
Utility of next-generation sequencing technologies for the efficient genetic resolution of...
10
Clin Genet 2015 Printed in Singapore. All rights reserved © 2015 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd CLINICAL GENETICS doi: 10.1111/cge.12573 Original Article Utility of next-generation sequencing technologies for the efficient genetic resolution of haematological disorders Zhang J., Barbaro P., Guo Y., Alodaib A., Li J., Gold W., Adès L., Keating B.J., Xu X., Teo J., Hakonarson H., Christodoulou J. Utility of next-generation sequencing technologies for the efficient genetic resolution of haematological disorders. Clin Genet 2015. © John Wiley & Sons A/S. Published by John Wiley & Sons Ltd, 2015 Next-generation sequencing (NGS) has now evolved to be a relatively affordable and efficient means of detecting genetic mutations. Whole genome sequencing (WGS) or whole exome sequencing (WES) offers the opportunity for rapid diagnosis in many paediatric haematological conditions, where phenotypes are variable and either a large number of genes are involved, or the genes are large making sanger sequencing expensive and labour-intensive. NGS offers the potential for gene discovery in patients who do not have mutations in currently known genes. This report shows how WES was used in the diagnosis of six paediatric haematology cases. In four cases (Diamond – Blackfan anaemia, congenital neutropenia (n = 2), and Fanconi anaemia), the diagnosis was suspected based on classical phenotype, and NGS confirmed those suspicions. Mutations in RPS19, ELANE and FANCD2 were found. The final two cases (MYH9 associated macrothrombocytopenia associated with multiple congenital anomalies; atypical juvenile myelomonocytic leukaemia associated with a KRAS mutation) highlight the utility of NGS where the diagnosis is less certain, or where there is an unusual phenotype. We discuss the advantages and limitations of NGS in the setting of these cases, and in haematological conditions more broadly, and discuss where NGS is most efficiently used. Conflict of interest The authors declare that they have no conflict of interest. J. Zhang a,b† , P. Barbaro c,d† , Y. Guo e† , A. Alodaib f,g,h , J. Li b , W. Gold f,g , L. Adès g,i,j , B.J. Keating e,k,l , X. Xu b,m,n , J. Teo c‡ , H. Hakonarson e,k,l‡ and J. Christodoulou f,g,j‡ a T-Life Research Center, Fudan University, Shanghai 200433, China, b Department of BioMedical Research, BGI-Shenzhen, Shenzhen 518083, China, c Haematology Department, The Children’s Hospital at Westmead, Sydney, Australia, d Cancer Research Unit, Children’s Medical Research Institute, Westmead, Australia, e The Center for Applied Genomics, The Children’s Hospital of Philadelphia, Philadelphia, PA, USA, f Genetic Metabolic Disorders Research Unit, Western Sydney Genetics Program, The Children’s Hospital at Westmead, Sydney, Australia, g Discipline of Paediatrics & Child Health, Sydney Medical School, University of Sydney, Sydney, Australia, h Department of Genetics, King Faisal Specialist Hospital and Research Centre, Riyadh, Saudi Arabia, i Clinical Genetics Department, Western Sydney Genetics Program, The Children’s Hospital at Westmead, Sydney, Australia, j Discipline of Genetic Medicine, Sydney Medical School, University of Sydney, Sydney, Australia, k Department of Pediatrics, The Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA, l Department of Human Genetics Graduate School of Public Health, University of Pittsburgh, Pittsburgh, PA, USA, m Shenzhen Key Laboratory of Genomics, Shenzhen, China, and n The Guangdong Enterprise Key Laboratory of Human Disease Genomics, Shenzhen, China † These authors contributed equally to this research. ‡ Co-last authors. 1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Utility of next-generation sequencing technologies for the efficient genetic resolution of...

Clin Genet 2015Printed in Singapore. All rights reserved
© 2015 John Wiley & Sons A/S.Published by John Wiley & Sons Ltd
CLINICAL GENETICSdoi: 10.1111/cge.12573
Original Article
Utility of next-generation sequencingtechnologies for the efficient genetic resolutionof haematological disorders
Zhang J., Barbaro P., Guo Y., Alodaib A., Li J., Gold W., Adès L., KeatingB.J., Xu X., Teo J., Hakonarson H., Christodoulou J. Utility ofnext-generation sequencing technologies for the efficient genetic resolutionof haematological disorders.Clin Genet 2015. © John Wiley & Sons A/S. Published by John Wiley &Sons Ltd, 2015
Next-generation sequencing (NGS) has now evolved to be a relativelyaffordable and efficient means of detecting genetic mutations. Wholegenome sequencing (WGS) or whole exome sequencing (WES) offers theopportunity for rapid diagnosis in many paediatric haematologicalconditions, where phenotypes are variable and either a large number ofgenes are involved, or the genes are large making sanger sequencingexpensive and labour-intensive. NGS offers the potential for gene discoveryin patients who do not have mutations in currently known genes. This reportshows how WES was used in the diagnosis of six paediatric haematologycases. In four cases (Diamond–Blackfan anaemia, congenital neutropenia(n= 2), and Fanconi anaemia), the diagnosis was suspected based onclassical phenotype, and NGS confirmed those suspicions. Mutations inRPS19, ELANE and FANCD2 were found. The final two cases (MYH9associated macrothrombocytopenia associated with multiple congenitalanomalies; atypical juvenile myelomonocytic leukaemia associated with aKRAS mutation) highlight the utility of NGS where the diagnosis is lesscertain, or where there is an unusual phenotype. We discuss the advantagesand limitations of NGS in the setting of these cases, and in haematologicalconditions more broadly, and discuss where NGS is most efficiently used.
Conflict of interest
The authors declare that they have no conflict of interest.
