
Translational control of tumor necrosis factor-related apoptosis-inducing ligand death receptor...
10
Translational Control of Tumor Necrosis Factor-related Apoptosis- inducing Ligand Death Receptor Expression in Melanoma Cells* Received for publication, July 28, 2003, and in revised form, December 19, 2003 Published, JBC Papers in Press, December 19, 2003, DOI 10.1074/jbc.M308211200 Xi Yi Zhang, Xu Dong Zhang, Jodie M. Borrow, Tam Nguyen, and Peter Hersey‡ From the Immunology and Oncology Unit, David Maddison Building, Corner of King & Watt Streets, Newcastle, New South Wales 2300, Australia In the present study, it was found that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)-R2 protein expression did not correlate with mRNA expres- sion in melanoma cell lines. In particular, early passage primary cultures from patients had low TRAIL-R2 pro- tein expression compared with later passage cultures although TRAIL-R2 mRNA expression was similar in early and late passages. Similarly, cell lines made resist- ant to TRAIL by cultures in TRAIL had low TRAIL-R2 protein expression but normal levels of mRNA for TRAIL-R2. Expression from a luciferase reporter gene construct with the 3-untranslated region (UTR) (but not the 5-UTR) of TRAIL-R2 was suppressed when trans- fected into the TRAIL-selected (resistant) melanoma lines compared with that seen in the parental (sensitive) lines. Similar results were seen in early passage (resist- ant) cultures compared with late passage (sensitive) pri- mary melanoma cultures. RNA gel shift assays demon- strated protein(s) binding to the 3-UTR of TRAIL-R2 mRNA that were more evident in TRAIL-resistant cul- tures with low TRAIL-R2 protein expression. A 23-base fragment of the 3-UTR inhibited binding of the proteins to the 3-UTR, and a probe using this fragment bound to proteins in TRAIL-selected melanoma lines and early passage isolates of melanoma. Binding of the 3-UTR probe to the cytosolic protein(s) was induced by expo- sure to TRAIL and was lost from the TRAIL selected lines 2–3 days after withdrawal of TRAIL from the cul- tures. These results are consistent with post-transcrip- tional regulation of TRAIL-R2 expression by cytosolic proteins induced by TRAIL that bind to the 3-UTR re- gion of TRAIL-R2 mRNA. TRAIL 1 is a member of the TNF family that, like TNF- and Fas ligand, is a type II membrane protein that can induce apoptotic cell death in a variety of cell types (1–3). TRAIL appears to be particularly important because it can induce apoptosis in a wide range of cultured malignant cells but not in normal tissues (4 –9), with the possible exception of human liver cells (10). Normal human liver cells were reported to be sensitive to TRAIL, but this is believed to be due to the partic- ular form of TRAIL used in those studies (10). Induction of apoptosis by TRAIL is believed to be mediated by interaction with two death receptors on cells referred to as TRAIL-R1 and TRAIL-R2 (see review in Ref. 3 for alternate nomenclature). Normal cells were postulated to be protected from TRAIL- induced apoptosis by their expression of TRAIL-R3 and TRAIL- R4, which lack cytoplasmic death domains and act to sequester TRAIL (decoy receptors) or to mediate antiapoptotic signals (6, 12). We have shown previously that TRAIL was able to induce varying degrees of apoptosis in approximately two-thirds of the melanoma cell lines tested (4, 5). Sensitivity of melanoma cells to TRAIL-induced apoptosis showed an overall correlation with the level of death receptors, and in particular TRAIL-R2 ex- pression, but did not correlate with the level of expression of the decoy receptors, TRAIL-R3 and TRAIL-R4 (5). Resistance of some cell lines to TRAIL was due to the absence of all receptors for TRAIL. Exposure of melanoma cells to TRAIL resulted in down-regulation of death receptors due to internal- ization in endosomes, and re-expression was from the trans- Golgi network. In contrast, the decoy receptors were located in the nucleus and underwent relocation to cytosol and cell mem- branes due to signals from the death receptors (13). Factors that determine the level of TRAIL-R expression are not well understood. In some cell types, chemotherapy and irradiation was shown to up-regulate TRAIL-R2 expression by activation of p53 (14), whereas in others, p53-independent mechanisms were involved (15). Up-regulation of TRAIL-R2 by dexamethasone and interferon-gamma was independent of p53 (15). TRAIL-R1 (DR4) and TRAIL-R3 (DcR1) appeared to be also regulated by p53 (16, 17). The DNA binding sites for p53 were found to be located at three sites in the genomic locus of TRAIL-R2, either upstream of the ATG site or within intron 1 or intron 2 (18). The promoter region of the TRAIL-R2 gene was found to have transcription start sites 122 and 137 base pairs upstream of the initiation codon. Two Sp1 sites were responsi- ble for the basal transcriptional activity (19), and it was spec- ulated that agents binding to the Sp1 sites (such as certain histone deacetylase inhibitors) may up-regulate TRAIL-R2 ex- pression. The promoter region for TRAIL-R1 (DR4) contained several AP-1 binding sites, which is a target for the c-Jun N-terminal kinase pathway that can be activated by several chemotherapeutic agents (20). In addition to the variability of TRAIL-R2 expression be- tween different melanoma lines, we reported that primary cul- tures established from patients commonly have low TRAIL death receptor expression but on successive culture passages acquire higher expression levels of TRAIL-R2 and sensitivity to TRAIL-induced apoptosis comparable with that seen in estab- lished lines (21). We also found that exposure to TRAIL over * This work was supported by the Melanoma and Skin Cancer Re- search Institute, Sydney, New South Wales; the New South Wales State Cancer Council, New South Wales; and the Hunter Melanoma Founda- tion, Newcastle, New South Wales, Australia. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact. ‡ To whom correspondence and reprint requests should be addressed. Tel.: 61-2-49236828; Fax: 61-2-49236184; E-mail: Peter.Hersey@ newcastle.edu.au. 1 The abbreviations used are: TRAIL, tumor necrosis factor-related apoptosis-inducing ligand; TNF, tumor necrosis factor; TRAIL-R, TRAIL receptor; PE, phycoerythrin; RACE, rapid amplification of cDNA ends; UTR, untranslated region; ARE, AU-rich element. THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 11, Issue of March 12, pp. 10606 –10614, 2004 © 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A. This paper is available on line at http://www.jbc.org 10606 by guest on May 5, 2016 http://www.jbc.org/ Downloaded from
Transcript of Translational control of tumor necrosis factor-related apoptosis-inducing ligand death receptor...

Translational Control of Tumor Necrosis Factor-related Apoptosis-inducing Ligand Death Receptor Expression in Melanoma Cells*
Received for publication, July 28, 2003, and in revised form, December 19, 2003Published, JBC Papers in Press, December 19, 2003, DOI 10.1074/jbc.M308211200
Xi Yi Zhang, Xu Dong Zhang, Jodie M. Borrow, Tam Nguyen, and Peter Hersey‡
From the Immunology and Oncology Unit, David Maddison Building, Corner of King & Watt Streets,Newcastle, New South Wales 2300, Australia
In the present study, it was found that tumor necrosisfactor-related apoptosis-inducing ligand (TRAIL)-R2protein expression did not correlate with mRNA expres-sion in melanoma cell lines. In particular, early passageprimary cultures from patients had low TRAIL-R2 pro-tein expression compared with later passage culturesalthough TRAIL-R2 mRNA expression was similar inearly and late passages. Similarly, cell lines made resist-ant to TRAIL by cultures in TRAIL had low TRAIL-R2protein expression but normal levels of mRNA forTRAIL-R2. Expression from a luciferase reporter geneconstruct with the 3�-untranslated region (UTR) (but notthe 5�-UTR) of TRAIL-R2 was suppressed when trans-fected into the TRAIL-selected (resistant) melanomalines compared with that seen in the parental (sensitive)lines. Similar results were seen in early passage (resist-ant) cultures compared with late passage (sensitive) pri-mary melanoma cultures. RNA gel shift assays demon-strated protein(s) binding to the 3�-UTR of TRAIL-R2mRNA that were more evident in TRAIL-resistant cul-tures with low TRAIL-R2 protein expression. A 23-basefragment of the 3�-UTR inhibited binding of the proteinsto the 3�-UTR, and a probe using this fragment bound toproteins in TRAIL-selected melanoma lines and earlypassage isolates of melanoma. Binding of the 3�-UTRprobe to the cytosolic protein(s) was induced by expo-sure to TRAIL and was lost from the TRAIL selectedlines 2–3 days after withdrawal of TRAIL from the cul-tures. These results are consistent with post-transcrip-tional regulation of TRAIL-R2 expression by cytosolicproteins induced by TRAIL that bind to the 3�-UTR re-gion of TRAIL-R2 mRNA.
