
1-s2 0-S1381514814001588-main
12
New epoxy thermosets modified with amphiphilic multiarm star polymers as toughness enhancer Carles Lagunas a , Xavier Fernández-Francos b , Francesc Ferrando c , Marjorie Flores b , Àngels Serra b , Josep M. Morancho a , Josep M. Salla a , Xavier Ramis a,⇑ a Thermodynamics Laboratory, ETSEIB Universitat Politècnica de Catalunya, Av. Diagonal 647, 08028 Barcelona, Spain b Department of Analytical and Organic Chemistry, Universitat Rovira i Virgili, C/ Marcellí Domingo s/n, 43007 Tarragona, Spain c Department of Mechanical Engineering, Universitat Rovira i Virgili, C/ Països Catalans, 26, 43007 Tarragona, Spain article info Article history: Received 25 April 2014 Received in revised form 23 July 2014 Accepted 28 July 2014 Available online 6 August 2014 Keywords: Star polymers Miktoarm Hyperbranched Pegylation Epoxy resin Thermosets abstract The synthesis and characterization of two novel amphiphilic multiarm star polymers with linear polyeth- ylene glycol (PEG) and poly(e-caprolactone) (PCL) arms and their use as toughening modifiers of epoxy anhydride thermosets are reported. The new star polymers were obtained by partial pegylation of a hyperbranched polyester and subsequent growth of PCL arms. The curing process was studied by calo- rimetry and thermomechanical analysis, demonstrating the accelerating effect and the influence on gela- tion of the hydroxyl terminal groups. The curing kinetics was analyzed by model-free and model-fitting methods. The final properties of the resulting materials were determined by thermal and mechanical tests. The addition of the star-like modifiers led only to notable improvement on impact strength in the material containing a 10% of the star with PCL and PEG arms, without compromising glass transition temperature and thermal stability. The morphology of the resulting materials depended on the structure of the toughness modifier used, as demonstrated by electron microscopy, but all modified thermosets obtained showed phase-separated morphologies with nanosized particles. Ó 2014 Elsevier B.V. All rights reserved. 1. Introduction Epoxy resins are widely used as coatings, electrical and elec- tronic materials because of their high adhesion and other attractive properties such as thermal stability and electrical insulation [1–3]. However, in some applications their high crosslinking density leads to fragility, and sometimes damage of the substrate occurs because of the apparition of voids and cracks due to the generation of stresses that are originated by shrinkage during curing and pro- cessing. These drawbacks can be reduced by using dendritic poly- mers as reactive modifiers [4–11]. By adding a conveniently tailored dendritic polymer to the epoxy formulation, the impact resistance of the material can be enhanced many folds without compromising processability and thermomechanical characteristics and even the internal stresses can be reduced [8,12,13]. By this strategy, the best results are achieved when the modifier phase-separates during curing and there is good interfacial adhesion between phases. Enhancement of interfacial adhesion can be reached via chemical incorporation of the modifier into the epoxy matrix or by establishment of effec- tive physical interactions. In a previous work, we reported the use of partially modified Boltorn H30 polyester with nonpolar 10-undecenoyl moieties in epoxy/anhydride thermosets. Concretely, when the degree of mod- ification of hydroxyl groups with 10-undecenoyl moieties was 76%, a phase separated material was obtained and more than a 4-fold increase in impact strength was reached [8]. On the other hand, it is known that the use of block copolymers, with blocks of different miscibility, capable of self-assembling upon blending with uncured precursor, can lead to materials with phase-separated morphologies and a significant toughness enhancement. Mülhaupt et al. reported the modification of epoxy resin with a branched polysiloxane–polycaprolactone block copolymer [14]. It was found that in the modified thermosets, spherical particles of about 20 nm in diameter were uniformly dis- persed in the continuous matrix. Meng et al. used star-shaped block copolymers and obtained phase separation in epoxy resin, and even increasing glass transition temperature of the resulting thermosets [7]. Amphiphilic dendrimers, whose surfaces have two distinct phys- ical properties, prepared by coupling of tailored hydrophilic and hydrophobic segments, have shown the ability of self-assembling http://dx.doi.org/10.1016/j.reactfunctpolym.2014.07.022 1381-5148/Ó 2014 Elsevier B.V. All rights reserved. ⇑ Corresponding author. Tel.: +34 934016592; fax: +34 934017389. E-mail address: [email protected] (X. Ramis). Reactive & Functional Polymers 83 (2014) 132–143 Contents lists available at ScienceDirect Reactive & Functional Polymers journal homepage: www.elsevier.com/locate/react
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of 1-s2 0-S1381514814001588-main

Reactive & Functional Polymers 83 (2014) 132–143
Contents lists available at ScienceDirect
Reactive & Functional Polymers
journal homepage: www.elsevier .com/ locate / react
New epoxy thermosets modified with amphiphilic multiarm starpolymers as toughness enhancer
http://dx.doi.org/10.1016/j.reactfunctpolym.2014.07.0221381-5148/� 2014 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Tel.: +34 934016592; fax: +34 934017389.E-mail address: [email protected] (X. Ramis).
Carles Lagunas a, Xavier Fernández-Francos b, Francesc Ferrando c, Marjorie Flores b, Àngels Serra b,Josep M. Morancho a, Josep M. Salla a, Xavier Ramis a,⇑a Thermodynamics Laboratory, ETSEIB Universitat Politècnica de Catalunya, Av. Diagonal 647, 08028 Barcelona, Spainb Department of Analytical and Organic Chemistry, Universitat Rovira i Virgili, C/ Marcel�lí Domingo s/n, 43007 Tarragona, Spainc Department of Mechanical Engineering, Universitat Rovira i Virgili, C/ Països Catalans, 26, 43007 Tarragona, Spain
a r t i c l e i n f o a b s t r a c t
Article history:Received 25 April 2014Received in revised form 23 July 2014Accepted 28 July 2014Available online 6 August 2014
Keywords:Star polymersMiktoarmHyperbranchedPegylationEpoxy resinThermosets
The synthesis and characterization of two novel amphiphilic multiarm star polymers with linear polyeth-ylene glycol (PEG) and poly(e-caprolactone) (PCL) arms and their use as toughening modifiers of epoxyanhydride thermosets are reported. The new star polymers were obtained by partial pegylation of ahyperbranched polyester and subsequent growth of PCL arms. The curing process was studied by calo-rimetry and thermomechanical analysis, demonstrating the accelerating effect and the influence on gela-tion of the hydroxyl terminal groups. The curing kinetics was analyzed by model-free and model-fittingmethods. The final properties of the resulting materials were determined by thermal and mechanicaltests. The addition of the star-like modifiers led only to notable improvement on impact strength inthe material containing a 10% of the star with PCL and PEG arms, without compromising glass transitiontemperature and thermal stability. The morphology of the resulting materials depended on the structureof the toughness modifier used, as demonstrated by electron microscopy, but all modified thermosetsobtained showed phase-separated morphologies with nanosized particles.
� 2014 Elsevier B.V. All rights reserved.
1. Introduction of the modifier into the epoxy matrix or by establishment of effec-
Epoxy resins are widely used as coatings, electrical and elec-tronic materials because of their high adhesion and other attractiveproperties such as thermal stability and electrical insulation [1–3].However, in some applications their high crosslinking densityleads to fragility, and sometimes damage of the substrate occursbecause of the apparition of voids and cracks due to the generationof stresses that are originated by shrinkage during curing and pro-cessing. These drawbacks can be reduced by using dendritic poly-mers as reactive modifiers [4–11].
By adding a conveniently tailored dendritic polymer to theepoxy formulation, the impact resistance of the material can beenhanced many folds without compromising processability andthermomechanical characteristics and even the internal stressescan be reduced [8,12,13]. By this strategy, the best results areachieved when the modifier phase-separates during curing andthere is good interfacial adhesion between phases. Enhancementof interfacial adhesion can be reached via chemical incorporation
tive physical interactions.In a previous work, we reported the use of partially modified
Boltorn H30 polyester with nonpolar 10-undecenoyl moieties inepoxy/anhydride thermosets. Concretely, when the degree of mod-ification of hydroxyl groups with 10-undecenoyl moieties was 76%,a phase separated material was obtained and more than a 4-foldincrease in impact strength was reached [8].
On the other hand, it is known that the use of block copolymers,with blocks of different miscibility, capable of self-assemblingupon blending with uncured precursor, can lead to materials withphase-separated morphologies and a significant toughnessenhancement. Mülhaupt et al. reported the modification of epoxyresin with a branched polysiloxane–polycaprolactone blockcopolymer [14]. It was found that in the modified thermosets,spherical particles of about 20 nm in diameter were uniformly dis-persed in the continuous matrix. Meng et al. used star-shapedblock copolymers and obtained phase separation in epoxy resin,and even increasing glass transition temperature of the resultingthermosets [7].
