
Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation
11
Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation Eric K. Long,* Tonya C. Murphy,* Laura J. Leiphon,* John Watt, Jason D. Morrow,à Ginger L. Milne,à Jocelyn R. H. Howardà and Matthew J. Picklo Sr* , § *Department of Pharmacology, Physiology, and Therapeutics, University of North Dakota, Grand Forks, North Dakota, USA Department of Anatomy and Cell Biology, University of North Dakota, Grand Forks, North Dakota, USA àDivision of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA §Department of Chemistry, University of North Dakota, Grand Forks, North Dakota, USA Owing to the enrichment of polyunsaturated fatty acids (PUFA’s) in the CNS, lipid peroxidation is of particular importance. The formation of lipid peroxidation products is elevated in multiple neurodegenerative diseases (e.g. Alz- heimer’s disease, Parkinson’s disease) and following acute injury (Yoritaka et al. 1996; Sayre et al. 1997; Reich et al. 2001; Picklo et al. 2002). Peroxidation of PUFA’s leads to the formation of cytotoxic lipid–aldehyde species (Esterbauer et al. 1990). Of these, 4-hydroxy-trans-2-nonenal (HNE), derived from the peroxidation of linoleic acid (18:2; n-6) and arachidonic acid (20:4; n-6) has gained the most attention as a neurotoxin (Keller et al. 1997, 1999; Kruman et al. 1997; Picklo et al. 2002). Less appreciated, however, is that docosahexaenoic acid (DHA; 22:6; n-3) undergoes significant peroxidation in the brain. DHA content in many brain regions is 30–50% higher than that of arachidonic acid (Pawlosky et al. 2001; Salem et al. 2001; Lim et al. 2005). Elevated levels of the DHA- derived lipid peroxidation products F 4 -neuroprostanes (F 4 -NPs) are observed in diseased regions of brain in patients with Alzheimer’s disease, animals treated with kainic acid, and animals undergoing ethanol withdrawal (Reich et al. 2001; Montine et al. 2002; Musiek et al. 2004; Milne et al. 2006). These data suggest that DHA-derived Received October 26, 2007; revised manuscript received November 26, 2007; accepted November 26, 2007. Address correspondence and reprint requests to Matthew J. Picklo, Sr, PhD, Department of Pharmacology, Physiology, and Therapeutics, University of North Dakota School of Medicine, 501 N, Columbia Rd, Grand Forks, ND 58203, USA. E-mail: [email protected] Abbreviations used: ALDH5A, aldehyde dehydrogenase class 5A; BSA, bovine serum albumin; CM-H 2 DCFDA, 5-(and-6)-chloro- methyl-2¢,7¢-dichlorodihydrofluorescein diacetate, acetyl ester; DHA, docosahexaenoic acid; F 4 -NP, F 4 -neuroprostane; HHE, trans-4-hydroxy- 2-hexenal; HNE, trans-4-hydroxy-2-nonenal; KLH, keyhole limpet hemocyanin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; NOH, hydroxylamine; PBS, phosphate-buffered saline; PUFA, polyunsaturated fatty acid; ROS, reactive oxygen species; TBS, Tris- buffered saline; TBST, Tris-buffered saline Tween. Abstract Lipid peroxidation of docosahexaenoic (22:6; n-3) acid (DHA) is elevated in the CNS in patients with Alzheimer’s disease and in animal models of seizure and ethanol withdrawal. One product of DHA oxidation is trans-4-hydroxy-2-hexenal (HHE), a six carbon analog of the n-6 fatty acid derived trans-4- hydroxy-2-nonenal (HNE). In this work, we studied the neurotoxic potential of HHE. HHE and HNE were toxic to primary cultures of cerebral cortical neurons with LD 50 ’s of 23 and 18 lmol/L, respectively. Toxicity was prevented by the addition of thiol scavengers. HHE and HNE depleted neuronal GSH content identically with depletion observed with 10 lmol/L of either compound. Using an antibody raised against HHE–protein adducts, we show that HHE modified specific proteins of 75, 50, and 45 kDa in concentration- and time-dependent manners. The time-dependent formation of HHE differed from that of F 4 -neuroprostanes following in vitro DHA oxidation likely as a result of the different oxidation pathways involved. Using purified mitochondrial aldehyde dehydrogenase ALDH5A, we found that HHE was oxidized 6.5-fold less efficiently than HNE. Our data demonstrate that HHE and HNE have similarities but also differences in their neurotoxic mechanisms and metabolism. Keywords: 4-hydroxyhexenal, 4-hydroxynonenal, docosa- hexaenoic acid, glutathione, lipid peroxidation, neuropros- tanes. J. Neurochem. (2008) 105, 714–724. d JOURNAL OF NEUROCHEMISTRY | 2008 | 105 | 714–724 doi: 10.1111/j.1471-4159.2007.05175.x 714 Journal Compilation ȑ 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724 ȑ 2008 The Authors
-
Upload
vanderbilt -
Category
Documents
-
view
3 -
download
0
Transcript of Trans-4-hydroxy-2-hexenal is a neurotoxic product of docosahexaenoic (22:6; n-3) acid oxidation

Trans-4-hydroxy-2-hexenal is a neurotoxic product ofdocosahexaenoic (22:6; n-3) acid oxidation
Eric K. Long,* Tonya C. Murphy,* Laura J. Leiphon,* John Watt,� Jason D. Morrow,�Ginger L. Milne,� Jocelyn R. H. Howard� and Matthew J. Picklo Sr*,§
*Department of Pharmacology, Physiology, and Therapeutics, University of North Dakota, Grand Forks, North Dakota, USA
�Department of Anatomy and Cell Biology, University of North Dakota, Grand Forks, North Dakota, USA
�Division of Clinical Pharmacology, Vanderbilt University School of Medicine, Nashville, Tennessee, USA
§Department of Chemistry, University of North Dakota, Grand Forks, North Dakota, USA
Owing to the enrichment of polyunsaturated fatty acids(PUFA’s) in the CNS, lipid peroxidation is of particularimportance. The formation of lipid peroxidation products iselevated in multiple neurodegenerative diseases (e.g. Alz-heimer’s disease, Parkinson’s disease) and following acuteinjury (Yoritaka et al. 1996; Sayre et al. 1997; Reich et al.2001; Picklo et al. 2002). Peroxidation of PUFA’s leads tothe formation of cytotoxic lipid–aldehyde species (Esterbaueret al. 1990). Of these, 4-hydroxy-trans-2-nonenal (HNE),derived from the peroxidation of linoleic acid (18:2; n-6) andarachidonic acid (20:4; n-6) has gained the most attention asa neurotoxin (Keller et al. 1997, 1999; Kruman et al. 1997;Picklo et al. 2002).
Less appreciated, however, is that docosahexaenoic acid(DHA; 22:6; n-3) undergoes significant peroxidation in thebrain. DHA content in many brain regions is 30–50% higherthan that of arachidonic acid (Pawlosky et al. 2001; Salemet al. 2001; Lim et al. 2005). Elevated levels of the DHA-derived lipid peroxidation products F4-neuroprostanes
(F4-NPs) are observed in diseased regions of brain inpatients with Alzheimer’s disease, animals treated withkainic acid, and animals undergoing ethanol withdrawal(Reich et al. 2001; Montine et al. 2002; Musiek et al. 2004;Milne et al. 2006). These data suggest that DHA-derived
Received October 26, 2007; revised manuscript received November 26,2007; accepted November 26, 2007.Address correspondence and reprint requests to Matthew J. Picklo, Sr,
PhD, Department of Pharmacology, Physiology, and Therapeutics,University of North Dakota School of Medicine, 501 N, Columbia Rd,Grand Forks, ND 58203, USA. E-mail: [email protected] used: ALDH5A, aldehyde dehydrogenase class 5A;
BSA, bovine serum albumin; CM-H2DCFDA, 5-(and-6)-chloro-methyl-2¢,7¢-dichlorodihydrofluorescein diacetate, acetyl ester; DHA,docosahexaenoic acid; F4-NP, F4-neuroprostane; HHE, trans-4-hydroxy-2-hexenal; HNE, trans-4-hydroxy-2-nonenal; KLH, keyhole limpethemocyanin; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide; NOH, hydroxylamine; PBS, phosphate-buffered saline; PUFA,polyunsaturated fatty acid; ROS, reactive oxygen species; TBS, Tris-buffered saline; TBST, Tris-buffered saline Tween.
