
3830.full.pdf - The Journal of Immunology
12
of July 24, 2022. This information is current as THP-1 Human Macrophages in Differentiated Mycobacterium tuberculosis IL-32 Is a Host Protective Cytokine against Chan D. McGibney, Hua Huang, Charles A. Dinarello and Edward Xiyuan Bai, Soo-Hyun Kim, Tania Azam, Mischa T. http://www.jimmunol.org/content/184/7/3830 doi: 10.4049/jimmunol.0901913 February 2010; 2010; 184:3830-3840; Prepublished online 26 J Immunol References http://www.jimmunol.org/content/184/7/3830.full#ref-list-1 , 44 of which you can access for free at: cites 85 articles This article average * 4 weeks from acceptance to publication Fast Publication! • Every submission reviewed by practicing scientists No Triage! • from submission to initial decision Rapid Reviews! 30 days* • Submit online. ? The JI Why Subscription http://jimmunol.org/subscription is online at: The Journal of Immunology Information about subscribing to Permissions http://www.aai.org/About/Publications/JI/copyright.html Submit copyright permission requests at: Email Alerts http://jimmunol.org/alerts Receive free email-alerts when new articles cite this article. Sign up at: Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved. Copyright © 2010 by The American Association of 1451 Rockville Pike, Suite 650, Rockville, MD 20852 The American Association of Immunologists, Inc., is published twice each month by The Journal of Immunology by guest on July 24, 2022 http://www.jimmunol.org/ Downloaded from by guest on July 24, 2022 http://www.jimmunol.org/ Downloaded from
-
Upload
khangminh22 -
Category
Documents
-
view
4 -
download
0
Transcript of 3830.full.pdf - The Journal of Immunology

of July 24, 2022.This information is current as
THP-1 Human Macrophages in DifferentiatedMycobacterium tuberculosis
IL-32 Is a Host Protective Cytokine against
ChanD.McGibney, Hua Huang, Charles A. Dinarello and Edward
Xiyuan Bai, Soo-Hyun Kim, Tania Azam, Mischa T.
http://www.jimmunol.org/content/184/7/3830doi: 10.4049/jimmunol.0901913February 2010;
2010; 184:3830-3840; Prepublished online 26J Immunol
Referenceshttp://www.jimmunol.org/content/184/7/3830.full#ref-list-1
, 44 of which you can access for free at: cites 85 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists, Inc. All rights reserved.Copyright © 2010 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on July 24, 2022
http://ww
w.jim
munol.org/
Dow
nloaded from

The Journal of Immunology
IL-32 Is a Host Protective Cytokine against Mycobacteriumtuberculosis in Differentiated THP-1 Human Macrophages
Xiyuan Bai,* Soo-Hyun Kim,† Tania Azam,‡ Mischa T. McGibney,* Hua Huang,x
Charles A. Dinarello,‡ and Edward D. Chan*,{,‖
Macrophages provide a first line of defense againstMycobacterium tuberculosis. However, in instances wheremacrophage activation
for killing is suboptimal,M. tuberculosis is capable of surviving intracellularly. IL-32 is a recently described cytokine induced byM.
tuberculosis in a variety of cell types including human monocytes and macrophages. In this study, we investigated the biological
significance of IL-32 in an in vitro model of M. tuberculosis infection in differentiated THP-1 human macrophages in which IL-32
expression was silenced using stable expression of short hairpin RNA (shRNA). Inhibition of endogenous IL-32 production in THP-1
cells that express one of three distinct shRNA-IL-32 constructs significantly decreasedM. tuberculosis induction of TNF-a by∼60%,
IL-1b by 30–60%, and IL-8 by 40–50% and concomitantly increased the number of cell-associated M. tuberculosis bacteria
compared with THP-1 cells stably expressing a scrambled shRNA. In THP-1 cells infected with M. tuberculosis and stimulated
with rIL-32, a greater level of apoptosis was observed compared with that withM. tuberculosis infection alone. Obversely, there was
significant abrogation of apoptosis induced byM. tuberculosis and a concomitant decrease in caspase-3 activation in cells depleted of
endogenous IL-32. rIL-32g significantly reduced the number of viable intracellularM. tuberculosis bacteria, which wasmodestly but
significantly abrogated with a caspase-3 inhibitor. We conclude that IL-32 plays a host defense role against M. tuberculosis in
differentiated THP-1 human macrophages. The Journal of Immunology, 2010, 184: 3830–3840.
Tuberculosis (TB) remains a leading cause of suffering anddeath worldwide (1). Although alveolar macrophagesprovide a first line of defense against Mycobacterium tu-
berculosis, macrophages may also provide a safe intracellular nichefor mycobacteria if host cells are unable to kill them (2, 3). Whetherthe initial infection leads to complete elimination, a progressiveinfection, or a state of latent infection ultimately depends on theeffectiveness of both the innate immune system and the cell-mediated immune system, orchestrated by macrophages and den-dritic cells. Presentation of mycobacterial Ags by macrophages anddendritic cells to lymphocytes in regional lymph nodes, resulting inthe differentiation, activation, and efflux of effector T cells to sitesof infection is a major pillar in the effective host response againstM. tuberculosis. Known killing mechanisms against intracellularM. tuberculosis include phagosome–lysosome fusion, apoptosis,autophagy, direct toxic effect of NO, and antimicrobial peptides,such as cathelicidin, defensins, and granulysin (4–15).The collaboration between APCs and T cells culminating in
their mutual activation and participation in the granulomatous
response are mediated in large part by the release of cytokines,chemokines, and other effector molecules by both phagocytes andT cells. Cytokines important in host defense against M. tuber-culosis and other pathogenic mycobacteria in both experimentalanimals and humans include TNF-a, IFN-g, IL-8, IL-12, IL-17,IL-18, and IL-23 (16–27). By contrast, overexpression of Th2 orTh2-like cytokines, such as IL-4, IL-10, and TGF-b, is generallyconsidered to predispose the host to M. tuberculosis (28–34),although controversies exist (35–37). Categorizing a cytokine asbeing helpful or harmful to the host in the context of TB is likelyan oversimplistic view. For example, although TNF-a is criticalin maintaining granuloma formation and containing mycobacte-rial infections, in the presence of IL-4, TNF-a is also responsiblefor many of the deleterious clinical manifestations, such as fever,tissue necrosis, anorexia, and weight loss (29, 38). Similarly, IL-4, IL-10, and TGF-b may be necessary to the host in the latterparts of the host immune response to prevent relentless in-flammation and limit tissue damage. More recently, with micewith genetic disruption of IL-27R, IL-27 was shown to have theseemingly paradoxical effect of increasing M. tuberculosis bur-den and yet reducing cellular inflammation and lung pathology,resulting in a net effect of increased survival in the infectedmice (39, 40).IL-32 is a recently described cytokine produced by T lympho-
cytes, NK cells, epithelial cells, endothelial cells, monocytes,macrophages, and dendritic cells (41–45). IL-32 is also produced byhuman pancreatic periacinar myofibroblasts, pancreatic ductalcells, synovial fibroblasts, and marrow stromal cells in patients withmyelodysplastic syndrome (46–49). IL-32 has proinflammatoryproperties in that it can induce production of TNF-a, MIP-2, IL-6,and IL-8 (43, 50). In turn, cytokines such as IFN-g, IL-1b, IL-12,and IL-18 can induce IL-32 production (43). Six different isoformsof IL-32 have been identified, namely, IL-32a, b, g, d, ε, and z (43,51). Depending on the cell type and stimulus, different isoformsmay predominate (44). IL-32g is the most biologically active
*Division of Pulmonary Sciences and Critical Care Medicine and ‡Division of In-fectious Diseases, University of Colorado Denver at AnschutzMedical Center, Denver,CO 80045; ||Division of the Mycobacterial and Respiratory Infections, Department ofMedicine and xDepartment of Immunology, National Jewish Health, Denver, CO80206; {Denver Veteran Affairs Medical Center, Denver, CO 80220; and †Laboratoryof Cytokine Immunology, Institute of Biomedical Science and Technology, Konkuk Uni-versity, Seoul, South Korea
Received for publication June 18, 2009. Accepted for publication January 15, 2010.
This work was supported in part by the Office of Research and Development, Vet-erans Health Administration, Department of Veterans Affairs (to E.D.C.) and Na-tional Institutes of Health Grant AI15614 (to C.A.D.).
Address correspondence and reprint requests to Dr. Edward D. Chan, Neustadt Build-ing, D509, National Jewish Health, 1400 Jackson Street, Denver, CO 80206. E-mailaddress: [email protected]
Abbreviations used in this paper: COPD, chronic obstructive pulmonary disease;shRNA, short hairpin RNA; siRNA, small interfering RNA; TB, tuberculosis; WT,wild-type; z-DEVD-fmk, benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone.
www.jimmunol.org/cgi/doi/10.4049/jimmunol.0901913
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

isoform in inducing cytokine production, perhaps because thisisoform has no exonic deletions (52).Although the receptor for IL-32 has not been found, IL-32 can
induce activation of the MAPK and NF-kB signaling pathways (43,47, 53). Interestingly, during the search for the receptor for IL-32, itwas found that proteinase 3, independent of its serine protease ac-tivity, binds to IL-32 and cleaves it into fragments that aremore active,in terms of induction of cytokines, than the parent molecule (54, 55).We previously showed that IL-32 is induced by M. tuberculosis
and that in PBMCs induction of IL-32 byM. tuberculosis occurredin an IL-18– and IFN-g–dependent mechanism (56). However, thebiological significance of endogenous IL-32 in M. tuberculosis in-fection remains unknown. Because IL-32 is induced by cytokinesand itself induces cytokines that are of immense importance in thecontrol of M. tuberculosis, we hypothesized that IL-32 may playa host defense role against M. tuberculosis. Because mouse homo-logue of IL-32 has not yet been found, murine models could not beused to study the role of IL-32 in host defense against M. tubercu-losis. Therefore, we used small interfering RNA (siRNA) technol-ogy to reduce the level of endogenous IL-32 to study its role in M.tuberculosis infection of a human monocyte/macrophage cell line.We chose to create stable cell lines that express short hairpin RNA(shRNA) sequences. The human myeloid cell line THP-1 was usedas a model for human macrophages in in vitro infection studies fortwo reasons. First, THP-1 cells are amenable to stable transfectionwithDNA fragments that encode shRNAsequences. Second, THP-1cells have been shown to behave similarly to primary human mac-rophages in their immune responses to M. tuberculosis (57, 58).
