
RING CLEAVAGE OF 3-HYDROXY-2,2,4-TRIMETHYL
173
RING CLEAVAGE OF 3-HYDROXY-2,2,4-TRIMETHYL- 3-PENTENOIC ACID ~-LACTOijE BY THE ANION OF DIISOPROPYL KETONE By ROBERT BURTON HANSON Bachelor of Arts Oklahoma State·University Stillwater, Oklahoma 1963 Submitted to the faculty of the Graduate College of the Oklahoma State University in partial fulfillment of the requirements for the degree of DOCTOR OF.PHILOSOPHY July, 1967
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of RING CLEAVAGE OF 3-HYDROXY-2,2,4-TRIMETHYL

RING CLEAVAGE OF 3-HYDROXY-2,2,4-TRIMETHYL-
3-PENTENOIC ACID ~-LACTOijE BY THE ANION
OF DIISOPROPYL KETONE
By
ROBERT BURTON HANSON
Bachelor of Arts
Oklahoma State·University
Stillwater, Oklahoma
1963
Submitted to the faculty of the Graduate College of the Oklahoma State University
in partial fulfillment of the requirements for the degree of
DOCTOR OF.PHILOSOPHY July, 1967

RING CLEAVAGE OF 3-HYDROXY- 2,2,4 - TRIMETHYL-
3-PENTENOIC ACID S-LACTONE BY THE ANION
OF DIISOPROPYL KETONE
Thesis Approved :
Thesis Adviser
~
El58797
ii
OKlANOiA STAT£ ti fVERSift LIBRAR'f'
JAN 10 l988 -' r

ACKNOWLEDGMENTS
The author is indeed deeply grateful for the encouragement, counsel
and guidance provided by Dr. K. Darrell Berlin during the course of this
study, not to mention his invaluable assistance which made possible the
herein documented research.
The author also wishes to express appreciation to Dr. Claude H.
Dungan, Monsanto Chemical Company, St. Louis, Missouri, for determina
tion of some high resolution nuclear magnetic resonance spectra and for
several invaluable molecular weight determinations. Also deepest thanks
are extended to those herein unnamed fellow graduate students of Dr.
Berlin's with whom I had the pleasure to work.
Gratitude is also extended to the Oklahoma State University Depart~
ment of Chemistry, the Research Foundation, and to the National Science
Foundation for financial assistance during the course of my studies.
iii

TABLE OF CONTENTS
Page
INTRODUCTION •ooooeooooooeoooo111000•011000 X
.chapter
I. HISTORICAL
Recent Chemistry of the ~-Lactone Dimer of Dimethyl-ketene ••••••
~-Lactones and Their Reactions with Grignard Reagents • Recent Carbanion Enolate Chemistry ••••••••••• Dicarbanions of Monoketones, 1,3-Diketones and ~-Keto-
aldehydes • • • • • • • • •••• Poly-~-Carbonyl Compounds •••••
i
1 4 6
12 27
IL .DISCUSSION OF RESULTS AND CONCLUSIONS. 31
. III. EXPERIMENTAL • • 59
Preparation of Phenyl Magnesium Bromide 59 Reaction (1:0.8) of Phenyl Grignard Reagent and Lactone
I - Run 1 • • • • • • • • • • • • • • • • • • • • • • 6 9 Reaction (1:1) of Phenyl Grignard Reagent and Lactone
I - Run 2 " c o o o ., o o o " o o o o o o o o o " o o 81 Reaction (1:1.2), (1:1.3). (1:1.5) and (1:2) of Phenyl
Grignard Reagent and Lactone I - Runs 3, 4, 5,and 6 • 81 Carbonation of 1:1.2 Reaction Mixture of Phenyl Grig-
nard Reagent and Lactone I. • • • • • • • • • • • • . 82 Esterification of Acid Material (Page 84) Obtained From
the Carbonation of the Reaction Mixture of Phenyl Grignard Reagent and Lactone I. • • • • • 85
Reaction (1:1.2) of Phenyl Grignard Reagent and Lactone ·· I in~Tetrahydrofuran - Run 7. • • • • • • • • • • • • 86 Carbonation of 1:1.2 Reaction Mixture of Phenyl Grig-
nard Reagent and Lactone I in Tetrahydrofuran. • • • 87 Esterification of Acid Material (Page 87) Obtained From
the Carbonation of the Reaction Mixture of Phenyl Grignard Reagent and Lactone I in THF • • • . • • 8~1
Reaction (1:1.9) of Methyl Grignard Reagent and Lactone !0000000•
Reaction (0.3:1) of Ethyl Grignard Reagent and Lactone I o o ., • 0 8 o 0
Preparation of !~Butyl Grignard Reagent ••••••••
iv
90
90 91

Chapter
Reaction (1:1.15) of ! - Butyl Grignard Reagent and Lac-tone I. . . . . . . . . o o • •
Sublimation of Ketol XXXII •••• Attempted Preparation of Carbonyl Derivatives of Ketol
Page
91 92
XXXII and Triketone XXXIII. A. 2,4-Dinitrophenylhydrozone. B. Oxime. C. Semicarbezone • • • • • • 93
Attempted Acylat i on of Ketol XXXII With Acetic Anhy-dride in Pyridine. • • • • • • • • • 94
Acid Hydrolysis of Ketol XXXII. • • • • • • • • • • • • 95 Dehydrat:ion cif Ketol XXXII. • • • • • • • • • • • • • • 96 Attempted Dehydration of Ketol XXXII With Dimethyl Sul-
foxide. • • • • • • • • • • • . • • • • • • • • • 98 Thermolysis of Ketol XXXII. • • • • • • • • • • • • • • 98 Reaction of Ketol XXXII With Li thium Aluminum Hydride • 99 Reaction of Triketone XXXII I With Lithium Aluminum Hy-
dride O O O O O O O O O O O O O O O O O O O O O O O o ioo The (1 : 1) Reaction of Triketone XXXIII With Potassium
Hydroxide in Ethyl Alcohol • . • . ••• •. ••••••• 101 The (1 : 1) Reaction of Ketol XXXII With Potassium Hy-
droxide i n Ethyl Alcohol ••••••• .•• •••• ·• 103 Preparation of 2,2,4-Trimethyl- 3- oxovaleryl Chloride
(XL~) . .. o o • • • • o o • • • • • • • • • • • • • • • 1 OS Preparation of Di isopropylcadmium and its Reaction With
2,2,4 - Trimethyl-3-oxovaleryl Chloride (XLIX). • • 106 Preparation of Methyl 2,2,4 -Trimethyl - 3-oxovalerate
(XXXV I) • • • • • • . • • • • • • . • . . . • • . 10 7 Preparation of Dimethyl 2,2,4,4-Te tramethyl - 3- oxoglu-
tarate (XXXVII) • • • • • • • • • • • • • • • • • • • 108 Attempted Claisen Condensation of Methyl 2,2,4-Tri
methyl - 3-oxovalerate (XXXVI) With Diisopropyl Ketone (XLIV). A. Sodium Methoxide as t he Condensing Agent. B. Sodium Hydride as the Condensing Agent. C. Sodium Amide as the Condensing Agent ••••••• 109
Reaction of Diisopropyl Ketone (XLIV) With Sodium Hy-d ride • . . . . . . . . . . . . . . . . . . . . . 111
Attempted Ring-Closure of Triketone XXXIII. • • • 111 Preparation of Hexamethyl - 1,3,5- cyclohexanetrione
(II). . . . . . . . . . .· . . . o • • • • • • • • 118 Preparation of Isopropyl Grignard Reagent • • • • • 113 Attempted Condensation of Isopropyl Grignard Reagent
With Hexamethyl-1,3,5- cyclohexanetriorie (II ) in the Presence of Magnesium Bromide • • • • • • 114
Preparation of Li thium Dust • • • • • • • • • • • • 115 Preparation of Isopropyllithium. • • • • • • • • • 115 Reaction of Isopropyllithium With Hexamethyl- 1,3,5-
cyclohexanetrione (II). . . . . . . . . . . . . • • • • • • • 116 Condensation of Methyl Grignard Reagent With Hexa -
methyl-1,3,5-cyclohexanetrione (II) •• • • 117
BIBLIOGRAPHY 0 0 0 0 0 0 O O O O O O O O O O O O 158
V

LIST OF TABLES
Table Page
I. Acidy Constants for Pseudo Acids at 25° in Water. 11
II. GLC Column Specifications ·"· ••• 61
III. Physical Properties of Products 63
IV. -1
Infrared Absorption (cm. ) Bands of Products • 66
v. NMR Chemical Shifts and Coupling Constants of Products, 71
vi

Plate
LIST OF ILLUSTRATIONS
Page
Infrared Spectra
I. 3-Hydroxy=2,2,4-trimethyl-3-pentenoic Acid ~-Lactone (I), Film on NaCl Plates. • • • • • • • • • • • • • • • -118
II. 5-Hydroxy-2,2,4,4,6,6-hexamethyl-5-isopropyl-l,3-cyclohexanedione (XXXII), KBr Pellet •••••••••••• 119
III. 5-Phenyl-2,2,4,4-tetramethyl-3,5-dioxovaleric Acid (XXXIV), KBr Pellet............... 120
IV. l-Phenyl-2,2,4-trimethyl-l,3-pentanedione (XXXIX), Film on NaCl Plates •••••••••••••• 121
V. 2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxonanoic Acid (XXXV), KBr Pel.let • • • • • • • • • • • • 122
VI. Methyl 2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxonanoate (XXXVIII), Film on NaCl Plates • • • • • • • • • • 123
VII. 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9,nonanedioic Acid (XLI), KBr Pellet • . • • • • • • • • • . • • • • 124
VIII. Dimethyl 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-nonanedioate (XLIII), KBr Pellet ••••••••••• 125
IX. 2, 2 ,4-Trimethyl-3-oxovaleramide (XL), KBr Pellet • 126
X. Unknown E, Film on NaCl Plates • • • • • • • • • 127
XI. 2,4,4,6,6,8-Hexamethyl-3,5,7-nonanetrione :(XXXIII), Film on NaCl Plates • • • • • • • • 128
XII. Unknown F, KBr Pellet. • • 129
XIII. Unknown G, KBr Pellet. 0 0 , 0 'o G 130
XIV. Ethyl 2,2,4-Trimethyl-3-oxovalerate (XLV), Film on NaCl Plates •••••••••••••• 0 'e 0 131
XV. 2,4,4,6-Tetramethyl-3,S=heptanedione (XLVI), Film on NaCl Plates •••••••••••• 132
vii

Plate Page
XVI. 2,2,4-Trimethyl,..,3-oxovaleryl Chloride (XLIX), Film on NaC 1 Plates. • . • • • • • • . . . • • • • • . . 133
XVII. Methyl 2,2,4·-Trimethyl-3-oxovalerate (XXXVI), Film on NaCl Plates. • • . • • • • • • . . • . • . . • • 134
XVIII. Dimethyl 2,2,4,4-Tetramethyl-3-oxoglutarate (XXXVII), Chloroform Solution ••••• , • . . . . . • • . • • 135
XIX. 5-Hydroxy-2,2,4,4,5,6,6-heptamethyl-l,3-cyclohexanedione (L), KBr Pellet .••..•...•.••••..••. 136
Nuclear Magnetic Resonance Spectra
XX. 3-Hydroxy-2, 2 ,4-trimethyl-3-pentenoic Ac id ~-lac tone (I). 13 7
XXI. 5-Hydroxy-2 ,2 ,4 ,4·,6 ,6-hexamethyl-5-isopropyl-1,3-cyclohexanedione (XXXII) •..•••••.. , ••.••.•• 138
XXII. 5-Hydroxy-2,2,4,4,6,6-hexamethyl-5-isopropyl-3,5-cyclohexanedione (XX.XII). • • • • • • • • • • • • • • • • • 139
XXIII. 5-Hydroxy-2, 2, 4 ,4, 6, 6-hexamethyl 0-5-isopropyl-3, 5, -eye lo-hexanedione (XXXII). • • • • . . • • • . • • 140
XXIV. 5-Phenyl-2 ,2 ,4 ,4-tetramethyl.;,3 ,5-trioxovaleric Acid (XXX IV ) o o o o o o o o o o o o o o o o o o o 141
XXV. l-Phenyl-2, 2 ,4-trimethyl-l, 3-pentanedione (XXXIX). • . 142
XXVI. 2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxononanoic Acid (XXXV) o o (I • o " o o o o o o " o ,, o • o • 143
XXVII. Methyl 2 ,2 ,4 ,4, 6, 6, 8-Heptamethyl-3, 5, 7-trioxononanoate (XXXVI I I) o o o o o o o o o o o o o • o o o o o o o o • 144
XXXVIII. 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-nonanedioic Ac id (XLI) . " o o • o o ., o o • " o o • o (I o o " o o 145
XXIX. Dimethyl 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-( ) 146 nonanedioate XLIII ........................................ .
XXX. Dimethyl 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-nonanedioate (XLIII) • • . • . • . • • • • . 147
XX.XI. 2,2,4-Trimethyl-3-oxovaleramide (XL) 148
XXXII. Unknown E. . • • • • • • • • • • • • 149
XXXIII. 2,4,4,6,6,8-Hexamethyl-·3,5,7-nonanetrione (XXXIII) • 150
viii

Plate Page
XXXIV. Ethyl 2,2,4-Trimethyl-3-oxovalerate (XLV) ••• 151
XXXV. 2,4,4,6-Tetramethyl-3,5-heptanedione (XLVI). 152
XXXVI. 2 ,2 ,4-Trimethyl-3-oxovaleryl Chloride (XLIX) •• 153
XXXVII • Methyl 2 ,2 ,4-Trimethyl-3-oxovalerate · (XXXVI) • 154
. XXXVIII. · Dimethyl-2, 2 ,4 ,4-Tetramethyl-3-oxoglutara te (XXXVII) • • 155
.. XXXIX. . 5-Hydroxy-2 ,2 ,4 ,4, 5, 6, 6-heptarnethyl-l, 3-cyclohexanedione . (L) o o o o o o o o o a o a o o o o o o a o o o o o o o 15 6
XL. 5-Hydroxy-2,2,4,4,5,6,6-heptamethyl-1~3-cyclohexanedione (L) 0 0 0 0 0 0 0 D O O O O O O O O O O O O O O O O O O 15 7
ix

INTRODUCTION
Ring cleavage of ~-lactones by certain anionic reagents is well
known, although ring opening by Gr.ignard reagents is not well documented.
Recent experimental evidence presented in this Laboratory revealed the
possibility of the presence of a dicarbanion form of diisopropyl ketone
in conde.nsa tion of phenyl Grignard reagent and 3-hydroxy-2, 2 ,4-trimethyl-
3-pentenoic acid ~-lactone. Furthermore the production (in fair yields)
of two compounds of highly unusual structure, a cyclic ketol and an
acyclic triketone, suggested dicarbanion intermediates. Since from
previous mechanistic considerations the condensation of the lactone with
phenyl Crignard reagent was believed to involve anion species which con
densed with ~dditional lactone, the use of excess lact~ne might possibly
increase the yields of the ketol and triketone. Moreover, the anion
species could be carbonated giving acids if the ketone-encl equilibrium
favored the ketone form. In addition, spectral data and chemical and
elemental analysis might lead to a structural assignment for the ketol
and the triketone.
X

CHAPTER I
HISTORICAL
Recent Chemistry of the ~-Lactone Dimer of Dimethylketene
.Since the preparation of the ~-lactone dimer I of dimethylketene,
many reactions of this lachromatory, moderately stable liquid compound
have been reported. Examples are: aminolysis, hydrolysis and alcoholy-
sis and reactions with anions such as me·thoxide and those from active
methylene compounds. Reactions with other nucleophiles and polymeriza-
11 b d d b h b · d · b Z 88 tion wi not e iscusse ut ave een reviewe in part y augg.
Various trimers, tE!tramers and polymers of dimethylketene have been
prepared. A stable trimer of dimethylketene, 2,2,4,4,6,6-hexamethyl-l,
3 ,5-cyclohexanetrione (TI) is formed by a base-catalyzed reaction of the
15 65 ketene, its dimers or its polymers.·· ' ·
I
A less stable trimer 0£ dimethylketene, 5-hydroxy-2,2,4,4,6-penta-
methyl-3-oxo-5-heptenoic acid 5-lactone, has been synthesized by base-
catalyzed disproportionation of the ~-lactone dimer of dimethylketene,
1 ) 14 3-hydroxy-2, 2 ,4-trimethyl-3-pentenoic acid ~- lac tone '.,l; .• ·•In the
1

2
11
absence of labile hydrogen atoms, lactone I reacts with strong bases
42 such as a sodiu~ alkoxide to form an enolate anion. .With less than
equivalent amounts of base, other products are formed. A polyester con-
ceivably is obtained from lactone I by simple ring opening; propagation
is reported to proceed through the enolate anion rather than the car-
boxylate anion. Reaction at the oxygen atom of the enolate anion pro-
duces a polyester, whereas reaction at the carbon atom would possibly
yield a polyketone. This latter type of attack would lead to triketone
II. With a strong base such as sodium methoxide and a low temperature
(boiling ether), only the polyester is formed. With a slightly weaker
base such as sodium hydroxide, the polyester is reported to form exo-
thermally in boiling lactone I. Further heating of the mixture to 200-
250° without removal of lactone I converts the polyester into a mixture
of the tr imers.
I I

~)c (CH3 )2IH3 II ..,_~-c -C
3
~H3 --+
51 Recently, Martin and co-workers· '.have reported that lactone I con-
denses with cyclic enamines to give highly substituted carbocyclic ring
compounds. 1- (1-Cyc lohexen-1-yl) 'pyrrolidine · and lac tone I when heated
for several hours on a steam bath under nitrogen, gave hexahydro-2,2,4,
4-tetramethyl-4a- (1-pyrrolidinyl)-1,3-(2H,4H)-naphthalenedione in good
yield (80%). This adduct was reported to decompose slowly when stored
at room temperature. 1-Ethyl-N,N-dimethylpropenylamine and lactone I
reacted to give a cyclic, unsaturated diketone and an amide.

4
I -t I I
S-Lactones and Their Reactions With Grignard Reagents
Gresham and co-workers in 1949 reported the first evidence of ring
I 27 cleavage of a ~-lactone by Grignard reagents. When ~-propiolactone and
0 phenyl Grignard reagent (1:1) were allowed to conde~se at -6 , products
from 0-alkyl and 0-acyl ring opening were observed. A small amount of
polymer (9.6%), ~-bromopropionic acid (43%) and phenyl vinyl ketone
(21.2%) were isolated. Phenyl vinyl ketone may have arisen through
spontijneous dehydration of the intermediate, ~-hydroxypropiophenone.
-1:: CH2-cH2 + polymer
I I Br co2H
Methylmagnesium iodide condensed with- ~-propiolactone (1 : 1) to give~ -
iodopropionic acid, [a neutral oil which exhibited a large boiling range,
29-78°/25-28mm], a small a~ount of polymer and methyl vinyl ketone.
Benzylmagnesium chloride and ~-propiolactone gave polymer (22.4%), ~-
chloropropionic acid (16.1%) and 4-phenylbutanoic acid (32.4%). How -
ever the remaining 20% of the reaction mixture was not completely iden-
tified.
Stuckwisch and Bailey75 found that allylmagnesium bromide and~-
propiolactone gave 5-hexenoic acid in fair yield (54%). This product
was the only compound isolated, however.

5
When phenyllithium and ~-propiolactone were allowed to condense at
0°, the diaddition product [l,l-diphenyl-1,3-propanediol (40%)] and
phenyl vinyl ketone (45%) were the only compounds isolated. When two
equivalents of E_-butyllithium and one equivalent of the lactone were
used, the corresponding diaddition product was the sole product isolated
from the reaction mixture.
Many investigations of the reactions of aldoketene dimers have been
carried out, but only a few have dealt with their reactions with Grig-
nard reagents. Reid and Croszos have reported that solid ethylketene
dimer, which is stated to have the structure of 2,4-diethyl-1,3-cyclo-
butanedione, behaved abnormally in a Grignard machine yielding 1.3
66 active hydrogens along with less than two additions to carbonyl groups.
61 Nightingale and Turley found that when a two-mole excess of
phenyl Grignard reagent was added to the ~-lactone dimer of E_-butylke-
tene, three products were isolable, these arising from 0-acyl ring
cleavage of the lactone. Benzophenone (16%) (IV,R 1 = c6H5 ), 6-unde
canone (III, R = E_-C4H9) and 5-(diphenylhydroxymethyl)-6-undecanone (V,
R = E_-C4H9) were found.
0 0
RCR::DHR + 2R uMgBr
0 ~ "0
II a. RCH2CCH2R
III
II + R 1CR 1
IV V
E,-Hexylketene dimer (~-lactone) reacted with excess of phenyl Grig-
nard reagent to give 7-(diphenylhydroxymethyn-s-pentadecanone(V,,R = n-
c6H13 ) as the only isolated product. The remainder of the reaction mix-
ture was thought to contain 8-pentadecanone and benzophenone which could
not be separated. The ~-lactone dimer of E_-octylketene and excess phenyl
Grignard reagent gave :products analogous to those reported for

6
.!!_-butylketene dimer and phenyl Grignard reagent •
. An excess of the aliphatic Grignard reagents (ethyl, !!_-butyl, .!!_
octyl) condensed with the aldoketene dimers to yield the simple ketones
• III and IV, but nort.e of the dtaddition, product V could be isolated from
the high-boiling residues •. The mechanism of the formation of the two
ketones has been formulated as involving a simple retrograde aldo
2R'MgBr RCH =c -rCH-C(R')
I • I I · 2
BrMgO i R OMgBr
II IH RCH2C,HC (R' )2
a II + R CR I
R
V III IV
reaction of V. either· during the reaction or during the hydrolysis of the
· Grignard complex.
5 .. It was reported from this Laboratory · that an excess of phenyl,
£-tolyl and 1-naphthyl Grignard reagents condensed with th~ ~-lactone
dimer I, and high-boiling ~-diketones were produced in yields of less
than 50%. The remaining portion of the reaction mixture in each case
was complex, with more than 15 components detectable by gas liquid
chromatography (GLC).
Recent Carbanion Enolate Chemistry
.Becquse of the insurmountable task of sifting through the litera-
ture and extracting all information on enolate carbanion chemistry, this

7
discussion will be limited to important work which has been publmhed
within the past decade.
The use of acid chlorides and anhydrides for the acylation of ke-
tones having an a-hydrogen atom has been reported as a convenient route
to many ~-diketones.48 ,lO With acid chlorides, the condensation is
usually effected by means of basic reagents such as sodium alkoxides,
sodium hydride and sodium amide; with acid anhydrides, a Lewis acid such
as boron trifluoride is generally employed. 0-Acyl derivatives are
formed in these reactions, but the conditions can be controlled so that
favorable yields of 1,3-diketones can be obtained. Several authors have
· proposed one-stage mechanisms, 51 , 52 , 63 while other data has led to a
proposed circular transfer mechanism. 79
The acylation of enolate carbanions formed from the reaction of an
aliphatic ketone and sodium amide has been found to be sensitive to the
60 molar ratio of acid halide (or anhydride) to ketone-base. Benzoyl
chloride,sodium amide and acetone, when combined in a 1:2:2 ratio,
respectively, gave chiefly the 1,3-diketbne, but when a 2:1 : 1 ratio of
reactants was employed, the major product ~as the enol ester. With a
more sterically hindered ketone, such as 2,6-dimethyl-4-heptanone, enol
ester formation predominated. Increased hindrance in a substituted ke-
24 tone tends to stabilize the enol structure.
52 House and Kramer have performed extensive investigations on enol-
ate anion compositions of reaction mixtures obtained f rom acyclic un-
symmetrical ketones. The base-solvent system used for the conversion of
the ketones was triphenylmethylpotassium in 1,2,-dimethoxyethane. Two
methods were used for measuring the enolate co~centrations. One con-
sisted of quenching part of the reaction mixture with a solution of

8
deuterioacetic acid-deuterium oxide followed by analysis of the formed
monodeuterated ketones (mixture) by Illl.SS spectrometry. In all cases, the
recovered ketones contained no significant amount of dideuterated
species. The second method used to measure the composition of the enol-
ate solutions consisted of adding solutions of the enolates to an excess
of acetic anhydride with the formation of enol acetates. In all cases
the enol acetates were the major products of the reaction along with
small.amounts of recovered ketone. The authors reported no evidence for
the existence of C-acylated products. Several generalizations were made:
(1) acyclic ketones of the type R2CHCOCH2R form the less highly substi-
r tuted enolates R2CHC==CHR as the predominate structural isomer in the
equilibrium mixture; (2) ketones of the type RCH2COCH3 form approxi-
mately equal amounts of the structurally isomeric enolates if R is an
unbranched-alkyl group and form mostly the less highly substituted
r enolate RCH2C==CH2 if Risa branched alkyl group; (3) cyclic ketones,
. 1-methylcyclohexanone and 1-methylcyclopentanone, afford approximately
. equal amounts of the structurally isomeric enolates; (4) where stereo-
isomeric enolates of types VI and VII are possible, the latter structure
R R'
\ I c:==c ·
R (/2) \ I
C ==: C
I \ H Of;
I \ H R'
VI VIII
will predominate. An enhanced preference for the less highly substi~
tuted enolate in equilibrium mixtures involving lithium enolates was
also reported.

Alkylation of enolate anions has been found to be improved by the
90 use of dimethyl ~thers of ethylene glycol. The activity of the car-
banion was increased by coordination of the solvent with the cation.
9
Solutions of this type have been found to be conductors of electricity,
whereas solutions of carbanions associated with metallic cations as
.aggregates in aromatic hydrocarbons or diethyl ether have been found
not to conduct an electric current.
1 h d . 73,67,85 k l h h In severa ot er stu ies, etone eno ate anions w ic had
been generated in the absence of either a relatively acidic proton-
donating solvent or an excess of the ketone appeared not to produce
equilibrium mixtures of enolate anions. This failure to equilibrate was
notable when lithium enolates were involved. Further studies along this
53 same line have been done by House and Trost. It was found that the
potassium enolates of several cyclic ketones equilibrated much faster
Q
II c:::(~ 0
CP 9'co: 9cnH 0 H O H
than did the corresponding lithium derivatives in a 25-100 mole% ex-
cess of the ketone. Triphenylmethylpotassium or the lithium analogue
were used to form the enolates.
The base-solvent system lithium in liquid ammonia has been used ex-
73 74 tensively for the conversion of a,~-unsaturated ketones ' and ster-
. 84 56 89 86 . . oids ' ' ' into the corresponding enolate anions. These latter
species have been trapped by alkylation and carbonation.
Theoretically, anions derived from acyclic ~-dicarbonyl compounds
can exist in any one or all of three plane~parallel molecular orbital
systems which can be termed U-shaped, W-shaped and sickle-shaped.

e ~
0 0
Vi ii -c .. c-
"=o a';il' C
10
/
Representative examples unequivocally possessing the latter structure are
not known. Ultraviolet spectroscopy was used to determine that acyclic
89 enolates primarily exist in the U-shaped ion pairs.
Infrared spectroscopy studies have been made on the free enolate
ions of acetylacetone and diacetylacetonitrile, Diacetylacetonitrile in
contrast to acetylacetone was shown to exist completely as the enol
86 form.
Acids and bases catalyze enolization of ketones by similar mechan-
isms. In the presence of strong acids, a ketone is converted reversibly
to its conjugate acid; loss of an alpha proton occurs by reaction with a
nucleophile in the rate~etermining step to give the corresponding enol.
Enolization by strong bases involves reaction of a base and a ketone with
slow.., ..--
fast
OH R 1: \ . C~C-R /:
H • . B •••H
OH
I ~ BHEB + RCH=CR .....-
assistance of weak electrophiles present to yield the enolate anion.

