
DNA Gyrase and Topoisomerase IV on the Bacterial Chromosome: Quinolone-induced DNA Cleavage
11
J. Mol. Biol. (1996) 258, 627–637 DNA Gyrase and Topoisomerase IV on the Bacterial Chromosome: Quinolone-induced DNA Cleavage Chang-Rung Chen 1 , Muhammad Malik 1 , Michael Snyder 2 and Karl Drlica 1 * DNA gyrase, the bacterial enzyme that supercoils DNA, is trapped on 1 Public Health Research chromosomal DNA by the 4-quinolone compounds, as drug–gyrase Institute, 455 First Avenue complexes that contain DNA breaks. Examination of chromosomal DNA New York, NY 10016, USA extracted from Escherichia coli indicated that bacteriostatic concentrations of 2 Departments of Biology and oxolinic acid trap gyrase and block DNA synthesis without releasing of Molecular Biophysics and broken DNA from gyrase–DNA complexes. Release, detected as free Biochemistry, Yale University rotation of DNA in the presence of an intercalating dye, occurred only at New Haven, CT 06520-8103 high, bactericidal oxolinic acid concentrations. Release of DNA breaks and USA cell death were both blocked by chloramphenicol, an inhibitor of protein synthesis, suggesting that synthesis of additional protein activity is required to free the DNA ends. Ciprofloxacin, a more potent quinolone, released DNA breaks and killed cells even in the presence of chloramphenicol. It is proposed that this second, chloramphenicol-insensi- tive mode for release of DNA breaks and cell killing arises from dissociation of gyrase subunits. Ciprofloxacin also killed a gyrase (gyrA) mutant resistant to the prototype of quinolone, nalidixic acid, and created complexes on DNA detected by DNA fragmentation. This lethal effect of ciprofloxacin was eliminated by additional mutations mapping in parC, one of the two genes encoding topoisomerase IV. Thus, the fluoroquinolone compounds have two intracellular targets. In the absence of the gyrA mutation, the parC (Cip R ) allele did not by itself confer resistance to ciprofloxacin, indicating that gyrase is the major quinolone target in E. coli . These findings provide a molecular explanation for quinolone action in bacteria and a new way to study topoisomerase IV–chromosome interactions. 7 1996 Academic Press Limited Keywords: gyrase; topoisomerase IV; Escherichia coli; ciprofloxacin; lexA *Corresponding author gene Introduction Bacterial DNA topology is controlled by three enzymes: DNA gyrase introduces negative super- coiling (Gellert et al ., 1976a,b; Drlica & Snyder, 1978); DNA topoisomerase I counters the action of gyrase to prevent the accumulation of excess supercoiling (DiNardo et al ., 1982; Pruss et al ., 1982); and DNA topoisomerase IV plays a central role in the resolution of interlinked, replicated daughter chromosomes (Kato et al ., 1990; Adams et al ., 1992). Gyrase and topoisomerase IV are related, having amino acid sequence similarity and similar double- strand passage modes of action. In addition, both are sensitive to the 4-quinolone class of antibacterial compound in vitro (Gellert et al ., 1977; Sugino et al ., 1977; Peng & Marians, 1993b; Hoshino et al ., 1994). We have been particularly interested in quinolone sensitivity because the drug–topoisomerase inter- action appears to be marked by DNA breakage that can be monitored for study of topoisomerase– chromosome relationships (Drlica et al ., 1990). It has been known for many years that gyrase is a physiological target of quinolone compounds in Escherichia coli . Resistance mutations map in the gyrase genes (Hane & Wood, 1969; Gellert et al ., 1977; Sugino et al ., 1977), and the quinolone compounds trap gyrase on DNA (Gellert et al ., 1977; Sugino et al ., 1977) in parallel with rapid inhibition of DNA synthesis (Snyder & Drlica, 1979). Indeed, Present address: C.-R. Chen, Department of Biology, New York University, Washington Square, New York, NY 10003, USA. 0022–2836/96/190627–11 $18.00/0 7 1996 Academic Press Limited
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of DNA Gyrase and Topoisomerase IV on the Bacterial Chromosome: Quinolone-induced DNA Cleavage

J. Mol. Biol. (1996) 258, 627–637
DNA Gyrase and Topoisomerase IV on the BacterialChromosome: Quinolone-induced DNA Cleavage
Chang-Rung Chen 1, Muhammad Malik 1, Michael Snyder 2 andKarl Drlica 1*
DNA gyrase, the bacterial enzyme that supercoils DNA, is trapped on1Public Health Researchchromosomal DNA by the 4-quinolone compounds, as drug–gyraseInstitute, 455 First Avenuecomplexes that contain DNA breaks. Examination of chromosomal DNANew York, NY 10016, USAextracted from Escherichia coli indicated that bacteriostatic concentrations of2Departments of Biology and oxolinic acid trap gyrase and block DNA synthesis without releasing
of Molecular Biophysics and broken DNA from gyrase–DNA complexes. Release, detected as freeBiochemistry, Yale University rotation of DNA in the presence of an intercalating dye, occurred only atNew Haven, CT 06520-8103 high, bactericidal oxolinic acid concentrations. Release of DNA breaks andUSA cell death were both blocked by chloramphenicol, an inhibitor of protein
synthesis, suggesting that synthesis of additional protein activity isrequired to free the DNA ends. Ciprofloxacin, a more potent quinolone,released DNA breaks and killed cells even in the presence ofchloramphenicol. It is proposed that this second, chloramphenicol-insensi-tive mode for release of DNA breaks and cell killing arises fromdissociation of gyrase subunits. Ciprofloxacin also killed a gyrase (gyrA)mutant resistant to the prototype of quinolone, nalidixic acid, and createdcomplexes on DNA detected by DNA fragmentation. This lethal effect ofciprofloxacin was eliminated by additional mutations mapping in parC, oneof the two genes encoding topoisomerase IV. Thus, the fluoroquinolonecompounds have two intracellular targets. In the absence of the gyrAmutation, the parC (CipR) allele did not by itself confer resistance tociprofloxacin, indicating that gyrase is the major quinolone target in E. coli.These findings provide a molecular explanation for quinolone action inbacteria and a new way to study topoisomerase IV–chromosomeinteractions.
7 1996 Academic Press Limited
Keywords: gyrase; topoisomerase IV; Escherichia coli; ciprofloxacin; lexA*Corresponding author gene
Introduction
Bacterial DNA topology is controlled by threeenzymes: DNA gyrase introduces negative super-coiling (Gellert et al., 1976a,b; Drlica & Snyder,1978); DNA topoisomerase I counters the action ofgyrase to prevent the accumulation of excesssupercoiling (DiNardo et al., 1982; Pruss et al., 1982);and DNA topoisomerase IV plays a central role inthe resolution of interlinked, replicated daughterchromosomes (Kato et al., 1990; Adams et al., 1992).Gyrase and topoisomerase IV are related, havingamino acid sequence similarity and similar double-
strand passage modes of action. In addition, bothare sensitive to the 4-quinolone class of antibacterialcompound in vitro (Gellert et al., 1977; Sugino et al.,1977; Peng & Marians, 1993b; Hoshino et al., 1994).We have been particularly interested in quinolonesensitivity because the drug–topoisomerase inter-action appears to be marked by DNA breakage thatcan be monitored for study of topoisomerase–chromosome relationships (Drlica et al., 1990).