Materials and MethodsMaterials
The human promonocytic cell line THP-1 was obtained from the AmericanTypeCultureCollection (TIB-202;Manassas,VA).RPMI1640was obtainedfrom Cambrex (East Rutherford, NJ). FBS was purchased from AtlantaBiologicals (Norcross, GA) and heat-inactivated at 56˚C for 1 h. THP-1 cellswere cultured in RPMI 1640 supplemented with 10% FBS and 2 mM glu-tamine and were maintained at a concentration between 2 and 103 105 cellsper milliliter. PMA was purchased from Sigma-Aldrich (St. Louis, MO).TRIzol reagents, Superscript II Reverse Transcriptase First-Strand cDNA,and PCRPlatinumTaqDNApolymerasewere purchased from InvitrogenLifeTechnologies (Carlsbad, CA). Apoptosis In Situ Detection Kit was purchasedfrom Roche Diagnostic Systems (Somerville, NJ). rIL-32a and rIL-32gproteins were expressed in Escherichia coli as hexahistadine-tagged N-terminal fusion proteins, followed by affinity purification on a TALON affinitycolumn (Invitrogen Life Technologies) (43, 56). Human rIFN-g, anotherpreparation of rIL-32g (confirmed to be free from LPS), the caspase-3 in-hibitor benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone (z-DEVD-fmk), and ELISA kits for human active caspase-3 (Human active Caspase-3Immunoassay), IL-10, and TGF-b were purchased from R&D Systems(Minneapolis, MN). Rabbit polyclonal anti-human BcL-2 Ab, Bax Ab,cytochrome cAb, and Phototope-HRPWestern Blot Detection System werepurchased from Cell Signaling Technology (Beverly, MA). Goat anti–rIL-32a polyclonal Ab was produced by Rockland (Gilbertsville, PA) and pu-rified according to the manufacturer’s protocol. All primers and oligonu-cletide products were obtained from GeneLink (Hawthorne, NY).
Mycobacterial culture and reagents
The H37Rv strain ofM. tuberculosis was obtained from the American TypeCulture Collection (27294).M. tuberculosis was grown to late log phase inMiddlebrook 7H9 supplemented with albumin dextrose complex. After in-cubation by rotation at 37˚C, aliquots were frozen at 270˚C in 1.0 McFar-land standard stocks. Middlebrook 7H9, 7H10, and Middlebrook albumindextrose complex enrichment were obtained from Difco (Detroit, MI).
Stable expression of shRNA–IL-32 sequences in THP-1 cells
Tocreate stable clones ofTHP-1 cells that inhibit IL-32protein expression bysiRNA technology, three separate plasmids were generated in which senseand antisense sequences to the IL-32 gene are separated by a 9-bp spacer ofa nonhomologous sequence (Table I). These DNA constructs were cloned
downstream of the H1 promoter within the pSUPER.puro RNAi vector thatalso contains the PUROMYCIN-RESISTANT gene (OligoEngine, Seattle,WA). The sense, antisense, and spacer DNA sequences encode shRNAs thatcontain antisense sequences to IL-32 mRNA (shRNA–IL-32). More spe-cifically, the DNA sequences contain a 19-nt sense sequence of IL-32cDNA, separated by a 9-mer spacer (TTCAAGAGA) from a 19-nt antisensesequence complementary to the sense sequence, followed by TTTTT as thetranscriptional terminator. These three DNA sequences (Table I) werechosen from ten sequences provided by the RNAi Design Program fromDharmacon (Boulder, CO) based on the greatest homology to the three mainisoforms of the IL-32 gene (a, b, and g). Circular pSUPER.puro RNAivector was linearized by BglII and HindIII restriction enzymes. The threechosen DNA sequences that contain sense and antisense sequences to IL-32were ligated into the linearized pSUPER.puro RNAi vector. The insertion ofthe aforementioned DNA sequences was confirmed by DNA sequencingusing the T7 primer prior to transfection into humanmonocytic THP-1 cells.In addition, a scrambled DNA sequence was also inserted into the pSUPER.puro RNAi vector to serve as a negative control.
The empty pSUPER.puro RNAi vector, one containing the scrambledDNA sequence, or each of the pSUPER.puro IL-32 siRNA plasmids weretransfected into THP-1 cells by electroporation using the Cell Line Nucle-ofector Kit V (Amaxa, Gaithersburg, MD) according to the manufacturer’sinstructions. In brief, 2 mg highly purified plasmid DNAwas added to 2 3106 THP-1 cells, followed by addition of 100 ml Nucleofector Solution V.Themixturewas transferred to an Amaxa certified cuvette. After the optimalnucleofector program for transfection by electroporation was used, the cu-vette was rinsed with medium, and the cells were immediately transferredfrom the cuvette into the culture dish. Clones of THP-1 cells stably ex-pressing shRNA–IL-32 were selected with 1 mg/ml puromycin for 12 d,followed by 0.5 mg/ml puromycin for an additional 12 d, with the mediumchanged every 3 d. Prior to experimentation, the THP-1 cells were treatedwith 15 ng/ml PMA overnight to allow differentiation into macrophages.The PMA-containing medium was removed and replaced with fresh me-dium just prior to exposing the cells to the experimental conditions.
RNA isolation and RT-PCR
After treatment of THP-1monocytes/macrophages on six-well tissue cultureplates, total RNA was extracted using the TRIzol reagent, and cDNA wasprepared using reverse transcriptase, according to the manufacturer’s in-structions (InvitrogenLife Technologies). Briefly, 3mg sample of total RNA,dissolved in diethyl pyrocarbonate-treated water, was heated at 65˚C for 10min and then cooled on ice for 10min. Then 1ml of 10 mM dNTPsmix, 1mloligo(dT)12–18 primer, and enough diethyl pyrocarbonate-treated water wereadded to the sample for a total volume of 10ml. After brief centrifugation, thesamples were incubated at 65˚C for 5 min, incubated on ice for 1 min, andfollowed by addition of a reaction mixture that contained 2 ml 103 reversetranscriptase buffer, 4 ml 25 mM MgCl2, 2 ml 0.1 M DTT, and 1 ml RNa-seOUT Recombinant Ribonuclease Inhibitor. The samples were mixedgently, collected by brief centrifugation, and then incubated at 42˚C for2 min. One microliter (50 U) of SuperScript II Reverse Transcriptase wasadded, continuously incubated at 42˚C for 50 min, and then inactivated at70˚C for 15min. The samples were treated with 1ml RNaseH to digest RNAat 37˚C for 30 min. The single-stranded cDNA products were used as tem-plates in a 30 ml PCR amplification reaction according to the instructions ofthe PCR kit. Amplifications were done by initial denaturation at 94˚C for4 min and 20 or 25 cycles of denaturation at 94˚C for 30 s, annealing at 60˚Cfor 45 s, extension at 72˚C for 1 min, and final extension at 72˚C for 4 min.The resulting PCR products were resolved by 1.5% agarose gel electro-phoresis. The following primers were used: IL-32 (549 bp) sense, 59-CTGAAG GCC CGA ATG CAC CAG-39; antisense, 59-GCA AAG GTG GTGTCAGTATC-39; IL-32 (314 bp) sense, 59-TGAGGAGCAGCACCCAGAGC-39; antisense, 59-CCGTAGGACTGGAAAGAGGA-39; GAPDH sense,59-TCC ATG ACA ACT TTG GTA TCG TG-39; antisense, 59-TGT CGCTGT TGA AGT CAG AGGA-39.
Western blot analysis
Briefly, THP-1monocytes/macrophageswere lysed using a phosphotyrosinelysis buffer that contains 50mMTris-HCl (pH8.0), 137mMNaCl, 10% (v/v)glycerol, 1% (v/v) NP-40, 2 mMNa3VO4, 1 mMNaF, 1 mMPMSF, 2mg/mlleupeptin, and 2 mg/ml aprotinin. The cell lysates were vortexed, sonicated,and centrifuged at 10,000 3 g for 10 min at 4˚C. Protein concentrations ofthe cell lysates were determined using the Bradford protein assay (Bio-Rad,Hercules, CA). The supernatants of the centrifuged lysates were mixed withequal volumes of 23 Laemmli sample buffer containing 40 mM DTT andboiled for 5 min. Twenty micrograms of protein for each condition wereresolved by 12% SDS-PAGE and transferred onto polyvinylidene difluoridemembrane (Immobilon P; Millipore, Bedford, MA). After the membranes
The Journal of Immunology 3831
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

were blocked in 5% milk for 1 h, they were probed with Abs to IL-32 ata dilution of 1:500 (v/v), Bcl-2 (1:1000), Bax (1:1000), cytochrome c(1:1000), or b-actin (1:1000), followed by detection with HRP-conjugatedanti-rabbit IgG in a 1:2000 dilution. Bands were visualized by chem-iluminescence using the HRP Western Blot Detection System (Cell Sig-naling Technology) as described in the manufacturer’s protocol.
In vitro infection of differentiated THP-1 human macrophages
THP-1 cells were infected with M. tuberculosis H37Rv at a multiplicity ofinfection of ten bacilli to one macrophage. Briefly, THP-1 cells were seededin 6- or 24-well flat-bottom tissue culture plates and allowed to adhere anddifferentiate at 37˚C in humidified, 5% CO2 atmosphere for 20 h in thepresence of 15 ng/ml PMA.After overnight incubation, the PMA-containingmedium was replaced with fresh RPMI 1640 plus 10% FBS and 2 mMglutaminewithout PMA. For day 0 cells, the supernatants were collected andfiltered after 1 h of infection. The cells were washed twice with a solutioncontaining 50% RPMI 1640 and 50% 13 PBS, followed by lysis of theadherent cells using 500ml 0.25%SDS solution in eachwell. After 5min, thecells were viewed under the inverted microscope to confirm cell lysis. Then500 ml 7H9 plating medium was added to each well. For day 4 and day 8conditions, the cellswerewashed twice after 1 h of infection, and themediumwas replaced with fresh RPMI 1640 containing 10% FBS and 2 mM gluta-mine. The cells were then incubated for an additional 4 or 8 d prior to fil-tration of the supernatants, lysis of the THP-1 cells, and culture of theliberatedM. tuberculosis.
Determination of CFUs
For the macrophage lysates, a series of 6-fold dilutions were prepared bycombining 50 ml cell lysate (500 ml 0.25% SDS plus 500 ml 7H9 medium)with 450 ml 7H9 medium dilution broth after 1 h, 4 d, or 8 d of infection.
TUNEL assay
Cell suspensions (0.5 ml) were grown on four-well slides, followed by dif-ferentiation with PMA and infection withM. tuberculosis as described pre-viously (59). For each condition, triplicate slides were prepared. The in situcell death detection kit was used to assay for apoptosis at the single-cell levelbased on labeling of DNA strand breaks and followed according to themanufacturer’s instructions. Briefly, postinfection with M. tuberculosis inglass-chambered slides, the medium was removed, the cells were washedwith 50% medium and 50% 13 PBS, and fixed in 4% paraformaldehydesolution (pH 7.4) for 1 h at room temperature, followed by incubation witha blocking solution containing 3% H2O2 in methanol for 10 min at roomtemperature. The slides were rinsed with 13 PBS and incubated with a per-meabilization solution (0.1% Triton X-100 in 0.1% sodium citrate solution)for 10min. They were rinsed twicewith 13 PBS, and 50ml TUNEL reactionmixture was added to each sample and allowed to incubate for 1 h in a hu-midified chamber located in a 37˚C incubator. The slides were rinsed threetimes with 13 PBS, and 50 ml Converter-POD substrate (Roche DiagnosticSystems, Indianapolis, IN) was added to each sample and incubated for 30min in a dark, humidified atmosphere at room temperature. After the slideswere washed with 13 PBS three times, 100 ml diaminobenzidine substratewas added and incubated for 2–5min at room temperature until a brown colordeveloped and then visualized by a light microscope. The slides were rinsedwith 13 PBS and dehydrated, and a cover slip was mounted using Permount(Fisher Scientific, Pittsburgh, PA). Enumeration of apoptotic cells was per-formed by light microscopy, counting ∼5000 cells per condition per exper-iment. The positive control for apoptosis was prepared from fixed andpermeablized cells that were then treated with a DNase solution to inducestrand breakage for 10 min. The negative control was incubated with labelsolution (a nucleotide mixture in reaction buffer) for 30 min.