Amphiphilic dendrimers, whose surfaces have two distinct phys-ical properties, prepared by coupling of tailored hydrophilic andhydrophobic segments, have shown the ability of self-assembling

C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 133
to form Janus particles [15]. Recently, Samuel and Ramakrishnan[16] have demonstrated that hyperbranched polyesters, bearingalkyl and polyethylene glycol segments randomly placed on theirperiphery, can reconfigure so as to self-segregate the hydrophobicand hydrophilic segments to form Janus-type structures. It has beenfound that the immiscibility of both segments, the strong tendencyof the alkyl segments to crystallize and the conformational flexibilityof the hyperbranched core, are the thermodynamic gradientsresponsible of the reconfiguration of the polymer backbone.
Miktoarm star polymers are a relatively new and versatile classof macromolecules due to their intriguing properties, which can betailored by varying the nucleus and/or their arms. This kind ofpolymers, where any number of various types or polymer armsemanate from the core, shows as relevant property their self-assembly in solution and in the bulk [17]. The arms of this typeof star can have different molecular weights and/or chemical nat-ures. It is expected to obtain nano-structured thermosets, causedby a reaction-induced phase separation process or by self-assem-bly, when miktoarm polymers containing arms of different hydro-philicity/hydrophobicity are used as epoxy modifiers. The initialmiscibility between the resin and the modifier, the viscosity ofthe mixture and the temperature of curing are the factors that con-trol the morphologies obtained.
Taking all of this into account, in the present work, we proposethe use of two new amphiphilic polymers with hyperbranchedpolyester core and poly(ethylene glycol) (SP-PEG) or poly(ethyleneglycol)/poly(e-caprolactone) arms (SP-PEGPCL), as toughness mod-ifiers of an epoxy/anhydride formulation. Hyperbranched polyesterand poly(e-caprolactone) were separately selected as the hydro-phobic core and shell and poly(ethylene glycol) as the hydrophilicshell. The selection of the core and arms aims to reach differentphase-separated morphologies using star polymers with differentflexibility and amphiphilicity. Moreover, the hydroxyl terminalsgroups of the hyperbranched polyester and the OH groups at theend of the poly(e-caprolactone) arms can react with epoxy/anhy-dride thermoset precursors, allowing the chemical incorporationof the modifier into the network structure, enhancing the interfa-cial adhesion between phases, if they are present. The materialsobtained were characterized by means of thermogravimetry, ther-momechanical analysis and the fracture surface explored by SEMmicroscopy. Impact resistance was evaluated by Izod impact tests.
2. Experimental section
2.1. Materials
Diglycidylether of bisphenol A (DGEBA) with an epoxy equiva-lent of 187 g/eq (Epikote 828, Hexion Speciality Chemicals B.V.)was dried in vacuum before use. N,N’-dicyclohexyl carbodiimide(DCC), tin (II) 2-ethyl hexanoate, monomethyl ether of poly(ethyl-ene glycol) (Mn = 550 g/mol) (CH3OPEG-OH), methyl hexahydroph-thalic anhydride (MHHPA), benzyl dimethyl amine (BDMA),succinic anhydride and pyridine were purchased from Sigma–Aldrich and were used as received. e-Caprolactone (e-CL)(Sigma–Aldrich) was distilled under vacuum before use. 4-(N,N-dimethylamino) pyridinium p-toluenesulfonate (DPTS) was pre-pared as described in the literature [18]. The hyperbranched Bol-torn H30 (Mw = 3500 g/mol, 32 OH) was donated by Perstorp andwas used as received.
2.2. Synthesis of the acid terminated monomethyl ether ofpoly(ethylene glycol) (CH3OPEG–COOH)
In a 100 mL two-necked round-bottom flask 10 g (0.018 mol) ofmonomethyl ether of poly(ethylene glycol) (CH3OPEG-OH) weredissolved in 150 mL of chloroform (Scheme 1). Then, 9 mL of
pyridine were added and finally 9 g (0.090 mol) of succinic anhy-dride were added in portions. The mixture was heated up to60 �C and allowed to react at this temperature for 72 h on stirring.After that, the solution was cooled down to room temperature andthe solvent was eliminated under vacuum. The crude product wasdissolved in 15 mL of 1 N HCl, washed with ether, to eliminate theexcess of succinic anhydride, and then extracted with chloroformand dried. The solvent was removed under vacuum.
1H NMR (CDCl3): d (in ppm) 4.20 (–CH2OOC–); 3.60–3.70 (–O–CH2CH2O–); 3.30 (OCH3); 2.50–2.60 (–OOC–CH2CH2–COO–).
13C NMR (CDCl3): d (in ppm) 175.5 (COOH); 172.2 (COOCH2);71.9, 70.6, 69.0 (OCH2CH2); 63.9 (terminal PEG CH2); 59.1(OCH3); 29.2, 28.8 (CH2 succinic).
2.3. Synthesis of the multiarm star with poly(ethylene glycol) arms(SP-PEG)
In a 250 mL round bottom two necked flask provided with mag-netic stirrer and Ar inlet, 1 g of Boltorn H30 (9 meq OH), 0.54 g(1.5 mmol) of 4-(N,N-dimethylamino) pyridinium p-toluenesulfo-nate (DPTS) and 3 g (4.6 mmol) of CH3OPEG-COOH were dissolvedin 30 mL of DMF (Scheme 1). Then 2.26 g (11 mmol) of dicyclo-hexyl carbodiimide (DCC) were added and the mixture allowedreacting at room temperature for 48 h. Then, the precipitate wasfiltered off and the volume of DMF was reduced to 10 mL. Thecrude product was dialyzed against the same solvent.
1H NMR (CDCl3): d (in ppm) 4.20 (–CH2OOC (A + c)); 3.60–3.70(–CH2CH2OCH2– (B + d + e + f)); 3.30 (OCH3 (g)); 2.50–2.60 (–OOC–CH2CH2–COO– (a + b)); 1.15–1.20 (–CH3 (C)) (Fig. 1).
Mn = 11,300 g/mol (by 1H NMR analysis). 12 PEG arms/multiarmstar molecule (from 1H NMR).
2.4. Synthesis of the miktoarm star with poly(ethylene glycol) andpoly(e-caprolactone) arms (SP-PEGPCL)
2 g of SP-PEG (0.18 mmol, 3.6 meq OH) and 4.11 g of e-CL(36 mmol) were placed in a two necked flask at room temperatureequipped with a magnetic stirrer and Ar inlet (Schemes 1 and 2).Then, four drops of Sn(oct)2 were added to the mixture and thereaction was carried out at 130 �C during 72 h in a thermostatizedoil bath. Then, the solid product was dissolved in chloroform andisolated by reprecipitation in 10-fold cold diethyl ether. Finally,the polymer was filtered and dried at 45 �C under vacuum for2 days.
1H NMR (CDCl3): d (in ppm) 4.20 (–CH2OOC (A + c)); 4.0 (–CH2–OOC–, 5); 3.60–3.70 (–CH2CH2OCH2– (B + d + e + f) and –CH2–OH,50); 3.30 (OCH3 (g)); 2.50–2.60 (–OOC–CH2CH2–COO– (a + b));2.20 (–CH2–COO–, 1 and 10); 1.70–1.50 (–CH2–, 3, 30 and 4, 40);1.40–1.30 (–CH2–, 2 and 20); 1.15–1.20 (–CH3 (C)) (Fig. 2)
Mn = 29,000 g/mol (by 1H NMR analysis). 12.2 PEG arms and 9PCL arms by multiarm star molecule (from 1H NMR). Each PCLarm has a degree of polymerization of 17.
2.5. Samples preparation
Mixtures containing MHHPA and the selected proportion of thestar polymer were heated mildly until the modifier was dissolvedand the solution became clear. The mixture was then cooled downto room temperature and added to the corresponding amount ofDGEBA and catalyst. Finally, samples were carefully stirred anddegassed under vacuum (at 40 �C) during 15 min to prevent theappearance of bubbles during curing. The catalyst, BDMA, was usedat a concentration of 1 phr (1 part of catalyst per hundred parts ofDGEBA/MHHPA mixture). The modifier was added at concentra-tions of 5 and 10% with respect to DGEBA/MHHPA/BDMA mixture.

H3C O CH2 CH2 OHn
CH3OPEG-OH
O OO
Chloroform/Pyridine60ºC/72h
H3C O CH2 CH2 O C
O
CH2 CH2 C
O
OH
n
1) Synthesis of the acid terminated monomethyl poly (ethylene glycol)
O O
O
O
OH
OH
O
OH
O
OH
OH
OH
OHO
O
O
O
O
O
HBP Boltorn H30
CH3OPEG-COOH
Ar,DPTS, DMF, DCCroom temperature 48h
O O
O
O
O
O
O
OH
O
OH
O
OH
OHO
O
O
O
O
O
OO
CH3
O
O
O OCH3O
On
n
OO
CH3
O
O
n
2) Synthesis of SP-PEG
SP-PEG
O O
O
O
O
O
O
OH
O
OH
O
OH
OHO
O
O
O
O
O
OO
CH3
O
O
O OCH3O
On
n
OO
CH3
OO
n
OO
Sn(oct)2 130ºC, 72hO O
O
O
O
O
O
OH
O
O
O
O
OHO
O
O
O
O
O
O
O
CH3
OO
OO
CH3
OO
n
nO O CH3
O
On
O
O
O OH
m
O OO
OHm
3) Synthesis of SP-PEGPCL
CH3OPEG-COOH
m
SP-PEG SP-PEGPCL
Scheme 1. Synthetic steps for the preparation of SP-PEG and SP-PEGPCL.