0
II HA B + RCH2CR ~
O ••• H-A R f" ' : c~c -R
I:
11
+
Bromine titration, infrared (IR) and ultraviolet (UV) spectroscopy and
12 more recently NMR analysis of ~-dicarbonyl compounds have been used to
determine the percent of enolization.
The K values found in Table I are important to the understanding a
of some of the work described in the Discussion section.
TABLE I
64,50 ACIDITY CONSTANTS FOR PSEUDO ACIDS AT 25° IN WATER.
Carbonyl Compound Ka'a i
Carbonyl Compound
CH3COCH3 ..,,;10-20 CH3COCH2COCF3
CH2 (COCH3 ) 2 1.0 X 10- 9 c6H5COCH2COCF 3
· CH(COCH3 ) 3 1.4 X 10-6 o=O 10- 9 CH3COCH2COCH3 1.0 X COCH3
CH3 COCH(CH3) COCH3 1.0 X 10- 11 o=(] CH3COCH2coc6H5 4 X 10-lO COCH3
CH3COCH2CHO 1.2 X 10 -6 · CH2 (CH0) 2
aThis is gross acid constant uncorrected for enol content.
K. .a -a
8 1 10-11 • X
-8 1.5 X 10
-5 1.0 X 10
Potentiometric methods have also been used for the determination of
p~ values for several substituted ~-diketones at different a

12
19 temperatures. The acidity values were found to decrease with increas..;
ing temperature •
. In order for there to be any contribution of structures (shown be-
low) to the total hybrid of an enolate anion, it has been found that the
' e /C===C--0 system and the three atoms attached directly thereto lie at
50, 1'7 least very nearly in the same plane. · ·
· Proton removal from an asymmetric carbon atom to form a carbanion
results in racemization. The charge of the carbanion (ambident carban-
ions) is known to be largely concentrated on the oxygen of the carbonyl
group (the more electronegative element), and proton capture undoubtedly
20 occurs there much faster than on carbon.
Dicarbanions of Monoketones, 1,3-Diketones and 13-Ketoaldehydes
Intermediates in which two negative charges have been generated on
two vicinal or nonvicinal atoms in the same mplecule have been formed
from a variety of compounds. When the charges are on carbon atoms, the
anions are more precisely labeled dicarbanions •.
. The first ·dicarbanion of an acyclic ketone reported in the litera-
45 ture was prepared by Hauser and Harris. They found that the reaction
of dibenzyl ketone with two equivalents of potassium amide. in liquid
ammonia produced dark red dicarbanion VIII •. The dark red color of
species VIII suggested that the hybrid possessed properties
. o oe 0g
elle Cf I. ® I -0 @ c6H5CHCCHC6H5 , 2~+-+C 6H5CH==CCHC6H5 ,. 2K .-.c6H5CH=C-CH- G), 2K VIII -

13
characteristic of not only an enolate anion resonance form, but it is
likely that the phenyl group makes significant contributions.47
Dicarbanion VIII, when treated with one molar equivalent of benzyl
chloride, gave a high yield of the monoalkylated product whereas the
monovalent precursor species of VIII under the same conditions gave a
mixture of products. Supposedly the dicarbanion VIII exhibits much
greater basic and nucleophilic strength than the monovalent species.
Essentially pure dibenzylated derivative was obtained when dicarbanion
VIII was treated with two equivalents of benzyl chloride. A monocon-
jugate addition product was obtained in excellent yield when VIII was
condensed with ethyl cinnamate. The monocarbanion precursor of VIII
failed to give any appreciable conjugate addition product.
Benzalacetophenone was found to undergo rapid reductive metalation
with two equivalents of potassium amide in liquid ammonia to form black
35 dicarbanion IX.- The dipotassio salt of IX when allowed to react with
benzyl chloride (one equivalent) gave the ~-alkylated product in good
0
e e II . c6H5CHCHCC 6H5 ,
IX
2K~

14
yield. This product may be further metalated and the resulting carban-
ion treated with benzyl chloride to yield the dibenzylated derivative.
Ay-hydroxy ketone was formed when IX was condensed with benzophenone.
Acetylacetone undergoes conversion to a dicarbanion when treated
with two equivalents of potassium amide in liquid ammonia. 46 The species
was monoalkylated with several primary halides, a secondary and tertiary
58,29 halide to give terminal alkylated products. Along with the mono-
alkylated derivative a small amount of a dialkylated product was
0 0
lie 110 CH3 CCHCCH2 ,
2~ ··~1-·~RX~'~l-i_q_._N_H_J_.
; 2. NH4Cl / H20
detected in the reaction mixtures. When analyzed structurally, the di-
alkylated products were observed to contain the alkylated group on the
terminal ends of the ketone. The mechanism of formation of the di-
alkylated compounds has been postulated to involve metal-hydrogen ex-
28 change. When a secondary halide,. isopropyl bromide, combined with IX
the yield of the monoalkylated ~-diketone was 27%; with !_-butyl bromide
the alkylation failed.
In contrast to the typical primary halides, ~-phenylethyl chloride,
which has a relatively reactive:~-hydrogen, failed to alkylate
1. 0 0 II II
IX c6H5CH==CH2 +.CH3CCH2CCH3
2.
dicarbanion IX. Instead the halide underwent ~-elimination to form
styrene (89%) with most of the ketone being recovered.
Benzhydryl chloride, which has a more reactive O:'-hydrogen than
benzyl chloride, also failed to alkylate dicarbanion IX. The halide

15
instead underwent self-alkylation accompanied by ~-elimination to form
tetraphenylethylene (98%).
In addition, the dicarbanion of acetylacetone, IX, has been re-
ported to undergo phenylation with bromobenzene (benzyne intermediate)
to form l-phenyl-2,4-pentanedione in low yields (13%). 46 However the
terminal derivative was formed (92%) when disodioacetylacetone and di-
phenyliodonium chloride were brought together in a 2:1 molar ratio,
respectively. The mechanism of the reaction has been postulated to in-
valve the formation of a radical anion and the phenyl radical which
30 combine to give the terminal product.
Benzoylacetone, not unlike acetylacetone, has undergone analogous
reactions •. When treated with two equivalents of potassium amide in
liquid ammonia, conversion to a dicarbanion was realized. The divalent
species has been treated with one equivalent of benzyl chloride to give
.. the terminal benzylated derivative Xa (77%). Triketone Xb was obtained
0 0 0 0
11 e ~ Q 2 ff) II II . c6H5C-CH0 -CH2 ' K c6H5CCH2CCH2R
Xa, R = CH2C6H5
b, R "" COC6H5
c, R C02H
d, R "" -CHC6H5 I OH
(58%) by condensing the dicarbanion with methyl benzoate; terminal acid
Xe was afforded (58%) by adding the dicarbanion to a slurry of pulver-
ized Dry Ice. It was added that carbonation possibly occured at C-3 but
decarboxylation took place during workup of the reaction mixture. Ketol
,Xd was prepared by condensing the dicarbanion with benzaldehyde and the

16
product was isolated as its copper chelate in an overall yield of 28%.'46
Aroylations of ~-diketones to form 1,3,5-triketones has been per-
formed; structure proof of the compounds was afforded by elemental
. . 55 32 . analyses, enol tests and cyclizations to 4-pyrones and 4-pyridones. '
.The general procedure for effecting the aroylations involved the addition
of the ~-diketone to two molar equivalents of potassium amide in liquid
ammonia followed by the addition of one-half of an equivalent of an
appropriate ester. In all cases, yields of the triketones were reported
b. R = CH3 , R' = P""C1C6H4-
c: R = C6H5, R' = C6H5
d. R = C6H5' R' = 11-ClC H -- ', 6 4
e. R = C6H5' R' = p~cH3oc6H -. . . 4
f. R = C6H5, R' =3 - pyridyl
to.be fair to good. (40-62%). In addition, 2-acetylcyclqhexanone, 2-
acetylcyclopentanone and £-hydroxyacetophenone were converted to their
corresponding dicarbanions and aroylated at the terminal position with
an appropriate ester; although the yields were lower than for the
acylic triketones •. A mechanism (three-step) was offered to explain the
formation of the triketones which was an adaptation of the method used
in the acylations of ketones with ~sters (by alkali amides) to form~
·48 diketones,

17
. The dilithio derivatives of acetylacetone and benzolacetone have
87 also been aroylated. · The dilithio salt of benzolacetone was studied
.as. the attempted condensation of either the dipotassio or disodio salt
failed to give a condensation product when treated with phenyl propio-
nate. Apparently the dicarbanions merely removed the QI-hydrogen of the
ester •. However by using the dilithio salts, condensation products
· (> 40%) with ethyl acetate and higher esters were obtained; with the
disodio or dipotassio salts the reactions failed •
. Excess sodium hydride has also been used for the·prepatation of
59 1,3,5-triketones from several ~-diketones and an appropriate ester.
A comparison of the methods of arylation using excess sodium hydride in
boiling monoglyme and potassium amide in liquid ammonia to effect the
condensations, revealed that in most cases the sodium hydride method
was superior, as yields .of the triketones were far higher than for the
potassium amide experiments. However for carbonation, benzylation.and
aldol-type condensations the pbtassium amide in liquid .ammonia method
is reported to be superior. Although four possible mechanisms involving
. ini tia 1 formation. o f a monocarbanion were postulated for the condensa-
tions using sodium hydride in boiling monoglyme, no explanation for the
. increased yields of the triketones was provided •
. In contrast to the situation of acetylacetone and benzoylacetone,
two different dicarbanions XI and XII canarise-from certain unsymmetri-
cal ~-diketones depending on whether a.methyl or 5-methylene hydrogen
d h d . . . 39,31 un ergoes t e secon ary 1on1zat1on. It was shown by Meyer and

XI
2 6:) M
18
e"9u ffi RCHCCHCCH3 , . 2W
XII
58 Hauser· that 6-phenyl-2,4-hexanedione can be benzylated through a di-
carbanion of type XI,: yieldiri.g 1, 7-diphenyl-3 ,5-heptaneefone in 65%
yield. No products corresponding to alkylation of dicarbanion-XII were
obtained. The l3-diketone;2,4-tridecanedione, was converted to its
2 KNH2 fle ?le 6:) 1. RX ~ ~ liq• NH3• RCH2CCHCCH2 ,, 2K ----RCH2CCH2CCH2R
2. NH4Cl
XI Ila,. R = R 1 = CH2C6H5
dicarbanion with two molar equivalents of potassium amide in liquid
ammonia.and alkylated with one equivalent of ~-octyl bromide to give 13-
diketone XIIIb (71%). The dicarbanion of 6-phenyl-2,4-hexanedione
underwent butylation with ~-butyl,.broniide to give !3-diketone XIIIc · (67%).
Structure proof of the compound was established by IR analysis of its
copper chelate and an independent synthesis involving the condensation
of methyl hydrocinnamate·with methyl.amyl ketone.
Further studies of alkylation of unsymmetrical !3-diketones have
.•,, ·~ ..
shown that structural effects have an influence on the position alkyl-
31 ated.

19
0 0 a II llb CH3CCH2CCH2CH3
0 0
a II II CH3CCH2CCH(CH3 ) 2
XIV xv
0 0 0 0
a II llb CH3cH2CCH2CCH(CH3 ) 2
a II llb CH3CCH2CcH2C6H5
XVI XVII
Compound XIV was converted to the dicarbanion with two equivalents
of sodium amide in liquid ammonia and the disodio salt treated with one
equivalent of methyl iodide. Gas chromatographic analysis of the reac-
tion mixture indicated that terminal alkylation at position~ occurred
to a far greater extent than at position.£.• The yield of the terminal
methylated diketone was 89% while the product arising from alkylation at
b was 11%. The dicarbanions arising from ketones XV and XVI, when methy-
lated under the same conditions as for the dicarbanion of XIV, formed the
methylated product f rom alkylation at position a in 99% yield. The pro-
duct ratios of the alkylated products obtained from the dicarbanion of
XVI were not analyzed by GLC as the isomers could not be separated. How-
ever, analysis by GLC of the products obtained from alkaline cleavage of
the components present in the reaction mixture gave only traces of pina-
colone. The authors surmi sed that pinacolone would have been formed from
base degradation of the reaction mixture if XVI had been alkylated at
position b. Under identical conditions, the dicarbanion derived from
compound XVII underwent exclus i ve methylation, ~-butylation and benzy-
lation at position.£. • The disodio salts of the ~-diketones were used in
preference to the potassio salts as the latter were found to undergo
rapid metal-hydrogen exchange with the monopotassio salts of the alky-
lation products.

20
Terminal bis-~-diketones have been prepar ed by treating the dicar-
banions derived from acetylacetone, benzoylacetone and other related~
diketones ·with 1,3-dibromopropane and higher methy le ne halides. 34 A
0 0 0
" 0 0
II II II II II RCCH2CCH2 (cH2 )nCH2CCH2CR ~ (CH2) 2CcH2cc6H5
a• R = c6tt5 ; n = 3 (CH2 ) 2cctt2cc6H5
II ir 0 0
b. R = c 6H5 ; n =4 0 0
c. R = CH3 ; n = 3 II II (CH2 ) 2CCH2cc6H5
d. R CH3 ; n = 4
~ 2!C~ ~C 6H5 e . R CH3 ; n = 9
two-fold alkylation of benzoylacetone was accompl ished by condensing
the dicarbanion with a,&-dibromo-£-xylene and a ,a '-dichloro-2-xylene.
The reaction was reported to fail with ethy lene and methylene halides,
The dicarbanion derived f rom 3-phenyl - 2, 4- pentanedione has been
shown to be less reactive than the d i carbanion derived from acetylace-
tone, as benzylation atte~pts on the forme r failed to give any appreci-
62 able benzylated product. This relative slugg i shness toward alkylation
has been attributed to lower solubility of the d icarbanion in liquid
ammonia and/or a lower nucleophilicity of the d i ca r banion because of
contributing structures in which the phenyl group participates in de-
localizing part of the charge. Although the terminal benzylated deriva-
tive of 3-phenyl-2,4- pentanedione was obta i ned (16%) when its dicarban-
ion (in liquid ammonia) was treated with be nzyl chloride, the yield was

21
increased to 50-60% when the solvent system was changed to pyridine con-
taining a small amount of liquid ammonia. No explanation was given as
to the function of pyridine in increasing the yield of the terminal
derivative • . Preformed benzylpyridinium chloride, when .added to the .di-
carbanion,gave no benzylated product • . Aroylation of the dicarbanion in
liquid ammonia with methyl benzoate gave the corresponding triketone in
a yield of 14%, but the yield of the triketone was increased to 47% when
the reaction was run in pyridine containing a little liquid ammonia.
Carbonation of the dicarbanion was accomplished by pouring a solution of
the dicarbanion in ether over a large excess of pulverized Dry Ice. The
terminal acid was obtained in 26% yield.
0 0 0 0
11 e lie EB 1. CO2 II II cH3c-7-ccH2 , 2M CH3CCHCCH2co2H
C6HS 2. Acid
i6HS
The monosodio salt of acetoacetaldehyde has been converted to a di-
carbanion with one equivalent of potassium with one equivalent of po-
tassium amide in liquid ammonia and the resulting species treated with
benzyl, methyl, E_-butyl and E_-octyl halides giving the corresponding
36 alkylated monocarbanions which were converted to their copper chelates.
0 0 0 0
II c:.. II KNH e lie II .,p '+' c, NaEB 2 w · CH3CCHCH, ----- CH2CCHCH, , Na
RCH2 liq• NH3 l ~-o 0-CH Rx
~H \I JH R P. mbol' __ '-o-_. ~11
A cu(OAc) 2 lie II EB ... RCH2 CCHCH, Na
CWR 2
In addition, the monosodio salts obtained from the reaction of the di -
carbanion with benzyl and methyl halides were converted to cyanopyri-
dones by treatment with cyanoacetamide.

22
a-Benzylacetoacetaldehyde was converted to a dicarbanion with two
equivalents of potassium amide in liquid ammonia and alkylated with
benzyl and E_-butyl halides to yield the corresponding terminal alkylated
derivative in yields of 51 and 27% respectively. 36 That the alkylation
0 0 1. RX II II 2. Ac id
RCH2CIHCH
CH2C6H5
products of the dicarbanion were the terminal derivatives, was supported
by positive enol tests with ferric chloride and by IR spectral. data.
A benzyne-dicarbanion intermediate was reported to arise when 5:..-
chlorophenyl ~-diketone XVIII was slowly treated with excess potassium
.amide in liquid ammonia. 40 The intermediate formation of the dicarbanion
liq. NH3
cyclization acidification
in the reaction mixture was indicated as a transient green color was ob-
served. Indirect evidence that the two-fold ionization to form the di-
carbanion preceded dehydrohalogenation was the lack of any appreciable
amination which would have been expected if a benzyne were produced
first. Similarly a-substituted derivatives XIX and XX when converted to

23
corresponding qicarbanions were cyclized to six-membered ring compounds.
XIX xx Dicarbanions of compounds other than acyclic ones have been pre-
39,9 pared from a-formyl cyclic ketones .. The monosodio salt of 2-formyl-
6-methylcyclohexanone was changed to the dicarbanion with an equivalent
of potassium amide in liquid ammonia. Benzylation of the dicarbanion and
Oo NaEH I~ ., e e CH-3 CHO KNH2 CH3 r1- CHO
liq. N'3 V ' EfJ E9 K, Na RX
0
R II 8 CH3~HO
·• V subsequent deformylation with aqueous sodium hydroxide gave 2-benzyl-2-
methylcyclohexanone (50%). Methylation and ~-butylation of the dicar-
banion offered analogous products after deformylation. All three of the
2-alkyl-2-methylcyclohexanones were detected by GLC and NMR to be uncon-
taminated with the isomeric 2,6-disubstituted ketone. This method of
angular alkylation has been reported to be superior to direct alkylation
of a 2-substituted cyclohexanone, which produces a mixture of the 2- and
6-alkylation products as well as higher alkylated derivatives. Likewise,
alkylation of the dicarbanion of 2-formyl-1-decalone with methyl,~-
butyl and benzyl halides offered only the 9-alkyldecalones uncontaminated
with any of the 2-alkylated product. Single diastereoisomers were
0 0
II ~ ~CHO 1. RX co

24
reported for the benzyL and !!:-butyl derivatives, but GLC indicated a
46:54 mixture of the.cis and trans isomers from the methylation experi-
ment. The butyl derivative was shown to contain a cis-fused ring system
by reduction of its ethylene thioketal under Wolff-Kishner conditions to
9-butyl-cis-decalin. Direct alkylation of 1-decalone has.been reported
to give chiefly the 2-alkyl derivative rather than the 9-alkyl deriva-
tive. -~ure 2-alkylated products, as indicated by GLC, have been prepared
from the dicarbanions of cyclopentanone and cyclohexanone. The method is
0
II eoe '.2~
0
e.A_ 1 e LJ '2~
1. RX .,2 •. Acid
1. RX
2 •. Acid
reported to be superior to direct alkylation of these systems by means
of strong bases.
Certain bis-~-diketones have been converted by four molar equiva-
lents of sodium amide in liquid ammonia to their tetrasodio salts which

n = 5,6
2. Acid
0 0 @ IIQ II CH2CCHC
I (CH2) , I n
· CH y.CHC e 211e II 0 0
n = 5,6
0 0
II II c 6H5CH2CH2CCH21
4 Er> Na
(CH2 ) I n
c 6H5CH2CH211cH2B 0 0
n = 5 ,6
25
33 were condensed with certain electrophilic reagents. Benzyl chloride
when condensed with the tetrasodio salt gave a 6i% yield of the diterm-
inal derivative. Polymer formation was observed when 1,3-dibromopropane
was condensed with the tetrasodio salt. The polymer was reported to be
0 0
e lie II CH2CCHC
I (CH2 ) , I n
4Na®
CH fiCHC e 211e II
0 0
poly(l,3-octanedione) with a molecular weight of 1800 indicating an
average value of n to be 12 to 13.
Bis-~-diketones XXIa and b were reported to form the tetrasodio
salts when the compounds were treated with four equivalents of sodium
amide in liquid ammonia. The diadducts obtained from the condensation

0 0
II II C6H5CCH2CIH2
(CH2 ) I n
C6H511CH211CH2
0 0
ttel~e C6H5CGHCIH
, "(OH') 4NaE8 ---, 2 ,n'
C6H5rHf~H .. 0 0
a, n = 3; R,R' = c6H5
b, n = 4; R = H, R 1 = C6H4 0CH3 -..e.
26
of the tetrasodio salt with benzophenone and anisaldehyde were reported
to be difficult to crystallize and were thus treated with methanolic
hydrochloric acid to give the corresponding bisdihydro pyrones,
Carboxylations of dicarbanions derived from a large number of~-
diketones have been carried out by the addition of lumps of Dry Ice to
a.
b.
C,
d.
e,
f.
g.
·h.
R = c6H5
R = CH 3
NH 8 lls ie ---2-•• RCCHCCH2 liq, NH3
R = c6H5CH2CH2
R = (CH3 ) 2CHCH2
R = c6HfH=CH
R = (C H ) C=CH 6 5 2
R = H
1.
2.
0 0 0 co2 , ether II II II
RCCH2 CCH2 COH Acid
a.
b.
C •
d.
e.
f.
g.
h.
R = c6H5
R = CH 3
R = c6HfH2CH2
R = (CH3 ) 2CHCH2
R = c6H5CH=CH
R = (C 6H5 ) 2C=GH
R = H

27
a mixture containing the dicarbanion. The yields of terminal acids in
37 all cases were below 55%. ·
Poly-[3-Carbonyl Compounds.
Linear ~-polyketones and analogous compounds, although not exten-
sively investigated, have been shown to undergo condensation to more
complex sturctures, some of which are related in type to substances
found in nature. The ease of some of these reactions led Collie in
16 h h d h b ' d' 1907 to suggest tat sue compoun s mig t e interme iates in some
biosynthetic processes. and. might be derived from acetic acid.
Birch and co-workers, 6 in a reinvestigation of Collie's earlier
work, condensed two equivalents of the barium salt of 2,4,6..;heptanetri..;
one under weakly acidic conditions to form a yellow compound c14H14o3
Acid
0
II
XXII XXIII XXIV
assigned structure XXIV. In addition, two intermediate compounds were
identified by UV data. Similarly, natural compounds containing hydroxy-
0
II cyclohexenone structures XXII and XXIII (RC= oleoyl) were readily
aromatized by dehydration.

28
Chirnaphilline (XXV) from Ericaceae species is qn example of a
natural product which has been postulated as arising by intermolecular
condensations between ~-polyketones or,poly keto acids.
CH 3
CIL, :J
XXVI
It was suggested that the flavonoid constituents of pines could be
correlated with the pinosylvin(XXVI) derivatives on the basis of a·com-
man origin from ci:i:mamic acid and three acetate. units through the inter-
mediate (XXVII; R = co2H). However, no indication of the formation of
0 0
II 11 C 6 Hf H=CHCCH2 CCH2 CCH2R
XXVII
pinosylvin ~XXVl) was found when compound XXVII was treated with alkali
. . 1 under a variety of conditions.
Ozonolysis has also been found to lead to poly-~-carbonyl compounds.
The ozonide of ketal XXVIIIa was hydrogenated to give the tetraketone
R :) 0 0 0 ('"'-l' DH . o c:> (CHJHJ)2 1. 3: L.•3'- c ,<0
?. • p-1 fr. • H ,._L ., 2
XXVHia, R = R' = H XXIXa, R :; RI H
b, R = R' = CH b '
R "'R' - CH3
· XXIXa •. Similarly hydrogenation of the ozonide of ketalXXVIIIb gave
8 tetraketone XXIXb.
Tasmanone, a constituent of the essential oils of Eucalyptus
ta-s.manica Blakely and E. camfieldi Maiden, apparently exists as a

29
tautomeric mixture of two enolic forms since its titration curve showed
s l ow
-a pronounced hysteresis effect on back-titration, and in its NMR spec -
trum the signals of most of the groups of protons were doubled, with the
intensity of one of each pair half that of the other. Addition of a
small amount of diethylamine to a chloroform solution containing tasma-
none produced a coalescence of the signals attributed to the two tasma~
6 none tautomers.
The NMR spectrum of syncarpic acid, in deuterochloroform, reveals
that the acid exists almost entirely in the keto form. However, in
acetone-d 6 solution the substance is almost entirely in the enol form
(92%) •6
23 In the case of unsic acid, it has been shown by NMR analysis that
only one enolic form exists which is either XXX or XXXI. Form XXX has
been reported to be likely the more stable one, not only because it
. -/CH3
O·_ ., • ..
e....._ 0
XXX
. . c=o ,-CH3
XX I

30
contains a conjugated system, but also because the hydrogen bonding in
XXXI might be expected to be less stable than in XXX in which all three
hydroxy groups are bonded. to indivi.dual carbonyl groups.
Recently, several trispiro triketones were synthesized by base
catalysis of the dimer of trimethyleneketene, tetramethyleneketene
dimer, and pentamethyleneketene dimer. Alcoholysis, in the presence of
a catalytic amount of sodium ethoxide, of the trispiro compounds led to
acyclic ~, 0-diketo esters. 65

CHAPTER II
DISCUSSION OF RESULTS AND CONCLUSIONS
A study of the condensation of 3-hydroxy-2,2,4-trimethyl-3-penten-
oic acid ~-lactone (!)with phenyl Grignard reagent was undertaken be-
5 cause previous experiments performed in this Laboratory gave support
for the presence of the dicarbanion of diisopropyl ketone in the reac-
tion mixture. In addition, this dianion intermediate was surmised to be
involved in the formation of a ketol XXXII and a triketone XXXIII.
CH~
CH :--·toJ CH3 3 ' · CH 3 1.
0 ~ 2. 0
H o® 3
+
+ XXXII
0 0 0 II II II other
(CH3 ) 2CHCC (CH3 ) 2CC (CH3 ) 2CCH(CH3 ) 2 + products
XXXIII
' . Since one mole of lac tone in theory is consumed by two moles of
Grignard reagent, excluding secondary processes such as cleavage or re-
duction of products, a 1:1 mole ratio of lactone I and Grignard reagent
should provide 0.5 mole of the anion or dianion of diisopropyl ketone,
which in turn may react with the remaining 0.5 mole of lactone.r to give
a new anion or dianion. This has been accomplished although an excess
31

32
of lactone I was used because iniufficient reaction (diaddition) was 6b-
served, and an increase in the yield of ketol was noted. The reaction
mixture, was found to contain, in addition to benzophenone and the ketol
XXXII, more than a dozen minor components including the triketone·XXXIII
(GLC analysis).
Carbonation of the reaction mixture of phenyl Grignard reagent and
lac tone I in ether gave acid XXXIV, XXXV and an oil. The oil was
0 0 0
C 6HJC (CH3 ))c (CH3 )z~OH
XXXIV ·XXXV
XXXVI ,xxxvrr
XXXVIII XXXIX
XL
treated with diazomethane; esters XXXVI,,XXXVII, and XXXVIII1 diketone
· XXXIX, amide XL and trike tone· XXXIII were found int he reaction mixture.
Carbonation of the reaction mixture of phenyl Grignard reagent and
lactone I in tetrahydrofuran gave acids XLI,·XLII and an oil. The oil
ij ·~ ij HOCC (CH3 ) 2 CC (CH3 ) 2 COH
XLI XLII

33
when treated with diazomethane gave esters XXXVI, ,XXXVII, ·and XXXVIII, ·
diketone'.XJP(IX, amid~:LXL~d ·triketone XXXIII. A mixture of acids iLXI ·and
'XLII treated with diazomethane gave diesters ·XLII, :XLIII, and ,XXXVII.
-XLIII
Lactone I with methyl and ethyl Grignard reagents produced complex
reaction mixtures but good yields of triketone XXXIII. Lactone I and t-
butyl Grignard reagent gave a complex reaction mixture,with amide XL
being isolated and triketone XXXIII detected in the reaction mixture.
Acid hydrolysis of ketol'XXXII gave triketone· XXXIII and unknown,E,
-while.heating.k~tol'XXXII in DMSO yielded ttiketone·XXXIII and a trace
of unknown:E. Thermolysis of XXXII gave·XXXIII in quantatative yield.
XXXII acid or 6 in DMSO
Unknown F
•• xxx:1:1 + Unknown E
LiAlH 4
Unknown 1G
Ketol XXXII and lithium aluminum hydride gave a solid product, unknown F.
The same reducing agents converted triketone-XXXIII to a 1folid. compound,
unknown.G.
Base degradation in ethyl alcohol of triketone·xxx:111 gave diiso-
propyl ketone ((XLIV); ethyl 2 ., 2 ,4-trimethyl-3-oxova le rate '(XLV)? 2 ,4 ,4, 6-
tetramethyl-3, 5-heptanedione 1(XLV!},. potassium carbonate, ethyl isobuty-
rate (Xt.Vtt) and isobutyric acid '.(XLVII!) .• Base degradation of ketol, XXXII

34
XLIV . XLV
XLVI
XLVII XLVIII
in ethyLalcohol gave rise to the same products as from triketone XXXIII
except that triketone XXXIII was detected in the reaction mixture,
Diketone XLVI was prepared by condensing 2,2,4-trimethyl-3-oxo-
valeryl chloridei'(:XLDOwith diisopropylcadmium. Dimethyl 2,2,4,4-
XLIX XLVI
, tetramethyl-3-oxoglutarate (:XXXvn') was prepared by the reacti.fln of
" I ClCOCH3
NaOCH3 , THF
XXXVII

35
lac tone I ,with methyl. chloroformate in the presence of sodium methoxide.
Hexamethyl-l ,.3, 5-cyclohexanetrione ~(]J:') and isopropyl Grignard
reagent, in the presence of magnesium.bromide and is0propyllithium,
II (CH3 !zCHMgBr (in MgBr)
or (CH3 hCHI.i .. XXXIII + other products
·respectively, gave triketone XXXIII and several other products. Methyl
Grignard reagent and II gave hydroxy diketone L.
1.
2. Ho® 3
L
The original condensation of lactone I and phenyl Grignard reagent
was·performed utilizing various molar ratios of the lactone I to the
Grignard reagent. In all cases except where the molar ratio .of lac tone
·Ito phenyl Grignard reagent was 0.8:1, ketol XXXII could be crystallized
directly from the·reaction mixture and the compound easily purified (see
· Experimental section). . Evidently, XXXII was present in the O. 8: 1 reac-
tion of land phenyl Grignard reagent in too· low. yield to crystallize
directly. Removal of most of the products,.excluding ketollXXXII, was
effected by trec1tment of the reaction residue with Girard 's ·:T .. reagent.

36
This process-resulted in the eventual isolation of several grams of pure
ketol xxkn.
A molar ratio of 1:1.2 for phenyl Grignard reagent and lactone I
was found to give the maximum amount (30.1%) of ketol XXXII. When the
molar concentration of I was raised above 1.2, the yield of ketol XXXII
dropped slightly. The general procedure used for effecting the conden
sation was to,atid the lactone I to the chilled.Grignard !rolution, stir
the mixture and then heat at reflux. Hydrolysis of the mixture was per
formed at room temperature with aqueous ammonium chloride solution be
cause at ice-salt bath temperatures, _the. mixture solidified to·a large
extent and stirring became difficult. _ In general, workup of the reac
tion mixture proceeded smoothly unless the reaction mixture, after hy
drolysis, was stirred vigorously and then extracted immediately. Separ-
·ation.of layers :was-exceedingly slow when.this latter process was tried.
Since the 1:1.2 mole condensation of lactone I-and phenyl Grignard
reagent was frequently used for the· preparation of ketol .XXXII, the
reaction residue left after the removal of this compound was analyzed by
GLC. Benzophenone (40.1%) was found to be the-major component, in addi
tion to-more than.a dozen other components including diisopropyl ketone
(XLIV) (4%), l-phenyl-2 ,2 ,4-trimethyl-1,3-pentanedione ((XXXDO (8%) and
triketone XXXIII (4%).
Both methyl. and ethyl Grignard reagents, when condensed with lac
tone I, gave complex reaction mixtures, with only the ethyl Grignard·run
providing_ketol,XXXII. Both reaction mixtqres, .via GLC ·analysis :wel:"e .. ,_
shown to contain triketone XXXIII in good yields •. No explanation is
offered as to the high yield (60%) of the triketone XXXIII and :the trace
amount of ketolXXXII (ethyl Grignard run) •. Thus, the condensation of

31
ethyl and methyl Grignard reagents with lac tone I needs further investi
gation. !-Butyl Grignard reagent and lactone I also gave a complex reac
tion mixture (GLC analysis). A small amount of 2,2,4-trimethyl-3-oxova
leramide~XLJ)was obtained from the reaction mixture, and the structure
of the compound was supported by NMR (Plate XXXI) and IR (Plate IX)
analyses. Triketone XXXIII was also identified as one of the products •
. Amide XL::is postulated to arise from the reaction of aqueous ammonium
chloride solution on lactone I during hydrolysis of the reaction mixture,
The mechanism of formation of XXXII and XXXIII and several other
products from the reaction of I and phenyl Grignard reagent is believed
to involve initial cleayage of the lactone I by one mole of the phenyl
Grignard reagent. A second mole of Grignard may add to give an inter
mediate which undergoes retrograde aldol-type cleavage to yield benzo
phenone and an .anion of diisopropyl ketone, species LI, LII and/or LIII.
These carbanion-enolate or dicarbanion intermediates may react with
additional lactone I giving rise to new anion intermediates LIV, LV and
LVI. Which of the species LI, LII or LIII condenses with the lactone I
to form anions LIV, LV or LVI cannot be determined from the present data 9
but LI and LIII are perhaps better candidates than LII because of the
presence of two negative charges on LII. Hydrolysis of the reaction
mixture with aqueous ammonium chloride solution gives ketol XXXII and
triketone XXXIII in relatively low yields. The structure LVI may be
favored over LIV and LV because ring closure of the latter two species

R
R~ (R)z
Q+ 0
I
c6H5MgBr · Et2o
XLIV
!H30E9
....
R R R
R-yvc6Hs OM II 0
H3o l EB
XXXIX
c6H5MgBr ... R R ,.R,
l.X/c6Hs ·1:1 ~6H5 JM (OM
I - - -- ~------,- , R .R
M;YR ll,,,.
.,
0
LI
i ,; ~~R
O• •M
f 1! R~
·OM
LII LIII
+ ·(C6H5 ) 2c=O· • ·M
I !r I R
R ....
- -R R R
OM 0 o. 0 0, •M
LIV ' '·
I LV -·--------------t·~~~~~~_J LVI 1 ' ·@ I (CH )
oll o ~o R . (C~l32 O''CH OH (CH3L (c~c3;)CXCM o" '"H ) 11 ii ,.. • z 'V 3 2
(CH3)2CHCC(CH)iC(CH ) CCH(CH ) f9' .:::,.0 ll:JOB; 0~ ·· 3 2 3 2 (CH ) 3 2 (CH3)2
M = MgBr, R = CH3 XXXIII XXXII XXXIIa (.,.)
OJ

39
R R R R R R
~ R~
R . e
0 (Rh . . (R)z -
OM II R 0
LIV
LV
. R = CH3
would give a tertiary carbanion the stability of which may be less than
that of XXXIIa. The stability of the enol forms LIII and LIV compared
·to keto forms :LI,,LV and LV.I cannot be evaluated from the present data;
thus a definite designation of LIV, LV or LVI as the precursor of xxtu.a
is not possible •. Since GLC indicates a complex reaction mixture, the
anions :LI, LII, LIII, LIV, LV,and LVI may be involved in other reaction
se(luences.
When.tetrahydrofuran was.used as a-solvent for the condensation of
lactone·I and phenyl Grignard reagent, salvation of the Grignard complex
was observed •. Initially upon heating,. the reaction m-ixtµre became a
dark red-brown solution. The yield of ketol.XXXII in this reaction .was
.found to be lower than in the condensation of phenylGrignard reagent
and lactone I in ethyl ether •
. A cyclization similar to. tha.t shown for XXX.IIa has. been observed in
the formation of 5-dimethylamino-2,2,4,4 1 6,6-hexamethyl-l,3-cyclohexane
dione (LVII).44 Cyclization of the carbanion intermediate in this case