TRAIL1 is a member of the TNF family that, like TNF-� andFas ligand, is a type II membrane protein that can induceapoptotic cell death in a variety of cell types (1–3). TRAILappears to be particularly important because it can induceapoptosis in a wide range of cultured malignant cells but not innormal tissues (4–9), with the possible exception of human
liver cells (10). Normal human liver cells were reported to besensitive to TRAIL, but this is believed to be due to the partic-ular form of TRAIL used in those studies (10). Induction ofapoptosis by TRAIL is believed to be mediated by interactionwith two death receptors on cells referred to as TRAIL-R1 andTRAIL-R2 (see review in Ref. 3 for alternate nomenclature).Normal cells were postulated to be protected from TRAIL-induced apoptosis by their expression of TRAIL-R3 and TRAIL-R4, which lack cytoplasmic death domains and act to sequesterTRAIL (decoy receptors) or to mediate antiapoptotic signals(6, 12).
We have shown previously that TRAIL was able to inducevarying degrees of apoptosis in approximately two-thirds of themelanoma cell lines tested (4, 5). Sensitivity of melanoma cellsto TRAIL-induced apoptosis showed an overall correlation withthe level of death receptors, and in particular TRAIL-R2 ex-pression, but did not correlate with the level of expression ofthe decoy receptors, TRAIL-R3 and TRAIL-R4 (5). Resistanceof some cell lines to TRAIL was due to the absence of allreceptors for TRAIL. Exposure of melanoma cells to TRAILresulted in down-regulation of death receptors due to internal-ization in endosomes, and re-expression was from the trans-Golgi network. In contrast, the decoy receptors were located inthe nucleus and underwent relocation to cytosol and cell mem-branes due to signals from the death receptors (13).
Factors that determine the level of TRAIL-R expression arenot well understood. In some cell types, chemotherapy andirradiation was shown to up-regulate TRAIL-R2 expression byactivation of p53 (14), whereas in others, p53-independentmechanisms were involved (15). Up-regulation of TRAIL-R2 bydexamethasone and interferon-gamma was independent of p53(15). TRAIL-R1 (DR4) and TRAIL-R3 (DcR1) appeared to bealso regulated by p53 (16, 17). The DNA binding sites for p53were found to be located at three sites in the genomic locus ofTRAIL-R2, either upstream of the ATG site or within intron 1or intron 2 (18). The promoter region of the TRAIL-R2 gene wasfound to have transcription start sites 122 and 137 base pairsupstream of the initiation codon. Two Sp1 sites were responsi-ble for the basal transcriptional activity (19), and it was spec-ulated that agents binding to the Sp1 sites (such as certainhistone deacetylase inhibitors) may up-regulate TRAIL-R2 ex-pression. The promoter region for TRAIL-R1 (DR4) containedseveral AP-1 binding sites, which is a target for the c-JunN-terminal kinase pathway that can be activated by severalchemotherapeutic agents (20).
In addition to the variability of TRAIL-R2 expression be-tween different melanoma lines, we reported that primary cul-tures established from patients commonly have low TRAILdeath receptor expression but on successive culture passagesacquire higher expression levels of TRAIL-R2 and sensitivity toTRAIL-induced apoptosis comparable with that seen in estab-lished lines (21). We also found that exposure to TRAIL over
* This work was supported by the Melanoma and Skin Cancer Re-search Institute, Sydney, New South Wales; the New South Wales StateCancer Council, New South Wales; and the Hunter Melanoma Founda-tion, Newcastle, New South Wales, Australia. The costs of publication ofthis article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
‡ To whom correspondence and reprint requests should be addressed.Tel.: 61-2-49236828; Fax: 61-2-49236184; E-mail: [email protected].
1 The abbreviations used are: TRAIL, tumor necrosis factor-relatedapoptosis-inducing ligand; TNF, tumor necrosis factor; TRAIL-R,TRAIL receptor; PE, phycoerythrin; RACE, rapid amplification ofcDNA ends; UTR, untranslated region; ARE, AU-rich element.
THE JOURNAL OF BIOLOGICAL CHEMISTRY Vol. 279, No. 11, Issue of March 12, pp. 10606–10614, 2004© 2004 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in U.S.A.
This paper is available on line at http://www.jbc.org10606
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

several weeks resulted in down-regulation of TRAIL receptorsand that re-expression occurred over several weeks in the ab-sence of TRAIL. These are referred to as TRAIL-selected re-sistant cells (21). The basis for down-regulation of TRAIL-R2 inthe latter cell lines and in primary cultures is unknown, but wehypothesized that it may be due to transcriptional down-regu-lation of TRAIL-R2. To answer these questions, we examinedTRAIL-R2 mRNA expression by real time PCR and correlatedthis with TRAIL-R2 protein expression. The results suggestthat post-transcriptional events may be more important thantranscriptional events in regulation of TRAIL-R2 expression.
EXPERIMENTAL PROCEDURES
Cell Lines—Human melanoma cell lines Me4405, Mel-FH, Mel-RM,Mel-CV, MM200, Me1007, Mel-RMu, IgR3, Mel-LT, Mel-AT, Me10538,and SK-Mel-28 have been described previously (4, 5). The TRAIL-selected lines of Mel-FH, Mel-RM, and MM200 are described elsewhere(21). The cell lines were cultured in Dulbecco’s modified Eagle’s mediumcontaining 5% fetal calf serum (Commonwealth Serum Laboratories,Melbourne, Australia).
Antibodies, Recombinant Proteins, and Other Reagents—Recombi-nant human TRAIL (lot 6321-19) was supplied by Immunex (Seattle,WA). The preparation was supplied as a leucine zipper fusion protein,which required no further cross-linking for maximal activity. The mono-clonal antibodies against TRAIL-R1 (IgG2a hu TR1-M271; lot 7136-07),TRAIL-R2 (IgG1 hu TRAIL-R2-M413; lot 5274-96), TRAIL-R3 (IgG1 huTR3-M430; lot 7313-217), and TRAIL-R4 (IgG1 hu TR4-M444; lot 7136-15) were supplied by Immunex and have been described previously (22).
Flow Cytometry—Analysis was carried out using a Becton Dickinson(Mountain View, CA) FACScan flow cytometer, as described previously(5). The percentage of antigen-positive cells was calculated as the dif-ference in positive area between the positive and negative controlhistograms. The positive area was that to the right of the intersection ofthe two curves (23).
Apoptosis—Apoptotic cells were determined by the propidium iodidemethod described elsewhere (4, 5). In some experiments, apoptosis wasmeasured by staining with PE-conjugated annexin V according to themanufacturer’s instructions. In brief, cells with or without pretreat-ment with TRAIL were washed twice with cold phosphate-bufferedsaline and then resuspended in binding buffer at a concentration of 1 �106 cells/ml. One hundred �l of the resulting solution (1 � 105 cells)were transferred to a 5-ml culture tube, and 5 �l of annexin V-PE wasadded. After incubation at room temperature for 15 min in the dark, anadditional 400 �l of binding buffer was added to each tube, and cellswere analyzed by flow cytometry within 1 h.
