
Preparation and characterization of sulfonated polyethersulfone for cation-exchange membranes

European Journal of Soil Science, June 1997, 48, 249-262
Release of Al by hydroxy-interlayered vermiculite and hydroxy-interlayered smectite during determination of cation exchange capacity in fine earth and rock fragments fractions
G. C O R T I , A. A G N E L L I & F.C. U G O L I N I Dipartimento di Scienza del Suolo e Nutrizione della Pianta, Universitci degli Studi di Firenze, Piazzale delle Cascine 28, 50 I44 Firenze, Ita Iy
Summary
The fine earth ( < 2 mm) and rock fragments (> 2 mm) fractions of two soils derived from Oligocene sandstone have been examined to assess the origin of the discrepancies between cation exchange capacity (CEC) and effective CEC (ECEC). The soils differ in terms of acidity: soil A is more acid than soil B. When the A samples are treated with BaC12, the solution became sufficiently acid (pH < 4.5) to dissolve and to maintain A1 in solution. From these samples more A1 is released than base cations. Aluminium was continuously replenished even after 192 h, so that the ECEC was always larger than the CEC. Samples from soil B contain less H and A1 ions, and the BaC12 solution could not lower the pH below 5.0. In these samples little A1 is released, and the base cations dominate the exchangeable pool of ions. This A1 can be considered to be exchangeable, and a good agreement exists between the ECEC and the CEC. The source of non-exchangeable A1 in the A samples is the OH- A1 polymers of the hydroxy-interlayered vermiculite (HIV) and hydroxy- interlayered smectite (HIS) that tend to dissolve during the BaC12 treatments. In the less acid B samples the A1 polymers are not affected by BaC12 treatment. Different results were obtained when the clays, extracted from an Na-dispersed suspension, were treated with BaCI2 solution. Because the clays are no longer acid, no H+ is released, and the OH-A1 polymers are not dissolved. Therefore, the saturating ions play an important role in the dissolution of the OH-A1 polymers and cause differences between the CEC and ECEC. We discount organic matter and specifically Al- organo complexes as a source of non-exchangeable Al. Both A and B soils contain very similar pyrophosphate-extractable Al, but show substantial differences in the amount of exchangeable Al.
Introduction
Discrepancies between cation exchange capacity (CEC) determined by compulsive methods (Cecconi & Polesello, 1956; Gillman, 1979; Gillman & Sumpter, 1986) and by summation of the exchangeable cations including A1 and H- the effective CEC or ECEC-have been widely reported (e.g. Rabenhorst et al., 1982; Gillman & Sumpter, 1985; Matsue & Wada, 1985). These discrepancies are found in soils that differ considerably in their genesis and properties, suggesting that many processes are involved. In Andosols, for example, the use of the compulsive method, where the counter ion SO:- is used, overestimates the CEC because of
Correspondence: G. Cork E-mail: corti @cscs.fi.cnr.it Received 18 June 1996; revised version accepted 19 November 1996
the specific adsorption of SO$- on allophane or imogolite or both (Masue & Wada, 1985). In soils derived from ultrabasic rocks, primary minerals dissolve when they are saturated with Ba and then washed with MgS04. In this case the solubiliza- tion of Ca and Mg from the mineral structures causes the underestimation of the CEC (Corti, 1993). In other cases, the discrepancies are due to an overestimation of the sum of cations caused by the presence of soluble A1 only in part derived from the exchange sites (Arai, 1975; Amedee & Peech, 1976). Ponette et al. (1996) also had these problems and listed the factors that might affect the extraction of non- exchangeable Al.
In general, in the determinations of cation exchange capacity much attention has been given to the methodology, to the physical characteristics of the interaction between soil and solution, and to contact time. Less frequently, other
6 1997 Blackwell Science Ltd. 249

250 G. Corti et a1
properties have been taken into account. When these are con- sidered, the soil pH and the organic matter content have been regarded as being responsible for the release of the non- exchangeable A1 (e.g. Amedee & Peech, 1976; Bloom et al., 1979; Oates & Kamprath, 1983; Ponette et al., 1996). Only a few authors have considered the mineral structure important for the release of non-exchangeable A1 during cation exchange (e.g. Sivasubramaniam & Talibudeen, 1972; Juo & Kamprath, 1979; Gillman & Sumpter, 1985). Juo & Kamprath (1979) proposed the interlayered hydroxy-Al in vermiculite and smectite as sources of reactive A1 in acid soils. Until now, however, only Lee et al. (1985) considered the dioctahedral vermiculite with the interlayered Al and Al-chlorite as the minerals giving incorrect values of ECEC because non- exchangeable A1 was released. Nevertheless, as far as we know, no one has reported on the specific role of hydroxy- interlayered vermiculite (HIV) and hydroxy-interlayered smectite (HIS) as the main sources of non-exchangeable A1 when soil samples are treated with unbuffered BaC12 solutions. However, some authors have observed that, during the displacement by unbuffered solution, a decreasing pH causes the release of non-exchangeable A1 that could be wrongly attributed to the exchangeable pool (Ayres et al., 1965; Juo & Kamprath, 1979; Ponette et al., 1996).
In the course of characterizing stony soils, we have found that rock fragments are not inert. In some cases, amongst other features, the rock fragments display a CEC approaching that of the fine earth (Ugolini et al., 1996). In the present study we have compared the CEC of the fine earth with that of the rock fragments in two soil profiles of different acidity, and we have found that the non-exchangeable A1 derives from dissolution of the interlayered OH-AI polymers of HIV and HIS. The release of non-exchangeable Al occurred specifically when Ba2+ is used as the replacing ion.
Materials
The soils examined come from two sites in the Vallombrosa Forest, 50 km E of Firenze, Italy. The two sites are: (i) Cavalla, at 1100 m above sea level in a plantation of Abies alba Mill., and (ii) Termine, at 1250 m above sea level in a plantation of Fagus sylvatica L. Both plantations are 70 years old, and the soils beneath them are Typic Haplumbrepts fine loamy, mixed, mesic (Soil Survey Staff, 1994). Profile descriptions of the two sites are given in the Appendix. The soils are developed on the ‘Arenaria del Falterona’, a sand- stone (Oligocene) consisting of coarse turbidites intercalated with siltstone.
The soil material used in these experiments consisted of the fine earth ( < 2 mm) and the skeleton or rock fragments (> 2 mm) obtained by sieving air dried samples. The washed rock fragments were classed according to lithology (sandstone and siltstone) and the degree of alteration (highly altered, moderately altered and slightly altered); the details of the
procedure for separation and classing are given in Ugolini et al. (1996). The classification adopted allows for a correlation between the degree of alteration and the size of the clasts. The highly altered fraction represents the smallest, the slightly altered the coarsest and the moderately altered the intermediate size fraction. The largest clasts in the slightly altered class had a maximum diameter of about 24-25 cm, whilst the highly altered clasts ranged from 2 to 8 mm at Cavalla and from 2 to 25 mm at Termine.
The samples analysed were obtained from the A2, Bwl and the BCxb2 horizons of the Cavalla profile and the A, Bw2 and BC2 horizons of the Termine profile. For each horizon we report on the fine earth and on the highly altered and slightly altered sandstone rock fragments.
Methods
Representative subsamples of the rock fragments of each horizon were ground until all the material passed through a 2-mm sieve. On these ground samples and fine earth the pH was measured in water, 0.01 M CaC12 and M KCl (so1id:liquid ratio I : 2.5); the bulk pH was determined potentiometrically using a combined glass-calomel electrode immersed in the suspension. Subsamples of fine earth and rock fragments were further ground until all the material passed through a 0.5-mm sieve and, on these samples, carbon was determined by the Walkley -Black method, without heating (McLeod, 1975). These ground samples were treated with Na-oxalate-oxalic acid at pH 3 (Blakemore et al., 1981) to assess the presence of amorphous Al-silicate. For this purpose, the same samples were dispersed at pH 4 and the suspended material submitted to IR analyses by a Perkin Elmer FT IR 1710 spectrometer. Gentle disintegration of clasts and fine earth was used to obtain specimens for X-ray diffraction. Mineralogy was determined with a Philips PW 1710 diffracto- meter using Fe-filtered Co-Kn radiation at 35 KV and 25 mA. Porosity, P, was calculated knowing the bulk density (Db) and the specific density (D,) of the samples from the equation P = (1 - Db/Ds).