Activated caspase-3 assay
During the terminal stages of the apoptotic pathway, caspase-3 zymogen iscleavedbyother caspases into three components: a proregion, a large subunit,and a small subunit. Activated caspase-3 was measured by a commerciallyavailable kit that uses a biotinylated caspase inhibitor (biotin-ZVKD-fluoromethylketone) that covalently modifies only the large subunit but notthe inactive caspase-3 zymogen. With a caspase-3 mAb coated on themicrotiter plate to capture total caspase-3 and the biotinylated caspase in-hibitor that binds to the large subunit added sequentially followed by HRP–streptavidin, only activated caspase-3 is detected.
Cytokine measurements
Electrochemiluminescence assays for IL-32, TNF-a, IL-1b, and IL-8 wereperformed as described previously (43). Human TGF-b and IL-10 levelswere measured by ELISA kits (R&D Systems) according to the manu-facturer’s instructions.
Statistical analysis
Replicate experiments were independent, and where appropriate, summaryresults are presented as mean 6 SD. Differences were considered signif-icant for p , 0.05. Group means were compared by repeated-measuresANOVA using Fisher’s least significance difference test.
ResultsStable expression of siRNA–IL-32 sequences in THP-1monocytes
Each of the three DNA sequences shown in Table I was cloned intothe pSUPER.puro RNAi vector and transfected into THP-1 cells.The transfected cells were selected by culture in the presence ofpuromycin. Through the actions of RNA polymerase III (60),shRNAs are synthesized, comprised two identical 19-nt sequencemotifs in an inverted orientation (forming the stems of the hair-pin), separated by a 9-nt loop of nonhomologous sequences (theloop of the hairpin). Double-stranded shRNA then undergo en-zymatic cleavage by DICER to ultimately yield single-strandedsiRNA fragments that are complementary and bind to the targetIL-32 mRNA.To determine whether this DNA vector-based approach can be
successfully transferred to human monocytic THP-1 cells, we firsttransfected the pmaxGFP vector by electroporation. By fluorescentmicroscopy, transfection efficiency was determined to be ∼30%(data not shown). Each of the three DNA fragments encodingshRNA–IL-32, referred to in this study as shRNA–IL-32 (clone 5),shRNA–IL-32 (clone 6), and shRNA–IL-32 (clone 7)—wastransfected separately into wild-type (WT) THP-1 cells. Thetransfected cells were cultured and selected in the presence ofpuromycin as described in Materials and Methods.
Confirmation of knockdown for IL-32 expression
Inhibitionofendogenous IL-32production in thepuromycin-selected,siRNA–IL-32–containing cells was confirmed by three methods.First, RT-PCR for IL-32 transcripts was performed after stimulatingTHP-1 cells expressing one of the three shRNA–IL-32 sequences or
Table I. DNA fragments that encode shRNAs that target IL-32 mRNAs
Construct 59-Antisense Sequence/Hairpin Loop/Sense Sequence/Termination-39
psiRNA-IL-32-5 59-agatctAGACAGTGGCGGCTTATTAttcaagagaTAATAAGCCGCCACTGTCTtaaA-3939-aTCTGTCACCGCCGAATAATaagttctctATTATTCGGCGGTGACAGAattttcgaa-59
psiRNA-IL-32-6 59-agatctGACAGTGGCGGCTTATTATttcaagagaATAATAAGCCGCCACTGTCtaaA-3939-aCTGTCACCGCCGAATAATAaagttctctTATTATTCGGCGGTGACAGattttcgaa-59
psiRNA-IL-32-7 59-agatctAGAGGGCTACCTGGAGACAttcaagagaTGTCTCCAGGTAGCCCTCTaaaA-3939-aTCTCCCGATGGACCTCTGTaagttctctACAGAGGTCCATCGGGAGAtttttcgaa-59
pScramble-siRNA 59-agatctATCGAACGATTCGATGTAAttcaagagaTTACATCGAATCGTTCGATaaaA-3939-aTAGCTTGCTAAGCTACATTaagttctctAATGTAGCTTAGCAAGCTAtttttcgaa-59
Shown are three dsDNA constructs that encode three distinct shRNA–IL-32 clones or a scrambled shRNA sequence. Each DNA construct wasannealed and ligated into the pSuper.puro-shRNA vector with the HindIII and BglII restriction sites underlined. The underlined sequences at the 59 and 39ends are for directional cloning into the pSuper.puro-shRNA vector. See Materials and Methods for additional details.
3832 ROLE OF IL-32 IN TUBERCULOSIS
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

a scrambled shRNA sequence with 10 U/ml IFN-g for 18 h. For thePCR reactions, two different sets of sense and antisense primersshown in Materials and Methods were used. As shown in Fig. 1A(lanes 1–4), IL-32 transcripts were not detected in any of the THP-1clones under basal conditions. In THP-1 cells expressing thescrambled shRNA sequence (shRNA–IL-32 Scr), the IL-32 mRNAlevel was increased following stimulation with 10 U/ml IFN-g for18 h (Fig. 1A, lane 5). As can be seen, two separateRT-PCRproductswere present, corresponding to the two different primer sets used.By contrast, IFN-g–stimulated IL-32 mRNA production was sig-nificantly inhibited in the three THP-1 cell lines expressing shRNA–IL-32 sequences, with the greatest inhibition in the cell line stablyexpressing the shRNA–IL-32 (clone 7) (Fig. 1A, lanes 6–8). RT-PCR forGAPDH revealed equal loading of samples (Fig. 1A,bottompanel).Second, we confirmed these RNA studies by determining IL-32
protein expression by Western blot analyses. WT THP-1 cells andTHP-1 cells expressing scrambled shRNA or one of the threeshRNA–IL-32 sequences were stimulated with IFN-g or infectedwithM. tuberculosisH37Rv. After 24 h, the cells were lysed, and 20mg nuclear-free whole-cell lysates from each condition was sepa-rated by SDS-PAGE. The proteins were then transferred to nitro-cellulose membranes and immunoblotted with an Ab directedagainst IL-32. This Ab recognizes all known isoforms of IL-32 buthas the greatest affinity for IL-32a. As shown in Fig. 1B (lane 1, leftpanel and right panel), there was no detectable IL-32 protein inunstimulated WT THP-1 cells. Stimulation of WT THP-1 cells orTHP-1 cells expressing the scrambled shRNAwith 10 U/ml IFN-gfor 24 h resulted in a significant increase in the level of IL-32–aprotein (Fig. 1B, left panel, lanes 2 and 3). However, IFN-g stim-ulation of each THP-1 cell line expressing one of the three shRNA–IL-32 sequences showed significant inhibition of IL-32a pro-duction (Fig. 1B, left panel, lanes 4–6). As was seen for IL-32mRNA expression, THP-1 cells expressing shRNA–IL-32 (clone 7)exhibited the greatest reduction in IL-32 protein levels. Infection ofWT THP-1 cells or THP-1 cells expressing the scrambled shRNAwith M. tuberculosis H37Rv for 24 h resulted in a significant in-
crease in the level of IL-32a (Fig. 1B, right panel, lanes 2 and 3).By contrast, there was significantly less IL-32 in THP-1 cells ex-pressing shRNA–IL-32 sequences following M. tuberculosis in-fection (Fig. 1B, right panel, lanes 4–6). Immunoblotting forb-actin revealed roughly equal loading of proteins.Third, after stimulation with IFN-g, the cells were lysed, and the
lysates were assayed for IL-32a by electrochemiluminescence us-ing the sameAbused for theWestern blot analyses (43). As shown inFig. 1C, THP-1 clones expressing one of the three shRNA–IL-32sequences showed significant inhibition of IFN-g–induced IL-32aproduction compared with that of cells that expressed the scrambledshRNA. Little or no IL-32a was detected in the supernatant. Weconclude from these studies that stable expression of shRNA–IL-32is an efficient method to inhibit IL-32 production induced by IFN-gorM. tuberculosis.
Knockdown of IL-32 inhibits M. tuberculosis induction ofproinflammatory cytokines
We next determined whether cytokines induced byM. tuberculosiswere affected by knockdown of IL-32. WT THP-1 cells or THP-1cells expressing scrambled shRNA or one of the three clones ofshRNA–IL-32 sequences were either uninfected or infected withM.tuberculosis H37Rv for 6 and 24 h. After the indicated period ofincubation, cell culture supernatants were sterile-filtered to removelive M. tuberculosis bacteria and assayed for cytokine levels. Asshown in Fig. 2A, in the three clones of THP-1 cells that exhibitedreduced IL-32 levels, M. tuberculosis induction of TNF-a wassignificantly decreased by ∼35 and 60% after 6 and 24 h of in-fection, respectively, compared with those of WT THP-1 cells andTHP-1 cells expressing the scrambled shRNA. Similarly, pro-duction of IL-1b and IL-8 were substantially less (by ∼30–60 and∼40–50%, respectively) at 6 and 24 h following infection with M.tuberculosis H37Rv in cells with reduced IL-32 compared withthose ofWT THP-1 cells and THP-1 cells expressing the scrambledshRNA (Fig. 2B, 2C). There was little or no induction of TGF-bwith M. tuberculosis infection by THP-1 cells expressing eitherscrambled shRNA (open bars) or shRNA–IL-32 (clone 7) (closed
FIGURE 1. Inhibition of endogenous IL-32 production in THP-1 cells by siRNA. A, THP-1 cells expressing the scrambled shRNA or shRNA–IL-32
(clones 5, 6, or 7) were left unstimulated or stimulated with 10 U/ml IFN-g for 18 h. RT-PCR was performed with two separate primer sets for IL-32 as well
as for GAPDH. The data shown are representative of three independent experiments. B, THP-1 cells expressing the scrambled shRNA or shRNA–IL-32
(clones 5, 6, or 7) were left unstimulated, stimulated with 10 U/ml IFN-g for 24 h, or infected with M. tuberculosis for 24 h. Whole-cell lysates were
prepared and sterile-filtered, and Western blot analyses were performed for IL-32. The blots were also probed for b-actin. The data shown are representative
of three independent experiments. C, THP-1 cells expressing the scrambled shRNA or shRNA–IL-32 (clones 5, 6, or 7) were left unstimulated or stimulated
with 10 U/ml IFN-g for 24 h. The cells were then lysed, and the amount of IL-32a protein in the nuclear-free whole-cell lysates was measured by
electrochemiluminescence. The data shown are the mean 6 SD of three independent experiments. pppp , 0.001, compared with bar 2.
