
UNIVERSIDAD AUTÓNOMA DE CIUDAD JUÁREZ INSTITUTO DE CIENCIAS BIOMÉDICAS DEPARTAMENTO DE CIENCIAS...
63
UNIVERSIDAD AUTÓNOMA DE CIUDAD JUÁREZ INSTITUTO DE CIENCIAS BIOMÉDICAS DEPARTAMENTO DE CIENCIAS QUÍMICO-BIOLÓGICAS ACADEMIA DE BIOQUÍMICA MANUAL DE PRACTICAS DE BIOQUÍMICA GENERAL NUTRICIÓN Y MEDICINA (PLAN VIEJO) FECHA DE ACTUALIZACIÓN AGOSTO 2011 M. EN C. GLORIA E. MEJIA CARMONA
-
Upload
independent -
Category
Documents
-
view
1 -
download
0
Transcript of UNIVERSIDAD AUTÓNOMA DE CIUDAD JUÁREZ INSTITUTO DE CIENCIAS BIOMÉDICAS DEPARTAMENTO DE CIENCIAS...

UNIVERSIDAD AUTÓNOMA DE CIUDAD JUÁREZ INSTITUTO DE CIENCIAS BIOMÉDICAS
DEPARTAMENTO DE CIENCIAS QUÍMICO-BIOLÓGICAS ACADEMIA DE BIOQUÍMICA
MANUAL DE PRACTICAS DE BIOQUÍMICA GENERAL NUTRICIÓN Y MEDICINA (PLAN VIEJO)
FECHA DE ACTUALIZACIÓN AGOSTO 2011 M. EN C. GLORIA E. MEJIA CARMONA

Contenido PRACTICA #. 1 ................................................................................................................................................ 4
EXPLICACION Y MANEJO DEL MATERIAL DE LABORATORIO......................................................................... 4
Practica # 2 .................................................................................................................................................... 7
BIOSEGURIDAD ............................................................................................................................................. 7
PRÁCTICA #3 ............................................................................................................................................... 10
USO, CALIBRACIÓN Y MANEJO DEL POTENCIÓMETRO ............................................................................... 10
PRÁCTICA #4 ............................................................................................................................................... 12
TITULACIÓN ACIDO-BASE ............................................................................................................................ 12
PRACTICA # 5 ............................................................................................................................................... 14
REACCIONES DE IDENTIFICACION DE AMINOACIDOS ................................................................................. 14
PRACTICA # 6 ............................................................................................................................................... 18
SEPARACIÓN EN CAPA FINA DE UNA MEZCLA DE AMINOÁCIDOS. ............................................................ 18
Practica # 7 .................................................................................................................................................. 21
REACCIONES DE IDENTIFICACION DE PROTEINAS ...................................................................................... 21
Practica # 8 .................................................................................................................................................. 25
DETERMINACIÓN DE LAS PROPIEDADES QUÍMICAS DE LAS PROTEÍNAS ................................................... 25
Práctica #. 9 ................................................................................................................................................. 30
FOTOCOLORIMETRIA .................................................................................................................................. 30
Práctica #. 10 ............................................................................................................................................... 35
ANALISIS ENZIMATICO CUALITATIVO.......................................................................................................... 35
Práctica #. 11 ............................................................................................................................................... 37
CINETICA ENZIMATICA 1: EFECTO DE LA CONCENTRACION DEL SUSTRATO .............................................. 37
SOBRE LA ACTIVIDAD ENZIMATICA ............................................................................................................. 37
Práctica #. 12 ............................................................................................................................................... 40
CINETICA ENZIMATICA 2: EFECTO DEL inhibidor sobre la actividad enzimática ........................................ 40
Práctica #. 13 ............................................................................................................................................... 42
CINETICA ENZIMATICA 3: EFECTO DEL PH Y TEMPERATURA ...................................................................... 42
Práctica #. 14 ............................................................................................................................................... 46
CUANTIFICACION DE COLESTEROL SERICO ................................................................................................. 46
Práctica #. 15 ............................................................................................................................................... 48
EXTRACCIÓN DE GLUCOGENO Y REACCIONES DE IDENTIFICACION DE CARBOHIDRATOS ......................... 48
Práctica #. 16 ............................................................................................................................................... 52

3
EFECTO DE LAS COENZIMAS SOBRE LA ACTIVIDAD ENZIMATICA ............................................................... 52
Práctica #. 17 ............................................................................................................................................... 56
EFECTO DE LOS INHIBIDORES SOBRE LA ACTIVIDAD ENZIMATICA ............................................................. 56
Práctica #. 18 ............................................................................................................................................... 60
DETERMINACION DE UREA Y CREATININA SERICA ..................................................................................... 60

4
PRACTICA #. 1 EXPLICACION Y MANEJO DEL MATERIAL DE LABORATORIO
OBJETIVO
Desarrollar destreza en el manejo tanto de material como de instrumentación aplicable a técnicas de
experimentación en el área bioquímica.
PRERREQUISITOS
1.- Conocimientos sobre diluciones, soluciones y mezclas. 2.- Definición de ácido, base y amortiguador. 3.- Conocimiento básico del manejo del potenciómetro y el fotocolorímetro. 4.- Que es una solución Molar, Normal y % w/v
INTRODUCCION
Para llevar a cabo con éxito un experimento en cualquier laboratorio es necesario que se manejen
técnicas básicas de experimentación tales como:
- Técnicas de medición de líquidos. - Técnicas de pesado de sólidos. - Técnicas de mezclado de líquidos y sólidos.
Todas estas técnicas se relacionan con frecuencia con las actividades que cotidianamente se llevan a
cabo en el laboratorio de Bioquímica. A continuación se dan algunos datos sobre cada una de estas
técnicas.
Medición de líquidos: Los instrumentos de medición de líquidos más utilizados son: pipetas, probetas y
matraces volumétricos. Las pipetas sirven para medir volúmenes de hasta 25 ml. En este rango de
volumen, las pipetas tienen un uso muy frecuente en el laboratorio por su exactitud y confiablilidad. hay
dos tipos generales de pipetas: las serológicas y las volumétricas.
La pipetas serológicas miden volúmenes diversos debido a que están graduadas en diferentes medidas,
por ejemplo, una pipeta de 1.0 ml. esta graduada en décimas de mililitro y por lo tanto sirve para la
medición de cualquier volumen menor de 1.0 ml. Las pipetas más frecuentemente usadas son de 1.0,
2.0, 5.0 y 10.0 ml..
Las pipetas volumétricas sólo miden volúmenes fijos ya que están graduadas para medir un solo
volumen, por ejemplo, una pipeta volumétrica de 1.0 ml. solo servirá para medir este volumen.
Algunas pipetas están diseñadas para que al momento de liberar el líquido que se está midiendo quede
una pequeña porción en la punta de las mismas, esta porción debe dejarse contenida en la pipeta
porque de otra manera se introducirá un error en la medición. La manera de reconocer este tipo de
pipetas es por las letras TD ( To Deliver ) que se encuentran en la parte superior de la pipeta. Otras
pipetas están diseñadas para liberar todo el volumen incluyendo la porción que se queda en la punta,
este tipo de pipetas se reconocen por las letras TC ( To Contain ), en este caso, si no se libera el total del
líquido medido se incurre en un error de medición.

5
Las buretas varían en tamaño, generalmente de 1 a 100 ml. El flujo del líquido es controlado por una
llave de vidrio o de teflón. Si es de vidrio requiere lubricación. Las buretas deben llenarse por arriba de
la marca de cero y posteriormente abrir la llave cuidadosamente para que fluya el líquido y el menisco
se ajuste a cero. Dentro de los usos indicados para las buretas se encuentran las titulaciones y para
medición de volumenes de líquidos tóxicos, ácidos, sustancias corrosivas, etc.
Las probetas son cilindros graduados que miden volúmenes por arriba de los 25 ml. todas ellas son
usadas para medir volúmenes diversos, por ejemplo, con una probeta de 50 ml. se pueden medir
volúmenes de 26, 38 o 42 ml etc.. Estos volúmenes se denotan por marcas que se encuentran entre una
graduación y otra. las probetas más frecuentemente utilizadas son: 50, 100, 250, 500 y 1000 ml.. Para
medir volumen en una probeta se tiene que tomar en cuenta la siguiente regla: La superficie de un
líquido dentro de una probeta siempre forma una curva o menisco. El diseño de la probeta esta hecho
para medir volúmenes exactos al igualar la parte inferior del menisco con la graduación. Si se mide el
volumen con la parte superior del menisco, se incurrirá en un error.
Los matraces volumétricos, tambien llamados de aforación, sirven para medir volúmenes exactos y son
usados cuando se hacen soluciones porcentuales y molares. Los matraces más frecuentemente usados
son de 50, 100, 500 y 1000 ml.. El uso de estos matraces sigue la misma técnica de medición que las
probetas.
Todos los recipientes arriba mencionados son material de vidrio, por lo tanto, están expuestos a
cambios de tamaño en altas y bajas temperaturas y esto puede modificar drásticamente la exactitud de
la medición. El diseño de este material de cristalería está hecho para medir volúmenes a una
temperatura promedio de 25°C.
Medición de sólidos: La balanza granataria y la balanza analítica son las más utilizadas en la medición de
reactivos químicos de textura sólida. La granataria tiene una exactitud de aproximadamente una
décima de gramo, se utiliza para pesar cantidades mayores de un gramo y cuando la precisión no sea
menor de 0.1 g.
La balanza analítica se utiliza cuando el peso deseado requiera una precisión menor de 0.1 g. Es uno de
los instrumentos más sensibles y permite pesar hasta décimas de miligramo.
Sin embargo, el uso de ambos tipos de balanzas requiere de cuidados básicos comunes tales como:
limpieza completa de todas las partes de la balanza; mantener la tara específica de la balanza antes de
cualquier pesada y hacer pesadas racionales con respecto a la capacidad de la balanza.
Las balanzas de torción son útiles en el laboratorio de bioquímica como una herramienta para el buen
uso de la centrífuga ya que para equilibrar los tubos de centrífuga siempre debe hacerse en base al peso
y no al volumen del contenido.
La centrífuga es un aparato que permite separar sólidos de líquidos. Es un instrumento que separa
suspensiones a alta velocidad. El material sólido se queda en la base del recipiente (sedimento) y la
porción líquida en la parte superior del recipiente (líquido sobrenadante).

6
Los recipientes utilizados en la centrífuga generalmente son tubos de ensayo y estos siempre deben
balancearse en cuanto a peso y al ser colocados en la centrífuga siempre debe ser en direcciones
opuestas.
Mezclado de líquidos y sólidos: Las soluciones normales, molares y porcentuales así como los diferentes
tipos de diluciones son las mezclas más utilizadas en el laboratorio. Frecuentemente, las soluciones
involucran mezclas de sólidos en líquidos o de líquidos en líquidos en tanto que las diluciones solo
involucran mezclas líquidos en líquidos. De la destreza con que se lleven a cabo estas mezclas depende
el éxito de la experimentación en el laboratorio.
El fotocolorímetro permite cuantificar concentración de sustancias en base al color desarrollado. La
concentración de una sustancia en solución generalmente es determinada por una reacción de color,
siendo el principio general que a mayor concentración de la sustancia en la solución mayor será la
intensidad del desarrollo del color. Los fotocolorímetros son instrumentos que nos sirven para hacer
estas determinaciones.

7
PRACTICA # 2 BIOSEGURIDAD
OBJETIVO
Que el alumno se familiarice con el material Peligroso Biologico infeccioso, su manejo y desecho
adecuado, de acuerdo a las normas mexicanas NOM-087 y NOM 052.
INTRODUCCION
Residuos peligrosos biológicos infecciosos
¿Qué son los residuos peligrosos biológicos infecciosos?
Los residuos peligrosos biológicos infecciosos en lo sucesivo (RPBI), son aquellos que se generan durante
las actividades asistenciales a la salud de humanos o animales en los centros de salud, laboratorios
clínicos o de investigación, bioteros, centros de enseñanza e investigación, principalmente; que por el
contenido de sus componentes puedan representar un riesgo para la salud y el ambiente.
¿Cuáles son considerados los RPBI?
De acuerdo a la Norma Oficial Mexicana NOM-087-SEMARNAT-SSA1-2002, son considerados los
siguientes:
La sangre
La sangre y los componentes de ésta, sólo en su forma líquida, así como los derivados no comerciales,
incluyendo las células progenitoras, hematopoyéticas y las fracciones celulares o acelulares de . la
sangre resultante (hemoderivados).
Los cultivos y cepas de agentes biológico-infecciosos
Los cultivos generados en los procedimientos de diagnóstico e investigación, así como los generados en
la producción y control de agentes biológico-infecciosos.
Utensilios desechables usados para contener, transferir, inocular y mezclar cultivos de agentes
biológico-infecciosos.
Los patológicos
Los tejidos, órganos y partes que se extirpan o remueven durante las necropsias, la cirugía o algún otro
tipo de intervención quirúrgica, que no se encuentren en formol.
Las muestras biológicas para análisis químico, microbiológico, citológico e histológico, excluyendo orina
y excremento.