40
involves attack on a carbon atom of.an enamine salt rather than a car-
bony'l carbon atom.
In order to demonstrate the intermediacy of species LI, LII, LIII,
LIV, ·LV and LVI the reaction mixture of phenyl Grignard reagent and lac-
tone -I was carbonated. .Carbon dioxide gas was bubbled through the
ethereal reaction mixture. No noticeable reaction was observed, but the
mixture _had the appearance of caramel candy with layers of white
material. Workup of the reaction afforded a monoacid, 5-phenyl-2,2,4,4-
tetramethyl~3 ,5-dioxovaleric acid ·(XXX:IV), in low yield and a brown
XXXIV
I}. or pyridine ., -CO2
-XXXIX
viscous oil. The structure of monoacid XXXIV .was supported by IR, NMR
(Plates III and XXIV) and elemental analyses ·(see Tables III, IV and V).
The NMR. spectrum (pyridine-d5) had to be taken quickly as compound XXXIV

41
rapidly (10-15 minutes) decomposed into diketone XXXIX. Thermolysis of
acid XXXIV at its melting point gave the diketone XXXIX in quantitative
yield. IR and NMR..analyses (Plates IV and XXV - Tables IV and V)
further aSUpport the known structure of diketone XXXIX. 5 The aforemen-
tioned oil was treated with diazomethane and afforded a complex reaction
mixture. GLC analysis revealed the presence of methyl 2,2,4-trimethyl-
3-oxova lera te {XXXVI), dimethyl 2 ,2 ,4 ,4-te tramethyl-3-oxoglutara te •
(XXXVII), l-phenyl-2, 2 ,4-trimethyl- l, 3-pentaned ione({XXXIX)., 2, 2, 4-tri-
methyl-3-oxovaleramide((XL), triketone XXXIII and methyl 2,2,4,4,6,6,8-
heptamethyl-3, 5, 7-trioxononanoa te (XXJ.CV!Ilh
c13 HCl gas
I
~ Cl 1. c6HllgBr
XLIX 2. CO2 gas
3. H30<±J
XXXIV + oil + XXXV
XXXVI XXXVIII
TffF
fl ClCOCIL
5
XXXVII
Ester XXXVI was prepared by treating lactone I with anhydrous hy
drogen chloride gas, 79 followed by esterification of 2,2,4-trimethyl-3-
oxovaleryl chloride r(XLIX) with methanol in the presence of triethylamine4

42
to give the ester XXXVI in good yields. The s.tructure of acid chloride
XLIX was supported by NMR.and IR data (Plates.XXX:VI and XVI) •. NMR. and
. IR.spectral data (Plates XXXVII and XVII) authenticated the structure of
ester XXXVI. Methyl chloroformate and sodium methoxide with lactone I
in tetrahydrofuran gave ester·XXXVII in fair yield. 78 Authentication of
its·. structure was• affo~ded by physical data, including. IR (Plate ·XVIII)
and NMR (Plate·XXXVIII) analyses (Tables IV and.V). Ester·XXXVIII was
obtained in.the pure form by preparative GLC and its structure elucidated
by NMR (Plate XXXVII), IR (Plate VI) and elemental analyses (see.Tables
·III, ·IV, and.V). The presence of triketone XXXIII in the reaction
·XXXVIII XXXV
.mixture is.attributed to decarboxylation of acid XXXV, probably during
,workup of the reaction.mixture •
. Acid mv ;\las ptoduced ~httt the oil obtained fr·om tn identical car•
. bomition experiment was. allowed to stand for several weeks. The compound
precipitated:and was identified as•2,2,4,4,6,6,8-heptamet:hyl-3,5,7-,tri
oxononanoic acid (XXXV) by elemental analysis, IR (Plate·V) and NMR.
(Plate XXVI) spectral data ('l'al>les. III,. IV. and. V).
When the condensation of phenyl Grignard reagent and lactone·I was
performed in ethyl ether and the solvent displaced by tetrahydrofuran,

43
carbonation of the resulting red-brown solution gave a mixture of acids
HO
OH
H C CH 3 3
HO
r9'
CH3
''cH3 n
XLI XLII
OR
XLI, XLII and an oil. Although the mixture of XLI and XLII had a rela
tively sharp melting point (134-135.5° dee.), both NMR and GLC analyses
of a sample of the mixture revealed the presence of two compounds. The
GLC chromato·gram · revealed two peaks which were identified as arising
from diisopropyl ketone (XLIV).and triketone XXXIII. Apparently the acids
decarboxylated on the column. A sample of pure XLI was obtained by re-
peated recrystallization of a sample of the acid mixture from benzene.
Elemental analysis, NMR (Plate XXVIII) and IR (Plate VII) spectral data
support the structure of this acid, 2,2,4,4,6,6,8,8-octamethyl-3,5,7-
trioxo-1,9-nonanedioic acid 1(XLI'), (see Tables III, IV and V).
A sample of a mixture of acids XLI and XLII was treated with diazo-
methane. Diester XLIII precipitated from the resulting golden-colored
oil as a white solid. NMR (Plates XXIX and XXX), IR (Plate VIII) and
elemental analyses support the proposed structure of dimethyl 2,2,4,4,6,
6,8,8-octamethyl-3,5,7-trioxo-l,9-nonanedioate~(XLIIJ'.). GLC analysis of

44
0
I
XXXVII XLIII
the filtrate obtained from the removal of diester XLII showed only one
peak; mixed injection with an authentic sample identified it as dimethyl
2 ,2 ,4 ,4-tetramethyl-3-oxoglutarate (XXXVII};
The NMR spectra of diesters XXXVII and XLIII (Plates XXXIX, XXX and
XXXVIII) are unusual, since magnetic nonequivalence of the ester methyl
protons in the ester group and of the internal gem-dimethyl protons is
observed. Both compounds seem to have a preferred conformation.and one
can only speculate as to the true stereochemistry of both molecules.
However, a conformation in which the carbonyl groups are trans-or·iente·d
and the gem-dimethyl groups are staggered would imply less dipole-dipole
interactions between these groups and represents one possible conforma-
tion. 0
At a temperature above 50, the difference in the field position
of the ester methyl protons in dieSter XLIII increased 2 c·.p.s. ·Perhaps
at a lower temperature,. say 0°, these.two singlets might coalesce.
The equilibrium that is postulated between LI, LIIP.rid LIII, LIV,.
LV and LVI is based upon the fact that the rat.io of monoacid (precursor
of ester XXXVI) to diacid XLII (precursor of diester XXXVII) remained

R
R~
XXXVII
l CH2N2
XLII
J 1. co2 ©
2. H30
r- 1 R
\ Ir~ M,~
II . R R 0
:0 R II ~-
0· •M
XXXVI
f cH2N
H,~R RR 2 ,, .
R I II 0 0
OH
r 1. co 2. H ~©
R 3
RYR OM
LII LIII LI I l 1
I
R R R R R R R i...
R R OM O
LIV
I:· co~ J;• H30
XXXV
icH2N2
XX.XVIII M = MgBr, R = CH3
LV
1. co 2r.p f• H30\D
XLI
!CH2N2
XLIII
LVI
R
R
~
+:u,

46
fairly constant in .all runs in ethjl ether. -This suggests that the
anions have a common precursor and that variation in the efficiency of
carbonation is minimal. It is not possible to establi~h whether LJ or
LII is the precursor of the dicarboxylic acid XLII, but it is not un-
reasonable to suggest that LIII is carbonated to give the monoacid which
esterified gives ester XXXVI. Likewise, whether species LV or LVI is
the precursor of diacid XLI cannot be evaluated with the present data;
again it is not unreasonable to assume that LIV is.carboxylated to give
monoacid XXXV and esterification of this compound gives ester XXXVIII.
When the carbonation of the reaction mixture of lactone I and phenyl
Grignard reagent was performed in tetrahydrofuran, an increase in the
ratio of diacid,XLII (precursor of diester.XXXVII) to monoacid (precur-
sor of ester·XXXVI) was noted. This could be a result of increased sol-
vation of all reactants or a shift in the equilibrium between LI and LII
or LIII. These arguments .receive some support from experimental data.
Carbonation of the original condensation product (in ethyl ether) was
performed in a heterogeneous reaction mixture, but use of tetrahydro-
furan i.e., permitted carbonation in.a homogeneous system. The conden-
sation of lactone I and phenyl Grignard reagent in tetrahydrofuran at
0 68 gave a lower yield of ketol XXXII than when performed in ethyl ether.
If a shift in the equilibrium .between LI and LII or LIII and LVI, LIV or
LV occurred at this.higher temperat re, ketol XXXII should have been
formed in a higher yield. As suggested earlier, dianion species LVI
cyclizes giving XXXIIa which upon hydrolysis yields XXXII. However,
the original condensation in tetrahydrofuran was performed only once,
and because of insufficient data, this one condensation cannot be ex-
pected to disprove our arguments. Furthermore many condensations (1:1.2)

47
between phenyl Grignard reagent and lactone I were effected in order to
prepare ketol XXXII. Although the lactone I was distilled before using
and the yields of phenyl Grignard reagent were generally above 95%, the
yield of ketol XXXII varied from 15-30.1%. Many explanations can be
adduced for such a range of yields; thus the lower yield of ketol XXXII
obtained when tetrahydrofuran was used as the solvent cannot be a basis
for stating that this is the highest possible yield of ketol XXXII with
this solvent system.
A maximum yield (30.1%) of ketol, 5-hydroxy-2,2,4,4,6,6-hexamethyl-
5-isopropyl-1, 3-cyclohexanedione 1(XXXII}, was obtained unce when phenyl
Grignard reagent and lactone I were combined in a molar ratio of 1:1.2.
The compound was easily crystallized directly from the reaction mixture;
purification was relatively simple. Elemental analysis (Table III) in-
dicated an empirical formula of c15H26o3 with a molecular weight of 253
(Monsanto Chemical Company, Inc., St. Louis, Missouri). Infrared ab-
sopptions (Plate II) occurred at 3460 (hydroxyl group) 1692 (carbonyl
-1 group:shoulder at 1718), 1390 and 1378 cm. (isopropyl group) with
several other sharp bands as found in Table IV. The NMR spectrum (100
M.c.) displayed signals in benzene (Plate XXI).at ol.90 (singlet), 1.43
(doublet:J = 7 c.p.s.), 1.19 (doublet:J = 6 c.p.s.) and 0.91 (doublet:
J = 6.5 c.p.s.) in a ratio of 1:6:12:6 respectively. At a sweep width
of 500 c.p.s., the peaks representing the proton of the tertiary hydro-
gen were easily discernible but no assignment in regard to their rela-
tive field position .was made. When the NMR spectrum (60 M.c.) of ketol
XXXII was determined in pyridine-d5 (Plate XXIII), an enhanced chemical
shift placed the heptet of proton~ at 02.38 clearly outside the area of
the methyl resonances. Further NMR studies [100 M.c., at a sweep width

. , CH3 (g and h) (bJ H~ I
~c CH.-
3
XXXII
of 250 c.p.s. and a spectrum amplitude of 6000] of ketol XXXII (Plate
XXII) revealed the finer points of the previous spectrum (Plate XXI).
48
In benzene, five of the theoretical seven peaks of the expected heptet
for proton~ on XXXII were revealed. The doublet at ~1.43 has been
assigned to methyl group protons£ and i, while the doublet at 51.19 is
due to methyl protons~ and i and the doublet at 50.91 to the methyl
protons of the isopropyl group~ and~. Decoupling protons~ and~ from
the tertiary proton E. (-273.3 c.p.s.). (and the reverse decoupling] re-
sulted in the collapse of the doublet due to protons ~ and h and the
heptet due to the proton.!?.~ respectively, to singlets. These decoupling
experiments show that the protons a and~ are the true protons on an
isopropyl group •. Splitting of the gem-dimethyl res.onances may result
from a preferred conformation imposed upon the system by steric require-
ment of the isopropyl group and geometric stipulations of the 1,3-di-
carbonyl system. It is rather interesting to note that~the isOpropyl
group of ketol XXXII apparently experiences only on! environment.
Drieding models infer that a chair conformation for ketol XXXII is rigid
and has imposing 1,3-interactions of axial methyl groups. However, a

49
twisted-chair form can be constructed, in which interactions of non-
bonded groups are reduced (structure shown on page 48). Although the
twisted-chair form may imply a rigid system for ketol XXXII, an NMR
analysis in a suitable solvent at low temperature would resolve whether
the molecule is rigid or is flipping through all possible forms faster
than the sweep rate of the instrument (2 c.p.s./sec.). An X-ray analy-
sis of ketol XXXII would provide knowledge of the true conformation of
this molecule in the solid state. Because of a lack of a model similar
to XXXII, assignment of the proton resonances c and d and e and f to
individual methyl groups could not be determined. This situation is not
met in the NMR spectrum of II, which displays two sets of magnetically
equivalent, internal gem-dimethyl groups as one signal. Dipole moment
studies indicate that triketone II probably exists in a twisted flexible
boat conformation. This form shows an equivalency of all methyl
18 groups.
Treatment of ketol XXXII with aqueous hydrochloric acid gave two
products which were detected in the reaction mixture by GLC analysis •
. Attempted distillation of this mixture did not afford any pure compound.
However, both products were obtained in the pure form by preparative
GLC (see Experimental section). The first peak collected from the
Autoprep unit was labeled unknown E. Although the mass spectrum of this
compound indicated a molecular weight of 236 [thus probably a dehydra-
tion product of ketol XXXII], elemental analysis did not lead to the
molecular formula c15H24o2 as expected (see Table III). The IR spectrum
(Plate X) indicates the presence of a vinylic methylene group and possi-
bly a five-membered ring (carbonyl absorptions at 1722 - shoulder at
-1 1748 cm. ). The NMR spectrum (Plate XXXII) also shows a terminal

50
methylene group (see Table V).but the rest of the spectrum sugge~ts a
rather complex system. A 100 M.c. spectral analysis and decoupling ex-
periments might be helpful in resolving the structure of unknown E. The
second peak collected from the Autoprep unit was identified by spectral
data as triketone XXXIII. Dehydration of ketol XXXII with potassium
acid sulfate likewise gave unknown E and triketone XXXIII. Attempted
solvolytic dehydration of XXXII with dimethyl sulfoxide 80 at 178° for
nine hours resulted in a 98% conversion to triketone XXXIII. The trace
product accompanying XXXIII was identified by GLC analysis as unknown E.
0 Thermolysis of ketol XXXII at 185 for one hour gave triketone
XXXIII in quantitative yield. Although pyrolysis of this type is rare
in the literature, an analogy may be found in the pyrolysis of ~-hydroxy
olefins, which is believed to involve a cyclic mechanism. 2
R'
H \1
H H
"' 500° ~ RCR" + R CH2CH=CH2
~R
As stated previously, models imply that nonbonded interactions are
minimal in the twist conformation of XXXII but the hydroxyl proton~ can
rotate close to an oxygen atom of a ~-carbonyl group. One rationale for
the apparent lower activation energy for thermal conversion of XXXII to

51
Tautomerization XXXII
XXXII
triketoneXXXIII compared to the analogous pyrolYsis of ~"'.hydroxy ole-
fins.is perhaps found in the conformational rigidity of the ring in
XXXII, which increases the strain.
In attempts to prepare ketol XXXII, hexamethyl-1 13,5-cyclohexane-
trione {II) was treated with isop:rnpyl Grignard reagent :(in the presence
of magnesium bromide) 76 and isopropyllithium, respectively. The reaction
mixture obtained from the isopropyl Grignard reaction was shown to con-
tain [GLC analysis] triketone XXXIII in low yield; starting material
[triketone II] was largely recovered. A slightly improved yield of
triketone XXXIII was observed by GLC analysis of the reaction mixture of
isopropyllithium and triketone II. Several other products including
triketone II were·also present in the reaction mixture.
Ring closure of triketone XXXIII was attempted using sodium meth-
oxide and sodium hydride as bases. It could not be established from GLC
analysis of the two reaction mixtures obtained, that ketol,XXXII was not
one of the many products present. Triketone·XXXIII was identified in the
aqueous portion (after hydrolysis of the reaction mixture and workup of
the a.queous layer) of the reaction mixture and its presence can be

52
. explained in sever a 1 ways: . inferior procedure for processing the reac
tion mixture, carbanion formation of triketone XXXIII and subsequent hy
drolysis yielding XXXIII, or cyclization of triketone·XXXIII giving ke
tol XXXII, after hydrolysis, with subsequent decomposition .of XXXII on
the GLC column. When an ethereal solution of pure ketol.XXXII was
placed on a GLC column, a chromatogram representative of a decomposi
tion pattern was observed with a large peak having the same retention
. time as triketone XXXIII was noticed.
By using an excess .of potassium hydroxide in absolute ethanol, ke
tol XXXII was degraded into three compounds, diisopropyl ketone (XLIV),
isobutyric acid (XLVIII) and potassium carbonate. Because .of the rather
large amount of potassium carbonate that precipitated, it was antici
pated that using.equtmolar quantities of ketol'XXXII and base would pro
duce intermediary products that might be isolated. - This was verified;
the 1:1 reaction of XXXII with potassium hydroxide in absolute ethanol
gave (after hydrolysis) triketone XXXIII, ethyl isobutyrate (XLVII),
ethyl 2,2,4-trimethyl-3-oxovalerate (XLV), diisopropyl ketone (XLIV), 2,
. 2 ,4 ,4-tetramE;!thyl-3 ., 5-heptanedione (XLVI), potassium carbonate and a
trace of isobutyric acid (XLVIII). Since XXXIII was present in the re
action mixture.the mechanism of the reaction is postulated.as.involving
initial ring.opening of ketol XXXII to yield the monocarbanion of XXXIII •
. since hydroxide ion would be tbtally consumed in the.initial step, eth
oxide ion is believed to form and become a competitive base. -Although
attack by base at the carbonyl carbon atom in.XXXII could OCGUr, no·sim
ple mechanism can be written which would involve the formation.of trike
tone. XXXIII. Diisopropyl ketone (XLIV), triketone XXXIII, isobutyric
acid (XXXVIII), and ethyl isobutyrate (XLVII)·weredetected by GLC
0

53
a'nalysis of the reaction mixture. Diketone XLVI and ~-oxoester XLV were
isolated in, pure_ form by preparative GLC. The structure of diketone
XLV:I was supported by IR (Plate XV), and NMR (Plate XXV). analyses.
OK
XLIV
r HzO
0
R R R
II l',~'H
+ 0 0
R II II m~o8iz®.
R R R
0
11. KOH 2. H2o
R
KOH
0
R,A )-, HI/ \~H +
R R
II H~OH
R XLVIII XLIV
R = CH 3
XXXII
KOH, !::.
XLIV
r,~o
0 0
R II II lf'~OCH / RI\ . 2 5
XLV
@ K +
fl -~~OCH ]'f)I/ ' 2 5
R
XLVII
H2o '--~~~~~~ ...... XLVI
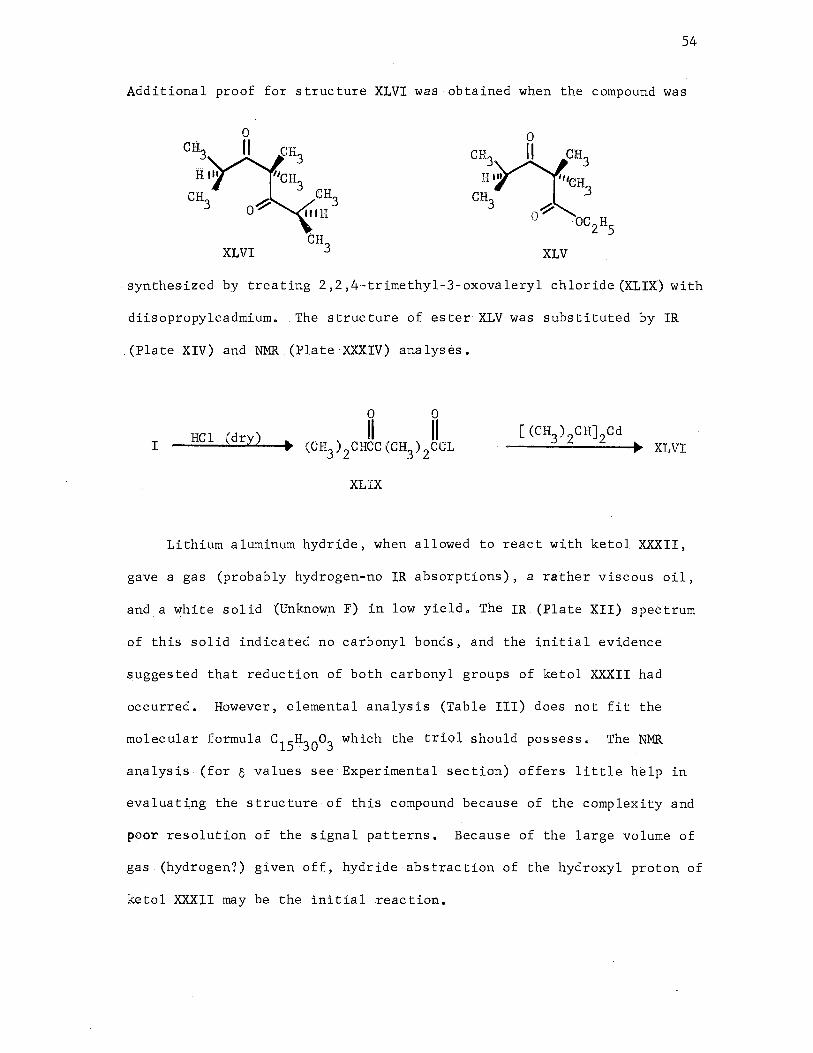
54
Additional proof for structure·XLVI was obtained when the compound was
XLV
synthesized by treating 2,2,4-trimethyl-3-oxovaleryl chloride (XLIX) with
diisopropylcadmium. The structure of ester·XLV was substituted by IR
. (Plate XIV) and NMR (Plate XXXIV) analyses.
I HCl (dry)
XLIX
[ (CH3 ) 2CHJ 2Cd ~~~~~~~•~ XLVI
Lithium aluminum hydride, when allowed to react with ketol XXXII,
gave a gas (probably hydrogen-no IR absorptions), a rather viscous oil,
and a white solid (Unknow.n F) in low yield. The IR (Plate XII) spectrum
of this solid indicated no carbonyl bonds,.and the initial evidence
suggested that reduction of both carbonyl groups of ketol XXXII had
occurred. However, elemental analysis (Table III) does .not fit the
molecular formula c15H30o3 which the triol should possess. The NMR
analysis. (for o values see Experimental section) offers little help in
evaluati,ng the structure of this compound because of the complexity and
poor resolution of the signal patterns. Because of the large volume of
gas (hydrogen?) given off, hydride abstraction of the hydroxyl proton of
ketol XXXII may be the initial reaction.

55
In an effort to prepare a model similar to XXXII, hexamethyl-1,3,5-
cyclohexanetrione (II) was allowed to condense wi'trh: methyl Grignard rea-
gent •. The solid product L obtained had an IR (Plate XIX) spectrum which
bore a striking resemblance to that of ketol XXXII (see Table IV). The
NMR spectrum (Plates X:XXIX and XL) further support the structure of this
-?o
II
compound, 5-hydroxy-2 ,2 ,4 ,4, 5, 6, 6-heptamethyl-l, 3-cyclohexanedione (l~).
70 Although ketol XXXII possesses carbonyl groups, normal procedures
to prepare an oxime, 2,4-DNP or semicarbazone derivatives failed. In
contrast, a compound such as LVII, which could be envisioned as an al--
ternate structure for ketol.XXXII, might be expected to form carbonyl
LVII LVIII
derivatives as does diketone LVIII. 83 Attempted esterification of ketol
·XXXII with acetic anhydride in pyridine11 was also unsuccessful, with
most of XXXII being recovered.

56
Good evidence in favor of the six-membered diketone structure XXXII,
is afforded by· IR.data. -1 A band at 1779 cm. was recorded for the car-
bonyl groups44 in LIX while ·LVIII exhibits a doublet at 1745 and 1721
-1 1 cm. , triketone II has a broad band at 1698 cm.- For comparison,
LIX LX
-1 XXXII has a peak at 1694 cm. with a sharp band of lower intensity at
-1 1718 cm. (see Table· IV). Most convincing was the similarity of the
carbonyl absorptions of LX to those of XXXII, as twin carbonyl peaks
-1 21 were reported for the former at 1718 and 1688 cm. in chloroform •
. The spectrum of XXXII in potassium bromide or chloroform contained the
two bands mentioned previously for the carbonyl groups in nearly identi-
cal positions to those in LX.
In previous discussion in this chapter, it was noted that the trike-
tone, 2,4,4,6,6,8-hexamethyl-3,5,7-trioxononanetrione (XXXIII) was ob-
tained initially by preparation GLC of the reaction mixture from acid-
olysis and dehydration of ketol XXXII •. Elemental analysis (Table III)
of triketone XXXIII indicates·an empirical formula of c15H26o3 and a
molecular weight of 254 (Monsanto Chemical Company, Inc., St. Louis,
,Missouri) nearly identical to that of ketol XXXII. -Infrared absorptions
(Plate· XI) are visible at 1700 · (carbonyl group:shoulder at 1722), 1390
-1 and 1378 cm. (gem-dimethyl groups) along with several other sharp

57
intense bands at 1018 and 988 cm. -l (Table IV). The NMR srectrum (Plate
·XXXIII) revealed a simple pattern in carbon tetrachloride at 03.08
(multiplet;J = 7 c.p,s.), 1.34 (singlet) and 1.05 (doublet:J = 7 c.p.s.),
When triketone XXXIII was boiled in a three-mole excess of 2,4-DNP
reagent for 17 hours, recovery of·XXXIII was quantitative. This is un-
usual since diisopropyl ketone(XLIV) gives a derivative although the
49 reaction is slow. Attempted preparation of an oxime and a semicarba-
zone also failed. A solid derivative was obtained when XXXIII was
treated with phenyldrazine but the product could not be purified.
Triketone XXXIII and lithium aluminum hydride, when combined at 0°
gave a white solid unknown G, and a viscous yellow oil. Like unknown F,
obtained from the lithium aluminum hydride-ketol XXXII experiment, un-
known G has an IR spectrum (Plate XIII) containing no carbonyl absorp-
tion. Also like unknown F, elemental analysis (Table III) of unknown G
did not correspond to a molecular formula of c15H32o3 • The NMR spectrum
in pyridine-d5 was poorly resolved (see Experimental section for 6
values) and offered no help in elucidating the structure of unknown G.
Trike tone XXXIII was prepared in low yield (4%) by the Claisen con-
dens a tion of methyl 2, 2 ,4-tr imethyl-3-oxova lera te (XXXVI) and d iisopropyl
ketone (XDIV) in the presence of sodium methoxide. Unfortunately, using
sodium amide and sodium hydride as bases with XXXVI did not give trike-
tone XXXIII as indicated by GLC analysis of the reaction mixtures,
Sodium hydride was found to have converted diisopropyl ketone(XLIV) into
diisopropylmethanol, This latter reaction is rather unusual as it has
77 been reported that the ketone evolves hydrogen gas (80%) when treated
with sodium hydride in toluene at 120° for 5 hours. Most of the ketone

58
XLIV was recovered after workup of the reaction mixture.
A 1: 1 mole· reaction of XXXIII with ethanolic potassium hydroxide
gave products identical to those found from the same reaction with ketol
XXXII except that XXXIII was totally consumed in the former case. Using
an excess· of potassium hydroxide only diisopropyl ketoneJKtt:V),, isobuty- ·
ric acid '(XXVIII) and potassium carbonate were isolated. A mechanism for
formation of products is representative of a classic attack on the car
bonyl carbon atom;which is known to occur in 1,3-diketone systems82 as
-summarized on page 23.
0

CHAPTER III
a-e EXPERIMENTAL
Preparation of Phenylmagnesium Bromide, A 2-1. three-necked flask
was equipped with a mechanical stirrer, a condenser with drying tube
(CaC12), a thermometer, and a pressure-equalizing funnel connected to
72 an anhydrous, deoxygenated nitrogen supply. The entire system was
swept with nitrogen for 30 minutes and 24.00 g. (1 g.-atom) of magnesium
was placed in the flask with just enough anhydrous ether to cover the
metal. The addition funnel was charged with 157 .02 g. (LO mole) of
bromobenzene and approximately 10 ml. was added to the flask. When the
reaction had been initiated by gentle crushing of the metal with a
stirring rod, 400 ml, of anhydrous ether was quickly added to the
aAll melting points are corrected; all boiling points are uncorrected. Skelly Solvent F boiled at 30-60°.
b . The infrared spectra were determined on a Beckman IR-SA as films
on sodium chloride cells or as potassium bromide pellets unless otherwise specified.
cThe nuclear magnetic resonance spectra were determined on a Varian A-60 high resolution spectrometer with a field-sensing stabilizer ("Super-Stabilizer"), Tetramethylsilane was used as an internal standard,
dGas chromatographic analyses were performed using an Aerograph HyFi Model A-500, Varian-Aerograph Hy-Fi Model 1520 with a hydrogen flame ionization detector and an Aerograph Model A-700 Autoprep unit from Wilkens Instrument and Research, Inc., Walnut Creek, California,
eThe microanalyses were performed by Galbraith Laboratories, Knoxville, Tennessee and Midwest Microlab, Inc,, Indianapolis, Indiana.
59

60
react.ion flask and 150.ml. of ether added to the bromobenzene. With
stirring, under nitrogen, the bromobenzene-ether solution was added over
a period of 2 hours. When the addition was complete, 200 ml. of ether
was added and the reaction mixture was heated under gentle reflux for 3
hours. After cooling, the mixture was filtered through a glass wool
plug into a graduated cylinder continuously flushed with nitrogen. The
volume was adjusted to 850 ml. with ether, and two· 1-ml. aliquots were
withdrawn and added to 50.00 ml. of aqueous hydrochloric acid (0.1029
meq./1 ml.). The acid mixture was titrated to a phenolphthalein end
26 point with aqueous sodium hydroxide (0. 1024 meq. / 1 ml.). The yield
was 0.989 mole (98.9%).
Reaction (1~0.8) of Phenyl Grignard Reagent and Lactone I - Run 1.
One mole of phenylmagnesium bromide (98%) was prepared by the method
described in the previous experiment. The Grignard solution was placed
in a 2-1., three-necked flask equipped with a mechanical stirrer, con-
denser with drying tube (CaC12 ) a thermometer, and a pressure-equalizing
f funnel •. A solution of lactone I (118.3 g., 0.84 mole) dissolved in 175
ml. of anhydrous ether was added dropwise with stirring under dry nitro-
gen. The flask was kept in an ice-salt bath during the 2-hour addition
period to keep the reaction mixture below 15°. The brown mixture was
allowed to warm to room temperature, and the·resulting orange mixture
was stirred overnight. The lemon-orange mixture, which now contained a
pale-white granular pirecipitate, was heated at reflux for 5 hours,
allowed to come to room .temperature and then hydrolyzed by slow dropwise
(to control foaming) addition of 300 ml. of 2.0% aqueous ammonium chloride
fEastman Chemical Products, Inc., Kingsport, Tennessee, b.p. 170°/ 730 mm •. See IR (Plate I) and NMR. (Plate XX) spectra.

