
Translation Series No. 608 - Fisheries and Oceans Canada ...
14
FISHERIES RESEARCH BOARD OF CANADA Translation Series No. 608 -• !, .e- - ; Reaction of methyl 10-undecenoate with mercuric acetate By ,Kyo Takaoka and Yuji Izumisawa From: Chemical Society of Japan, Industrial Chemistry Section, Vol. 67, No. 8, pp. 1244-1247, 1964. . Translated by M. Chiba, Bureau for -- Translations, Foreign Languages Division . , • Department of the Secretary of.State of Canada Fisheries Research Board of Canada Technological Research Laboratory, Halifax, N. S. 1965- RSI-IERIESRESEARCHBO.UDOFCUNDA . Library • 17b7 LOVr.:R WATER STREEt *F.--;:7;0TIA f,1 ,
-
Upload
khangminh22 -
Category
Documents
-
view
0 -
download
0
Transcript of Translation Series No. 608 - Fisheries and Oceans Canada ...

FISHERIES RESEARCH BOARD OF CANADA
Translation Series No. 608
-• !,
.e- -;
Reaction of methyl 10-undecenoate with mercuric acetate
By ,Kyo Takaoka and Yuji Izumisawa
From: Chemical Society of Japan, Industrial Chemistry Section, Vol. 67, No. 8, pp. 1244-1247, 1964. .
Translated by M. Chiba, Bureau for -- Translations, Foreign Languages Division. , •
Department of the Secretary of.State of Canada
Fisheries Research Board of Canada Technological Research Laboratory,
Halifax, N. S.
1965-
RSI-IERIESRESEARCHBO.UDOFCUNDA . Library •
17b7 LOVr.:R WATER STREEt
*F.--;:7;0TIA f,1 ,

4.
Reaction of -gethy71 10-urd oelloat,p, with Mercuric .',cetato.
(Received reb. 27,, 1964) -
Kyo Takaoka . Yuji Izemisawa
'n9.thyl 10-undocencate wa3 treated with mereuric acetate in
acetic .acid at 120 00 for 10-15 hrs. to Ç2ive the methyl este .r.s of
9-acetoxy 10-undecenoic acid and Il-acetoxy 9-undecenoic acid.
Unsaturated oxycarboxylic acids were obtPined in good yield by the
saponification of . these teo methyl undecenoates.
2) The reaction of methyl 10-undecenoate with mercuric acetate was ,
found to be second order. K = 111 x 10-° sec-1 . It seemed reasonable
to assume that the reaction is a bi-molecular reaction.
3) Pyrolysis of an addition product of methyl 10-undecenoate and .
mercuric acetate was studied by differential.thermal analysis,
thermogravimetric analysis and gas chromatography. Methyl 10-undecenoate
was found as the main product of decomposition-and-as a side product
methyl 9-acetoxy 10-undecenoate was obtained between room temp. and •
3300 C. Methyl 11-acetoxy 9-undecenoate was obtained. between 220°C- -
- 3300C as a side product.
1. Introduction
Addition product of unsaturated fatty acid with mercuric acetate - -
. - • is Used for the analysis, separation and purificationl of fatty acids.
-.7lowever, little attention has been paid to the reaction with heating of
the unsatarated fatty acid with mercuric acetate in acetic acid.
A number of papers 3) have been published about the reaction of
unsaturated hydrocarbons with mercuric acetate in acetic acid.
. since Treios2) et. al. reported that substitution of an acetoxy group

