
Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity
6
Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity Yong-Jie Zhang a , Ya-Fei Xu a , Casey Cook a , Tania F. Gendron a , Paul Roettges a , Christopher D. Link b , Wen-Lang Lin a , Jimei Tong a , Monica Castanedes-Casey a , Peter Ash a , Jennifer Gass a , Vijayaraghavan Rangachari a,1 , Emanuele Buratti c , Francisco Baralle c , Todd E. Golde a , Dennis W. Dickson a , and Leonard Petrucelli a,2 a Department of Neuroscience, Mayo Clinic College of Medicine, 4500 San Pablo Road, Jacksonville, FL 32224; b Institute for Behavioral Genetics, University of Colorado, Campus Box 447, Boulder, CO 80309; and c International Centre for Genetic Engineering and Biotechnology, 34012 Trieste, Italy Edited by Solomon H. Snyder, Johns Hopkins University School of Medicine, Baltimore, MD, and approved March 19, 2009 (received for review January 21, 2009) Inclusions of TAR DNA-binding protein-43 (TDP-43), a nuclear protein that regulates transcription and RNA splicing, are the defining his- topathological feature of frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-Us) and sporadic and familial forms of amyotrophic lateral sclerosis (ALS). In ALS and FTLD-U, aggregated, ubiquitinated, and N-terminally truncated TDP-43 can be isolated from brain tissue rich in neuronal and glial cytoplasmic inclusions. The loss of TDP-43 function resulting from inappropriate cleavage, translocation from the nucleus, or its sequestration into inclusions could play important roles in neurodegeneration. How- ever, it is not known whether TDP-43 fragments directly mediate toxicity and, more specifically, whether their abnormal aggregation is a cause or consequence of pathogenesis. We report that the ectopic expression of a 25-kDa TDP-43 fragment corresponding to the C-terminal truncation product of caspase-cleaved TDP-43 leads to the formation of toxic, insoluble, and ubiquitin- and phospho-positive cytoplasmic inclusions within cells. The 25-kDa C-terminal fragment is more prone to phosphorylation at S409/S410 than full-length TDP-43, but phosphorylation at these sites is not required for inclusion formation or toxicity. Although this fragment shows no biological activity, its exogenous expression neither inhibits the function nor causes the sequestration of full-length nuclear TDP-43, suggesting that the 25-kDa fragment can induce cell death through a toxic gain-of-function. Finally, by generating a conformation-dependent antibody that detects C-terminal fragments, we show that this toxic cleavage product is specific for pathologic inclusions in human TDP-43 proteinopathies. caspase inclusion amyotrophic lateral sclerosis cell death frontotemporal lobar degeneration with ubiquitin-positive inclusions T he aggregation and deposition of ubiquitinated proteins in neuronal and glial cells is a characteristic feature of a number of neurodegenerative diseases, like Alzheimer’s disease, Parkin- son’s disease, frontotemporal lobar degeneration (FTLD) and amyotrophic lateral sclerosis (ALS). Recently, TAR DNA-binding protein of 43-kDa molecular mass (TDP-43) was identified as the major component of the predominately cytoplasmic inclusions observed in ALS and FTLD with ubiquitin-positive inclusions (FTLD-U), the most common form of FTLD (1, 2). Normally, TDP-43 is a nuclear protein but neurons with cytoplasmic inclu- sions have a substantial loss of nuclear TDP-43, suggesting it redistributes to the cytoplasm (1). In addition to the abnormal distribution and aggregation of TDP-43 in disease, the molecular signature of pathologic TDP-43 includes ubiquitination, phosphoryla- tion and proteolytic cleavage generating C-terminal fragments (1). TDP-43 is a 414-amino acid protein and the primary amino acid structure has strong homology in domain composition to members of the heterogeneous ribonucleoprotein (hnRNP) family (3, 4). Like other members of this family, TDP-43 has 2 highly conserved RNA recognition motifs (RRM1 and RRM2) flanked by the N-terminal and the C-terminal tail. The C- terminal tail contains a glycine-rich region often found to mediate protein–protein interactions. This region is reported to bind members of the hnRNP protein family, such as hnRNP A1, A2/B1 and A3 (5). The N-terminal region contains 2 nuclear localization signals (NLS), and 3 potential caspase-3 cleavage consensus sites span the TDP-43 molecule (6). Rare mutations in genes encoding the major protein component of inclusions in familial neurodegenerative diseases have provided unequivocal evidence that the proteins in the inclusions are causally related to neurodegeneration. For example, mutations in MAPT cause tau-positive FTLD (7) and mutations in the amyloid precur- sor protein (APP) lead to Alzheimer’s disease, which is character- ized by amyloid deposits (8). Extensive mutation analysis of TAR- DBP, the gene encoding TDP-43, lead to the discovery of missense mutations in sporadic and familial ALS (9–14). All but 1 of the 14 reported mutations cluster in exon 6 of TARDBP, which encodes the highly-conserved glycine-rich domain of TDP-43. Mutant forms of TDP-43 form fragments more readily than wild-type TDP-43 in Chinese hamster ovary cells and cause neural apoptosis and devel- opmental delay in chick embryos (12). In lymphoblastoid cell lines derived from patients carrying TARDBP mutations, the accumu- lation of detergent-insoluble TDP-43 cleavage products of 25 and 35-kDa is observed (10, 11), providing additional indication for a pathological role of TDP-43 fragments in disease. We have shown that the proteolytic cleavage of TDP-43 by caspases generates C-terminal fragments (25 and 35 kDa) similar to those observed in FTLD-U brains (6). Caspase-cleavage of TDP-43 appears to be a prerequisite for its redistribution from the nucleus to the cytosol in H4 neuroglioma cells (6), and C-terminal TDP-43 fragments are associated with enhanced aggregation potential and toxicity in yeast (15). Whether TDP-43 C-terminal fragments are prone to aggregate in mammalian cells has not yet been determined, nor has the mechanism of toxicity of these C-terminal fragments been examined. Thus, this study compared the aggregation and toxicity of C-terminal TDP-43 fragments and full-length TDP-43 in human cell lines and investigated the mechanisms of toxicity induced by the 25-kDa C-terminal TDP-43 fragment. Results C-Terminal Fragments of TDP-43 Corresponding to Caspase-Cleavage Products Form Cytoplasmic, Ubiquitin-Positive Inclusions. The patho- logical inclusions in ALS and FTLD-U are composed of TDP-43, Author contributions: Y.-J.Z. and L.P. designed research; Y.-J.Z., Y.-F.X., C.C., T.F.G., P.R., C.D.L., W.-L.L., J.T., M.C.-C., P.A., J.G., and V.R. performed research; E.B. and F.B. contributed new reagents/analytic tools; Y.-J.Z. and L.P. analyzed data; and Y.-J.Z., T.F.G., T.E.G., D.W.D., and L.P. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. Freely available online through the PNAS open access option. 1 Present address: Department of Chemistry and Biochemistry, 118 College Drive #5043, University of Southern Mississippi, Hattiesburg, MS 39406-0001. 2 To whom correspondence should be addressed. E-mail: [email protected]. This article contains supporting information online at www.pnas.org/cgi/content/full/ 0900688106/DCSupplemental. www.pnas.orgcgidoi10.1073pnas.0900688106 PNAS May 5, 2009 vol. 106 no. 18 7607–7612 NEUROSCIENCE