Abstract
Lipid peroxidation of docosahexaenoic (22:6; n-3) acid (DHA)
is elevated in the CNS in patients with Alzheimer’s disease
and in animal models of seizure and ethanol withdrawal. One
product of DHA oxidation is trans-4-hydroxy-2-hexenal (HHE),
a six carbon analog of the n-6 fatty acid derived trans-4-
hydroxy-2-nonenal (HNE). In this work, we studied the
neurotoxic potential of HHE. HHE and HNE were toxic to
primary cultures of cerebral cortical neurons with LD50’s of
23 and 18 lmol/L, respectively. Toxicity was prevented by the
addition of thiol scavengers. HHE and HNE depleted neuronal
GSH content identically with depletion observed with
10 lmol/L of either compound. Using an antibody raised
against HHE–protein adducts, we show that HHE modified
specific proteins of 75, 50, and 45 kDa in concentration- and
time-dependent manners. The time-dependent formation of
HHE differed from that of F4-neuroprostanes following in vitro
DHA oxidation likely as a result of the different oxidation
pathways involved. Using purified mitochondrial aldehyde
dehydrogenase ALDH5A, we found that HHE was oxidized
6.5-fold less efficiently than HNE. Our data demonstrate that
HHE and HNE have similarities but also differences in their
neurotoxic mechanisms and metabolism.
Keywords: 4-hydroxyhexenal, 4-hydroxynonenal, docosa-
hexaenoic acid, glutathione, lipid peroxidation, neuropros-
tanes.
J. Neurochem. (2008) 105, 714–724.
d JOURNAL OF NEUROCHEMISTRY | 2008 | 105 | 714–724 doi: 10.1111/j.1471-4159.2007.05175.x
714 Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
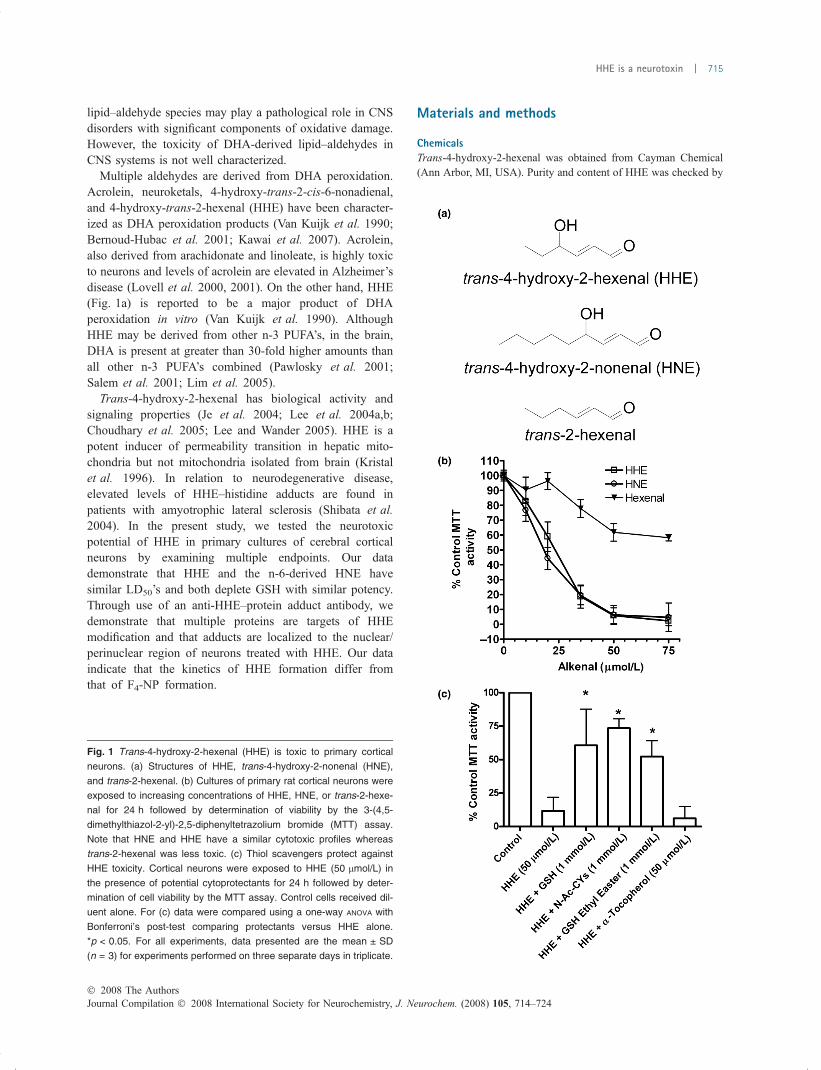
lipid–aldehyde species may play a pathological role in CNSdisorders with significant components of oxidative damage.However, the toxicity of DHA-derived lipid–aldehydes inCNS systems is not well characterized.
Multiple aldehydes are derived from DHA peroxidation.Acrolein, neuroketals, 4-hydroxy-trans-2-cis-6-nonadienal,and 4-hydroxy-trans-2-hexenal (HHE) have been character-ized as DHA peroxidation products (Van Kuijk et al. 1990;Bernoud-Hubac et al. 2001; Kawai et al. 2007). Acrolein,also derived from arachidonate and linoleate, is highly toxicto neurons and levels of acrolein are elevated in Alzheimer’sdisease (Lovell et al. 2000, 2001). On the other hand, HHE(Fig. 1a) is reported to be a major product of DHAperoxidation in vitro (Van Kuijk et al. 1990). AlthoughHHE may be derived from other n-3 PUFA’s, in the brain,DHA is present at greater than 30-fold higher amounts thanall other n-3 PUFA’s combined (Pawlosky et al. 2001;Salem et al. 2001; Lim et al. 2005).
Trans-4-hydroxy-2-hexenal has biological activity andsignaling properties (Je et al. 2004; Lee et al. 2004a,b;Choudhary et al. 2005; Lee and Wander 2005). HHE is apotent inducer of permeability transition in hepatic mito-chondria but not mitochondria isolated from brain (Kristalet al. 1996). In relation to neurodegenerative disease,elevated levels of HHE–histidine adducts are found inpatients with amyotrophic lateral sclerosis (Shibata et al.2004). In the present study, we tested the neurotoxicpotential of HHE in primary cultures of cerebral corticalneurons by examining multiple endpoints. Our datademonstrate that HHE and the n-6-derived HNE havesimilar LD50’s and both deplete GSH with similar potency.Through use of an anti-HHE–protein adduct antibody, wedemonstrate that multiple proteins are targets of HHEmodification and that adducts are localized to the nuclear/perinuclear region of neurons treated with HHE. Our dataindicate that the kinetics of HHE formation differ fromthat of F4-NP formation.
Materials and methods
ChemicalsTrans-4-hydroxy-2-hexenal was obtained from Cayman Chemical
(Ann Arbor, MI, USA). Purity and content of HHE was checked by
Fig. 1 Trans-4-hydroxy-2-hexenal (HHE) is toxic to primary cortical
neurons. (a) Structures of HHE, trans-4-hydroxy-2-nonenal (HNE),
and trans-2-hexenal. (b) Cultures of primary rat cortical neurons were
exposed to increasing concentrations of HHE, HNE, or trans-2-hexe-
nal for 24 h followed by determination of viability by the 3-(4,5-
dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
Note that HNE and HHE have a similar cytotoxic profiles whereas
trans-2-hexenal was less toxic. (c) Thiol scavengers protect against
HHE toxicity. Cortical neurons were exposed to HHE (50 lmol/L) in
the presence of potential cytoprotectants for 24 h followed by deter-
mination of cell viability by the MTT assay. Control cells received dil-
uent alone. For (c) data were compared using a one-way ANOVA with
Bonferroni’s post-test comparing protectants versus HHE alone.
*p < 0.05. For all experiments, data presented are the mean ± SD
(n = 3) for experiments performed on three separate days in triplicate.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724
HHE is a neurotoxin | 715

HPLC following purchase. HNE was synthesized in our laboratory
as previously described (Honzatko et al. 2005). NAD+, GSH, GSH-
ethyl ester, and trans-2-hexenal were purchased from Sigma-Aldrich
(St Louis, MO, USA). 5-(and-6)-chloromethyl-2¢,7¢-dichlorodi-hydrofluorescein diacetate, acetyl ester (CM-H2DCFDA) was
purchased from Invitrogen (Grand Island, NY, USA). DHA was
purchased from Nu-Chek-Prep, Inc. (Elysian, MN, USA). Hydrox-
ylamine (NOH) hydrochloride was purchased from Acros Organics
(Geel, Belgium). Bovine serum albumin (Fraction V, BSA) was
purchased from Fisher Scientific (Pittsburgh, PA, USA). Purified
recombinant human a-synuclein was the kind gift of Dr Julia M.