The Journal of Immunology 3833
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from
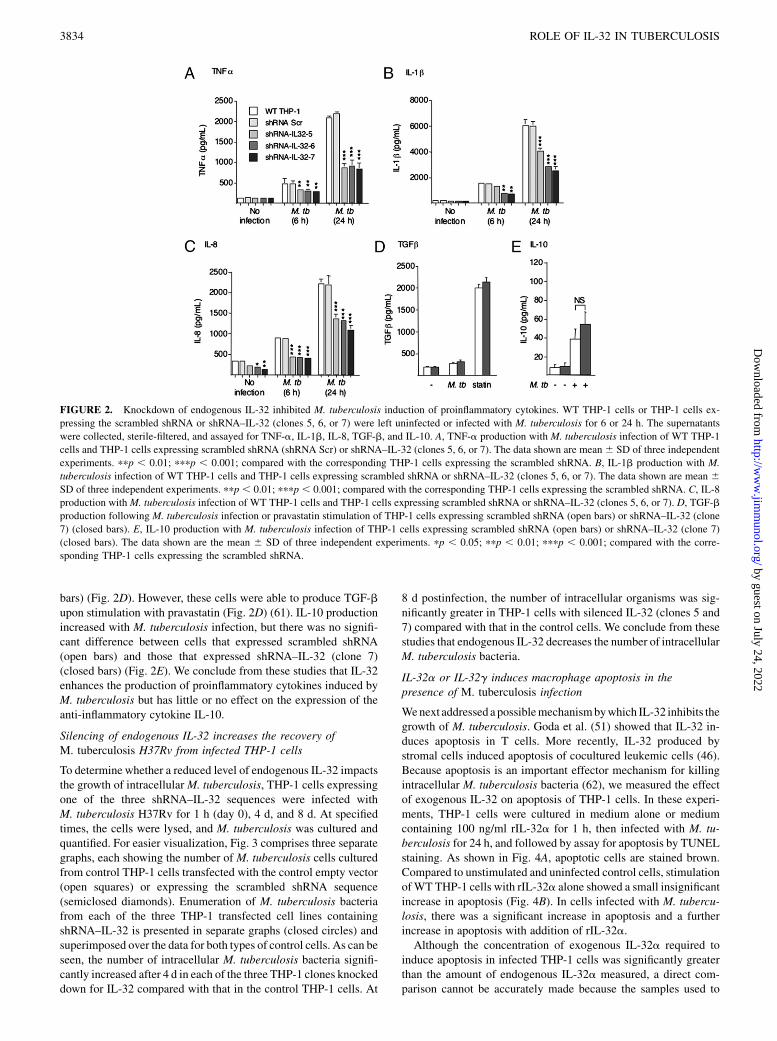
bars) (Fig. 2D). However, these cells were able to produce TGF-bupon stimulation with pravastatin (Fig. 2D) (61). IL-10 productionincreased with M. tuberculosis infection, but there was no signifi-cant difference between cells that expressed scrambled shRNA(open bars) and those that expressed shRNA–IL-32 (clone 7)(closed bars) (Fig. 2E). We conclude from these studies that IL-32enhances the production of proinflammatory cytokines induced byM. tuberculosis but has little or no effect on the expression of theanti-inflammatory cytokine IL-10.
Silencing of endogenous IL-32 increases the recovery ofM. tuberculosis H37Rv from infected THP-1 cells
To determine whether a reduced level of endogenous IL-32 impactsthe growth of intracellularM. tuberculosis, THP-1 cells expressingone of the three shRNA–IL-32 sequences were infected withM. tuberculosis H37Rv for 1 h (day 0), 4 d, and 8 d. At specifiedtimes, the cells were lysed, and M. tuberculosis was cultured andquantified. For easier visualization, Fig. 3 comprises three separategraphs, each showing the number of M. tuberculosis cells culturedfrom control THP-1 cells transfected with the control empty vector(open squares) or expressing the scrambled shRNA sequence(semiclosed diamonds). Enumeration of M. tuberculosis bacteriafrom each of the three THP-1 transfected cell lines containingshRNA–IL-32 is presented in separate graphs (closed circles) andsuperimposed over the data for both types of control cells. As can beseen, the number of intracellular M. tuberculosis bacteria signifi-cantly increased after 4 d in each of the three THP-1 clones knockeddown for IL-32 compared with that in the control THP-1 cells. At
8 d postinfection, the number of intracellular organisms was sig-nificantly greater in THP-1 cells with silenced IL-32 (clones 5 and7) compared with that in the control cells. We conclude from thesestudies that endogenous IL-32 decreases the number of intracellularM. tuberculosis bacteria.
IL-32a or IL-32g induces macrophage apoptosis in thepresence of M. tuberculosis infection
Wenext addressed apossiblemechanismbywhich IL-32 inhibits thegrowth of M. tuberculosis. Goda et al. (51) showed that IL-32 in-duces apoptosis in T cells. More recently, IL-32 produced bystromal cells induced apoptosis of cocultured leukemic cells (46).Because apoptosis is an important effector mechanism for killingintracellular M. tuberculosis bacteria (62), we measured the effectof exogenous IL-32 on apoptosis of THP-1 cells. In these experi-ments, THP-1 cells were cultured in medium alone or mediumcontaining 100 ng/ml rIL-32a for 1 h, then infected with M. tu-berculosis for 24 h, and followed by assay for apoptosis by TUNELstaining. As shown in Fig. 4A, apoptotic cells are stained brown.Compared to unstimulated and uninfected control cells, stimulationofWT THP-1 cells with rIL-32a alone showed a small insignificantincrease in apoptosis (Fig. 4B). In cells infected with M. tubercu-losis, there was a significant increase in apoptosis and a furtherincrease in apoptosis with addition of rIL-32a.Although the concentration of exogenous IL-32a required to
induce apoptosis in infected THP-1 cells was significantly greaterthan the amount of endogenous IL-32a measured, a direct com-parison cannot be accurately made because the samples used to
FIGURE 2. Knockdown of endogenous IL-32 inhibited M. tuberculosis induction of proinflammatory cytokines. WT THP-1 cells or THP-1 cells ex-
pressing the scrambled shRNA or shRNA–IL-32 (clones 5, 6, or 7) were left uninfected or infected with M. tuberculosis for 6 or 24 h. The supernatants
were collected, sterile-filtered, and assayed for TNF-a, IL-1b, IL-8, TGF-b, and IL-10. A, TNF-a production with M. tuberculosis infection of WT THP-1
cells and THP-1 cells expressing scrambled shRNA (shRNA Scr) or shRNA–IL-32 (clones 5, 6, or 7). The data shown are mean6 SD of three independent
experiments. ppp , 0.01; pppp , 0.001; compared with the corresponding THP-1 cells expressing the scrambled shRNA. B, IL-1b production with M.
tuberculosis infection of WT THP-1 cells and THP-1 cells expressing scrambled shRNA or shRNA–IL-32 (clones 5, 6, or 7). The data shown are mean 6SD of three independent experiments. ppp , 0.01; pppp , 0.001; compared with the corresponding THP-1 cells expressing the scrambled shRNA. C, IL-8
production withM. tuberculosis infection of WT THP-1 cells and THP-1 cells expressing scrambled shRNA or shRNA–IL-32 (clones 5, 6, or 7). D, TGF-b
production following M. tuberculosis infection or pravastatin stimulation of THP-1 cells expressing scrambled shRNA (open bars) or shRNA–IL-32 (clone
7) (closed bars). E, IL-10 production with M. tuberculosis infection of THP-1 cells expressing scrambled shRNA (open bars) or shRNA–IL-32 (clone 7)
(closed bars). The data shown are the mean 6 SD of three independent experiments. pp , 0.05; ppp , 0.01; pppp , 0.001; compared with the corre-
sponding THP-1 cells expressing the scrambled shRNA.
3834 ROLE OF IL-32 IN TUBERCULOSIS
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

measure IL-32a concentrations by electrochemiluminescence werecell lysates that reflected intracellular IL-32 levels subsequentlydiluted by the lysis buffer. Nevertheless, because the IL-32g iso-form is significantly more active than IL-32a in regards to cytokineproduction (52), we determined whether lower concentrations ofrIL-32g could recapitulate the findings seen with rIL-32a. Asshown in Fig. 4C, 50 ng/ml rIL-32g or M. tuberculosis induceda significant increase in apoptosis. Similar to the data with IL-32a,there was a further increase in macrophage apoptosis when rIL-32gwas added to the cells infected with M. tuberculosis. With an evenlower concentration of rIL-32g (10 ng/ml), therewas a trend towardincreased apoptosis in the cells stimulated with IL-32, but this didnot reach statistical significance (Fig. 4D).
Knockdown of IL-32 abrogates apoptosis induced byM. tuberculosis
Tovalidate the apoptotic response to rIL-32, the level of endogenousIL-32 was reduced by siRNA, and apoptosis in THP-1 cells infectedwith M. tuberculosis was evaluated. THP-1 cells expressing
scrambled shRNA or shRNA–IL-32 (clone 7) were uninfected orinfected with M. tuberculosis for 24 h. TUNEL staining was thenperformed, and the number of apoptotic cells was quantified. Asshown in Fig. 5, reduction of the level of endogenous IL-32 partly,but significantly, abrogated the apoptosis induced by M. tubercu-losis (compare bar 4 versus bar 2). We conclude from these ex-periments that endogenous IL-32 contributes to apoptosis in THP-1cells infected with M. tuberculosis.