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity

Aberrant cleavage of TDP-43 enhances aggregationand cellular toxicityYong-Jie Zhanga, Ya-Fei Xua, Casey Cooka, Tania F. Gendrona, Paul Roettgesa, Christopher D. Linkb, Wen-Lang Lina,Jimei Tonga, Monica Castanedes-Caseya, Peter Asha, Jennifer Gassa, Vijayaraghavan Rangacharia,1, Emanuele Burattic,Francisco Barallec, Todd E. Goldea, Dennis W. Dicksona, and Leonard Petrucellia,2
aDepartment of Neuroscience, Mayo Clinic College of Medicine, 4500 San Pablo Road, Jacksonville, FL 32224; bInstitute for Behavioral Genetics, University ofColorado, Campus Box 447, Boulder, CO 80309; and cInternational Centre for Genetic Engineering and Biotechnology, 34012 Trieste, Italy
Edited by Solomon H. Snyder, Johns Hopkins University School of Medicine, Baltimore, MD, and approved March 19, 2009 (received for reviewJanuary 21, 2009)
Inclusions of TAR DNA-binding protein-43 (TDP-43), a nuclear proteinthat regulates transcription and RNA splicing, are the defining his-topathological feature of frontotemporal lobar degeneration withubiquitin-positive inclusions (FTLD-Us) and sporadic and familialforms of amyotrophic lateral sclerosis (ALS). In ALS and FTLD-U,aggregated, ubiquitinated, and N-terminally truncated TDP-43 can beisolated from brain tissue rich in neuronal and glial cytoplasmicinclusions. The loss of TDP-43 function resulting from inappropriatecleavage, translocation from the nucleus, or its sequestration intoinclusions could play important roles in neurodegeneration. How-ever, it is not known whether TDP-43 fragments directly mediatetoxicity and, more specifically, whether their abnormal aggregationis a cause or consequence of pathogenesis. We report that the ectopicexpression of a �25-kDa TDP-43 fragment corresponding to theC-terminal truncation product of caspase-cleaved TDP-43 leads to theformation of toxic, insoluble, and ubiquitin- and phospho-positivecytoplasmic inclusions within cells. The 25-kDa C-terminal fragment ismore prone to phosphorylation at S409/S410 than full-length TDP-43,but phosphorylation at these sites is not required for inclusionformation or toxicity. Although this fragment shows no biologicalactivity, its exogenous expression neither inhibits the function norcauses the sequestration of full-length nuclear TDP-43, suggestingthat the 25-kDa fragment can induce cell death through a toxicgain-of-function. Finally, by generating a conformation-dependentantibody that detects C-terminal fragments, we show that this toxiccleavage product is specific for pathologic inclusions in human TDP-43proteinopathies.
caspase � inclusion � amyotrophic lateral sclerosis � cell death �frontotemporal lobar degeneration with ubiquitin-positive inclusions
The aggregation and deposition of ubiquitinated proteins inneuronal and glial cells is a characteristic feature of a number
of neurodegenerative diseases, like Alzheimer’s disease, Parkin-son’s disease, frontotemporal lobar degeneration (FTLD) andamyotrophic lateral sclerosis (ALS). Recently, TAR DNA-bindingprotein of 43-kDa molecular mass (TDP-43) was identified as themajor component of the predominately cytoplasmic inclusionsobserved in ALS and FTLD with ubiquitin-positive inclusions(FTLD-U), the most common form of FTLD (1, 2). Normally,TDP-43 is a nuclear protein but neurons with cytoplasmic inclu-sions have a substantial loss of nuclear TDP-43, suggesting itredistributes to the cytoplasm (1). In addition to the abnormaldistribution and aggregation of TDP-43 in disease, the molecularsignature of pathologic TDP-43 includes ubiquitination, phosphoryla-tion and proteolytic cleavage generating C-terminal fragments (1).
TDP-43 is a 414-amino acid protein and the primary aminoacid structure has strong homology in domain composition tomembers of the heterogeneous ribonucleoprotein (hnRNP)family (3, 4). Like other members of this family, TDP-43 has 2highly conserved RNA recognition motifs (RRM1 and RRM2)flanked by the N-terminal and the C-terminal tail. The C-terminal tail contains a glycine-rich region often found to
mediate protein–protein interactions. This region is reported tobind members of the hnRNP protein family, such as hnRNP A1,A2/B1 and A3 (5). The N-terminal region contains 2 nuclearlocalization signals (NLS), and 3 potential caspase-3 cleavageconsensus sites span the TDP-43 molecule (6).
Rare mutations in genes encoding the major protein componentof inclusions in familial neurodegenerative diseases have providedunequivocal evidence that the proteins in the inclusions are causallyrelated to neurodegeneration. For example, mutations in MAPTcause tau-positive FTLD (7) and mutations in the amyloid precur-sor protein (APP) lead to Alzheimer’s disease, which is character-ized by amyloid deposits (8). Extensive mutation analysis of TAR-DBP, the gene encoding TDP-43, lead to the discovery of missensemutations in sporadic and familial ALS (9–14). All but 1 of the 14reported mutations cluster in exon 6 of TARDBP, which encodesthe highly-conserved glycine-rich domain of TDP-43. Mutant formsof TDP-43 form fragments more readily than wild-type TDP-43 inChinese hamster ovary cells and cause neural apoptosis and devel-opmental delay in chick embryos (12). In lymphoblastoid cell linesderived from patients carrying TARDBP mutations, the accumu-lation of detergent-insoluble TDP-43 cleavage products of �25 and35-kDa is observed (10, 11), providing additional indication for apathological role of TDP-43 fragments in disease.
We have shown that the proteolytic cleavage of TDP-43 bycaspases generates C-terminal fragments (25 and 35 kDa) similarto those observed in FTLD-U brains (6). Caspase-cleavage ofTDP-43 appears to be a prerequisite for its redistribution fromthe nucleus to the cytosol in H4 neuroglioma cells (6), andC-terminal TDP-43 fragments are associated with enhancedaggregation potential and toxicity in yeast (15). Whether TDP-43C-terminal fragments are prone to aggregate in mammalian cellshas not yet been determined, nor has the mechanism of toxicityof these C-terminal fragments been examined. Thus, this studycompared the aggregation and toxicity of C-terminal TDP-43fragments and full-length TDP-43 in human cell lines andinvestigated the mechanisms of toxicity induced by the 25-kDaC-terminal TDP-43 fragment.
ResultsC-Terminal Fragments of TDP-43 Corresponding to Caspase-CleavageProducts Form Cytoplasmic, Ubiquitin-Positive Inclusions. The patho-logical inclusions in ALS and FTLD-U are composed of TDP-43,
Author contributions: Y.-J.Z. and L.P. designed research; Y.-J.Z., Y.-F.X., C.C., T.F.G., P.R.,C.D.L., W.-L.L., J.T., M.C.-C., P.A., J.G., and V.R. performed research; E.B. and F.B. contributednew reagents/analytic tools; Y.-J.Z. and L.P. analyzed data; and Y.-J.Z., T.F.G., T.E.G., D.W.D.,and L.P. wrote the paper.
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.