" DEPARTME.NT OF THE SECRETARY OF STATE BUREAU FOR TRANSLATIONS
ON CANADA
SECRÉTARIAT D'ÉTAT
BUREAU DES TRADUCTIONS
•
DMS1ON DES LANGUES e V ei
R AN S L A "r D FRON - TRADUCTION DE INTO A
Japanese English
W- I 0
1--- .77- 4"A p tz. r _2_
cd(cAscut .v‘c 1-4c14 o
CITY - VILLE
Tokyo DATE
August e 1964 PAGES
12441247
17
SUBjECT - SUJET
Organic Chenistry
AUTHOR - AUTEUR
Kyo Takaoka and Yuji Izumisawa
TITLE IN ENGLISH - TITRE ANGLAIS
Reaction of Methyl 10-undecenoate with Mercuric acetate
TITLE IN FOREIGN LANGUAGE - TITRE EN LANGUE ETRANGERE
REFERENCE RdFÉRENCE (NAME OF BOOK OR PUBLICATION - NOM DU LIVRE ou PUBLICATION)
Chemical Society of Japan, Industrial Chemistry Section y 67 (1964)
PUBLISHER - EDITEUR
REQUEST RECEIVED FROM OUR NUMBER
REQUIS PAR NOTRE DOSSIER N°
ivl DEPARTM2NT Department of Fisheries MiNISTER
YOUR NUMBER
VOTRE DOSSIER N°
TRANSLATOR Chiba TRADUCTEUR
DATE COMPLETED July 12 1965.
REMPLIE LE
DATE RECEiVED 14.P. 21'. 1965 REÇU LE
•

