
A one-carbon modification of protein lysine associated with elevated oxidative stress in human...
11
A one-carbon modification of protein lysine associated with elevated oxidative stress in human substantia nigra Erik Floor,* Anne M. Maples,* Carolyn A. Rankin,* Vamsee M. Yaganti,* Sylvan S. Shank,* Grant S. Nichols,* Michael O’Laughlin,* Nadezhda A. Galeva and Todd D. Williams * Department of Molecular Biosciences and Mass Spectrometry Laboratory, University of Kansas, Lawrence, Kansas, USA Abstract We describe for the first time a naturally occurring lysine modification that is converted to methyllysine by reduction with sodium borohydride. This modification is 1.7 times as abundant in soluble proteins from human substantia nigra pars compacta as in proteins from other brain regions, possibly as a result of elevated oxidative stress in the nigra. Proteins from cultured PC12 cells exposed to oxidative stress conditions also contain elevated levels of this lysine modification. The abundance of the naturally occurring modification is roughly 0.08 nmoles/mg protein in either unstressed brain or PC12 cells. Modification levels remain stable in isolated proteins incubated for 2 h at 37ŶC in pH 7 buffer. We propose that the endogenous modification is the lysine Schiff base, e-N-methylenelysine, and that lysine modifications may result from a reaction with formaldehyde in vivo. Rat brain contains 60 nmoles/g wet weight of for- maldehyde, which probably includes both free and reversibly bound forms. Adding 35 lM HCHO to PC12 cell growth medium introduces methylenelysine modifications in cell proteins and impairs cell viability. The existence of this post- translational modification suggests new mechanisms of oxi- dative stress that may contribute to tissue degeneration, including loss of nigral dopamine neurons during normal aging and in Parkinson’s disease. Keywords: aging, brain, formaldehyde, oxidative stress, protein oxidation, substantia nigra. J. Neurochem. (2006) 97, 504–514. Mitochondria generate small quantities of reactive molecular species that modify proteins, DNA and lipids and, in addition, modulate gene expression (Balaban et al. 2005; Giorgio et al. 2005; Schriner et al. 2005). These modifications may have beneficial effects by triggering antioxidant responses that protect cells from low levels of reactive oxidants. On the other hand, more extensive oxidative stress may result in either excessive macromolecular oxidation or aberrant cellular signaling that contributes to cell and tissue degeneration (Halliwell and Gutteridge 1999; Lee et al. 2004). The substantia nigra pars compacta is a small midbrain region containing dopaminergic neurons that are critical for voluntary movement. Human substantia nigra appears to be subjected to greater oxidative stress than other brain regions as indicated by elevated oxidative modification of proteins and DNA (Floor and Wetzel 1998; Lezza et al. 1999; McCormack et al. 2005). This chronic stress may contribute to the loss of 5% of nigral dopamine neurons per decade during normal aging (Ma et al. 1999). These dopaminergic losses correlate with declines in motor control and certain cognitive functions (Emborg et al. 1998; Volkow et al. 1998). Additional sources of oxidative stress in Parkinson’s disease may accelerate the degeneration of vulnerable dopaminergic neurons (Jenner 2003). In a study designed to detect carbonyl modifications generated by oxidative stress, we radiolabeled proteins by reduction with tritiated sodium borohydride ([ 3 H]NaBH 4 ) and, after acid hydrolysis, analyzed the resulting 3 H-labeled amino acids by HPLC (Amici et al. 1989). Unexpectedly, a 3 H-labeled amino acid derivative from rat brain proteins was Received September 13, 2005; revised manuscript received December 12, 2005; accepted December 21, 2005. Address correspondence and reprint requests to Dr Erik Floor, Department of Molecular Biosciences, 2045 Haworth Hall, 1200 Sunnyside Avenue, Lawrence, KS 66045–7534, USA. E-mail: fl[email protected] Abbreviations used: ADH x , alcohol dehydrogenase x ; BSO, l-buthio- nine-sulfoximine; CID, collision-induced dissociation; DMEM, Dul- becco’s modified Eagle’s medium; DTNB, 5,5¢-dithiobis(2-nitrobenzoic acid); ESI, electrospray ionization; FDH, formaldehydedehydrogenase; FlNH2, fluoresceineamine; GSH, reduced glutathione; GSSG, glutathi- one disulfide; HACA, 6-hydroxy-2-aminocaproic acid; HNE, trans-4- hydroxy-2-nonenal; MW, molecular weight; PBS, phosphate-buffered saline; PITC, phenylisothiocyanate; PTC, phenylthiocarbamyl; SDS, sodium dodecyl sulfate. Journal of Neurochemistry , 2006, 97, 504–514 doi:10.1111/j.1471-4159.2006.03768.x 504 Journal Compilation ȑ 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514 ȑ 2006 The Authors
Transcript of A one-carbon modification of protein lysine associated with elevated oxidative stress in human...

A one-carbon modification of protein lysine associated withelevated oxidative stress in human substantia nigra
Erik Floor,* Anne M. Maples,* Carolyn A. Rankin,* Vamsee M. Yaganti,* Sylvan S. Shank,*Grant S. Nichols,* Michael O’Laughlin,* Nadezhda A. Galeva� and Todd D. Williams�*Department of Molecular Biosciences and �Mass Spectrometry Laboratory, University of Kansas, Lawrence, Kansas, USA
Abstract
We describe for the first time a naturally occurring lysine
modification that is converted to methyllysine by reduction
with sodium borohydride. This modification is �1.7 times as
abundant in soluble proteins from human substantia nigra
pars compacta as in proteins from other brain regions,
possibly as a result of elevated oxidative stress in the nigra.
Proteins from cultured PC12 cells exposed to oxidative
stress conditions also contain elevated levels of this lysine
modification. The abundance of the naturally occurring
modification is roughly 0.08 nmoles/mg protein in either
unstressed brain or PC12 cells. Modification levels remain
stable in isolated proteins incubated for 2 h at 37�C in pH 7
buffer. We propose that the endogenous modification is the
lysine Schiff base, e-N-methylenelysine, and that lysine
modifications may result from a reaction with formaldehyde
in vivo. Rat brain contains �60 nmoles/g wet weight of for-
maldehyde, which probably includes both free and reversibly
bound forms. Adding �35 lM HCHO to PC12 cell growth
medium introduces methylenelysine modifications in cell
proteins and impairs cell viability. The existence of this post-
translational modification suggests new mechanisms of oxi-
dative stress that may contribute to tissue degeneration,
including loss of nigral dopamine neurons during normal
aging and in Parkinson’s disease.
Keywords: aging, brain, formaldehyde, oxidative stress,
protein oxidation, substantia nigra.
J. Neurochem. (2006) 97, 504–514.
Mitochondria generate small quantities of reactive molecularspecies that modify proteins, DNA and lipids and, in addition,modulate gene expression (Balaban et al. 2005; Giorgio et al.2005; Schriner et al. 2005). These modifications may havebeneficial effects by triggering antioxidant responses thatprotect cells from low levels of reactive oxidants. On the otherhand, more extensive oxidative stress may result in eitherexcessive macromolecular oxidation or aberrant cellularsignaling that contributes to cell and tissue degeneration(Halliwell and Gutteridge 1999; Lee et al. 2004).
The substantia nigra pars compacta is a small midbrainregion containing dopaminergic neurons that are critical forvoluntary movement. Human substantia nigra appears to besubjected to greater oxidative stress than other brain regionsas indicated by elevated oxidative modification of proteinsand DNA (Floor and Wetzel 1998; Lezza et al. 1999;McCormack et al. 2005). This chronic stress may contributeto the loss of �5% of nigral dopamine neurons per decadeduring normal aging (Ma et al. 1999). These dopaminergiclosses correlate with declines in motor control and certaincognitive functions (Emborg et al. 1998; Volkow et al.1998). Additional sources of oxidative stress in Parkinson’s
disease may accelerate the degeneration of vulnerabledopaminergic neurons (Jenner 2003).
In a study designed to detect carbonyl modificationsgenerated by oxidative stress, we radiolabeled proteins byreduction with tritiated sodium borohydride ([3H]NaBH4)and, after acid hydrolysis, analyzed the resulting 3H-labeledamino acids by HPLC (Amici et al. 1989). Unexpectedly, a3H-labeled amino acid derivative from rat brain proteins was
Received September 13, 2005; revised manuscript received December12, 2005; accepted December 21, 2005.Address correspondence and reprint requests to Dr Erik Floor,
Department of Molecular Biosciences, 2045 Haworth Hall, 1200Sunnyside Avenue, Lawrence, KS 66045–7534, USA.E-mail: [email protected] used: ADHx, alcohol dehydrogenasex; BSO, l-buthio-
nine-sulfoximine; CID, collision-induced dissociation; DMEM, Dul-becco’s modified Eagle’s medium; DTNB, 5,5¢-dithiobis(2-nitrobenzoicacid); ESI, electrospray ionization; FDH, formaldehydedehydrogenase;FlNH2, fluoresceineamine; GSH, reduced glutathione; GSSG, glutathi-one disulfide; HACA, 6-hydroxy-2-aminocaproic acid; HNE, trans-4-hydroxy-2-nonenal; MW, molecular weight; PBS, phosphate-bufferedsaline; PITC, phenylisothiocyanate; PTC, phenylthiocarbamyl; SDS,sodium dodecyl sulfate.