1Present address: Department of Chemistry and Biochemistry, 118 College Drive #5043,University of Southern Mississippi, Hattiesburg, MS 39406-0001.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/cgi/content/full/0900688106/DCSupplemental.
www.pnas.org�cgi�doi�10.1073�pnas.0900688106 PNAS � May 5, 2009 � vol. 106 � no. 18 � 7607–7612
NEU
ROSC
IEN
CE
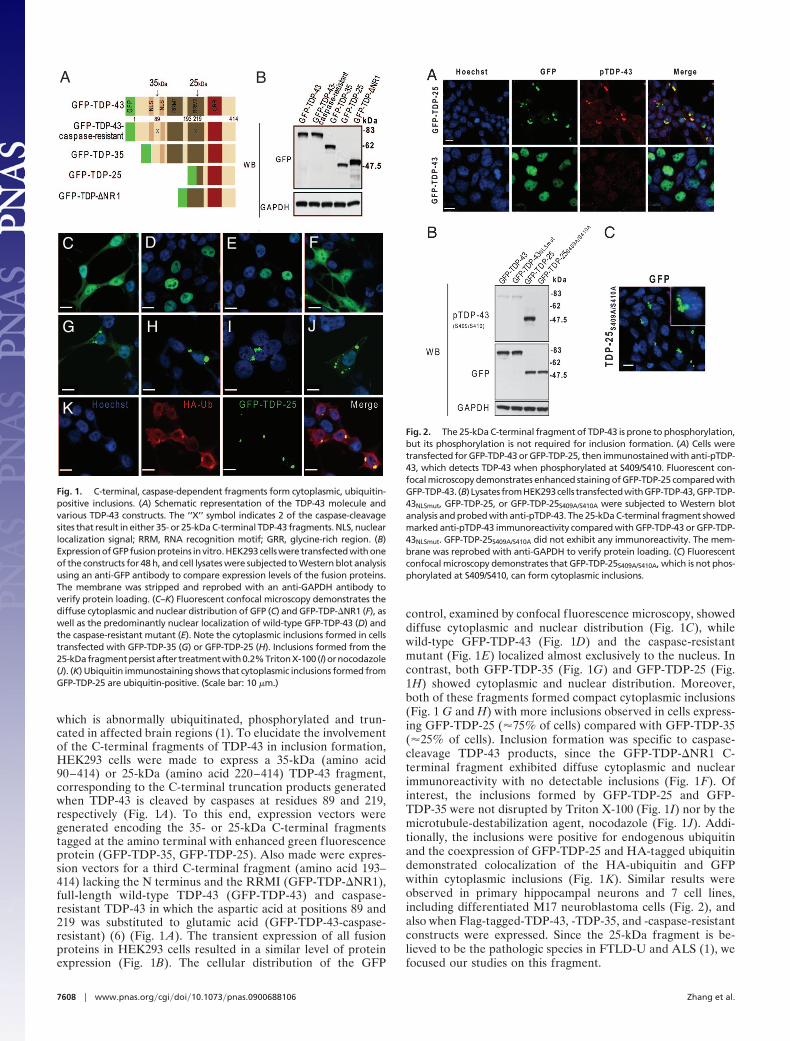
which is abnormally ubiquitinated, phosphorylated and trun-cated in affected brain regions (1). To elucidate the involvementof the C-terminal fragments of TDP-43 in inclusion formation,HEK293 cells were made to express a 35-kDa (amino acid90–414) or 25-kDa (amino acid 220–414) TDP-43 fragment,corresponding to the C-terminal truncation products generatedwhen TDP-43 is cleaved by caspases at residues 89 and 219,respectively (Fig. 1A). To this end, expression vectors weregenerated encoding the 35- or 25-kDa C-terminal fragmentstagged at the amino terminal with enhanced green fluorescenceprotein (GFP-TDP-35, GFP-TDP-25). Also made were expres-sion vectors for a third C-terminal fragment (amino acid 193–414) lacking the N terminus and the RRMI (GFP-TDP-�NR1),full-length wild-type TDP-43 (GFP-TDP-43) and caspase-resistant TDP-43 in which the aspartic acid at positions 89 and219 was substituted to glutamic acid (GFP-TDP-43-caspase-resistant) (6) (Fig. 1 A). The transient expression of all fusionproteins in HEK293 cells resulted in a similar level of proteinexpression (Fig. 1B). The cellular distribution of the GFP
control, examined by confocal f luorescence microscopy, showeddiffuse cytoplasmic and nuclear distribution (Fig. 1C), whilewild-type GFP-TDP-43 (Fig. 1D) and the caspase-resistantmutant (Fig. 1E) localized almost exclusively to the nucleus. Incontrast, both GFP-TDP-35 (Fig. 1G) and GFP-TDP-25 (Fig.1H) showed cytoplasmic and nuclear distribution. Moreover,both of these fragments formed compact cytoplasmic inclusions(Fig. 1 G and H) with more inclusions observed in cells express-ing GFP-TDP-25 (�75% of cells) compared with GFP-TDP-35(�25% of cells). Inclusion formation was specific to caspase-cleavage TDP-43 products, since the GFP-TDP-�NR1 C-terminal fragment exhibited diffuse cytoplasmic and nuclearimmunoreactivity with no detectable inclusions (Fig. 1F). Ofinterest, the inclusions formed by GFP-TDP-25 and GFP-TDP-35 were not disrupted by Triton X-100 (Fig. 1I) nor by themicrotubule-destabilization agent, nocodazole (Fig. 1J). Addi-tionally, the inclusions were positive for endogenous ubiquitinand the coexpression of GFP-TDP-25 and HA-tagged ubiquitindemonstrated colocalization of the HA-ubiquitin and GFPwithin cytoplasmic inclusions (Fig. 1K). Similar results wereobserved in primary hippocampal neurons and 7 cell lines,including differentiated M17 neuroblastoma cells (Fig. 2), andalso when Flag-tagged-TDP-43, -TDP-35, and -caspase-resistantconstructs were expressed. Since the 25-kDa fragment is be-lieved to be the pathologic species in FTLD-U and ALS (1), wefocused our studies on this fragment.
A B
C D E F
G H I J
K
Fig. 1. C-terminal, caspase-dependent fragments form cytoplasmic, ubiquitin-positive inclusions. (A) Schematic representation of the TDP-43 molecule andvarious TDP-43 constructs. The ‘‘X’’ symbol indicates 2 of the caspase-cleavagesites that result in either 35- or 25-kDa C-terminal TDP-43 fragments. NLS, nuclearlocalization signal; RRM, RNA recognition motif; GRR, glycine-rich region. (B)Expression of GFP fusion proteins in vitro. HEK293 cells were transfected with oneof the constructs for 48 h, and cell lysates were subjected to Western blot analysisusing an anti-GFP antibody to compare expression levels of the fusion proteins.The membrane was stripped and reprobed with an anti-GAPDH antibody toverify protein loading. (C–K) Fluorescent confocal microscopy demonstrates thediffuse cytoplasmic and nuclear distribution of GFP (C) and GFP-TDP-�NR1 (F), aswell as the predominantly nuclear localization of wild-type GFP-TDP-43 (D) andthe caspase-resistant mutant (E). Note the cytoplasmic inclusions formed in cellstransfected with GFP-TDP-35 (G) or GFP-TDP-25 (H). Inclusions formed from the25-kDafragmentpersistafter treatmentwith0.2%TritonX-100 (I)ornocodazole(J). (K) Ubiquitin immunostaining shows that cytoplasmic inclusions formed fromGFP-TDP-25 are ubiquitin-positive. (Scale bar: 10 �m.)