8
Los cadáveres y partes de animales que fueron inoculados con agentes enteropatógenos en centros de
investigación y bioterios.
Los residuos no anatómicos
Son residuos no anatómicos los siguientes:
Los recipientes desechables que contengan sangre líquida.
Los materiales de curación, empapados, saturados, o goteando sangre o cualquiera de los siguientes
fluidos corporales: líquido sinovial, líquido pericárdico, líquido pleural, líquido Céfalo-Raquídeo o líquido
peritoneal.
Los materiales desechables que contengan esputo, secreciones pulmonares y cualquier material usado
para contener éstos, de pacientes con sospecha o diagnóstico de tuberculosis o de otra enfermedad
infecciosa según sea determinado por la SSA mediante memorándum interno o el Boletín
Epidemiológico.
Los materiales desechables que estén empapados, saturados o goteando sangre, o secreciones de
pacientes con sospecha o diagnóstico de fiebres hemorrágicas, así como otras enfermedades infecciosas
emergentes según sea determinado por la SSA mediante memorándum interno o el Boletín
Epidemiológico.
Materiales absorbentes utilizados en las jaulas de animales que hayan sido expuestos a agentes
enteropatógenos.
Los objetos punzocortantes
Los que han estado en contacto con humanos o animales o sus muestras biológicas durante el
diagnóstico y tratamiento, únicamente: tubos capilares, navajas, lancetas, agujas de jeringas
desechables, agujas hipodérmicas, de sutura, de acupuntura y para tatuaje, bisturís y estiletes de
catéter, excepto todo material de vidrio roto utilizado en el laboratorio, el cual deberá desinfectar o
esterilizar antes de ser dispuesto como residuo municipal.
¿Por qué representan un riesgo a la salud o al ambiente?
Por sus características, ya que pueden contener agentes biológicos infecciosos que se definen como
“cualquier microorganismo capaz de producir enfermedades cuando esta presente en
concentraciones suficientes (inóculo), en un ambiente propicio (supervivencia), en un
hospedero susceptible y en presencia de una vía de entrada)”.
¿Existe regulación para los RPBI?
Con la finalidad de prevenir alguna situación de emergencia debida al mal manejo de los RPBI, la
autoridad Ambiental (SEMARNAT) publicó el 7 de noviembre de 1995 la Norma Oficial Mexicana NOM-

9
087-SEMARNAT-1995, Que establece los requisitos para la separación, envasado, almacenamiento,
recolección, transporte, tratamiento y disposición final de los residuos peligrosos biológico-infecciosos
que se generan en establecimientos que presten atención médica.
Debido a que esta Norma presentó algunos problemas de interpretación como en su aplicación, se
gestiono en coordinación con la Secretaría de Salud, su modificación, derivándose el 1 de noviembre de
2001 el Proyecto de Norma Oficial Mexicana NOM-087-SEMARNAT-SSA1-2000, Protección Ambiental -
Salud Ambiental – Residuos Peligrosos Biológico-Infecciosos – Clasificación y especificaciones de
manejo. De este proyecto se derivaron 370 comentarios, mismos que fueron contestados y publicados
en el Diario Oficial de la Federación el día 20 de enero de 2003.
Finalmente el 17 de febrero de 2003 se publica en el Diario Oficial de la Federación la Norma Oficial
Mexicana NOM-087-SEMARNAT-SSA1-2002, Protección Ambiental – Salud Ambiental – Residuos
Peligrosos Biológico-Infecciosos – Clasificación y especificaciones de Manejo.
¿A quines aplica la NOM-087-SEMARANT-SSA1-2002?
Con base en el campo de aplicación de esta norma es de observancia obligatoria a todos aquellos que
por sus actividades generen los RPBI descritos en la Norma, sin importar el volumen de sus generación,
aclarando que aquellos que su generación sea menor a 25 kilogramos al mes, podrán ubicarse como
generadores de Nivel I, esto para el tiempo de almacenamiento de sus RPBI.
¿Existe otra Norma Oficial Mexicana donde aparezcan los RPBI?
Existe la Norma Oficial Mexicana NOM-052-SEMARNAT-2005 que establece las características, el
procedimiento de identificación, clasificación y los listados de los residuos peligrosos, sin embargo esta
es de carácter General, por lo que para los RPBI prevalecen las condiciones establecidas en la NOM-
087-SEMARNAT-SSA1-2002.
¿Cuál es la infraestructura para el tratamiento de los RPBI?
A partir de la publicación de la Norma Oficial Mexicana NOM-087-SEMARNAT-1995, se desarrollo la
infraestructura para el tratamiento de este tipo de residuos, con la condición de que dicha norma
establece que los residuos denominados patológicos sean incinerados, por ese motivo se incremento el
número de hornos para el tratamiento de los residuos, sin embargo han sido muchos equipos que han
cerrado por no cumplir con los parámetros establecidos en la Norma Oficial Mexicana NOM-098-
SEMARNAT-2002, Protección ambiental-Incineración de residuos, especificaciones de operación y
límites de emisión de contaminantes. Actualmente la incineración es uno de los procesos de tratamiento
más observados por las asociaciones no Gubernamentales por ese motivo se concluye que las empresas
que actualmente cuentan con esta autorización son capaces de cumplir con los parámetros establecidos
en la NOM-098.

10
PRÁCTICA #3 USO, CALIBRACIÓN Y MANEJO DEL POTENCIÓMETRO
OBJETIVO:
Que el alumno conozca el instrumento de medición, principios y fundamento para aplicarlo en las
características ácido base de las soluciones.
INTRODUCCIÓN:
El potenciómetro, es un sensor utilizado en el método electroquímico como equipo de medición, en la
cuantificación del potencial Hidrógeno (pH) de cualquier disolución.
La determinación del pH, consiste en medir el potencial que se desarrolla a través de una fina
membrana de vidrio que separa dos soluciones, con diferente concentración de protones. En
consecuencia, se conoce la sensibilidad y la selectividad de las membranas de vidrio ante el pH.
Un electrodo para medir el pH consiste en un par de electrodos, uno de calomel (mercurio, cloruro de
mercurio) y otro de vidrio, sumergidos en la disolución en la que queremos encontrar el pH. El soporte
del electrodo es de vidrio común y no es conductor, mientras que el bulbo sensible se encuentra
formado por un vidrio polarizable (vidrio sensible de pH). Se llena el bulbo con la solución de HCl 0.1N
saturado con AgCl. El voltaje en el interior del tubo es constante (pH 7) de manera que la diferencia de
potencial solo depende del pH del medio externo. El alambre que se sumerge al interior (normalmente
Ag/AgCl) permite conducir este potencial hasta un amplificador.
El pH metro es relativamente inmune a las interferencias de color, turbidez, material coloidal, cloro
libre, substancias oxidantes y/o reductoras. La medición es afectada solo cuando la superficie de la
membrana está sucia con grasa o material orgánico insoluble en agua, que le impide hacer contacto con
la muestra, por lo que se recomienda la limpieza escrupulosa de los electrodos. Los electrodos deben ser
enjuagados con agua destilada entre muestras (el uso adecuado de la pizeta es de vital importancia),
posteriormente deberá secarse el electrodo preferentemente con papel klenex o papel sanitario de
textura suave pero que no deje pelusa, nunca debe secarse con tela porque podría cargase
electrostáticamente.
Como los electrodos de vidrio de pH cuantifican la concentración de H+ relativa a sus referencias, deben
ser calibrados periódicamente para asegurar la precisión, es por eso que se utilizan soluciones
específicas llamadas buffers de calibración (disoluciones reguladoras de pH conocido). Estas soluciones
son: solución buffer (fosfato) de pH 7 color amarillo, solución buffer (biftalato) de pH 4 color rojo,
solución buffer (borato) de pH 10 color azul.

11
El pHmetro digital portátil Conductronic pH 10, modelo 1024, electrodo y batería cuadrada. Debe
mantenerse siempre humedecido.
Para su calibración a un punto:
1. Conecte el electrodo al instrumento y encienda la tecla ON.
2. Introduzca el electrodo en la solución patrón de pH 7 y permita la lectura que la lectura se estabilice (aproximadamente 30 seg).
3. Ajuste la perilla de compensación de temperatura temp oC a la temperatura de la solución patrón (buffer).
4. Ajuste la perilla de calibración calíbrate, hasta que el medidor indique el valor pH 7.0 de la solución patrón (buffer).
5. Retire el electrodo de la solución patrón (buffer) y enjuáguelo muy bien con agua destilada (pizeta), pero no seque el electrodo.
6. Ajuste la perilla de compensación de temperatura temp. oC a la temperatura a la temperatura de la solución a medir.
7. Introduzca el electrodo en la solución a medir y lea el valor pH del medidor. Si el valor de la solución no está entre ± 3 unidades de pH de la solución patrón (7.0 pH), se necesita hacer una calibración a dos puntos (con dos buffers de diferente pH, 7 y 4, o 7 y 10 dependiendo de las soluciones que vaya a manejar).
8. Después de cada medición, retire el electrodo y enjuáguelo muy bien con agua destilada (pizeta)
Calibración a dos puntos
1. Siga los pasos de calibración a un punto hasta el paso 5
2. Sumerja el electrodo en la segunda solución patrón de pH 4.00 o pH 10.00. La temperatura de esta solución patrón debe ser idéntica a la de la primera
3. Ajuste el control de pendiente slope, hasta que el medidor indique el valor del pH de la segunda solución patrón.
4. Retire el electrodo, enjuáguelo con agua destilada y proceda a efectuar las mediciones. No olvide enjuagar el electrodo muy bien con agua destilada (pizeta) después de cada medición.
Precauciones
El pHmetro debe siempre mantenerse en su estuche si no se está utilizando, con respecto al electrodo
deberá mantenerse en la solución buffer de pH 4.0, misma que se encuentra en el pequeño recipiente
en el que descansa el electrodo.

12
PRÁCTICA #4 TITULACIÓN ACIDO-BASE
OBJETIVO
Observar una reacción acido-base fuerte utilizando un indicador de pH y comprobar el pH de la solución
mediante el uso del potenciómetro.
INTRODUCCION
Es una técnica o método de análisis cuantitativo muy usada, que permite conocer la concentración
desconocida de una disolución de una sustancia que pueda actuar como ácido o base, neutralizándolo
con una base o ácido de concentración conocida.1 Es un tipo de valoración basada en una reacción
ácido-base o reacción de neutralización entre el analito (la sustancia cuya concentración queremos
conocer) y la sustancia valorante.
Un indicador de pH es una sustancia que permite medir el pH de un medio. Habitualmente, se utilizan
como indicador sustancias químicas que cambian su color al cambiar el pH de la disolución. El cambio de
color se debe a un cambio estructural inducido por la protonación o desprotonación de la especie. Los
indicadores Ácido-base tienen un intervalo de viraje de unas dos unidades de pH, en la que cambian la
disolución en la que se encuentran de un color a otro, o de una disolución incolora, a una coloreada.
Los más conocidos son el naranja de metilo, que vira en el intervalo de pH 3,1 - 4,4, de color rojo a
naranja, y la fenolftaleína, que vira desde un pH 8 hasta un pH 10, transformando disoluciones incoloras
en disoluciones con colores rosados / violetas.
Los indicadores de pH tienen una constante de protonación, K, que informa sobre el desplazamiento de
la reacción de protonación de la forma básica del indicador.
Se dice que el cambio de color de un indicador es apreciable cuando la concentración de la forma ácida
o de la forma básica es superior o igual a 10 veces la concentración de la forma básica o la forma ácida
respectivamente.
MATERIALES
2 buretas 50 mL 2 Matraz erlenmeyer 150 ml HCl 0.1N NaOH 0.1N Fenolftaleina
METODOLOGIA
1. Verter NaOH y HCl en buretas graduadas.
2. Tomar 20 mL de HCl y colocarlo en un matraz erlenmeyer.

13
3. Medir el pH de la solucion un potenciómetro
4. Agregar 4 gotas de fenolftaleína y mezclar
5. Agregar gota por gota NaOH a los 20 mL de HCl siempre mezclando
6. Observar cambios de coloración.
7. Una vez obtenido el cambio de coloración del indicador de pH medir una vez más el pH de la solución ahora titulada.
RESULTADOS
Volumen de NaOH utilizado durante la titulación de HCl_____________
Concentración de NaOH y de HCl en la solución final ______________
Establecer la relación entre el indicador de pH y el pH obtenido con el potenciómetro.

14
PRACTICA # 5 REACCIONES DE IDENTIFICACION DE AMINOACIDOS
OBJETIVO
Al término de esta practica el alumno sera capaz de diferenciar a los aminoácidos utilizando técnicas
colorimétricas mas comunes para identificarlos, así cómo, relacionar la estructura de los aminoácidos
con su reactividad y sus funciones en el organismo.
PREREQUISITOS
1.- Conocer las caracteristicas diferenciales de la estructura de los aminoácidos.
2.- Identificar las diferencias y semejanzas de los aminoácidos con otros monómeros (por ejemplo :
carbohidratos y lípidos).
3.- Identificar las propiedades químicas en base a su polaridad.
INTRODUCCION
Los aminoácidos son los monómeros que dan la estructura de los polímeros conocidos como proteínas o
polipéptidos, 20 tipos de aminoácidos, son los principales componentes de las proteínas, los cuales
poseen las siguientes caracteristicas estructurales:
Contienen un carbón asímetrico (con excepción de la glicina) llamado carbono alfa. Al cual se le unen
cuatro radicales que son:
- un grupo carboxilo (COO-)
- un grupo amino (+NH3)
- un átomo de hidrógeno (H)
- un radical de composición variable que permite diferenciar a los aminoácidos (R). Llamado cadena
lateral.
Debido a las caracteristicas fisicoquímicas de la cadena lateral, es posible relacionar su posición, dentro
de la estructura proteíca ya que si es de caracteristicas no polares (hidrofóbico), al contacto con el agua
tenderá a esconderse; o en su defecto, si es de caracteristicas polares (hidrofílico), tenderá a
interaccionar con el agua. Desde luego, para saber cual es la estructura de la cadena lateral debemos de
conocer cual es el pH de la solución ya que dependiendo del pH la cadena lateral estará protonada (H+) o

15
desprotonada, este parámetro se puede conocer utilizando los pKa de cada aminoácido y la ecuación de
Henderson-Hasselbach lo cual permite determinar cual es la estructura predominante a un pH dado.
Los cambios de carga observados en el aminoácido mostrado, dependen del pH en que se encuentre el
aminoácido, ya que como se observa en la figura anterior, bajo condiciones ácidas el aminoácido está
totalmente protonado y en condiciones alcalinas está totalmente desprotonado, esto se debe a que los
aminoácidos son ácidos débiles y por lo tanto tienen una constante de disociación por cada grupo o
radical capaz de comportarse como ácido o base débil. Esto permite conocer el punto isoelétrico de un
aminoácido, el cual se define como el pH en el cual la carga neta es cero.
Los aminoácidos que son utilizados para la síntesis de proteínas son 20, los cuales se les ha identificado
como aminoácidos naturales y los no naturales son los que aún cuando siendo aminoácidos, no forman
parte estructural de las proteínas (no están codificados en el código genético).
El cuerpo humano es incapaz de sintetizar los 20 aminoácidos requeridos para la formación de todas las
proteínas y por lo cual partiendo de esta base, los aminoácidos se dividen en : esenciales; porque es
necesario que el individuo ingiera en la dieta y no esenciales; los cuales son producidos por el propio
organismo y por lo tanto no es crítico si el individuo no los consume en su dieta. Cabe hacer notar, que
los 20 aminoácidos son importantes para el buen desarrollo del organismo.
MATERIAL.-
1.-15 tubos de ensaye 5.-pipetas de 5.0 ml 2.-gradilla 6.-pipeta de 10.0 ml 3.-pipetas de 1.0 ml 7.-Bureta 4.-pipetas Pasteur
METODOLOGIA
Nota.- A menos que se especifique, todos los volúmenes a añadir están dados en mililítros (ml)
Experimento No. 1
Prueba de sullivan
REACTIVOS BLANCO PATRON PROBLEMA
AGUA 0.5 ----------- --------
CISTEINA ---------- 0.5 ---------
NH4OH 2 gotas 2 gotas 2 gotas
NaCN 0.5 0.5 0.5
NITROPRUSIATO DE 2 gotas 2 gotas 2 gotas