Journal of Neurochemistry, 2006, 97, 504–514 doi:10.1111/j.1471-4159.2006.03768.x
504 Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors

identified as [3H]methyllysine. A naturally occurring proteinmodification that can be reduced to methyllysine has not, toour knowledge, been described previously. Moreover, detec-tion of this lysine modification by a reductive strategy raisedthe possibility that it was generated by oxidative stress. Herewe describe the initial identification and characterization ofthis modification in proteins from rat brain, human substantianigra and other regions, and cultured PC12 cells.
Materials and methods
Materials
Sodium borotritide was purchased as a crystalline solid (15 Ci/
mmole; PerkinElmer-NEN, Boston, MA, USA), dissolved in 10 mM
NaOH under N2 at a nominal concentration of 35 mM, dispensed
into aliquots, and stored under N2 at )80�C. Different batches of
[3H]NaBH4 contained 30–130% of the stated 3H content and, thus,
the actual [3H]NaBH4 content of a given batch was determined
retrospectively by calculating the total quantity of 3H-dpm collected
in experiments conducted with that batch.
L-[3H]Glutamate (30 Ci/mmole) was from American Radiolabe-
led Chemicals (St Louis, MO, USA) and [14C]HCHO (53 mCi/
mmole) from Moravek Biochemicals (Brea, CA, USA). 4-Hydroxy-
2-nonenal (HNE) was from EMD/Calbiochem (San Diego, CA,
USA). Acetonitrile (HPLC Grade) and reagent grade inorganic salts
and buffers were from Fisher Scientific (Pittsburgh, PA, USA). HCl
(6 M, constant boiling), phenol, phenylisothiocyanate (PITC) and
triethylamine were from Pierce (Rockford, IL, USA). HEPES was
from Research Organics (Cleveland, OH, USA). All other reagents,
biochemicals and culture media components were from Sigma-
Aldrich (St Louis, MO, USA).
Cells and tissues
Clone A of PC12 rat adrenal pheochromocytoma cells obtained
from Dr Erik S. Schweitzer (Brain Research Institute, UCLA, Los
Angeles, CA, USA) synthesizes and releases dopamine. Undiffer-
entiated PC12 cells were grown at 37�C in 10% CO2/air in
Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 mg/
mL glucose, 25 mM HEPES, 44 mM NaHCO3, 100 U/mL penicillin,
100 lg/mL streptomycin, 10% horse serum and 5% fetal bovine
serum.
Sprague–Dawley rats (200–400 g; 2–4-month-old) were
stunned by a blow to the head, killed by decapitation and the
heads placed immediately in a saline/ice bath to cool the tissue.
Brains were removed and placed in iced saline buffer prior to
homogenization. Rats were housed under the supervision of a
veterinarian in the Animal Care Unit of the University of Kansas
in strict adherence to the principles stated in the NIH Guide forthe Care and Use of Laboratory Animals. Protocols for their use
were approved by the Institutional Animal Care and Use
Committee.
Postmortem human brain samples from three neurologically
normal (control) cases were obtained from the Harvard Brain Tissue
Resource Center, McLean Hospital, Belmont, MA, USA. Tissue
samples were frozen in liquid N2 and stored under liquid N2 in this
laboratory for less than 4 months prior to analysis.
Preparation of soluble proteins
All procedures were performed at 0–2�C. Brain samples were
homogenized in a Teflon/glass tissue grinder on ice in phosphate-
buffered saline (PBS; in mM, 137 NaCl, 2.7 KCl, 10 Na2HPO4, 1.8
K2HPO4, 0.04% NaN3, pH 7.2), containing the following protease
inhibitors: 1 mM EDTA, 1 mM EGTA, 1 lg/mL aprotinin, leupeptin
and pepstatin, and 0.3 mM phenylmethylsulfonyl fluoride (Smith
et al. 1991). Homogenates were centrifuged at 1000 g for 5 min and
the resulting supernatant at 100 000 g for 1 h to yield a supernatant
fraction containing crude soluble proteins. Proteins were stored in
liquid N2 until analyzed.
Exponentially growing PC12 cells were washed twice with ice-
cold PBS, dislodged by pipetting, pelleted at 3000 g for 5 min at
2�C, and resuspended in 2 mL of iced PBS with protease inhibitors.
Cells were disrupted by sonication for 10 s and soluble proteins
prepared.
Borotritide labeling
Proteins were concentrated to 16 mg/mL by centrifugation at 2�C in
an Ultrafree-0.5 centrifugal filtration unit with 10 000 molecular
weight (MW) cut-off Biomax filter from Millipore (Billerica, MA,
USA). The borotritide reaction mixture (40 lL, pH 8) contained
20 lL concentrated protein (320 lg), 10 lL 0.1 M HEPES, 4 mM
EDTA, pH 7.8, and 10 lL of [3H]NaBH4 in 10 mM NaOH. EDTA
was added to prevent 3H-labeling of aromatic residues catalyzed by
Mn2+ (Levine et al. 1990). The mixture was incubated at 37�C for
30 min and dialyzed for 20 min three times against 500 mL of
50 mM ammonium acetate, pH 7.2, at 2�C in a Slide-A-Lyzer unit
with 10 000 MW cut-off from Pierce (Rockford, IL, USA). Control
proteins to test the specificity of 3H-labeling were prereduced by
adding 10 lL of 1 M NaBH4 (250 mM), incubated as above and
dialyzed against 0.1 M HEPES, 1 mM EDTA, pH 7.8 at 4�C prior to
reaction with [3H]NaBH4. To test whether modifications were
located on proteins, 3H-labeled material was incubated with
�50 lg/mL proteinase K for 2 h at 37�C after the ammonium
acetate dialysis step and then subjected to acid hydrolysis.
Tritium gas production during these procedures is a potential
safety concern and thus, initially, was monitored by performing
either the reaction or dialysis in an air-tight box flushed with N2.3H2
was collected on two glass-wool PtO2 (Adams’ catalyst)/CaSO4
traps in series in the exhaust gas. In reduction reactions, the first trap
collected �0.2% of the added 3H (�2 lCi) and the second, about
half as much. During dialysis, < 0.05% of added 3H was recovered
from the traps. Since evolution of 3H2 gas was minimal during these
procedures, subsequent reactions were carried out in a conventional
fume hood and dialysis performed in a 4�C cold room.
Protein hydrolysis and PITC labeling
Gas-phase hydrolysis of up to 320 lg protein was performed in
13 · 100-mm acid-washed Pyrex culture tubes that were dried
under vacuum and placed in a 160-mm inner diameter Pyrex
desiccator containing 2 mL 6 N HCl and 20 lL phenol (Meltzer
et al. 1987). The desiccator was evacuated until HCl started to boil,
filled with N2 at �10 psi, evacuated again and filled with N2 two
more times, and finally evocuated and placed in an oven at 105�Covernight. After hydrolysis, the tubes were dried under vacuum,
dried again after adding 40 lL of H2O : ethanol : triethylamine
(4 : 4 : 1), incubated at room temperature (�22�C) for 20 min after
One-carbon protein lysine modification 505
� 2006 The AuthorsJournal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514

adding 30 lL of a 7 : 1 : 1 : 1 solution of ethanol : H2O : triethyl-
amine : PITC and then dried under a vacuum of < 300 mtorr.
Methyllysine is stable during 6 M HCl hydrolysis at 105�C for 16 h
(Taborsky 1973).
The accuracy of amino acid analysis was checked by assessing
errors in the determination of the composition of horse heart
myoglobin. Our average error was 13% (absolute percentage error in
amino acid content averaged over all amino acids), which is
comparable to the mean accuracy of 11% obtained in a large survey
of analytical facilities (Mahrenholz et al. 1996).
HPLC amino acid analysis
Phenylthiocarbamyl (PTC)-amino acids were separated on a
4.6 · 250-mm Spherisorb 5 lm ODS2 C18 column from Waters
(Milford, MA, USA) at 50�C as described previously (Scholze
1985). Briefly, samples were injected in buffer A (10 mM K2HPO4,
pH 6.5) and run at 1 mL/min on a 55 min, triphasic linear gradient
of buffer B (70% acetonitrile, 30% buffer A). PTC-amino acids
were detected by absorbance at 254 nm with an ISCO V4 UV/vis
detector and 3H-labeled compounds with a Radiomatic 500TR Flow
Scintillation Analyzer from PerkinElmer (Boston, MA, USA). The
detectors were connected in series, and data were collected and
analyzed with FLO-ONE software from PerkinElmer. Absorbance and3H data displays were synchronized by chromatography of PTC-L-
[3H]glutamate to determine the delay correction between detectors.