Fig. 2. The 25-kDa C-terminal fragment of TDP-43 is prone to phosphorylation,but its phosphorylation is not required for inclusion formation. (A) Cells weretransfected for GFP-TDP-43 or GFP-TDP-25, then immunostained with anti-pTDP-43, which detects TDP-43 when phosphorylated at S409/S410. Fluorescent con-focal microscopy demonstrates enhanced staining of GFP-TDP-25 compared withGFP-TDP-43. (B) Lysates fromHEK293cells transfectedwithGFP-TDP-43,GFP-TDP-43NLSmut, GFP-TDP-25, or GFP-TDP-25S409A/S410A were subjected to Western blotanalysis and probed with anti-pTDP-43. The 25-kDa C-terminal fragment showedmarked anti-pTDP-43 immunoreactivity compared with GFP-TDP-43 or GFP-TDP-43NLSmut. GFP-TDP-25S409A/S410A did not exhibit any immunoreactivity. The mem-brane was reprobed with anti-GAPDH to verify protein loading. (C) Fluorescentconfocal microscopy demonstrates that GFP-TDP-25S409A/S410A, which is not phos-phorylated at S409/S410, can form cytoplasmic inclusions.
7608 � www.pnas.org�cgi�doi�10.1073�pnas.0900688106 Zhang et al.
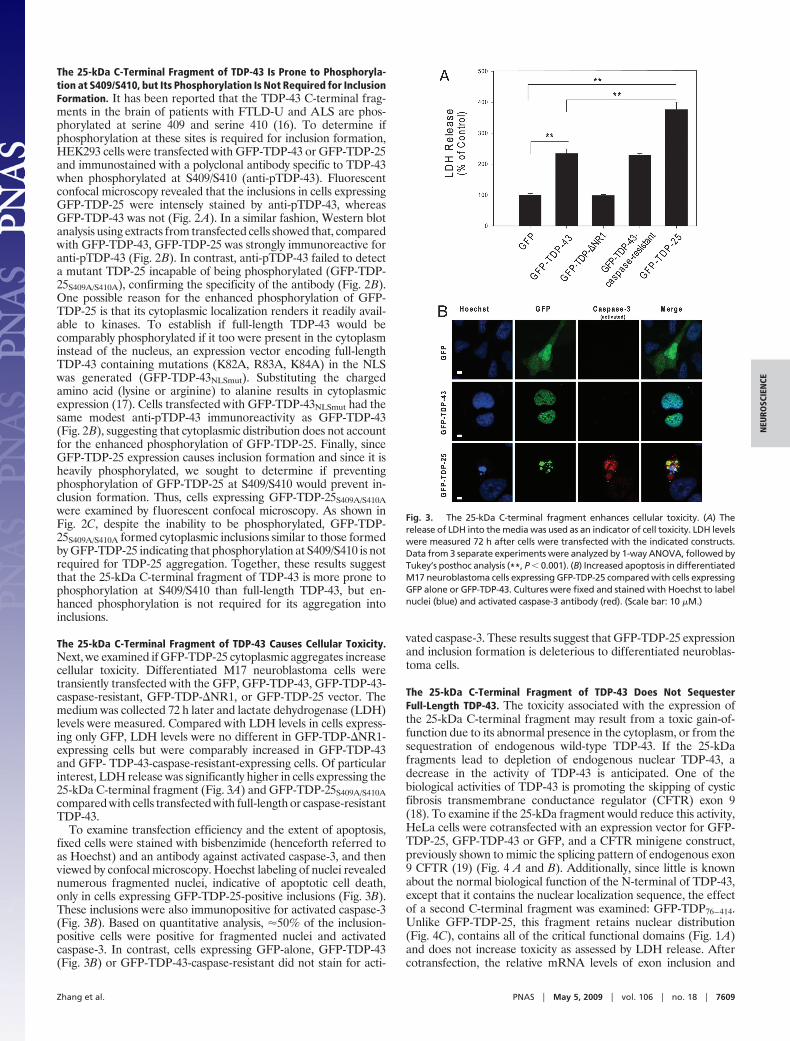
The 25-kDa C-Terminal Fragment of TDP-43 Is Prone to Phosphoryla-tion at S409/S410, but Its Phosphorylation Is Not Required for InclusionFormation. It has been reported that the TDP-43 C-terminal frag-ments in the brain of patients with FTLD-U and ALS are phos-phorylated at serine 409 and serine 410 (16). To determine ifphosphorylation at these sites is required for inclusion formation,HEK293 cells were transfected with GFP-TDP-43 or GFP-TDP-25and immunostained with a polyclonal antibody specific to TDP-43when phosphorylated at S409/S410 (anti-pTDP-43). Fluorescentconfocal microscopy revealed that the inclusions in cells expressingGFP-TDP-25 were intensely stained by anti-pTDP-43, whereasGFP-TDP-43 was not (Fig. 2A). In a similar fashion, Western blotanalysis using extracts from transfected cells showed that, comparedwith GFP-TDP-43, GFP-TDP-25 was strongly immunoreactive foranti-pTDP-43 (Fig. 2B). In contrast, anti-pTDP-43 failed to detecta mutant TDP-25 incapable of being phosphorylated (GFP-TDP-25S409A/S410A), confirming the specificity of the antibody (Fig. 2B).One possible reason for the enhanced phosphorylation of GFP-TDP-25 is that its cytoplasmic localization renders it readily avail-able to kinases. To establish if full-length TDP-43 would becomparably phosphorylated if it too were present in the cytoplasminstead of the nucleus, an expression vector encoding full-lengthTDP-43 containing mutations (K82A, R83A, K84A) in the NLSwas generated (GFP-TDP-43NLSmut). Substituting the chargedamino acid (lysine or arginine) to alanine results in cytoplasmicexpression (17). Cells transfected with GFP-TDP-43NLSmut had thesame modest anti-pTDP-43 immunoreactivity as GFP-TDP-43(Fig. 2B), suggesting that cytoplasmic distribution does not accountfor the enhanced phosphorylation of GFP-TDP-25. Finally, sinceGFP-TDP-25 expression causes inclusion formation and since it isheavily phosphorylated, we sought to determine if preventingphosphorylation of GFP-TDP-25 at S409/S410 would prevent in-clusion formation. Thus, cells expressing GFP-TDP-25S409A/S410Awere examined by fluorescent confocal microscopy. As shown inFig. 2C, despite the inability to be phosphorylated, GFP-TDP-25S409A/S410A formed cytoplasmic inclusions similar to those formedby GFP-TDP-25 indicating that phosphorylation at S409/S410 is notrequired for TDP-25 aggregation. Together, these results suggestthat the 25-kDa C-terminal fragment of TDP-43 is more prone tophosphorylation at S409/S410 than full-length TDP-43, but en-hanced phosphorylation is not required for its aggregation intoinclusions.