2Q
was found at the alpha position in the reaction of cycloolefine with
::ercuric acetate, but very little study was reported . on the reaction of •
unsaturated aliohatic hydrocarbons with mercuric acetate. Matsuda et.
studied the unsaturated aliphatic hydrocarbons octene-1„ octené-2 (cis and
ans) with mercuric acetate and obtained a single substitution product with
substitution of the acetoxy group at the alpha position with a shift of tha .
double bond.
In this paper the following two subjects were studied in an effort to
- easily Obtain the unsaturated oxycarboxylic acid by the reaction of
unsaturated carboxylic acid with mercuric acetate* _
(1)The characteristics of the reaction product, which was Obtained
by the reaction with heating of methyl 10-undecenoaté„ which has one double
bond in the omega-position, and mercuric acetate in acetic .acid was. examined.
(2)The pyrolysis of the addition product of methyl 10-undecenoate and
mercuric acetate was studied.
2. R.c.sults and Discussion
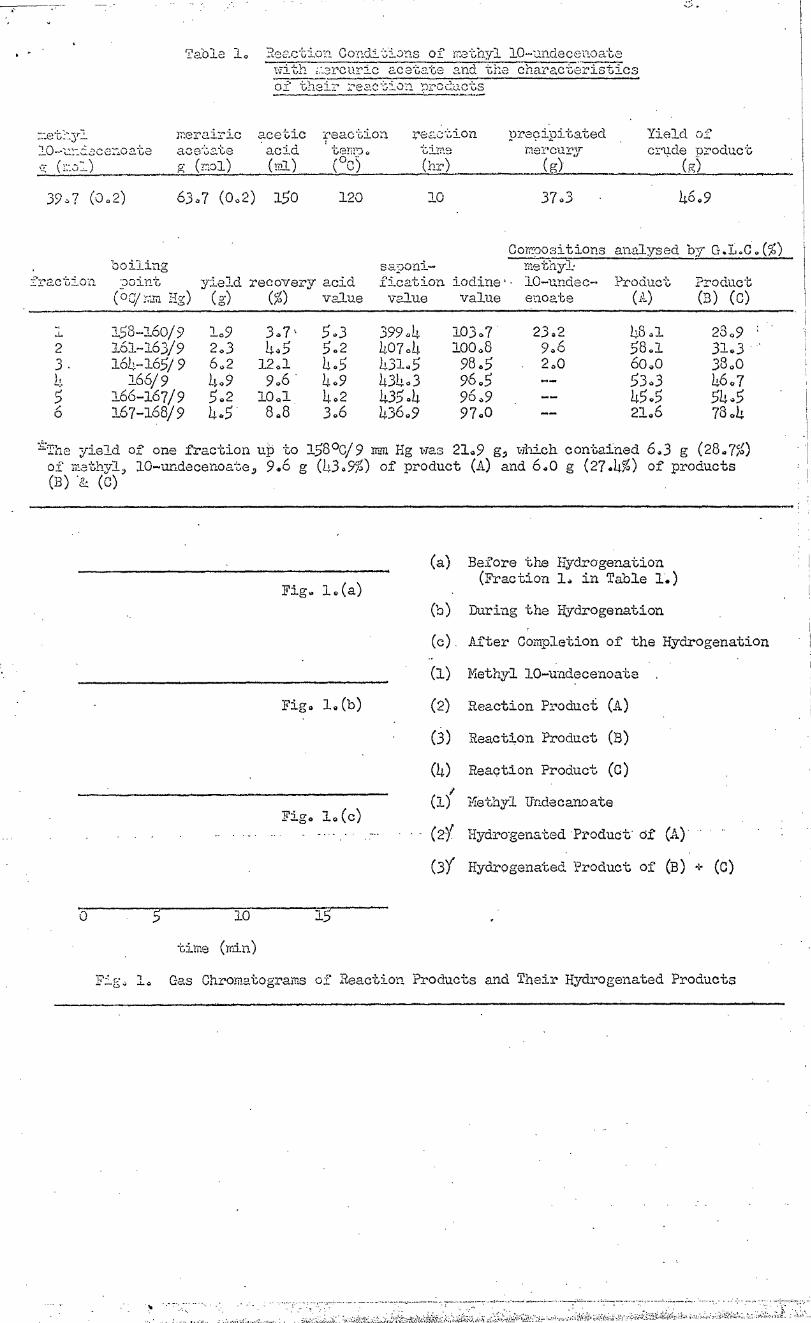
2.1. Reaction Conditions and Identification of Reaction Products•
Reaction conditions and characteristics of the distilled fractions of
the reaction products are shown in Table 1. Fig. .1 (a) shows the gas -
chromatogram of the first fraction (in Table 1 ) fraction 1) of the reaction
products,
-
..•.•, ,
39.7 (0.2) 63 47 (0 42) 150 1 0 190 37.3 46 .9
boiling -point yield
( °q/mm HZ) ( .3')
saponi- recovery acid fication
(%) value value fraction
158-160/9 161-163/9 164-165/9
166/9 166-167/9 167-168/9
3 . 4 2 6
11.41 eae
(a) old
Before the Hydrogenation (Fraction 1 4 in Table 1.)
Fig. 1.(a)
During the Hydrogenation
After Completion of the Hydrogenation
5- 10
rPble. 1. P,,,, action Conditions of methyl 10-undecenoate with ;:ercuric acetate and the characteristics of their reaction products
reaction tem,D. 1 0-und:.,cenoate
(no1)
nerairic acetic acetate acid g (mol) (m1)
reation tine (hr)
precipitated mercury
(g) •
Yield of crude product
(g) _
Compositions analysed by 0.14C4(%) methyl.
iodine. 10-undec• Product Product value enoate (L) (B) (C)
23 4 2 1,8 31 23 49
9 4 6 58.1 3143
2.0 60 90 38 40
53.3 46.7
45.5 54.5
21.6 73.4
1.9 3.7' 5.3 399.4 103 47 2 4 3 4,5 5 4 2 407 4 11 100 48 6 42 12 4 1 4.5 431.5 98.5 4.9 9.6 4.9 434.3 96.5 5 4 2 10 41 4 4 2 435.4 96 49 4.5 8 48 3.6 436.9 97 40
yield of one fraction up to 158°C/9 mm Hg was 21 49 g, which contained 6 4 3 g (2847%) of methyl, 10-undecenoate, 946 g (43 4 9%) of product (A) and 6 40 g (2744%) of products (B) & (c)
'The
Fig. 1.(b)
Fig. 1.(c)
(b)
(c)
(1) Methyl 10-undecenoate
(2) Reaction Product (à)
(3) Reaction Product (B)
(4) Reaction Product (C)
(1) Methyl Uhdecanoate
(2 )' Hydrugenated*Product« of
Hydrogenated Product of (B) + (0)
(A)'
(3)e
time (min)
Fig. 1 4 Gas Chromatograms of Reaction Products and Their Hydrogenated Products