Real Time PCR—Real time PCR was performed using the ABI Prism7700 sequence detection system (PerkinElmer Life Sciences). A totalvolume of 25 �l was used, made up of 5-�l aliquots of the following:cDNA sample (0.2 �g/ml); 300 nM TRAIL R2 forward primer (CGCTG-CACCAGGTGTGATT); 300 nM TRAIL R2 reverse primer (GTGCCTT-CTTCGCACTGACA); TRAIL R2 probe (6-carboxyfluorescein-CCCTGC-ACCACGACCAGAAACACAG-6-carboxy-N,N,N�,N�-tetramethylrhoda-mine) (Sigma); and 9 mM MgCl2 (Brilliant QPCR Kit; Integrated Scien-ces, Willoughby, New South Wales, Australia). The same samples wereanalyzed simultaneously for their �-actin content using �-actin forwardprimer (CTCGCAGCTCACCATGGAT), �-actin reverse primer (ATGC-CGGAGCCGTTGTC), and �-actin probe (6-carboxyfluorescein-TGATA-TCGCCGCGCTCGTCGT-6-carboxy-N,N,N�,N�-tetramethylrhoda-mine). PCR was performed using the following conditions: 2 min at50 °C; 10 min at 95 °C; 15 s at 95 °C, 1 min at 60 °C, 40 cycles. Thethreshold cycle values (Ct), a fractional cycle number at which thefluorescence passes the fixed threshold, for TRAIL R2 expression weretaken and subtracted from the Ct for �-actin expression level from thesame individual sample (� �CT). These were then subtracted from the�CT of the control sample in each assay (� ��CT). Relative abundancewas then calculated as 2���CT. Each experiment was performed intriplicate and repeated three times. These methods are as described inUser Bulletin 2 (1997) of Applied Biosystems ABI Prism 7700 detectionsystems.
RNA Rapid Amplification of cDNA Ends (RACE)—Total RNA wasextracted from melanoma cell lines with the spin or vacuum total RNAisolation system (Promega, Madison, WI). The 5� and 3� RACEs wereprocessed according to protocol of First Choice RLM-RACE kit (Ambion,Austin, TX). Briefly, for 5� RACE, total RNA was treated with calfintestinal phosphatase to remove free 5�-phosphates from moleculessuch as ribosomal RNA, fragmented mRNA, tRNA, and contaminating
genomic DNA. The RNA is then treated with tobacco acid pyrophophos-phatase to remove the cap structure from full-length mRNA to producea 5�-monophosphate end. A 45-base RNA adapter containing two for-ward primer sequences (see the protocol supplied by the manufacturer)is ligated to the RNA population using T4 RNA ligase. A random primerreverse transcriptase-PCR and a nested PCR then amplified the 5�-endof the specific RNA. Two reverse primers for the nested PCR areGGAGATGCAATCTCTACCGTCTTC (outer reverse primer) and TT-GGGTGATCAGAGCAGACTCA (inner reverse primer). For 3� RACE,the first strand cDNA was synthesized from above total RNA using asupplied 3�-end adapter containing two reverse primer sequences (asper the protocol supplied by the manufacturer). Sequentially, nestedPCR was processed to the resulting cDNA with other two primers,which were specific to the R2 3�-end. The two forward nesting primersare GTGGCTGTGTTTGTTTGCAA (outer primer) and GACAGAAGCT-CACAACGA (inner primer).
Plasmid Construction—The above RACE products were purifiedwith QIAEX II gel extraction kit (Qiagen Pty. Ltd.) and directly ligatedinto a T-tailed vector pGEM-T (Promega, Madison, WI) to form con-structs of pGEM-T-5�-end and pGEM-T-3�-end. The inserts of 3�- and5�-ends of TRAIL-R2 were verified by sequencing with the above prim-ers without restriction enzyme sequences.
For assay of luciferase expression, the firefly luciferase vector pGL3-promoter (Promega, Madison, WI) was digested with HindIII and NcoI,which allowed the 5�-UTR to be inserted immediately downstream ofthe SV40 promoter in order to produce the luciferase mRNA withTRAIL-R2 5�-UTR. The inserts of 5�-UTR were produced by PCR witha primer pair of TGGGCGAAGCTTGCGTTTGCTGGGTTTGAT (con-taining HindIII) and GTTGTCCATGGGCGGTGGGAAGGCTCTTAT(containing NcoI) and the above pGEM-T-5�-end as a template. Theabove insert was ligated into the linearized pGL3-promoter to formpGL3–5�-UTR.
Similarly, the 3�-UTR and 3�-UTR-invert inserts were cloned by PCRwith pGEM-T-3�-end as a template but with invert restriction sites.Then TRAIL-R2 3�-UTR and 3�-UTR-invert were ligated to the positionof the deleted 3�-UTR and poly(A) (from 1934 to 2196) of the luciferasegene of pGL3-promoter by digestion with restriction enzymes of XbaIand BamHI, and thus pGL3–3�-UTR and pGL3-R2-invert were formed.Primers used were as follows: 3�-UTR forward primer (containing SpeI),GAAGCACTAGTTTGAGGACCACTTGTTGAGC; 3�-UTR reverseprimer (containing BamHI), CGCGGATCCGCGAGCACAGAATTAAT-ACGA; 3�-UTR-invert forward primer (containing BamHI), GAAGCG-GATCCTTGAGGACCACTTGTTGAGC; 3�-UTR-invert reverse primer(containing SpeI), TGGATATCTAGAGCGAGCACAGAATTAATACGA.
The AU-rich element (ARE) insert was synthesized commercially(Sigma). Sense and antisense sequences were annealed by heating at95 °C and cooled down to room temperature in annealing buffer (100mM Tris-Cl, 100 mM NaCl) in 1 h. The formed double strand DNA wasligated into the 3�-UTR and poly(A)-deleted pGL3-promoter as above.The mutant ARE (M-ARE) and mutant 3�-UTR (M-3�-UTR) insertswere created by the same methods except that Ts in the ARE regionwere changed into Cs. All of the above inserts in the pGL3-promoterwere verified by sequencing with the above primers.
The above 3�-UTR and ARE inserts were also cloned into pGEM-Tand pcDNA3 as above for the purpose of synthesis of the mRNA probeand transfection to Mel-RM.
Dual Luciferase Reporter Assay—The firefly luciferase reporter con-structs were co-transfected in melanoma cells with Renilla luciferasereporter pRL-SV40 vector. Two million melanoma cells were electropo-rated with 30 �g of each vector. Forty-eight h after transfection, thecells were analyzed by the Dual Luciferase Reporter Assay System(Promega, Annandale, New South Wales, Australia). Briefly, the cellswere harvested with passive lysis buffer. The assays for firefly lucifer-ase activity and Renilla luciferase activity were performed sequentiallyin a Lumat LB 9501/16 luminometer (Bertold, Germany) after addingtheir substrates. The relative light units from firefly vectors werenormalized with those from the Renilla vector according to the man-ufacturer’s (Promega, Annandale, New South Wales, Australia)instructions.
Preparation of 32P-Labeled RNA Probes—Transcription in vitro wasperformed in a reaction mix containing 40 mM Tris-HCl (pH 7.9), 6 mM
MgCl2, 2 mM spermidine, 10 mM NaCl, 10 mM dithiothreitol, 0.5 mM
each ATP, GTP, and CTP, 12 �M UTP, 50 �Ci of [�-32P]UTP (10 �Ci/�l),1 �g of plasma DNA (pGEM-T-3UTR or pGEM-T-5UTR), and 20 unitsof T7 RNA polymerase using Riboprobe In Vitro Transcription Systems(Promega, Annandale, Australia). An irrelevant Escherichia coli cRNAwas used for a negative control, and unlabeled competitor cRNA syn-thesized under the same conditions was used as a specificity control.