George (University of Illinois, Urbana, IL, USA) (Perrin et al. 2000,2001).
Neuron preparationExperimental protocols were in accordance with the NIH guidelines
for the use of live animals and were approved by the University of
North Dakota Institutional Animal Care and Use Committee. Timed-
pregnant rats (E18) were anesthetized with an i.p. injection of
ketamine (100 mg/kg + 0.1 mL) and xylazine (13 mg/kg) followed
by cervical dislocation. Rat pups were harvested and placed in ice-
cold dissecting media [0.002% of 0.5 mmol/L EDTA, 0.01% of
0.1 mmol/L EGTA, 1 mg/mL glucose in phosphate-buffered saline
(PBS), pH 7.4]. Brains were removed and placed in ice-cold
dissecting media. The cerebral cortex was dissected and the
meninges removed. Cortical pieces were minced in ice-cold
dissecting media and incubated in pre-warmed (37�C) 0.25%
trypsin for 15–20 min at 37�C. Trypsin was inactivated with fetal
bovine serum, and the tissue was added to 10 mL of plating media
(Neurobasal media, 10% L-glutamine, 1% penicillin/streptomycin,
10% B-27 supplement) and triturated to break up clumps. Neurons
were counted and plated on poly-d-lysine coated plates at a density
of 30 000 cells/cm2. Neurons were used on days 7–10. Immuno-
chemical staining of cells with anti-bIII tubulin (Sigma) demon-
strated that the cultures were 90–95% neurons. Neurons were grown
in a humidified incubator at 37�C under normal atmosphere
maintained with a final concentration of 5% CO2. Under these
conditions, O2 content is approximately 20%.
MTT activityCell viability was determined by the cellular reduction of 3-(4,5-
dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) to its
formazan product (Mosmann 1983). For protection studies, cells
were briefly rinsed with PBS before addition of MTT to remove
thiol protectants from the media that interfered with the MTT assay.
Culture medium containing 5 mg/10 mL MTT was added to each
well. Cells were incubated for 45–60 min at 37�C. The MTT
solution was carefully aspirated and 1 N HCl : isopropanol (4 : 96
v/v) was added. The plate was placed on a rocking shaker for
30 min. The absorbance of the dissolved formazan reduction
product was quantified at 570 nm using a SpectraMax 384Plus
UV/VIS plate reader (Molecular Devices, Sunnyvale, CA, USA).
GSH depletionNeurons were exposed to HHE or HNE at different concentrations
and time for characterization of GSH depletion. GSH content was
measured according to a method described by Dringen et al. (1999)with slight modifications. Following exposure, neurons were rinsed
with 1 mL PBS, lysed in 600 lL of 1% sulfosalicylic acid, scraped
from the plate and centrifuged 13 600 g, 15 min, 4�C. The
supernatant was used for GSH determination and pellets were
retained for protein quantification. GSH standards were prepared in
the same buffer as samples (1% sulfosalicylic acid); 10 lL of
supernatant or GSH standard was added to 90 lL of water in a 96-
well plate; 100 lL of reaction mixture (0.3 mmol/L 5,5¢-dithiobis-(2-nitrobenzoic acid), 1.0 mmol/L EDTA, 0.4 mmol/L NADPH,
1 U/mL GSH reductase in 0.1 mol/L sodium phosphate buffer, pH
7.5) was added. Samples were mixed and read immediately using a
SpectraMax 384Plus UV/VIS plate reader (Molecular Devices) at
405 nm for 2.5 min with readings at 15 s intervals. For protein
quantification, pellets were allowed to dry briefly followed by the
addition of 200 lL of 0.5 mol/L NaOH. The pellet was vortexed to
resuspend the protein. Protein content was determined using Protein
Assay Reagent (BioRad, Inc; Hercules, CA, USA).
Measurement of neuronal ROSNeurons were pre-treated with 7 lmol/L CM-H2DCFDA (Chi et al.2007). After 30 min, HHE (10, 20, 35, and 50 lmol/L) or H2O2
(250 lmol/L) as a positive control was added. Neurons were
incubated for an additional 3 h at 37�C. Fluorescence was measured
using SpectraMax Gemini XS plate fluorimeter (Molecular Devices)
with an excitation of 485 nm, emission of 540 nm and a cutoff of
515 nm. All procedures were performed under dim lighting to
preserve fluorescent signal.
Oxidation of DHADocosahexaenoic acid was oxidized using the method developed by
Kawai et al. (2006). DHA (10 mmol/L) was oxidized at 37�C using
50 lmol/L ammonium iron(II) sulfate hexahydrate and 1 mmol/L
ascorbic acid in sodium phosphate buffer at pH 7.4 and 6.5. Aliquots
(2 mL) were taken at 0, 2, and 6 h and added to 200 lL 10% 2,6-di-
tert-butyl-4-methylphenol (w/v). Samples were immediately deriv-
atized with pentafluorobenzyl NOH (see below) for subsequent
analysis.
Gas chromatographic-mass spectrometric analysis of HHEDocosahexaenoic acid oxidation samples and standards were
derivatized for 1 h by adding an equal amount of 50 mmol/L
O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine (Alfa Aesar, Ward-
hill, MA, USA) in 50 mmol/L sodium acetate buffer, pH 5.0 (van
Kuijk et al. 1986). Benzaldehyde (50 lmol/L) was added as an
internal standard prior to derivatization. The pH of the final reaction
was approximately 6.0. After 1 h, the reaction was acidified using
three drops of concentrated sulfuric acid to precipitate excess
O-(2,3,4,5,6-pentafluorobenzyl) hydroxylamine. Samples were
extracted twice into methylene chloride, dried over sodium sulfate,
evaporated under nitrogen and raised in the appropriate volume of
methylene chloride. Samples and standards were placed in auto-
sampler vial inserts. The trimethylsilyl derivative of the 4-hydroxy
group was formed by diluting the sample twofold in N,O-bis(trimethylsilyl)trifluoroacetamide with trimethylchlorosilane (99 : 1;
Supelco-Sigma) and allowed to derivatize for 12 h at 22�C.For the analysis of HHE content, gas chromatography with
negative-ion chemical ionization mass spectrometry was used. The
gas chromatographic-mass spectrometric system consisted of a
Trace GC, a PolarisQ ion trap mass spectrometer, and an AS3000
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
716 | E. K. Long et al.

autosampler (Thermo-Finnigan Corp.). All analyses employed a
1 lL splitless injection. The initial temperature was held at 50�Cfor 1 min followed by a 20�C per minute increase to 320�C, whichwas held for 10 min. Total ion current was measured from 6 to
24 min. Methane was used as a reagent gas at a flow rate of 1.5 mL/
min. HHE was quantified using the characteristic m/z 241 fragment
and benzaldehyde was quantified using the characteristic m/z 281
fragment.
HPLC analysis of HNE and HHEWhen appropriate, HNE and HHE content were determined by
HPLC using a Shimadzu HPLC system, an ODS2 (4.6 mm ·250 mm) column and 220 nm detection (Murphy et al. 2003a,b).The solvent consisted of 30% acetonitrile : 70% water (v/v) in
isocratic mode.
Analysis of F4-neuroprostanes
F4-neuroprostane content was quantified using gas chromatography
with negative-ion chemical ionization mass spectrometry with
selected ion monitoring utilizing deuterated 17-F4-NeuroP
(1.19 ng/sample) as the internal standard (Musiek et al. 2004).
Production of anti-HHE–protein adduct antiserumMariculture keyhole limpet hemocyanin (KLH; Pierce Chemical,
Rockford, IL, USA) was diluted with water to 10 mg/mL and
dialyzed versus 0.1 mol/L sodium phosphate (pH 8.0) with
0.15 mol/L NaCl overnight at 4�C. HHE was added at a final
concentration of 13 mmol/L (1.3 lmol/mg KLH) and incubated for
6 h at 37�C. After 6 h an aliquot was removed for determination of
protein-bound carbonyls using dinitrophenylhydrazine (Levine
et al. 1990). Protein-bound carbonyl content following HHE
incubation was 0.43 lmol/mg KLH. To the remainder of the
reaction, NOHÆHCl (Acros Organics) was added to a final
concentration of 20 mmol/L and incubated at 37�C for 30 min to
derivatize any free aldehydes of HHE–protein Michael adducts to an
oxime. This process prevents cyclic hemiacetal formation bet-
ween the 4-hydroxy group of HHE and the aldehyde. NaBH4
(100 mmol/L final concentration) was then added to reduce any
remaining HHE and potentially to reduce oximes and imines and the
reaction was incubated 1 h at 37�C. NOH and NaBH4 were removed
by dialysis twice versus a 1000-fold excess of 0.1 mol/L sodium
phosphate (pH 7.2) with 0.15 mol/L NaCl. The HHE–KLH
derivative was sent to Chemicon, Inc. (Temecula, CA, USA) for
immunization of three rabbits to produce polyclonal rabbit antisera.
