
thesis final 20171117 for macbook version 6
154
Cover Page The handle http://hdl.handle.net/1887/57798 holds various files of this Leiden University dissertation Author: Ren, Baoyan Title: Bone marrow transplantation in mice as a tool to study M2 macrophage activation in atherogenesis Date: 2017-12-14
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of thesis final 20171117 for macbook version 6

Cover Page
The handle http://hdl.handle.net/1887/57798 holds various files of this Leiden University dissertation Author: Ren, Baoyan Title: Bone marrow transplantation in mice as a tool to study M2 macrophage activation in atherogenesis Date: 2017-12-14

BonemarrowtransplantationinmiceasatooltostudyM2macrophageactivationin
atherogenesis
Baoyan(Olive)Ren
78


BonemarrowtransplantationinmiceasatooltostudyM2macrophageactivationin
atherogenesis
PROEFSCHRIFT
terverkrijgingvan
degraadvanDoctoraandeUniversiteitLeiden,
opgezagvandeRectorMagnificusprof.mr.C.J.J.M.Stolker,
volgensbesluitvanhetCollegevoorPromoties
teverdedigenopdonderdag14december2017
teklokke10.00uur
door
BaoyanRen
GeborenteDingzhou,China
in1985

Promotor: Prof.dr.M.vanEck
Promotiecommissie:
Prof.dr.H.Irth LACDR,Leiden (voorzitter)
Prof.dr.J.A.Bouwstra LACDR,Leiden (secretaris)
OverigeLeden: Prof.dr.J.A.P.WillemsvanDijk LUMC,Leiden
Prof.dr.E.A.L.Biessen MUMC,Maastricht
Prof.dr.M.P.J.deWinther AMC,Amsterdam
The studies presented in this thesis were performed at the Division ofBiopharmaceutics, Leiden Academic Centre for Drug research (LACDR), LeidenUniversity,Leiden,TheNetherlands.
Financial support by the Dutch Heart Foundation and the Leiden UniversityFoundationforthepublicationofthisthesisisgratefullyacknowledged.

�
�
3 4 C 0
-.- 3 2
SixtyyearsagoIkneweverything;nowIknownothing;educationisaprogressivediscoveryofourownignorance.
--WillDurant
Formyfamily
B
� �

�
�
�
�
�
�
�
�
�
�
�
BonemarrowtransplantationinmiceasatooltostudyM2macrophageactivationinatherogenesis/BaoyanRen
Cover� Schematicdiagramof arterial atherosclerotic lesion (front) originallyderivedfromahealthyartery(back),paintedbyMengyinAn
Layout: BaoyanRen
Printing: RidderprintBV,Ridderkerk,TheNetherlands
ISBN: 978-94-6299-810-0
ProefschriftLeiden
Metliteratuuropgave–MetsamenvattinginhetNederlands
©2017BaoyanRenNopartofthisthesismaybereproducedortransmittedinanyformorbyanymeans,withoutwrittenpermissionfromtheauthor.

Tableofcontents
Chapter1Generalintroduction......................................................................................9
Chapter2HematopoieticArginase1deficiencyresultsindecreasedleukocytosisandincreasedfoamcellformationbutdoesnotaffectatherosclerosis.................................41Atherosclerosis256(2017):35-46.........................................................................................41
Chapter3HematopoieticAkt2restorationenhancesfoamcellformationbutdoesnotaffectatherosclerosisinAkt2/LDLreceptordoubleknockoutmice.................................63Manuscriptinpreparation....................................................................................................63
Chapter4MacrophageMKP2deficiencyisassociatedwithanM2-drivenfoamcellphenotypeandincreasesatherosclerosissusceptibilityofLDLreceptorknockoutmice..79Submittedforpublication......................................................................................................79
Chapter5EnhancedatheroscleroticlesiondevelopmentinLDLreceptorknockoutmicelackingUpstreamStimulatingFactor1(Usf1)inbonemarrow-derivedcells...................97Submittedforpublication......................................................................................................97
Chapter6Summaryandperspectives.........................................................................119
6.1 Englishsummary......................................................................................................120
6.2 Nederlandsesamenvatting......................................................................................129
Abbreviations...............................................................................................................145
Listofpublications.......................................................................................................147
CurriculumVitae..........................................................................................................149
PhDPortfolio................................................................................................................151