Post-transcriptional Regulation of TRAIL Death Receptors 10607
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

The size and integrity of cRNA transcript was verified by electrophore-sis on polyacrylamide gel containing 6 M urea.
Preparation of Cytoplasmic Extracts (S-100 Fraction)—Cytoplasmicextracts were prepared from Mel-FH, Mel-FH-select, Mel-RM, and Mel-RM-select cells using a protocol modified from Ref. 24. Briefly, the cellswere collected by a scraper from three 175-cm2 flasks, washed twicewith phosphate-buffered saline at 4 °C, and resuspended in two packedvolumes of buffer A (10 mM HEPES (pH 7.9), 1.5 mM MgCl2, 10 mM KCl,0.5 mM dithiothreitol, 0.2 mM phenylmethylsulfonyl fluoride, and amixture of other proteinase inhibitors (Protease inhibitor mixture;Roche Applied Science)) at 4 °C, lysed on ice by 20 strokes in a Douncehomogenizer. The nuclei were removed by centrifugation at 14,000 � gfor 5 min in a bench microcentrifuge. The crude cytoplasmic extractswere mixed with 0.11 volume of buffer B (0.3 M HEPES (pH 7.9), 1.4 M
KCl, and 0.0 M MgCl2) at 4 °C and centrifuged for 60 min at 80,000 at4 °C using a SW 50.1 rotor in a Backman XL-90 ultracentrifuge. Thehigh speed supernatants were dialyzed against buffer D (20 mM HEPES(pH 7.9), 20% glycerol, 0.1 M KCl, 0.2 mM EDTA, 0.5 mM dithiothreitol,and 0.5 mM phenylmethylsulfonyl fluoride). The supernatant was re-covered and stored at �80 °C after the concentration was measured bythe Bradford assay method (Bio-Rad).
RNA Gel Retardation—This was performed according to a modifica-tion of the protocol of Walker et al. (25). Ten �g of the above S-100protein extracts were incubated with the indicated complementaryRNA probes in a buffer containing 10 mM HEPES (pH 7.2), 3 mM MgCl2,5% glycerol, 1 mM dithiothreitol, rRNA 0.5 mg/ml, tRNA 0.5 mg/ml, 100mM KCl, and 1 �l of 32P-labeled cRNA probes (5� 1010 cpm/0.6 ng) ina total volume of 20 �l. To determine RNA-binding specificity, 2 �l (0.2�g) of unlabeled, nonspecific, 3�-UTR and ARE probes were incubatedwith cell extract before 32P-labeled probes were added for RNA gelretardation. The reaction was carried out at room temperature for 30min. RNase T1 was added at 1 unit per action to remove all labeled RNAnot protected by bound protein and incubated for 15 min. One �l of 20mg/ml heparin was added and incubated for a further 10 min. Thesamples were then resolved on 4% polyacrylamide gel (60:1 acrylamideto bisacrylamide) using 0.5� TBE as a running buffer. Gels were driedand exposed to Kodak film at �80 °C.
RNase T1 mapping was performed as described by Leibold andMunro (26). Briefly after RNA gel was isolated by electroelution fromthe native gel in 45 mM Tris-HCl, 45 mM boric acid, 1 mM EDTA, pH 8.3,at 150 V for 3 h. Then the current was reversed for the last 5 min ofelectroelution. The electroelution buffer was followed by extraction withphenol after the addition of tRNA (2.5 �g/ml) and precipitation withethanol. The isolated RNAs were digested to completion with 10 units ofRNase T1 (Ambion). For comparison, intact 3�-UTR and a small fractionof 3�-UTR were also digested with RNase T1. The samples were dena-tured at 95 °C for 5 min with 7 �l of RNA gel loading buffer II (Ambion)and electrophoresed on a 20% polyacrylamide, 7 M urea sequencing gel.The Decade marker system (Ambion), which ranged from 10 to 150nucleotides, was used after being labeled with [�-32P]ATP according tothe manufacturer’s protocol. The gel was dried and autoradiographed.
Actinomycin D Chase Study—The 3�-UTR-pcDNA3 and pcDNA3were transfected to Mel-RM with LipofectAMINE 2000 (Invitrogen)according to the manufacturer’s instructions and selected by G418 (300�g/ml). The transfected cells then were treated with actinomycin D (0.5�g/ml) for 0, 1, 2, 3, and 2 h. The total RNA was extracted at differenttime points, and real time PCR was performed for TRAIL-R2 andglyceraldehyde-3-phosphate dehydrogenase mRNA.
RESULTS
TRAIL-R2 mRNA Expression in Relation to TRAIL-R2 Pro-tein Expression and Sensitivity to TRAIL-induced Apopto-sis—We examined the relation between TRAIL-R2 mRNA,TRAIL-R2 protein, and apoptosis in a panel of nine melanomalines that exhibited a range of sensitivity to TRAIL-inducedapoptosis. As reported previously (5), there was an overallcorrelation between TRAIL-R2 protein expression and sensi-tivity to TRAIL-induced apoptosis (Fig. 1a). There was, how-ever, very little correlation between TRAIL-R2 protein expres-sion and mRNA levels (e.g. in Me4405, there was highTRAIL-R2 protein and apoptosis but relatively low mRNA lev-els, whereas Mel007 had high TRAIL-R2 mRNA levels andrelatively low TRAIL-R2 protein). The latter was also the pat-tern shown in studies on melanocytes.
Dissociation of Protein and mRNA Expression for TRAIL-R2in Early Versus Late Passage Primary Melanoma Cultures fromPatients—Fresh isolates of melanoma cells from patients fre-quently had relatively low TRAIL receptor expression, but ex-pression of the latter increased with successive passages inculture (21). We examined mRNA levels for TRAIL-R2 in rela-tion to TRAIL-R2 protein in successive passages from fourpatients. As shown in Fig. 1b, TRAIL-induced apoptosis and
FIG. 1. Comparison of apoptosis, TRAIL-R2 expression, andTRAIL-R2 mRNA levels in melanoma cells. a, dissociation ofTRAIL-R2 protein expression and TRAIL-R2 mRNA levels in culturedmelanoma cells is shown. TRAIL-R2 mRNA levels in a panel of humanmelanoma cell lines and cultured melanocytes are shown. mRNA levelswere assessed by real time PCR, and TRAIL-R2 protein expression wasassessed by flow cytometry. TRAIL-R2 protein expression was stronglycorrelated to TRAIL-induced apoptosis (correlation coefficient r2 �0.725, p � 0.02 � 0.05) but not TRAIL-R2 mRNA (r2 � �0.082). Theerror bars indicate 1 S.D.; n � 5. b, TRAIL-R2 mRNA and proteinexpression in successive passages of three primary isolates of mela-noma. RW, KC, and MC were from lymph node metastases. mRNA waspresent in early passages of the cultures although TRAIL-R2 proteinwas absent or at low levels of expression. (Correlation of apoptosisversus R2 expression r2 � 0.882, p � 0.001; correlation of R2 expressionversus R2 mRNA � 0.090). The error bars indicate 1 S.D.; n � 5. c,TRAIL-R2 mRNA in melanoma cell lines before (Mel-FH, Mel-RM, andMM200) and after selection by culture in TRAIL (Mel-FH-select, Mel-RM-select, and M200-select). Assays were carried out within 1 day afterculture in the absence of TRAIL. TRAIL-R2 protein expression was atlow levels in the selected lines, but mRNA levels were little changed.(Correlation of apoptosis versus R2 expression r2 � 0.096). The errorbars indicate 1 S.D.; n � 5.