It has been known for many years that gyrase isa physiological target of quinolone compounds inEscherichia coli. Resistance mutations map in thegyrase genes (Hane & Wood, 1969; Gellert et al.,1977; Sugino et al., 1977), and the quinolonecompounds trap gyrase on DNA (Gellert et al., 1977;Sugino et al., 1977) in parallel with rapid inhibitionof DNA synthesis (Snyder & Drlica, 1979). Indeed,
Present address: C.-R. Chen, Department of Biology,New York University, Washington Square, New York,NY 10003, USA.
0022–2836/96/190627–11 $18.00/0 7 1996 Academic Press Limited

Topoisomerase–Chromosome Interactions628
for several quinolone compounds, inhibition ofDNA synthesis occurs at the minimum inhibitoryconcentration for growth (Chow et al., 1988), whichhas been thought to equal minimum bactericidalconcentration. Inhibitors of RNA or protein syn-thesis prevent the prototype quinolone, nalidixicacid, from quickly killing cells (Deitz et al., 1966);consequently, rapid killing appears to arise from anactive process subsequent to the passive block ofDNA replication. Breakage of DNA would seem tobe involved, since quinolone–gyrase–DNA com-plexes contain broken DNA (Gellert et al., 1977;Sugino et al., 1977; Snyder & Drlica, 1979). However,a lethal role for DNA breaks has been unclearbecause they have been observed only afterextraction of DNA under conditions of proteindenaturation. Nucleoid DNA remains supercoiledwhen isolated from cells treated with concentrationsof oxolinic acid that fully inhibit DNA synthesis(Snyder & Drlica, 1979) and apparently kill cells(Chow et al., 1988). Thus, it has not been obvioushow quinolone compounds cause cell death. Belowwe show that the release of DNA breaks fromgyrase–DNA complexes, not inhibition of DNAsynthesis, correlates with killing of E. coli by oxolinicacid.
A related issue concerns the ability of inhibitorsof RNA and protein synthesis to interfere withkilling by quinolone compounds. Rifampicin andchloramphenicol are very effective at inhibiting thelethal effect of nalidixic acid arising from nalidixicacid–gyrase–DNA interactions. However, the lethaleffect of the potent fluoroquinolone ciprofloxacin isonly partially blocked by either rifampicin or thepresence of a nalidixic acid resistance (NalR) alleleof gyrA, one of the two genes encoding gyrase(Lewin et al., 1991). Since rifampicin completelyblocks the lethal effect of ciprofloxacin on a gyrA(NalR) mutant, protein synthesis-independentkilling involves gyrase. To account for the inabilityof rifampicin to fully block the killing effect ofciprofloxacin on wild-type cells, we postulate thatdissociation of gyrase subunits in ciprofloxacin–gyrase–DNA complexes releases DNA ends, anevent expected to be lethal. Since the gyrA (NalR)allele confers only partial protection from cipro-floxacin, this quinolone also has a non-gyrase target.Below we present evidence that topoisomerase IVis the non-gyrase target of ciprofloxacin. Thisaccounts for the susceptibility of gyrA (NalR)strains to ciprofloxacin and provides a new way tostudy topoisomerase IV–chromosome interactions.
Results
Oxolinic acid–gyrase–DNA complex formationis insufficient to kill cells
To connect chromosomal changes and lethaleffects caused by oxolinic acid, we first examinedthe action of chloramphenicol, an inhibitor ofprotein synthesis. When added to liquid cultures ofE. coli shortly before oxolinic acid, chloramphenicol
Figure 1. Effect of chloramphenicol on viability andDNA synthesis in the presence of oxolinic acid. A, Viablecells. Aliquots of E. coli strain NI747, growing exponen-tially, were treated with the indicated concentrations ofoxolinic acid with (w) or without (W) treatment with20 mg/ml chloramphenicol. Cells were then incubated for2.5 hours at 37°C, diluted into cold LB lacking antibiotics,and plated. Viable colonies were determined afterovernight incubation at 37°C. B, DNA synthesis. Aliquotsof E. coli strain NI747 were treated with the indicatedconcentrations of oxolinic acid with (w) or without (W)treatment with 20 mg/ml chloramphenicol. At varioustimes after addition of oxolinic acid, samples wereremoved for a three minute labelling period with3H-labelled thymidine. Incorporation of radioactivity intoan acid-insoluble form drops within a few minutes afterquinolone treatment and reaches a plateau (Snyder &Drlica, 1979). DNA synthesis rate at the plateau isexpressed as a percentage of the untreated control.
almost eliminated cell death (Figure 1A). However,chloramphenicol had little effect on inhibition ofDNA synthesis by oxolinic acid (Figure 1B). Inaddition, inhibition of DNA synthesis was moresensitive to oxolinic acid than was survival. A drugconcentration sufficient to inhibit DNA synthesis by90% in ten minutes had little effect on survivalduring a 2.5 hour treatment (Figure 1A and B).

Topoisomerase–Chromosome Interactions 629
Figure 2. Sedimentation of quinolone-generated DNAfragments. Exponentially growing E. coli cells (strainNI747) were treated with 3H-labelled thymidine for 30minutes, and then oxolinic acid was added for tenminutes. Cells were lysed, treated with 0.5% (w/v)sodium dodecyl sulfate, and centrifuged into sodiumdodecyl sulfate-containing sucrose density-gradients(Snyder & Drlica, 1979). Radioactivity in each fraction hasbeen normalized to the maximum value observed foreach gradient. A, 1 mg/ml oxolinic acid; B, 30 mg/mloxolinic acid. DNA from untreated cells sedimented about2.5 times faster than that from cells treated with oxolinicacid. Arrows indicate the position of 14C-labelledbacteriophage T4 DNA included in each gradient.Sedimentation was from right to left.
Quinolone-induced release of DNA supercoils
Treatment of cells with bacteriostatic concen-trations of oxolinic acid (1 mg/ml) has little or noeffect on the sedimentation properties of isolatednucleoids (Snyder & Drlica, 1979). To examine theeffect of high, bactericidal concentrations of oxolinicacid, E. coli DNA was radioactively labelled bygrowth in [3H]thymidine, cells were treated with 0,5, 10, or 20 mg/ml oxolinic acid for ten minutes priorto gentle lysis, and nucleoids were sedimented intoneutral sucrose density-gradients lacking detergent.In each case discrete, rapidly sedimenting nucleoidswere observed, and little radioactivity was recov-ered as small DNA fragments at the tops of sucrosedensity-gradients (Figure 3). Thus, the nucleoidsmaintained much of their integrity even at highquinolone concentration. However, oxolinic acid atbactericidal concentrations (5 to 10 mg/ml) reducedthe nucleoid sedimentation rate from 1750 S toabout 1300 S. With twice as much drug, a shoulderof more slowly sedimenting material could be seen(Figure 3D). At very high concentrations of oxolinicacid (50 to 75 mg/ml), the shoulder, whichsedimented at about 800 S, became more prominent(Figure 4). Thus, treatment of cells with increasing,bactericidal concentrations of oxolinic acid causedtwo changes in nucleoid structure, conversion to a1300 S form at intermediate drug concentrations andto an 800 S form at high drug concentrations.
