCS and IκBαWT- or IκBαMT- expressing cells with DOX treatment were statistically significant (P<0.01). Highlighting statistical significance was omitted for DOX treated samples to avoid making graphs too busy. (D) The copy number of HPV16 genomes was measured by qPCR with indicated gene expression. An asterisk indicates a p-value <0.05. (E) The number of cells staying attached to tissue culture plates is shown. An asterisk indicates a p-value < 0.05. The averages of at least three independent experiments are shown.
TNFα IL6
A HCK1T-HPV16 tetON E1+E2 clones
2
4
6
8
10
12
14
MCS IκBαMTIκBαWT
HP
V16
gen
ome
copy
/cel
l
Vehicle +Dox(24h)
*
*
Cell N
umbe
r (x1
05 )
IκBαWT IκBαMTMCS
Vehicle
+DOX (24h)
0.5
1.0
1.5
2.0
2.5 * * *
120
B
TNF α
+ MC
S
IkBα
WT
IkBα
MT
- + - + - +
Vinculin
HA(E1)
tetON E1+E2 clone B2
--DOX(24h)
1 2 3 4 6 7 85
MCS IκBαWT IκBαMT
IL8
10
100
110
130
290
300
10
280
270
260
MCS IκBαWT IκBαMT
C
D E(x103)
1
11
12
HP
V16
gen
ome
copy
/cel
l
A42 A26B3 A5B2
2
4
10
(x103)
3
5
13
14 Vehicle +DOX (24h)
Rel
ativ
e E
xpre
ssio
n 62
64
68
60
12
MCS IκBαWT IκBαMT
E1^E4 spliced VehicleDOX(24h)10
2
4
6
8
MCS IκBαWT IκBαMT
1
- + - + - + - + - +
A42
B3 A26
B2 A5
DOX(24h)
IκBα
αtubulin
HA(short)
1 2 3 4 6 7 85 109
HA(long)
E1
E2
E1
- +
Par
enta
l po
p
E1HA(short)
Vinculin11 12
** ** ** ** ** **
** **
HCK1T-HPV16
IκBα

Figure 5. Effects of inhibitors of DDR on activation of NFκB, E1-dependent replication of HPV16 genomes and cell proliferation. (A) Western blot analysis for activation of ATM and ATR mediated DDR pathways. The left panel (lanes 1 to 8) shows the results of original cell populations of HCK1T-HPV16 with the indicated gene expression. The right panel (lanes 9 to 18) shows the results for representative clones of HCK1T-HPV16 tetON E1+E2 cells. Vinculin or α-tubulin indicates a loading control in each panel. White arrowheads indicate a slower migration pattern of NBS1 and CHK2. (B) A time course experiment for DDR activation and the level of IκBα in the presence of DMSO as a control, 10 μM of Wortmannin or 2.5 μM of KU55933. (C) Transcriptional activation of NFκB target genes and the viral early transcripts was examined by RT-qPCR in the presence or absence of the inhibitors incubated with or without DOX for 24 hours. An asterisk indicates a p-value < 0.05 for the comparison of the mRNAs in between DMSO treated and Wortmannin treated cells in the absence of DOX. The mRNAs of NFκB target genes but not the viral early transcripts in Wortmannin or KU55933 treated cells compared to those in DMSO treated cells in the presence of DOX were significantly reduced (P<0.05). Highlighting p-values less than 0.05 was omitted for DOX treated samples. (D) The copy number of HPV16 genomes was measured by qPCR at 24 hours after DOX addition in the presence of the indicated inhibitors. (E) The number of cells staying attached to tissue culture plates in the presence or absence of inhibitors was counted. An asterisk indicates a p-value < 0.05. (F) Relative mRNA levels of HA-E1 were analyzed by RT-qPCR in the presence or absence of Wortmannin following incubation with DOX or vehicle for 24 hours. (G) Western blot analysis for solubility of E1 in the presence of Wortmannin or vehicle. S and I indicate soluble (lanes 1, 3 and 5) and insoluble (lanes 2, 4 and 6) fractions, respectively. The averages or representative images of at least three independent experiments are shown.