J. Zhanga,b†, P. Barbaroc,d†,Y. Guoe†, A. Alodaibf,g,h, J. Lib,W. Goldf,g, L. Adèsg,i,j,B.J. Keatinge,k,l, X. Xub,m,n,J. Teoc‡, H. Hakonarsone,k,l‡ andJ. Christodoulouf,g,j‡
aT-Life Research Center, FudanUniversity, Shanghai 200433, China,bDepartment of BioMedical Research,BGI-Shenzhen, Shenzhen 518083,China, cHaematology Department, TheChildren’s Hospital at Westmead, Sydney,Australia, dCancer Research Unit,Children’s Medical Research Institute,Westmead, Australia, eThe Center forApplied Genomics, The Children’sHospital of Philadelphia, Philadelphia, PA,USA, fGenetic Metabolic DisordersResearch Unit, Western Sydney GeneticsProgram, The Children’s Hospital atWestmead, Sydney, Australia, gDisciplineof Paediatrics & Child Health, SydneyMedical School, University of Sydney,Sydney, Australia, hDepartment ofGenetics, King Faisal Specialist Hospitaland Research Centre, Riyadh, SaudiArabia, iClinical Genetics Department,Western Sydney Genetics Program, TheChildren’s Hospital at Westmead, Sydney,Australia, jDiscipline of Genetic Medicine,Sydney Medical School, University ofSydney, Sydney, Australia, kDepartmentof Pediatrics, The Perelman School ofMedicine, University of Pennsylvania,Philadelphia, PA, USA, lDepartment ofHuman Genetics Graduate School ofPublic Health, University of Pittsburgh,Pittsburgh, PA, USA, mShenzhen KeyLaboratory of Genomics, Shenzhen,China, and nThe Guangdong EnterpriseKey Laboratory of Human DiseaseGenomics, Shenzhen, China
†These authors contributed equally to thisresearch.
‡Co-last authors.
1

Zhang et al.
Key words: congenital neutropenia –Diamond–Blackfan anaemia – Fanconianaemia – juvenile myelomonocyticleukaemia – macrothrombocytopenia –whole exome sequencing
Corresponding author: Prof JohnChristodoulou, Western SydneyGenetics Program, Children’s Hospitalat Westmead, Locked Bag 4001,Westmead, NSW 2145, Australia.Tel.: +61 2 9845 3452;fax: +61 2 9845 1864;e-mail:[email protected] Hakon Hakonarson, Center forApplied Genomics, Children’s Hospitalof Philadelphia, Philadelphia, PA 19104,USA.Tel.: +1 267 426 0088;fax: +1 267 426 0363;e-mail: [email protected] Juliana Teo, HaematologyDepartment, Children’s Hospital atWestmead, Locked Bag 4001,Westmead, NSW 2145, Australia.Tel.: +61 2 9845 3296;fax: +61 2 9845 6854;e-mail: [email protected]
Received 9 November 2014, revisedand accepted for publication 12February 2015
Next-generation sequencing (NGS) has evolved overthe last decade to now, becoming a relatively affordableand reliable, high-throughput method for pathogenicmutation detection (2), and it affords the opportunityfor genetic diagnosis, gene discovery and allows for theexpansion of known phenotypes of many disorders (1,3–5). NGS, in the form of whole genome sequencing(WGS) or whole exome sequencing (WES), is ideal forthe diagnosis of many paediatric haematological con-ditions which have complex genotypic and phenotypicvariations. Diamond–Blackfan anaemia (DBA) andFanconi anaemia (FA) are two such conditions. DBAclassically presents in early childhood as a pure red cellaplasia; however, phenotypic variability is wide, rangingfrom mild macrocytosis to severe transfusion-dependentanaemia associated with additional congenital anomalies(6–8). There are a number of genes implicated in DBA,all of which are involved in ribosome genesis (9). Themost common gene (RPS19) accounts for a quarter ofpatients, while the remaining genes are relatively evenlyspread across the remaining patients where mutations areidentified. In a significant proportion of patients, thereis no mutation found in currently known genes, and inthese patients, NGS has been increasingly used for genediscovery (5). This in turn leads to improved understand-ing of normal cellular mechanisms and pathobiology,
which has the potential for more effective and targetedtherapies. FA classically presents in childhood with shortstature, facial dysmorphism, radial ray anomalies andbone marrow failure. However, it is now recognised thatFA has a broader phenotype with many non-classicalcases now being diagnosed (10, 11). Numerous genesinvolved in DNA repair have been implicated in itspathogenesis (12). Now, 16 genes are implicated in FAand the gene most commonly involved is large withmany exons, making Sanger-sequencing this gene rela-tively expensive. In genetically heterogeneous disorders,sequential sequencing of multiple genes can be time andlabour intensive as well as costly. Moreover, dependingon the strategy used, the mutation underlying the diseasemay not be identified.
WES and WGS also offer the chance of diagnosisin patients with atypical presentations, or who havehaematological manifestations as part of a more com-plex syndromic phenotype, particularly of geneticallyheterogeneous disorders (4). In other conditions, suchas cyclic neutropenia, or severe congenital neutrope-nia (SCN), where there are only a few, small genesknown to be involved, standard Sanger sequencingusing small panels may still have advantage overNGS, because of the low cost and relative ease of dataanalysis.
2

NGS for genetic diagnosis of haematological disorders
Here, we present six cases where NGS technologyhas been used to confirm the diagnosis of a number ofpaediatric haematological disorders and then discuss thebenefits and limitations of this approach.
Methods
Patient selection
Patients treated at the Children’s Hospital at Westmeadwith haematological disorders were selected to undergoWES where there was a high clinical suspicion of anunderlying genetic cause for their conditions. Consentwas obtained from the family members to undergo WESand also to be involved in publication. Clinical informa-tion was collected through review of medical records andpathology databases. Two additional families, includinga family with two siblings with macrothrombocytopeniaand craniocynostosis, and a family with an autosomaldominant short telomere syndrome, were also analysedusing WES. A novel disease gene, ACD, was identi-fied in the family with short telomere syndrome, whichhas been described elsewhere (13). In the siblings withmacrothrombocytopenia and craniosynostosis, a previ-ously reported heterozygous MYH9 mutation has beenidentified, as well as a heterozygous variation in a novelpotential candidate gene that could be the cause of thecraniosynostosis, which is currently under further inves-tigation. In addition, there were seven further familieswith various haematological conditions analysed usingWES, with no mutations identified. These families areundergoing further analysis using WGS and have notbeen included in this series.
Exome capture and sequencing
The Agilent SureSelect Human All Exon Kit [(in solu-tion) Agilent Technologies, Santa Clara, CA] was usedto carry out exome capture for the affected individualsand unaffected relatives in all the families included inthis study, following the manufacturer’s protocols. TheCovaris AFA (Covaris, Woburn, MA, USA) was used torandomly break genomic DNA samples into fragmentswith a fragment size of 150–200 bp, and adapters wereadded to both ends. The Agencourt AMPure SPRI beads(Beckman Coulter, Brea, CA) were used to purify theadapter-ligated templates with an insert size of around250 bp. Ligation-mediated polymerase chain reaction(LM-PCR) and SureSelect Biotinylated RNA Library[(BAITS) Agilent Technologies, Santa Clara, CA] wereused to amplify, purify and hybridise the DNA forenrichment. The non-hybridised fragments were washedout after 24 h. The capture LM-PCR products were usedto estimate the magnitude of enrichment using an Agi-lent 2100 Bioanalyzer, (Agilent Technologies) and thenpaired-end sequencing was performed on the Hiseq2000platform (Illumina, San Diego, CA), with read lengthsof 90 bp. Illumina base-calling software V.1.7 set at itsdefault parameters was used to process raw image filesfor base calling.