Determination of effective cation exchange capacity (ECEC)
Representative subsamples (10-30 g) of the intact rock fragments and fine earth were placed into centrifuge tubes with 0.2 M BaC12 solution (so1id:liquid ratio 1 : 10). The use of BaC12 is recommended by Wada & Harada (1969) because it does not cause the collapse of 2: 1 expandable minerals while the Ba ion is not preferentially adsorbed. The samples were shaken gently for 5 min, left to stand for 3 min and shaken again for 5 min; after 20 min the suspensions were shaken for a few seconds and centrifuged for 5 rnin at about 450 g. The first extraction occurred 40 rnin after the mixing of solid and liquid phases. Successive extractions were made after 1.5, 2,4, 6,24,48,72,96, 120, 144, 168 and 192 h. Each extraction was
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

Release of A1 by hydroxy-interlayered vermiculite and smectite 25 1
preceded by shaking for a few seconds to re-suspend the sediments before centrifuging. Shaking and centrifuging were gentle to avoid the breaking of the clasts. Despite this pre- caution, a small amount of fine material was produced from the rock fragments. Each extracted solution was filtered through Whatman 42 filter paper, discarding the first 20% of filtrate, and analysed for Ca, Mg, Na, K, Mn, A1 and Fe. An unfiltered aliquot of this solution was used to measure the pH potentiometrically. Another set of samples was put in contact with distilled water; the pool of cations in water is called ‘soluble cations’. All analyses were done in triplicate.
The cations Ca, Mg, Na, K, Mn and Fe were measured by atomic absorption with a Perkin Elmer 1 IOOB Spectrometer. Aluminium was measured on a Varian SpectrAA 250 Plus equipped with graphite furnace (GTA 97). The cmol( +)kg-’ of exchangeable H was calculated from the difference between the pH of BaCI2 solution and the pH of water, both obtained from the suspension at each extraction time. Because of the presence of weak organic acids, some of the H+ liberated by the soil matrix by the salt effect may be readsorbed by the dissociated functional groups of the organic acids. Conse- quently, some of the liberated protons might not have contributed to the lowering of the pH. The ECEC might therefore be underestimated. Our analytical data show, however, that even if the H + values calculated are multiplied by a factor of 5 , the increase on the ECEC would be at maximum 2.5%. The values of ECEC were obtained by the summation of exchangeable Ca, Mg, Na, K, Mn, Al, Fe and H.
CEC meusurenzents
Both fine earth and rock fragment samples used for the determination of the exchangeable cations were considered Ba-saturated after 192 h of immersion in 0.2 M BaC12 solution and used to measure the CEC. The Ba-samples were washed twice with distilled water. The Ba was replaced by a single 15 min shaking treatment with 0.2 M MgS0, solution (solid: liquid ratio 1 : 10). After centrifuging for 3 min at about 450 g, an aliquot (10-20 ml) of solution was extracted, filtered and analysed for Mg using 0.05 N EDTA solution in presence of eriochrom black T buffered at pH 10 with NH4CI- NH40H. Cation exchange capacity was calculated after correction for the dilution caused by the wetting solution.
Many tests were conducted for verifying the validity of the procedure. For example, we decreased and increased the concentration of MgS04 and changed the shaking time and the centrifuging acceleration. For this purpose the Ba-saturated specimens were treated with 0.1 M and 0.4 M MgS04 solution and, in both cases, shaken for different lengths of time, the longest being 120 h, without centrifuging. Other aliquots of the same samples were treated in the same manner and centrifuged 10 min at about lo00 g. Some crushed rock frag- ment samples previously saturated with Ba for 192 h were also treated with 0.2 M MgS04 solution. The Mg was then
measured by 0.1 N EDTA and by atomic absorption. Other treatments were made to control the possibility that the Mg of the MgS04 solution could be responsible for the liberation of Ca, Mg or Al from the samples during the displacement of Ba. In these cases we used 0.2 M CuSO4 and BeS04 solutions. With these reagents the anion that forces the dislodgement of Ba is still SO,, but the replacing cation differs. All these experiments were run in triplicate.
Mineralogical investigations and behaviour of the HIV/HIS
Samples of unground fine earth and rock fragments were treated with distilled water and 0.2 M BaC12 solution, solid:liquid ratio I : 10. Portions of solid sample were separated from the suspension at different times (24, 48, 96, 192 h) by centrifuging. For each portion, a subsample was washed with distilled water, dried and powdered. Another subsample was used to obtain clay specimen for X-ray diffraction. The clay was retrieved from rock fragments through fragmentation by using gentle percussion with a pestle. The collected fine material was sonicated for 3 min at 15 MHz and sieved at 0.053 mm. The aim of this operation was to dislodge the clay minerals present in the clasts. Our intent was to examine specifically the clay minerals, regardless of whether other primary minerals were reduced to clay size. Consequently, if during the fragmentation other minerals were powdered it was not important for our purpose. The fraction <0.053 mm was suspended to separate the clay fraction in 0.001 M NaOH. On the clays obtained the following diagnostic treatments were made: saturations with Mg, K and glycerol and heating at 300 and 550°C.
To control the effects of the different treatments on the mineral particles, aliquots of the Ba-saturated fine earth and rock fragments were treated with 0.2 M MgS04 and BeS04 solutions for 15 min and washed twice with distilled water. The MgS04 and BeS04 solutions were combined with the washing solutions and analysed for Ca, Mg, Na, K, Mn, A1 and Fe. The clay fractions were separated and prepared in oriented form; then they were submitted to the diagnostic treatments mentioned above and analyzed at the X-ray diffractometer.
On the clays separated from the samples maintained in water for 192 h the following tests were made:
1 boiled in M KOH + KCI solution for 5 h (Brown, 1953); 2 boiled in 0.5 M NaOH for 5 min (Hashimoto & Jackson,
1960); 3 heated at 400°C and boiled in 0.5 M NaOH for 15 min
(Dixon & Jackson, 1962).
Furthermore, the clays were treated with:
0.2 M BaC12, 0.2 M MgS0, and 0.2 M BeS04 solutions; Na-hypochlorite (Lavkulich & Wiens, 1970); Na-dithionite-citrate-bicarbonate -DCB- (Mehra & Jack- son, 1960); Na-oxalate-oxalic acid at pH 3 (Blakemore et al., 1981).
t1 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

252 G. Corti et al.
Table 1 (Vallombrosa, Italy)
Selected chemical, physical and mineralogical properties for fine earth and rock fragments of Cavalla and Termine profiles
pH(H20) pH(CaC12) pH(KCI) Organic C Porosity Clay mineralogya 1% I% by volume
Cavalla A2
Bwl
BCxb2
Termine A
Bw2
BC2
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
4.59 4.36 4.64 4.75 4.60 4.80 5.06 4.7 1 4.83
5.53 6.15 5.89 6.30 5.97 5.74 6.55 6.50 6.3 I
4.02 4.27 4.61 4.14 4.23 4.52 4.28 4.46 4.44
4.89 4.96 5.05 5.59 5.62 5.31 5.77 5.78 5.93
3.74 4.03 4.5 I 3.67 4.14 4.40 3.74 4.09 4.12
4.55 4.29 4.44 4.98 5.02 4.77 4.94 4.84 4.82
3.36 1.72 0.24 1.29 0.59 0.10 0.26 0.07 0.06
5.21 0.32 0.22 0.90 0.10 0.08 0.30 0.07 0.04
13.2 43.3 21 .o 67.2 32.8 18.4 42.6 36.8 35.0
64.3 22.6 18.1 61.7 15.0 15.4 38.5 22.4 11.1
HIV, K, M, C, K-HIV, M-V HIV, K, M, C, M-V, K-HIV M,HIV,K,C,MV-V,K-HIV HIS, C, K, M, HIV, K-HIV, M-V M, C, HIS, K, M-V, V, HIV, K-HIV M, HIS, K, C, M-V, HIV, K-HIV HIS, M, K, C, M-V, V, K-HIV HIS, M, K, C, M-V, V, C-S, M-S HIS, M, K, C, M-V, C-S, M-S
HIV, K, M, M-V, K-HIV, C M, K, HIV, C, M-V, K-HIV M, K, HIV, C, K-HIV K, M, HIV, C, M-V, C-S, K-HIV K, M, HIV, C, M-V, C-S, K-HIV K, M, HIV, C, C-S, K-HIV K, M, HIV, V, C, M-V, K-HIV M, HIV, K, C, M-V M, HIV, K
"The order of the symbols represents the relative amount, the first phyllosilicate being the dominant one. high. alt., highly altered; sligh. alt., slightly altered; SS, sandstone; HIV, hydroxy-A1 interlayered vermiculite; HIS, hydroxy-Al interlayered smectite; K, kaolinite; M, micaceous minerals; C, chlorite; M-V, mica-vermiculite interlayered; M-S, mica-smectite interlayered; C-S, chlorite-smectite interlayered; K-HIV, kaolinite-HIV interlayered.