16
SODIO
Sol. Problema ------------ ---------- 0.5
Experimento No. 2
Reacción de identificación de fenoles para-sustituidos (tirosina)
REACTIVOS BLANCO PATRON PROBLEMA
AGUA 1.0 --- ---
TIROSINA --- 1.0 ---
PROBLEMA --- --- 1.0
NITROSONAFTOL 2 gotas 2 gotas 2 gotas
Mezclar y calentar en baño a ebullición durante 5 minutos.
ACIDO NITRICO 2 gotas 2 gotas 2 gotas
La aparición de un color rosa violáceo es positiva.
Experimento No. 3
Reacción de identificación de aminoácidos con ninhidrina
La ninhidrina (hidrato de tricetohidrindeno) es un agente oxidante que reacciona con todos los alfa
aminoácidos dando un color purpura. La prolina e hidroxiprolina da un color amarillo.
REACTIVOS BLANCO PATRON PROBLEMA
AMINOACIDO --- 1.0 ---
PROBLEMA --- --- 1.0
NINHIDRINA 1.0 1.0 1.0
AGUA 1.0 --- 1.0
Mezclar y calentar en baño de agua hirviendo durante 10 min.
La aparición de un color azul violáceo es positiva con excepción de la prolina y la hidroxiprolina.
RESULTADOS
En la siguiente tabla indicar si los resultados del problema fueron positivos o negativos.
Tubo EXP. No. 1 EXP. No. 2 EXP. No. 3

17
1
2
3
4
Por lo cual concluímos que el o los aminoácidos presentes en la muestra son:______________
CUESTIONARIO
1.- Mencione 5 diferencias fundamentales tanto químicas como bioquímicas que permiten diferenciar a
los aminoácidos de los carbohidratos y de los lípidos.
2.- Existe un enorme grupo de aminoácidos no naturales, mencione 10 ejemplos e indique su
importancia bioquímica.
3.- Cómo se calcula el punto isoeléctrico de un aminoácido?
4.- Cual es el fundamento de la reacción de Pauly.
5.- Que importancia médica tiene la identificación de los aminoácidos.?
6.- Cuál es el mecanismo de identificación de aminoácidos al utilizar ninhidrina.
7.- Cuál es la función del cianuro de sodio en la reacción del nitroprusiato?

18
PRACTICA # 6 SEPARACIÓN EN CAPA FINA DE UNA MEZCLA DE AMINOÁCIDOS.
OBJETIVO
Al termino de esta practica el alumno será capaz de identificar por lo menos dos técnicas de separación
de aminoacidos, así como relacionar las caracteríasticas fisicoquímicas de la estructura en función de la
técnica de separación. Además describirá las ventajas y desventajas de la tecnica utilizada,
comparándola con otras.
PRE-REQUISITOS
1. Cuáles son las características diferenciales de los aminoácidos naturales en base a su estructura
2. Cuál es el fundamento de la reacción de ninhidrina con los aminoácidos.
INTRODUCCIÓN
Se llama cromatografía al método que se sigue para la resolución o separación de mezclas de
componentes en zonas concentradas o en diferentes fases de aquellas en que se encontraban
inicialmente. Esto es independiente, de las fuerzas que provocan el paso de dichas sustancias de una
fase a otra.
Debido a lo anterior, la cromatografía presenta una gran versatilidad, eficiencia y conveniencia que
permite su aplicación en un gran número de técnicas en el laboratorio.
- Las técnicas de cromatografía permiten separar mezclas de:
Gases
Iones simples
Moléculas orgánicas e inorgánicas (aminoácidos, azúcares, vitaminas, drogas, lípidos, esteroides)
Macromoléculas (proteínas, polisacáridos, ácidos nucleícos)
Partículas (componentes subcelulares, bacterias)
- Las técnicas cromatografícas permiten conocer:
El peso molecular de la sustancia de interés
Identificar una o mas sustancias de una mezcla
La concentración de las moléculas presentes
- La cromatografía basa su separación en:
La diferencia en velocidad de migración de las sustancias presentes en la mezcla a través de los poros del soporte llamado adsorbente.
La migración se efectua por el flujo de un gas o un liquido llamado fase
móvil, el cual viaja a través del adsorbente llamado fase estacionaria
- La separación de los componentes se lleva a cabo por una combinación de procesos, los cuales son:
Partición del soluto (componente a separar) entre la mezcla de solventes (fase móvil)

19
Adsorción de los solutos con la fase estacionaria (interacciones superficiales)
Atracción electrostática de solutos (iones) con carga opuesta.
- La clasificación de las técnicas de cromatografía en base al método en
Cromatografía en columna
Cromatografía en capa fina
Cromatografía en papel
Cromatografía de gases
En el caso de la cromatografía en capa fina, su separación se basa en una mezcla se procesos: adsorción
y partición. Ésta técnica de separación de aminoácidos es utilizada para determinar fallas metabólicas o
renales (aminoaciduria), y la muestra que se utiliza es un concentrado de orina.
MATERIAL
-Equipo para cromatografía en capa fina - Horno a 80 °C -Pipetas pasteur -Papel
METODOLOGÍA
NOTA. No tocar con las manos la placa cromatográfica porque se contamina por la presencia de
aminoácidos en las manos.
1. Hacer unas marcas con lápiz sobre la placa a un centímetro de la base de la placa. No raye a todo lo largo de la placa, únicamente marque en las orillas
2. A esta altura colocar una gota de la solución de aminoácidos conocidos separados a un centímetro cada uno y colocar una muestra problema.
3. Dejar secar al aire, sin soplar.
4. Añadir fase móvil a la cámara cromatografica: 40 mL butanol: 10 mL ácido acético: 50 mL agua y cerrar
5. Colocar las placas de cromatografía en la cámara cuidando de no empalmar una con otra. Las placas deben quedar con la muestra de aminoácidos en la parte inferior para hacer una cromatografía ascendente.
6. Cerrar la cámara y dejar que ascienda el solvente por la placa hasta un poco antes que salga alrededor de 1 hr. Sacar las placas y marcar con lápiz hasta donde avanzó el solvente.
7. Sacar las placas y marcar con lápiz hasta donde avanzó el solvente.
8. Secar por 5 minutos en horno a 80°C
9. Rociar con la solución de ninhidrina toda la placa y calentar en el horno por 5 min.
10. Medir los frentes del solvente y del soluto para cada mancha y calcular sus respectivos relación de frentes con la siguiente fórmula:
Rf= Frente de soluto
Frente de solvente

20
11. Compare el Rf de los aminoácidos conocidos con los obtenidos para el problema para identificar el o los aminoácidos presentes en la mezcla.
RESULTADOS
Problema No.___________
La muestra contiene:_____________
CUESTIONARIO
1. Si el Rf para la L-glicina es de 0.45 cual seria el rf para la D-glicina. Explique su respuesta.
2. En base a los resultados obtenidos por todo el grupo, indique de manera ascendente como se separarían los siguientes aminoácidos, utilizando la misma mezcla de solventes.
Ala, Met, Phe, Tyr, His, Glu, Gln.
3. En caso de que se estuviera analizando una mezcla de aminoácidos presentes en una muestra de orina, que componentes de ésta además de los aminoácidos se teñirán con la ninhidrina.
4. Por qué los Rf son específicos de la técnica de separación de los solventes utilizados.

21
PRACTICA # 7 REACCIONES DE IDENTIFICACION DE PROTEINAS
OBJETIVO:
Al término de esta practica, el alumno deberá ser capaz de describir algunas de las técnicas de detección
cualitativa de proteínas en base a los componentes de su estructura.
PR-ERREQUISITOS:
1.- Definición de Péptido, Polipéptido y Proteína. 2.- Clasificación de las Proteínas. 3.- Niveles de complejidad estructural de las Proteínas.
INTRODUCCION: La palabra Proteína proviene del griego preeminente, que significa primero; fué inicialmente propuesta
por Berzeluis en 1838 para referirse a unas serie de compuestos orgánicos nitrogenados con cierta
complejidad que se encuentran altamente distribuidos en la naturaleza y son la base para actividades
tanto estructurales y funcionales de todo organismo vivo.
Las proteínas estas compuestas por aminoácidos naturales ordenados en una secuencia discreta y
unidos entre sí por enlaces peptídicos que siempre involucran los radicales amino y carboxilo de cada
aminoácido.
La variabilidad de las proteínas se debe a una serie de aspectos tales como: complejidad estructural,
residuos aminoacídicos que la forman, propiedades químicas y físicas y funciones específicas de cada
proteína en el organismo que la contiene.
Se concluye, por lo tanto, que el análisis químico de las proteínas en base a sus propiedades es muy
amplio y su aplicación en el campo médico, clínico y de investigación es muy importante.
Basándose en los análisis físicos y químicos de ellas, se han planteado cuatro niveles estructurales de
conformación, se han postulado las posible formas estructurales , su naturaleza electrostática , su
capacidad de solubilidad así como sus funciones dentro del organismo.
MATERIAL.-
1.- 12 tubos de 20 x 150 mm
2.- 3 pipetas de 1.0 ml.
3.- 1 pipeta de 10.0 ml.
4.- 1 Gradilla
5.- Sol. de albúmina 0.1%
6.- Sol. de peptona 0.1%
7.- Sol Gelatina 0.1 %
8.- Sol. Tirosina 0.1 %
9.- Sol. NaOH 10.0 %
10.- Sol. Sulfato de Cu 0.5 %
11.- Ac. nítrico concentrado
12.- Hidróxido de amonio

22
METODOLOGIA.-
Experimento No. 1
Reacción de Biuret: Existen diversas técnicas de detección y cuantificación de proteínas algunas más complicadas que otras
y algunas más confiables que otras. Una de las reacciones más utilizadas es la de Biuret; en esta
reacción, el sulfato de cobre alcalino reacciona con compuestos que contienen dos o más enlaces
peptídicos, ya que el radical carbamilo que se forma en el enlace peptídico es muy afín a los iones de
cobre. Se le da el nombre de Biuret porque este es un compuesto que da una reacción típicamente
positiva con el sulfato de cobre alcalino. La formación del color violeta es directamente proporcional al
número de enlaces peptídicos que contenga la proteína y no es sensible a péptidos.
Preparar una serie de tubos de acuerdo al siguiente cuadro:
Tubo 1 2 3 4 5
Agua destilada 1 mL -- -- -- --
Albúmina -- 1 mL -- -- --
Peptona -- -- 1 mL -- --
Gelatina -- -- -- 1 mL --
Tirosina -- -- -- -- 1 mL
NaOH 2 mL 2 mL 2 mL 2 mL 2 mL
CuSO4 0.5 mL 0.5 mL 0.5 mL 0.5 mL 0.5 mL
Agitar y dejar reposar los tubos durante 15 min. Observar la coloración y su intensidad.
Reportar los resultados de la siguiente forma: (++) = violeta intenso, (+) = Ligeramente coloreado, (-) =
no formación de color.
Experimento No. 2
Reacción Xantoproteíca: Esta reacción afecta la integridad estructural de los residuos aminoacídicos de las proteínas. En este
caso, Los aminoácidos que contienen un núcleo aromático, forman nitroderivados de color amarillo
cuando se calientan con ácido Nítrico concentrado. Al añadir una solución alcalina se forman sales de
estos derivados que son de color naranja.
Preparar una serie de tubos de acuerdo al siguiente cuadro:
Tubo 1 2 3 4 5

23
Agua destilada 1 mL -- -- -- --
Albúmina -- 1 mL -- -- --
Peptona -- -- 1 mL -- --
Gelatina -- -- -- 1 mL --
Tirosina -- -- -- -- 1 mL
HNO3 1 mL 1 mL 1 mL 1 mL 1 mL
Mezclar y calentar a baño de agua a ebullición durante 5 min. y enfriar a temperatura ambiente durante
10 min.
Agregar por estratificación y SIN MEZCLAR 0.5 mL de Hidróxido de Amonio a todos los tubos.
La formación de un anillo color naranja en la interfase es resultado positivo. Reportar con un signo positivo (+) los tubos donde se haya formado el color naranja
RESULTADOS
Experimento Testigo Algumina Peptona Gelatina Tirosina
Reacción de Biuret
Reacción Xantoproteica
DISCUSION:
1.- Describir y explicar mediante un esquema la reacción entre el Reactivo de Biuret y los enlaces peptídicos. 2.- Correlacionar los pesos moleculares y los componentes aminoacídicos de la albúmina, peptona, gelatina y tirosina. 3.- Explique porque cada proteína se comporta de diferente manera en las reacciones de Biuret y Xantoproteica. 4.- Analice las dos reacciones y diga si son confiables para una determinación a nivel clínico. CUESTIONARIO:
1.- Mencione y describa dos reacciones que sean análogas a la xantoproteica. 2.- Mencione dos reacciones que sirvan para cuantificar proteínas 3.- Describa el metodo de Sanger y mencione que papel juega en la identificación de proteínas. 4.- Defina el término Secuenciación protéica. 5.- Mencione tres reacciones de identificación que se utilicen en la secuenciación de proteínas. 6.- Analice detalladamente una técnica que se utilice para secuenciar a las proteínas.

24
7.- Describa la tecnica de Millon y especifique el tipo de proteínas que puede identificar.