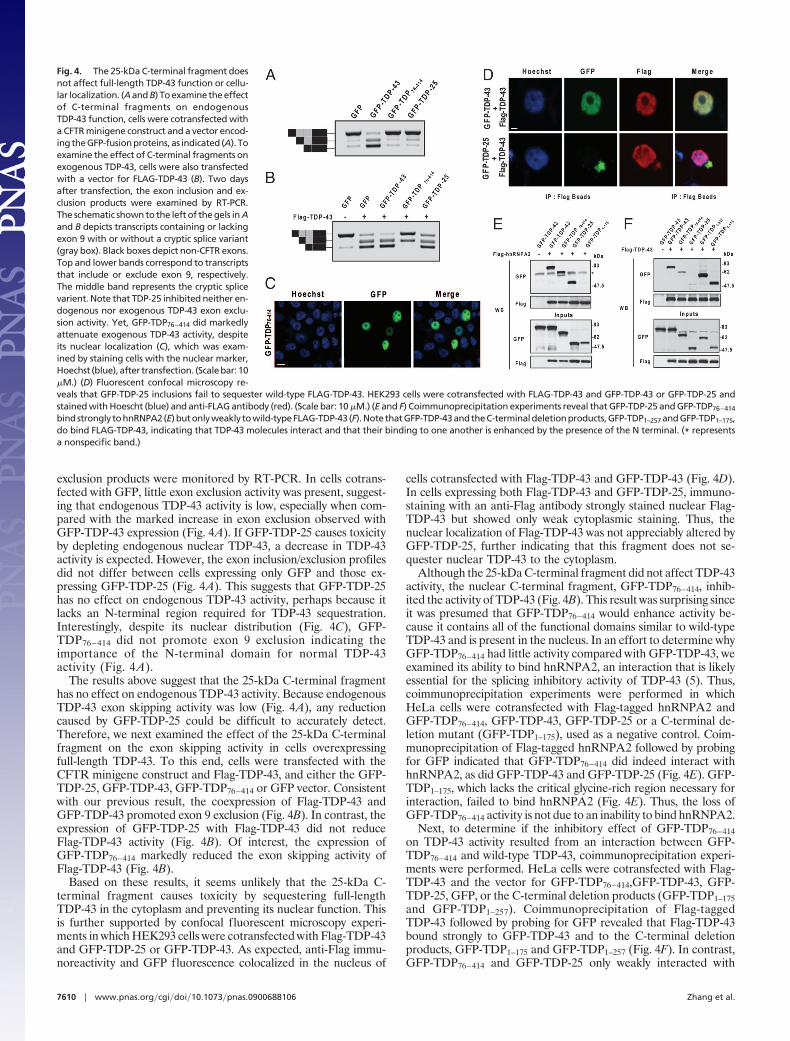
The 25-kDa C-Terminal Fragment of TDP-43 Causes Cellular Toxicity.Next, we examined if GFP-TDP-25 cytoplasmic aggregates increasecellular toxicity. Differentiated M17 neuroblastoma cells weretransiently transfected with the GFP, GFP-TDP-43, GFP-TDP-43-caspase-resistant, GFP-TDP-�NR1, or GFP-TDP-25 vector. Themedium was collected 72 h later and lactate dehydrogenase (LDH)levels were measured. Compared with LDH levels in cells express-ing only GFP, LDH levels were no different in GFP-TDP-�NR1-expressing cells but were comparably increased in GFP-TDP-43and GFP- TDP-43-caspase-resistant-expressing cells. Of particularinterest, LDH release was significantly higher in cells expressing the25-kDa C-terminal fragment (Fig. 3A) and GFP-TDP-25S409A/S410Acompared with cells transfected with full-length or caspase-resistantTDP-43.
To examine transfection efficiency and the extent of apoptosis,fixed cells were stained with bisbenzimide (henceforth referred toas Hoechst) and an antibody against activated caspase-3, and thenviewed by confocal microscopy. Hoechst labeling of nuclei revealednumerous fragmented nuclei, indicative of apoptotic cell death,only in cells expressing GFP-TDP-25-positive inclusions (Fig. 3B).These inclusions were also immunopositive for activated caspase-3(Fig. 3B). Based on quantitative analysis, �50% of the inclusion-positive cells were positive for fragmented nuclei and activatedcaspase-3. In contrast, cells expressing GFP-alone, GFP-TDP-43(Fig. 3B) or GFP-TDP-43-caspase-resistant did not stain for acti-
vated caspase-3. These results suggest that GFP-TDP-25 expressionand inclusion formation is deleterious to differentiated neuroblas-toma cells.
The 25-kDa C-Terminal Fragment of TDP-43 Does Not SequesterFull-Length TDP-43. The toxicity associated with the expression ofthe 25-kDa C-terminal fragment may result from a toxic gain-of-function due to its abnormal presence in the cytoplasm, or from thesequestration of endogenous wild-type TDP-43. If the 25-kDafragments lead to depletion of endogenous nuclear TDP-43, adecrease in the activity of TDP-43 is anticipated. One of thebiological activities of TDP-43 is promoting the skipping of cysticfibrosis transmembrane conductance regulator (CFTR) exon 9(18). To examine if the 25-kDa fragment would reduce this activity,HeLa cells were cotransfected with an expression vector for GFP-TDP-25, GFP-TDP-43 or GFP, and a CFTR minigene construct,previously shown to mimic the splicing pattern of endogenous exon9 CFTR (19) (Fig. 4 A and B). Additionally, since little is knownabout the normal biological function of the N-terminal of TDP-43,except that it contains the nuclear localization sequence, the effectof a second C-terminal fragment was examined: GFP-TDP76–414.Unlike GFP-TDP-25, this fragment retains nuclear distribution(Fig. 4C), contains all of the critical functional domains (Fig. 1A)and does not increase toxicity as assessed by LDH release. Aftercotransfection, the relative mRNA levels of exon inclusion and
Fig. 3. The 25-kDa C-terminal fragment enhances cellular toxicity. (A) Therelease of LDH into the media was used as an indicator of cell toxicity. LDH levelswere measured 72 h after cells were transfected with the indicated constructs.Data from 3 separate experiments were analyzed by 1-way ANOVA, followed byTukey’s posthoc analysis (**, P � 0.001). (B) Increased apoptosis in differentiatedM17 neuroblastoma cells expressing GFP-TDP-25 compared with cells expressingGFP alone or GFP-TDP-43. Cultures were fixed and stained with Hoechst to labelnuclei (blue) and activated caspase-3 antibody (red). (Scale bar: 10 �M.)
Zhang et al. PNAS � May 5, 2009 � vol. 106 � no. 18 � 7609
NEU
ROSC
IEN
CE
exclusion products were monitored by RT-PCR. In cells cotrans-fected with GFP, little exon exclusion activity was present, suggest-ing that endogenous TDP-43 activity is low, especially when com-pared with the marked increase in exon exclusion observed withGFP-TDP-43 expression (Fig. 4A). If GFP-TDP-25 causes toxicityby depleting endogenous nuclear TDP-43, a decrease in TDP-43activity is expected. However, the exon inclusion/exclusion profilesdid not differ between cells expressing only GFP and those ex-pressing GFP-TDP-25 (Fig. 4A). This suggests that GFP-TDP-25has no effect on endogenous TDP-43 activity, perhaps because itlacks an N-terminal region required for TDP-43 sequestration.Interestingly, despite its nuclear distribution (Fig. 4C), GFP-TDP76–414 did not promote exon 9 exclusion indicating theimportance of the N-terminal domain for normal TDP-43activity (Fig. 4A).
The results above suggest that the 25-kDa C-terminal fragmenthas no effect on endogenous TDP-43 activity. Because endogenousTDP-43 exon skipping activity was low (Fig. 4A), any reductioncaused by GFP-TDP-25 could be difficult to accurately detect.Therefore, we next examined the effect of the 25-kDa C-terminalfragment on the exon skipping activity in cells overexpressingfull-length TDP-43. To this end, cells were transfected with theCFTR minigene construct and Flag-TDP-43, and either the GFP-TDP-25, GFP-TDP-43, GFP-TDP76–414 or GFP vector. Consistentwith our previous result, the coexpression of Flag-TDP-43 andGFP-TDP-43 promoted exon 9 exclusion (Fig. 4B). In contrast, theexpression of GFP-TDP-25 with Flag-TDP-43 did not reduceFlag-TDP-43 activity (Fig. 4B). Of interest, the expression ofGFP-TDP76–414 markedly reduced the exon skipping activity ofFlag-TDP-43 (Fig. 4B).