Methyllysine was baseline-resolved from neighboring peaks in all
experiments. Synthetic e-N-monomethyllysine was added in all
HPLC runs.
The following calculations were performed to determine the
abundance of [3H]methyllysine in proteins. First, 3H-cpm were
converted to a molar basis by integrating HPLC 3H-cpm peak areas
with FLO-ONE software and then calculating molar quantities of
labeled methyllysine from the specific radioactivity of [3H]NaBH4
after correcting for the counting efficiency of the flow scintillation
analyzer. It was assumed that one tritium atom was incorporated per
labeled monomethyllysine and that there was no isotope effect
(Taborsky 1973). The quantity of protein injected in an HPLC
analysis was obtained from the quantity hydrolyzed as measured by
an amido black assay (Schaffner and Weissmann 1973). When either
brain regions or treatment conditions were compared, quantities of
protein in each HPLC analysis were normalized using the sum of
PTC-amino acid peak areas.
Purification and identification of e-N-methyllysine
Rat brain soluble proteins (4 mg) were reduced with 250 mM
NaBH4 and a tracer quantity of 3H-labeled protein (6 lg,300 000 cpm) was added. After hydrolysis and derivatization with
PITC, amino acid derivatives were fractionated by anion-exchange
HPLC followed by two cycles of HPLC on a C18 column using the
amino acid analysis protocol. Anion-exchange HPLC was per-
formed on a 4.6 · 150-mm Macrosphere WAX 7 lm column from
Alltech (Deerfield, IL, USA) in 5 mM acetic acid, pH 3.5, at 1 mL/
min eluted with a 20-min linear gradient of 1 M HOAc, pH 3.0. A
sharp peak of 3H-cpm comigrating at 29 min with synthetic (PTC)2-
e-N-methyllysine was collected for C18 purification, and a sharp
peak with retention time of �48 min that was well resolved from
adjacent peaks in both 3H-cpm and OD254 channels was collected
for mass analysis.
Mass spectrometry
For initial mass analysis of the tritiated product purified from brain
proteins, electrospray ionization (ESI) experiments were performed
on a VG AUTOSPEC-Q instrument (Micromass Ltd, Manchester,
UK) equipped with a Mark III electrospray source and MiniPAL
autosampler from CTC Analytics AG (Zwinger, Switzerland). ESI
conditions were as follows: capillary voltage 3000 V, source
temperature 110�C and cone voltage 60 V. Samples of PITC-
labeled protein hydrolysate were prepared in 1% formic acid,
trapped on a 320 lm · 5 cm polymeric bead column with 0.4 lmpores from Michrom Biosciences (Auburn, CA, USA), eluted into
the ESI source with 90% methanol, 1% formic acid at 15 lL/min
through a 130 lm (inner diameter) stainless steel needle and
nebulized with a coaxial gas flow of 10 L/h N2.
For LC/MS/MS quantitation of methyllysine, collision-induced
dissociation (CID) mass spectra in positive ion mode were
acquired on a Q-TOF 2 instrument (Micromass). Proteins were
concentrated on an Ultrafree-0.5 filter unit and reduced with either
NaBD4 or NaBH4 (250 mM) followed by dialysis against ammo-
nium acetate. PITC-labeled hydrolysates of these proteins were
desalted on a 1.5 cm · 1 mm inner diameter column hand-packed
with Zorbax SB-C18, 5 lm from Agilent Technologies (Wilming-
ton, DE, USA), washed with 1% acetic acid and eluted directly
into the ESI source with 90% MeOH and 0.5% formic acid.
Spectra were acquired with the Q-TOF 2 operated at maximum
resolution (10 000), Ar in the collision cell and a cone voltage of
40 V. Precursor MH+ ions with a m/z of either 417.1 for lysine
quantitation or 431.1 for methyllysine quantitation were selected in
quadrupole 1 and fragmented with a collision energy of 17 eV.
Ions transmitted into the pusher region of the TOF analyzer were
scanned over m/z 100–500 with a 1-s integration time and data
acquired in continuum mode until acceptable averaged data were
obtained (5–10 min). MS data were deconvoluted using MAXENT I
(Micromass).
The molar abundance of methyllysine in proteins reduced with
NaBD4 was calculated as follows. The ratio of [D]methyllysine/
lysine was calculated from the peak intensities of the 339-Da
daughter fragment of (PTC)2-e-N-[D]methyllysine and the 324-Da
fragment of (PTC)2-lysine. This ratio was corrected for natural
heavy isotopomers and for possible interfering fragments by
subtracting the 339/324-Da intensity ratio for proteins reduced
with NaBH4. The abundance of [D]methyllysine was also corrected
for the 60% lower detection efficiency of methyllysine as
determined from analyses of mixtures of methyllysine and lysine.
Finally, the mole ratio of [D]methyllysine/protein was calculated
using the lysine content of this protein preparation (28.2 Lys/mole
protein) and an estimate of 50 000 for the average protein MW.
Proteins from quadruplicate reduction reactions with either NaBD4
or NaBH4 (250 mM) were subjected to four MS/MS analyses per
reaction.
Postmortem human brain samples
Coronal sections (5–6-mm thick) containing putamen rostral to the
anterior commissure, putamen immediately posterior to the com-
missure, caudate nucleus, and cerebellum as well as horizontal
mesencephalic sections containing substantia nigra or tangential
sections of prefrontal cortex (Brodmann area 9) were obtained for
each of the three control cases. Gray matter from regions of interest,
506 E. Floor et al.
Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors

or pigmented portions of substantia nigra, primarily pars compacta
(Parent 1996), were dissected on an ice pack at )15�C. The mean
age of subjects (1 female and 2 males) was 59 ± 8 years and the
postmortem intervals were 20 ± 1 h. Causes of death were either
heart attack or unknown.
Fluoresceineamine assay
Carbonyl groups were assayed by modification of a published
procedure (Climent et al. 1989). A 100-lL protein sample was
pipeted into microtubes, dried in a vacuum centrifuge and
redissolved in 100 lL of 0.25 M Mes buffer, pH 6.0, containing
1% sodium dodecyl sulfate (SDS) and heated at 100�C for 1 min.
Then 12.8 lL of 0.25 M FlNH2 in 0.52 M NaOH and 10 lL of
freshly prepared NaCNBH3 in 0.25 M Mes buffer, pH 6.0, were
added and the volume adjusted to 160 lL with 0.25 M Mes buffer
pH 6 without SDS. After incubation at 37�C for 1 h, 1 mL of 10%
trichloroacetic acid was added and allowed to stand for 10 min. The
samples were then centrifuged for 5 min at 16 000 g, the superna-
tant discarded, and the residual reagent removed by washing the
pellet four times with ethanol : ethylacetate (1 : 1) containing
10 mM HCl. The precipitate was dissolved in 0.3 mL of 6 M
guanidine pH 12, insoluble material removed by centrifugation for
5 min at 16 000 g, and absorbance measured at 490 nM. Samples
were assayed in quadruplicate, and duplicate tissue blanks incubated
with 0.52 M NaOH not containing FlNH2 were included for each
sample. Total protein was assayed in duplicate by the amido black
method in the final guanidine suspension.
Stability of [14C]formaldehyde-lysine adducts
To prepare 14C-labeled proteins, rat brain soluble proteins (16 mg/
mL) in 40 lL PBS were mixed with 1 lL [14C]HCHO at a final
concentration of 650 lM and incubated at 37�C for 30 min. Free
[14C]HCHO was then removed by low-speed centrifugation for
1 min on a fresh 1-mL Sephadex G-25 spin-column at 2�C. Afterreduction with NaBH4, monomethyllysine was the only 14C-labeled
compound detected by HPLC amino acid analysis.
To measure stability, 14C-labeled proteins were diluted to 2 mg/
mL in PBS containing protease inhibitors used in soluble protein
preparation (above) and incubated at 0 or 37�C. 14C-Labeled
proteins were reduced with freshly prepared 250 mM NaBH4
immediately after each time sample was collected, and [14C]meth-
yllysine was assayed after protein hydrolysis and HPLC analysis.
Hydrolysis half-times were obtained with ORIGIN 7.0 software from
OriginLab (Northampton, MA, USA) by fitting decay data to the
equation, y ¼ y0 + Y1e(–t/t1) + Y2e
(–t/t2), where y ¼ the quantity of
[14C]methyllysine at time t, Y ¼ the initial quantity of [14C]meth-
yllysine and y0 ¼ stable ‘14C adducts’ (possibly residual free
[14C]HCHO). Data for [14C]HCHO adducts at 37�C were fitted
(R2 ¼ 0.998) by biphasic exponential decay kinetics in which
�54% of lysine adducts were lost with a half-life of 3.9 min and the
remainder with t1/2 ¼ 20 min.
Stability of the endogenous lysine modification
Rat brain soluble proteins (1.8 mg/mL) were incubated at 37�C in
PBS containing protease inhibitors (above). Immediately after each
timepoint was taken, proteins were concentrated to 16 mg/mL at
2�C and labeled with [3H]NaBH4. [3H]Methyllysine content was
then determined by amino acid analysis.