25
PRACTICA # 8 DETERMINACIÓN DE LAS PROPIEDADES QUÍMICAS DE LAS PROTEÍNAS
OBJETIVOS
Al término de la práctica, el alumno será capaz de describir el efecto de los solventes sobre las
propiedades electroquímicas de las proteínas de diferente PM de acuerdo a las siguientes variables.
a) Solubilidad de las proteínas en diferentes soluciones de sales.
b) Propiedades electroquímicas de las proteínas.
c) Efecto del acido acético sobre el punto isoeléctrico de una proteína.
PRERREQUISITOS INVESTIGAR LOS SIGUIENTES CONCEPTOS
A) Salting In y Salting Out. B) Agentes precipitantes de proteínas. C) Propiedades fisicoquímicas del agua. D) Ecuación de Henderson/Hasselbach.
INTRODUCCIÓN
Las proteína son macromoléculas compuestas por unidades de alfa –aminoácidos que se unen entre sí
mediante enlaces peptídico y que alcanzan un peso molecular igual o superior a 5000 D.
El enlace peptídico, característico de estos compuestos, resulta de la reacción del grupo carboxilo de un
aminoácido con el grupo amino del otro aminoácido, lo cual da lugar a un enlace covalente de tipo
carbamida y que tiene algunas características peculiares como:
a) El enlace C-N tiene cierto carácter de doble enlace, por lo cual le confiere rigidez a la molécula.
b) El oxigeno y el nitrógeno quedan en posición trans.
c) Todos los elementos que componen el enlace se encuentran ubicados en el mismo plano.
d) La rotación de la molécula formada queda restringida a los carbonos alfa.
El gran número de aminoácidos que componen la molécula proteica determina que su estructura sea
extraordinariamente compleja, y que para poder estudiarla nos veamos precisados a dividirla
artificialmente en los llamados <<niveles de organización de la estructura proteica >>, de los cuales se
describen habitualmente 4, aunque algunos autores llegan a describir 5 o más.
Por supuesto que toda organización estructural que implique determinado orden requiere de la
presencia de una fuerza estabilizadora, que en el caso de las proteínas estaría constituida por
diferentes tipos de enlaces o interacciones físicas y químicas que se enuncian en el cuadro siguiente:
Nivel de organización Enlaces que lo mantienen

26
Primario Enlace peptídico.
Secundario Puentes de hidrogeno entre los elementos del enlace peptídico.
Terciario Puentes de hidrógeno entre las cadenas laterales de los aminoácidos,
interacción hidrofóbica, interacciónes ectrostatica, puentes disulfuro, enlace
éster.
Cuaternario Los mismos que para el nivel terciario más las fuerzas de Van der Walls.
La complejidad estructural de las proteínas se manifiesta desde el punto de vista funcional en una gran
diversidad de funciones biológicas.
En las proteinas existen aminoacidos hidrofobicos e hidrofilicos. Luego del doblamiento en solución
acuosa, los aminoácidos hidrofobicos usualemente se encuentran en areas protegidas, mientras que los
aminoácidos hidrofilicos interactúan con las moléculas del solvente, permitiendo la formación de puentes
de hidrógeno con las moléculas de agua del medio. Si hay suficiente superficie hidrofilida, la proteína
puede ser disuelta en agua.
Cuando la concentración de sales aumenta, algunas de las moleculas de agua son atraidas por los ions
salinos, lo cual disminuye la cantidad de moleculas de agua disponibles para su interaccion con la
protein. Como resultado de esto, las interacciones proteína-proteina se hacen mas fuertes que las
interacciones proteína-solvente por lo que las proteínas coagulan formando interacciones hidrofobicas
entre ellas, proceso al cual se le conoce como Salting-out.
MÉTODOS
Experimento No. 1
Efecto de la solubilidad de las proteínas por la adición de sales.
Fundamento químico.
La adición de una sal a una solución proteica modifica la constante dieléctrica del solvente, permitiendo
una mayor solubilidad de la proteína. Sin embargo, cuando se añade exceso de una sal se neutralizan las
cargas de la proteína y esta tiende a precipitar. Este mismo efecto ocurre al añadir sales metálicas
cargadas electropositivamente. Existe variante entre diferentes sales metálicas de la misma valencia, a
la misma concentración, y de masa atómica diferente.
También se demostrara, la diferencia existente tratándose con diferentes tipos de proteínas.
Prepara tres series de tubos como se indica.
REACTIVO Albúmina 1% (1ml) Gelatina 1% (1ml) Peptona 1% (1ml)
Testigo H2O 0.5 ml 0.5 ml 0.5 ml
ZnSO4 2 % 0.5 ml 0.5 ml 0.5 ml
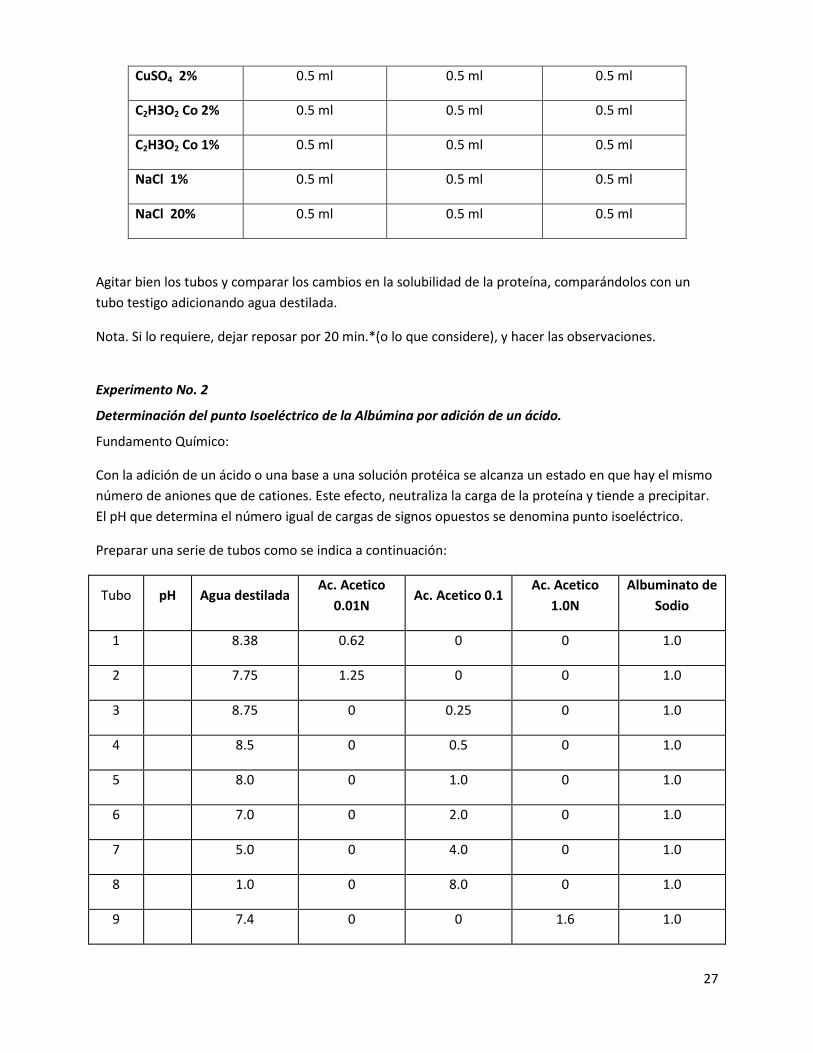
27
CuSO4 2% 0.5 ml 0.5 ml 0.5 ml
C2H3O2 Co 2% 0.5 ml 0.5 ml 0.5 ml
C2H3O2 Co 1% 0.5 ml 0.5 ml 0.5 ml
NaCl 1% 0.5 ml 0.5 ml 0.5 ml
NaCl 20% 0.5 ml 0.5 ml 0.5 ml
Agitar bien los tubos y comparar los cambios en la solubilidad de la proteína, comparándolos con un
tubo testigo adicionando agua destilada.
Nota. Si lo requiere, dejar reposar por 20 min.*(o lo que considere), y hacer las observaciones.
Experimento No. 2
Determinación del punto Isoeléctrico de la Albúmina por adición de un ácido.
Fundamento Químico:
Con la adición de un ácido o una base a una solución protéica se alcanza un estado en que hay el mismo
número de aniones que de cationes. Este efecto, neutraliza la carga de la proteína y tiende a precipitar.
El pH que determina el número igual de cargas de signos opuestos se denomina punto isoeléctrico.
Preparar una serie de tubos como se indica a continuación:
Tubo pH Agua destilada Ac. Acetico
0.01N Ac. Acetico 0.1
Ac. Acetico
1.0N
Albuminato de
Sodio
1 8.38 0.62 0 0 1.0
2 7.75 1.25 0 0 1.0
3 8.75 0 0.25 0 1.0
4 8.5 0 0.5 0 1.0
5 8.0 0 1.0 0 1.0
6 7.0 0 2.0 0 1.0
7 5.0 0 4.0 0 1.0
8 1.0 0 8.0 0 1.0
9 7.4 0 0 1.6 1.0

28
10 5.8 0 0 3.2 1.0
Mezclar bien y observar los cambios que presente la solubilidad de la caseína en cada tubo.
Calcular el pH de cada tubo utilizando la ecuación de Hendersosn-Hasselbach.
RESULTADOS:
Experimento No.1
Efecto en la solubilidad de las proteínas por adición de sales.
Sal utilizada Solubilidad de la
Albumina
Solubilidad de la
Peptona
Solubilidad de la
Gelatina
Acetato de Cobalto
Cloruro de sodio 1.0%
Cloruro de Sodio 20.0%
Sulfato de Zinc 5.0%
Sulfato de zinc al 20 %
Experimento No. 2
Determinación del punto Isoeléctrico de la caseína por adición de un ácido.
Tubo pH Solubilidad
1
2
3
4
5
6
7
8

29
9
10
Cuál es el punto isoeléctrico de la proteína ?
DISCUSION:
1.-Cual es el efecto de la adición de un sal metálica a una solución proteínica.
2.-De que factores depende la solubilidad de una proteína en el agua.
3.-Explique en términos fisicoquímicos el efecto que provoca la adición de un ácido o una base fuerte.
4.-Explique como afecta un cambio en la constante dieléctrica del agua a la solubilidad de una proteína.
5.-Qué es el Salting in y que tipo de compuestos pueden promoverlo.
6.-Qué es el Salting out y que tipo de compuestos pueden promoverlo.
7.-Qué relación existe entre el punto isolelectrico y la solubilidad de una proteína.
8.-Qué es la ecuación de Henderson-Hasselbach y en que casos está indicado utilizarse.
9.-Diga si es posible tener dos proteínas con puntos isoeléctricos muy alejados solubles en un mismo
solvente. Porqué?
CUESTIONARIO:
1.- Las proteínas Bence Jones precipitan a una temperatura mayor de 60°C. Las Crioglobulinas precipitan
a una temperatura de 4°C. La albúmina de huevo precipita a una temperatura mayor de 90°C y tambien
precipita a un pH menor de 4.5. Explique el comportamiento de cada una de estas proteínas tomando
en cuenta los siguientes parámetros: Fuerza iónica del solvente, interacciones electrostáticas soluto-
solvente, agentes desacoplantes de la interacción soluto-solvente.
2.- Mencione que propiedades fisicoquímicas de la albúmina le permiten interaccionar y transportar
cualquier compuesto incluyendo hidrofóbicos a través de un medio acuoso.
3.- El punto Isoeléctrico de la Gelatina es de 4.7 y el de la colágena es de 8.4. Se sabe que la colágena es
una proteína fibrilar precursora de la gelatina. Explique porque hay una diferencia tan grande entre los
puntos isoeléctricos de ambas.
4.- El Ac. tricloroacético, el Ac. sulfosalicílico y el Ac. fosfotúngstico tienen la propiedad de precipitar
proteínas. Cuál es el mecanismo fisicoquímico que ocurre en esta precipitación.
5.- Especifique cual es el efecto del plomo sobre las propiedades electroquímicas de las proteínas de la
sangre.
6.- Determine el efecto del litio cuando se encuentra en altas concentraciones sobre las proteínas de las
células nerviosas.

30
PRÁCTICA #. 9 FOTOCOLORIMETRIA
OBJETIVO
a) Determinar los espectros de absorción de 2 sustancias coloridas.
b) Determinar la longitud de onda óptima para la cuantificación de cada sustancia (máxima
absorbancia).
c) Desarrollar curvas de calibración para cada una de las sustancias.
d) Comprobar si se sigue la Ley de Lambert-Beer para cada una de las sustancias y cuales serán los
límites.
PRERREQUISITOS
1.- Definición de los siguientes términos:
a) Longitud de onda
b) espectro de absorción
c) absorbancia
d) transmitancia.
2.- Longitud de onda del espectro visible, ultravioleta e infrarrojo.
INTRODUCCION
La utilización de fotocolorímetros y espectrofotómetros se basa en la determinación de un parámetro,
esto es, medir la luz absorbida por una sustancia que se encuentra en el seno de un líquido y esto se
relaciona directamente con la concentración de la sustancia en el líquido. Estos instrumentos que sirven
para medir la intensidad de la luz transmitida o absorbida por una solución, consisten prácticamente de
una fuente de luz, filtros (fotocolorímetros) o un monocromador (espectrofotómetro ), una hendidura
de salida de luz seleccionada, un receptáculo para la muestra ( celdilla ), fotocelda y galvanómetro (
figura 1 ).
Lámpara ------> Filtro ------> Celdilla ---> Fotocelda ------> Galvanómetro
Figura No. 1.- Diagrama de flujo de luz en un fotocolorímetro o espectrofotómetro.
La diferencia entre un fotocolorímetro y un espectrofotómetro es a nivel del separador de las diferentes
longitudes de onda que componen la luz, siendo más sensible el monocromador del espectrofotómetro,
ya que los límites son más estrechos, que los filtros del fotocolorímetro en cuál los límites están más
separados.
Las medidas fotocolorimétricas están basadas en 2 leyes:

31
a) La Ley de Lambert
b) La Ley de Beer
La Ley de Lambert nos indica que la proporción de luz incidente absorbida por un medio es
independiente de su intensidad, pero no del espesor de la capa líquida ni de las características del
medio.
La Ley de Beer nos indica que la intensidad de la luz que pasa a través de una solución a una
concentración c y un grosor l es la misma cuando la concentración es 2c y el grosor l/2 o cuando la
concentración es c/2 y el grosor 2l, de tal manera que la absorción de luz es proporcional al número de
moléculas del material absorbente a través de las cuáles pasa la luz. Así, si la sustancia absorbente está
disuelta en un solvente transparente, la absorción de la solución es proporcional a la concentración
molar.(1)
Matemáticamente la ley de Lambert-Beer se expresa como sigue:
Los materiales que cumplen con la ley de Lambert-Beer, o sea donde hay proporcionalidad entre la
Absorbancia (As) y la concentración dentro de ciertos límites de concentración, pueden ser estudiados
por este método, trabajando una curva de calibración que permita la interpolación dentro del margen
de cumplimiento de la ley de Lambert-Beer o utilizando un estándar.
MATERIAL
Solución de H2SO4 0.5 M.
Solución de KMnO4 0.0004 M.
Solución de K2Cr2O7 0.0016 M.
11 Tubos de ensaye de 15x160 mm
2 Pipetas de 10 ml.
2 Pipetas de 5 ml.
2 Celdillas para Fotocolorímetro.
1 Gradilla.
Fotocolorímetro
METODOLOGIA.-
Experimento No. 1
DETERMINACION DE LOS ESPECTROS DE ABSORCION.
1. Determinar los espectros de absorción de cada una de las soluciones tipo, leyendo las absorbancias (As) a cada una de las longitudes de onda (nanómetros, nm) del Fotocolorímetro; calibrando a cero con la solución de H2SO4 0.05 M.
2. Seleccionar las longitudes de onda de máxima absorbancia para cada una de las soluciones tipo.
3. En el reporte incluir una tabla con las absorbancias de cada sustancia tipo y la longitud de onda utilizada. Con estos datos construir una gráfica para cada espectro de absorción, colocando en las ordenadas las absorbancias y en las abcisas las longitudes de onda. Debe indicar cual es la longitud