Bcl-2 is upregulated and Bax is downregulated by shRNA–IL-32
Bcl-2 and Bax are mitochondrial membrane proteins that promotecell survival or induce apoptosis, respectively. Bax helps to initiateapoptosis by the intrinsic pathway through the release of cytochromec from the mitochondria into the cytoplasm. Although the extrinsicpathway is considered to be the principal mechanism by whichapoptosis serves a host defense function against M. tuberculosis,there is cross-talk between the two canonical apoptotic pathwaysbecause caspase-8 of the extrinsic pathway is able to activate up-stream mediators of the intrinsic apoptotic pathway. Thus, becauseIL-32 enhanced apoptosis of infected macrophages, we determinedwhether reduction in the level of endogenous IL-32 affected ex-pression of Bcl-2, Bax, and cytochrome c. THP-1 cells expressingscrambled shRNA or shRNA–IL-32 (clone 7) were either left un-infected or infected with M. tuberculosis H37Rv for 1–96 h. Afterthe indicated times, the cells were lysed and sterile-filtered, andnuclear-free lysates (20 mg per condition) were separated by SDS-PAGE. After transfer of the proteins onto nitrocellulose mem-branes, the membranes were immunoblotted with anti–Bcl-2, anti-Bax, or anti-cytochrome c Abs. As shown in Fig. 6A, uninfectedTHP-1 cells expressing scrambled shRNA had increased expres-sion of Bcl-2 with increasing times of incubation, peaking at ∼72 hof incubation. In THP-1 cells knocked down for IL-32 (shRNA–IL-32, clone 7), there was a similar increase in Bcl-2 expression, al-though the increase was qualitatively greater than that in the controlcells. However, semiquantitative analysis of Bcl-2 expressionnormalized for b-actin, as shown by bar graphs beneath the im-munoblots, revealed that Bcl-2 was only increased in cells silencedfor IL-32 at 1 and 48 h postinfection. In both control THP-1 cellsand cells with reduced IL-32 levels infected with M. tuberculosis,there was a noticeable decrease in Bcl-2 expression, particularlywith longer times of infection, although in the cells with reducedIL-32 levels there was an increase in Bcl-2 expression after 1 h ofinfection compared with that of the control THP-1 cells.As shown in Fig. 6B, Bax expression was similar in uninfected
THP-1 cells that expressed either scrambled shRNA or shRNA–IL-32 (clone 7). With M. tuberculosis infection, there wasa qualitative increase in Bax expression early (i.e., #24 h of in-fection), although there were no significant differences betweenTHP-1 cells that expressed scrambled shRNA and those that ex-pressed shRNA–IL-32 (clone 7).Cytochrome c expression was also analyzed in cells expressing
shRNA–IL-32 (clone 7) and control cells. As shown in Fig. 6C, inuninfected THP-1 cells expressing either scrambled shRNA orshRNA–IL-32 (clone 7), cytochrome c expression was qualita-tively similar, although semiquantitative analysis revealed thatcytochrome c expression was reduced at later time points of in-fection in the THP-1 cells silenced for IL-32. In cells infected withM. tuberculosis H37Rv, cytochrome c expression was modestlydecreased at the earlier times following infection in cells withreduced IL-32 levels compared with that of the control cells. Weconclude from these experiments that reduction of the IL-32 levelmarginally increased the expression of Bcl-2 and slightly de-creased the expression of cytochrome c in THP-1 cells infectedwith M. tuberculosis.
FIGURE 3. Knockdown of endogenous IL-32 production increased the
number of intracellular M. tuberculosis bacteria in THP-1 cells. THP-1
cells stably transfected with the control empty vector, scrambled shRNA,
or shRNA–IL-32 (clones 5, 6, or 7) were infected withM. tuberculosis, and
the number of cell-associatedM. tuberculosis bacteria were quantified after
1 h (0 d), 4 d, and 8 d postinfection. A, CFUs recovered from infected
THP-1 cells expressing the control vector (open squares), scrambled
shRNA (semiclosed diamonds), or shRNA–IL-32 (clone 5) (closed cir-
cles). B, CFUs recovered from infected THP-1 cells expressing the control
vector (open squares), scrambled shRNA (semiclosed diamonds), or
shRNA–IL-32 (clone 6) (closed circles). C, CFUs recovered from infected
THP-1 cells expressing the control vector (open squares), scrambled
shRNA (semiclosed diamonds), or shRNA–IL-32 (clone 7) (closed cir-
cles). Values are means6 SD from five to six independent experiments. pp
, 0.05; ppp , 0.01; pppp , 0.001; compared with scrambled shRNA at
the corresponding time points.
The Journal of Immunology 3835
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

Caspase-3 activation by M. tuberculosis is enhanced byendogenous IL-32
Caspase-3 is one of the major effector caspases common to bothintrinsic and extrinsic pathways of apoptosis. To corroborate therole of IL-32 in inducing apoptosis in macrophages infected withM. tuberculosis, we quantified the level of caspase-3 activation incontrol and IL-32–silenced cells with and without M. tuberculosisinfection. The level of activated caspase-3 in THP-1 cells infectedwith M. tuberculosis was assessed by ELISA. As shown in Fig. 7,M. tuberculosis infection of control THP-1 cells (shRNA Scr)induced caspase-3 activation with increasing time of infection(open circles). By contrast, THP-1 cells with reduced levels of IL-32 (shRNA–IL-32, clone 7) produced significantly less activatedcaspase-3 at 6 and 24 h following M. tuberculosis infection(closed squares). We conclude from these experiments that withM. tuberculosis infection of THP-1 cells IL-32, directly or in-directly, is an important activator of caspase-3.
rIL-32g reduced viability of intracellular M. tuberculosis,which was modestly abrogated by a caspase-3 inhibitor
Commercially available rIL-32g from R&D Systems, rIL-32g(R&D), in the absence or presence of a caspase-3 inhibitor, wasused to further corroborate the IL-32 knockdown studies. THP-1
cells were infected withM. tuberculosisH37Rv and simultaneouslyincubated with 50 ng/ml rIL-32g (R&D). At 1 h and 2 d post-infection, the cells were washed and lysed, and serially dilutedlysates were cultured for M. tuberculosis on 7H10 solid agar. Asshown in Fig. 8A, rIL-32g significantly reduced the number of vi-able M. tuberculosis bacteria by nearly 50% (p , 0.01). Co-incubation with 10 mM caspase-3 inhibitor (z-DEVD-fmk), whichfully inhibited caspase-3 activation (data not shown), modestly butsignificantly abrogated the inhibition in growth (Fig. 8A, p, 0.05).To determine whether rIL-32g (R&D) induced apoptosis of theinfected cells, TUNEL staining was performed, and apoptosis wasquantified. As shown in Fig. 8B, rIL-32g (R&D) and M. tubercu-losis individually and additively induced apoptosis. In the presenceof 10 or 20 mM caspase-3 inhibitor (z-DEVD-fmk), there wasa significant decrease in apoptosis induced with M. tuberculosisplus rIL-32g (R&D) (p, 0.01), although the level of apoptosis didnot return to baseline. We conclude from these studies that exog-enous rIL-32g (R&D) can inhibit the growth of intracellularM. tuberculosis through induction of caspase-dependent and pos-sibly caspase-independent apoptotic pathways.
DiscussionIL-32 is increasingly recognized to have pleiotropic functions. Asoriginally reported by Kim et al. (43), IL-32 can activate in-flammatory signaling pathways and induce cytokine production.Consistent with its proinflammatory effects, IL-32 has been im-plicated in the pathogenesis of autoimmune disorders such asrheumatoid arthritis and Crohn’s disease (63–65). Whereas IL-32 isnot seen in the joints of osteoarthritic patients, it is expressed in theinflamed synovia of patients with rheumatoid arthritis (65). InCrohn’s disease, IL-32 contributes to inflammation by synergizingwith bacteria-derived ligands (e.g., muropeptides of peptidoglycan)that bind cytosolic sensors (nucleotide-binding oligomerizationdomains 1 and 2) to augment the production of IL-1b and IL-6 (64).More recently, the IL-32 level was reported to be increased in thelung tissues of patients with chronic obstructive pulmonary disease(COPD), where it localized with TNF-a and correlated with thedegree of airflow obstruction (66).In contrast to its pathogenic role in the aforementioned diseases,
IL-32 is an antagonist for HIV (67, 68) and influenza (69, 70).
FIGURE 4. IL-32a or IL-32g increased apoptosis in
infected THP-1 cells. THP-1 cells were stimulated with
100 ng/ml IL-32a, infected with M. tuberculosis, or
both for 24 h. TUNEL staining was then performed. A,
Representative microscopy of the cells after TUNEL
staining (magnification 3100). B, THP-1 cells were
stimulated with 100 ng/ml IL-32a, infected with M.
tuberculosis, or both, and the amount of apoptosis was
quantified and reported as the percentage of apoptotic
cells of a total of 5000 cells counted per condition from
three separate experiments. pp , 0.05; ppp , 0.01;
compared with bar 1. C, THP-1 cells were stimulated
with 50 ng/ml IL-32g, M. tuberculosis infection, or
both, and the amount of apoptosis was quantified and
reported as the percentage of a total of 5000 cells
counted per condition from three separate experiments.
pp , 0.05; ppp , 0.01; pppp , 0.001; compared with
control cells. D, THP-1 cells were stimulated with 10
ng/ml IL-32g, M. tuberculosis infection, or both, and
the amount of apoptosis was quantified and reported as
the percentage of a total of 5000 cells counted per
condition from three separate experiments. pppp ,0.001 compared with control cells.
FIGURE 5. Apoptosis induced by M. tuberculosis was abrogated in
THP-1 cells knocked down for IL-32. THP-1 cells expressing the scram-
bled shRNA or shRNA–IL-32 (clone 7) were infected with M. tuberculosis
for 24 h, followed by TUNEL staining and quantification of apoptosis
reported as the percentage of apoptotic cells of a total of 5000 cells
counted per condition from three independent experiments. pp , 0.05
compared with bar 2.
3836 ROLE OF IL-32 IN TUBERCULOSIS
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

Transient transfection of HIV-infected PBMCs or U1 cells withsiRNA that targeted IL-32 mRNA increased p24 production, in-dicating that IL-32 may be a natural inhibitor of HIV (68). In theA549 human type 2 epithelial cell line, influenza induced IL-32production (70). Interestingly, a selective cyclooxygenase 2 in-hibitor (NS398) or aspirin inhibited influenza-induced IL-32production, suggesting that the PGs may play an intermediary role
in the induction of IL-32. We showed previously that IL-32 is alsoinduced by M. tuberculosis infection of various primary humancells, including PBMCs, monocyte-derived macrophages, mono-cyte-derived dendritic cells, and blood lymphocytes (56).In seeking the biological significance of IL-32 in TB, we found
that stable expression of siRNA fragments to silence IL-32 proteinproduction in THP-1 cells decreased production of the proin-flammatory cytokines TNF-a, IL-1b, and IL-8 induced by M.tuberculosis, indicating that IL-32 is involved in a complex net-work of cytokine production with M. tuberculosis infection. Werecently showed that silencing of IL-32 also reduced LPS or PMAinduction of proinflammatory cytokines (71). Furthermore, inTHP-1 cells with IL-32 levels reduced by siRNA, there was anincreased number of M. tuberculosis bacteria recovered. Thissignificant finding implicates IL-32 as a host protective cytokinein the in vitro infection model. Interestingly, the THP-1 clone thathad the greatest inhibition of IL-32 protein expression was alsoassociated with the greatest recovery of M. tuberculosis.We demonstrated that a plausible mechanism by which IL-32
inhibited recovery of M. tuberculosis is via induction of apoptosisof the infected THP-1 cells. Although the concentration of rIL-32a required to induce apoptosis in THP-1 cells infected with M.tuberculosis was more than that produced endogenously, this islikely due to the fact that the measured IL-32 concentration is anunderestimation of the intracellular levels due to dilution by thecell lysis buffer. We therefore corroborated these apoptotic studiesby showing that a lower concentration of rIL-32g had a similar
FIGURE 6. Intrinsic pro- and antiapoptotic gene expression in THP-1 cells knocked down for IL-32 and infected with M. tuberculosis. A, THP-1 cells
expressing the scrambled shRNA (shRNA Scr) or shRNA–IL-32 (clone 7) were left unstimulated or infected with M. tuberculosis for variable lengths of
time. After the indicated times of infection, the supernatant was removed, the cells were lysed, and the whole-cell lysates were separated by SDS-PAGE,
transferred onto a nitrocellulose membrane, and immunoblotted for Bcl-2. B, THP-1 cells expressing the scrambled shRNA or shRNA–IL-32 (clone 7) were
similarly treated as in A, and the cell lysates were immunoblotted for Bax expression. C, THP-1 cells expressing the scrambled shRNA or shRNA–IL-32
(clone 7) were similarly treated as in A, and the cell lysates were immunoblotted for cytochrome c expression. The membranes were also immunoblotted for
b-actin. The bar graphs below each set of immunoblots are mean relative density measurements for the Bcl-2, Bax, and cytochrome c bands normalized for
the densities of their corresponding b-actin band. The data shown are representative of three independent experiments. pp , 0.05; ppp , 0.01; pppp ,0.001; compared with the corresponding condition with THP-1 cells transfected with scrambled shRNA.