134 C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143
Table 1 collects the notation and composition of the different for-mulations studied.
Fully cured samples for DMTA, TMA, TGA and DSC assays wereprepared by isothermal curing at 100 �C for 2 h followed by a post-curing at 180 �C for two hours.
2.6. NMR Spectroscopy
1H NMR and 13C NMR measurements were carried out at400 MHz and 100.6 MHz, respectively, in a Varian Gemini 400spectrometer. CDCl3 was used as solvent. For internal calibration thesolvent signals were used: d (13C) = 77.16 ppm and d (1H) = 7.26 ppmfor CDCl3.
2.7. Gel permeation chromatography (GPC)
Size exclusion chromatography (SEC) analysis was carried outwith an Agilent 1200 series system with PLgel 3 lm MIXED-E,PLgel 5 lm MIXED-D, and PLgel 20 lm MIXED-A columns in series,and equipped with an Agilent 1100 series refractive-index detec-tor. Calibration curves were based in polystyrene standards having
low polydispersities. THF was used as the eluent at a flow rate of1.0 ml/min, the sample concentrations were 5–10 mg/ml, andinjection volumes of 100 ll were used.
2.8. Calorimetric measurements (DSC)
Calorimetric analyses were carried out on a Mettler DSC-822ethermal analyzer. Samples of approximately 10 mg were placedin aluminium pans under nitrogen atmosphere. The calorimeterwas calibrated using an indium standard (heat flow calibration)and an indium–zinc standard (temperature calibration).
Non-isothermal curing was performed from 0 to 300 �C at heat-ing rates of 2, 5, 10 and 15 �C/min to determine the reaction heatassociated with the complete conversion of all reactive groupsand study the curing kinetics. In a non-isothermal curing process,the degree of conversion by DSC is calculated as follows:
a ¼ DhT
Dhdynð1Þ
where DhT is the heat released up to a temperature T, obtained byintegration of the calorimetric signal up to this temperature, and

Fig. 1. 1H NMR spectra of SP-PEG multiarm star.
OO
O O
O
OOH
O
OH
O
OO
O
O
O
O
OO
O O
O
OOHO
O
OO
O
O OH
O
O
O
O
OH
OH
OO
O
OO
OO
OO
OO
HOO
O
HO
O
O
HO
HO
O
O
O
O
HO
O
O
O
OH
OO
O
O
O
OO
CH3
OO
n
O OO CH3
O
O n
O
O
OCH3
O
O
nO
O
OCH3
O
O
n
O
O OCH3
O
O
n
O
O
O OH
O
OO
O
OHmO
O
OO
OHm
O
O
OO
OH
m
O
OO
H3C
OO
n
O
OOH3C O
O
O
OO
O
HO
m
O
O
OO
HO
m
OO
OH3C
O
O
n
OOOH3C
O
O
n
O
O
O
OHO
OOOH3C
O
O
n
m
m
n
Scheme 2. Idealized structure of SP-PEGPCL multiarm star.
C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 135
Dhdyn is the total reaction heat associated with the complete con-version of all reactive groups.
The glass transition temperatures (Tg1s) of the obtainedthermosets, uncured materials and star polymers were deter-mined by means of a scan at 10 �C/min, as the temperature ofthe half-way point of the jump in the heat capacity when thematerial changed from glassy to the rubbery state under N2
atmosphere and the error is estimated to be approximately±1 �C.
2.9. Curing kinetics (DSC)
The linear integral isoconversional, model-free method ofKissinger–Akahira–Sunose (KAS) was used for the determinationof the activation energy based on the non-isothermal curing curves[19]
lnb
T2
� �¼ ln
A � RgðaÞ � E
� �� E
RTð2Þ
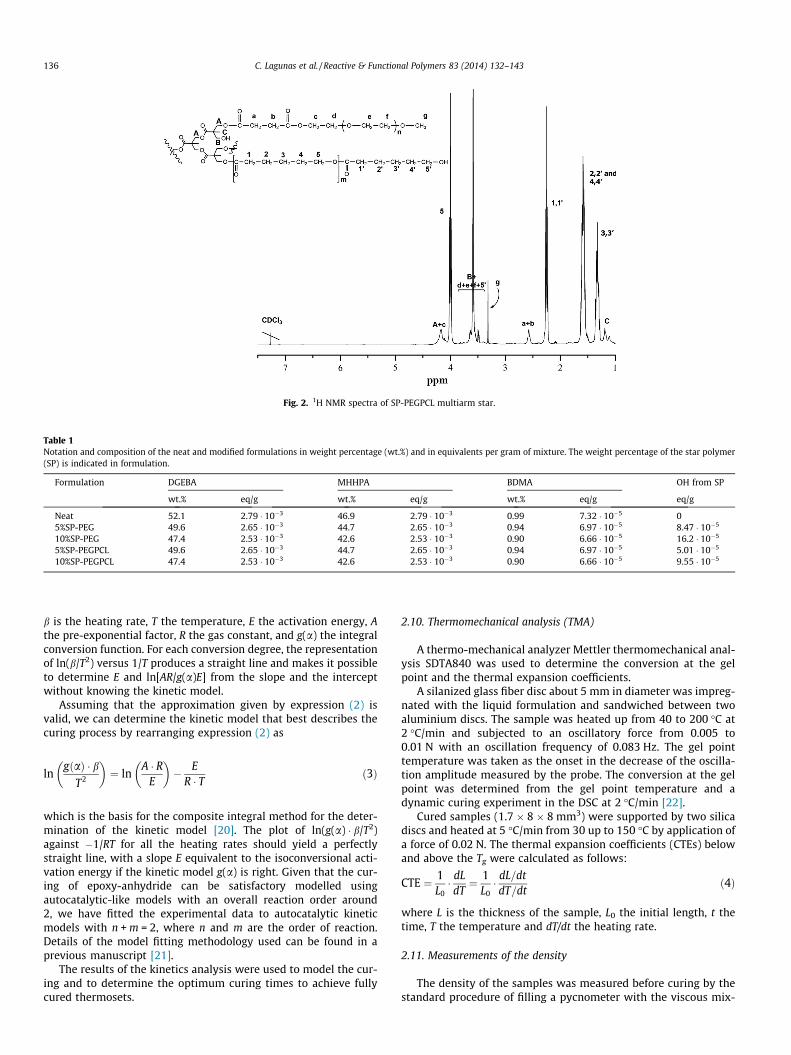
Fig. 2. 1H NMR spectra of SP-PEGPCL multiarm star.
Table 1Notation and composition of the neat and modified formulations in weight percentage (wt.%) and in equivalents per gram of mixture. The weight percentage of the star polymer(SP) is indicated in formulation.
Formulation DGEBA MHHPA BDMA OH from SP
wt.% eq/g wt.% eq/g wt.% eq/g eq/g
Neat 52.1 2.79 � 10�3 46.9 2.79 � 10�3 0.99 7.32 � 10�5 05%SP-PEG 49.6 2.65 � 10�3 44.7 2.65 � 10�3 0.94 6.97 � 10�5 8.47 � 10�5
10%SP-PEG 47.4 2.53 � 10�3 42.6 2.53 � 10�3 0.90 6.66 � 10�5 16.2 � 10�5
5%SP-PEGPCL 49.6 2.65 � 10�3 44.7 2.65 � 10�3 0.94 6.97 � 10�5 5.01 � 10�5
10%SP-PEGPCL 47.4 2.53 � 10�3 42.6 2.53 � 10�3 0.90 6.66 � 10�5 9.55 � 10�5
136 C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143
b is the heating rate, T the temperature, E the activation energy, Athe pre-exponential factor, R the gas constant, and g(a) the integralconversion function. For each conversion degree, the representationof ln(b/T2) versus 1/T produces a straight line and makes it possibleto determine E and ln[AR/g(a)E] from the slope and the interceptwithout knowing the kinetic model.
Assuming that the approximation given by expression (2) isvalid, we can determine the kinetic model that best describes thecuring process by rearranging expression (2) as
lngðaÞ � b
T2
� �¼ ln
A � RE
� �� E
R � T ð3Þ
which is the basis for the composite integral method for the deter-mination of the kinetic model [20]. The plot of ln(g(a) � b/T2)against �1/RT for all the heating rates should yield a perfectlystraight line, with a slope E equivalent to the isoconversional acti-vation energy if the kinetic model g(a) is right. Given that the cur-ing of epoxy-anhydride can be satisfactory modelled usingautocatalytic-like models with an overall reaction order around2, we have fitted the experimental data to autocatalytic kineticmodels with n + m = 2, where n and m are the order of reaction.Details of the model fitting methodology used can be found in aprevious manuscript [21].
The results of the kinetics analysis were used to model the cur-ing and to determine the optimum curing times to achieve fullycured thermosets.