Exome data analysis
Bioinformatics for WES data analysis procedures aredescribed elsewhere (13). Specifically, we harnessedtwo independent procedures to analyse the exomedata. Pipeline 1 aligned fast files to the human refer-ence genome (NCBI build 37.1, UCSC hg19) with theBurrows–Wheeler alignment (BWA) (14), made variantcalls with Genome Analysis Tool Kit (version 1.4;MIT, Cambridge, MA) (15), and completed functionalannotation of the variants with Annovar (16) and SnpEff(17). In pipeline 2, the SOAPaligner 2.20 (18) was usedto align sequence reads in each individual to the hg19reference human genome sequence with a maximum oftwo mismatches. Reads that had duplicated start siteswere removed and the remaining reads which mappedon or near targets were collected for subsequent analysisand variant calling. Soapsnp software (19) was set at itsdefault parameters to call genotypes in target regions.Variants with the following requirements were extractedfor further analysis: phred-like quality ≥20, sequencingdepth of 4–200, and a distance between two adjacentSNPs of no less than 5. These variants were functionallyannotated and categorised into missense, nonsense,splice-site mutations, and synonymous and non-codingvariations.
The variants identified through both pipelineswere then further filtered to eliminate benign vari-ants with a frequency of >0.5% in dbSNP132(http://www.ncbi.nlm.nih.gov/SNP/) (20, 21), 1000Genomes (http://www.1000genomes.org/) or ESP6500(http://evs.gs.washington.edu/EVS/) databases. Then,the pathogenicity of candidate variants was predictedusing PolyPhen2 (22), SIFT (23), MutPred (24) andPanther (21), and finally the candidate variants werechecked against HGMD (2014 Quarter 3 version) toidentify published mutations. A flow chart describingthe approaches used is outlined in Figure 1.
PCR and Sanger sequencing was used to validate themutations identified through WES, and the informationon the method for these techniques can be found in theTable S1 (Supporting Information).
Results
Table 1 summarises the molecular genetic findings inthese six cases, and pedigrees can be found in Figure S1.
Case 1
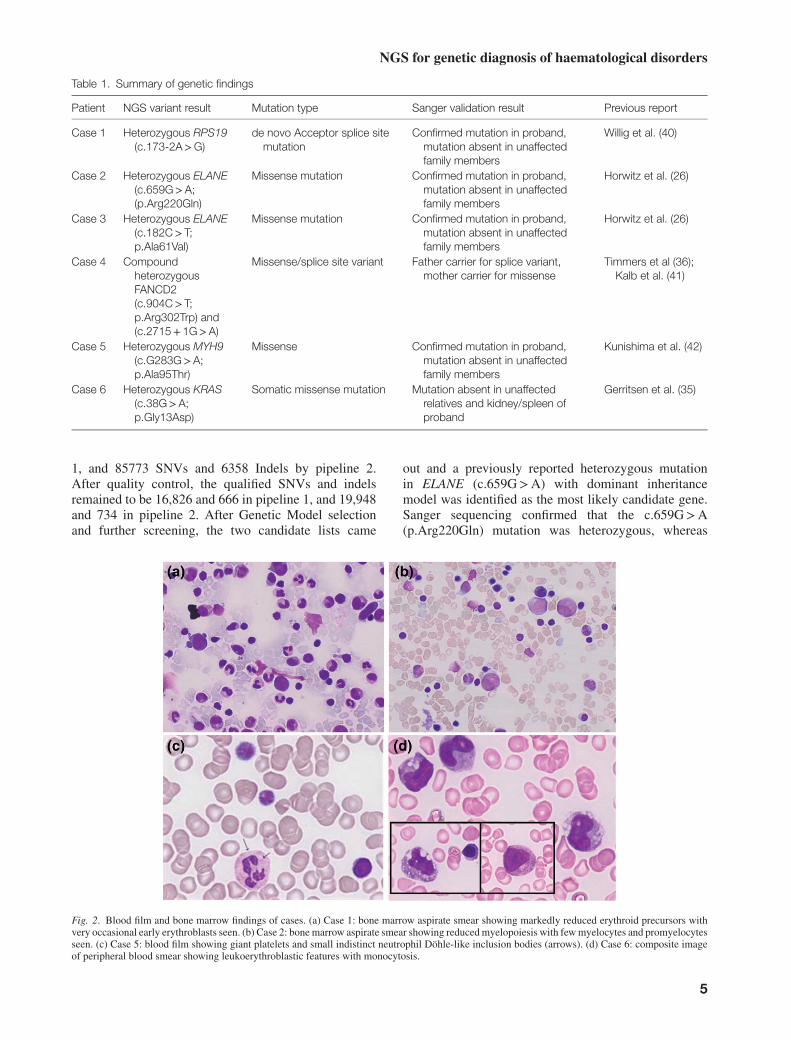
The patient was a female and the youngest of threechildren born to non-consanguineous parents ofBurmese decent. She presented at 7 months of age withnon-regenerative macrocytic anaemia (haemoglobin59 g/l, MCV 99 fl, reticulocyte 0.6%) and failure tothrive. Bone marrow findings of pure red cell aplasia(Figure 2a), elevated red cell adenosine deaminaseof 3.8 IU/gHb (reference range 0.6–1.6 IU/gHb) andsteroid-responsiveness led to the diagnosis of DBA.Normal haemoglobin level has been maintained onlow-dose prednisone.
3

Zhang et al.
Fig. 1. Flow chart indicating pathway used for WES analysis. Two independent pipelines were used to analyse the exome data. Pipeline 1 applied bwa,gatk, Annovar and SnpEff softwares, while pipeline 2 using Soapaligner, Soapsnp and in-house script to do the alignment, variants calling andannotation. Then, intronic and synonymous variants were eliminated. The variants identified through both pipelines were further filtered by frequencyof >0.5% in dbSNP, 1000 Genomes Project and ESP6500 database. The pathogenicity of candidate variants was predicted using PolyPhen2, SIFT,MutPred and Panther and finally the candidate variants were checked against HGMD to identify reported mutations.