The decrease in sedimentation rate from 1750 S to1300 S (Figure 3) was similar to that seen whensupercoils are relaxed by ethidium bromide or byDNA nicks (Worcel & Burgi, 1972). To determinewhether supercoils were eliminated by bactericidalconcentrations of oxolinic acid, nucleoids weresedimented into a series of sucrose gradients con-taining various concentrations of ethidium bromide.Increasing concentrations of ethidium titrate nega-tive supercoils and reduce the sedimentationcoefficient. At a critical ethidium concentration, allsupercoils are removed; further increases in dyeconcentration introduce positive supercoils andincrease sedimentation rate. Although bacteriostaticconcentrations of oxolinic acid sufficient to inhibitDNA synthesis by 95% (1 mg/ml) relaxed supercoil-ing slightly, as shown by a shift in the sedimentationminimum to lower ethidium concentrations (Fig-ure 5A), high concentrations of ethidium clearlyintroduced positive supercoils, as shown by theincrease in sedimentation rate (Figure 5A, opencircles). These data indicate that DNA breaks,presumably generated during quinolone–gyrase–DNA complex formation and observed upontreatment with sodium dodecyl sulfate (Figure 2),were constrained and unable to release superhelicaltension. In contrast, bactericidal quinolone concen-trations rendered nucleoid sedimentation indepen-dent of ethidium concentration (Figure 5B to D).These are the results expected if DNA breakscreated by oxolinic acid at high concentration werereleased and allowed free rotation of the DNAstrands, since treatment with DNase creates a
These data demonstrate that inhibition of DNAsynthesis is not by itself responsible for thebactericidal effect of oxolinic acid.
Since inhibition of DNA synthesis parallelsformation of quinolone–gyrase complexes withchromosomal DNA (Snyder & Drlica, 1979), wedetermined whether the same number of complexesform at the low drug concentrations sufficient toinhibit DNA synthesis as form at the highconcentrations needed for killing. No difference wasobserved in the size of DNA fragments generatedby treatment of cells with oxolinic acid at 1 and30 mg/ml when followed by protein denaturationto release DNA ends constrained in drug–gyrase–DNA complexes (Figure 2). Thus, quinolone–gyrase–DNA complex formation, as detected bysodium dodecyl sulfate-dependent DNA fragmen-tation, is not sufficient to kill cells.

Topoisomerase–Chromosome Interactions630
Figure 3. Effect of oxolinic acid on nucleoid sedimen-tation. 3H-labelled nucleoids were obtained from strainNI747 treated with oxolinic acid at 0 (A), 5 mg/ml (B),10 mg/ml (C), or 20 mg/ml (D) for ten minutes by cell lysisin the absence of ionic detergents (Drlica & Snyder, 1978).Nucleoids were then sedimented into 10% to 30% sucrose
similar response (Worcel & Burgi, 1972). It isunlikely that the flattening of the ethidium titrationcurves seen in Figure 5 arose from heterogeneitywithin or among nucleoids, since plasmid DNAknown to be topologically heterogeneous shows adistinct increase in sedimentation rate at 4 mg/mlethidium bromide (Drlica et al., 1988). The experi-ment shown in Figure 5D showed no nucleoidsedimentation increase even at 6 mg/ml ethidiumbromide (data not shown).
Since chloramphenicol blocked the lethal effect ofoxolinic acid (Figure 1A), we determined whetherparallel changes in chromosome structure occurredin the presence of chloramphenicol. Nucleoids fromcells treated with both chloramphenicol and oxolinicacid displayed a distinct sedimentation minimum(Figure 6A, open circles), indicating that chloram-phenicol treatment blocked the release of supercoilconstraint associated with oxolinic acid treatment.Since the same result would be seen if inhibition ofprotein synthesis eliminated gyrase–chromosomeinteractions, cells were treated with chlorampheni-col for 30 minutes, oxolinic acid was added foranother ten minutes and cells were lysed in thepresence of sodium dodecyl sulfate to release DNAbreaks from gyrase-mediated constraint. When theDNA fragments were sedimented into sucrosedensity-gradients, they exhibited only a modest(14%) increase in average sedimentation rate relativeto fragments from cells treated with oxolinic acidalone (data not shown). Thus, oxolinic acid-inducedcomplexes are formed in the presence of chloram-phenicol (they formed in undiminished numbersafter inhibition of protein synthesis by 75 minutesof starvation for essential amino acids (data notshown)). Taken together, these data support the ideathat intracellular release of DNA ends, not simplyformation of quinolone–gyrase–DNA complexes,accounts for the lethal effect of oxolinic acid.
The more recently discovered fluoroquinolonecompounds, such as ciprofloxacin, possess anadditional mode of killing that does not require on-going protein synthesis (Howard et al., 1993a,b).Since gyrase can participate in quinolone-stimulatedsubunit exchange (Ikeda, 1994), it seemed possiblethat the second mode of ciprofloxacin actionmight involve direct dissociation of quinolone–gyrase–DNA complexes. As a test for this idea,E. coli was treated with 1 mg/ml ciprofloxacin andsupercoil constraint was examined in nucleoids.Ethidium bromide titration curves of nucleoidswere flat, whether or not cells were treated withchloramphenicol (Figure 6B). At this concentrationof ciprofloxacin, chloramphenicol protected fewerthan 1% of the cells from killing by ciprofloxacin(Figure 7). Thus, cell death parallels the freedom ofDNA strands to rotate, which is presumably due to
density-gradients at 7000 rpm, and gradients were frac-tionated from the bottom of the centrifuge tubes. Thebroken line indicates the position of 14C-labelled bacterio-phage T4B added to each gradient as a sedimentationmarker. Sedimentation was from right to left.

Topoisomerase–Chromosome Interactions 631
Figure 4. Slow-sedimenting species of nucleoidgenerated by treatment of cells with oxolinic acid at highconcentration. 3H-labelled nucleoids were obtained fromstrain NI747 treated for ten minutes with oxolinic acid at0 (A), 50 mg/ml (B), or 75 mg/ml (C) as described forFigure 3. Nucleoids were then sedimented into 10% to30% sucrose density-gradients at 7000 rpm. The positionof 14C-labelled bacteriophage T4B is indicated by arrows.Sedimentation is from right to left.