32
de onda de máxima absorbancia para cada una de las soluciones tipo. Comparar los espectros de absorción de cada solución tipo y discutir porque tienen diferentes espectros de absorción.
Experimento No. 2
PREPARACION DE CURVA DE CALIBRACION PARA UNA SOLUCION TIPO.
1. Preparar 4 diluciones (1:2, 1:4, 1:8, 1:16) para una de las soluciones tipo, utilizando la solución de H2SO4 como diluyente.
2. Leer la absorbancia de cada dilución a la longitud de onda seleccionada en el inciso anterior.
3. En el reporte incluir las curvas de calibración para la solucion tipo, graficando absorbancia vs. concentración molar. Definir la relación que existe entre la concentración y la absorbancia obtenida.
Experimento No. 3 DETERMINACION DE LAS CONCENTRACIONES MOLARES DE LOS COMPONENTES EN UNA MEZCLA.
1.- Preparar las siguientes mezclas:
Tubo K2CRsO7 KMnO4 H2SO4 H2O
1 5.0 1.0 0.2 3.8
2 2.5 2.5 0.2 3.8
3 1.0 5.0 0.2 3.8
2.- Medir las absorbancias de cada mezcla a las longitudes de onda seleccionadas en el experimento No.
1. Utilice la misma solución tipo del experimento 2. Determinar la concentración del analito deseado.
3.- Medir un tubo problema a la misma longitud de onda para determinación de la concentración
desconocida, en base a una lectura patrón de la solución tipo.
RESULTADOS:
EXPERIMENTO No. 1
1.- Llenar la siguiente tabla:
Longitud de onda
Abs K2CRsO7
Abs KMnO4
415
445
460
490

33
520
535
550
580
610
640
En el reporte incluir una tabla con las absorbancias de cada sustancia tipo y la longitud de onda
utilizada. Con estos datos construir una gráfica para cada espectro de absorción, colocando en las
ordenadas las absorbancias y en las abcisas las longitudes de onda. Debe indicar cual es la longitud de
onda de máxima absorbancia para cada una de las soluciones tipo.
EXPERIMENTO No. 2
Longitud de onda seleccionada para el K2Cr2O7 = __________ =As1
Longitud de onda seleccionada para el KMnO4 = __________ =As2
K2Cr2O7 ó KMnO4
Dilución Absorbancia Concentración
1 : 2
1 : 4
1 : 8
1 : 16
En el reporte incluir la curva de calibración para la solucion tipo, graficando absorbancia vs.
concentración molar.
EXPERIMENTO No. 3
Llenar la siguiente tabla:
Determina la concentración de K2Cr2O7 ó KMnO4 en las mezclas y en el tubo problema utilizando un
tubo patrón de solución tipo.
Mezcla Absorbancia Conc. Practica

34
1
2
3
Problema

35
PRÁCTICA #. 10 ANALISIS ENZIMATICO CUALITATIVO
OBJETIVO:
Al término de esta práctica, el alumno será capaz de definir el efecto fisicoquímico que generan las
enzimas sobre diferentes sustratos. Indicando además, las propiedades químicas que permiten que un
enzima, catalize la modificación de un sustrato
PREREQUISITOS:
1. Definición de enzima, substrato, apoenzima, holoenzima, proenzima, zimógeno, coenzima, cofactor.
2. Determine lo que significa: especifidad enzimática y alosterismo. 3. Defina el efecto de una hidrolasa, oxidoreductasa, transferasa, liasa, ligasa e isomerasa.
INTRODUCCION:
Todos los organismos vivos pueden obtener y gastar la energía muy rápidamente, debido a la presencia
de catalizadores biológicos llamadas enzimas. Al igual que los catalizadores inorgánicos; las enzimas
modifican la velocidad de una reacción química sin afectar el equilibrio final y sólo se requieren
pequeñas cantidades para efectuar la transformación de un gran número de moléculas de sustrato. Sin
embargo, a diferencia de la mayoría de los catalizadores inorgánicos las enzimas son bastante
específicas ya que catalizan un número comparativamente pequeño de reacciones; en algunos casos,
tan solo una reacción , así mismo las enzimas funcionan solo bajo condiciones muy definidas de pH,
temperatura, concentración de sustrato, coenzimas etc.
Estas condiciones se ilustran en algunos de los experimentos que se tienen determinados en este tema.
Las enzimas se designan y se clasifican de acuerdo con el tipo de reacción que catalizan; los 6 grupos
principales de enzimas son: Oxidoreductasas, Transferasas, Hidrolasas, Liasas, Isomerasas, Ligasas o
Sintetasas.
MATERIAL:
1.- 6 tubos de ensaye 2.- 3 pipetas de 1.0 ml 3.- 2 pipetas de 5.0 ml 4.- 2 pipetas Pasteur 5.- 2 gradillas 6.- Tapones de algodon
METODOLOGIA.-
Experimento No. 1
EFECTO DEL CALOR SOBRE LA ACTIVIDAD DE LAS ENZIMAS. "ACTIVIDAD DE LA UREASA"
Tubo 1 2 3
Semilla de Vegetal pizca pizca pizca

36
Agua 0 5.0 5.0
Mezclar para tratar de homogenizar el polvo de sandia y enseguida coloque un tapón de algodón a los
tubos 1 y 2 y colóquelos en baño a ebullición durante 20 minutos. (cuidar que no se acabe el agua del
baño,)
Agua 5.0 0 0
Azul de Bromotimol 2 Gotas 2 Gotas 2 Gotas
Añadir ácido débil (GOTAS) en caso de que la solución tenga un color azul.
Urea 5.0 5.0 5.0
Mezclar y tomar el tiempo que transcurre hasta alcanzar el vire de color amarillo a azul en cada uno de
los tubos.
Experimento No. 2
DETERMINACION DE LA ACTIVIDAD DE RENINA.
Tubo 1 2 3
Leche 3.0 3.0 3.0
Oxalato de Amonio 0 0.5 0.5
Solución renina 3 Gotas 3 Gotas 3 Gotas
Mezclar e incubar a 37°C durante 10 minutos y anotar los cambios observados en cada uno de los tubos.
Calentar el tubo No. 2 a ebullición y enfriar.
Cloruro de Calcio 0 10 Gotas 10 Gotas
Mezclar y comparar los cambios observados en los tubos No 2 y 3 con el 1.
CUESTIONARIO:
1.- Deacuerdo a la clasificación enzimática a que clase pertenecen las enzimas utilizadas (renina y
ureasa) y porqué?
2.- Mencione 3 bacterias que contengan la enzima ureasa:
3.- Qué grupos químicos participan en el sitio activo de las enzimas ?
4.- Las enzimas cambian la constante de equilibrio de las reacciones que catalizan ? si, no ; porqué?
5.- Porqué algunas proteínas actúan como catalizadores (enzimas) de reacciones con alta especificidad,
mientras que otras proteínas que también contienen aminoácidos en su estructura no lo hacen?

37
PRÁCTICA #. 11 CINETICA ENZIMATICA 1: EFECTO DE LA CONCENTRACION DEL SUSTRATO
SOBRE LA ACTIVIDAD ENZIMATICA
OBJETIVOS:
El alumno observará el comportamiento de la actividad enzimática de acuerdo a los parámetros de
concentración tanto del sustrato determinando los valores de:
a) Vmax ( velocidad máxima de la reacción ). b) 1/2 Vmax c) Km ( constante de Michaelis ). d) [S] óptima ( concentración óptima de sustrato ).
PRE-RREQUISITOS:
1. Describir el mecanismo de acción de las enzimas en las reacciones químicas. 2. Cuál es la relación que existe entre la velocidad de una reacción catalizada y la concentración de
enzima. 3. Definir la relación entre la concentración de sustrato y la velocidad de la reacción enzimática. 4. Interpretar la cinética de Michaelis-Menten y la de Lineweaver-Burk.
INTRODUCCION:
La cinética química es el estudio de la velocidad de cambio que existe entre el estado inicial de
reactantes y productos y el estado final. La velocidad es expresada en términos de cambios de
concentración de sustrato o producto por unidad de tiempo, esto se puede seguir, determinando la
desaparición de reactantes o aparición de productos en función del tiempo. Es particularmente
importante hacer notar que constantemente hay cambios en la velocidad de reacción conforme procede
la reacción hasta llegar al equilibrio, una vez alcanzado éste, los cambios de velocidad son iguales a cero.
El determinar la velocidad de una reacción no revela nada acerca de la estequiometría de los reactantes
y productos ni del mecanismo de la reacción.
En la mayoría de las reacciones enzimáticas al seguir su comportamiento cinético se pueden obtener
generalmente varios órdenes de reacción; al examinar la curva de velocidad respecto a la concentración
de sustrato, se encuentran tres regiones distintas donde la velocidad responde en forma característica a
los aumentos de concentración de sustrato:
a) A muy bajas concentraciones de sustrato el comportamiento de velocidad con respecto a la
concentración de sustrato es esencialmente lineal, esto es, la velocidad es directamente
proporcional a la concentración de sustrato y en esta región se presenta la cinética de primer
orden.
b) En la etapa final de la curva observamos que la velocidad es esencialmente independiente de la
concentración de sustrato, aquí la cinética es de orden cero.

38
c) En la parte intermedia de la curva se presenta una mezcla de órdenes de reacción, ya que se
observan cambios en la velocidad de reacción pero no siguen una constante de
proporcionalidad.
Este tipo de estudio cinético fue desarrollado por primera vez por Michaelis-Menten y formularon la
siguiente ecuación:
V =Vmax [S] Km + [S]
donde las constantes Vmáx y Km son características para cada enzima bajo ciertas condiciones.
En forma práctica es difícil determinar el valor de Km de una curva de saturación de sustrato, pero se
puede recurrir a la gráfica de Lineweaver-Burk que utiliza los recíprocos de velocidad y sustrato, ésta
tiene la ventaja de dar valores de Km y Vmáx estadísticamente más significativos con tan sólo 6 a 8
datos experimentales.
En los experimentos a realizar se van a observar los efectos de la concentración de sustrato y
concentración de enzima sobre la actividad enzimática en una reacción.
MATERIAL
1.- 8 Tubos de ensaye 2.- 1 Gradilla 3.- 3 Pipetas de 1.0 ml 4.- 2 Pipetas de 5.0 ml 5.- 1 Baño de agua a 37°C 6.- 1 Baño de agua en ebullición
METODOGIA:
La invertasa es una enzima hidrolítica que actúa sobre sacarosa ( un disacárido ) liberando glucosa y
fructosa. La actividad de la enzima se detiene al añadir una solución alcalina y el poder reductor de los
productos se mide haciéndolos reaccionar con el reactivo de 3,5-dinitrosalicilato.
Tubo
1 2 3 4 5 6
Invertasa 1.0 1.0 1.0 1.0 1.0 1.0
Amortiguador 0.5 0.5 0.5 0.5 0.5 0.5
Sacarosa 0.02 M 1.0 0 0 0 0 0
Sacarosa 0.03 M 0 1.0 0 0 0 0
Sacarosa 0.05 M 0 0 1.0 0 0 0
Sacarosa 0.10 M 0 0 0 1.0 0 0
Sacarosa 0.30 0 0 0 0 1.0 0
Agua 0 0 0 0 0 1.0
Mezclar e incubar a 35° C durante 20 min.
Agregar 1 mL de Dinitrosalicilato a todos los tubos.

39
Mezclar y calentar en baño de agua a ebullición durante 5 min. Enfriar y leer a 540 nm ajustando a cero
con el tubo No. 6
RESULTADOS:
1.- Graficar los valores de As vs. [S] 2.- Graficar los valores de 1/As vs. 1/[S] 3.- Determinar los valores de:
Gráfica de M-M Gráfica de L-B
Vmáx
Km
½ Vmáx
[ S ] óptimo
CUESTIONARIO:
1.- La velocidad inicial de una reacción enzimática es dependiente exclusivamente de la cantidad de
sustrato presente? si, no por qué?
2.- Cual es la diferencia entre los cambios de energía libre que existen entre un proceso catalizado por
una enzima y el mismo proceso no catalizado.
3.- Por qué a concentraciones de sustrato mucho mayores que el valor de la Km la velocidad de la
reacción es independiente de la concentración de sustrato?
4.- La Km de una enzima varía con la concentración de enzima? si, no, por qué?

40
PRÁCTICA #. 12 CINETICA ENZIMATICA 2: EFECTO DEL INHIBIDOR SOBRE LA ACTIVIDAD ENZIMÁTICA
OBJETIVO
El alumno observará el comportamiento de la inhibición de la actividad enzimática de acuerdo a los
parámetros de concentración tanto del sustrato como de inhibidor determinando los valores de:
a) Vmax ( velocidad máxima de la reacción ). b) 1/2 Vmax c) Km ( constante de Michaelis ).
INTRODUCCIÓN
Los inhibidores enzimáticos son moléculas que se unen a enzimas y disminuyen su actividad. Puesto que
el bloqueo de una enzima puede matar a un organismo patógeno o corregir un desequilibrio
metabólico, muchos medicamentos actúan como inhibidores enzimáticos. También son usados como
herbicidas y pesticidas. Sin embargo, no todas las moléculas que se unen a las enzimas son inhibidores;
los activadores enzimáticos se unen a las enzimas e incrementan su actividad.
La unión de un inhibidor puede impedir la entrada del sustrato al sitio activo de la enzima y/u
obstaculizar que la enzima catalice su reacción correspondiente. La unión del inhibidor puede ser
reversible o irreversible. Normalmente, los inhibidores irreversibles reaccionan con la enzima de forma
covalente y modifican su estructura química a nivel de residuos esenciales de los aminoácidos
necesarios para la actividad enzimática. En cambio, los inhibidores reversibles se unen a la enzima de
forma no covalente, dando lugar a diferentes tipos de inhibiciones, dependiendo de si el inhibidor se
une a la enzima, al complejo enzima-sustrato o a ambos.
Los inhibidores reversibles se unen a las enzimas mediante interacciones no covalentes tales como los
puentes de hidrógeno, las interacciones hidrofóbicas y los enlaces iónicos. Los enlaces débiles múltiples
entre el inhibidor y el sitio activo se combinan para producir una unión fuerte y específica. Al contrario
de lo que ocurre con el sustrato y los inhibidores irreversibles, los inhibidores reversibles generalmente
no experimentan reacciones químicas cuando se unen a la enzima y pueden ser eliminados fácilmente
por dilución o por diálisis.
Existen tres tipos de inhibidores reversibles. Se clasifican en base al efecto producido por la variación de
la concentración del sustrato de la enzima en el inhibidor.
En la inhibición competitiva, el sustrato y el inhibidor no se pueden unir a la misma enzima al mismo
tiempo, como se muestra en la figura de la derecha. Esto generalmente ocurre cuando el inhibidor tiene
afinidad por el sitio activo de una enzima en el que también se une el sustrato; el sustrato y el inhibidor
compite para el acceso al sitio activo de la enzima. Este tipo de inhibición se puede superar con
concentraciones suficientemente altas del sustrato, es decir, dejando fuera de competición al inhibidor.
Los inhibidores competitivos son a menudo similares en estructura al sustrato verdadero (ver ejemplos
expuestos más abajo).

