
UNIVERSITÉ LAVAL - CiteSeerX
263
DAO WEI ZHU Thèse Présentée à la Faculté des études supérieures de IIUniversité Laval pour l'obtention du grade de philosophiae Doctor (Ph. D.) Département de physiologie (Endocrinologie moléculaire) FACULTÉ DE MÉDECINE UNIVERSITÉ LAVAL QUÉBEC O Dao Wei Zhu, 1997
-
Upload
khangminh22 -
Category
Documents
-
view
3 -
download
0
Transcript of UNIVERSITÉ LAVAL - CiteSeerX

DAO WEI ZHU
T h è s e P r é s e n t é e
à la Faculté des études supérieures de IIUniversité Laval
pour l'obtention du grade de philosophiae Doctor (Ph. D.)
Département de physiologie (Endocrinologie moléculaire)
FACULTÉ DE MÉDECINE UNIVERSITÉ LAVAL
QUÉBEC
O Dao Wei Zhu, 1997

National Library 1*1 of Canada Bibliothèque nationale du Canada
Acquisitions and Aquisitions et Bibliographic Services services bibliographiques 395 Wellington Strwt 395. rue Wellington Ottawa ON K1A ON4 Ottawa ON K1A ON4 Canada canada
The author has granted a non- exclusive licence allowing the National Library of Canada to reproduce, loan, distribute or sell copies of this thesis in microfonn, paper or electronic formats.
The author retains ownership of the copyright in this thesis. Neither the thesis nor substantial extracts fiom it may be printed or otherwise reproduced without the author's permission.
L'auteur a accorde une licence non exclusive permettant à la Bibliothèque nationale du Canada de reproduire, prêter, distriher ou vendre des copies de cette thèse sous la forme de microfichelfh, de reproduction sur papier ou sur format électronique.
L'auteur conserve la propriété du droit d'auteur qui protège cette thèse. Ni la thèse ni des extraits substantiels de celle-ci ne doivent être imprimés ou autrement reproduits sans son autorisation.

Résumé Court
La 17B-hydroxystéroïde déshydrogénase estrogénique (17B-HSDI) responsable de la synthèse des œstrogènes actifs qui stimulent le canc du sein. Afin d'étudier cette enzyme importante, nous avons cristallisé déterminé la structure tridimensionnelle de cette dernière. En utilisant "Fast Protein Liquid Chromatography", nous avons purifié l'enzyme de fraction cellulaire soluble du placenta humain. Les différentes propriéi pour I'apoenzyme et l'holoenzyme de Ia 1713-HSDl ont été identifiées E des méthodes spectrophotométriques et fluorométriques. Nous avo utilisé la technique de diffusion en vapeur et avons obtenu des crista de 17B-HSD1 native de formes différentes. La structure de la 178-HSDI été par la suite déterminée. Ceci est la première réussite dans cristailisation et Ia détermination de la structure tridimensionneIle d enzymes humaines responsables de la conversion des stéroïdes. No avons déterminé la structure des cristaux du complexe 17%-HSD oestradiol et nous sommes présentement en train de compléter celle complexe 17B-HSD1-EM-139. Ces résultats nous aident à élucider 1 interactions enzyme-substrat et enzyme-inhibiteur ainsi que la relatic structure-fonction essentielle à la conception de ces inhibiteurs pour u utilisation thérapeutique contre le cancer du sein. De plus, nous avo développé I'étude de la cristallogénèse des protéines au cours des étud ci-haut mentionnées. Ces résultats montrent les mécanismes i
développement des cristaux de façon détaillée pouvant ainsi aider à cristallisation des autres déshydrogénases stéroïdiennes.

A bstract
Abstrac t
Human estrogenic 17B-hydroxysteroid dehydrogenase (178-HSD 1 ) responsible for the synthesis of active estrogens that stimulate the brea cancer. To study this pivota1 enzyme, we have crystallized 178-HSD1 ai
determined its three-dimensional structure. Using Fast Protein Liqu Chromatography, we have purified the I7B-HSD1 in the solub subcellular fraction of human placenta. Different optical properties of tl apoenzyme and holoenzyme were identified by spectrophotometry ai fluorometry. Using the vapor diffusion technique, we have obtainc crystals of the native 17B-HSDI in different forms. The structure of tl 17B-HSD1 has now been determined. This is the first report on tl successful crystallization and determination of the three-dimension structure of any steroid-converting enzyme from a human source. Tl structure of the complexe 178-HSDl/estradiol have now bet determined. At present, the structure of 17B-HSD 1 -EM-139 deterrnining. These results will g ive direct evidence for the interactic between enzyme-substrate, enzyme inhibitor and structure-functic relationships of the enzyme, contributing potentially to breast canc therapy. In the meantirne, these results show the detailed mechanism 1
crystals growth and are also useful for establishing a new method fl crystallization of other steroid dehydrogenases.

Résumé
La fraction cellulaire soluble du placenta humain constitue la sou naturelle la plus abondante de la 17B-hydroxystéroïde déshydrogén type 1 (170-HSD1). Cette enzyme est responsable de l'interconversion l'œstrone et l'œstradiol et, moins spécifiquement, du le DHEA et du le A
diol, un autre œstrogène découvert très récemment qui stimule développement du cancer du sein. Nous avons projeté de faire cristallisation de la 17B-HSD 1 afin d'obtenir sa structi tridimensionnelle.
En utilisant la "Fast Protein Liquid Chromatography", nous avc purifié la 170-HSD1 de la fraction cellulaire soluble du placenta h u m Les différentes propriétés pour l'apoenzyme et I'holoenzyme de la 1' HSDl ont été identifiées par des méthodes spectrophotométriques fluorométriques. Nous avons utilisé la technique de diffusion de vapl et avons obtenu des cristaux de 17B-HSDl native sous forme complexes enzyme-cofacteur, enzyme-substrat, enzyme-inhibiteur apoenzyme. Les cristaux ont le "groupe d'espace" CS et diffractent en 1.8 A et 2.2 A. Ceci est la première cristallisation une enzyme huma responsable de la conversion des stéroïdes.
En Janvier 1994, nous avons aussi obtenu des cristaux de la 1' HSDl complexée avec différents ligands (NADP+, œstradiol) microgravité 3 bord de la station spatiale Russe (MIR).
Nous avons également obtenu des diagrammes phase de pour cristallisation de la 170-HSD 1. Ces derniers permettent de compreni plus en profondeur le mécanisme de cristallogénèse. En utilisant diagrammes de phase, nous avons pu améliorer la qualité des cristaux obtenir des cristaux de la 178-HSDI complexée avec différents ligands.
Prochainement, nous allons déterminer les structures des cristaux la 176-HSD1 et des différents complexes. La structure tridimensionnel1

Résumé
de la 176-HSDl montre que la structure complète de cette enzyme es
similaire aux autres enzymes appartenant à la famille d déshydrogénases à courte chaîne contenant la séquence conservte Tyr-: X-X-Lys et possédant un résidu sérine dans le site actif. Cette structi diffère cependant des autres structures rencontrées chez 1 déshydrogénases à courte chaîne par l'insertion de deux motifs hélic tour-hélice.
Ces résultats vont nous aider à élucider les interactions enzyrr substrat et enzyme-inhibiteur ainsi que la relation structure-foncti essentielle à l'élaboration d'inhibiteurs de ca 17B-WSD1 pour u utilisation thérapeutique contre le cancer du sein.

Summary
Summary
The soluble fraction of human placenta is the richest natural souri of 178-hydroxysteroid dehydrogenase type 1 ( 17B-HSD 1 ). This enzyme responsible for the interconversion of estrone and estradiol and has lower activity for that of D E A and AS-diol, the latter is another estrogc discovered recently that stimulates the development of breast cancer. 1 study this pivotal enzyme, we have crystallized 178-HSD1 in order resolve its three-dimensional structure.
Using Fast Protein Liquid Chromatography, we have purified tl 170-HSD1 in the soluble subcellular fraction of human placenta. Differe optical properties of apoenzyme and holoenzyme were identified € spectrophotometry and fluorometry. Using the vapor diffusion techniqu we have obtained crystals of the native L7B-HSDI in different complt forms (enzyme-cofactor, enzyme-substrate, enzyme-inhibitor ar apoenzyme). The crystals belong to the space group CS and diffractic from 1.8 A to 2.2 A. This is the first report on the successf crystallization of any steroid-converting enzyme from a human source.
We have also crystallized the 1713-HSD1 under microgravity. ! January 1994, we have obtained crystals of 178-HSD 1 which complexf with different ligands (NADP+, estradiol) aboard the Russian MIR spac station.
The phase diagrams for the crystallization of 178-HSD1 wei
determined. These results are very useful for improving the cryst quality. Using the phase diagram, higher quality crystals were obtaine for complex of 17B-HSDl with different ligands.
Then the structure of the complexes of 178-HSD1 and with estradic have been determined. The 3D-structure of 17B-HSDl has shown that tl
overall structure of the enzyme is similar to the other enzymes in th

Summary
short-chain dehydrogenase family, with a conserved Tyr-X-X-X-L sequence and a serine residue in the active site. It is distinguished frc the other known structures reported for short-chain dehydrogenases the insertion of two helix-turn-helix motifs.
These results have given direct evidence for the enzyme-substra enzyme-inhibitor interactions and structure-function relationshi provide a strong of potent inhibitors for breast cancer therapy.

Acknowledgments
Acknowledgments It is rny pleasure to acknowledge my supervisor, Dr. S.-X. Lin, for h
competent direction, encouragement and enthusiasm in the whole proce, of my study. With Dr. S.-X. Lin's personal instructions and support, 1 w; able to accomplish this thesis and complete my Ph,D. studies in tï Laboratory of Molecular Endocrinology at the CHUL Research Centre ar Laval University.
1 would like to thank the Laboratory Director, Dr. F. Labrie, for h interest in my project of Ph. D.
1 would like to thank Dr. Xavier Lee for his patient direction c crystallization during my stay in National Research Council of Canada.
In addition, 1 would also like to acknowledge my collaborators: M F.Yang , for her aid in the rapid purification of 17B-HSDl; Dr. D. Ghosh, fc the crystal of 17B-HSD1-NADP+ analysis; Ms. J.-Z. Jin, for the work c study of optical properties of 17B-HSDl NADP+; Dr. M. Zhou and Mr. 1 Mao, for their collaboration in the crystallization of 17B-HSDl undi microgravity; Mr. Arezki Azzi and Dr. Peter H. Rehse, for their work c analysis of crystals; Dr. R. L. Campbell for his aid in crystallogenesis an X-ray analysis. Their help and advice during the experiments are greatl appreciated. Many other people deserve recognition for their usefi discussions. In particular, 1 would like to thank Dr. J. Y. Wang, Mr. Q. Ha; W. Qiu, and Dr. P. M. Rong.
1 would like to thank Ms. Isabelle Pineau for her diligent correctic of the French language version of the summary. 1 also thank Mr. B.-X. Xi for his diligent correction of English language version of the Summar: Introduction and Conclusion of my thesis as well as for his kind help i typing. 1 wish also to extend my thanks to Ms. Joyce Gardiner and othi secretaries, Medam Aline Douville, Elaine Leclerc, Josée Poulin, Hélèn Rodrigue and Lise Theriault, for al1 their kind collaboration during th past five years.
When completing my Ph.D. studies, 1 would especially like to expre! my sincerest appreciation to my wife Mrs. 2. J. Zhao, for her energeticall support during my graduate studies.
Finally, 1 would like to acknowledge the Laval University Foundatioi for a fellowship during the past three years of my Ph.D. study.

Publications
Publ icat ions
1. Refereed papers: (1) S.-X. Lin, F. Yang, J.-Z. Jin, R. Breton, D.-W. Zhu, V.-Lue. The, and Labrie (1992). Subunit identity of the dimeric 178-hydroxys teroi dehydrogenase from Human placenta. J. Biol. Chem. 267: 1618: 16187.
(2) F. Yang, P.-W. Zb, J.-Y. Wang, and S.-X. Lin (1992). Rap purification yielding highly active 178-hydroxysteroid dehydrogenas :application of hydrophobic interaction affinity fast protein liqui chromatogrophy. J. Chromatogr. 582: 7 1-76.
(3) J L W . Zhu, X. Lee, R. Breton, D. Ghosh, W. Pangborn, W. L. Dua and S.-X. Lin (1993). Crystallization and pretiminary X-Ray diffractio analysis of the complex of Human placental 178-hydroxysteroi dehydrogenase with NADP'. J. Mol. Biol., 234: 242-244.
(4) P.-W. Zhu, X. Lee, F. Labrie, and S.-X. Lin (1994). Crystal growi of human estrogenic l7B-hydroxysteroid dehydrogenase. A c ta Crystallogr. D 50: 550-555.
(5) p.-W. Zhu, T. Dahrns, K. WiIIis, A.G. Szabo, and X. Lee (1994 Crystallization and the preliminary crystallographic studies of th Azurin Pseudomonas fluorescens. Archives of Biochemistry an Biophysics 308: 469-470.
(6) D-, J.-2. Jin, and S.-X. Lin (1995). Human Placenta1 171 hydroxysteroid dehydrogenase: Optical properties of its complex witl N ADP'. J. Steroid Biochem. Molec. Biol. 52: 77-81.

Publications
(7) D. Ghosh, V. 2. Pletnev, D.-W. Zhu, 2. Wawrzak, W. L. Duax, \;
Pangborn, F. Labrie, and S.-X. Lin (1995). Structure of Huma Estrogenic 178-Hydroxys teroid De hydrogenase and Mechanism c
Estradiol Formation. Structure 3: 503-5 13.
(8) D.-W. Zhu, M. Zhou, Y. Mao, and S.-X. Lin (1995). Crystallizatic of Human estrogenic 17B-Hydroxysteroid Dehydrogenase und€ microgravity. J. Crystal Growth 15611 -2: 108- 1 1 1.
(9) D,-W. Zhu, Azzi, A., Rehse, P. H. and S.-X. Lin (1996). TI crystallogenesis of a human estradiol dehydrogenase-substrat cornplex J. Crystal Growth 168: 272-276.
(10) S.-X. Lin, p.-W. Zhu, A. Azzi, R. Campbell, R. Breton, F. Labrie, 1 Ghosh, V. Pletnev, W. L. Duax, and W. Pangborn (1996). Studies O
the 3D-sructure of estrogenic 176-hydroxysteroid dehydrogenase. J
EndocrinoC. 150: 5 13-520.
(1 1) A. Azzi, P. H. Rehse, D.- W. Zhy, R. L. Campbell, F. Labrie, and S X. Lin (1996). Crystal structure of human estrogenic 1713 Hydroxysteroid Dehydrogenase cornplexed with 17B-estradiol at 2. A resolution (1 996). Nature Structural Biology 3: 665-668.
(12) p.-W. m, R. L. Campbell, F. Labrie, and S.-X. Lin (1996 Preliminary study of different methods for crystallization of huma estradiol dehydrogenase-inhibitor complexes (in preparation).
(13) &W. Zhw, G.-J. Xu, P. Rehse, A. Azzi, F. K. Zhao, and S.-X. Li (1996). CrystalIization and prelirninary crystallographic analysis a the Snake muscle Fructose 1,6-bisphosphatase (in preparation).

Publications
2. Abstracts:
( 1 ) A. Azzi, D.-W. Zhu, P. Rehse, R. Campbell, F. Labrie, S.- (1996). Structure of estrogenic 17B-estradiol complex at 2.3 resolution. 10th International congress of Endocrinology in S: Francisco, USA, 12- 15 June.
(2) D.-W. Zhu, Q. Hun and S.-X. Lin (1995). Phase Diagrarn for t
crystallization of human 17B-Hydroxysteroid Dehydrogenase. The 6 International Conference on Crystallization of Biologic Macromolecules in Hiroshima, 12- 17 November.
(3) D.-W. Zhu, A. Azzi, and S.-X. Lin (1995). The crystallogenesis Human estradiol Dehydrogenase-substrate complexe. The 61 International Conference on Crystallization of Biologic, Macromolecules in Hiroshima, 12- 17 November.
(4) S.-X. Lin, D.-W. Zhu, A. Azzi, R. Breton, F. Labrie, D. Ghosh, V. Pletnev, Z. Wawrzak, W. L. Duax, and W. Pangborn (1995). 31 structure of estrogenic I7B-HSD. International Symposium on DHE Transformation into Androgens and Estrogens in Target Tissue Intracrinology. Quebec City, Canada, September 13- 15, 95.
(5) V. 2. Pletnev, B. Burkhart, W. L. Duax, D. Gosh, D.-W. Zhu, Labrie, and S.-X. Lin, (1995). Structure of human estrogenic 171 hydroxysteroid dehydrogenase as a basis for inhibitor design i breast cancer therapy. ACA.
(6) D.-W. Zhu, et S.-X. Lin (1995). La cristallisation de la 17 hydroxystéroïde déshydrogénase humaine native sous différente formes. 63e Congrès de I'ACFAS, UQAC. Chicoutimi, 22 au 26 mai. 37.

Publications
(7) A. Azzi, P. Rechse, D.-W. Zhu, and S.-X. Lin (1995). Structure the Human Estrogenic 1713-Hydroxysteroid Dehydrogenase cornpIexe with a steroid substrat. Ninth Symposium of the protein societ! Boston, MA, July 8-12. P91, 156-M.
(8) D. Ghosh, Z. Wawrzak, V. Pletnev, W. L. Duax, D.-W. Zhu, F. Labr and S .-X. Lin (1995). Human estrogenic 17B-Hydroxysteroi Dehydrogenase as a target for drug design in breast cancer therapj Ninth Symposium of the protein society, Boston, MA, July 8-12. Pl12 280-S.
(9) D.-W. Zhu, F. Labrie and S.-X. Lin (1995). Human estradil dehydrogenase-inhibitor complexes: structure cancer therapy.9t Symposium of the protein society, Boston, MA, July 8-12. P132, 397 T.
( 1 0) D.-W. Zhu, et S.-X. Lin (1994). Cristallisation de la 171 hydroxy stéroïde déshydrogénase dans le gel d' ag arose. 62e Congre de I'ACFAS, de Montréal (Québec) mai Vo1.62.
(11) D. Ghosh, Z. Wawrzak, V. Pletnev, W. L. Duax, S.-X. Lin, D.-W. Zh and F. Labrie (1994). The three-Dimensional Structure of 178 hydroxysteroid dehydrogenase. 76th Annual Meeting of the Endocrin Society, Abst. No. 1393, p. 524.
(12). D. Ghosh, 2. Wawnak, V. Pletnev, W. L, Duax, D.-W. Zhu, X., Lei R., Breton, F. Labrie and S.-X, Lin (1994). Structure of human placenta 17B-hydroxysteroid dehydrogenase: insig h t in to rnolecular mechanism of estrone to 178-estradiol conversion. IX Int. Congress on Hormona Steroids, Abst. No. B119, p. 98.
(13) D. Ghosh, Z, Wawrzak, V. Pletnev, W. L. Duax, S.-X. Lin, D.-W. Zh and F. Labrie (1994). The three - Dimensional Structure of humai

Publications
178-hydroxysteroid dehydrogenase. XVI Int. Congress of Biochemisu and Molecular Biology.
(14) D. Ghosh, Z. Warwzak, V. Pletnev, M. Ermen, W. L. Duax, , '
Pangborn, D.-W. Zhu,F. Labrie, and S.-X. Lin, (1994). Molecu mec hanism of inhibition of steroid dehydrogenase by licorice-deriv steroid analogues in modulation of steroid reception function. The New York Acad. of Sciences, Steroid receptor antihormones, Dallas TX, USA.
(15) S.-X. Lin, D.-W. Zhu, Y. Mao, et M. Zhou (1994). Cristallogénè de la 17B-hydroxystéroïde déshydrogénase (1 7B-HSD) en micrc gravité. 62e Congrès de I'ACFAS, de Université Montréal (Québec) mm Vol. 62.
(16) D.-W. Zhu, 1.-Z. i n , et S.-X. Lin (1993). Propriét spectrophotométriques et fluorornétriques de la 178-hydroxystéroïdr déshydrogénase. 61e Congrès de I'ACFAS, UQAR, Rimouski, 17-21 mi
Vol. 61, p. 34.
(17) D.-W. Zhu, X. Lee, and S.-X. Lin (1993). Human 17
hydroxysteroid dehydrogenase NADP+ Complex: crystallization an
preliminary suuetural study. The summer conference for Youn Chines Researchers in Biochemistry and Molecular Biology, Shangha CHINA, August 9-14, p. 148-151.
(18) S.-X. Lin, .Dm-W. Zhu, W.-L. Duax. and D. Ghosh (1993). Study i the structure of human 1713-hydroxysteroid dehydrogenase. 7t Symposium of the Protein Society., Abst. No. 439-T, p. 131.
(19) S.-X. Lin, D.-W. Zhu, and R. Breton,(1993) The Crystallogenesis human placenta1 176-Hydroxysteroid Dehydrongenase. Internation: Symposium on Microgravity Science and Application, Beijing, Chini May 10-13, p.116-117.

Publications
(20) S.-X. Lin, D.-W. Zhu, and X. Lee (1993). Human 171 Hydroxysteroid Dehydrogenase: cristallogenesis on ground and i space. Space Bound Meeting of Canada., May 16-18, p. 63-64.
(21) S.-X. Lin, D.-W. Zhu, et R. Breton, (1993) La cristallogénèse de
17B-hydroxystéroïde déshydrogénase humaine. Proc. 6 1 Congrés d I'ACFAS, UQAR, Rimouski, 17-21 mai Vol. 61, p. 28.
(22) S.-X. Lin, D.-W. Zhu, W.-L. Duax, and D. Ghosh, (1993). Study c the structure of human 17B-Hydroxysteroid Dehydrogenase, 7t: Symposium of the Protein Society, San Diego, USA, July 24-28, p. 131.
(23) S.-X. Lin, D.-W. Zhu,, X. Lee, R. Breton, R. Duax, and D. Ghosl (1993) The crystallogenesis of human 178-Hydroxysteroic Dehydrogenase in the presence of its cofactor and substrate. 5tl Intermational Conference on Crystallization of Biologica Macromolecules., San Diego, USA, August 8-13, p. 43.
(24) S.-X. Lin, F. Yang, J . - 2 . Jin, R. Breton, et D.-W. Zhu (1992 Démonstration de l'identité des deux sous-unités de la 178 hydroxystéroïde déshydrogénase. 6oe Congrès de I'ACFAS. Universitc de Montréal (Québec), 11-15 mai, p 25.
(25) S.-X. Lin, J.-Z. Jin, F. Yang, R. Breton, and D.-W. Zhu (1992 Subunit structure and asymmetric kinetics of human placental 17B hydroxysteroid dehydrogenase. 6th Symposium of the Protein Society San Diego, USA July 25-29, p. 107.
(26) J.-Z. Jin, D.-W. Zhu, F. Yang, R. Breton, et S.-X. Lin (1992: Cinétique asymétrique de la 17B-hydroxystéroïde déshydrogénase di
placenta humain. 6oe Congrès de I'ACFAS, Université de Montréa (Québec), 11-15 mai,. p. 24.

Contents
Contents
Résumé court P a g i
. . 11
A b s t r a c t *. . 111
R é s u m é iv
S u m m a r y v
Acknowledgments v i
Publications vii
List of Figures xxi
List of Tables xxii
List of Abbreviations xxil
Chapter 1. Introduction 1
1.1 Current state of knowledge in 17B-HSDl 2 1.1.1 The major function 3 1.1.2 Purification of 178-HSD 1 5 1.1.3 Crystallization of 1713-HSD 1 6
1.1.3.1 Protein crystallization 6 1.1.3.2 Methodology of crystallization 7 1.1 -3.3 Protein crystallization in microgravity 8 1.1.3.4 Phase diagram for crystallization 9 1.1.3.5 CrystaIIization of hydroxysteroid dehydrogenase 1 0 1.1.3 -6 Crystallization of 2 7B-HSD 1 1 0
1.1.4 Structure studies of 17B-HSDl 1 1 1.1.4.1 176-HSD 1 genes expression 1 1 1.1.4.2 Subunit structure of 17B-HSD 1 1 2

Contents 1.1.4.3 Study of the active or binding site of 178-HSD1 1 2 1.1.4.4 3D-structure of bacteria1 3a,2013-HSD and rat Iiver
DHPR 1.1.5 Study of 178-HSD1 inhibitors
1.2 Major results 1.2.1 Rapid purification of 1713-HSD 1 1.2.2 Identification of 170-HSDl-NADP+, I7B-HSDI-
estradiol and1 7B-HSD 1 inhibitor (EM- 139) 1.2.3 Using 0-octyl glucoside to increase solubility
of 17B-HSD 1 1.2.4 Crystallization of 17B-HSD1 1.2.5 Structure study of 17B-HSD1 1.2.6 Phase diagram for crystdlization of 178-HSD1
1.3 Thesis outline 1.4 References
Chapter 2 Preparation of 178-HSD1 for crys tallization
2.1 Subunit identity of the dimeric 176-HSD1 from Human placenta 2.1.1 Introduction 2.1.2 Materials and methods 2.1.3 Results 2.1.4 Discussion 2.1.5 References
2.2 Rapid purification yielding highly active 17B-HSD1: application of hydrophobic interaction and affinity FPLC
2.2.1 Introduction 2.2.2 Experimental
2.2.2.1 Materials 2.2.2.2 I7B-HSD1 assay 2.2.2.3 SDS-PAGE 2.2.2.4 Protein concentration measurements 2.2.2.5 Purification steps

Contents
2.2.2.6 Placental homogenization and ce11 extract 2.2.3 Results
2.2.3.1 Hydrophobic interaction chromatography 2.2.3.2 Affinity chromatography 2.2.3.3 Concentration, specific activity and storage 2.2.3.4 SDS-PAGE and immunoblotting
2.2.4 Discussion 2.2.5 References
Chapter 3 Human 17B-HSD1: Optical properties of its complex with NADP+ 7 3
3.1 Introduction 7 6 3.2 Materials and methods 7 6
3.2.1 Materials 7 6 3.2.2 Enzyme assay 7 7 3.2.3 Enzyme purification 7 7 3.2.4 Protein concentration determinations 7 8 3.2.5 Absorption measurements 7 8 3.2.6 Fluorescence measurement 7 8
3.3 Results 7 9 3.3.1 Different preparations of 1713-HSD 1 leading to
different A 280lA 260 ratio 7 9 3.3.2 Absorption spectra of 1713-HSD 1 apoenzyme
and its complex with NADP+ 7 9 3.3.3 Fluorescence emission of NADP+ following an
excitation at 350 nm 8 1 3.4 Discussion 8 1 3.5 References 8 3
Chapter 4 Crystallization of 17R-HSD1 4.1 Crystal growth of human estrogenic 17B-HSD
4.1.1 Introduction 4.1.2 Materials and Methods
4.1.2.1 Chernicals

Contents
4.1.2.2 Methods 9 6 4.1.3 Results 9 8
4.1.3.1 Rapid preparation of homogeneous and highly active 17B-HSD 9 8
4.1.3.2 S tabilization of 1713-HSD 9 9 4.1.3.3 Detergent search to increase 178-HSD solubility 9 9 4.1.3.4 Screening and crystal growth of 17B-HSD-
NADP+ compIex 1 0 0 4.1.3.5 Crystals obtained in the presence of different salts
1 0 1 4.1.4 Discussion 1 0 2 4.1.5 References 1 03
4.2 Crystallization and preliminary X-ray diffraction analysis of the complex of human placental 1713-HSD with NADP+ 1 0 8
4.2.1 Introduction 1 1 0 4.2.2 Purification of 17B-HSD 1 1 0 4.2 3 170-HSD crystallization 1 1 1 4.2.4 Crystal characterization, Data collection and Analysis 1 1 2 4.2.5 References 1 1 3
4.3 The crystallogenesis of a human estradiol dehydrogenase- su bstrate complex 115
4.3.1 Introduction 1 1 7 4.3.2 Materials and Methods 1 1 8
4.3.2.1 Chemicals 1 1 8 4.3.2.2 Methods 1 1 8
4.3.3 Results and Discussion 1 2 0 4.3.3.1 Enzyme preparation 1 2 0 4.3.3.2 Crystal growth of 178-HSD 1 -estradio1 complex 1 2 2 4.3.3.3 Preliminary X-ray diffraction analysis 1 2 2
4.3.4 References 1 2 3 4.4 Preliminary study of different methods for crystallization
of human estradiol deh ydrogenase-in hibitor complexes 1 2 7 4.4.1 Introduction 1 2 9 4.4.2 Experimental 1 3 0

Contents
4.4.3 Results and discussion 4.4.3.1 Preparation of 178-HSD1-EM139 4.4.3.2 Co-crystallization and Soak method 4.4.3.3 Preliminary X-ray results
4.4.4 References 4.5 Crystallization of human esuogenic 17B-HSD under
microgravity 4.5.1 Introduction 4.5.2 Materials and Methods
4.5.2.1 Chemicals 4.5.2.2 Purification of 17B-HSD 4.5.2.3 Crystallization under microgravity
4.5.3 Results and discussion 4.5.4 References
Chapter 5 Structure of human estrogenic 17B-HSD at 2.20 A resolution
5.1 Introduction 5.2 Results and Discussion
5.2.1 Description of the structure 5.2.2 Architecture of the active site 5.2.3 Substrate recognition and the transition state 5.2.4 Possible membrane association 5.2.5 Other isozymes of 17B-HSD
5.3 Biological implications 5.4 Materials and Methods
5-4.1 Data collection 5.4.2 Structure solution and refinernent
5.5 References
Chapter 6 The phase diagram for crystallization of 17B-HSD1 181
6.1 Introduction 182 6.2 Materials and methods 183

Contents
6.3 Results and discussion 6.4 References
Chapter 7 Conclusion 1 9 8 7.1 The high quality of enzyme is the first important
step for crystailization 199 7.2 The new method for enzyme-ligand complex preparation is
very useful for structure studies of various members of 17B-HSD farnily and other steroid enzymes 200
7.3 An improvement on screening method for crystallization of new protein 20 1
7.4 Further the understanding for structure-function relationship of I7B-HSDl 202
Appendix 205
1. Crystallization and the preliminary crystallographic studies of the Azurin Pseudornonas fluorescens 207
2. Crystal structure of human estrogenic 17B-Hydroxysteroid Dehydrogenase complexed with 178-estradiol at 2.3 A resolution
3 Crystallization and preliminary crystallographic analysis of the Snake muscle Fructose 1,6- bisphosphatase

List of Figures
List of Figures
Figure 1.1 The essential roles of different types of 178-HSD 4 Figure 1.2 Structure of representative novel compounds acting
as pure anti-estrogens and inhibitors of 178-HSDI activity 1 5
Figure 2.1.1 Phenyl-Superose (HR 1011 0) chromatography of the 176-HSD1 5 5
Figure 2.1.2 SDS-PAGE of 17B-HSD1 from three different sources: 5 5 Figure 2.1.3 Superose-12 Gel filtration of 17B-HSD1 5 6 Figure 2.1.4 Native gel electrophoresis 5 6 Figure 2.2.1 Hydrophobic interaction chrornatography 7 O Figure 2.2.2 Affinity chromatography 7 0 Figure 2.2.3 SDS-PAGE of different 17B-HSD 1 fractions 7 1 Figure 2.2.4 Apparent molecular mass evaluation of 17B-HSD 1
60m SDS-PAGE 7 1 Figure 3.1 Absorption spectra of 178-HSD1 8 6 Figure 3.2 The hypochromic effect of NADPH absorption in the
presence of 170-HSD 1 8 7 Figure 3.3 Fluorescence spectra of 170- H S D 1 8 7 Figure 4.1.1 SDS-PAGE of 17B-HSD directly purified from human
placenta or from dissolved crystals 106 Figure 4.1.2 17B-HSD-NADP' crystals grown in the presence
of MgCl2 106 Figure 4.1.3 Crystals grown in the presence of LiCl and NaCl 1 0 7 Figure 4.3.1 The crystals of 17B-HSD 1 -estradio1 1 2 6 Figure 4.4.1 Structure of representative novel compounds acting
as pwe anti-estrogens and inhibitors of 178-HSDl activity 1 3 9 Figure 4.4.2 Co-crystallization: the crystals of 1713-HSD 1 -EM 139 1 3 9 Figure 4.4.3 Soak method: the crystals of 17B-HSD 1-EM139 1 3 9 Figure 4.5.1 Crystallization geometry used in the MIR space
from Payload System Inc 1 4 9 Figure 4.5.2 The space crystals 1 4 9

List of Figure
Figure 5.1 Stereo ribbon diagram of a monomer of 170-HSD Figure 5.2 A(2Fobs-Fcaic) electron-density map of the helix aGO Figure 5.3 A ribbon diagram of 175-HSD structure with the
substrate- binding domain Figure 5.4 Stereo ribbon representation of the dimer of 17B-HSD Figure 5.5 A stereodiagram of superimposed Car chains of
bacterial 3a,208-HSD and 170-HSD Figure 5.6 Close-up stereoview of the active site of 178-HSD Figure 5.7 (a) Stereoview of the atomic mode1 of the proposed
transition state of estrone to estradiol interconversion Figure 5.7 (b) The proposed mechanism of estrone to estradiol
interconversion Figure 5.8 A dotted Connolly surface of the active-site cavity Figure 5.9 Helices(a) a G O and (b) aH viewed dong the helical axis Figure 6.1 The setting for micro-batch method Figure 6.2 Precipitation, nucleation, metastable and
undersaturation zones Figure 6.3 Crystallization of 178-HSD1 in one control Figure 6.4 Crystallization was accompanied by the progressive
dissolution of the precipitate around the growing crystals Figure 6.5 Crystals grown near the edge of precipitation zone
diffract to high resolution Figure 6.6 The solubility curve determination

List of Tables
List of Tables
Table 2.1.1 Purific ation of 178-HSD1 from hum ian pl acenta Table 2.2.1 178-HSD purification by two chrornatographic steps Table 3.1 Absorption characteristics of 178-HSD I complexes
with NADP' Table 4.4.1 Data collection Table 5.1 Surnmary of data collection for native and derivative
crystals Table 5.2 Isomorphous replacement phasing statistics Table 5.3 Statistics from phase combination Table 5.4 Refinement statistics Table 5.5 Distances (in A) between pairs of Ca atorns Table 6.1 Conditions and diffraction data for obtained crystals Table 7.1 Crystallization and Preliminary X-ray Diffraction
Analysis of 178-hydroxysteroid dehydrogenase type 1

List of A bbreviations
List of Abbreviations
170-HSD 1 DHEA A5-di01 A$-dione FPLC HSDS 3D-structure 3a-HSD SCAD 3a,20B-HSD DHPR 1 10-HSD 0-OG CMC
E2
2001-OH-P PEG PMSF SDS PAGE AcNPV 0-SH rn Hepes
ADA Fru- 1,6-Pase CHUL
170-Hydroxys teroid Dehydrogenase dehydroepiandrosterone 5-androstene-3B, 2 7B-di01 4-androstene 3,17-dione fast protein liquid chromatography Hydroxysteroid Dehydrogenases three- dimensional structure 3a-Hydroxysteroid Dehydrogenase short-chain-alco ho1 de h ydrogenase 3a.208-Hydroxysteroid Dehydrogenase dihydropteridine reductase 1 18-Hydroxysteroid dehygenase 8-octyl glucoside critical micelle concentration estradiol 20a-dihydroxysteroid progesterone
polyethylene glycol phenylmethanesulfonyl fluoride sodium dodecyt sulfate polyacrylamide gel electrophoresis Autographa californica nuclear polyedross virus 17B-estradiol, 2-mercaptoethanol dithiothreitol N-(2-hydroxyethyl) piperazine-N'-2-ethanesulfor acid N-(2-acetamido) iminodiacetic acid Fructose- 1 $6-bisphosphatase Centre Hospitalier de l'université Laval

Chapter 1
I n t r o d u c t i o n

Chapter I . Introduction
Hormone-dependent cancers (breast, prostate, endometrial an ovarian) represent 30% of al1 cancers. About one in nine women wi develop a breast cancer over their entire lifetime (Wingo et al., 1995). 1 Canada 17,700 new cases of breast cancer were estimated for 1995 an 5400 deaths were expected (Wingo et al., 1995). Breast cancer is th second cause of cancer-related mortality after lung cancer. Further moi 30-40% of al1 breast cancers are estrogen dependent (Edery et al., 1981 The incidence of breast cancer has increased at an annual rate of 1% ove the past 50 years. In the U.S. in - 1995, it was estimated th; approximately 46,000 women would die from breast cancer (Feuer et al 1993). Post menopausal women are more susceptible to develop brea! carcinoma because they have a high blood concentration of estroge precursors such as sulfated steroids (Hobkirk et al., 1993; Franz et al 1979; Santner et al., 1993). In human breast tissue, estrogenic 17f hydroxysteroid dehydrogenase (1 70-HSD) (E.C. 1.1.1.62) is responsible fc the production of the most active estrogen 17B-estradiol. The activity c 17B-HSD is hormonally regulated (Adams et al., 1988; Couture et al 1993) and appears to be higher in breast tumor cells than in surroundin normal mammary tissue (McNeill et al., 1986; Vermeulen et al., 1986: Because 178-estradiol enhances breast ce11 proliferation, 170-HS D is a attractive target for the design of inhibitors that may be effective antj tumor agents. So, the enzyme is important for both endocrinolog research and cancer therapy.
1.1. Current s t a t e of knowledge in 178 hydroxysteroid dehydrogenases, taking type 1 (171 HSD1) as the major example
170-HSDs are responsible for the biosynthesis and interconversioi of the principal sex-hormones that stimulate the proliferation of breas and prostate cancers (Fig.1 .l) (Labrie et al., 1986; Poulin et al., 1986; Lii et al 1996). Both estrogens and androgens are more active in the 170 hydroxy configuration than the corresponding 17-keto steroids. In bot1 gonadal and peripheral tissues, 170-HSD catalyses the interconversion of

Chapter I . Introduction
testosterone and androstenedione, as well as estradiol and estron (Martel et al., 1992). It is now clear that several isozymes of 17B-HSI: are involved in the oxidation and reduction of different estrogens an androgens in distinct tissue locdizations (Descomps et al., 1968; Jarabal 1969; Chin and Warren, 1973; Murdock e t al., 1986; Luu The et al., 198! Lin et al., 1992 b; Wu et al., 1993; Geissler et al., 1994; Labire et a 1996). Human 17B-HSD1 is the form responsible for the synthesis of t h
active estrogens which stimulate the proliferation of breast cancer ce11 (Mouridsen et al., 1978; Poulin et al., 1986; C. Labrie et al., 1992; Lin (
al., 1992 b).
1.11 The major function
Recently, five types of human 176-HSDs were identified and th€ primary structures elucidated (Luu et al., 1989; Peltoketo et al., 1988; V 1993; Casey 1994; GeissIer 1994; Adamski & de Launoit; Labrie et i
1996). Human estradiol dehydrogenase, or 17B-HSDl (EC 1.1.1.62), is sohble enzyme found in great quantity in the human placenta. Th estrogenic form of 17B-HSD catalyses the conversion of estrone to 17 estradiol and to a lesser extent, dehydroepiandrosterone (DHEA) to androstene-38, 178-di01 (A5-did) (Dumont et al., 1992). The human tyl 2 isozyrne (from the rni~rosomal fraction), or 17B-HSD2, which h, recentiy been cloned, is responsible for the oxidation of testosteron estradiol and dihydrotestosterone (Wu et al., 1993). SubsequentIy the, more types of human 17B-HSD have been identified: type 3, which responsible for the conversion of androstenedione to testosterone, pIa: an important role in the formation of active androgens (this type al! catalyses the reduction of DHEA and EI); 17B-HSD4, homologous to porcii ovarian 178-HSD, has also been identified (Adamski & de Launoit); ar 178-HSD5 These 170-HSD isozymes (Fig.l.1) catalyse important p a t h w ~ in the biosynthesis ,of estrogens and androgens and thus have i
important impact on the therapy of breast and prostate cancers.

Chapter 1. Introduction
H
,DH IOL (Estrogens)
-&:=""", Testosterone
Fig. 1 . 1 The essential roles of different types of 17o-HSD (indicated by numbers) in the formation of sex hormones in gonadal and peripheral t issues.