Glutathione assay
Cells were rinsed with basal DMEM, pelleted, resuspended in 5%
5-sulfosalicylic acid to precipitate proteins and extract glutathione,
centrifuged 5 min at 16 000 g, and the resulting supernatant
assayed. Total glutathione [reduced glutathione (GSH) + glutathi-
one disulfide (GSSG)] was assayed following 5,5¢-dithiobis(2-nitrobenzoic acid) (DTNB) reduction at 412 nm by GSSG reductase
and NADPH (Anderson 1985). GSSG was assayed after blocking
GSH by reaction with 2-vinylpyridine.
Treatment of PC12 cells with l-buthionine-sulfoximine (BSO)
and formaldehyde
Cells in 15-cm culture dishes at a surface coverage of �50% were
treated with 10 lM BSO and incubated for 24 h to deplete GSH.
HCHO was then added to the culture medium, the cells incubated at
37�C for 2 h, and soluble proteins harvested and methyllysine
precursor assayed by [3H]NaBH4 reduction and amino acid analysis.
To measure cell viability by acid phosphatase assay, cells in
6-well plates at a surface coverage of �50% were treated with
10 lM BSO, incubated for 24 h, HCHO added and the plates
incubated for an additional 48 h. The wells were washed with PBS,
cells lysed in 2 mL assay buffer without p-nitrophenyl phosphateand containing 0.1% Triton X-100 at room temperature for 10 min,
and then 200 lL of lysate was assayed by absorbance at 420 nm as
described (Si et al. 1999).
Formaldehyde and H2O2 assays
HCHO was assayed by following the reaction of formaldehyde
dehydrogenase spectrophotometrically at 340 nm as described by
Nudelman et al. (2005). Rat brain soluble proteins prepared as
described above were filtered through an Ultrafree-MC centrifugal
unit with 10 000 MW cut-off Biomax filter (Millipore). The filtrate
was made 40 mM NaxHxPO4, pH 8, and 8 mg/mL NAD+, 0.13 U/
mL of FDH added to start the reaction, the absorbance recorded at
3-s intervals for 1.5 min at room temperature, and the initial slope
obtained by linear regression through these data points to measure
enzyme activity. Controls with FDH omitted and a standard curve
with added HCHO were included in every experiment. [HCHO] in
samples was determined from the activity for brain filtrate after
subtracting the value for the control lacking FDH. Results with
boiled brain filtrate were within experimental error of results with
unboiled samples.
H2O2 was assayed by measuring the absorbance at 440 nm of the
oxidation product of o-dianisidine by horseradish peroxidase as
described by Desagher et al. (1996).
Statistical methods
Data are given as mean ± SEM. Statistical significance was assessed
by t-tests calculated using ORIGIN 7.0 software (OriginLab).
Results
Reduction with borohydride yields methyllysine
To obtain sufficient 3H-labeling for subsequent HPLCanalysis, we optimized the conditions for borotritide reduc-tion. Reactions of [3H]NaBH4 in aqueous solution are limited
One-carbon protein lysine modification 507
� 2006 The AuthorsJournal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514
by rapid and extensive 3H exchange between [3H]NaBH4 andwater. Indeed, although borohydride is maximally stable atpH > 10, approximately 98% of tritium from [3H]NaBH4
stock solutions in 10 mM NaOH was converted to a volatileform, presumably 3HOH, within 15 min as determined byscintillation counting of liquid versus air-dried samples. Tocompensate for this loss, high specific activity [3H]NaBH4
(15 Ci/mmole) was used, and reduction was carried out inconcentrated protein solution (8 mg/mL).
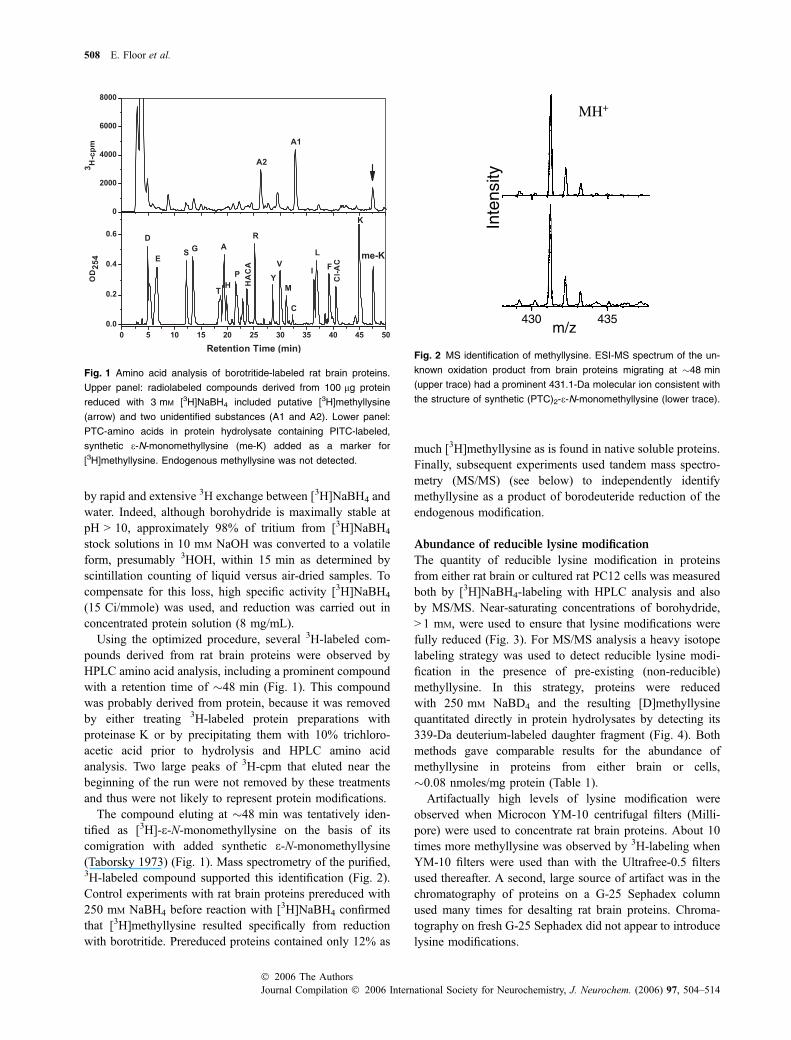
Using the optimized procedure, several 3H-labeled com-pounds derived from rat brain proteins were observed byHPLC amino acid analysis, including a prominent compoundwith a retention time of �48 min (Fig. 1). This compoundwas probably derived from protein, because it was removedby either treating 3H-labeled protein preparations withproteinase K or by precipitating them with 10% trichloro-acetic acid prior to hydrolysis and HPLC amino acidanalysis. Two large peaks of 3H-cpm that eluted near thebeginning of the run were not removed by these treatmentsand thus were not likely to represent protein modifications.
The compound eluting at �48 min was tentatively iden-tified as [3H]-e-N-monomethyllysine on the basis of itscomigration with added synthetic e-N-monomethyllysine(Taborsky 1973) (Fig. 1). Mass spectrometry of the purified,3H-labeled compound supported this identification (Fig. 2).Control experiments with rat brain proteins prereduced with250 mM NaBH4 before reaction with [3H]NaBH4 confirmedthat [3H]methyllysine resulted specifically from reductionwith borotritide. Prereduced proteins contained only 12% as
much [3H]methyllysine as is found in native soluble proteins.Finally, subsequent experiments used tandem mass spectro-metry (MS/MS) (see below) to independently identifymethyllysine as a product of borodeuteride reduction of theendogenous modification.