These treated clays were prepared in oriented form and submitted to the diagnostic treatments (saturations with Mg, K and glycerol and heatings at 300 and 550°C).
Results and discussion
Before discussing the cation exchange experiments, we should consider some of the physical and chemical properties of the rock fragments of the two profiles at Cavalla and Termine. These properties are in turn compared with those of the fine earth. The measurements for these fractions for both profiles are presented in Table 1.
The bulk pH shows considerable differences in the two soils. Rock fragments and the fine earth of Cavalla are more acid than those of Termine. The lowest pH(KCI), less than 4, is in the fine earth of Cavalla. Rock fragments are less acid than the fine earth in the A horizon of Termine, but more so in the Bw2 and BC2 horizons. At Cavalla the pH(H20) of the rock fragments was equal to or less than that of the fine earth. The bulk pH of the rock fragments is a function of weathering which in turn controls porosity and degree of alteration of the primary minerals (Ugolini et al., 1996).
The organic C content decreases with depth, both in the fine earth and in the rock fragments, in the two profiles. The C content in the highly altered clasts of Cavalla and Termine is
surprisingly large; the clasts in the A horizons contain respectively 1.72% and 0.32% C. As expected, the skeletal material is less porous than the fine earth, but some of the rock fragments are unexpectedly porous.
The HIV and HIS are present in the rock fragments, but tend to be more abundant in the fine earth.
The pH trend, the organic C content, the porosity and the mineralogical composition of the rock fragments indicate that these properties approach those of the fine earth; consequently the rock fragments are not inert diluents of the fine earth, hence they could play a role in the cation exchange of these soils.
Long term exchange experiments
The pH measured in H20 and BaCI2 solution for the fine earth and rock fragments shows a general tendency to decrease as the time increases over 192 h (Fig. 1). The samples of Terrnine are less acid, and the pH tends to decrease with time less markedly than in those of Cavalla. The separation between the pH curves obtained in H20 and BaCI2 is less pronounced at Terrnine than at Cavalla (Fig. 1). It appears that the soil samples from Cavalla contain more exchangeable acidity than those from Termine. We do not have a definite explanation for the discrepancy between the pH values measured in water reported in Table 1 and Fig. 1. One possibility is the difference
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

Release of A1 by hydroxy-interlayered vermiculite and smectite 253
Cavalla profile
A2 horizon 7 -
I, 5'5 5 1 4 4 1 , 3 5
0 7 1 5 2 4 6 24 48 72 96 120 144 168 192
Bwl horizon
7
6.5 1 \
4:11, 3 5
0 7 1 5 2 4 6 24 48 72 96 120 144 168 192
BCxb2 horizon
6.5 7x 1
35 '0.7 '1.5 I 2 ' 4 I 6 I 24 ' 4 8 ' 7 2 ' 96 '120'144116811921
Tirne/h
Termine profile
A horizon 7 7
6 24 48 72 96 120 144 168 192 4.5
Bw2 horizon
6.5 ' 1 1
0.7 '1.5 2 ' 4 6 24 48 72 96 120 144 168 192 4.5 ! , 1 1 1 1 1 1 1 1 1
BC2 horizon
6.5
6 24 48 72 96 120 144 168 192 4.5
Time/h
Fig. 1 pH curves of H 2 0 and BaCI2 solutions for fine earth and rock fragments as a function of time, Cavalla and Termine profiles (Vallombrosa, Italy). 0, BaC12 fine earth; A, HzO fine earth; 0. BaCIz highly altered SS; m, BaCI2 slightly altered SS; V, H 2 0 highly altered SS; 1, H 2 0 slightly altered SS.
in solid : liquid ratios of the suspensions. The pH values reported in Table 1 were measured in 1 :2.5 soil : water ratio, while the pH values shown in Fig. 1 were obtained in 1 : 10 soil :water ratio.
The ECEC for the four extraction intervals 40 min, 48 h, 120 h and 192 h is reported in Table 2. The table shows that the values of ECEC increase with time of contact. Summation
of cations including Ca, Mg, Na, K, Mn, Fe and H as function of duration of the extractions for both Cavalla and Termine profiles are presented in Fig. 2. In the same graphs the exchangeable A1 has been plotted separately. From each of the values plotted the amount of water-soluble cations has been subtracted. At Cavalla it appears that: (i) the exchangeable A1 exceeds the sum of cations; (ii) in the fine earth, the
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

254 G. Corti et al.
Cavalla A2
Bwl
BCxb2
Termine A
Bw2
BC2
Time intervals
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
40 min
5.57 (0.10) 2.67 (0.05) Q.65 (0.03) 1.39 (0.10) 2.66 (0.03) 0.59 (0.02) 4.56 (0.10) 2.70 (0.03) 1.00 (0.03)
13.46 (0.17) 1.46 (0.03) 0.68 (0.02) 6.22 (0.07) 1.17 (0.02) 0.94 (0.05) 6.26 (0.09) 1.79 (0.05) 1.17 (0.05)
48 h 120 h
cmol(+)kg- ' 6.83 (0.12) 4.82 (0.09) 1.30 (0.05) 5.03 (0.12) 3.78 (0.05) 1.68 (0.03) 5.89 (0.12) 4.55 (0.07) 3.45 (0.09)
13.42 (0.11) 3.88 (0.04) 1.38 (0.02) 6.47 (0.07) 2.84 (0.03) 3.06 (0.07) 6.44 (0.07) 4.31 (0.10) 3.98 (0.05)
6.93 (0.10) 5.35 (0.05) 1.59 (0.03) 5.07 (0.06) 4.32 (0.05) 2.20 (0.05) 6.14 (0.10) 5.09 (0.07) 4.11 (0.10)
13.53 (0.17) 3.82 (0.04) 1.61 (0.05) 6.59 (0.05) 2.86 (0.05) 3.20 (0.07) 6.47 (0.07) 4.53 (0.09) 4.10 (0.05)
192 h
7.21 (0.12) 5.39 (0.07) 1.81 (0.05) 5.21 (0.12) 4.33 (0.07) 2.38 (0.05) 6.12 (0.10) 5.1 1 (0.07) 4.20 (0.09)
13.59 (0.1 1) 3.90 (0.07) 1.68 (0.02) 6.62 (0.05) 2.91 (0.05) 3.23 (0.07) 6.60 (0.09) 4.49 (0.09) 4.30 (0.05)
high. alt., highly altered; sligh. alt., slightly altered; SS, sandstone.
Table 3 Values of organically bound Al extracted by pyro- phosphate (Alp) and exchangeable Al at 192 h of immersion in 012 M BaClz solution for fine earth and rock fragments of Cavalla and Termine (Vallombrosa, Italy)
Exchangeable Al Alp at 192 h
Cavalla A2
Bw I
BCxb2
Termine A
Bw2
BC2
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS fine earth high. alt. SS sligh. alt. SS
/mg kg-' /cmol(+)kg-'
3.2 5.50 3.3 4.28 1.5 1.54 4.6 4.10 3.2 3.32 1.8 1.81 2.6 3.80 1.1 3.21 I .5 2.63
4.7 0.38 1.8 0.05 I .5 0.05 3.0 0.05 1.9 0.04 1.8 0.02 1.6 0.01 1.3 0.04 1.1 0.02
high. alt., highly altered; sligh. alt., slightly altered; SS, sandstone.