41
En la inhibición mixta, el inhibidor se puede unir a la enzima al mismo tiempo que el sustrato. Sin
embargo, la unión del inhibidor afecta la unión del sustrato, y viceversa. Este tipo de inhibición se puede
reducir, pero no superar al aumentar las concentraciones del sustrato. Aunque es posible que los
inhibidores de tipo mixto se unan en el sitio activo, este tipo de inhibición resulta generalmente de un
efecto alostérico donde el inhibidor se une a otro sitio que no es el sitio activo de la enzima. La unión del
inhibidor con el sitio alostérico cambia la conformación (es decir, la estructura terciaria o la forma
tridimensional) de la enzima de modo que la afinidad del sustrato por el sitio activo se reduce.
La inhibición no competitiva es una forma de inhibición mixta donde la unión del inhibidor con la enzima
reduce su actividad pero no afecta la unión con el sustrato. Como resultado, el grado de inhibición
depende solamente de la concentración de inhibidor
MATERIALES
METODOLOGÍA

42
PRÁCTICA #. 13 CINETICA ENZIMATICA 3: EFECTO DEL PH Y TEMPERATURA
OBJETIVOS:
El alumno analizará el efecto del pH y de la temperatura sobre la actividad enzimática en una reacción. Basado en:
a. Discutirá el efecto sobre el comportamiento catalítico de una enzima al variar el pH de la mezcla de reacción.
b. Discutirá los cambios que se presentan en la cinética enzimática al variar la temperatura de reacción.
c. Determinará las condiciones óptimas de reacción en función de estos dos factores ( pH, temperatura).
PRE-RREQUISITOS:
1. Describir el efecto del pH sobre la estabilidad y actividad de una enzima. 2. Describir el efecto de la temperatura sobre la estabilidad y actividad de una enzima. 3. Describir el sitio activo y sitio de enlace de una enzima. 4. Describir los agentes desnaturalizantes y su mecanismo de acción sobre una enzima. 5. Explicar los conceptos de pH y temperatura óptimos de una enzima en una reacción catalizada.
INTRODUCCION:
Es lógico pensar que el pH de una solución influye sobre la velocidad de una reacción catalizada por
enzimas. El sitio activo y el sitio de enlace de una enzima generalmente están compuestos por grupos
ionizables que deben presentarse con una forma iónica apropiada o específica, de tal manera, que se
pueda mantener la conformación adecuada de estos sitios en la enzima y pueda llevarse a cabo la unión
del sustrato y su transformación hacia productos.
Igualmente puede existir un sustrato con grupos ionizables y debe contener una forma iónica específica
para que pueda unirse a la enzima y posteriormente se realize la catálisis. Los efectos de pH sobre la
estabilidad de una enzima se debe tomar en cuenta en cualquier estudio de efecto de pH sobre la
catálisis de sustrato.
Se observa que el pH óptimo de la reacción enzima -sustrato es a 6.8 según la gráfica de la curva A, sin
embargo, no nos indica porque declina la velocidad por arriba y abajo de este valor.
En la curva B se observa que la preincubación de la enzima entre pH de 5.0 - 8.0 no tiene efecto sobre la
actividad medida a pH 6.8. Por lo tanto, la interpretación de las 2 gráficas nos indica que la caída en la
actividad entre 6.8 - 8.0 y entre 6.8 - 5.0 es el resultado de la formación de una estructura iónica
impropia de la enzima y/o sustrato o los dos.
Cuando la enzima es preincubada a pH entre 5.0 - 8.0, se mantiene la actividad completa de la enzima a
6.8. Por arriba o abajo de este valor de pH resulta en inactivación de la enzima.
La estabilidad de la enzima depende de varios factores, por ejemplo, temperatura, fuerza iónica,
naturaleza del amortiguador, concentración de iones metálicos, concentración de sustratos o cofactores
y concentración de enzima.

43
El último factor es muy importante ya que existen algunas enzimas que a bajas concentaciones se
disocian en monómeros y que son menos estables.
Otro de los factores que afectan la velocidad de una reacción es la temperatura, de tal suerte que un
incremento de temperatura proporciona mayor energía cinética a las moléculas reactantes, dando como
resultado choques más productivos por unidad de tiempo. La actividad catalítica de las enzimas resulta
de una estructura altamente ordenada. Si la molécula absorbe mucha energía, la estructura se rompe y
la enzima se desnaturaliza y por lo tanto pierde su actividad catalítica.
Si se grafica velocidad vs. temperatura se obtiene una curva en forma de campana, en el cual el pico se
conoce como temperatura óptima. La estabilidad a la temperatura, de una enzima depende del pH,
fuerza iónica del medio, así como de la presencia o ausencia de metales. La mayoría de los sustratos
ejercen un efecto de protección a la enzima contra la desnaturalización por temperatura. Las enzimas de
bajo PM compuestas de cadenas sencillas y que poseen enlaces disulfuro son más estables al calor que
las de alto PM. Las enzimas obtenidas de un extracto crudo libre de células son más estables al calor por
la alta concentración de otras proteínas (efecto protector).
MATERIAL :
1.- 14 tubos de ensaye 2.- 1 gradilla 3.- Celdas de fotocolorímetro 4.- Fotocolorímetro 5.- Baños de agua de temperatura controlada 6.- 3 Pipetas de 1.0 ml 7.- 2 pipetas de 5.0 ml METODOLOGIA :
Efecto del pH
Tubo 1 2 3 4 5 6 7
8
Sacarosa 0.3 M 1.0 1.0 1.0 1.0 1.0 1.0 1.0 1.0
Amortiguador pH = 2.5 0.5 0 0 0 0 0 0 0
Amortiguador pH = 2.5 0 0.5 0 0 0 0 0 0
Amortiguador pH = 3.5 0 0 0.5 0 0 0 0 0
Amortiguador pH = 4.5 0 0 0 0.5 0 0 0 0
Amortiguador pH = 5.5 0 0 0 0 0.5 0 0 0
Amortiguador pH = 6.5 0 0 0 0 0 0.5 0 0
Amortiguador pH = 7.5 0 0 0 0 0 0 0.5 0
Agua 0 0 0 0 0 0 0 1.0
Mezclar e incubar a 37° C durante 2 min.
Agregar 0.5 mL de Enzima (Invertasa) a todos los tubos.
Mezclar e incubar a 25°C durante 20 min.
Agregar 1 mL de Dinitrosalicilato a todos los tubos.

44
Mezclar y calentar en baño de agua a ebullición durante 5 min. Enfriar y leer a 540 nm ajustando a cero
con el tubo No. 8
Efecto de la Temperatura
Tubo 1 2 3 4 5 6 7
Sacarosa 0.3 M 1.0 1.0 1.0 1.0 1.0 1.0 1.0
Amortiguaor pH = 4.7 0.5 0.5 0.5 0.5 0.5 0.5 0.5
Invertasa 1.0 1.0 1.0 1.0 1.0 1.0 0
Agua 0 0 0 0 0 0 1.0
Temperatura °C 4 26 37 45 60 100 25
Mezclar e incubar 5 min a la temperatura indicada.
Agregar 1 mL de Dinitrosalicilato a todos los tubos.
Mezclar y calentar en baño de agua a ebullición durante 5 min. Enfriar y leer a 540 nm ajustando a cero
con el tubo No. 7
RESULTADOS :
1.- Grafique los resultados obtenidos de velocidad vs pH.
2.- Grafique los resultados obtenidos de velocidad vs temperatura.
3.- Indique cuales son los valores de pH y temperatura óptimos obtenidos.
CUESTIONARIO :
1.- La pepsina presenta su máxima actividad a un pH » 1. Mencione cuáles podrían ser los grupos
funcionales presentes en el sitio activo y sobre cuales residuos de aminoácidos rompe los enlaces
peptídicos.
2.- Si la concentración de sustrato en una reacción enzimática es igual a un 1/3 de Km. Cuál será la
velocidad inicial de la reacción.
3.- De acuerdo a la clasificación de las enzimas a que clase pertenece una enzima que participa en la
conversión de un azúcar de tipo aldosa a un azúcar de tipo cetosa.
4.- En esta práctica se ha observado como influye el pH en la actividad de invertasa, a una concentración
de sacarosa dada. La estaquiosa es un tetrasacárido y es un sustrato para esta enzima y se ha observado
que es hidrolizado a una velocidad igual al 5% de la sacarosa, bajo las mismas condiciones de
concentración y pH. Cómo explicaría esta velocidad máxima tan baja en relación al pH de la reacción. ( a-
Gal-a-Gal-a-Glu-ß-Fru ).
5.- Cuando las soluciones enzimáticas son calentadas, hay una pérdida progresiva de la actividad
catalítica con el tiempo. Así, una solución de hexocinasa incubada a 45°C durante 12 minutos pierde el
50% de su actividad; pero cuando la hexocinasa se incuba a 45°C, en presencia de altas concentraciones
de glucosa durante el mismo tiempo, solamente se pierde un 3% de su actividad. Explique el porque de

45
la variación desnaturalizante de la hexocinasa por efecto de la temperatura en ausencia y presencia del
sustrato.

46
PRÁCTICA #. 14 CUANTIFICACION DE COLESTEROL SERICO
OBJETIVO:
Determinar la concentración de colesterol sérico y analizar las implicaciones clínicas de niveles
anormales de colesterol.
PRE-REQUISITOS.
1. Estructura, función y tipos de colesterol 2. Relación metabólica entre el colesterol y las lipoproteínas 3. Análisis de la digestión del colesterol esterificada y no esterificado 4. Relación de las Lipoproteínas plasmáticas con la Aterogénesis.
INTRODUCCION.
El colesterol es uno de los lípidos que se encuentran en mayor concentración en la sangre, se puede
afirmar que la fracción lipídica del colesterol, es la segunda en cuanto a concentración y ésta se localiza
principalmente formando parte de las lipoproteínas. Químicamente el colesterol es un compuesto
cíclico con estructura básica de perhidrofenantreno con un total de 27 carbones, un radical OH en el
carbono 3 y un doble enlace entre los carbonos 5 y 6.
El colesterol existe en dos formas: como colesterol libre en muy pequeñas cantidades y como colesterol
esterificado mediante la unión de un ácido graso no saturado de cadena media al carbono 3. Este tipo
de colesterol es el más frecuente tanto en sangre como en tejidos.
La mayor parte de colesterol que existe en el organismo es sintetizado en el hígado, ésto implica que el
colesterol exógeno tenga menor importancia metabólica. La síntesis de colesterol endógeno es activada
en primera instancia ante el excedente de Acetil-S-CoA que haya en el hepatocito, ya que éste funciona
como metabolito precursor y en segunda instancia ante la presencia de efectores hipercolesterolémicos
que aumentan la síntesis de colesterol o bién disminuyen el catabolismo del colesterol ya formado. El
colesterol exógeno se reduce en el intestino por medio de enzimas bacterianas para dar lugar al ß
colestanol y así entra a la sangre en pequeñas concentraciones, otra parte del colesterol exógeno se
convierte en un producto de excresión llamado coprostano que se libera con las heces.
El colesterol endógeno con frecuencia se cataboliza en el hígado y en otros órganos. El 7-
dihidrocolesterol se obtiene por oxidación del colesterol; éste compuesto predomina en la piel y en
presencia de radiación ultravioleta se convierte en colecalciferol que es una de las formas de vitamina D.
Otros productos del catabolismo del colesterol son los ácidos biliares que en general se deben a una
carboxilación de la cadena lateral del colesterol y ésta reacción sucede en el hígado. Entre los ácidos
biliares más comunes está el ácido cólico, el ácido desoxicólico, ácido quenodesoxicólico y el ácido
litocólico. Por último, otros metabolitos son la progesterona y los esteroides corticales, así como
hormonas androgénicas y estrogénicas.

47
METODOLOGIA
Fundamento químico : Esta técnica se basa en utilizar los derivados polienos del colesterol como
elementos cromóforos( que dan reacción de color). En presencia de ácido sulfúrico concentrado y
anhídrido acético ( reactivo de colesterol) se producen diferentes tonalidades de color verde cuya
tonalidad es proporcional a la cantidad de colesterol presente en la muestra. Los polienos son
estructuras cíclicas que resultan de la oxidación del colesterol.
La cuantificación de HDL colesterol se determina por métodos de precipitación en los cuales la
formación de complejos insolubles se debe a interacciones entre los grupos cargados negativamente de
los polianiones y los grupos positivos de las subunidades proteicas de las lipoproteínas
TECNICA: preparar una serie de tubos de acuerdo al siguiente cuadro.
REACTIVOS Colesterol HDL Patron o Estandar Blanco
SUERO 0.05 ml. -- -- --
SOL. PATRON -- -- 0.05 ml. --
AGUA -- -- -- 0.05 mL
SOBRENADANTE* -- 0.05 ml -- --
REACTIVO DE COLESTEROL
2.0 ml. 2.O ml 2.0 ml. 2.0 ml
*NOTA: Para adquirir sobrenadante: A 0.5 ml de suero, agregar 0.05 ml de Reactivo precipitante de
HDL-COLESTEROL, mezclar, reposar 5 minutos y centrifugar 5 min a 3000 rpm.
Mezclar y dejar que se lleve a cabo la reacción durante 20 minutos a temperatura ambiente y añadir
en forma directa sobre la superficie del líquido 0.20 ml. de Acido sulfúrico concentrado.
Colocar los tubos por separado inmediatamente despues de la adición del ácido sulfúrico en un baño de
agua a temperatura ambiente y agitar suavemente. Se deben mantener los tubos en baño de agua
mínimo 5.0 minutos.
Leer la absorbancia del problema y el patrón ajustando a cero con el tubo blanco a una longitud de onda
de 640nm.
CALCULOS:
[Colesterol Total] = Abs. problema / Abs. del Patrón X [ conc. patrón mg/dl ]
[ HDL-COLESTEROL ] = Abs problema / abs patrón x [ conc. Patrón]