Abundance of reducible lysine modification
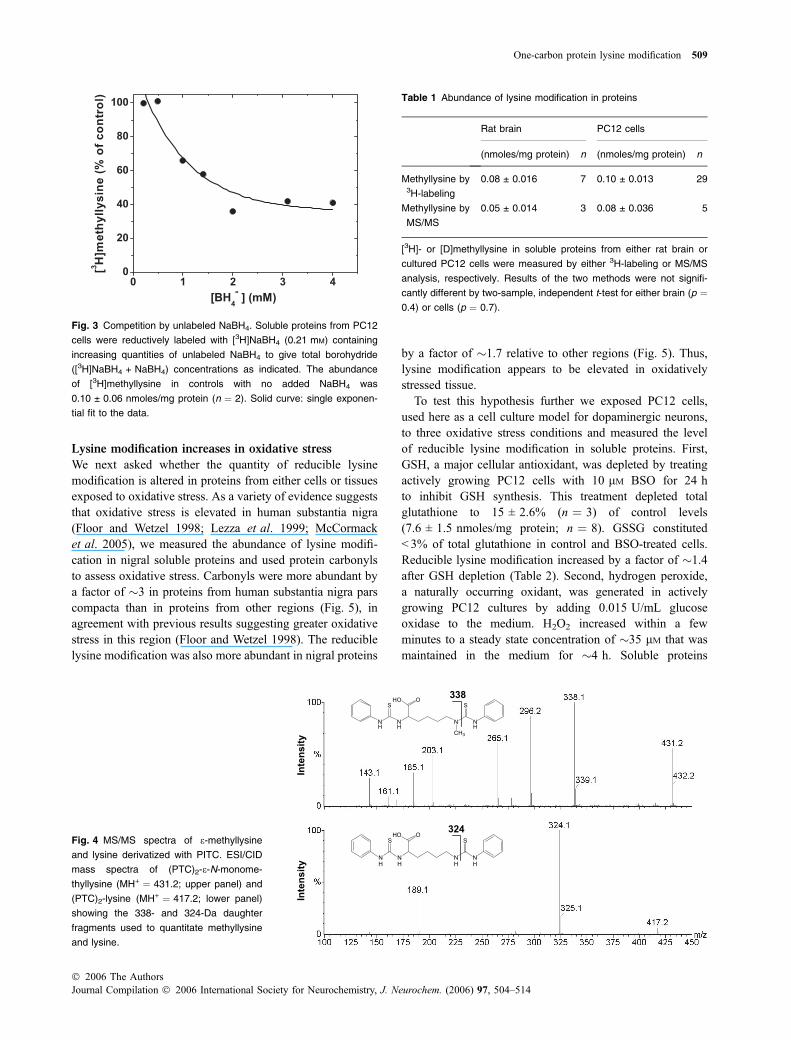
The quantity of reducible lysine modification in proteinsfrom either rat brain or cultured rat PC12 cells was measuredboth by [3H]NaBH4-labeling with HPLC analysis and alsoby MS/MS. Near-saturating concentrations of borohydride,> 1 mM, were used to ensure that lysine modifications werefully reduced (Fig. 3). For MS/MS analysis a heavy isotopelabeling strategy was used to detect reducible lysine modi-fication in the presence of pre-existing (non-reducible)methyllysine. In this strategy, proteins were reducedwith 250 mM NaBD4 and the resulting [D]methyllysinequantitated directly in protein hydrolysates by detecting its339-Da deuterium-labeled daughter fragment (Fig. 4). Bothmethods gave comparable results for the abundance ofmethyllysine in proteins from either brain or cells,�0.08 nmoles/mg protein (Table 1).
Artifactually high levels of lysine modification wereobserved when Microcon YM-10 centrifugal filters (Milli-pore) were used to concentrate rat brain proteins. About 10times more methyllysine was observed by 3H-labeling whenYM-10 filters were used than with the Ultrafree-0.5 filtersused thereafter. A second, large source of artifact was in thechromatography of proteins on a G-25 Sephadex columnused many times for desalting rat brain proteins. Chroma-tography on fresh G-25 Sephadex did not appear to introducelysine modifications.
Fig. 1 Amino acid analysis of borotritide-labeled rat brain proteins.
Upper panel: radiolabeled compounds derived from 100 lg protein
reduced with 3 mM [3H]NaBH4 included putative [3H]methyllysine
(arrow) and two unidentified substances (A1 and A2). Lower panel:
PTC-amino acids in protein hydrolysate containing PITC-labeled,
synthetic e-N-monomethyllysine (me-K) added as a marker for
[3H]methyllysine. Endogenous methyllysine was not detected.
ytisnetnI
m/z430 435
MH+
Fig. 2 MS identification of methyllysine. ESI-MS spectrum of the un-
known oxidation product from brain proteins migrating at �48 min
(upper trace) had a prominent 431.1-Da molecular ion consistent with
the structure of synthetic (PTC)2-e-N-monomethyllysine (lower trace).
508 E. Floor et al.
Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors
Lysine modification increases in oxidative stress
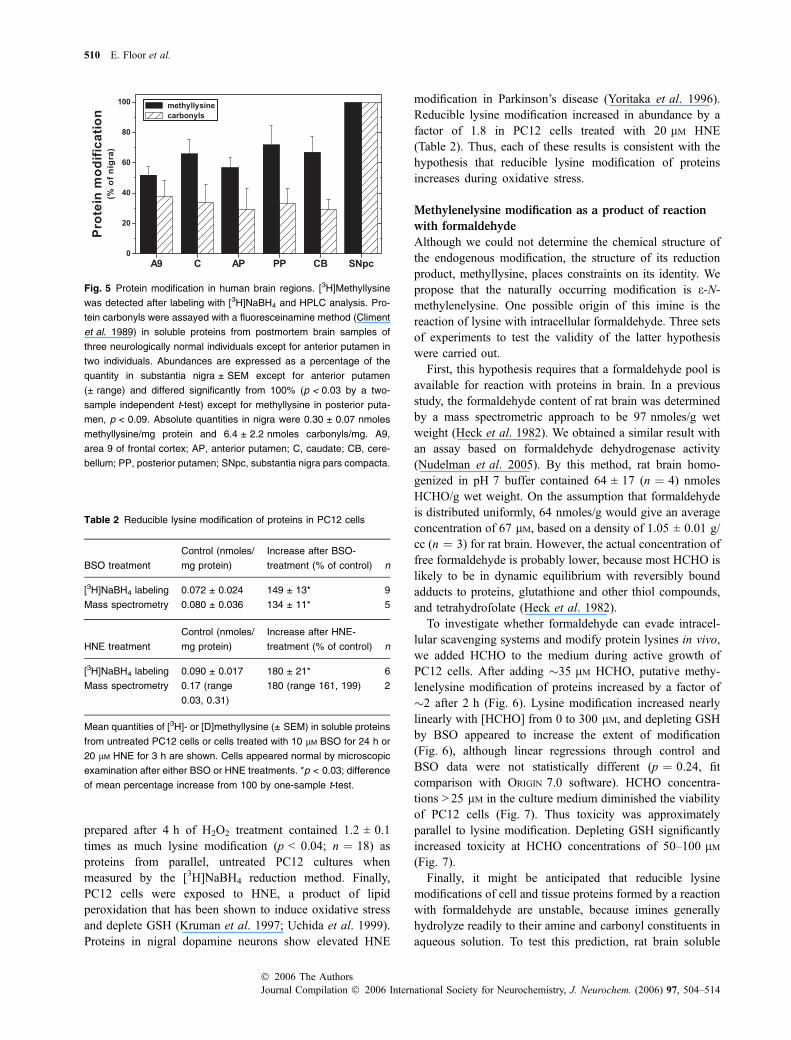
We next asked whether the quantity of reducible lysinemodification is altered in proteins from either cells or tissuesexposed to oxidative stress. As a variety of evidence suggeststhat oxidative stress is elevated in human substantia nigra(Floor and Wetzel 1998; Lezza et al. 1999; McCormacket al. 2005), we measured the abundance of lysine modifi-cation in nigral soluble proteins and used protein carbonylsto assess oxidative stress. Carbonyls were more abundant bya factor of �3 in proteins from human substantia nigra parscompacta than in proteins from other regions (Fig. 5), inagreement with previous results suggesting greater oxidativestress in this region (Floor and Wetzel 1998). The reduciblelysine modification was also more abundant in nigral proteins
by a factor of �1.7 relative to other regions (Fig. 5). Thus,lysine modification appears to be elevated in oxidativelystressed tissue.
To test this hypothesis further we exposed PC12 cells,used here as a cell culture model for dopaminergic neurons,to three oxidative stress conditions and measured the levelof reducible lysine modification in soluble proteins. First,GSH, a major cellular antioxidant, was depleted by treatingactively growing PC12 cells with 10 lM BSO for 24 hto inhibit GSH synthesis. This treatment depleted totalglutathione to 15 ± 2.6% (n ¼ 3) of control levels(7.6 ± 1.5 nmoles/mg protein; n ¼ 8). GSSG constituted< 3% of total glutathione in control and BSO-treated cells.Reducible lysine modification increased by a factor of �1.4after GSH depletion (Table 2). Second, hydrogen peroxide,a naturally occurring oxidant, was generated in activelygrowing PC12 cultures by adding 0.015 U/mL glucoseoxidase to the medium. H2O2 increased within a fewminutes to a steady state concentration of �35 lM that wasmaintained in the medium for �4 h. Soluble proteins
N
H
N
H
N
H
N
H
SS
OHO324
NH
NH
N
CH3
NH
SS
OHO338
ytisnet
nII
ytisnet
n
Fig. 4 MS/MS spectra of e-methyllysine
and lysine derivatized with PITC. ESI/CID
mass spectra of (PTC)2-e-N-monome-
thyllysine (MH+ ¼ 431.2; upper panel) and
(PTC)2-lysine (MH+ ¼ 417.2; lower panel)
showing the 338- and 324-Da daughter
fragments used to quantitate methyllysine
and lysine.