We sequenced the exomes of five individuals from thefamily. An average of 60.3 Mb of sequence reads wasgenerated per individual. A total of 3.0 Gb of mappablesequence with an average 66-fold coverage was achievedafter discarding duplicated reads. On average, ∼45.576SNVs and ∼7705 indels were called for each exome bypipeline 1, while ∼86926 SNVs and ∼6389 indels werecalled by pipeline 2. After filteration, ∼16580 SNVs and618 indels were retained as qualified for further analysisin pipeline 1 and over 23780 SNVs and ∼755 indels inpipeline 2. We implemented further screening strategyand generated a list of candidate variants. Finally, weidentified a previously reported heterozygous acceptorsplice site mutation in RPS19 (c.173-2A>G) with denovo model of inheritance. RPS19 was selected as themost likely causal gene.
Sanger sequencing confirmed a heterozygous acceptorsplice site mutation in the patient, with the unaf-fected parents and sisters having only the wild-typesequence. This acceptor splice site mutation was pre-dicted to cause three amino acid deletion in exon 4.Sequencing of cDNA from the patient confirmed thedeletion (p.Ala58Thr60del) located at the start of exon4 (Fig. S4). In addition, the RPS19 mRNA expression
level was analysed from the patient’s cDNA using quan-titative reverse transcription PCR (RT-PCR) and wasfound to be no different from a normal control and herunaffected mother (data not shown).
Case 2
This male patient was the only child of unrelated Cau-casian parents. He presented with recurrent fevers,impetigo and mouth ulcers from 4 months of age asso-ciated with severe neutropenia. Twice weekly full bloodcounts showed persistent non-cyclical severe neutrope-nia, ranging from 0.2 to <0.05 x 109/l). Bone marrowexamination revealed reduced and markedly left-shiftedgranulopoiesis (Figure 2b), without cytogenetic abnor-mality. His clinical course was consistent with thediagnosis of severe congenital neutropenia (SCN) andhe commenced treatment with granulocyte-colony stim-ulating factor (GCSF) at 4 years of age with an excellentresponse.
An average of 63.3 Mb of sequence data was gen-erated with each exome covered by at least 74 timesafter sequencing the patient and his parents. On average,44865 SNVs and 7519 indels were identified by pipeline
4
NGS for genetic diagnosis of haematological disorders
Table 1. Summary of genetic findings
Patient NGS variant result Mutation type Sanger validation result Previous report
Case 1 Heterozygous RPS19(c.173-2A>G)
de novo Acceptor splice sitemutation
Confirmed mutation in proband,mutation absent in unaffectedfamily members
Willig et al. (40)
Case 2 Heterozygous ELANE(c.659G>A;(p.Arg220Gln)
Missense mutation Confirmed mutation in proband,mutation absent in unaffectedfamily members
Horwitz et al. (26)
Case 3 Heterozygous ELANE(c.182C>T;p.Ala61Val)
Missense mutation Confirmed mutation in proband,mutation absent in unaffectedfamily members
Horwitz et al. (26)
Case 4 CompoundheterozygousFANCD2(c.904C>T;p.Arg302Trp) and(c.2715+ 1G>A)
Missense/splice site variant Father carrier for splice variant,mother carrier for missense
Timmers et al (36);Kalb et al. (41)
Case 5 Heterozygous MYH9(c.G283G>A;p.Ala95Thr)
Missense Confirmed mutation in proband,mutation absent in unaffectedfamily members
Kunishima et al. (42)
Case 6 Heterozygous KRAS(c.38G>A;p.Gly13Asp)
Somatic missense mutation Mutation absent in unaffectedrelatives and kidney/spleen ofproband
Gerritsen et al. (35)
1, and 85773 SNVs and 6358 Indels by pipeline 2.After quality control, the qualified SNVs and indelsremained to be 16,826 and 666 in pipeline 1, and 19,948and 734 in pipeline 2. After Genetic Model selectionand further screening, the two candidate lists came
out and a previously reported heterozygous mutationin ELANE (c.659G>A) with dominant inheritancemodel was identified as the most likely candidate gene.Sanger sequencing confirmed that the c.659G>A(p.Arg220Gln) mutation was heterozygous, whereas
Fig. 2. Blood film and bone marrow findings of cases. (a) Case 1: bone marrow aspirate smear showing markedly reduced erythroid precursors withvery occasional early erythroblasts seen. (b) Case 2: bone marrow aspirate smear showing reduced myelopoiesis with few myelocytes and promyelocytesseen. (c) Case 5: blood film showing giant platelets and small indistinct neutrophil Döhle-like inclusion bodies (arrows). (d) Case 6: composite imageof peripheral blood smear showing leukoerythroblastic features with monocytosis.
5

Zhang et al.
sequencing of his parents showed only wild-typesequence.
Case 3
This male patient presented with recurrent neutropenia,mouth ulcers and bacterial infections including otitismedia, cellulitis and pneumonia in the first 3 years oflife. Twice weekly full blood counts showed cyclicalneutropenia every 3–4 weeks (Figure S2). Family historywas significant with his mother suffering recurrent mouthulcers in childhood and a maternal great-aunt dyingfrom methicillin-resistant Staphylococcal aureus sepsisassociated with mastitis and agranulocytosis at 21 yearsof age. Case 3 was treated with GCSF from 8 years ofage with good clinical response.
After sequencing the exomes of four individualsin the family, we obtained an average of 61.4 Mb ofsequence data and 67-fold sequencing depth. The totalSNVs and indels were 46,588 and 7913 for pipeline 1,while 88,648 and 6263 for pipeline 2 on average. Afterquality filtering, there were 16,915 SNVs and 643 indelsfor pipeline 1, and 23,770 SNVs and 765 indels forpipeline 2. A reported heterozygous mutation in ELANE(c.182C>T; p.Ala61Val) with dominant inheritancemodel was found in our candidate lists after screening.Sanger sequencing confirmed that the mutation washeterozygous, whereas sequencing of both parents andunaffected sibling showed only the wild-type sequence.
Case 4
This male patient was the only child of non-consanguineous Caucasian parents. There was no sig-nificant family history although the patient’s father hasunilateral thenar hypoplasia. Bilateral radial ray abnor-malities were identified on antenatal ultrasound. At birth,his weight and length were below the 1st percentile, andhe had facial dysmorphism (small ears, hypotelorism),narrow external auditory canals and chordee in additionto absent radii and thumbs (Figure S3). No cardiac orrenal tract abnormalities were identified, and the fullblood count was normal. Conventional karyotype wasnormal, however, lymphocyte cultures had significantlyincreased chromosome breakage in the presence ofclastogenic drugs (mitomycin C and diepoxybutane).These findings together with the clinical features wereconsistent with the diagnosis of FA.