Based on these results, it seems unlikely that the 25-kDa C-terminal fragment causes toxicity by sequestering full-lengthTDP-43 in the cytoplasm and preventing its nuclear function. Thisis further supported by confocal fluorescent microscopy experi-ments in which HEK293 cells were cotransfected with Flag-TDP-43and GFP-TDP-25 or GFP-TDP-43. As expected, anti-Flag immu-noreactivity and GFP fluorescence colocalized in the nucleus of
cells cotransfected with Flag-TDP-43 and GFP-TDP-43 (Fig. 4D).In cells expressing both Flag-TDP-43 and GFP-TDP-25, immuno-staining with an anti-Flag antibody strongly stained nuclear Flag-TDP-43 but showed only weak cytoplasmic staining. Thus, thenuclear localization of Flag-TDP-43 was not appreciably altered byGFP-TDP-25, further indicating that this fragment does not se-quester nuclear TDP-43 to the cytoplasm.
Although the 25-kDa C-terminal fragment did not affect TDP-43activity, the nuclear C-terminal fragment, GFP-TDP76–414, inhib-ited the activity of TDP-43 (Fig. 4B). This result was surprising sinceit was presumed that GFP-TDP76–414 would enhance activity be-cause it contains all of the functional domains similar to wild-typeTDP-43 and is present in the nucleus. In an effort to determine whyGFP-TDP76–414 had little activity compared with GFP-TDP-43, weexamined its ability to bind hnRNPA2, an interaction that is likelyessential for the splicing inhibitory activity of TDP-43 (5). Thus,coimmunoprecipitation experiments were performed in whichHeLa cells were cotransfected with Flag-tagged hnRNPA2 andGFP-TDP76–414, GFP-TDP-43, GFP-TDP-25 or a C-terminal de-letion mutant (GFP-TDP1–175), used as a negative control. Coim-munoprecipitation of Flag-tagged hnRNPA2 followed by probingfor GFP indicated that GFP-TDP76–414 did indeed interact withhnRNPA2, as did GFP-TDP-43 and GFP-TDP-25 (Fig. 4E). GFP-TDP1–175, which lacks the critical glycine-rich region necessary forinteraction, failed to bind hnRNPA2 (Fig. 4E). Thus, the loss ofGFP-TDP76–414 activity is not due to an inability to bind hnRNPA2.
Next, to determine if the inhibitory effect of GFP-TDP76–414on TDP-43 activity resulted from an interaction between GFP-TDP76–414 and wild-type TDP-43, coimmunoprecipitation experi-ments were performed. HeLa cells were cotransfected with Flag-TDP-43 and the vector for GFP-TDP76–414,GFP-TDP-43, GFP-TDP-25, GFP, or the C-terminal deletion products (GFP-TDP1–175and GFP-TDP1–257). Coimmunoprecipitation of Flag-taggedTDP-43 followed by probing for GFP revealed that Flag-TDP-43bound strongly to GFP-TDP-43 and to the C-terminal deletionproducts, GFP-TDP1–175 and GFP-TDP1–257 (Fig. 4F). In contrast,GFP-TDP76–414 and GFP-TDP-25 only weakly interacted with
Fig. 4. The 25-kDa C-terminal fragment doesnot affect full-length TDP-43 function or cellu-lar localization. (A and B) To examine the effectof C-terminal fragments on endogenousTDP-43 function, cells were cotransfected witha CFTR minigene construct and a vector encod-ingtheGFP-fusionproteins,as indicated (A). Toexamine the effect of C-terminal fragments onexogenous TDP-43, cells were also transfectedwith a vector for FLAG-TDP-43 (B). Two daysafter transfection, the exon inclusion and ex-clusion products were examined by RT-PCR.The schematic shown to the left of the gels in Aand B depicts transcripts containing or lackingexon 9 with or without a cryptic splice variant(gray box). Black boxes depict non-CFTR exons.Top and lower bands correspond to transcriptsthat include or exclude exon 9, respectively.The middle band represents the cryptic splicevarient. Note that TDP-25 inhibited neither en-dogenous nor exogenous TDP-43 exon exclu-sion activity. Yet, GFP-TDP76–414 did markedlyattenuate exogenous TDP-43 activity, despiteits nuclear localization (C), which was exam-ined by staining cells with the nuclear marker,Hoechst (blue),after transfection. (Scalebar:10�M.) (D) Fluorescent confocal microscopy re-veals that GFP-TDP-25 inclusions fail to sequester wild-type FLAG-TDP-43. HEK293 cells were cotransfected with FLAG-TDP-43 and GFP-TDP-43 or GFP-TDP-25 andstained with Hoescht (blue) and anti-FLAG antibody (red). (Scale bar: 10 �M.) (E and F) Coimmunoprecipitation experiments reveal that GFP-TDP-25 and GFP-TDP76–414
bindstrongly tohnRNPA2(E)butonlyweakly towild-typeFLAG-TDP-43 (F).NotethatGFP-TDP-43andtheC-terminaldeletionproducts,GFP-TDP1–257 andGFP-TDP1–175,do bind FLAG-TDP-43, indicating that TDP-43 molecules interact and that their binding to one another is enhanced by the presence of the N terminal. (* representsa nonspecific band.)
7610 � www.pnas.org�cgi�doi�10.1073�pnas.0900688106 Zhang et al.

Flag-TDP-43, despite comparable expression levels (Fig. 4F). Thus,the ability of GFP-TDP76–414 to inhibit Flag-TDP-43 activity doesnot occur through the interaction of these 2 proteins. It is note-worthy that these results indicate that the interaction of TDP-43with other TDP-43 molecules appears enhanced by the presence ofan intact N-terminal.
A Conformation-Dependent TDP-43 Antibody Detects C-TerminalTDP-43 Fragments and Specifically Labels the Pathologic Inclusions inHuman TDP-43 Proteinopathies. Since the caspase-derived C-terminal fragments aggregate into cytoplasmic inclusions, wehypothesized that they may be the major pathological species inFTLD-U and ALS. Previous studies have shown that C-terminalspecific TDP-43 antibodies detect TDP-43 pathology in FTLD-Uand ALS (20); however, all current commercially availableTDP-43 antibodies detect both normal nuclear TDP-43 andTDP-43-positive pathological structures, like neuronal cytoplas-mic inclusions (NCI), dystrophic neurites (DN) and neuronalintranuclear inclusions (NII). In some cases, extensive nuclearlabeling makes it difficult to detect mild TDP-43 pathology.Hasegawa et al. (16) have shown that antibodies to TDP-43phospho-peptides appear to be lesion-specific. Hence, we rea-soned that an antibody that would detect the neoepitope formedupon caspase cleavage of TDP-43 might also be lesion specific.We thus generated a polyclonal TDP-43 antibody (MC2085) toa peptide sequence in the 25-kDa C-terminal fragment. Based oncomposite analysis of the TDP-43 aa sequence and 3-dimen-sional modeling (10) (Fig. 5A), we chose the amino acid residues,VFIPKPFR, C-terminal to the putative caspase cleavage site, asthe peptide antigen. This region is buried within the TDP-43molecule and so is predicted to be detected only when exposedby proteolytic cleavage or under denaturing conditions.