4.'
In the gns chromatogram, peaks (1); methyl 10-2undecenoate, (2); re-
action product (A) and (4); reaction product (0) are Observed as sharp
peaks but (3); reaction product (E) is observed only as a small shoulder.
(2ig0 1. (a)).
The fraction 1. (Table 1) was then hydroenated with a platinum
catalyst. Samples obtained during (Fig. 1. (b)) and after (Fig. 1 (c))
hydrogenation were gas chromatographed. Fig. 1. (c) shows the following
results.
(1)Methyl 10-undecenoate was saturated byhydrogenationi to methyl
. . undecanoate.'
(2)product (A); (peak (2)) was hydrogenated to a_new - product (.e)..;
(oeak (2)e , in Fig. 1. (c)).
(3)The mixture of products (B) and (C) became one produce; (111').
It is assumed that (B) and (C) were cis and trans isomers..:
-1rDepartment of Chemistry, Musashi Institute of Technology: Todoroki, Tamagawa, Setagaya, Tokyo
1) H.B. White, jr., F0W0 Quakenbush, J. Am. Oil Chemists' Soc., 39, 511, 517 (1>'o2) etc.
2) Same as original copy.
3) Same as original cooy.
4) Matsuda, Moritake„ Ihara and Akiyoshi. Journal' of the Chemical Society of Japan, 82, 1570 (1961) --(Translation).
The reaction products were separated to product (A) and a mixture Page 2 '(1) . 151)
of - (B) and (C) by fraCtional diStillationS'and then by preParative gas :..(*R.124-1;;)
chromatograch.
The characteristics of these two fractions are shown in Table II. and
their infrared soectra ara shown in Fig. 2. (A,E,C and D).

molecular refraction
specific gravity (e) 4
eler.ental analysis
carbon (%) hydrogen (%)
saponi-- acid fication acetyl
.froo.uct yite..\Ld value value value lg)
(B) and Inst lu
(2„) r r 7 .7
8.5
Tra
nsp
aren
cy
(%)
Table II ■
Characteristics of Product (A) and .a mixture of (B) and (C).
1.5 1436,6 214,8 0.9797 69.91 65.99 9.18
(437.8) (21809) (69.89) (b5.60)e
1.8 436.0 (218.0) 0.9840 70.05 65.42 9.14
(437.8) 2; (218.9)u (69.89) -1c (66.60) (9.44)
"( ) shows the theoretical value of methyl acetoxy undecenoate.
• i
0 o
ca
Fig. 2..A infrared Spectrum of Reaction -Product (A)
Ca
Fig. 2. B Infrared Spectrum of Saponified (A)
Fig. 2. C Infrared Spectrum of Reaction Product (B)+(C)
• • . , , .•:.. •