TABLE II
GLC COLUMN SPECIFICATIONS
Title · Length (ft.) Diameter (in.) Packing Mesh · Substrate a
CAb-1 6 1/8 6% Silicone-30 80/100 G
CA-2 6 1/8 6%. Silicone-30 60/80 w
·cA-3 6 1/8 5% Silicone-30 60/80 G
CA-4 6 1/8 6% SE-30 80/100 w
CA-5 6 1/8 6% SE-30 80/100 G
.CA-6 6 1/8 4% Silicone-30 80/100 G
CA-7 ¢ 1/8 5% UCON Polar 80/100 w
CA-8 5 1/4 5% Silicone Rubber 80/100 G
CA-9 8 1/4 5% Silicone-30 60/80 G
CA-10 6 1/4 5% Silicone-30 80/100 .G
. CA-11 6 1/8 10% Silicone Rubber 80/100 G
CA-12 6 1/8 5% SE-30 60/80 ·G
CPC-1 20 3/8 30% PEGS 60/80 w "' t-'

TABLE II (CONTINUED)
Title Length (ft.) Diameter (in.) Packing
CP-2 10 3/8 15% DEGS
a G = DMCS - treated Chromosorb G; W = Chromosorb W
bAnalytical columns used on the hydrogen flame detection unit
cRefers to columns used on the Autoprep unit
Mesh Substrate
60/80 w
a
0\ N

Compound
H CH, ~1 j CH -~C
3 \ OH
' H3C CH3 XXXII
0 0 0
c6H5~C(CH3) 2~C(CH3 ) 2~0H
XXXIV
0 0 0 0
(CH3) 2CH~C(CH3) 2~C(CH3) 2~C(CH3)2~0H
XXXV
TABLE III
PHYSICAL PROPERTIES OF PRODUCTS
0 m. p.' C
147 .5-149
148.5-150
74.5-75.5
Yield
30.1 (max .)
3
20
Analysis (percent) Calculated Found C H C H
70.86 10.23 70.83 10.30 71.18 10.32
68.68 6. 92 68.61 6.80 68.03 6.87
64.43 8. 72 64.73 8.80
0\ I.,.)

Compound
0 0 0 0
II II II II (CH3)2CHCC(CH3) 2cc(CH3) 2CC(CH3) 2COCH3
XXXVIII
.Q O O O 0
HO~C(CH3) 2~C(CH3)2~C(CH3) 2~C(CH3) 2~0H
XLI
fl 11 ~ 'tt fl cu3occ (CI;Ij )2cc (Cll3)2CC(Cij3) 2cc (CH3)2COClf3
XLIII
Unknown ·.E
H fl fl (CH3)2(JHCC (ca3) 2cc (CH3)2CCH(Cll3) 2
·XXXIII
TABLE·III (CONTINUED)
0 m.p.,.c
121-122
74-:75
19-:21
Yield
18
2.5
3 .5
4
Analysis· (percent) Calculated Found C H C H
65.38 8.97
59.64 7.60
61.60 8.10
70.86 10.23
65.17
59.35
61.64
75 .14
70. 94 71.64
8. 77
7.20
8.13
. 10 •1d1
10.24 10.08
"' +:"

TABLEIII (CONTINUED)
Compound
Unknown ··.F
·unknown G
CH
H3C .. CH3
L
aYields are.based on lactone I
0 · m.p., C
196-198
121-122
97.5-98.5
bRefractive index of compound XXXVIII is n23n 1.4641
Yield
. . •. .
. . . .
.
.
. Analisis !eercenq Calculated .,Found C H C H
. . . . . . . 68.60 11.46 68.68 11.56
. . . . . . . 67.05 11. 75
69.03 9.73 68.64 9.60
°' V1

Compound
CH,) H I .J ~
CH3---C'- OH
H3c CH5
XXXII
Q O 0
II II 11 c6H5CC(CH3 ) 2CC(CH3 ) 2COH
XXXIV
0 0
II II c6H5CC(CH3) 2CCH(CH3) 2
XXXIX
TABLE IV
INFRARED ABSORPTION.BANDS (cm.-l) OF PRODUCTS
Hydroxyl Carbonyl
3460 1692 (shoulder at 1718)
3300 1720 (shoulder at 1746)
••• ' • 0 •• 16 72 (shoulder at 1718)
Gemdimethyl
1390, 13 78
g • • •
1390, 1370
Additional Bands
1071, 1000, 982, 960
1390, 1305, 1175, 1095, 979, 768, 707
1252, 1045, 1000, 754, 706
CJ"\ CJ"\

Compound
0 0 0 0
11 II II II (CH3) 2CHCC(CH3) 2CC(CH3) 2CC(CH3) 2coH
XXXV
0 0 0 0
II II II II (CH3) 2CHCC(CH3) 2CC(CH3) 2CC(CH3) 2COCH3
XXXVIII
0 0 0 0 0
II II II II II HOCC (CH3) 2cc (CH3)2CC (CH3 )2CC (CH3) 2COH
XLI
0 0 0 0 0
II II II ll ti CH30CC(CH3) 2CC(CH3) 2CC(CH3) 2C!'C(CH3) 2~0CH3
XLIII
TABLE·IV (CONTINUED)
Hydroxyl Carbonyl
3000 (broad) 1738, 1703
Gemdimethyl
Additiona 1 Bands
1273, 1243, 1115-1130, 1038, 996
1757, 1721, 1699 1390, 1372 1265, 1155, 983
2900 (broad) . 1755, 1700
1752, 1733, 1700 1390, 1379
1305, 1120, 1070, 1043, 1007, 935, 690
1278, 1060, 988, 837
O'\ -...J

TABLE IV (CONTINUED)
Compound Hydroxyl Carbonyl
0
~ n 3430, 3218a (CH3 ) 2CHCC (CH3 ) 2CNH2 . 1675 (broad)
XL
Unknown E . . . . 1722 (shoulder at 1748)
0 0 0
II II II (CH3 ) 2CHCC (CH3 ) 2cc (CH3 ) 2CCH(CH3 ) 2 • • 0 • 1700 (shoulder
at 1722)
XXXIII
Unknown F 3400 (broad) . . . .
Unknown G 3035 (broad) 0 0 D Cl
0 0
II II (CH3 ) 2cHCC (CH3 ) lDClis 0 0 0 . 1718 (shoulder
at 1737)
XLV
Gem-dimethyl
. . . .
. . . .
1390, 1378
. . . .
1396, 1378
1390, 1370
Additional Bands
1397, 1268, 1097, 1043, 810, 768
1639, 1266, 1070, 995, 902
1351, 1096, 1002, 989
1490, 1393, 1290, 950, 874
1470, 1113, 1025, 795
1265, 1148, 1041
(j\
00

Compound
fl w (CH3 )2CHCC(CH3 )2CCH(CH3) 2
XLVI
0 0
II II (CH3 ) 2CHCC (CH3) 2CCL
XLIX
0 0
II II (CH3) 2CHCC(CH3) 2COCH3
·XXXVI
0 0 0
II II II CH30CC(CH3 ) 2CC(CH3) 2COCH3
XXXVII
TABLE IV (CONTINUED)
Hydroxyl Carbonyl
e O O 0 1703 (shoulder at 1721)
• • 0 • 1788, 1725
• 0 1!11 0 1725 (shoulder at 1742)
• 0 • • 1775 (shoulder at 1723)
Gem-dimethyl
1390, 13 77
1390, 1373
e • e C
e • C e
Additional Bands
1097, 1019, 1000
1099, 1046, 945, 754
1269, 1157, 1043
1246, 1144, 1037, 1024, 790-760 (broad)
0\ I.O

Compound
H3C CH3
L
TABLE IV (CONTINUED)
Hyd r mcy 1
3510
Carbonyl
1690 (shoulder at 1720)
Gemdimethyl ·~
Compounds XXXII, XXXIV, XXXV,. XLI,, XLIII,, XL, unknown F, unknown G, and. L, (KBr pellets) •
. Compounds XXXIII,·XXXVI, ,XXXVIII,.XXXIX, unknown E, XLV, XLVI and XLIX, (NaCl cells) •
. Compound XXXVII, (CHC13 solution).
aBands at 3430, 3212 cm9-l are due to amide linkage.
Additional Bands
1328, 1289, 1204, 1025, 995, 913
...... 0

Compound
H~/CH3 (b)
CH-C 3 \. .o-. B
CH3 'CH (a)
3 ~o
' H3c CH3 (c)
XXXII
cn3 (a)
,....
XXXIV
TABLE V
NMR CHEMICAL SHIFTS AND COUPLING CONSTANTS OF PRODUCTS
Plate Solvent
XXI
XXIII benzene
XXII pyridine-d5
XXIV pyridine-d 5
OH
o (values) (p.p.m.)l
1.90 s
1.43 d
1.19 d
0.91 d
2.38 h
9.80 s
7.83-7.21
1.66 s
1.38 s
J(c.p.s.) Assignment
. . .. . OH
7.0 CH3 (c)
6.0 CH3 (a)
6.5 ·CH3 (b)
6.5 H
. . . . OH
. . . . C6H5
. . . . CH3 (a)
. . . . CH3 (b) -...J I-'

Compound Plate
XXV
XXXIX
CH3 (b)
()
XXVI
XXXV
TABLE V (CONTINUED)
Solvent
CC14
CDC13
o(values) (p.p.m.)1
7.71, 7.38(m)
2. 73 (h)
L44(s)
0.90(d)
11. 66 s
3.19 h
1.59 s (split)
1.28 s
1.06 d
J(c.p.s.) Assignment
. . . C6H5
7.0 H
" • {> • CH3 (a)
7.0 CH3 (b)
. . . . OH
6.5 H
2.5 CH3 (c)
• .. fl • CH3 (b)
6.5 CH3 (a)
........ N

.Compound
. c~,/'J.-.. 3 ,-:
XXXVIII
CH3 (a) HO'
'(,cH 0 3 II /"'U /h \
OH
XLI
TAaLE V (CONTINUED)
Plate Solvent
. XXVII CDC13
XLI CDC13
o(values) (p.p.m.)1
3.69 s
3.09 h
1.41 s
1.38 s
1.06 d
11. 96 s
1.57 s
1.54 s
1.45 s
1.39 s
J(c.p.s.) Assignment
0 e g 0 OCH3
6.5 H
Q 18 0 e CH3 (c)
19 • • • CH3 (b)
6.5 CH3 (a)
. 0 . . OH
. 0 . 0 CH3 (a)
. . . . CH3 (a)
. . . . CH3 (b)
. . . . CH3 (b) ...... w

0
I H3C
0/
I H3C
Compound
CH~(a)
H3C CH3 (a)
XLIII
XLIII
TABLE V (CONTINUED)
Plate Solvent
XXIX benzene
0
I CH3
XXX CDC13
0
I CH3
o(values) (p.p.m.)l
3.39 s 3.39 s2
3.37 s 3.36 s
1.46 s
1.42 s
3.79 s
3.70 s
1.56 s
1.54 s
1.45 s
1.34 s
J(cop.s.)
. . 0 . ' . 0 . . . ' . . . 0 . . . . . . . . .
Assignment
OCH3
OCH3
CH3 (a)
CH3 (b or c)
OCH3
OCH3
CH3 a
CH3
CH3 b or c
CH3 " .p,.

TABLE V (CONTINUED)
Compound Plate Solvent
XXXI
H· 2
XL
Unknown.E .XXXII CDC13
o(values) (p.p.m.)l
6.56, (broad)
3.19 h
1 .44 s
1.09 d
5.07 s
4 .86 s
2.21 h
L79 s
1.35 s
1.26 s
1.15 s
0.99 d
o. 71 d
J(c.p.s.) Assignment
. . . . NH2
7.0 H
. . . . CH3 (b)
7.0 CH3 (a)
. . . 0 . . . .
. . . . 6.5
e O • •• 0 •. 0 • .
0 • • . . . . 0
. . . . . . . .
. . . . . . . . 6.5 . . . . 7.0 . . . . .......
Vl

TABLE V (CONTINUED)
. Compound Plate ,Solvent
CH3
CH3 (a)
1'''H XXXIII CC14
CH3
XXXIII
CH .A vu3 3...._ , (1,\
XXXIV CC14
XLV
I) (values) (p.p.m.)l
3.08 h
1.34 s
1.04 d
4.17 g
2.84 h
1.29 s
1.25 t
1.05 d
J(c.p.s.)
6.5
6.5
7.0
6.5
• Cl • •
7.0
6.5
Ass ignmep.t
H
CH3 (b)
CH3 . (a)
-CH -2
H
CH3 (b)
CH3 (c)
CH3 (a) '-I
"'

TABLE V (CONTINUED)
Compound Plate Solvent
CH 3..._ ~ ...... , -
XXXV cs2
---j
XLVI
0
II _CH-:2
XXXVI neat
Cl
XLIX
0
CFk Jl l!H3
XXXVII neat
XXXVI
6 (values) (p.pcm.)l
3c86 h
1.33 s
1.03 d
3.05 h
1.52 s
1.13 d
3.73 s
2.92 h
1.32 s
1.03 d
J(c.p.s.)
7.0
0 e O •
7.0
7.0
0 e • 0
7.0
0 e O Cl
6.5
0 • • •
6.5
Assignment
H
CH3 (b)
CH3 (a)
H
CH3 (b)
CH3 (a)
OCH3
H
. CH3 (b)
CH3 . (a) ....... .......

TABLE V (CONTINUED)
Compound Plate Solvent
n3c o9""')('" - 0 - I
XXXVIII CC14
CH3
XXXVII
-H
CH~H (b) 3 3 'b) CH H \.
3 3 0~ ~ XXXIX pyridine-d5
H3C CH3 (c)
L
El (values) (p.p.mo)l
3.79 s
3.67 s
1.54 s
1.30 s
6 .49 s
1.54 s
1.38 s
1.33 s
1.17 s
J(c.p.s.)
G • • D
. . . . . . . D e e 0
. . . .
. . . .
. . . .
. . . .
. . . .
Assignment
OCH 3
·OCH 3
CH3 (b)
CH3 (b)
OH
CH3 (c)
CH3
CH3 b
CH3 (a)
"' 00

Comp0ound
(a)CH3 CH
(b) .
CH3
L
TABLE V (CONTINUED)
Plate Solvent
XL benzene
6 (values) (p.p.m.)l,
2.30 s
1.38 d
1.15 s
0,91 s
0.88 s
J(c.p.s.)
6.5
Assignment
OH
CH3 (c)
CH3
CH b 3
CH3 (a)
1The multiplicity of each peak is indicated as follows: singlet, s; doublet, d; triplet, t; quartet, q; and heptet, h.
2 0 Delta (6) values for ester methyl group protons when sample was heated above 50 •
---i \0

80
solution. When approximately 150 ml. of the ammonium chloride solution
had been added, the reaction subsided and the rest of the ammonium
chloride solution was added rapidly. The two-layered mixture (yellow
green organic layer) was poured into an additional 100 ml. of the ammon
ium chloride solution. The mixture was allowed to stand overni&ht and
then extracted with three 200-ml. portions of ether. The yellow organic
extracts were combined, dried (Mgso4) and filtered under suction; the
ether was removed under aspirator pressure giving approximately 112 ml •
. of a yellow oily liquid. No precipitate formed when the liquid was
chilled overnight.
1n an attempt to separate the components of the previous reaction
mixture a 1-liter, three-necked flask equipped as usual, except for an
addition funnel was charged with 50.0 g. of the reaction residue,
50.0 g. (0.38 mole) of Girards I T reagent, 50 ml. of glacial acetic acid
and 500 ml. of methanol. The solution (yellow in color) was heated un
der reflux for 3.5 hours, allowed to cool to room temperature and then
treated with 5% aqueous sodium hydroxide solution until a pH of about 8
was obtained in the solution. Water (100 ml.) was added and the mix
ture was extracted with 300 ml. of ether. The organic layer was dried
(Mgso4 ) and filtered; removal of the solvent under aspirator pressure
gave an oily liquid. Chilling the liquid overnight resulted in the
precipitation of a white solid. The solid was filtered from the mixture,
washed with hot water (100 ml.) and recrystallized from a minimum amount
of hot benzene-cyclohexane (1:1) to give 2.84 g. (1.8%) of rectangular
crystals. From the infrared (IR) (Plate II), nuclear magnetic resonance
.(NMR) (Plates XX.I, XXII and XXIII) spectra, and physical data, the
structure of this compound was assigned as that of ketol .XXXII,the

81
properties of which are found in Tables III, IV and V.
Reaction (1:1) of ~henyl Grignard Reagent and Lactone I - Run 2.
The apparatus and procedure used in this experiment were identical to
those described in the previous experiment. The lactone I (140.1 g.,
1.0 mole) was added to the chilled Grignard solution, After the reac-
tion period, workup of the reaction mixture afforded a yellow liquid
which deposited a white solid when chilled; 7.26 g. (4.7%) of ketol·
XXXII was obtained, mixture m.p. 147 .5-149° with a sample from Run 1
(benzene-_g_-heptane solution).
Reaction (1: 1.2) of Phenyl Grignard .Reagent and Lac tone I ... Run 3.
One mole of phenylmagnesium bromide was prepared (96%) as before. The
0 Grignard solution.was cooled to O, and 168.12 g. (1.2 mole) of lactone
I in 175 ml. of ether (anhydrous) was added as described in the previous
experiments. The reaction mixture was treated as previously outlined
for Run 1. The oily yellow-brown liquid (approximately 110 ml.), when
chilled overnight, afforded a white solid which was washed as before
and recrystallized from benzene-_g_-heptane solution (1:1) to give 45.95
g. (yield, 30.1%) of ketol XXXII; mixture m.p. 148-149° with the sample
obtained from Run 1.
The mother liquor obtained from the removal of ketol XXXII was
analyzed by GLC (CA-1). The following compounds were identified and
their yields, based on lactone I, were obtained by repeated injections
with known standard solutions and calculations of peak area ratios.
Compounds identified were: diisopropyl ketone '.(XLIV) (4%), lac tone I
(13%), 2,2,4-trimethyl-3-oxovaleramide {Xt) (1.5%), biphenyl (3.5%), 1-
phenyl-2 ,2 ,4-trimethyl-1,3-pentanedione (XXXUO (8%), triketone XXXIII
(4%) and benzophenone (40.1%). Retention times at 139° were:

82
diisopropyl ketone[(XLIV), 2 minutes; lactone I, 2.5 minutes; amide XL,
3 minutes; biphenyl, 6 minutes; diketone·XXXIX, 36 minutes; triketone
XXXIII, 41 minutes; benzophenone, 48 minutes.
In an.experiment practically identical to that described above,
lactone I (182.1 g., 1.3 mole) was added to one mole of phenyl Grignard
reagent (Run 4). Normal reaction conditions and workup (see page 60)
of the reaction mixture gave (after purification), 38.42 g. (23.6%) of
ketol XXXII, mixture m.p. 148-149° with a sample obtained from Run 1.
The reaction of one mole of phenyl Grignard reagent and lactone I
(210.2 g., 1.5 mole - Run 5) gave 38.79 g. (23.6%) of ketol XXXII, mix
ture m.p. 148-149° with a sample obtained from the previous run.
Condensing one mole of phenyl Grignard reagent (98%) with 280.2 g.
(2.0 mole) of lactone I (run 6) gave 39.00 g. (23.9%) of ketol XXXII;
mixture m.p. 148Cl49° with a sample obtained from the previous run.
Carbonation of 1:1.2 Reaction Mixture of Phenyl Grignard Reagent
and Lactone I. One mole of phenylmagnesium bromide was prepared (97.5%)
and combined with 168.12 g. (1.2 mole) of lactone I as previously de
scribed. The brown mixture was allowed to warm to room temperature and
then heated at reflux for 3 .5 hours. When the contents d£ the flask had
attained room temperature, gaseous carbon dioxide was bubbled into the
stirred orange mixture for 27 hours. During the first 2 hours of car
bonation, the mixture became increasingly difficult to stir because of a
very viscous brown material which formed on the bottom of the reaction
vessel. During the remainder of the carbonation, however, the brown sub
stance solidified and stirring became easier. A .lemon-colored reaction
mixture finally resulted; this was decomposed at room temperature with
aqueous ammonium chloride as usual. The resulting golden colored,

83
jelly-like mixture was stirred at room temperature for 1 hour and then
extracted with three 300-mlrportions of 1:1 ether-benzene solution.
The combined yellow organic extracts (T) were now extracted with three
300-ml. portions of saturated sodium bicarbonate solution; the bicar-
bonate extracts were washed with two 100-ml. ·portions of ether-benzene
(1:1) solution, and the organic washings were added to the original or-
ganic layer (T). After the sodium bicarbonate extracts were acidified
to a pH of 1 with aqueous 6!i hydro!=!hloric acid (130 ml.), the mixture
was e~tracted with three 150 ml.-portions of ether-benzene solution
(1:1). These combined organic extracts (brown in color) were dried
(Mgso4 ), and filtered under suction. When about 75% of the solvent had
been removed from the filtrate (under aspirator pressure), a white solid
formed in the flask. The flask was chilled and the solid filtered out.
After being washed successively with water (75 ml.) and benzene
(75 ml.), the solid was dissolved in hot acetone and reprecipitated by
the addition of cold water. A snow-white solid was obtained - 8.36 g.
0 (3% - based on lactone I); m.p. 148.5-150. Physical properties (Table
III) and IR .(Plate III) and NMR (Plate XXIV) spectral data (see Tables
IV and V led to the structure assignment of acid XXIV for this compound.
The purity analysis of the acid was demoristrat'ed by GLC [(CA.:.2 -
Table II] analysis. Only one peak was observed on the chromatogram;
this was found to have the same retention time as l-phenyl-2,2,4-tri-
methyl-1,3-pentanedione :(XXXIX),. This was varified by a mixed injection
with a known sample of the diketone XXXIX, and peak reinforcement was
observed. Apparently decarboxylation of the acid occurred on the column.
A 1.0-g. (0.004 mole) sample of the acid was placed in a large test
tube having a side-arm outlet. The test tube was fitted with a

thermometer which extended into the solid and with a rubber hose
connected to a small piece of glass tubing immersed in .a solution of
10% aqueous calcium hydroxide. The test tube was slowly heated oh a
sand bath to 155° where the temperature was held for 1 hour •. Smoky
vapors were observed to rise in the test tube with hubbies emerging
84
from the end of the glass .tube in the calcium hydroxide solution. After
1 hour of heating, the calcium hydroxide solution contained a milky
white precipitate. When the test tube was cool (room temperature), the
IR spectrum. (Plate IV) and the NMR (Plate XXV) spectrum of the contents
in the tube were identical to those of an authentic sample of l-phenyl-
2, 2 ,4-trimethyl-l, 3-pentanedione (XXXIX),. The mother liquor obtained
from the removal of acid XXXIV was further concentrated on a rotary
evaporator to give 25.02 g. of a viscous light brown oil. This acid
mixture was treated with diazomethane as described in a later experi
ment (page 85).
The original organic extracts (T) were concentrated under aspirator
pressure to yield 90 ml. of a yellow-colored oil. GLC (CA-11) analysis
revealed the presence of benzophenone (44%), and small amounts of lac
tone I, biphenyl, and bromobenzene as the only components present. Re
tention times at 161° were: bromobenzene, 2 minutes; lactone I, 2.7
minutes; biphenyl, 4.5 minutes; and benzophenone, 30 minutes •
. In another essentially identical carbonation experiment, the or
ganic layer obtained from the acidified bicarbonate extracts was
. stripped of solvent under aspirator pressure and the oily• liquid wa:s
bottled and stored. Weeks later it was observed that the bottle con
taine·d a white precipitate. The solid was filtered from the mixture
and wa~hed with 10 ml. of Skelly Solvent F and 10 ml. of warm water •. A

85
solution of the compound in hot acetone yielded a white solid on the
addition of cold water; 125 mg. of the solid was obtained, m.p. 74.5-
0 75.5 • Elemental analysis and IR (Plate V) and NMR (Plate XXXVI) spec-
tral data identified the compound as monoacid XXXV, (see Tables III, N,
and V).
Esterification of Acid Material (Page 84) Obtained From the Carbon-
ation of the Reaction Mixture of Phenyl Grignard Reagent and Lactone I.
A 1-liter one-necked flask, equipped with a 12-inch modified Claisen U-
tube, a thermometer and a condenser, was charged with a solution of
4.4 g. (0.11 mole) of sodium hydroxide pellets dissolved in 35 ml. of
water, 85 ml. of capitol and 275 ml. of ether. The flask was cooled
to o0 by means of an ice-salt bath. EXR-lOl(N,N'-dinitroso-N,N'-di-
methylterephthalamide), 12.0 g. (containing inert filler), was funneled
into the flask rapidly. The mixture (two layers) was allowed to come
to room temperature and then was slowly heated until the ether-diazo-
methane distilled slowly. The distillate was caught in a 250-ml. flask
(chilled by Dry Ice) containing.25.0 g. of the acid material obtained
from carbonation of reaction mixture of phenyl Grignard reagent and lac-
tone I - (page 84). Distillation was continued until the distillate
was water-white. Heating was discontinued, and the apparatus was
allowed to stand for 1 hour. The ether was removed from the receiver
flask under aspirator pressure to give 24.97 g. of a light yellow oil.
GLC analysis (CA-3) of the residue revealed the presence of a many-
component reaction.mixture. Compounds were identified by mixed injec-
tions with authentic samples and yields were based on lactone I: methyl
2,2,4-trimethyl-3-oxovalerate (XXXVI) (6%), 2,2,4-trimethyl-3-oxovaler-
amide (XL) (2%), dimethyl 2, 2 ,4 ,4-tetramethyl-3-oxoglutarate (XXXVII) (1%),

86
triketone XXXIII (16%), and 1-phenyl-2,2,4-trimethyl-1,3-pentanedione
(XXXIX) (2%). Retention times at 161° were: ester XXXVI, 0.5 minute;
amide XL, 1.3 minutes; ester XXXVII, 2.1 minutes; diketone XXXIX, 1.4
minutes; triketone XXXIII, 16.3 minutes. Seven other small peaks ob;-
served in.the Ghromatogram were not identified. However, the compound
representing the largest peak (18% - from peak area ratios) and having
a retention time on the column of 49 minutes was isolated in the pure
state by injecting twenty 165-ml.· samples of the reaction residue (dis-
solved in 15 ml. of ether) onto a column.in the Autoprep GLC unit (CP-
1) h h h f 11 C 1 1890, • Settings on t e c romatograp were as. o ows: .. o umn, ,
0 0 helium flow,> 600 cc./min.; injector, 265 ; collector, 270 ; detector,
310°, Retention time for the compound on the prep.column was 72 min-
utes. -The small amount of a viscous, light brown-colored liquid ob-
tained was analyzed for purity by GLC (CA-4). The chromatogram ~hawed
only one peak present;.retention time at 150° was 19 minutes. The
structure of this compound was assigned as that of ester XXXVIII from
. :IR (Plate VI), NMR (Plate -XXVII) and elemental analyses. (See Tables
III, IVand V).
Reacti.on .(1:1.2) of.Phenyl Grignard Reagent and Lactone I in Tetra-
hydrofuran - Run 7. One mole of phenyl Grignard reagent was prepared
(99%) and combined with 168.12 g •. (1.2 mole) of lactone I in a manner
similar to that previously described. The brown mixture was allowed to
warm to room temperature and the addition funnel was removed. A Claisen
head with a thermometer was attached to the flask, and the ether was
slowly distilled out through a short path condenser until a slurry
formed in the distillation flask. Dry anhydrous tetrahydrofuran (THF)
(750 ml.) was added, and the solution was distilled until the

87
temperature of the contents in the flask reac.hed 68°. The brownish-red
solution was heated at reflux for 10 hours, then allowed to cool to room
temperature and was further stirred for 12 hours. The mixture was de-
composed at room temperature by the dropwise addition of 350 ml. of 20%
aqueous ammonium chloride solution with violent foaming. The resulting
yellow-green mixture was extracted with three 300-ml. -portions of ether;
the organic extracts were combined, dried (Mgso4 ) and filtered with·suc
tion. When the solvent was removed by distillation, approximately 100
ml. of a yellow oil remained. The oil was dissolved in.100 ml. of
Skelly Solvent F and chilled overnight with the ~ubsequent formation of
a white precipitate. The white solid was filtered from the solution and
washed with hot water (75 ml.). and cold Skelly Solvent F (75 ml.).
Solution of the solid occurred in hot benzene from which precipitation
was effected by the addition of cold ~-heptane, giving 22.07 g •. (15%) of
ketolXXXII, mixture m. p. 148-149° with a sample obtained from Run 1.
Carbonation of 1:1.2 Reaction Mixture of Phenyl Grignard Reagent
and Lac tone· I in .Tetrahydrofuran. One mole of phenylmagnes.ium bromide
was prepared (97%) and combined with· lac tone I (168 .12 g., 1 .2 mole) in
a ·rrtanner ·:as previously described in Run .3. The ether was "distilled from
the· flask as described. in the previous experiment (page 86) and dry THF
(750 ml.) added to the slurry until the temperature of the contents in
' 0 the·flask reached 68. The brownish-red solution was heated under re-
.flux for an additional 7 hours and allowed to cool to room temperature;
then carbon·dioxide gas was bubbled into the solution for 24 hours.
After 15 minutes of carbonation, the temperature of the solution rose
14° and remained at 41° for 0.5 hour. The temperature of the solution
0 then slowly fell to room temperature (27·) v.ihere it remained for the

88
duration of the carbonation, The brown-red mixture containing a small
amount of chunky white precipitate was hydrolyzed at room temperature
by the dropwise addition.of 300 ml. of 20% aqueous ammonium chloride
solution (as before), Workup of the reaction mixture was practically
the same as that described for the experiment cited on page 82, Filtra
tion under suction, of the organic extracts obtained from.the acidified
bicarbonate extracts, followed by removal of most of the solvent by dis
tillation, gave a white precipitate. The mixture was chilled overnight
and the solid was filtered out and washed with·warm water (50 ml.) and
then cold Skelly,Solvent F (50 ml.). The solid (8.60 g.) had a melting
point of 134-135.5 (dee.); its purity was checked by GLC analysis (CA-
5) and the presence of two peaks was observed. These peaks were identi
fied as arising from diisopropyl ketone (XLIV) and triketone XXXIII in a
ratio· of 1:9 respectively. Retention times at 147° were: diisopropyl
ketone<(XLIV), 2.5 minutes; triketone XXXIII, 17 minutes. The IR spec
trum of the solid contained absorptions (both hydroxyl and carbonyl
regions) which indicated the presence of a carboxylic acid group. A
small amount of the solid (4 g.) was repeatedly recrystallized from
benzene and gave some solid (100 mg.) which melted sharply (121-122~).
GLC analysis of this solid material (same column and tempe1tature as
cited above) showed only one peak that corresponded to the retention
time of triketone XXXIII. Apparently decarboxylation occurred on the
column. Structure proof of this diacid XLI was further obtained from
elemental analysis '(Table III), NMR. (Plate ·XXVIII) and IR. (Plate· VII)
spectral data (Tables IV and V).
The mother liquor (filtrate obtained from the removal of the above
acid mixture) was further concentrated on a rotary evaporator giving

89
39.05 g. of a light brown oil which was treated with diazomethane in a
later experiment (page 89).
The remaining 4.0 g. of the acid mixture was treated with 1.2 g. of
diazomethane (prepared as before - page 85). A light golden-colored oil
was obtained and was dissolved in 8 ml. of Skelly Solvent F. The solu-
tion, when chilled, deposited a white solid which was filtered out and
washed with cold.Skelly Solvent F (10.ml.). Dissolving the precipitate
in hot benzene and then allowing the solution to cool gave clear plate-
0 lets (2.64 g.), m.p. 74-75 • IR (Plate VIII) and NMR (Plates XXXIX and
XlCX) spectral data. and elemental analysis (Tables III,. IV and V). sub-
stantiate its proposed structure of diester XLIII •. Analysis (GLC, CA-
· 5). of the mother liquor obtained from the removal of diester. XLIII re-
vealed only one peak on the chromatogram. It was identified as diester
·xxxvrr by mixed injection·with an authentic sample •. The original or-
ganic extracts were stripped of solvent under aspirator pressure to give
· 109 ml. of a golden-colored oil. GLC analysis (same column and tempera-
ture as cited above - page 89).showed the presence of only benzophenone
(45%), and small amounts of bromobenzene, biphenyl, and lactone I. Re-
tention time was: benzophenone, 29 minutes. The yield of benzophenone
was .based on lactone I •
. Esterification of. Acid Material (Page 87) · Obtained From the Car-
bonation.of the Reaction Mixture of Phenyl Grignard Reagent and Lactone
.I in THF •. Diazomethane (12.0 g.) was prepared (as previously described
- page 85) and slowly distilled into a solution of 25.0 g. of the acid
material (page 87) dissolved in 75 ml. of ether cooled in Dry Ice. The
solution in the receiver flask was allowed to attain room temperature;
then the ether was.removed.under aspirator pressure to yield 24.83 g. of

90
a red-colored oil. GLC analysis (CA-3) .. revealed the presence of many
components. Compounds identified by mixed injections with authentic
samples were: methyl 2, 2 ,4-trimethyl-3-oxovalera te '(XXXVI) (4%), di-
methyl 2 ,2 ,4 ,4-tetramethyl-3-oxoglutarate (XXXVI:t') (8%), 2 ,2 ,4-trimethyl-
3-oxovaleramide :(XL) (2%), triketone · XXXIII (13%), 1-phenyl-2 ,2 ,4-tri-
methyl-1,3-pentanedione (XXXIX) (2%), and ester XXXVIII (12%). Yields
.are based on lactone I and retention times.on the column.are the same
as those o.btained from the treatment of the acid mixture received from
the carbonation of the reaction mixture of phenyl Grignard Reagent and
lactone·I - page 82,,with diazomethane.
h .Reaction (1:1.9) of Methyl.Grignard Reagent and Lactone•I. The
apparatus, procedure and workup used for this experiment were identical
to those used for the reaction of lactone I With phenyl Grignard rea-
gent. Lactone I (91.07 g., 0.65 mole) was added to methyl Grignard
reagent (0.33 mole by titration). The amber-colored oil (approximately
55 ml.) was chilled but no precipitate formed. Analysis of the oil by
GLC (CA-6) disclosed the presence of more than a dozen components.
Triketone · XXXIII (70%) was identified by mixed injection with· an auth
entic standard solution of this compound. Retention time at 178° was:
triketone XXXIII, 8 minutes. No attempt was made to identify any of the
other components present in the reaction mixture.
h . Reaction (0.30:1.0) of Ethyl Grignard Reagent and Lactone I. The
procedure used for this experiment is identical to that described for
the reaction of phenyl Grignard reagent and lactone I •. Ethyl Grignard
reagent (0.15 mole) was chilled to 0° and lactone I (70.0 g., 0.5 mole)
hArap9 hoe·Chemicals Inc., Boulder, Colorado, concentration (in' ether), 3 molar ..