2.10. Thermomechanical analysis (TMA)
A thermo-mechanical analyzer Mettler thermomechanical anal-ysis SDTA840 was used to determine the conversion at the gelpoint and the thermal expansion coefficients.
A silanized glass fiber disc about 5 mm in diameter was impreg-nated with the liquid formulation and sandwiched between twoaluminium discs. The sample was heated up from 40 to 200 �C at2 �C/min and subjected to an oscillatory force from 0.005 to0.01 N with an oscillation frequency of 0.083 Hz. The gel pointtemperature was taken as the onset in the decrease of the oscilla-tion amplitude measured by the probe. The conversion at the gelpoint was determined from the gel point temperature and adynamic curing experiment in the DSC at 2 �C/min [22].
Cured samples (1.7 � 8 � 8 mm3) were supported by two silicadiscs and heated at 5 �C/min from 30 up to 150 �C by application ofa force of 0.02 N. The thermal expansion coefficients (CTEs) belowand above the Tg were calculated as follows:
CTE ¼ 1L0� dLdT¼ 1
L0� dL=dtdT=dt
ð4Þ
where L is the thickness of the sample, L0 the initial length, t thetime, T the temperature and dT/dt the heating rate.
2.11. Measurements of the density
The density of the samples was measured before curing by thestandard procedure of filling a pycnometer with the viscous mix-

C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 137
ture in a thermostated bath at 30 �C. For the cured samples, thedensity was determined indirectly by the flotation method, usingKBr solutions with densities lower and higher than that of thesample.
2.12. Dynamomechanical analysis (DMTA)
DMTA was carried out with a TA Instruments DMA Q800. Threepoint bending of 10 mm at 1 Hz and 0.05% strain was performed at3 �C/min from �150 to 250 �C on prismatic rectangular samples(ca. 1.7 � 11.5 � 23 mm3).
2.13. Thermogravimetric analysis (TGA)
Thermogravimetric analysis was carried out with a MettlerTGA/SDTA 851e/LF/1100 thermobalance. Samples with an approx-imate mass of 8 mg were degraded between 30 and 800 �C at aheating rate of 10 �C/min in a nitrogen atmosphere (50 cm3/minmeasured in normal conditions).
2.14. Impact resistance
The impact test was performed at room temperature by meansof an Izod 5110 impact tester according to ASTM D 4508-10 (2010)using rectangular samples (1.7 � 11.5 � 23 mm3). The pendulumemployed had a kinetic energy of 0.5 J. For each material, ninedeterminations were made. The impact strength (IS) was calcu-lated from the energy absorbed by the sample upon fracture as
IS ¼ E� E0
Sð5Þ
where E and E0 are the energy loss of the pendulum with and with-out sample respectively, and S is the cross-section of the samples.
2.15. Electron microscopy analysis (SEM)
The fracture area of samples were metalized with gold andobserved with a scanning electron microscopy (SEM) Jeol JSM6400 with a resolution of 3.5 nm. The samples examined were pre-viously fractured by impact at room temperature.
3. Results and discussion
3.1. Synthesis and characterization of multiarm stars
There are two main methodologies for the synthesis of multi-arm stars: arm first and core first [23]. The attachment of thepoly(ethylene glycol) to the core to obtain SP-PEG was done as pre-viously described in the synthesis of stars with an aromatic–ali-phatic hyperbranched polyester core by esterification with amonomethyl poly(ethylene glycol) previously reacted with succi-nic anhydride [24]. A similar attachment of PEG arms wasdescribed by Chen et al. [25] but they firstly reacted succinic anhy-dride to hydroxyl groups in the dendritic structure and then thecarboxylic groups formed were condensed with the commercialmonomethyl poly(ethylene glycol). Because of the multifunctional-ity of the HBP polyester, we prefer the previous modification ofPEG with succinic anhydride and the condensation with hydroxylHBP terminal units to avoid any type of star–star coupling. It wasalso described a procedure of pegylation of Boltorn polyesters byketalization of a 4-hydroxybenzaldehyde derivatized monomethylpoly(ethylene glycol), but this method offers in our case no advan-tage over the esterification procedure [26]. The polymeric star pre-pared was characterized by 1H NMR spectroscopy.
Signals A, B and C correspond to the structure of Boltorn H30.During the modification, B is transformed into A and the signal C
is split reflecting not only the proportion of dendrimeric, linearand terminal units but also the modified ones. Because of the over-lapping of the signals corresponding to the poly(ethylene glycol)arms with A and B of the polyester core, the degree of esterificationcould not be evaluated as described in a previous paper [8]. Thedegree of esterification allows evaluating the number of PEG armsper molecule. Thus, by comparing the integrals of the methyl sig-nals of the core C and the methoxy protons (g) at the end of thearms, and knowing that the average number of methyl groups inBoltorn H30 is 87, we calculated that the average number of armsper star molecule is 12.2.
In the preparation of the miktoarm star SP-PEGPCL we haveused both synthetic methodologies in a two-step procedure. Tak-ing the SP-PEG star, previously synthesized as the core moleculewe grew PCL arms from the unmodified OH groups of the polyestercore, by the Sn(oct)2 catalyzed ring-opening polymerization of e-CL. This core-first methodology allows in principle to grow PCLarms of a selected length, because the living character of the poly-merization mechanism [9]. Thus, we selected a molar proportion(e-CL/OH) of 10 in order to get PCL arms with an average degreeof polymerization of 10. Fig. 2 shows the 1H NMR spectrum ofthe miktoarm star obtained with the corresponding assignments.
The average degree of polymerization of PCL is usually evalu-ated from the signal 5 and 50, corresponding to the methylene pro-tons attached to the oxygen ester and hydroxyl groups,respectively. However, it can be seen in the spectrum that the sig-nal 50 partially overlaps with signal B of the core and signals d, eand f of the PEG arms. To evaluate accurately the DP of the PCLarms we substract from the value of the integral of the signal1,10 in the spectrum the integral of the signal 5, obtaining indirectlythe area of the signal 10 which should be the same that the integralof the signal 50. Then, by dividing the integrals of the signals 5 by 10
and adding one terminal unit we obtain the average degree of poly-merization of PCL, which in this case resulted to be 17. As weexpected a DP of 10, we get to the conclusion that less PCL armshas been formed. In fact, only 9 PCL arms in average were formedand therefore 11 hydroxyl groups of the Boltorn core remainsunreacted. The non-reactivity of all the OH groups of the Boltorncore molecule agrees with previously reported results [25,27,28].By GPC a monomodal curve was obtained for this star polymerindicating that no linear PCL was formed.
The thermal behaviour of the synthesized multiarm stars wasstudied by calorimetry. As can be seen in Table 2, the Tg of theamorphous part of the multiarm stars and the linear carboxy ter-minated PEG is quite similar and occurs at very low temperature.In addition to the amorphous phase, all these materials show melt-ing phenomena. It is well known that PCL and PEG are crystallinepolymers with reported enthalpies of fusion of pure crystals ofPEG and PCL of 203 J/g [29] and 139.5 J/g [30], respectively. Severalstudies have been done in the calorimetric behavior of linear PCL-PEG copolymers [31,32], but little information can be found in thecrystallization behavior of PCL-PEG miktoarm stars [33].
As we can see in the table, the three polymers containing PEGunits present a melting endotherm between 6 and 11 �C and quitedifferent melting enthalpies, which reflects the proportion of PEGcrystalline phase in these materials. In the miktoarm star withPCL arms, in addition to the little endotherm of PEG crystallites,there is a second melting endotherm at 53 �C with an enthalpy ofabout 55 J/g related to the crystalline PCL rich phase.