Three individuals from the family were subjected toexome sequencing. An average of 61.0 Mb of sequencedata was generated for each individual with ∼70-foldcoverage. There were totally 44,748 SNVs and 7421indels for pipeline1 and 88,852 SNVs and 6324 indelsfor pipeline 2. When the low-quality variants werediscarded and the numbers changed to 16,962 SNVsand 623 indels for pipeline 1 and 24,191 SNVs and 723indels for pipeline 2. A candidate list was generatedafter further screening and reported that compoundheterozygous mutations in FANCD2; (c.904C>T;p.Arg302Trp) and (c.2715+ 1G>A) were identified.
Sanger sequencing confirmed the segregation of thecompound heterozygous mutations with the father car-rying the splicing mutation, whereas the mother was acarrier for the missense mutation.
Case 5
This 16-year-old female was the only child of anunrelated Caucasian/Indonesian couple. Both parentshave normal platelet counts and morphology, and werephenotypically normal. The patient has macrothrombo-cytopenia (platelet count. 40–50× 10^9/l) with largeand giant platelets on blood films and small, Döhle-likeinclusion bodies in her neutrophils (Figure 2c). Inaddition, she had facial dysmorphism (dolichocephaly,thick curly hair, upslanting palpebral fissures, high nasalbridge, broad globular nasal tip, short philtrum andpointed chin), short stature, microcephaly, epilepsy,severe global and cognitive retardation. Expression ofplatelet glycoproteins Ib (CD42b) and secretion of denseand alpha granules were found to be normal using flowcytometry. Her bleeding phenotype was clinically milduntil menarche at 9.5 years of age, when menorrhagiawas difficult to manage despite platelet transfusions,antifibrinolytics and hormonal therapy. She eventuallyunderwent a hysterectomy at 13 years of age.
Subtelomere FISH and CGH microarray identifieda previously reported benign copy number variant,shared with her phenotypically normal father, whichhas not been reported in association with an abnormalphenotype.
The patient and her mother underwent WES (pater-nal DNA was not available at the time of WES). Foreach individual, an average 65-fold sequencing depthwas achieved. A total of 42,038 SNVs and 6837 indelswere identified for pipeline 1, and 85,134 SNVs and6086 indels for pipeline 2. Following quality control,16,865 SNVs and 625 indels were qualified for pipeline1, and 21,004 SNVs and 719 indels for pipeline 2. Wethen followed the dominant inheritance screening strat-egy and found a reported heterozygous missense muta-tion in MYH9 (c.283G>A; p.Ala95Thr). This mutationin MYH9 was confirmed in the patient using Sangersequencing, with only the wild-type sequence found inboth parents.
Case 6
This female patient presented at 5 months of agewith petechiae, organomegaly and thrombocytopenia(platelet count 64x109/l) during a viral illness. Overthe next 12 months she had a number of brief hospi-tal admissions with fevers (without infective focus),persistent splenomegaly, anaemia (Hb 85 – 115 g/l),thrombocytopenia (35 – 108 x 109/l), intermittentmonocytosis (0.6–2.9× 109/l) and variable neutropenia(0.6–1.5× 109/l). She had one prolonged admissionwith a pericardial effusion, which was suspected tobe infective in origin, although no organism was iden-tified. Bone marrow examination showed left shifted
6

NGS for genetic diagnosis of haematological disorders
granulopoiesis and cytogenetic analysis was normal.There was no increased chromosomal breakage notedin response to clastogenic drugs in cultured peripherallymphocytes and foetal haemoglobin was 7% (withinnormal limits for age). Skin biopsy of the petechialrash was non-diagnostic, showing very mild perivas-cular lymphohistiocytic inflammatory infiltrate in thesuperficial dermis. Multiple immune and metabolicinvestigations did not yield a diagnosis.
At 20 months of age, she was admitted with feverand diarrhoea. Hepatosplenomegaly had progressed,with the tip of the spleen reaching below the level ofthe umbilicus. Anaemia and thrombocytopenia weremore pronounced (Hb, 65 g/l; platelet count, 37x109/l),but total white cell, neutrophil and monocyte countswere normal. A bone marrow aspirate was blooddilute; however, the trephine biopsy was hypercel-lular with histiocytic infiltrate (CD68 positive) andreduced megakaryocytes. Lymph node biopsy wasnon-diagnostic although phagocytosing histiocytes werenoted. A diagnosis of haemophagocytic lymphohistio-cytosis (HLH) was entertained, and corticosteroids werecommenced. She developed a coagulopathy requiringtransfusion support, serum ferritin was elevated (414ug/l; normal range, 10–150), but liver enzymes andplasma lipids remained normal. Natural killer (NK)cell function was slightly reduced in comparison withnormal controls. The monocyte count increased duringthe admission to a peak of 7.8× 109/l, associated witha leukoerythroblastic blood picture (Figure 2d). Shedeveloped rapid cardiorespiratory deterioration precipi-tated by pulmonary haemorrhage, and despite maximumsupport, died on the 10th day of the admission. Duringautopsy, there was massive hepatosplenomegaly andextensive lymphohisitocytic infiltrate (CD163 and CD68positive) with erythrophagocytosis in the liver and lymphnodes. There was also extensive fibrosis in the liver, andextramedullary haematopoiesis in the liver and spleen.
Genetic testing for primary HLH did not identify muta-tions in PRF1, UNC13d or STX11. Exome sequencingwas performed on four family members, with an aver-age of 62-fold depth after removing duplicated reads.Following quality control, 16,524 qualified SNVs and635 qualified indels were retained from pipeline 1, and21,894 SNVs and 726 indels from pipeline 2. Apply-ing a dominant inheritance screening strategy, we iden-tified a previously reported heterozygous mutation inKRAS (c.38G>A) as the most promising candidate. Theheterozygous c.38G>A (p.Gly13Asp) KRAS missensemutation was confirmed using Sanger sequencing in thepatient and homozygous wild type in the parents andunaffected sister. This KRAS mutation was absent in theDNA extracted from post-mortem kidney and spleen tis-sues from the proband, indicative of somatic mosaicismfor the mutation.
Discussion
These six cases highlight the potential clinical appli-cation of NGS as a diagnostic tool in haematology. Intotal, 8 of 15 families were identified with mutations
using WES. This rate of detection is very acceptableconsidering many of these patients had atypical pheno-types or had undergone previous genetic testing with nomutation identified. Patients with no mutation identifiedusing WES are currently undergoing WGS with thehope of increasing mutation detection even further. Thisapproach has both advantages and also limitations.