G
0
Fig. 2. D Infrared Spectrum of Saponified (B)+(C)
(1)Saponification value, acetyl value, molecular refraction value
and the values of elemental analysis of product (A) Were essentially
identical to the theoretical values of methyl acetoxy undecenoate.
(2)The presence of CH9 ...CH absorption5) peaks in the infrared spectrum'
(ig. 2A) at 3005 . Cm-1, - 1638 cf-, 987 cm71 and 916 cm71 eliminated the
possibility of shift of double bond.
(3)Product was saponified by the usual method, the rate of estari-
fication of this saponified sample was 24.09%'oy Mirrahashi Method ,. (In
secondary alcohols, the rate is usually 20-30%).
(4)In the infrared spectrum (Fig. 2B) a broad and strong hydroxy groUp
-1 • absorption at 3500 cm-1 and a secondary alcoholic absorption at 1100 cm -
• • were displayed.
From a consideration of these four facts, it Seems reasonable to conlude -
that the product (A) is methyl 9-acetoxy 10-undecenoate.
As it was almost impossible to separate (B) and (C) bymeans of prepara-
tive gas chromatography, several measurements were'carried.out on the -
• mixture, as follows:
(1)Saponification value, acetyl value, molecular refraction value and
values of elemental analysis of this mixture.were essentially identical to
the theoretical values of methyl acetoxy undecenoatc.
(2)In the infrared spectrum (Fig. 2 0) CHr.CH -:double bond absorption •
is not displayed, but trans type absorbance is displayed at 963 . cm71 From
these facts, it is assumed that there has been a'shift of a double bond in
each compound.
(3) To determine the position of double bond, ozone 'oxidation waS
-- ar,-7 ied. Resulting ozonide wa decomposed With hydrogen peroXide, then
methylated and gas chromatogranhed. Since the final product was identical

7 . to the dimathyl ester of azelaic acid in the gas chromatography, it is
considered that the final product has a double bond at C-9 position which
was at C-10 position before the reaction*
(4)The mixture of (3) and (C) was saponified. by the usual method and the
esterification rate of the resulting oxy acid was 51.2% by MUrahashi method6)
.
(In primary alcohol, the rate is usUally more than 50%).
(5)P bro;d and strong hydroxylic .group absorption at 3500 cm l and
primary alcoholic absorption at 1070 cm - are displayed in the infrared
spectrum (Fig. 2D).
From a consideration - of these five facts it seems' most reasonable to
conclude that the mixture (B) .and (C) is a mixture of cis and trans isomers of
methyl 11-ace-boxy 9-undecenoate„ although these two compds.'were not separated.
5) Same as original reprint 6) Mnrahashi, Riken Th5 (Report of the Institute of Physical and Chemical
Research.--Translation) 15, 1186 (1936), 16, 548 (1937).
2.2 Pyrolysis of addition-products of methyl 10-undecenoate and mercuric acetate
• -Page 3- _ (P.152) .
(P.12 )46) At the beginning of the reaction of methyl 10-undecenoate with mercuric
acetate in acetic acid, it was confirmed that an addition, product is formed
between these two compounds. Following experiments were carried out in order
to check whether methyl acetoxy undecenoate is obtained from the pyrolysis
of this addition product. . The addition product mas obtained, after a long
treatment. of methyl 10-undecenoate with mercuric acetate in acetic acid at a
low temperature. Then the. product was analysed by differential thermal analysis„ .
thermogravimetric analysis and gas.chromatography. Differential thermal analysis.
curve (D.T.A.), thermogravimetric analysis curve (nA).and rate of weight loss
curve are shown in Fig. 3.

'CA
• 4-)
0
0
8. 1
s 1
L.
1 I .
1
Pyrolysis Pràducts of Addition Product of Methyl 10-undecenoate
: and Mel-sou-Ho Acetate
Fig., 5
s.
1;'ig0 3 DTA ard TGAof Addition Product of Methyl 10-undecenoate and • ercuric Acetate
In the DTA curve three main peaks are displayed; one is attributed to •
endothermic fusion, Which shows a sharp peak at 65°C e and the other two Peaks
are attributed to endothermic decomposition; one is a definite peak at 190 °C
and the other is rainer broad around 330°C 0
in
becomes a maximum at 185-190°C and then-slows dom add a second maximum peak
appears at 240°C4 To check the decomposition products, two groups of the
decorosition materials were collected using the collecting tube shown in
Fig. 4. •
LJ Coolirc
Fig. 4 Decomposition Products Collecting Tube in Dri'A
One fraction was collected between - room tempo to 220°C arid the other was
collected between 220-330 0C. These.two fractions were then analysed be gas
chromatography (Fig. 5).
TG A weight loss starts.around 120°C and the rate of weight loss -
(1)acetic acid (2)methyl 10-undecenoate (3)methyl 9-aceo:,:y 10 undecenoato
:t:-y1 11-acetoxy 9-undecaroate
7-1decomwsition products between 30-220°C 7,- decomposition products between 220-330 °C