FIGURE 7. M. tuberculosis induction of activated caspase-3 is inhibited
in cells knocked down for IL-32. THP-1 cells expressing the scrambled
shRNA (shRNA Scr) or shRNA–IL-32 (clone 7) were infected with M.
tuberculosis at the indicated times, the cells were lysed, and activated
caspase-3 was quantified by ELISA. Data shown are the mean 6 SD of
three independent experiments. ppp , 0.01 between scrambled shRNA
and shRNA–IL-32 (clone 7).
The Journal of Immunology 3837
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

apoptotic effect as the higher concentration of IL-32a and thatTHP-1 cells knocked down for IL-32 had partial but significantabrogation of apoptosis induced by M. tuberculosis. To furtherconfirm these findings and to delineate the role of apoptosis asa host defense mechanism of IL-32, we used commerciallyavailable rIL-32g (R&D) with or without the presence of a cas-pase-3 inhibitor in our THP-1 infection model. Although rIL-32gsignificantly reduced intracellular viability of M. tuberculosis andinduced apoptosis and caspase-3 inhibitor significantly abrogatedboth effects, the degree of abrogation was modest. These findingssuggest that the host defense functions of IL-32 are complex andlikely involve both caspase-dependent and caspase-independentapoptotic pathways as well as possibly nonapoptotic mechanisms.IL-32 has also been shown to induce apoptosis in other experi-mental conditions. For example, Marcondes et al. (46) showed thatKG1a leukemic cells underwent apoptosis when cocultured withHS5 stromal cells and that apoptosis was due to IL-32 producedby the stromal cells. The abundant expression of IL-32 in lungs ofCOPD patients (66) may indirectly indicate increased apoptosisbecause apoptosis is considered to be an important mechanism inthe pathogenesis of COPD. However, apoptotic effects of IL-32are not universally seen because IL-32 is constitutively expressedin various cancer cell lines or cancer tissues, suggesting that IL-32may not only be antiapoptotic but may also be involved in thepathogenesis of some tumors (48, 72, 73).Apoptosis is becoming well-established as an important killing
mechanism for intracellular M. tuberculosis, although the precisemeans by which it occurs in killing mycobacteria is complex (2).The extrinsic apoptotic pathway is activated by avirulent M. tu-berculosis H37Ra to a greater degree than by the more virulentM. tuberculosis H37Rv strain, accounting in part for the lowervirulence in the former strain (74). TNF-a is considered to be anessential mediator of this classical apoptotic pathway in killing of
M. tuberculosis and other mycobacteria (2, 6). However, other
apoptotic mechanisms and nonapoptotic cell death that have been
shown to affect killing of M. tuberculosis or bacillus Calmette-
Guerin, including that induced by the interaction of FasL ex-
pressed on cytotoxic T lymphocytes with Fas on macrophages
infected with M. tuberculosis (75), NO (7), or ATP ligation to the
P2X7R receptor (76). Apoptosis is also considered to have both
direct and indirect antimicrobial activity against M. tuberculosis.
Although direct antimicrobial activity was demonstrated against
various mycobacterial strains (74, 77), the mechanism has not
been elucidated. One killing mechanism of apoptosis occurs when
apoptotic cells containing M. tuberculosis are ingested by un-
infected macrophages, resulting in a greater killing ability because
this two-step phagocytic process allows the host cells to override
the ability of M. tuberculosis to inhibit phagosome acidification or
phagosome–lysosome fusion. However, macrophage necrosis,
seen when the number of infecting organisms overwhelms the
number of phagocytes or when there are insufficient phagocytes to
ingest the infected apoptotic cells, is considered to be detrimental
to the host through suboptimal intracellular killing and permissive
spread of organisms from cell to cell (2, 78, 79).Although the extrinsic pathway of apoptosis is considered to be
an important mechanism by which intracellular M. tuberculosis
bacteria are killed (2, 6), our findings of a marginal increase in
Bcl-2 and a modest decrease in cytochrome c in THP-1 cells in-
fected with M. tuberculosis knocked down for IL-32 suggest that
the intrinsic pathway may play a minor role. Although inhibition
of casapse-3 significantly abrogated the inhibitory effects of rIL-
32g on the growth of M. tuberculosis and on apoptosis, the re-
versal was modest. Because caspase-3 is a common pathway for
both intrinsic and extrinsic pathways of classical apoptosis, it
remains to be seen whether nonapoptotic mechanisms and/or
caspase-independent apoptotic pathways, such as the less well
characterized autophagic mechanism of cell death, caspase-
independent apoptosis involving apoptosis-inducing factor and
endonuclease G, or those involving sequential apoptosis–necrosis
through the lysosomal release of proteases such as cathepsin B,
cathepsin D, cathepsin L, and/or the calpains (79–81), may also be
mechanistically involved in the antituberculous effects of IL-32.
However, necrotic cell death of phagocytes does not kill in-
tracellular M. tuberculosis and may in fact serve to propagate the
mycobacteria (82–84). It is also likely that the host defense role of
apoptosis may be more substantial in vivo because efferocytosis of
apoptotic bodies and subsequent facilitation of Ag presentation
can accelerate activation of the adaptive immune response.The known biological effects of IL-32 suggest that it may also
antagonizeM. tuberculosis indirectly by other means in vivo. For
example, we found that with M. tuberculosis infection knock-
down of IL-32 also inhibited M. tuberculosis production of TNF-
a, IL-1b, and IL-8, cytokines shown to be important in the host
defense against TB (17, 25, 85). Thus, in the in vivo setting, in
the presence of numerous other cell types and a network of both
immunostimulatory and immunosuppressive cytokines, the role
that IL-32 plays in TB infection is likely to be significantly more
complex.In summary, using an in vitro model of infection of human
macrophages that were knocked down for IL-32 expression, we
showed that IL-32 is an important host defense molecule againstM.
tuberculosis. Although induction of caspase-dependent apoptosis
is likely an important mechanism for the anti-TB effect of IL-32,
caspase-independent forms of cell death and nonapoptotic mech-
anisms may also be involved.
FIGURE 8. rIL-32g (R&D) reduces intracellular M. tuberculosis and
induces apoptosis. A, THP-1 cells were simultaneously infected with M.
tuberculosis H37Rv and stimulated with rIL-32g. Intracellular M. tuber-
culosis was enumerated at 1 h and 2 d following infection. B, THP-1 cells
were incubated with 50 ng/ml rIL-32g, M. tuberculosis H37Rv, or both
with and without 10 or 20 mM caspase-3 inhibitor (z-DEVD-fmk) for 24 h,
followed by TUNEL assay. Data shown are mean 6 SD of three in-
dependent experiments. pp , 0.05; ppp , 0.01; pppp , 0.001.
3838 ROLE OF IL-32 IN TUBERCULOSIS
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

AcknowledgmentsWe thank Dr. Robert Winn at University of Colorado Denver School of
Medicine for helpful discussions.We are grateful to Dr. Mark Geraci at Uni-
versity of Colorado Denver School of Medicine, Dr. Greg Downey at Na-
tional Jewish Health, and Dr. Edward Dempsey at the Denver Veterans
Affairs Medical Center for their unwavering support for this project.
DisclosuresThe authors have no financial conflicts of interest.
References1. Dolin, P. J., M. C. Raviglione, and A. Kochi. 1994. Global tuberculosis incidence
and mortality during 1990-2000. Bull. World Health Organ. 72: 213–220.2. Lee, J., M. Hartman, and H. Kornfeld. 2009. Macrophage apoptosis in tuber-
culosis. Yonsei Med. J. 50: 1–11.3. Rubin, E. J. 2009. The granuloma in tuberculosis—friend or foe? N. Engl. J.
Med. 360: 2471–2473.4. Kusner, D. J., and J. A. Barton. 2001. ATP stimulates human macrophages to kill
intracellular virulent Mycobacterium tuberculosis via calcium-dependent phag-osome-lysosome fusion. J. Immunol. 167: 3308–3315.
5. Gutierrez, M. G., S. S. Master, S. B. Singh, G. A. Taylor, M. I. Colombo, andV. Deretic. 2004. Autophagy is a defense mechanism inhibiting BCG and My-cobacterium tuberculosis survival in infected macrophages. Cell 119: 753–766.
6. Keane, J., M. K. Balcewicz-Sablinska, H. G. Remold, G. L. Chupp, B. B. Meek,M. J. Fenton, and H. Kornfeld. 1997. Infection by Mycobacterium tuberculosispromotes human alveolar macrophage apoptosis. Infect. Immun. 65: 298–304.
7. Rojas, M., L. F. Barrera, G. Puzo, and L. F. Garcia. 1997. Differential inductionof apoptosis by virulent Mycobacterium tuberculosis in resistant and susceptiblemurine macrophages: role of nitric oxide and mycobacterial products. J. Im-munol. 159: 1352–1361.
8. Chan, J., K. Tanaka, D. Carroll, J. Flynn, and B. R. Bloom. 1995. Effects of nitricoxide synthase inhibitors on murine infection with Mycobacterium tuberculosis.Infect. Immun. 63: 736–740.
9. MacMicking, J. D., R. J. North, R. LaCourse, J. S. Mudgett, S. K. Shah, andC. F. Nathan. 1997. Identification of nitric oxide synthase as a protective locusagainst tuberculosis. Proc. Natl. Acad. Sci. USA 94: 5243–5248.
10. Liu, P. T., S. Stenger, H. Li, L. Wenzel, B. H. Tan, S. R. Krutzik, M. T. Ochoa,J. Schauber, K. Wu, C. Meinken, et al. 2006. Toll-like receptor triggering ofa vitamin D-mediated human antimicrobial response. Science 311: 1770–1773.