Chapter 1. Introduction
1.1.2 Purification of 17R-HSD1
17B-HSDl has been studied since the 1950s and has been purifie partially or to apparent homogeneity by several groups. After man
unsuccesful attempts, researchers have found that the enzyme can b stabilized during purification procedure either by its substrate, estradiol or by a high cencentration of glycerol (Langer and Engle, 1958). Lange and Engle (1958) obtained a 50-fold purification of 178-HSD b: ammonium sulfate fractionation, using glycerol as a stabilizing ageni Four years later, Jaxabak et al. reported that they obtained a 2500-fol( purification of the enzyme, using ion exchange chromatography. II 1966, KaravoIas et al. introduced chrornatographic separation of estradia dehydrogenase from the estradiol-activated transhydrogenase which i also present in placenta1 cytoplasm. A hydroxyapatite column was alsc used by Jarabak (Jarabak, 1969), who modified his 1962 procedure. II 1968, Descomps et al. markedly shortened the procedurefor isolatin; pure enzyme by using successive chromatography over DEAE-Sephade: and Sephadex (3-150. They reported a 500-fold purification with ai overall yield of 14%. A substrate analogue was used during some step and glycerol was used throughout the procedure as stabilizing agents Karavolas et al. (1970), taking advantage of the efficiency O
chromatography on hydroxyo-patite and DEAE-cellulose, obtainec homogeneous enzyme in four steps. Relatively high specific activity ha: been obtained only in somc reports. It was Burns et al. (1972) whc developed a procedure more suitable for large-scale preparations an( who obtained sufficient quantities of enzyme. The homogenate O
placenta, prepared in a buffer containing 20% of glycerol, wai precipitated with ammonium sulphate. The material collected betweei 30 to 50% saturation of ammonium sulphate was centrifuged to removc microsornes and could then be stored indefinitely in 50% glycerol a -80°C (Lewis et al., 1974). An important advance was made by Nicolas e al. (1972), when they developed an affinity column that enabled them tc achieve a 100-fold purification of the enzyme in a single step. Chin et al (1973) and Murdock et al. (1986), reported that an apparently

Chap ter 1 . Introduction
homogeneous enzyme with a specific activity of 2.5-3.7 U per mg w, obtained.
The conventional chromatography procedures used by these autho are generally long, limiting scale-up and improvement of the purificatic of this enzyme. Since the early 1980s, fast protein liqu: chromatography (FPLC) has been demonstrated to be very efficient preparing high-quality proteins (Markey 1984; Tarn et al. 1984; Lin al., 1988; Lin et al. 1992 a). For example, using the technique of FPL( Lin et al. (1992) reported a method of rapid purification. The E.co glutamyl-tRNA synthetase was highly (>99.5%) purified in about hours from cells of an overproducing E.coli strain. The enzyme preparc by this rapid procedure was 3 times more active than when obtained t conventional methods.
The FPLC methods provide a technique for rapid purification th, w i l be very useful for the purification of 178-HSD1 of high specif activity. in 1992, Lin et al. simplified and accelerated the purification (
this labile enzyme from human placenta using FPLC. A homogeneous an highly active preparation was obtained and the specific activity is aboi 2-3 fold higher than that reported in the Iiterature (Lin et al., 1992 b). 1 fact, the preparation of a highly homogeneous enzyme is the fir important step for crystallization of 178-HSD 1 and subsequei determination of its 3D-structure.
1.1.3 Crystallization of 170-HSD1
1.1.3.1 Protein crystallization
In the study of the structure-function relationships of the 17B-HSD the study by structural biology is critical. Of al1 the modem methods fc the determination of the three dimensional structures of macromolecule crystallography still remains the most important (Lattman, 1994). It is mature and powerful technology that has contributed immeasurably t

Chapter 1 . Introduction
Our understanding of biological processes through the knowledge protein structure.
In crystallography, the presence of high quaiity crystals is vei important. Although crystallization is one of the oldest sciences (Schee 1993), the first published observation of the crystallization of a prote: appeared to be by Hunefeld in 1840 (McPherson, 1991). In the ear: days, crystallization was a tool for protein purification an characterization, and before the advent of X-ray crystallograph- researchers were able to establish definitively that the biologie, catalysts (enzymes) are proteins only through crystallizatic experiments (Dounce & Allen, 1988). In 1934, Dorothy Hodgkin mac the first X-ray studies of protein crystals, and she obtained the fir diffraction pattern of pepsin crystals (Berna1 & Crowfoot, 1934). In tï early days of crystallography, the main problerns were in the X-ra methods and not in the crystallization of proteins. But as soon as th, methods for solving crystal structures became better established, a: molecular biology gave access to more sophisticated molecuIes, t situation has changed. More basic biological questions could then addressed (e.g. molecular understanding of metabolic pathways, genetic mechanisms, etc.) and a great dernand f ~ r differe macromolecule crystals has been raised (Giegé et al., 1994). Throu, recent advancement in molecular biology and protein crystallizatior several thousand soluble proteins, belonging to about 300 families proteins, have been crystallized, giving X-ray diffraction at hi; resolution (Michel, 1995). i n contrast, the number of crystallizf membrane proteins or membrane protein complexes, only about a doze belonging to seven farnilies (Michel, 1995).
1.1.3.2 Methodoiogy of crystallization
Among methods for macromolecule crystallization, vapour diffusir techniques are probab1.y the most widely used throughout the worl Recently, important new developments in the methodology crystallization have been realized by many scientists studying differe types of proteins. The crystallization methods in capillaries and those b:

Chapter 1. Introduction
interface diffusion (Salemme, 1 WZ), are widely used. Other metha which are less universaily employed include, for instance, crystallizati in gels, in electric fields, under pressure; under micro- or supergravi under levitation, or methods that conuol or alter parameters as function of time, pH or temperature (Giegé et al., 1994). It can expected that some of them will be very useful for crystallization of 17 HSD1.
In spite of recent innovations and an expanded base of experient the growth of single crystaIs of macromolecules generally rernains empirical and frequently tedious process. The variable set over whi successful crystallization conditions must be sought is vast while t quantity of rnacromolecule available may be extremely limit (McPherson, 1990). Nonetheless, the number of macromolecul crystallized during the past decade has increased at a near exponent: rate (Gilliland & Bickham, 1990). A major contribution to this increase success has been the development of novel screening protocols ai optimization strategies which have provided investigators with expanded portfolio of effective crystallization tools (Carter & Carter, 197 Jancarik & Kim, 1991; McPherson, 1992; Samudzi & Fivash, 1992; Stwa al. 1992; Weber, 1990). At present, the sparse manix screen (Jancar & Kim, 1991) provides a highly effective and rapid screening method f the crystallization of macromolecuIes and is widely used in numero universities and private research laboratory.
1. f .3.3 Protein crystallization in microgravity
Protein crystaIlography requires crystals of suitable size and quali for high-resolution diffraction analyses. A new development in proteit crystal growth involves studies of crystal growth processes in tl microgravity environment obtainabIe in space (Bugg, 1986; Delucas Bugg 1988). The major motivation behind these space experiments is diminate the density-driven convective flow that accompanies cryst growth in gravitational fields (Kroes et al., 1984; Pusey et al., 1988). addition, sedimentation of growing crystals, which can interfere with tl formation of single crystais, is eliminated in the absence of gravir

Chapter 1. Introduction
(Lawrence e t al., 1989). The first microgravity protein crystal growi experiments were conducted on the U. S. Space Shuttle in 1984 (Litth 1984). Littke and John indicated that the space-grown crystals from liquid-liquid diffusion system were larger than crystals obtained by tt same experimental system on Earth. Since that time, more than ont hundred different macromolecules (proteins and nucleic acids) have bee flown in space usin a variety of space vehicles. Results from thes experiments demonstrate that Iarger andlor higher quality crystals ca be produced in rnicrogravity using a variety of crystallization technique, These techniques include vapour diffusion, liquid diffusion, dialysis an temperature-induced crys tallization.
1.1.3.4 Phase diagram for crystallization
Although crystallization of proteins has been used repeatedly in various epoch-making works in biochemistry and molecular biolog (Sumner, 1948; Perutz, 1969; Michel, 1983), the physics of protein crysti growth remains Iargely unknown. Systematic efforts started only in th 1980s toward an understanding of the underlying mechanisms c protein crystal growth (Kam et al., 1978; Feigelson, 1986; Giegé, 1988: Micrornethods to establish phase diagrams have been worked out i severaï laboratories (Chayen et al., 1988; Mikol & Giegé, 1989; Caciopp c
al., 1991). Some of these diagrams have illustrated the possibility c
changing the solubility of proteins by pH or temperature, but have als shown how complex and unpredictable solubility can be when varyin conditions (Giegé et ai. 1994). Its successful establishment required the precise determination of each crystallization parameter and carefu; sample preparation. The results enable the growth of crystals to be understood within a general physicochemical framework, provide s justification for hitherto empirically selected crystallization conditions, and offer rational guidelines for their improvement. By studying phase diagrams for the crystallization of 178-HSD1, we can further modify the crystallization conditions, leading to further improvement of 170-HSD1

Chapter 1. Introduction
crystals and for the crystallization of 17B-HSD1 complexed with variou: ligands.
1.1.3.5 Crystallization of Hydroxysteroid Dehydrogenases
The hydroxysteroid dehydrogenases (HSDs) belong to a group O
pyridine nucleotide-dependent enzymes which catalyze thr oxidoreduction of alcohols and carbonyls in a positional and stereospecific rnanner on the steroid thereby synthesising or degrading active steroic hormones (Trevor 1991). At present, the mammalian enzymes of interes include 3a-, 3B-, 11B-, 178-, and 20a-HSDs.
178-HSDl is a member of the short-chain alcohol dehydrogenast (SCAD) family. At least 57 different enzymes, belong to this familj showing only 15-30% sequence identities but exhibiting a commor tertiary structure. Al1 of these enzymes are thought to bind thr coenzyme via a classical "Rossman fold" (Hans et al., 1995). In thi! family, the 3a,20B-hydroxysteroid dehydrogenase ( 3 ~ ~ 2 0 5 - H S D ; EC 1.1.1.53) from streptomyces hydrogenans and the dihydropteridinr reductase (DWR) from rat liver have been crystallized. The bacteria ho10 3a,SOB-HSD complex with NADH was crystallized and its three- dimensional structure determined at 2.6 A (Ghosh et al., 1991). The crystals, grown in the presence of 4 mM NADH, belong to the space group P432 ,2 having unit ce11 dimensions a = 106.2 A and c = 203.8 A anc contain one full tetramer (106 kDa) in the asymmetric unit (Ghosh et al.. 1991). This group also reported the crystallization of a bacterial holc 3a,20B-HSD-NADH- inhibitor (a licorice compound) complex. Rat liver
DHPR was also crystallized. They were of the space group C2221, the unii ce11 dimensions being: a = 50.10 A; b = 139.13 A; c = 61.29 A and its 3D- structure determined at 2.3 A (Varughese et al., 1992).
1.1.3.6 Crystallization of 178-HSD1
Crystallization of the human estrogenic 17B-HSD has been attempted

Chapter 1 . Introduction
since the 1970s, when Chin et al. reported a crystallization of 178-HSI by an electrophoretic diffusion method (Chin et al., 1976). A solution the enzyme (specific activity 7.1 units/mg) in 1.5 ml of Tris-barbitui acid buffer, pH 7.0, containing 20% glyceroI as stabilizer, was placed in electrophoresis tube and the tube was closed at both ends with a dialy: membrane which permits the passage of substances of molecular weig iess than 18,000. The tube was pIaced in a gel electrophoresis apparat and the reservoirs filled with the Tris-barbitunc acid buffer. A potentj of 100 V was applied for 12 hours, then raised to 200 V for another hours, and finally to 300 V until opalescence appeared at the bottorn the tube. Activity measurements showed that more than 90% of tl enzyme had concentrated in the bottom 0.15 ml portion of the solutio When this section of the solution was removed and kept overnight at 4O1 gross and microscopic examination revealed a heavy crop of crysta which possessed a specific activity of 7.2 units/mg. The specific activi remained constant throughout t hree recrystallizations. The crys tallii enzyme displayed a single band by analytical and sodium dodec sulfate-polyacrylamide gel analysis. Crystals of enzyme of high specif activity could also be obtained from an enzyme sample initiai possessing a specific activity of only 4.5 units/mg (Chin et al., 1976). spite of this crystallization report, no X-ray diffraction data were ev published by this laboratory.
1.1.4 Structure studies of 178-HSD I
The importance of this enzyme is also demonstrated by the extensil studies of this enzyme since the 1950's. The knowledge of the structu of I7B-HSD1 is critical to the understanding of its function, and mi result in markedly advancing the human knowledge to a new level endocrinology, as it is a representative enzyme contributing to the la step of formation of active estrogens. This is certainly of major impact (
the development of therapies for treating hormone-dependent cancer.
1.1.4.1 178-HSD 1 genes expression and overproduction

Chapter I . Introduction
Isolation of the type 1 17B-HSD cDNA led to the identification of tl
corresponding HSD1701 gene, and that of the highly homologous HSD17I pseudo-gene, both of which lie on chromosome 17q21 in close proximi to the BRCAI locus (Peltoketo et al., 1988, 1992; Luu-The et al., 198 1990; TrembIay e t al., 1989; Simard et al., 1993). The 170-HSD1 gene contained within a genomic DNA fragment of 3.3 kb and consists of s exons that encode a protein of 327 amino acids (Luu et al., 1989). : 1992, Breton et al. overproduced the protein using an expression syste based on the infection of insect cells (Spodoptera frugiperda) by baculovirus which canied the cDNA of 17B-HSD1 (Lin et d.,1992b; Bretc et al., 1994). The specific activity of 17B-HSDl (0.26 U/mg) in the ce extract reached a level 60 to 70 fold higher than that in human placenta,
1.1 A.2 Subunit structure of 178-HSD1
The dimeric nature of 178-HSD1 was not elucidated until the 197( (Burns et al., 1971; Jarabak and Street, 1971). Contradictory opinions c the identity of the subunits of this isozyme have been reported. Jarabz and Street (Jarabak and Street, 1971) and Burns et al. (Burns et al., 1971 Burns et al., 1972) proposed that the two subunits were probabl identical but Engel and Groman suggested the existence of three differei monomers that could interact with each other to form 6 dimers (Enge and Groman, 1974; Inano and Tamaoki, 1986). Lin, et al. (1992b) studie the subunit structure of 17B-HSDl with a highly active preparation froi placenta. The subunit mass was determined to be 34.5 kDa by SD polyacrylamide gel electrophoresis. The molecular mass was determine to be 68 kDa by native gradient gels and verified by Superose-12 gi filtration. Similar results have been obtained for the enzyme recombinant forms produced from a single cDNA encoding a protein (
34.5 kDa. The placenta enzyme and the recombinant ones are als identical at the levels of steady-state kinetics (Lin et al., 1992b). It concluded that 17B-HSD1 is formed by two identical subunits.
1.1.4.3 Study of the active or binding site of 17B-HSD1

Chapter 1. Introduction
The SCAD super family has a characteristic Tyr-X-X-X-Lys (where X any amino acid) sequence of residues at the active site (Hoog et al., 1 9 9 ~ Some structural studies of the active or binding site of 170-HSD1 usii conventional methods in protein chernistry, have been reported. F example, a 20-residue amino acid sequence including this site and tv histidine residues in the active site had been identified (Chin et al., 198 Murdock e t al., 1986) and the essential role of lysine and cysteii residues in the active site had been demonstrated by chemic modification (Inano, 1988). Krozowski reported that, in hurnan 17 HSD1, the amino acid residues 155-172 make up a region called the 1 domain, which is one of the six conserved regions (A, B, C, D, E, and among the SCAD members (Krozowski, 1992). Among these s regions, those suggested to be involved in the binding of the cofact [NAD(P)/NAD(P)H] and those suggested to have a role in the conformatic of the secondary and tertiary structure of the SCADs are also conserved human 17B-HSD1. The tyrosine of the Tyr-X-X-X-Lys sequence entirely conserved in al1 the members of the SCAD family. This TI residue (Tyr155 in human 178-HSD1) has been localized in tl substrate binding region in glucose dehydrogenase (Jany et al., 1984). is also one of the amino acid residues which line the steroid-bindir pocket of holo-3a, 208-HSD (Ghosh et al., 1991).
Although the above-mentioned information was still insufficient define the 17B-HSD1 structure, these results and methods will be usef in the study of the crystallization of 170-HSD1 and in the analysis on tl 3D-structure of 17B-HSD1.
1.1.4.4 3D-structure of bacterial 3a,20B-HSD and rat liver DHPR
Among the SCAD family, 3D-structures had been reported onIy fc bacterial ho10 3a,208-HSD (Ghosh et al., 1991) and rat liver DHP
(Varughese et al., 1992;) before we obtained the structure of 170-HSD Each of the four identical subunits of bacterial ho10 3a,200-HSD has parallel a/B structure with a classic doubly wound B-sheet (Richardson

Chapter 1. Introduction
1981). It f o m s a single domain structure consisting of a 0-sheet fomr by seven parallel strands having three parallel a-helices on each sic (Ghosh e t al., 1991). 178-HSD1 shares less than 15% sequence identi with the bacterial ho10 3a,206-HSD. The amino acid sequence of 171
HSDl also differs from that of bacterial 3a,208-HSD by two insertions ((
11 and 14 residues) and 52 additional residues at the C terminus. Despi these differences, the structure of bacterial ho10 3a,200-HSD was used i
a search mode1 for structure deterrnination of 178-HSDl.
1.1.5 Study of 1713-HSD1 inhibitors
Many studies have been devoted to finding inhibitors of 17B-HSD1 Some affinity label inhibitors for human placenta1 170-HSD have bee: reported (Sweet et al., 1991; Saurabh et al., 1990; Richard et ai., 1989 Murdock et al., 1986: Inano et al., 1983; Thomas et al., 1983). Twl important disadvantages were associated with these compounds: thei low selectivity and their unsuitable estrogenic activity which virtuall: eliminated their therapeutic use. Recently, a series of dual-actioi inhibitors (e.g., EM-139, EM-221, EM-140, and EM-123) werl synthesized in the Laboratory of Molecular Endocrinology at CHU1 (Fig.l.2) (Lévesque et al., 1991; Labrie et al., 1992). They possess ai estrogen nucleus and cm block the formation of active estradiol by 170 HSDI, as well as the estrogen action via its receptor. EM-139 is a 7a- alkyl omide estradiol derivative. It was tested for its antiestrogenil activity as well as its potential 17B-HSD-inhibitory activity il ovarectomized mice treated with E l , the immediate precursor of E: (Labrie et al., 1992). Experiments using kinetic methods in Our grou] also showed that EM-139 is a reversible and cornpetitive inhibitor fo 17B-HSDI. A ki = 6.6 f 0.6 pM was calculated from the slope of th1 linear curve in this study. It binds with free I7B-HSD1 to preven substrate (estradiol) binding (Wang et al unpublished). In order tc obtain better inhibiting activity and to elucidate the mechanism O
inhibition, the crystallization of the 17B-HSD 1 -inhibitor (EM-139 complex will provide a good opportunity to optimize that type of

Chapter 1. Introduction
Ez (estradiol) R7 = H Rla = H
EM 139: R7 = (CHz)ioCONnBuMe, R16 = CI. EM 221: R7=(CH2)ioCONnBuMe, R16 = F.
Fig. 1.2 Structure of representative novel compounds acting as pure anti-estro and inhibitors of 1713-HSD 1 activity. l6a-Halogenated compounds: EM- 139 and -22 1. D-ring unsaturated compounds: EM- 140 and EM- 123.

Chapter 1. Introduction
inhibitor from the direct demons tration of protein inhibitor interactions.
1.2 Project and major results
As mentioned above, 170-HSD1 is responsible for the synthesis of tk active estrogen which stimulates the proliferation of breas t cancer ce11 Detailed study of the mechanisms of action of this enzyme is thus c major importance for the inhibitor design to improve endocrine therap! Although the enzyme from human placenta was purified about thirt years ago, its mechanism of action has not been clearly elucidated at t h beginning of 1990's. Determination of the three-dimensional structure c 170-HSD1 would contribute to the elucidation of the moleculr mechanism of action and permit the design of therapeutic agents that wi inhibit the enzyme and modulate endogenous estrogen levels.
We have chosen to study 170-HSD1 in the soluble subcellula fraction of human placenta. We have proposed to purify 170-HSD1 ti homogeneity and high specific activity . We attempted to cry stallize 1713 HSDl in order to resolve its three-dimensional structure. We have alsc planned to crystallize 17B-HSD1 complexes with different ligands anc inhibitors. These results will lead to the understanding of the: similarities and differences in structure-function. Such information i important to the knowledge of the enzyme-ligand interaction and henci their physiological roles, that will have a major impact on bot1 endocrinology research and hormone-dependent cancer therapy.
1.2.1 Rapid purification of 17B-HSD1
We used a new purification procedure for this labile enzyme frorr human placenta using the FPLC technique (Fast Protein Liquic Chromatography). We obtained a homogeneous and highly activr preparation, probably due to the elimination of microheterogeneities thai are often caused by in vitro modifications, such as oxidation-reduction oi partial proteolysis during conventional chromatography. One milligram

Chaprer I . Introduction
of pure 176-HSD1 catalyses the formation of 7 to 8 pmol estrone fron estradiol in one minute. The reaction mixture contained 0.5mM NAD+, 21 pM estradiol in 50 mM NaHC03-Na2C03 buffer, pH 9.2 at 23OC f 1°C. Thi: value is about 2-3 fold higher than that reported in the literature (Lin e al., 1992; Yang et al., 1992).
1.2.2 Identification of 17B-HSD1-NADP+, L7R-HSD1-estradi a n d 17B-HSDl-inhibitor(EM-139)
In order to study the interactions between the enzyme and ii cofactors, the apoenzyrne and holoenzyrne forms of 17B-HSDI wex prepared by modifying the purification procedure (Jin et al., 1993). The different optical properties were identified by spectrophotometer an fluorometry (Zhu et al., 1995 a). The apoenzyme had a higher Azso/A,, ratio than the holoenzyme carrying one cofactor. Using this method, we could easily quantify the cofactor binding of the enzyme. It is also ver) useful for the crystallization of different complexes of 17B-HSDl.
Using a special procedure, we saturated a high concentration of th enzyme in solution with the substrate estradiol (E2). We succeeded i making the 1:2 176-HSDI-estradio1 complex at high concentration fc crystailization, as venfied by determining the arnount of Cl4-labeled estradiol bound to the enzyme (Zhu et al., 1996). The high quality of th€ resulting electron density for the substrate supports the efficacy oj this method. The establishment of such a new technique provides 2
strong basis for the crystallogenesis of other steroid-dehydrogenase complexes.
The formation of 178-HSD1-inhibitor (EM-139) complex was showr by an optical method. The result showed that when 176-HSD1 (dimei concentration of 220 PM, for Our experimental conditions) is mixed with EM-139 (> 440 PM), each subunit of 17B-HSDI is complexed with one EM-139. The hydrophobic EM-139 was bound to 170-HSD1 and il saturated the latter in a special repeated "dialysis", gradually reaching a

Chapter 1. Introduction
stoicheometric binding to the enzyme.
1.2.3 Using fi-octyl glucoside to in crease solubility of 17B-HS:
As the human 170-HSD1 is only soluble up to 2-3 mg/ml, w attempted to use different detergents to increase its solubility whil maintaining its activity. Satisfactory results were obtained in th presence of 0.06% fi-octyl glucoside (0-OG), with which we obtained 170-HSD1 solution at more than 40 rnglrnl (Zhu et al., 1994). The B-O( concentration used here is well below its CMC (critical micell concentration) of 1 %.
1.2.4 Crystallization of 17B-HSD1
In 199 1, we successfully crystallized 17B-HSD 1 yielding diffractio: data with high resolution, the first example of any steroid-convertin, enzyme from a human source (Zhu et al., 1993). Crystals of 178-HSD1 N A D P + were obtained with the vapour diffusion method. The crystal were of the space group C2 with the following ce11 parameters: a :
123.03 A; b = 45.03 A; c = 61.29 A; B = 99.1". The first crystals diffracte, up to 2.7 A resolution. Later we obtained bigger crystals and of bette quality. A native data set was obtained to a resolution of 2.2 A on rotating anodeX-ray source. Recently, using phase diagrams of 178 HSD 1, weobtained high quality crystals of 170-HSD 1 -NADP+ and a nativl data set was increased to a resolution of 2.0 A. At present, nine othe different forms of crystals of human 17B-HSD1 with various ligands havc also been obtained [l . 17B-HSD 1 -estradiol; 2. 17B-HSD 1 -NAD+; 3. 178 HSD1-EM139 (inhibitor); 4. apoenzyme; 5. 178-HSD1-estrone; 6. 178 HSD1 -testosterone; 7. 170-HSD1 -(17a-E2)-NADP+; 8. 17B-HSDl-20a-OH-P 9. 170-HSD1-(17a-Methyl-E2)-NADP+]. Native crystals diffracted X-ray from a synchrotron radiation source to 1.8 - 2.3 A resolution.
Crystallization under microgravity

Chapter I . Introduction
To eliminate multiseeding, formation of multicrystals and to obtai higher quality crystals, we carried out the crystallization aboard th Russian MIR Space station and crystaIs were recovered in January, 1994 The space experiments showed better resd ts i n nucleation numbei crystal size and morphology than the ground ones, yielding crystals wit resolutions between 2.5-2.7 A (Zhu et al., 1995 b). At that time, crystal grown in the laboratory diffracted no better than this, but subsequen improvements in purification and crystallization conditions have resulte' in crystals that diffract to higher resolution.
1.2.5 Structure studies of 17R-HSD1 and 17B-HSD1-estradiol
Recently, the structure determination of 17B-HSD1 has bee completed with Our 1713-HSD1 crystals (Ghosh et al., 1995). The 2.20 resolution structure of 1713-HSD 1, the first mammalian steroidogen. enzyme studied by X-ray crystailographic techniques, reveals a fol( characteristic of the short-chain dehydrogenases. The active site contai] a Tyr-X-X-X-Lys sequence (were X is any amino acid) and a serir residue, features that are conserved in short-chain steroi dehydrogenases. The core of the structure is composed of the sevei stranded parallel B-sheet (BA to 13G), surrounded by six parallel a-helicr (aB to a G), three on each side of the 13-sheet. The basic fold thesegment BA to BF segment is the classic 'Rossmann foIdl, associate with nicotinamide adenine dinucleotide binding, the BD to BG segment, i addition to being partly in the 'Rossrnann fold', governs quaterna1 association and substrate binding. 178-HSD i differs significantly frorr the reported structures of other short-chain dehydrogenases by th presence of two insertions, each comprising a helix-turn-helix moti These helices have an amphiphilic nature and are located at one end (
the substrate-binding cleft away from the catalytic triad, restrictin access to the active site and influencing substrate specificity. One (
more of these helices may also be involved in the reported association (
the enzyme with membranes (Lin et al., 1996).

Chaprer 1. Introduction
More recently, the crystal structure of the 17B-HSD1-E, complex has been determined. The initial structure of this complex was built into a difference Fourier map for which the FCaI, and phase vlues were calculated from the 1713-HSD1 structure. The structure of the complex has been refined to give an R-factor of 0.194 with data to 2.3 A (Azzi el al., 1996). The structure of the complex demonstrates in detail the interactions between the substrate and residues Ser 142, Tyr 155, His221 and Glu282 of the enzyme. These interactions and the complementary of the substrate with the hydrophobic binding pocket make criticaI contributions to the enzyme specificity. The above results provide a strong basis for the design of potent inhibitors of this pivota1 steroid dehydrogenase.
1-2.6 Phase diagram for crystallization of 17R-HSD1
Using micro-batch method, phase diagrams for the crystallization of 1713-HSDl were determined, as a function of the polyethylene glycol (PEG 4000) and protein concentrations of human estradiol dehydrogenase. While the protein concentration decreased from 15 mglm1 to 0.84 mg/ml, the solubility curve was determined with increasing PEG (4000) concentration from 6% to 15% (wlv). The precipitation, nucleation, metastable and undersaturation zones were further determined. These results have shown an important feature for crystallization of 1713-HSD1. Crystals of 178-HSD1 can be obtained from the precipitation zone. These results are very useful for improving the crystal quality and for the crystallization of 178-HSD1 complexed with various ligands, including cofactors, substrates, and inhibitors. More recently, combined with the soaking method, higher quality crystals were obtained for 1713-HSDI complexed with estrone, testosterone, 20a-OH-progesterone, 17a-methyl- estradiol-NADP+. These results are also useful to establish a new methud for crystallization of other types of human 17B-HSDs.
1.3 Thesis outline

Chapter I . Introduction
The subsequent chapters of the thesis are organized in the followii way. Chapter 2 provides a detailed description of the enzyn preparation for crystallization of 1713-HSD1, including a basic method f rapid purification of 178-HSDI from human placenta. Chapter 3 presen different optical properties of the apoenzyme and holoenzyme forms 178-HSD 1 by spectrophotometer and fluorometry. In chapter cry stallization of 176-HSD 1 is described, including the search for detergent to increase 176-HSD 1 solubility, screening and crystal grow of 17B -HSD-NADP+, the crystallization of a human estradil dehydrogenase-substrate(estradio1) cornplex, prelirninary study i
different methods for crystallization of 178-HSD1-EM-139 ar crystallization of 178-HSD 1 under microgravity . Chap ter 5 presents tl structure determination of 178-HSD1. Chapter 6 reports a further stuc of the phase diagram for crystallization of 178-HSD1, including ti precipitation, nucleation, metastable and undersaturation zones. Usir these results, combined with the soaking method, seven forms of crystal in new complexes were obtained. The main concIusion of this thesis ar prospective work are summarized in chapter 7. The last chapter is 2
appendix, including three papers in crystallization and crystallography.
It should be noted that, apart from chapters 1, 6, 7, each of the othe chapters constitutes the body of a publication in an internationa refereed journal since 1992. Chapter 2 is consist of two papers "Subuni identity of the dimeric 178-hydroxysteroid dehydrogenase from Humai placenta." (Lin et al., 1992 b) and "Rapid purification yielding highl: active 178-hydroxysteroid dehydrogenase: application of hydrophobi~ interaction affinity fast protein liquid chromatography" (Yang et al. 1992).
Chapter 3 is consist of a paper "Human Placentai 170-hydroxysteroii dehydrogenase: Optical properties of its cornplex with NADP+-" (Zhu et al, 1995 a).
Chapter 4 is consist of five papers:

Chapter I . Introduction
1). "Crystal growth of human estrogenic 17B-hydroxysteroi~ dehydrogenase." (Zhu et al., 1994)- 2). "Crystallization and pretiminary X Ray diffraction analysis of the complex of Human placenta1 17B hydroxysteroid dehydrogenase with NADP+." (Zhu et al., 1993)- 3). "Th1 crystallogenesis of a human estradiol dehydrogenase-substrate " (Zhu e al., 1996)- 4). "Preliminary study of different methods for crystallizatioi of human estradiol dehydrogenase-inhibitor complexes" (Zhu et al., 199' a). 5). "Crystallization of Human estrogenic 17B-Hydroxysteroic Dehydrogenase under microgravity" (Zhu et al., 1995 b).
Chapter 5 is consist of the paper "Structure of human estrogenii 170-hydroxysteroid dehydrogenase at 2.20 A resolution" (Ghosh et al. 1995).
The appendix is consist of three papers: "Crystal structure of huma estrogenic 178-Hydroxys teroid Dehydrogenase complexed with 17E estradiol" (Azzi et al., 1996). "Crystallization and the preliminar crystallographic studies of the Azurin Pseudomonas fluoresces." (Zhu i
al., 1994 b) and "Preliminary Crystallographic analysis of the Snak muscle Fructose 1,6-bisphosphatase" (Zhu et al., 1997 b).
1.4 References
Adams E. F., Coldham N. G. & James V. H. (1988) Journal of Endocrinolog: 1 18: 149- 154.
Agacwal, A. K., Monder, C., Eckstein, B. & White, P. C. (1989) J. Biol. Chem. 264: 18939-18943.
Azzi A., Rehse P. H., Zhu D.-W., Campbell R. L., Labrie F., and Lin S.-X (1996). Nature Structure Biology 3: 665-668.
Bernal, J. D. & Crowfoot, D. (1934) Nature (London), 133, 794-795.
Breton R., Yang F., Jin J.-Z., Li B., Labrie F. & Lin S.-X. (1994) J. Steroic; Biochem. Molec. Biol. 50, 275-282.
Bugg C. E. (1986). J. Crystal Growth. 76: 535.

Chapter 1. Introduction
Burns D. J. W., Engel, L. L., and Bethune J. L. (1971) Biochem. Biophy. Res. Comm. 44: 786-791.
Burns D. J. W., Engel, L. L., and Bethune J. L. (1972). Biochemistry, 11: 2699.
Cacioppo, E., Munson, S. and Pusey, M. L, (1991) 3. Crystal Growth, 110 66-7 1.
Carter, C. W. Jr & Carter, C. W. (1979) J. Biol. Chem. 254: 12219-12223.
Casey M. L., Macdonald P. C., and Crsson S. (1994) J . Clin. Invest. 94: 213: -2141.
Chayen, N. E., Akins, J., Campbell-Smith, S. and Blow, D. W. (1988) J Cryst. Growth, 90: 112-1 16.
Chin C. C., Dence J. B. and Warren J. C. (1976) J. Biol. Chem. 12: 3700- 3705.
Chin, C.C., Warren, J.C. (1973) Steroid , 22: 373-378.
Chin, C.C., Murdock, G.L., Warren, J. (1982) Biochemistry 21: 3322-3326.
Couture, P, Theriault, C, Sirnard, J. & Labrie, F. (1993) Endocrinology. 132: 179-185.
Delucas, L. J. and Bugg, C. E. (1988) Trends Biotechnol. 76: 188.
Descomps, B., Nicolas, J.-C., Crastes De PauIet, A. (1968) Bull. Soc. Chim. Biol. 50: 1681-1692.
Dounce, A. t. & Allen, P. T. (1988) Trends Biochem. Sci. 13: 317-320.
Dumont, M., Luu-The, V., de Launoit, Y., Labrie, F. (1992) J. Steroi~ Biochem. Mol. Biol., 41: 605-608.
Edery, M., Goussard, J., Dehennin, L., Scholler, R., Reiffsteck, J., Drosclowsky, M. A., Eur. J. Cancer (1981) 17: 115-120.
Engel, L. L., Groman, E. V. (1974) Rechent Progress in Hormone Researcl 30: 130-169.
Feigekon, R. S., (1986) J. Crystal Growth. 76: 529-532.

Chapter 1 . Introduction
Feuer, E.J., Wun, L.M., Boring, C C , Flanders, W.D., TirnmeI, M.J., Tong, ? (1993) J . Natl. Cancer Inst ., 85: 892-897.
~ran t , C., Watson, D., Longcope, C.. Steroids (1979), 34: 563-573.
Geissler, W.M., Davis, D.L., Wu, L., Bradshaw, K.D., Patel, S., Mendonca, B.B, Elliston, K.O., Wilson, J.D., Russell, D.W., Andersson, S. (1994). Nat. Gener 7: 34-39.
Giegé, R., Lorber, B., and Théobald-Dietrich, A. (1994) Acta Cryst. DSO 339-350.
Giegé, R. (1988). J. Cryst. Growth. 90: xi-xiv.
Ghosh D., Week C. M., Grochulski P., Duax W. L., Errnan M., Rimsay R. 1 and Orr J. C. (1991) Proc. Natl. Acad. Sci. U.S.A. 88: 10064-10068.
Ghosh, D., Erman, M., Pangborn, W., Duax, W. and Baker, M. E. (1992) J Steroid Biochern. Molec. Biol. 8: 849-853.
Ghosh, D., Wawrzak, Z., Weeks, C. M., Duax, W. L. & Erman, M. (1994) Structure 2: 629-640.
Ghosh, D., Pletnev, V. Z., Zhu, D.-W., Wawrzak, Z., Duax, W. L., Pangborn W., Labrie, F. and Lin, S.-X. (1995) Structure 5: 503-513.
Gilliland, G. L. & Bickharn, D. M. (1990) Methods: a companion to Method, in Emzymology, Vol. 1 , New York: Academic Press, pp. 31-37.
Hans Jïrnvall, Bengt Persson, Maria Krook, Silvia Atrian, Rose Gonzàle; Duarte, Jonathan Jeffery, and Debashis Ghosh (1995) American Chernical Society 34: 6003-6013.
Hobkirk R. (1993) Trends in Endocrinology and Metabolism 4: 69-74.
Inano et al., (1983) Eur. J. Biochem., 129: 691.
Inano, H. and Tamaoki, B. (1986) Steroids 48: 1-26
Inano, H. (1988) Biochern. Biophy. Res. Comm. 152: 789-793
Jany, K. D., Ulmer, W., Froschle, M. and Plleiderer, G. (1984) FEBS Lett.

Chapter 1. Introduction
Jancarik, J. & Kim, S. H. (1991) J. Appl. Crysf. 24: 409-411.
Jarabak, J., Adams, J, A., Williams-Ashxnan, H. G., and Talalay, P. (1962) J Biol. Chem. 237: 345.
Jarabak, J. (1969) Methods Enzymol 15: 746-752.
Jarabak, J. , Street, M. A. (1971) Biochemistry 10: 3831-3834.
James L. Thomas., Marie C. Larochelle., Douglas F. Covey and Roneld (
Strickler(l983) J. Biol. Chem. 258:11500.
Jiu-Zhen Jin, Arezki Azzi, Jing-Yu Wang and Sheng-Xiang Lin (1993) J Chrornatogr. 614: 159-163.
Karn et al., (1978) J. Moi. Biol. 123539-555.
Karavolas, H. J., and Engel, L. L. (1966) J. Biol. Chem. 241, 3454.
Karavolas, H. J., Orr, J. C., and Engel, L. L. (1969) J. Biol. Chem. 244: 4413.
Karavolas, H. J., Baedecker, M. L., and Engel, L. L. (1970) J. Biol. Chem 245: 4948.
Kroes R. L. and Reiss, D. (1984) ibid. 69: 414.
Krozowski, Z. (1 992) Moi. Cell. Endocrinol. 84: C25-C3 1
Labrie C., Martel C., Dufour, J.-M., Lévesque, C., Mérand Y., and Labrie F (1992) Cancer Res., 52: 610-615
Labrie, F., Dupont, A., Bélanger, A. (1985) Important advances i, oncology. (eds). J . B. Lippincott: pp . pp 193 -21 7.
Labrie, F., Dupont, A., Bélanger, A. (1986) Endocrine Rev. 7: 67-74.
Langer, L. J., and Engel, L. L. (1958) J. Biol. Chem. 233: 583,
Lattman, E. E. (1994) Protein: Structure, Function and Genetics, 18: 101 106.
Lawrence J. D. et al- (1989) Science 246: 652-654.

Chapter 1. Introduction
Lévesque, C,, Mérand, Y., Dufour, J. M., Labrie C. and Labrie F. (1991: J.Med. Chem., 34: 1624-1 630.
Lewis L. Engel and Ernest V. Groman (1974) Recent Progress in Hormonc research 30: 139-169.
Littke W. and C. John, (1984) Science 225: 203
Lin, S.-X., Shi, 1.-P., Cheng X.-D. and Wang, Y.-L. (1988) Biochernistry,, 27: 6343.
Lin, S.-X., Brisson, A., Liu, J.-H., Roy, P. H. and Lapointe J. (1992 a), Protein Expression PuriJ, 3: 41.
Lin S.-X., Yang, F., Jin, J.-Z., Breton, R., Zhu, D.-W., Luu The, V., Labne, F, (1992 b) J Bi01 Chem 267: 16182-16187.
Lin S.-X., Zhu D.-W., Azzi A., Campbell R., Breton R., Labrie F., Ghosh D., Pletnev V., Duax W. L., Pangborn W. (1996) J. Endocrinol. .150 supplemat 513-520.
Luu The, V., Labrie, C., Zhao, H.F., Couët, J., Lachance, Y., Simard, J., Leblanc, G., Côté, J., Bérubé, D., Gagné, R., Labrie, F. (1989) Mol Endocrinor 3: 1301-1309.
Luu The, V., Labrie, C., Simard, J. Lachance, Y., Zhao, H.F., Couët, J., Leblanc, G. and Labrie, F. (1990) Mol. Endocrinol. 4: 268-275.
Marekov, V., Krook, M. & Jornvall, H. (1990) Fed. Eur. Biochem. Soc. 266: 5 1-54.
Markey, (1984). FEBS Lett., 167: 155.
Martel, C., Rhéaume, M., Takahashi, C., Trudel, J., Couët, J., Luu-The, V., Simard, J., and Labtie, F. (1992). J. Steroid Biochem. Mdec . Biol .41: 597- 603.
McNeill, L.M., Reed, M. J., Beranek, P. A., Booney, R. C., Ghilchik, M. W., Robinson, D. J. & James V. H. (1986) International Journal of Cancer 38 193-196.
McPherson, A. (1990) Eur. J . Biochem. 189: 1-23.
McPherson, A. (1991) 3. Cryst. Growth, 110: 1-10.

Chapter 1. Introduction
McPherson, A. (1992) J. Cryst. Growth, 122: 161-167.
Michel, H. (1983) Trends Biochem. Sci. 8: 56-59.
Michel, H. (1995) The Sixth International Conference on Crystallization Biological Macromolecules. P-2, 60.
Mikol, V. & Giegé, R. (1989) J. Cryst. Growth, 97: 324-332.
Mouridsen, H., Palshof, T., Patterson, J., and Batlersby, L. (1978) Canc Treat. Rev. 5, 131-141.
Murdock, G.L., Chin, C.C., Warren, J.C. (1986) Biochemistry 25: 641-646.
Nicolas, J. C., Pons, M., Descomps, B., and Crastes de Paulet, A. (1971 FEBS Lett. 23: 175.
Pasteur L. ( 19 86) Recherches sur la Dissymetrie Moltculaire ( 1861 1883). edited by C. BOURGEOIS. Paris, France.
Peltoketo, H., Isomaa, V., Maentausta, O., and Vikho, R. (1988) FEBS Let1 239: 73-77.
Peltoketo, H., Isomaa, V., and Vikho, R. (1992) J. Biol. Chern. 209: 45! 46 6.
Perutz, M. F. (1969) Eur. J. Biochem. 8: 455-466.
Poulin R. and Labrie F. (1986) Canace Res. 46: 4933-4937.
Puranen, T. J., Poutanen, M. H., Peltoketo, H. E., Vihko, P. T., Vihko, R. 1 (1994) Biochem J. 304: 289-293.
Pusey, M., Witherow, W., Naumann, R. (1988) ibid. 90: 105.
Reichert, E. T. & Brown, A. P. (1909) The differentiation and Specificity t
Corresponding Proteins and Other Viral Substances in Relation Biological Classification and Evolution: the Crystalliza fion of Hernoglobin Washington, DC: Carnegie Institution.
Richard J. Auchus., James O. Palmer, H. L. Carrell, and Douglas F. Covey
(1989) Steroids, 53:77.

Chapter I . Introduction
Richardson, J. S. (1981) Adv. Protein Chem. 34: 167-339.
Salemme, F. R. (1972) Arch. Biochem. Biophys. 151: 533-539.
Samudzi, C. T. & Fivash, M. J. (1992) J. Cryst-al Growth, 123: 47-58
Santner S. J.. Santen R. J. (1993) J. Steroid Biochem. Molec. Biol. 45:383 390.
Saurabh S. Lawate., Douglas F. Covey (1990) J. Med. Chem., 33: 2319.
Scheel, H. J. (1993) Handbook of Crystal Growth, Vol. 1, edited by D. T. 1 Hurle, Amsterdam: North Holland,,pp. 1 -42.
Simard, J., Feunteun, J., Lenoir, G., Tonin, P., Normand, T., Luu-The, V, Vivier, A., Lasko, D., Morgan, K., RouIeau, G., Lynch, H., Labrie, F., an1 Narod, S. (1993) Hum. Mol. Genet. 2: 1193-1199.
Stura, E. A., Nemerow, R. & Wilson, 1. A. (1992) J. Crystal Growth, 122 273-285.
Sumner, J. B. (1948) J. Washington Acad. Sci. 38: 113-117.
Susan S. Hoog, John E. Pawiowski, Pedro M. Alzari, Trevor M. Penning and Mitchell Lewis. (1994) Proc. Natl. Acad. Sci. USA. 91: 2517-2521.
Sweet ,F., Boyd, J. , Medina O., Konderski, L., Murdock, G. L. (1991 Biochem. Bioph. Res. Commun., 180: 1057
Talalay, P. (1963) Enzymes, 2nd Ed. Vol. 7. New York. Acadcemic Press., pp. 177-202.
Tarn, M., Taylor A and Giri, L. (1984) Presented at the 4th Internafiont Symposium on HPLC of Proteins, Peptides and Polynucleotides, Baltimore
MD, Tarn, M. , Taylor A and Gin, L. Wingo, B.A., Tony, T., Bolden, S (1995) CA Cancer J Clin 45: 8-30.
Thomas et al., (1983) J. Biol. Chem. 258:11500.
Tremblay, Y., Ringler, G. E., Morel, Y., Mohandas, T. K., Labrie, F., Strauss, J . F., and Miller, W. (1989) J. Biol. Chem. 264: 20458-20462.

Chapter 2 . Introduction
Trevor M., Penning and Joseph W., Ricigliano (1991) J. Enzyme lnhibiti~ 5: 165-198.
Varughese, K.I., Skinner, M. M., Whiteley, J. M., Matthews, D. A. &Xuon N. H. (1992) Proc. Natl. Acad. Sci. USA. 89: 6080-6084.
Vermeulen A., Deslypere J. P., Paridaens R., Leclerq G., Roy F. & Heusent I. C. (1986) European Journal of Cancer and Clinical Oncoligy 22: 515-52
Weber, P. (1990) Methods: a Cornpanion to Methods in Enzymology, Vc 1, pp. 31-37. New York: Academic Press.
Wingo, P. A., Tong, T., Bolden, S. (1995) Cancer statistics, Ca-A Canc Journal for Clinicians 45: 8-30.
Wu, L., Einstein, M., Geissler, W.M., Chan, H.K., Elliston, K.O., Andersson, : (1993) J. Biol. Chern. 268: 12964-9.
Yang, F., Zhu, D.-W., Wang, J.-Y., Lin, S.-X. (1992) J. Chromatogr. 582: 71 76.
Zeppenzauer, M. (1971) Methods Enzymol. 22: 253-266.
Zhu, D.-W., Lee, X., Breton, R., Ghosh, D., Pangborn, W., Duax, W. L. and Lin S.-X. (1993) J. Mol. Biol., 234: 242-244.
Zhu, D.-W., Lee, X., Labne, F. and Lin, S.-X. (1994) Acta. Crystallogr. D 5C 550 -555 .
Zhu, 0.-W., Dahms, T., Willis, K., Szabo, A.G., Lee, X. (1994). Archives oj Biochemistry and Biophsics 1:469-470.
Zhu, D.-W., Jin, J.-Z., and Lin, S.-X., (1995 a). J. Steroid Biochem. Molec Biol. 1 : 77-81.
Zhu, D.-W., Zhou, M., Mao, Y. and Lin, S.-X. (1995 b). J. Crystal Growth 156 108-111.
Zhu, D.-W., Arezki, A., Peter, R., and Lin, S.-X. (1996). J. Crystal Growt,

Chapter 2
Preparation of 17R-HSD1 for crystallization

Chapter 2 . Preparation of 17J-HSDI for crystallization
In the crystallization of biological macromolecufes the quality a quantity of the required material is important. Although experimenti may have no choice, difficulties in crystal growth may be linked to 1
nature or source of the biological material. Purification, stabifizatii storage, and handling of macromolecules are therefore essential ste p i o r to crystallization (Ducruix and Giegé, 1992). As a general ri purity and homogeneity are regarded as conditions sine qua nt however, quality of the macromolecules will depend upon the way t h are prepared. Once crystals can be produced which are suitable for X-I analysis, additional material will be needed to improve their quality a size and to prepare heavy-atom derivatives. For these reasons it essentiai that isolation procedures are able to supply enough fresh l î HSDl of reproducible quality. In fact, Our experiments have also shol that the preparation of a highly homogenous enzyme is the fi. important step for crystallization of 1713-HSD1 and obtaining its 3 structure.
Chapter 2 is consist of two papers "Subunit identity of the dime: 178-hydroxysteroid dehydrogenase from human placenta." and "Rar purification yielding highly active 17f3-hydroxys teroid dehydrogena: Application of hydrophobic interaction affinity fast protein liqu chromatogrophy". The purification method described in the first paper used in principle for al1 the enzymes' purification for crystallization later experiments. In the second paper, we have shown an improv purification that NADP+ elution was very efficient in an improv purification resulting in a homogeneous preparation of high yield.
During the first p e n d of my Ph. D. study, 1 had a chance to join those experiments. It is very important for me to master the techniq of FPLC system and purification of protein. Combining the methods us in above two papers by using three FPLC chromatographies: Q-Sephara ion exchange, Blue-Sepharose CL-6B affinity (with NADP+ elution) a. phenyl-Superose hydrophobic interaction chromatographies, we ha obtained a homogeneous and highly active preparation from huma

Chapter 2. Preparation of I7J-HSDl for crystallization
placenta. This resuIt provides a sound basis for crystalllogenesis study (
17B-HSDI.

Chapter 2. Preparation of 2 7J-HSD 2 for crystallizatiun
2.1 Subunit identity of the dimeric 178-hydroxystei dehydrogenase from Human placenta
For this paper, 1 have participated in purification of 170-HSDl froi human placenta, including assay of 17B-HSD 1, gel electrophoresii determination of protein concentration and purification.
* 2.1 is adapted €rom the paper of "Subunit identity of the dimeric 170 hydroxysteroid dehydrogenase from Human placenta." by S.-X. Lin, F Yang, J.-2. lin, R. Breton, D.-W. Zhu, V.-L. The, F. Labrie in J. Biol. Cherr (1992) 267: 16182-16187.