Figure 5. Effect of oxolinic acid on supercoiling inbacterial nucleoids. 3H-labelled nucleoids were obtainedfrom cells treated with oxolinic acid for ten minutes at 0(W) or 1 mg/ml (w) (A), 5 mg/ml (B), 10 mg/ml (C), or20 mg/ml (D). Sedimentation coefficients, relative to14C-labelled bacteriophage T4B, were estimated fromcentrifugation into sucrose density gradients similar tothose shown in Figure 3 but containing the indicatedconcentrations of ethidium bromide (EBr; Hsieh et al.,1991).
the release of DNA breaks from constraint bygyrase. Figure 7 also revealed that ciprofloxacin atlow concentration behaves much like the less potentoxolinic acid at high concentration, since chloram-phenicol was very effective at blocking the bacteri-cidal action at low concentrations of ciprofloxacin.Thus, increasing the concentration or potency of aquinolone appears to shift its mode of action fromone requiring protein synthesis to one that does not.
DNA topoisomerase IV is an intracellulartarget of ciprofloxacin
Since quinolone–resistant mutants of Staphylococ-cus aureus contain mutations in a gene encoding
topoisomerase IV (Ferrero et al., 1994), we con-sidered the possibility that topoisomerase IV mightbe a second target of the quinolone compounds. Totest this idea, we prepared E. coli mutants andanalyzed them by transduction of parC, one of thegenes encoding topoisomerase IV. Two strains, CR1and CR3, were isolated by plating the gyrA-307(NalR) strain GP201 on agar containing 0.5 mg/mlciprofloxacin, a concentration that prevented growth
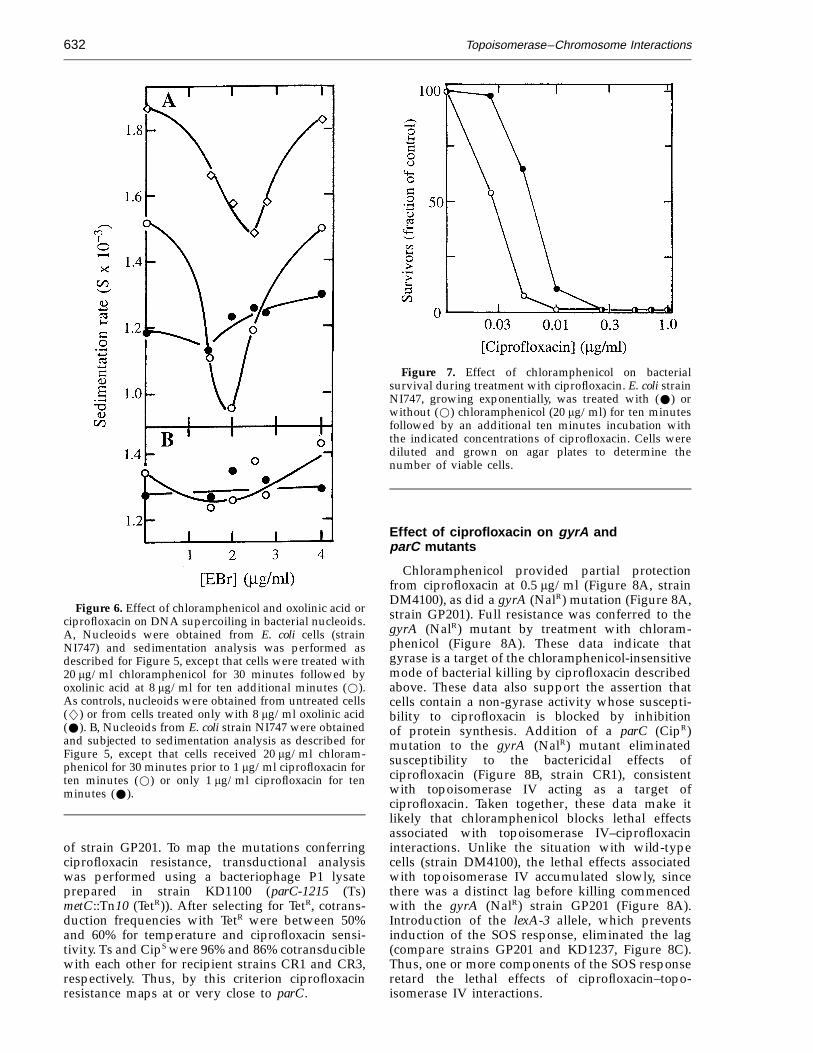
Topoisomerase–Chromosome Interactions632
Figure 6. Effect of chloramphenicol and oxolinic acid orciprofloxacin on DNA supercoiling in bacterial nucleoids.A, Nucleoids were obtained from E. coli cells (strainNI747) and sedimentation analysis was performed asdescribed for Figure 5, except that cells were treated with20 mg/ml chloramphenicol for 30 minutes followed byoxolinic acid at 8 mg/ml for ten additional minutes (w).As controls, nucleoids were obtained from untreated cells(w) or from cells treated only with 8 mg/ml oxolinic acid(W). B, Nucleoids from E. coli strain NI747 were obtainedand subjected to sedimentation analysis as described forFigure 5, except that cells received 20 mg/ml chloram-phenicol for 30 minutes prior to 1 mg/ml ciprofloxacin forten minutes (w) or only 1 mg/ml ciprofloxacin for tenminutes (W).
Figure 7. Effect of chloramphenicol on bacterialsurvival during treatment with ciprofloxacin. E. coli strainNI747, growing exponentially, was treated with (W) orwithout (w) chloramphenicol (20 mg/ml) for ten minutesfollowed by an additional ten minutes incubation withthe indicated concentrations of ciprofloxacin. Cells werediluted and grown on agar plates to determine thenumber of viable cells.
Effect of ciprofloxacin on gyrA andparC mutants
Chloramphenicol provided partial protectionfrom ciprofloxacin at 0.5 mg/ml (Figure 8A, strainDM4100), as did a gyrA (NalR) mutation (Figure 8A,strain GP201). Full resistance was conferred to thegyrA (NalR) mutant by treatment with chloram-phenicol (Figure 8A). These data indicate thatgyrase is a target of the chloramphenicol-insensitivemode of bacterial killing by ciprofloxacin describedabove. These data also support the assertion thatcells contain a non-gyrase activity whose suscepti-bility to ciprofloxacin is blocked by inhibitionof protein synthesis. Addition of a parC (CipR)mutation to the gyrA (NalR) mutant eliminatedsusceptibility to the bactericidal effects ofciprofloxacin (Figure 8B, strain CR1), consistentwith topoisomerase IV acting as a target ofciprofloxacin. Taken together, these data make itlikely that chloramphenicol blocks lethal effectsassociated with topoisomerase IV–ciprofloxacininteractions. Unlike the situation with wild-typecells (strain DM4100), the lethal effects associatedwith topoisomerase IV accumulated slowly, sincethere was a distinct lag before killing commencedwith the gyrA (NalR) strain GP201 (Figure 8A).Introduction of the lexA-3 allele, which preventsinduction of the SOS response, eliminated the lag(compare strains GP201 and KD1237, Figure 8C).Thus, one or more components of the SOS responseretard the lethal effects of ciprofloxacin–topo-isomerase IV interactions.
of strain GP201. To map the mutations conferringciprofloxacin resistance, transductional analysiswas performed using a bacteriophage P1 lysateprepared in strain KD1100 (parC-1215 (Ts)metC::Tn10 (TetR)). After selecting for TetR, cotrans-duction frequencies with TetR were between 50%and 60% for temperature and ciprofloxacin sensi-tivity. Ts and CipS were 96% and 86% cotransduciblewith each other for recipient strains CR1 and CR3,respectively. Thus, by this criterion ciprofloxacinresistance maps at or very close to parC.