C
A HCK1T-HPV16 tetON HCK1T-HPV16 tetON E1+E2 clones
10
DMSO Wortmannin KU55933
p<0.05
Vehicle
+DOX (24h)
D
D1
2
3
4
5
HP
V16
gen
ome
copy
(x10
3 ) /c
ell
DMSO, VehicleWort 10µM, VehicleDMSO, +DOX (24h)Wort 10µM, +DOX (24h)
Rel
ativ
e Ex
pres
sion
Wortmannin
10
20
30
40
TNFα IL6 IL8 E1^E4 spliced
B
TNFα IL6 IL8 E1^E4 spliced
DMSO, VehicleKU 2.5µM, VehicleDMSO, +DOX (24h)KU 2.5µM, +DOX (24h)
KU55933
10
20
30
40
50
60
+
ATM
pCHK1(S345)CHK1
pCHK2(T68)
-
109 11 12 13 14 15 16 17 18
+ - + - + - + - +
A42
B3
A26
B2
A5
pNBS1
pCHK2
pATM(S1981)
NBS1CHK2
Vinculin8
E1+
E2
E1K
483A
+E2
E1
E2
- + - + - + -DOX(24h)
1 2 3 4 5 6 7αtubulin
pCHK2
pNBS1
NBS1
CHK2
Vinculin
CHK1
IκBα
Vinculin
HA(E1)
HCK1T-HPV16 tetON E1+E2 clone B2
0 8 16 24 0 8 16 24
DMSO Wortmannin
pCHK2(T68)
pCHK1(S345)
0 8 16 24
KU55933+DOX(h)
pNBS1
1 2 3 4 5 6 7 8 9 10 11 12
* * *
F
DMSO Wortmannin
0.1
0.2
0.3
Rel
ativ
e Ex
pres
sion
Vehicle +DOX (24h)E
DMSO Wortmannin KU55933
* **
*
0.2
0.4
0.6
0.8
1.2
1.0
1.4
1.6
* <0.05
1.8
Cel
l Num
ber (
x105 )
Vehicle +DOX (24h)
Figure 5
GHCK1T-HPV16 tetON E1+E2 clone B2
S I S I S IHA(E1)
MCM2
Histone H3
Vinculin
DOX(24h)Wortmannin
- + + + ++ +
2xSDS buffer
-
lysis buffer
1 2 3 4 5 6 7 8 9

Figure 6. ATR, but not DNA-PKcs and p38MAPK, is involved in the E1-dependent reduction of IκBα. (A) Activation of p38MAPK was examined in HCK1T-HPV16 populations with the indicated gene expression. (B) Effects of Wortmannin and KU55933 on activation of p38MAPK and DNA-PKcs in clone B2. Cells lysate were prepared at 0 and 24 hours after DOX addition following pre-incubation with the inhibitors for 2 hours. Vinculin was detected as a loading control. (C) Western blot analysis for the levels of IκBα and E1 at 24 hours after incubation with DOX in the presence of indicated inhibitors. A bottom graph shows intensities of E1 with inhibitors relative to that with a vehicle control. The intensities of E1 were normalized to that of vinculin. Concentrations of inhibitors used were 10 μM for Wortmannin, 0.5 nM for VE-822 (ATR inhibitor), 20 μM for Nu7026 (DNA-PKcs inhibitor), 2.5 μM for KU55933 and 10μM for SB203580 (p38MAPK inhibitor). (D) Western blot analysis for a time course experiment in the presence of Wortmannin (lanes 5 to 8), VE-822 (lanes 9 to 12) or a vehicle control (lanes 1 to 4). (D) The copy number of HPV16 genomes was measured by qPCR and (E) the number of cells staying attached was examined after incubation with DOX or vehicle in the presence of DMSO or VE-822 for 24 hours. Astarisks indicate p<0.05. (G) Western blot analysis for the stability of E2 in the presence of indicated inhibitors as described in Fig.5. The averages or representative images of at least three independent experiments are shown.