To determine if MC2085 detects the 25-kDa C-terminal frag-ment, wild-type TDP-43 or both, we transfected cells with GFP,GFP-TDP-43, or GFP-TDP-25 for Western blot analysis andimmunofluorescent confocal microscopy. By Western blot analysis,
both GFP-TDP-43 and GFP-TDP-25 were immunoreactive forMC2085 (Fig. 2B). In contrast, immunostaining with MC2085showed it to react only with the inclusions in GFP-TDP-25 trans-fected cells and not with nuclear TDP-43 in GFP-TDP-43 trans-fected cells (Fig. 2C). Of interest, the inclusions were not detectedby a TDP-43 N-terminal-specific antibody (MC2079), suggestingthey are composed mainly of C-terminal fragments (Fig. S1). Next,immunohistochemistry of FTLD-U and ALS tissue was conductedusing MC2085. Neuronal cytoplasmic inclusions in FTLD-U (Fig.5D) and ALS (Fig. 5E) cases stained positively for MC2085 butminimal or no staining of normal neuronal TDP-43 and glial nucleiwas observed (Fig. 5 D and E). These findings suggest that someinclusions in both FTLD-U and ALS contain caspase-derivedC-terminal fragments of TDP-43.
DiscussionOur data highlight the pathologic relevance of caspase-derivedC-terminal TDP-43 fragments. Using molecular and biochemicalassays in various cell culture systems, we demonstrate that: (i) the25-kDa fragment forms cytoplasmic inclusions that are ubiquiti-nated and phosphorylated at S409/S410; (ii) phosphorylation ofthese fragments at S409/S410 is not required for their aggregation;and (iii) inclusion formation is associated with enhanced cellulartoxicity, likely through a toxic gain-of-function. Moreover, using anantibody that selectively detects caspase-derived C-terminal frag-ments in human tissue, we provide evidence that these fragmentsare diagnostic characteristics of TDP-43 proteinopathies.
Herein, we show that the expression of the 25-kDa C-terminalfragment, corresponding to a caspase-cleavage product of TDP-43,causes inclusion formation and cellular toxicity. Interestingly, just asthe inclusions in FTLD-U and ALS are immunoreactive to phos-phorylation-specific antibodies toward pS409/S410 (16, 21), so arethe inclusions formed by the 25-kDa fragments (Fig. 2). In fact,GFP-TDP-25 was much more prone to phosphorylation at S409/S410 than full-length TDP-43. This is consistent with the findings ofHasegawa et al., who show by Western blot that a �25-kDafragment in sarkosyl-insoluble, urea soluble fractions of FTLD-Uand ALS cases is more intensely stained with an antibody topS409/S410 than full-length TDP-43 (16). This group also reportsthat treating recombinant full-length TDP-43 with casein kinase Ileads to TDP-43 phosphorylation and facilitates filament formationin vitro (16). While phosphorylation may expedite the aggregationof full-length TDP-43 in vitro, we show that S409/S410 phosphor-ylation is not required for inclusion formation nor toxicity resultingfrom GFP-TDP-25 expression. These results strongly suggest thatcleavage is a prerequisite for TDP-43 hyperphosphorylation, ag-gregation, and toxicity, and highlight the importance of the 25-kDaC-terminal fragment as a pathological hallmark of disease. Indeed,the antibody generated to detect C-terminal fragments, MC2085,strongly stained inclusions in histological sections from FTLD-Uand ALS brains, making it a powerful tool for the detection ofTDP-43 pathology by IHC, especially since it does not stain normalneuronal or glial nuclei.
Although our results establish the toxicity of the 25-kDafragment, the exact mechanism by which this occurs is notknown. Toxicity could result from the aggregation of the frag-ments and their interference with normal cellular activitythrough a toxic gain-of-function. Alternatively, it may resultfrom the sequestration of normal TDP-43 into inclusions, thusdepriving the cell of the normal biological functions of TDP-43.For example, Winton and colleagues have shown that theexogenous expression of full-length TDP-43 with NLS mutationsleads to the accumulation of cytoplasmic aggregates and thesequestration of endogenous TDP-43, thereby depleting normalnuclear TDP-43 (22). Yet, this seems not to be the case withcaspase-cleaved TDP-43 since the expression of GFP-TDP-25,while toxic, did not reduce the exon skipping activity of eitherendogenous or exogenous TDP-43, nor did it alter the nuclear
Fig. 5. A conformation-dependent TDP-43 antibody that detects C-terminalfragments is specific for pathologic inclusions in human TDP-43 proteinopa-thies. (A) Three-dimensional modeling of wild-type TDP-43 (amino acids 101–264). The positions of the 8 amino acid residues following the caspase cleavagemotif, selected to generate a polyclonal antibody, are indicated. Note thataspartate 219, which is part of the caspase cleavage motif, is buried within themolecule (arrow). (B) Western blot analysis of lysates from HEK293 cellstransfected with GFP only, GFP-TDP-43, or GFP-TDP-25 and probed withMC2085. *, Endogenous TDP-43. (C) MC2085 immunostaining of HEK293 cellstransfected with GFP-TDP-43 or GFP-TDP-25. Note the absence of staining incells expressing wild-type TDP-43 compared with those expressing the 25-kDaC-terminal fragment. (Scale bars: 10 �m.) (D and E) Immunostaining of tissuefrom FTLD-U brain (D) and ALS brain (E) with MC2085 shows cytoplasmicinclusions with little or no nuclear TDP-43. (Magnification: 40�.)
Zhang et al. PNAS � May 5, 2009 � vol. 106 � no. 18 � 7611
NEU
ROSC
IEN
CE

distribution of Flag-TDP-43 (Fig. 4). Also, the inclusions werenot detected by the N-terminal specific antibody, MC2079,further suggesting that full-length TDP-43 is not sequestered intothe inclusions (Fig. S1). Rather, the toxicity associated withC-terminal fragment inclusions appears to occur independentlyof their effect on normal TDP-43 function. This is consistent withthe work by Johnson and colleagues who examined the moleculardeterminants of TDP-43 aggregation and its cellular conse-quences in yeast models. They concluded that aggregation iscritical for toxicity, with the most toxic fragments containing boththe C terminus and RRM2, similar to the 25-kDa fragment ofthe present study (15). As observed in yeast cells (15), TDP-43overexpression was toxic to M17 cells, albeit to a lesser extentthan GFP-TDP-25. While the mechanism of cell death is notyet clear, it may result from the burden of high TDP-43 levelsin the nucleus and the dysregulation of TDP-43 activity.Indeed, TDP-43-caspase-resistant, which also localized pre-dominately to the nucleus, was comparably toxic, whereasGFP-TDP-�NR1, which exhibited both diffuse cytoplasmicand nuclear staining, was not and neither was GFP-TDP76–414which did localize to the nucleus but had no biological activity.Just as depletion of TDP-43 results in apoptosis in human cellslines (23), so may elevated TDP-43 levels. Until more is knownabout TDP-43 function, it will be difficult to ascertain howchanges in nuclear TDP-43 levels will affect cell survival.