The fact that PEG and PCL arms which are distributed randomlyin the shell of the star form crystalline phases could be explainedby the formation of phases or Janus-type particles. In a previousstudy [16], on hyperbranched modified polymers with hydrophilic(PEG) and hydrophobic (docosyl groups) structures, it was demon-strated for the first time that the perfection in the placement ofthese different structures was not essential for the appearance of
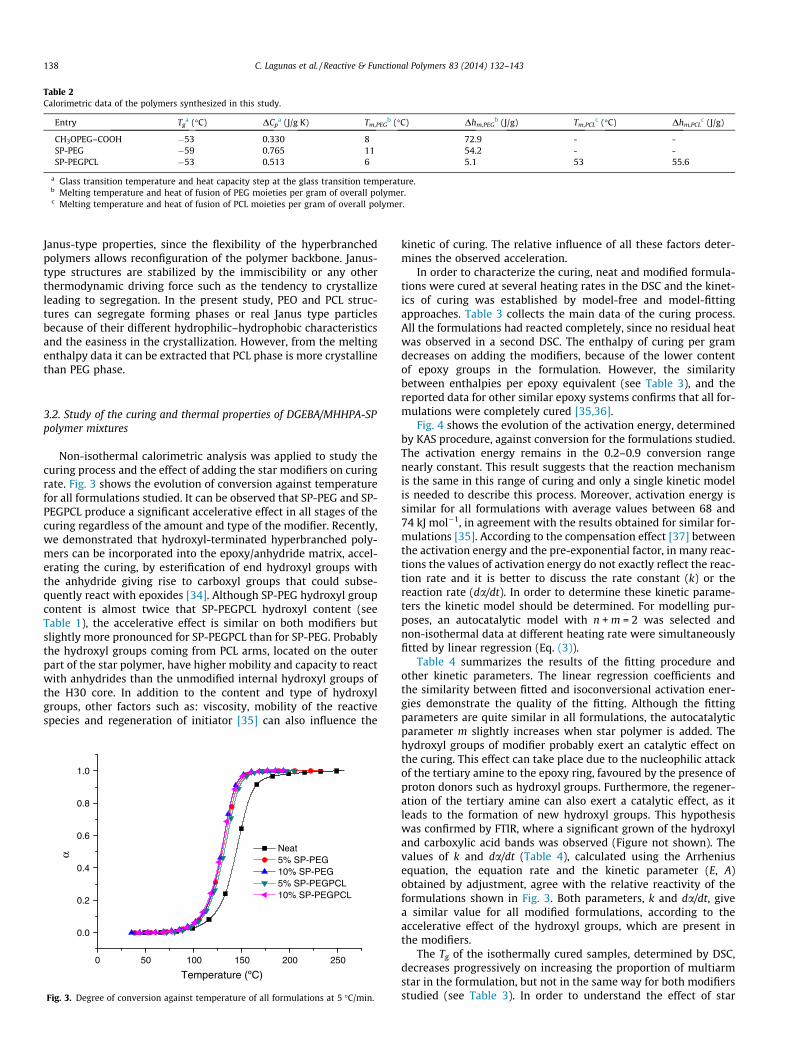
Table 2Calorimetric data of the polymers synthesized in this study.
Entry Tga (�C) DCp
a (J/g K) Tm,PEGb (�C) Dhm,PEG
b (J/g) Tm,PCLc (�C) Dhm,PCL
c (J/g)
CH3OPEG–COOH �53 0.330 8 72.9 - -SP-PEG �59 0.765 11 54.2 - -SP-PEGPCL �53 0.513 6 5.1 53 55.6
a Glass transition temperature and heat capacity step at the glass transition temperature.b Melting temperature and heat of fusion of PEG moieties per gram of overall polymer.c Melting temperature and heat of fusion of PCL moieties per gram of overall polymer.
138 C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143
Janus-type properties, since the flexibility of the hyperbranchedpolymers allows reconfiguration of the polymer backbone. Janus-type structures are stabilized by the immiscibility or any otherthermodynamic driving force such as the tendency to crystallizeleading to segregation. In the present study, PEO and PCL struc-tures can segregate forming phases or real Janus type particlesbecause of their different hydrophilic–hydrophobic characteristicsand the easiness in the crystallization. However, from the meltingenthalpy data it can be extracted that PCL phase is more crystallinethan PEG phase.
3.2. Study of the curing and thermal properties of DGEBA/MHHPA-SPpolymer mixtures
Non-isothermal calorimetric analysis was applied to study thecuring process and the effect of adding the star modifiers on curingrate. Fig. 3 shows the evolution of conversion against temperaturefor all formulations studied. It can be observed that SP-PEG and SP-PEGPCL produce a significant accelerative effect in all stages of thecuring regardless of the amount and type of the modifier. Recently,we demonstrated that hydroxyl-terminated hyperbranched poly-mers can be incorporated into the epoxy/anhydride matrix, accel-erating the curing, by esterification of end hydroxyl groups withthe anhydride giving rise to carboxyl groups that could subse-quently react with epoxides [34]. Although SP-PEG hydroxyl groupcontent is almost twice that SP-PEGPCL hydroxyl content (seeTable 1), the accelerative effect is similar on both modifiers butslightly more pronounced for SP-PEGPCL than for SP-PEG. Probablythe hydroxyl groups coming from PCL arms, located on the outerpart of the star polymer, have higher mobility and capacity to reactwith anhydrides than the unmodified internal hydroxyl groups ofthe H30 core. In addition to the content and type of hydroxylgroups, other factors such as: viscosity, mobility of the reactivespecies and regeneration of initiator [35] can also influence the
0 50 100 150 200 250
0.0
0.2
0.4
0.6
0.8
1.0
α
Temperature (ºC)
Neat 5% SP-PEG 10% SP-PEG 5% SP-PEGPCL 10% SP-PEGPCL
Fig. 3. Degree of conversion against temperature of all formulations at 5 �C/min.
kinetic of curing. The relative influence of all these factors deter-mines the observed acceleration.
In order to characterize the curing, neat and modified formula-tions were cured at several heating rates in the DSC and the kinet-ics of curing was established by model-free and model-fittingapproaches. Table 3 collects the main data of the curing process.All the formulations had reacted completely, since no residual heatwas observed in a second DSC. The enthalpy of curing per gramdecreases on adding the modifiers, because of the lower contentof epoxy groups in the formulation. However, the similaritybetween enthalpies per epoxy equivalent (see Table 3), and thereported data for other similar epoxy systems confirms that all for-mulations were completely cured [35,36].
Fig. 4 shows the evolution of the activation energy, determinedby KAS procedure, against conversion for the formulations studied.The activation energy remains in the 0.2–0.9 conversion rangenearly constant. This result suggests that the reaction mechanismis the same in this range of curing and only a single kinetic modelis needed to describe this process. Moreover, activation energy issimilar for all formulations with average values between 68 and74 kJ mol�1, in agreement with the results obtained for similar for-mulations [35]. According to the compensation effect [37] betweenthe activation energy and the pre-exponential factor, in many reac-tions the values of activation energy do not exactly reflect the reac-tion rate and it is better to discuss the rate constant (k) or thereaction rate (da/dt). In order to determine these kinetic parame-ters the kinetic model should be determined. For modelling pur-poses, an autocatalytic model with n + m = 2 was selected andnon-isothermal data at different heating rate were simultaneouslyfitted by linear regression (Eq. (3)).
Table 4 summarizes the results of the fitting procedure andother kinetic parameters. The linear regression coefficients andthe similarity between fitted and isoconversional activation ener-gies demonstrate the quality of the fitting. Although the fittingparameters are quite similar in all formulations, the autocatalyticparameter m slightly increases when star polymer is added. Thehydroxyl groups of modifier probably exert an catalytic effect onthe curing. This effect can take place due to the nucleophilic attackof the tertiary amine to the epoxy ring, favoured by the presence ofproton donors such as hydroxyl groups. Furthermore, the regener-ation of the tertiary amine can also exert a catalytic effect, as itleads to the formation of new hydroxyl groups. This hypothesiswas confirmed by FTIR, where a significant grown of the hydroxyland carboxylic acid bands was observed (Figure not shown). Thevalues of k and da/dt (Table 4), calculated using the Arrheniusequation, the equation rate and the kinetic parameter (E, A)obtained by adjustment, agree with the relative reactivity of theformulations shown in Fig. 3. Both parameters, k and da/dt, givea similar value for all modified formulations, according to theaccelerative effect of the hydroxyl groups, which are present inthe modifiers.
The Tg of the isothermally cured samples, determined by DSC,decreases progressively on increasing the proportion of multiarmstar in the formulation, but not in the same way for both modifiersstudied (see Table 3). In order to understand the effect of star

Table 3Calorimetric data of the formulation studied in this work.
Formulation Dha (J/g) Dhb (kJ/ee) Tg0c (�C) DCp0
c (J/g�K) Tg1d (�C) Tg1,Fox
e (�C) DCp1d (J/g�K) kf
Neat 317 114 �39 0.529 152 152 0.304 0.5745%SP-PEG 306 117 �38 0.538 132 131 0.332 0.61710%SP-PEG 277 109 �39 0.571 119 117 0.356 0.6245%SP-PEGPCL 313 118 �39 0.541 133 133 0.317 0.58510%SP-PEGPCL 303 121 �39 0.551 126 115 0.328 0.595
a Enthalpies per gram of mixtures.b Enthalpies per equivalent of epoxy groups.c Glass transition temperature and heat capacity step at the glass transition temperature before curing.d Glass transition temperature and heat capacity step at the glass transition temperature after curing.e Calculated glass transition temperature, using DSC data and Fox equation.f Relationship between the segmental mobilities of the crosslinked and noncrosslinked polymers, calculated as k = DCp1/DCp0.
0.0 0.2 0.4 0.6 0.8 1.00
20
40
60
80
100
E (
kJ·m
ol-1)
α
Neat 5% SP-PEG 10% SP-PEG 5% SP-PEGPCL 10% SP-PEGPCL
Fig. 4. Apparent activation energy, E, during curing of all formulations using KASintegral method.
C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 139
polymers on the matrix, the Fox equation was applied to predictthe Tg of the fully cured materials. Up to 5% of content, the calcu-lated values of Tg for both types of formulations are similar thanthe experimental ones. Formulations containing 10% of modifierpresent experimental Tgs above the theoretical for fully misciblematerials. This result suggests that a part of the modifiers, espe-cially in the case of miktoarm SP-PEGPCL, forms a separate phase.