Chapter1
Chapter
Generalintroduction

Chapter1
10
Tableofcontents
Chapter1Generalintroduction.........................................................................................9
1 Atherosclerosis...........................................................................................................11
1.1 Lipoproteins:metabolismandassociationwithatherosclerosis............................111.2 Generalpathogenesisunderlyingatheroscleroticlesiondevelopment..................14
2 Theroleofmonocytesandmacrophagesinthepathologyofatherosclerosis..........15
2.1 Monocyteheterogeneity........................................................................................152.2 M1/M2macrophagesandatherosclerosis.............................................................162.3 Macrophagephenotypeandfoamcellsusceptibility.............................................172.4 Macrophagephenotypeswitch..............................................................................17
3 Experimentalmousemodelsandstrategiesforstudyingatherosclerosis.................17
3.1 Miceandatherosclerosissusceptibility..................................................................173.2 ApoEKOmiceandLDLrKOmice.............................................................................183.3 Bonemarrowtransplantation.................................................................................18
4 Signalingpathwaysandatherosclerosis.....................................................................19
4.1 Nitricoxide/L-argininepathwayinatherosclerosis.................................................204.2 ProteinkinaseB(Akt)inatherosclerosis................................................................224.3 MAPKs/MKPspathwayinatherosclerosis...............................................................234.4 Usftranscriptionfactorsandatherosclerosis.........................................................25
5 OutlineofthisThesis..................................................................................................26
6 References..................................................................................................................26

Chapter1
11
1 Atherosclerosis
therosclerosis is a blood vessel narrowing and hardening disease characterized by thedeposition of cholesterol locally in the arterial wall, leading to a low-grade chronic
inflammation.1 Atherosclerotic lesions take decades to become large enough as to have asignificant effect and cause cardiovascular complications in humans.2 Atherosclerosis caneventuallyleadtoseriousproblemssuchaschestpain(angina),heartfailure,heartattack,stroke,ischemicattack,aneurysms,orevendeath.3Nowadays,atheroscleroticcardiovasculardiseaseistheleadingcauseofmortalityworldwide,accountingfor31%ofglobaldeathsin2015.4Althoughitiscloselyrelatedtocontemporarylifestyles,atherosclerosisisnotonlyfoundinmodernhumanbeings.5 In contrast, the disease has been found in 4000 years oldmummies and the earliestliteraturethatdescribedthepathologicalchangesofatherosclerosiscanbetracedbackto442years ago. In the year 1575, the Italian anatomist Gabriel Fallopius had already describedcalcificationofthearterialwall,apathologicalphenomenonofatheroscleroticplaques.6Lateron,several researchers andmedical doctors had observed atherosclerosis in large arteries,7-9 andproposed a connection of atherosclerosis with angina and ischemic heart disease.10 The firstdescriptionofplaquerupturewasreported in1844.10 In1829, JeanLobstein for the first timeintroducedtheword“arteriosclerosis”inhisunfinishedbook“Traitéd’AnatomiePathologique”whichwasultimatelypublishedintheyear1933,11,12almost100yearsafterhisdeath.However,ingeneraltheGermanpathologistFelixMarchand(1904)isrecognizedasthefirstusingtheterm“atherosclerosis”,stemmingfromtheGreekwordsofporridge“athero”andhardening“sclerosis”,todescribethefat-richmaterialsthataccumulatedinsideahardenedartery.13,14
Since its discovery, researchers have been trying to uncover the etiopathogenesis ofatherosclerosis.Attheendof18thcentury,theoriesunderlyingatherosclerosisdevelopmentwereproposed by Carl von Rokitansky and Rudolf Virchow15who both recognized the presence ofinflammation.However, itwasunclearwhether inflammationplayedacausativerole.CarlvonRokitansky considered atherosclerosis to be the result of the buildup of fibrin or other bloodelements,whichsubsequentlywasmodifiedtoalipid-richplaqueinthearterialwall.14Incontrast,RudolfVirchowsuggestedthattheinflammatoryresponsetolipidinsudationorintimalinjuryisthecauseofatherosclerosis.15Theexactpathogenesisofatherosclerosisremainedunclearuntilthe“responsetoinjuryhypothesis”,initiallyproposedbyRudolfVirchowandrevivedbyRussellRossin1999,becameawidelyacceptedtheory.16
1.1 Lipoproteins:metabolismandassociationwithatherosclerosis
Hyperlipidemiaplaysa leadingrole intriggeringandpromotingatherosclerosisdevelopment.17Lipoproteins,being themaincarrierofcholesterol in thecirculation,were firstlyassociated tocardiovasculardisease in1949by JohnGofmanandhis colleagues.They found that increasedlevelsoflow-densitylipoprotein(LDL)cholesterolareassociatedwithanincreasedcardiovascularrisk,andthatpatientswithfamilialhypercholesterolemiaarepredisposedtothedevelopmentofprematureatherosclerosis.18Thesepatientshaveanoverallcholesterolelevationintheirplasma,whichcanmainlybeattributedtoanincreaseinLDLandintermediate-densitylipoprotein(IDL).14Thehypothesisthathighplasmalipidlevelsareassociatedwithincreasedcardiovascularriskwasfurthersupportedbyacooperativestudyperformedinthe1950sand1960s,whichconfirmedtheconnectionofcardiovascularrisktoplasmacholesterol levelsusingpatientcohortsfromsevendifferentcountries.19Circulatinglipidsaretransportedbylipoproteins,particlescomposedofashellofamonolayerofphospholipidswithfreecholesterolandapolipoproteins(Apo)andalipid-
A