11. Kisich, K. O., L. Heifets, M. Higgins, and G. Diamond. 2001. Antimycobacterialagent based on mRNA encoding human b-defensin 2 enables primary macro-phages to restrict growth of Mycobacterium tuberculosis. Infect. Immun. 69:2692–2699.
12. Tan, B. H., C. Meinken, M. Bastian, H. Bruns, A. Legaspi, M. T. Ochoa,S. R. Krutzik, B. R. Bloom, T. Ganz, R. L. Modlin, and S. Stenger. 2006.Macrophages acquire neutrophil granules for antimicrobial activity against in-tracellular pathogens. J. Immunol. 177: 1864–1871.
13. Stegelmann, F., M. Bastian, K. Swoboda, R. Bhat, V. Kiessler, A. M. Krensky,M. Roellinghoff, R. L. Modlin, and S. Stenger. 2005. Coordinate expression ofCC chemokine ligand 5, granulysin, and perforin in CD8+ T cells provides a hostdefense mechanism against Mycobacterium tuberculosis. J. Immunol. 175:7474–7483.
14. Stenger, S., D. A. Hanson, R. Teitelbaum, P. Dewan, K. R. Niazi, C. J. Froelich,T. Ganz, S. Thoma-Uszynski, A. Melian, C. Bogdan, et al. 1998. An antimicrobialactivity of cytolytic T cells mediated by granulysin. Science 282: 121–125.
15. Laube, D. M., S. Yim, L. K. Ryan, K. O. Kisich, and G. Diamond. 2006. An-timicrobial peptides in the airway. Curr. Top. Microbiol. Immunol. 306: 153–182.
16. Cooper, A. M., and S. A. Khader. 2008. The role of cytokines in the initiation,expansion, and control of cellular immunity to tuberculosis. Immunol. Rev. 226:191–204.
17. Flynn, J. L., M. M. Goldstein, J. Chan, K. J. Triebold, K. Pfeffer,C. J. Lowenstein, R. D. Schreiber, T. W. Mak, and B. R. Bloom. 1995. Tumornecrosis factor-a is required in the protective immune response against Myco-bacterium tuberculosis in mice. Immunity 2: 561–572.
18. Flynn, J. L., M. M. Goldstein, K. J. Triebold, J. Sypek, S. Wolf, and B. R. Bloom.1995. IL-12 increases resistance of BALB/c mice toMycobacterium tuberculosisinfection. J. Immunol. 155: 2515–2524.
19. Flynn, J. L., J. Chan, K. J. Triebold, D. K. Dalton, T. A. Stewart, andB. R. Bloom. 1993. An essential role for interferon g in resistance to Myco-bacterium tuberculosis infection. J. Exp. Med. 178: 2249–2254.
20. Kinjo, Y., K. Kawakami, K. Uezu, S. Yara, K. Miyagi, Y. Koguchi, T. Hoshino,M. Okamoto, Y. Kawase, K. Yokota, et al. 2002. Contribution of IL-18 to Th1response and host defense against infection by Mycobacterium tuberculosis:a comparative study with IL-12p40. J. Immunol. 169: 323–329.
21. Sugawara, I., H. Yamada, H. Kaneko, S. Mizuno, K. Takeda, and S. Akira. 1999.Role of interleukin-18 (IL-18) in mycobacterial infection in IL-18-gene-dis-rupted mice. Infect. Immun. 67: 2585–2589.
22. Smith, S., D. Liggitt, E. Jeromsky, X. Tan, S. J. Skerrett, and C. B. Wilson. 2002.Local role for tumor necrosis factor alpha in the pulmonary inflammatory re-sponse to Mycobacterium tuberculosis infection. Infect. Immun. 70: 2082–2089.
23. Bekker, L. G., S. Freeman, P. J. Murray, B. Ryffel, and G. Kaplan. 2001. TNF-acontrols intracellular mycobacterial growth by both inducible nitric oxide syn-
thase-dependent and inducible nitric oxide synthase-independent pathways. J.Immunol. 166: 6728–6734.
24. MacMicking, J. D., G. A. Taylor, and J. D. McKinney. 2003. Immune control oftuberculosis by IFN-g-inducible LRG-47. Science 302: 654–659.
25. O’Kane, C. M., J. J. Boyle, D. E. Horncastle, P. T. Elkington, and J. S. Friedland.2007.Monocyte-dependent fibroblast CXCL8 secretion occurs in tuberculosis andlimits survival of mycobacteria withinmacrophages. J. Immunol. 178: 3767–3776.
26. Cooper, A. M., D. K. Dalton, T. A. Stewart, J. P. Griffin, D. G. Russell, andI. M. Orme. 1993. Disseminated tuberculosis in interferon g gene-disruptedmice. J. Exp. Med. 178: 2243–2247.
27. Cooper, A. M., J. Magram, J. Ferrante, and I. M. Orme. 1997. Interleukin 12 (IL-12) is crucial to the development of protective immunity in mice intravenouslyinfected with Mycobacterium tuberculosis. J. Exp. Med. 186: 39–45.
28. Buccheri, S., R. Reljic, N. Caccamo, J. Ivanyi, M. Singh, A. Salerno, and F. Dieli.2007. IL-4 depletion enhances host resistance and passive IgA protection againsttuberculosis infection in BALB/c mice. Eur. J. Immunol. 37: 729–737.
29. Hernandez-Pando, R., D. Aguilar, M. L. Hernandez, H. Orozco, and G. Rook.2004. Pulmonary tuberculosis in BALB/c mice with non-functional IL-4 genes:changes in the inflammatory effects of TNF-a and in the regulation of fibrosis.Eur. J. Immunol. 34: 174–183.
30. Ordway, D. J., L. Costa, M. Martins, H. Silveira, L. Amaral, M. J. Arroz,F. A. Ventura, and H. M. Dockrell. 2004. Increased interleukin-4 production byCD8 and gd T cells in health-care workers is associated with the subsequentdevelopment of active tuberculosis. J. Infect. Dis. 190: 756–766.
31. Hickman, S. P., J. Chan, and P. Salgame. 2002. Mycobacterium tuberculosisinduces differential cytokine production from dendritic cells and macrophageswith divergent effects on naive T cell polarization. J. Immunol. 168: 4636–4642.
32. Murray, P. J., and R. A. Young. 1999. Increased antimycobacterial immunity ininterleukin-10-deficient mice. Infect. Immun. 67: 3087–3095.
33. Hirsch, C. S., J. J. Ellner, R. Blinkhorn, and Z. Toossi. 1997. In vitro restorationof T cell responses in tuberculosis and augmentation of monocyte effectorfunction against Mycobacterium tuberculosis by natural inhibitors of trans-forming growth factor b. Proc. Natl. Acad. Sci. USA 94: 3926–3931.
34. Wilkinson, K. A., T. D. Martin, S. M. Reba, H. Aung, R. W. Redline,W. H. Boom, Z. Toossi, and S. A. Fulton. 2000. Latency-associated peptide oftransforming growth factor b enhances mycobacteriocidal immunity in the lungduring Mycobacterium bovis BCG infection in C57BL/6 mice. Infect. Immun.68: 6505–6508.
35. Jung, Y.-J., R. LaCourse, L. Ryan, and R. J. North. 2002. Evidence inconsistentwith a negative influence of T helper 2 cells on protection afforded by a domi-nant T helper 1 response against Mycobacterium tuberculosis lung infection inmice. Infect. Immun. 70: 6436–6443.
36. Fortsch, D., M. Rollinghoff, and S. Stenger. 2000. IL-10 converts human den-dritic cells into macrophage-like cells with increased antibacterial activityagainst virulent Mycobacterium tuberculosis. J. Immunol. 165: 978–987.
37. North, R. J. 1998. Mice incapable of making IL-4 or IL-10 display normal re-sistance to infection with Mycobacterium tuberculosis. Clin. Exp. Immunol. 113:55–58.
38. Rook, G. A. W., R. Hernandez-Pando, K. Dheda, and G. Teng Seah. 2004. IL-4in tuberculosis: implications for vaccine design. Trends Immunol. 25: 483–488.
39. Holscher, C., A. Holscher, D. Ruckerl, T. Yoshimoto, H. Yoshida, T. Mak,C. Saris, and S. Ehlers. 2005. The IL-27 receptor chain WSX-1 differentiallyregulates antibacterial immunity and survival during experimental tuberculosis.J. Immunol. 174: 3534–3544.
40. Pearl, J. E., S. A. Khader, A. Solache, L. Gilmartin, N. Ghilardi, F. deSauvage,and A. M. Cooper. 2004. IL-27 signaling compromises control of bacterialgrowth in mycobacteria-infected mice. J. Immunol. 173: 7490–7496.
41. Nishimoto, K. P., A. K. Laust, and E. L. Nelson. 2008. A human dendritic cellsubset receptive to the Venezuelan equine encephalitis virus-derived repliconparticle constitutively expresses IL-32. J. Immunol. 181: 4010–4018.
42. Nold-Petry, C. A., M. F. Nold, J. A. Zepp, S.-H. Kim, N. F. Voelkel, andC. A. Dinarello. 2009. IL-32-dependent effects of IL-1b on endothelial cellfunctions. Proc. Natl. Acad. Sci. USA 106: 3883–3888.
43. Kim, S.-H., S.-Y. Han, T. Azam, D.-Y. Yoon, and C. A. Dinarello. 2005. In-terleukin-32: a cytokine and inducer of TNFa. Immunity 22: 131–142.
44. Dinarello, C. A., and S.-H. Kim. 2006. IL-32, a novel cytokine with a possiblerole in disease. Ann. Rheum. Dis. 65: iii61–iii64.
45. Kobayashi, H., and P. C. Lin. 2009. Molecular characterization of IL-32 in hu-man endothelial cells. Cytokine 46: 351–358.
46. Marcondes, A. M., A. J. Mhyre, D. L. Stirewalt, S.-H. Kim, C. A. Dinarello, andH. J. Deeg. 2008. Dysregulation of IL-32 in myelodysplastic syndrome andchronic myelomonocytic leukemia modulates apoptosis and impairs NK func-tion. Proc. Natl. Acad. Sci. USA 105: 2865–2870.
47. Mun, S. H., J. W. Kim, S. S. Nah, N. Y. Ko, J. H. Lee, J. D. Kim, K. Kim,H. S. Kim, J. D. Choi, S. H. Kim, et al. 2009. Tumor necrosis factor a-inducedinterleukin-32 is positively regulated via the Syk/protein kinase Cd/JNK path-way in rheumatoid synovial fibroblasts. Arthritis Rheum. 60: 678–685.
48. Nishida, A., A. Andoh, O. Inatomi, and Y. Fujiyama. 2009. Interleukin-32 ex-pression in the pancreas. J. Biol. Chem. 284: 17868–17876.
49. Nishida, A., A. Andoh, M. Shioya, S. Kim-Mitsuyama, A. Takayanagi, andY. Fujiyama. 2008. Phosphatidylinositol 3-kinase/Akt signaling mediates in-terleukin-32a induction in human pancreatic periacinar myofibroblasts. Am. J.Physiol. Gastrointest. Liver Physiol. 294: G831–G838.