91
added over a period of 1.5 hour. Reaction conditions and workup of the
reaction .mixture were essentially the same as described for the reac-
tion of phenyl Grignard reagent and lactone I (page 60). Chilling the
yellow oil (50.09 g.) gave a white solid. The precipitate was filtered
from the mixture and recrystallized from benzene-_g-heptane solution to
give 123.mg. of ketol XXXII, mixture m.p. 148-149°. GLC analysis (CA-3)
of the filtrate revealed a complex reaction mixture with nearly. 20
components observed on the chromatogram. A major component of the reac-
tion mixture was identified as triketone XXXIII (60%) by mixed injection
0 with an authentic standard solution. Retention time at 162 was:
triketone XXXIII, 8 minutes. No attempt was made to identify any of
the other peaks indicated on the chromatogram.
54 Preparation of ~-Butyl Grignard Reagent. A l~liter three-
necked flask equipped as usual for Grignard formation was charged with
12.00 g. (0.5 g.-atom) of magnesium shavings; and the metal was covered
with 90 ml. of anhydrous ether. !-Butyl bromide (68.51 g., 0.50 mole)
was placed in the addition funnel and approximately 5 ml. was added to
the flask. When the reaction had been initiated by gentle crushing of
the metal with a stirring rod, ether (250 ml.) was quickly introduced
into the flask and ether (250 ml.) was added to the addition funnel.
The proc~dure from this point was nearly identical to that used for the
preparation of phenyl Grignard reagent (page 59), Titration led to a
calculated yield of 82% (0,41 mole).
Reaction (1:1.15) of !,-~dtyl Grignard Reagent and Lactone I. The t-
butyl Grignard reagent (0.41 mole) was placed in a 2~1iter three-necked
flask (same apparatus as described in the previous experiment) and
chilled to o0 using an ice-salt bath. Lactone I (91.07 g., 0.65 mole)

92
diluted with 250 ml. of anhydrous ether was added dropwise with stirr-
ing, under nitrogen over a period of 1.5 hours •. The addition was not
exothermic. The resulting.dark-green mixture was allowed to come to
room temperature; heat was applied and the mixture held at reflux for
12 hours, without noticeable color change. The flask was chilled in an
ice-salt bath to 0° and the reaction mixture decomposed by adding 500
ml. of aqueous 20% ammonium chloride solution; The resulting yellow-
green mixture was extracted with three 250-ml. portions.of ether. The
. solvent extracts were dried (Mgso4J and filtered under suction; the
ether was removed under water aspirator pressure to give 97.12 g. of a
yellow oil. A solid was deposited when the oil was chilled, When
filtered from solution and recrystallized from benzene-E,-heptane solu-
tion (1:1) there was obtained 1.52 g. of 2,2,4-trimethyl-3-oxovaleramide
(XL), mixture m.p. 107-109° (with an authentic sample). IR (Plate IX)
and NMR (Plate ·XXXI) spectral data further support this known structure
(see Tables IV and V). GLC analysis (CA-3) of the mother liquor ob-
tained from the filtration of this compound disclosed the presence of
more than 15 components. Mixed injection (authentic standard solution)
identified one of the components as· trike tone· XXXIII (no yield
measured •. Retention time at 167° was: triketone XXXIII, 8 minutes •
. Sublimation of Ketol XXXII. Ketol XXXII,. 5.0 g. (0.02 mole) was
placed in a 10-inch sublimation gun.and heated at 150°. Vacuum was then
0 applied and heating was continued until the temperature reached 190 /
0.45 mm. Heating was maintained to hold this temperature and pressure,
with a white solid coating the cold finger at the end of 30 minutes •
. Heating was discontinued and the tube was allowed to cool to room
temperature under vacuum •. The vacuum.was then slowly removed and the

93
cold finger carefully withdrawn from the apparatus. The very hard and
very nicely crystalline solid had the appearance 0£ small uncut diamonds.
The melting point (148-149°), and the sharp bands.observed in.the IR
spectrum identified the material as ketol XXXII.
Attempted Preparation of Carbonyl Derivative/0 of KetoL XXXII and
Triketone XXXIII •. A. 2,4,-Dinitrophenylhydrazone. Ketol,XXXII (1.0 g.,
0.004 mole) was placed in a large test tube along with 20 ml. of 2,4-
68 DNP reagent. The test tube was stoppered and vigorously shaken for
15 minutes. When no precipitate formed, the test tube was attached to
a water condenser artd heated for 6 hours .on a steam ,bath. ,The _orange
solution was poured into a 1:1 mixture of water and cracked ice. An
oil formed on top of the ice mixture but co~ld not be made to crystal-
lize, even when allowed to stand for 1 week.
B. Oxime. Hydroxylamine hydrochloride (0.25 g., 0.003 mole) dis-
solved in 15 ml. of water was placed in a large test tube. Ketol XXXII
(1.0 g., 0.004 mole) and 10 ml. of 10% aqueous sodium hydroxide were
.added along with 2 ml. of ethanol until a clear solution formed •. A
condenser was attached, and the contents of the tube·were heated on.a
steam.bath for 3 hours. The tube was chilled in an ice bath, but crys-
tallization did not occur and could not. be induced by scratching the
sides of the test tube with a stirring rod .
. C •. semicarbazone. Ketol XXXII (1.0 g., 0.004 mole) was dissolved
in 10 ml. of ethanol in a ·large test tube. Water was then added until
the solution became faintly turbid. Semicarbazide hydrochloride (1.0 g.,
0.001 mole) and 1.5 g. of sodium .acetate were then added and the mix~,
ture vigorously shaken for 30 minutes. Because no precipitate formed,
the contents of the tube were heated in a beaker of boiling.water for 10

94
minutes; when cool no precipitate formed. The tube was chilled in a
_beaker containing ice, but crystallization did not occur •
. Identical procedures as described above were followed in attempts
to prepare derivatives of triketone XXXIII, In all cases, attempts to
induce the formation of crystals were of no avail. A solid product was
obtained, however, when 1.0.g. (0.004 mole) of triketone·XXXIII and 3.0
g. (0.028 mole) of phenylhydrazine, dissolved in 25 ml. of ethanol, were
heated at reflux in.a test tube for 12 hours; however, the solid product
qbtained cquld not be purified, .Recrystallii~tion,using benzene,
ethanol, acetone and chloroform as solvents.failed to give a pure
product.
Attempted.Acylation of Ketol XXX1l With Acetic Anhydride in.Pyri--1
d . 11 1ne. Ketol XXXII 4.86 g. (0.02 ~ale) was placed in a 100-ml. one-
necked flask fitted with a mechanical stirrer and a condenser with dry-
ing tube (CaC12) •. Pyridine (23 ml.) and acetic anhydride (7 ml.) were
added in that order and the mixture was heated at reflux for 15 hours.
The reaction mixture turned brown after 1.5.hours. The mi~ture was
then chilled in an ice bath for 1 hour and poured into ·100 ml. of dis-
tilled water .. The brown solution was treated with powdered sodium car-
bonate (9.78 g.) .until evolution of carbon dioxide ceased. Ether-
benzene solution (250 ml.-1:1 ratio) was.used to extract the mixture.
The. solvent extracts were· washed successively with two. 200-rol. ·portions
.of 1~ hydrochloric acid, 170 ml. of 10% sodium bicarbonate solution and
finally 100 ml .. of water. The organic layer was then dried (Mgso4 ) and
filtered under suction. When most of the solvent had been removed by
distillation, a white precipitate formed •. The solid was filtered out
and washed successively with SO ml.· of water and Skelly Solvent F

95
(60 ml.). Dissolving the material in hot benzene and precipitating it
by the addition of cold E:-heptane gave 4. 79 g. (9'8% recovery) of snowy
white crystals, mixture m.p. 148-149° with a sample of ketol XXXII from
Run 1 - page 60 •
. Acid Hydrolysis of Ketol XXXII. A 250-ml. single-necked flask
equipped with a mechanical stirrer and reflux condenser with- drying;- tube
(Cacl 2 ) was charged with 150 ml. of aqueous 6~ hydrochloric acid ap.d 10 g."
(0.039 mole) of ketol XXXII. The mixture was boiled for 6 hours. The
ketol seemed to dissolve after the mixture had been.heated for 0.5
hour,. and a yellow liquid formed on top of the solution. The mixture
was allowed to cool to room temperature and was then extracted with
250 ml. of ether. The yellow organic layer was dried (MgS04 ), and fil
tered; the ether was removed to give 9.83 g. of a yellow liquid. GLC
analysis (CA-4) revealed the presence of two components. Retention
0 times at 167 were: unknown E, 12 minutes; triketone XXXIII, 16 min-
utes. Two grams of the residue were dissolved in an equal volume of
ether and chromatographed on the Autoprep unit (CP-2) •. settings on
the chromatograph were as follows: column, 190°; helium flow, 300 cc./
0 0 0 min.; injector, 255 ; c0llector, 280; detector, 305 • Twenty-five
100-ml. injections of the ether solution of the reaction residue were
placed on the column and both unknown E (retention time,.26 minutes)
and triketone XXXIII (retention time, 30 minutes) were collected in
small glass collector tubes immersed in a Dry Ice - :acetone bath. During
the first few injections it was found that both materials readily
yielded aerosols when emerging from the collection po~t. By using a
stainless steel L-shaped tube, wrapped with an electric heating tape,
connected to a number 19 hypodermic needle, the amount of aerosol was

96
reduced and the efficiency of collection greatly increased,
After the twenty-fifth injection, the tubes were allowed to warm to
room temperature and the partially solidified material present in each
tube was washed into separate vials with ether •. Analysis of both solu-
0 tions by GLC (CA-4 - temperature 167) indicated only one peak from the
solution containing triketone XXXIII. However, the solution containing
unknown E was shown to be contaminated with approximately 2% of trike-
tone XXXIII. It was evident that peak separation was not large enough
or the technique for collection was poor. So, the column temperature
was dropped to 185° and twenty 100-ml. injections were placed on the
col~mn, care being taken to wash the needle in.acetone before each
collection. GLC analysis (same column and temperature as cited above)
indicated that both compounds collected were pure, as only one peak was
observed for each sample. Elemental analysis and IR (Plate·X) and NMR
(Plate XXXII) spectral data are given in Tables III, IV and V for un-
known E. The structure of triketone XXXIII was deduced from data like
that given above .. IR (Plate XI, NMR (Plate XXXIII) data and results of
analysis are also shown in Tables·. III, IV and V.
Dehydration of Ketol,XXXII. An intimately ground mixture of 20.0
g. (0.078 mole) of ketol XXXII and 20!0 g .. (0.14 mole) of fre~hly fused
potassium acid sulfate was placed in a 250-ml. one-neck flask equipped
as in the previous experiment. The mixture was heated slowly by means
f db h db 1 1050. o a san at an egan to met at When the temperature reached
0 llO , the mixture had entirely liqui!fied and had turned purple. Slow
heating was continued until the temperature of the mixture reached 155°
where it was thus held for 1 hour. At this temperature, droplets of
w.ater could be seen condensing on the sides of the flask and the

97
condenser. The melt was allowed to cool to room temperature; 250 ml. of
ether was added and the mixture was filtered, The filtrate was treated
with solid sodium bicarbonate until the mixture was neutral to litmus.
Extraction of the mixture with 300 ml. of ether followed. The ether ex-
tract was dried (Mgso4 ) and filtered. Removal of the ether from the fil-
trate on a rotary evaporator gave 19.39 g. of a brown oil. GLC analysis
(CA-7) of the oil revealed two components present. Retention times at
0 163 were: unknown E, 18 minutes; triketone XXXIII, 20 minutes.
Approximate area ratio~ of the components showed the yields of these
to be about 40 and 60% respectively.
Distillation of 18 g. of the brown oil using a short-path Kontes
distillation apparatus appeared to be unsuccessful. Fractions were
0 0 0 0 taken at ll5-ll8 , ll8-120 , 120-126 and 126-129 /3.5 mm. Each frac-
tion consisted of about 4 ml. of a clear colorless·liquid •. GLC analy-
sis (same column and temperature as cited ab:ove) of each fraction still
showed the presence of two components, but the peak attributed to un-
known E decreased steadily in the higher-boiling fractions .and was
present to the extent of only about 5% in the highest-boiling fraction •
. Each fraction was dissolved in 5 ml. of Skelly Solvent F and
chilled overnight. An oily solid was deposited in each· fraction and was
removed by filtration. The precipitate obtained from the highest-boil-
ing fraction was washed with 10 ml. of cold Skelly Solvent F and
analyzed by GLC (same column.and temperature as cited above). Again two
components were observed, but the amount of unknown E_was:estimated-to·
be only about 3%.
A quantitative analysis was performed on the reaction residue by
making repeated ip,jections of the residue (1.0027 g. in 25 ml. of ether)

98
alternately.with a prepared standard solution (1.0080 g. of triketone
XXXIII in 25 ml. of ether) •. From the molar concentration of triketone
XXXIII obtained from the standard solution,.and from the observed peak
area .of this component obtained from the standard solution and from the
reaction mixture, the.yield of triketone XXXIII was found to be 62%.
The.yield of unknown.E was•38% ·since it was the only other component de-
tee ted by GLC •
. Attempted.Dehydration of Ket0l XXXII with·Dimethyl Sulfoxide. 81
Ketol XXXII (13.90 g., 0.055 mole) was placed in a 200-ml. three-
necked flask equipped as. in the previous experiment •.. Dimethyl sulfoxide
(30.03 g.,. 0.385 mole) was funneled into the flask and the solution was
heated in an oil bath to 178°,and held at this temperature for 11 hours.
The brown solution was allowed to cool to room temperature and was then
poured into· 150 ml. of water. The solution was extracted with three
100-ml. portions of ether-benzene solution (1:1). Filtration of the
dried (MgS04) extracts gave a light yellow solution which was concen
trated under aspirator pressure to give 13.79 g. of a yellow liquid.
GLC analysis (CA-8) disclosed the presence of only one peak along with
a trace impurity. By mixed injection with a standard solution, the peak
was identified as that corresponding to triketone·XXXIII (98%). The
0 impurity was.identified as unknown E •. Retention times at 160 were:
trike tone XXXIIL, 5 minutes; unknown E, 3 minutes.
Thermolysis of. Ketol XXXII. A 50-ml. flask equipped like that
described for the experiment cited on page 95 was charged with 5.0 g.
t~0.02 mole).· of ketol XXXII. The flask was slowly. heated and the ketol
liquified at 151°. Further heat was.applied until the temperature of

99
. 0 the contents in the flask reached 185 • The light brown.liquid was
heated at this temperature for 1 hour, then allowed to cool to room
temperature. The IR spectrum of the liquid (4. 96 g.) was identical to
that of triketone XXXIII. GLC analysis (CA-4) disclosed only one peak';:
on the chromatogram •. By a mixed injection with an authentic sample,
the compound was identified as triketone XXXIII. Retention time at
165° was 17 minutes.
Reaction of Ketol XXXII with Lithium .Aluminum Hydride. A 1-liter
three-necked flask equipped similarly as that described for phenyl Grig-
nard preparation with the exception of a glass tube outlet connected to
a gas .buret (filled with water), was charged with 200 ml. of anhydrous
ether and 1.74 g. (0.044 mole) of lithium.aluminum hydride; the: grey
suspension.was chilled to o0 in an ice-salt bath with stirring, under
nitrogen. After passing nitrogen through the system for 15 minutes, the
nitrogen flow was stopped and 25.00 g. (0.098 mole) of ketol XXXII dis-
.solved in 100 ml. of anhydrous ether was added dropwise. With each
drop of the solution of ketol XXXII a large volume of gas was liberated
as bubbles were observed to,rise to the top of the gas buret •. After the
addition (1 hour), which was quite exothermic, the grey mixture was
O· stirred for 1 hour at O, then decomposed at this temperature by adding
dropwise 100 ml. of ethyl.acetate followed by water (60 ml.). The
white, gelatinous slurry was filtered (difficult) and the filtrate ex-
tracted with two 200-ml,· portions of ether-benzene solution (1:1). The
solvent extracts were combined, dried (MgS04 ) and filtered •. Solvent
.was stripped from the mother liquor under aspirator pressure until
approximately. 40 ml. of solution remained. The solution was chilled to
cause deposition of a white solid. The precipitate was filtered out and

100
washed with 50 ml. of Skelly Solvent F. followed by 20 ml. of chloroform,
0 The solid (4.57 g.), unknown F, m.p. 196-198 ·was shown to be pure by
GLC analysis (CA-9). Only one compound was observed with a retention
time at 204° of 12 minutes.
The NMR spectrum (pyridine-d5) was poorly resolved but indicated a
singlet at 05.47 superimposed on a very broad hump at 55.25, three
singlets at 53.75, p,58, 1)3.10, two more singlets at 62,90 and 62,80,
a multiplet (hept~t) centered at 60.94 (J = 6 c.p.s.) and finally a
doublet centered 50.78 (J = 7 c.p.s.). The IR spectrum (Plate ·XII) was
simple but poorly resolved (see Table IV for analysis).
The filtrate obtained from the removal of unknown F was further con-
centrated on a rotary evaporator and gave 20.33 g. of a thick brown oil.
No attempt was made to analyze this residue. I~ analysis of the gas
evolved from the residue showed no peaks - it was probably hydrogen.
Reaction of Trike tone· XXXIII with ·Lithium Aluminum Hydride. . The
apparatus used for this experiment was identical to that described in
the previous experiment. Lithium aluminum hydride (1.59 g., 0.042
mole) suspended in 200 ml •. of anhydrous ether was placed in the flask.
The addition of triketone XXXII (10,0 g., 0.04 mole), diluted with 100
ml. of anhydrous ether, the reaction conditions, and the method of work-
up of the reaction mixture were the same as those described in the
previous experiment. No noticeable gas evolution was observed •. A
solid was obtained from the chilled, partially.concentrated organic
layer. When recrystallized from benzene, a white solid, unknown.G
(3.62 g.) was isolated, m.p. 121-i22°. · The IR spectrum (Pl1:1te XIII)
ana analysis are shown in Tables· III and IV •. All peaks observed in the
N:MR spectrum (pyridine-d5 ) were very poorly resolved. Broad singlets

101
were observed at 67,55, 66.12, 65,77, 54.03, 03.79, 63.39, a multiplet
centered at 02.13 (J = 6 c.p.s . ), another singlet at ol.33 and a singlet
at 61.13 with shoulders at ol.25 and ol.06.
The mother liquor obtained from the removal of this compound was
further concentrated under aspirator pressure and gave 6 . 29 g . of a
yellow viscous oil. The residue was not analyzed further.
The (1:1) Reaction of Triketone XXXIII with Potassium Hydroxide in
Ethyl Alcohol. A 100-ml. three-necked flask equipped like that des
cribed for Grignard preparations (page 59) was charged with 3.00 g.
(0.012 mole) of triketone XXXIII diluted with 40 ml. of absolute ethanol.
The solution was stirred under nitrogen for 15 mihutes; a solution of
0 . 672 g. (0.012 mole) of potassium hydroxide dissolved in 20 ml. of
absolute ethanol was added over a period o f 45 minutes. During the
addition of the first few drops of the potassium hydroxide solution,
the color of the contents in the flask became yellow-brown, but the
temperature did not rise. Heat was applied and the solution heated to
reflux, and the rest of the potassium hydroxide solution added to the
boiling solution. After the addition was complete, the mixture was
further heated at reflux an additional hour . The flask, which now con
tained a white precipitate, was allowed to cool to room temperature ,
then chilled for 1 hour in an ice-salt bath. The mixture was filtered
and the filter cake was washed with 10 ml. of ether and the washings
added to t he fi ltrate. After the solid (0.53 g.) was dried in air, it
was identified as potassium carbonate by comparing its IR spectrum to
that of an authentic sample. The mother liquor was poured into 50 ml.
of distilled water, and the brown mixture was extracted with two 125-ml .
portions of ether. The ether was distilled from the solution until

102
22.5 ml. (measured) of liquid remained. GLC analysis of this solution
(CA-3) disclosed the presence of a 4-component system. The first two
peaks, identified by mixed injecti6ns with authentic samples (standard
solutions) were due to: ethyl isobutyrate ((XLVIJ;_)_ (10%) and diisopropyl
ketone ':(XLIV) (43%); retention times at 86° were : ethyl isobutyrate
(XLVII), 0. 8 minutes; diisopropyl ketone (XLIV), 0. 95 minutes. Two
peaks representing 24 and 19% (peak area ratios) of the reaction mix
ture had retention times at 86° of 16 . 8 and 23 minutes, respectively.
The solution containing the residue was f urther c oncentrated by dis-
tillation to a yellow-brown oil which was dissolved in 5 ml. of ether.
Chromatography of the solution was effected on the Autoprep unit (CP-2).
Temperature settings on the unit were as follows : 0
column, 152 ; in-
. 0 0 0 Jector, 233 ; collector, 295 ; and detector, 312 . The helium flow rate
was 93 cc./min . Twenty-five 250-ml. injections of the ether solution
were placed on the column and materials corresponding to both of the
unknown peaks, having r e t ention time s of 7.3 and 10 minute s r e spec -
tively, were collected in the same manner as that described previously
for the collection of unknown E and triketone XXXIII, pa ge 95 . Spec tral
data (Tables IV and V) [ IR (Plate XIV), NMR (Pla t e XXXIV) ana l ys is j
identified one of the compounds as ethyl 2,2,4-trimethyl-3-oxovalerate
(XLV). Spectral data (Tables IV and V) [IR (Plate XV) and NMR (Plate
XXXV) ] also identified the o t her compound as 2,4 , 4,6 - t e t r ame t hyl - 3 ,5 -
heptanedione :(XLVl).
The aqueous layer was acidi f ied with 2 ml. of aqueous 6!i hydro-
chloric acid, and the r esult i ng brown s o lut ion wa s extr acted with 100
ml. of ether. The or ga n ic l ayer was dr i ed (Mgso4 ), fi l tered under suc
tion and the ether was removed by distillation until 7 ml . (measured)

103
of solution remained. GLC analysis (same column as used above) re-
vealed the presence of only isobutyric acid '(XLVII~) (3.5% - based on
lactone I obtained by alternate injections with standard solution). Re-
0 tention time at 89 was: isobutyric ac::(d'. :(XLVIiI) 1.0 minute.
In another experiment, triketone XXXIII (2.35 g., 0.009 mole) and
20 ml, of a solution containing 20.0 g. (0.36 mole) of potassium hy-
droxide dissolved in 80 ml. of methanol were heated at reflux for 4 '
hours. Workup of the reaction mixture from this point was identical to
that described in the previous run. Potassium carbonate (0.54 g.) was
obtained and identified by comparing its IR spectrum to that of an
authentic sample. GLC analysis (same column and temperature as above)
of the residue obtained from the distillation of the filtrate revealed
the presence of only diisopropyl ketone(XLIV). Analysis (CA-3) of the
residue obtained from the distillation of the organic extracts received
from the acidified aqueous layer showed isobutyric acid ' (XLVIII) as the
only compound present.
The (1:1) Reaction of Ketol XXXII with Potassium Hydroxide in Ethyl
Alcohol. The procedure and apparatus for this reaction [ using 21.50 g.
(0.084 mole) of ketol XXXII and 4.50 g. (0.084 mole) of potassium hy-
droxide] were essentially identical to those described in the previous
experiment. Filtration of the reaction mixture gave 3.61 g. of potas-
sium carbonate. The filtrate was poured into 225 ml . of water, and the
resulting mixture was extracted with three 150-ml. portions of 1:1
ether-benzene solution. The solvent extracts were combined, dried
(MgS04 ) and filtered under suction. Distillation of the organic layer
afforded an oily residue wh ich was diluted to 25 ml. with ether. GLC
analysis (same column as cited in the previous experiment) revealed a

104
5-component system. Compounds identified were: ethyl isobutyrate
(XLVII) (5%), d iisopropyl ketone (XLIV) (30%), ethyl 2, 2 ,4-trime thyl-1, 3-
oxova lera te (XLV) (18%), 2,4,4,6-tetramethyl-3 ,5-heptanedione (XLVI)
(11%), and triketone XXXIII (35%). Retention times at 86° were: ethyl
isobutyrate (XLVII), 0.8 minutes; diisopropyl ketone (XLIV), 0.95 min
utes; ethyl 2,2,4-trimethyl-3-oxovalerate (XLV), 16.8 minutes; 2,4,4,6-
tetramethyl-3,5-heptanedione (XLVI) 23 minutes . Retention time at 165°
was: triketone XXXIII, 17.3 minutes.
Workup (see previous experiment) of the aqueous layer revealed only
a trace of isobutyric acid (XLVIII) when analyzed by GLC (same column
and temperature as previously described for this part of the experiment
cited earlier).
In a similar experiment as described for the (1:1) reaction, ketol
XXXII (20.0 g., 0.08 mole) was combined with a solution composed of
40.0 g. (0 . 71 mole) of potassium hydroxide dissolved in 160 ml. of
methanol. Reaction conditions and workup of the r eact i on mixture were
essentially identical to those described in the previous experiments.
The reaction mixture when filtered gave 10.0 g . of potassium carbonate.
The filtrate obtained (containing ether) was dried (Mgso4 ) and suction
filtered . Removal of the solvent by distillation gave a light yellow
liquid. Further distillation using a short-patch apparatus gave one
main fraction (4.95 g.) b.p. 124-126/744 mm. which was identified as
diisopropyl ke tone (XLIV) by compar ing its IR spectrum with that of an
authentic sample . Distillation of the ether extracts obtained from the
acidified aqueous layer gave 3.90 g. o f a foul-s melling liquid, b.p.
138-140°/744 mm. The IR spectrum of this liquid was identical to that
of an authentic sample of isobutyric acid (XLVIII). The small amount of

105
dark residue accompanying each distillation was not analyzed.
8 Preparation of 2,2,4-Trimethyl-3-oxovaleryl Chloride(XLIX). Lac-
tone I (195.0 g., 1.39 mole) was placed in a 500-ml . three-necked flask
equipped with a mechanical stirrer, a thermometer, and a Dry Ice con-
denser. Hydrogen chloride gas was bubbled slowly into the stirred lac-
tone through a glass frit. Any excess hydrogen chloride vapors which
were not liquified by the Dry Ice condenser passed through the side arm
of the condenser and into a 1-liter flask containing approximately 500
ml. of water. The contents in the reaction flask turned red in color
after 30 minutes of reaction , and the temperature of the solution rose
7° during that time. The hydrogen chloride gas was passed through the
lactone I for 24 hours with the temperature of the contents iri the
flask slowly falling to room temperature (26°). The maroon-colored
solution was distilled through a 50-cm. Vigreux column, and gave a small
amount of forerun (5 ml.) between 100-170°, with two large fractions
taken at 171-180° and 181-184°/744 . rrnn. Both latter fractions were
shown to be contaminated with lactone I as the IR spectra of both cuts
indicated absorptions in the carbonyl region that were observed in the
spectrum of the lactone I. All fractions and the small amount of dark
residue in the distillation flask were combined and hydrogen chloride
gas passed through the solution for an additional 16 hours .
During this additional reaction period, a 1-liter three-necked
flask, equipped with a wate r inlet tube, a glas s stopper, and a Claisen
U-tubeiwas filled halfway with \ -inch glass helices • . A funnel was
placed on top of a 24-inch glass column bearing a side arm and the
apparatus arranged so that the Claisen U-tube extended hal fway into the
funnel. The water was turned on and its rate of flow controlled so that

106
only one or two helices· at-. a time passed through the U-tube and into
the column. The total time to pack the column was 5.5 hours. The col-
umn was washed with acetone and allowed to drip-dry. After the 16-
hour reaction period, the red solution~as distilled through the col-
umn. The column was_heated just enough that vapors rose to the top of
the column but did not distill. After this preliminary equilibration
period, heating was increased and the liquid allowed to distill. Three
0 0 clear, colorless fractions were taken at 169-179 , 180-185 and 186-
o O 79 187 /735 mm. The main fraction, b.p. 186-187 /735 mm. [lit. 85-86/
23 mm.] - yield 110 g. (49%), was shown to contain no,lactone I. [See
IR (Plate· XVI) and NMR (Plate XXXVI) spectra, Tables IV-and V for data
on _acid chloride XLIX].
Preparation of Diisopropylcadmium69 and-its Reaction Wfrh 2,2,4-
Trimethyl-3-oxova leryl Chloride {XLIX)~. Isopropyl Grignard reagent (0. 5
mole; 87% by titration), prepared in essentially the same manner as
those Grignard reagents previously described, was chilled to -10° in
a Dry Ice - acetone bath. Pulverized cadmium chloride (49.0 g., 0.27
mole) which was previously dried at 120° for 1 week was added in 2-3 g.
portions over a period of 25 minutes, under nitrogen with stirting.
The contents of the flask (grey in color) were allowed to warm to room
. tell).pera ture and turned brown in color and finally to dark. brown (almost
black) •. The dark mixture was heated at reflux for 45 minutes and then
ether was rapidly distilled .from the flask_ through a shor.t- path conden-
ser. When a slurry formed in the distilling flask, 350 ml. of anhydrous
benzene was added and an additional 75 ml. of solution distilled from
the reaction flask. 0 The dark mixture was then cooled to -7 (Dry Ice-
acetone bath) and 83.25 g. (0.5 mole) of 2,2,4-trimethyl-3-oxovaleryl

107
chloride (XLIX) dissolved in 50 ml. of dry benzene was added over a per-
iod of 0.5 hour. The resulting black mixture was boiled for 3.5 hours,
cooled to 10° in an ice-bath and decomposed by the dropwise addition of
250 ml . of 20% aqueous ammonium chloride . The resulting mixture (two
layers) was extracted with three 200-ml. portions of benzene. The or-
ganic extracts were combined and dried (Mgso4 ), and the solvent was re
moved under aspirator pressure to give 82.96 g . of a light brown liquid .
GLC analysis (CA-10) revealed 8 peaks on the chromatogram, 6 of which
were not identified. Compounds identified were: 2,4,4,5-tetramethyl-
3,5-heptanedione (XLVI) (29% - peak area ratio) and unreacted acid
chloride (XLIX) (43% - obtained from alternate injections with a standard
solution) . Retention times at 66° were: 2,2,4-trimethyl-3-oxovaleryl
chloride (XLIX), 15 minutes; 2,4,4,6-tetramethyl~3,5-heptanedione (XLVI),
32 minutes . The residue (10 ml . ) was diluted with 5 ml. of ether and
chromatographed on the Autoprep unit (CP-2). Temperature settings on
the unit were : column, 150°; injector, 245°; co llector , 250°; detector,
305° . Helium flow rate was 95 cc./min. Ten 85-µl. injections of the
ether solution were placed on the column. The compound which repre-
sented the peak having a retention time of 11 minutes we re caught in a
small collector tube (in a manner like that described previously - page
95) ~. The IR spectrum of this compound was identical to that of an
authentic sample of 2,4,4,6-tetramethyl-3,5-heptanedione (XLVI) •
. pre paration of Me thyl 2,2,4-Trimethyl-3-oxovalerate (XXXVI) . The
procedure used for the preparation of this sample was similar to the
method of Be rlin and Austin for t he pre para tion of diphenylphosphinic
9 esters . A 1-liter flask, equipped like that described on page 59 f or
Grignard formation, was purged with nitrogen and charged with 80.75 g.