Chapter 2. Preparation of I 7J-HSD 1 for crystallization
Subunit Identity o f the Dimeric 178-Hydroxyste Dehydrogenase from Human Placenta
Sheng-Xiang Lin, Fu Yang, Jiu-Zhen Jin, Rock Breton, Dao Zhu, Van Luu-The, and Fernand Labrie
From the Medical Research Council Group in Molecular Endocrinolog Centre Hospitalier de 1'Université Laval Research Center and Lav University, Quebec G1V 4G2, Canada
Human placenta 17B-hydroxysteroid dehydrogenase has bec purified with a new rapid procedure based on fast protein liqu: chromatography, yielding quantitatively a homogeneous preparation wi high specific activity catalyzing the oxidation of 7.2 pmo1 i
estradiol/min/mg of enzyme protein at 23'C, pH 9.2. This preparatic was shown to have a subunit mass of 34.5 kDa by sodium dodec: sulfate-polyacrylamide gel electrophoresis while having a moIecular mai of 68 kDa by both Superose-12 gel filtration and native pore gradient g electrophoresis. When 170-hydroxysteroid dehydrogenase wz expressed in HeLa ce1Is or overproduced in insect cells using tt baculovirus expression system, both from its cDNA encoding a protein t
34 kDa, the enzyme had the same migration in native and sodium dodec: sulfate-gel electrophoresis as the purified one from human placenta an eluted from the Superose-12 column at the same elution voIumi Moreover, al1 the above forms of this enzyme have similar specif activity. These results clearly demonstrate the identity of the thrc enzyme foms. The enzyme produced from the cDNA is expressed as dimer, and its two subunits are identical. 1713 - Hydroxysteroid

Chapter 2. Preparation of I7J-HSDI for crystallization
dehydrogenase subunit identity is thus proved. The NH2- t e r m i n analysis revealed a unique sequence of Ala-Arg-Thr-Val-Val-Leu-Ile f the purified enzyme from placenta, further confirming the abo, conclusion.
2.1.1 In troduct ion
178-hydroxysteroid dehydrogenase type 1 (17B-HSD1) (E.C. 1 .l. 1.6 found in the soluble subcellular fraction of the human placenta responsible for the interconversion of estrone and estradiol, and to less extent, dehydroepiandrosterone to 5-androstene-30,178-di01 (Dumont al., 1992). The cDNA and gene(s) (Luu-The et al., 1989, 1990) encodii this enzyme have been isolated. Mapping of the gene by in si hybridization shows that it is localized in the q.121 band of chromoson 17. Recent studies by Hall et al. (1990) indicate that the q region chromosome 17 is associated with the candidate gene(s) responsible f the early-onset familial breast cancer. Since it is well known th estradiol can stimulate the proliferation of mammary tumor cells, th observation strenghtened the potential for 1713-HSDl to be responsib for early onset of breast tumors (Reed et al., 1991; Reed, 1991). Hum; placental 17B-HSDl has been purified partialfy or to appare homogeneity by several groups (Langer and Engei, 1958; Descomps et a 1968; Jarabak, 1969; Karavolas et al., 1970), however, relatively hil specific activity has been achieved only in some reports (e.g. Burns et a 1972; Chin and Warren, 1973; Murdock et al., 1986). The convention column procedures used during purification are usually long, th1 limiting to scale-up and irnprovement of the purification for furthl mechanism and crystallization studies. 17B-HSD has been studied sint the late fifties, but its dimeric nature was not elucidated until tt seventies (Langer et al., 1959; Jarabak and Street, 1971; Burns et al 197 1,1972). Nevertheless, Jarabak and Street (1 97 1) concluded th although their results would suggest that the subunits are identica;

Chapter 2 . Prepararion of I7J-HSDI for crystallization
further studies would be necessary to confirm this possibility; whil Burns et al. (1 97 1.1 972) pointed out that 17B-estradiol dehydrogenase i a dimer of probably identical subunits.
Here we report a rapid purification protocol based on FPLi chromatography, in which an analytical system has been adapted fc preparative purposes. The whole process now takes 3-4 days as oppose to severd weeks for other protocols using conventional columns. Thi method yields a pure 170-HSD1 preparation which exhibits a specifi activity much higher than previously reported. The enzyme from tw human placentas is purified to more than 95% homogeneity (10-15 mg by a two-step chromatographic procedure: anion-exchange and affinit: chromatography. An additional hydrophobic interact io chromatography step resulted in more than 99% pure preparation. W then investigated the dimeric structure of the enzyme and demonstratel its subunit identity with the aid of 1713-HSD1 expressed in HeLa ce11 andjor overproduced in insect cells with the baculovirus expressioi system using the same placental cDNA (Luu-The et al., 1989; Dumont e al. 1992).
2.1.2 Materials and methods
Reagents - 1713-Estradiol, estrone, LI-mercaptoethanol, and DTT we: from Aldrich. NAD+, NADH, PMSF, glycerol, nitro blue tetrazolium, ar phenazine methosulfate were purchased from Sigma. Protein marke (low rnolecular weight) for SDS-PAGE and native pore gradient gels we: obtained from Bio-Rad and Sigma, respectively, while those for Superos1 12 gel filtration were from Pharmacia LKB Biotechnology Inc. TI Sepharose Fast Flow and Blue-Sepharose CL-6B columns were packed t ourselves with Pharmacia chromatographic media and columns, while tl Superose-12 HR 10130, phenyl-Superose HR 10110, and Mono-Q HR 5, columns were bought from Pharmacia LKB. Al1 reagents were the highe grade available. [4-14C]-Estradiol (56.4 mCi/mmolo) was a Du Pont-Ne England Nuclear product. Centricon and Centri-prep concentrators fc

Chapter 2. Preparation of 17J-HSDI for crysfallization
buffer exchange and sample concentration were from Amicon. Thin- layer chromatography plates (60f254) were obtained from Merck.
Assay of I 7 J - H S D I - When the enzyme was assayed by specsophotometric measurement of the reduction of the NAD+ indicated by the absorbance increase at 340 nm, the mixture contained 25 pM estradiol, 0.5 mM NAD+ in 50 mM NaHC03-Na2C03 buffer, pH 9.2, similar to (Langer and Engel, 1958) but with some modifications. For example, a lower estradiol concentraiton than reported was used to maintain transparency of the assay mixture (compare Jarabak, 1969; Karavolas et al., 1970). This measurement was used throughout the purification procedure and a blank value obtained using the same reaction mixture but containing no estradiol was substracted.
- Estradiol + NAD+ - Estrone + NADH + El?
A direct assay of the formation of estrone from estradiol was carried out using [14C] estradiol (30 pCi/pmol) under similar conditions . The reaction was initiated by addition of 1713-HSD1. At different time intervals, aliquots were taken and mixed immediately with 2 ml of chilied CH2Cl2 to stop the reaction. Nonradioactive estradiol (20 ng) and estrone (20 ng) were added to each sample as radioinert tracers. The steroids were extracted twice with CH2C12 and the solvent was evaporated under nitrogen. Estradiol and estrone were separated by thin-Iayer chromatography on 60f254 gel plates using a 4:l (v:v) mixture of benzene and acetone. The steroid positions were identified by localization of nonradioactive steroids after development with iodine vapor, then cut and counted in scintillation liquid (Formula 989, Du Pont- New England Nuclear) containing 1% ethanol (Wahawsan and Gorell, 1980; Simard et al., 1991).
Another method was used to easily detect the 17B-HSD1 activity

Chapter 2 . Preparation of I7J-HSD I for crystallization
peak after gel filtration with high sensitivity (Fig. 2.1.3). This was base on the increase in fluorescence due to NADH formation. NAD fluorescence was detected using 350 and 450 nm for excitation an emission wavelengths (both dits were 8 nm), respectively, with an SLI 8000 or Hatachi F 2000 fluorometers.
Al1 assays were carried out at 23 f 1°C. One unit of enzyme :
defined as the amount of enzyme that catalyzes the formation of pmol product in one minute under the above conditions (se "Discussion").
Gel electrophoresis - Native or SDS gel electrophoreses were tank out in the Laemmli discontinuous buffer system (Laemmli, 1970) usin the Bio-Rad Mini-PROTEAN II (or PROTEAN II) or Pharmaci PhastSystems. The samples contain 2-4 pg of each protein in the Min: PROTEAN system (4-10 pg for PROTEAN II) and 0.2-0.5 pg i PhastSystem. The latter accelerates analysis during purificatio procedure. Native gels were run in 8-25% acrylamide gels to cornpa the three 17B-HSD1 froms in the PhastSystem while SDS-PAGE in 125 gels were used in both types of apparatus. The samples contained les than 10% glycerol and 0.5-1% SDS in SDS-PAGE. The gels were staine with Coomassie Blue (Bio-Rad, 1990) or "High biue staining" fror Pharmacia for higher sensitivity, where 0.05% Coomassie Blue, 10$ methanol, 9% acetic acid and 7% ammonium sulphate were used in th staining step.
The molecular weight of 17B-HSDl was determined in 4.5-10% por gradient gels under nondenaturing conditions similar to Sigma (1986: except with use of Bio-Rad slab gels. The logarithm of the relativ mobility (RF) of each protein was plotted against percent of ge concentration. The dope of such a plot is the retardation coefficient. Th logarithm of the negative slope was then plotted against the logarithm O
the molecular weight of each marker. This established the calibratioi

Chapter 2 . Preparation of 1 7J-HSDZ for crystallization
curve from which the molecular weight of 170-HSD1 was determined.
Specific Enzyme Staining for 171-HSD 1 - One of the duplicate native gels after migration was immediately incubated in a reaction mixturl containing 100 m M tris-HCI, pH 8, 25 p.M estradiol, 0.5 mM NAD+* O.: mg/ml nitro blue tetrazolium and 0.1 mg/ml phenazine methosulfate The incubations were carried out in the dark at 37OC until the dark blul bands developed which indicated the active enzyme (about 0.5-3 h) The resulting specific enzyme staining was compared with the proteii bands shown by Coomassie Blue on another native gel. The gels werc finally treated with a preservative solution containing 5% glycerol anc 10% acetic acid, then dned and stored.
lmmunoblot analysis - 178-HSD1 samples were subjected to SDS O:
native gel electrop horesis and then electroblotted ont0 nitrocellulose lose. The blots were then washed and treated with rabbit polyclona anti-17B-HSD1 serum and 1251-labeled goat anti-rabbit immunoglobulii G as described elsewhere (Ausubel et al., 1987; Luu-The et al., 1989).
Determination of Protein Concentration - The optical method c Warbung and Christian (1942) [protein (mglml) = 1.55 A280-0.76 A260 was used. The optical density was determined with a Beckman DU-7( spectrophotometer. A Microcell which requires only 50 pl samples wai used tu determine protein concentration. The bicinchoninic acid proteii assay (Pierce, 1989) give similar results.
Superose-12 Gel filtration - The Superose-12 HR 10130 column wa equilibrated with buffer A (see below) containing 0.15 M NaCl both fo the protein standard andlor the 17B-HSD1 sample. The sample volume! were 100 to 200 pl while a flow rate of 0.4 mllmin and a fraction size O
200 pl were used.
NHz-terminal Sequence Analysis - 100 to 200 pmol of purified 178- HSD1 from placenta (referred to as native enzyme hereafter) wa3

Chapter 2 , Preparation of 17J-HSDI for crystallization
equili brated with a buffer containing 10 m M ammonium bicarbonate carbonate at pH 7.8, 0.15 mM DTT and 0.04% n-octyl glucoside using i
Centricon concentrator. The sample was applied to a 12-mm diamete trifluoroacetic acid-treated GFC filter. The filter was initially treatei with BioBreneTM plus (polybrene) and then cycled through one sequena of Edman chemistry, which improves the efficiency of subsequen sequencing. The sample was sequenced in an AppIied Biosystems, Mode 473 A instrument.
Purification steps - The chrornatographic steps utilized a Pharmaci FPLC system at room temperature. The active enzyme fractions werc moved as soon as possible to 4OC in the presence of 20% glyccrol. Whei preparations were to be stored overnight, they always contained 504 glycerol and were kept at -20°C. During the homogenization an( ammonium sulfate fractionation, a buffer containing 40 mM Tris-HCI, pE 7.5, 0.25 M sucrose, 5 mM EDTA, 7 mM 8-mercaptoethanol, and 1 mk PMSF was used. In al1 chromatographies, a buffer solution containing 4( mM Tris-HCI, pH 7.5, 1 m M EDTA, 0.2 m M DTT and 20% glycerol wa: used as the low ionic strength buffer which is referred to herein a! buffer A. During Q-sepharose chromatography, 0.5 mM PMSF was addet to the gradient buffer while in later chromatographies, only 0.2 mN PMSF was included.
Construction of Expression Vector and Transient Expression il
mammalian HeLa cells. A fragment of human placental 170-HSD1 cDNA containing the entire coding region and the 3'-untranslated region wa! inserted downstream to the methallothionein promoter of the modifiec pHs 1 vector. Recombinant plasmids (pHs- 170-HSD) were prepared bj the alkaline lysis procedure (Maniatis et al., 1989) and purified by twc cesium chloride - ethidiurn bromide density gradient centrifugations. Fifteen pg of vector was used to transfect human cervical carcinomr cells (HeLa) using the calcium-phosphate procedure (Kingston, 1987),

Chapter 2. Preparation of 17J-HSDI for crystallization
Cells were plated initially at 104 cells/cm2 in 175 cm2 Falcon culturc flasks and grown in Dulbecco's modified Eagle's medium containing 104 (v/v) fetal bovine serum supplemented with 2 mM L-glutamine, 1 mh sodium pyruvate, 100 IU penicillin/ml and 100 pg streptomycii sulfate/mI. Mock transfections were carried out with pHs1 vector alone Cells were harvested by scraping with a rubber spatula and suspendet in 0.5 ml of buffer containing 50 m M potassium phosphate, pH 7.4, 20% glycerol and 1 mM EDTA.
Extraction of Expressed I7J-HSDI from HeLa cells - Each lot of 20 x
106 cells were suspended in 3 ml buffer A in the presence of 1 mh, PMSF and disrupted by sonication (3 x 0.5 min at 0.5 min intervals) ir an ice-bath with a sonicator from Sonics & Materials Inc., CT, with outpu control at 4. The ce11 disruption was controlled by microscopie observation. No intact cells could be detected after sonication.
The suspension was centrifuged at 10,000 x g for 30 min to removc the ce11 debris. The supernatant was either made 0.82 M in ammoniun sulfate prior to loading on a phenyl-Superose column or concenuatec 10-fold with a Centricon concentrator to be used directly for moleculai weight estimation by gel filtration.
The expressed 178-HSD 1 was partially purified on a phenyl, Superose column using the fast purification of 17B-HSDl of the higt specific activity method described under "Results" and thereaftei concentrated. This preparation was used to compare the Stokes radii oi different 17B-HSD forrns in this study by Superose-12 gel filtration oi gel electrophoresis.
Overproduction of Recombinant 17J-HSDl in Insect Cells Using Baculovirus Expression system - A 1.2-kilobase pair cDNA encoding the human placenta 17B-HSD1 was cloned into the transfer vector pVL1393 Recombinant plasmid (pVL/17B-HSDl), purified by two cycles of CsCl

Chapter 2 . Preparation of I7J-HSD I for crysra llization
density gradient centrifugation, and Autographa californica nuclea polyhedrosis virus (AcNPV) genomic DNA were used to cotransfec momolayer Spodoprera frugiperda (Sf9) cells by the calcium-phosphatl precipitation method (Summer and Smith, 1987). Recombinan baculovirus was cloned by plaque purification. Sf9 cells in suspensior culture were infected with recombinant virus a t a multiplicity oi infection of 10 and incubated a t 28 OC for 60h. Infected cells wen harvested by centrifugation (10 min at 1000 x g), resuspended in 2
buffer (0.25 mM Tris-HC1, pH 7.5, 20% (vlv) glycerol, 0.1 mM EDTA, 0.2 rnM DTT, 20 pM NAD+, 0.2% (w/v) sodium cholate, 0.5 pg/ml pepstatin A, 5 pg/ml Ieupeptin, 5 p.g/mI chyrnostatin, 5 pg/ml antipain, 5 pg/ml aprotinin, 5 m M benzamidine, and 1 m M phenylmethanesuIfony1 fluoride), and frozen at -70°C.
Purification of Overproduced 17J-HSDI in Insect Cells The infected ceIls were disrupted by sonication in the same way as
the above-mentioned HeLa cells and the debris was removed by centrifugation at 10,000 x g during 30 min. The homogenate was partially purified by a Mono-Q FPLC column or a blue-Sepharose affinitj column. Both the homogenate and the purified 17B-HSDl were analyzed to confirm the molecular mass of the enzyme.
2.1.3 RESULTS
Fast Purification of 171E-HSDI of High Specific Activity
Homogenization and Ammonium Sulfate Fractionation
Two placentas were homogenized with a Waring blender and about 0.6 kg of tissue was obtained and submitted to a two-step ammonium sulfate fractionation. Fractions precipitating between 30 and 55% saturation were collected and diluted with buffer A to a conductivi ty

Chapter 2 . Prepararion of I7J-HSDI for crystaiiization
close to that of the same buffer containing 0.09 M NaCl. This sample wa then loaded on Q-Sepharose. The sample volume was about 1200 m (this purification protocol was used for three or four placentas).
Q - S e p harose chromatography. - The above sample was loade directly on a Q-Sepharose column (50 mm x 170 mm) and washed wit: buffer A containing 0.5% Triton in the presence of 0.09M NaCl tl separate other proteins during sample loading. A gradient of 0.09-0.5 h NaCl was run in the sarne buffer. The 170-HSDl activity eluted at abou 0.22M NaCI. A flow rate of 12 ml/min could be used in sarnple loadin; (with a joint function of two P3500 pumps) and 6 ml/rnin in the elution Fractions with four times or more of the enzyme specific activity thai that of the applied sampie were collected (Table 2.1).
Blue-Sepharose CL-6B chromatography - The above fractions werl diluted 4-fold with buffer A (to about 500 ml) to be layered directly oi a blue-Sepharose column (16 x 90 mm) at a flow rate of 3-4 ml/min About 40% of the proteins passed directly through the column an4 possessed no steroid dehydrogenase activity. The column was thel washed with 80 m M NaCl in buffer A and about 15% contaminath] protein was rernoved. Finally, the enzyme activity was eluted by 4 mh NAD+ in the same buffer at a fiow rate of 2-3 ml/min (adapted for Lin e al., 1988). After this step, the enzyme was about 95% homogeneous, a judged by SDS-PAGE and gel scanning (Amersham PAS system and GL 1000 software).
Pheny 1-Superose chromatography - The trace impurity detected i , the blue-Sepharose fractions was easily separated on a phenyl-Superosi hydrophobic interaction column (HR 10/10). The fractions were madi 0.82M in ammonium sulfate in buffer A and loaded ont0 this column a about 1 mllmin. Most pruteins and NAD+ were not retained under thesi conditions. The column was then washed with 0.74 M arnmoniun sulfate, followed by a reverse gradient of 0.74-0 M ammonium sulfate il
the same buffer. The enzyme activity eluted at the very end of thi

Chapter 2 . Preparation of I7J-HSD I for crystallization
gradient, when the ammonium sulfate concentration reached O (Fig.2.1.1).
Concentration and storage - M o s t fractions from the main peal (excluding the final trailing region) showed only one band on SDS-PAGE which accounted for more than 99% of the applied protein as judged by gel scanning. The above homogeneous fractions of 17B-HSD1 were then pooled and concentrated with Centticon o r Centri-prep concentrators to about 2-3 mg/ml. The buffer was adjusted to 50% glycerol, 0.4 rnM DTT and 40 mM Tris, pH 7.5. This preparation was stored at -20°C and the high specific activity (see below) was maintained for many months.
Placenta1 17J-HSDI specific activity - The homogeneous phenyl- Superose fraction can catalyze the oxidation of about 7.2 i 0.3 pmol of estradiol/mg of enzyme within I min at pH 9.2 and 23*C, or with an apparent turnover nurnber of 8 . 2 1 ~ ~ the specific activity being 2-3-fold that of preparations obtained by conventional methods (See "Discussion") The high specific activity may be mainly due to the elimination of in vitro modifications (See "Discussion").
Recombinant 17J-HSDI Preparations and Their Specific Activity - The phenyl-Superose fraction of expressed 178-HSD 1 in HeLa cells revealed about 30% purity by gel scanning (Fig. 2.1.2). As this fraction has an activity of 2.2 units/rng, the corresponding pure enzyme activity is extrapolated to be 7.4units/mg, similar to that of native 17B-HSD1 purified directly from human placenta. The overproduced and partially purified 176-HSDl (Mono-Q chromatography) afforded about 60% pure enzyme (Fig. 2.1.4). The homogeneous blue-Sepharose fraction of the overproduced form had a specific activity of 7.5 units/mg. Thus al1 the native or recombinant 1713-HSDs are identical in specific activity.
SDS-PAGE and lmmunoblotting - In SDS-PAGE, the phenyl- superose fraction from placenta showed a unique band on Coomassie Blue and

Chapter 2. Preparation of 2 7P-HSD 1 for crystallizarion
silver staining, indicating a high purity of > 99%, as dernonstrated by g scanning. The two recombinant foms of 178-HSD1 showed the san band after rnigrated (Fig.2.1. 2). In immunoblotting with SDS-PAGE, a three forms of 17B-HSD1 exhibited a unique hybridized band, whic migrated at the sarne distance. A plot of the relative mobility versus I( molecular mass, using protein standards, gave an apparent molecul mass of 34.5 kDa for ail f o m s (data not shown).
Superose-12 gel filtration - 170-HSD1 activity of human plancen from our prepration eluted in a syrnmetric peak (not shown in Fig. 2.1.3 at an elution volume very close to that of the bovine serum album: standard (66 kDa). A standard cuve relating Kav, i .e . , the fractions of tI
stationary gel volume which is available for diffusion of a given solu species (Pharmacia Fine Chemicals, 1982) and the logarithm of molecul, mass of each protein standard was established (Fig. 2.1.3B). Since the Ki for I7B-HSD1 is 0.297I 0.020, its molecular mass was estimated at f kDa. The apparent molecular mass determined by SDS-PAGE is 34.5 kD demonstrating that the 1713-HSD1 is composed of two subunits, similar i
the results reported by Jarabak and Street (1971) and Burns et a (1971,1972). 17B-HSD1 expressed in HeLa celIs either in the ce11 extra( or partially purified with a phenyl-Superose column had exactly tt same elution volume of its activity as the pIacentat enzyme in the gi filtration (Fig. 2.1.3A). Moreover, both the ce11 extract or the Mono-1 fraction of overproduced 1713-HSDl in Sf9 cells demonstrated the sam elution volume on this column. The same Stokes radius indicates th; 17B-HSD1 frum three sources is identical in molecular mass. The tw subunits are identical because the two recombinant forms wei expressed from a single cDNA which encodes a 34-kDa protein.
Native Gel Electrophoresis and Speciflc Enzyme Staining - The pur placental 176-HSD1 migrated as a single band in native gc electrophoresis. The other forms of recombinant 17 B-HSD 1 also had protein band at the same level (Fig. 2.1.4A). Al1 three forms showed a active protein band at the same migration level after specific enzyrr

Chapter 2. Preparafion of 17J-KSDI for crysfallization
staining, thus further indicating their identity (Fig. 2.1.4B) and verifyir the equivalence of the two subunits of 170-HSDl (the upper band of 2 Fig. 2.1.4A was also shown to respond to the 17B-HSDI antibody immunoblotting).
In pore gradient gels, identical dopes of RF versus gel concentratic were found for the native and overproduced 17B-HSDI. A molecul; mass of 68 kDa was deduced (Fig, 2.1.4C), further confirming the resul of Superose-12 gel filtration. It is clear that the enzyme is composed 4
two identical subunits.
N H a -terminal Sequence - 100-200 pmol 17B-HSDI purified froi human pIacenta was sequenced. The intial yield was 47 pmo1 and tt repetitive 94%. The analysis clearly showed a unique NHz-termin; sequence of Ala-Arg-Thr-Val-Val-Leu-Ile, thus confirming the identii of the two subunits in 178-HSD1. This result is in cornplete agreemei with the published DNA sequence of Luu-The et al. (1989), but differs i the second residue reported by Burns et al. (1972) where Glu was foun instead of Arg.
2.1.4 Discussion
Due to the crucial importance of 170-HSDl in the conversions (
hormone steroids rnany studies have been devoted to its mechanism (
action (Jarabak and Street, 1971; Burns et al., 1971, 1972; Murdock et al 1986) and inhibitors (Chin and Warren, 1975; Tobias et a . 198: Thomas et al., 1983). However, its structure-function relationships hav not been totally ciarified. For example, its subunit structure and th
relation between this structure and enzyme activity have not bee clearly elucidated.
In the present study, we verified the subunit and molecular mass (:
this enzyme, and dernonstrated the subunit identity by cornparin enzyme preparations purified from placenta and expressed in HeLa ce11

Chapter 2 . Preparation of 17J-HSD 1 for crystallization
or overproduced in Sf9 cells. Al1 preparations had identical properties ix different gel electrophoretic systems, gel filtration, and specific activit] studies. Since recombinant 170-HSD1 is expressed from a single cDNA encoding a protein of 34 kDa and shows a molecular mass of 68 kDi exactly Iike the placenta enzyme, the subunits of 178-HSD1 must bc identical. The unique NHz-terminal sequence obtained from the purifiec placenta enzyme further supports this concfusion. The kinetic behavioi and the interactions of the two subunits are presently undei investigation.
2713-HSD1 has been purified partially or to apparent homogeneity bq several groups. Different specific activities have been reported ever when precautions were taken to protect the enzyme from cola inactivation (Descomps et al., 1968; Jarabak, 1969; Karavolas et al., 1970: Burns et al., 1972; Chin and Warren, 1973; Murdock et al., 1986). We suggest some general comparisons. First, the definition of the unit of the enzyme should be clarified. In most papers dealing with 178-HSD1, activity is related to the absorption increase in a 3 ml cuvette e.g. Jarabak (1969) defined one unit as the amount of enzyme reducing 1 pmol of pyridine nucleotidelmin; initial rates obtained from assays were converted to enzyme units by dividing the change i n absorbance/min by 2.07 for the systems containing NAD+ or NADP+; Karavolas et al. (1970) defined 1 unit as the amount of enzyme thal catalyzes the conversion of 1 pmol of substrate/min/3 ml, similar ta that of Jarabak (1969). Therefore, they obtained values in micromoles of substrate conversion per min in 3 ml and the final enzyme unit values they derived would be 3 times those of pmollrnin for the substrate conversion. We suggest that it would be much clearer to divide the experimentally derived change in absorbancelmin by 6.22 to obtain the value in m mollrnin (as the molar extinction coefficient for NADH at 340 nm is 6220), whichis equivalent to pmol/ml/min or unitlml, taking
pmol/min as the enzyme unit (we can subsequentIy multiply the former by the volume to get the total units or divide the former by the enzyme

Chapter 2. Preparation of I7J-HSDI for crystailization
concentration in mglml to get the specific activity in unitsimg). The uni should correspond to a certain amount of enzyme and its value shoulc not be affected by the assay volume. It is now possible to compare tht different specific activities from different preparations in pmol/min/ml enzyme protein (pmol in substrate or cofactor conversion). Indeed Jarabak and Street (1971) stated that the enzyme specific activitie! obtained by two methods (Jarabak, 1969; Karavolas et al., 1970) werc between 1.5 and 2.0 unitslmg of protein (25OC). The actual value ir pmol/min/mg of protein could be still lower taking into account thc coefficient they used in calculating the units. Similarly, the real specific activity reported by Burns et al. (1972) could be 2.6-2.7 pmol/min/mg of enzyme protein at 22 + 1°C. We also obtained 2-3 pmoliminlmg ir this laboratory using conventional chromatography with the same assay.
As rnost of the previous work and Our present study used the conditions very similar to those of Langer and Engel (1958), (pH 9.2, anc saturating concentrations of NAD+ and substrate), we can Say that the 7.2 pmol/min/mg (23 -t. 1°C) we obtained from Our present fast purificatior is about 2-3-fold higher than most reported values using conventional chromatography. With our rapid procedure we also deduced higei specific active in other purifications such as that of glutamyl-tRNA synthetase (Lin et al., 1992). The high specific activity obtained by fasi purification may be due to the elimination of protein rnicroheterogeneitj which is often caused by in vitro modifications, such as oxidation. reduction eifects or partial proteolysis (Giegé et al., 1986; Giegé, 1987) The rapidity is due to the high flow rate and high resolution of FPLC columns and also to the reduction in the number of procedural steps, e . g , the sample dialyses was completely avoided between steps which usually requires substantial periods of time and is often accompanied by partial enzyme inactivation, Chin and Warren (1973) developed a simple and relatively quick preparation with a specific affinity column, but the standardized purification protocol presently used in theii laboratory is based on reactive blue 2-agarose and gel filtration columns

Chapter 2. Preparation of I7J-HSDI for crystallization
(Murdock et al., 1986), probably due to the difficulty to obtain estrio 16 hemisuccinate affinity media. Rapid purification plays an importai role in Our physico-chemicai study of this enzyme and especially for i crystallization in which a highly homogeneous protein is required.
A c k n o w l e d g e m e n t s
We thank Dr. J. Côté very much for aid in placenta hornogenatic and fractionation and Dr. P. Savigne (Pharrnacia) for helpfd discussior on PhastSystem gel electrophoresis. We are very grateful to Drs. Jentoft and A. Rosler for creful reading of the manuscript and to Elaj Leclerc for manuscript preparation. We also thank Drs. J. Lagueux an S. Bourassa for help in NH2-terminal sequence analysis of 17B-HSD1.
2.1.5. References
Ausubel, F. M., Kingston, R.E., Moore, D.D., Seidman, T.G., Smith, T.A., ar Struhl, K. (1987) (eds) in Currenr Protocols in Molecular Biology, pp. 67 675, Wiley and Sons, New York.
Bio-Rad,(l990) Mini-prorein II Dual Slab Cell, Instruction Manual, Bi Rad.
Burns, D.J.W., Engel, L. L., and Bethune, J.L. (1971) Biochem. Biophys. Re: Commun. 44, 786-792.
Burns, D.J.W., Engel, L. L., and Bethune, J.L. (1972) Biochernistry f l 2699-2703.
Chin, C.C. and Warren, J.C. (1973) Steroids 22, 373-378.
Chin, C.C. and Warren, J.C. (1975) J . Biol. Chem. 250, 7682-7686.
Descomps, B., Nicolas, J.-C., and Crastes de Paulet, A. (1968) Bull. Soc Chim. Biol. 50, 1681-1692.
Dumont, M., Luu-The, V., de Launoit, Y., and Labrie, F. (1992) J. Stero. Biochem. Mol. Biol., 41, 605-608.

Chapter 2 . Preparation of I 7J-HSD I for crystallization
Giegé, R. (1987) in Crystallography in Molecular Biology (Moras, D. et a eds), vol. 126: 15-26. Nato Asi Series Plenum Press, New York.
Giegé, R., Dock, AC., Kern, D., Lorbere, B., Thierry, J.C., and Maras, : (1986) J. Crystall. Growth 7 6 , 554-561.
Hall, J.M., Lee, M.K., Newman, B., Morrow, J.E., Anderson, LA. , Huey, E King, M.C. (1990) Science 250: 16841-1689.
Jarabak, J. (1969) Methods Enzymol. 15, 746-752.
Jarabak, J. and Street, M.A. (1971) Biochemistry 10, 383 1-3834.
Karavolas, S., Baedecker, M.L. and Engel, L.L. (1970) J. Biol. Chem. 24: 4948-4952.
Kingston, R.E. (1987) In Current Prorocols in Molecular Biology (Ausubc F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, T.G., Smith, T.A. Struhl, K., eds), pp. 9.1.1-9.1.4, Wiley & Sons, New York.
Laernmli, U.K. (1970), Nature, 227, 680-685.
Langer, L. J. and Engel, L.L. (1958) J . Biol, Chem. 233, 583-588.
Langer, L.J., Alexander, J.A. and Engel, L.L. (1959) J . Biol. Chem. 231 2609-26 14.
Lin, S.-X., Shi, J.-P., Cheng, X.-D., and Wang, Y.-L (1988) Biochemistry 2' 6343-6348.
Lin, S.X., Brisson, A., Liu, J.H., Roy, P.H., Lapointe, J. (1992) Prote Expression and Purification, 3, 41 -44.
Luu The, V., Labrie, C., Zhao, H.F., Couët J., Lachance, Y., Simard, 1 Leblanc, G., Côté, J., Bérubé, D., Gagné, R., and Labrie, F. (1989), M t Endocrinol. 3, 1301-1309.
Luu-The, V., Labrie, C., Simard, J., Lachance, Y., Zhao, H. F., Couët, :
Leblanc, G., Labrie, F. (1990) Mol. Endocrinol. 4: 268-275.
Maniatis, T., Fritsch, E.F. & Sambrook, J. (1989) Molecular cloning, laboratory manual. Coid Spring Harbor Laboratory, Cold Spring Harbc New York.

Chapter 2 . Preparation of 17J-HSDI for crystallization
Murdock, G.L., Chin, C.C. and Warren, J.C. (1986) Biochernistry 25, 64 646.
Pharmacia Fine Chemicals (1982) Gel Filtration, Theory and Practi Pharmacia Fine Chemicals, Uppsala, Sweden.
Pierce,(1989) BCA Protein Assay Reagent, Instructions, Pierce Chernic Co., Rockford, IL.
Reed, M.J. (199 1) J . Endocrinol. 129, 163-1 65.
Reed, M.J., Singh, A., Ghilchick, M.W., Coldham, N.G., Purohit, A. (1991) Steroid Biochem. Mol. Biol. 39: 791 -798.
Sigma (1986) Nondenatured protein molecular weight marker k Technical Bulletin No. MKR- 137, Sigma Chernical Co.
Simard, J., de Launoit, Y., and Labrie, F. (1991) J. Biol. Chem. 266, 2484 2 8 8 4 5 .
Summer, M.D. and Smith, G.E. (1987) A ManuaI of Methods f Bacculovirus Vectors and lnsect Ce11 Culture Procedures (Texas A and University, College Station, Bulletin no. 1555.
Thomas, J.L., LaRochelle, M.C., Covey, D.F. and Strickler, R.C. (1983) J . Bi, C h e m . 258 , 11500-
Tobias, B., Covey, D.F. and Strickler, R.C. (1982) J. Biol. Chem. 257, 278 2794.
Wahawsan, R. and Gorell, T.A. (1980) Steroids 36, 115-129.
Warboung, O., and Christian, W. (1942) Biochem. J . 310, 384-421.

Chapter 2. Preparaf ion of 17J-HSD I for crystallization
Fig, 2 , l . l . Pheny I-Superose (HR 10110) chromatography of 178-HSD1. The sample was loaded in the presence of 0.82M ammonium sulfate an a reverse-phase gradient eluted 17B-HSDl at O M ammonium sulfat (peak II). Peak 1 is NAD+ contaminant with A260 k i n g greater than A280
Fig. 2.1.2. SDS-PAGE of 17B-HSD1 from three different sourcl 1. Expressed in Hela cells and partially purified; 2. Purified directly from human placenta; 3. overproduced in a baculovirus expression system and purified b blue-Sepharose CL-6 Baffinity chromatography ; 4. Protein standards of 97.4, 66.2, 45, 31, and 21.5, kDa from top t
bottom.
Fig. 2.1.3. Superose -12 Gel filtration of 1 7 4 HSD1 : A, elution Profile. Samples of 0.1-0.2 ml were loaded on an HR 10/31 column and eluted at 0.4 mllmin. , Protein standards (667 kDa and 6' kDa) ; *, activity of purified enzyme from placenta measured by spectrophotometer; O, activity of expressed enzyme in HeLa cells an1 partially purified, measured by a spectrofluorometer. B, molecular mass evaluation from Kav (see Superose- 12 gel fil tratio: under "Resufts"). Standards were thyroglobulin (667 kDa), bovine serur: albumin (66 kDa), ovalbumin (43 kDa), B-lactoglobulin (35 kDa] chromatoglobin A (25 kDa), and potassium chromate (194 kDa).
Fig. 2.1.4, Native gel electrophoresis shown by Coomassie BI staining (A) and active enzyme staining (B) of the placer 17B-HSD1 from three ' sources: 1. partially purified 176-HSD1 expressed in HeLa cells; 2. overproduced in bacculovirus expression system and partially purifiec on Mono-Q; 3.178-HSD 1 purified from human placenta.

Chapter 2. Preparaîion of I7J-HSD 2 for crystallization
C . evaluation of 17B-HSD1 molecular mass by pore gradient gels. TI standards are: a-lactalbumin (14.2 kDa), carbonic anhydrase (79 k D a chicken egg albumin (45 kDa), bovine serum albumin (66 kDa fi monomer and 132 kDa for dirner), and urease (272 kDa for trimer) fro Sigma.

Chapter 2 . Preparation of 17J-HSD I for crystallization
Table 2.1.1 Purification of 17B-HSD1 from human placentas
Process Total Total S pecific Yield Purificatic protein activity activity
m g unit s units/mg % -folc
Ce11 extract 30,860 108 0.0035 100 1
50% Ammonium su1 4,15 4 9 8 0.024 9 1 7.4 -fate precipitation
Blue-Sepharose 8.4 4 6 5 .5 4 2 1,74
Phenyl-Superose 5.0 3 6 7.2 3 2 1,84
a This is a purification from about 600 g of human placenta tissue.

Chapter 2. Preparation of 1 7J- HSD I for crystallization
Fig. 2.1.1
-
-
-
. . . . . . . . . . . . . . . . 1 I l 1
.- 0.0 20 40 60 80
ELUTION VOLUME (ml)
. . . . . .
1
0.4
- 0.2
2

Chnpter 2. Prepnration of 17J-HSDI for crysrallizntion
Fig. 2.1.3
VOLUME (ml) M O ~ C U L A R WEiGHT ( x IO
Fig. 2.1.4
1; i 1 IO MOLECULAR WEIGKT 100 (X la3) iaaa
C

Chapter 2 . Preparation of I7P-HSD 1 for crystallization
2.2. Rapid purification yielding highly active 1 hydroxystero id dehydrogenase : a p p l i c a t i o n hydrophobie interaction and affinity fast protein Li c h r o m a t o g r a p h y
I have participated in most processes of purification, includin placenta homogenization, ce11 extract , 17B-HSDI assay, gt electrophoresis, protein concentration measurements and purification.
* 2.2 is adapted from the paper of "Rapid purification yielding high active 176-hydroxysteroid dehydrogenase: application of hydrophob interaction affinity fast protein liquid chromatogrophy." by Fu Yang, Da< Wei Zhu, Jing-Yu Wang and Sheng -Xiang Lin in J. Chromatogr. (199: 582, 71-76.

Chapter 2 . Preparafion of I7J-HSD I for ctystallization
Rapid purification yielding highly active 170-hydroxyst dehydrogenase: application of hydrophobic interaction ai fast protein liquid chromatography
Fu Yang, Dao-Wei Zhu, Jing-Yu Wang and Sheng-Xiang Lin
MRC Group in Molecular Endocrinology, CHUL Research Center and La University, Ste-Foy, Quebec GI V 4G2 (Canada)
Homogeneous human placenta 17B-hydroxysteroid dehydrogena was obtained by a procedure consisting of two fast protein liql chromatographic (FPLC) steps using Phenyl-Sepharose hydrophot interaction and Blue-Sepharose affinity columns. In the fi: chromatography, the enzyme eluted only when an additional decrease ionic strength was inserted after the ammonium suIfate concentrati had reached zero, thus enhancing the separation. In the affin: chromatography, separation of contaminating proteins occurred different stages of loading and washing. The specific elution of t
enzyme by the CO-factor NADP+ is very efficient in obtaining homogeneous preparation in high yield. The rapidity of FPLC was furti increased by a maximum simplification of the intermediate steps, and i
whole procedure lasted only two days. This preparation has a yield more than 50% and a high specific activity, catalyzing the formation 7.9 pmol of estrone from estradiol per minute at pH 9.2 and 23OC. It k an apparent molecular mass of 35000. This provides an efficie candidate for the purification of other membrane-associated proteins.

Chapter 2. Preparation of I7J-HSD I for crystallization
2.2.1 Introduction
The development of protein structure studies requires high homogeneous and active proteins. Since the early 1980s, fast prote liquid chromatography (FPLC) has been demonstrated to be very efficie in preparing high-quality proteins (Markey, 1984; Tam et al., 1984; Lin ai., 1988; Orbe11 et al,, 1988; Franke et al., 1990). We have demonstrati the importance of the rapid purification to enzyme activity (Lin et a 1991; Lin et al., 1992), which has been shown to be extremely importa for crystal growth and other physicochemical studies of proteins (Orbe et al., 1988; Franke et al., 1990; Lin et al., 1992; Giegé et ai., 1988; Ruff al., 1988; Ruff et al., 1991).
A further need in the application of FPLC is the maximu simplification of the preparative steps. 17B-Hydroxystero, dehydrogenase type 1 (170-HSD1, EC 1.1.1.62) is responsible for tl conversion of most active androgens and estrogens, thus being of cntic importance both in basic research and in the therapy of breast ar prostate cancers (Beaulieu, 1960; Labne et al., 1982; Labrie et al., 198 Hall, 1990; Reed, 1991; Reed et al., 1991). In this work, we used tl hydrophobicity of 170-HSD 1 from human placenta, and purified efficiently first using a Phenyl-Sepharose hydrophobic interactic colurnn. Combined with an affinity column (Blue-Sepharose CL-6B), H
were able to purify 170-HSD1 within two days, yielding highly actii enzyme. With this method, the dialysis process is completely avoide and the sample volume for loading is markedly reduced. This protoci may also be used for other membrane-associated proteins.
2.2.2 Experimental
2.2.2.1 Materials
NAD+, NADP+, glycerol, phenylmethylsulphonyl fluoride (PMSF) an protein standards for gel permeation chromatography were obtained

Chapter 2. Preparation of 2 73-HSDI for crystallization
from Sigma (S t, Louis, MO, USA). 17fbEstradio1, 2-mercaptoethanol (B-SI and dithiothreitol (DTT) were purchased from Aldrich (Milwaukee, W USA). Protein markers (low molecular mass) for sodium dodecyl sulpha polyacrylarnide gel electrophoresis (SDS-PAGE) were supplied by Bio-Rz Labs. (Richmond, CA, USA). Phnyl-Sepharose and Blue-Sepharose CL-6 columns were packed in the laboratory; the empty columns wei obtained from Pharmacia LKB (Montreal, Canada). Al1 reagents were i
the best grade availabIe. Centricon-30 and Centri-prep-30 concentrato were purchased from Amicon (Beverly, MA, USA).
2.2.2.2 17B-HSDI assay
The enzyme was assayed by measuring NAD+ reduction monitore by the increase in absorption at 340 nm with a Beckman DU-7 spectrophotometer. The reaction mixture contained 25 pM estradiol, O mM NAD+ in 50 mM sodium hydrogencarbonate buffer (pH 9.2). A enzyme unit (U) is defined as the amount of enzyme protein th; catalyses the reduction of 1 pmol of NAD+ in I min under the ab01 conditions. The absorbance change per minute is divided by 6.22 to yiel the reduction of NAD+ in mM/min, which is equivaient to the number i
enzyme units per ml (mM/min = U/ml). A blank value using the sarr reaction mixture but containing no estradiol was subtracted durin assays throughout the purification. The reaction is as follows:
L
Estradiol + NAD+ - Estrone + NADH + I-??
The resulting homogeneous 176-HSDl activity was checked with a direc assay of the formation of [14C]estrone from [14C] estradiol under simila conditions. At different time intervals, aliquots were taken and th1 reaction was stopped with cold dichlorornethane. Estradiol and estront

Chapter 2 . Preparation of I7J-HSD I for ciystullization
were extracted and separated by thin-tayer chromatography, then c and counted in a scintillation liquid, as described by Wahawsan a;
Gorell, 1980.
2.2.2.3 SDS-PAGE
SDS-PAGE was carried out in the Laemmli discontinuous buff system (Laemmli U.K., 1970) using a Bio-Rad Mini-Protean II Pharmacia Phast S ystem. The gels were 12% polyacrylamide-N',t bismethyl-eneacrylamide (37:l) and the samples contained 0.5% SDS ai 10% glycerol. When doing immunoblotting, duplicate gels were run: o. underwent Coomassie Brilliant Btue staining and the other w eiectroblotted with nitrocellulose. The blots were was hed and treati with polyclinal anti-17B-HSD 1 serum from rabbit and 1251-labelled an: rabbit immunoglobulin G from goat, similarly to the method of St. Joh 1987.
2.2.2.4 Protein concentration measurements
We used the optical method of Warburg and Christian (1942) and tl
measurements were made with a Beckman DU-70 spectrophotomete For determination of homogeneous 1713-HSD 1, a microcuvette requirii 50 pi of sample was used.
2.2.2.5 Purification steps
Unless mentioned otherwise, buffer A, which contained 40 mM Tri HCl (pH 7.9, 1 mM EDTA, 0.2mM DTT and 20% glycerol, was used as tl principle buffer of low ionic strength after placenta fractionation. Pharmacia FPLC system was used, consisting of two P3500 pumps, a U7 M monitor and an LCC-500 controller.
2.2.2.6 Placenta1 homogenization and ce11 extract

Chapter 2 . Preparation of I7J-HSDI for crystallization
The placenta were refrigerated on ice immediately after delivr and treated within 45 min. The umbilical cord, blood clots a membranes were removed and the placenta were washed with O.! sodium chloride Irrigation USP (neutralized). About 260 g of wet tiss obtained per placenta, which was then cut into pieces and mixed w 520 ml of buffer containing 50 mM Tris-HC1 (pK 7.2), 7 m M 0-SH, 0.25 sucrose, 5 mM EDTA and 1 mM PMSF. The tissue was then ground in liquefier-blender (Osterizer) three times for 10 s at 1-min intervals. 1
the above treatments were carried out in a cold room (4OC). T homogenate was centrifuged for 30 min at 800 g in a Sorvall RC centrifuge. The resulting supernatant was decanted and recentrifuged 10000 g in a Sorvall RC-5 centrifuge. The above supernatant was fina recentrifuged at 100 000 g for 60 min in a Beckman L5-65 ulti centrifuge. The centrifugations were also carried out at 4OC.
The supernatant after the ultracentrifugation was submitted ammonium sulfate fractionation. During the fractionation, the saml was always kept neutral with ammonia solution. The enzyme fractia precipitating between 30 and 50% saturation were collected a dissolved in buffer A made 0.85 M in ammonium sulfate. The saml volume at this stage was about 250 ml per placenta.
2.2.3 Results
2.2.3.1 Hydrophobic interaction chromatography
The sample obtained as described above was loaded directly on Phenyl-Sepharose column (65 mm x 26 mm 1.D) and washed with buffi A made 0.6 M in ammonium sulfate. A Iarge amount of contaminatir proteins was removed during the loading. Then a reverse gradient i
0.6-0 M ammonium sulfate in buffer A (Fig. 2.2.1) was generated by t1
LCC-500 controller, which decreased slowly at the beginning to separa well many contaminating proteins. The decrease in ammonium sulfa

Chapter 2 . Preparation of 17J-HSD I for ctystallization
concentration was fastest between 0.45 and 0.15 M during the gradier where few proteins were eluted. 17B-HSDI activity was eluted only aft the ammonium sulfate concentration had reached zero and when tl column was further washed with a buffer similar to buffer A b, containing less Tris-HCl (10 mM), yielding fractions containing up to 20' of 178-HSD1 protein, as revealed by gel scanning using an Amersha PAS system and GL-1000 software (Fig. 2.2.3, lanes 1 and 2). OnIy tl fractions with four times or more 178-HSDI specific activity than that :
the applied sample, i.e., fractions between 35 and 52 ml elution volun after 10 mM Tris buffer was applied, were collected. In this way, a 24 fold purification of the enzyme could be obtained with this column. 1 this step, a flow-rate of 10 mllmin was easily achieved with the joii functioning of two P3500 pumps. Here, 0.5 m M PMSF was added to tl buffers.
2.2.3.2 Affinity chromatography
The above-collected fractions (only about 27 ml) were applie directly to a Blue-Sepharose CL-6B column (100 mm x 16 mm I.D.) at flow-rate of 3-4 mllmin. About 45-50% of the proteins passed direct1 through the column and possessed no 17B-HSD1 activity. The coIum was then washed with 0.1 M sodium chloride in buffer A, sepstratin further about 10% of interfering proteins (over the total applied proteir and then re-equilibrated with buffer A. Stepwise washing of NADP+ w; carried out at a flow-rate of 2-3 mllrnin and the enzyme activity efute at 35 PM NADP+ in the same buffer (Fig. 2.2.2) where most fractior were hornogeneous, as verified by SDS-PAGE (Fig. 2.2.3). A furthr increase in NADP+ did not elute more 178-HSDI activity. The yield wz about 80% in this step. PMSF (0.2 mM) was included in al1 buffers durin the affinity chromatography.
2.2.3.3 Concentration, specific activity and storage

Chapter 2. Preparation of 2 7J-HSDI for crystallization
The homogeneous fractions resulting from the affinity column wei collected, then concentrated and equilibrated in a buffer containing 4 rnM Tris-HCl (pH 7.5). 1 mM EDTA, 0.4 mM DTT' and 50% glycerol wii Centricon (or Centriprep) concentrators using centrifugation at 4500 ; This preparation has a high specific activity which can catalyse th oxidation of nearly 8 nmol of estradiol per minute and per milligram (
enzyme protein (Descomps et al., 1968; Jarabak J., 1969: Karavola et al 1970; Murdock et al., 1986). When stored 'at -20°C in the above buffer i the presence of 50% glycerol, the activity can be maintained for man months.
2.2.3.4 SDS-PAGE and immunoblotting
Our preparation showed a unique band on SDS-PAGE (Fig. 2.2.3, lant 3-5). This band hybridized with polyclonal anti-17B-HSD1 serum froi rabbit in immunoblotting (data not shown). The plot of the relativ migration versus the logarithm of the molecular mass of protein standar determined the apparent molecular mass of 1713-HSD1 as 35000 (Fil 2.2.4).
2.2.4 Discussion
Placental 1713-HSD 1 is a hydrophobic and relative unstable enzyme and therefore has been relatively difficult and long to prepare to higl homogeneity by conventional chromatography (Descomps et al., 1968 Jarabak, 1969; Karavola, et al., 1970; Murdock et al., 1986). Using ITLC we have substantially simplified the chrornatographic steps to the use O
two columns. The conventional dialysis, often a time-consuming anc destabilizing step for enzyme activity, is completely avoided and thc sample volume is minimized by making use of Phenyl-Sepharose ii FPLC. On this column, the enzyme is well separated because of its higl hydrophobicity. When the enzyme is eluted at nearly the lowest ionic strength possible, it is ready to be loaded on a second column, avoidinl any dialysis or dilution. In this way, the sample volumes are

Chapter 2. Preparation of 17J-HSDI for crystallization
substantially reduced.
Because of the rapidity of FPLC and the simplicity of the procedur we can obtain large amounts of 178-HSDl with high specific activic This is probably due to the elimination of protein microheterogeneii from in vivo modifications, e .g ., oxidation-reduction effects or parti; proteolysis (Giegé et al., 1987; Giegé et al., 1986) using the fast proceduri
Such a procedure may also be applied to many other hydrophob; and membrane-associated proteins. T h e laboratory-packed FPL coIurnns are fairly inexpensive and can provide quantitative and fa, preparations because of their high flow-rates and large diameters. Tt column size mentioned is suitable for purification using two placenta, bi large-scale purification can be easily achieved by increasing the colum size. For example, a Phenyl-Sepharose column of 130 mm x 26 mm 1.1
combined with a Blue-Sepharose column of 100 mm x 26 mm 1.1
afforded good separations for three or four placenta, aithough the elutio profile was slightly changed, i-e., 17B-HSDl eluted from the first colum slightly before the end of the reverse gradient, earlier than in the wor described here. After the Phenyl-Sepharose chromatography more tha a 200-fold purification was achieved.
Hydrophobic interaction chromatography is suitable for man membrane-associated proteins, e.g., human placenta1 38-hydroxysteroi dehydrogenase (Luu-The e t al., 1989), while the second step can b alternated with another high-resolution FPLC column, such as ior exchange (anionic or cationic, e.g., Pharmacia Q or S series), affinit columns other than Blue-Sepharose or geI permeation. It is important t note that the present procedure yields very reproducible result! Application of the above procedure to several other hydrophobic protein is in progress to develop a more general purification method fc membrane-associated proteins whose biological importance has bee increasingl y recognized in recen t years.