Post-transcriptional Regulation of TRAIL Death Receptors10608
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from
TRAIL-R2 protein was low or absent in passage 1 cultures andshowed small to moderate increases with successive passages.mRNA values for TRAIL-R2 were, however, relatively constantthroughout. In patient RW, mRNA levels were highest in pas-sage 2 cells and then decreased with successive passages,whereas TRAIL-R2 protein was highest in passage 8 cells. Inpatient MC, TRAIL-R protein was only detected in the latepassage 7 cells, whereas TRAIL-R2 mRNA was relatively con-stant in successive passages. In melanoma cultures from pa-tient KC, mRNA for TRAIL-R2 showed an increase from pas-sage 1 to passage 6 cells, but no significant TRAIL-R2 proteinwas detected until passage 6. There was no significant variationin the expression of HLA class I antigens detected by the mono-clonal antibody W6/32 on melanoma. The percentage expressionwas 96.5% on RW passage 3 and 97.7% on RW passage 8.
Cultures from RW were sensitive to TRAIL-induced apopto-sis. Those from MC, DC, and KC were relatively resistant. MCmelanoma cells expressed relatively few receptors, but culturesfrom DC and KC were resistant even in late passage culturesexpressing modest levels of TRAIL-R2 protein.
TRAIL-R2 mRNA Levels in Melanoma Cells Selected forResistance to TRAIL—Culture of melanoma cells in TRAIL forprolonged periods results in generation of TRAIL-resistant cul-tures that have low TRAIL-R expression (21). Culture of theresistant lines in the absence of TRAIL resulted in partialrecovery of both receptor expression and sensitivity to apopto-sis. We examined the association between TRAIL-R2 proteinand mRNA in these cells. The results in Fig. 1c indicate thatmRNA from Mel-RM was at similar levels in the parental andresistant TRAIL-selected lines despite relatively lowTRAIL-R2 protein. In the Mel-FH and MM200 lines, there wasa reduction in both TRAIL-R2 mRNA and protein, but thereduction in TRAIL-R2 protein was relatively greater. Therewas no significant variation in the expression of the HLA classI antigen detected by monoclonal antibody W6/32 on Mel-FH,Mel-FH-select (99.1%, 99.4%) and Mel-RM, Mel-RM-select(98.5%, 98.5%).
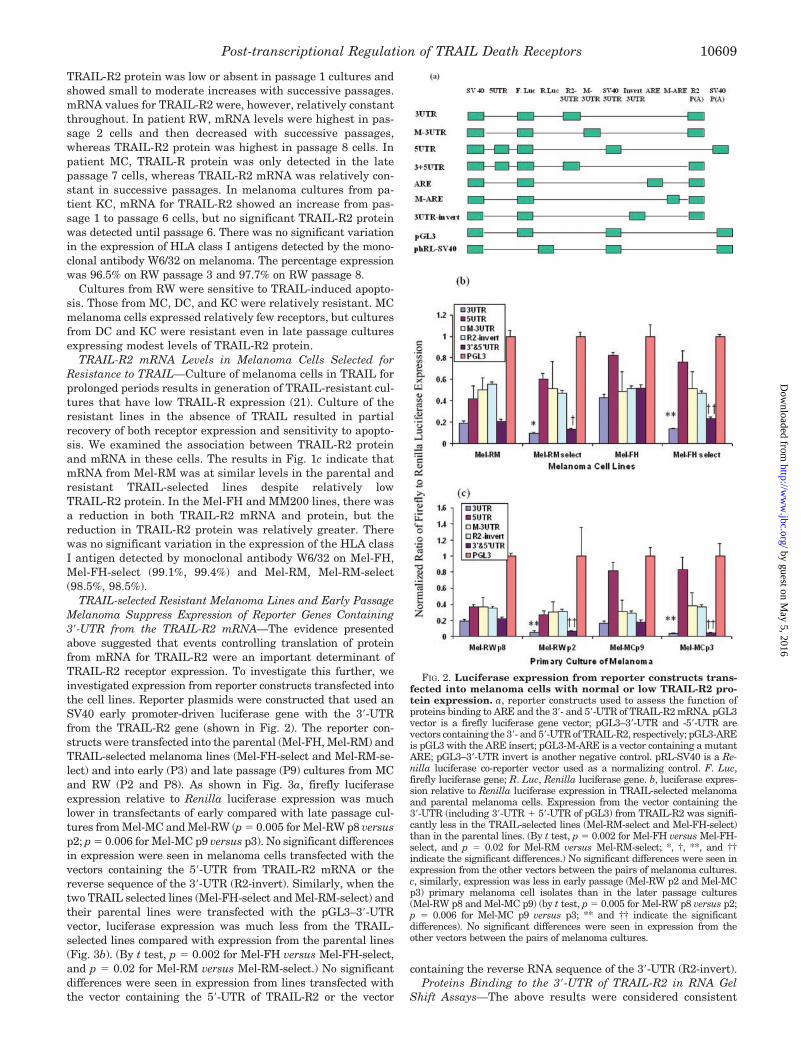
TRAIL-selected Resistant Melanoma Lines and Early PassageMelanoma Suppress Expression of Reporter Genes Containing3�-UTR from the TRAIL-R2 mRNA—The evidence presentedabove suggested that events controlling translation of proteinfrom mRNA for TRAIL-R2 were an important determinant ofTRAIL-R2 receptor expression. To investigate this further, weinvestigated expression from reporter constructs transfected intothe cell lines. Reporter plasmids were constructed that used anSV40 early promoter-driven luciferase gene with the 3�-UTRfrom the TRAIL-R2 gene (shown in Fig. 2). The reporter con-structs were transfected into the parental (Mel-FH, Mel-RM) andTRAIL-selected melanoma lines (Mel-FH-select and Mel-RM-se-lect) and into early (P3) and late passage (P9) cultures from MCand RW (P2 and P8). As shown in Fig. 3a, firefly luciferaseexpression relative to Renilla luciferase expression was muchlower in transfectants of early compared with late passage cul-tures from Mel-MC and Mel-RW (p � 0.005 for Mel-RW p8 versusp2; p � 0.006 for Mel-MC p9 versus p3). No significant differencesin expression were seen in melanoma cells transfected with thevectors containing the 5�-UTR from TRAIL-R2 mRNA or thereverse sequence of the 3�-UTR (R2-invert). Similarly, when thetwo TRAIL selected lines (Mel-FH-select and Mel-RM-select) andtheir parental lines were transfected with the pGL3–3�-UTRvector, luciferase expression was much less from the TRAIL-selected lines compared with expression from the parental lines(Fig. 3b). (By t test, p � 0.002 for Mel-FH versus Mel-FH-select,and p � 0.02 for Mel-RM versus Mel-RM-select.) No significantdifferences were seen in expression from lines transfected withthe vector containing the 5�-UTR of TRAIL-R2 or the vector
containing the reverse RNA sequence of the 3�-UTR (R2-invert).Proteins Binding to the 3�-UTR of TRAIL-R2 in RNA Gel
Shift Assays—The above results were considered consistent
FIG. 2. Luciferase expression from reporter constructs trans-fected into melanoma cells with normal or low TRAIL-R2 pro-tein expression. a, reporter constructs used to assess the function ofproteins binding to ARE and the 3�- and 5�-UTR of TRAIL-R2 mRNA. pGL3vector is a firefly luciferase gene vector; pGL3–3�-UTR and -5�-UTR arevectors containing the 3�- and 5�-UTR of TRAIL-R2, respectively; pGL3-AREis pGL3 with the ARE insert; pGL3-M-ARE is a vector containing a mutantARE; pGL3–3�-UTR invert is another negative control. pRL-SV40 is a Re-nilla luciferase co-reporter vector used as a normalizing control. F. Luc,firefly luciferase gene; R. Luc, Renilla luciferase gene. b, luciferase expres-sion relative to Renilla luciferase expression in TRAIL-selected melanomaand parental melanoma cells. Expression from the vector containing the3�-UTR (including 3�-UTR � 5�-UTR of pGL3) from TRAIL-R2 was signifi-cantly less in the TRAIL-selected lines (Mel-RM-select and Mel-FH-select)than in the parental lines. (By t test, p � 0.002 for Mel-FH versus Mel-FH-select, and p � 0.02 for Mel-RM versus Mel-RM-select; *, †, **, and ††indicate the significant differences.) No significant differences were seen inexpression from the other vectors between the pairs of melanoma cultures.c, similarly, expression was less in early passage (Mel-RW p2 and Mel-MCp3) primary melanoma cell isolates than in the later passage cultures(Mel-RW p8 and Mel-MC p9) (by t test, p � 0.005 for Mel-RW p8 versus p2;p � 0.006 for Mel-MC p9 versus p3; ** and †† indicate the significantdifferences). No significant differences were seen in expression from theother vectors between the pairs of melanoma cultures.