108
(0.50 mole), of 2 ,2,4-trimethyl-3-oxovaleryl chloridei ((XDIX;) dissolved in
200 ml. of. anhydrous ether; triethylamine (50.60 g., 0.50 mole). diluted
·with 150 ml •. of anhydrous ether was added dropwise to· the stirred solu-
tion of acid chloride over a period of 1 hour. Methanol (22.40 g., 0.70
mole)·was then added dropwise to the·white, slightly opaque mixture over
a period of 45 minutes •. Stirring became difficult during the addition
of the methanol owing to the formation of the amine hydrochloride. Heat
was applied to the flocculent heterogeneous mixture which was kept under
reflux for 1.5 hours. The mixture was allowed to cool to room tempera-
ture and stand overnight and then filtered under suction. The filtrate
was washed with a total of 750 ml. of 10% aqueous sodium.bicarbonate so-
lution. The.organic layer was washed with two.700-ml. portions.of water
and then dried (MgS04 ). : .. ])'i,ltrat:Jon followed by removal of solvent on
a rotary evaporator gave 54 ml. of a light yellow liquid. -Distillation
through an 18-inch silver-jacketed tantalum wire-packed column. gave
forerun fractions at 65-90° (3 ml.) and 90-180° (4 ml.). The principal
0 43 fraction.distilled at 188-189.5 /729 mm •. [lit. 88-91/22 mm~]: yield
59.42 g. (76%) I~20n 1.4215; lit. 10 ~20n 1.4244] •. It was checked for
purity by GLC analysis.(CA-11) •. Only one peak was observed •. Retention
time.at 138°,was: methyl 2,2,4-trimethyl-3-oxovalerate_(XXXVI), 0.9 min-
utes. IR (Plate XVII) and NMR (Plate XXXVII) spectral data (Tables. IV
and V) further substantiate this known structure.
Preparation of Dimethyl.2,2,4,4-Tetramethyl-3-oxoglutarate(XXXVII).
The procedure used for the preparation of this compound was identical to
79 that given .in Eastman Technical Data Report No •. X-129. The quantities
.of materials used were: lactone.I (46.65 g., 0.34 mole), sodium methox-
ide ·(18.60 g; 0.34 mole).and 31.50 g. (0.33 mole) of methyl

109
chlo:toformate. The ester XXXVII was isolated (47.00 g., 62%), b.p. 110-
o .20 . 20 112/6 mm 9 n .D 1.4481, [ht. b.p. 97-98/3 mm., Ii D 1.4470 - 1.4480],
. as a clear colorless liquid. The IR (Pia te XVIlI) . and mm.·· (Fla te
·XXXVIII) · spectra are consistent with the structure of this known com.:. ··
pound (see· Ta,bles IV and. V) •
. Attempted:claisen Condensation of Methyl 2,2,4-Trimethyl-3-oxova-
lerate (XXXVI). with· Diisopropyl Ketone (XLIV) •. A •. Sodium Methoxide as
78 .· the Condensing Agento Sodium methoxide (25.056 g., 0.464 .mole) and
. 100 ml •. of dry methanol .were· placed in a 500-ml. flask equipped ·like
that for Grignard formation (page 59). Diisopropyl ketone((XLIV) (12.104
g., 0.116 mole) was added to the stirred mixture over a period of 45
minutes. . Methyl 2, 2 ,4-trimethyl-3-oxova lera te 1(XXXVJ;) (20. 00 .g., . 0 .116
mole) was added dropwise over a period of 1 hour. Neither the addition
of the ketone XLIV nor the addition of ester XXXVIwas exothermic, so
heat was applied .and the mixture was heated under reflux for. 18 hours •
. After standing overnight, the mixture was chilled .in an .ice bath and
. 350 ml. of ice water was added. The mixture was extracted with three
125-ml. · portions of ether and the ether extracts combined and dried
(MgS04 ). . The. organic extracts were filtered and the solvent was re
moved by distillation. The residue, very light tan in color,,was•analy-
' zed by GLC (CA-3). Compounds·identified by mixed injections with·solu-
tions of known .samples· were: diisopropyl ketone ((XLIV) (near qu~nt:Lta-
tive recovery),and a small amount of methyl 2,2,4-trimethyl-3-oxovaler-
at~ ~(XXXV!) .•. Retention times at 160° ·were: methyl.2, 2 ,4-trimethyl-3-
oxovalerate (XXXV.I)., 0.4 minutes; diisopropyl ketone (XLIV), 0.6 ·minutes.
The aqueous layet was.acidified and extracted with ether •. After
drying (Mgso4 ), and filtering, the ether was distilled .until 50 ml.

110
(approximately) of solution remained. GLC analysis (same column as
above) showed that methyl isobutyrate was the major component al6hg with
trace amounts of six other compounds. One of these was identified by
mixed injection with a known standard as triketone XXXIII, but this
compound was present to,an extent of only 4% in the reaction mixture •
. Retention time at 160° was: triketone XXXIII, 9 minutes. The other
peaks observed in the chromatogram.were not identified.
B. Sodium Hydride as Condensing Agent. 71 The apparatus was iden-
tical to that used in the preceding experiment. The quantities used in
this run were identical to that of the previous experiment except that
sodium hydride (3.84 g., 0.116 mole) was used in place of sodium methdx-
ide. . GLC analysis (same column and temperature as in part. A) of the
initial organic solution revealed the presence·of diisopropyl ketone
(XLIV) {35% recovery - from alternate injections with known standard
solution), methyl 2 ,2 ,4-trimethyl-2-oxovalerate (XXXVl) (near quantita
tive recovery), and diisopropylmethanol (50% - from peak area ratios).
Retention time at 160° was: diisopropylmethanol, 0.3 minutes. No
triketohe XXXIII was detected by GLC •
. C. Sodium Amide as. the Condensing Agent. 41 Same procedure and
apparatus was used in this e~periment as in the previous experiment ex-
cept that 3.12 g. (0.081 mole) of sodium amide, 4.56 g. (0.040 mole) bf
diisopropyl ketone (:iCUIV} and 14. 00 g. (O .081 mole) of methyl 2 ,2 ,4-tri-
methyl-3-oxova le rate ((XXX:V:[) were the quantities used. GLC analysis
(same column and temperature as cited in parts A and B) of the initial
organic solutions revealed. the presence of only diisopropyl'ketone (XLI:V,)
and methyl 2,2,4-trimethyl-3-oxovalerate (XXXV]J .• GLC analysis of the
extracts (ether) obtained from the acidified aqueous lqyer showed no

111
trike tone XXXIII present, There were several• sma 11 peaks observed in
the chromatogram but no attempt to identify any of them was made.
R . f D . . 1 K '(X IV) · h S d · H d · d · 7 T · n • · eaction o 11sopropy etone. L , wit o ium. y ra:: e •. -. 1:1.1-
isopitopyl ketone :(XLIY) (4.00 g., 0.035 mole) dissolved in 25 mL of an-
hydrous ether along with L68 g. (0,070 mole) of: sodium Hy;droxide were
placed in a 50-ml. one-necked flask equipped like that described for
the thermolysis of ketol XXXII (page 98). The mixture was heated at
reflux for 16 hours .under nit'rogen; wi'.th. the color of the contents in
the flask becoming dark brown •. After the flask had cooled to room
temperature, the mixture was poured into 100 ml. of ice water, Normal
workup followed,
The aqueous layer was acidified with 6.5 ml. of 6~ hydrochloric
acid, with the brown color of the solution vanishing. The other ex-
tracts (150 ml.) were dried (Mgso4 ), filtered and distilled until
approximately 5 ml. of solution remained. Analysis of this solution by
GLC (CA-10) revealed diisopropy1methanol. as. the major c.omponent (55% -
peak area ratio) along with a trace amount of diisopropyl ketone (XL:CV:) .•
0 Retention time at 91 was: diisopropylmethanol~ 0.8 minutes~"
Attempted Ring-Closure of Triketone · XXXIII. Trike tone ·XXXIII
(1.00 g., 0.004 mole) was placed in a 50-mlo one-necked flask equipped
like that described in the previous experiment, Sodium methoxide
(0.432 go, 0.008 mole) was added along with 20 ml. of anhydrous ether
to the flask and the suspension heated .under reflux for 11 hours" The
brown-colored mixture was allowed to attain room temperature, then
slowly poured into 75 mL of ice water" Extraction with three 75 .. ml,·
portions of ether was followed by combining the organic layers, drying
(MgS04 ) and distillation until approximately 5 mlo of solution remained,

112
GLC analysis (CA-12) revealed the presence of only triketone·XXX:Illo
Retention time at 169° was: triketone ·XXX:III, 7 minutes.
The aqueous layer was acidified with 5.6 mlo of aqueous 6~ hydro-
. chloric acid and the colorless solution was extracted with three 75-ml.-
portions of ethero The combined, dried (MgS04 ) organic iayers were dis-
tilled until approximately 5 ml. of solution remained. No precipitate
formed when the solution was chilled. GLC analysis (same column and
temperature as above) disclosed the presence of many components in the
reaction mixture. One peak was identified, by mixed injection with a
standard solution, .as arising from triketone·XXX:III. This compound may
have arisen by poor techniques in working up the reaction mixture, or
ketolXXX:II may have been present in the reaction mixture and produced
triketone XXXIII as a decomposition product when GLC analysis was per-
formed on the reaction mixture. Ketol XXXII when analyzed for purity
0 by GLC (CA-11), column temperature 158, gave a chromatogram repre-
sentative of a decomposition pattern.
Ring closure of triketone XXXIII (1.00 g., 0.004 mole) was also
attempted using sodium hydride (0.192 g. 1 0.008 mole) as the base. The
reaction mixture was worked up exactly like that from the sodium
methoxide run - page 111. GLC analysis (same column and temperature as
described above) revealed the presence of triketone·XXX:III in the
initial organic extracts. Analysis by GLC of the organic extracts ob-
tained from the acidified aqueous layer showed a multi-component sys-
tern. Triketone XXXIII was also identified as present in the reaction
mixture, and the same explanation for its appearance may also apply
here as in the sodium methoxide run - page 111.

113
Preparation of Hexamethyl-1,3 ,5-cyclohexanetrione. (':cI),.. The pro-
cedure used for the preparation of this sample was essentially the same
79 as that given in Eastman Technical Data Report No •. X-129. A 500-ml.
one-necked flask equipped like that cited previously for the acid hy-
drolysis of ketol XXXII (page 95) was charged with 140. 1 g. (1.0 mole)
of lactone I and 2.28 g. (0.042 mole) of sodium methoxide •. Heat was
applied until the contents of the flask reached a temperature of 90°.
At this temperature an exothermic reaction began and the reaction mix-
ture boiled violently for 10 minutes. When the reaction began to sub-
side, heat was applied and the brown-colored reaction mixture held at
reflux for 1 hour. After the contents in the flask had cooled to 100°,
the mixture was poured into 1 liter of water and the heterogeneous mix-
ture was rapidly stirred, Filtration gave a solid material consisting
of white needle-shaped crystals and many pieces of a brown polymeric-
like material. The brown substance was removed from the white crystals
with a pair of tweezers (tedious) and discarded. A solution of the
white solid in hot ethanol gave a white precipitate upon the addition
of cold water. The precipitate was washed with hot water (100 ml.) and
dried (atmosphere) to give 94.32 g. (68%) of hexamethyl-1,3,5-cyclo
hexanetrione{II)~ mixture m.p. 79-80° with an authentic sample.
13 Preparation of Isopropyl Grignard Reagent. ·A 300-ml. three-
necked flask equipped for Grignard formation (see page 59) was charged
with·2.40 g. (0.10 g.-atom) of magnesium shavings and enough ether to
cover the metal. .Isopropyl bromide (12.30 g., 0.10 mole) was placed in
the addition funnel. From this point the procedure and reaction con-
d~tions were identical to that for the preparation of phenyl Grignard
reagent. The yield was 0.078 mole (78%).

114
.·Attempted.Condensation of Isopropyl Grignard · Rea.gent with, Hexa-
,methyl-1,3,5-cyclohexanetrione (II) in t:he Presence of Magnesium Brei-
'd 76 ·mi e. A 1-liter three-necked flask equipped for Grignard formation
(see· page 59) was· charged with magnesium E;havings (2 .40 g., 0.10 mole)
,and 85 ml. of anhydrous ether and bromine (16.00 g., 0.10 mole) was
added dropwise to the stirred mixture. A vigorous, exothermic reaction
occurred after the addition of each drop of bromine •. After the addi-
tion (1.5 hours), hexamethyl-1,3,5-cyclohexanetrione(II) (20.00 g.,
. 0.10.mole) dissolved in 90 ml. of anhydrous ether was added .to the
brown magnesium bromide solution, under nitrogen, over a period of 1
hour. The resulting mixture (two layers) was stirred for 15 minutes,
then isopropylmagnesium bromide (0.078 mole; prepared in the previous
.experiment- page 113) was added slowly to the mixture over a period of 2
hours. The addition of the Grignard solution was exothermic as, evi-
denced by the gentle boiling of the mixture. After the addition, the
yellow mixture was stirred overnight at room .temperature, then heated
under reflux for 12 hours. The mixture was cooled to 0° in an ice-salt
bath and decomposed by the dropwise addition (to control foaming) of
125 ml. of 20% anueous ammonium chloride solution; the ether extracts
,were combined,. dried (Mgso4 ) and filtered under suction~ .· Distillation
of the organic layer afforded 19.86 g. of a .yellow liquid •. The liquid,
.when analyzed by GLC (CA-10) was found to contain starting material
(ketone II) ·-92% - peak.area ratio, .and three other components •. One of
the peaks was .identified by mixed injection with an authentic sample as
triketone XXXIII (4%). The other three peaks,were not identified. Re-
0 tention times at 165 were: hexamethyl-1,3,5-cyclohexanetrione (II), 9
minutes; trike tone XXXIII, 15 .5 .minutes. Chilling the. yellow liquid

115
afforded a white precipitate which could not be further purified by
- recrystallization •
. Preparation of• Lithium Dust. The procedure - is essentially the same
as _that of Bartlett, Swain arid Woodward. 3 A 500-ml. three-necked flask
equipped with a high-speed mechanical $tirrer, a condenser with drying
tube (CaC12), and a nitrogen inlet tube was charged with 110.ml. of
mineral oil (Fisher paraffin oil, white and heavy), and 2.0 g. (0.28
mole) of freshly chipped lithium. The contents of the flask were heated
until the lithium began to melt; the mixture was stirred vigorously un-
til the metal dispersed into droplets. Heating and stirring.were dis-
continued and_the contents were allowed to-cool to room temperature.
The. mineral oil was removed by sucking.it from the flask (by means of
a .vacuum pump) through a glass frit. The lithium dust was washed with
four portions. of Skelly Solvent F (100 ml. each)' and the Skelly Solvent
F removed through the glass frit. _ The lithium dust was finally sus
pended in 100 ml. of Skelly Solvent F and used in the next experiment.
,Preparation of Isopropyllithium. 25 Using the same apparatus as
described .in the previous experiment, isopropyl chloride (9.8 g., 0.125
mole) dissolved in 100 ml; of dry Skelly· Solvent F was placed in the
addition funnel and added dropwise to the suspension of lithium dust
(0.28 mole) held at reflux in the Skelly Solvent F. After the addition
(45 minutes), the contents of the flask which were charcoal-black in
-color,. were heated .at reflux an additional 0.5 hour. At the end of this
- period, an exothermic reaction began and the mixture boiled gently.with
out any external heating for 0.5 hour. _After the reaction had ·sub-
sided, the mixture-was stirred for 1 hour. Titration (as in experiment
1) indicated a yield of 72% (0.072 mole).

116
Reaction of Isopropyllithium with Hexamethyl-1,3,5-cyclohexane
trione {II).,. A 500-ml. three-necked flask (equipped as . in the previous
experiment) was charged with 20.00 g. (0.10 mole) of hexamethyl-1,3,5-
cyclohexanetrione (II) and 100 ml. of dry Skelly Solvent F; the contents
of the flask~ere thilled to -30° in a Dry Ice - acetone bath .. The iso
propyllithium (0.072 mole - 206 ml.) solution prepared in the previous
experiment was added to the contents of the flask over a period of 1
hour. The addition was slightly exothermic, and the color cif the mix
ture became grey .. After the addition, the mixture was allowed to warm
to room temperature and was further stirred for 12 hours. Water (175
ml.) was added dropwise to decompose the grey mixture (not exothermic)
followed by an extraction of the mixture with two 150-ml. ·portions of
ether - Skelly Solvent F solution (1:1), The organic extracts were
combined, dried (Mgso4 ), filtered under suction and stripped of solvent
on a rotary evaporator to give 19.90 g. of a light yellow liquid.
Chilling the liquid gave a precipitate, but recrystalliz~tion attempts
using a number of solvents (benzene, ether, acetone, chloroform) gave
no pure product. GLC analysis (CA-10) of the reaction residue showed
starting material (ketone II) present (53%), triketone XXXIII (8%) and
two other peaks which·were not identified. Retention times at 162°
were: hexamethyl-1,3,5-cyclohexanetrione (IT), 10 minutes; triketone
XXXIII, 16.5 minutes. Yields of triketones II and XXXIII were based on
a comparison of peak area ratios of the compounds present in the reac~
tion mixture to those obtained from standard_solutions of these com
pounds.

117
Condensation of Methyl Grignard Reagenth with Hexamethyl-1,3,5-
Cyclohexanetrione (II). Hexamethyl-1,3,5-cycQohexanetrione (II) (20.00
g., 0.10 mole) dissolved in 125 ml. of anhydrous ether was added drop
wise to the stirred solution over a period of 2.5 hours. The addition
was slightly exothermic, and the color of the contents in the flask
initially were lemon colored, but at the end of the addition had changed
to milky white. After the mixture, containing a white precipitate, had
been allowed to warm to room temperature, the mixture was heated to a
boil and was maintained thus for 12 hours. The flask was chilled to 5°
by means of an ice bath, and the.mixture decomposed by the dropwise
addition of 100 ml. of 20% aqueous ammonium chloride solution. The
ether extracts (300 ml.) were combined, dried (MgS04 ) and filtered.
Distillation (removal of solvent) gave 19.91 g. of a yellow liquid.
Chilling the liquid deposited a white solid which was filtered out. The
precipitate was recrystallized from benzene-E_-heptane solution (1: 1) to
give (after drying in air), 14.84 g. (69%) of a fine white powder, IR
(Plate XIX) and NMR (Plates XXXIX and XL) spectral analyses allowed the
compound to be assigned the structure of new ketol L (see also Tables
III, IV and V). GLC analysis (CA-10) of the filtrate obtained from the
removal of this compound disclosed only starting material (ketone II)
present. Retention time of ketone II at 160° was 10.5 minutes.
hibid, page 90.

5000 4000
100~
90
80
70
60
50
40
30-
20-
10
z:
3000
Plate I
WAVENUMBER CM·1
2500 2000 1500 lAOO 1300 1200 1100 1000 900 800 700 650
a:x'g:c'W:i'J#i_gJ;tlg~!ffiw~1Jt£-1'1m --,--f= ~-=;-E"j=:J:..:::i=j.....-4-- -!-f-,,,:._,
"cE';--. ~.=:~ T:;:=i= ~ TI ~:,~~~1~;~:~· ~~ ~~::::4?t~~=1 1· :_~-~~~-~1--~-~--~ -:;2.~-~~~-~~~_; - ~:-::d±~~-P=t--:_[!~:=~_:.i::!-~ -··tt-~-::i===:c:=J~I~~- ·--l::~1:; -- 1 '··::t=~-r-
:\----:'.-:.::i-::i:..... 90
l=i=:3,.::~~:1=:+a:¥ ~~ ~, :;:::j=r __ ;:_ -4-=:r:'.~__q:::::::l._~---l-::;:: __ ,·*3± -------~---~-~-:-~=i ---,...~.=r+-~ .. FL-:--'-+ -~L..;--=
----1:..:;= ·-±_.,. ~ ~ _,_._ ~--,=__.- -- _g:::_J~~-3= J~::;~33=~:· .::i :- ~ .. r·I-· ~- ---~ -~~ ~::L-:t.. EE§E-~-=---t=E :·~: _,~-=±-::.~~- ~ ~3: 80
~--; ;~~~il~t!;i~ii~ii~§ail: E- -==~-ITfi ta==1-ffj !!!~~= ~~~r!~~~ ~;;r:~:{!~t~;~'~tci~~:j~!~ r:1;=.;:_:--t:~t~~!?=:=~~ 50
=:.--- ::;~ := ~~) : ~ =~~:; ~ :~H- ,<:=~~I~~~~~-~ Ji~ :'-1;~~- ~'~ - =~"F-~~ ~~='.= ~1j~l 40
-c---±::=-~ -=-~-:=4- ~1. ~- ~t=:=J=-Ff:'.= · ~~~~ i± =f-t~~'= :. $- .::;±..~-
----y+ :::::;::;
·;=.'=1 ~'=:=!=fc; ,__._f:::: r~.
:'='=:! F 30
=1=l 20
~
~ 10
10 11 12 13 14 15 16
WAVELENGTH IN MICRONS
3-Hydroxy-2,2,4-trimethylm3-pentenoic Acid 6-Lactone (I), Film on NaCl Plates
I-' I-' 00

Plate II
,.( WAVENUMBER CM 1
5000 •IOOO 2000 1500 l.400 1300 1200 · 1100 1000 900 BOO 700 650
'):':' 'j':c':cJl lj!Lf ~!:!ff l;j;I} lf':!_}/Jtt~~-IJ_:lc',I, ~}~~~~~~
-,--i P1~:~e=·~tai=li;~~~~i,,3 t,-· l ~--j~,--
~1$ r~·o .~~ •o § <--~- IPlflil :=b-~ft§_ -~~~=R--
.~--.... ~ 100
= -~__,_.....,_-t··~
T- •• ~~---~r:3c-,~-~= == ;:~==--==. -"'-. ;,~~ ccc,~~~".c:= __ =?=~ .. ~:--~~'~:tl-""F~=-~;:~,•o .:..-:a:-~ ·t.==.~~- -ic-i---±=1-=:::±J=:=..+::::l::::
, - '§o=G~~ :_ - ,~ _. j/_t~;_:;:~ ~-:: - -~:~----'_:~=::•, <:'- - -•~ -,-~2-~:,::·C~c'::::~;:;~ ~;:}~t~ E~:: ,.~:~~;,~:·~:;_\ :;~ 70
• 0 ~-: J~;t~l li~~ ~=~: ~ct:=, L~n- '1~ !:::2 t;r-~~··:; ~~!:}: ,f:!' f;I ?1: .:;t) ~~;·;.• ft~t:Jf;: 1J::I' t;!~ ~:~r~:t ~:~: :!;] 1\I -): ~ .. 1:t;t 00
so -,-'~ ~-_::·~- ,~ "F.' -::- : __ :-c-,:c:'_ ~~ ij{:_ ~[~ ~~ il' _·. :l 7 1 ~f · ;, ~• } ~r~ •:_!_.:- ·t;!;. ,~~ ... : \\ ?J~I :!: ~?~:;:: :=~~~ ~::=;z.~::, t~~: -~~1~I1"l:-~~ so·
,_ -" - -. ~ i::~~~LJ~. -~ -~~. v~~:~: ~~l~: ~~~~~;;:~~~ ~: ~--~,!; -~;~ ,_ t~;~~~~:~=~~:~~:~:,~::,~~~:if ~~;, :: 40 =~=t= =c_~=,-
·---- ---- r---E¥.2:=t=:~ ~~=<1.: ' +Jic;"f; 30 g~==J~ ,
10 ~
.cc-'-==t· ---
20[~-~ ~ . --· =='!C:,:c,c' ::---:u-1~:~·.:::: .U,.c=J::.-c=-= ==-:E..l____i:::::::: ~
·.?d,- ~~c:-:..:.:::j.:::=_:: :1:1~·:"'::::-~~===:: __ "= 0 ',·cbcc±_: ~-~,t~=f!-
20
·-··- 3 2$=!-:-_ fF~ ,, --"-~:±:dc:~'~F~l~~~ ===t
.,t .C1-;-
~i- ~---- ··~-=~=, t:: ~~J£+~-~-1ii~11-r~~-1 <~:?I, <~~ ·.·:±-1--- ·t-:::r:::::::::.--, 10
ot=F'c=: -- . ·--H.=:-=-~~,- - :-..::1=1 ~=i~--· =-'I= £=~~;:~;:~~.;,:!~' ··~-~;:, .. _-=::r.:
10 11 12 13 ,. 15 16
WAVELENGTH IN MICRONS
5-Hydroxy-2, 2 ,4 ,4, 6, 6-hexamethyi-5-isopropyl-l, 3-cyc lohexaned ione (XXXII), KBr Pellet
t-' t-'
"'

Plate III
WAVt:NUMBER CM· 1
5000 4000 3000 2-soo t200 1100
1:3c,1c!:_c!fl 100
90
80
70
60
so
40
30
20
10
10 11 12 13
WAVELENGTH IN MICRONS
5-Phenyl-2,2,4,4-tetramethyl-3,5-dioxovaleric Acid (XXXIV), KBr Pellet
14 15
650
lTFflf] 100
90
80
70
60
so
40
30
20
10
I 0
16
t-' N 0