50. Shoda, H., K. Fujio, Y. Yamaguchi, A. Okamoto, T. Sawada, Y. Kochi, andK. Yamamoto. 2006. Interactions between IL-32 and tumor necrosis factor alphacontribute to the exacerbation of immune-inflammatory diseases. Arthritis Res.Ther. 8: R166.
The Journal of Immunology 3839
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from

51. Goda, C., T. Kanaji, S. Kanaji, G. Tanaka, K. Arima, S. Ohno, and K. Izuhara.2006. Involvement of IL-32 in activation-induced cell death in T cells. Int.Immunol. 18: 233–240.
52. Choi, J.-D., S.-Y. Bae, J.-W. Hong, T. Azam, C. A. Dinarello, E. Her, W.-S. Choi,B.-K. Kim, C.-K. Lee, D.-Y. Yoon, et al. 2009. Identification of the most activeinterleukin-32 isoform. Immunology 126: 535–542.
53. Mabilleau, G., and A. Sabokbar. 2009. Interleukin-32 promotes osteoclast dif-ferentiation but not osteoclast activation. PLoS One 4: e4173.
54. Kim, S., S. Lee, E. Her, S. Bae, J. Choi, J. Hong, J. Jaekal, D. Yoon, T. Azam,C. A. Dinarello, and S. Kim. 2008. Proteinase 3-processed form of the re-combinant IL-32 separate domain. BMB Rep 41: 814–819.
55. Novick, D., M. Rubinstein, T. Azam, A. Rabinkov, C. A. Dinarello, andS. H. Kim. 2006. Proteinase 3 is an IL-32 binding protein. Proc. Natl. Acad. Sci.USA 103: 3316–3321.
56. Netea, M. G., T. Azam, E. C. Lewis, L. A. Joosten, M. Wang, D. Langenberg,X. Meng, E. D. Chan, D.-Y. Yoon, T. H. M. Ottenhoff, et al. 2006. Mycobac-terium tuberculosis induces interleukin-32 production through a caspase-1/IL-18/interferon-g-dependent mechanism. PLoS Med. 3: e277.
57. Riendeau, C. J., and H. Kornfeld. 2003. THP-1 cell apoptosis in response toMycobacterial infection. Infect. Immun. 71: 254–259.
58. Stokes, R. W., and D. Doxsee. 1999. The receptor-mediated uptake, survival,replication, and drug sensitivity of Mycobacterium tuberculosis within themacrophage-like cell line THP-1: a comparison with human monocyte-derivedmacrophages. Cell. Immunol. 197: 1–9.
59. Bai, X., S. E. Wilson, K. Chmura, N. E. Feldman, and E. D. Chan. 2004.Morphometric analysis of Th1 and Th2 cytokine expression in human pulmonarytuberculosis. Tuberculosis (Edinb.) 84: 375–385.
60. Paule, M. R., and R. J. White. 2000. Survey and summary: transcription by RNApolymerases I and III. Nucleic Acids Res. 28: 1283–1298.
61. Baccante, G., G. Mincione, M. C. Di Marcantonio, A. Piccirelli, F. Cuccurullo,and E. Porreca. 2004. Pravastatin up-regulates transforming growth factor-b1 inTHP-1 human macrophages: effect on scavenger receptor class A expression.Biochem. Biophys. Res. Commun. 314: 704–710.
62. Perskvist, N., M. Long, O. Stendahl, and L. Zheng. 2002. Mycobacterium tu-berculosis promotes apoptosis in human neutrophils by activating caspase-3 andaltering expression of Bax/Bcl-xL via an oxygen-dependent pathway. J. Im-munol. 168: 6358–6365.
63. Shioya, M., A. Nishida, Y. Yagi, A. Ogawa, T. Tsujikawa, S. Kim-Mitsuyama,A. Takayanagi, N. Shimizu, Y. Fujiyama, and A. Andoh. 2007. Epithelialoverexpression of interleukin-32a in inflammatory bowel disease. Clin. Exp.Immunol. 149: 480–486.
64. Netea, M. G., T. Azam, G. Ferwerda, S. E. Girardin, M. Walsh, J.-S. Park,E. Abraham, J.-M. Kim, D.-Y. Yoon, C. A. Dinarello, and S. H. Kim. 2005. IL-32 synergizes with nucleotide oligomerization domain (NOD) 1 and NOD2 li-gands for IL-1b and IL-6 production through a caspase 1-dependent mechanism.Proc. Natl. Acad. Sci. USA 102: 16309–16314.
65. Joosten, L. A., M. G. Netea, S. H. Kim, D. Y. Yoon, B. Oppers-Walgreen,T. R. Radstake, P. Barrera, F. A. van de Loo, C. A. Dinarello, and W. B. van denBerg. 2006. IL-32, a proinflammatory cytokine in rheumatoid arthritis. Proc.Natl. Acad. Sci. USA 103: 3298–3303.
66. Calabrese, F., S. Baraldo, E. Bazzan, F. Lunardi, F. Rea, P. Maestrelli, G. Turato,K. Lokar-Oliani, A. Papi, R. Zuin, et al. 2008. IL-32, a novel proinflammatorycytokine in chronic obstructive pulmonary disease. Am. J. Respir. Crit. CareMed. 178: 894–901.
67. Rasool, S. T., H. Tang, J. Wu, W. Li, M. M. Mukhtar, J. Zhang, Y. Mu, H. X. Xing,J. Wu, and Y. Zhu. 2008. Increased level of IL-32 during human immunodefi-ciency virus infection suppresses HIV replication. Immunol. Lett. 117: 161–167.
68. Nold, M. F., C. A. Nold-Petry, G. B. Pott, J. A. Zepp, M. T. Saavedra, S.-H. Kim,and C. A. Dinarello. 2008. Endogenous IL-32 controls cytokine and HIV-1production. J. Immunol. 181: 557–565.
69. Li, W., F. Yang, Y. Liu, R. Gong, L. Liu, Y. Feng, P. Hu, W. Sun, Q. Hao,L. Kang, et al. 2009. Negative feedback regulation of IL-32 production by iNOSactivation in response to dsRNA or influenza virus infection. Eur. J. Immunol.39: 1019–1024.
70. Li, W., Y. Liu, M. M. Mukhtar, R. Gong, Y. Pan, S. T. Rasool, Y. Gao, L. Kang,Q. Hao, G. Peng, et al. 2008. Activation of interleukin-32 pro-inflammatorypathway in response to influenza A virus infection. PLoS One 3: e1985.
71. Hong, J., S. Bae, Y. Kang, D. Yoon, X. Bai, E. D. Chan, T. Azam,C. A. Dinarello, S. Lee, E. Her, et al. 2009. Suppressing IL-32 in monocytesimpairs the induction of the proinflammatory cytokines TNFa and IL-1b. Cy-tokine 49: 171–176.
72. Ko, N. Y., S. H. Chang, J. H. Lee, N. W. Kim, Y. M. Kim, W. S. Choi, J. D. Choi,S. Y. Bae, J. W. Hong, J. Jaekal, et al. 2008. Unique expression of a small IL-32protein in the Jurkat leukemic T cell line. Cytokine 42: 121–127.
73. Seo, E. H., J. Kang, K. H. Kim, M. C. Cho, S. Lee, H. J. Kim, J. H. Kim,E. J. Kim, D. K. Park, S. H. Kim, et al. 2008. Detection of expressed IL-32 inhuman stomach cancer using ELISA and immunostaining. J. Microbiol. Bio-technol. 18: 1606–1612.
74. Keane, J., H. G. Remold, and H. Kornfeld. 2000. Virulent Mycobacterium tu-berculosis strains evade apoptosis of infected alveolar macrophages. J. Immunol.15: 2016–2020.
75. Oddo, M., T. Renno, A. Attinger, T. Bakker, H. R. MacDonald, and P. R. Meylan.1998. Fas ligand-induced apoptosis of infected human macrophages reduces theviability of intracellular Mycobacterium tuberculosis. J. Immunol. 160: 5448–5454.
76. Fairbairn, I. P., C. B. Stober, D. S. Kumararatne, and D. A. Lammas. 2001. ATP-mediated killing of intracellular mycobacteria by macrophages is a P2X7-dependent process inducing bacterial death by phagosome-lysosome fusion. J.Immunol. 167: 3300–3307.
77. Molloy, A., P. Laochumroonvorapong, and G. Kaplan. 1994. Apoptosis, but notnecrosis, of infected monocytes is coupled with killing of intracellular bacillusCalmette-Guerin. J. Exp. Med. 180: 1499–1509.
78. Park, J. S., M. H. Tamayo, M. Gonzalez-Juarrero, I. M. Orme, and D. J. Ordway.2006. Virulent clinical isolates of Mycobacterium tuberculosis grow rapidly andinduce cellular necrosis but minimal apoptosis in murine macrophages. J. Leu-koc. Biol. 79: 80–86.
79. Lee, J., H. G. Remold, M. H. Ieong, and H. Kornfeld. 2006. Macrophage apo-ptosis in response to high intracellular burden of Mycobacterium tuberculosis ismediated by a novel caspase-independent pathway. J. Immunol. 176: 4267–4274.
80. Kroemer, G., and S. J. Martin. 2005. Caspase-independent cell death. Nat. Med.11: 725–730.
81. O’Sullivan, M. P., S. O’Leary, D. M. Kelly, and J. Keane. 2007. A caspase-in-dependent pathway mediates macrophage cell death in response to Mycobacte-rium tuberculosis infection. Infect. Immun. 75: 1984–1993.
82. Gil, O., E. Guirado, S. Gordillo, J. Dıaz, G. Tapia, C. Vilaplana, A. Ariza,V. Ausina, and P. J. Cardona. 2006. Intragranulomatous necrosis in lungs of miceinfected by aerosol with Mycobacterium tuberculosis is related to bacterial loadrather than to any one cytokine or T cell type. Microbes Infect. 8: 628–636.
83. Russell, D. G., P. J. Cardona, M. J. Kim, S. Allain, and F. Altare. 2009. Foamymacrophages and the progression of the human tuberculosis granuloma. Nat.Immunol. 10: 943–948.
84. Divangahi, M., M. Chen, H. Gan, D. Desjardins, T. T. Hickman, D. M. Lee,S. Fortune, S. M. Behar, and H. G. Remold. 2009. Mycobacterium tuberculosisevades macrophage defenses by inhibiting plasma membrane repair. Nat. Im-munol. 10: 899–906.
85. Master, S. S., S. K. Rampini, A. S. Davis, C. Keller, S. Ehlers, B. Springer,G. S. Timmins, P. Sander, and V. Deretic. 2008. Mycobacterium tuberculosisprevents inflammasome activation. Cell Host Microbe 3: 224–232.
3840 ROLE OF IL-32 IN TUBERCULOSIS
by guest on July 24, 2022http://w
ww
.jimm
unol.org/D
ownloaded from




