Post-transcriptional Regulation of TRAIL Death Receptors 10609
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

with regulation of translation by binding of proteins to the3�-UTR of the mRNA for TRAIL-R2. To examine this, RNAprobes were synthesized by in vitro transcription and labeledwith [�-32P]CTP. The probes were then mixed with cytosolicextracts of the melanoma cells and examined using RNA elec-trophoretic mobility shift assays.
Analysis of extracts from the Mel-RM and Mel-FH lines,together with their matching TRAIL-selected lines with lowTRAIL-R2 protein (Mel-RM-select and Mel-FH-select) areshown in Fig. 4. The extracts from RM-select and FH-selectcontained protein(s) that bound to the 3�-UTR probe that wasnot detectable in the parental lines that had normal TRAIL-R2protein expression. The probe was also bound by a protein(s)common to all of the cells (lower band) and a protein (upperband). The latter could not be inhibited by an excess of theunlabeled 3�-UTR probe and represented nonspecific binding.Fig. 4 shows similar studies on early and late passages from theprimary culture from MC and RW and melanocytes. In eachcase, extracts from cells with low expression of protein forTRAIL-R2 had protein(s) that bound to the 3�-UTR probe. Thisincluded normal cultured melanocytes. Similar studies with32P-labeled 5�-UTR probes did not show proteins binding to the5�-UTR (not shown).
Identification of a 23-Base RNA Fragment from the 3�-UTRof TRAIL-R2 mRNA That Binds to Protein(s) in MelanomaCells with Low TRAIL-R2 Protein Expression—RNase T1 map-ping methods (26) shown in Fig. 5a were used to identify theRNA sequence in the 3�-UTR that bound the protein(s) shownin Fig. 4. A 23-base ARE was identified with the sequenceTAAATGCTTTATTTATTTATTTG (Fig. 4b). This was synthe-sized by in vitro transcription and tested in RNA gel shiftassays with mixtures of cytosolic extracts of melanoma cellsand the 32P-labeled 3�-UTR probe. As shown in Fig. 5b, the23-base ARE specifically inhibited binding of the protein(s) inrow 2 (B2) to the 32P-labeled 3�-UTR. This contrasted with the
unlabeled whole 3�-UTR, which inhibited binding of the pro-tein(s) also in the lower band (B3). The 23-base ARE waslabeled with 32P and used as a probe. It was shown (Fig. 5c) tobind only to proteins in row 2 (B2) of cytosolic extracts fromTRAIL-selected lines (FH-select, RM-select) and early passageprimary cultures MCp3 and RWp3.
To further validate the functional significance of protein(s)binding to the 23-base ARE, it was used in place of the whole3�-UTR in the luciferase reporter gene construct shown in Fig.2. When transfected into the TRAIL-selected and parentalMel-RM and Mel-FH melanoma lines, luciferase expressionfrom the construct with the 23-base ARE was shown to besuppressed in the selected lines relative to the parental lines(Fig. 3c) (by t test, Mel-FH versus Mel-FH-select, p � 0.001;Mel-RM versus Mel-RM-select, p � 0.01).
Protein Binding to ARE Is Related to Exposure of the TRAIL-selected Lines to TRAIL—We have shown previously thatTRAIL-selected lines recover TRAIL-R2 expression severaldays after culture in the absence of TRAIL (21). We thereforeexamined whether this may be associated with loss of ARE-binding protein(s) after withdrawal of TRAIL. The study in Fig.6, a and b, shows an increase in TRAIL-R2 protein expressioncommencing on day 1 and maximal by day 3 for both Mel-FH-select and Mel-RM-select lines. The ARE binding activity wasapparent up to day 1 for Mel-FH-select and day 2 for Mel-RM-select. (Faint binding was evident on day 2 and day 3 forMel-FH-select and Mel-RM-select, respectively.) Conversely,when the TRAIL-selected lines were grown in the absence ofTRAIL for 5 days and then re-exposed to TRAIL, there was arapid reduction in TRAIL-R2 protein expression evident by 2 h(Fig. 6, c and d). Reappearance of protein binding to the labeledARE was evident by 6 h. We have reported previously thatTRAIL receptors undergo rapid endocytosis within 30 min ofexposure to TRAIL (13), which may account for the apparentlag between reduction in TRAIL-R2 protein expression and theappearance of the protein binding to the ARE.
We examined whether expression of the ARE may changethe stability of the mRNA for TRAIL-R2. To answer this, wemade permanent transfectants with the vector containing theARE in the Mel-RM and Mel-RM-selected melanoma cells andtested stability by measuring mRNA levels by real time PCRafter pulsing the melanoma cells with actinomycin D. As shownin Fig. 6, the half-life of the mRNA for TRAIL-R2 was increasedin the cells transfected with the ARE compared with cellstransfected with the mutant ARE or the vector alone. t formRNA in Mel-RM was 1.50 h in the untreated cells, 1.49 h inthe control vector-transfected cells, and 2.30 h in the ARE-transfected cells. Similarly, in the Mel-RM TRAIL-selectedcells the t was 1.10 h for the untreated cells, 1.10 h in thecontrol vector-transfected cells, and 2.15 h for the ARE-trans-fected cells. t for the control glyceraldehyde-3-phosphate dehy-drogenase mRNA was 9.90 h in both the parental and TRAIL-selected cell lines.
DISCUSSION
We have reported previously that sensitivity of melanomacells to TRAIL-induced apoptosis is determined predominantlyby the level of TRAIL-R2 death receptor expression (5).TRAIL-R1 expression on melanoma cells may also mediateTRAIL-induced apoptosis but was not expressed as frequentlyas TRAIL-R2 (5). In the present study, we have thereforefocused on TRAIL-R2 expression. We show that the level ofTRAIL-R2 protein expression was not closely associated withthe level of TRAIL-R2 mRNA expression. In particular, freshisolates of melanoma cells and melanocytes with low levels ofTRAIL-R2 protein expression and low sensitivity to TRAIL-induced apoptosis, had normal TRAIL-R2 mRNA levels. With
FIG. 3. Identification of proteins binding to the 3�-UTR ofmRNA for TRAIL-R2. RNA gel retardation assay using 32P-labeled3�-UTR of R2 and cytosolic extracts from Mel-FH, Mel-FH-select, Mel-RM, Mel-RM-select, Mel-MC passage 3 and 9, RW passage 3 and 9, andcultured melanocytes. Three RNA-protein complexes were detected.The upper band was not inhibitable by the unlabeled 3�-UTR probe andrepresents nonspecific binding to cytosolic proteins. The middle (row 2)complex was only detectable in the TRAIL-insensitive cells having lowTRAIL-R2 protein expression, including Mel-FH-select, Mel-RM-select,Mel-MC passage 3, RW passage 3, and melanocytes. The binding ofprotein(s) shown in row B3 is inhibitable by unlabeled 3�-UTR andexists in all of the cell lines.