HA(E1)
Vinculin
p-p38MAPK
p38MAPK
+DOX(h)
DMSO
Wor
tman
nin
KU55
933
0 24 0 24 0 24
1 2 3 4 5 6
pDNA-PKc(S2056)
DNA-PKc
HCK1T-HPV16 tetON E1+E2Clone B2A B C
D
FEVehicle DOX(24h)
1
2
3
4
5
6x103
HP
V16
gen
ome
copy
/cel
l
DMSO VE-822
*
**
2
Cel
l Num
ber (
x105
)
DMSO VE-822
0.5
1
3.5
1.5
2.5
3
** *
Vehicle DOX(24h)
G
HCK1T-HPV16 tetON E1+E2Clone B2
IκBαVinculin
- + - + - + - + - + - +DOX(24h)HA(E1)
DM
SO
Wor
tman
nin
KU
5593
3
VE
-822
Nu7
026
SB
2035
80
1 2 3 4 5 6 7 8 9 11 1210
Rel
ativ
e in
tens
ity(E
1) 2
1
HCK1T-HPV16 tetON
p-p38MAPK(T180/Y182)p38MAPK
IκBαVinculin
E1+E
2
E1K4
83A+
E2E2 E1
- + - + - + - +
1 2 3 4 5 6 7 8
DOX(24h)
0 8 16 24 0 8 16 24 0 8 16 24HA(E1)
pCHK1(S345)
CHK1
Vinculin
IκBα
+DOX(h)DMSO Wortmannin VE-822
1 2 3 4 5 6 7 8 9 10 11 12
HCK1T-HPV16 tetON E1+E2 Clone B2
VinculinFLAG(E2)
HA(E1)0 8 16 24 0 8 16 24 0 8 16 24
DMSO Wortmannin VE-822
1 2 3 4 5 6 7 8 9 10 11 12
DOX(h)
HCK1T-HPV16 tetON HA-E1+FLAG-E2
Figure 6

Figure 7
Figure 7. Activation of NFκB mediates destabilization of E1 and promotes polyubiquitination. (A) Time course experiments for clone B2 transduced with MCS (lanes 1 to 4), IκBαWT (lanes 9 to 12) or IκBαMT (lanes 5 to 8) following incubation with DOX. A white arrowhead indicates a slower migrating pattern of NBS1. Vinculin was detected as a loading control. (B) RT-qPCR analysis for HA-E1 in the indicated cells upon DOX incubation. (C) Western blot analysis for the levels of E1 and IκBα in clone B2 incubated with vehicle (lanes 1 and 2) or 10nM of Epoximicin (lanes 3 and 4) for 12 hours. (D) An ubiquitin conjugation assay was performed as described in the Materials and Methods. The left panel (lanes 8 to 13) shows results of Western blot analysis for input cell lysates subjected to immunoprecipitation (IP). The right panel (lanes 14 to 19) shows results of IP-WB to detect His-Ub conjugation to HA-E1. The cell lysates from 293FT cells transfected without a His-Ub expression vector (lanes 1 to 6) were also included. Wor and TNF indicate treatment with 10 μM of Wortmannin and 20 ng/ml of TNFα, respectively, from 20 hours post-transfection. IkB indicates co-transfection with an expression vector of IκBαMT.
0 8 16 24 0 8 16 24 0 8 16 24NBS1
pCHK(S345)
CHK1
IκBα
HA(E1)
Vinculin
+DOX(h)MCS IκBαWTIκBαMT
1 2 3 4 5 6 7 8 9 10 11 12
pNBS1
Vinculin
1 2 3 4
A HCK1T-HPV16 tetON E1+E2 clone B2
+-+-DOX(12h)HA(E1)
IκBα
Vinculin
EpoxVehicle
C
MCS
IκBαW
T
IκBαM
T
0.6
0.4
0.2
Rel
ativ
e E
xpre
ssio
n
Vehicle +DOX (24h)B
D
12119 10
+++ + +- - - - -
+++ + +Wor TNF IkB TNF
+IkB-
+-
--
HA(E1)
IκBα
GFP
Vinculin
HA-E1His-UbGFP
1 2 3 4 5 6 7 8
treatment
+++ + + ++++ + + -+++ + + +
Wor TNF IkB- -
WB (input)
TNF+IkB
GFP
HA-E1
75
75
150
250
100
WB:His
WB:HA
(M.W)
1914 15 16 17 18
+++ + + ++++ + + ++++ + + -
Wor TNF IkB- -
IP(with HA)
TNF+IkB
His-Ub
Treatment
+
-
+-
13

200
100
Cel
l Cou
nt
200 400 600 800 200 400 600 800 200 400 600 800 200 400 600 800 200 400 600 800 200 400 600 8000 0 0 0 0 0
200 400 600 8000200 400 600 8000200 400 600 8000200 400 600 8000200 400 600 8000
200
100
Cel
l Cou
ntFL2 (PI)
FL2 (PI)
0h 8h 16h 24h 32h 40h
+DOX
Vehicle
70
60
50
40
30
20
10
% o
f tot
al c
ell c
ount
s
FL2 (PI)
G0/G1S
G2/Msub-G1
Vehicle +DOX
0 8 16 24 32 40 8 16 24 32 40Time (h) Time (h)
G1 S G2/M
subG1
Figure 8
Figure 8. E1 and E2 expression induces accumulation of cells in S and G2 phases.Clones B2 were collected at indicated time points after a vehicle (above) or DOX (bottom) addition and cell cycle profiles were analyzed by flow cytometry. Representative profiles are shown. The graphs indicate the percentages of cells in the cell cycle phases.