The relationship between segmental mobilities of the cross-linked and uncrosslinked materials, k, follows a related trend tothat of Tg. The segmental mobility of the materials increases withthe content of modifier, showing SP-PEGPCL formulations lower kvalues than SP-PEG formulations, in agreement with the fact thata larger amount of SP-PEGPCL may be segregated in a separatephase. Taking into account all these results, it is expected a higherdensity of crosslinking in mixtures containing SP-PEGPCL than inSP-PEG formulations.
The results of the thermogravimetric analysis are summarizedin Table 5. In general, formulations containing modifier show lower
Table 4Kinetic parameters of curing process.
Formulation Ea (kJ/mol) ln Aa (min�1) na ma
Neat 71.8 19.99 1.52 0.485%SP-PEG 72.1 20.81 1.51 0.4910%SP-PEG 70.0 20.28 1.48 0.525%SP-PEGPCL 74.0 21.17 1.51 0.4910%SP-PEGPCL 67.5 19.56 1.46 0.54
a Kinetic parameters obtained by model-fitting using an autocatalytic model with n +b Values of rate constant at 150 �C using the Arrhenius equation and the parametersc Values of reaction rate at 150 �C obtained using the equation rate da/dt = k � f(a)=k �d Average isoconversional activation energy.
T5% than neat formulation, due to the presence of a higher contentof thermolabile ester groups. We can also see that thermosets con-taining a 10% of SP-PEG or SP-PEGPCL show a higher stability thanformulations containing a 5% of the same modifiers. This behaviourcan be attributed to the phase separated character of the rich-mod-ifier thermosets (10%) and to the interfacial adhesion betweenphases. The SP segregated particles are surrounded by the epoxy/anhydride matrix, which may prevent the elimination of volatilefragments. 10% SP-PEGPCL is more thermally stable than 10% SP-PEG, probably due to it higher degree of crosslinking, as it will beseen in the thermomechanical properties section.
3.3. Gelation and shrinkage
The reduction of the shrinkage and the stress generated duringcuring is one of the goals to improve the applicability of epoxythermosetting as coatings and encapsulants. Because the appear-ance of internal stresses during curing only take places after gela-tion, it is desirable to increase the conversion at the gel point, agel.
Table 5 collects the densities before and after curing and theshrinkage calculated from them for all the formulations studied.It can be seen that both star polymers reduce the contraction dur-ing curing and a near-zero shrinkage is reached. This behaviour hasbeen reported previously [34,38] and can be related with theincrease of unoccupied spaces in the cured network when SP isadded [39]. The decrease in H-bond interactions, when SP reactsthrough hydroxyl groups and is incorporated into the network,can also explain the behaviour observed [37]. In addition, Eomet al. demonstrated that it is possible to produce void-freeepoxy/HBP laminates by tailoring the cure cycle in order to facili-tate molecular rearrangements that allow the local stress redistri-bution [40].
The agel values determined by non-isothermal DSC/TMA com-bined experiments are collected in Table 5. As can be seen the addi-tion of SP-PEG and SP-PEGPCL produces a significant progressiveincrease of agel. This behaviour can be related fundamentally withchain-transfer reactions between propagating carboxylate and car-
ra k150�Ca,b (min�1) (da/dt)0.5
c (min�1) Eisod
0.999 0.66 0.164 72.30.999 1.35 0.337 72.40.999 1.43 0.360 70.20.999 1.16 0.290 74.10.999 1.44 0.360 67.8
m = 2.E and ln A.(1 � a)n�am at 50% conversion.
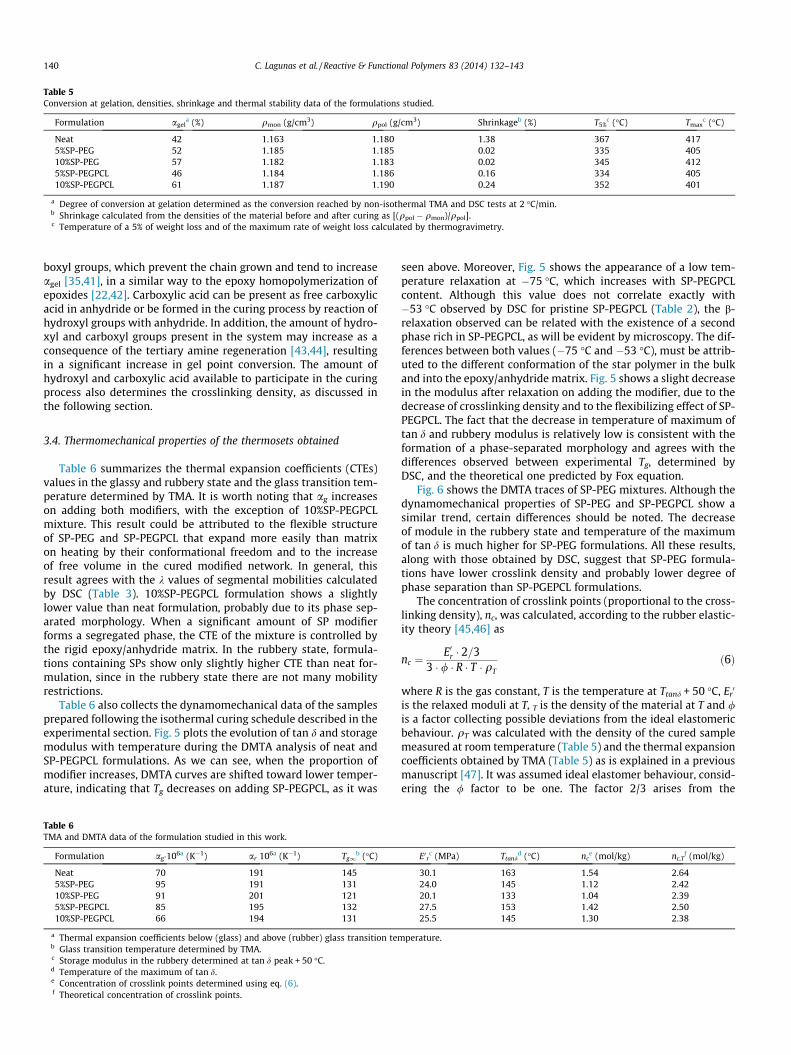
Table 5Conversion at gelation, densities, shrinkage and thermal stability data of the formulations studied.
Formulation agela (%) qmon (g/cm3) qpol (g/cm3) Shrinkageb (%) T5%
c (�C) Tmaxc (�C)
Neat 42 1.163 1.180 1.38 367 4175%SP-PEG 52 1.185 1.185 0.02 335 40510%SP-PEG 57 1.182 1.183 0.02 345 4125%SP-PEGPCL 46 1.184 1.186 0.16 334 40510%SP-PEGPCL 61 1.187 1.190 0.24 352 401
a Degree of conversion at gelation determined as the conversion reached by non-isothermal TMA and DSC tests at 2 �C/min.b Shrinkage calculated from the densities of the material before and after curing as [(qpol � qmon)/qpol].c Temperature of a 5% of weight loss and of the maximum rate of weight loss calculated by thermogravimetry.
140 C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143
boxyl groups, which prevent the chain grown and tend to increaseagel [35,41], in a similar way to the epoxy homopolymerization ofepoxides [22,42]. Carboxylic acid can be present as free carboxylicacid in anhydride or be formed in the curing process by reaction ofhydroxyl groups with anhydride. In addition, the amount of hydro-xyl and carboxyl groups present in the system may increase as aconsequence of the tertiary amine regeneration [43,44], resultingin a significant increase in gel point conversion. The amount ofhydroxyl and carboxylic acid available to participate in the curingprocess also determines the crosslinking density, as discussed inthe following section.
3.4. Thermomechanical properties of the thermosets obtained
Table 6 summarizes the thermal expansion coefficients (CTEs)values in the glassy and rubbery state and the glass transition tem-perature determined by TMA. It is worth noting that ag increaseson adding both modifiers, with the exception of 10%SP-PEGPCLmixture. This result could be attributed to the flexible structureof SP-PEG and SP-PEGPCL that expand more easily than matrixon heating by their conformational freedom and to the increaseof free volume in the cured modified network. In general, thisresult agrees with the k values of segmental mobilities calculatedby DSC (Table 3). 10%SP-PEGPCL formulation shows a slightlylower value than neat formulation, probably due to its phase sep-arated morphology. When a significant amount of SP modifierforms a segregated phase, the CTE of the mixture is controlled bythe rigid epoxy/anhydride matrix. In the rubbery state, formula-tions containing SPs show only slightly higher CTE than neat for-mulation, since in the rubbery state there are not many mobilityrestrictions.
Table 6 also collects the dynamomechanical data of the samplesprepared following the isothermal curing schedule described in theexperimental section. Fig. 5 plots the evolution of tan d and storagemodulus with temperature during the DMTA analysis of neat andSP-PEGPCL formulations. As we can see, when the proportion ofmodifier increases, DMTA curves are shifted toward lower temper-ature, indicating that Tg decreases on adding SP-PEGPCL, as it was
Table 6TMA and DMTA data of the formulation studied in this work.