A previously reported splicing mutation in the RPS19gene (c.173-2A>G) (9) detected using WES confirmedthe clinical diagnosis of DBA in case 1. Mutationsare most commonly found in RPS19 accounting for25% of patients with DBA (6, 9). In this case, WESmay not have been the most efficient means of geneticdiagnosis, as RPS19 would have been among the first tobe sequenced using a standard panel sequencing strategy.However, in the remaining patients, there are at least tenadditional genes implicated, with no mutations detectedin these known genes in ∼40% of patients. Thus, WESor WGS offers advantages over sequential sequencing inthe majority of patients with DBA and also the potentialof novel disease gene discovery.
For cases 2 and 3 with SCN and cyclic neutropenia, nomutations were detected using routine Sanger sequenc-ing of the ELANE gene performed through a researchlaboratory. However, previously reported heterozygousELANE mutations in case 2 (c.659G>A: p.Arg220Gln)(25) and in case 3 (c.182C>T: p.Ala61Val) (26) weredetected in both patients by WES, but not in either set ofparents, suggesting that these were de novo mutations,although germline mosaicism cannot be excluded. Itis unclear why the research laboratory missed thesemutations, but we cannot exclude pre-analytical (samplemix-up), analytical (performance of the PCR or sequenc-ing) or post-analytical (interpretation of the sequencingresults) errors. Sanger sequencing is appropriate formost patients as ELANE is the most common geneimplicated in both SCN and cyclic neutropenia. In con-genital neutropenic patients without ELANE mutations,WES or WGS offer benefits, as more genes are nowimplicated in SCN, and there is still a subset of patientswhere no gene is identified on sequencing known genes.
In case 4, a routine sequencing strategy prioritising themore common genes was undertaken with no mutationidentified in the FANCA, FANCC, FANCE, FANCFand FANCG genes. Previously reported compoundheterozygous mutations (c.904C>T: p.Arg302Trp)(27) and (c.2715+ 1G>A) (28) in the FANCD2 genewere detected using WES. FANCD2 is one of the rarergenes implicated in FA, found in ∼3% of patients (12).Over 95% of patients with FA will have a mutation inone of five known genes – FANCA, FANCC, FANCE,FANCF and FANCG. With WES or WGS, these genescan be analysed simultaneously and offers advantagesin sequencing the most common gene, FANCA, whichis a large gene with 43 exons. Thus, Sanger sequenc-ing of these five genes may be comparable in termsof cost and resources with that of WES with currenttechniques; however, WES offers the added advantageof finding rare mutations in the remaining 11 genesimplicated in FA. As an example, one commercialcompany offers an NGS panel for FA involving 15
7

Zhang et al.
genes at a cost of $3290, while the cost of Sangersequencing the four most common genes totals $5460(https://www.preventiongenetics.com). Panel sequenc-ing is another option, and may prove more time effectiveand cost-efficient if the mutation is found in one of themore common genes involved. In FA, genetic diagnosisis preferable to complementation analysis for a numberof reasons. First, it allows carrier detection in parents,which enables more informed family-planning decisionsto be made. Second, complementation analysis usingcell fusions and retroviral vectors is time-consuming,and requires specialised laboratory services, whichare not routine in most diagnostic laboratories today.Finally, NGS in FA is also useful in the small number ofpatients who have somatic mosaicism or who undergohaematopoietic reversion, where the ‘gold standard’chromosome fragility analysis may not be reliable(29, 30).
The complex phenotype of case 5 was not diagnos-tic of a previously recognised dysmorphic syndrome,nor was it typical of the clinical features associatedwith macrothrobocytopenia. The finding of a previ-ously reported mutation (c.283G>A: p.Ala95Thr) inMYH9 gene (31) through WES explains the patient’smacrothrombocytopenia. Further analysis of the WESdata is ongoing, which may lead to the identification ofa candidate gene to account for the non-haematologicalphenotype. This raises a potential limitation of WES, inthat mutations identified may be interpreted as causing abroad and unusual phenotype, where in fact, there maybe more than one gene effected. In this case, it wouldseem inappropriate to attribute the non-haematologicalphenotypic features (dysmorphism, epilepsy and severemental retardation) to the MYH9 mutation, as similarphenotypic features have, thus far, not been associ-ated with MYH9. Explanation of these features maylay with variants detected in other genes using WES;however, those variants require very careful scrutinyin order to confirm pathogenicity or rule them out asmerely single-nucleotide polymorphic variants (SNPs).Mutations in MYH9 are a frequent cause of macrothrobo-cytopenia with neutrophil inclusions, as part of the MayHegglin anomaly, or other associated disorders. Thus,sequencing MYH9 in this clear phenotype is an appro-priate first step in obtaining a genetic diagnosis. As withFANCA, MYH9 is a large gene with many exons (32);thus, the challenges and expense of Sanger sequenc-ing this gene may tip the balance in favour of WES,especially as the cost of this technique falls with time.Again as an example, the cost of sequencing this genealone would be $1750 through a commercial company(https://www.preventiongenetics.com).
Case 6 presented a diagnostic dilemma through-out her clinical course. While there was histiocyticinfiltration with evidence of haemophagocytosis inlymph nodes (antemortem) and more extensively onpost-mortem examination, she did not fulfil the diag-nostic criteria for HLH. A previously reported mutationin the KRAS gene (c.38G>A: p.Gly13Asp) (33) wasdetected post-mortem by WES in DNA extracted fromblood, but not in DNA extracted from kidney or spleen
tissue collected post-mortem, indicative of somaticmosaicism, and lending support to a diagnosis of juvenilemyelomonocytic leukaemia (JMML) rather than HLH.The diagnosis of JMML and HLH rely on a constellationof non-specific clinical and laboratory features, makingdiagnosis difficult in atypical presentations. Monocy-tosis generally accompanies leucocytosis in JMML,and is a key diagnostic feature; however, it was onlyintermittently noted throughout the clinical course ofthis patient. The finding of haemophagocytosis in tissuesamples, while helpful, is not specific for HLH, and it hasbeen reported in a number of cases of JMML (34, 35).