Resulting antisera were screened by immunoblot and ELISA for
specificity (see specific figure legends).
Immunodetection of HHE–protein and HNE-protein adductsFor immunoblot analysis of HHE– or HNE–protein adducts,
neurons were plated in either 25 or 75 cm2 plates. Neurons were
exposed to HHE or HNE as described in the specific figure legends.
After incubation, the medium was aspirated and the neurons washed
with cold PBS and harvested in 3 mL of PBS. For detection of
HHE–modified proteins, cells were pelleted by centrifugation, and
the resulting pellet was resuspended in 0.5 mL of ice-cold buffer
(20 mmol/L sodium phosphate, 1 mmol/L diethylenetriaminepenta-
acetic acid, 50 lmol/L 2,6-di-tert-butyl-4-methylphenol, 0.5% v/v
Triton X-100, 2 mmol/L EDTA) with 25 mmol/L NOH and
mammalian protease inhibitor cocktail (Sigma), according to
manufacturers instructions. The lysate was sonicated for three, ten
second bursts with a Sonic Dismembrator Model 100 (Fisher
Scientific) at power level 3. The samples were then incubated on ice
for 30 min for NOH derivatization of free aldehydes. For detection
of HNE-modified proteins, lysis buffer without NOH was used and
following sonication the samples were incubated with a final
concentration of 10 mmol/L NaBH4 (Neely et al. 1999).Proteins were separated by sodium dodecyl sulfate–polyacryl-
amide gel electrophoresis using 10% polyacrylamide gels and
transferred to an Immobilon Transfer Membrane (Millipore, Inc.,
Bedford, MA, USA) for 55 min at 90 V. The blot was blocked with
3% (w/v) non-fat dry milk in Tris-buffered saline (TBS) for 2 h at
22�C. Blots were incubated overnight at 4�C with either anti-HHE
antiserum (1 : 4000) or anti-HNE antibody (1 : 2000; Calbiochem,
San Diego, CA, USA) in blocking solution containing 0.2% Tween
20 (TBST). The rabbit anti-HNE–protein adduct antibody recog-
nizes adducts reduced with NaBH4. The blot was washed four times
at 10 min each with TBST and incubated with a goat anti-rabbit
conjugated to horse radish peroxidase (1 : 5000; Promega, Madison,
WI, USA) for 2 h at 22�C. After washing three times at 10 min each
with TBST and three times for 10 min each with TBS, the blot was
developed using Pierce Detection Reagent and Pierce CL-Xposure
film (Pierce Biotechnology, Inc.).
Immunolocalization of anti-HHE protein adducts was performed
on neurons plated onto poly-D-lysine coated coverslips. Neurons
were plated and used as described above except that they were plated
onto poly-D-lysine coated coverslips set into six-well plates. Neurons
were treated for 6 h with or without HHE followed by washing with
PBS and fixation for 25 min at 22�C with 4% (v/v) formaldehyde
diluted in PBS. Following fixation the coverslips were washed with
PBS three times. For NOH derivatization, coverslips were treated
with 25 mmol/L NOH in 0.1 mol/L sodium phosphate (pH 7.4) for
30 min at 22�C. Following derivatization, coverslips were rinsed
three times with TBS. Permeabilization and quenching of endo-
genous horseradish peroxidase activity was accomplished by
incubating coverslips with 0.1% Triton X-100/0.3% H2O2 in TBS
for 30 min at 22�C. Following three rinses with TBS, coverslips
were blocked with 3% non-fat dry milk (w/v) in TBS for 2 h at 22�C.Primary anti-HHE–protein adduct antiserum (R68) was diluted
1 : 2000 in 3% non-fat dry milk (w/v) in TBST and incubated 4�Covernight. Coverslips were rinsed three times with TBST, incubated
for 2 h at 22�C with goat anti-rabbit conjugated to horse radish
peroxidase (1 : 5000; Promega) diluted in 3% non-fat dry milk (w/v)
in TBST. Coverslips were washed three times with TBST. Coverslips
were developed using the DAB peroxidase substrate kit (Vector
Laboratories, Inc., Burlingame, CA, USA) and placed onto slides
using Vectashield Hardset mounting medium with DAPI (Vector
Laboratories, Inc.). Following immunocytochemical preparation, all
slides were viewed and photographed using an Olympus BX-51
fluorescent microscope with attached Olympus DP-71 color camera
with dedicated software. Areas of interest were digitized and
prepared for page-reproduction using Adobe Photoshop (v.6.0).
Preparation and activity of recombinant rat aldehydedehydrogenase class 5ARecombinant rat aldehyde dehydrogenase class 5A (ALDH5A)
possessing an N-terminal poly-histidine tag was prepared as
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724
HHE is a neurotoxin | 717

described previously (Brichac et al. 2007). Assays were performed
in sodium phosphate buffer (40 mmol/L, pH 7.4) at 37�C with
1 mmol/L NAD+ as cofactor. ALDH activity was measured as the
increase in NADH formation at A340 nm following the addition of the
aldehyde substrate.
Statistical analysesData analysis was performed using one-way and two-way ANOVA as
appropriate with Prism 4.0 software (GraphPad, Inc., San Diego,
CA, USA). Data are presented as the mean ± SD.
Results
Our initial experiments were designed to test the neurotoxicpotential of HHE. For comparison, we also tested the toxicityof the nine-carbon HNE and the non-hydroxylated analog,trans-2-hexenal (Fig. 1a). As shown in Fig. 1, HHE andHNE had similar LD50’s of 23 lmol/L for HHE and18 lmol/L for HNE (Fig. 1b). trans-2-Hexenal was lesspotent than HHE and HNE indicating that the C4-hydroxylmoiety enhances a,b-unsaturated aldehyde toxicity. Protec-tion against HHE toxicity was effected by co-incubation withthe thiol-based scavengers N-acetyl cysteine, GSH, and GSHethyl ester (Fig. 1c). The antioxidant a-tocopherol was noteffective, suggesting that secondary, toxic lipid peroxidationevents were not occurring.
Glutathione depletion is a common toxic effector of a,b-unsaturated aldehydes (Neely et al. 2005; Arakawa et al.2006; Malone and Hernandez 2007). HHE significantlydepleted GSH content in a concentration-dependent manner(Fig. 2a). A similar concentration-dependent GSH depletionwas observed for HNE. The data in Fig. 2(b and c) show thatdepletion of GSH occurred within 15 min of HHE (20 lmol/L) exposure. While our GSH detection method monitorsGSH as well as GSSG, likely, HHE reacts only with GSH.This suggests that the amount of free GSH depletion weobserved is an underestimation in the loss of the totalneuronal GSH pool.
Because HHE depleted GSH levels, we predicted thatHHE would cause subsequent accumulation of reactiveoxygen species (ROS). We tested this hypothesis throughmeasuring the oxidation of the ROS-sensitive dye, CM-H2DCFDA (Chi et al. 2007). Exposure of neurons toincreasing concentrations of HHE led to an increase inROS content (Fig. 3). However, significant increases in DCFfluorescence were not observed until concentrations of HHEexceeded levels needed to deplete GSH content.
Our next series of experiments were designed to studythe formation of HHE–protein adducts. Since the proteinadducts of a,b-unsaturated aldehydes are immunogenic, wedeveloped antibodies against HHE–protein adducts. Immu-nogen was prepared by incubating KLH with HHE. Toincrease the specificity of the antibodies, we incubated theresulting HHE–KLH with NOH such that the aldehyde
component of any HHE–Michael adducts would beconverted to an oxime derivative. The antiserum fromone rabbit (R68) recognized HHE–protein adducts thatneeded to be derivatized with NOH but not NaBH4
(Fig. 4a) indicating that the antiserum recognized HHE–protein Michael adducts converted to the oxime derivativewith NOH (Fig. 4b). The R68 antiserum was specific forHHE–protein adducts but not those against HNE or
Fig. 2 Trans-4-hydroxy-2-hexenal (HHE) exposure depletes neuronal
GSH. (a) Cortical neurons were treated with increasing concentrations
of HHE or trans-4-hydroxy-2-nonenal (HNE) for 6 h before washing
and harvesting for GSH content. (b and c) Measurement of GSH loss
at increasing timepoints from 15 min to 6 h demonstrate that HHE
(20 lmol/L) caused rapid depletion of neuronal GSH. Neurons were
treated with HHE (20 lmol/L) and were harvested at increasing times
for determination of GSH content. Data presented are the mean ± SD
(n = 3) for three separate experiments.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
718 | E. K. Long et al.