Table 1 Abundance of lysine modification in proteins
Rat brain PC12 cells
(nmoles/mg protein) n (nmoles/mg protein) n
Methyllysine by3H-labeling
0.08 ± 0.016 7 0.10 ± 0.013 29
Methyllysine by
MS/MS
0.05 ± 0.014 3 0.08 ± 0.036 5
[3H]- or [D]methyllysine in soluble proteins from either rat brain or
cultured PC12 cells were measured by either 3H-labeling or MS/MS
analysis, respectively. Results of the two methods were not signifi-
cantly different by two-sample, independent t-test for either brain (p ¼0.4) or cells (p ¼ 0.7).
Fig. 3 Competition by unlabeled NaBH4. Soluble proteins from PC12
cells were reductively labeled with [3H]NaBH4 (0.21 mM) containing
increasing quantities of unlabeled NaBH4 to give total borohydride
([3H]NaBH4 + NaBH4) concentrations as indicated. The abundance
of [3H]methyllysine in controls with no added NaBH4 was
0.10 ± 0.06 nmoles/mg protein (n ¼ 2). Solid curve: single exponen-
tial fit to the data.
One-carbon protein lysine modification 509
� 2006 The AuthorsJournal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514
prepared after 4 h of H2O2 treatment contained 1.2 ± 0.1times as much lysine modification (p < 0.04; n ¼ 18) asproteins from parallel, untreated PC12 cultures whenmeasured by the [3H]NaBH4 reduction method. Finally,PC12 cells were exposed to HNE, a product of lipidperoxidation that has been shown to induce oxidative stressand deplete GSH (Kruman et al. 1997; Uchida et al. 1999).Proteins in nigral dopamine neurons show elevated HNE
modification in Parkinson’s disease (Yoritaka et al. 1996).Reducible lysine modification increased in abundance by afactor of 1.8 in PC12 cells treated with 20 lM HNE(Table 2). Thus, each of these results is consistent with thehypothesis that reducible lysine modification of proteinsincreases during oxidative stress.
Methylenelysine modification as a product of reaction
with formaldehyde
Although we could not determine the chemical structure ofthe endogenous modification, the structure of its reductionproduct, methyllysine, places constraints on its identity. Wepropose that the naturally occurring modification is e-N-methylenelysine. One possible origin of this imine is thereaction of lysine with intracellular formaldehyde. Three setsof experiments to test the validity of the latter hypothesiswere carried out.
First, this hypothesis requires that a formaldehyde pool isavailable for reaction with proteins in brain. In a previousstudy, the formaldehyde content of rat brain was determinedby a mass spectrometric approach to be 97 nmoles/g wetweight (Heck et al. 1982). We obtained a similar result withan assay based on formaldehyde dehydrogenase activity(Nudelman et al. 2005). By this method, rat brain homo-genized in pH 7 buffer contained 64 ± 17 (n ¼ 4) nmolesHCHO/g wet weight. On the assumption that formaldehydeis distributed uniformly, 64 nmoles/g would give an averageconcentration of 67 lM, based on a density of 1.05 ± 0.01 g/cc (n ¼ 3) for rat brain. However, the actual concentration offree formaldehyde is probably lower, because most HCHO islikely to be in dynamic equilibrium with reversibly boundadducts to proteins, glutathione and other thiol compounds,and tetrahydrofolate (Heck et al. 1982).
To investigate whether formaldehyde can evade intracel-lular scavenging systems and modify protein lysines in vivo,we added HCHO to the medium during active growth ofPC12 cells. After adding �35 lM HCHO, putative methy-lenelysine modification of proteins increased by a factor of�2 after 2 h (Fig. 6). Lysine modification increased nearlylinearly with [HCHO] from 0 to 300 lM, and depleting GSHby BSO appeared to increase the extent of modification(Fig. 6), although linear regressions through control andBSO data were not statistically different (p ¼ 0.24, fitcomparison with ORIGIN 7.0 software). HCHO concentra-tions > 25 lM in the culture medium diminished the viabilityof PC12 cells (Fig. 7). Thus toxicity was approximatelyparallel to lysine modification. Depleting GSH significantlyincreased toxicity at HCHO concentrations of 50–100 lM(Fig. 7).
Finally, it might be anticipated that reducible lysinemodifications of cell and tissue proteins formed by a reactionwith formaldehyde are unstable, because imines generallyhydrolyze readily to their amine and carbonyl constituents inaqueous solution. To test this prediction, rat brain soluble
Fig. 5 Protein modification in human brain regions. [3H]Methyllysine
was detected after labeling with [3H]NaBH4 and HPLC analysis. Pro-
tein carbonyls were assayed with a fluoresceinamine method (Climent
et al. 1989) in soluble proteins from postmortem brain samples of
three neurologically normal individuals except for anterior putamen in
two individuals. Abundances are expressed as a percentage of the
quantity in substantia nigra ± SEM except for anterior putamen
(± range) and differed significantly from 100% (p < 0.03 by a two-
sample independent t-test) except for methyllysine in posterior puta-
men, p < 0.09. Absolute quantities in nigra were 0.30 ± 0.07 nmoles
methyllysine/mg protein and 6.4 ± 2.2 nmoles carbonyls/mg. A9,
area 9 of frontal cortex; AP, anterior putamen; C, caudate; CB, cere-
bellum; PP, posterior putamen; SNpc, substantia nigra pars compacta.
Table 2 Reducible lysine modification of proteins in PC12 cells
BSO treatment
Control (nmoles/
mg protein)
Increase after BSO-
treatment (% of control) n
[3H]NaBH4 labeling 0.072 ± 0.024 149 ± 13* 9
Mass spectrometry 0.080 ± 0.036 134 ± 11* 5
HNE treatment
Control (nmoles/
mg protein)
Increase after HNE-
treatment (% of control) n
[3H]NaBH4 labeling 0.090 ± 0.017 180 ± 21* 6
Mass spectrometry 0.17 (range
0.03, 0.31)
180 (range 161, 199) 2
Mean quantities of [3H]- or [D]methyllysine (± SEM) in soluble proteins
from untreated PC12 cells or cells treated with 10 lM BSO for 24 h or
20 lM HNE for 3 h are shown. Cells appeared normal by microscopic
examination after either BSO or HNE treatments. *p < 0.03; difference
of mean percentage increase from 100 by one-sample t-test.
510 E. Floor et al.
Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors

proteins were reacted with 650 lM [14C]HCHO to introduce0.2 nmoles [14C]methylenelysine/mg protein as detected byreduction to [14C]methyllysine with NaBH4. As expected,[14C]methylenelysine hydrolyzed rapidly at 37�C, although itwas stable at 0�C (Fig. 8). By contrast, endogenous lysinemodifications of rat brain proteins appeared to be stable at37�C (Fig. 8) when analyzed under similar conditions, i.e.0.1–0.2 nmoles lysine modification/mg protein and incuba-tion at �2 mg protein/mL at pH 7.2. This stability ofendogenous modifications reveals a difference from formal-dehyde-protein adducts formed in vitro.
Discussion
Biochemical origin of the lysine modification
The fact that a physiological stimulus, in this case oxidativestress, increased the abundance of reducible lysine modifi-cations strongly argues that they are produced in vivo.Moreover, their occurrence under stress conditions suggeststhat they are very likely to occur at a lower level inunstressed cells. In addition to these biological origins, wecannot exclude the possibility that some modifications wereproduced by sources of artifact during protein preparation oranalysis. Therefore, the level of lysine modification measuredin proteins from either unstressed cells or tissues must beconsidered as an upper estimate of the endogenous level.Two mechanisms for the physiological origin of borohy-dride-reducible lysine modifications, which are not mutuallyexclusive, will be considered here.
The formaldehyde hypothesisThe hypothesis that reducible lysine modifications derivefrom methylenelysine produced by metabolically generatedHCHO is supported by the occurrence of formaldehyde inbrain at concentrations that probably are sufficient to formlysine imines in proteins and, in addition, our demonstrationthat micromolar concentrations of extracellular HCHO mod-ify lysines in PC12 cell proteins. Formaldehyde arises fromnumerous metabolic sources including the microsomal oxi-dation of glycols (Rashba-Step et al. 1994), HO• attack onproteins (Headlam and Davies 2002), oxidative demethyla-tion of nucleic acids (Aas et al. 2003), methylamine oxidationby semicarbazide-sensitive amine oxidase (Gubisne-Haberle
Fig. 7 HCHO toxicity to PC12 cells. BSO-pretreatment (d) signifi-
cantly decreased viability in comparison to control PC12 cells (j)
measured by acid phosphatase assay 48 h after adding 50–100 lM
HCHO (p < 0.03, one-sample t-test). Data are the means from two
experiments (error bars, range).
Fig. 8 Stability of lysine modifications. [14C]Methylenelysine hydro-
lyzed rapidly in rat brain soluble proteins incubated at pH 7.2 and 37�C(d) but was stable at 0�C (j). Endogenous lysine modification of rat
brain soluble proteins was assayed by [3H]NaBH4 reduction immedi-
ately after each time sample (s). Error bars denote range (n ¼ 2)
except for the 0, 30 and 60 min points for the endogenous modifica-
tion, ±SEM (n ¼ 3).