Chapter 2. Prepuration of 17J-HSDl for crystallizution
A c k n o w l e d g e m e n t s The authors thank Dr. A. Rosler for his careful reading of t
manuscript and Elaine Leclerc for its preparation.
2.2.5 References
Franke, A. E., Danley, D. E., Kaczmarek, F. S., Hawrylick, S. J., Gerard, R. 1 Lee, S. E. and Geoghegan, F. Biochim. Biophys. Acta, 1037 (1990) 16.
Descomps, B. Nicolas, J. C. and Crastes, A. de Paulet, Bull. Soc. Chim. Bio, 50 (1968) 1681.
Beaulieu, E. E., C. R. Acad Sci., 25 1 (1 960) 1421.
Markey, F. FEBS Lett., 167 (1984) 155.
Labrie, F. , Dupont, A., Bélanger, A., Cusan, L., Lacourcière, Y. Monfette, ( Laberge, J. G., Emond, J., Fazekas, A. T. A., Raynaud, J. P. and Husson, J. J. Clim. Invest. Med., 5 (1982) 267.
Labrie, F., Bélanger,A. and Dupont,A. in V. T. De Vita, S. Hellman and S. Rosenberg (Editors), Important Advances in Oncology, J. B. Lippinco, Philadelphia. PA, 1985, p. 193.
Murdock, G. L., Chin, C. C. and Warren, J. C. Biochemistry, 25 (1986) 642.
ûrbell, J. D., Guddat, L. W., Machin, K. J. and Isaacs, N. W. Anal. Biochem 170 (1988) 393.anley, F. S. Kaczmarek, S. J. Hawrylick, R. D. Gerard, S.
Jarabak, J. Methods Enzymol., 15 (1969) 746.
Hall, J. M., Lee, M. K., Newman, B., Morton, J. E., Anderson, L. A., Huey, and King, M. C. Science, 250 (1990) 15841.
Reed, M. J. J. Endocrinol., 129 (1991) 163.
Reed, M. J., Singh, A., Ghilchick, M. W., Coldham, N. G. and Purohit, A. Steroid Biochem. Mol. Biol., 39 (1991) 791.

Chapter 2. Preparation of I7J-HSD I for crystallization
Ruff, M., Mitschler, A., Cavarelli, I., Giegé, R., Mikol, V., Thierry, J. C LorbeqB. and Moras, D. J. Mol. Biol., 201 (1988).
Ruff, M., Kirshnaswamy, S., Boeglin, M., Poterszman, A., Mitschler, A Podjarny, A., Rees, B., Thierry, J. C. and Moras, D. Science, 252 (1991 1982.
Tarn, M., Taylor, A. and Giri, L. presented at the 4th Internationr Symposium on HPLC of Proteins, Peptides and Polynucleotides, Baltimort MD, 1984
Warburg, O. and Christian, W. Biochem. J., 310 (1942) 384.
Giegé, R., Lorber, B., Milkol, V., Moras, D., Ruff, M., Thobald A. an Thierry,J. C. Bull. Inst. Pasteur Paris, 86 (1988) 9.
Giegé, R., Moras, D., Drenth, J., Strandberg, B., Suck, D. and Wilson, K (Editors), Crystallography in Molecular Biology (NATO AS1 Series A, No. 126), Plenum Press, New York, 1987, p. 15.
Giegé, R., Dock, A. C., Kern, D., Lorbere, B., Thierry, J. C. and D. Moras, 1 Crystall Growth, 76 (1986) 554.
Wahawsan, R. and GorelI, T. A. Steroids, 36 (1980) 115.
KaravoIa, S., Briedcker, M. L. and Engel, L. L. J. Biol. Chem., 245 (1970 4948.
Lin, S.-X., Yang, F., Jin, J.-2. Brisson, A., Liu, J.-H. and Lapointe, J. presente~ at FASEB Meeting, Atlanta, GA, April 1 99I.
Lin, S.-X., Brisson, A., Liu, J.-H., Roy, P. H. and Lapointe, J. P r o t e i Expression Purif., 3 (1992) 41.
Lin, S.-X., Shi, J.-P., Chenga, X.-D. and Wang, Y.-L. Biochemistry, 27 (1988 6343.
T. P. St. John, in F. M. Ausubel, R. Brent, R. E. Kingston, D. D. Moore, T. G Seidman, T. A. Smith and K. Struhl (Editors), Current Protocols i Molecular Biology, wi1ey. New York, 1987, 6.7.1-6.7.4.

Chapter 2. Preparation of 174-HSDI for crystallization
U. K . Laemmli, Nature (London), 227 (1970) 680.
Luu-The, V., Takahashi, M. and Labrie, F. in L. Castagnetta, S. d'Aquino, F Labrie and H . Bradlow (Editors), Steroid Format ion, Degradation al Action in Peripheral Tissues, Academy of Sciences, New York, 1989, pl 386-388.

Chapter 2. Preparation of 17J-HSDI for crystallization
Fig.2.2.1 Hydrophob ic in terac t ion chromatography.
A 176-HSDl sample after ammonium sulfate fractionation w dissolved in the presence of 0.6 M (NH&S04 and loaded directly on Phenyl-Sepharose column (65 mm x 26 mm 1. D.). The sample was on eluted when the ammonium sulfate concentration reached zero and wi a further decrease in Tris-HCI concentration yielding fractions containii up to 20% of 178-HSD1 protein.
Fig.2.2.2 Affinity chromatography.
The Phenyl-Sepharose fractions of 178-HSD1 were loaded directly (
a Blue-Sepharose CL-6B column (100 mm x 16 mm I.D.). The column w
first washed with buffer A of low NaCl content (0.1 M) and r equilibrated with the same buffer ("O M NaCl"), then eluted with NADP Homogeneous 178-HSD1 fractions eluted at 35 pM NADP+. The fractii size is 2 ml each.
Fig.2.2.3 SDS-PAGE of different 17B-HSDl fractions.
Lanes 1-2 = fractions eluted from Phenyl-Sepharose column. Lane = protein markers; from top to bottom: phophorylase b (M,974000 serum albumin (M ,662000). ovalbumin (M, 45000). bovine carbon anhydrase (M , 3 1 OOO), main soybean trypsin inhibitor (M ,2 15 OO), ar lysozyme (M, 14400). Lanes 3-5 = 1713-HSDI fractions eluted from tl Blue-Sepharose affinity chromatography.
Fig. 2.2.4 A p p a r e n t molecular mass evaluation of 17fl-H from SDS-PAGE.
Protein markers ( 0 ) are of M , 66000, 45000, 3 1000, 21500, 144( from top to bottom.

Chaprer 2. Prepararion of I7J-HSDI for crystnllizarion
Fig. 2.2.1
ELUTION VOLUME (ml)

Chnpter 2. Prepcmrtion of I 7J-HSDI for crystcrllizntion
Fig. 2.2.4
0.4 0.8
RELATIVE MIGRATION

Chaprer 2 . Preparation of 174-HSDI for crystallizatiun
T a b l e 2.2.1 1 7 R - H S D 1 (placenta) purification by chrornatographic steps
S tep To ta1 Total Specific YieId purificatic protein activity
units {mg) (U/mg) (W ( f o ~ d )
Ce11 extract 5 0 1223 8 0.0040 100 1
50% CNH,), S04 40.6 97 1 0.0418 8 1 10.5
Phenyl-Sepharose 3 4.5 34.2 .O1 6 9 253
Blue-Sepharose 26.9 3.4 7.9 5 4 1980

Chapter 3
Human 17R-Hydroxysteroid Dehydrogenase:
Optical Properties of its Complex with NADP+

Chapter 3 Optical properiies of 17J-HSDI
In order to study the interactions between the enzyme and i cofactors, the apoenzyme of the human placental 17%-hydroxystero dehydrogenase type 1 (17B-HSDl) and its complex with NADP+ (ho1 enzyme) were prepared by modifying the purification procedure. The different optical properties were identified by spectrophotometry ai fluorometry. The optical analyses and the homogeneous apo- or ho1 enzyme preparations are important in the study of the enzyme's functic and crystallization. Using this method, we could easily identify tl cofactor binding of the enzyme. We can assay the state of 17B-HSD NADP+. It is also very useful for the crystallization of differe
complexes of 17LHSD1.
For this paper, 1 was responsible for optical analyses, includi~ protein concentration determination, absorption measurement fluorescence measurement.

Chapter 3 Optical properties of 17J-HSDl
Human 17B-Hydroxysteroid Dehydrogenase:
Optical Properties of its Complex with NADP+
Dao-Wei Zhu, Jiu-Zhen Jin and Sheng-Xiang Lin
The Medical Research Council Group in Molecular Endocrinology, CHUL
Research Center and Laval University, Quebec G1V 4G2, Canada.
* This i s adapted from the paper of "Human 17B-Hydroxysteroi Dehydrogenase: Optical Properties of its Complex with NADP+" by Dao Wei Zhu, Jiu-Zhen Jin and Sheng-Xiang Lin in J. Steroid Biochem. Molec Biol. (1995) 52: 77-81.

Chaprer 3 Optical properties of I7J-HSDI
3.1 Introduction
17B-Hydroxysteroid dehydrogenase type 1 (178-HSD 1) [ECl .1.1.62 from the soluble subcellular fraction of human placenta is responsible fa the formation of active estrogens, thus stimulating the development a breast tumors (Reed et al., 1991; Labrie et al., 1992; Lin et al. 1992). 1 is pivota1 both in endocrinology and cancer therapy. It has bee proposed that 17B-HSD1 has a dimeric structure, but different opinion were reported concerning the subunit identity (Jarabak et al., 1971 Burns et al., 1971; Burns 1972; Engel e t al., 1974). Our recent result proved that 178-HSDl is a homodimer using a combined study O
enzymology and molecular biology (Lin et al., 1992). As 170-HSDl i efficiently purified using affinity chromatography with coenzyml elution, it is important to know the coenzyme content in differen preparations (Lin et al., 1992; Murdock et al., 1986; Mendoza 1985; Yan; et al., 1992). This is very useful in the further study of subuni interactions, ligand binding and crystallization of this enzyme.
In the present work we demonstrate the marked influence of th1 coenzyme on the absorption spectra of 1713-HSD1 and the influence of th1 enzyme on the fluorescence of reduced coenzyme. This can be used tc determine different stoichiometries of coenzyme association with 178 HSD1. The apoenzyme and holoenzyme forrns are important for furthe binding and structural studies of 178-HSD 1.
3.2 Materials and methods
3.2.1 Materials.
NAD', NADP+, NADPH, phenylmethane sulfonyl fluoride (PMSF glycerol and Tris-base were purchased from Sigma. Estradiol an dithiothreitol (DTT) were from Aldrich. The Q-Sepharose Fast Flow an blue - Sepharose CL - 6B columns were packed by ourselves wit

Chapter 3 Optical properties of I7J-HSD1
Pharrnacia-LKB chromatographic media and XK columns, while tl phenyl-Superose HR10110 column was directly from the same compan Centricon and Centri-prep concentrators for sample concentration ai buffer exchange were from Amicon. Al1 reagents were of the highe grade available.
3.2.2 Enzyme Assay.
Activity of 17B-HSDl was assayed by spectrophotometr measurement of NADt reduction indicated by the absorbance increase 340 nm at 23 + 1°C. The reaction mixture contained 0.5 m M NAD+, : p M estradiol in 50 mM NaHC03-Na2C03 buffer, pH 9.2. At this pH tl
maximum activity of estradioï oxidation is obtained. A blank va11 lacking estradiol was obtained under the same condition and su btracted
- Estradiol + NAD+ b Estrone + NADH +
One unit of enzyme is defined as the amount of enzyme that catalyzc the formation of 1 pmol of product in 1 min under the above conditions
3.2.3 Enzyme Purification.
Placenta1 17B-HSD was purified in two alternative ways. The fir procedure involves three fast protein liquid chromatography (FPL( chromatographies af ter ce11 extraction and ammonium sulfa, fractionation: Q-Sepharose ion exchange, blue-Sepharose affinity (wii N A D + elution) and phenyl-Superose hydrophobic interactic chromatographies (Lin et al., 1992). Alternatively, the second procedui consists of only the first two chromatographies, in which the bluc Sepharose chromatography was canied out with NADP+ elution (Jin et ai 1993). These preparations yield homogeneous 176-HSD1 as revealed t sodium dodecyl sulfate (SDS) and native gel electrophore. They ha1 similar high specific activities catalyzing the formation of 7.5 - 8.0 pmc

Chapter 3 Optical properties of I7J-HSDI
estrone from estradiol per min per mg enzyme protein under the abov mentioned assay conditions ('Lin et ai., 1992).
3.2.4 Prote in Concentration Determinations.
The bicinchoninic acid protein assay (Pierce Chernical Co., 1989) w used to determine the protein concentrations. Protein samples we precipitated with deoxycholate and trichloroacetic acid to elimina different types of interference (Bensadoun et al., 1976; Brown et a 1989). The optical method of Warburg and Christian (Warburg et a 1941) gave slightly lower value (- 12%) than the bicinchoninic ac assay. This is similar to the results of Jarabak and Street (Jarabak et a 1971) where they found that this optical method gave prote concentration values of about 10% less than the values deduced from tl weights of the constituent amino acids (after correction for water r
hydrolysis).
3.2.5 Absorpt ion Measurements.
Absorption spectra were measured with a Beckman DU-; Spectrophotometer. When necessary, a micro-cuvette of 50 pl was use1 Spectra were stored each time after scanning. A molar extinctic coefficient of 6220 is used for the reduced form of NADP+ at 340 nm. buffer containing 40 mM Tris-HCI, pH 7.5, 1 m M EDTA, 0.2 rnM DTT, ar 20% glycerol was used as the principal buffer and is referred to herein ;
buffer A. The background contributed by buffer is subtracted for a spectra.
3.2.6 Fluorescence measurement.
Fluorescence spectra were obtained using an SLM-8000 fluoromete NADPH fluorescence emission was measured at 450 nm following a excitation at 350 nm. The slit width for excitation was 8 nm while th, for emission was 16 nm.

Chapter 3 Optical properties of 17J-HSDI
3.3 Results
3.3.1 Different preparations of 17B-HSDl leading to diffei A 280/A260 ratio.
Using the three step chromatography procedure (see Methods), w were able to obtain a preparation with an A2801A260 absorption ratio o 1.65 f O. 1. This preparation, collected from the phenyl-Superosl fractions, is referred to as form 1 which will be identified as th1 apoenzyme below. The A280/A260 of form 1 remained constant after 3 tf
4 times Centricon centrifuge to "dialyse" the free cofactor. Alternatively when blue-Sepharose affinity chromatography and NADP+ elution arl used in the last step, the A2801A260 ratio is 1.10 f 0.05, and repeatec centricon centrifuge cannot increase this ratio. This preparation i referred to as form II that will be identified as the holoenzyme below Both preparations have the same specific activity, catalyzing th4 formation of 7.5 - 8.0 pmol estrone from estradiol per min per mi enzyme protein under the aforementioned assay conditions.
3.3.2 Absorption spectra of 17B-HSD1 apoenzyme and complex with NADP+.
Form 1 exhibited a spectrum with a maximum at 278-279 nm [Fi$ 3.1 (A,a); Table 3.11, while form II exhibited a relatively broad pea between 268 and 278 nm which may be constituted by two smal maxima at 268 and 278 nm [Fig. 3.1 (A,d)]. Upon addition of estradic (20 pM final concentration) to Form II (4.57 pM final concentration), i exhibited an important absorption increase at 340 nm due to NADP- reduction. It is equivalent to 4 p M NADPH formation or a stoichiometr of 0.87 f 0.05 of the enzyme concentration [Fig. 3.1 (B, d)] as th constant A280/A260 value indicated almost al1 NADP+ are tightly boun~ under the experimental conditions. While under the same conditions form 1 exhibited a minute absorption increase at 340 nm upon estradiol

Chapter 3 Optical properties of 174-HSDI
addition [corresponding to about 0.1 stoichiornetry of the enzyn concentration, Fig. 3.1 (B, b)]. This lies within the limit of experimeni errors.
When an equal concentration of NADP+ was added to form 1, tl
A 2gojA 260 ratio shifted to 1.10 i 0.05, while the absorption spect demonstrated a broad peak region between 268-278 nm [Fig. 3.1 (A,b: Again, this ratio is constant during repeated centricon centrifuge. Th was very similar to the behavior of form II. Moreover, upon addition I
a saturating estradiol concentration as above, a 340 nm absorptic increase indicated 0.70 t 0.05 stoichiometry of enzyme concentration N A D P + reduction, the same as in the case of fonn II (data not shown When a two-fold concentration of NADP+ was added to form I solutio the absorption maximum shifted to 268 nm and the ratio A280/A2t
became 0.88 t 0.05 [Fig. 3.1 (A,c)]. This value cannot be used estimate the true stoichiometry, as it is not constant in the course I
repeated centricon centrifugations. it is however a useful vaIue to veril the existence of extra-NADP of form 1. This is also why the valui reported in Table 3.1 are very useful to indicate different NADP+ conte of a 178-HSD1 sample. The above results demonstrated that where; form 1 was free of coenzyme, the form II 17B-HSD1 molecule bour strongly one NADP+ molecule. Therefore the forms 1 and 11 ar respectively, the apoenzyme and holoenzyme. The somewh, hypochromic effect is similar to the results reported by Velick et a (1963) and Murdock and Koeppe (1964) which is further studied i
follows.
A cornparison of NADPH absorption in the absence and in tt
presence of 178-HSDl was camed out. The absorbance contribution i
NADPH at 340 nm in the presence of 4 p M 17B-HSD1 ([NADPH]/[171 HSDl] = 2) is obtained after subtracting the enzyme contribution (Fi, 3.2b). We can see clearly that the absorption contributions of NADPH i the presence or absence of 178-HSD1 are comparable. A hypochrom: effect, which accounts for about 19% of free NADH absorption, howevei

Chapter 3 Optical properties of I7J-HSD I
is shown in repeated experiments when the enzyme is present. This similar to many reports in the literature ( g Bensadoun et al 1976; Brown et al., 1989). So, the 0.87 stoichiometry of NADP+ in tl complex reflects a corrected value close to 1.
3.3.3 Fluorescence emission of NADPH foltowing an excitat at 350 nrn.
The apoenzyme of 17B-HSD1 has no fluorescence emission at 45 nm following an excitation at 350 nm. Nevertheless, the complex wii NADP+ exhibited a slight emission at 450 nm [Fig. 3.3 (b)]. Upon additio of estradiol (final concentration 20 PM) to an apoenzyme solution of relatively high concentration (3 PM), the resulting fluorescence increas was minute and within the limit of experimental errors (data not shown The addition of the same concentration of estradiol to the form : complex solution (1 PM) resulted in an important fluorescence increasi due to NADPH formation [Fig. 3 (d)]. These results are in good agreemel with the above-mentioned experiments in absorption. When cornparin Fig. 3 (d) with Fig. 3 (c), we can see that the presence of 27%-HSD enhances NADPH fluorescence and shifts the maximum from about 46 to 440 nm. This is also verified by direct addition of 178-HSDl to fre NADH solution (data not shown).
3.4 D i s c u s s i o n
Using the above mentioned optical methods, we identifie homogeneous apo- and holoenzyme forms. With these preparation! further study on enzyme-ligand interactions are being carried out an 1713-HSD1 is successfully crystallized (Zhu et al., 1993). This is the fin report on crystals suitable for complete X-ray structure determination c a human steroid-converting enzyme. The optical rnethod is simple, cleai and useful for the study of other dehydrogenases. The specific activity c 17B-HSDl reported have (7.5 - 8.0 Ulmg) is slightly higher than

Chapter 3 Optical properties of I7J-HSDI
before (e.8. 7.2 Ulmg in Lin et al., 1992). That may be due to a m a i n acceleration and simplification of the purification, which can furthc eliminate the microheterogeneity of 170-HSD1 (Lin et al., 1992; Yang al., 1992). Such higher specific activity has been obtained in repeate enzyme preparations.
The minimal affinity of NADP+ to 178-HSDI can be estimated i
follows: As mentioned in Results, the A280/A260 ratio remained constai for form II after several "dialyses", i.e., the free NADP+ concentratic should be much smaller than the bound NADP+ concentration, [NADP+] < [E-NADP+], (or [E-NADP+]/[NADP+] > 10). As when NADW is added to e equal concentration of form 1, the A28o/A260 ratio and the absorptic spectra are identical to those of form II, the molecular compositic should be the same in the two cases. As the total enzyme concentratic is equal to the total NADP+ concentration, the enzyme molecules shoul also exist mostly in the bound state, Le., free [El << [E-NA D P+]. Tt minimal affinity can be estimated:
as [El < 1/10 [E-N A D P + ] < 1/10 [Ela, while the lowest [El0 (toti concentration of 17B-HSD) in centricon was about 1.5 PM.
That is to Say, 17B-HSD1 has a strong binding site to NADP+, with K D < 0.015 PM. This is coincident with the strong elution property (
NADP+ in the enzyme purification (Jin et al., 1993). The present resuli not only helped in the purification of different enzyme forms, but als helped in 170-HSD1 crystallization. In fact, due to the strong binding c NAD P+ and the need of enzyme-coenzyme interaction study, 178-HSD 1 NADP+ complex was recently crystallized (Zhu e t al 1993). This is th first report on the successful crystallization of any steroid-convertin enzyme from human source.

Chapter 3 Optical properties of 174-HSDI
Acknowledgements
We thank Dr. F. Labrie for his interest in this work. We also think Dr J.-Y. Wang for his help in computer prograrnming, We thank Dr. J Lapointe for his careful reading of the manuscript. This work i: supported by the Medical Research Council of Canada and a Scholarshi~ award to S.-X. L. from "Le Fonds de la Recherche en Santé du Québec".
3.5. References
Bensadoun, A. and Weinstein, D. (2976) Anal. Biochem. 70, 241-250.
Brown, R.E., Jarvis, K.L., and Hyland, K.J. (1989) Anal. Biochem. 180, 136- 139.
Burns, D.J.W., Engel, L.L. and Bethune, J.L. (1971) Biochem. Biophys. Res. Commun. 4 4 , 786-792.
Burns, D.J.W., Engel, L.L. and Bethune, J.L. (1972) Biochemis try 11, 2699-2703.
Engel, L.L. and Groman, E.V. (1974) . Rec. Progr. Horm. Res. 30, 139-169.
Jarabak, J. and Street, M.A. (1971) Biochemistry 10 , 383 1-3834.
Jin, J.-Z., Azzi, A., Wang, J.-Y. and Lin, S.-X. (1993) J. Chromatogr. 614 159-163.
Labrie, C., Martel, C., Dufour, J.M., Lévesque, C., Mérand, Y. and Labrie, F. (1992) Cancer Res. 52, 610-615.
Lin, S.-X., Yang, F., Jin, J.-Z., Breton, R., Zhu, D.-W., Luu-The, V. and Labrie, F. (1992) J. Biol. Chem. 267, 16182-16187.
Mendoza-Hernandez, G., Randon, J.L. and Diaz-Zagoya, J-C. (1985) Biochem. Biophys. Res. Commun. 126, 477-48 1.
Murdock, G.L., Chin, C.C., and Warren, J.C. (1986) Biochemistry 25, 641 -646.
Murdock, A.L. and Koeppe, O.J. (1964) J. Biol. Chem. 239, 1983-1988.

Chapter 3 Optical properties of 17P-HSDI
Pierce Chemical Co. (1 989) BCA Protein Assay Reagent, Instruction: Pierce Chemical Co., Rockford, IL, USA.
Reed, M.J., Singh, A., Ghilchick, M.W., Coldharn, N.G. and Purohit, A. (199: J. Steroid Biochem. Mol. Biol. 39, 791-798.
Velick, S.F., and Furfine, C., In The Enzymes, (Edited by P . D. Boyer, H Lardy and K. Myrback). Academic Press, New York, Vol. 7, (1963) 24: 273.
Warburg, O. and Christian, W. (1941) Biochem. J., 310 , 384-421.
Yang, F., Zhu, D.-W., Wang, J.-Y. and Lin, S.-X. (1992) J . Chromatograpl 5 8 2 , 71-76.
Zhu, D.-W., Lee, X., Breton, R., Ghosh, D., Pangborn, W., Duax, W.L. and Lii S.-X. (1993) J . Mol. Biol. 234, 232-244.

Chapter 3 Optical properties of I7J-HSDI
Legends to Figures
Fig. 3.1. Absorption spectra of 178-RSD1. (A). UV absorption spectra. (a) The apoenzyme (4.57 PM) in buffer 1
(b) same as (a), plus 4.57 p M NADP+; (c) same as (a), plus 9.14 p. NADP+; (d) the holoenzyme (5 ph4) in buffer A, prepared as mentiont in the text, with a peak between 268 and 278 nm. (B). Spectra near NADPH absorption region. (a) The apoenzyme (4.57 ph in buffer A; (b) saturating estradiol (20 p M final) was added to tI
sample in (a); (c) the haloenzyme (4.57 PM) in buffer A, prepared ;
mentioned in the text; (d) saturating estradiol (20 pM final) was addr to the sample in (c) the created peak at 340 nm denotes that NADP reduction being apparently 4 FM calculated from fiee NADP+ absorptic coefficient; (e) the insert is the difference between (d) and (c), indicatir the contribution of NADPH+ formation with a maximun at 340 nm.
Fig. 3.2. The hypochromic effect of NADPH absorption in presence of 17B-HSD1.
(a) NADPH (8 PM) in buffer A in the absence of 178-HSDl and (1 NADPH (8 PM) in the presence of 17B-HSDI apoenzyme (4 PM) (Tt apoenzyme contribution is subtracted).
Fig. 3.3. Fluorescence spectra of 17B-HSD 1. (a) The apoenzyme (1 FM) in buffer A which does not fluoresce at 42
nm. (b) The addition of 1 pM NADP' to the apoenzyme. (c) Free NADP (1 PM) in the same buffer. (d) Form II complex solution (1 pM saturated with estradiol (the contribution of 178-HSD 1 and estradiol ai subtracted). Spectra (c) and (d) indicate that the presence of 170-HSD enhances NADPH fluorescence and shifts the maximum from about 46 to 440 nm.

Ctiapter 3 Optical properties of I7P-NSDl
Fig. 3.1.
250 280 310 340 370 400 WAVELENGTH hm1
O m o I I
WAVELENGTH (nm)

Chapter 3 Opfical properties of 17J-HSDI
Fig. 3.2
Fig. 3.3
200 300 400 500 WAVELENGTH h m 1
I I 1 I I 400 425 450 475 500
WAVELENGTH (nm)

Table 3.1 Absorption characteristics of 17B-HSD1 complexes with NADP+
l Characteristics
l Absorption
1 maxima (nm)
Nurnbzrs o f NADP+
apoenzyme molecuk
addcd pcr
Holoenzymc prepg
2 (see methods)
0.88 f 0.05 2.10 f 0.05
268 268-278

Chapter 4
Crystallization of 17B-HSD1

Chapter 4 Crystallization of I7J-HSDl
The crystailization of human 17B-HSD1 has been attempted since i
1970's. Dr. Chin's group (1976) had reported the crystallization of i
17B-HSDI , but there was no further report, even preliminary, on 1
structure of this crystal. Using Fast Protein Liquid Chromatography, first purified the placenta1 17B-HSDl to homogeneity and high speci activity. We increased 170-HSD1 solubility in the presence of O.Ot (w/v) octylgfucoside. Using optical method, we succeeded in analyzi complex of 178-HSD1 with NADP+ or EM-139 (inhibitor). The complex 178-HSD 1 -estradio1 has been also confirmcd by using a special procedi to saturate a high concentration of the enzyme in solution with t
substrate. These results provide a sound basis for crystallization of 17 HSDI.
Using various crystallization methods, we obtained crystals suitat for diffraction studies. This is the first diffraction quality crystals of a steroid-converting enzyme from a human source. Since 1992, thr different forms of crystals of human 170-HSDI with various ligands ha been obtained [a. 17B-HSD f -NADP+; b. 17B-HSD1 -estradid; c. 170-HSC EM139 (inhibitor)]. To eliminate rnultiseeding, formation of multicrysti and to obtain higher quality crystals, we carried out the crystallizati abroad the Russian MIR Space station and crystals were recovered January, 1994. The results provided some important information for t
space crystallization in 1994.
Depend on above results, the structures of 178-HSD1 and its compl with estradiol were determined (Ghosh et al., 1995; Azzi et al., 199( These are the first mammalian steroidogenic enzyme studied by X-r crystallographic techniques, which reveals a fold characteristic of t short-chain dehydrogenases. These results lead to the understanding 170-HSD I -ligand interaction and structure-function relationships of t enzyme. They will provide a strong basis for the design of pote inhibitors of this steroid dehydrogenase. The crystallization of 1713-HSI:

Chapter 4 Cryslallization of I7P-HSDI
provide usefu1 experience for the successful crystal growth of 0th members in the 17B-HSD family and other hydrophobic or rnembrani bound protein.

4.1. Crystal growth of Human estrogenic 17B-hydroxyst d e h y d r o g e n a s e
The paper reports the critical steps in 17B-HSD crystallizatic stabilization of the enzyme activity, detergent search to increase lt solubility, and combined screening for its crystallization. The information are very useful for the future screening for t
crystallization of 176-HSD1 and its complex with various ligani including substrates and inhibitors. In this paper, the experimcnts wt completed by myself.

Chapter 4 Crystallization of I7J-HSDI
Crystal growth of human estrogenic 17B- hydroxysteroid dehydrogenase
D.-W. Zhu, X. Lee, F. Labrie and S.-X. Lin
The Medical Research Council Group in Molecular Endocrinology, CHUL Research Center and Laval University, Quebec G1V 4G2, Canada
This is adapted from the paper of "Crystal growth of human estrogenic 17B-hydroxysteroid dehydrogenase" by D.-W. Zhu, X. Lee F. Labile and S.- X. Lin in Acta. Crystallogr. (1994). 50, 550-555.

Estrogenic 178-hydroxysteroid dehydrogenase from humai placenta, an enzyme of low solubility, has been crystallized in thc cornplex form with its cofactor NADP+. These are the first crystals witi X-ray diffraction quality for structure analysis from any human steroid, converting enzyme. The crystals were grown by vapor diffusion in tht presence of 0.06% 8-octylglucoside, using polyethylene glycol 4000 a! the precipitant (27-28%) and one of several different sdts at pH 7.5 anc room temperature. Crystals grown with magnesium chloride diffract ul to 2.4 A. The most important steps leading to the rapid success of thc crystallization of this labile enzyme were the following: preparation of : highIy active and homogeneous enzyme protein using a rapid procedure the choice of a suitable enzyme buffer system and a detergent favorablc to maintaining high activity and solubility for the enzyme; and s combined screening procedure. The present study could be useful foi the successful crystal growth of other hydrophobic or membrane-bounc proteins.
4.1.1 Introduction
The crystallization of human 1713-hydroxysteroid dehydrogenase (178-HSD) has been attempted since the 1970s (Chin et al., 1976), Because this enzyme plays an important role in estrogen conversion and breast cancer proliferation, determination of its three-dimensional structure is critical to the drug design in breast cancer therapy.
Chin and colleagues (Chin et al., 1976) obtained crystals of 178-HSI with a technique of "electrophoretic diffusion" using tris-barbituric aci~ buffer, pH 7.0, containing 20% glycerol. This technique combines th pnnciples of zone electrophoresis and membrane dialysis. In fact, the: had first attempted ammonium sulfate fractionation, equiIibrium dialysis and vapor diffusion in their laboratory to crystallize estradiol 178 HSD, bat without success. They then reported obtaining "a heavy crop CI
crystals" from the electrophoretically concentrated enzyme solution kept

Chapter 4 Crystallization of 174-HSDI
at 277 K overnight using electrophoretic diffusion. However, to ou knowledge, no X-ray diffraction data were ever published.
Kt should be mentioned that 17B-HSD has been studied since thc 1950's. Because of its liability, difficulties were encountered in thc development of a satisfactory procedure for its homogenization unde conditions commonly used for enzyme purification; the presence of iti cofactors, 178-estradiol, or glycerol stabilizes the enzyme (Langer an( Engel, 1958; Jarabak et al., 1966; Jarabak, 1969; Chin et al., 1976; Lin e al., 1992). In fact, 17B-HSD is rapidly inactivated at 273 K in the absenct of glycerol and is stable in the presence of 50% glycerol at thii temperature for many months. A homogeneous and highly active 178 HSD protein was successfully prepared from a modification of a rapic purification procedure (Lin et al., 1992, and for comparison of thc enzyme specificity from various preparations, please refer to tht Discussion section of this reference). Contradictory opinions about thc identity of the subunits of 170-HSD have been reported. Jarabak anc Street (1971) and Burns et al. (1971, 1972) proposed that the twc subunits are probably identical, but Engel and Groman (1972) suggestec the existence of three different monomers that can interact with eack other to form six dimers. Our recent study using a combined method oi enzymology and molecular biology concluded for the first time that 178. HSD is formed by two identical 34.5 kDa subunits (Lin et al., 1992) Besides the subunit structure, some studies on the enzyme active site oi binding site using chemical modification have been reported (Chin et al. 1982; Murdock et al., 1986). Nevertheless, the information above is stil: incomplete and determination of the enzyme structure is required for 2
proper understanding of its functions.
Thus, the crystallization and determination of the three-dimensiona structure becomes critical for further study of this enzyme and for the synthesis of therapeutic inhibitors. We report here the following critica steps in 1713-HSD crystallization: stabilization of the enzyme, detergen search to increase its low solubility, and the combined screening for it!

crystallization.
4.1.2 Materials and Methods
4.1.2.1 Chernicals
NADP', NAD+, glycerol, B-OG (O-octyl glucoside), MgCI2, LiCI, NaCl, Na K tartarate, PEG (polyethylene glycol) 4000, Tris-base [Tris = Tris. (hydroxymethyl) aminomethane], Hepes [N-(2-hydroxyethyl) piperazine. NI-2-ethanesulfonic acid] , AD A [N-(2-acetamido) iminodiacetic acid] diethanolamine, EDTA (ethylenediaminetetraacetic acid), and PMSE (phenylmethanesulfonyl fluoride) were purchased from Sigma (St. Louis MO, USA); 17B-estradiol and DTT (dithiothreitol) were obtained frorr Aldrich (Milwaukee, WI, USA); Q-Sepharose Fast Flow and Blue. Sepharose CL-6B columns were packed in Our laboratory using media from Pharrnacia Biotech. (Montreal, Canada), and the phenyl-Superose Hi? 10/10 column was from the same Company. Al1 reagents were of the best grade available. Centricon-30 and Centri-prep-30 concentrators were bought from Amicon (Beverly, MA, USA).
4.1.2.2 Methods 179-HSD assay The enzyme was assayed by monitoring the absorption increase ai
340 nrn from the NAD+ reduction following the oxidation of estradiol. The reaction mixture contained 0.5 m M NAD+ and 25 pM estradiol in 50 mM diethanolamine buffer, pH 9.1. One unit of enzyme is the amouni required to catalyze the formation of 1 pmol of estrone in 1 min undex the above conditions [295 (1) KI.
SDS-PAGE Sodium dodecyl sulfate polyacrylamide gel electrophoresis wa
carried out in the Laemmli discontinuous buffer system (Laemmli, 1970 using the Bio-Rad Mini-PROTEAN II ( or PROTEAN II ). The sarnples

Chapter 4 Crystallization of I7J-HSDI
contained 2-4 l g of each protein for Mini-PROTEAN II (or 4-10 pg fa PROTEAN II), 0 5 1 % SDS, and less than 10% glycerol. The gels wer stained with Coomassie Blue (Bio-Rad, 1990) or "High blue staining" fror Pharmacia for higher sensitivity, in which 0.05% Coomassie Blue, 109 methanol, 7% ammonium sulfate, and 9% acetic acid were used fo staining.
To check the protein integrity in the crystals, the crystals werl washed rapidly several times in the reservoir solution with the help O
capillaries before being dissolved and applied on the gel.
Protein concentration measurements Protein concentration was determined by the optical method O
Warburg and Christian (1942), expressed by protein concentratioi (mglml) = 1.55A280 - 0.76A260. The optical density was determined witl a Beckman DU-70 spectrophotometer in which microcells of 50 pl weri used. The bicinchoninic acid assay (Pierce, 1989) gave similar bu slightly higher readings.
Preparation of 17J-HSD The purification of 176-HSD from the soluble subcellular fraction c
human placenta was a modification of a procedure described previousl (Lin et al., 1992) that consisted of three chromatographic steps: C Sepharose anion exchange, Blue-Sepharose affinity, and phenyl-Superos hydrophobic interaction columns. A11 chromatûgraphic steps wer carried out using a fast-protein liquid chromatography (FPLC) syster consisting of two P3500 pumps, a UV-M monitor, and an LCC-50 controller from Pharmacia Biotech. A Pharmacia peristaltic pump P5 was used instead of the p l pump previously used during the sampl loading in the Q-Sepharose chromatography. This resufted in a notabI acceleration of the purification, as a sarnple nearly 2000 ml needs to bi applied on this column from three placentas. NADP+ replaced NAD+ in th elution of the affinity chromatography, and the latter was employed in s

Chapter 4 Crystallization of 17J-HSD I
previously reported procedure (Lin e t al., 1992). Our recer spectrometric and fluorometric studies indicated that the homodimer c 170-HSD molecule binds two NADP+ in the presence of the cofactor at O. mM (unpublished results).
Crystallization of I7J-HSD The crystallizations were carried out with the vapor-diffusio
technique in hanging drops. The reservoir contained 0.9 ml of sohtio including the precipitant (PEG 4000), the salt, and sodium azide (2 mM: at pH 7.5. The enzyme solution is prepared in a buffer containing 201 glycerol, 1 mM EDTA, 0.4 mM DIT, 0.5 mM PMSF, and 40 m M Tris-HC pH 7.5, hereafter referred to as buffer A. B-OG (0.06%) was added to th enzyme sample just before crystallization via centricon buffer changr and at the same time the protein was concentrated to 20-22 mg/m Finally, NADP' was added to the sample to 0.6 mM concentration, an equal volumes (2-3 pl) of the sample and the reservoir solutions wer mixed to initiate the crystallization.
4.1.3 Results
4.1.3.1 Rapid preparation of homogeneous and highly active 1 7 B - H S D
Our preparation was based on FPLC. The procedure is very rap because of the high flow rate of FPLC media and also because of t1 optimal simpIification of intermediate steps. Moreover, NADP+ elution affinity chromatography is very specific, thus improving both ti homogeneity and the yield of the purification. The total recovei increased to about 40% from 30% using the former procedure (Lin et a 1992). The enzyme preparation has a high specific activity, whic catalyzes the oxidation of more than 8 pmol estradiol /mg of 178-HSI protein ( i . e . > 8 U/mg), which could be due to the elimination 4
microheterogeneity caused in vitro by oxidation-reduction effects oi

Chapter 4 Crystallization of I7P-HSDl
partial proteolysis (Giegé et al., 1986; Lorber e t a1.,1987). This preparation showed a single band at 34.5 kDa on SDS-PAGE, similar ta Lin et al., 1992 (Fig. 4.1.la).
4. i .3.2 Stabilization of I7B-HSD
As noted in the Introduct ion, the presence of 20% glycerol in the buffer system is critical to the stabilization of 17B-HSD. In fact, this glycerol content is necessary for crystallization (see Screening ana Crystal Growth of I7J-HSD-NADP+ Complex, below). In the repeated purification of the enzyme, we also found that inclusion of EDTA is important to yield a highly active preparation, as some metal ions have inhibitory effects on 178-HSD (unpublished results). A principal buffer (buffer A, see Materials and Methods) containing EDTA, glycerol, and PMSF at pH 7.5, was used for the purification procedure and for handling the protein sample for crystallization. The near neutral pH is also important to the enzyme's stability as suggested in the literature, and verified in our experiments (Jarabak, 1969; Chin et al., 1976; Lin et al., 1992). Enzyme activity is stable when stored in buffer A at room temperature for more than 10 d, within this period, 178-HSD crystals are reproducible. Furthermore, when the crystals of 176-HSD-NADP+ obtained up to one month earlier were washed several times with the reservoir solution, dissolved, and electrophoresed on SDS-PAGE, a single band was observed, indicating the protein integrity (Fig. 4.1.1 b). Enzyme samples from the dissolved crystals were assayed at the same time, and no significant loss of activity was found. The above control showed that the enzyme is stable in its crystalline form in buffer A in the presence of NADP+ and B-OG (see Screening and Crystal Growth oj I7J-HSD -NAD P+ Complex below).
4.1.3.3 Detergent search to increase 178-HSD solubility

Chapter 4 Crystallization of 174-HSDI
Our highly active 17B-HSD preparation revealed a solubility about 3 mg/ml in buffer A at 295 (1) K. This enzyme had a roor temperature solubility of about 1 to 2 mg/ml in 0.05 M potassiu phosphate buffer containing 0.005 M EDTA and 20% glycerol, pH 7.0, Chin et al., 1976).
When assayed with the inclusion of 6-OG, a mild non-ioni detergent, at various concentrations, the specific activity of the enzym remained unaffected in the presence of the detergent from 0.02 to 0.15: in buffer A. Within that range of B-OG concentration, 178-HSD solubilit was increased to >40 mg/ml, or more than tenfold higher than in th absence of the detergent.
4.1.3.4 Screening and crystal growth of 17B-HSD-NADP+ complex
As 170-HSD has a low solubility that can be increased many fold i the presence of the detergent 13-OG. We first used the PEG/NaCl/B-O( mode1 system to screen the complex ( Garavito and Picot, 1990). Th initial conditions consisted of 5 1 5 % ( w l v ) PEG 4000, 0.1-0.3M NaCl 0.02-0.15% 0-OG, and 6-9 mgfml as the final concentration for 17B-HSC Tiny crystals were obtained, and the best ones were grown in th presence of 0.06% I3-OG and 7 4 % PEG (4000). To complement the abov screening, we performed a parallel search with the sparse matri sampling method (Jancarik and Kim, 1991). Only those with pH > 6.5 c their 50 conditions were used, since 17B-HSD is unstable a t lower pl levels. In the hanging drops, the final concentration of 170-HSD was 1 mg/ml. Very fine, long needles were obtained in the presence of 1M Ns K tartarate, and 0.1 M ADA, pH 6.5. Some cubic but multiple crystal were obtained in the presence of 30% PEG 4000, 0.6 mM NADP+, 0.2 h MgC12. and 0.1 M Hepes, pH 7.5. These results were similar to thos from PEG/ NaCl/ 8-OG system, confirming that we were close to optima crystallization conditions. In al1 subsequent crystallizations, 0.06% B-O( was included.