trans-2-hexenal modified protein as determined by ELISAand immunoblot (Fig. 4c and d). The antiserum from onerabbit (R67) was reactive towards HHE-modified andHNE–modified proteins derivatized with NOH (not shown).As shown by ELISA using primary neurons treated withHHE, the NOH derivatization step was necessary forantibody recognition in whole cells (Fig. 4e).
The R68 antiserum was used to determine the localizationof HHE–protein adducts following HHE treatment. Neuronswere treated with HHE (0, 20, and 50 lmol/L) for six hoursfollowed by washing and fixing. For most but not allneurons, staining was heaviest in the nuclear/perinuclear area(Fig. 5a–c). For comparison, neurons were also stained forbIII tubulin as a cytoplasmic protein (Fig. 5d). No staining oronly faint immunoreactivity was observed in cells receivingHHE but not derivatized with NOH (Fig. 5e) and controlcells without HHE treatment (Fig. 5f). This localization isdifferent from the cytoplasmic localization noted for HNE–protein adducts in diseased regions of brain of patients withAlzheimer’s disease (Montine et al. 1997, 1998).
We then used the R68 antiserum to assess the multi-plicity of HHE–modified proteins accruing after HHEexposure and the time-course of adduct formation. Neuronswere exposed for 6 h to increasing concentrations of HHE(0–50 lmol/L). Immunoblot analysis of the neuron lysatesshows that HHE adducts accumulated in a dose-dependentmanner and that specific proteins of approximately 75, 50,and 45 kDa were targeted (Fig. 6a). A clear elevation inHHE–protein adduct formation was noted after exposure to5 lmol/L HHE. To define a time course of adductformation, neurons were exposed to 20 lmol/L HHEfollowed by harvesting at increasing timepoints. Immuno-
blot analysis demonstrated that these proteins were adductedfollowing a 30 min exposure to 20 lmol/L HHE (Fig. 6b).
Although HNE and HHE had similar cytotoxic and GSHdepletion characteristics, it is possible that HHE and HNEpossess different cellular protein adduction targets or differ-ent reactivity towards the same target. To test this possibility,neurons were exposed to either HNE or HHE (20 lmol/L)for 6 h followed by harvesting for immunoblot analysis. Thedata in Fig. 7a show that although similar sized proteins wereadducted by HHE and HNE, the apparent amounts of thoseproteins varied, suggesting that there exist different reactiv-ities towards the same proteins. The extent that HNE and
(a) (b)
(c)
(d) (e)
Fig. 4 Characterization of anti-trans-4-hydroxy-2-hexenal (HHE)–
protein adduct antiserum. (a) The antiserum (R68) recognizes HHE–
protein adducts derivatized with hydroxylamine (NOH). Following
binding of the bovine serum albumin (BSA) or HHE–modified BSA to
the ELISA plate selected wells were treated with NOH or NOH fol-
lowed by NaBH4. The data in (a) indicate that the antiserum recog-
nized an HHE–Michael protein adduct derivatized to an oxime with
NOH as drawn in (b). (c) The R68 antiserum has high specificity for
BSA–HHE adducts by immunoblot compared with HNE–BSA or trans-
2-hexenal–BSA adducts. Proteins were treated with NOH prior to
electrophoresis; 20 ng of each modified protein was loaded in their
respective lanes. (d) The R68 antiserum recognizes the HHE–BSA
adducts by ELISA versus other aldehyde derivatives. Samples were
derivatized with NOH prior to blocking. (e) The R68 antibody recog-
nizes HHE adducts on fixed neurons treated with HHE. Note that NOH
treatment was necessary for antibody recognition. Neurons were
treated with HHE (50 lmol/L) for 6 h, washed with PBS, and fixed with
formalin. Wells were treated with or without NOH prior to incubation
with the R68 antibody.
Fig. 3 Trans-4-hydroxy-2-hexenal (HHE) exposure elevates neuronal
reactive oxygen species (ROS) production. Neurons were pre-incu-
bated for 30 min with the ROS sensitive dye DCFDA (7 lmol/L)
followed by exposure to HHE at increasing concentrations for 3 h.
Exposure to hydrogen peroxide (250 lmol/L) increased DCF fluores-
cence by 284 ± 65% above control (not shown). Data are the
mean ± SD (n = 6) for experiments performed on two separate days
in triplicate. Data were compared using a one-way ANOVA with Bon-
ferroni’s post-test comparing cells receiving HHE versus control
(0 lmol/L HHE) cells, *p < 0.05.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724
HHE is a neurotoxin | 719

HHE had different reactivities with the same protein wasassessed by incubating HNE or HHE (50 lmol/L individu-ally) with BSA (50 lmol/L) or recombinant humana-synuclein (50 lmol/L) and measuring the loss of HHEand HNE as an indicator of protein alkylation. Because aprevious report demonstrated that HNE-modified a-synucleinwas neurotoxic a comparison of HHE and HNE modificationof a-synuclein was performed (Qin et al. 2007). HNEalkylated BSA at a faster rate than HHE; however wedetected no loss of either HNE or HHE when incubated witha-synuclein (Fig. 7b). The lack of HNE loss during incuba-tion with a-synuclein may be attributed in part to the 1 hincubation period we performed versus the overnight incu-bation described by Qin et al. (2007). There was no loss ofthese alkenals when BSA was omitted, indicating that non-specific loss did not occur.
To further clarify the disposition of HHE, we furtherexamined HHE formation resulting from DHA oxidation.HHE formation occurred in a time-dependent manner. Wetested the extent to which the amount of HHE generatedwas dependent upon the pH of the incubation using pH 7.4and 6.5 to mimic control and the acidotic condition thatoccurs in ischemia (Nemoto and Frinak 1981; Paschenet al. 1987; Hoffman et al. 1998). No significant differencein HHE generation was observed when comparing the pHof the reaction (Fig. 8a). As F4-NP formation results from
DHA oxidation, the same samples also were analyzed forF4-NP content. F4-NP formation increased with time;however, the marked increase in HHE content at 6 h wasnot mirrored by a large increase in F4-NP content(Fig. 8b). Similar to HHE formation, there was no effectof pH.
Mechanisms exist for the detoxification of 4-hydroxy-alkenals like HHE and HNE. In isolated brain mitochondria,detoxification of HNE is mostly through oxidation to trans-4-hydroxy-2-nonenoic acid by ALDH5A. We examinedwhether HHE was an efficient substrate for ALDH5Acompared with HNE (Fig. 9). HHE had a 13-fold higherKm (388 ± 40 lmol/L) than HNE (28.5 ± 2.6 lmol/L).However, the Vmax of ALDH5A for HHE was about twofoldhigher (3.52 ± 0.2 lmol/min/mg; kcat = 12.89 s)1) com-pared with HNE (1.7 ± 0.1 lmol/min/mg; kcat = 6.22 s)1).A comparison of kcat/Km shows that ALDH5A is 6.5-foldmore efficient towards utilization of HNE.
Discussion
Lipid peroxidation of n-3 PUFA’s occurs under basalconditions and is elevated in disease states. Multiple studieshave shown that CNS levels of the DHA oxidation productsF4-NP’s are elevated in Alzheimer’s disease, ethanol with-drawal, and inflammation. In this work, we examined the
(a) (b)
(c) (d)
(e) (f)
Fig. 5 Trans-4-hydroxy-2-hexenal (HHE)–
protein adducts are localized to the nuclear
region of neurons. Cultures were treated
with 0, 20, and 50 lmol/L HHE for 6 h prior
to washing and fixing with formalin. (a and
b) Neurons treated with HHE (20 lmol/L).
Following fixation coverslips were treated
with NOH. (a) is a low magnification of the
stained cultures with an increased magnifi-
cation shown in (b). Note the heavy locali-
zation to nuclei (N) versus cytoplasmic (c)
staining observed in cultures treated
with 50 lmol/L HHE in (c). (d) Untreated
neurons were stained for bIII-tubulin to
show cytoplasmic (c) staining with lighter
nuclei (N). Neurons in (e) were treated with
HHE (50 lmol/L) but not derivatized with
NOH and in (f) were not treated with HHE,
demonstrating the specificity of staining.