Fig. 6 Exogenous HCHO increases lysine modification. HCHO was
added to the medium 2 h before soluble proteins were extracted from
either control PC12 cells (j) or cells pretreated with 10 lM BSO for
24 h (d). Lysine modification was measured by the [3H]NaBH4
method. Cells appeared normal after HCHO treatment. Data are the
means of three experiments.
One-carbon protein lysine modification 511
� 2006 The AuthorsJournal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514

et al. 2004), protein demethylases (Shi et al. 2004), andspontaneous decomposition of N5,N10-methylenetetrahydro-folic acid (‘active formaldehyde’; Kucharczyk et al. 1984).Oxidative stress could augment formaldehyde production byseveral of these mechanisms.
Cells contain several detoxification systems for formalde-hyde. HCHO can be metabolized to either methanol byalcohol dehydrogenase (ADH1) or to formate by aldehydedehydrogenase (ALDH2) and can be oxidized by cyto-chrome P-450 enzymes (Teng et al. 2001). In addition,HCHO can be metabolized by incorporation into the C1 poolvia tetrahydrofolate pathways. Finally, in a GSH-dependentpathway that is widespread in mammalian tissues (Edenbergand Bosron 1997), HCHO reacts non-enzymatically withGSH to yield S-hydroxymethyl-GSH, which is then conver-ted to S-formylglutathione by alcohol dehydrogenasex
(ADHx; ‘formaldehyde dehydrogenase’, EC 1.2.1.1). Asecond enzyme, S-formylglutathione hydrolase, yields for-mate and regenerates GSH.
The HCHO detoxification capacity of the ADHv systemappears to be limited, because GSH depletion had anegligible effect on lysine modification induced by externalHCHO (Fig. 6). The small increase in HCHO toxicityobserved in ADHx knock-out mice also indicates a limitedcapacity of this system for HCHO detoxification (Molotkovet al. 2002). Interestingly, however, ADHx is the ancestralmember of the ADH family, which suggests that HCHOscavenging may have been an early metabolic necessity(Kaiser et al. 1993).
The demethylase hypothesisMethylenelysine could also be produced by enzymaticdemethylation of proteins. A demethylase has recently beenidentified that removes methyl groups from methyllysines onhistones (Shi et al. 2004). The enzyme is an amine oxidasethat forms H2O2 during the demethylation reaction. Mono-methyl lysine groups are oxidized to methylenelysine, whichthen hydrolyzes spontaneously to formaldehyde and lysinewith a half-time of minutes. Perhaps the modification wedetected represents methylenelysine intermediates in ourprotein preparation that were reduced to methyllysine byNaBH4. This hypothesis predicts that reducible lysinemodifications will be found specifically on proteins that areenzymatically methylated. Increases in methylenelysinemodification during oxidative stress would imply that proteinmethylation, or the activity of the demethylase, increaseunder such conditions.
Stability of the lysine modification
The stability of these modifications implies that they may belocated on lysines in unique protein microenvironments.Imines can be stabilized by either electrostatic interactions orhydrogen bond networks in hydrophobic protein domains(Mure et al. 2002; Janz and Farrens 2004). Moreover, lysine
amines at such locations would be more reactive with eitherformaldehyde or other neutral electrophiles because ofdiminished protonation, and therefore may be modifiedpreferentially in vivo. These considerations lead to theprediction that naturally occurring lysine modifications arelocated on reactive lysines that may, in some cases, modulateprotein function. Examples of such reactive lysines are anactive site lysine in bovine pancreatic ribonuclease A that ishighly reactive with HCHO, which inactivates the enzymeafter reduction with NaBH4 (Means and Feeney 1968).Activity of the imine modification was not studied. Similarly,a Lys that binds pyridoxal phosphate in aspartate amino-transferase reacts preferentially with HCHO. After reducingthe resulting Schiff base with NaCNBH3, the methylatedenzyme is catalytically inactive (Roberts et al. 1988).
In addition to these implications for protein function, therelative stability of endogenous lysine modifications in eitherbrain or cell proteins has two practical consequences for theiranalysis. First, the observed stability argues that they werenot introduced by the reaction with HCHO during eitherprotein preparation or analysis, because putative methylen-elysine produced in vitro was highly labile. Secondly, thestability of the endogenous modifications indicates that theycan be detected in good yield even in either tissues or extractsthat have been exposed to room temperature for severalhours.
Methylene lysine modifications and cell viability
Oxidative stress in either brain tissue or PC12 cells appearedto increase putative methylenelysine modification by a factorof roughly 1.7. A similar increase resulted from adding�25 lM HCHO to PC12 cell cultures. This concentration ofexternal HCHO also impaired cell viability. Although bothtoxicity and formaldehyde levels in dopamine neurons ofhuman substantia nigra are likely to differ from those inPC12 cells, the similarity in order of magnitude of the HCHOconcentrations at which protein modification and cytotoxicityoccur raises the possibility that formaldehyde concentrationsin midbrain dopamine neurons reach toxic levels as a resultof oxidative stress in that region.
Formation of methylene imines could affect lysine func-tion by converting the highly basic e-amine group to a weakbase with pKa � 7 (French and Edsall 1945). To determinethe physiological significance of such modifications, it willbe necessary to identify proteins affected by lysine iminemodification and determine the functional consequences ofthis alteration.
Methylenelysine modification of a-synuclein could poten-tially contribute to its pathological aggregation in eitherParkinson’s disease or other synucleinopathies. The highlyconserved Glu46 in a-synuclein is mutated to Lys in anautosomal dominant form of Parkinson’s disease (Zarranzet al. 2004). Although a-synuclein contains 15 lysines, mostin imperfect KTKEGV repeats that mediate binding to
512 E. Floor et al.
Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors

phospholipid membranes, change of a single charged groupin one of these repeats in the E46K mutation greatlyincreases a-synuclein lipid binding and aggregation intofilaments (Choi et al. 2004). Thus it is conceivable thataltered basicity caused by the formation of imines on eitherone or more lysines could similarly alter the functionalproperties and pathogenicity of a-synuclein. In addition,modification of lysines could reduce ubiquitination and leadto pathological accumulation of a-synuclein.
In conclusion, our results suggest that reducible, one-carbon modification of protein lysine is a previouslyunexplored consequence of oxidative stress. Elevated methy-lene lysine modification and, possibly, associated increases inintracellular formaldehyde may impair the viability of cellsand tissues, including dopamine neurons in the substantianigra, and heighten their vulnerability to degeneration.
Acknowledgements
This work was supported by NIH Grant PO1 AG12993. Thanks to
Dr Robert P. Hanzlik, Department of Medicinal Chemistry,
University of Kansas, for advice and to Dr Hanzlik and Dr Mary
L. Michaelis, Department of Pharmacology & Toxicology, for
critical reading of the manuscript. Postmortem human brain was
provided by the Harvard Brain Tissue Resource Center, which is
supported in part by PHS grant R24 MH068855.
References
Aas P. A., Otterlei M., Falnes P. Ø. et al. (2003) Human and bacterialoxidative demethylases repair alkylation damage in both RNA andDNA. Nature 421, 859–863.
Amici A., Levine R. L., Tsai L. and Stadtman E. R. (1989) Conversionof amino acid residues in proteins and amino acid homopolymersto carbonyl derivatives by metal-catalyzed oxidation reactions.J. Biol. Chem. 264, 3341–3346.
Anderson M. E. (1985) Determination of glutathione and glutathionedisulfide in biological samples. Meth. Enzymol. 113, 548–555.
Balaban R. S., Nemoto S. and Finkel T. (2005) Mitochondria, oxidants,and aging. Cell 120, 483–495.
Choi W., Zibaee S., Jakes R., Serpell L. C., Davletov B., Crowther R. A.and Goedert M. (2004) Mutation E46K increases phospholipidbinding and assembly into filaments of human a-synuclein. FEBSLett. 576, 363–368.
Climent I., Tsai L. and Levine R. L. (1989) Derivatization of c-glutamylsemialdehyde residues in oxidized proteins by fluoresceinamine.Anal. Biochem. 182, 226–232.
Desagher S., Glowinski J. and Premont J. (1996) Pyruvate protectsneurons against hydrogen peroxide-induced toxicity. J. Neurosci.17, 9060–9067.
Edenberg H. J. and Bosron W. F. (1997) Alcohol Dehydrogenases, inComprehensive Toxicology (F. P. Guengerich, ed.), Vol. 3, pp.119–131. Pergamon Press, New York.
Emborg M. E., Ma S. Y., Mufson E. J., Levey A. I., Taylor M. D., BrownW. D., Holden J. E. and Kordower J. H. (1998) Age-relateddeclines in nigral neuronal function correlate with motor impair-ments in rhesus monkeys. J. Comp. Neurol. 401, 253–265.
Floor E. and Wetzel M. G. (1998) Increased protein oxidation in humansubstantia nigra pars compacta in comparison with basal ganglia
and prefrontal cortex measured with an improved din-itrophenylhydrazine assay. J. Neurochem. 70, 268–275.