48
PRÁCTICA #. 15 EXTRACCIÓN DE GLUCOGENO Y REACCIONES DE IDENTIFICACION DE CARBOHIDRATOS
OBJETIVO.
a) Desarrollar una técnica de extracción de glucógeno a partir de tejido hepático de conejo o rata y
determinar la calidad del glucógeno obtenido por medio de reacciones cualitativas de
caracterización.
b) Conocer las técnicas de análisis cualitativo que permiten identificar carbohidratos y su
utilización como herramienta de trabajo en ciertos experimentos.
PREREQUISITOS.
1. Identificar la composición del glucógeno y sus diferencias estructurales y funcionales con el almidón.
2. Diferenciar los efectos sobre el metabolismo del glucógeno de las siguientes hormonas : Insulina, Glucagón y Adrenalina
3. Identificar las características de por lo menos tres enfermedades por almacenamiento de glucógeno.
4. Definir el término carbohidrato. 5. Diferenciar los tipos de carbohidratos y mencionar ejemplos. 6. Identificar las propiedades químicas de los carbohidratos.
INTRODUCCIÓN. Los carbohidratos son derivados aldehídicos o cetónicos de alcoholes superiores polivalentes ( más de
un OH). se clasifican en base al número de moléculas que los componen. Monosacáridos (azúcares
simples) no se pueden hidrolizar en moléculas más sencillas, pueden subdividirse en triosas, tetrosas,
pentosas, hexosas etc. según el número de átomos de carbono que tengan. Las siguientes reacciónes
permiten identificar de manera cualitativa la presencia de cierto tipo de azucares en una muestra
problema y se utilizan en la práctica tanto clínica como vegetal.
Muchas de las pruebas que se estudiarán en el laboratorio requieren tiempo para que aparezca un
precipitado o se desarrolle un color. La mayaría de las reacciones son muy sensibles, por lo que el uso de
reactivos en cantidades mayores pueden conducir a interpretaciones erróneas.
El glucógeno es un polisacárido ramificado constituído de moléculas de alfa d-glucosa unidas por enlaces
glucosídicos. Es el único polisacárido que se produce y se almacena en el humano, siendo el hígado el
principal promotor de la glucogénesis que es la vía metabólica de dicho polisacárido.
Todas las células del organismo son capaces de producir glucógeno y la capacidad de síntesis de cada
célula estará dada por la cantidad de enzima glucógeno sintetasa que presenta. El hepatocito, por lo
tanto, es la célula que mayor cantidad de glucógeno sintetasa presenta.
Desde el punto de vista energético, éste polisacárido es importante ya que por ser una fuente
almacenadora de glucosa es factible que se pueda utilizar cuando el organismo así lo requiera mediante
una vía de hidrólisis del glucógeno que se conoce como glucogenolisis.

49
Tanto en la glucogénesis como la glucogenolisis son vías reguladas hormonal y alostéricamente. La
insulina promueve la actividad de la glucogénesis y en forma directa sobre la glucógeno sintetasa y la
adrenalina activa en forma indirecta a la glucogenólisis e inhibe de la misma forma a la glucogenesis esto
es , induce la formación de AMP-Cíclico para que este a su vez active a la glucogeno-fosforilasa e inhiba
a la glucógeno sintetasa.
El mecanismo de regulación es complejo y algunas veces puede fallar a tal grado que altera el
almacenamiento de glucógeno y generar estados patológicos llamados glucogénosis, algunos de los
cuales pueden llegar a ser fatales tales como las enferedades de Von-Gierke, Andersen, de Pompe, Mc.
Ardle, Hers, etc. Estas enfermedades se caracterizan por presentarse debido a una deficiencia congénita
de algunas de las enzimas arriba mencionadas y otras relacionadas con ellas.
MATERIAL.
1. 6 Tubos de ensaye 15 X 150 mm 2. 1 Mortero con pistilo 3. 2 Pipetas de 1.0 ml. 4. 1 Pipeta de 5.0 ml. 5. 1 vaso de precipitados 6. Gradilla. 7. Vidrio de reloj
8. Balanza granataria 9. Probeta de 100 ml. 10.- Baño de agua a ebullición 11 Tubos de ensaye 12.- 2 Pipetas de 1.0 ml 13.- Pipeta de 5.0 ml 14.- Gradilla
METODOLOGIA:
Extracción de Glucógeno
1. Pesar 5g de hígado y cortarlo en trozos pequeños (utilizar el vidrio de reloj). 2. Colocar unos trozos en un mortero frío al cual se le añade TCA al 10 % en proporción de 1.0 ml
por cada gramo de tejido y una pizca de arena lavada. 3. Centrifugar 5 min. a 2000 r.p.m. 4. Recuperar el sobrenadante en un tubo de ensaye grande y medir el volúmen. 5. Añadir 2 volúmenes de alcohol al 95 % por volúmen de sobrenadante agitando lentamente
hasta que el glucógeno flocule. 6. Centrifugar a 2 000 r.p.m. 5 Min. 7. Desechar el sobrenadante y resuspender el precipitado en 4.0 ml. de agua destilada. 8. Utilizado la suspensión de glucógeno como muestra problema, desarrolle las técnicas de Fehling
,Lugol y Molish al mismo tiempo que los carbohidratos proveídos por el profesor.
EXPERIMENTO # 1
REACCION DE FEHLING (IDENTIFICACION DE COMPUESTOS REDUCTORES ) Esta reacción nos identifica carbohidratos con capacidad reductora, se basa en que algunos azúcares en
un medio fuertemente alcalino y en presencia de oxígeno o de diversos agentes oxidantes ej. Cu, Ag dan
las reacciones de reducción que dependen de la existencia de un grupo carbonilo libre como en la
glucosa, galactosa, fructosa, maltosa y lactosa. Si la prueba se efectúa con una solución que contenga
cloroformo como conservador, puede obtenerse un resultado positivo, sin que exista azúcar, las sales de
amonio también interfieren en la reacción. Si la solución problema es ácida debe neutralizarse antes de
efectuar la prueba.

50
Fundamento: si calentamos una solución de Cu(OH)2 en un medio alcalino formamos oxido cúprico
(negro) CuO y en presencia de sustancias reductoras se precipita como oxido cuproso Cu2O de color
café rojizo pardo .
Solucion A contiene sulfato de cobre .
Solucion B contiene tartrato de sodio y potasio e Hidroxido de potasio .
METODO
MUESTRA (carbohidratos señalados y glucógeno) 1.0 ml. SOL. DE FEHLING A 0.5 ml SOL. DE FEHLING B 0.5 ml. MEZCLAR Y CALENTAR A BANO DE EBULLICION 5 MIN. COLOR ROJO ES POSITIVO.
EXPERIMENTO # 2
REACCION DE MOLISH-UDRANSKY. Esta prueba es positiva con aldehidos y algunos ácidos como el fórmico, oxálico, láctico, cítrico, etc. Esta
reacción es muy sensible , dicha sensibilidad para la glucosa es de hasta 0.001% y para sacarosa de
0.0001%.
METODO
MUESTRA (carbohidratos señalados y glucógeno) 1.0 ml Reactivo de Molish 3 GOTAS
Mezclar y añadir 0.5 ml de ácido sulfúrico concentrado ( resbalando por la pared del tubo ) NO
mezclar.
La aparición en la interfase de un anillo violáceo es indicativo de que la prueba resultó positiva.
EXPERIMENTO # 3
REACCION DE SELIWANOFF Está reacción permite diferenciar Aldosas de Cetosas. Anote el tiempo en el cual aparece el color en
cada uno de los tubos. Una reacción positiva está dada por la aparición de un color rojo. Es utilizada
para diferenciar Cetosas de Aldosas ( Reacción de Seliwanoff )
METODO
MUESTRA (carbohidratos señalados y glucógeno) 1.0 ml REACTIVO DE SELIWANOFF 5.0 ml
Mezclar y calentar en baño de ebullición durante 1 minuto, anote el resultado.
Las cetosas dan un color rojo y las aldosas dan la prueba débil y lentamente.
EXPERIMENTO # 4
REACCION DEL YODO O LUGOL

51
Esta reacción identifica los polisacáridos como Amilosa, Amilopectina, Glucogeno, Almidon formando
un complejo adsortivo entre el Iodo y las cadenas helicoidales del polisacárido (enlaces alfa 1,4
glucosídicos y enlaces alfa 1,6 glucosídicos de por lo menos 8 residuos de glucosas ) .
METODO.
MUESTRA (carbohidratos señalados y glucógeno)………… 1.0 ml. LUGOL ………… 3 GOTAS
Mezclar y si es necesario calentar 2 segundos en baño de ebullición.
La aparición de un color azul o violeta indica la presencia de un polisacarido.
RESULTADOS
FEHLING LUGOL MOLISH
MUESTRA
GLUCÓGENO
AGUA
CUESTIONARIO
1. Mencione las diferencias en las propiedades químicas de los monosacáridos con los aminoácidos
y los lípidos.
2. Mencione 3 funciones fundamentales de los carbohidratos en la célula, ejemplificando en cada
caso.
3. Mencione 2 homopolisacaridos y 3 heteropolisacaridos y su composición, indicando también los
tipos de enlaces que presentan.
4. Defina y ejemplifique con carbohidratos los siguientes conceptos: Isomería óptica, Anomerismo,
Epimerismo.
5. Explique el mecanismo de acción de los segundos mensajeros en la regulación hormonal y
enzimática del metabolismo del glucógeno.
6. Describa las implicaciones bioquímicas que se presentan en la enfermedad de Von Gierke
7. Describa dos eventos bajo los cuales se puede activar la vía de la glucogenolisis.

52
PRÁCTICA #. 16 EFECTO DE LAS COENZIMAS SOBRE LA ACTIVIDAD ENZIMATICA
OBJETIVO:
Determinar cualitativamente, mediante la observación de los grados de decoloración; las relaciones existentes entre la actividad de la deshidrogenasa láctica (LDH) y la presencia de nicotinamida adenin dinucleótido (NAD), en función de los siguientes aspectos: a) Determinar si la actividad de LDH depende de la presencia de NAD. b) Especificar la actividad de indicador redox del azul de metileno. c) Aportar pruebas experimentales que demuestren que la coenzima no se modifica estructuralmente
durante la actividad de la enzima.
PRERREQUISITOS:
1.- Definir los términos: cofactor, coenzima y metaloenzima. 2.- Mencionar las coenzimas que actúan en oxidorreducciones. 3.- Explicar como afectan las coenzimas a la cinética enzimática.
INTRODUCCION:
La mayoría de las manifestaciones metabólicas de un organismo, se relacionan con cambios de energía,
la cual se obtiene por la oxidación sistemática de los nutrientes y esto es debido a que en los procesos
de oxidorreducción hay transferencias de energía generadas por diferencias en el potencial
elecroquímico. Un sistema de potencial elevado, oxida al de más bajo potencial con la consiguiente
liberación de energía para las necesidades vitales.
En todas las reacciones de oxidorreducción, existe transferencia de electrones. La oxidación se define
como: la pérdida de electrones por parte de un compuesto, mientras que la reducción es la ganancia de
dichos electrones por otro compuesto. Por lo tanto, la oxidación y la reducción son fenómenos que se
realizan en forma simultánea.
La oxidación biológica implica transferencia de hidrógenos y electrones a través de una serie de sistemas
enzimáticos que actúan como intermediarios, para llegar al aceptor final que es el oxígeno. El proceso
oxidativo se inicia por la oxidación de una deshidrogenasa específica que no reacciona directamente con
el oxígeno, sino que requiere intermediarios como el NAD, las flavoproteínas y los citocrómos.
Piruvato Lactato

53
En el presente experimento, se pretende realizar un estudio cualitativo, donde se utilizará la
deshidrogenasa láctica de músculo de rata; que sólo es activa en estado de anaerobiosis y requiere NAD
como intermediario para completar su actividad, la cual se manifiesta en la siguiente reacción:
Con este experimento se pretende demostrar que: Si la LDH es una enzima que funciona normalmente,
en base a la concentración de NAD y anerobiosis promoviendo la reducción del piruvato en presencia de
NADH + H+; entonces, aumentando la concentración de NAD+ podemos revertir la reacción para oxidar
al lactato y generar NADH + H+ el cual reccionará con el azul de metileno y provocará su decoloración. Si
esta premisa se cumple se habrá demostrado que la concentración de la coenzima puede regular la
dirección de la actividad de una enzima reversible.
MATERIAL :
1. 4 tubos de ensayo de 20 x 150 mm. 2. 3 pipetas de 5.0 ml. 3. 1 pipeta de 2.0 ml. 4. 1 pipeta de 1.0 ml. 5. 2 vasos de precipitado de 150 ml. 6. 1 matraz erlenmeyer de 125 ml. 7. 1 mortero con pistilo. 8. 1 baño María ajustado a 37°C. 9. Arena lavada. (agente triturante). 10. NaCl al 0.9% (solución isotónica para suspender células) 11. KCN al 0.5% (inhibidor de las deshirogenasas mitocondriales). 12. Lactato de sodio al 1.0%.(substrato de la LDH). 13. Azul de metileno 0.002 M.(indicador redox que reacciona con NAD). 14. Aceite mineral. (compuesto que al sellar los tubos promueve la anaerobiosis).
METODOLOGIA :
Preparación de la enzima LDH:
1. Colocar 5 gr de músculo de res en un mortero previamente sumergido en hielo y añadir NaCl al
0.9% (10 ml. por gramo de tejido) y agregar un poco de arena. Triturar perfectamente. 2. Dejar reposar la mezcla en el mortero por 15 minutos. 3. Centrifugar a 3000 rpm por 15 minutos y recuperar el sobrenadante etiquetándolo como LDH.
Preparación de la coenzima NAD:
1. Obtener otros 3 gr. de tejido muscular. 2. Cortar el músculo en fragmentos más pequeños y ponerlo en agua a ebullición por 5 minutos en
un matraz Erlenmeyer de 125 ml. en una proporción de 8 ml. de agua por gramo de músculo. 3. Pasar la preparación a un mortero con un poco de arena y triturar perfectamente. 4. Centrifugar a 3000 rpm por 15 minutos. Recuperar el sobrenadante y etiquetarlo como NAD.
Preparación de la enzima DSC a partir de músculo cardiáco
1. Tomar de 2 a 3 g de músculo de corazón cortarlo en fragmentos pequeños. 2. Colocar el tejido en un mortero previamente sumergido en hielo y añadir NaCl al 0.9% en
una proporción de 10 ml. por gramo de músculo, agregar un poco de arena lavada y triturar perfectamente.