Table 2 Summation of exchangeable cations including Al and H (ECEC, effective cation exchange capacity) for different time intervals of fine earth and rock fragments, Cavalla and Termine profiles (Vallombrosa, Italy). Values in parentheses are the standard errors calcu- lated on three replicates
exchangeable A1 does not reach equilibrium at the end of the 192 h, whereas the sum of cations does so after only a few hours; and (iii) in the rock fragments the exchangeable Al reaches equilibrium only after the sum of cations. Other authors have obtained similar results with acid soils and exchangeable Al. For exampie, Amedee & Peech (1976) and Grove et al. ( 1 982) recognized the predominance of A1 among the exchangeable cations in acid soils, and they and others attributed this fact to the decrease in pH caused when a neutral salt is added to an acid soil (Yuan & h k e l l , 1959; Dewan & Rich, 1970). According to Ayres et al. (1965) and Gillman & Sumpter (1 985) the exchangeable A1 becomes predominant on the exchange complex when the pH of soil-solution falls below 5. In soils containing crystalline and amorphous Fe and A1 oxyhydroxides and organic matter the addition of concentrated solutions of neutral salts enhances the negative charges on variable charge surfaces as a result of proton release. These protons are responsible for the Al-oxyhydroxides dissolution and of the solubilization of any other solid-phase A1 (Amedee & Peech, 1976). In the light of these previous findings we offer the following interpretation of our data. Firstly, let us examine the possible sources of exchangeable Al. One could be the A1 released from the Al-organo complexes. If this is the case, a correlation must exist between the pyrophosphate extractable A1 (Al,) and the exchangeable Al. The lack of this correlation can be promptly seen by comparing the data of Alp with those of the exchangeable A1 after 192 h of contact time (Table 3). Whereas the amount of Alp in the samples of Cavalla and
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262
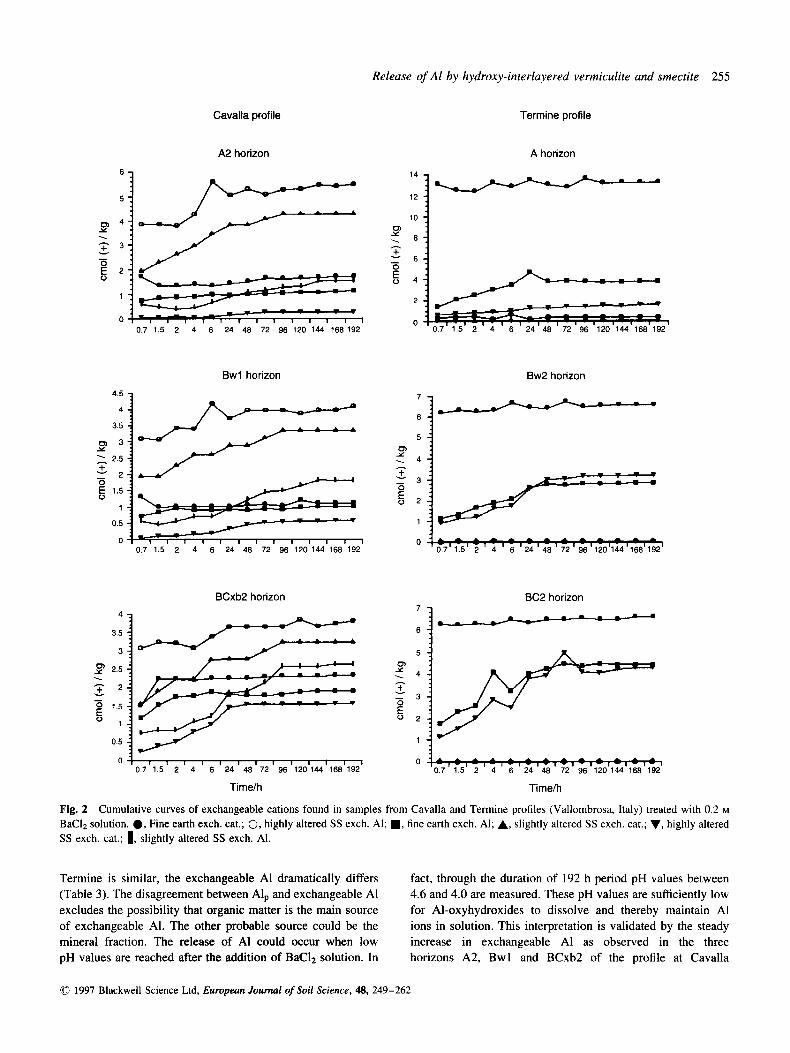
Release of A1 by hydroxy-interlayered vermiculite and smectite 255
Cavalla profile Termine profile
A2 horizon A horizon
0.7 1.5 2 4 6 24 48 72 96 120 144 168 192
2
0 0.7 1.5 2 6 24 48 72 96 120 144 168 192
Bw l horizon Bw2 horizon 4.5
4
Y 1 2.5 + 6 1.5
1
0.5
0 v -
0 1 , 1 1 ~ 1 1 1 , 1 1 1 1 1
0.7 1.5 2 4 6 24 48 72 96 120 144 168 192
B h 4 4
0
BCxb2 horizon BC2 horizon 4
3.5
3
2 2.5 . h v + * -
1.5
0 1
0.5
0 1 I 1 1 1 l l 1 1 1 1 1 1 I 1
0.7 1.5 2 4 6 24 48 72 96 120144 168 192
Time/h
5 0) Y . 4 h
v + - 3 0
6 2
1
0 0.7 1.5 2 4 6 24 48 72 96 120 144 166 192
Time/h
Fig. 2 Cumulative curves of exchangeable cations found in samples from Cavalla and Termine profiles (Vallombrosa, Italy) treated with 0.2 M
BaClz solution. 0, Fine earth exch. cat.; 0, highly altered SS exch. Al; W , fine earth exch. Al; A, slightly altered SS exch. cat.; V, highly altered SS exch. cat.; 1, slightly altered SS exch. Al.
Termine is similar, the exchangeable A1 dramatically differs fact, through the duration of 192 h period pH values between (Table 3). The disagreement between Alp and exchangeable A1 4.6 and 4.0 are measured. These pH values are sufficiently low excludes the possibility that organic matter is the main source for Al-oxyhydroxides to dissolve and thereby maintain A1 of exchangeable Al. The other probable source could be the ions in solution. This interpretation is validated by the steady mineral fraction. The release of A1 could occur when low increase in exchangeable Al as observed in the three pH values are reached after the addition of BaC& solution. In horizons A2, Bwl and BCxb2 of the profile at Cavalla
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

256 G. Corti et al.
(Fig. 2) . Consequently, we believe that the measured A1 in solution is derived mostly from the dissolution of the solid phase rather than from the exchange sites.
The data reveal that in Temine the exchangeable A1 never exceeds the sum of exchangeable cations (Fig. 2), while the Al liberated does not increase with time. In fact, the A1 in solution reaches equilibrium immediately after the solid-liquid contact
Table 4 Values of CEC for fine earth and rock fragments of Cavalla and Termine profiles (Vallombrosa, Italy). The Ba-saturated samples were treated with 0.2 M MgS04 solution and the excess of Mg measured by black eriochrom T and by atomic absorption. Values in parentheses are the standard errors (calculated on three replicates) which takes into account the CEC calculated measuring the Mg by titration and atomic absorption
(Fig. 2). Exchangeable cations reach equilibrium ab initio for the fine earth, but it takes about 48 h of contact for the rock fragments to reach this condition. The small quantity of A1 extracted from Termine samples, mineralogically similar to those of Cavalla, could arise because the pH of Termine suspension is never more acid than pH 5 (Fig. 1). At this hydrogen activity the Al is scarcely soluble and thus the concentration of Al-ions in solution is small (Lindsay, 1979).
In both profiles the sum of cations of the rock fragments reaches equilibrium more slowly than the fine earth. The delay is due to the slower diffusion of cations inside the clasts compared with their rapid diffusion in the fine earth. In spite of the slow diffusion, rock fragments reach equilibrium in about 48 h (Fig. 2).
The lowering of pH by the addition of a neutral salt (0.2 M
BaC12) could also have an effect on the release of cations from the minerals. This could cause an increase in cations in solution and thus an overestimation of the exchangeable cations. In view of this, we cannot exclude that in some cases a portion of the measured .base cations are not exchangeable (Fig. 2).
CEC measurements
The values of the CEC (Table 4) were obtained from the same samples used for the determination of exchangeable cations. The CEC for Cavalla fine earths gives data close to those of the highly altered clasts. The CEC of the highly altered sandstone clasts in the Bwl horizon exceeds that of the fine earth (3.68 against 1.13 cmol( +)kg-'). We offer no explanation for the small CEC of the fine earth. In the BCxb2 horizon the slightly altered rock fragments had a CEC as large as that of the fine earth. In this buried horizon the clasts have weathered for a long time (Magaldi, 1993), hence they have acquired many physico-chemical characteristics similar to those of fine earth (Corti et al., 1995).
In the Termine profile the largest CEC (14.21 cmol (+)kg-') is in the fine earth of the A horizon. The highly altered rock fragments have CECs similar to those of Cavalla profile.
Cation exchange capacity values presented in Table 4 show standard errors that agree with those obtained, using the same method, by Hendershot & Duquette (1986) and with those obtained with BaC12 leaching by Rasmussen et al. (1991).
The differences between ECEC (Table 2 ) and CEC (Table 4) measurements are considerable even if the standard errors are taken into consideration. The fairly small standard errors seem to indicate that the different results obtained are due to a
Fine earth
Cavalla A2 Bw 1 BCxb2
Termine A Bw2 BC2
4.21 (0.09) 1.13 (0.16) 3.61 (0.12)
14.21 (0.31) 7.43 (0.16) 5.68 (0.16)
Highly altered SS Slighly altered SS
/cmol( +)kg- ' 3.78 (0.07) 0.44 (0.14)
0.85 (0.14) 3.68 (0.07) 3.07 (0.07) 3.23 (0.12)
3.53 (0.07) 3.29 (0.09) 3.11 (0.10) 0.67 (0.12) 3.77 (0.09) 0.85 (0.16)
high. alt., highly altered; sligh. alt., slightly altered; SS, sandstone.