5000 4000 3000 2500
~ 100
90
80
70
60
50
40
30
20 :t:·-----+---
10
0
Plate·IV
WAVENUMBER CM-1
2000 1500 1400 1300 1200 1100 1000 900 800 700 650
+m=·=,-
~!_::i
~~~~~-+-+-++-H.=t+e,
.::::iI.::.::r:11---+::'..:::r..::
::::;:=:I~
"' §
::::i
--
10 11 12 13 14 15
WAVELENGTH IN MICRONS
l-Phenyl-2,2,4-trimethyl-l,3-pentanedione (XXXIX), Film on NaCl Plates
90
16
.1-' N I-'

Plate V
WAVENUMBER CM· 1
5000 4000 3000 2500 2000 1500 1300 1200 1000
100
90
80
70
60
50
40
30
20
10
0
l=c,c~::+"-=='C'~'C---l,c :·.~c.'1--' : '"4 '"'='~t'~c:c=~+-",', '1 '' :~J: ' ' ' 1 ''=_~...i.:_..::._.;::.:..ii'·:-- --l'-"'==f=·' ':C'.f=~=--:J-=="'=3.c'cc"'''tc:-=:::ij -- -- <_l_: : : --~J.L_..:.; '.I 10 11 12 13
WAVELENGTH IN MICRONS
2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxononanoic Acid (XXXV), K.Br Pellet
650
14 15 16
100
90
80
70
60
so
40
30
I-' N N

Plate VI
WAVENUMBER CM-1
5000 4000 3000 2500 2000 1500 1400 1300 1200 1100 1000 900 800 700 650
CT'F,lT~i" ,w.,-.c l_';c'J'J' lB'J}: IJ-P' -i'?!'i- f]I;•i i'JlfL 'i,l'l ?'tn :f'r:'llf f=l;iJ=i'i :':. -· -~;1~gT :fft.1c , · '" '';jJ: .:jj '. " ;_c:cj_cj ; LU~ ;;Ti U=i:1 "-.J1=1, Jin= , ±i -,:' 1 c:r.~· = 100 100
90
-.:i-::-;
= '""~ 70
so
,-:..- _,.;_;_r_;.}'~3-_-'·:..::<--:.:~[.;c:"=:' '"t~,.t~:~:c::c:-;Sc_,--!E'-'tb:·" \l!~ ''"' ''Li , .. ·;' , ' i',' ;,,, j;jf, li'i ;;
30 ;; ~c;J± ~:~;~~t:fti0:i+i=~-}~~:J·:; -~~ifl~~l--, ~-~::; i:~,~-~\_;~~~L1(~~ ; (i , u: : : · i 1 '. '.>~·1:_; 1-:~: ;. ,= ,;JHJ ~Jr+ 1itt~] ·0
40
20 tc·==l= ~~~!~ 1ft'. "~~" ::r:!:~~~~ t=~:\2::~ -t;;~.i:~~ Ef=.~~~~-~~--~ =~:}-, :1, ,:,;:· r~l r~:t: ;r:; Ji l f , l !X' '.:· r :. ~,1itJ:1~ifc: :,-=r= 20
10 =! ~~ ~:=;f ;j~~~~;~j_ ~i ~~ ~~~~ ~?..:c~:~~~-:=_~ ~~=-~ :~_; -~-, ;; ~.:-;;'. tEl- ·jj j I- ; ; ;_; IJU I iHIA~-lYJ&~~ ~~~~:: ~~~= ~- ··.:c-~ ~~-- -~·,=i'·· ---·-" ·:·:::;·.·:·, - .. ,.; c: .. :,::::::: .. "~ .~ 0 c."T'::;,,,c:.:· =-'•, ''i·~tb_•;HLi-J~.j~T]dlE'·t=IC,=!;;,J=;=!+tFi=t"l=t-+'i"
10
±:E? =A..c;::~-.:r~ 1---±±-'--'- ~~--- • c. c:..""l-=L:c:c"' ,_. ~ ·-· I ''i ~~ u.n:1 J+-:;~--> = 0
10 11 12 13 14 15 15
WAVELENGTH IN MICRONS
Methyl 2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxononanoate (XXXVIII), Film on NaCl Plates
t-' N w

Plate VII
WAVENUMBER CM·1
sooo ,oao 3000 2soo 2000 1soo Moo 1300 1200 1100 1000 900 aoo 700 650
100 ~?~ ~u:~:~~'-"!'."';;{1 ·~·~:tY~t['(.~ )I; 1~-~L~ Itt'l'UJ I~-;~/lt'J 'J~I ,;;~·L;] lltLtJI; :i!;tI;~;. 'Jl[X _;\~\ ~r; _~1 ~l- ~t~.::ic=l i~ui· rF 100
90 9~;1;:"=:-:::--i~J~ :;~_,'-~Ti- '.:;/-~}ft1f_:~i--,,1c;~~,:~:-t_I{;{}}~t~~:~t-~: '}~{,l}ff _:ns-;_i t-Jt~-;:tJl- ,;:tt: i~:~! I:~ftr!~J1~: 90
: : '1;i~Lfil, c,,f ;'"' ..... rt!\ ;m ;;;: :;r, Il1 rnr Tl: ~it -1:~i}t~~ iJff r:3, ';2 m: .f if 111Lt181 :: 60 ~,- ~~~: ' 'j : if: ~: i, ' ' L,: : ; ; ; ! ! r I : : ii /:_' :_:: ! : ; : : : !;l }z I~ :1 /' i ~~ ! :,:,: : : : : ) : 60
50 L'I_'_ 1~t\L\, <~,_ ,, ii.i-_:_ -,, "'-)-., _, __ 1:i-h ,'.1: '. ,- ;_~, n:_:- -· ,, -----L-r·-·- '1JJL/iJ11,j.! },~_ ~!{: .:-i/i, }.=.~:--= ,_, 1' ::,::, L J .1_:: i \-: c 50
: ri1:rnjf ;.;;f'.'t1 ·: ,nr n: 'J ';;: 'Tr~::~.''!,} ]!1~~:Il! rr:£r L ,n; ;m ~111:fll fl~t:
:}\~~fu~f~tiit";}i::~jJ ,; :~.;);'Cllft;:;~~i~-;/~tlffff}}' ·rti:;)j;~: 5 6 7 8 9 10 11 12 13 1.4 15 16
WAVELENGTH IN MICRONS
2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-nonanedioic Acid (XLI), KBr Pellet
t-' N ~

Plate VIII
WAVENUMBER CM· 1
5000 4000 3000 2500 2000 1500 1400 130"0 1200 1100 1000 900 800 700 650
----~= ~ ~:-cct~'il~:~~'"':r~;: :}:::t?~r~-~ !t? /L'i-lJ/!'; f:'-.111i t: '.';:': T0\.tnl. nt~ ii~I n~t= ,oo
--,c=~=~_-c,,=~~;~c;,;-:t:~:t;~:==~:~~~f:~~~:1:1~:,~2:J :.~-- ,,"ri~=~ "., ,; ., . :'Ji''.:! '.;:'.1;;;-;~TT~J~~:-1~·0
- ,< ,~-,.-: '.'-rv~::;~.J±]~[i:Jr~: .,... j't~Tµ C)J?;H? 1mm iJ: 335"'--'-"f 100 ~-~
I I
+---'--+-~----,-~
90 --•-+ --=±::::::-i-=-i:
BO
70
-~::t,,:- ";_': s.~ -~;:-'tt;JS?i i~~; iI;-r td ' 1-. t • , i' " ' " : : !" : ;:; l ! F : fr:: 60 :£U+
50
40 l:L- ~/_"~~3~~ ~;-"Ii j'.:;_~-:~~- !,.~l~~i;:;~~:'.___::tt ~\i < • -, •·•·• ••.• • : ]: ::: 1? LL; ;-;]}o ,~--'=,,r -·:.:;·~~~-:·
30 ~J,::;:-e,,.::~~;:-~1~-~~;l~.;l~:!=--,.,:~=--~-.1; ==,-~=tc:;;/-''•:ci ;. <-,1
\ _\ ::,: :::; :;;: ]J;; 1] :~J,o · :t =~j '~'.~~,--~-'1.:~," ~.r>~~l/~ :~-~=~ -~. 1~ ! ~~ ~=- -~=:~1 :: :·i__ · -· · .;t;ttr Jt:: f t::t D11 1 El~ 20 20
·=+-~~.::::::!. r
~-¥- -~ -~'1,,-o= ,_ :==~:~~-~==,~::-: _ _ --= _ " ,; .;~.~t :.;;.is,~~;~ -:i]Ir ~!JI 1:+L~ 10
~,_.,_ ·==t=F _,.... ~~~"- - - -- -,,- 0 ,_ :,:~,- ::=-n -i,h T'=Lr --•,' cgi~ li,;)=;=j=f!-.'b, n
~~••· "-'l• _ ~ t:1=_:-=- _ __.._ -=·~·~,_- :_j::-~~~:,-:--·,:>',•q:cee3g::~,:-jJl}_;{J'jj_;_j,;:::.=-lj-+1ti41±:l=f- -
,o •• ~~~ -:- _,______::-. -::'.=-+f-t-=~S=cc r=::- =i::------±::::=:::=:::::::::~-::.
'~-=t--=c
0 -+:.:==: :-:c.::::::;: -~
.--:-:~~-=+- :,:~ .;:_-::::. --~::::r=-· ---"~c,1=::.:,c,~c:c--,,;.c
10 11 12 13 14 IS 16
WAVELENGTH IN MICRONS
Dimethyl 2,2,4,4,6,6,8,8-0ctamethyl-3,5,7-trioxo-l,9-nonanedioate (XLIII), KBr Pellet
I-' N u,

5000 4000 3000 2500
~ 100
90
80
70
~ . 60
50
40
30
20
10 +--....:::::
Plate IX
WAVENUMBER CM·'
2000 1500 1400 1300 1200 1100 1000 900 BOO 700 650
ic-.J=R ]"~ icll~3.'fcl -:1 ---1~·~ ~~~-g; ~$= ~~-~~ =lffi_ =i''-.i'h---= ~~, =i=E ~-- ___;
~i--:---.--l-, :_::·---~--:- ·-· ~ .:::~~--±-~-·+:==:~-= ~·::¥1::~:=:± ~f :_:!:3f:"-=.=E7-~:.=.~- '. ~~~~ ~ a~-+--+-+- t-~-..--- -r-::'§t-~:-~.-=t::+~EF.-1~·.:-..... ; .:.::E~--:f--~~ ~:q.:_~±~;=±-·::-:-:t:i-.:] -t 1-fJ···]::-~ ... _ ~- R=!_--:~~~~J__ .. j~:pj~~:=!=J~
::i--f= ~~-i=B~; -=i·-=f=t=t=~-:f.? --~=-~:~£ :.-::-:-:;-=~~- ::r-~~-- -~~:.:'.~: ---t;.--:~: .-=-:-t"t:1: ·~~-~~-:i: -=~-::J-:;-::= ~~- -~1·. l_ -=;~,: ~t~=¥ .1::r-1.: -::Ee:=-:-;=~= ·-,-a=i 90
:""~~ == -~,-+---~~~~~;~;'~~s;~~ ~I\;:~~~~z:~/{;-~:;;,:~t~:'.=:-!=~r:~,~~ct-~* BO
" h-'.-Cc~;~ ;ft~'- ,~:::~;~~tt~rt~I:r-- ~~/·~ _1-:;~1~ 't)}r-;:~r'~~;~Jt ~:~,~~aj:;;:;~-:~; 70
~~~~l~~:t )tf ~]~"~ :r:: ~:g;- .}]: t c;~ ''.t,\'.~>''~~::t;: ):f-}; 1;r1~;~J, J,t~t~~:\ ~tr;; ~~t~!~ li~~ ~:~:f; 60
,~~~ ~~ ~:i~-~ ~~· ,~~ · ):: ; ; -:::~1:~ tlI~~~.::;~ ~~~~~=~~: :_;:; ~J\~.~t~tL;~I so
~' =----Hl~--~~'-~;S~ :~-i~ ~~--;k=~~·=t=~;~-~;--- ±~-~!~~t~=~ '0
Fl~_:~-:-:~':.\===-!=~~ ~=+-, ·---1. ~~ =x=:-:i::=·~::~~=2:-' -~-~~ -~~ -~ --!=: -I--~ 30
.. 53~- ..t:: ---~
-~-cfil§~=cc-, "~~~:_:- . '----=-=Fee, -·· EE ,=l, 20
--
10 ~1---=--J=ifil4§f!b'.:.cfil~=~W-"=j41.+'
~ ·'+r:::
~"F..."I= F_t, S.~tlll¥Sfil£f&n~ =i=r-.:::::..r. ,-ec5:r=?_
~F~~ic-~rc ~~ ~:
10 11 12 13 14 15 16
WAVELENGTH IN MICRONS
2,2,4-Trimethyl-3-oxovaleramide (XL), KBr Pellet
..... N
°'

Plate X
WAVENUMSER CM·1
5000 .4000 3000 ·1100 1000 900 800 700 650 f400 1300 . 1200 ~-~,-;-,r.n...,- 11 ·111 I 1·11: : I • t I I'-• I :':-:l :··1 TITIIE:C"·,' l ''H Er'-! "u : : : -~ ··i)~_ i: _ .i :t.j: ~: .. -.i--=-~ :i~j. 100
70
···,•; . ' .· ].\ ' JI A I . ,, -. ., - ' -60.
0 [ii=i{F, . ·.1 ... J · · •vi· :-I . • 8-±-b · l ! I, : 4t·: •·I• · -•:I: •:: tf r: I:: id ;/~~'.+~~tli~ifiii 10
'I. 10~
J.· ..cc :Jt::.:d ,_ <:ht':--± 7· 10 11 12 ll ,. 15 "
WAVELENGTH IN MICRONS
Unknowri E, Filni on NaCl Plates
I-' N .......

5000 ,ooo 3000
Fff'ttt!l' I 'i Ii' l'!f-#-!-i 100
90
80
70
60
50
40
30 ~·~~~
' -~-~--J#j=I= 20
10
Plate XI
WAVENUMBER CM·1
2500 2000 1500 1400 1300 1200 1100 1000 900 800 700 650
@tf'4[3:c4 ~ ~ ~~~ ·-1-t'±:-t
,_· ::::p::::t - _=i--~~~=b--;--8=:i_~ ~ +-~ :~ _, ...... ~1- l±k -~·r--"-=t--~-.:i p::!
~F +-"t--r-. _ _,_ 1-:;_._r---:..-:::-~-~-~--r:-~:2f~~~--·_.· -~~--i_i:::::t:-tE -:--ft-J~;f~.ff~±-:i:3 j~ ~~~1E-~~~?r ~~~ -2:.· !2 · =i:~1-=~3 :··-~::; .. :·-~=tf-L~-.--~::-:E-=IE~-- -'.--j=~-::r:_g=:-Ef= '~¥:::.:=+±: --=-±
20
""j~ _ _,_1"_:-·_ ---t--. -=-i • ·-. .:;_-;~~rt:E~;·-~---::..~-~:·:;:-~-'.--.~-.-ir-:-'·;_; ______ -~::_::_:· -.:=~ ~ .r.-:: .1• : ~ -- ~ ---- --,- ~: ~ : ~~ ·--~-. ,-~ ~ ~::t~i-;i--.-:~: ··:::.::::i~~- z~~-~ ':. s:---:-.::~~-- :"!:33~ --,~1=£: . "-:.;:::j:..:t=-+-• _±::p:.-:
80
·sct~:; :· - -~ ~;;!'-'.:""' ~-:=":er. ~c~:- 1=l~~~rc~i · ;~~'c~j:~:;: _::_~~::r ~-;:;:~" ±± - S- -~· __ ;; = ,0
~~:,;: ~- i,~: ?'=:: ~ ~;~~:~: :~1-t :li:Jr::~:Jf;f ~}]:;t }f;I :::j;~~t~ {I~~~~~~~~~- 60
--==C... ~t~=i-_icl:fcc '=t 3=ic:c=·fcl= ~ ~.:"f=s~±,~~C-g=j,ic~!=ic .:~r~ ~- C: :?'c-~~::~;~~~,;~10:£i~~~::-....::_:_ ~· --{ '~- ~::'=~I~~~ 50 f::i
'1='~
~~~-;::t"--=-=-1·
~~~.1=s: ~=f=t=:~~t ·E ~~-==. ... ~--=-i.:::t- ------:
~. ~~- -•-+- i_ ,_, ·'--:i= _;:~. ~ ~ ... __: ~.~ ---~~-·- ---+--"-er
·E:.-:d ....,_ ' _. ~· ~--' . -,-·;____.-:::J;=:q:::.:i:
'' . ' - -:~ ~ ;·- ~;-4.=:14~ ~- 1-+- -r~-~ ~~-:'. 1: :=i::'. +:_~i=l=± -:-1-~::_~ ~ ~:-:-$-~:-_:_ B=-=±=~- ;_;:-.,~ili~ :·j·.~1-:Jf~_;- r.::.
~~-?%1=~~-=S=i::-:.-+ 4~f.~::=-:~~--~:~-=~~~ ~:;:~~=~ -~J~;:~'! :;;t;:: -- -cr~...d±t;::::
10 11 12
WAVELENGTH IN MICRONS
13
2,4,4,6,6,8-Hexamethyl-3,5,7-nonanetrione (XXXIII), Film· on NaCl Plates
§§cc, 40
30
20
10
o.
14 15
I-' N 00

Plate XII
WAVENUMBER CM'
5000 4000 3000 2500 2000 ,500 1400 1300 1200 1100 1000 900 800 700 650
60 './
1-J. ,_·; t ;7 . • ... ·. ': • . : • /fl , • · ·:. [7- ~ r"'- _ · I · T ; I;~ i' ~
10 11 12 13 14 15 16
WAVELENGTH IN MIQlONS
Unknown F, KBr Pellet
I-' N
'°