Formulation ag�106a (K�1) ar 106a (K�1) Tg1b (�C)
Neat 70 191 1455%SP-PEG 95 191 13110%SP-PEG 91 201 1215%SP-PEGPCL 85 195 13210%SP-PEGPCL 66 194 131
a Thermal expansion coefficients below (glass) and above (rubber) glass transition temb Glass transition temperature determined by TMA.c Storage modulus in the rubbery determined at tan d peak + 50 �C.d Temperature of the maximum of tan d.e Concentration of crosslink points determined using eq. (6).f Theoretical concentration of crosslink points.
seen above. Moreover, Fig. 5 shows the appearance of a low tem-perature relaxation at �75 �C, which increases with SP-PEGPCLcontent. Although this value does not correlate exactly with�53 �C observed by DSC for pristine SP-PEGPCL (Table 2), the b-relaxation observed can be related with the existence of a secondphase rich in SP-PEGPCL, as will be evident by microscopy. The dif-ferences between both values (�75 �C and �53 �C), must be attrib-uted to the different conformation of the star polymer in the bulkand into the epoxy/anhydride matrix. Fig. 5 shows a slight decreasein the modulus after relaxation on adding the modifier, due to thedecrease of crosslinking density and to the flexibilizing effect of SP-PEGPCL. The fact that the decrease in temperature of maximum oftan d and rubbery modulus is relatively low is consistent with theformation of a phase-separated morphology and agrees with thedifferences observed between experimental Tg, determined byDSC, and the theoretical one predicted by Fox equation.
Fig. 6 shows the DMTA traces of SP-PEG mixtures. Although thedynamomechanical properties of SP-PEG and SP-PEGPCL show asimilar trend, certain differences should be noted. The decreaseof module in the rubbery state and temperature of the maximumof tan d is much higher for SP-PEG formulations. All these results,along with those obtained by DSC, suggest that SP-PEG formula-tions have lower crosslink density and probably lower degree ofphase separation than SP-PGEPCL formulations.
The concentration of crosslink points (proportional to the cross-linking density), nc, was calculated, according to the rubber elastic-ity theory [45,46] as
nc ¼E0r � 2=3
3 � / � R � T � qTð6Þ
where R is the gas constant, T is the temperature at Ttand + 50 �C, Er0
is the relaxed moduli at T, T is the density of the material at T and /is a factor collecting possible deviations from the ideal elastomericbehaviour. qT was calculated with the density of the cured samplemeasured at room temperature (Table 5) and the thermal expansioncoefficients obtained by TMA (Table 5) as is explained in a previousmanuscript [47]. It was assumed ideal elastomer behaviour, consid-ering the / factor to be one. The factor 2/3 arises from the
E0rc (MPa) Ttand
d (�C) nce (mol/kg) nc,T
f (mol/kg)
30.1 163 1.54 2.6424.0 145 1.12 2.4220.1 133 1.04 2.3927.5 153 1.42 2.5025.5 145 1.30 2.38
perature.

40 60 80 100 120 140 160 180 20010
100
1000
E' (
MP
a)
Temperature (ºC)
Neat
5% SP-PEGPCL
10% SP-PEGPCL
-100 -50 00.01
0.02
0.03
0.04
0.0
0.2
0.4
0.6
0.8
1.0
1.2
tan
δ
Fig. 5. Storage modulus (empty symbols) and tan d (filled symbols) curves against temperature for neat DGEBA/anhydride and DGEBA/SP-PEGPCL modified materials. Theinset shows a detail at low temperatures.
40 60 80 100 120 140 160 180 20010
100
1000
E' (
MP
a)
Temperature (ºC)
Neat
5% SP-PEG
10% SP-PEG
-100 -50 00.01
0.02
0.03
0.04
0.0
0.2
0.4
0.6
0.8
1.0
1.2
tan
δ
Fig. 6. Storage modulus (empty symbols) and tan d curves (filled symbols) against temperature for neat DGEBA/anhydride and DGEBA/SP-PEG modified materials. The insetshows a detail at low temperatures.
C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 141
consideration that every crosslink point is trifunctional and elasticstrands are shared by two crosslinks, A theoretical concentrationof crosslink points, nc,T, was calculated as the number of epoxyequivalent per mass minus twice the number of initiator groupsper mass.
Table 6 collects the experimental and theoretical concentrationof crosslink points for all formulations. It can be observed that thecrosslinking density decreases when the proportion of both modi-fier increases. The presence of SP with a large number of hydroxylgroups that can participate in the polymerization process by reac-tion with anhydride groups and chain transfer reactions can resultin a certain loosening of crosslink the network structure, asreported previously [47]. Moreover, the minor presence of epoxy-crosslinkable groups can also contribute to the decrease of cross-linking. The minor decrease of nc in SP-PEGPCL formulations couldbe related to the higher degree of phase separation achieved duringcuring for these mixtures. There was a remarkable difference
between the theoretical and experimental nc values, mainly attrib-utable to the occurrence of regeneration to some extent, which canlead to more chain ends than expected [47]. The differencesobserved can also be partially explained by the presence of freecarboxylic acid which can act as chain-starting points and chain-transfer agents.
3.5. Mechanical characterization and morphology
The toughness behaviour of neat and modified thermosets wasinvestigated by impact test, and the morphology was analyzed bySEM on the fracture surface. Fig. 7 shows the results of the impacttests for all materials prepared. It can be observed that the additionof a 5% of either modifier to the formulation barely increases theimpact strength. A further increase in impact strength is achievedwhen 10% of the modifiers is used, but the effect is specially rele-vant in the case of SP-PEGPCL. It can be hypothesized that, when

Neat
5% SP-PEG
10% SP-PEG
5% SP-PEGPCL
10% SP-PEGPCL
0
2
4
6
Impa
ct s
tren
gth
(kJ·
m-2)
Fig. 7. Impact strength for neat DGEBA/anhydride and thermosets with SP-PEG andSP-PEGPCL.
142 C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143
only a 5% of the modifiers is used, the major part of the star poly-mer is solubilized or incorporated in the epoxy/anhydride matrix,resulting in a discrete toughening effect by enhanced matrix shearyielding caused by a loosening of the network structure and inter-nal plasticization. When 10% of the modifiers are used, one mayexplain the differences in terms of the structures of both star poly-mers. In the case of SP-PEG, with a reduced number of arms and ahigher amount of unmodified hydroxyl groups, the compatibilitywith the epoxy-anhydride matrix may be too high, thus makingit difficult to phase-separate. In the case of SP-PEGPCL, the pres-ence of hydrophobic/hydrophilic arms may allow the polymer toself-assemble and form separated particles well dispersed withinthe matrix. The hydrophobic PCL arms may contribute to the goodinterfacial adhesion between the hydrophobic matrix and SP-PEG-PCL particles. This results in a higher toughness enhancement,because stresses acting on the matrix can be efficiently transferredto the soft particles across the matrix–particle interphase. Thiseffect is similar to that observed in epoxy-anhydride thermosets
Neat 10%SP-PEGPCL
Neat 10%SP-PEGPCL
100 μm 100 μm
2 μm 2 μm
Fig. 8. SEM micrographs at 500 and 20,000 magnifications for f
modified with hyperbranched polyesters bearing undecenoyl sidechains [8].
Fig. 8 shows some SEM micrographs of the fractured surfaces ofthe materials after the impact tests. The fracture surface of the neatthermoset is relatively smooth and glassy, showing that little plas-tic energy dissipation has taken place during the process of frac-ture. The fracture surface of modified thermosets was different,showing a multi-planar nature of the surface with thicker cracks,resulting from plastic deformation upon fracture. This feature ismore remarkable for the formulation containing 10% of SP-PEGPCL,in agreement with its higher impact strength shown in Fig. 7. In themicrographs with higher magnification it can be observed thepresence of some nanometric structures homogenously distributedin the matrix of the modified thermosets. Formulation containing10% of SP-PEG presents a nanograined morphology with small par-ticles, but large enough to show a b-relaxation in the DMTA spectraat low temperature related to the phase separated structure.10%SP-PEGPCL formulation show more clearly a phase-separatemorphology with two kinds of nanosized particles. By comparisonwith SP-PEG thermosets, it can be hypothesized that the clear anddark particles correspond respectively to miktoarm star-rich phasewith their self-assembled PEG or PCL arms. Probably, the confor-mational flexibility of the miktoarm star-polymer core permitsthe reconfiguration of the polymer backbone and the segregationof the randomly placed PEG and PCL arms of the shell.
The appearance of voids, due to debonding of the particles,reveals that the toughening effect in SP-PEGPCL formulations couldbe controlled by a plastic void growth mechanism. The occurrenceof certain tails and regions of plastic deformation [48] suggeststhat other mechanisms as crack pinning, crack deflection or immo-bilised polymer could also contribute to the toughening effectobserved.
4. Conclusions
Two new amphiphilic multiarm star polymers with linear poly-ethylene glycol and poly(e-caprolactone) arms were synthesizedby partial pegylation of a hyperbranched polyester and subsequentgrowth of poly(e-caprolactone) arms. The multiarm star polymerswere characterized and used in different proportions as modifiers
10%SP-PEG
10%SP-PEG
100 μm
2 μm
racturated surfaces of the some impacted samples studied.