(some) repeatable process(es) that occur(s) during the determinations and invalidate the data. In fact, causes of errors might be due to the concentration of MgS04 and to the fragmentation of the samples, the latter depending on the duration of shaking and the acceleration during centrifuging. To investigate these causes we made many tests (see Methods, CEC measurements). The displacement of Ba from Ba- saturated samples with MgS04 solutions at different concen- trations gives results (Table 5) in agreement with those obtained with the 0.2 M MgS04 solution (Table 4). This agreement tends to disregard the shaking time and the centrifuging acceleration (Table 5). Cation exchange capacity of crushed rock fragments was slightly larger than those of intact samples (Table 6). We attribute the increase of CEC to new exchangeable sites created during the crushing.
The results in Tables 5 and 6 confirm the CEC values in Table 4 and indicate that the internal surfaces and exchange sites are available in a fairly short time when the second displacement occurs on a wet sample.
The reliability of the CEC values is also confirmed by the treatments of Ba-saturated samples with 0.2 M CuS04 and BeS04 solutions. In both solutions, separated by centrifuging after 15 min of solid-liquid contact, Ca2+ and Mg2+ were present in negligible amounts. The occurrence of these cations could invalidate the CEC measurements with black eriochrom T, because the indicator does not distinguish Ca from Mg, and the CEC value is obtained through a back titration. Therefore, the solubilization of these cations would have led to an erroneous (smaller) value of CEC.
Mineralogy
The experiments on Cavalla samples showed that a solution of BaClz can release non-exchangeable A1 from the fine earth and
@ 1997 BhEkwell Science Ltd, European Journal of Soil Science, 48, 249-262

Release of A1 by hydroxy-interlayered vermiculite and smectite 257
M
3 4
m $ c
0
Table 6 Values of CEC for natural and crashed rock fragments of Cavalla and Termine profiles (Vallombrosa, Italy) obtained using 0.2 M MgS04 as washing solution. Values in parentheses are the standard errors calculated on three replicates
Natural Crushed specimen specimen Difference
/cmol( +)kg- I
Cavalla A2 high. alt. SS 3.78 (0.07) 4.58 (0.07) 0.80 Bwl sligh. alt. SS 0.85 (0.14) 1.37 (0.03) 0.52
Termine Bw2 high. alt. SS 3.1 1 (0.10) 3.95 (0.05) 0.84 BC2 sligh. alt. SS 0.85 (0.16) 1.52 (0.03) 0.67
high. alt., highly altered; sligh. alt., slightly altered; SS, sandstone.
rock fragments. The question can then be posed: where is this A1 coming from? Because of the lack of evidence that the Al-organo complexes are responsible for the difference between the exchangeable A1 in the two soils, we investigated the mineral composition to find the mineral(s) phase(s) respon- sible for the release of AI.
In this investigation we failed to identify gibbsite or other distinct Al-mineral(s) by X-ray diffraction. While both the acid oxalate extraction and the IR analyses of the fraction dispersed at pH 4 indicated that amorphous Al-silicates such as allophane and imogolite are absent. Evidently, other minerals are the source of the non-exchangeable A1 in solution.
We established that the primary minerals of the fine earth and rock fragments in both profiles consist of quartz, plagio- clase, chlorite, mica and kaolinite. The X-ray diffraction patterns of several samples that were subjected to different treatments (water, BaCI2, BaC12 + MgS04 and BaC12+ BeS04) showed that none of the first-order peaks of these primary minerals were affected. Of course, we cannot exclude the possibility that there was some dissolution. If it did occur, however, it did not affect the position, the shape and the relative intensities of the peaks, within the limits of the X-ray apparatus used.
When the clay fraction separated from the fine earth and rock fragments was examined different results were obtained. In these diffractograms we observed structural changes in the HIV or HIS or both. Figure 3 shows the X-ray patterns of the clays obtained from fine earth and highly altered sandstone of Cavalla A2 horizon that have undergone different treatments. For reasons of space, we selected this horizon because the diffractograms are more evident, and only the HIV are present (Table 1). Diffractograms of the clay extracted from samples treated for 192 h in wafer indicate that the 1.4 nm peak in Fig. 3a and 3b is mostly due to the presence of HIV with interlayer filled by OH-A1 polymers. Positive identification of HIV is based on the disappearance of the 1.4 nm peak, concomitant with the appearance of a shoulder between 1.4
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262
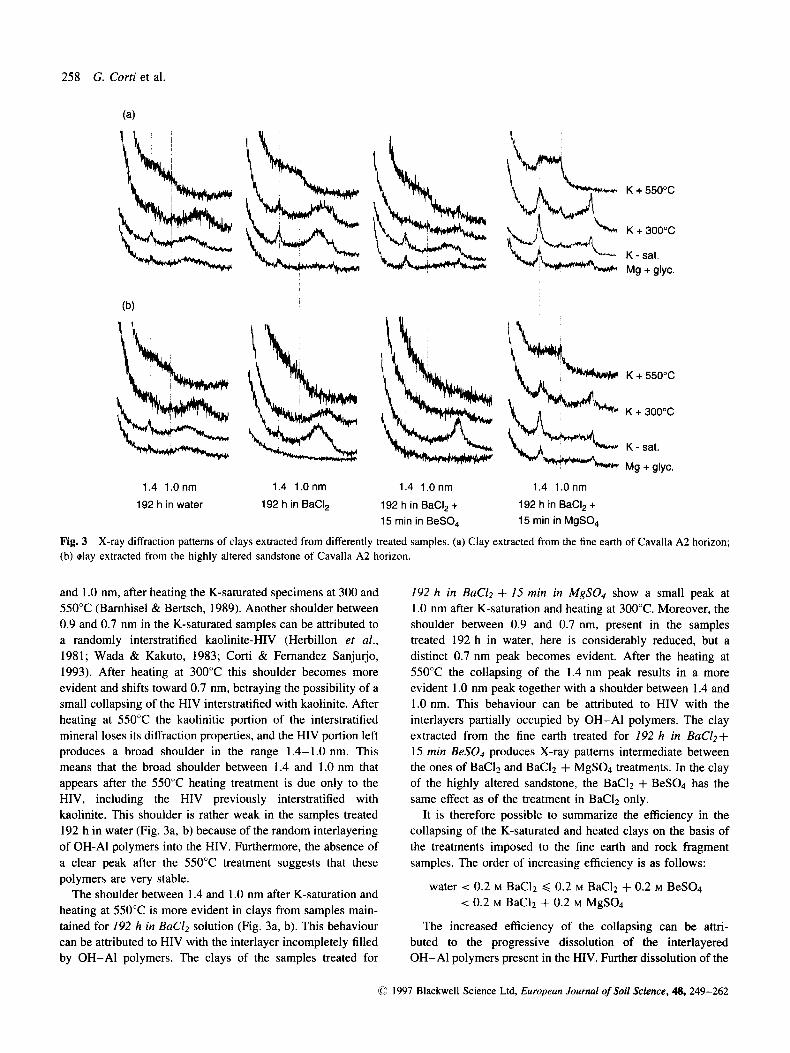
258 G. Corti et al.
1.4 1.0nm
192 h in water
1.4 1.0nm
192 h in BaCI,
K + 550°C
K + 300°C
K - sat. Mg + glyc.
1.4 1.0nm 1.4 1.0nm
192 h in BaCI, + 15 min in BeSO,
192 h in BaCI, + 15 rnin in MgSO,
K + 550°C
K + 300°C
K - sat.
Mg + glyc.
Fig. 3 (b) day extracted from the highly altered sandstone of Cavalla A2 horizon.