Topoisomerase–Chromosome Interactions 633
Figure 8. Protection from the bactericidal action of ciprofloxacin by chloramphenicol and topoisomerase mutations.Exponentially growing E. coli cells were treated with 0.5 mg/ml ciprofloxacin. Chloramphenicol treatment, whenadministered, began five minutes prior to ciprofloxacin. At the indicated times, cells were diluted and grown on agarplates to determine the number of viable cells. A, Effect of a gyrA (NalR) mutation. E. coli strains were DM4100 (wildtype (w); DM4100 treated with 20 mg/ml chloramphenicol (W); GP201 (gyrA-307 (q); GP201 (gyrA-307 treated with20 mg/ml chloramphenicol (Q)). B, Effect of gyrA (NalR) and parC (CipR) mutations. E. coli strains were GP201 (gyrA-307(q)) and CR1 (gyrA-307 parC-2101 (r)). C, Effect of lexA-3 (Ind−) and gyrA (NalR) mutations. E. coli strains were GP201(gyrA-307 (q)) and KD1237 (gyrA-307 lexA-3 (R)). D, Effect of a parC (CipR) mutation. E. coli strains were DM4100(wild type (w)); DM4100 treated with 20 mg/ml chloramphenicol (W); and CR104 (parC CipR (r)). E, Effect ofchloramphenicol and a parC (CipR) mutation. E. coli strains were DM4100 (w); DM4100 treated with 20 mg/mlchloramphenicol (W); (GP201 (gyrA-307 (q)); and CR104 (parC CipR) treated with 20 mg/ml chloramphenicol (R).
By itself, the parC (CipR) allele showed little or noprotective effect against ciprofloxacin (Figure 8D,strain CR104), consistent with the hypothesis thatgyrase is the primary target of the antibiotic. Sincechloramphenicol provided more protection than theparC mutation (Figure 8D, compare strain CR104with strain DM4100 plus chloramphenicol), inhi-bition of protein synthesis does not confer protec-tion only from ciprofloxacin–topoisomerase IV–DNA complexes. It is likely that most of theprotective effect of chloramphenicol arises frominterference with the release of DNA breaks fromciprofloxacin–gyrase–DNA complexes, as is the casefor oxolinic acid–gyrase–DNA complexes.
Shortly after administration of ciprofloxacin,strain CR104 (parC CipR), when also treated withchloramphenicol, was less susceptible to cipro-floxacin than the wild-type strain DM4100 treatedwith chloramphenicol (Figure 8E). After about 60minutes, however, strain CR104 was killed. Asimilar response, but with a longer lag, was seenwith the gyrA (NalR) mutant (Figure 8E). If gyraseand topoisomerase IV are the only targets of cipro-floxacin, as suggested by Figure 8B, then theresponse seen with strain CR104 (parC CipR) treatedwith chloramphenicol reflects the putative gyrasedissociation mode of killing. The response observedwith the gyrA (NalR) mutant GP201 would representthe topoisomerase IV-mediated effect. It appearsthat potentially lethal events due to ciprofloxacinaccumulate more quickly with gyrase than with
topoisomerase IV, consistent with gyrase being themajor target for this drug.
Sedimentation analysis revealed that DNA frag-ments generated by treatment of wild-type cellswith 1 mg/ml ciprofloxacin were smaller than thoseobserved with oxolinic acid (Figure 9), consistentwith ciprofloxacin trapping both gyrase andtopoisomerase IV on the chromosome. As expected,the size of the DNA fragments created byciprofloxacin with the parC (CipR) gyrA+ mutantCR104 was the same as that following treatment ofwild-type cells with oxolinic acid (data not shown).For the gyrA (NalR) strain GP201, preliminarysedimentation analysis showed that the numberaverage molecular weight of ciprofloxacin-inducedfragments was twice that seen following oxolinicacid treatment of the wild-type strain DM4100 (datanot shown). Thus, topoisomerase IV appears to betrapped at more widely dispersed sites on thechromosome than is gyrase.
Discussion
Once it became clear that higher oxolinic acidconcentrations were required to kill cells than toblock DNA synthesis, connections emerged be-tween lethal action and DNA lesions due to thisdrug. Our current view is sketched in Figure 10.Moderate concentrations of oxolinic acid (1 mg/ml)trap gyrase on DNA such that the ends of brokenDNA are constrained by gyrase (Figure 10, step b).

Topoisomerase–Chromosome Interactions634
Figure 9. Sedimentation of ciprofloxacin-generatedDNA fragments. Exponentially growing E. coli strainNI747 was treated with 3H-labelled thymidine for 30minutes and then 0.5 mg/ml ciprofloxacin (W) or30 mg/ml oxolinic acid (w) for 10 minutes. Cells werelysed, treated with 0.5% (w/v) sodium dodecyl sulfate,and centrifuged into separate sodium dodecyl sulfate-containing sucrose density-gradients (Snyder & Drlica,1979). Each density-gradient contained 14C-labelledbacteriophage T4B DNA as a sedimentation marker thatallowed the gradients to be superimposed in the Figure.DNA radioactivity in each fraction has been normalizedto the maximum value observed for each gradient. Arrowindicates the position of bacteriophage T4 DNA.Sedimentation was from right to left.
Figure 10. Schematic representation of events involvedin quinolone-mediated cell death. Gyrase and DNAinteract (a) in such a way that 4-quinolone compounds cantrap (b) a reaction intermediate that blocks DNA synthesisand cell growth. DNA in the reaction intermediate isbroken, but the ends are restrained by gyrase. The DNAends can be released by two pathways, both of which leadto cell death. Pathway c is inhibited by chloramphenicol.It is postulated that this pathway is mediated by a repairfactor that removes quinolone–gyrase complexes fromDNA. Such a factor has not been identified. At highconcentrations of potent quinolone compounds the DNAends can be released by a chloramphenicol-insensitivepathway (d). It is postulated that this pathway involvesgyrase subunit dissociation. The presence of gyrase onDNA ends has not been demonstrated. A similar series ofcomplexes and broken DNA products may form whenquinolone compounds interact with topoisomerase IV.
Supercoils remain, and only after treatment withsodium dodecyl sulfate is fragmentation revealed(Figures 2A and 5A; Snyder & Drlica, 1979). Sinceremoval of quinolone reverses inhibition of DNAsynthesis (Deitz et al., 1966) and DNA fragmentation(data not shown), moderate (1 mg/ml) quinoloneconcentrations are only bacteriostatic. Raising theoxolinic acid concentration to 5 to 20 mg/ml had noeffect on the number of drug–gyrase–DNA com-plexes on chromosomal DNA (Figure 2). It did,however, lead to a decrease in nucleoid sedimen-tation rate (Figure 3), to the release of DNA breaks,as seen by loss of supercoil constraint (Figure 5) andto cell death. Additional support for the presence ofDNA ends in nucleoids obtained from cells treatedwith oxolinic acid emerged from the sensitivity ofnucleoid sedimentation rate to rotor speed: nucle-oids sedimented at 800 S rather than 1300 S whenthe rotor speed was increased from 7000 rpm to17,000 rpm (data not shown). This type of rotorspeed effect is characteristic of long, extended DNAcontaining ends (Zimm, 1974; Zimm & Schumaker,1976).