Post-transcriptional Regulation of TRAIL Death Receptors10610
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

successive passages, the freshly isolated melanoma cells ac-quired TRAIL-R2 protein expression and increased their sen-sitivity to TRAIL. The present studies show that there wasrelatively little change in mRNA expression during culture,
suggesting that post-transcriptional events were key determi-nants of TRAIL-R2 expression. Similar findings were made instudies on melanoma cells made resistant to TRAIL by pro-longed culture in TRAIL. The latter had low TRAIL-R2 expres-
FIG. 4. Identification of a 23-basebinding sequence of the 3�-UTR ofTRAIL-R2 mRNA and its bindingproperties. a, cytosolic extracts fromMel-RM select were complexed with the32P-labeled 3�-UTR of TRAIL-R2 mRNA.The protein RNA complex in the middleband was excised, and a 23-base RNAfragment was eluted. This was then di-gested with RNase T1, and two segments(17 and 6 bases, respectively; lane 5) wereformed (lane 1, RNA marker; lane 2, 3�-UTR; lane 3, segments of 3�-UTR ofTRAIL-R2 RNA after RNase T1 digestion;lane 4, 23-base fragment bound by proteinin row 2; lane 5, 17- and 6-base fragmentsfrom RNA in lane 4. NT, nucleotides. b,the sequence of the 3�-UTR cloned byRNA RACE is shown with arrows indicat-ing guanosine residues cleaved by RNaseT1. The numbers below indicate sizes ofthe predicted oligonucleotide fragments.Boxed oligonucleotides are the predictedfragment obtained from the eluted bands.c, RNA gel retardation assay using the32P-labeled 3�-UTR of TRAIL-R2 and thecytosolic extracts from Mel-FH-select,Mel-FH, Mel-RM-select, and Mel-RM.The unlabeled transcripts at a molar ex-cess of 330-fold were incubated with thecytoplasmic extracts before the labeledtranscripts were added. The unlabeled 3�-UTR inhibited binding to proteins in row2 and 3, whereas the 23-base ARE frag-ment identified in a (lane 4) (ARE) onlyinhibited binding of proteins in the mid-dle row. d, RNA gel retardation assay us-ing labeled 3�-UTR of TRAIL-R2 and the23-base binding sequence shown in a,lane 4. Cytosolic extracts were from Mel-FH-select, Mel-FH, Mel-RM-select, Mel-RM, Mel-MC passages 3 and 9, andMel-RW passages 3 and 8. The identifiedbinding sequence (ARE) probe showedonly RNA-protein complex, whereas the3�-UTR showed three RNA-protein com-plexes. e, luciferase expression from plas-mid constructs containing a 23-base AREfragment from the 3�-UTR. The same pat-tern of reduced expression was seen in theTRAIL-selected lines, RM-select andFH-select (* and † indicate significantdifferences).
Post-transcriptional Regulation of TRAIL Death Receptors 10611
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

sion and sensitivity to TRAIL-induced apoptosis but normallevels of mRNA for TRAIL-R2. We reported previously thatnormal melanocytes appear to be protected from TRAIL-in-duced apoptosis because of low TRAIL death receptor expres-sion (9). The existence of normal TRAIL-R2 mRNA levels butlow TRAIL-R2 protein expression in melanocytes suggests thatthis may be a physiological control mechanism adapted bymelanoma cells for their survival.
Regulation of translation has been shown to be important inmany systems including production of ferritin (25) and up-regulation of p21 by epithelial growth factor (27, 28). In the
case of TNF-�, it was shown that a protein of �50,000 daltonsbound to the 3�-UTR of the mRNA for TNF-� and regulatedTNF-� protein production. The protein was increased in re-sponse to TNF-� and bound to AU-rich elements in the3�-UTR (29).
In view of these studies, we examined whether similarevents may be involved in regulation of TRAIL-R2 proteinexpression. Our results indicate that lysates of the TRAIL-resistant fresh isolates of melanoma and the TRAIL-selectedmelanoma lines had protein(s) that bound to 32P-labeled 3�-UTR from TRAIL-R2 mRNA in gel shift assays. These proteins
FIG. 5. Binding of proteins in TRAIL-selected lines to 3�-UTR of TRAIL-R2 mRNA. a–d, proteins binding to the middle row in the RNAgel (the ARE segment) disappeared after the removal of TRAIL from the cultures approximating the kinetics of reappearance of the TRAIL-R2protein shown in e. p, parental lines. f–j, re-exposure of the melanoma cells to TRAIL was associated with reappearance of the proteins binding tothe ARE fragment shown in the middle row. j, there was rapid disappearance of the TRAIL-R2 protein from the cell surface. The histogram ofprotein-RNA complexes in a, c, f, and h were shown in b, d, c, g, and i, respectively. (Our previous studies have shown that the early disappearanceof the receptor is likely to be due to endocytosis (13).
Post-transcriptional Regulation of TRAIL Death Receptors10612
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

were not detectable in matching melanoma cells with normalTRAIL-R2 protein expression. Furthermore, it was shown thatexpression from luciferase reporter constructs containing the3�-UTR of TRAIL-R2 mRNA was inhibited when transfected
into the TRAIL-resistant cells with low TRAIL-R2 protein com-pared with expression from the reporter constructs transfectedinto melanoma cells with normal TRAIL-R2 protein expression.We identified the binding site of the protein in the 3�-UTR ofTRAIL-R2 by RNase T1 mapping and showed that a 23-basefragment identified by this method inhibited binding of one ofthe proteins to the whole 3�-UTR probe. A probe made from thissequence bound to proteins in melanoma cell lysates in thesame pattern as the whole 3�-UTR probe, and luciferase ex-pression from reporter constructs containing this sequence wasinhibited similar to that seen from constructs containing thewhole 3�-UTR. These results support the view that TRAIL-R2protein expression was regulated by protein(s) binding to a23-base region from the 3�-UTR of the mRNA for TRAIL-R2.Negative regulatory effects may be mediated by the 5�-UTR(reviewed in Ref. 28), but we could find no evidence for involve-ment of this region in regulation of TRAIL-R2 expression.
The sequence identified in the 3�-UTR of TRAIL-R2 mRNA isrich in AU elements, and a number of proteins have beenidentified by others that might bind to this region. Multipleproteins were shown to bind to the 3�-UTR of p21. These in-cluded HuR, a member of the Drosophila ELAV family, andpoly(C)-binding protein (CP1) (27). Both HuR and HuD areknown to bind to AU-rich elements in the 3�-UTR and to sta-bilize mRNA (30). Studies on TNF-� production in macro-phages found that two proteins (TIA-1 (T cell intracellularantigen) and TIAR) bind to the 3�-UTR of mRNA for TNF-� andsilenced its translation. Another protein, tristetrapslin, boundto the 3�-UTR of mRNA for TNF-� in activated macrophagesand destabilized the mRNA (31). In the present studies, super-shift assays with antibodies to HuR and HuD did not affect themigration of proteins binding to the ARE identified in thepresent study (data not shown). Actinomycin D chase studieson Mel-RM indicated that the half-life of mRNA for TRAIL-R2was shorter in the TRAIL-selected lines than the parental orvector control lines and was increased in lines transfected withthe ARE. These results are consistent with destabilization ofthe mRNA by the protein(s) binding to the ARE sequence in the3�-UTR of TRAIL-R2.
Another question relates to the pathways that may be in-volved in regulation of the protein(s) binding to the 3�-UTR ofTRAIL-R2. The substrate of p38 mitogen-activated protein ki-nase, mitogen-activated protein kinase-activated protein ki-nase (MK2), was reported to target AU-rich elements, perhapsvia poly(A)-binding protein and thereby to regulate productionof TNF-� and interleukin-6 in response to lipopolysaccharide(32, 33). Whether similar pathways are involved in regulationof TRAIL-R2 protein expression is as yet unknown. We foundthat removal of TRAIL from cultures of TRAIL-selected mela-noma cell lines was associated with loss of the 23-base AREbinding activity in cytosolic extracts from the cells with kinet-ics that approximated increased expression of TRAIL-R2 pro-tein. Conversely, re-exposure of the TRAIL-selected lines toTRAIL resulted in reappearance of the proteins binding to theARE. It is therefore possible that TRAIL itself may be one ofthe factors regulating the appearance of the ARE binding ac-tivity. Studies on this aspect are continuing.