X-ray diffraction patterns of clays extracted from differently treated samples. (a) Clay extracted from the fine earth of Cavalla A2 horizon;
and 1 .O nm, after heating the K-saturated specimens at 300 and 550°C (Barnhisel & Bertsch, 1989). Another shoulder between 0.9 and 0.7 nm in the K-saturated samples can be attributed to a randomly interstratified kaolinite-HIV (Herbillon et al., 1981; Wada & Kakuto, 1983; Corti & Fernandez Sanjurjo, 1993). After heating at 300°C this shoulder becomes more evident and shifts toward 0.7 nm, betraying the possibility of a small collapsing of the HIV interstratified with kaolinite. After heating at 550°C the kaolinitic portion of the interstratified mineral loses its diffraction properties, and the HIV portion left produces a broad shoulder in the range 1.4-1.0 nm. This means that the broad shoulder between 1.4 and 1.0 nm that appears after the 550°C heating treatment is due only to the HIV, including the HIV previously interstratified with kaolinite. This shoulder is rather weak in the samples treated 192 h in water (Fig. 3a, b) because of the random interlayering of OH-AI polymers into the HIV. Furthermore, the absence of a clear peak after the 550°C treatment suggests that these
192 h in BaC12 + 15 min in MgSO4 show a small peak at 1 .O nm after K-saturation and heating at 300°C. Moreover, the shoulder between 0.9 and 0.7 nm, present in the samples treated 192 h in water, here is considerably reduced, but a distinct 0.7 nm peak becomes evident. After the heating at 550°C the collapsing of the 1.4 nm peak results in a more evident I .O nm peak together with a shoulder between 1.4 and 1.0 nm. This behaviour can be attributed to HIV with the interlayers partially occupied by OH-A1 polymers. The clay extracted from the fine earth treated for 192 h in BaCl2+ 1 5 min BeS04 produces X-ray patterns intermediate between the ones of BaC12 and BaC12 + MgS04 treatments. In the clay of the highly altered sandstone, the BaC12 + BeS04 has the same effect as of the treatment in BaCI2 only.
It is therefore possible to summarize the efficiency in the collapsing of the K-saturated and heated clays on the basis of the treatments imposed to the fine earth and rock fragment samples. The order of increasing efficiency is as follows:
polymers are very stable. The shoulder between 1.4 and 1 .O nm after K-saturation and
heating at 550°C is more evident in clays from samples main-
water < 0.2 M BaC12 < 0.2 M BaC12 + 0.2 M BeS04 < 0.2 M BaC12 + 0.2 M MgS04
tainedfor 192 h in BaClz solution (Fig. 3a, b). This-behaviour The increased efficiency of the collapsing can be attri- can be attributed to HIV with the interlayer incompletely filled buted to the progressive dissolution of the interlayered by OH-A1 polymers. The clays of the samples treated for OH-AI polymers present in the HIV. Further dissolution of the
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262
Release of A1 by hydroxy-interlayered vermiculite and smectite 259
K + 550°C
K + 300°C
K - sat.
Mg + glyc.
K + 550°C
K + 300°C
K - sat.
Mg + glyc.
1.4 1.0nm 1.4 1.0nm
192 h in water + Na-hypochlorite
192 h in water + DCB
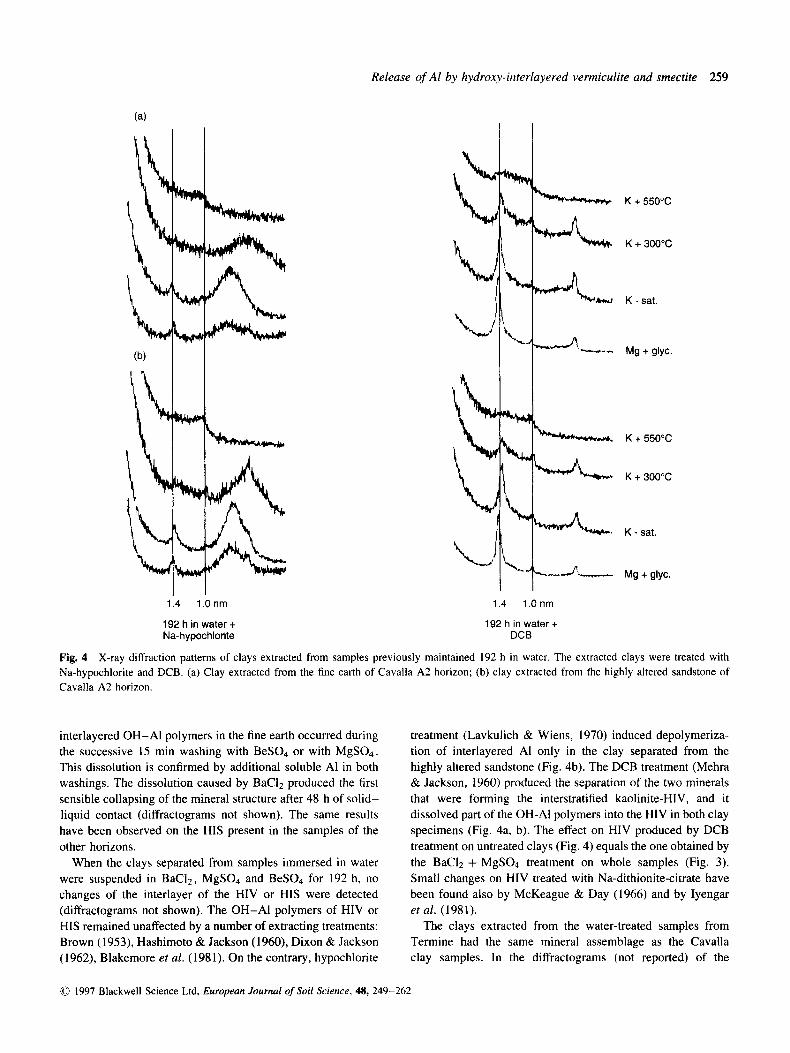
Fig. 4 X-ray diffraction patterns of clays extracted from samples previously maintained 192 h in water. The extracted clays were treated with Na-hypochlorite and DCB. (a) Clay extracted from the fine earth of Cavalla A2 horizon; (b) clay extracted from (he highly altered sandstone of Cavalla A2 horizon.
interlayered OH-A1 polymers in the fine earth occurred during the successive 15 min washing with BeS04 or with MgS04. This dissolution is confirmed by additional soluble A1 in both washings. The dissolution caused by BaClz produced the first sensible collapsing of the mineral structure after 48 h of solid- liquid contact (diffractograms not shown). The same results have been observed on the HIS present in the samples of the other horizons.
When the clays separated from samples immersed in water were suspended in BaCl2, MgS04 and BeS04 for 192 h, no changes of the interlayer of the HIV or HIS were detected (diffractograms not shown). The OH-A1 polymers of HIV or HIS remained unaffected by a number of extracting treatments: Brown (1953), Hashimoto & Jackson (1960), Dixon & Jackson (1962), Blakemore et al. (1981). On the contrary, hypochlorite
treatment (Lavkulich & Wiens, 1970) induced depolymeriza- tion of interlayered A1 only in the clay separated from the highly altered sandstone (Fig. 4b). The DCB treatment (Mehra & Jackson, 1960) produced the separation of the two minerals that were forming the interstratified kaolinite-HIV, and it dissolved part of the OH-A1 polymers into the HIV in both clay specimens (Fig. 4a, b). The effect on HIV produced by DCB treatment on untreated clays (Fig. 4) equals the one obtained by the BaC12 + MgS04 treatment on whole samples (Fig. 3). Small changes on HIV treated with Na-dithionite-citrate have been found also by McKeague & Day (1966) and by Iyengar et al. (1981).
The clays extracted from the water-treated samples from Termine had the same mineral assemblage as the Cavalla clay samples. In the diffractograms (not reported) of the
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

260 G. Corti et al.
K-saturated clays, the appearance of a shoulder between 1.4 and 1.0 nm, when the clays were heated at 550°C, led us to believe that OH- A1 polymers in the Termine HIV/HIS had the same degree of stability as those in Cavalla HIV/HIS. How- ever, the clays extracted from fine earth and rock fragment samples of Termine treated with BaCI2, MgS04 and BeS04 solutions produced X-ray diffraction patterns similar to the clays derived from the water treated samples. In addition, the MgS04 and BeS04 solutions used after BaC12 treatments did not contain Al. The clays separated from samples suspended in water and subsequently treated with neutral, acid and alkaline reagents did not show any influence on the OH-AI polymers of HIV or HIS. Only the DCB treatment produced modifica- tions of the HIV or HIS similar to those observed for the Cavalla samples.
These results indicate that OH-AI polymers of HIV or HIS are so stable that only the DCB treatment can induce their dissolution. When samples of fine earth and rock fragments of an acid soil (Cavalla) are treated with a neutral salt solution such as BaC12 and washed with MgS04 we observed the dissolution of the interlayered-Al. The dissolution of the HIV or HIS may be further helped by additional protons liberated by the hydrolysis of the Al ions in solution. An additional comment could be that the 2 : 1 minerals containing interlayered hydroxy-Al-bridges, when subjected to low pH, lose more A1 because the equilibrium between the interlayer ions and the outer solutiod is favoured by the absence of collapse (Lee et al., 1985). Also, when HIV or HIS are saturated with Ba, Mg and Be, the OH-AI polymers or the saturating cations maintain sufficiently wide spacing (1.24- 1.42 nm) to allow exchanges between the inner part of interlayers and the outer solution. If the liquid phase has a pH 6 4.5, then the dissolution of the interlayered OH-AI polymers is further helped.