The bactericidal action of nalidixic and oxolinicacids occurs primarily through a pathway that isinhibited by chloramphenicol (Figure 10, step c).Chloramphenicol prevented the oxolinic acid-in-duced loss of supercoil constraint (Figure 6A) andcell death (Figure 1A), but not inhibition of DNAsynthesis (Figure 1B; Deitz et al., 1966). We postulatethat a repair factor is involved in this pathway. The
putative factor is independent of the SOS response,since the lethal effects of nalidixic acid areunaffected by the lexA-3 mutation (Lewin et al.,1989), which prevents the SOS response (Rinken &Wackernagel, 1992). Since chloramphenicol rapidlyblocks the lethal effect of nalidixic acid even whenadded two to three hours after the quinolone (Deitzet al., 1966), it is likely that the putative factor turnsover rapidly.
Fluoroquinolone compounds; such as cipro-floxacin, also have gyrase as a target and behave ina way similar to nalidixic and oxolinic acids;however, ciprofloxacin causes two additional eventsto occur. One is a chloramphenicol-insensitive modeof killing, which we postulate to involve dis-sociation of the gyrase subunits that constrain DNAends in ciprofloxacin–gyrase–DNA complexes (Fig-ure 10, step d). The idea of subunit dissociationemerged from Ikeda’s observation that certainclasses of illegitimate recombination can be ex-plained by gyrase subunit exchange and thatquinolone compounds stimulate this exchange(reviewed by Ikeda, 1994). Support for the gyrasedissociation idea is provided by the inability ofchloramphenicol to block either the lethal action ofciprofloxacin (Figure 7) or the ciprofloxacin-depen-

Topoisomerase–Chromosome Interactions 635
dent flattening of nucleoid ethidium titration curves(Figure 6B). Evidence for gyrase involvement comesfrom the ability of a gyrA (NalR) mutation toeliminate the chloramphenicol or rifampicin-insen-sitive mode of killing by ciprofloxacin (Figure 8A;Lewin et al., 1991). Repair or prevention of DNAdamage due to subunit dissociation appears toinvolve the SOS response, since a lexA-3 mutant ishypersensitive to ciprofloxacin (Howard et al., 1993)but not to nalidixic acid (Lewin et al., 1989).
Oxolinic acid may also participate in the putativesubunit dissociation mode, since this compoundstimulates a gyrase-dependent form of illegitimaterecombination and deletion formation that is mosteasily interpreted in terms of gyrase subunitdissociation and exchange (Ikeda et al., 1981, 1982;Miura-Masuda & Ikeda, 1990). If this interpretationis correct, a physical manifestation of subunitdissociation may be the change in nucleoidstructure that decreases sedimentation rate from1300 S to 800 S (Figure 4). This possibility is beingexamined.
The second event seen with ciprofloxacin istrapping of topoisomerase IV in complexes on DNA.Transductional analysis, in which the recipientstrain was a gyrA (NalR) mutant that had acquiredadditional, spontaneous resistance to ciprofloxacin(Figure 8B), showed that the additional cipro-floxacin resistance could be eliminated by replace-ment of parC, one of the two genes encodingtopoisomerase IV. A similar conclusion was reachedin concurrent work using strains constructed tocontain parC mutations (Khodursky et al., 1995). Thehypothesis of dual ciprofloxacin targets explainswhy DNA cleavage fragments generated byciprofloxacin were smaller than those produced byoxolinic acid (Figure 9). A chloramphenicol-inhib-ited mode of killing by ciprofloxacin–topoisomeraseIV interactions would also explain why chloram-phenicol protects gyrA (NalR) mutants from thelethal action of ciprofloxacin (Figure 8A).
Primary resistance to ciprofloxacin maps in gyrArather than in parC (gyrA NalR mutations typicallyreduce susceptibility by 30 to 40-fold while a parCCipR allele by itself provided no protection fromciprofloxacin; Figure 8D). Why this occurs is notobvious, since gyrase from E. coli is sometimes onlyslightly more sensitive to fluoroquinolone com-pounds in vitro than is topoisomerase IV (Peng &Marians, 1993b; Hoshino et al., 1994; Khodurskyet al., 1995). The two enzymes probably play verydifferent roles in chromosome topology. Gyrase isassociated with replication forks (Drlica et al., 1980)and has access to most of the chromosome (Snyder& Drlica, 1979; Franco & Drlica, 1988), presumablyto maintain appropriate levels of superhelicaltension. Topoisomerase IV is involved in decatenta-tion (Adams et al., 1992), and it may be membrane-bound (Kato et al., 1992). Thus, the enzymes, whenbound to the chromosome, may differ in theiraccessibility to the quinolone compounds and to therecA-mediated recombinational processes import-ant for survival following quinolone treatment
(Urios et al., 1991; Howard et al., 1993). Explanationsof the differences that focus on lethal, gyrase-medi-ated cleavage at replication forks (Khodursky et al.,1995) are less likely because inhibition of DNAsynthesis is reversible (bacteriostatic) and fails tocorrelate with lethal events. Cases in whichtopoisomerase IV is the primary target, such as seenwith Staphylococcus aureus (Ferrero et al., 1994, 1995),probably represent situations in which gyrase isnaturally resistant to quinolone compounds.
We currently favor the idea that the quinolonecompounds act on gyrase and topoisomerase IV ina similar way and that the scheme shown in Fig-ure 10 applies to both. According to this idea, thebactericidal effect of ciprofloxacin with strain GP201(gyrA NalR) is due to an interaction withtopoisomerase IV. After an initial delay, cell deathoccurs at the same rate as seen with wild-type cells.Thus ciprofloxacin–topoisomerase IV interactionsare bactericidal. Chloramphenicol eliminates thebactericidal effect (Figure 8A), indicating thatmuch of the topoisomerase IV-mediated effect isby pathway c (Figure 10). Since induced repairfunctions contribute to bactericidal kinetics (Fig-ure 8C), a complete explanation of the kineticresponse is likely to be complex.
According to the scheme in Figure 10, pathway dis responsible for the lethal effect of ciprofloxacinwith strain CR104 (parC CipR) when also treatedwith chloramphenicol. In this case the target isgyrase. Since inhibition of protein synthesis blocksinduced repair functions, the biphasic response(Figure 8E) suggests that the putative gyrasesubunit dissociation mode kills cells after a delay.Explaining this delay requires additional exper-imentation.