Patient 6 was an example of an atypical clinicalpresentation where a correct diagnosis was not madein life despite multiple investigations. The decision tocontinue searching for a genetic diagnosis post mortemwas made to exclude familial causes of HLH whichwould have major family planning implications for theparents. In terms of cost, the three genes tested, wouldhave costed $2610 if performed commercially, whilean NGS panel involving seven genes involved in HLHwould have costed $1690. Indeed, in this case, both ofthese approaches would not have yielded a diagnosis,yet, depending on the NGS technique used, other genesmay have been available for analysis from the initialdata, which may have identified the KRAS mutation.Mutations in the RAS pathway have been found inover 70% of patients with JMML and are extremelyuseful for diagnostic purposes, particularly in those withatypical presentations. Inclusion of these mutations isbeing considered in the revised WHO diagnostic criteriafor JMML (36, 37). Early identification of the KRASmutation would have confirmed the diagnosis of JMMLand led to appropriate treatment decisions for the patient.WES also has potential to identify secondary mutationsinvolved in leukemogenesis, disease progression andprognosis, which impact overall and transplant-freesurvival and may be important in management decisions(38). These findings will help to better understand themolecular mechanisms of JMML and may influencetreatment decisions.
In haematology, there are a number of examples wherenovel mutations in known genes and mutations in newgenes have been identified using NGS (1, 13, 39).Examples of these include mutations in XRCC2 foundin FA patients (32) and GATA1 mutations in DBA.The identification of a GATA1 mutation in two siblingswith the DBA phenotype who did not have mutationsin known ribosomal proteins has expanded the geneticheterogeneity of this disorder, which had been thoughtto only involve genes that are involved in ribosomehomeostasis (5).
Some limitations of WES include ‘incidental’ iden-tification of potentially pathogenic variants in genesunrelated to the disease phenotype in question, and theidentification of novel or private mutations, which havenot been previously reported. The discovery of BRCA2mutations when searching for other disorders is oneexample of unrelated incidental findings which wouldhave future implications for the individual and theirrelatives. Deciding how to manage these incidentally
8

NGS for genetic diagnosis of haematological disorders
detected variants raises many ethical issues surroundingconsent for this type of testing and which variants shouldor should not be disclosed to patients and their fami-lies. The process of investigating novel variants foundby NGS techniques may be another limitation of thisapproach. This process is labour-intensive, involvingfunctional studies which are often only available inresearch laboratories. This can foster stronger linksbetween clinicians and basic science researchers, andcan yield novel findings when these collaborations exist(13). However, these efforts may be difficult due to thelack of existing relationships or competing time interestsof researchers and clinicians. The process of investigat-ing new variants may become simpler in the future withadvances in bioinformatics software and expansion ofwell-curated SNP and locus specific mutation databases.
In conclusion, we report the successful implementationof NGS technologies for the diagnosis of a range ofgenetic haematological disorders. This was a researchproject; therefore, a comprehensive cost-benefit analysisof WES is beyond the scope. In many conditions,where the phenotype is clear and the number of genesare limited, for example cyclic neutropenia, Sangersequencing using a standard Panel approach remainsthe most clinically useful genetic technique, this isespecially true if the laboratory performing these panelshas extensive experience, with reliable results. However,it is our experience that in disorders such as FA andDBA, where there are large numbers of genes involved,where common genes are large or have multiple exons, orin patients with an atypical phenotype, the developmentof diagnostic algorithms coupling WES or WGS withexisting technologies, such as chromosomal fragilitystudies for the marrow failure syndromes, will result inmore efficient genetic diagnosis, with major implicationsfor guiding disease-specific management and accurategenetic counselling.
Supporting Information
Additional supporting information may be found in the onlineversion of this article at the publisher’s web-site.
Acknowledgements
This research was supported by a PhD Scholarship to A. A.provided by the Academic and Training Affairs at King FaisalSpecialist Hospital and Research Center (KFSHRC) and theMinistry of Higher Education (MOHE) in Riyadh, Saudi Ara-bia, and by grants from the Shenzhen Municipal Governmentof China (NO.CXZZ20130517144604091), the Shenzhen KeyLaboratory of Genomics (NO.CXB200903110066A) and theGuangdong Enterprise Key Laboratory of Human Disease Genomics(NO.2011A060906007). The study was also supported by Insti-tutional Development Funds to Dr. Hakon Hakonarson in theCenter for Applied Genomics (CAG) at the Children’s Hospital ofPhiladelphia (CHOP).
Author Contributions
J. Z., P. B., and Y. G. were equally involved in writingthe manuscript. J. C., J. T., Y. G., H. H. and L. A.
were equally involved in revising it critically and to theacquisition, analysis and interpretation of data. A. A.,J. L., W. G., L. A., and B. K. Substantially contributedto research design, or the acquisition, analysis andinterpretation of data.
Synopsis
We demonstrate that the use of NGS technologies isa rapid strategy enabling the resolution of the geneticbasis of a range of Mendelian haematological disordersfor which there may be multiple genetic causes, therebyfacilitating disease-specific management and more accu-rate genetic counselling.
Compliance with ethics guidelines
All procedures followed were in accordance with theethical standards of the responsible committee on humanexperimentation (institutional and national) and with theHelsinki Declaration of 1975, as revised in 2000 (1).Informed consent was obtained from all patients forbeing included in the study.
References1. Zheng Z, Geng J, Yao RE et al. Molecular defects identified by whole
exome sequencing in a child with Fanconi anemia. Gene 2013: 530:295–300.
2. Bamshad MJ, Ng SB, Bigham AW et al. Exome sequencing as a tool forMendelian disease gene discovery. Nat Rev Genet 2011: 12: 745–755.
3. Le Guen T, Jullien L, Touzot F et al. Human RTEL1 deficiency causesHoyeraal-Hreidarsson syndrome with short telomeres and genome insta-bility. Hum Mol Genet 2013: 22: 3239–3249.
4. Cullinane AR, Vilboux T, O’Brien K et al. Homozygosity mapping andwhole-exome sequencing to detect SLC45A2 and G6PC3 mutations ina single patient with oculocutaneous albinism and neutropenia. J InvestDermatol 2011: 131: 2017–2025.
5. Sankaran VG, Ghazvinian R, Do R et al. Exome sequencing identifiesGATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest2012: 122: 2439–2443.
6. Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia inthe UK: clinical and genetic heterogeneity. Br J Haematol 2004: 125:243–252.
7. Kim SK, Ahn HS, Back HJ et al. Clinical and hematologic manifestationsin patients with Diamond Blackfan anemia in Korea. Korean J Hematol2012: 47: 131–135.
8. Pospisilova D, Cmejlova J, Ludikova B et al. The Czech NationalDiamond-Blackfan Anemia Registry: clinical data and ribosomal proteinmutations update. Blood Cells Mol Dis 2012: 48: 209–218.