Conclusions
Despite their similar organic and mineral composition, the soils from Cavalla and Termine behave differently in terms of their cation exchange. This difference can be explained by the intrinsic nature of these two profiles: Cavalla, because of different pedological history, is more acid than Termine.
In fine earth and rock fragments of Cavalla the strongly acid pH in water further decreases in the presence of a neutral salt. In these samples the BaC12 treatment lowers the pH to 64.5 , a value sufficient to induce dissolution and to maintain Al ions in solution. The pH curves in Fig. 1 provide evidence that besides dissolution, hydrolysis of the Al is also expected. Both processes determine the release of OH-A1 polymers from the HIV or HIS and thus the discharge of non-exchangeable Al. Consequently, the A1 released from samples treated with BaC12 solution even for 192 h did not reach a plateau, and the ECEC is always greater than the CEC. In these samples, the large amount of Al in the extracting solution is only partly exchangeable Al.
Samples from Termine are less acid, and the BaC12 solution cannot depress the pH below 5.0. Consequently, there is little exchangeable A1 in the fine earth and rock fragments, and the base cations dominate the pool of exchangeable ions. For Termine samples the measurements of ECEC agree well with those of CEC, and the A1 in the extracting solution could be considered really exchangeable Al.
The HIV or HIS clay minerals present in the fine earth and rock fragments are the main source of the non-exchangeable Al in the extracts of Cavalla samples. These minerals have OH-AI polymers that remain stable when the clays obtained from natural fine earth and rock fragments are submitted to numerous extracting treatments. Only the DCB treatment can dissolve part of these polymers. On the other hand, the OH-A1 polymers of the HIV or HIS become unstable and dissolve when the fine earth and rock fragments are treated initially with BaC12 (or BaC12 + MgS04). In this case the H+ present on the exchangeable complex is displaced by the Ba2+ and causes a drop in pH below 4.5. This low pH solubilizes the OH-A1 polymers from the HIV or HIS. A lowering of the suspension pH can also be reached by adding acid solution, as in the case of the 0.2 M BeS04 (pH 3.03). When clays are extracted from natural samples, and the exchangeable H is replaced by Na during the dispersion, despite the clays being derived from an acid soil, they are no longer acid clays but Na-clays. Consequently, when further treated with BaC12, exchangeable H is not present and, therefore, pH does not drop. Therefore, only in the cases when the pH of the liquid phase has a value 64.5 does the dissolution of the OH-AI polymers occur. This dissolution vitiates the ECEC and creates the discrepancy between the sum of cations (including A1 and H) and the CEC. In our study this discrepancy is more evident in the fine earth because of the abundance of HIV or HIS in this fraction. The rock fragments display to a lesser degree the difference between the ECEC and CEC because they are less acid and have a smaller content of OH-AI polymers than the fine earth.
Acknowledgements We thank the Centro di Studio per i Colloidi del Suolo (CNR) for the use of their instruments; the European Union, Direct- orate General XII, Science, Research and Development (Contract No EV5V-CT93-0280) and the CNR (indagini sulle caratteristiche fisiche e chimiche della frazione grossolana del suolo, particelle maggiori di 2 mm) for financial support.
References Amedee, G. & Peech, M. 1976. The significance of KCI-extractable
AI(II1) as an index to lime requirement of soils of the humid tropics. Soil Science, 121, 227-233.
Arai, S. 1975. Extraction of active aluminium from acid soils in Japan with different reagents. Genderma, 14, 63-74.
Ayres, A.S., Hagihara, H.H. & Stanford, G. 1965. Significance of extractable aluminum in Hawaiian sugarcane soils. Soil Science Society of America Proceedings, 29, 387-392.
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

Release of A1 by hydroxy-interlayered vermiculite and smectite 261
Barnhisel, R.I. & Bertsch, P.M. 1989. Chlorites and hydroxy- interlayered vermiculite and smectite. In: Minerals in Soil Environ- ments, 2nd edn (eds J.B. Dixon & S.B. Weed), pp. 729-788. Soil Science Society of America, Madison, WI.
Blakemore, L.C., Searle, P.L. & Daly, B.K. 1981. Soil Bureau Laboratory Merhods: A. Methods for Chemical Analyses of Soils. Department of Scientific and Industrial Research, Lower Hutt, New Zealand.
Bloom, P.R., McBride, M.B. & Weaver, R.M. 1979. Aluminum organic matter in acid soils: buffering and solution aluminum activity. Soil Science SocieQ> of America Journal, 43, 488-493.
Brown, G. 1953. The dioctahedral analogue of vermiculite. Clay Minerals Bulletin, 2, 64-70.
Cecconi, S. & Polesello, A. 1956. Metodi rapidi per la determinazione della capacith di scambio cationico (CSC) del terreno. Annuli di Sperimentazione Agraria, 10, 127- 132.
Corti, G. 1993. Influenza dell’alterazione dei minerali primari nella determinazione della capacith di scambio cationico nei suoli su rocce otiolitiche. Asrochimica, 37, 388-399.
Corti, G. & Fernandez Sanjurjo, M.J. 1993. Trasformazione pedo- genetica di smectite in caolinite in un suolo acido della Galizia (Spagna). Quaderni di Scienza del Suolo, 5, 5-22.
Corti, G., Piccardi, F., Agnelli, A. & Ugolini, F.C. 1995. Carbonio e azoto nella frazione grossolana ( > 2 mm) del suolo: quali le implicazioni? In: Atti X f f Convegno Nazionale Societa ftaliana Chimica Agraria, pp. 8 1-88. Patron Publications, Piacenza.
Dewan, H.C. & Rich, C.I. 1970. Titration of acid soils. Soil Science Society qf America Proceedings, 34, 38-44.
Dixon, J.B. & Jackson, M.L. 1962. Properties of intergradient chlorite-expansible layer silicates of soils. Soil Science Society of
America Proceedings. 26, 358 -362. Gillman, G.P. 1979. A proposed method for the measurement of
exchange properties of highly weathered soils. Aurtralian Journal qf Soil Reseurch, 17, 129- 139.
Gillman, G.P. & Sumpter, E.A. 1985. KCI-extractable aluminium in highly weathered soils. Is it exchangeable? Communications in Soil Science and Planr Analysis, 16, 561 -568.
Gillman, G.P. & Sumpter, E.A. 1986. Modification to the compulsive exchange method for measuring exchange characteristics of soils. Australian Journal of Soil Research, 24, 61 -66.
Grove, J.H., Fowler, C.S. & Sumner, M.E. 1982. Determination of the charge character of selected acid soils. Soil Science Society of America Journal, 46, 32-38.
Hashimoto, I. & Jackson, M.L. 1960. Rapid dissolution of allophane and kaolinite-halloysite after dehydration. In: Proceedings of the 7th National Conference on Clays and Clay Minerals (ed. E. Ingerson), pp. 102- 113. Pergamon Press, London.
Hendershot, W.H. & Duquette, M. 1986. A simple barium chloride method for determining cation exchange capacity and exchangeable cations. Soil Science Sociery of America Journal, 50, 605-608.
Herbillon, A.J., Frankart, R. & Vielvoye, L. 1981. An occurrence of interstratified kaolinite-smectite minerals in a red-black soil topo- sequence. Clay Minerals, 16, 195-201.
lyengar, S.S., Zelazny, L.W. & Martens, D.C. 1981. Effect of photolytic oxalate treatment on soil hydroxy-interlayered vermicu- lites. Clays and Clay Minerals, 29, 429-434.
Juo, A.S.R. & Kamprath, E.J. 1979. Copper chloride as an extractant for estimating the potentially reactive aluminum pool in acid soils. Soil Science Sociery of America Journal, 43, 35-38.
Lavkulich, L.M. & Wiens, J.H. 1970. Comparison of organic matter destruction by hydrogen peroxide and sodium hypochlorite and its effects on selected mineral constituents. Soil Science Society of America Proceedings, 34, 755-758.
Lee, R., Bache, B.W., Wilson, M.J. & Sharp, G.S. 1985. Aluminium release in relation to the determination of cation exchange capacity of some podzolized New Zealand soils. Journal of Soil Science, 36,
Lindsay, W.L. 1979. Chemical Equilibria in Soils. John Wiley & Sons, New York.
Magaldi, D. 1993. Indizi di paleopedogenesi in un suolo Bruno Acido della foresta di Vallombrosa (Firenze). I1 Quaternario, 6,205-212.
Matsue, N. & Wada, K. 1985. A new equilibration method for cation- exchange capacity measurement. Soil Science Society of America Journal, 49, 574-578.
McKeague, J.A. & Day, J.H. 1966. Dithionite- and oxalate-extractable Fe and Al as aids in differentiating various classes of soils. Canadian Journal of Soil Science, 46, 13-22.