Preliminary examination of DNA fragmentsobtained following ciprofloxacin treatment of a gyrA(NalR) mutant suggests that topoisomerase IVinteracts with the chromosome at widely dispersedsites. This would allow topoisomerase IV todecatenate DNA during replication (Peng &Marians, 1993a). Through its association withmembrane (Kato et al., 1992), topoisomerase IVmight also serve to attach the chromosome to themembrane (Drlica et al., 1978). Gyrase would thenspecialize in maintaining superhelical tension(Gellert et al., 1976a; Drlica & Snyder, 1978) and inrelaxing torsional tension arising from replicationfork movement (Drlica et al., 1980). Whether gyrasenormally supplements the decatenating activity oftopoisomerase IV is not known (Steck & Drlica,1984; Bliska & Cozzarelli, 1987; Kato et al., 1990;Adams et al., 1992).
Materials and Methods
Bacteriological methods and chemicals
The Escherichia coli K-12 strains studied were AQ8300(lexA-3 (Ind−) malF3089::Tn10) obtained from T. Kogoma,DM4100 (Sternglanz et al., 1981), NI747 (Gellert et al.,1976a), GP201 (gyrA-307 NalR (Pruss et al., 1986)), and

Topoisomerase–Chromosome Interactions636
NK6027 (metC::Tn10, obtained from B. Bachmann). CR1and CR3 were spontaneous ciprofloxacin-resistant mu-tants of strain GP201; CR104 was a gyrA+ zeg-298::Tn10transductant of CR1 isolated by screening for sensitivityto nalidixic acid. For DNA cleavage studies, cells weregrown in M9 medium (Miller, 1972) supplemented with40 mg/ml thymine, 1 mg/ml vitamin B1, and 60 mg/mlleucine, isoleucine, valine, and threonine for strain NI747;the only amino acid supplement for GP201 was cysteine.For all other experiments growth was in LB medium(Miller, 1972). Bacteriophage P1-mediated transduction(Wall & Harriman, 1974) was used to construct ametC::Tn10 parC-1215 (Kato et al., 1988) derivative ofDM4100 (KD1100), a lexA-3 derivative of GP201(KD1237), and to map ciprofloxacin resistance. Cells weretreated with chloramphenicol (20 mg/ml) five minutesbefore the addition of quinolone, unless otherwiseindicated, and this was continued for the duration of thequinolone treatment. Radiolabelled compounds wereobtained from Amersham Chemical Co. Chlorampheni-col, nalidixic acid, oxolinic acid, and ciprofloxacin wereproducts of Sigma Chemical Co.
Detection of quinolone-induced DNA cleavage
Release of chromosomal DNA breaks was monitoredby sedimenting 3H-labelled nucleoids in 10% to 30%(w/v) sucrose density-gradients containing variousconcentrations of ethidium bromide (Hsieh et al., 1991) inthe absence of sodium dodecyl sulfate. Breakage ofchromosomal DNA in the presence of sodium dodecylsulfate was monitored by sedimentation of DNA into 5%to 20% (w/v) sucrose gradients containing sodiumdodecyl sulfate, following treatment of cell lysates withsodium dodecyl sulfate (Snyder & Drlica, 1979).Radioactive labelling, centrifugation conditions, gradientfractionation, and radioactive counting were as described(Drlica & Snyder, 1978; Snyder & Drlica, 1979). Centri-fugation was performed with Beckman SW50.1 rotors.
AcknowledgementsWe thank Drs R. Burger, M. Gennaro, and S. Kayman
for critical comments on the manuscript, J.-I. Kato andT. Kogoma for providing strains carrying parC-1215and lexA-3 alleles, respectively, and N. Cozzarelli forcommunicating experimental results prior to publication.A portion of the work was performed at the University ofRochester. Support was from NIH grant AI35257.
ReferencesAdams, D., Shekhtman, E., Zechiedrich, E., Schmid, M. &
Cozzarelli, N. (1992). The role of topoisomerase IV inpartitioning bacterial replicons and the structure ofcatenated intermediates in DNA replication. Cell, 71,277–288.
Bliska, J. & Cozzarelli, N. (1987). Use of site-specificrecombination as a probe of DNA structure andmetabolism in vivo. J. Mol. Biol. 194, 205–218.
Chow, R., Dougherty, T., Fraimow, H., Bellin, E. &Miller, M. (1988). Association between early inhi-bition of DNA synthesis and the MICs and MBCs ofcarboxyquinolone antimicrobial agents for wild-typeand mutant [gyrA nfxB(ompF) acrA] Escherichia coliK-12. Antimicrob. Agents Chemother. 32, 1113–1118.
Deitz, W. H., Cook, T. M. & Goss, W. A. (1966).Mechanism of action of nalidixic acid on Escherichiacoli. III. Conditions required for lethality. J. Bacteriol.91, 768–773.
DiNardo, S., Voelkel, K., Sternglanz, R., Reynolds, A. &Wright, A. (1982). Escherichia coli DNA topoiso-merase I mutants have compensatory mutations inDNA gyrase genes. Cell, 31, 43–51.
Drlica, K. & Snyder, M. (1978). Superhelical Escherichiacoli DNA: relaxation by coumermycin. J. Mol. Biol.120, 145–154.
Drlica, K., Burgi, E. & Worcel, A. (1978). Association ofthe folded chromosome with the cell envelope ofEscherichia coli: characterization of membrane-associ-ated DNA. J. Bacteriol. 134, 1108–1116.
Drlica, K., Manes, S. H. & Engle, E. C. (1980). DNA gyraseon the bacterial chromosome: possibility of twolevels of action. Proc. Natl Acad. Sci. USA, 77,6879–6883.
Drlica, K., Franco, R. & Steck, T. (1988). Rifampicin andrpoB mutations can alter DNA supercoiling inEscherichia coli. J. Bacteriol. 170, 4983–4985.
Drlica, K., Pruss, G., Burger, R., Franco, R., Hsieh, L.-S.& Berger, B. (1990). Roles of DNA topoisomerases inbacterial chromosome structure and function. In TheBacterial Chromosome (Drlica, K. & Riley, M., eds),pp. 195–204, American Society for Microbiology,Washington, DC.
Ferrero, L., Cameron, B., Manse, B., Lagneaux, D.,Crouzet, J., Famechon, A. & Blanche, F. (1994).Cloning and primary structure of Staphylococcusaureus DNA topoisomerase IV: a primary target offluoroquinolones. Mol. Microbiol. 13, 641–653.
Ferrero, L., Cameron, B. & Couzet, J. (1995). Analysis ofgyrA and grlA mutations in stepwise-selectedciprofloxacin-resistant mutants of Staphylococcusaureus. Antimicrob. Agents Chemother. 39, 1554–1558.
Franco, R. & Drlica, K. (1988). DNA gyrase on thebacterial chromosome: oxolinic acid-induced DNAcleavage in the dnaA-gyrB region. J. Mol. Biol. 201,229–233.
Gellert, M., O’Dea, M. H., Itoh, T. & Tomizawa, J.-I.(1976a). Novobiocin and coumermycin inhibit DNAsupercoiling catalyzed by DNA gyrase. Proc. NatlAcad. Sci. USA, 73, 4474–4478.