Chapter 4 Crystallization of 17J-HSDI
We cal1 this approach a combined screening, as the parallel search i the two Iines can increase the probability of success. In fact, in the cas of 170-HSD, the screening dong the line of PEG/NaCI/ 8-OG and that a sparse matrix sampling complemented each other and improve th results. Based on the above preliminary data, further refinement wa carried out with the PEG-MgCl2 system. . A series of crystals wer obtained under similar conditions, but the best ones were obtained in th presence of 0.15 M MgCl2 and 22 mg/ml protein as the fina concentrations in the droplet after equilibrium. Although quite smal (typically - 0.05 x0.05 x 0.15 mm3) (Fig. 4.1.2a). the crystals diffracte( remarkably well. to a resolution of 2.7 A. The best crystals using MgCl: as salt were recently obtained in the presence of 27-28% PEG, and the: grew to a larger typical size of 0.07 x 0.12 x 0.25 mm3 (maximum 0.15 >
0.18 x 0.36 mm3) than using other conditions (Fig. 4.1.2b). Thesi crystals appeared 3-4 d after setting the drop and grew to the full sizi in 3 to 4 weeks. They were in monoclinic space group C2 with unit ce1 parameters a = 123.03 A, b = 45.03A. c = 61.29& and b = 99.1' (Zhu e al., 1993). The more recent crystals diffracted to 2.4 A.
The glycerol content (20%) is necessary for 178-HSD crystallization No crystals have ever been obtained under similar conditions but witl reduced glycerol concentration, e .g ., at 15%. This requirement might bc closely related to the stabilization of the enzyme activity (sec Stabilization of 17J-HSD) .
4.1.3.5 Crystals obtained in the presence of different salts
Crystals of 170-HSD-NADP+ were also obtained in the presence O
LiCl (0.2M) instead of MgC12. 26% (wlv) PEG 4000, and 40 rnM Tris-HC1, pH 7.2. After setting up hanging drops, precipitates formed in 1-2 d anc crystals appeared in about 10 d (- 0.05 x 0.06 x 0.08 mm3) precipitate These crystals grew to a typical size of 0.2 x 0.2 x 0.3 mm3 in 3-! weeks (Fig. 4.1.3a and 4.1.3b).

Chapter 4 Crystallization of 17J-HSDI
The same complex was also crystallized in the presence of NaC (0.16M). 26% PEG, and 40 rnM Tris-HC1, pH 7.2 (Fig. 4.1.3~). Cïysti growth was sirnilar to that observed in the presence of LiCl, but crystal appeared more slowly than that in the presence of MgC12.
4.1.4 Discussion
The rapid preparation of homogeneous and highly active enzym protein played an important role in the crystal growth of 17B-HSD. Ou preparation can catalyze the oxidation of about 8 pmol estradiol in min per mg enzyme protein, which is about 2 to 3-fold higher than mo! reported values for 170-HSD using conventional chromatography. A
detailed comparison of various preparations of 178-HSD was given b Lin et al. (1992).
178-HSD has a much higher solubility (> 40 mg/ml) in the presenc of 0.06% 0-OG than in its absence (-3 mgfml) while maintaining its hig specific activity. The somewhat strong hydrophobicity of 178-HSD wa also demonstrated in phenyl-Superose chromatography (Lin et al., 1992: although the enzyme was isolated from the soluble subcellular fractio of human placenta. The 8-OG concentration used here is well below it CMC (critical micelle concentration). This mild nonionic detergent ma play a role to reduce non-specific hydrophobic interactions an1 encourage electrostatic interactions. That may result in eliminatin, different States of aggregation of 170-HSD and favor crystalIization similar to observations with a nurnber of soluble proteins (McPherson e al., 1986). We are studying 1713-HSD's overall ce11 localization an1 solubility further, to see whether it has hydrophobic interactions witi membranes or other proteins. We have also found that the addition of cofactor is critical to the enzyme's crystallization.
Using the steps above, crystals of this labile and relativel: hydrophobic enzyme were readily obtained. The crystals diffracted to a

Chapter 4 Crystailization of I7J-HSDI
high resolution of 2.4-2.5 A. The preliminary X-ray diffraction analysi suggested that the enzyme's molecular twofold axis is coincident with th crystallographic twofold axis along the b axis, confirming Our previou enzymology study (Zhu et al, 1993; Lin et al., 1992). Further screenin for the crystaIlization of 17B-HSD complexed with various ligands including substrates and inhibitors, is being carried out. The structur determination of enzyme-ligand crystals is important for a bette understanding of 17B-HSD-ligand interactions and eventually will help i the design of efficient inhibitors. As mentioned above, 17B-HSD i essential for the synthesis of the most active estrogen, namely 178 estradiol. This enzyme is present not only in the ovary, but also i; peripheral target tissues, especially the mammary gland, where it i responsible for the IocaI formation of estrogen (Labrie, 1991; Martel e al., 1992). As another experirnent, 178-HSD samples have been sent fo crystallization under microgravity on board the Russian MIR station with the expection of further improving the crystal quality unde reduced convection.
Acknowledgments. This work was supported in part by the Medic; Research Council of Canada, EndoRecherche, and more recently by th4 Canadian Space Agency in the refinement of the crystals. The X-ra: diffraction studies are being carred out in collaboration with the Medica Foundation of Buffalo.
4.1.5 References
Bio-Rad (1990) Mini-PROTEAN II Dual Slab Cell, Instruction Manual. Bic Rad, Richmond, California, USA.
Burns, D. J. W., Engel, L. L., and Bethune, J. L. (1971) Biochem. Biophys. Res. Commun. 44, 786- 792.
Burns, D. J. W., Engel, L. L. and Bethune, J. L.(1972) Biochemistry 11, 2699-2703.

Chapter 4 Crystallization of 174-HSDI
Chin, C.-C., Dence, J. B. and Warren J. C. (1976)J- Biol. Chem. 251, 3700- 3705 .
Chin, C.-C., Murdock, G.L. and Warren, J. C. (1982) Biochemistry 21, 3322- 3326.
Engel, L.L., and Groman, E.V. (1972) Recent Prog. Horm. Res. 30, 139-169.
Garavito, R.M. and Picot, D. (1990) Methods: A cornparison to Methoh in Enzymology 1, 57-69.
Giegé, R., et a1.(1986) J. Crystal Growth 76, 554-561.
Jancarik, J. and Kim, S.-H. (1991) J . Appi. Cryst., 24, 409-411,
Jarabak, J. (1 969) Methods Enzymoi. 15, 746-752.
Jarabak, J., and Street, M.A. (1971) Biochemistry 10, 3831-3834.
Jarabak, J., Seeds, A.E., and Talalay, P. (1966) Biochemistry 5, 1269-1278.
Labrie, F. (1991) Mol. Endocrinol. 78, 113-1 18.
Laemmli, U. K. (1970) Nature 227, 680-685.
Langer, L. J., and Engel, L. L. (1958) J. Biol. Chem. 233, 583-588.
Lin, S.-X., et a1.(1992) J. Biol. Chern. 267, 16182-16187.
Lorber, B. et al., (1987) European J. Biochem. 165, 409-417.
McPherson, A., et al., (1986) J. Biol. Chem., 261, 1969-1975.
Martel, C., et al., ( 1992) J . Steroid Biochem. Molec. Biol. 41, 597-603.
Murdock, G.L., Chin,C.-C. and Warren, J.C. (1 986) Biochemistry 25, 641 - 646.
Pierce Chemicai Co. (1989) BCA Protein Assay Reagent, Instructions, Pierce Chernical Co., Rockford, IL.
Warburg, O., and Christian, W. (1942) Biochem. J. 310, 384-421.
Zhu, D.-W. et al. (1993) J. Mol. Biol., 234, 242-244.

Chapter 4 Crystallization of I7j3-HSD 2
LEGEND TO FIGURES:
Figure 4.1.1. SDS PAGE of 17B-HSD directly purified f human placenta or from dissolved crystals.
a. Lanes 1,2,3: phenyl-Sepharose fractions from the purification. b. Lanes 4,5,6: Samples obtained from dissolved crystals of 178-HSI NADP+ complex. In both a. and b., M indicates the protein standards of 97.4, 66.2, 45, 3 21.5 and 14.1 kDa from top to bottorn.
Figure 4.1.2. 17B-HSD-NADP+ crystals grown in the presence M W 2 .
(a ) : A large number of small cubic crystals were obtained in ti
presence of 30% PEG (4000), 0.15 M MgC12, and 0.1M Hepes (fin concentrations), pH 7.5. The final protein concentration was 22 mg/m The crystals have a typiçai size of 0.05 x 0.05 x 0.15 mm3 and diffract I
2.7 A. (b): The same type of crystals were grown under similar conditions b in the presence of 27-28% PEG. The typical size is 0.07 x 0.12 x 0.2 mm3 and the crystals diffracted to 2.4 A.
Figure 4.1.3. Crystals grown in the presence of LiCl and NaC
The same complex crystaIs were obtained in the presence of 0.2 M Li( or 0.16 M NaCl, 26% (wlv) PEG (4000), at pH 7.2. (a): Crystals with a size of 0.05 x 0.06 x 0.08 mm3 appeared in tk
presence of LiCl in about 10 days on the background of precipitants. (b): The above crystals grew to a typical size of 0.2 x 0.3 mm3 (rnaxim; 0.25 x 0.40 x 0.45 mm3) in 3 to 5 weeks. (c): Crystals grew to about 0.15 x 0.26 x 0.55 mm3 in the presence (
NaCl in 3 to 5 weeks after drop setting.

Chapter 4 Crystallization of I 74-HSD I
Figure 4.1.1
a) Fig. 4.1.2

Chapter 4 Crystallization of 171-HSDI
Fig.4.1.3

4.2. Crystallization and Preliminary X-ray Diffrac Analysis of the Complex of Human Placental 1 Hydroxysteroid Dehydrogenase with NADP+
Single crystals of human placental 176-hydroxysteroic dehydrogenase, an enzyme that plays an important role in th( interconversion of estrogens, were obtained as a complex ligated witl N A D P+. These are the first crystals suitable for complete X-ra: structural analysis ever reported for a steroid-converting enzyme from i
human source. The crystals were grown by vapor diffusion at pH 7.' with polyethyleneglycol (4K) as the precipitating agent. They have ;
monoclinic space group C2 and unit ce11 parameters are a =1 23.03& b = 45.03A. c = 61.29A, and 8 = 99.1'. A complete set of diffraction data tc 2.9A has been collected on native crystals.
In this paper, 1 was responsible for enzyme preparation an crystallization.

Chapter 4 Crystallization of I7J-HSDI
C RYSTALLIZATION AND PRELIMINARY X-R AY DIFFRACTION ANALYSIS OF THE COMPLEX OF HUMAN PLACENTAL 17B- HYDROXYSTEROID DEHYDROGENASE WITH NADP+
D.-W. ~ h u f ' , X. ~ e e s , R. ~retonf ' , D. ~hosh ,* , W. ~ a n ~ b o r n * , W.: ~ u a x * and S.-X. ~ i n f p *
t The Medical Research Council Group in Molecular Endocnnology, CHUL Research Center and Laval University, Quebec GlV 4G2, Canada. $ The Medical Foundation of Buffalo, Inc., Buffalo, NY14203, USA § The Cleveland C h i c Foundation, Cleveland, OH44 195, USA
This is adapted from the paper of "CRYSTALLIZATION AND PRELIMINARY X- RAY DIFFRACTION ANALYSIS OF THE COMPLEX OF HUMAN PLACENTAL 178- HYDROXYSTEROID DEHYDROGENASE WITH NADP+ " by D.-W. Zhu, X. Lee, R. Breton, D. Ghosh, W. Pangbom, W.L. Duax and S.-X. Lin in J. Mol. Biol. (1 993) 234, 242-244.

Chap fer 4 Crystallization of I 7p- H S D 1
4.2.1 Introduction
Hurnan placental 170-hydroxysteroid dehydrogenase (1713-HSD EC1.1.1.62) governs the formation of active estrogens that stimulati breast cancer (Mouridsen et al., 1978; Poulin & Labrie, 1986; Lin et al. 1992). Determination of the three-dimensional structure of the enzymc will contribute to the elucidation of the molecular mechanism of actioi and permit the design of therapeutic agents that will inhibit the enzymc and modulate endogenous estrogen levels. 176-HSD from the solubli subcellular fraction of the human placenta was purified to homogeneitj with high specific activity using Fast Protein Liquid Chromatographl (FPLC), the homodimeric structure of the enzyme (2~34.5 kDa) has beer demonstrated (Lin et al., 1992; Yang et ai., 1992).
178-HSD belongs to a superfamily of enzymes referred to as "short. chain" dehydrogenases (Persson et al., 1991). The only steroic dehydrogenase for which the X-ray crys tallographic structurt determination has been reported is bacterial 3a ,2013 h ydrox y steroic dehydrogenase, another member of this super family (Ghosh et al. 1991). 3a,208-HSD is active as a tetramer and has 255 residues in eack subunit. Crystdlization of human I7B-HSD was reported by Chin et al (1976); however, to our knowledge, no detailed X-ray diffraction datl were ever pubIished. Here we report the crystallization of an active form of 17B-HSD and compare the preliminary diffraction data with thai of bacterial 3a,20L3-hydroxysteroid dehydrogenase.
4.2.2 Purification of 17R-HSD
The enzyme was purified with a modification of a previouslj described procedure (Lin et al., 1992). Briefly, placentas were freshlj homogenized with a Waring blender and the tissue obtained was

Chapter 4 Crystallization of 17J-HSDI
submitted to ammonium sulfate fractionations. The fractioi precipitating between 30 and 55% (w/v) ammonium sulfate was dilutec and loaded on a Q-Sepharose column. 170-HSD activity eluted at 0.22 h,
NaCl in buffer A (40 mM Tris HC1, pH 7.5, 1 mM EDTA, 0.2 mh,
dithiothreitol, and 20% glycerol). NADP+ substituted for NAD+ in the blui Sepharose CL-6B chromotographic step previously described (Lin et al. 1992). 17B-HSD eluted sharply at 35 pM NADP+ (in buffer A) with i
yield of 75% to 80%. A trace impurity in the blue Sepharose fraction; was easily separated on a phenyl-Superose column. The final samplc had high specific activity, catalyzing the conversion of 8 pmol O
estradiol into esuone per mg of enzyme per minute. The homodimer O
17B-HSD binds two molecules of the cofactor in the presence of 0.6 mh,
NADP+ (unpublished resul ts).
4.2.3 17B-HSD Crystallization
The enzyme fractions from the phenyl-Sepharose column wa! concentrated at >20 rng/ml in the presence of 0.06% octylglucoside Vapor diffusion techniques were used for crystallization. The reservoii contained 0.15 M MgC12, 0.1 M Hepes (N-(2-hydroxyethyl) piperzine-N', 2-ethanesulfonic acid) and 28% polyethyleneglycol (4K) as thc precipitant. Hanging drops of 3 pl of the above-concentrated proteir solution (20 mg/ml, in the presence of 20% glycerol) and 3 pl of thc reservoir solution were prepared. Monoclinic crystals appeared in threc to four days at room temperature and grew to a typical size of 0.07 mrr x 0.12 mm x 0.25 mm in about three weeks. Rapid purification anc immediate crystallization account for the good quality of the crystals.
The ability of 17B-HSD to bind NADP' was confirmed by the l o ~ A280fA260 ratio of the preparation which becomes I .1 (I 0.05) when one N A D P+ is bound and 0.88 (I 0.05) when two NADP+ are bound to the apoenzyme. This ratio is 1.65 (i 0.1) for the apoenzyme. Complex

Chapter 4 Crystallization of f 7J-NSDI
formation is demonstrated by a significant fluorescence increase at 45 nm following an excitation at 350 nm (unpublished results).
4.2.4 Crystal Characterization, Data Collection and Analysis
The X-ray diffraction analysis of crystals of 178-HSD was performe at the Medicai Foundation of Buffalo using an R-AXIS IIc image plai area detector and Rigaku RU-200 rotating anode generator operating i
50 kV and 90 mA. Graphite-monochromated CuKa (A = 1.541 8A radiation was used. The detector was placed at a distance of 165 ml from the crystal. The crystal was mounted in a glass capillary and seale with mother liquor. The unit ce11 dimensions and crystal orientatio angles were determined by least-squares fitting of a lattice of parti; reflections recorded on the image plate ac three fixed positions of th crystal, 45 O apart in spindle rotation. The crystal belongs to the spac group C2. The unit ce11 parameters are a = 123.03 A, b = 45.03 A, c : 61.29 A and p = 99. Io (volume=335,276 A3). Taking a 34.5 kDa monome as the subunit in the asymmetric unit, the specific volume is 2.43 A 3 / ~ a
a number which is near the midpoint of the range found for protei crystals (Matthews, 1968).
The diffraction data were collected on 22 frames, each containin data for a 6' oscillation of the crystal. The exposure time for each fram was 60 minutes. The data were processed using the R-AXIS prograr package, version 2.12, from Rigaku. The crystal diffracted well for it small size. Measurable diffraction was recorded up to a resolution of 2. A. Although the crystal survived for more than 24 hours, the highe resolution data deteriorated rapidly. A data set of 6,644 reflections an1 17,572 total observations were measured to a 2.93 A resolution. Th Rmerge on intensities was 9.5%. The average I/o(I) value for the shel
between 3.00 A and 2.93 A resolutions was 2.91. The data represen 92% of the theoretically possible number of unique reflections to tha resolution.

Chapter 4 Crystailizatiun of 17P-HSDI
Since the specific volume of the crystal with a full dimer in tht asymmetric unit is unusually low and inconsistent with Matthew*! caIculation, the determined space group C2 and the dimeric biochemica nature of the enzyme (Lin et al., 1992) suggest that the molecular two fold symmetry-axis of the enzyme is coincident with the crystallographic two-fold axis along the b-axis, the shortest dimension of the unit ce11 From the cornparison of amino acid sequences of 1713-HSD and 3a,20B HSD (Persson et al., 1991) and assignments of secondary structurc elements to the sequence of 17B-HSD from the known three-dimensions: structure of 3ct,20B-HSD, it appears likely that the additional 72 aminc acids in 178-HSD are distributed primarily in two loop regions and at the C-terminal end of the enzyme (Persson et al., 1991; Ghosh et al., 1991) The so-called Q-axis dimer of the tetrameric 3a.208-HSD, the dimer witt the most intimate subunit-subunit association, has approximate dimensions of 33 A x 62 A x 54 A, the shortest dimension being along the direction of the molecular diad (Ghosh et al., 1991). It is plausibk that a molecular two-fold axis similar to the Q-axis diad in 3a,20i3-HSC crystal structure also exists in the crystal structure of 178-HSD.
Acknowledgements We thank Dr. F. Labrie for his interest in thi work. This work is supported by the Medical Research Council of Canada to the MRC Group in Molecular Endocrinology and the National Institutes of Health, USA, gan t no. DK26546 to the Medical Foundation of Buffalo.
4.2.5 References
Chin, CC., Dence, J.B. and Warren, J.C. (1976) J. Biol. Chem. 251, 3700- 3705.
Ghosh, D., Weeks, CM., GruchuIski, P., Duax, W.L., Ekman, M., Rimsay, R.L., and Orr, J.C. (1991). froc. Narf Acad Sci. USA 88, 10064-10068.
Lin, S.-X., Yang, F., Jin, J.., Breton, R., Zhu, D.-W., Luu-The, V. and Labrie, F. (1992) J. Biol. Chem. 267, 16182-16187.

Chapter 4 Ctystallization cf I7J-HSDI
Matthews, B.W. (1968) Solvent content of protein crystals. J . Mol. Biol. 37 49 1-497.
Mouridsen, H., Palshof, T., Patterson, J., and Battersby, L. (1978) Cance Treat. Rev. 5, 131-141.
Persson, B., Krook, M., and Jornvall, H. (1991) Eur. J. Biochem. 200, 537 5 4 3 .
Poulin, R., and Labrie, F. (1986) Cancer Res. 46, 4933-4937.
Yang, F., Zhu, D.-W., Wang, J.-Y. and Lin, S.-X. (1992) J . Chrornatogr. 58: 7 1 - 7 6 .

Chapter 4 Crystallization of 174-HSD1
4.3. The crystallogenesis of a human estradi dehydrogenase-substrate complex
In this paper, we report the preparation of a stoichiometric 170- HSDI-estradiol cornplex sample at a much higher concentration than the solubility of the free substrate, using a gradua1 concentration of the enzyme-substrate mixture starting at low concentration. The complex is successfully crystallized by vapor diffusion a t pH 7.5 with polyethyleneglycol 4000 as the precipitating agent. The space group is C2 with a = 123.56 A, b = 45.21 A, c = 61.30 A, and B = 99.06'. There is one monomer in the asymrnetric unit and two molecules of the enzyme in a unit cell. A diffraction data set to 2.5 A has k e n collected to 86% completeness on native crystals. The high quality of the electronic density map of estradiol supports the full occupancy of the binding site, thus confirming the efficiency of the cornplex preparation. This method will also be useful in crystallizing other steroid-dehydrogenase complexes.
For this paper, 1 was responsible for most of the experiments, including enzyme purification, preparation of stoichiometric of 170- HSD 1 -estradio1 complex and crystallization.

Chapter 4 cr-ystallizarion of 17J-HSDI
The crystallogenesis of a Human estrad dehydrogenase-substrate eomplex
DAO-WEI ZHU, AREZKI AZZI, PETER REHSE AND SHENG-XIANG LIN*
T h e Medica l Research Council G r o u p in Molecu Endocrinology, CHUL Research Center and Laval Univei Quebec G1V 4G2, Canada
* This is adapted from the paper of "The crystallogenesis of a Hum estradiol dehydrogenase-substrate complex" by Dao-Wei Zhu, Arezki Az Peter Rehse and Sheng-Xiang Lin in J. Crystai Growth (1996), 168: 27 276 .

Chapter 4 Crystallization of 17J-HSDI
4.3.1 Introduction
Breast cancer is the most incidental cancer in woman in North America with 182,000 projected cases in 1995 (Wingo et al., 1995). Human estradiol 17B-hydroxysteroid dehydrogenase type 1 (176-HSD1, EC. 1.1.1.62) is responsible for the synthesis of the active estrogens which stimulate the proliferation of breast cancer cells (Mouridsen, et al., 1978; Poulin, et al., 1986; Labrie, et al., 1992; Lin et al., 1992). Detailed study of the mechanisms of action of 178-HSD1 is thus of major importance for the inhibitor design to improve endocrine therapy.
Human placenta1 178-HSDl has been studied since the 1950s (Langer, et al., 1958; Descomps et al., 1968; Karavola et al., 1970). The cDNA and gene coding 178-HSD1 had been isolated and characterized (Luu-The et al., 1989). In 1991, Lin et al. (1992) concluded that estrogenic 170-HSD1 is formed by two identical subunits using a combined method of enzyrnology and molecular biology. Human 178- HSDl crystallization was studied since the 1970's (Chin et al., 1976). Chin et al. (1976) reported the obtaining of some crystals of this enzyme, but to our knowledge, no preliminary X-Ray diffraction data has ever been published. In Our laboratory, the crystallization of 170- HSDl has been attempted since 1991. In 1992, it has been crystallized in the presence of NADP+ (Zhu et al., 1993). The structure of this crystal has been determined (Ghosh et al., 19951, but the electronic density corresponding to the cofactor could not be reliably identified, and the estradiol binding has been modeled. Here we report a special effort to obtain the 17B-HSDl-estradiol sample at high concentration (1:2 complex with estradiol up to 600 FM). This made possible the crystallization and the preliminary crystallographic study of this complex. The corresponding structure and the enzyme-substrate interactions are being determined.

Chrrpter 4 Crystallization of I7J-HSDI
4.3.2 Materials and Methods
4.3.2.1 Chernicals
NAD+, glycerol, l3-OG (B-octyl glucoside), NaCl, LiC1, MgC12. NaHC03- Na& 0 3 , PEG (polyethylene glyco1)4000, Tris [tris-(hydroxymethyl)
aminomethane] , Hepes [N-(2-hydroxyethy1)piperazine-Nt-2- ethanesulfonic acid], EDTA (ethylenediaminetetraacetic acid) and PMSF (phenylmethanesulfonyl fluoride) were purchased from Sigma (St. Louis, MO, USA); 17B-estradiol, DTT (dithiothreitol) were obtained from Aldrich (Milwaukee, WI, USA); [4-14c] Estradiol (55.5 mci/mrnol) was a Du Pont-New England Nuclear produc t. Bio-Rad Protein Assay was purchased from Bio-Rad (Montreai, Canada). Q-Sepharose Fast Flow and Blue-Sepharose CL-6B columns were packed in Our laboratory using media from Pharmacia Biotech. (Montreal, Canada), and the phenyl- Superose KR 10/10 column was from the same Company. Al1 reagents were of the best grade available. Centricon-30 and Centri-prep-30 concentrators were bought from Arnicon (Beverly, MA, USA).
4.3.2.2 Methods
Enzyme Assay. 1713-HSD 1 was assayed by spectrop hotometric measurement of
N A D + reduction indicated by the absorbance increase at 340 nm at 23 f 1°C. The reaction mixture contained 0.5 m M NAD+, 25 p M estradiol in 50 m M NaHC03-Na2C03 buffer, pH 9.2, one unit is defined as the
amount of the enzyme protein that catalyzes the formation of 1 pmole of estrone in 1 min under the above conditions.
Protein concentration measurements. Protein concentration was determined by the method of Bradford,
M. (Bradford 1976). This is measured by absorption at 595 nm, using BSA (bovine serum albumin) as a standard. The optical density was determined with a Beckman DU-70 S pectrophotometer.

Chapter 4 Crystallization of I7P-HSDI
Enzyme purification. Placental 170-HSD1 was purified with a modification of a
previously described procedure (Lin et al., 1992; Zhu et aI., 1993). Briefiy, enzyme purification consisted of three chrornatographic steps: Q-Sepharose anion exchange, Blue-Sepharose affinity, and phenyl- Superose hydrophobic interaction columns. Al1 chrornatographic steps were carried out using a fast protein liquid chromatography (FPLC) sys tem.
Stoichiometiy of 17J-HSDI -estradio1 cornplex. This is evaluated using [4-14c] estradiol. Just before crystailization
via ceniricon buffer change for three times. Each time, 0.06% (w/v) 8- OG and estradiol (25 PM) were added to the enzyme sample (c 12 pM). The sample was centrifuged at 4451 g in a Sorvall RC-5 centrifuge, and at the final, the concentration was increased to 14.1 mg/ml. A parallel binding assay to check the stoichiometry was performed using [4-14c] estradiol counted with scintilation liquid (Formula 989, Du Pont-New England Nuclear) in a Wallac 1400 DSA (PROT14 Smin).
Crystallization of 174-HSD 1 -estradiol. The crystallizations were carried out with the vapor-diffusion
technique in hanging drops. The reservoir contained lm1 of solution including the precipitant (PEG 4000), salt and sodium azide (2 mM), at pH 7.5. The enzyme solution was prepared in a buffer containing 20% glycerol, 1 mM EDTA, 0.4 mM DTT, 0.5 mM PMSF, and 40 mM Tris-HCL, pH 7.5, hereafter referred to as buffer A. Equal volumes (3 pl) of the sample and the reservoir solutions were mixed to initiate the crystallization.
Crystal characterization and data collection. Data collection and crystal analysis of 176-HSD1-estradio1 was
perforrned using an R-AXIS IIc image plate and a Rigaku RU-300 rotating anode. The crystal was mounted in a glass capillary and sealed

Chapter 4 Crystallization of 2 7J-HSDI
with mother liquor. The detector was placed at a distance of 120 mm from the crystal and the beam was collimated to 0.3 mm. The unit ce11 dimensions and crystal orientation angles were determined by least- squares fitting with a lattice of partial reflections recorded on the image plate at three fixed positions of the crystaI, 45" apart in spindle rotation. The collected data were processed using the HKL software package.
4.3-3 Results and Discussion
4.3.3.1 Enzyme preparation
Placental 170-HSD1 is a hydrophobic and labile enzyme, and therefore it has been difficult in getting a homogeneous preparation by conventional chromatography (Descomps et al., 1968; Karavolas et al., 1970; Jarabak 1969; Murdock et al., 1986). Using Fast Protein Liquid Chromotography (FPLC), we were abIe to obtain an enzyme preparation with a high specific activity and homogeneity, which catalyzes the oxidation of more than 8 pmol esuadiol per mg of 17B-HSD1 protein (Lin et al., 1992; Zhu et al., 1994).. The high quality of our preparation helped us in obtaining crystals and has been proven to be critical in crystal reproduction.
As the human 178-HSD1 is only soluble up to 2-3 mglml, we tried to use various concentration of B-OG to increase its solubility. Satisfactory result was obtained and 170-HSD1 solubility was increased to > 40 mglml in the presence of 0.06% (wfv) 8-OG (Zhu et al., 1994). 0-OG is a mild non-ionic detergent, which may play a role in reducing non-specific hydrophobic interactions and encourage electrostatic interactions. These may result in elirninating different States of 1713- HSDI and may favor crystallization, similar to the observations with a number of other proteins (Zhu et ai., 1994; Mcpherson A. et al., 1986).
Complex formation

Before crystallization the formation of 170-HSD1-estradiol cornplen was verified by a radioactive labeling of estradiol. It is well knowr that steroids have very low aqueous solubility, for example about 3C p M for estradiol under Our experirnental conditions a t roon temperature. The 170-HSD 1 concentration used in crystallization is usually higher than 200 PM. If each subunit of 170-HSD1 is bound with one estradiol (lin et al, unpublished), the total estradiol concentration should be more than 400 p M in the complex. We have had to develop a special method, to gradually saturate the enzyme bj estradiol starting at a low protein concentration. In the course of 178- HSDl concentration in the presence of estradiol, we observed that the total estradiol concentration could be higher and higher than its solubility, due to its gradua1 binding to the enzyme molecule. This result is very probably due to the engagement of estradiol in the hydrophobic binding pocket, while permitting a stable free steroid solubility in this sample. This process allowed continued binding of the steroid to the still free enzyme molecules. For example, in one control the protein concentration was increased from 11.7 p M to 207 pM using centricont-30, the radioactive estradiol gave 7445 cpm representing the bound substrate after su bstrating the blank obtained from the filtrate with 3p1 sample. Because the specific activity of [4- 14c] estradiol is 6 cpm/pmoi, the cornplexed estradiol concentration was evaluated to be 413 PM, indicating that a stoichiometric binding of 2:l (estradiol : enzyme) has been reached. The estradiol concentration in the complex is fifteen fold higher than the solubility of the free steroid. The 170-HSD 1 -estradio1 sample was thus obtained at elevated concentration, indicating that each enzyme subunit is complexed with one estradiol. The complex solution at 300:600 pM stoicheometry has been obtained using this method. The preliminary analysis of crystals obtained with such prepared complex sample showed that the electronic density corresponding to estradiol bound to the enzyme could be reliably identified. The high quality of such a density supports the full occupation of 17B-HSD1 binding sites. The above method will

Chapter 4 Crystallization of I7J-HSD I
be useful for obtaining compiexes of other steroid-converting enzymes with their substrates.
4-3-3.2 Crystal growth of 17B-HSDl-estradiol complex
Because of some hydrophobic nature of 170-HSDl (Lin et al., 1992 Zhu et al., 1993; Breton et al. 1994), in al1 crystallization 6-OG was used t increase the enzyme's solubiIity. We studied the crystallization of th complex of human placentai 17B-HSD 1 with estradiol using a combine! method. We first used the PEG/NaCI/B-OG mode1 system fo crystallization screen. The initial conditions consis ted of 7- 16% (w/v PEG 4000, 0.1-0.3 M NaCI, 0.06% B-OG (w/v) and 4-12 mg/ml as the fina concentration for 1713-HSDI . Tiny crys tals were first obtained unde these conditions (Fig. 4.3.1 a). To complete the screening, we performec a parallel search with the sparse matrix sampling method (Jancarik et al, 1991). Fine, long needles were obtained in the presence of 0.1M LiCI, 41 mM Tris-HC1 pH 7.2 (Fig.4.3.f b). Some multiple crystals were obtained il the presence of 30% (w/v) PEG 4000, 0.2 M MgC12 , 0.06% B-OG (w/v) an4
0.1 M Hepes, pH 7.5 (Fig. 4.3.1 c). Based on the above preliminar results, further refinement was carried out with the PEG/salt/13-O( system. A series of crystals were obtained with different proteii concentration, but the best ones were obtained in the presence of 0.16 h MgC12, 28% (w/v) PEG (4000), 0.06% 6-OG (w/v) and 0.1 M Hepes pH 7.5
with 14 mglrnl protein as the final concentrations after equilibrium Monoclinic crystals appeared in 24 h at room temperature and grew to ;
typical size of 0.15 mm x 0.25 mm x 0.48 mm in about four weeks (Fig 4.3.1 d).
4.3.3.3 Pre l iminary x-ray d i f f rac t ion analysis
The diffraction data was collected at room temperature, on 3' frames each containing 2.5' osciIlation of the crystal. The exposure timc for each frame was 30 minutes. The crystal survived for more than 2( hours of diffraction with no evident loss in the higher resolution data

Chapter 4 Crystallization of I7J-HSDI
The initial data set was processed, using the HKL package program (Dtwinowski 1993), to 2.5 A although many diffraction were observed at 2.3 A. Based on the systematic absences from the collected data the space group was determined to be C2 with unit ce11 dimensions of a = 123.56 A, b = 45.21 A, c = 61.30 A, and D = 99.06'. being close to the former reported 17B-HSD1 crystal grown in the presence of NADP+ (Ghosh e t al., 1995). The processed data yielded 12890 unique refiections from 33053 independent measurements with an Rmerge based on structure factors of 6.20%. The data were 86.1% complete within the 8.0 to 2.5 A shell and showing a complete elecnon density around the estradiol substrate (unpublished result). Taking the ce11 dimension mentioned above we determined a Matthews coefficient of 2.4 A 3 / ~ a . This is consistent with the presence of one subunit in the asymmetric unit and two molecules of the enzyme in a unit cell, as previously found for the 1713-HSD crystal grown in the presence of NADP (Zhu et al., 1993).
Ac kn O w 1 ed gmen t s : This work was supported by the Medical Resear Council of Canada by a grant for the MRC group in Molecul; Endocrinology, and the Canadian Space Agency. The authors would like i
thank Dr. F. Labrie for his interest in this work.
4.3.4. References
Bradford, M. (1976) Anal. Biochem., 72: 248-254.
Breton, R,, Yang F., Jin, J. -Z., Li, B., Labrie, F., Lin, S.-X (1994). JSteroi Biochem. Mol. Biol., 50, 275-282.
Chin, C.C., Dence, J.B. and Warren, J.C. (1976) J. Biol. Chem., 251, 3700 3705.
Descomps B., Nicolas J-C., Crastes De Paufet A. Bull. Soc. Chim. Giol., 1968 50, 1681-1692. Dtwinowski, 2. in Data Collection and Processing (eds Sawyer, L., Lsaacs, N

Chapter 4 Crystallization of 174-HSD I
W. & Bailey S.) 55-62 DL/SCI/R34 (Daresbury Laboratory, Wamngto: 1993).
Ghosh, D., Pletnev, V. Z., Zhu, D. -W., Wawnak, Z., Duax, W. L., Pangborn W., Labrie, F., and Lin, S.-X. (1995). Structure, 5, 503-513.
Jancarik, J. & Kim, S.-H. (1991). J. Appl. Cryst. 24, 409-41 1.
Jarabak, J. (1969) Methods Emzymol., 15, 746-752.
Karavola H. J., Baedecker M. L., Engel L. L. (1970) J. Biol. Chem., 245 4948-4952.
Labrie, C. et al. (1992) Cancer Res. 52:610-615.
Langer, L. J. and Engel, L. L., (1958) J. Biol. Chem., 233, 583-588.
Lin, S.-X., Yan, F., Jin, J.-Z., Breton, R., Zhu, D.-W., Luu-The, V., and Labrie F. (1992) J.Biol.Chem. 267, 16182-16187.
Luu The V., Labrie C., Zhao H. F., Couet J. Lachance Y., Simard J., Leblancc G., Côté J., Bémbé D., Gagné R., Labrie F. (1989) Mol. Endocrinol, 3, 1301 1309.
Mcpherson, A., Koszelak, S., Axelrod, H., Day, J., Williams, R., Mcgrath, M. Robinson, J. & Cascio, D. (1986). J. Biol. Chem 261, 1969-1975.
Mouridsen, H., Palshof, T., Patterson, J., and Batlersby, L. (1978) Cance Treat. Rev. 5, 131-141.
Murdock, G. L., Chin, C. C. and Warren, J. C. (1986). Biochemistry, 25, 641. 646.
Poulin, R., and Labne, F. (1986) Cancer'Res. 46, 4933-4937.
Wingo, P. A., Tong, T., Bolden, S. (1995) Cancer statistics, Ca-A Cance Journal for Clinicians 45: 8-30.
Zhu, D.-W., Lee, X., Breton, R., Ghosh, D., Pangborn, W., Duax, W. L. & Lin S.-X. (1993). J. Mol. Biol. 234,242-244.
Zhu D.-W., Lee, X., Labrie, F. and Lin S.-X. (1994) Acta Cryst. D50, 550. 555.

Chapter 4 Ctystallizarion of 17J-HSD I
Legend to figures
Fig.4.3.1 The crystals of 170-HSD1-estradiol
a) The crystals were obtained in the presence of 20% glycerol, 0.1 N NaCl, 0.06% fi-OG, 7.5% (w/v) PEG (4000) at pH 7.2. The final protein concentration is 6 mg/mi. The typical size is 0.04 mm x 0.04 mm x 0.1 mm.
b) The crystals wert obtained in four days in the presence of 0.1 M LiCl, 0.06% 0-OG, 16% (wfv) PEG (4000), at pH 7.2. The final protein concentration is 10.7 mgfml.
c). Some multipIe crystals were obtained in the presence of 30% (wfv) PEG (4000), 0.2 M MgCl2 and 0.1 M Hepes, pH 7.5. The typical size is 0.053 mm x 0.053 mm x 0.2 mm.
d) Crystal grows in the presence of 0.16 M MgC12, 0.1 M Hepes, pH 7.5 and 28% (w/v) PEG (4000) as the preciptant. Final protein concentration was 13 mg/ml. Crystal grew to 0.15 mm x 0.25 mm x
0.48 mm in 4 weeks after hanging drop initiation.

Chapter 4 Crystallization of 17J-HSDI
Fig. 4.3.1

Chapter 4 Crystallization of 17P-HSDI
4.4 Preliminary study of different methods f crystallization of Human estradiol dehydrogena! inhibitor complexes
Human 17B-hydroxysteroid dehydrogenase is responsible for th synthesis of al1 active androgens and estrogens which stimulate brea; and prostate cancers. Detailed study of the structure of the 171 hydroxysteroid dehydrogenase-in hibi tor (EM- 139) complex is thus (
major importance for the development of breast cancer therapies. Usin CO-crystallization and soaking method, the complex of human 171 hydroxysteroid dehydrogenase with an inhi bitor (EM- 139) has bee crystallized in the presence of 0.06% (w/v) 8-octylglucoside, wii polyethyleneglycol 4000 as the precipitating agent. The crystals grow i the a monoclinic space group C2 with unit ce11 parameters of about a 124 A, b = 45 A, c = 61 A , with B = 99'. Native crystals diffracted 1 rays from a synchrotron radiation source to 1.8 A resolution.
For this paper, 1 was in charge of purification, preparation of 171 HSD 1 -inhibitor and crystallization.

Chapter 4 Crystullization of I 7J-HSDI
Preliminary study of different methods f crystallization of human estradiol dehydrogena, inhibitor complexes
Dao-Wei Zhu, Robert L. Campbell, Fernand Labrie and Shi Xiang Lin
The Medical Research Council Group in Molecul Endocrinology, CHUL Research Center and Laval Univer! Quebec, Canada G l V 4G2
* 4.4 is adapted from the paper of "Preliminary study of different methods for crystallization of human estradioi dehydrogenase-inhibitor complexes" by Dao-Wei Zhu, Robert L. Campbell, Fernand Labrie and Sheng-Xiang Lin in submission.

Chapter 4 Crystallization of I7J-HSD I
4.4.1 Introduction
176-hydroxysteroid dehydrogenase type 1 (178-HSD 1, EC1.l. 1.62) expressed in breast-cancer tissue (Luu-The et al., 1990; Poutanen et al 1992), and the enzyme might therefore be responsible for the relative1 high intracellular estradiol concentrations detected in breast-canct tissues of post-menopausal women (Beranek et al., 1985; Bonney et al 1986). So, many studies have been devoted to inhibiting of 170-HSD activity . Some affinity label inhibitors for human placenta1 176-HSD have been reported (Sweet et al., 1991; Lawate et al., 1990; Auchus et al 1989; Murdock et al., 1986; Inano et al., 1983; Thomas et al., 1983). Bi two important disadvantages were associated with these compoundi their low selectivity and unsuitabIe estrogenic activity which, virtuall eliminated their therapeutic use. Recently, a series of dual-site actio inhibitors (EM-139, EM-221, EM-140, and EM-123) were synthesized i Our laboratory (Fig.4.4.1) (Lévesque et al., 1991; Labrie et al. 1992 They possess an estrogen nucleus and can block the formation of activ estradiol by 17B-HSD, as well as the estrogen action with its withoi estrogenic activity receptor. EM139 is a 7a-alkyl estradiol derivative. was tested for its antiestrogenic activity as well as its potential 176 HSD1 -inhibitory activity in ovarectomized mice treated with E l , th immediate precursor of E2 (Labrie et al. 1992). At present, kineti studies also showed that EM-139 is a reversible and competitiv inhibitor for 178-HSD1 in our group (Wang et al, unpublished). In orde to elucidate the mechanism of inhibition and to improve the inhibitior the crystallization of the complex 176-HSD 1 -inhibitor (EM- 139) wi provide a good opportunity to optimize that kind of inhibitor with th direct demonstration of protein inhibitor interactions. We successfull crystallized different complex of 17B-HSD 1 -NADP+ and 17B-HSD 1 -E (estradiol) yielding diffraction data with high resolution, the firz example of any steroid-converting enzyme from a human source (Zhu r al., 1993; 1996 ). The structure determination of 178-HSD 1 was

Chapter 4 Crystallization of 17J-HSDI
determined (Ghosh e t al., 1995). RecentIy, in our group, the structure O:
the 170-HSD1-E2 complex has been also determined (Azzi . et al 1996) The high quality of density for the substrate supports the full occupatior of 17B-HSDI binding sites. Here we report using co-crystailization anc soaking methods, crystaliization of the complex of human 178-HSDl witl inhibitor (EM-139). The preliminary crystailographic study showed tha, co-crystailization and soak methods give similar quality data while witl a higher mosaicity for the later. In spite of this, high-resolution data wa! obtained at a synchrotron beam line from the soaked crystals.
4.4.2 E x p e r i m e n t a l
Chernicals Di thiothreitol (DTT) was obtained from Aldrich (Milwaukee, WI.
USA). NAD+, glycerol, O-OG (6-octyl glucoside), MgC12. NaHCOs-Na2CO3, PEG (polyethylene glycol) 4000, Tris-base[Tris = tris-(hydroxymethyl: a m i n o m e t h a n e ] , Hepes [N-(2-hydroxyethy1)piperazine-N'-2- ethanesulfonic acid], EDTA (ethylenediaminetetraacetic acid) and PMSF (phenylmethanesuIfony1 fluoride) were purchased from Sigma (St. Louis, MO, USA). Inhibitor (EM-139) was obtained from our laboratory. Q- Sepharose Fast Flow and Blue-Sepharose CL-6B columns were packed in our laboratory using media from Pharrnacia Biotech. (Montreal, Canada), and the phenyl-Superose HR 10/10 column was from the same Company. Al1 reagents were of the best grade availabk. Centricon-30 and Centri- prep-30 concentrators were bought from Amicon (Beverly, MA, USA).
Assay of 1 73-HSD I Measuremen t of 1713-HSD 1 activity was performed as described
previously (Lin e t al., 1992). When the enzyme was assayed by spectrophotometer rneasurement of the reduction of NAD+ indicated by the absorptance increase at 340 nm and 23 f 1' C. The mixture contained 25 pM estradiol, 0.5 mM NAD+ in 50 m M NaHCO, buffer, pH 9.2. At this pH the maximum activity of estradiol oxidation is obtained. A blank value lacking estradiol was obtained under the same

Chapter 4 Crystallization of I 7 j - H S D I
condition and subtracted.
- Estradiol + NAD+ - Estrone + NADH + l?
One unit of enzyme is defined as the amount of enzyme that catalyze the formation of 1 p o l of product in 1 min under the above conditions,
Purification of I7J-HSD A Pharmacia FPLC (Fast Protein Liquid Chrornatography) systen
was used in a rapid purification of enzyme. 178-HSDl is from the solubl~ subcellular fraction of human placenta. The purification has beei described previously (Lin et al 1992; Zhu et al., 1993). A high specifii activity of the purified human 17B-HSD1 at room temperature, whicl was catalyzed the formation of 8 pmol estrune from estradiol per rnii per mg enzyme protein, at pH 9.2.
Protein concentration determinations The concentration of 178-HSD1 was deterrnined by the Bradford
(Bradford, 1976). This is determined by absorption at 595 nm, using BSI (bovine serum albumin) as a standard. The optical density was measurec with a Beckman DU-70 spectrop hotometer.
Preparation of apoenzyrne and I7J-HSDl -inhibitor (EM-139) The sample was from the phenyl-Sepharose Chromatography an(
prepared in a buffer containing 20% glycerol, 1 mM EDTA, 0.4 mM DTT 0.5 mM PMSF, and 40 mM Tris-HCl, pH 7.5, hereafter referred to ai buffer A. Before crystallization, 0-OG (0.06%) was added to thc apoenzyme sample (c 12 PM) by centricon buffer change. The samplc was centrifuged at 4500 g in a Sorvall RC-5 centrifuge and the fina concentration was 15 mg/rnl. The formation of 17B-HSD1 -inhibitor (EM 139) complex was shown by an optical method. During the buffe;
change, inhibitor (EM-139, 25 PM) was added to the apoenzyme sample.