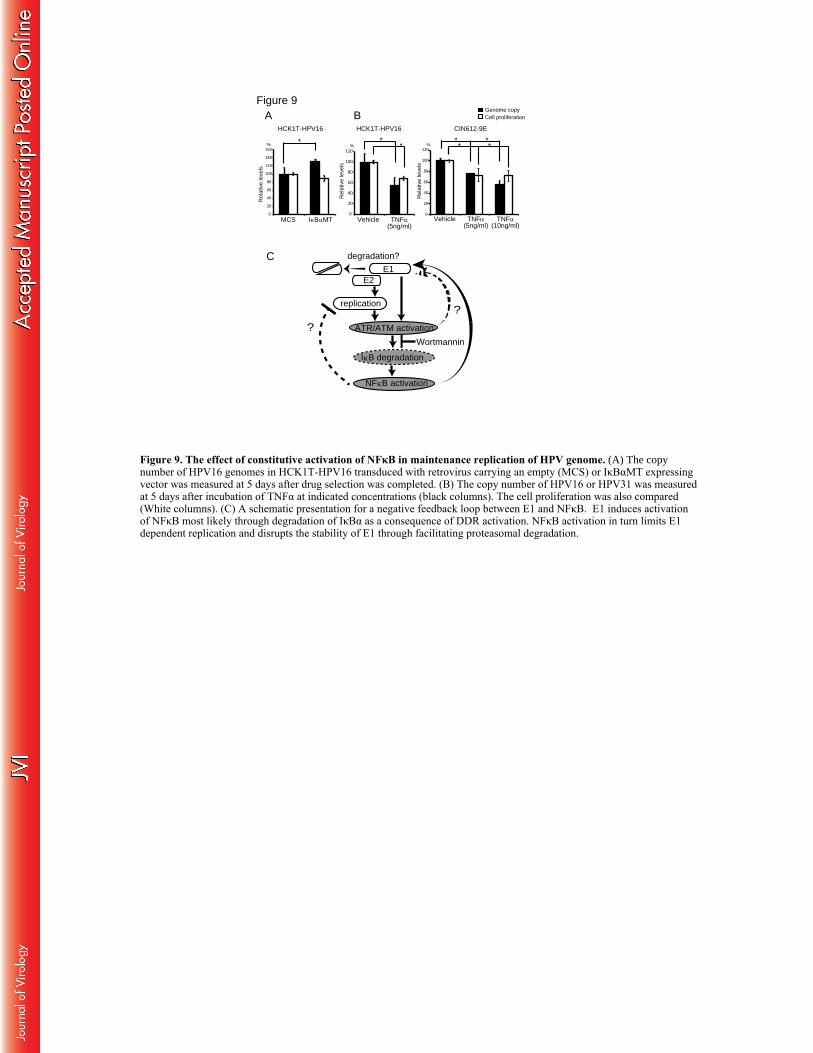
0
20
40
60
80
100
120
140
160
IκBαMTMCS
Rel
ativ
e le
vels
%
0
20
40
60
80
100
120
Rel
ativ
e le
vels
0
20
40
60
80
100
120
Rel
ativ
e le
vels
% %
HCK1T-HPV16
Vehicle TNFα(5ng/ml)
Cell proliferationGenome copy
Vehicle TNFα(5ng/ml)
TNFα(10ng/ml)
CIN612-9E
* **
***
*
E1
ATR/ATM activation
replication
IκB degradation
NFκB activation
degradation?
?Wortmannin
E2
?
A
C
Figure 9BHCK1T-HPV16
Figure 9. The effect of constitutive activation of NFκB in maintenance replication of HPV genome. (A) The copy number of HPV16 genomes in HCK1T-HPV16 transduced with retrovirus carrying an empty (MCS) or IκBαMT expressing vector was measured at 5 days after drug selection was completed. (B) The copy number of HPV16 or HPV31 was measured at 5 days after incubation of TNFα at indicated concentrations (black columns). The cell proliferation was also compared (White columns). (C) A schematic presentation for a negative feedback loop between E1 and NFκB. E1 induces activation of NFκB most likely through degradation of IκBα as a consequence of DDR activation. NFκB activation in turn limits E1 dependent replication and disrupts the stability of E1 through facilitating proteasomal degradation.
Copyright © 2022 FDOKUMEN