54
3. Dejar reposar la mezcla en el mortero por 15 min. 4. Centrifugar el homogeneizado a 3000 rpm por 15 min. Recuperar el sobrenadante y
etiquetarlo como DSC.
Detección de la actividad enzimática de LDH:
Preparar una serie de cuatro tubos de ensayo como se indica a continuación:
NOTA: se debe tener precaución al utilizar el KCN ya que es altamente tóxico. utilice bureta.
TUBO 1 2 3 4
KCN 0.5% 1.0 ml 1.0 ml 1.0 ml 1.0 ml
Lactato de Sodio al
1.0%
1.0 ml 1.0 ml --------- 1.0 ml
Azul de metileno
0.002 M
0.3 ml 0.3 ml 0.3 ml 0.3 ml
Agua destilada 1.0 ml ---------- 1.0 ml 1.0 ml
Coenzima ( NAD ) -------- 1.0 ml 1.0 ml 1.0 ml
Enzima ( LDH ) 1.0 ml 1.0 ml 1.0 ml ---------
Mezclar bien el contenido de cada tubo y agregar 1.0 ml. de aceite mineral, evitando mezclarlos.
Incubar en baño de agua a 37°C los tubos MANTENIENDOLOS INMOVILES y observar los grados de
decoloración de cada solución. Hacer estas observaciones en intervalos de 15 minutos durante 45
minutos.

55
RESULTADOS :
TUBO 1 2 3 4
15 min
30 min
45 min
DISCUSION:
1.- Relacione el efecto de cada reactivo con los resultados obtenidos.
2.- Determine si los resultados que obtiene son acordes con los esperados.
3.- Explique el fundamento químico de la reacción entre el azul de metileno y el NAD.
4.- Mencione los errores, si es el caso, en el planteamiento o en el manejo del experimento que
pudieran modificar los resultados.
CUESTIONARIO:
1.- Mencione las diferencias entre una enzima y un cofactor.
2.- Explique cual es el efecto del oxígeno en la actividad de las oxidasas.
3.- Mencione cinco coenzimas que actúen con transferasas.
4.- Explique la relación que existe entre algunas coenzimas y las vitaminas del complejo B.
5.- Describa la función del fosfato de piridoxal en la reacción que cataliza la enzima transaminasa
glutámico oxaloacética (GOT).

56
PRÁCTICA #. 17 EFECTO DE LOS INHIBIDORES SOBRE LA ACTIVIDAD ENZIMATICA
OBJETIVO:
Analizar el efecto inhibitorio de los compuestos químicos malonato de sodio y oxalato de sodio, sobre la
actividad enzimática de lactato deshidrogenasa y succinato deshidrogenasa, en base al tipo de inhibición
que generan.
PRERREQUISITOS:
1. Explicar el término actividad enzimática. 2. Describir el fenómeno de inhibición competitiva. 3. Describir el fenómeno de inhibición no competitiva. 4. Enumerar al menos cinco inhibidores que actúen en cada tipo deinhibición. 5. Explicar el comportamiento cinético de una enzima cuando esta bajo el efecto de ambos tipos
de inhibición.
INTRODUCCION:
Algunos compuestos reaccionan con las enzimas disminuyendo su actividad catalítica. Este efecto se
utiliza para preparar drogas o insecticidas que inhiben selectivamente ciertas enzimas en la bacteria o
insecto infectante y que no afecte al hospedero.
La inhibición es una de las formas más útiles para regular la actividad de una enzima. Los estudios
logrados en base a inhibidores, han contribuido a la información actual sobre cinética enzimática y
mecanismos tanto metabólicos como clínicos.
Los dos tipos principales de inhibición son: La inhibición competitiva: donde el compuesto inhibidor
interacciona con el sitio catalítico, compitiendo con el sustrato por la unión con dicho sitio. En este caso
el grado de inhibición depende directamente de la concentración del inhibidor, con respecto al sustrato
específico, por lo tanto, si la concentración de sustrato es mayor que la del inhibidor, no se presentará el
efecto inhibitorio. Más aún, si a una enzima bajo el efecto de un inhibidor competitivo se le adiciona
mayor cantidad de sustrato, casi siempre desaparecerá el efecto inhibitorio.
La mayoría de los inhibidores competitivos tienen una estructura química semejante a la del sustrato
natural por lo tanto son muy específicos. Como el inhibidor competitivo se une al sitio catalítico, la
enzima pierde afinidad con el sustrato original y esto modifica la constante de Michaelis (Km).
La inhibición no competitiva es aquella, donde el inhibidor se combina con la enzima, pero no con el sitio
catalítico de manera que la enzima puede unirse al sustrato y al inhibidor simultáneamente. La
inhibición ocurre porque al unirse el inhibidor provoca cambios moleculares en la enzima que
disminuyen su actividad catalítica. El aumento de la concentración de sustrato no tiene efecto sobre el
inhibidor.
Los inhibidores no competitivos son inespecíficos o sea que un mismo inhibidor puede afectar a varias
enzimas a la vez. Existen inhibidores muy complejos como el pentacloruro de mercurio, el benzoato de

57
sodio o muy simples como los iones metálicos plata, cobre, plomo y cianuro. La inhibición no
competitiva no afecta la afinidad de la enzima por el sustrato por los que la Km se mantiene intacta.
En este caso, el complejo enzima-sustrato-inhibidor no puede disociarse y ocurre una disminución en la
cantidad de enzima activa disponible.
MATERIAL :
1. 14 tubos de ensayo de 20 X 150 mm. 2. Cuatro pipetas de 1.0 ml. 3. Dos pipetas de .0 ml. 4. Dos pipetas Pasteur. 5. Una gradilla. 6. Solución de succinato de sodio 0.1 M. 7. Solución de malonato de sodio 0.1 M. 8. Solución de azul de metileno 0.002 M. 9. Preparación de la enzima deshidrogenasa succinica (DSC). 10. Solución de cloruro de sodio al 0.9%.
METODOLOGIA:
En este experimento se probará el efecto inhibitorio del malonato de sodio mediante un diseño en
donde, se aumenta gradualmente la concentración de inhibidor con respecto al sustrato, manteniendo
constante la concentración de la enzima. La verificación de la actividad se determinará, en referencia a
la actividad de un redox biológico (azul de metileno) ya que las enzimas que utiliza el diseño son
oxidorreductasas.
Preparación de DSC a partir de músculo cardiáco de rata.
1.- Tomar de 2 a 3 g de músculo de corazón cortarlo en fragmentos pequeños. 2.- Colocar el tejido en un mortero previamente sumergido en hielo y añadir NaCl al 0.9% en una proporción de 10 ml. por gramo de músculo, agregar un poco de arena lavada y triturar perfectamente. 3.- Dejar reposar la mezcla en el mortero por 15 min. 4.- Centrifugar el homogeneizado a 3000 rpm por 15 min. Recuperar el sobrenadante y etiquetarlo como
DSC.
Efecto del malonato de sodio sobre la actividad de DSC:
Preparar una serie de tubos de acuerdo al siguiente cuadro:
TUBO 1 2 3 4 5 6 7

58
Succinato de Sodio 0.1M 0.5 ------- 0.5 0.5 0.5 0.5 1.0
Azul de metileno 0.002M 0.3 0.3 0.3 0.3 0.3 0.3 0.3
Malonato de sodio 0.1M ------ ----- ----- 1.5 0.5 0.25 0.25
Agua destilada 1.0 1.5 3.0 ----- 0.5 0.75 0.25
Enzima DSC 2.0 2.0 ---- 2.0 2.0 2.0 2.0
Mezclar bien el contenido de cada tubo.
Incubar en baño de agua a 37° C y observar los grados de decoloración que genera cada tubo a los 15,
30, 45 y 60 min de incubación.
RESULTADOS
TUBO 1 2 3 4 5 6 7
15 min
30 min
45 min
60 min
DISCUSION:
1.-Discuta el tipo de inhibición que genera el malonato de sodio.
2.-Determine si el diseño experimental que se plantea en esta practica es el adecuado para la
diferenciación de una inhibición competitiva y no competitiva.
3.-Explique los cambios moleculares que genera el inhibidor no competitivo sobre la estructura de la
enzima.
CUESTIONARIO:
1.- Describa el comportamiento cinético de una enzima bajo el efecto de un inhibidor competitivo.
2.- Haga la misma descripción para una inhibición no competitiva.
3.- Analice las diferencias entre un inhibidor y un efector alostérico negativo.
4.- Qué tipo reacciones específicas inhiben los siguientes compuestos:

59
a.- Yodoacetato b.- Cianuro c.- Alopurinol d.- Tioguanina e.- Fluoruros f.- 2,4-dinitrofenol g.- Metotrexato

60
PRÁCTICA #. 18 DETERMINACION DE UREA Y CREATININA SERICA
OBJETIVO:
Al finalizar esta práctica, el alumno deberá tener los fundamentos para poder interpretar desde el punto
de vista clínico, los resultados obtenidos de la cuantificación de urea y creatinina de una muestra de
suero de un paciente.
PREREQUISITOS:
1.- Describir los puntos principales del ciclo de la urea. 2.- Describir los mecanismos de producción de la cretinina. 3.- Identificar los principales tejidos productores de urea y creatinina. 4.- Interpretación de aumento y disminuciónes de los analitos anteriormente analizados.
INTRODUCCION:
La urea es el principal producto de excreción, proveniente del catabolismo de las proteínas, el cual se
origina a partir de los grupos amino de los aminoácidos. Esta vía, es el principal medio de excreción de
nitrógeno por el organismo. La urea es sintetizada en el hígado por medio del ciclo de la urea (descrito
en 1932 por Sir Hans Krebs y Kurt Henseleit) también llamado ciclo de la ornitina, cuya función
fundamental es convertir el amoniáco y el bióxido de carbono en urea. Fig. 1
La urea es el producto final del metabolismo protéico; sintetizada por el hígado, transferida al torrente
circulatorio y excretada por el riñon.
La urea contiene en su estructura dos nitrógenos cuyo peso atómico es 28, los cuales relacionados con el
peso molecular de la urea que es 60 se obtiene una constante de 2.14 que es utilizado para convertir en
valores de nitrógeno ureico y viceversa.
Los aumentos del BUN están relacionados con los siguientes trastornos tales como: PREREANAL en los
que se encuentra un flujo sanguíneo reducido como en la insuficiencia cardíaca congestiva, shock,
depleción de agua y sal así como tambieén vómito, diarrea, diuresis, sudoración etc.
En trastornos POSTRENALES como obstrucciones del tracto urinario, hemorragia digestiva, infarto al
miocardio, stress etc.
Y clásicamente en insuficiencia renal y otras entidades tales como dieta alta en proteínas y pancreatitis.
El nitrógeno uréico sanguíneo (BUN) esta elevado en toda lesión renal, que perturbe su función
excretora. La urea también se eleva en casos de azoemia prerrenal (elevación de los productos
nitrogenados del metabolismo orgánico POR CAUSAS NO RENALES), que depende un bajo volumen
plasmático circulante, como ocurre en la hemorragia masiva, deshidratación.
Está también elevada en los casos que cursen con catabolia proteínica elevada, acompañada de baja
eliminación renal.

61
Cuando los niveles de urea pasan de 214 mg/100 ml, aparece depresión mental, somnolencia y
desequilibrio hidroelectrolítico, y si los niveles siguen aumentando, aparece el coma urémico.
La creatinina se forma en los músculos a partir del fosfato de creatina. Un 2% de esta sustancia se
convierte diariamente en esta sustancia. Es excretada por los riñones, y en pequeña cantidad por las
heces. La creatinina libre que aparece en la sangre y orina no vuelve a ser utilizada. Esta se excreta en la
orina de manera constante. En condiciones fisiológicas normales la creatinina urinaria representa la
filtración glomerular y la excreción tubular activa, formándose a partir de la creatina en cantidades
constantes. La creatinina normal del suero no se modifica ni con la dieta, edad, sexo, catabólia
proteínica ni ejercicio. Sin embargo, con una ingesta proteíca sustancial, que incluye una cantidad
considerable de carne y con ejercicios intensos durante largos períodos, se han observado en sujetos
jóvenes sanos, excreciones urinarias de creatinina del orden del 2.5 a 2.7 g/24 hrs., pero con niveles
sanguíneos normales; por lo tanto, se deduce que los niveles sanguíneos de creatinina en los sujetos
normales son más constantes que los niveles en la orina.
La cifra normal dependiendo de la técnica esta comprendida entre 0.5 y 1.6 mg/100 ml de suero o
plasma, y es proporcional a la masa muscular. Sus aumentos generalmente van parejos con los de la
urea, aunque la creatinina tarda más en aumentar.
Se presentan aumentos:
62
1.BUN/Creatinina Mayor 10:1 en aporte excesivo de proteínas, excesivo catabolismo tisular
(quemaduras, fiebre, tratamiento con corticosteroides), obstrucción postrenal, shock hipovolémico y en
la insuficiencia renal aguda y crónica.
2. BUN/Creatinina Menor 10:1 Escaso aporte de proteínas, dialisis, deshidratación (diarrea, vómito),
insuficiencia hepática.
MATERIAL:
1.- Fotocolorímetro 2.- Baño de agua a ebullición 3.- 2 Pipetas 5.0 ml 4.- 2 Pipetas 1.0 ml 5.- 2 Pipetas 0.1 ml 6.- 3 Tubos de ensaye 13 X 150 mm 7.- 2 Tubos de ensaye 10 X 100 mm 8.- 2 celdas de fotocolorímetro
METODOLOGIA :
Tomar una muestra de sangre pos punción venosa (3.0 ml aproximadamente) de uno de sus
compañeros, (revisar el Apéndice I) con o sin anticoagulante, centrigugar la muestra ysepare el suero o
plasma según sea el caso.
Experimento No. 1
Cuantificación de urea sérica.
Reactivos Blanco Patron Problema
Diacetilmonoxima 3 ml. 3 ml. 3 ml.
Agua 0.05 ml ----- ------
Sol. patrón o std. ----- 0.05 ml. ------
Suero o plasma ----- ----- 0.05 ml
Reactivo ácido 3 ml. 3 ml. 3 ml
Mezclar y calentar en baño a ebullición durante 10 minutos. Enfriar al chorro de agua y leer las
absorbancias ajustando con el blanco a cero a una longitud de onda de 520 nm.
CALCULOS

63
Experimento No. 2
Cuantificación de creatinina sérica
REACTIVOS PATRON PROBLEMA BLANCO
Agua 3.5 3.5 4.0
Tungstato de sodio 10% 0.5 0.5 0.5
Acido sulfúrico 0.5 0.5 0.5
Suero o plasma (ml.) --- 0.5 0.5
Sol. Patrón o estandar (ml.) 0.5 --- ---
Centrifugar los tubos Patron y Problema a 2,500 rpm por 5 min
Tomar sobrenadante y realizar lo siguiente:
REACTIVOS PATRON PROBLEMA BLANCO
Sobrenadante 3.0 3.0 3.0
NaOH 0.75 N 1.0 1.0 1.0
Acido Picrico 1.0 1.0 1.0
Reposar por 15 min a temperatura ambiente y leer absorbancia a 490 nm del tubo patrón y problema,
ajustando a cero de absorbancia con el tubo blanco.
CALCULOS




