The results would appear to have implications for therapeu-tic interventions that might increase translation of TRAIL-R2protein (e.g. immunomodulatory peptides were reported to in-hibit binding of proteins to the 3�-UTR mRNA for TNF-�) (34).Inhibitors of signal pathways involved in regulation of theprotein binding to the 3�-UTR may also up-regulate theTRAIL-R2 protein. Whether such approaches might up-regu-late receptors on normal tissue is as yet unknown.
FIG. 6. R2 mRNA stability in Mel-RM (RM) and Mel-RM select(RM-select) lines transfected with ARE pCDNA3 (RM-ARE),mutant-ARE-pCDNA (RM-M-ARE), and vector pCDNA3 (RM-pCDNA3) after inhibition of transcription with actinomcyin D(0.5 �g/ml). Total RNA was extracted at the time points shown afterthe addition of actinomycin D (0.5 �g/ml), and relative R2 mRNA wasmeasured by real time PCR. Glyceraldehyde-3-phosphate dehydrogen-ase mRNA was measured at the same time periods by real time PCR inthe parental (c) and Mel-RM selected lines (d). There was no differencein mRNA levels for glyceraldehyde-3-phosphate dehydrogenase be-tween cell lines transfected with the different constructs. t forTRAIL-R2 mRNA in parental cells (a) was significantly longer than t inTRAIL-selected cells (b) (by t test; p � 0.042). t in cells transfected withARE was significantly longer than t in parental (a) or TRAIL-selectedcells (b) transfected with vector alone or vector with mutant ARE (by ttest, p � 0.01). The relative abundance data were plotted and calculatedby statistics software Sigma Plot and Sigma Stat (Hearne ScientificSoftware, Australia).
Post-transcriptional Regulation of TRAIL Death Receptors 10613
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

REFERENCES
1. Wiley, S. R., Schooley, K., Smolak, P. J., Din, W. S., Huang, C.-P., Nicholl,J. K., Sutherland, G. R., Smith, T. D., Rauch, C., Smith, C. A., and Goodwin,R. G. (1995) Immunity 3, 673–682
2. Pitti, R. M., Marsters, S. A., Ruppert, S., Donahue, C. J., Moore, A., andAshkenazi, A. (1996) J. Biol. Chem. 271, 12687–12690
3. Griffith, T. S., and Lynch, D. H. (1998) Curr. Opin. Immunol. 10, 559–5634. Thomas, W. D., and Hersey, P. (1998) J. Immunol. 161, 2195–22005. Zhang, X. D., Franco, A., Myers, K., Gray, C., Nguyen, T., and Hersey, P.
(1999) Cancer Res. 59, 2747–27536. Griffith, T. S., Chin, W. A., Jackson, G. C., Lynch, D. H., and Kubin, M. Z.
(1998) J. Immunol. 161, 2833–28407. Rieger, J., Naumann, U., Glaser, T., Ashkenazi, A., and Weller, M. (1998)
FEBS Lett. 427, 124–1288. Phillips, T. A., Ni, J., Pan, G., Ruben, S. M., Wei, Y.-F., Pace, J. L., and Hunt,
J. S. (1999) J. Immunol. 162, 6053–60599. Zhang, X. D., Nguyen, T., Thomas, W. D., Sanders, J. E., and Hersey, P. (2000)
FEBS Lett. 482, 193–19910. Jo, M., Kim, T.-H., Seol, D.-W., Esplen, J. E., Dorko, K., Billiar, T. R., and
Strom, S. C. (2000) Nat. Med. 6, 564–56711. Ashkenazi, A. (2002) Nat. Rev. Cancer 2, 420–43012. Gura, T. (1997) Science 277, 76813. Zhang, X. D., Franco, A. V., Nguyen, T., Gray, C. P., and Hersey, P. (2000)
J. Immunol. 164, 3961–397014. Sheikh, M. S., Burns, T. F., Huang, Y., Wu, G. S., Amundson, S., Brooks, K. S.,
Fornace, A. J., Jr., and el-Deiry, W. S. (1998) Cancer Res. 58, 1593–159815. Meng, R. D., and el-Deiry, W. S. (2001) Exp. Cell Res. 262, 154–16916. Guan, B., Yue, P., Lotan, R., and Sun, S. Y. (2002) Oncogene 21, 3121–312917. Sheikh, M. S., and Fornace, A. J., Jr. (2000) Leukemia 14, 1509–151318. Takimoto, R., and el-Deiry, W. S. (2000) Oncogene 19, 1735–174319. Yoshida, T., Maeda, A., Tani, N., and Sakai, T. (2001) FEBS Lett. 507, 381–38520. Guan, B., Yue, P., Clayman, G. L., and Sun, S. Y. (2001) J. Cell. Physiol. 188,
98–10521. Nguyen, T., Zhang, X. D., and Hersey, P. (2001) Clin. Cancer Res. 7, Suppl. 3,
966–97322. Griffith, T. S., Rauch, C., Smolak, P. J., Waugh, J. Y., Boiani, N., Lynch, D. H.,
Smith, C. A., Goodwin, R. G., and Kubin, M. Z. (1999) J. Immunol. 162,2597–2605
23. Sharrow, C. O. (1996) in Current Protocols in Immunology (Coligan, J. E.,Kruisbeek, A. M., Margulies, D. H., Shevach, E. M., and Strober, W., eds)pp. 5.2.1–5.2.2, John Wiley & Sons, New York
24. Dignam, J. D., Lebovitz, R. M., and Roeder, R. G. (1983) Nucleic Acids Res. 11,1475–1489
25. Walker, J., de Melo Neto, O., and Standart, N. (1998) in Methods in MolecularBiology (Martin, R., ed) Humana Press Inc., Totowa, NJ
26. Leibold, E. A., and Munro, H. N. (1988) Proc. Natl. Acad. Sci., U. S. A. 85,2171–2175
27. Giles, R. M., Daly, J. M., Beveridge, D. J., Thomson, A. M., Voon, D. C.,Furneaux, H. M., Jazayeri, J. A., and Leedman, P. J. (2003) J. Biol. Chem.278, 2937–2946
28. Cazzola, M., and Skoda, R. C. (2000) Blood 95, 3280–328829. Wang, E., Ma, W.-J., Aghajanian, C., and Spriggs, D. R. (1997) Cancer Res. 57,
5426–543330. Kasashima, K., Sakashita, E., Saito, K., and Sakamoto, H. (2002) Nucleic
Acids Res. 30, 4519–452631. Zhang, T., Kruys, V., Huez, G., and Gueydan, C. (2002) Biochem. Soc. Trans.
30, 952–95832. Bollig, F., Winzen, R., Gaestel, M., Kostka, S., Resch, K., and Holtmann, H.
(2003) Biochem. Biophys. Res. Commun. 301, 665–67033. Neininger, A., Kontoyiannis, D., Kotlyarov, A., Winzen, R., Eckert, R., Volk,
H. D., Holtmann, H., Kollias, G., and Gaestel, M. (2002) J. Biol. Chem. 277,3065–3068
34. Iyer, S., Kontoyiannis, D., Chevrier, D., Woo, J., Mori, N., Cornejo, M., Kollias,G., and Buelow, R. (2000) J. Biol. Chem. 275, 17051–17057
Post-transcriptional Regulation of TRAIL Death Receptors10614
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from

Xi Yi Zhang, Xu Dong Zhang, Jodie M. Borrow, Tam Nguyen and Peter HerseyDeath Receptor Expression in Melanoma Cells
Translational Control of Tumor Necrosis Factor-related Apoptosis-inducing Ligand
doi: 10.1074/jbc.M308211200 originally published online December 19, 20032004, 279:10606-10614.J. Biol. Chem.
10.1074/jbc.M308211200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/279/11/10606.full.html#ref-list-1
This article cites 32 references, 17 of which can be accessed free at
by guest on May 5, 2016
http://ww
w.jbc.org/
Dow
nloaded from




