Chapter 4 Crystallization of I7J-HSDI
Through three times of change buffer A (It contained 0.06% 0-OG and 2. pM EM-139.), the stoicheometric 17B-HSD 1 - inhibitor (EM1-39) wa obtained. A parallel binding assay to check for the presence O
EM-139 was performed using a Beckman DU-70 Spectrophotometer a 280 nm.
Crystallization of 17J-HSD-EM-139
The CO-crystallization experiment was carried out using the vapou diffusion method at room temperature with polyethyleneglycol (4000 26% w/v) as the precipitant. The reservoir contained 0.15 M MgCl2 am was buffered to pH 7.5. The hanging drop was formed of 3 pl of th1 concentrated complex solution (15 mglml) and 3 pl of the solution fron the reservoir. Using CO-crystallization, we also obtained apoenzymc crystals at pH 7.4. The soaking method was based on crystallizing th1 apoenzyme using the above conditions. After the apoenzyme crystal: stopped growing (about 4-6 weeks), 1 mM EM-139 was added to drop which is soluble in the presence of PEG.
Crystal characterizatian, data collection and analysis The X-ray diffraction analysis of crystals of 17B-HSD1-EM-139 wa:
performed using an R-AXIS-IIc image plate detector on a Rigaku RU-20( rotating anode with a normal focus O.5xlOmm filament. Data were ais( collected using a MAR 30 cm image plate at beam line X12C at thc National Synchrotron Light Source, Bookhaven National Laboratory Upton, NY. Data coIlection on the R-AXIS IIc was performed at ambien temperature (-23OC) with the crystals mounted in glass capillaries sealer with mother liquor. The R-AXIS IIc detector was at 100 mm from tht crystal and the beam was collimated to 0.5 mm. Data collection at thi
synchrotron was performed at a temperature of 123 K and the detectoi was placed 197 mm from the crystal. The wavelength used was 1.15 A In both cases, data were collected using the oscillation method (1.5' oscillations) about the axis. At least 58" worth of data were collectec on the R-AXIS IIc from each crystal before excessive radiation-induced

decay was observed. A full 175.5' of data were collected at the synchrotron. Al1 data sets were processed with the HKL software package.
4.4.3 Results and discussion
4.4.3.1 Preparation .of 17B-HSDLEM-139 complex
Using Fast Protein Liquid Chromatography (FPLC), 176-HSDl with a high specific activity and homogeneity was obtained. The enzyme catalyzes the oxidation of more than 8 pmol esbadiol in 1 min per mg enzyme protein (Zhu et al., 1994). The high quality of enzyme protein played an important role in the crystal growth of 17B-HSDI.
The preparation possesses a solubility of 2-3 mg/rnI in the absence of detergent. When the mild detergent 8-OG was added, 170- HSDl solubility was increased to > 40 mg/mI. The non-ionic detergent plays a role in reducing non-specific hydrophobie interactions (Zhu et al., 1994).
EM-139 is a 7u-alkyl amideestradiol derivative, so its structure is
sirnilar ru that of estradiol. Recently, the cornplex of 17B-HSDI-E2 has been successfully crystallized by using a special procedure to saturate the enzyme at a high concentration with the substrate (Zhu et al., 1996). Before crystallization, the formation of 1713-HSD 1 -EM-139 complex was verified by an optical rnethod. I t is well known that steroids are very low aqueous solu bility . Under our experimental conditions at room temperature, about 30 pM of EM-139 could be soluble in enzyme. Using a special method, to gradually saturate the enzyme, by EM-139 starting at a low 178-HSD1 concentration. We observed that the total EM-139 concentration could be higher and higher than its solubility, due to its gradua1 binding to the enzyme molecule. The 1713-HSD 1 concentration used in crystalIization is usually

Chapter 4 Crystallization of 174-HSDl
higher than 220 PM. If each subunit of 170-HSD1 is bound with one EM-139, the total EM-139 concentration should be more than 440 pM in the complex. For example in one control, before crystallization via centricon buffer change, EM-139 was added to enzyme sample (V1+V2= 2.5 ml) and the concentration ([EM-139Io) was 25 PM. Measuring the absorptance at 280 nm, we can find that the absorption of sarnple buffer was 0.3995 ([EM-I39Io). After centrifuged for 60 min, the volume of 178-HSD 1 -(EM-139) sample (VI) was decreased for more than 78 fold (32 pi). The absorption of centricon buffer (V2 = 2.468 ml) was decreased to 0.2588, and the centricon buffer contained 16 p M EM-139 ([EM-139], = 25 pM/0.3995 x 0.2588 = 16 m). The high concentration of 176-HSD 1 -(EM-139) sample can be expressed by an equation: (V1+V2) x [EM-I39I0 = Vl x [EM-13911 + V2 x [EM-13912. The result showed that 178-HSD1 is complexed with EM-139 (> 440 pM). The hydrophobic EM-139 was bound to 17B-HSDI and i t saturated the Iatter in a special repeated "dialysis", gradually reaching a s toicheometric binding to the enzyme.
4.4.3.2 Co-crystallization and soaking
Based on the former results of crystallization of other 17B-HSD forms (Zhu et al., 1993; I996), further refinement was carried out witl the PEGIMgC12IB-OG system. A series of crystaIs of 170-HSD I -EM 13! were obtained with different PEG (4000) and protein concentrations, bu the best ones were obtained in the presence of 0.16 M MgC12, 26%(w/v PEG (4000) and 0.1M Hepes pH 7.5, with 15 mg/ml protein (with 0.069 6-QG.) as the final concentration after equilibrium. Single crystal appeared in 48 h at room temperature and grew to a typical size O
0.375 mm x 0.27 mm x 0.15 mm in about three weeks (Fig. 4.4.2).
Using PEG/MgCI2/B-OG system some crystals of apoenzyme were obtained in the presence of 0.15 M MgC12, 26% (w/v) PEG (4000) and 0.1 M Hepes pH 7.4, with 15 mg/ml protein (with 0.06% 8-OG) as the final concentration after equilibrium. In six weeks, single crystals grew

Chapter 4 Crysta Nization of I 7J-HSD I
to a typical size of 0.675 mm x 0.255 mm x 0.15 mm (Fig. 4.4.3). Thc EM-139 solution was added to drop (the final concentration is 1 mM) an( the crystals were soaked for 10 days. EM-139 has very limite( solubility which the 170-HSDl concentration used in crystallization (1' mg/ml) corresponds to a concentration of about 220 pM for the enzymc dimer. If each subunit of 178-HSD1 is bound with one EM-139, the tota EM-139 concentration should be more than 440 PM. At 26% PEG (4000, and 20% glycerol, 600 ph4 of EM-139 could be dissolved.
The preliminary analysis of crystals obtained with the above twc methods of crystallization s howed that the electronic densi tj corresponding to inhibitor (EM-139) bound to the enzyme could bt reliably identified. The above methods will be useful for obtainini complexes of other steroid-converting enzymes with their inhibitors 01
subs trates.
4.4.3.3 Preliminary X-ray results
The crystals of the 178-HSDl-(EM-139) cornplex grow in the space group C2 with approximate dimensions of a = 123& b = 45A, c = 61A anc 0=9g0 being isomorphous to the 178-HSD1-E2 cornplex. Two data sel were collected: one using the cocrystaIlization and one using the soaking Neither data set was comptete, but they were integrated, scaled anc merged using the HKL program package to determine the mosaicitj present in each crystal type. The Table 4.4.1 summarizes the results.
Analysis of enzyme-inhibitor interactions wilI permit the prediction of the structure of more potent inhibitors that utilises al1 possible binding energies. The resulting inhibitor may thus contribute to breasi cancer therapy.
Ac kno w 1 e d g me n t s : This work was supported by a collaborative grant from the Natural Science and Engineering Research Council of Canada and the Medical Research Council of Canada.

Chapter 4 Crystallization qf I7J-HSDI
Table 4.4.1 Data collection
Data set R-AXS : R-AXIS: Synchrotron: crystal with soaked crys ta1 soaked crystal
cocrystallization Unit ce11 a=123.86A a=i23.17A a=122.30A
b=45.09A b=44.87A b=43.64A c=61.61A c=60.95A c=60.43A 0=99.16" B=99.38O 8=99.58"
Resolution 2.1 A 2.1 A 1.8 A limit
Data completion(%) 5 8
Mosaicity 0.76 0.7 1 0.93
4.4.4 Reference
Azzi, A., Rehse, P. H., Zhu, D.-W., CarnPbell, R. L., Labrie, F. and Lin, S.-J (1 996) Nature Structure Biology 3, '665-668.
Auchus, et al., (1989) Sreroids, 53:77.
Beranek, P. A., Folkerd, E. J., Newton, C. J., Reed, M. J., Chilchik, M. W an James, V. H. T. (1985) Inr. J. Cancer 36, 685-687.
Bonney. R. C., Reed, M. J., Beranek, P. A., Ghilchik, M. W. and James, V. E T. (1986) J. Steroid Biochem. 24, 361-364.
Bradford, M. (1976) Anal. Biochem., 72, 248-254.

Chupter 4 Crystullizution of 17J-HSD I
Ghosh, D., Pletnev, V. S., Zhu, D. -W., Wawrzak, Z., Duax, W. L., Pangborn, W., Labrie, F., and Lin, S.-X. (1995) Srrucfure, 503-513.
Inano, et ai., (1983) Eur. J, Biochem., 129: 691.
Labrie, C. Martel, C., Dufour, J.-M., Lévesque, C., Mérand Y., and Labrie 1 (1992) Cancer Res., 52, 610-615.
Lawate, et ai., (1990) J . Med. Chem., 33: 2319.
Lévesque, C., Mérand, Y., Dufour, 1. M., Labrie, C. and Labrie, F. (1991) . Med. Chem., 34, 1624-1630.
Lin, S.-X., Yan, F., Jin, J.-Z., Breton, R., Zhu, D.-W., Luu-The, V., and Labrie, F. (1992) J , Biol. Chern. 267, 16182-16187.
Luu-The, V., Labrie, C., Simard, J., Lachance, Y., Zhao, H. F., Couet, J LebIanc, G. and Labrie, F. (1990) Mol. Endocrinul. 4, 268-275.
Poutanen, M., Lsomaa, V., Lehto, V.-P. and Vihko, R., (1992) Int. J. Cancr 50, 386-390.
Sweet, et al., (1991) Biochem. Bioph. Res. Commun., 180, 1057-1058.
Murdock, G.L., Chin, C. C., Warren, J. C., (1986) Biochemisrry, 25: 641-646,
Thomas, et al., (1983) J . Biol. Chem. 258: 11500.
Zhu D.-W., Lee, X., Breton, R., Ghosh, D., Pangborn, W., Duax, W. L & Lin. S. X. (1993) J . Mol. Biol. 234, 242-244.
Zhu, D.-W., Lee, X., LabrIe, F. and Lin S.-X.(1994).Acta C v s t . DSO, 550 555.
Zhu, D.-W., Azzi, A., Rehse, P. and Lin, S.-X. (1996) J. Crystal Growth. 161 272-276.

Chapter 4 Crystallization of I7J-HSDI
Legends to Figures
Fig. 4.4.1 Structure of representative noveI compounds ac as pure antiestrogens and inhibitors of 17B-HSD1 activity. 1 6a-halogenated compounds: EM- 139 and EM-221. D-ring unsaturatec compounds: EM- 140, and EM- 123.
Fig. 4.4.2 Co-crystallization: the crystals of 178-HSDl-EM-1 The crystals were obtained in the presence of 20% glycerol, 0.16 M MI Cl2, 0.1 M Hepes, pH 7.5 and 26% (w/v) PEG (4000) as the precipitani Final protein concentration was 15 mgfml, the total EM-13! concentration should be more than 440 p M in the compIex. Crystal grev to 0.375 mm x 0.27 mm x 0.15 mm in three weeks.
Fig. 4.4.3 Soak method: the crystals of 17fi-HSD1-EM-139. The crystals of apoenzyme were obtained in the presence of 26% (w/v PEG (4000), 0.15 M Mg Clz, 20% glycerol and 0.1 M Hepes. pH 7.4. Fina protein concentration was 15 mg/ml. After six weeks. EM-139 solutioi added to drop (the final concentration was 1 mM). The crystals werc soaked for ten days. The crystal size is 0.675 mm x 0.255 mm x O. 11 mm.

Fig. 4.4.1 OH
-0 Ris
HO &OH R7

Chapter 4 Crystallization of 17J-HSDI
4.5 Crystal l ization of human estrogenic 1 7 hydroxysteroid dehydrogenase under microgravity
The main problems for protein crystallization on the ground art multinucleation and formation of rnulticrystals. Thus, we were trying tc: improve crystal quality via crystallization under microgravity. On othei hand, this is a first attempt of space crystalIization for membranc associated or hydrophobic proteins. In this paper, 1 was responsible foi most purification, deterrnining condition of space test and al1 sirnulatior test on ground.

Chapter 4 Crystallization of 171-HSDI
CRYSTALLIZATION OF HUMAN
ESTROGENIC 17B-HYDROXYSTEROID
DEHYDROGENASE UNDER MICROGRAVITY
Dao-Wei Zhu, Ming Zhou, Ying Mao,
Fernand Labrie and Sheng-Xiang Lin*
Medical Research Council Group in Molecular
Endocrinology,
CHUL Research Center and Laval University,
Quebec, Canada, G1V 4G2
* 4.5 is adapted from the paper of "Crystallization of human estrogenic
170-hydroxysteroid dehydrogenase under rnicrogravity" by Dao-Wei
Zhu- Ming Zhou, Ying Mao, Fernand Labrie and Sheng-Xiang Lin in J,
Crystal Growth (1995) 156, 108-1 11.

Chupter 4 Crystallization of 17J-HSDI
Abstract
Human 178-hydroxysteroid dehydrogenase had been crystallized O
the ground in the complex form with NADP+ and a complete data set c the crystal was primarily collected at 2.9 A (Zhu et a1.,1993). T eliminate multiseeding, formation of multicrystals and to obtain highe quality crystals, we camed out the crystallization aboard the Russia MIR space station and crystals were recovered in January, 1994. Crystal of the enzyme were forrned in 9 of the total 12 sitting drops in the spac mission, in the presence of NADP+ or estradiol. This is a first attempt c crystallization of a membrane associated protein under microgravity i the presence of a detergent. The space experiments showed bette results in nucleation number, crystal size and morphology than th ground ones, obtaining crystals diffracting to resolutions between 2.5-2. A. The too early ground mixing has limited a more importan improvement of the crystallization.
4.5.1 Introduction
The critical importance of human 178 - h y d r O x y s t e r O i dehydrogenase (1 7B-HSD 1 ) (EC 1.1.1.62) in the conversion of se hormones makes it a significant target for breast cancers drug therap: Determination of its three-dimensional structure will facilitate the searc for inhibitors. In fact, Dr. Chin's group reported the crystallization (
human placenta1 17B-HSD (Chin et al., 1976), however, to Our knowledgi about the crystal structure, even preliminary data has never bee published before Our study (Lin et al., 1992a).
Using fast protein liquid chromatography, we first purified placent; 170-HSDl to homogeneity and to high specific activity, which was aboi two- to three times those reported in the literature (Lin et al., 1992a, 1 Jarabak 1969). We increased the solubility of human 178-HSD1 using E octylglucoside and crystallized it in the presence of a saturatin1

Chapter 4 Crystaliization of I 7 j -HSD I
concentration of NADP+ and 30% PEG-4K. We obtained crystals complexi with NADP+, the complete data set was to a resolution of 2.9 A (Zhu et a 1993). These are the first crystals suitable for complete X-Ray structu analysis ever reported for a steroid-converting enzyme from a hum; source.
The main problems f o r our ground crystallization we multinucleation and the formation of multicrystals, which reduce bo crystal size and quality. Thus, it was important to improve the cryst quality via crystallization under microgravity where the convectic effects and the sedimentation of growing crystals could be minimize and the differences in the density of protein, salt, and buffer sohtioi could also be avoided (see De Lucas et al., 1989; Littke and John 1984 This will Iead to a high resolution in the structural determination of 17, HSD, helping in the search for inhibitors in the enzyme therapy of brea and prostate cancers. On the other hand, our results from this study wi enable us to understand the crystallogenesis of membrane-associated 1
hydrophobic proteins.
4.5.2 Materials and Methods
4.5.2.1 C hemicals
NADP+, NAD+, glycerol, B-OG, MgC12, LiC1, PEG-4K, Hepes, Tris we: purchased from Sigma (St. Louis, Mo, USA); 1713-Estradiol was fro Aldrich (Milwaukee, WI, USA); Blue-Sepharose CL-6B colurnns wei packed in the laboratory using media from Pharmacia Biotech. (Montrea Canada). A l reagents were of the best grade available.
The equipment used for 17B-HSD 1 crystal growth experiments c Russian MIR station was from the Payload System Inc which emplo] vapor diffusion geometry, as illustrated in Fig.4.5.1.
4.5.2.2 Purification of 17B-RSD1

Chapter 4 Crystallization of I7J-HSDI
Native 17B-HSD1 possessing a low aqueous solubility of 2-3mg/d, from the soluble subcellular fraction of human placenta. The purificatic is similar to previously described procedures (Lin et al., 1992b; Zhu et a 1993): NADP+ has been used to elute protein from Blue-Sepharo! column because of its higher affinity to 178-HSD1 than NAD+. Tl purified 170-HSDI has a high specific activity catalyzing the formation i
8 pmol estrone from estradiol per min per mg enzyme protein, at pH 9.. After purification, 170-HSD1 is in the apoenzyme form which is the mixed wi th its cofactor and/or substrate to form the correspondir; complex(es) according to the requirements of crystallization. Protei concentrations were measured by the optical method of Warburg Christian (1 942).
4.5.2.3 Crystall ization under microgravi ty
We attempted to crystailize two protein complexes, 178-HSD-NADP and 17B-HSD-estradio1 in twelve wells in total. As a ground preparatioi 20 pl of 17B-HSDI solution and 20 pl of resewoir solution were adde and well mixed in cups A and equilibrated with 500 pl correspondin well solutions in B (Fig. 4.5.1). We used 12 wells ranging from Wells Ni 13 to 24 of a crystallization device.
The 17B-HSD1 solution contained 20 mglml of enzyme, 0.06% 8-01 and 20% glycerol, in the presence of 0.6 m M NADP+ (form-1) or 25 pl estradiol (form-2). In wells No. 13 to 16 and 19 to 22 (form-1), th reservoirs contained MgCl2 (0.15 or 0.16 M), Hepes (from 0.06 to 0.1 M pH 7.5, and PEG-4K (frorn 26 to 29%). In wells No. 17 and No. 18, thel were 0.2 M LiCl and 0.2 M NaCl respectively, both including 0.04 M Tri! Cl pH 7.2, and 26% PEG-4K. In Nos. 23 and 24 (form-2). the reservoii contained 0.2 M NaCl and 0.2 M LiCl respectively, both in the presence c
0.04 M Tris-HCl, pH 7.2 and 26% PEG-4K. A screw-plunger on the to plate of the device was closed down un the protein cup before the fligh When screwed down, the plunger presses a teflon and silicone rubix septum against the edges of the protein cup, thus sealing the protei solution away from the welI solution (launch, as shown in Fig. 4.5.1)

Chapter 4 crystalIization of 17P-HSDI
After activation in space, the septum is raised, establishing an air path between the solutions, to allow vapor phase equilibration (activation, as shown in Fig. 4.5.1). The cup can be re-sealed with the septum, to stop the experiments. The temperature of the crystallization was controlled at 19 f 1°C. A duplicate set of experiments which was activated and deactivated at approximately the sarne time as the apparatus on MIR served as a control on ground.
4.5.3 Results and discussion
Before the space experiments, we made various ground preparations including assaying crystallization conditions with hanging drop or sitting drop, and reproduced the crystallization experiments many times. Considering that the absence of gravity will decrease the differences in sitting and hanging drop methods, we used conditions similar to hanging drops.
The space mission: The system that carried out the 178-HSD1 crystal growth under microgravity was launched on board the Russian MIR station in October 1993 with schedule modifications. This mission had a duration of 88 days in space, which was twice as long as expected, and crystals were recovered in January 1994. The I7B-HSD crystals, mostly grown in the presence of the cofactor NADP+, had been formed in 9 of the total 12 wells.
(A) In Wells No. 13 to 16 and No. 20 and 21, the space experiments resulted in fewer and somewhat larger sized crystals. For example, in Well No. 13, the ground experiment had about 80 crystals, but the space experirnent had only about 20. In Well No.15, the crystals were about 40 in space and about 100 on the ground. In Well No. 20, the ground expenment resulted in more than 100 crystals with a mean size of 0.1 x
0.1 x 0.25 mm3, and the space experiment had about 20 crystals at 0.2 x

Chapter Crystallization of I7J-HSD
0.2 x 0.35 mm3. The reduction of nucleation in space crystallizatio happened for almost al1 drops in which we had crystals.
(B) In most wells, e.g. No. 13 and 15, there were more single crystals i the space experïment than those in the ground.
(C) In several wells such as No. 20 and 15, the space-grown crystal possessed much better morphology (and a smaller number of crystai than mentioned above) than the ground control (Fig. 4.5.2).
(D) In well No. 24, we had 5 crystals from the microgravity experimen but no crystals on the ground.
In some of the drops, e.g. No. 15 of the flight, the crystals diffracte to 2.5 A, which is somewhat higher than the values of about 2.7 A fror most of the ground crystals we analyzed.
The results showed less nucleation, better morphology and relative1 larger size of crystals for the space mission. Under certain conditions, e.g No. 24, crystals were only obtained in space indicating that th microgravity condition is favorable for ordered packing of th macromoiecuIes in the absence of convection. The too early mixing a protein and precipitant solutions (8 days before the activation of th crystallization device) can cause nucleation and even micro-crysta development before the flight, thus hindering more pronounced result for this space mission. This occurred because of the sudden schedul change in October 1993, a consequence of circumstances in Russia at tha moment. In fact, our post-flight control showed that several days afte the rnixing and before equilibrating with the well solution, certain srnal crystals started to form under some conditions. This was probably th1 major hindrance to further improving crystal quality in this spacl mission. More rigid control is absolutely necessary and is being used fo Our current space crystallization.

Chapter Crystallization of I 7J-HSD
Our experiments are also of the first attempts to crystallize membrane associated protein: 170-HSD1 (Breton e t al., 1994) in tl
presence of a detergent. Our present results in the above missic showed that microgravity could facilitate the crystallization for sui proteins which possess many important biologicai functions.
A c k n o w l e d g m e n t
This work was supported by the Canadian Space Agency. We thar Drs. D. Ghosh for carrying out the major part of X-ray analysis in tl Medical Foundation of Buffalo. We like to thank Ms. Naiini Warriar f i
her contribution on language correction.
4.5.4 References
Breton, R. Yang, F, Jin, J.-2. B. Li, B. Labrie, F.and Lin, S.-X. J . Stero Biochem. Molec, Biol. 50 (1994), 275-282.
Chin, C.C. Dence, J.B. and Warren, J.C. J. Biol. Chem. 25l(f W6), 3700-3701
De Lucas L.J. et al., Science, 246 (19891, 651-654.
Jarabak, J. Methods Enzymol. 15 ( 1 969). 746-752.
Lin, S.-X. Sailofsky, B. Lapointe J. and Zhou, M. J. Crystal Growth, 1: (1992),242- 245.
Lin, S.-X. Yang, F. Jin, J.-2. Breton, R. Zhu, D.-W. Luu-The V. and Labrie, 1 J.Bio1. Chem. 267 (1992) , 16 182-1 6 187.
Littke W. and John, C. Science 225 (1984), 203-204.
Warburg 0. and Christian, W. Biochem. (1942) J . 310, 384-421.
Zhu, D.-W. Lee, X. Breton, R. Ghosh, D. Pangborn, W. Duax, W.L. and Lin, S X. J . Mol. Biol., 234 (1993), 242-244.

Chapter 4 Crystallization of I7J-HSD
Legends to figures:
Fig. 4.5.1. Crystallization geometry used in the MIR space Payload System Inc.
Each protein solution is mixed with a low initial concentration precipitant in A, with a total volume of 50 pl; precipitant solutions 51 pl were in B. Here the left and right positions are for launch ai activation respectively, and the operations were mentioned in the tel To deactivate the experiment, the cup is re-sealed with the septum. TI cosmonauts wiIl activate and deactivate the space flight experiments.
Fig. 4.5.2.
In the Well No. 20, the crystals from space (left) were fewer, b somewhat larger, and possessing much better morphology than tho. from ground experiments (right). The space crystals were mostly single.

Fig. 4.5.1
stop pin k washer
UW( rrmA1111

Chapter 5
Structure of 17B-HSD1

Chapter 5 Structure of I7J-HSDI
The structures of 178-HSDl was determined. This is the fir mammalian steroidogenic enzyme studied by X-ray crystallograph techniques, which reveals a fold characteristic of the short-cha: dehydrogenases, The results lead to the understanding of the structu: of 178-HSD 1 -ligand complex and structure-function relationships. Th( will provide a strong basis for the design of potent inhibitors of th s teroid dehydrogenase.
This chapter is adapted from paper "Structure of human estrogenic 171 hydroxysteroid dehydrogenase at 2.20 A resolution" by Ghosh et al. :
Structure (1995).

Chapter 5 Structure of 17J-HSDl
Structure of human estrogenic l7B-hydroxystei dehydrogenase at 2.20 A resolution
D. Ghosh, V. Z. Plemev, D.-W. Zhu, 2. Wawnak, W. L. Duax, W. Pangbon F. Labrie and S.-X. Lin
My contribution to this paper was the crystailization of 178-HSD1 NADP+, providing crystals for data collection. Data collection and crysti analysis was performed at the Medical Foundation of Buffalo, USA.
* This is adapted from the paper of "Structure of human estrogenic 17 hydroxysteroid dehydrogenase at 2.20 A resolution" by D. Ghosh, V. Pletnev, D.-W. Zhy 2. Wawnak, W. L. Duax, W. Pangborn, F. Labrie and ! X. Lin in Structure (1995) 503-513 .

Chapter 5 Structure of I7J-HSDI
5.1 Introduction
Steroids, including 178-estradiol and androst-5-ene-30,17B-diol (A! diol), are potent stimulators of breast cancer ce11 growth (Dickson Lippman., 1987; Asselin & Labrie, 1978; Horwitz & McGuire, 1987; Pouli & Labrie, 1986; Spinola et al., 1986; Dauvois et al., 1989). Because huma estrogenic 176-hydroxysteroid dehydrogenase (type 1 178-HSD) catalyzt the last step in the biosynthesis of the most active estrogen, 171 estradiol, from estrone (Engel & Groman, 1974; Strickler & Tobias, f98( Luu-The et al., 1989)- it is an attractive target for the design of inhibitoi of estrogen production and tumor growth (Labrie et al., 1992). The ger encoding type 1 human 178-HSD and a tandem pseudogene are located a the ql l -q12 region of chromosome 17 (Luu-The e t al., 1989; Luu-The t
al., 1990), near the BRCAl gene associated with hereditary breast an ovarian cancers (Simard et al 1993).
Estrogenic 17B-HSD was purified from hurnan placenta (Luu-The 1
al., 1989), where it is present in soluble and membrane-associated form We have expressed type 1 17B-HSD cDNA in insect cells, where it is als distributed between soluble and membrane fractions (Breton e t al 1994). The fact that salt extraction of 178-HSD in aqueous mediui requires the addition of sodium c holate, and crystailization requires th presence of octyl glucoside detergent (Zhu et al., 1993; Lin et al., 1992 indicates the hydrophobic nature of the surface residues and the affinit of the enzyme for ce11 membranes.
After purification to homogeneity (Luu-The et al., 1989; Zhu et al 1993; Lin et al., 1992), to homodimeric nature of 178-HSD wa demonstrated (Lin et al., 1992). Mechanisms of enzyme action and th composition of the active site have been probed by biochemical method (Bhatnagar et al., 1978; Tobias et al., 1986; Murdock et al., 1986; Inan 1988). Sequence alignment analysis and homology studies (Presson et al 1991; Ghosh et al., 1 994; Baker et al., 1991; Urozowski et al., 1992:

Chapter 5 Structure of 174-HSDI
reveaIed that 17B-HSD belongs to the short-chain dehydrogena: superfarnily, and has a characteristic Tyr-X-X-X-Lys sequence of residut at the active site. The invariant tyrosine and lysine residues have bee shown to be catalytically important by site-directed mutagenesis (Chen (
al., 1993; Ensor e t al., 1991; Obeid et al., 1992) and are found at the actil sites of bacterial 3a, 20B-HSD (Ghosh et al., 1994; Ghosh et al., 1991) an rat liver dihydropteridine reductase (DHPR) (Vanighese et al., 1992), tw members of the family whose three-dimensional structures have bee reported. Here we report the three-dimensional structure of human t y ~ 1 170-HSD, describe the active site of the enzyme in detail and propose binding mode for the steroid and a mode1 of the transition state of th estrone to estradiol reaction.
5.2 Results and discussion
5.2.1 Description of the s t ruc tu re
The structure of the type 1 178-HSD from human placenta wz determined at 2.20 A resolution by a combination of isomorphous heavj atom derivative and molecular replacement techniques. The structure c
bacterial 3a,20B-HSD was used as the search model. Tables 5.1-5.
sumrnarize data collection, heavy-atom phasing, phase combination an refinement results, respectively. The refinernent was also carried ot with a 2.5 A data set collected on a crystal of the recombinant enzymt isomorphous to that of the native enzyme. These two refined structure are identical.
Forty-three residues at the C terminus of the protein (285-325 could not be located and residues 192-199 had very weak density. Th amino acid side chains for the rest of the protein were clearly defined i electron-density maps and the agreement between the cDNA-derive sequence (Luu-The et al., 1989) and the electron density of the amin

Chapter 5 Structure of 174-HSDI
acid side chains was excellent. The ribbon diagram of a monomer of 17 HSD (Fig. 5.1 a) reveais the same arrangement of B-sheet and a-helices has been observed in structures of bacteriai 3a,20B-HSD (Ghosh D. et al 1994; Ghosh et al., 1991) and rat liver DHPR (Varughese et al., 199: There are, however. some structural features in 17B-HSD that are n observed in the other two enzymes. There are 327 amino acid residu in 17B-HSD (Luu-The et al., 1989), compared with 255 and 240 3a,20B-HSD and DHPR, respectively. Of the 76 additional residues in 17
HSD (the N terminus of 170-HSD is four residues shorter than in 3a,20 HSD), 52 are at the C terminus and 14 are inserted at position 200 (bas1 on 3a,20B-HSD numbering) and reside near the active site. Togethr these additions alter the topography of one end of the active site of 17 HSD relative to 3u,SOO-HSD and DHPR.
The core of the structure is the seven-stranded parallel B-sheet (1! to BG), surrounded by six parallel a-helices (uB to aG), three on each si,
of the 0-sheet (Fig. 5.1 a, b). The basic fold of the segment BA to BF is doubly wound a/B motif, with alternating 0-strands and a - h e t i c e Whereas the 13A to BF segment is the classic 'Rossrnann fold', associati with nicotinamide adenine dinucleutide binding, the BD to BG segment, addition to being partly in the Rossmann fold, governs quaterna association and substrate binding (Ghosh et al., 1994). In 3a,2OB-HSD ai
DHPR (Ghosh et al., 1994; Varughese et al., 1992), the cofactor and tl substrate-binding sites are combined in a single domain. In 17B-HSD, long helix, aG' (Fig. 5.1,2), is inserted in the Ioop between BF and aG. TV
additional helices, aG" (residues 200-206) and aG' (residues 209-221 create a helix-turn-helix (HTH) motif preceding the helix aG. Helices al
(residues 260-266) and aH (residues 273-284) from another HTH moi that is not present in 3a,20B-HSD or DHPR. Together, these segments 1
the polypeptide chain (residues 190-229 and residues 259-284 containing helices aG", aG' aH' and a H , fiom one end of the 'substrat binding domain' (Fig. 5.3). Mass spectrometric determination of tf

Chapter 5 structure of 17#-HSDI
molecular weight of the crystalline enzyme indicates that al1 32 residues are present in the crystal (S.-X. Lin, unpublished data However, the a-carbon chain cannot be traced beyond residue 21 because the electron density is so diffuse. The missing 43-residue (
terminal segment is rich in alanine, glycine and proline residues, and hi repeating Ala-Gly sequence which may account for its lack of secondai structure and or high thermal motion.
Monomers related by the crystallographic two-fold axis form tl closest dimeric association in the crystal (Fig. 5.4), an associatic analogous to the Q-axis dimerization in tetrameric 3a.206-HSD (Ghosh I
al., 1994) and dimeric DHPR (Varughese et al., 1992). At the dimc interface, helices aE and aF from each monomer form a four-helix bundll The second interface in 3 a ,200-HSD, the P-axis interface, which a l l o ~ antiparallel association of the two OG strands and the two aG helict about a non-crystallographic two-fold axis, is accessible in the crysti structure of 17B-HSD, but not used. This contrasts with the DHP structure, where an eighth B-strand, BH, antiparallel to BG exists so th; no P-axis interface analogous to that in 3a,200-HSD could be formed. 1
as has been reported (Lin et al., 1992), 17B-HSD is active as a dimer, it likely to take the form illustrated in Figure 5.4. A postulated tetrameri assembly of 178-HSD monomers (GuIler et al., 1986) could be achieve through formation of a similar P-axis-type association of two of th dimers shown in Figure 5.4.
5-2-2 Architecture of the active site
The catalytic site comprises a cofactor-binding cleft and a steroic binding cleft. Although 1 mM NADP+ was present in the crystallizatio medium, density corresponding to the cofactor could not be reliabl identified. The mode1 of the cofactor position in the binding cleft of 17L HSD was derived from a superposition of the structures of 17B-HSD and ;

Chapter 5 Sfrudure of 1 7J-HSDI
complex of 3a,20B-HSD with NAD+ using a least-squares fitting procedur with 20 Ca atoms that were found to have highly similar relativ positions in the two enzymes (Table 5.5). These C a atorns (including thos of the catalytic niads) were within 1 A of one another in the initi; fitting. The average root rnean square (rms) deviation for these atom was 0.7 A. Figure 5.5 is a stereodiagram of the superirnposed C a cha in of 17B-HSD and the 3a,20B-HSD-NADP+ complex. Differences may exi5 between the binding modes for NAD(H) to 3a,20B-HSD, and for NADP(H to 17B-HSD. Asp37 in 3a,2OB-HSD, which forrns hydrogen bonds to th S'-and 3'-hydroxyls of the ribose at the adenine end, is replaced b Leu36 in 170-HSD. The analogous residue is a valine in glutathion reductase, another NADP+-specific enzyme with a Rossrnann fold. From cornparison of a large number of oxido-reductase structures, Wierenga e al. concluded that NADP(H) and NAD(H) bind similarly to the 0aB-fold Because 178-HSD also binds NAD(H) (Zhu et al., 1993), the position of th 2'-phosphate may be occupied by another ion, as has been proposed fa glutathione reductase (Scrutton et al., 1990).
In the cataIytic cleft (Fig. 5.6) the adenine-ribose end of th! modeled cofactor surrounded by five segments of the protein chain residus Gly9-Serll of the BA to aB turn, Arg37 from the BB to aC turn residus Asp65-Arg67 from the end of OC, residus Ala91-Leu93 of GD, anc Va113 of aE. The S'-phosphate oxygen atoms form hydrogen bonds witl the side-chain hydroxyl of S e r l l , with main-chah amide groups anc with the side chain of Arg37. The nicotinamide-ribose end of the cofacto is surrounded by Asn90, Thr140-Ser142, Tyr155, Lys156 and Va11 88 Met193. The p r o 4 hydride face (B-face) of the nicotinamide ring i directed towards the steroid-binding cleft, which is lined by thc conserved catalytic triad Tyr 15 5-Lys 159-Ser 142 on the side and th( helix a G 1 on the other. It has been shown that a steroid-like enzymi inhibitor binds competitively at the analogous catalytic site in 3a, 20B HSD and forms a hydrogen bond with the catalytic tyrosine hydroxy (Ghosh et al., 1992; Ghosh et al., 1994).

Chapter 5 Structure of I7Q-HSDI
5.2.3 Substrate recognition and the transition state
A 17B-estradiol molecule has been modeled into the active site ti mirnic the transition state of the estrone to 17B-estradiol reaction (Fig 5.7 a. b). In the proposed transition state, the pro-S hydride from th1 nicotinamide is transferred to the a-face of the steroid at the Cl7 atom whereas the keto oxygen at C l7 forms a strong hydrogen bond (2-2.5 A with the hydroxyl of Tyr155. The hydride transfer could occur over ;
short distance of -2 A (2.7 A between C4 of the nicotinamide ring an, C l 7 of the steroid in the model shown). A proton-transfer interactioi between the 17-keto oxygen and the Tyr155 hydroxyl could bi facilitated by the close proximity of the protonated Lys159 side chain Although no direct interaction between Lys159 and Tyr155 is o b s e ~ e i in the crystal structure (Tyr155 OH ... NC Lys159, 4.4 A), the transitioi state (shown in Fig. 5.7 b) might be arrived at by a small conformations change. The Ser142 hydroxyl could also donate a proton to Tyr155 either to stabilize any oxyanion intemediate, or to replenish the missinl proton on Tyr155 from the solvent network, or both. One or more O
these three side chains (Ser142, Tyr15.5, Lys159) should then bc undergoing dynamic exchange of protons with solvent molecules. Wt found that the Lys159 side chain, buried in the active-site cleft, ii hydrogen bonded to a solvent water molecule. This is not surprising, as i
protonated and buried Lysine is likely to be solvated. However, tht observation of a water molecule bound to Lys159 may also indicate tht participation of this side chain in the proton-relay network.
A color-coded Connolly surface of the active-site cavity, as it i! observed in the crystal structure deterrnination (Fig. 5.8). shows that th( 3-hydroxyl end of the A ring of 178-estradiol has to fit into a relative11 narrow cavity and make specific binding interactions in order to attain : suitable stereochemical orientation for catalysis to occur at the Cl7 end 01
the molecule. The 3-hydroxyl oxygen of the steroid molecule, a! positioned in this model, would be 3.4 A from Nt, of the His221 side chain

Chapter 5 Structure of 17J-HSDI
located on the aG' helix (Fig. 5.7 a). The side chains of a few 0th residues, including Tyr218, Ser222, Va1225, Met279 and Va1283 froi helices aG' and aH, which border the active site, are located within tl steroid-binding cavity. Therefore, the new domain composed of helict aG", aG', aH' and aH (gold colored in Fig. 5.3), may determine recognitic and selectivity of substrates and influence the stereospecificity of tf reaction. The shape-recognition of the substrate at this end (Le. the f l i
aromatic A-ring of estrogens) is achieved through steric interactions wii atoms bordering the narrow cavity, primarily from helices not found i the structures of 3a,208-HSD and DHPR.
A few cysteines, three histidines and a tyrosine residue, ail believe to be in the active site, were tagged with bromoacetoxy steroid inhibitoi in affinity-labeling experiments (Bhatnagar et al., 1978; Tobias et al 1981; Murdock et al., 1986; Karavolas et al., 1970; Chin et al., 1973). A oxidized cofactor protected the essential Iysine and a cysteine residu from being labeled by trinitrobenzene sulfonate (Inano 1988). 0i structural results suggest that the labeled lysine was probably Lys15 (Fig. 5.6), which would be protected by NADP+. Cysl85, which binds Hg2 producing the isomorphous heavy-atom derivative used here fc structure solution (see the Materials and methods section), would als have been protected from bromoacetoxy tagging by NADP*. Two othc cysteines are found in and around the active-site cleft: Cys156 near th steroid-binding site and CyslO near the cofactor site (Fig. 5.6). Helix aC contains two other histidine residues, His2lO and His213, that are farthe away from the active site than His221. The labeled peptides isolate from human placental and porcine testicular 17B-HSDs in histidine labeling experiments are identical to each other in amino acid sequenc and correspond .to the segment Leu204-Lys223 of 178-HSD, most a which belongs to aG'. His22l is the most likely candidate for having bee labeled, as clearly shown in Figure 5.7 a. A recent site-directe mutagenesis study (Puranen et al., 1994) showed that Tyr155 and His22 are essential for the activity of the enzyme.