The decomposition products Obtained from the first fraction e which were
obtained between room tempe and 220°C, were identified as acetic acides
methyl 10-undecenoate and a small amount of methyl 9-acetoxy L._-undecenoate. -
From the second fraction, which was Obtained between 220 -330 0C e acetic acid,
methyl 10-undecenoate and small amounts of methyl 9-acetoxy 10-undeceno: - te
and methyl 11-acetoxy 9-undecenoate were identified.
The difference found between the tuo fractions was the presence of
methyl 11-aceto:7 9-undecenoate in the second although the amount was very
small. The main products in the second fraction were still acetic acid and
methyl 10-undecenoate, which were the same as in the first fraction. From
- these results, it is concluded that it is possible to get two kinds of
methyl acetoxy undecenoate by the pyrebsis of the addition product of methyl
10-undecenoate and mercuric acetate, but the compounds are obtained only as
side products, not as the main products. . *-- • • -
2.3 .Change of amount of three compounds in the reaction of methyl 10-undecenoate and mercuric acetate in acetic acid (Kinetic Study)
Methyl 10-undecenoate was treated with mercuric acetate (1:1 mol/mol)
in acetic acid at 120 00 for 10 hrs0 with continuous stirring. The sangles
were taken from the mixture every 15 mine during the first 2 hrs and then
every 60 mins0 thereafter up to 10 hrs0 and were gas.chromatographed. The
proportions each of the three components; methyl 10-undecenoate„ methyl
9-acetoxy 10-undecenoate and methyl 11-acetoxy 9-uudecenoate are shown'
in Fig. 6A 0 •
Fig. 6 A Change of Amount of Three Comonents in the eaction of Methyl 10-undecenoate and Mercuric Acetate
•
(a) methyl 10-undecenoato (b) methyl 9-acetoxy 10-undecenoate (..,) methyl 11-acetoxy 9-undecenoate

Fig. 6 B
-page 4 - (P.153) (P.1247)
10.
Ti;hen the original amount of methy'l 10-undecenoate is expressed as
100 and the decrease in the amount of mathe 10-undecenoate at any
time is C (%), the reaction between t (time) and -L/o is shown in Fig. 6E,
which shows that t and 1/0 are in lineal relationshiD and that the reaction
is second order and reaction velocty constant K is 5 K=1 0 11 x 10-6 sec -
C=the decrease in the amount of methyl 10-undecenoate (%)
From these data it seems reasonable to assume that the methyl acetoxy
undecenoate is formed by a direct reaction of methyl 10-undecenoate with
mercuric acetate.
3 0 Experimental
• 3.1. Reagents •
1) Methyl 10-undecenoate; 10-undecenoic acid (obtained commercially) was
methylated by the usual method and fractionated . at 124°Q/8emHg (saponifica-
tion value is 282.0 (theoretical value is 282.9) and iodine value is
127..4 (1280)).
2) Mercuric-acetate; commercial product was recrystallized from 70% ethanol
solution.
3.2 Reaction of methyl undecenoate and • mercuric acetate
95 06g (003 mol) of mercuric acetate was dissolved in 150 ml of acetic •
and placed in a 300 mi three-neck, round-bottom flask, which uas equipped
with reflux condensor, thermometer and stirrer and then 59.5É .(0,3 mol) - of
methyl 10-undecencate was added. The reaction uaS carried out at 120oC with
1-> ci