McLeod, S. 1975. Studies on wet oxidation procedures for the determination of ‘organic C’. In: Notes on Soil Techniques, No 2, pp. 73-79. CSIRO Division of Soils, Adelaide.
Mehra, 0.P. & Jackson, M.L. 1960. Iron oxide removal from soils and clays by a dithionite-citrate system buffered with sodium bicarbonate. Proceedings of the 7th National Conference on Clays and Clay Minerals (ed. E. Ingerson), pp. 317-342, Pergamon Press, London.
Oates, K.M. & Kamprath, E.J. 1983. Soil acidity and liming: I. Effect of the extracting solution cation and pH on the removal of aluminum from acid soils. Soil Science Society of America Journal,
Ponette, Q., Andre, D. & Dufey, J.E. 1996. Chemical significance of aluminium extracted from three horizons of an acid forest soil, using chloride salt solutions. European Journal of Soil Science, 47,89-95.
Rabenhorst, M.C., Foss, J.E. & Fanning, D.S. 1982. Genesis of Maryland soils formed from serpentinite. Soil Science Society of America Journal, 46, 607-616.
Rasmussen, P.E., Schiff, S.L. & Nesbitt, H.W. 1991. The determina- tion of exchangeable cations in acid soils: errors caused by weathering reactions during neutral salt extraction. Canadian Journal of Soil Science, 71, 155- 163.
Sivasubramaniam, S. & Talibudeen, 0 . 1972. Potassium-aluminium exchange in acid soils. I. Kinetics. Journal of Soil Science, 23,
Soil Survey Staff 1994. Keys to Soil Taxonomy. Sixth edition. US Department of Agriculture, Soil Conservation Service, US Government Printing Office, Washington, DC.
Ugolini, F.C., Corti, G., Agnelli, A. & Piccardi, F. 1996. Mineral- ogical, physical and chemical properties of rock fragments in soil. Soil Science, 161, 521 -542.
Wada, K. & Harada, Y. 1969. Effects of salt concentration and cation species on the measured cation exchange capacity of soils and clays. In: Proceedings of the fnternational Clay Conference, Tokyo, September 5-f0 1969, Volume I (ed. L. Heller), pp. 561-571. Israel University Press, Jerusalem.
Wada, K. & Kakuto, Y. 1983. Intergradient vermiculite-kaolin mineral in a Korean Ultisol. Clays and Clay Minerals, 31, 183-190.
Yuan, T.L. & Fiskell, J.G.A. 1959. Aluminum studies: 11. The extraction of aluminum from some Florida soils. Soil Science Society of America Proceedings, 23,202-205.
239-253.
47,686-689.
163-176.
0 1997 Blackwell Science Ltd, European Journal of Soil Science, 48, 249-262

N
Q\
N a
App
endi
x 9
3. 2 C
AV
ALL
A
2
Tab
le
Des
crip
tions
of
Cav
alla
and
Ter
min
e pr
ofile
s (V
allo
mbr
osa,
Ital
y). F
or s
ymbo
ls s
ee l
egen
d.
Alti
tude
: 1 I
00 m
-Exp
osur
e:
N-N
E-Sl
ope:
i5
%-P
aren
t m
ater
ial:
‘Are
naria
del
Fal
tero
na’
(Olig
ocen
e)-V
eget
atio
n:
plan
tatio
n of
Abi
es a
lba
abou
t 70
year
s ol
d U
nder
stor
y: H
iera
cium
mur
orum
, Pre
nanr
es p
urpu
rea,
Luz
ula
nive
a, S
anic
ula
aeur
opea
, Sen
ecio
fich
sii,
Rub
us id
aeus
, Fra
gari
a ve
sca,
Ger
aniu
m ro
bert
ianu
m, C
arda
min
e sp.
, Gal
ium
sp.,
seed
lings
of A
bies
alb
a So
il: T
ypic
Hap
lum
brep
t, fin
e lo
amy,
mix
ed, m
esic
(So
il Su
rvey
Sta
ff,
1994
)
g R 9
Hor
izon
s O
e A
1 A
2 B
wl
Bw
2 B
Cxb
l B
Cxb
2
Dep
th
Mun
sell
cm
Col
o?
1-0
0-10
lO
YR
3/2
10-2
5 10
YR
3/3
25-6
2 10
YR
5/4
62-
108
10Y
R5/
5 10
8-13
0 IO
YR
5/4
130-
168+
IO
YR
4/4
Text
ureb
St
ruct
ure‘
(U
SDA
)
cl
2-3
f-m
cr
gsil
3-2
m-f
sbk
gsil
2-3
m-f
abk-
sbk
gsil-
gl
2-3
f sb
k vs
tl 2
c-m
abk
st
l-sl
2 c-
m a
bk
Con
sist
ency
d Pl
astic
itye
Roo
ts‘
- m
fr, w
ss
WPS
3f
, 2m
m
fr, w
ss
WPS
2f
m
fr-m
fi, w
ss
wps
If
m
fi, w
ss
WPS
If
m
fi, w
ss
WPS
If
m
fi, w
ss
wps
Oth
er o
bser
vatio
nsh
Bou
ndar
yg
cw
parti
ally
dec
ompo
sed
need
les
of f
ir cw
cw
ci
ci
ci
w
d si
lt ca
ps; f
r pr
w
d si
lt ca
ps; f
r pr
; som
e cl
asts
are
sap
rolit
ic
wd
silt
caps
; fr
pr;
som
e cl
asts
are
sap
rolit
ic
TER
MIN
E A
ltitu
de:
1250
m-E
xpos
ure:
N
-E-S
lope
: ab
out
10%
-Par
ent
mat
eria
l: ’A
rena
ria d
el F
alte
rona
’ (O
ligoc
ene)
-Veg
etat
ion:
pl
anta
tion
of F
agus
syl
vatic
u ab
out
70 y
ears
old
U
nder
stor
y: v
irtua
lly a
bsen
t So
il: T
ipic
hap
lum
brep
t, fin
e lo
amy,
mix
ed, m
esic
(So
il Su
rvey
Staff, 1
994)
Dep
th
Mun
sell
Text
ureb
St
ruct
ure‘
C
onsi
sten
cyd
Plas
ticity
e R
oots‘
B
ound
aryg
O
ther
obs
erva
tions
h H
oriz
ons
cm
Col
oP
(USD
A)
Oi
6-0
2mi&
vf&
f cw
de
ad le
aves
of
fagu
s A
0-
11
10Y
R3/
2 si
l 1
f-m
cr
mfr
, w
ss
wps
3m
i&vf
&f,
lco
cw
Bw
2 33
-53
10Y
R4/
4 vg
l 2
c-m
sbk
m
fr, w
ss
WPS
2f
cs
vw
silt
cap
s B
C 1
53-7
9 10
YR
5/4
vsts
cl
2 m
-c a
bk-s
bk
mfr
-mfi,
wss
w
p Im
cs
w
d si
lt ca
ps; p
e fr
pr;
clas
ts m
ostly
alte
red
pe f
r pr
; m
ottle
s (1
0YR
4.5/
4); l
arge
cla
sts
BC
2 79
-107
+ 10
YR
5.5/
4 st
scl
2 c
pl
mfi,
wss
aMoi
st an
d cr
ushe
d.
‘1,
wea
k; 2
, mod
erat
e; 3
, stro
ng, f
, fin
e; m
, med
ium
; c, c
oars
e; p
l, pl
aty;
abk
, ang
ular
blo
cky;
sbk
, sub
angu
lar b
lock
y; c
r, cr
umb
dm, m
oist;
w, w
et; f
r, fr
iabl
e; fi
, firm; ss
, slig
htly
stic
ky.
e~
,
wet
; ps,
sligh
tly p
last
ic; p
, pla
stic.
gc, c
lear
; s, s
moo
th; w
, wav
y; i,
irre
gula
r. h
~~
,
very
wea
k; w
d, w
ell d
evel
oped
; pe, p
oorly
exp
ress
ed; f
r, fr
agic
pro
perti
es.
Bw
l 11
-33
10Y
R3.
5/3
gsil
2 m
sbk
m
fr, w
ss
wps
2v
f&f,
1 co
cs
WP
very
; g,
grav
elly
; st,
ston
y; c,
cla
y; si
, silt
; s, s
andy
; I, l
oam
.
1, fe
w; 2
, ple
ntifu
l; 3,
abu
ndan
t; m
i, m
icro
; vf,
very
fin
e; f,
fine
; m, m
ediu
m; c
o, c
oars
e.
Copyright © 2022 FDOKUMEN




