9. Boria I, Garelli E, Gazda HT et al. The ribosomal basis ofDiamond-Blackfan anemia: mutation and database update. HumMutat 2010: 31: 1269–1279.
10. Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosisof Fanconi anemia in patients without congenital malformations: aninternational Fanconi Anemia Registry Study. Am J Med Genet 1997:68: 58–61.
11. Cavenagh JD, Richardson DS, Gibson RA, Mathew CG, Newland AC.Fanconi’s anaemia presenting as acute myeloid leukaemia in adulthood.Br J Haematol 1996: 94: 126–128.
12. de Winter JP, Joenje H. The genetic and molecular basis of Fanconianemia. Mutat Res 2009: 668: 11–19.
13. Guo Y, Kartawinata M, Li J et al. Inherited bone marrow failureassociated with germline mutation of ACD, the gene encoding telomereprotein TPP1. Blood 2014: 124: 2767–2774.
14. Li H, Durbin R. Fast and accurate short read alignment withBurrows-Wheeler transform. Bioinformatics 2009: 25: 1754–1760.
9

Zhang et al.
15. McKenna A, Hanna M, Banks E et al. The genome analysis toolkit: aMapReduce framework for analyzing next-generation DNA sequencingdata. Genome Res 2010: 20: 1297–1303.
16. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation ofgenetic variants from high-throughput sequencing data. Nucleic AcidsRes 2010: 38: e164.
17. Cingolani P, Platts A, Wang le L et al. A program for annotating andpredicting the effects of single nucleotide polymorphisms, SnpEff: SNPsin the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly2012: 6: 80–92.
18. Li R, Yu C, Li Y et al. SOAP2: an improved ultrafast tool for short readalignment. Bioinformatics 2009: 25: 1966–1967.
19. Li R, Li Y, Fang X et al. SNP detection for massively parallelwhole-genome resequencing. Genome Res 2009: 19: 1124–1132.
20. Kitts A, Phan L, Ward M, Bradley Holmes J. The database of shortgenetic variation (dbSNP), 2nd edn. Bethesda, MD: National Center forBiotechnology Information, 2013.
21. Mi H, Lazareva-Ulitsky B, Loo R et al. The PANTHER database ofprotein families, subfamilies, functions and pathways. Nucleic Acids Res2005: 33: D284–D288.
22. Adzhubei IA, Schmidt S, Peshkin L et al. A method and server for pre-dicting damaging missense mutations. Nat Methods 2010: 7: 248–249.
23. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affectprotein function. Nucleic Acids Res 2003: 31: 3812–3814.
24. Li B, Krishnan VG, Mort ME et al. Automated inference of molecularmechanisms of disease from amino acid substitutions. Bioinformatics2009: 25: 2744–2750.
25. Germeshausen M, Deerberg S, Peter Y, Reimer C, Kratz CP, BallmaierM. The spectrum of ELANE mutations and their implications in severecongenital and cyclic neutropenia. Hum Mutat 2013: 34: 905–914.
26. Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC. Mutationsin ELA2, encoding neutrophil elastase, define a 21-day biological clockin cyclic haematopoiesis. Nat Genet 1999: 23: 433–436.
27. Timmers C, Taniguchi T, Hejna J et al. Positional cloning of a novelfanconi anemia gene, FANCD2. Mol Cell 2001: 7: 241–248.
28. Smetsers S, Muter J, Bristow C et al. Heterozygote FANCD2 mutationsassociated with childhood T cell ALL and testicular seminoma. FamCancer 2012: 11: 661–665.
29. Lo Ten Foe JR, Kwee ML, Rooimans MA et al. Somatic mosaicism inFanconi anemia: molecular basis and clinical significance. Eur J HumanGenet 1997: 5: 137–148.
30. Dokal I, Chase A, Morgan NV et al. Positive diepoxybutane test in onlyone of two brothers found to be compound heterozygotes for Fanconi’sanaemia complementation group C mutations. Br J Haematol 1996: 93:813–816.
31. Althaus K, Greinacher A. MYH9-related platelet disorders. SeminThromb Hemost 2009: 35: 189–203.
32. Shamseldin HE, Elfaki M, Alkuraya FS. Exome sequencing reveals anovel Fanconi group defined by XRCC2 mutation. J Med Genet 2012:49: 184–186.
33. Takagi M, Shinoda K, Piao J et al. Autoimmune lymphoproliferativesyndrome–like disease with somatic KRAS mutation. Blood 2011: 117:2887–2890.
34. Urs L, Qualman SJ, Kahwash SB. Juvenile myelomonocytic leukemia:report of seven cases and review of literature. Pediatric Dev Pathol 2009:12: 136–142.
35. Gerritsen A, Lam K, Marion Schneider E, van den Heuvel-Eibrink M.An exclusive case of juvenile myelomonocytic leukemia in associationwith Kikuchi’s disease and hemophagocytic lymphohistiocytosis and areview of the literature. Leuk Res 2006: 30: 1299–1303.
36. Loh ML. Recent advances in the pathogenesis and treatment of juvenilemyelomonocytic leukaemia. Br J Haematol 2011: 152: 677–687.
37. Loh ML. Childhood myelodysplastic syndrome: focus on the approachto diagnosis and treatment of juvenile myelomonocytic leukemia. Hema-tology Am Soc Hematol Educ Program 2010: 2010: 357–362.
38. Sakaguchi H, Okuno Y, Muramatsu H et al. Exome sequencing identifiessecondary mutations of SETBP1 and JAK3 in juvenile myelomonocyticleukemia. Nat Genet 2013: 45: 937–941.
39. Schuster B, Knies K, Stoepker C et al. Whole exome sequencing revealsuncommon mutations in the recently identified Fanconi anemia geneSLX4/FANCP. Hum Mutat 2013: 34: 93–96.
40. Willig T, Draptchinskaia N, Dianzani I et al. Mutations in ribosomalprotein S19 gene and diamond blackfan anemia: wide variations inphenotypic expression. Blood 1999: 94 (12): 4294–4306.
41. Kalb R, Neveling K, Hoehn H et al. Hypomorphic mutations in the geneencoding a key Fanconi anemia protein, FANCD2, sustain a significantgroup of FA-D2 patients with severe phenotype. Am J Hum Genet 2007:80 (5): 895–910.
42. Kunishima S, Matsushita T, Kojima T et al. Identification of six novelMYH9 mutations and genotype–phenotype relationships in autoso-mal dominant macrothrombocytopenia with leukocyte inclusions. HumGenet 2001: 46: 722–729.
10




