li
continuous stirring. As reaction proceeds, mercury was gradually precipi-
t::..ted at the bottom of the flask. After completion of the reaction, mercury
was removed by filtration, 100 n1 of ether was added to the filtrate, the
filtrate mixture was washed with water-and then dried with sodium sulfate
(anhydrous). After evaporation of ether, fractional distillation was repeated
under reduced pressure. .A fraction, op 165-1o8 C/9mmHg was obtained as a
- crude reaction product.
3.3. Separation of reaction products (A) and mixture • .of (B) and (C)
The crude products •ere separated into 20 fractions by fractional distil
lation (Daika industry, Model TA) and each fraction was analysed by gas
chromatography (Hitachi Mode]. KGL, column 10% polydiethylene glycol succinate.
on Daiya Solid S, 100-120 mesh, column tempo 2100C, carrier gas Be 35 24/min).
Two crude fractions, one containing more than 80% of (126--131°C/3mmHg)
and the other containing more than 80% of the mixture (B) and (C) • (143-147°C/
3mmHg) were further purified bymeans of preparative gas chromatograPhy.
(Shimadzu Model GC-10, column 25% succinËte polyester on Shimarite 30-60 mesh,
•column temp. 218 °C, carrier gas He 2400 ml/min).
. 3.4 Addition product of methyl 10-undecenoate and •mercuric acetate
158.4g (005mo1) of mercuric acetate was dissolved in 300 ml of acetic
acid and 99 00 g (0.5 mol) of methyl- 10-undecenoate was added, than the soln. -
was thoroughly stirred. The mixture was left standing for 150 hrs. at 5°C and
the Precipitate formed.with this treatment was separated by filtration, Washed
with acetic acid and ether and then vacuum dried and finally white crystals
were obtained. [-MD 60-61°C, the results of elemental analysis were C=37.76%
(theoretical value 37.17n e H=5.55% (5.46)e Hg=390% (38.8%)7, yield was
• 162.0 g (recovery 63.0%).

12.
3.5 Therrl ar ,I'lysis of addition product of methyl 10-undecenoate and mercuric acetate
Diffei-ential thermal analysis ecuipment is Shinadzu Model DP- 1 0 0 _à1Pha-
alumina was used as a standard material. 350 mg of sample was placed in a
platinum cell, and measured at a heating rate of 5°C/min0 Samples of the
products of decomposition were collected by passing the gaàes into ice cold
ether.
Thermal balance used is Shinadzu Model TE-10 0 350 mg of sample was
placed in a platinum cell and measured at a heating rate cif 5 °0/min 0
3.6 Changes of the annunts of three . components in the . reaction of methyl'10-undecenoate and mercuric acetate in acetic acid
200 ml of acetic acid soin. of 95.6 g (0.3 mol) of,marcuric acetate mas
placed in a 500 ml th.,-ee-neck round-:bottom flask equipped mith . refIux
condancer, thermometer and stirrer and then 59,5 g (0.3 mol) of methyl ,
10-undecenoate was added and the reaction mas carried out at 12000 for 10 hrs.
10 ml sales were taken from the mixture every 15 min a during the first
2 hrSo and thereafter every hour up to 10 hrs. To eaCh'sample mas added
20 ri of ethyl eeler and then acidified mith 6M-HC10 A small amount of the .
addition product of methyl 10-undecenoate and mercuric acetate was decomposed
to the original parent compounds. Then the ether layer was separated, washed
witb mater and dried. After evaporation of the solvent, the residual.
mate-ials were analysed by gas chro:.:_atograplw.
(This paper mus presented at the Oil Chemistry Symposium (at Nagoya) on .
the 7th of MoveMber„ 1963,)
We wish to thank Dr, So Toyama, (or Sotoyama) Professor of Dept. of
Technology, Toyo University.
We also thank Mr. So Maruchi, Depto of Technology, Chiba University, and
T. Kawamoto and T. Hasegawa of Shimadzu Seisakasho for assisting mith
the fractional distillation and preparative gas chromatography,,




