It is important to note that, while inclusions formed from the25-kDa fragment are toxic, it does not rule out the potential forTDP-43 loss-of-function to cause toxicity in TDP-43 proteinopa-thies. In fact, loss of TDP-43 in cells by RNAi causes cell death andaberrant nuclear morphology through the misregulation of theretinoblastoma protein (pRb) (23). We have previously shown thatcaspase-cleavage of endogenous TDP-43 results in the redistribu-tion of TDP-43 from the nucleus to the cytoplasm (6), perhaps dueto the loss of the NLS. This would result in the depletion of nuclearTDP-43 and, thus, loss of function. Indeed, the 25-kDa fragmenthas no biological activity (Fig. 4A). In the present study, the effectof C-terminal TDP-43 fragments on toxicity was examined byexogenously expressing the 25-kDa fragment and not by inducingthe cleavage of endogenous TDP-43, so it is expected (based on thefindings shown in Fig. 4) that endogenous remaining nuclearTDP-43 remains functional.
Our findings provide previously undescribed insights on TDP-43function. We show that the N-terminal region of TDP-43 is requiredfor normal TDP-43 exon 9 exclusion activity (Fig. 4A). The lack ofactivity of N-terminal deletion mutants, like GFP-TDP-25 and
GFP-TDP76–414, does not result from impaired hnRNPA2 binding,an interaction thought critical for the splicing inhibitory activity ofTDP-43 (5). Although it could be assumed that loss of activity forGFP-TDP-25 results from its cytosolic localization, this cannot besaid for GFP-TDP76–414 which is present in the nucleus. Indeed,when GFP-TDP76–414 is coexpressed with full-length TDP-43, itinhibits full-length TDP-43 function (Fig. 4B). This effect is notcaused by an interaction between wild-type TDP-43 and GFP-TDP76–414 since they seem to bind poorly to one another (Fig. 4F).Instead, it is possible that the functionally inert GFP-TDP76–414competes with wild-type TDP-43 for binding the substrate andpartners necessary for activity, like hnRNPA2. Finally, we showthat the binding of full-length TDP-43 to other TDP-43 moleculesis enhanced by the presence of an intact N-terminal.
Overall, these findings, along with previous biochemical, patho-logical and genetic studies, indicate that cleavage of TDP-43 resultsin the abnormal cytosolic localization of C-terminal TDP-43 frag-ments and the formation of toxic aggregates. Further study of thepathways that generate these toxic C-terminal fragments and theirmechanisms of toxicity may lead to the identification of newtherapeutic strategies to treat or prevent ALS, FTLD-U and otherTDP-43 proteinopathies.
Materials and MethodsPlasmids, Antibodies, and Reagents. For information about the materials used inthis study please refer to SI Materials and Methods.
Cell Culture and Treatments. Cell culture studies are detailed in SI Materials andMethods.
Immunofluorescence and Western Blot Analysis. Immunofluroscence and West-ern blot analysis are detailed in SI Materials and Methods.
Toxicity Assay. Toxicity assay is detailed in SI Materials and Methods.
RNA Extraction and Semiquantitative RT-PCR. CFTR splicing assay was followedaccording to a protocol adapted from ref. 18 and detailed in SI Materials andMethods.
Generation of Antibodies and Immunohistochemistry. Please refer to SI Materialsand Methods for details regarding antibodies and immunohistochemistry.
ACKNOWLEDGMENTS. This work was supported by the Mayo Clinic Founda-tion, National Institutes of Health/National Institute on Aging GrantsR01AG026251 and P01-AG17216-08, National Institutes of Health/NationalInstitute of Neurological Disorders and Stroke Grant R01 NS 063964-01, andthe Amyotrophic Lateral Sclerosis Association Grant 1736.
1. Neumann M, et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degenerationand amyotrophic lateral sclerosis. Science 314:130–133.
2. Arai T, et al. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusionsin frontotemporal lobar degeneration and amyotrophic lateral sclerosis. BiochemBiophys Res Commun 351:602–611.
3. Dreyfuss G, Kim VN, Kataoka N (2002) Messenger-RNA-binding proteins and themessages they carry. Nat Rev Mol Cell Biol 3:195–205.
4. Krecic AM, Swanson MS (1999) hnRNP complexes: Composition, structure, and func-tion. Curr Opin Cell Biol 11:363–371.
5. Buratti E, et al. (2005) TDP-43 binds heterogeneous nuclear ribonucleoprotein A/Bthrough its C-terminal tail: An important region for the inhibition of cystic fibrosistransmembrane conductance regulator exon 9 splicing. J Biol Chem 280:37572–37584.
6. Zhang YJ, et al. (2007) Progranulin mediates caspase-dependent cleavage of TAR DNAbinding protein-43. J Neurosci 27:10530–10534.
7. Hutton M, et al. (1998) Association of missense and 5�-splice-site mutations in tau withthe inherited dementia FTDP-17. Nature 393:702–705.
8. Goate A, et al. (1991) Segregation of a missense mutation in the amyloid precursorprotein gene with familial Alzheimer’s disease. Nature 349:704–706.
9. Gitcho MA, et al. (2008) TDP-43 A315T mutation in familial motor neuron disease. AnnNeurol 63:535–538.
10. Kabashi E, et al. (2008) TARDBP mutations in individuals with sporadic and familialamyotrophic lateral sclerosis. Nat Genet 40:572–574.
11. Rutherford NJ, et al. (2008) Novel mutations in TARDBP (TDP-43) in patients withfamilial amyotrophic lateral sclerosis. PLoS Genet 4:e1000193.
12. Sreedharan J, et al. (2008) TDP-43 mutations in familial and sporadic amyotrophiclateral sclerosis. Science 319:1668–1672.
13. Van Deerlin VM, et al. (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43neuropathology: A genetic and histopathological analysis. Lancet Neurol 7:409–416.
14. Yokoseki A, et al. (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. AnnNeurol 63:538–542.
15. Johnson BS, McCaffery JM, Lindquist S, Gitler AD (2008) A yeast TDP-43 proteinopathymodel: Exploring the molecular determinants of TDP-43 aggregation and cellulartoxicity. Proc Natl Acad Sci USA 105:6439–6444.
16. Hasegawa M, et al. (2008) Phosphorylated TDP-43 in frontotemporal lobar degener-ation and amyotrophic lateral sclerosis. Ann Neurol 64:60–70.
17. Ayala YM, et al. (2008) Structural determinants of the cellular localization and shut-tling of TDP-43. J Cell Sci 121:3778–3785.
18. Buratti E, Baralle FE (2001) Characterization and functional implications of the RNAbinding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9.J Biol Chem 276:36337–36343.
19. Pagani F, et al. (2000) Splicing factors induce cystic fibrosis transmembrane regulatorexon 9 skipping through a nonevolutionary conserved intronic element. J Biol Chem275:21041–21047.
20. Igaz LM, et al. (2008) Enrichment of C-terminal fragments in TAR DNA-bindingprotein-43 cytoplasmic inclusions in brain but not in spinal cord of fronto-temporal lobar degeneration and amyotrophic lateral sclerosis. Am J Pathol173:182–194.
21. Inukai Y, et al. (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-Uand ALS. FEBS Lett 582:2899–2904.
22. Winton MJ, et al. (2008) Disturbance of nuclear and cytoplasmic TAR DNA-bindingprotein (TDP-43) induces disease-like redistribution, sequestration, and aggregateformation. J Biol Chem 283:13302–13309.
23. Ayala YM, Misteli T, Baralle FE (2008) TDP-43 regulates retinoblastoma protein phos-phorylation through the repression of cyclin-dependent kinase 6 expression. Proc NatlAcad Sci USA 105:3785–3789.
7612 � www.pnas.org�cgi�doi�10.1073�pnas.0900688106 Zhang et al.




