Figures shown were from the same cul-
tures, treated and stained in parallel and
under the same conditions. The scale bar
indicates 50 lm.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
720 | E. K. Long et al.

neurotoxic potential of trans-4-hydroxy-2-hexenal (HHE), aproduct of oxidative damage to DHA. Our results demon-strate that HHE was toxic to primary cultures of rat cerebralcortical neurons, depleted neuronal GSH levels and elevatedneuronal ROS content. HHE protein adduction occurred inconcentration- and time-dependent manner. Our resultsindicate that the kinetics of HHE formation from DHAoxidation differ from that of F4-NPs.
Most studies examining the neurotoxicity of aldehydesproduced from lipid peroxidation have focused on theproduct HNE, generated from the n-6 PUFA’s arachidonicacid and linoleic acid. Yet, comparisons of the disposition ofHNE and HHE in neuronal systems have not beenperformed. Our studies demonstrated no difference in thecytotoxic potency (LD50 for HNE of 18 lmol/L and LD50 forHHE of 23 lmol/L) as measured by loss of MTT activity orby GSH depletion. This is in contrast to the lower potency ofHHE (LD50 of 60 lmol/L) versus HNE (LD50 of 40 lmol/L)towards inducing cell death in ARPE-19 retinal pigmentedepithelial cells (Choudhary et al. 2005). Similar to ourresults, the LD50 of HHE for YPEN-1 rat prostate cells areapproximately 20 lmol/L (Lee et al. 2004b) . In both these
other cell types, apoptosis was evident (Lee et al. 2004b;Choudhary et al. 2005). Our cytotoxicity data with trans-2-hexenal indicate that the neurotoxic potential of HHE andHNE in this system is governed by the 4-hydroxyalkenalmotif independent of the two carbon and five carbon tails incontrast to the data obtained using ARPE-19 cells (Choudh-ary et al. 2005).
Similar to studies examining protection against HNE-mediated toxicity, thiol scavengers such as N-acetyl cysteine,GSH, and GSH analogs provided protection against HHEtoxicity (Kruman et al. 1997; Neely et al. 2000; Arakawaet al. 2006).
The parallel toxicity data of HHE andHNE are in contrast tothe difference between the activities of these aldehydes withknown detoxification systems. Previous data by Hubatschet al. (1998) found that that HHE is a poor substrate (31-foldless active) versus HNE for conjugation by glutathione-S-transferases A4-4. Our data show that HHE is a poorersubstrate (�6.5 fold) than HNE for the mitochondrialALDH5A. On the other hand, HHE and HNE are detoxified
(a)
(b)
Fig. 7 Trans-4-hydroxy-2-hexenal (HHE) has adduction characteris-
tics different than trans-4-hydroxy-2-nonenal (HNE). (a) Two indepen-
dent neuron cultures were treated with either HHE (20 lmol/L) or HNE
(20 lmol/L) for 6 h before harvesting for immunoblot detection of
protein adducts. The black arrows demonstrate protein bands of similar
masses. The gray arrow denotes a difference in immunoreactive
protein banding between the two treatments. (b) HHE and HNE show
protein-dependent adduction. HNE or HHE, 50 lmol/L each, were
incubated with bovine serum albumin (BSA) (50 lmol/L) or human
recombinant a-synuclein (a-Syn) in 50 mmol/L phosphate buffer at pH
7.4 for increasing times followed by determination of remaining HHE
or HNE as an indicator of protein adduction. Incubations were termi-
nated by the addition of 1/10 volume of 6% phosphoric acid.
HHE (µmol/L)
20 µmol/L HHETime (h)
0.5 1 3 6
0150
(a)
(b)
100755037
25
10
10075
50
37
25
10
5 10 20 50BSA-HHE
BSA-HHE
0 HHE
Fig. 6 Trans-4-hydroxy-2-hexenal (HHE) forms adducts with selected
neuronal proteins. (a) Adducts are formed in a concentration-depen-
dent manner. Neuronal cultures were treated with increasing con-
centrations of HHE for 6 h. Major immunoreactive bands are found at
approximately 75, 50, and 45 kDa. (b) Adduct formation occurs in a
time-dependent manner. Neurons were treated with 20 lmol/L HHE
and harvested at increasing times. Immunoreactive proteins of the
same mass were found in unexposed cells suggesting the presence of
HHE–protein adducts under basal conditions.
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724
HHE is a neurotoxin | 721

by aldose reductase with similar efficiency (He et al. 1998).Thus, the two carbon tail of HHE has a significant effect uponits detoxification versus HNE.
Protein adduction is a major mechanism by which agentslike HHE, HNE, and acrolein can exert neurotoxicity.Although levels of HHE–protein adducts are elevated inamyotrophic lateral sclerosis (Shibata et al. 2004), our resultsare the first to demonstrate that HHE modifies specificproteins rather than modifying proteinaceous nucleophilicgroups non-discriminantly. Three major proteins weretargeted by HHE adduction in concentration- and time-dependent manners. Two proteins of approximately of 75 and45 kDa were primary targets at 5 lmol/L HHE and10 lmol/L HHE whereas a protein of approximately50 kDa became labeled more prominently at 20 lmol/LHHE. The finding that immunoreactive proteins of the samemasses were observed in untreated neurons, albeit lessintensely compared with HHE-treated neurons, suggests thatHHE–modified proteins exist within neurons under these
conditions. The selective modification of these proteins candepend upon multiple factors including the amount ofexpression of the protein and the interaction of HHE towardsone particular protein motif versus another. More detailedproteomic analysis is needed to clarify these issues.
Our experiments directly comparing HHE modification toHNE modification show that HNE modified three majorproteins of similar masses. Potentially these are the sameproteins although this needs to be confirmed using subsequentproteomic techniques. Comparison of banding intensity sug-gests that the HNE targeted these proteins in differing amountscompared with HHE. We cannot exclude however that theseproteins were adducted to the same extent possibly at differentsites or that different types of adducts (e.g.Michael adducts vs.Schiff bases) were formedwith differing or no reactivities withthe antibodies. These data, along with those demonstratingdivergent reactivities towards BSA and detoxification en-zymes, indicate that HHE has biochemical properties that areunique and others that are similar to HNE.
The mechanisms involving HHE formation from DHAhave not been explored. Based upon the mechanismsproposed for HNE, we propose that HHE formation is theresult of the extraction of the bis-allylic hydrogens of C18and C15 and subsequent peroxide formation at C20 and C16of DHA (Schneider et al. 2001, 2004). In vitro DHAoxidation studies indicate that the C20 and C16 hydroper-oxides combined are the most abundant hydroperoxidesformed (VanRollins and Murphy 1984). Whether the C20and C16 hydroperoxides of DHA are the most abundant onesformed in vivo under pathological or control conditions arenot known.
Fig. 8 Trans-4-hydroxy-2-hexenal (HHE) formation does not mirror
F4-neuroprostane (F4-NP) formation from docosahexaenoic acid
(DHA) oxidation. (a) DHA (10 mmol/L) was oxidized in the presence of
Fe2+ (50 lmol/L)/ascorbate (1 mmol/L) at either pH 6.5 or 7.4. Sam-
ples were taken at the timepoints shown and analyzed by gas chro-
matography with negative-ion chemical ionization mass spectrometry
(GC-NICI/MS) for HHE and F4-NP content. Data presented are the
mean ± SD, n = 3. Two-way ANOVA revealed no significant effect of pH
upon HHE or F4-NP production although the time of incubation was a
significant factor for both products.
Fig. 9 Trans-4-hydroxy-2-hexenal (HHE) is a less efficient substrate
for aldehyde dehydrogenase class 5A (ALDH5A) detoxification versus
trans-4-hydroxy-2-nonenal (HNE). HHE and HNE were compared as
substrates for recombinant rat ALDH5A. NADH formation (Abs
340 nm) was used as a measure of activity. Non-linear regression
analysis utilizing the Michaelis–Menten equation was used to deter-
mine Km and Vmax. Assays contained 3 lg of purified enzyme in a
0.3 mL final volume. The calculated Km’s were 388 ± 40 lmol/L for
HHE and 28.5 ± 2.6 lmol/L for HNE. The calculated Vmax’s were
3.52 ± 0.2 lmol/min/mg for HHE and 1.7 ± 0.1 lmol/min/mg for HNE.
Data are the mean ± SD for assays performed in triplicate.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
722 | E. K. Long et al.