Chapter 5 Structure of. I7J-HSDI
5.2.4 Possible membrane association
The amphiphilic nature of two of the helices that form one end of t
substrate-binding pocket, aG' and aH, is illustrated in Figure 5.9. A thii
shorter, helix in that region, EH', is primarily hydrophobic. Ma: amphiphilic helices are postulated to bind or insert into lipid bilaye (Tamm et al., 1994). These three helices, aG', aH and aH', are rough coplanar. The hydrophobic surface thus generated contains residu Phe215, Tyr21 8, Leu219, Va1225, Phe226, Leu260, Pro261, Leu26 Leu263, Met265, Tyr275, Va1276, Ala278, Met279, His280, Va1283 ai Phe284. Most of this hydrophobic surface is directed towards t steroid-binding cleft. The hydrophilic surface of the opposite face of t
helix cluster contains residues Thr213, Arg2 14, Tyr216, Gln217, Gln22 Lys223, Arg227, Arg264, Arg266, Ser273, Asn274, Thr277 and Arg28 al1 of which are capable of interacting with the phosphate-rich surface the phospholipid bilayer. From the topological distribution of the helices, it appears that the polar surface could interact with the bilay surface of the membrane, whereas some of the hydrophobic side chai might penetrate deeper into the bilayer. However, this polar surface al contains two glutamic acid side chains (228 and 282) which are not like to interact with the bilayer surface. The Glu282 side chah forms a si bridge with the His221 side chain in the crystal structure. The His2: side chain, postulated above to form a hydrogen bond with the hydroxyl group of the substrate as a mode of recognition, is rough intermediate in position between the two surfaces.
It is possible that molecules of estrogenic 178-HSD associate with membrane through interaction of one or more of the helices aG', aH' ai
a H with the lipid bilayer. Because these helices are distant from both tl Q-axis and the P-axis interface, catalytically active dimers or tetrame could form without disturbing the potential for membrane associatio Interestingly, the electron density for the C terminus, which is rich alanine, glycine, proline and other hydrophobic residues, is il1 define

Chapter 5 Structure of 17J-HSDI
beyond the end of aH at position 285. A stable conformation of the i
defined C-terminal residues may arise as a result of hydrophob interaction within the membrane.
5.2.5 Other isozymes of 17R-HSD
In addition to the type 1 178-HSD that is the subject of this prese: study, two I 7 B-HSDs with different substrate specificities have bef characterized (Geissler et al., 1994): the type II enzyme (capable 4
testosterone to androstenedione as well as estrone to estradii interconversion) and the type III enzyme (specific for testosterone I
androstenedione interconversion) have 80 and 46 additional N-termin, residues, respectively, relative to the type 1 enzyme (387 and 310 tot. amino acid residues, respectively). Because these N-terminal insertior are hydrophobic in nature they may enhance membrane association r
type II and type III enzymes. However, sequence comparison sugges that the overall structures of the two f o m s of 178-HSD are quite simili to that of type 1. Differing substrate specificities may arise froi sequence differences in the region 192-228 (of type 1), a significai portion of which is the helix aGr, which constitutes the propose substrate-recognition region of the active site. The equivalent substrat1 recognition regions in type II and III incorporate residues 269-313 an 235-26 1, respectively . Pairwise corn pansons between the three proteir reveal an overall sequence identity of 23% (Geissler et al., 1994), but th
identity in the substrate-recognition region is only ,IO%, which is nearl random. Moreover, residue His221, which is proposed to be critical fi substrate recognition by 17B-HSD type l, is not conserved in isozym type II and III. Despite the structural similarity in the overall folc differences in the active sites of the various short-chain steroi dehydrogenases must account for substrate specificity and should perm the design of specific inhibitors.
5.3 Biological implications

Chapter 5 Structure of I7J-HSDI
Human estrogenic 178-hydroxysteroid dehydrogenase (type I 171 HSD) is responsible for the last step of the biosynthesis of 170-estradic the most potent naturally occurring human estrogen and a pote stirnulator of breast cancer. 17B-HSD is a member of a superfamily 1
oxido-reductases, known as short-chain dehydrogenases, that ah includes bacterial 3a.200-HSD, human rend 11B-HSD, human placent 15-hydroxyprostaglandin dehydrogenase, rat liver dihydropteridir reductase (DHPR) and Drosophila alcohol dehydrogenase. X-ray structu analyses of DHPR and of bacterial 3a,208-HSD (in the presence of i NAD+ cofactor and an inhibitor derived from licorice) have identified tt basic fold of this class of enzymes and the location of cofactor ar substrate-binding sites. A catalytic mechanism involving an invariai Tyr-X-X-X-Ly s sequence has been proposed. Site-directed mutagenes studies have confirmed that the conserved tyrosine and lysine residuc are essential for enzyme function.
The present investigation constitutes the first X-ray structui determination of a mammalian steroidogenic enzyme. Our resul demonstrate that despite only about 15% sequence identity and tk presence of two major insertions and additional residues at the terminus, 176-HSD has the same overall fold as bacterial 3a,20B-HSD an mammalian DHPR. The additional residues in 17B-HSD form two heli~ turn-helix motifs, and these constitute a 'substrate-recognition domaii bordering the active site at the end of the steroid-binding cavity opposii the cofactor-binding site and the catalytic residues. The apparei amphiphilic nature of the additional helices and their topological locatia are consistent with the observed association of the enzyme wii rnicrosomal membranes. Modeling with 170-estradiol at the active si1 suggests that the 3-hydroxyl end of the substrate, which is at the end (
the molecufe opposite to where the oxido-reductive reactions occur, fil into a cavity in this dornain while forrning a specific hydrogen bond wii a histidine side chah. Comparable substrate-recognition features are nc present in the active site of the bacterial enzyme, allowing it to process

Chapter 5 Structure of I7J-HSDI
greater range of substrates. The proposed transition state of the enzyi is consistent with the relative locations of the recognition histidine histidine side chain that makes a specific hydrogen bond with the nt reactive 3-hydroxyl end of the substrate) and the catalytic Tyr-Ser-L triad in the active site. The mode1 of the active site of 17B-HSD deriv from the present structure analysis provides a template for the design specific inhibitors as drugs for breast cancer therapy.
5.4 Mater ials and methods
5.4.1 Data collection
Human placenta1 178-HSD was crystallized as described (Zhu et 2
1993). The space group is C2 and the unit ce11 parameters are a = 123. A, b = 45.05 A, c = 61.33 A, 0 = 99.02'. The data collection was carri
out on a Rigaku R-AXIS IIc image plate area detector receiving X-I from a Rigaku rotating anode, and processed with R-AXIS data processi program package (version 3.40). There native data sets were collectf two with placenta1 protein crystals and one with crystals prepared frt recombinant protein. Three data seta were collected for a mercu derivative, one set from each of three crystals socked with a solution HgC12. The placental enzyme crystals were used for heavy-atom soakir Al1 data sets were collected at ambient temperature. Table 1 summariz the data collection statistics.
5.4.2 S t ruc tu re solution and refinement
The structure of 1713-HSD was determined using a combination isornorphous heavy-atorn methods and molecular replaceme techniques. The bacterial 3a,20B-HSD structure was used for rotation a translation function searches. The program package X-PLOR (Brüng 1992) was used for molecular replacement searches and also for t Patterson correlation refinement of the rotation function results. Becau,

5 Structure of I7J-HSDI Chapter
of low potential Several
sequence identity (-15%) between 3a.20B-HSD and 176-HS1 molecular replacement solutions were interpreted with cautio potential solutions were selected, including the one th
generated the predicted four-helix bundle interface by ti crystallographic two-fold rotation. The intial phase sets from the: solutions were used to calculate difference Fourier maps with derivatil and native amplitudes. Only the phases from the correct molecul, replacement solution exhibited a large peak at the major heavy-ato binding site, independently determined by the difference Pattersc method. Furthermore, this peak was iocated close to the side cha: corresponding to Cys187 in 17B-HSD. By this cross-phasing techniqu the correct chirality of the heavy-atom and the cornmon origin choit between the two phase sets were also determined. The heavy ato positions were refined with HEAVY (Terwilliger T. C. et al., 1983). TI single isomorphous replacement (SIR) phase balculation, phaj combination with the model phases and solvent flattening were carrie out using the PHASES package (W Furey and S Swaminathan, Abstral PA33, Vol. 18, p. 73, American Crystailographic Association Meeting, Ne Orleans, July, 1990). Summaries of SIR phasing and phase combinatic statistics are provided in Tables 2 and 3, respectively.
The initial model of 17B-HSD was built into a phase-combine solvent-flattened rnap a t 3.20 A resolution. Mode1 building ws perforrned on a SGI Elan workstation using CHAIN, a modified version (
FRODO (Jones 1978). The model was refined with the X-PLOR packagl The new phases were calculated and combined with the SIR phases, an new annealed omit maps were calculated for the problem region Corrections to the model were made using these omit maps. Th resolution of the model was also gradually extended to 2.20 A, as th corrections became fewer and completely new areas of the molecul began to emerge in the electron-density maps. This process of correctin the existing model, refinement of the model and calculation of the ne> maps continued for many cycles. The final model was largely error-fen

Chapter 5 Structure of I7J-HSDI
The geometry and steresochemistry of the mode1 were checked with tl PROCHECK package (Laskowski et al., 1993). A modest number of wc defined water oxygens have been modeled as bound, first-sheI1, solve molecules. The recombinant enzyme structure was refined using tl partially refined coordinates of the placental 178-HSD. The refineme summary is provided in Table 5.4.
Atomic coordinates have been deposited with the Brookhavr Protein Data Bank.
A c k no w 1 e d g e m e n t s : We are pleased to acknowledge contributions this work by Xavier Lee in the screening of crystallization conditions fi 17B-HSD and Dr Rock Breton in setting up the overproduction of tl enzyme. We would also like to thank Drs JF Griffin and Y Osawa fc reading the manuscript, Melda Tugac and Gloria Del Bel for preparation 1
the manuscript. This work is supported by NIH grant number DK2654 and MRC Canada grant for the MRC group in molecular endocrinology.
5.5 References
Asselin, J. & Labrie, F. (1978). J. Steroid Biochem. 9, 1079-1082.
Baker, M. E. (1991). Steroids 56, 354-360.
Bhatnagar, Y. M., Chin, C. C. & Warren, J. C. (1978). J. Biol. Chem. 253, 81 1 815.
Breton, R., Yang, F., fin, J.-Z.,Li, Labrie, F. & Lin, S.-X. (1994) J . Steml Biochern. Mol. Biol. 50, 275-282.
Brünger, A. T. (1992). X-PLOR (Version 3.1) Manual. Yale University, Neï Haven, CT.
Chen, Z., Jiang, J. C., Lin, 2.-G., Lee, W. R., Maker, M. E. & Chang, S. E (1993). Biochemistry 32, 3342-3346.

Chapter 5 Structure of 179-HSD1
Chin, C. C. & Warren, J. C. (1973) Steroids 22, 373-378.
Dauvois, S. & Labrie, F. (1986). Cancer Res. 46, 4933-4937.
Dickson, R. B. & Lippman, M. E. (1987)- Endocrinol. Rev. 8, 29-43.
Engel, L. L, & Groman, E. V. (1974). Recent Prog. Horm. Res. 30, 139-169.
Ensor, C. M. & Tai, H. - H . (1991). Biochern. Biophys. Res. Commun. 17é 840-845.
Evans, S. V. (1993). J. Mol. Graphics 11, 134-138.
Geissler, W. M., et al., & Andersson, S . (1994). Nature Genet. 7 , 34-39.
Ghosh, D., Wawrzak, Z., Weeks, C. M., Duax, W. L. & Erman, M . (199L Structure 2, 629-640.
Ghosh, D., et ai., & Orr, J. C. (1991). Proc. Narl. Acad. Sci. USA 88, 10061 10068.
Ghosh, D., Erman, M., Pangborn, W., Duax, W. L. & Baker, M. E. (1992). Steroid Biochem. Mol. Biol. 42, 849-853.
Ghosh, D., Erman, M., Wawrzak, Z., Duax, W. L. & Pangborn, W. (199a Structure 2, 973-980.
Guller, S., Gravanis, A. & Gurpide, E. (1986). J. Sreroid Biochem. 24, 93: 944.
Horwitz, K. B. & McGuire, W. L. (1987). J . Biol. Chem. 253, 8185-8191.
Inano, H . (1988). Biochern. Biophys. Res. Commun. 152. 789-793.
Jones, T. A. (1978). J. Appl. Crystallogr. 11, 268-272.
Karavolas, H. J., Baedecker, M. L. & EngeI, L. L. (1970). J. Biol. Chem. 24: 4948-4952.

Chapter 5 Structure of 173-NSDI
Labrie, C., et al., & Labrie, F. (1992) Cancer Res. 52, 610-615.
Laskowski, R. A., Macarthur, M. W., Moss, D. S. & Thornton, J. M. (1993). Appl. Ctystallogr. 26, 283-291.
Lin, S. -X., et al Labrie, F. (1992) J. Biol. Chem. 267, 16182-16187.
Luu-The, V., et al., Br Labrie, F. (1990) Mol. Endocrinol. 4, 268-275.
Luu-The, V., et al., & Labrie, F. (1989) Mol, Endocrinol. 3, 1301-1309.
Murdock, G. L., Chin, C. C. & Warren, J. C. (1986) Biochemistry 25, 64 646.
Obeid, J. & White, p. c. (1992) Biochern. Biophys. Res. Commun. 188, 22: 227.
Poulin, R. & Labrie, f. (1986) Cancer Res. 46, 4933-4937.
Presson, B., Krook, M., & Jomvall, H. (1991) Eur. J . Biochem. 200, 537-74
Puranen, T. J., Pautanen, M. H., Peitoketo, H. E., Vihko, P. T. &Vihko, R. (1994) Biochem J. 304, 289-293.
Scrutton, N., Berry, A. & Perham, R. N. (1990) Nature 343, 38-43.
Simard, J., et al., & Narod, S. (1993) Human Mol. Genet. 2, 1193-1 199.
Spinola, P. G., Marchetti, B. & Labrie F. (1986) Breast Cancer Res. Treat. 241 -248.
Strickler, R. C. & Tobias, B. (1980) Steroids 36, 243-253.
Tamm L. K. (1994). Peptide-Bilayer interactions in Membrane Prori Structure. (White, S . H. , ed), pp.283-313, Oxford University Press, Nt York and Oxford.
Terwilliger, T. C. & Eisenberg, D. (1983) Acta Crystallogr. A 39, 813-817.
Tobias, B. & Stnckler, R. C. (1981) Biochemistry 20, 5546-5549.

Chapter 5 Structure of 17J-HSD1
Urozowski, Z. (1992) Mol. Cell. Endocrinol. 84, C25-C3 1 .
Varughese, K. I., Skinner, M. M., Whiteley, J. M., Matthews, D. A. & Xuor N. H. (1992) Proc. Natl. Acad. Sci. USA 89, 6068-6084.
Wierenga, R. K., De Maeyer, M. C. H. & Hol, W. G. J. (1985) Biochemistr 24, 1346-1357.
Zhu, D.-W., et al., & Lin, S.-X. (1993) J . Mol. Biol. 234, 242-244.

Chapter 5 Structure of I7P-HSDI
Table 5.1. Summary of data collection for native and derivative crystals.
Crystd Soaking No. of Maximum Total Unique FWs(F2) Completeness R(F2) R(l
type conditions Crystais resolution data &ta last 0.10A (%) % nati
(no. of sites) used (A) sheii merge &I
Native: purifid h m
placenta
1. 17RHSD3 2. 17gHSD5
3. Combined
Native:
recombinant 1. 17B-HSD9 1 2.52 28026 10367 2-01 90 8.59 --
Hg4:HgCI2 0.2 mM, 27 1 3.00 13539 5429 2.80 80 7.26 10.6
days; (1 major, 1 minor)
Hg10:HgC12 0.2 mM, 41 1 3.00 6612 4781 3.29 67 8.75 21.1
days; (1 major,
1 minor) Hgll:HgC12 0.2 mM, 42 1 3.00 5935 3968 6.12 56 5.45 15.7
days; (1 major, 1 minor)

Table 5.2. Isomorphous replacement phasing statistics.
Derivative Reflections Highest R-Centric* Phasing Phasing powerc phased resultion power+ in the last shell
* R-Centnc is CulIis for centric reflections and is defined as R=C IIIFpHIiIFpII -IfHll/ZllFpHI+IFpl[, where P and H refer to parent and heavy-atom derivative
data sets respectively and IF1 is the measured structure amplitudes. +Phasin power is defined as fH rms/lack of closure.
- -
Table 5.3. Statistics from phase combination.
Resolution (A) 3 -20
Number of reflections 4072
SIR* figure of merit (FOM) 0.49
Combined FOM (mode1 and SIR) O. 8 O
Correlation coefficient 0.97
inverse 0.23
*SIR: single isomorphous replacement

Chapter 5 Structure of t7J-HSDI
Table 5.4. Refinement statistics.
Protein souce
-
IsoIated from Recombinant human placenta
Protein atoms in mode1 (284 out of 327 residues)
Solvent atoms: water
Resolution range
Unique data used (FxF)
Free R-fac tor
Rms deviation: Bond distance (A) Bond angle(') Dihedral angle (O)
Planarity (O)
Ramachandran plot statistics: % residues out of a total of 234 non- glycine and non-proline residues in most favored regions % residues in disallowed regions Estimated random positional error (A) from Luzzati plot

Chapter 5 Structure of I7J-HSDI
Table 5.5. Distances (in A) between pairs of Ca atoms.
Ca pair C a pair
176-HSD 3a,200-HSD 17B-HSD 3a,20B-HSD
Lys7 I l e l 1 Gly13 Ala23 Va134 Asp 60 Asp82 Asn87 Val 107 Leu1 12
0.6 (A) 0.1 (A) 0.3 (A) 0.8 (A) 0.4 (A) 0.8 (A) 0.8 (A) 0.8 (A) 0.8 (A) 0.7 (A)
Leu126 Arg136 Ser 142 Gly145 A s n l 5 2 Tyr155 Lys159 ~ e u 1 6 9 Leu 182 His189
Ile 1 2 3 Serf 33 Ser139 Gly142 Thr149 Tyr152 Lys156 AIa166 S e r 1 7 9 Tyr186
0.9 (A) 0.4 (A) 0.8 (A) 0.5 (A) 0.8 (A) 0.9 (A) 0.1 (A) 0.9 (A) 0.5 (A) 0.9 (A)
The 20 C pairs used for least-squares superposition were selectc uniformty from structurally conserved residues along the polypeptic chain.

Chapter 5 Structure of I7J-HSDI
Legends to Figures
Fig. 5.1. (a) Stereo ribbon diagram of a monomer of human estrogenic 17 hydroxysteroid dehydrogenase (HSD). The course of the polypeptii chah is shown for residues 1-284. a-helices are drawn as magenta coi; 0-strands as blue arrows, and turns and loops as green ropes. The si1 chains for residues in the active site belonging to the catdytic tria Tyrf55-Lys159-Ser142, are shown in white. The view is almost para11 to the central 0-sheet. [Figure prepared using the program SETOR (Eva 1993).
(b) Folding topology of strands (triangles) and helices (circles) in 17 MD.
Fig. 5.2. A (2Fobs-Fcalc) electron-density map of the helix aG1, in stereo, caculati with the refined mode1 which is shown in standard atom colo (symmetry-related atoms are shown in white). Atoms in the neighbori~ regions have been removed for clariey. The map is contoured at 1.24 [Figure prepared using the program CHIN (Jones, 1978) 1.
Fig. 5.3. A ribbon diagram of 170-HSD structure with the substrate-bindir domain, comprising helices aG", aG', UN and aH, highlighted in gold. Tl
modeled substrate, 17B-estradiol, is shown in stick representation. Figure prepared using the program SETOR (Evans 1993)l.
Fig. 5.4. Stereo ribbon representation of the dimer of human estrogenic 176-HSI which is the biochemically active form of the enzyme. The two subuni are related by a crystallographic two-fold axis of symmetry. Tl interface generated by this rotation axis, parallel to the direction of view

Chapter 5 Structure of I7#-HSDI
at the center of the four-helix bundle (comprising the two aE and two a helices), is identical to the Q-axis interface in bacterid 3 a , 20 8 - H S 1 [Figure prepared using the program SETOR (Evans, 1 993)l.
Fig. 5.5. A stereodiagram of superimposed C a chain of bacterial 3a, 20 B - HS (magenta) and human estrogenic 17B-HSD (cyan). The superposition w achieved by ieast-squares fitting 20 C a atoms that are at similar positio in both structures. The cofactor, NAD, bound to 3a,206-HSD is shown yellow. Figure prepared using the program SETOR (Evans, 1993)l.
Fig. 5.6. .
Close-up stereoview of the active site of human estrogenic 178-HS showing the cofactor and the steroid-binding clefts. The modeled NA1 (H) and 170-estradiol molecules are shown in brown and gree respectively. Side chains of the residues constituting the catalytic tria Tyr155-Lys 159-Ser142, are shown in red. The steroid molecule positioned realative to Tyr155 and the nicotinarnide ring of the cofact( in accordance with the proposed transition state of estrone to estradi interconversion. Also ' shown (in white) are residues that Iine the cofacti and the steroid-binding clefts, namely Cys10, Ser l l , Asn90, Ala9 Va1113, Metl147, Leul49, Cys156, Cys185, Met193, TyrSl8, His22 Ser222, Va1225, Phe226, Leu262, Leu263, Met279, and Va1283. [Figu: prepared using the program SETOR (Evans, 1993)l.
Fig. 5.7. (a) Stereoview of the atomic mode1 of the proposed transition state estrone to estradiol interconversion. Al1 atomic positions shown a obtained from the present study, except those of the cofactor and tt
steroid molecules that were derived from modeling. The 8-face of tl steroid is oriented towards Tyr155 and the pro-S hydride from the C atom of the nicotinamide ring approaches close to Cl7 of 178-estradia [Figure prepared using the prograrn SETOR(Jones, 1 978)].

Chapter 5 Structure of I7J-HSDI
(b) The proposed mechanism of estrone to estradiol interconversic Lys159 and Ser142 side chains could also approach the Tyr155 hydrox in the transition state (see text for explanation). A few solvent wai molecules were located near the active-site cleft, although none is with hydrogen-bonding distance of the Tyr155 side-chain hydroxyl grou One water molecule was found in close interaction with the buri1 Lys159 side chain.
Fig. 5.8. A dotted Connolly surface of the active-site cavity, draw with a pro: radius of 1.35 A. Color coding as follows:carbon, green; oxygen, re nitrogen, blue; sulfur, yellow. Modeled 178-estradiol and part of tl
progra NADP+ molecule are also shown. [Figure SETOR (Evans, 1993)l.
prepared using the
Fig. 5.9. Helices (a) aG' and (b) aH viewed along the helica .1 axis. The side chains a color coded to illustrate their amphiphilicity, as follows: gree hydrophobic; red charged; and magenta, residues having a proto, donating functional group. [Figure prepared using the program SET0 (Evans, 993)l.

Chapter 5 Structure of 17J-HSDI
Fig.5.1

Chap
Fig.
Structure of I7J-HSDI
Fig.

Chap
Fig.
- 5 Structure of 17J-HSDI
14
Fig.

Chapter 5 Structure of 179-HSDI
Fig. 5.7

Chapter 5 Structure of I7J-HSDI
'ig. 5
ig. 5

Chapter 6
The phase diagram for crystallization of 1711-HSD1

Chapter 6 The phase diagram for crystallization of I7J-HSD I
The phase diagram for crystallization of 178-HSII
6.1 Introduction
Phase diagram in providing a design for crystallization of protein very important. In most experiments of the reported prote crystallization, the degree of supersaturation of a crystallization systei the condition for nucleation and crystal growth are not adequate described. The physics of protein crystal growth remains large unknown. It is only recently, that several laboratories reported son phase diagrams of protein crystal growth more systematically (Ataka al., 1986; Waber 1991; Ries et al., 1992; Mitsuo 1992; Ataka 199 Takayuki 1994). Human estradiol dehydrogenase (178-HSD 1, EC. 1. 1. 62) is an enzyme found in great quantity in human placenta. Th enzyme catalyses the interconversion of 178-estradioi and estrone, ar to a lesser extent, dehydroepiandros terone (DHEA) to 5-androstene-3 170-diol(A5-diol) (Dumont et al., 1992). 178-estradiol is the most actil estrogen while the estrogenic role of AS-di01 has been found on recently, both hormones stimulating the proliferation of breast cance The elucidation of the enzymes' structure-function relationship is thus i
critical importance for both the basic endocrine research and tI
hormone dependent cancer therapy. The crystallization of 178-HSD1 wi once reported in 1976 (Chin et al., 1976), but no structure informatio even prelirninary, has ever been reported. In our laboratory, ti crystalIization of 178-HSD1 has been attempted since 1991. It crystallized in different complex forms with its cofactor NADP+ (
estradiol (Zhu et al., 1993, 1996). or an inhibitor (EM-139, a duaI si inhibitor synthesized in our laboratory). This is the first report on tl successful crystallization of any steroid-converting enzyme from human source. The structures of 17B-HSD1 and its complex wii estradiol have now been determined (Ghosh et al., 1995; Azzi et al., 199i

Chapter 6 The phase diagram for crystallizafiun of I7J-HSDI
Here, we report the phase diagrams for the crystallization of hum 176-HSD1 that is very useful for improving the crystal quality and 1
crystallizing complexes of the enzyme with various tigands, includi cofactors, substrates and inhi bit ors. Its establishment required t
precise determination of each cry s tallization parameter and caret sample preparations. The results will lead to further improvement 176-HSD1 crystal quality and hence higher resolution structures.
6.2 Materials and methods
6.2.1 Chemicals
N AD P +, NAD+, glyceroI, O-OG (8-octyl glucoside), MgC12, PE (polyethylene glycol) 3500, Tris-base [Tris= Tris-(hydroxymethy aminomethane], Hepes [N-(2-hydroxyethyl) piperazine-Nt-: ethanesulfonic acid], EDTA (ethylenediaminetetraacetic acid), and PM5 (phenylmethanesulfonyl fluoride) were purchased from Sigma (St. Loui MO, USA); 17B-estradiol, estrone, testosterone, 17a-Met h y1 -es tradia 2 0 a - 0 H-P(20a-dihydroxysteroid progesterone) and DTT (dithiothreitc were obtained from Aldrich (Milwaukee, WI, USA); Inhibitor (EM13l was obtained from Our laboratory, Q-Sepharose Fast Flow and Blu Sepharose CL-6B columns were packed in our laboratory using med
from Pharmacia Biotech. (Montreal, Canada), and the phenyl-Superose H 10110 column was from the same Company. Al1 reagents were of tl best grade available. Centricon-30 and Centri-prep-30 concentrato were bought from Arnicon (Beverly, MA, USA).
6.2.2 Methods
Assay of I7J-HSD. When the enzyme was assayed by spectrophotometer measureme
the reduction of NAD+ was indicated by the absorption increase at 340 and 23 f 1" C. The mixture contained 25 p M estradiol, 0.5 mM NAD+ 50 mM NaHC03 buffer, pH 9.2. At this pH the maximum activity

Chapter 6 The phase diagram for crystallization of 17J-HSDI
estradiol oxidation is obtained. A blank value in the same reactic mixture but lacking estradiol was obtained under the same condition ar subtracted.
- Estradio1 + NAD+ b Fstrone + NADH + )-T+
One unit of enzyme is defined as the amount of enzyme th catalyzes the formation of 1 pmol of product in 1 min under the ab01 conditions.
The chromatographic steps use a Pharmacia FPLC system at roo temperature. 178-HSD1 is present in the soluble subcellular fraction human placenta with a aqueous solubility of 2-3 mg/ml. TI purification and the improvements have been described previously (L et al., 1992; Yang et al., 1992; Zhu et al., 1994). The purified human 17 HSD1 has a high specific activity catalyzing the formation of 8 pm estradiol per min per mg enzyme protein, at pH 9.2.
Protein concentration measurements.
Protein concentration was determined by the optical method Warburg & Christian (1942), using protein concentration (mg/ml) = 1.5 A 280 - 0.76 AMO. The optical density was determined with a Beckma DU-70 spectrophotometer in which a microcells of 50 pl can be used.
Phase diagrams determination
The crystal growth experiments were carried out using the mic, batch method at room temperature. The setting for this method indicated in Figure 6.1. Polyethylene glycol (3500) was used as tl

Chapter 6 The phase diagram for crystallization of I7J-HSDI
precipitant. The crystailization reagents contained 0.15 M MgC12, 2 n sodium azide, 20% glycerol and was buffered to pH 7.5. The enzyn sample was prepared in a buffer containing 20% glycerol, 1 mM EDTA, l rnM DTT, 0.5 mM PMSF, and 40 rnM Tris-HC1, pH 7.5. 0-OG (0.06%) 1
added to the enzyme sample just before crystallization via buffer char in Centricon. The drop was formed of 1 pl of the protein solution and of the crystdlizing agent. The starting concentrations of enzyme and Pl (3500) after mixing Vary correspondingly.
Phase diagrams were determined in identifying the precipitatic nucleation, metastable and undersaturation zones as a function of i
precipitant concentration and protein concentration. The intervals concentration were 0.5 mglm1 for protein and 0.25% (w/v) for PEG 351 respectively.
With different concentrations of protein and PEG the precipitati zone was determined by the appearance of amorphous precipit; corresponding to the minimun of initial protein concentration at a cert; PEG concentration which could induce amorphous precipitate. Dependi on the appearance of microcrystals, the nucleation zone was estimai similarly. In one control, a t a fixed PEG (3500) concentration of I I (w/v), the protein concentration was gradualiy decreased from 9mgiml 2.5 mg/ml. At 13% (w/v) PEG (3500), an amorphous precipitate form instantaneously in 9 mg/ml, 7.5 mg/ml, 6.5 mglml, 6 mglm1 and ! mg/ml. In about 6-24 hours nuclei were formed and the crystals began grow from the precipitate by slow dissolution. In 5 mglml, 4.5 mg/ml, mg/rnl, 3.5 mgfml and 3 mglml, nuclei were formed and the cryst; began to grow after 48 to 60 hours, but there was no appearance amorphous precipitate. So, 5.5 mglm1 protein and 13% PEG represented point on the precipitate curve on the right of which only precipit; happened, while 3 mg/ml protein and 13% PEG represented a point on i
nucleation curve, between that and the precipitate curve nucleation col take place. In determining the solubility curve, we could use a seedin

Chapter 6 The phase diagram for ctystallization of 17)-HSDI
method in which a single crystals were moved to the droplets that i
conditions were selected on the left-hand side of nucleation curve. Ni crystallization conditions are searched with varying concentrations protein in the presence of the sarne concentration of PEG (3500), or wi different concentrations of PEG (3500) while keeping 176-HSI concentration constant. Depending on the crystal growth or dissolutic the solubility curve was identified. In one control, under strictly fix protein concentration (12 mg/ml) with PEG (3500) concentrations 11.5% (w/v) (a), 10.5% (wlv) (b), 6.5% (wfv) (6). The crystals we formed in condition (a). After 72 hours, no crystals had appeared in (
and (c). A seeding method was used in which single crystals we moved from (a) to (b) and (c). After 5 hours, the crystals were dissolv in (c). After 48 hours we also found that the crystals had grown in ( I Then a single crystal was moved from (a) to another new crystallizatil condition (6.75% PEG, 12 mgfml protein). After 48 hours we found tE the crystal was not dissolved. So the point (6.5% PEG, 12 mg/ml protei was determined to be on the solubility curve. Between nucleation ai crystal dissolution curve is the metastable zone. On the left of the crysi dissolution curve is the undersaturation zone.
6.3 Results and Discussion:
The hanging-drop vapor-diffusion method, widely employed (Mich 1982; Allen et aI., 1984; Chang et al., 1985; Ducmix et al., 1987; Frank al., 1987). is not suitable for determining the phase diagrams (Odahara al., 1994), as the drop volume changes with time and al1 the componr concentrations Vary.
To avoid this problem the micro-batch method was used in tl study (Fig. 6.1). AIthough the drop volume is small, there is no detectal volume change within 6 days with good sealing. In the crystallization 178-HSD1, the crystals usually appeared within 6-48 hours, and thus t volume remained cons tant throughou t the experiment. Using micro-batc

Chapter 6 The phase diagram for crystallization of I7J-HSDI
method, a very smdl sample of protein was used. The concentrations 1
protein and precipitants were very easy to evaluate in our experiment.
Phase diagrams were thus deterrnined, as a function of the PI (3500) and protein concentrations of 178-HSDI (Fig. 6.2). For protc concentration values of 15 mglml, 12 mg/ml, 9 mglml, 7 mglml, mglml, 3.5 mg/ml and 2 mg/ml, precipitation started to appear when t relative value of PEG(3500) reached 11.5, 11.75, 12, 13, 13.5, 14 and 1: (w/v) respectively, thus identifying the precipitation curve. Precipitati occurs at high supersaturation (about 2.4 - 18 times the solubility val for 178-HSDl). Above this curve excess protein immediately precipitai from the solution in an amorphous state. Usually, crystallization does r take place in the precipitate region ( ~ u c r u i x and Giegé, 1992). Up addition of PEG (4000), further protein precipitation took place. In t
case of 178-HSD1 nuclei were formed and the crystals began to grc gradually from the precipitate by slow dissolution of the later, in 6- hours (Fig. 6.3). So, a high supersaturation is the driving force f crystallizing 178-HSD1. Because the nuclei of 178-HSDI can be form from the precipitates, the nucleation zone extends into the precipitatil zone. Although nuclei could be formed in the absence of the precipitate drop in 48-60 hours, these crystals were smaller, their diffraction quali were also lower than the crystals from the precipitation zone. The fi indicates that high quality crystals of 17B-HSD1 can be obtained frc the precipitation zone is an important feature of 178-HSD1 crystallizatic For HEW lysozyme the precipitation occurs at very high supersaturati! about 30-100 tirnes solubility value, and ' the nucleation range li between about 5-30 times the solubility value (Kantt et al., 1992). T result showed that the nucleation takes place at a lower supersaturatit than precipitation in the Lysozyme. In crys tallizing 178-HSD 1, t: precipitation occurs at only 2.4-18 times solubility value, this may '
related to the nucleation of 17B-HSDl from the precipitation zone. If t: condition of crystallization was near the edge of precipitation zone, t:
crystallization was accompanied by the progressive dissolution of tl precipitate around the growing crystals, indicating that growth occurs on

Chapter 6 The phase diagram for crystallization of I7J-HSDI
the expense of the precipitate, and the precipitate was greatly reduced the end (Fig. 6.4).
When the hanging-drop vapor-diffusion method for crystallization (
17B-HSD1 is used, similar phenornena also occurred. We obtained thri different forms of crystals of human 17B-HSD 1 with various ligands: 170-HSD1-NADP+; 6. 176-HSDl-estradiol; c. 176-HSD1-EM139 (inhibitoi Depending on diffraction analysis, we found that several crystals grov near the edge of precipitation zone diffract to high resolution. When ti
protein concentration is 5.8-7.5 mg/ml (after mixing) in the presence 13-14% (w/v) PEG (after mixing), the crystals of 170-HSD1 diffra resolutions from 2.2 A to 2.4 A with rotating anold X-ray source (Fig. 6.5 Conditions and diffraction data are listed in tabIe 6.1. The crystals beg; to grow gradually from the precipitate is an important feature in tl crystallization of 170-HSDI. These phenornena are in appare contradiction with the general belief that high supersaturations lead a great number of crystals or even' to amorphous precipitation. Recentl photographic monitoring and the time scales of these precipitatio crystallization, dissolution process as welI as kinetics of the cryst growth were reported (Ng et al., 1995).
In a metastable zone with a low supersaturation of solutic nucleation may not take place for a long period of time (two weeks). Th corresponds to the ideal conditions for the growth of seed crysta without new nucleation (Fig. 6.6). When a srna11 single crystal was movc from precipitation zone to metastable zone, crystals continued to gro and high quality crystals were obtained. This method is very useful :
obtaining crystals of human 1713-HSD1 complex with various ligand Using vapor-diffusion method and PEG/Mg/O-OG system, some crystals 4
apoenzyme were obtained in the presence of 13% PEG 3500 wii 7.5rngfml protein (after mixing). After two weeks, the single cryst, grew to a typical size of 0.06 nm x 0.1 nm x 0.18 nm and it was moved I
metastable zone condition (11.5% PEG, 7.5 mg/ml protein). The EM-11 solution was also added to drop. After four weeks, the single crysta

Chapter 6 The phase diagram for crystallization of 17J-Hm1
grew to a typical size of 0.1 nm x 0.16 nm x 0.32 nm. The crysi diffracted beyond 1.8 A with X-ray from a synchrotron radiation sourc More recentiy, combining with soaking method, we obtained anoth seven more crystaf forms of human 170-HSDl complex with vario1 ligands or inhibitors (estrone, testosterone, NAD+, 20a-OH-P, 17a-Methy E2 , 17a-Methyl-E2-NADP+ and 17a-estradiol-NADP+).
Using seeding method, the solubility curve was determined eithc by crystal growth or by dissolution of crystals. When the protei concentration decreased from 15 mg/ml to 0.84 mglml, the seedin crystals were dissolved with an increasing PEG (3500) concentratio from 6% to 15% (w/v) (Fig. 6.6). Between nucleation and cryst; dissolution curve is the metastable zone. On the left of the cryst: dissolution curve is the undersaturation zone.
With the phase diagram we can further modify the crystallizati conditions. The above studies will lead to further improvement of 17 HSDl crystals and to its structure determination at higher resolution. T crystallization and structure determination of enzyme-ligand crystals important for a better understanding of 178-HSD 1 -ligand interactions a will eventually help in the design of efficient inhibitors. These resu are also useful to establish a new method for crystallization of ott isozyrnes of human 170-HSDs.
6 . 4 References
Allen, J. P. & Feher, G. (1984). Proc. Nat1 Acad. Sci. USA, 81, 4795-4799.
Azzi A., Peter H. R., D.-W. Zhu, Robert L. C. Labrie, F., and S.-X. Lin (1991 Nature Structure Biology, 665-668.
Ataka, M. & Tanaka S. (1986) Biopolymers, 25, 337-350.
Ataka, M. (1993) Phase Transit., 45, 205-219. Chang, C. H., Schiffer, M., Tiede, D., Smith, V. & Nomsn, J. (1985) J. Mol.

Chapter 6 The phase diagram for crystallization of 173-HSDI
Biol. 186, 201-203.
Chin, C.-C., Dence, J. B.& Warren, J. C. (1976) J. Biol. Chem. 251, 3700-370
Dumont, M., Luu-The V, de Launoit, Y, Labrie, F. (1992) J. Steroid Bioch Mol. Biol . 41: 605-8.
Ducruix, A. & Reiss-Husson, F. (1987) J. Mol. Biol. 193, 419-421.
Frank, H. A., Taremi, S. S. & Knox, J. R. (1987) J. Mol. Bio1.198, 139-141.
Cryst. D50, 639-642.
Ghosh, D., Pletnev, V. Z., Zhu, D.-W., Wawrzak, Z., Daux, W. L., Pangbon W., Labrie, F., and Lin, S.-X. (1995). Structure, 503-5 13.
Lin, S.-X., Yan, F., Jin, J.-Z., Breton, G., Zhu, D.-W., Luu-The, V., and Labri F. (1992) J. Biol. Chem., 267, 16182-16187).
Michel, H. (1982). J. Mol. Biol. 158, 567-572.
Mitsuo, A., (1992) J. Gry. Growth 122, 60-65.
Ng, J. D., Lorber, B., ThéobaId-Dietrich A., and Giegé R.(1995) Collecte Abstracts ICCBM-6 in Japan, pl3 1
Ries, Kautt, M. and Ducruix, A. (1992) Crystallization of Nucleic Acids an proteins, edited by A. Ducruix & R. Giegé, Oxford: IRL Press, pp. 195-218
Takayuki O,, Mitsuo A. and Tatsuo K. (1994) Acta Cryst. D, 50, 639-642.
Waber, P. C. (1991). Adr. Protein Chem, 41, 1-36.
Zhu, D.-W., Lee, X., Breton, R., Ghosh, D., Dangborn, W., Duax, W. L & Lir S.-X. (1993) J. Mol. Biol., 234, 242-244.
Zhu, D.-W., Lee, X., Labrie, F., and Lin, S.-X. (1994). Acta. Cryst. D, 50, 5% 555.
Zhu, Da-W., Azzi, A., Rehse, P., and Lin, S.-X. (1996). J. Crystal Growth 168: 275-279.

Chapter 6 The phase diagram for crystallization of I7/-HSDI
Legend to figures
Fig. 6.1 The setting for micro-batch method The drop was made of 1 pl of the protein solution and 1 pl of t crystallization agent. There is no detectable volume change within 6 da when the cover slip is well sealed.
Fig. 6.2 178-HSD1 phase diagram as functions of protein PEG concentrations In 6-24 hours, nuclei were formed and the crystals began to grow frc the precipitation zone (m. In 48-60 hours, the crystals can be formed nucleation zone (no amorphous p rec ip i t a t e )m. So, the nucleation zo extends through the precipitation zone. In a metastable zone a, supersaturated solution may not nucleate for two weeks. In this zo corresponds ideally to the growth of seeding crystals. In undersaturatil zone (=, the condition would never crystallize and lead to dissolution the crystals.
Fig. 6.3 Crystallization of 17B-HSD1 in one control In different concentrations of protein and precipitant, the nucleation zt can be identified by the appearance of crystals and microcrystals. Uni the same conditions, the precipitation zone can also be identified by appearance of amorphous precipitate. In one control, at a fixed Pl (3500) concentration of 13% (w/v), the protein concentration v decreased from 9 rng/ml (a) to 2.5 rng/ml (h). Interestingly, upon addition of 13% (w/v) PEG (3500), an amorphous precipitate forrr instantaneously in a-e. In about 6-24 hours nuclei were forrned and crystals began to grow from the precipitate with slow dissolution of ,
Iater. In f and g, nuclei were also formed and the crystals began to gr after 48 to 60 hours, but there was no appearance of amorphoui precipitate. After 7 days, no crystals had appeared in drop h.

Chapter 6 The phase diagram for crystallization of I7P-HSDI
Fig. 6.4 Crystallization was accompanied by the progress dissolution of the precipitate around the growing crystaIs Photographs showing the crystallization was accompanied by t progressive dissolution of the precipitate around the growing crystal indicating that growth occurs on the expense of the precipitate, and tl precipitate disappeared completely in the final droplet. Crystals grown the presence of 13% (wjv) PEG (3500) and the concentration of prote was 6 mgfml,
Fig. 6.5 Crystals grown near the edge of precipitation z( diffract to high resolution Different cornplex forms of crystals of human 170-HSD 1 were obtained. 1) Apoenzyme (a, b, c, d). 2) 178-HSDI-NADP+ (e, f, g, h, i). 3) 17f HSD1 -estradio1 (i, j, k). 4) 170-HSD1 -inhibitor (b, 1). Depending on their conditions of crystallization, the high-resolution lim: for diffraction varied. When the protein concentration is 5.8 to 7. mg/mI with PEG (3500) 13 to 14% (wlv), the crystals of 17B-HSD diffracted to high resolutions (2.2 A - 2.4 A). The conditions an diffraction data are listed in Table 1.
Fig. 6.6 The solubility curve determination In two controls, under stnctly fixed protein concentration (12 mg/ml an 9 mglrnl) with PEG (3500) concentrations of 11.5% (w/v) (a), 10.54 (w/V) (b), 6.5% (w/v) (c), 12% (wlv) (d), 10.75% (wlv) (e) and 7.54 (w/v) (f). The crystals were formed in (a), (d). After 72 hours, n8 crystals had appeared in (b), (c), (e) and ( f ) . A seeding method was usel in which single crystals were moved from (a) to (b) and (c), and from (d to (e) and ( f ) . After 5 hours, the crystals were dissolved in (c) and (f: After 48 hours we also found that the crystals had grown in (b) and (e: So the points (6.5% PEG, 12 mglm1 protein and 7.5% PEG, 9 mg/m protein) were determined to be on the solubility curve. Betweei nucleation and crystal dissolution curve is the metastable zone. On th1 left of the crystal dissolution curve is the undersaturation zone.

Chapter 6. The phase diagram for crystallizatiun of I7J-HSDI
Fig. 6.1
The setting for micro-batch niethod
covers l ip
vacuum grease
M i c r o - B r i d g e
Fig. 6.2
8-Hydroxysteroid Dehydrogenase Phase Di agram 15
Preclpitntion zone
1 O a Nudeation zone
Predpitation and Nudealion
a MetastaMe zone
undersaturalion
pceapitalion curve
PEG concentration % @IV)
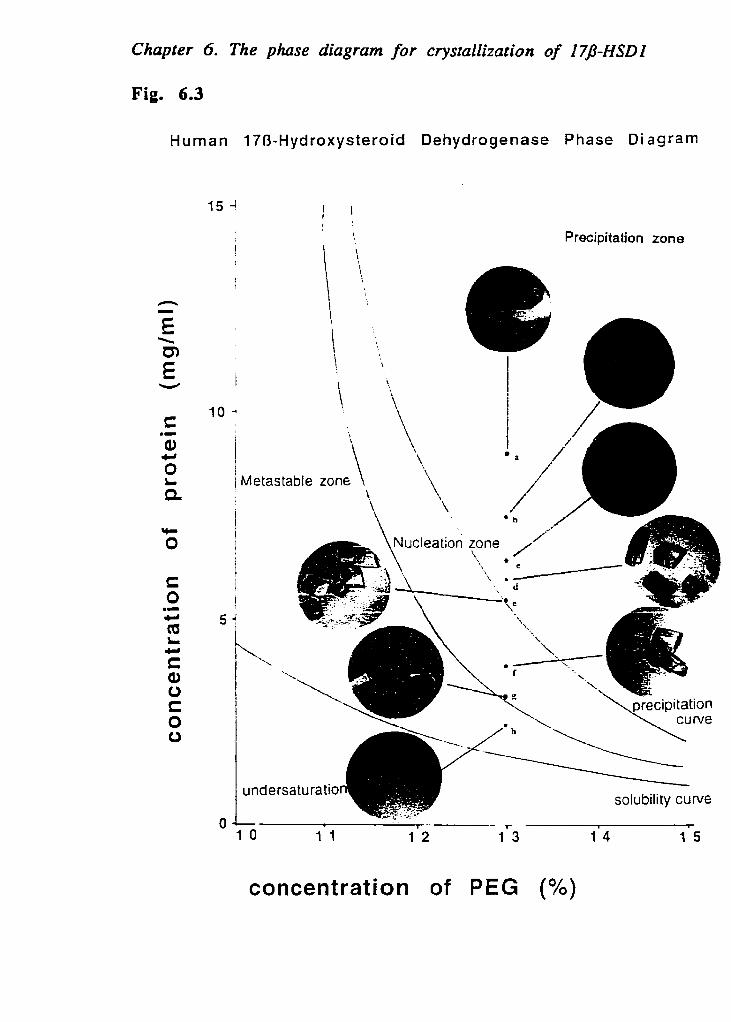
Chapter 6. The phase diagram for crystallizarion of I7J-HSDI
Fig. 6.3
Human 170-Hydroxysteroid Dehydrogenase Phase Diagram
Precipitation zone
\ \
bietastable zone '\
1 O 1 1 1.2 1.3 1 4 1 5
concentration of PEG (%)

Chapter 6. The phase diagram for crystallization of I7J-HSDI
5 days

Chapter 6. The phare diagram for crystallization of I7P-HSDI
Fig. 6.5 Human 170-HSD Phase Diagrme
PEG concentration (%)
Fig. 6.6
Human I7B-Hydroxysteroid Dehydrogenase Phase Diâgram
4 6 8 1 O 1 2 1 4
P E G concentration % (wlv)

Chapter 6. The phase diagram for crystallizarion of 174-HSDI
Table 6.1 Conditions and diffraction data for obtained cryst,
17B-HSD I 17B-HSD-NADP 17B-HSD- est
a Protein: 7.2 mglml
- PEG: 15% zs A 1 ~Proteih- 7 5 mglm1 2.4A - PEG: U X
Protein: 6 5 mglml 2+2 A
I - PEG: 14%
d Proteicc 5 mglml
I PEC: l u %
2.8.4 1 - e Protein: 15 mglml
PEG: 15% 1 3.zA -
f Protein: 10 mglm1 1 2.9A - PEG: 14%
g Protein: 8 5 mglml 1 UA - PEG: USY.
h Protein: 7 5 mglml 1 2.2 A - PEG: 135%
i Proteix 779 mglml 1 2.4 A 2.6A - PEC: 14%
Protein: 6.1 mglm1 I 2.3 A j PEG: 14% -
Protein: 4.7 mglml 22 A PEG: 1425%
- Protein: 5.8 mglml ' PEG: 13%
Some data are not complete data.