Gellert, M., O’Dea, M. H., Mizuuchi, K. & Nash, H.(1976b). DNA gyrase: an enzyme that introducessuperhelical turns into DNA. Proc. Natl Acad. Sci.USA, 73, 3872–3876.
Gellert, M., Mizuuchi, K., O’Dea, M. H., Itoh, T. &Tomizawa, J.-L. (1977). Nalidixic acid resistance: asecond genetic character involved in DNA gyraseactivity. Proc. Natl Acad. Sci. USA, 74, 4772–4776.
Hane, M. W. & Wood, T. H. (1969). Escherichia coli K-12mutants resistant to nalidixic acid: genetic mappingand dominance studies. J. Bacteriol. 99, 238–241.
Hoshino, M. W., Kitamura, A., Morrissey, I., Sato, K., Kato,J.-I. & Ikeda, H. (1994). Comparison of inhibition ofEscherichia coli topoisomerase IV by quinolones withDNA gyrase inhibition. Antimicrob. Agents Chemother.38, 2623–2627.
Howard, B. M., Pinney, R. J. & Smith, J. T. (1993a).4-Quinolone bactericidal mechanisms. Arzneimit-telforschung, 43, 1125–1129.
Howard, B. M., Pinney, R. J. & Smith, J. T. (1993b).Function of the SOS process in repair of DNAdamage induced by modern 4-quinolones. J. Pharm.Pharmacol. 45, 658–662.
Hsieh, L.-S., Burger, R. M. & Drlica, K. (1991). Bacterial

Topoisomerase–Chromosome Interactions 637
DNA supercoiling and [ATP]/[ADP]: changes associ-ated with a transition to anaerobic growth. J. Mol.Biol. 219, 443–450.
Ikeda, H. (1994). DNA topoisomerase-mediated illegiti-mate recombination. Advan. Pharmacol. 29A, 147–165.
Ikeda, H., Moriya, K. & Matsumoto, T. (1981). In vitrostudy of illegitimate recombination: involvement ofDNA gyrase. Cold Spring Harbor Symp. Quant. Biol.45, 399–408.
Ikeda, H., Aoki, K. & Naito, A. (1982). Illegitimaterecombination mediated in vitro by DNA gyraseof Escherichia coli: structure of recombinantDNA molecules. Proc. Natl Acad. Sci. USA, 79,3724–3728.
Kato, J.-I., Nishimura, Y. & Hirota, M. (1988). Geneorganization in the region containing a new geneinvolved in chromosome partition in Escherichia coli.J. Bacteriol. 170, 3967–3977.
Kato, J.-I., Nishimura, Y., Imamura, R., Niki, H., Hiraga,S. & Suzuki, H. (1990). New topoisomerase essentialfor chromosome segregation in E. coli. Cell, 63,393–404.
Kato, J.-I., Suzuki, H. & Ikeda, H. (1992). Purificationand characterization of DNA topoisomerase IV inEscherichia coli. J. Biol. Chem. 267, 25676–25684.
Khodursky, A. B., Zechiedrich, E. L. & Cozzarelli, N. R.(1995). Topoisomerase IV is a target of quinolones inEscherichia coli. Proc. Natl Acad. Sci. USA, 92,11801–11805.
Lewin, C., Howard, B., Ratcliffe, N. & Smith, J. (1989).4-Quinolones and the SOS response. J. Med. Microbiol.29, 139–144.
Lewin, C., Howard, B. & Smith, J. (1991). Protein- andRNA-synthesis independent bactericidal activity ofciprofloxacin that involves the A subunit of DNAgyrase. J. Med. Microbiol. 34, 19–22.
Miller, J. (1972). Experiments in Molecular Genetics. ColdSpring Harbor Laboratory Press, Cold Spring Harbor,NY.
Miura-Masuda, A. & Ikeda, H. (1990). The DNA gyraseof Escherichia coli participates in the formation of aspontaneous deletion by recA-independent recombi-nation in vivo. Mol. Gen. Genet. 220, 345–352.
Peng, H. & Marians, K. (1993a). Decatenation activity oftopoisomerase IV during oriC and pBR322 DNAreplication in vitro. Proc. Natl Acad. Sci. USA, 90,8571–8575.
Peng, H. & Marians, K. (1993b). Escherichia coli
topoisomerase IV: purification, characterization, sub-unit structure, and subunit interactions. J. Biol. Chem.268, 24481–24490.
Pruss, G. J., Manes, S. H. & Drlica, K. (1982). Escherichiacoli DNA topoisomerase I mutants: increasedsupercoiling is corrected by mutations near gyrasegenes. Cell, 31, 35–42.
Pruss, G. J., Franco, R., Chevalier, S., Manes, S. H. &Drlica, K. (1986). Effects of DNA gyrase inhibitors inEscherichia coli topoisomerase I mutants. J. Bacteriol.168, 276–282.
Rinken, R. & Wackernagel, W. (1992). Inhibition of therecBCD-dependent activation of chi recombinationalhot spots in SOS-induced cells of Escherichia coli.J. Bacteriol. 174, 1172–1178.
Snyder, M. & Drlica, K. (1979). DNA gyrase on thebacterial chromosome: DNA cleavage induced byoxolinic acid. J. Mol. Biol. 131, 287–302.
Steck, T. R. & Drlica, K. (1984). Bacterial chromosomesegregation: evidence for DNA gyrase involvement indecatenation. Cell, 36, 1081–1088.
Sternglanz, R., DiNardo, S., Voelkel, K. A., Nishimura, Y.,Hirota, Y., Becherer, A. K., Zumstein, L. & Wang, J. C.(1981). Mutations in the gene coding for Escherichiacoli DNA topoisomerase I affecting transcriptionand transposition. Proc. Natl Acad. Sci. USA, 78,2747–2751.
Sugino, A., Peebles, C., Kruezer, K. & Cozzarelli, N.(1977). Mechanism of action of nalidixic acid:purification of Escherichia coli nalA gene product andits relationship to DNA gyrase and a novelnicking-closing enzyme. Proc. Natl Acad. Sci. USA, 74,4767–4771.
Urios, A., Herrera, G., Aleixandre, V. & Blanco, M. (1991).Influence of recA mutations on gyrA dependentquinolone resistance. Biochimie, 73, 519–521.
Wall, J. D. & Harriman, P. D. (1974). Phage P1 mutantswith altered transducing abilities for Eschericia coli.Virology, 59, 532–544.
Worcel, A. & Burgi, E. (1972). On the structure of thefolded chromosome of Escherichia coli. J. Mol. Biol. 71,127–147.
Zimm, B. (1974). Anomalies in sedimentation. IV.Decrease in sedimentation coefficients of chains athigh fields. Biophys. Chem. 1, 279–291.
Zimm, B. H. & Schumaker, V. N. (1976). Anomalies insedimentation. V. Chains at high fields, practicalconsequences. Biophys. Chem. 5, 265–270.
Edited by M. Gottesman
(Received 24 July 1995; received in revised form 5 February 1996; accepted 19 February 1996)




