Our data demonstrate differences in the rates of formation ofHHE versus F4-NPs. Oxidation of DHA produced similarlevels of HHE and F4-NPs after 2 h, but far greater amounts ofHHE than F4-NPs after 6 h. The increase in HHE from 2 to 6 his consistent with a transition from the initiation to thepropagation phase of DHA oxidation (van Kuijk et al. 1995).However, the amounts of F4-NP formed after 6 h are notconsistent with a transition to the propagation phase, as levelsdo not appear to have increased exponentially. The mecha-nisms underlying the differences in formation are complex. F4-NPs are only a fraction of the total class of NP that are formedfrom DHA oxidation (Roberts et al. 1998). Furthermore, F4-NPs result from reduction of the bicyclic endoperoxide whoserearrangement can result in alternative products such asneuroketals (Roberts et al. 1999; Bernoud-Hubac et al. 2001).In contrast, formation of HHE from DHA likely occurs in afashion similar to formation of HNE from n-6 fatty acids andwould not require multiple rearrangements (Schneider et al.2001, 2004). As their kinetics of formation vary significantly,HHE and F4-NPs cannot be used interchangeably as biomar-kers of the magnitude of oxidative damage to DHA.
In summary, our data indicate that HHE is a neurotoxicaldehyde generated from DHA oxidation. Further study isneeded to determine the extent to which HHE levels may beelevated in disease conditions with increased F4-NP concen-trations like Alzheimer’s disease (Reich et al. 2001). Thesecurrent data along with those examining the neurotoxicity ofacrolein and HNE indicate that lipid peroxidation spawns theformation of multiple neurotoxic species with similar anddivergent effects (Keller et al. 1997, 1999; Kruman et al.1997; Lovell et al. 2000).
Acknowledgements
The authors gratefully acknowledge NIH grants P20 RR17699-01
COBRE (MJP) from the NCRR, AA15145-01 (MJP) GM15431
(JDM), ES131215 (JDM), ES000267 (JDM), and DK48831 (JDM).
References
Arakawa M., Ushimaru N., Osada N., Oda T., Ishige K. and Ito Y.(2006) N-acetylcysteine selectively protects cerebellar granulecells from 4-hydroxynonenal-induced cell death. Neurosci. Res.55, 255–263.
Bernoud-Hubac N., Davies S. S., Boutaud O., Montine T. J. and RobertsL. J. 2nd (2001) Formation of highly reactive gamma-ketoalde-hydes (neuroketals) as products of the neuroprostane pathway.J. Biol. Chem. 276, 30964–30970.
Brichac J., Ho K. K., Honzatko A., Wang R., Lu X., Weiner H. andPicklo M. (2007) Enantioselective oxidation of trans-4-hydroxy-2-nonenal is aldehyde dehydrogenase isozyme and Mg2+-dependent.Chem. Res. Toxicol. 20, 887–895.
Chi L., Ke Y., Luo C., Gozal D. and Liu R. (2007) Depletion of reducedglutathione enhances motor neuron degeneration in vitro and invivo. Neuroscience 144, 991–1003.
Choudhary S., Xiao T., Srivastava S., Zhang W., Chan L. L., VergaraL. A., Van Kuijk F. J. and Ansari N. H. (2005) Toxicity and
detoxification of lipid-derived aldehydes in cultured retinal pig-mented epithelial cells. Toxicol. Appl. Pharmacol. 204, 122–134.
Dringen R., Pfeiffer B. and Hamprecht B. (1999) Synthesis of theantioxidant glutathione in neurons: supply by astrocytes of CysGlyas precursor for neuronal glutathione. J. Neurosci. 19, 562–569.
Esterbauer H., Dieber-Rothneder M., Waeg G., Striegl G. and Jurgens G.(1990) Biochemical, structural, and functional properties of oxi-dized low-density lipoprotein. Chem. Res. Toxicol. 3, 77–92.
He Q., Khanna P., Srivastava S., van Kuijk F. J. and Ansari N. H.(1998) Reduction of 4-hydroxynonenal and 4-hydroxyhexenal byretinal aldose reductase. Biochem. Biophys. Res. Commun. 247,719–722.
Hoffman W. E., Charbel F. T., Edelman G., Misra M. and Ausman J. I.(1998) Comparison of the effect of etomidate and desflurane onbrain tissue gases and pH during prolonged middle cerebral arteryocclusion. Anesthesiology 88, 1188–1194.
Honzatko A., Brichac J., Murphy T. C., Reberg A., Kubatova A.,Smoliakova I. P. and Picklo M. J. Sr (2005) Enantioselectivemetabolism of trans-4-hydroxy-2-nonenal by brain mitochondria.Free Radic. Biol. Med. 39, 913–924.
Hubatsch I., RidderstromM. andMannervikB. (1998)Human glutathionetransferase A4-4: an alpha class enzyme with high catalytic effi-ciency in the conjugation of 4-hydroxynonenal and other genotoxicproducts of lipid peroxidation. Biochem. J. 330 (part 1), 175–179.
Je J. H., Lee J. Y., Jung K. J., Sung B., Go E. K., Yu B. P. and Chung H. Y.(2004) NF-kappaB activation mechanism of 4-hydroxyhexenal viaNIK/IKK and p38 MAPK pathway. FEBS Lett. 566, 183–189.
Kawai Y., Fujii H., Okada M., Tsuchie Y., Uchida K. and Osawa T.(2006) Formation of nepsilon-(succinyl)lysine in vivo: a novelmarker for docosahexaenoic acid-derived protein modification.J. Lipid Res. 47, 1386–1398.
Kawai Y., Takeda S. and Terao J. (2007) Lipidomic analysis for lipidperoxidation-derived aldehydes using gas chromatography-massspectrometry. Chem. Res. Toxicol. 20, 99–107.
Keller J. N., Mark R. J., Bruce A. J., Blanc E., Rothstein J. D., UchidaK., Waeg G. and Mattson M. P. (1997) 4-Hydroxynonenal, analdehydic product of membrane lipid peroxidation, impairs gluta-mate transport and mitochondrial function in synaptosomes.Neuroscience 80, 685–696.
Keller J. N., Hanni K. B. and Markesbery W. R. (1999) 4-hydroxy-nonenal increases neuronal susceptibility to oxidative stress.J. Neurosci. Res. 58, 823–830.
Kristal B. S., Park B. K. and Yu B. P. (1996) 4-Hydroxyhexenal is apotent inducer of the mitochondrial permeability transition. J. Biol.Chem. 271, 6033–6038.
Kruman I., Bruce-Keller A. J., Bredesen D., Waeg G. and Mattson M. P.(1997) Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J. Neurosci. 17, 5089–5100.
van Kuijk F. J., Thomas D. W., Stephens R. J. and Dratz E. A. (1986)Occurrence of 4-hydroxyalkenals in rat tissues determined aspentafluorobenzyl oxime derivatives by gas chromatography-massspectrometry. Biochem. Biophys. Res. Commun. 139, 144–149.
van Kuijk F. J., Siakotos A. N., Fong L. G., Stephens R. J. and ThomasD. W. (1995) Quantitative measurement of 4-hydroxyalkenals inoxidized low-density lipoprotein by gas chromatography-massspectrometry. Anal. Biochem. 224, 420–424.
Lee Y. S. and Wander R. C. (2005) Reduced effect on apoptosis of 4-hydroxyhexenal and oxidized LDL enriched with n-3 fatty acidsfrom postmenopausal women. J. Nutr. Biochem. 16, 213–221.
Lee J. Y., Je J. H., Jung K. J., Yu B. P. and Chung H. Y. (2004a)Induction of endothelial iNOS by 4-hydroxyhexenal through NF-kappaB activation. Free Radic. Biol. Med. 37, 539–548.
Lee J. Y., Je J. H., Kim D. H., Chung S. W., Zou Y., Kim N. D., Ae YooM., Suck Baik H., Yu B. P. and Chung H. Y. (2004b) Induction of
� 2008 The AuthorsJournal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724
HHE is a neurotoxin | 723

endothelial apoptosis by 4-hydroxyhexenal. Eur. J. Biochem. 271,1339–1347.
Levine R. L., Garland D., Oliver C. N., Amici A., Climent I., Lenz A.G., Ahn B. W., Shaltiel S. and Stadtman E. R. (1990) Determi-nation of carbonyl content in oxidatively modified proteins.Methods Enzymol. 186, 464–478.
Lim S. Y., Doherty J. D. and Salem N. Jr (2005) Lead exposure and (n-3)fatty acid deficiency during rat neonatal development alter liver,plasma, and brain polyunsaturated fatty acid composition. J. Nutr.135, 1027–1033.