Plate XIII
WAVENUMBER CM-1
5000 4000 3000 2500 2000 1500 1400 '1·300 1200 noo 1000
100
90
80
70
60 -==f'A~,-=,~""'-r "'~' "' 50 -~=.c;---'lt=±J
40 --- ~ ~ ::.:::::c:::~-.-
30
20 :Ef
10
0
10
WAVELENGTH IN MICRONS
Unknown G, KBr Pellet
900
cc= ..::==.:=
11 12
800
-::.+-+-H~:
~=-+
700
l:::;::::l~
650
90
~
ro
w
w
~~~~"'-3'~~3"'!3-'-2'±-'i'~-=i=~~~~'±-'i-ct.'~~ 40
w
cg~~cgc'-E:~~~:l§::~§3:r_l:33c~i:f-:i":120
i:.:=::t+:;:::::;::=I~ F.:::!:::l=t.:f:;: 10 ~-=t4=F 4~~ 0
= .=J.±.:=,-, 13 14 15 16
I-' w 0

Plate XIV
WAVENUMBER CM·1
5000 4000 3000 2500 2000 1500 1400 1300 1200 1100 1000 900 800 700· 650
·: ~! ~c:~ ;_""~=--=- "01 > i'~' "~;·· 1 ·"; ••••• ,~,,. 1· • ~::· ·::·." "": ,~rnr~ ;g;!r~~~~=: 80 ==±,+ _ ~c ~ ~~~.: __ :c =::'::~; ~-- -~-- ~~:--~:::' :-~~'~ ::~-. :--;_ ;:,'.:~: ~~ :. :-~'~-:~-->-;:~~ --· '":. - --~~~- • ,;; ·:--~~~~>\- :~·~r ~~= -:~; :;~ _;~~;~~ 80
70 t=1= ~· '·'". ~ ~ ,ec_--~-- ,"I~-_-·-~~~~'.~-~::_:~:'-~:' ?:}~:~~~--F;-';c __ :1~::'.f_).:,~:.:-.-_\;~~i ;: .;cs:,-~-~:::~ ~:~~~t~-,~~-~!'=1=~;'! ,o 60·;:=~:;::;; ~i::~,:_~:~~?:t:tt:{:_~:-~::~l~~:"J?':~t,.Y~) ~~:r :r::\~tZ'}:~rJ::/t~":~~::~~,::!:'~Y ~;:,- I,,.·- ~:.t:~ ---::-;t·• 60
50 ~,c:_f;~-~- __ :=: cc-•:::~~--\~~~~j~~~=~ cc_ :__c_ ='==~:. ~ __ ;::;.T~:;:,'.} _~~~-~-~/~ i-:~ -:: ::: __ -:: ::: _---~-,:~ -__ -_'-~:-~~~--~ -~-~~°-- .::~~ '- --~; ~~~-'.c'-~ ~;r,-~~ 50 -~~1-~;:~::;-= -f_F~~a-: -:•ell:: ~;~:=t j?t . :'.' :l /' .•.'c ;t~; ·.ri,: ;:!ft~~~:f--;'l'.~~;~ ~~~: ~~!;l-i~;::~ :~:.;l~ t'i:, !~~~1 u~~ }~ 40
• -= s.a:--cC - "> ~:_;r: )_cf: C ~ 00
-~ •. Jj c :.- ~--C i ·~~ ~- ~ < cc i -~- ~~ --;:rr; ;;;; : -=ftl==-=:c
30 . ·~%Jc.:;-·-:-:,·- ~---·
lO ~- ~ ~-~ --= 7 :-::';!~ ~ :,,,~~~::_\;{~:~::· --:--~-,~-:--,'- ~,-,: ---~:'-~2:::_-~.c.-_-:·--~:~-:.t~~:~::::;_2:\--::::;~! ~~~--;:s::iE::1=<- lO
......, : ~:.-~~-=i----:.::-:-~ .. -:=i:._:·:_ -=-:-~-------~:- h-_-:····.·~ ·.::::j/<-:::-_-- _ _:_;:.: :_:..::== -~~- .,._-::-:-:·-! --- 1-- . -~----,---:..::.-
10 11 12 13 14 15 16
WAVELENGTH IN MICRONS
Ethyl 2,2,4-Trimethyl-3-oxovalerate (XLV), Film on NaCl Plates
I-' l.,..)
I-'

5000 4000 3000 2500
§I 100 90 80 70 60 50 40 30
20 10
Plate XV
WAVENUMBER. CM-t
2000 1500 't400 1300 1200 1100 1000 900 800 700 ~50
90
-1-1:CX: a-s
i
-,,,JJ,.:CC
--~-=*' ·-
~=i-- ~-...;-:;__
.==;./'~a ;cl~=~
·=-·--·80 ;1;; 70
60 ------50 '""'
,:c~ B=ES'E=EEEEC:,C::="'EB":-"f±'H±c!-;-'c;...;...;=i":Efc!-'l-'T,'H''M'r!'c!'B 40_ =r=i:
~1 ~ 30
-+
=El:= i=t==;.· 20 ffi"~"±_t"mti~f:lffl=l=t'=§=f$$1:-=f=
111111111111111 10 0 10 11 12 13 14. 15 16
WAVELENGTH IN MICRONS
2,4,4,6-Tetramethyl-3,5-heptanedione (XLVI), Film on NaCl Plates
I-' w I',.)

ir. .. ··jll p., r'U J!l' III ii· -~ lij' I 'Ill d, ·"' 111 j UP \ ~I: :,'iii!;!~ 11 !il ill]' r r 11r i;~ I" II' 1 ! JJ!l ~, I I ~ >- · .. ,/ 1 Ir .u, 1 • -;1, 111, · , " I, •1 , , 1 • I , 1
B ' ut !!1: ~t !jji ;'1' ;!?: ~r i'li: ~,i :n1: :1i!J 'r1lr '1 I Jt: tJ,111 I II l~ ·1 · .- .. ti id 11·""1·11111 1 :111•· ·111 ! f-. : ·1 • 1 , n. Hb 1 l 1n 1 . t .. ·11: :11· ·!i , ii 11t :L· n11 ;·i· ·; ~ - ·111 ;h!' , ,,, i II Ht II r 1 :1
~;;:: ;!ii' nl: :111; rii ;iij' ~II' :·,1, tjfr :!!i ;;, $!~~1: !J!_' 1n111'1! 1111 iii ! Ir fr q ·ii J)l' •'1 ,J!, 11 ' . ·I, 'P' .,. <_:_.;,,; ~ """"l"LP ··~, ·II l1 l 11 I I I ·I ~ -·"' · '- ·'1' 1H· •, ,jn i 1 ·1i· + - 'Ill ill• !11· .,.. ; I I I i 1 , 1 I ..
>- ·, ,:1 ·11i 11ir :ilj 1·· .; r:l "iJi '11 ' ij''1 ij if;iJ !Ml I I ' I I ' f 11111 ~ ~: Ii :H! :~111r! T1 i~; ~, ;~ m iW ii' Ii:: ll~ n!! i l 'rt · 1 i i r : ji It
~ :: .,, ·111 "1i. ii i· :~,~~; :Ir '1;· ; ,i ll 1 , I I !t I I ~ ? i,I rli: :in i1 i r l i Ii ini nu ii ' ,;:! 1~1 · 1· • ~ ,1 i ~ ~ :tri ,~ m1 1 ,1 , , ; , ! , • : 1u 1 r1 , ~ :r:ir illJ1 l 1 1l ~ 11 '' H
1 I j
ij·
1i1
11
~ - -: I , . .c i)• 'I' "Ii l'I rJj, 'T'' -JI• ' ' ,;. .. , , ';I' j'r ... , .. ·u.i ~ ~ , n g >ii :· :~ i: -Iii rt1: ;!:; ;: ; t :1~ !-tr 1;: I 1 0
N .., .. lj 1 111.'. ·n; l'1l· •ii: ,i i' I rr''l'' f, -" 'lj'lil "" ll'lll"j'' 'I'
"J ii"';'!III' I" ·!1• "j'' ~ l' • 11111 4• • 1 j l •ifrl111 1 •• I
::"·· '11·1 ,·" ~I· Ii·· 1· d,·, 'I er· Ii,' ": ;; '.·, lf'jt' '111 )J!! :1 J :j;'. 1, I. I g I:": fr r ,I r I I 1,, 1111 I r ·II-· µ I
I l :e: p,; r ill; 1 :,; 1\!1~]' ::n:J:m' :J:;"t ·:1;·11 nrn 11m11m H. I ; 1 r i1~1r I . ·~:' r,· ! ,_. · , ·r I H 1 · , 1 ~. LT
~ · It· 1$ 8 .,, 'I
~ f¥ti l I g ~ lrft'tt IH,tt m]1]t ttl
0H IHlHtttttltl
11,, d1l
~ 2 0 ~ ~
I 11 1 I IT
lj
s: ~ 0 M
I , ,, 1 Ir II
I[
I' ,I
I'
~ 2
111 ·
I I
j
i I ~
I
I
~r
Q)
"Cl •.-! H 0
,-I
' ..c: u
H Q)
,-I
co :> 0 :< 0 I
('f) I
,-I
..c: .IJ Q)
8 •.-! H
H I
..;:t
N
N
133

. sooo 4000 3000 2500
100-
90
80 ,0-60
so
40
30
20
10 ·=;===1= ~--
Plate XVII
WAVENUMBER CM·1
2000 1500 1400 1300 1200 1100 1000 900 800 700 650
~
c:;-...:;_·
i~:r::~~ 3';~~ L-':'::~=-~r ±t ~#'i':j~E'.i!:·lljj=c,1::ol~i=_:C.~-~--'--<T,...:r_=_-·_·"-·-"-_,_~--+---+--""'"+-~----1-~....,--....,---=~ t---~- - - _:.._·-=-= · _____ .- ~~- ~ :~:?::::__:;:::::-·---·:;·-. ,~-~-- _··*--=~~- ~\~~-~~~- ~-·-:···- ;=i.: ---8==--i--d=,=2--p==f-~'
"--~· --=r~:==,~ - -1.-: ~- :.::J::::.:._--+--+--:
~·
~;;~;~~~~ ~---~::~~~-~:-~~~~~~--~L::~ ~·Z:;;1aj;.::~~~~J~~ ~~~: :;{_;;~~~.;~ ~J:~I ~~r~ei:t~ 90
'-----"'=~;:;_;Jc~:~~ :~-~~\:;"t:):::-!':= ;~;::~~~:~~~~' ~~:~~-~~-~~i~ = 80
~~;,.ci~~-~ 'tf:~~7; 0}'~,=-'':(j{';~ ,;~~-"\, = :~;c~ ~~ ~~,i~.!~f ~:;~; ~~;~!;1 70
c:lhccc,.cg=i- ~;!1!~0~~~ ~-~~c~f(~~~:·i:~~·:! i-;:~~c~n::- --tr-:_-~!~~!-~~'~:' c;~~~~: :~f.: :?~~ 60 1!1:c:~~~ =-----r~1---1-+0 1t 0-=r -==-=-±>11 ---·-· ~--~-~~-=1=-,--v----1-·~_--,·-~·--::1-
-~~=i~~~~so =~.....1=:.
-=r::..-:. "-\Ccc~+ .-=+'~- ,t---;=-=-=:c•- . -...,-1·.==-= _-_-,-
------:~-:_,~~ '~-d~-
40 - -:-g=s"=J'-'-,__l :=r=:=1=-'J---:~-:1=lc=1 ------1-~ :t="
30
·-----
1-----~· 20
·-:~=-;_;~ 1111)~~--o.
10 1S:Jalir~~~~?"~-~~,~=~~~ l=i.:::·:..:..._:_--!~
~::::::::L+:::1._, __ ,. -,---·- cc . _,,_
10 11 12 13 14 15 16
WAVELENGTH IN MICRONS
Methyl 2,2,4-Trimethyl-3-oxovalerate (XXXVI) Film on NaCl Plates
t-' w ~

Plate XVIII
WAVENUMBER c11-1
5000 4000 3000 2500 2000 1500 1400 1300 1200 1100 1000 900 800
~ 100
00
80
70
60
so
40
30
20
10
0
10 11 12 13
WAVELENGTH IN MICRONS
Dimethyl 2,2,4,4-Tetrarnethyl-3-oxoglutarate (XXXVII), Chloroform Solution
700 650
1111111 !1111111 !11 11 I
14 IS 16
100
,o
10
70
60
so
40
30
20
10
0
!-' l;.J V,

--" "
- 3000 2500 200~ 1500 1400 :300 1200
~ -'lm=tt±-x:
~ I~
Plate XIX
IMY!NUMftfl CM-'
1100 lOOO too 100 100 HO
cd---!tJ FH1V'=n !h~ i H ! M"i'd 1"1...1'r l' J ~:;- cj-_!=j - ,- -+-++- ~~- ~ -- --n ~E ~Jd:llifl; ~ ~~ -=-: r -!
: : : l
~~ ,~ -;c~;i=-1~:='1 ~ . ~ --i : --i : -.-.-.-. ~-= 4 j :·; ;1
Llcl~ ~~L--i-=tc J$Jlc~~ - ·:'c :.=t".fc lf'3i ]j ~ ' -- ['[f-1- 1-1--f, _ =,:- · 'F .E'B-- : [§'.
---t 1 ti J--j,--J--t =l=:Jc,j"· .c;._=!=1-- - Cl- · : d i1c1; :tci:c " -- --~d- -- ~~c.- -~:;~=-:;0 :~~~ ,/~ :~:,~~:~~~ ---- t:1 i~, ·:ur ,~ '"+J --~; i.~~ ·:r '.' J";f~~~£ ~rrn1i H
= .;c;~~~cl4 -r" ''
=~tffiJ~~E~;Hrtf ~'.;; nr~~&UFU s;:. rj:~ .:~·:: ~'.:!Mml-~~: 10 10
•• 30
20
10
•
"~ j';!,~§lsnrnrn~1i ~!, 1t1~J~~11 ii~tfJ~Tuti;:;: ~l tfo tii :fil ~it,," 20
1!=37~f:~ --·..;~ . ·- =£,~~ ~·; -hj--j· -
-~ ---- .-===--- - -~ ~-. ~-;=-
~-~ _ ,;-__
.::=i.::::-:-:.:.1-·i~=-=~ -1-4-4 • J.- -
~ --::::::.:: , 5
- -- -,~ :--=-=-£ :c:i-~c. ?; 1..:,- (\_' i ': i ' i ~J i :, i i [·i i 1 i f i t [ j · :='.3..i=, 4llffe!H ~~-H- t • •- =--:~ ~..I;-c : : ~ -=:. :
:...r:·=--c - -~ E :-:-: ·'-1~1-'---x':-.,'lci-";
i f-i -'LH -!°T.;t. .:;c; }--- , 10 11 12
WAVELEllfGTH JII MICRONS
'_L; j i1=l-i 1 i; 1 fj: ~t
~ : i i -~ i ~ -f. 1 +i ~
1 1 ~-d~-:tf13 i 1.:l~- I"3il ff1j -~-~
-~J,J..:-1 .:..c1c8-.f+¥£-13 M
~ 3--·• . EE1.::t.J-:::
15
5- Hydroxy- 2,2,4,4,5,6,6 -heptamethyl-l,3-cyclohexaned i one (I) , KBr Pel l et
IO
0
"
·t-' l,.)
°'

2;0 3.0 4.0
' _j I 1000
I i 400
250
I lllO
I 50
ctt•, ~l ~ o ~o
Plate- XX
.s'.o PPM{'T) 6.0 7.0 I I .l
300 200
8.0 9.0 I .l
100
. 1.h
HI~ 0 CPS
1---------___.;.----:---------·· V ~t--
I J t I
8.0 7.0
S0lvent ••••••••• CCl4 F.B •••••••••• 4.0 cps
I I .l I I I I I I
6.0 5.0 PPMTI) 4:0 3.0 2.0
3-Hydroxy-2,2,4-trimethyl-3-pentenoic acid S-Lactone (I)
R. F. Field •••••• 0.05 mG S.T •••••••••••• a.250 cps
s.w ••••..••..• 500 cps s.0 ••••.•••••• 000 cps
I I _J . I _J 1.0 0
S .A. e 4- & ,. o6 • a ti••"" Pe ir; • ,o • .3.,, 2 I.A ••••••••••••••••••• off
t-' L,.)
-..J

Ac.O
I 1-
.• OIi
j i 511
(a) CR, ca, (a)
,.o fo I
Plate XXI
1)0 2.00
300. ·200
9"
.$.O Z..t>
1.0 I
ICID
lo
c»,O
>-ti~ D OS
•o
5-Hydroxy-2 ,2 ,4 ,4, 6 ,6-hexamethyl-5-isopropyl-l ,3m.cyclohexanedione (XXXII)
Solvent •••••• Benzene F~R•o•••••••20.0 cps
R. F. Attn ••••••••• 26 db s.r ....••........ 250 sec
s.w •••.•...... 500 cps s.0 ••••••••••• 000 cps
L.S ••..•••••.•... eBenzene I-' w 00

2;0 1,0
·'·Y.JO
I 'j ,so I
100
I 50
a.o 7.0
.·. Plate XXII
4.0 s:o PflM(T 6.0
400 300•
C"9 II.'
(b) 1:1\~ ' pa
~ <ti, • (c)ca, \/ .
mi, . (9) c~, ai,
(0)
~
s.o·~~PPMm 4.0
7,0 8.0
2.00 100
3.0 2.0
9,0
1.0
m )-H~
0 CPs
0
5=Hydroxy-2,2,4,4,6,6~hexamethyl-5-isopropyl-l,~-cyclohexanedione (XXXI!)
Solvent •••••• Benzene F.R ••••••••• iO~O cps
R. F. Attn ••••••••• 17 db S.T •••••••••••••• 500 sec
s.w •••.•••.••• 250 cps s.0 •.••.••.••. 000 cps
S.A •••••••••••••••••• 6000 L.S ••••..•••.•.••• Benzene
t'-'· uJ \0

2.0 3.0
1000
I 500
I 250
I 100
I 50
I
4.0 5.0
400 300
(b) C"-J~,,~ OH
o~yW,;:;-" C!'J
'", (c)
Ot) Ctlj tHJ CllJ
(,>.) (:;)
Plate XXIII
PPM ('T) 6.0 7.0 8.0
200 100
i /1 1\ 11
Ii 11
I lij I J\ ~
I
9.0 1b
>--H~ 0 CPS
I-..._~~--..-.----.._--~----~--------~__,__._.-,,~ ...... . .
8.0 7.0 6.0 s.o PPMfbl 4.0 3.0 2.0 1.0 0
5-Hydroxy-2,2~4,4,6,6-hexamethyl-5-isopropyl-l,3-cyclohexanedione (XXXIJ)
Solvent •• Pyridine-d5 F.B ••••••••••• 4.0 cps
R. F. Field •••••• 0.05 mG S.T ••• ~~•••••••••250 sec
s.w •••••....•• 500 cps s.0 •••••.••••• 000 cps
s.A. Q. 61." 9 GO e Cl Q IC- •• 0" ~ Q 1.6 I.A ••••••••••••••••••• off
I-' .po 0

T T
J
u
u
-
7:0
Solvent •• ~yridine-d3. F • B ..... o ••• o • A .,0 cps
<LD
ca, ..., ~).
..
Plate XXIV
s:o ftlM(T
300
~
l, ... .t,
,..... 2CO IGO D OI
l.
· 5-Pheuyl-2,2,414~tetramethyl-3,5-dioxovaleric acid CXXXIV)
Ro F. Field •••••• 0.02 mG S.T •••••••••••••• 250 sec
s.w •••••••••• -500 cps s.o ••••••••••• 482 cps
S.A.•o••••••••••••••••6.3 I.A••••••••o••••••·oooeOff
$:'. I-'

2.0 3.0
;u~
I 000
I 250
I 100
I so
400
8.0 7.0
S0lvent ••••••••• ~cc14 F.B ••••••••••• 4.0 cps
Plate XXV
4.0 I
5.0 PPM{7" 6.0 7.0 8.0
300 200
(a)
CH3 (b)
CH3
~
6.0 5.0 PPMlo) 4.0 3.0 2.0
1-Phenyl-2,2,4-trimethyl-1,3-pentanedione (XXXI~)
R. F. Field •••••• 0.05 mG s.r ..••.......... 250 sec
s.w ••••.•••••• 500 cps s.0 ••••••••••. 000 cps
9.0
100
1.0
lQ
Ht~ 0 CPS
0
S.A.ei••••• .. ••e,. •••••• 1.0 I.A •••••••••••••••••• off ~
N

2.0 3.0
1000
I i 400
250 . r 'i 5D
8.0 7.0
Solvent ••••••••• CDC13 F.B ••••••••••• 4.0 cps
Plate XXVI
4.0 I
5.0 PPM(T) 6.0
300
ca,-/ ,ca,
'' {b)
e·
ce3 o'l
,JB3 {a)
ca, ca,
{c) rP
6.C 5.0 PPM(o 4.0
7.0
200
{c)
ce3
0 /"
3.0
8.0 9.0
100
2.0 1.0
1b
')-H~
0 CPS
0
2,2,4,4,6,6,8-Heptamethyl-3,5,7-trioxononanoic acid (XXXV)
R. F. Field •••••• 0.05 mG S.T •••••••••••••• 250 sec
s.w ••••..•..•• 500 cps s.0 ••••••••••• 000 cps
ScAo•••••o•••••eoe••••2•5 I.A ••••••••••••••••••• off
.....
.p,. w

2.0 3.0
1000
I i 400 -I '1111 I
11!1
1.0 7.0
Solvent.•o••••••CDC13 F .B •• •o•••••••4.0 cps
Plate XXVII
4.0 I
5.0 PPM('T'} 6.0
300
C"J
0/C"J
6.0 5.0 PPM~ 4.0
7.0 1.0
200 100
~ .·
3.0 2.0
9.0
1.0
1_b_
)-H~ o as
0
Methyl 2 ,2 ·,4 ,4, 6, 6, 8-Heptamethyl-3, 5, 7-trioxononanoate (XXXVIII)
R. - F . Field •••••• 0.05 mG S.T.o••••••••••••250 cps
s.w •••.••..••• 500 cps s.o ••••••.••.. 000 cps
S.A •••••..•••••. a ••••• 2.5 I.A ..• a•••••••••••••••off
~ ~ ~

2.0 3.0 4.0 s'.o -I i AOO 300
250
I 1tlD
I IO
HO
~~
11.0 7.0 6.0 5.0
Plate XXVIII
PPM(T) 6.0 7.0
200
(b)
CH3 CH3
(a)
PPM(o 4.0 3:0
1.0
100
I 11
I
2.0
9.0
1.0
th
)-H~ o CPS
0
2,2,4,4,6,6,8,8=0ctamethyl-3,5,7-trioxo-l,9-nonanedioic acid (XLI)'
S0lvent ••••••••• CDCl3 F.B ••••••••••• 1;0 cps
a. F. Field ••••• 0.018 mG s.r .............. 250 cps
s.w ••••. soo, 1000 cps s.o ••••....•.. 000 cps
S~Am o ~•so•• o • o o o o II mo•• .10 I.A ••••••••••••••••••• off
I-' ~ V1
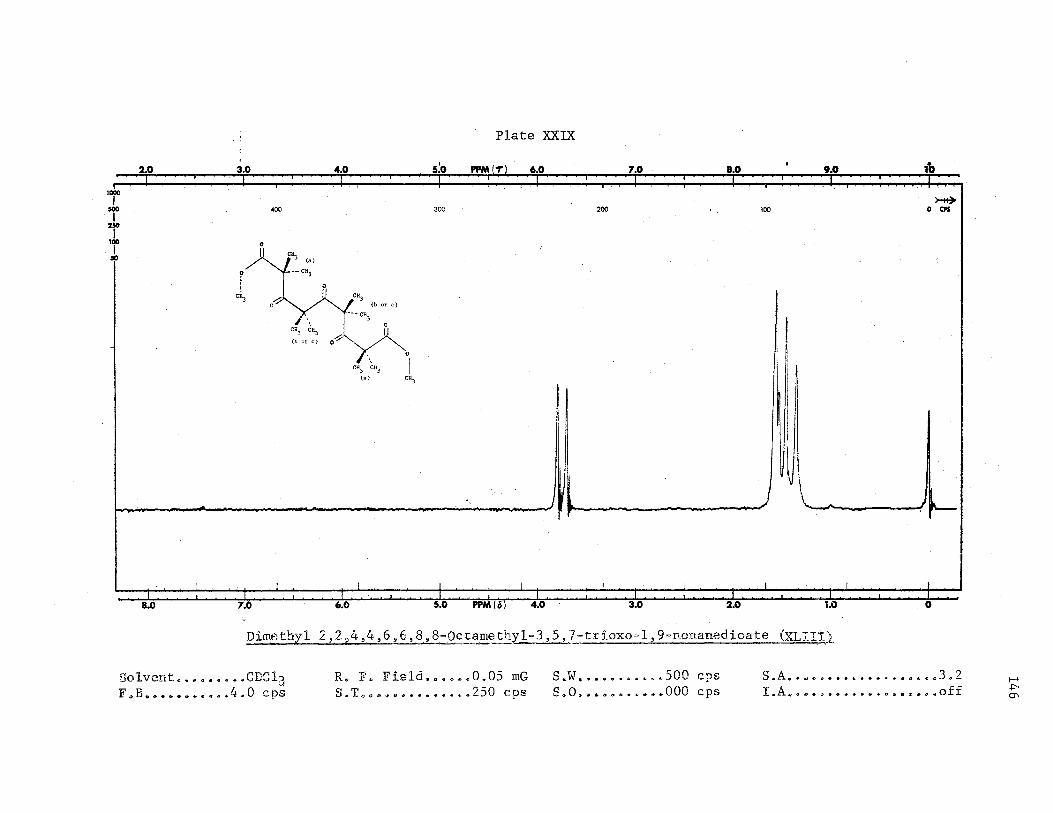
,_ I
I I
100 I ~
2.o_ 3.0
8.0 7.0
4.0 s:o
~00 300
ll
CH3 CH3 I (a) CH3
6.0 5.0
Plate XXIX
PPM('T) 6.0 1.1,
200
PPM(8) 4.0 3.0
f._Q t_.O
100
2.0 1.0
m HI~
O CPS
0
Dimethyl 2 ,2 ~4 ,4, 6, 6, 8, 8-0ctamethyl-3, 5, 7-trioxo-1, 9-nonanedioate (XLII_I)
S0lvent ••••••••• CDCl3 R. F. Field •••••• 0.05 mG F.B ••••••••••. 4.0 cps S.T •••••••••••••• 250 cps
s.w •.•.......• 500 cps s.o •••••....•. 000 cps
S~A •• o ....... ., ....... s ... v3 .. 2 I.A •••••••••••.••••••• off
I-' .p. a,

2.0 3l)
1000
I JOO I
400
250 I
11111 I
.50 I
0
I
8.0 7.0
4.0
(b .:,r c)
-ca3
I>)
0
I 1'ii3
I 5.0
300
Plate XXX
PPM (1' 6.0 7.0 8.0
200 100
,.... 2c.r.s
Ji~ j
.-. lc.p.s ..
~~ .. .U-4 -~t,. 4VtL ,._..... • fr 1'rt11P; t .,._.J ,., ..., r+ ~
6,0 5.0 PPM(o 4.0 3.0 2.0
9.0 1.b.
)-H~ 0 CPS
'~
1.0 ,o
Dimethyl 2, 2 j4 ,4 ~6 ~6 ,8 ~8-0ctamethyl=3 ,5, 7-trioxo-l, 9=nonanedioate OCLIID
Solvent •••••••• Benzene F.B •••••••••••• 4.0 cps
R. F. Field •••••• 0.03 mG S.T •• a•••••••••••250 sec
s.w •• e•••····-500 cps s.o ••••...•.•. 493 cps
S.Atl °' •• "'"'." Q • .,." o ... " •• 2ao6 I.A ••••••••••••••••••• off
I-" ~ --..J

Plate XXXI
2J) 3.0 co ... tt 7,1!
,:.x,
I . ..., I - - 2DD
'"' I ; (IO
I D so a,ry" ., ~,
..... .,
~ A I.O 7.D 6.0 u
2,2,4-Trimethyl-3-oxovaleramide (XL)
Solvent •••.• • •.~ •• CDCl.3 F • B ••••••• 2 • 0 ·, _: 0 • 4 cps
R. F. Field •••••• o.os inG S.T •••••••••••••• 250 sec
s.w ••• . .•••••• 500 cps s.o •••••••••••• 35 cps
u
2.cf
,._
~ !OD O CPS
0
S.A ••••••• 4 •••• ~.~2~0, · 24 I.A ••••••••••••.••••.. off
..... ~ 00

2.0 3.0_
t -
8.0 7.0
Solvent •••••••••• CDC10 F.B •••••••••••. l~O cps
Plate XXXII
4.0 .s_'.o PPM(T) 6.0 7.0 8.0
300 200
~~ ~
6.0 5.0 PPM( 6 l 4.0 3.0
Unknown E
R. F. Field •••••• 0.05 mG SeTeeOO$••••••e•u250 sec
s.w .•••....... 500 cps s.o ••• ~·······003 cps
2.0
9.0 fo
)-H~ 100 0 CPS
1.0 0
S.A111 • p e G> e o o II o e • • • • o • o .6 cs,3 I.A ••••••••••••••••••• off
I-' -P\0

i T i 1CIO
I 5!I
2.0 3.0
-
8.0 7.0
S0lvent ••••••••••• CCl4 F~B~$~O&e&9&•$e4s0 cps
Plate XXXIII
4.0 s'.o PPM('T'l ·6.0 7.0 8.0
300 200
ce, (b) <•>
ll 6.0 5.0 PPM 4.0 3.0 2.0
2 ,4 ,4, 6, 6, 8-Hexamethyl-3, 5, 7-nonanetrione (XXXI!'.[)
R. F. Field •••••• 0.05 mG s.w •.•........ 500 cps S.T •••••••••••••• 250 cps s.o .• ~ •.••.•.•• 49 cps
t..O
IOO.
1.0
10
>-H~ O CPS
'
.~
S lli'A~ Q. fj ". ~ 6 o ... °'.".,..,,,).., ,!/1, 1 ~ 0 I~A ••.••..•.•..•..••.• off
I-'-" u, 0

1000
I i i i 5P
:z.~
8.0
3.0
ADO
""3
7.0
S0lvent ••••••••••• cc14 F.B ••• oeo•a••••4.0 cps
0
u
Plate XXXIV
4.0 I
5.0 PPM('T} 6.0 7;0
300 ·200
CH,
o/",c"> (c)
A
6,0 5.0 PPM(o 4:0 3.0
Ethyl 2, 2 ,4-trimethyl-3-oxovalera te Q{LV)
R. F. Field •••••• 0.05 mG S.T ••• o••••••••••250 sec
s.w ••.•.•..•.. 500 cps s.o .••........ 000 cps
8.,
2.0
9.0
100
1.0
1b
>-H~ 0 CPS
0
S.A.,.•o•••••c,Gaec,oe•••1•6 I .A• ego e • 8 e 8 o D 8 8 O e O O a ct off
t-' Ul t-'

2,0_ 3.0
i -· .I olOO
2511
I 100
I J!I
ca,
8.0 7.0
Solvent •••••••••••• CS2 F.B ••••••• 2.0~ 0.4 cps
Plate XXXV
4.0 s:o PPM(T) 6.0 7.0
300 200
ca,
ca, (c)
6.0 5.0 PPM{o) 4.0 3.o
2,4j4,6-Tetramethyl-3,5.,,heptanedione (XLVI)
R. F. Field •••••• 0.05 mG S.W ••••••••••• 500 cps S.T •••••••••••••• 250 sec S.0 ••••••••••• 000 cps
8.0
2.0
9.0
100
1.0
lQ
)-.H~ O CPS
0
SeAe11ooesoDQO•oo~4,.Q, 20 I.A •••••••••••••••••• off-
I-' Vt N

2.0 3.0
1000 .
I i -400
250
I 100
I 50
cs,
8.0 7.0
Solvent.:~~ •• ~::;:Neat F:B:~::~:;~;.:~4;o cps
ca3
(a)
Plate XXXVI
4.0 I
5.0 PPM.('T) 6.0 7.0
300" 200
0
IJ C"3 / (b)
' °"' Cl
6.0 5.0 PPMTo 4.0 3.0
2,2~4-Trimethyl-3=oxovaleryl Chloride (XLIX)
R. F. Field •••••• 0.22 mG s~Tos••se~o•em•a~250 sec
s.w ••...•..... 500 cps s.0 .•.•....•.. 000 cps
8.0 9.0
100
1.0
1h
)-H~ 0 CPS
0
s.Ao GO 119"' 'I).~ Cl. Q e. 0 0 1111. a s0o8 I ,,,A$ SI 8. I> & e 9. Ill It O e e • 0 $ 41 9 off
f--' \JI (.,..)

2.0 3.0
1000
I 500 400
I 2liO
I 'IGD
I 5!I
a.o 7.0
Solvent •••••• " •••• Neat F.B~ ••••••••••• 2.0 cps
(•)
4.0
(b)
ce,
ce, 0/
I
5.0
300
Plate XXXVII
PPM(T) 6.0 7.0
200
11 di /1 ··~vu~ I I .
a.o 9.0
100
)b
>-ti~ 0 CPS
------.. /'VVV~
6'.0 s.o PPM(oJ 4.0 3.0 2.0
Methyl 2,2,4-Trimethyl-3-oxovalerate (X,'CXV]J
R. F. Field •••••• 0.02 mG SeT~o~~•oea~eoeoo250 sec
s.w •..•••..... 500 cps s;o •.•••.••••• 000 cps
1.0 .o
SaA-,G&O&i111&c;ioo&ae065, 2,;,Q I ,.Ae CO e 18 ,:;i O 51 Q a 09 0 0 0 t:; {) C, ~ soff
I-' V, +""

2.0 3.0
1000 I T AOO
250
I 111D 0
/' I 50
I° en, (•)
8.0 7.0
Solvent.o ••••••••• CCl4 F~B~&OQ•~a•~~".4.0 cps
Plate--XXXVIII
, 4.0 s'.o PPM{T 6.0 7.0 8.0
300 200
/~ C",
(b) (a)
6.C 5.0 PPM(o} 4.0 3.0 2.0
Dimethyl 2, 2 ,4 ,4-Tetramethyl-3-oxoglutara te (XXXVII)
Ro-F, Field •••••• 0.05 mG S.T •••••••••••.•• 250 sec
s.w .•......... 500 cps s.o ..••....... 002 cps
9.0
100
1.0
IQ
)-H~ 0 CPS
0
S 41Ae" " • ., co liJ"" <Si."" •• ~" ... .. 1 "6 I .. A. "o" .. o" Iii" o ... .,." c .o. "off
I-' l/1 l/1

Plate XXXIX
2.0 3.0 4.0 s'.o PPM(T) 6.0 7.0 8.0
1_
I
:c AOC 300 200 100
I 100
I ~o (a)
-70
(b) (b)
. l Ii
r-__ .._ ______ .._ __ ~~ i
8.0 7.0 6.0 5.0 PPM( 6) 4.0 3.0 2.0
5-Hydroxy-2,2,~,4,5,6,6-heptamethyl-1,3-cyclohexanedione (L)
9.0
1.0
fo
)-H~ 0 CPS
er
Solvent ••• ?yridine-d~~ R. F. Field •••••• 0.05 mG s.w .•........ ~soo cps S.O ••••••••••• 365 cps
S .A"'." •• "". o"' o .. ..,. ~""'. ~ 1 a 2 F.B ••.••••.•••• 4.0 cps S Cl T f) ii (I $ e "' G •• "'~ •••• 2 5 0 s e c I.A .. o o ••• '"' ....... ~ s ii,' •• & .off
I-' V,
°'

~. Plate XL .
,..o. 3.0 4.0 s!o JlllM ('I". M 7.0 I.O :t..O 1b
i -I. ,oo 300 200 100·
·~
0 OS.
2111 I
1GO 1 ID
<•> c·x· ·~·
X.. (cl 'c•, X .. c83 CB3 ' CB3 C83
(b) (b)
... 7.0 6.0 s:o PPM(o 4.0 3.0 2.0 1Jf 0.
5-Hydroxy-2.,.2"4,.4,.·5,. 6" 6-heptamethyl-1, 3-cyc lohexanedione ( L)
Solvent •••••••• l3enzene F.B •••••••••••• 4r.O cps
R. F. Field •••••• 0.05 mG S.T •••••••••••••• 250 sec
s.w .••..•••..• 500 cps s.0 ••••.••.••. 000 cps
SeA ........ " ...... 8 •••••• 2'°5 I.A ••••••••••••••••••• off
t-' Vl -.J

BIBLIOGRAPHY
·1. Anderton, K. and Richards, R. W., J.-Chem. ,Soc., 2543 (1965).
2. Arnold,. R. T. and Smolinsky, G., J ,- Am •. Chem. Soc., 82, 4918 (1960). :
3 •. Bartlett,, P •. D., ·.Swain,. C •.. G., and Woodward, R. B., J. Am •. Chem • . Soc., 63, 3230 (1941) •
. 4. Berlin, K.·D.,.Austin, T. H., and Nagabhushanam, M., J. Org •. Chem., 30, 1267 (1965).
5. Berlin, K. D. and Cooper,.M. H., J. Org •. Chem.,.29, 2057 (1964).
6. Bick, I.·R. C. and Horn, D. H. S.,.Aust. J. Chem.,~' 1405 (1965).
7. Birch,A. J.,.Cameron,.D. W., and Richards, R. W., J •. Chem •. Soc., 4395 (1960).
8. Birch, A. J., Fitton,.P., Smith, C.-C., Steere, D. E. and Stelfox, ,A.:R., J.~Chem •. Soc., 2209 (1963).
9. Boatman, S., Harris, T. M., and Hauser,,C. R., J.-Am. Chem •. Soc., 87, 82 (1965) •
. 10. Breslow,.D.·S. and Hauser,.C.-R., J •. Am. Chem.Soc., 62,.2385 (1940).
11. Buchis, G., Scharch, M., Witteran, V., and White,.D •. M., J. Am. Chem •. Soc., 81, 1968 (1959).
12. Burdett, J.-L. and-Rogers,.M •. T., J •. Am •. Chem •. Soc., 8,6, 2105 (1964).
13. Carlin,.R. B. and Smith, L. 0., Jr., J.·Am. Chem. Soc., 68, 2007 (1947).
14. Clark,.R.-D., J •. Qrg. Chem., 32, 399 (1966).
15. Clark,. R •. D •. (to· Eastman Kodak Co.), U .s. Patent 3,173,954 (March 16, 1965); Nations, R •. G. (to,Eastman.KodakCo.), U.S. Patent 3,091,642 (May 28, 1963) •.
16. . Collie, J •. N., J. Chem •. Soc., .21., 1806 (1907).
17. Cram, . D. J.,. "Fundamentals of Carbanion· Chemistry, 11 Academic· Press Inc.,.New York, .N. Y., 1965, pp. 86-94.
158

159
18. Dale, J., J. Chem •. Soc., 1028 (1963).
19. Diard, M. and Rumpf, P., Bull. Soc. Chim. France, 1645 (1962).
20. Eigen, M., Angew. Chem. (Intern. Ed. Engl.), 1_, 1 (1964).
21. Eistert, B. and Reiss, W., Chem. Ber., g, 92 (1954).
22. Erickson, J. L. E., Collins, F. E., Jr,, and Owens, B. L,, J, Org. Chem., l!_, 480 (1966).
23 •. Forsen, s., Nilson, M., and Wachtmeister,.c. A.,.Acta.Chem.,Scand., .!.§_, 583 (1962).
24. Fuson,.R. C., Chadwick, D. H., and Ward, M, L,, J. Am. Chem •. Soc., 6 8 , 3 8 9 (194 6 ) •
25. Gilman, H., Langham, W., and Moore, F., J. Am. Chem. Soc., 62, 232 7 (1940) •
. 26. Gilman, H., Zoellner, E., and Dickey, J., J. Am. Chem. Soc., .2.!_, 15 76 (1929) .•
27. Gresham, T. L., Jansen, J •. E., Sharer, F. W., and Bankert, R. A., J. Am. Chem,. Soc., .zl, 2807 (1949),
28. Hampton, K. G,, Harris, T. M., and Hauser, C.R., J. Org. Chem., 2 8' 194 6 ( 196 3) 0
29. Hampton, K. G., Harris, T. M., and Hauser, C.R., J. Org. Chem., 30, 61 (1965).
30. Hampton,.K. G., Harris, T. M., and Hauseri C. R,, J. Org. Chem., _;u_, 1035 (1966) 0
31. Hampton, K. G., Harris, T. M., and Hauser, G. R., J, Org. Chem., l!., 663 (196?).
32. Hampton, K. G., Harris, T. M., Harris, C. M., and Hauser,-C. R,, J •. Org.Chem., 30, 4263 (1965).
33. Hampton, K, G. and Hauser, C. R., J, Org. Chem., 30, 2934 (1965),
34. Hampton, K. G., Light, R •. J., and Hauser, C.R., J. Org. Chem., 3 0, 1413 (196 5) 0
35. Hamrick, P. J., Jr. and Hauser, C.R., J. Am. Chem. Soc., g, 493 (1959).
36. Harris, T. M., Boatman, S., and Hauser, C.R., J. Am. Chem •. Soc., 85, 3273 (1963),
37. Harris, T. M. and Harris, C. M., J. Org. Chem., l!., 1032 (1966),

160
38. Harris, T. M. and Ha user, c. R.,"J •. Am. Chem. Soc., g, 1160 (1959).
39. Harris, T. -M. and Hauser, c. . R. • J . Am. Chem. Soc., 84, 1751 (1962).
40. Harris, T •. M. and Hauser, - c. R.' J. Org. Chem.,. 29, 1391 (1964).
41. Harris, s. . R. and Levine, R. ' J. Am. Chem •.. Soc., l!, 1120. (1949).
42. Hasek,. R •. H., Clark, R. D.,,Elam, , E • u. ' and Nations,, R •. G., J • Org. . Chem., '!:]_, 3106 . (1962) 0
43. Hasek, R •. H.,-Elam,,E. V.,. Martin, J. C. ' and Nations, R. G •' J. Org. Chem., 26, 700 (1961.).
44. Hasek, R. H. and Martin, J. C., J •. Org. Chem., 28, 1468 · (1963).
45. Hauser, c. R. and Harris, T. M.' J. . Am •. Chem. Soc., 12, 6342 (1957).
46. Hauser, c. R. and Harris, T. M.' J ... Am. Chem. Soc., 80, 6360 (1958) 0
4 7 0 Hauser, c. . R. and Harris, T. M.' J •. Am. Chem. Soc., ~' 1154 -(1959).
48. Hauser, c. R., , Swarner, F. w.' and Adams, J. T., . "Organic Reactions, VIII, 59 (1954), J. Wiley and Sons, Inc.,, New York, N·, . . Y.
49. Herzog, H. L. and Buchman,.E. R., J.-Org. Chem., 1:§_, 99 (1951).
50. Hine, J.' "Physical Organic Chemistry, " McGraw-Hill Book Co., Inc., New York, N. y O' 1956, p. 237.
51. House, H. o. and Kramer, V •' J. · Org. Chem., '!:]_, 4146 (1962).
52. . House, H. o. and Kramer, V •' J. Org., Chem., 28, 3362 (1963) 0
53. House, H. O. and Trost, B. M., J. Org. Chem., 30, 1341 (1965).
54 •. Kharasch, M. · S., Hancock,. J. W., Nudenberg, W., and Tawney,. P. O., J. Org. Chem., ~' 322 (1956).
55. Light, R. J. and Hauser, C.R., J •. Org. Chem., 32_, 538 (1960),
56. Malhotra, S. K. and Ringold, H •. J., J. Am. Chem. Soc., 85, 1538 (1963).
II
57. Martin, J.C., Burpitt, R. D., and Hostettler, H. U., J. Org. Chem., 32, 210 (1967).
58. Meyer, R. B. and Hauser, C.R., J. Org. Chem., 25, 158 (1960).
59. Miles, M. L., Harris, T. M., and Hauser, C.R., J. Org •. Chem., 30, 1007 (1965).
60. Muir, W •. M., Ritchie, P •. D., and Lyman,-.D •. J., J. Org. Chem., I!_, 3790 (1966).

61. Nightingale, D. V. and Turley, R.H., J. Org. Chem., 26, 2656 (1961).
62. 0 'Sullivan,. W •. I. and Hauser, C. R.' J. Org. Chem., 25, 1110
63 • .. Parker, A. J. ' Quart. Rev. (London), Ji_, 163 (1963).
64. . Pearson, R. G. and Dillon, R. · L., J • Am. Chem •. Soc., ]2_, 2439 (1953).
161
(1960).
65 •. Pregaglia, G. F. and Binaghi,.M., J •. Org. Chem., 28, 1152 (1963).
66 •. Reid, E. B. and Groszos, S. J., J. Am. Chem. Soc., ]2_, 1655 (1963).
67 0 Schaub, R •. E. and Weiss, M •. J., Chem. :Ind. (London), 2003 (1961).
68 •. Schmutzler, R. and Reddy, G •. s., Inorg. Chem., i, 191.(1965).
69 •. Shichiro, A. and Nakazawa, Y.,.J. Pharm. Soc. Japan, 76, .1~01 (1956).
70. Shriner, R. L., Fuson, R. C., and Curtin, D. Y., "The· Systematic Identification of Organic Compounds," J. Wiley and Sons, New York,.N. Y., 1956.
71. Sprague, J.M., Beckham, L. J., and Adkins, H., J.-Am. Chem. Soc., 56, 2665 (1934).
72. Stone, H.-W. and Hume, D. N., Ind. Eng •. Chem.,,Anal..Ed., l.!., 598 (1939).
73 •. Stork, G., Rosen, .P., and Goldman, N.-L., J. Am. Chem. Soc., 83, 2965 (1961).
74. Stork, G., Rosen,,P., Goldman, N., Coambs, R. V., and Tsuji, T., J. Am. Chem. Soc., 87, 275 (1965).
75. Stuckwisch, C. G. and Bailey, J. V., J. Org. Chem., 28, 2362 (1963).
76. Swain, C •. G. and Boyler, H.B., J. Am. Chem •. Soc., ?'i, 870 (1951).
77. . Swarner, F. w. and Hauser,. c. R.' J. Am. Chem.· Soc., 68, 2647 (1946).
78. . Swarner, F. w. and Hauser, c . R.' J. Am •. Chem. Soc., 72, 1352 (1950).
79. "Technical Data Report No. X-129, II Eastman Chemical Products, Inc., . Kingsport, Tennessee, 1961 •
80. Toromanoff,. E., Bull •. Soc. .Chim. France, 799 (1961).
81. Traynelis, V. I., Hergenrather, W.-L., and in part with·Hanson, H. J., and Valicenti, J. A., J. Org. Chem., 29, 123 (1964).

162
82. Wasserman, H. H. ''Steric Effects in Organic Chemistry,''· Editor, Newmal), M. S., Chapt. 7, J. Wiley and. Sons,.Inc.,.New,York, N. Y., 1956.
83. Wedekind,.E. and Weisswange, W., Ber. ]2, 1631 (1906).
84. Weiss, M. J., Schaub,.R. E., Allen, G. R., Jr., Polleto,.J. F., Pidacks, C., Conrow, R .. B., and CascUa, C. J.,' .. 'Tetrahedron, 2 0' 3 5 7 (1964 ) 0
85. Weiss, M. J., Schaub, R. E., Polleto, .J. F., Allen, G. R., Jr., and Cascia, C •. J., Chem •. Ind. (London), 118 (1963).
86. Wierzchows ki, K. L. and Shugan, . D.,. Spee trochem. Ac ta. , ~' 943 (1965) 0
87. Work, S. D. and Hauser, C.R., J. Org. Chem., 28, 725 (1963).
88. Zaugg, H. E~, ''Organic Reactions," VIII, 322 (1954), J.-Wiley and Sons, Inc., New York, N. Y.
89. Zaugg, H. E. and Schaefer, A. D., J. Am. Chem. Soc.,~. 1857 (1965).
90. Zook, H. D. and Russo, T. J., J •. Am. Chem •. Soc., 82, 1258 (1960).

VITA
Robert Burton Hanson
Candidate for the Degree of
Doctor of Philosophy
Thesis: RING CLEAVAGE OF 3-HYDROXY-2,2,4-TRIMETHYL-3-PENTENOIC ACID~LACTONE BY TiiE·ANION OF DIISOPROPYL !\ETONE
. Major Field: Organic Chemistry
Biographical:
Personal Data: The author was born in Jackson Heights, New York on May 3, 1941, the son of Robert Andrew and Ruth Burton Hanson. On August 11, 1965, he married Barbara,Suzanne Logsdon.
Education: .The author graduated from Will Rogers High School in Tulsa, Oklahoma, in 1959. In September, 1959, he entered Oklahoma State University, Stillwater, Oklahoma, where he received a Bachelor of Arts Degree in Chemistry in August, 1963. In September, 1963, he was.admitted to the Graduate School of Oklahoma State University,. Stillwater, Oklahoma, where he completed the requirements for the Doctor of Philosophy degree in July, '1967; .
. Professional experience: The author served as a graduate teaching assistant from the fall of 1963 to January, 1967. He was a recipient of a Department Fellowship during the summer of 1965 and served as an NSF ''trainee" from September, 1966, to July, 1967 0
Membership in professional societies: The author is a member of Phi Lambda Upsilon.




