C. Lagunas et al. / Reactive & Functional Polymers 83 (2014) 132–143 143
of epoxy/anhydride mixtures catalyzed with benzyl dimethylamine.
The content and type of hydroxyl groups of the modifiers playedan important role in the curing process and influenced the finalproperties of the thermosets. Hydroxyl groups accelerated the cur-ing and promoted the incorporation of modifier into the networkstructure through esterification and chain-transfer reactions,increasing the conversion at gelation and decreasing the glass tran-sition temperature and the density of crosslinking, which is alsostrongly influenced by the regeneration of the initiator.
The resulting materials, due to the amphiphilic character of themultiarm star, showed phase-separated morphologies with nano-sized particles and a significant improvement on the impactstrength. The best improvement was obtained by using a miktoarmstar polymer containing poly(ethylene glycol) and poly(e-caprolac-tone) arms with different hydrophilic/hydrophobic character. Thismodifier has demonstrated a high self-assembling tendency,responsible of the improvement observed.
Acknowledgements
The authors would like to thank MICINN (Ministerio de Cienciae Innovación) and FEDER (Fondo Europeo de Desarrollo Regional)(MAT2011-27039-C03-01 and MAT2011-27039-C03-02) and tothe Comissionat per a Universitats i Recerca del DIUE de la Gener-alitat de Catalunya (2009-SGR-1512).
References
[1] C.A. May (Ed.), Epoxy Resins: Chemistry and Technology, Marcel Dekker, NewYork, 1988 (Chapter 1).
[2] E.M. Petrie, Epoxy Adhesive Formulations, McGraw-Hill, New York, 2006.[3] J.P. Pascault, R.J.J. Williams, Epoxy Polymers, Wiley VCH, Weinheim, 2010
(Chapter 1).[4] D. Ratna, R. Varley, G.P. Simon, J. Appl. Polym. Sci. 89 (2003) 2339–2345.[5] L. Boogh, B. Pettersson, J.-A.E. Månson, Polymer 40 (1999) 2249–2261.[6] M. Morell, A. Lederer, X. Ramis, B. Voit, A. Serra, J. Polym. Sci. Part A: Polym.
Chem. 49 (2011) 2395–2406.[7] F. Meng, X.H. Zhang, B.Y. Du, B.X. Zhou, X. Zhou, G.R. Qi, Polymer 52 (2011)
391–399.[8] M. Flores, X. Fernández-Francos, F. Ferrando, X. Ramis, A. Serra, Polymer 53
(2012) 5232–5241.[9] M. Morell, D. Foix, A. Lederer, X. Ramis, B. Voit, A. Serra, J. Polym. Sci. Part A:
Polym. Chem. 49 (2011) 4639–4649.[10] D. Foix, E. Jiménez-Piqué, X. Ramis, A. Serra, Polymer 52 (2011) 5009–5017.[11] M. Morell, X. Ramis, F. Ferrando, A. Serra, Polymer 52 (2011) 4694–4702.[12] C. Acebo, A. Picardi, X. Fernández-Francos, S. De La Flor, X. Ramis, A. Serra,
Prog. Org. Coat. 77 (2014) 1288–1298.[13] J. Karger-Kocsis, J. Frölich, O. Gryshchuk, H. Kautz, H. Frey, R. Mülhaupt,
Polymer 45 (2004) 1185–1195.
[14] L. Könczöl, W. Döll, U. Buchholz, R. Mülhaupt, J. Appl. Polym. Sci. 84 (1994)815–826.
[15] V. Percec, D.A. Wilson, P. Leowanawat, C.J. Wilson, A.D. Hughes, M.S. Kaucher,D.A. Hammer, D.H. Levine, A.J. Kim, F.S. Bates, K.P. Davis, T.P. Lodge, M.L. Klein,R.H. DeVane, E. Aqad, B.M. Rosen, A.O. Argintaru, M.J. Sienkowska, K. Rissanen,S. Nummelin, J. Ropponen, Science 328 (2010) 1009–1014.
[16] A.Z. Samuel, S. Ramakrishnan, Macromolecules 45 (2012) 2348–2358.[17] K. Khanna, S. Varsheney, A. Kakkar, Polym. Chem. 1 (2010) 1171–1185.[18] J.S. Moore, S.I. Stupp, Macromolecules 23 (1990) 65–70.[19] S. Vyazovkin, A.K. Burnham, J.M. Criado, L.A. Pérez-Maqueda, C. Popesco, N.
Sbirrazzuoli, Thermochim. Acta 520 (2011) 1–19.[20] A. Cadenato, J.M. Morancho, X. Fernández-Francos, J.M. Salla, X. Ramis X, J.
Thermal, Anal. Cal. 89 (2007) 233–244.[21] M. Flores, X. Fernández-Francos, X. Ramis, A. Serra, Thermochim. Acta 544
(2012) 17–26.[22] X. Fernández-Francos, W.D. Cook, J.M. Salla, A. Serra, X. Ramis, Polym. Int. 58
(2009) 1401–1410.[23] B. Voit, A. Lederer, Chem. Rev. 109 (2009) 5924–5973.[24] D. Foix, X. Fernández-Francos, X. Ramis, A. Serra, M. Sangermano, React. Funct.
Polym. 71 (2011) 417–424.[25] S. Chen, X-Z. Zhang, S-X. Cheng, R-X. Zhuo, Z-W. Gu, Biomacromolecules 9
(2008) 2578–2585.[26] C. Tu, L. Zhu, F. Qiu, D. Wang, Y. Su, X. Zhu, D. Yan, Polymer 54 (2013) 2020–
2027.[27] H. Claesson, E. Malmström, M. Johansson, A. Hult, Polymer 43 (2002) 3511–
3518.[28] Z.M. Miao, S.X. Cheng, X.Z. Zhang, Q.R. Wang, R.X. Zhuo, J. Biomed. Mater. Res.
81B (2007) 40–49.[29] L. Mandelkern, F.A. Quinn, P.J. Flory, J. Appl. Phys. 25 (1954) 830–839.[30] M. Aubin, R.E. Prud’homme, Macromolecules 21 (1988) 2945–2949.[31] V. Jacquier, C. Miola, M-F. Llauro, C. Monnet, T. Hamaide, Macromol. Chem.
Phys. 197 (1996) 1311–1324.[32] Z. Gan, J. Zhang, B. Jiang, J. Appl. Polym. Sci. 63 (1997) 1793–1804.[33] G. Maglio, F. Nicodemi, C. Conte, R. Palumbo, P. Tirino, E. Panza, A. Ianaro, F.
Ungaro, F. Quaglia, Biomacromolecules 12 (2011) 4221–4229.[34] M. Morell, M. Erber, X. Ramis, F. Ferrando, B. Voit, A. Serra, Eur. Polym. J. 46
(2010) 1498–1509.[35] X. Fernández-Francos, A. Rybak, R. Sekula, X. Ramis, A. Serra, Polym. Int. 61
(2012) 1710–1725.[36] J. Xu, M. Holst, M. Wenzel, I. Alig, Polym. Chem. 46 (2008) 2155–2165.[37] S. Vyazovkin, C.A. Wight, Annu. Ver. Phys. Chem. 48 (1997) 125–149.[38] X. Fernández-Francos, J.M. Salla, A. Cadenato, J.M. Morancho, A. Serra, A.
Mantecón, X. Ramis, J. Appl. Polym. Sci. 111 (2009) 2822–2839.[39] H.-P. Yang, Z.-K. Chen, G. Yang, S.-Y. Fu, L. Y8, Polymer 43 (2002) 3168–3175.[40] Y. Eom, L.B. Boogh, V. Michaud, J.-A. Månson, Polym. Comp. 23 (2002) 1044–
1056.[41] K. Dusek, S. Lunak, L. Matejka, Polym. Bull. 7 (1982) 145–152.[42] X. Fernández-Francos, Eur. Polym. J. 55 (2014) 35–47.[43] A.N. Mauri, N. Galego, C.C. Riccardi, R.J.J. Williams, Macromolecules 30 (1997)
1616–1620.[44] X. Fernández-Francos, X. Ramis, A. Serra, J. Polym. Sci. Part A: Polym. Chem. 52
(2014) 61–75.[45] J.P. Pascault, H. Sautereau, J. Verdu, R.J.J. Williams, Thermosetting Polymers,
Marcel Dekker, New York, 2002.[46] L.E. Nielsen, Macromol. Sci. Rev. Macromol. Chem. C3 (1) (1969) 69–103.[47] X. Fernández-Francos, A. Rybak, R. Sekula, X. Ramis, F. Ferrando, L. Okrasa, A.
Serra, J. Appl. Polym. Sci. 128 (2013) 4001–4013.[48] B.B. Johnsen, A.J. Kinloch, R.D. Mohammed, A.C. Taylor, S. Sprenger, Polymer 48
(2007) 530–541.




