Lovell M. A., Xie C. and Markesbery W. R. (2000) Acrolein, a productof lipid peroxidation, inhibits glucose and glutamate uptake inprimary neuronal cultures. Free Radic. Biol. Med. 29, 714–720.
Lovell M. A., Xie C. and Markesbery W. R. (2001) Acrolein is increasedin Alzheimer’s disease brain and is toxic to primary hippocampalcultures. Neurobiol. Aging 22, 187–194.
Malone P. E. and Hernandez M. R. (2007) 4-Hydroxynonenal, a productof oxidative stress, leads to an antioxidant response in optic nervehead astrocytes. Exp. Eye Res. 84, 444–454.
Milne G. L., Morrow J. D. and Picklo M. J. Sr (2006) Elevated oxidationof docosahexaenoic acid, 22:6 (n-3), in brain regions of ratsundergoing ethanol withdrawal. Neurosci. Lett. 405, 172–174.
Montine K. S., Olson S. J., Amarnath V., Whetsell W. O. Jr, GrahamD. G. and Montine T. J. (1997) Immunohistochemical detection of4-hydroxy-2-nonenal adducts in Alzheimer’s disease is associatedwith inheritance of APOE4. Am. J. Pathol. 150, 437–443.
Montine K. S., Reich E., Neely M. D., Sidell K. R., Olson S. J.,Markesbery W. R. and Montine T. J. (1998) Distribution ofreducible 4-hydroxynonenal adduct immunoreactivity in Alzhei-mer disease is associated with APOE genotype. J. Neuropathol.Exp. Neurol. 57, 415–425.
Montine T. J., Milatovic D., Gupta R. C., Valyi-Nagy T., Morrow J. D.and Breyer R. M. (2002) Neuronal oxidative damage from acti-vated innate immunity is EP2 receptor-dependent. J. Neurochem.83, 463–470.
Mosmann T. (1983) Rapid colorimetric assay for cellular growth andsurvival: application to proliferation and cytotoxicity assays.J. Immunol. Methods 65, 55–63.
Murphy T. C., Amarnath V. and Picklo M. J. Sr (2003a) Mitochondrialoxidation of 4-hydroxy-2-nonenal in rat cerebral cortex. J. Neu-rochem. 84, 1313–1321.
Murphy T. C., Amarnath V., Gibson K. M. and Picklo M. J. Sr (2003b)Oxidation of 4-hydroxy-2-nonenal by succinic semialdehydedehydrogenase (ALDH5A). J. Neurochem. 86, 298–305.
Musiek E. S., Cha J. K., Yin H., Zackert W. E., Terry E. S., Porter N. A.,Montine T. J. and Morrow J. D. (2004) Quantification of F-ringisoprostane-like compounds (F4-neuroprostanes) derived fromdocosahexaenoic acid in vivo in humans by a stable isotope dilu-tion mass spectrometric assay. J. Chromatogr. B Analyt. Technol.Biomed. Life Sci. 799, 95–102.
Neely M. D., Sidell K. R., Graham D. G. and Montine T. J. (1999) Thelipid peroxidation product 4-hydroxynonenal inhibits neurite out-growth, disrupts neuronal microtubules, and modifies cellulartubulin. J. Neurochem. 72, 2323–2333.
Neely M. D., Zimmerman L., Picklo M. J., Ou J. J., Morales C. R.,Montine K. S., Amaranth V. and Montine T. J. (2000) Congeners ofN(alpha)-acetyl-L-cysteine but not aminoguanidine act as neuro-protectants from the lipid peroxidation product 4-hydroxy-2-non-enal. Free Radic. Biol. Med. 29, 1028–1036.
Neely M. D., Boutte A., Milatovic D. and Montine T. J. (2005) Mech-anisms of 4-hydroxynonenal-induced neuronal microtubule dys-function. Brain Res. 1037, 90–98.
Nemoto E. M. and Frinak S. (1981) Brain tissue pH after globalbrain ischemia and barbiturate loading in rats. Stroke 12, 77–82.
PaschenW., Djuricic B., Mies G., Schmidt-Kastner R. and Linn F. (1987)Lactate and pH in the brain: association and dissociation in differentpathophysiological states. J. Neurochem. 48, 154–159.
Pawlosky R. J., Bacher J. and Salem N. Jr (2001) Ethanol consumptionalters electroretinograms and depletes neural tissues ofdocosahexaenoic acid in rhesus monkeys: nutritional consequencesof a low n-3 fatty acid diet. Alcohol. Clin. Exp. Res. 25, 1758–1765.
Perrin R. J., Woods W. S., Clayton D. F. and George J. M. (2000)Interaction of human alpha-synuclein and Parkinson’s diseasevariants with phospholipids. Structural analysis using site-directedmutagenesis. J. Biol. Chem. 275, 34393–34398.
Perrin R. J., Woods W. S., Clayton D. F. and George J. M. (2001)Exposure to long chain polyunsaturated fatty acids triggersrapid multimerization of synucleins. J. Biol. Chem. 276, 41958–41962.
Picklo M. J., Montine T. J., Amarnath V. and Neely M. D. (2002)Carbonyl toxicology and Alzheimer’s disease. Toxicol. Appl.Pharmacol. 184, 187–197.
Qin Z., Hu D., Han S., Reaney S. H., Di Monte D. A. and Fink A. L.(2007) Effect of 4-hydroxy-2-nonenal modification on alpha-syn-uclein aggregation. J. Biol. Chem. 282, 5862–5870.
Reich E. E., Markesbery W. R., Roberts L. J. 2nd, Swift L. L., MorrowJ. D. and Montine T. J. (2001) Brain regional quantification of F-ring and D-/E-ring isoprostanes and neuroprostanes in Alzheimer’sdisease. Am. J. Pathol. 158, 293–297.
Roberts L. J. 2nd,Montine T. J.,MarkesberyW.R., TapperA. R., Hardy P.,Chemtob S., Dettbarn W. D. and Morrow J. D. (1998) Formation ofisoprostane-like compounds (neuroprostanes) in vivo from docosa-hexaenoic acid. J. Biol. Chem. 273, 13605–13612.
Roberts L. J. 2nd, Salomon R. G., Morrow J. D. and Brame C. J. (1999)New developments in the isoprostane pathway: identification ofnovel highly reactive gamma-ketoaldehydes (isolevuglandins)and characterization of their protein adducts. FASEB J. 13, 1157–1168.
Salem N. Jr, Litman B., Kim H. Y. and Gawrisch K. (2001) Mechanismsof action of docosahexaenoic acid in the nervous system. Lipids 36,945–959.
Sayre L. M., Zelasko D. A., Harris P. L., Perry G., Salomon R. G. andSmith M. A. (1997) 4-Hydroxynonenal-derived advanced lipidperoxidation end products are increased in Alzheimer’s disease.J. Neurochem. 68, 2092–2097.
Schneider C., Tallman K. A., Porter N. A. and Brash A. R. (2001) Twodistinct pathways of formation of 4-hydroxynonenal. Mechanismsof nonenzymatic transformation of the 9- and 13-hydroperoxides oflinoleic acid to 4-hydroxyalkenals. J. Biol. Chem. 276, 20831–20838.
Schneider C., Porter N. A. and Brash A. R. (2004) Autoxidativetransformation of chiral omega6 hydroxy linoleic and arachidonicacids to chiral 4-hydroxy-2E-nonenal. Chem. Res. Toxicol. 17,937–941.
Shibata N., Yamada S., Uchida K. et al. (2004) Accumulation of protein-bound 4-hydroxy-2-hexenal in spinal cords from patients withsporadic amyotrophic lateral sclerosis. Brain Res. 1019, 170–177.
Van Kuijk F. J., Holte L. L. and Dratz E. A. (1990) 4-Hydroxyhexenal: alipid peroxidation product derived from oxidized docosahexaenoicacid. Biochim. Biophys. Acta 1043, 116–118.
VanRollins M. and Murphy R. C. (1984) Autooxidation of docosa-hexaenoic acid: analysis of ten isomers of hydroxydocosahexae-noate. J. Lipid Res. 25, 507–517.
Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E. R. andMizuno Y. (1996) Immunohistochemical detection of 4-hydroxy-nonenal protein adducts in Parkinson disease. Proc. Natl Acad. Sci.USA 93, 2696–2701.
Journal Compilation � 2008 International Society for Neurochemistry, J. Neurochem. (2008) 105, 714–724� 2008 The Authors
724 | E. K. Long et al.




