French D. and Edsall J. T. (1945) The Reactions of Formaldehyde withAmino Acids and Proteins, in Advances in Protein Chemistry(Anson M. L. and Edsall J. T., eds), Vol. II, pp. 277–335. Aca-demic Press, New York.
Giorgio M., Migliaccio E., Orsini F. et al. (2005) Electron transferbetween cytochrome c and p66Shc generates reactive oxygenspecies that trigger mitochondrial apoptosis. Cell 122, 221–233.
Gubisne-Haberle D., Hill W., Kazachkov M., Richardson J. S. and Yu P.H. (2004) Protein cross-linkage induced by formaldehyde derivedfrom semicarbazide-sensitive amine oxidase-mediated deaminationof methylamine. J. Pharmacol. Exper. Therapeut. 310, 1125–1132.
Halliwell B. and Gutteridge J. M. C. (1999) Free Radicals in Biologyand Medicine, 3rd edn. Oxford UP, Oxford.
Headlam H. A. and Davies M. J. (2002) Beta-scission of side-chainalkoxyl radicals on peptides and proteins results in the loss of side-chains as aldehydes and ketones. Free Radic. Biol. Med. 32, 1171–1184.
Heck H., d’A., White E. L. and Casanova-Schmitz M. (1982) Deter-mination of formaldehyde in biological tissues by gas chroma-tography/mass spectrometry. Biomed. Mass Spectrom. 9, 347–353.
Janz J. M. and Farrens D. L. (2004) Role of the retinal hydrogen bondnetwork in rhodopsin Schiff base stability and hydrolysis. J. Biol.Chem. 279, 55 886–55 894.
Jenner P. (2003) Oxidative stress in Parkinson’s disease. Ann. Neurol. 53,S26–S38.
Kaiser R., Fernandez M. R., Pares X. and Jornvall H. (1993) Origin ofthe human alcohol dehydrogenase system: implications from thestructure and properties of the octopus protein. Proc. Natl. Acad.Sci. USA 90, 11 222–11 226.
Kruman I., Bruce-Keller A. J., Bredesen D., Waeg G. and Mattson M. P.(1997) Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci. 17, 5089–5100.
Kucharczyk N., Yang J. T., Wong K. K. and Sofia R. D. (1984) Theformaldehyde-donating activity of N5,N10-methylene tetrahydro-folic acid in xenobiotic biotransformation. Xenobiotica 14, 667–676.
Lee C.-K., Pugh T. D., Klopp R. G., Edwards J., Allison D. B., WeindruchR. and Prolla T. A. (2004) The impact of a-lipoic acid, coenzymeQ10
and caloric restriction on life span and gene expression patterns inmice. Free Radic. Biol. Med. 36, 1043–1057.
Levine R. L., Garland D., Oliver C. N., Amici A., Climent I., Lenz A.G., Ahn B. W., Shaltiel S. and Stadtman E. R. (1990) Determin-ation of carbonyl content in oxidatively modified proteins. Meth.Enzymol. 186, 464–478.
Lezza A. M., Mecocci P., Cormio A., Beal M. F., Cherubini A., Can-tatore P., Senin U. and Gadaleta M. N. (1999) Mitochondrial DNA4977 bp deletion and OH8dG levels correlate in the brain of agedsubjects but not Alzheimer’s disease patients. FASEB J. 13, 1083–1088.
Ma S. Y., Ciliax B. J., Stebbins G., Jaffar S., Joyce J. N., Cochran E. J.,Kordower J. H., Mash D. C., Levey A. I. and Mufson E. J. (1999)Dopamine transporter-immunoreactive neurons decrease with agein the human substantia nigra. J. Comp. Neurol. 409, 25–37.
Mahrenholz A. M., Denslow N. D., Andersen T. T., Schegg K. M., MannK., Cohen S. A., Fox J. W. and Yuksel K. U. (1996) Amino acidanalysis- recovery from PVDF membranes. ABRF-95AAA Col-laborative Trial, in Techniques in Protein Chemistry (Marshak D.R., ed.), Vol. VII, pp. 323–330. Academic Press, New York.
McCormack A. L., Atienza J. G., Johnston L. C., Andersen J. K., Vu S.and Di Monte D. A. (2005) Role of oxidative stress in paraquat-induced dopaminergic cell degeneration. J. Neurochem. 93, 1030–1037.
One-carbon protein lysine modification 513
� 2006 The AuthorsJournal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514

Means G. E. and Feeney R. E. (1968) Reductive alkylation of aminogroups in proteins. Biochemistry 7, 2192–2201.
Meltzer N. M., Tous G. I., Gruber S. and Stein S. (1987) Gas-phasehydrolysis of proteins and peptides. Anal. Biochem. 160, 356–361.
Molotkov A., Fan X., Deltour L., Foglio M. H., Martras S., Farres J.,Pares X. and Duester G. (2002) Stimulation of retinoic acid pro-duction and growth by ubiquitously expressed alcohol dehydro-genase Adh3. Proc. Natl. Acad. Sci. USA 99, 5337–5342.
Mure M., Mills S. A. and Klinman J. P. (2002) Catalytic mechanism ofthe topa quinone containing copper amine oxidases. Biochemistry41, 9269–9278.
Nudelman A., Levovich I., Cutts S. M., Phillips D. R. and Rephaeli A.(2005) The role of intracellularly released formaldehyde and bu-tyric acid in the anticancer activity of acyloxyalkyl esters. J. Med.Chem. 48, 1042–1054.
Parent A. (1996) Carpenter’s Human Neuroanatomy 9th edn, pp. 557.Williams & Wilkins, Baltimore.
Rashba-Step J., Step E., Turro N. J. and Cedarbaum A. I. (1994) Oxi-dation of glycerol to formaldehyde by microsomes: are glycerolradicals produced in the reaction pathway? Biochemistry 33, 9504–9510.
Roberts W. J., Hubert E., Iriarte A. and Martinez-Carrion M., (1988)Site-specific methylation of a strategic lysyl residue in aspartateaminotransferase. J. Biol. Chem. 263, 7196–7202.
Schaffner W. and Weissmann C. (1973) A rapid, sensitive, and specificmethod for the determination of protein in dilute solution. Anal.Biochem. 56, 502–514.
Scholze H. (1985) Determination of phenylthiocarbamyl amino acidsby reversed-phase high-performance liquid chromatography.J. Chromatog. 350, 453–460.
Schriner S. E., Linford N. J., Martin G. M. et al. (2005) Extension ofmurine lifespan by overexpression of catalase targeted to mito-chondria. Science 308, 1909–1911.
Shi Y., Lan F., Matson C., Mulligan P., Whetstine J. R., Cole P. A.,Casero R. A. and Shi Y. (2004) Histone demethylation mediated bythe nuclear amine oxidase homolog LSD1. Cell 119, 941–953.
Si F., Shin S. H., Biedermann A. and Ross G. M. (1999) Estimation ofPC12 cell numbers with acid phosphatase assay and mitochondrialdehydrogenase assay: dopamine interferes with assay based ontetrazolium. Exp. Brain Res. 124, 145–150.
Smith C. D., Carney J. M., Starke-Reed P. E., Oliver C. N., Stadtman E.R., Floyd R. A. and Markesbery W. R. (1991) Excess brain proteinoxidation and enzyme dysfunction in normal aging and in Alz-heimer disease. Proc. Natl. Acad. Sci. USA 88, 10540–10543.
Taborsky G. (1973) Oxidative modification of proteins in the presence offerrous ion and air. Effect of ionic constituents of the reactionmedium on the nature of the oxidation products. Biochemistry 12,1341–1348.
Teng S., Beard K., Pourahmad J., Moridani M., Easson E., Poon R. andO’Brien P. J. (2001) The formaldehyde metabolic detoxificationenzyme systems and molecular cytotoxic mechanism in isolated rathepatocytes. Chem. Biol. Interact. 130–132, 285–296.
Uchida K., Shiraishi M., Naito Y., Torii Y., Nakamura Y. and Osawa T.(1999) Activation of stress signaling pathways by the end productof lipid peroxidation. J. Biol. Chem. 274, 2334–2342.
Volkow N. D., Gur R. C., Wang G. J., Fowler J. S., Moberg P. J., Ding Y.S., Hitzemann R., Smith G. and Logan J. (1998) Associationbetween decline in brain dopaminergic activity with age andcognitive and motor impairment in healthy individuals. Am. J.Psychiatry 155, 344–349.
Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E. R. andMizuno Y. (1996) Immunohistochemical detection of 4-hydroxy-nonenal protein adducts in Parkinson disease. Proc. Natl. Acad.Sci. USA 93, 2696–2701.
Zarranz J. J., Alegre J., Gomez-Esteban J. C. et al. (2004) The newmutation, E46K, of a-synuclein causes Parkinson and Lewy bodydementia. Ann. Neurol. 55, 164–173.
514 E. Floor et al.
Journal Compilation � 2006 International Society for Neurochemistry, J. Neurochem. (2006) 97, 504–514� 2006 The Authors




