Chapter 7
Conclusion

Chapter 7 Conclusion
Conclusion
The main conclusion of this thesis is summarized in this chapte Here, we have presented in detail the rapid preparation of 170-HSD1, tl methodology for crystallization of the enzyme and its various cornplex1 with ligands or inhibitors, the mechanism of its crystal growth, as well the structural features of this pivotal steroidogenic enzyme. Throuj these studies, it was shown that the preparation of a homogenous ar highly active protein is the first important step for its crystallization. was also shown that purification, stabilization, storage, handling 4
protein and increasing its solubility are essential steps prior '
crystallization. We have also established the method of enzyme-Iigar complex preparation. Using the improved crystallization methods, tc different complex forrns (Table 7.1) were crystallized.
7.1 The high quality of enzyme is the first important step f l
c r y s t a l l i z a t i o n
The need for high quality of an enzyme preparation includes a hi$ specific activity and high purity (> 97%). In chapter 2, two methods i
rapid purification using the technique of FPLC were detailed. Rencentl: we have combined those methods, using three FPLC chromatographies: (
Sepharose ion exchange, Blue-Sepharose CL-6B affinity (with NADF elution) and phenyl-Separose High performance chromatographies. Tt preparation obtained was 25% to 40% higher specific activity tha that reported in the Chapter 2.
These FPLC protocols will be useful for purification of othe members of the 176-HSD family.
The enzyme quality depends Iargely on its specific activitj Repeated experiments showed that the best crystals were usuall obtained from samples with the highest specific activity (8.5- 10 Ufmg:

Chapter 7 Conclusion
Repeated experiments have shown that with low specific activity (5 6.8 Ulmg), we usually obtained some rough multi-crystals anc sometimes we could not obtain crystals at al1 under the same conditions This may have been due to insufficient homogeneity. A high quality 01
the enzyme preparation is a sound basis for crystallization. For thi: protein, once crystals are produced, additional organic solvent wil improve their quality and size such that they will be suitable for X-raj analysis.
The degree of freshness of the enzyme also has a direct impact or the crystal quality. Usually, the more fresh the enzyme is. the highei quality will the crystals be. If the enzyme was kept for long time, multi- seeding and rnulti-crystals were produced. This may be due to somr proteoly sis. When 0.5 mM PMSF was present, repeated experimeni showed that the activity of enzyme was maintained for long time. Ir: order to decrease protein denaturation in solution, 10%-20% glycerol was introduced which was very useful for stabilizing the enzyme foi crystallization. Glycerol allows the total concentration of the protein tc be kept at a higher level and it also models the cellular environment. In the crystallization of Fructose Bisphosphatase, glycerol is also found to be useful. Without glycerol, the enzyme was easily hydrolyzed in the crystallization drop, and the nuclei were not formed in our experiment.
7.2 The new method for enzyme-ligand complex preparation very useful for structure studies of various members of 1 HSD family and other steroid enzymes
In this thesis, we have presented in detail the preparation oi enzyme-ligand complexes. In chapter 3, the experiments have showr that the apoenzyme had a higher A280/A260 ratio than the holoenzyme carrying one cofactor/dimer. Depending on this result, we can analyse the binding of 170-HSDl with NADP+. In chapter 4, before crystallizatior is carried out, we have verified the formation of 17B-HSD 1 -estradiol complex by studying the binding of radioactively labelled of estradiol. It

Chapter 7 Conclusion
is well known that steroids have very low aqueous solubility (often <5i FM). The concentration of 170-HSD1 used in crystallization is usuall higher than 15 mg/ml, corresponding to more than 220 FM in enzym concentration and 440 pM in estradiol binding sites. We first bound th steroid to the enzyme at low concentration and then concentrated th mixture using a Centricon filter system. When Es is bound to the enzyme new steroid molecules could be dissolved leading to further binding witl the free enzyme molecules. We have succeeded in making the 1:: enzyme-E2 complex at high concentration for crystallization, as verifiec by determining the amount of Cl4-labelled estradiol bound to th( enzyme. Using this method, we have also succeeded in obtaining th( 17B-HSD1 with inhibitor (EM-139). The establishment of such a neu technique provides a strong basis for the crystallogensis of other steroic dehydrogenase complexes and for further research on other types O
170-HSD.
7.3 A n improvement on the screening method for crystallization of new proteins
Since 1991, we have succeeded in crystallizing 17B-HSD1 (fron human placenta), Azurin (from Pseudomonas fluorescens) and Fru-1,6 Pase (from Snake muscle). In the recent crystallization of 178-HSD: which shows membrane association, we have accumulated mucl understanding in the crystallogenesis of hydrophobic and membrane associated proteins, especially in detergent use. Through this research an improvement on the screening method was established f o crystallizing new proteins. The first step is the preparation of thc enzyme and the understanding of its properties, including- the solubility, pH dependence ligand interactions, storage conditions and so on. For the second step, wc used Crystal ScreenTM and Crystal Screen IP'M (from Hampton Research, for preliminary screening. These products include novel screeniq protocols and optimization of a strategy which have providec investigators with an expanded portfolio of effective crystallization tools,

Chapter 7 Conclusion
Through preliminary screening, we have obtained some new informati (e.g. the degree of precipitant, appearance of some needle or mu1 crystals and so on). Depending on these results, we have made furtk adjustments of crystallization conditions for obtaining better crysta including concentration of precipitant, and salt, pH, temperature, a additives (e.g. organic solvent). Sometimes, the crystallization meth also had to be changed.. After we got initial batches of single crystals appropriate f o m s and sizes, we fixed the crystallization condition a adjusted the drop volume to further improve the quality and size of t crystals in order to produce crystals which are suitable for X-r anaiysis. The screening method is very useful in crystallization of 17 HSD family enzymes.
To study the crystallogensis of 170-HSD1, the major characterisi was showed that a high supersaturation is the driving force f crystallization of 17B-HSD1, and the nuclei of 17B-HSD1 can be forml from the precipitates, the nucleation zone extends through to t: precipitation zone. Through these studies, we have deepened O
understanding of the mechanism of crystallization.
7.4 Furt h e r u n d e r s t a n d i n g for s t r u c t u r e - f u n c t i ~ relationship of 17B-HSD1
Using advanced crystallization methods, ten different forms (
crystais of human 170-HSD1 cornpiex with various ligands or inhibitoi have been obtained. Depending on these results, the structures of 171 HSDl and its complex with estradiol were determined. This is the firi marnmalian steroidogenic enzyme studied by X-ray crystallographi techniques, and it reveals a fold characteristic of the short-chai dehydrogenases. It is shown that the overall structure of the enzyme i similar to the other enzymes in the short-chah dehydrogenase famil! with a conserved Tyr-X-X-X-Lys sequence and a serine residue in th active site. It is distinguished from the other known structures reporte for short-chah dehydrogenases by the insertion of two helix-turn- helix

Chapter 7 Conclusion
motifs that appear to govern quaternary associatin and substra binding. Recently, the complex of 17f3-HSD1 with estradiol has bec successfully crystallized and its structure determined. These results lei to the understanding of 178-HSD1-ligand interaction and structur function relationships. More recentiy, in Our group, the structures 1
176-HSD 1 -inhibitor (EM-139), I7B-HSD 1 -17a-Methyl-estradiol-NADP and 176-HSD 1 -20a-OH-P are being determined. These results wi
provide a strong basis for the design of potent inhibitors of this pivot steroid dehydrogenase and will have a major impact on bol endocnnology research and hormone-dependent cancer therapy.

Chapter 7 Conclusion Table 7.1 Crystallization and Preliminary X-ray Diffraction Anal; of 1711-hydroxysteroid dehydrogenase type 1
Form Space Unit ce11 Resolution Completeness Mc U o u P parameters (A) (%)
176-HSD1 (apoenzyme) Q a=124.30 A, b=44.64 A 2.1 9 0
c=6 1.1 19 A, 13=98.27O
170-HSD I - C2 a=122.8 A, b=43.92 A 2.3 * 7 2 estrone c=60.58 A, i3=100.29°
170-HSDI- C2 a=123.639 A, b=44.985 A 2.5 9 1.6 testosterone c=6 1.3 15 A, 8=99. 182'
* Native crystals diffracted X-ray from a synchrotron.

Appendices
A p p e n d i c e s

Appendices
A p p e n d i c e s
Appendices are consist of three papers: 1) "Crystallization and t preliminary crystallographic studies of the Azurin Pseudomon fluorescens". 2) "Crystal structure of human estrogenic 17 hydroxysteroid dehydrogenase complexed with 17B-estradiol at 2.3 resolution", and 3) "Crystallization and preliminary X-ray analysis of t
snake muscle Fructose 1-6-bisphosphatase". In 1991, 1 had a training I
the crystallization of protein at the Institute for Biological Science National Research Councif Canada. During this period, 1 crystallized a ne protein: Pseudornonas fluorescens holoazurin. This training helped me further experiments on the cryscallization of 178-HSD 1 and Fructose 1, bisphoshatase in the folfowing several years.

Appendices
1. Crystallization and the preliminary crysta~lographic s of the Azurin Pseudomonas fluorescens
The Azurin Pseudornones fluorescens has been crystallized in 1
presence of ammonium sulfate and Tris buffer at pH 7.5. The cryst diffract to 2.05 A using a FAST system. The space group is P2,2,2, w a = 31.95, b = 43.78, and c = 78.81 A.
* 1 is adapted from the paper of "Crystallization and the prelimina crystallographic studies of the Azurin Pseudomonas fluorescens" by Da Wei Zhu, Tanya Dahms, Kevin Willis, Arthur G. Szabo, and Xavier Lee Archives of Biochemistry and Biophsics (1994), Vol. 308, 469-470.

Appendices
1.1 Introduction
The azurins comprise a homologous class (Ambler et al., 1967; Baicd 1988) of blue copper-containing, low molecular weight (M, 14,00 proteins which function as redox partners in bacterial electron transf (Brunori e t al., 1974). The structural properties of these blue copp proteins have been the subject of a large number of studies (Canters al., 1984; Rosen et al., 1981). This interest is stimulated due to both thc electron transferase propertias and their unique spectroscopie propertie Azurin has been classified as a type 1 copper protein, owing to i characteristic blue absorption band in the 600-to 700-nm spectral regia The refined crystal structures of two different azurins have bec reported, one from Pseudomonas aeruginosa (Nar et al., 1991) and i
other from Alcaligenes denitrijYcans (Baker 1988). It is thought that tl characteristic blue absorption arises from the distorted tetrahedr configuration of the Cu ligands, composed of two histidines, one cystein and one methionine. Recently it has been suggested that the ligai structure is in fact trigonal bipyramidal with the two histidine residul and the cysteine residue a t the corners of the plane through the Cu ato and the methionine and an amide carbonyl are axial ligands on eith side of this plane. The timely review by Adman (1978) highligh structural features of the different azurins, a s well as reveals tl continuing intense interest in the structural determination of othl azurins.
The fluorescence spectra of the single tryptophan residue in both tl ho10 and apo f o m s of azurin from P. a e r u g i n o s a , and P s e ~ d o n i o n ~ fluorescens, have a spectral maximum at 308 nm, which is very unusu for tryptophan in a protein ( Grinvald A. et al., 1975). Time-resolvt fluorescence studies from our Iaboratory indicated that there is conformational heterogeneity reported by the tryptophan in the ho: form of these two azurins (Szabo et al., 1983; Szabo et al., 1989)

Appendices
The relative proportions of the fluorescence components were ver: different in the two azurins. In an effort to understand the structura differences between the two azurins, crystallographic studies of th1 azutin from the P. fluorescens are now being conducted. In this pape we report the crystallization and the preliminary crystallographic stud: of the protein.
The protein was prepared according to the previous report (Szabo e al., 1989). The copper to protein stoichiometry was determined b: atomic absorption and amino acid analysis to be 1:l. The spectral ratic (A620/A280 or A625/A280) was 0.55, indicating there is nc contamination of apo azurin. There was only one single band at 14.4 K ii SDS-PAGE and one band at pH 6.55 in the isoelectric focusini measurement. Al1 these assays indicated a pure and homogeneous azurii protein.
1.2 Crystal l izat ion
The concentration of protein used for crystallization was 6.2 mglm1 The method for crystallization was the conventional vapor diffusion O
hanging drop with a reservoir solution of 3 M ammonium sulfate buffered with 0.2 M Tris buffer at pH 7.5. The drops were made up of t 11 of protein solution and 6 pl of the reservoir solution. Crystalr appeared overnight. They became chunky bipyramids of 0.4 x 0.4 x O. ( mm3 after 1 week (Fig.l.1). The whole drop became colorless with only : few dark blue crystals. Ce11 parameters and the space group were determined first with precession camera and confirmed with the FASl area detector at the Institute of Structural Biology in Grenoble, France The space group is P 2 , 2 , 2 , with one molecule in one asymmetric uni and a packing density parameter Vrn=1.97 A 3 / ~ a (Matthews 1968). The ce11 parameters are: a=31.95 A, b=43.78 A, c=78.81. The crystal diffract: to 2.05 A. A complete set of data was collected with the FAST system anc with only one crystal. The Rsym was 5.0%.

Appendices
1.3 Molecular replacement
Azurin from P. fluorescens consists of 128 amino acid residuc There is 80% identity in arnino acid composition with either P. aerugino or A. dentrificatns ( Szabo et al., 1989). The primary sequence of t
molecule is presently being determined. The sequence of the first : residues is known. Despite this delay, molecular replacement has be started with the atomic coordinates of the A. denrrificans from t Protein Data Bank as a search mode1 and the experimental diffractil data obtained with the FAST system. The program MERLOT within t CCP4 package of the Daresbury Laboratory (Warrington, UK) has bel used to determine the location and orientation of the molecule in the ui cell. Rotation search was performed with various resolution ranges data and different lengths of vectors. A peak appeared constantly at t
a=89.00 8=78.50, y=191.50. It was about 6 sigmahigh and more than sigma higher than the second highest peak. This peak was refined to a=88.71, 8=78.52 and y=195.81 with the data between 3 and 8 A. TI translation search was carried out with this orientation of the molecu and with the same range of data. A unique solution was found at tl fractions of x=27.8/40, y=32.1/56, and z=60.4/100. This peak was 5.; sigma higher than the background and 1.6 sigma higher than the secoi highest peak. The correlation factor is 0.368. Graphics display was usc to check that the symmetrical relative molecules could be accommodati reasonably weIl without any overlap between molecules. The R factor (1
FOI-IFcll/CIFol) for this molecular replacement solution is 0.484 in tl resolution range of 3.0-8.0 A. An electron density map was calculatl and it was interpretable. The fitting has been carried out for the first : amino acid residues with a Silicon Graphics 4D-35 machine using tl program CHAIN kindly supplied by Professor Quiochio of the Bay11 College of Medicine, Houston. To Our knowledge al1 the azurin structurl were solved with isomorphous replacement except for the M121Q muta of A. denitrificans (Romero et al., 1993). The crystal form of azurin fluorescens reported here is the simplest with only one molecule in tl

Appendices
asymmetric unit. This rnay indicate why the structure can be determir just with molecular replacement alone.
Acknowledgments
We thank Dr. Claudine Cohen-Addad for allowing us to use the FA system at the Institute of Structural BioIogy in Grenoble, France, Thar also go to Dr. Manechen Shoham of Case Western Reserve Universi Cleveland, for his important suggestions during the preparation of tl paper.
1.4 References
Adman, E. T., Stankarnp, R. E., Sieker, L. C., and Jensen, L. H. (1978) J . M Biol. 123, 35-47.
Adman, E. T., (1991) in Advances in Protein Chemistry, Vol. L
Metalloproteins: Structural Aspects (C. B. Anfinsen, J. T. Edsall, F. Richards, and D. S. Eisenberg, Eds.), pp. 145-197, Academic Press, Nc York.
AmbIer, R. P., and Brown, L. W. (1967) Biochern. J. 104, 784-825.
Baker, G. N. (1988) J-Mol . Biol. 203, 1071-1095.
Brunori, M., Grenwood, C., and Wilson, M. T. (1974) Biochem. J . 145, 44 457.
Canters, G. W., HiII, H. A. O., Kitchen, N. A., and Adman, E. T. (1984) Eur. Biochern. 138, 141-152.
Corin, A. F., Bersohn, R., and Cole, P. G. (1983) Biochemistry 22, 203 2038.

Appendices
Grinvaid, A., Schlessinger, J., Pecht, I., and Steinberg. (1975) Biochemistr 14, 192 1 - 1929,
Matthews, B. W. (1968) J . Mol. Biol, 33, 491-497.
Nar, H., Messerschmidt, A., Huber, R., Van de Camp, M., and Canters, (1991) J. Mol. Biol. 221, 765-772.
Norris, G. G., Anderson, B. F., Baker, E. N., and Rumball, S. V. (1979) J. Mo Biol. 135, 309-312.
Noms, G. G., Anderson, B. F., and Baker, E. N. (1986) J. Am. Chem. Soc. 108 2784-2795.
Righetti, M., and Drysdale, J. W. (1971) Biûchern. Biophys. Acta 236, 17 28.
Rornero, A., Hoitink, C. W. G., Nar, H., Huber, R., Messerschrnidt, A., an Canters, G. W. (1993) J. Mol. Biol. 229, 1007-1021.
Rosen, P., SEgal, M., and Pecht, 1. (1981) Eur, J. Biochem. 120, 339-344.
Ryden, L., and Lundgren, J. 0. (1979) Nature 261, 344-346.
Szabo, A. G., Stepanik, T. M., Wagner, D., and Young, N. M. (1983) Biophys J . 41, 233-244.
Szabo, A. G., and Hutnik, C. M. (1989) Biochemistry 28, 3923-3934.
Wilson, M. T., Greenwood, C., Brunori, M., and Antonini, E. (1975) Biochem J. 145, 449-457.

Appendices
Legend t o figures
Fig.l.1 the azurin (from the P. f l u o r e s c e n s ) crystal
The crystds grown in the presence of 3 M ammonium sulfate, buffere with 0.2 M Tris buffer at pH 7.5. The final protein concentration was 6. m g f d . The crystal has a typical size of 0.4 x 0.4 x 0.6 mm and diffract t
2.05A.

Appe ndix
Fig. 1.1

Appendices
2. Crystal structure of human estrogenic 1; Hydroxysteroid Dehydrogenase complexed with 1 7 estradiol at 2.3 A resolution.
The complex of 178-HSD1 -estradio1 has been successfull crystallized by using a special procedure to saturate a high concentratic of the enzyme in solution with the substrate (Zhu et al., 1996). Tl crystal structure of the 170-HSD1-estradiol complex has now bee deterrnined by means of a difference Fourier map using the 178-HSC structure as the starting point and has been refined to give an R-factc of 0.194 with data to 2.3 A.
The structure data for 178-HSDl, especially that for the compfe between the enzyme and estradiol, provide a strong basis for the desig of potential inhibitors for this pivotal enzyme in both endocrinology an hormone sensitive cancer therapy.
In this paper, my contribution was the crystallization of 170-HSDI estradiol complex. 1 provided al1 crystals for X-ray analysis.
* 2 is adapted from the paper of " Crystal structure of human estrogeni 170-hydroxysteroid dehydrogenase complexed with 170-estradiol at 2. A resolution." by A. Aui , P. K. Rehse, D. -W. Zhu. R. L. Campbell., Labrie I and S. -X. Lin in Nature Structure Biology (1996). 3: 665-668.

Appendices
2.1 Introduction
Sir-Human estr ogenic 170-hydroxyst eroid dehydrogenas e (178. HSD1) plays a pivotal role in the formation of active estrogens in gonada and peripheral tissues (Martel et al., 1992). This enzyme convert! estrone to 170-estradiol the most active estrogen-in the presence of thc cofactor NADPH or NADH. Because estradiol is a mitogenic factor toward! breast epithelium, the interconversion of estrone to estradiol plays i
crucial role in stimulating the growth of hormone-dependent breas: carcinomas. It has been shown that this enzyme is expressed in breasi cancer tissue (Poutanen et al., 1992) and is responsible for maintainint the high intracellular estradiol concentration detected therein (Bonney ei al., 1986). Suppression of the 1713-HSD1 activity may provide a means oj reducing tumor estrogen levels and promoting tumor regression (Labrie et al., 1992).
The human 170-HSDI is a homodimer, each subunit having molecular mass of 34.5 kDa (Lin et al., 1992). It belongs to the shoi chain dehydrogenase famiIy (Persson et al., 1991) bearing the highl conserved and catalytically cruciaI Tyr-X-X-X-Lys sequence in the activ site (Puranen et al., 1994). In addition to its activity towards th interconversion of estrone and estradiol this enzyme also shows Som activity for the interconversion of dihydroepiandrosterone (DHEA) an1
5 A - diol, and of progesterone to 20a-hydroxy-4-pregnen-3-one (Tobias B et al., 1982) but shows no detectable activity for the interconversion o androstenedione and testosterone (V. Luu-The, persona1 communication] In the recently the structure of 178-HSD1 from crystals grown in th{ presence of saturating NADP+ has been deterrnined (Ghosh e t al., 1995) In that structure the electron density corresponding to the cofactor wa
not reliably identified but the location of the cofactor and estradiol wer~ inferred from molecular rnodeling. Here we report the 2.3 A-resolutioi crystal structure of 170-HSD1 complexed with estradiol (170 - HSDl - ES and describe the important intermolecular interactions relevant to th1 catalytic mechanism and substrate specificity.

Appendices
2.2 Results and Discussion
Data collection and refinement statistics are presented in Table 2. The overall structure of 170-HSD1 in the complex is essentially the saxr as the native enzyme stnicture (Ghosh D. et al., 1995) (root mean squai difference of 0.5A for al1 main-chain atoms) except for the segmei between residues 192 and 207. No electron density was observed fc the 43 C-terminai residues in either structure. The weak e1ectron densii for residues 192 to 297 in both structures preclude accurate refinemei of their position. While these residues are near the active site, t h regidly fixed end point prevent the residues in the flexible region froi making close interactions with the substrate. The weak electron densit reflects a high degree of flexibility of this segement that may perm access of ligands to the binding site. From previously kinetic studies, has been found that the binding of the cofactor and 17B-estradiol i unordered (Betz G. 1971), so the structures of the enzyme with eithe estradiol or cofactor are relevant, for example. While only small change in the structure of the enzyme are necessary to accommodate th binding of the steroid substrate, the complex structure reveals th interactions between the enzyme and the substrate that are importar for substrate specificity.
Estradiol binds close to the catalytically important residues Ser 141 Tyr 155, Lys 159, Bis 221 and Glu 282 (Fig 2.la). The clear electro density for estradiol (Fig. 2.lb) is consistent with only one bindin orientation. The binding site is a narrow hydrophobic tunnel showing high degree of cornplementarity to the whole substrate. The total buriel surface for estradiol is 229 A (Poutanen et al., 1992), which is 92% of th
surface area for free estradiol. The buned surface of 17B-HSD is 340 1
(Poutanen et al., 1992), 71.5% of which is hydrophobic (carbon atoms: Th i s Iarge hydrophobic surface likely contributes the mai thermodynamic force for binding. It is formed by hydrophobic an1 aromatic residues Val 143, Leu 149, Pro 187, His 221, Val 225, Phe 254,

Appendices
Phe 259, Leu 262, Leu 263, most of which are located in helices aH (257-271). aH (274-284) and aG' (209-227). There are 24 van de Waals contacts between estradiol and 178-HSD, most of them involvin rings A , B, C and the methyl group C l 8 (Table 2.2). In addition to th hydrophobic interactions, the 17-hydroxyl on the estradiol D ring form hydrogen bonds with the hydroxyls of conserved Ser 142 (3.1 A) an1 Tyr 155 (3.5 A) and the 3-hydroxyl on the A ring forms hydrogen bond with the NEZ atom of His 221 (3.1 A) and with a carboxyl oxygen of Gl. 282 (2.7 A) (Fig. 2.2a). This is in agreement with previous observation of the importance of His 221 in catalysis, based on mutagenesis an, modeling studies (Puranen et al., 1994; Ghosh et al., 1995). There is lûûO rotation of the ~2 angle of residue Glu 282 in comparison to th1 170-HSD1 native structure. The interaction of this residue with th1 substrate has not been reported previously.
The mechanism of the enzyme is believed to proceed through direct transfer of a hydride ion from the C4 position of the nicotinamidi nucleotide to the acceptor carbonyl C l 7 of estrone to produce 176 estradiol (Penning et a1.,1991). A proton abstracted from the bu11 solvent is provided to the 017 atom to compIete the reaction. It ha been proposed for other related members of the short chai1 dehydrogenase famiiy that the conserved tyrosine residue-Tyr 155 fo 170-HSDl-acts as a general acidlbase catalyst towards the 017 atom O
the substrate, and that conserved Ser 142 and Lys 159 facilitate thi, through hydrogen bonding and electrostatic effects with Tyr 151 respectively (Ghosh et al., 1995; Jornval et al., 1995; Varughese et al. 1994). The interaction between Tyr 155 and the 17-hydroxyl is weake than between Ser 142 and the 17-hydroxyl, but Tyr 155 is the mord likely candidate for the role of acidJbase catalyst, because it is on11 partly buried and its hydroxyl is somewhat accessible to the bu11 solvent, while Ser 142 is completely buried by the substrate, and alsr because its pKa of Tyr155 would be lower than that for Ser 142 Therefore the strong Ser 142-estradiol hydrogen bonds suggest that iti role is to orient estradiol in the binding cleft. It should be mentioned

Appendices
that mutations of Ser 142 to a cysteine or a glycine residue leads t
complete enzyme inactivation (S.-X. Lin, unpublished results).
There is no evidence of a hydrogen bond between the cataiyticall important Lys 159 and Tyr 155 in either the native structure (Ghosh t
al., 1995) or the estradiol bound structure of 178-HSD1, and the positio of Lys 159 does not change on binding estradiol. The positive charge c the lysyl amino group may still be close enough to lower the pKa of T j 155. Iaano reported that an oxidized cofactor prevented a lysine residu frorn k i n g labeled by trinitrobenzene sulfonate (Varughese et al 1994: These facts, together with the previously modeled position for NADP (Ghosh et al., 1995), suggest that Lys 159 may be primarily involved i the binding of the cofactor, since its amino group appears to be in position to form two hydrogen-bonds with the 02' and 03' hydroxyls c the nicotinamide ribose. This is further supported by Varughese (1994 et al. and Tanaka et al. (1996) who have shown that the conserve' lysines of dihydropteridine reductase, carbonyl reductase and 7a hydroxysteroid dehydrogenase, members of the short-chai dehydrogenase family, form a hydrogen bond with the nicotinamid ribose. This role in cofactor binding does not preclude the possibilit: that the lysine has an electrostatic effect that reduces the pK= of Tyr 155
The specificity for estradiol binding appears to be due to combination of hydrogen bonding interactions and the complementarit: of the hydrophobic surfaces and estradiol (Fig. 2.2a). The enzyme' preference for estradiol as substrate could be explained by th1 cornplementarity of the narrow cleft to the planar A ring and by th1 hydrogen bond between the 3-hydroxyl group of estradiol and th( carboxylate of Glu 282. In addition to having non-planar A ring testosterone and DHEA have a Cl9 methyl group. DHEA has the : hydroxy1 group while testosterone has a 3-keto group. These structuri features are in accord with the enzyme's substrate specificity.

Appendices
2.3 Methods
T o overcome the difficulties associated with the low solubility 01
17B-estradiol, 170-HSD 1 and estradiol were mixed at low concentratior followed by gradual centricon (Arnicon) concentration. An initial solutior of 10-12 pM protein and 25 pM estradiol was concentrated t o abou, 220 pM protein. By following ~ 1 4 - l a b e l e d estradiol, we determined tha: the final concentration of bound estradiol was 440 p M indicating i
stochiometry of 2:l. The great difference between this total E: concentration and the solubility of free estradiol (25-30 FM) indicatec the binding of the substrate into the hydrophobic binding pocket, Crystals were obtained in four weeks with 28% PEG (4000), 0.06% B. octylglucoside, 0.16 MgCl;, and O.1M Hepes pH 7.5 at room temperature. Typical crystal size is 0.15 x 0.25 x 0.48 mm. Data collection was carried
out on an R-Axis IIC irnaging plate area detector on a Rigaku RU30C rotating anode at room temperature and the data were processed using DENZO and SCALEPACK (Otwinowski 2. 1993).
The initial electron density map was calculated using the phases and the F,,l, magnitudes from the native crystal structure (Ghosh e t al., 1995). Fitting to the electron density was performed using the program O (Jones et ai., 1991). Al1 refinement was done with X-PLOR (Brünger . 1993). Water positions were determined using the program Peak (M. Cygler & .M. Desrochers, persona1 communication) and checked in the electron density map. The final refined mode1 contained 284 residues, 56 water molecules and the 178-estradiol molecule. The R-factor for this mode1 is 0.196 in the 8.0-2.3 A shell. The coordinates will be deposited in the Brookhaven Protein Data Bank, PDB a ccession code 1 loL.
2.4 References
Betz, G., J. Biol. Chem., 246, 2063-2068 (1971).

Appendices
Bonney, W.C., Reed, MJ., Beranek, P.A., Ghilchik, M.W. & James, V.H. . Steroid. Biochem. ,24, 361-364 (1986).
Brünger, A.T. X-PLOR (version 3.1) Manuai. Yale University, New Havel CT (1993).
Connolly, M. L. Science, 221, 709-713 (1983).
Connolly, M. L. QCPE Bull., 1, 75 (1981).
Evans, S. J. Mol. Graphics , 11, 134-138 (1993).
Ghosh, D., et al., & Lin, S-X. Structure , 3, 503-513 (1995).
Inano, H. Biochem. Biophys. Res. Commun. , 152, 789-793 (1988).
Jones, T.A., Zou, J.Y., Cowan , S.W. & Kjeldgaard, M. Acta Cryst. A 47:110 119 (1991).
Jornvall H., et al., & Ghosh, D. Biochemistry, 34, 6003-6013 (1995).
Labrie, C., Martel, C., Dufour, LM., Levesque, C., Merand, Y. & Labrie, E Cancer Res. ,52, 610-615 (1992).
Lin, S.-X., et al ., & Labrie, F. J . Biol. Chern. ,267, 16182-16187 (1992).
Luzzati, V . Acta. Crystallogr., 5, 802-8 10 (1 952).
Martel, C., et al., & Labrie, F. J . Steroid. Biochern. Mol. Biol., 41, 4597 4603 (2992).
Otwinowski, 2. Data Collection and Processing (eds. Sawyer, L., Issacs 1 W. &Bailey S .) 55-62 DL/S CI/R34. ( Daresbury Laboratory, Warrington 1993).
Penning, T. M., & Riciglianu, J. W. J . Enzyme inhibition., 5, 165-191 (1991).
Persson, B., Krook, M. & Jornvall, H. Eur. 3. Biochern. , 200, 537-54: (1991).
Poutanen, M., Isomaa, V., Lehto, V.P. & Vihko, R. Int. J. Cancer , 50, 38t 390 (1992).

Appendices
Puranen, T.J., Poutanen, M.H., Peltoketo, H.E., Vihko, P.T. & Vihko, R. Biochem. J . , 304, 289-293 (1994).
Sheriff, S., et al., & Davies, D. R. Proc. Nat. Acad. Sci. U.S.A., 84 , 807. 8079 (1987).
Tanaka, N., Nonaka, T., Nakanishi M., Deyashiki, Hara A. & Mitsui '
Structure, 4, 33-45 (1996).
Tobias, B., Covey, D.F. , Strickler, R.C. J. Biol. Chem. , 257, 2783-271 (1982).
Varughese K. I., Xuong N. H., Keifer, P. M., Matthews D. A. and Whiteley, M. Proc. Natl. Acad. Sci. USA, 91, 5582-5586 (1994).

Appendices
Legends to Figures
Figure 2.1: a) Cartoon representation of a monomer of 178-HSD with 17B-estradii positioned at its binding site. The estradiol molecule and side chains (
residues Ser 142, Tyr 155, Lys 159, His 221, and Glu 282 in the actii site are shown in white. This figure and figure 2b were prepared usin SETOR (Evans S. 1993)
b) Stereo view of the calculated final IFol-IFCI elecrron density omit ma contoured at the 2.8 o and the final refined mode1 for the 17B-estradic molecule. This figure and Fig. 2a were generated using the program (Jones et al., 1991).
Figure 2.2: a) Enzyme-substrate complementarity demonstrated by the burie molecular surfaces (Connolly M. L. 1983) of the active site and estradio The surfaces were generated with the program MS (ConnolIy 1981) usin a probe radius of 1.4 A. Carbons from estradiol are in cyan, carbon from the amino acid are white, and oxygens are red, nitrogens are gree for both molecules.
b). Detail of the active site stereochemistry of the estradiol bound t7l HSDl structure. Hydrogen-bonding interactions are represented by re dotted spheres and the water molecules are in blue.

Appendices
Table 2.1: Data collection, structural and refinement statis tit
Unit ceIl dimensions
Space group
Ref lec tions
Redundancy
Unique reflections with F>2sF
Rmerge
Resolution range
Resolution
Completness
R-value
Free R-value
R.m.s. deviation from ideal bond lenght (A)
0.007
ideal bond angles 1.53'
improper angles 1.17O
Estirnated average coordinat error (Luuati V. 1952) (A) 0.29

Appendices
Table 2.2: Interactions of 17B-estradiol with 17R-HSDI.
Type of Ligandparts Numberof Ligands 17g-HSD Distance (A) interaction interactions atom contact
residues and atoms
a.Van der Waais interactions*:
Ring A 7
Ring B
Ring C
Ring D Methyl
b. Hydrogen- bonds:*
Ring D 2
Ring A 2
PheZs9 CC Phe259 ~ € 1 Phe259 ~ 6 1 Phe259 CE^ Phe259 CM His221 ce1 &221 ca l Tyr218 O Sr222 OY Ser22î O'Y Tyr218 O Leu149 ~ 6 1 Va1143 cY1 Va11 43 C* Pr0187 ~6 Pro187 N Gly186 C Gly186 Ca Met193 SS sr142 OY Gly 144 Ca Gly 144 N Tyr155 c1 Leu149 ~ 6 1
Ser 142 O Tyr 1% O Glu282 O& His221 NQ
*Cutoffs of 3.4 A for hydrogen bonds and up to 4.1 A for van der Waal: contact were used (Sheriff S. et al., 1987).

Appendices
Fig. 2.1

Appendices
Fig. 2.2

Appendices
3 Crystallization and Preliminary Crystallographic analysi; the Snake muscle Fructose 1,6-bisphosphatase
This is a cooperative project with Dr. G.-J. Xu in Shanghai Institute (
Biochemistry, Academia Sinica, Shanghai, China. In this paper, 1 a responsible for crystallization of Fm-1,6-Pase.
* 3 is adapted from the paper of "Crystallization and Preliminar Crystallographic analysis o f the Snake muscle Fructose 1,f bisphosphatase" by P.-W. Z h b G.-J. Xu, P. Rehse, A. Azzi, F. K., Zhao an S.-X. Lin (in preparation)

Appendices
Crys ta l l i za t ion and Prel iminary Crystallograp analys is of the Snake muscle Fructose 1, b isphosphatase
D.-W. ~hu', G.-J. xu2, P. ~ e h s e ' , A. ~ z z i l , F.-K. 2hao2 and S., ~ i n l *
1 The Laboratoy of Molecular Endocnnology, CHUL Research Center anc Laval University Quebec G1V 4G2, Canada.
2 Shanghai Institute of Biochemistry, Academia Sinica, Shanghai, China.
3.1 Introduction
Fructose- 1.6-bisphosphatase (Fru- l,6-Pase; EC 3.1.3.1 l), a kej regulatory enzyme in gluconeogenesis, cataiyzes the hydrolysis ol fructose l,6-bisphosphate (Fru- 1,6-P2) to fructose 6-phosphate (F6p: and ortho-phosphate. The gluconeogenic roIe of the liver and kidnej enzymes has been extensively studied (Benkovic et al., 1977; Ke et al.. 1991; Pontremoli et al., 1971; Tejwani 1983; Van 1987). However, the physiological function of the muscle enzyme remains unclear (Black et ai, 1974; Krebs & Woodford 1965). Apart from some earlier reports on thai the enzyme from the bumble bee is responsible for the heat production (Storey 1978), most studies of mechanism of the muscle enzymes to date have been carried out on that from the snake, a hibernating animal. An understanding of the snake muscle Fru-l,6-Pase function, along with a comparison with the isoforms involved in gluconeogenesis, may provide important c h e s to the comprehension of the mechanism of hibernation, Such studies might eventually have medical impacts such as improving the transportation of patients. The snake muscle enzyme displays a cooperativity which has been studied in detail (Xu et al., 1982; Xu et al., 1985; Yu & Xu 1992; Zhao et ai., 1984; Zhao et al., 1995)- The authors of

Appendices
these references have found that the later enzyme catalyses th1 hydrolysis of Fru-1,6-P2 by a ping pong mechanism. Further more, AMI had a dual function being both an allosteric inhibitor at pH 7.5, and ai
activator of the same enzyme at pH 9.2. A pH shift in the presence O
AMP would alter the heat production through the cycle O
phosphofructokinase and fructose- 1,6-bisphosphatase.
The structural studies of Fru-1,6-Pase, especially the threc dimensional structures of the same enzyme complexed with differen inhibitors or activators, will contribute to the general understanding O
the enzyme cooperativity, as well as to the structure-functioi relationship of this specific enzyme and hence its physiological role Interesting cornparisons can be made with respect to crys tallographic studies of heart and kidney Fru-1.6-Pase from non-hibemating animal: (Anderson & Matthews 1977; Soloway & Mcpherson 1978). In recen years, the enzyme from pig kideny has been crystal1ized and iti structure determined. The interesting point is that the structures of thi unligated enzyme or its fructose bisphosphate complex showed largc quaternary and tertiary conformational differences (Ke et al., 1990), an( aiso that its allosteric transition has been dernonstrated in the tertiar! structure (Liang et al., 1993). Here we report the crystallization an( preliminary crystaliographic study of the snake (Zaoeys dhumnadesj muscle Fru- 1.6-f ase.
8.3.2. Exper imen ta l
Preparation of the sample The native form of the enzyme was prepared according to a previou!
report (Xu et al., 1982) and consists of four subunits with a moleculai mass of about 140 KDa (4 X 36 kDa). The optimum pH for activity was 7 The activity of the enzyme was determined either by a coupled enzyme assay measuring the formation of NADPH spectrophotometricall~ (Traniello et al., 1972) or by measuring the release of inorganic phosphate as described previously (Xu et al., 1982). The optical method

Appendices
was used in detennining the protein concentration, which (mg/ml) equals to A280 / 0.72. The absorbance was measured with a Beckm; DU-70 spectrophotometer in which a microcell of 50 pl can be used.
Crystallization The conventional vapor diffusion method using sitting drop wi
employed. The protein sample was at 20 mg/ml and prepared in a buff; containing 20% glycerol, 0.2 m M EDTA, 0.4 mM DTT, lOmM Tris-HCl pH 7.5 and 0.2 mM ATP added just before crystallization. The reservo solution contained 0.39 M magnesium chloride, and 27% (wb
polyethylenegiycol (3350) as the precipitant, and 100 mM Tris-HC1 pH 8.5. 10 pI of protein solution and 10 p1 of the reservoir solution w; initially mixed and equilibrated against the reservoir solution (for tl final concentrations of the components in the crystallization drop, pleai refer to the results section).
X-ray diffraction analysis The preliminary X-ray diffraction analysis of crystals of Fru-1,6-pa!
was performed at room temperature with an R-AXIS IIc image plai and a Rigaku rotating anode generator. The crystal was mounted in glass capillary and sealed wich a small amount of mother liquor at tk
end. The detector was placed at a distance of 160 mm from the cryst; and the beam was collimated to 0.3 mm. The unit ce11 dimensions an crystal orientation angles were determined by least-squares fitting of lattice of partial refiections recorded on the image plate at three fixe positions of the crystal, 45' apart in spindle rotation. The resultin imaging plates were processed using R-axis software.
8.3.3. Results and discussion
Crystal growth The Crystal Screen (lancarik & Kim, 1991) and Crystal Screen :
(from Hampton research) were used. Through preliminary screeninj

Appendices
some multiple crystaIs were obtained in the presence of magnesiu. chloride/Hepes/PEG (Fig. 3.1 a). Further refinement was carried out and a series of crystals were obtained with different magnesium chlorir and PEG concentrations, with the best ones in the presence of 19%(w/l PEG(4k)' 0.27M MgClz 0.14 mM ATP, 70 mM Tris-HC1 pH 8.5, 14' glycerol and 14 mg/ml protein in the drop after equilibrium. These fin; concentrations were evaluated using the final drop volume. The crysta appeared overnight at room temperature and grew to a typical size (
0.3 x 0.45 x 0.62 mm in four weeks (Fig.3.lb).
Data collection and analysis Based on the systematic absences from the collected data, the spac
group was determined to be P3(1)21 with unit ceil dimensions r a=b=83.7 A, c=202.41 A, a=8=90° and y=120°(An initial data set wi collected to 2.8 A although many diffraction points were obsewed at 2. A. The processed data yielded 16670 unique reflections from 4834 independent measurements with a Rmerge based on structure factors (
5.76%. The data were 86.3% complete between 12 and 2.87 A and 78.2: complete in the 3.0 to 2.87 A shell (Table 8.3.1). With the mar spectroscopy determined subunit mass of 36 kDa and assuming subunits per asymmetric unit, the Matthew's coefficient, Vm, wa calculated to be 2.84 A3/dalton. This is within the normal rang (Mattew, 1968).
In anticipation of the availability of the sequence of the enzyme (X et al., unpublished results), we have performed an initial moleculs replacement analysis using the fructose-l,6-biphosphatase from the pi adrenal cortex as mode1 (Ke et al., 1990) and the molecular replacemer package AMORE (Navaza, 1994). The 20 best rotation function solution had correlation functions ranging from 15.9 to 12.3 however only th third (15.8) and fourth peaks (15.7) yielded translation functio solutions significantly higher than the remainder of the peaks whe solved in P3(1)21. None of the rotation solutions yielded signifiar peaks in the translation search when the P3(2)21 enantiomorph was

Appendices
used. The two solutions were refined as individual rigid bodies withi AMORE and were found to be related to each other through two-fol symmetry. This confirmed the results previously obtained from a sel rotation function. Using the graphics program O (Jones, T. A., Bergdoll, h and Kjeldgaad, M., Uppsala University) the symmetry mate of the secon solution which was also related directly by the two-fold symmetry to té
first solution was output. The large number of contacts between th resultant two molecules lie mainly along this non-crystallographic twt fold axis which suggests the physiological importance of those subun interactions and could be closely related to the cooperative behavior (
fructose 1,6-phosphatase as demonstrated by kinetics studies. Ba contacts between atoms could be relieved through some min( manipulation of amino acid side chains. The resultant maps ar relatively clear but require the completed sequence of the protein befoi rebuilding can properly commence.
Acknowledgments : We thank Dr. M. Zhou for participating in thc discussion of crystallization strategy and Dr. R. Campbell for carefi reading of the manuscript.
3.4 REFERENCES
Anderson, W. F. & Matthews, B. W. (1977) J. Biol. Chem. 252, 5556-5557, Benkovic, S. J. & de Maine, M. M. (1982) Advan, Enzymol. 53, 45-82. Black, W. J., Van Tol, A., Fernando, J. & Horecker, B. L. (1972) Arc1 Biochem. Biophys. 151, 576-590. Jancarik, J., & Kim, S.-H. (1 991) J. Appl. Cryst. 24, 409-41 1. Ke, H., Zhang, Y. & Lipscomb, W. N. (1990) Proc. Natl. Acad. Sci. 87, 5241 5247. Ke, H., Zhang. Y., Liang. J. -Y. and Lipscomb W. N. (1989) Proc. Natl, Acac sci. USA. 88, 2989-2993. Krebs, H. A. & Woodford, M. (1965) J. Biochem. 94, 436-445. Liang J.-Y., Zhang, Y., Huang, S., and Lipscomb, W. N. (1993) Proc. Nat. Acad. Sci. 90, 2132-2136.

Appendices
Pilkis, S. J., Clans, T. H., Kountz, P. D & EL-Maghrabi, M. R. (1987) in d Enzymes, eds. Boyer, P.D. &Krebs, E. G. (Acadernic, New York), 3rd Ed., 1 3 -46. Pontremoli, S. & Horecker, B. L. Fructose 1,6-diphosphatase. Tl Enzymes, Boyer, P.D. (ed.) 3rd. Ed., 1971, 4, 612. Soloway, B. & Mcpherson. A. (1978) J. Biol. Chem. 253, 2461-2462. Storey, K. B. (1978) Acta. Biochim. Biophys. 523, 443. Tejwani, G. A. (1983) Advan. Enzyrnol. 54, 121-194. Traniello, S., Pontremoli, S., Tashima, Y. and Horecker, B. L. (1972) Arc Biochem. Biophys. 145, 160-166. Van Schaftingen, E. (1987) Advan. Enzymol. 59, 315-395. Xu, G.-J., Shi, J.-P. and Wang, Y.-L. (1982) Methods Enzymol. 90, 349-351 Xu, G.-J., Shi, J.-P. Zhao, F.-K. and Wang, Y.-L. (1985) Molecul; Architecture of proteins and Enzymes, Bradshaw. R. A. and Tang, J. ed Academic Press, New York, 91985) pp. 51-63. Yu, 2.-B. & Xu, G.-J. (1992) Acta Biochim. Biophys. Sic. 24, 428-433. Zhao, F.-K., Shi J.-P. and Xu, G.4, (1984) Acta Biochim. Biophys. Sic. 1 564-607 Zhao, F.-K., Xu, S. -Q. and Xu, G . 4 . (1995). Acta Biochim. Biophys. 27, 7: 81.

Appendices
Figure 3.1 : Crystai growth of Fm-l,6-Pase a. The first crystais grown in the presence of 21% (w/v) PEG(400), 0.1 M MgC12, 0.14 rnM ATP, 70 mM Hepes at pH 7.5, 14% glycerol with protein concentration of 14 mg/ml after equilibrium. b. Fm-1,6-Pase crystals grown in the presence of 19%(w/v) PEG(4k 0.27M MgClz 0.14 m M ATP, 70 mM Tris-HC1 pH 8.5, 14% glycerol ar 14 mg/ml protein in the drop after equilibrium. The crystal has typical size of 0.3 mm x 0.45 mm x 0.62 mm and diffracts to 2.3A.

Appendices
Fig.3.1 Crystal growth of Fru-1,6-Pase

Appendices
Table 3.1: Completeness of a data set from a Fm-1,6-Pase crystal.
Resolution Range (A) % Completeness accumulated

TEST TARGET (QA-3)
APPLIED IMAGE. lnc - = 1653 East Main Street - -. , , Rodiester. NY 14609 USA -- -- , Phone: 7161482-0300 -- -- - - F~x: 71 61288-5969




