Chapter1
12
richcoreconstitutedofesterifiedcholesterolandtriglycerides.20Basedontheproportionofeachcomponent as well as particle density, lipoproteins are classed into 5 groups: high-densitylipoprotein(HDL),low-densitylipoprotein(LDL),intermediate-densitylipoprotein(IDL),very-low-density lipoprotein (VLDL), and chylomicrons. Except for HDL, all these subclasses containapolipoprotein B (ApoB) as their major apolipoprotein and are considered athero-promotinglipoproteins.Incontrast,HDLhasapolipoproteinAasitsprimaryapolipoproteinandisconsideredtoactasanathero-protectivelipoprotein.
1.1.1 ApoB-containinglipoproteinsandatherosclerosis
Chylomicrons,ApoB48-containinglipoproteins,arethemajorcarriersforlipidsabsorbedfromthediet in the intestine, and represent amajor source of triglycerides (TG) for various tissues. InadditiontoTG,chylomicronstransportcholesterol,butonlya limitedamount.21Therefore, forquitea longtimechylomicronswerebelievednottocontributetoatherogenesis.22,23However,lateronApoB48wasfoundinatheroscleroticplaques24,25andanApoB48-specificreceptorwasdetected in human and murine macrophages,26,27 thereby highlighting the contribution ofchylomicronstoatherosclerosisdevelopment.Ingeneral, increasedchylomicronlevelsseemtogenerateapro-atherogenicprofile,however,thereisstillnotmuchevidenceshowingadirectlinkbetweenhighchylomicronlevelsandatherogenesis.28
Inhumans,andspecifically inwomenwithelevatedTGlevels, increasedriskforcardiovasculareventswasshownina11.4yearsfollow-upstudyinAmerica.29Interestingly,bothfastingandnon-fastingTGlevelsareassociatedwithcardiovasculardisease,withpostprandialTGlevelsshowingthe strongest association with future cardiovascular risk.29-31 Interestingly, the strong linearcorrelationbetweenplasmaTGandatherosclerosisismostlikelygender-dependent,ofwhichTGlevelsinfemaleshaveshowntobethebestpredictorforcardiovascularriskinbothhumanandmice.29,32
Fasting TG levels are determinedby the amount of TG transportedbyVLDLparticles. VLDL issynthesizedbyhepatocytes,33andservesastheprecursorforIDLandLDL.Similartochylomicrons,VLDLisalsoaTG-richlipoprotein.Incontrasttochylomicronsthatcarryexogenous(dietary)lipids,VLDLtransportsendogenouslipidproductstoperipheraltissues.HighVLDLlevelsareconsidereda risk factor for coronary artery disease. Likely the lipid composition of the VLDL particledeterminescardiovascularrisk.Forexample,VLDLisastrongpredictorforcardiovascularriskinfemaleswithasignificantlyhigherVLDLcholesterol/TGratiothanmales.32
IDL, as the remnant of VLDL and precursor of LDL, is also considered a causal factor for thedevelopmentofatherosclerosis.34TheIDLconcentrationhasbeenassociatedwiththeincidenceofcoronaryarterydisease,especiallyinpatientswithnormalcholesterollevels.35-38However,asanintermediateformbetweenVLDLandLDL,theexactroleofIDLinatherosclerosisisless-welldefined. The circulating IDLparticles arequickly takenupby the liver,or converted to LDLbyundergoingtriglyceridehydrolysisinperipheraltissues.
Inhumans,LDListheprimarycarrierofcholesterol,accountingfor70-80%ofthetotalcholesterolconcentration in the circulation. Importantly, each LDL particle contains a single copy ofApoB100.39 Hence, by analyzing the ApoB100 concentration, LDL particle numbers in thecirculation can be calculated. Plasma LDL cholesterol is highly associatedwith atherosclerosisdevelopment. Noteworthy, not only the concentration of LDL cholesterol, but also the

Chapter1
13
heterogeneityoftheLDLparticles,playsanimportantroleinatherogenesis.LDLiscomprisedofmultiple subclasses that differ in size and density and each contribute distinctly to thesusceptibility for cardiovascular disease.40,41 The size and density of LDL varies with its lipidcontent.42SmallanddenseLDLparticlesaremoreatherogenic,duetotheirhigherpenetrationcapabilitiesoftheendothelialbarrier43,44andgreateroxidationpotentialcomparedtothelarger,lessdenseLDLparticles.45EpidemiologicalstudiesshowedthatthesmallanddenseLDLparticlesareassociatedwithaclusterofcardiovasculardisease risk factors, includingelevated levelsofplasma TG and ApoB, reduced concentrations of HDL cholesterol, and impaired insulinsensitivity.46,47Thus,atherogenesisisnotonlyaffectedbytheamountofcholesteroltransportedbyLDL,butalsobythecharacteristicsandheterogeneityoftheLDLparticles.48
1.1.2 High-densitylipoproteinsandatherosclerosis
IncontrasttoLDLthatpromotesatherogenesis,high-densitylipoprotein(HDL)isconsideredtoprotectagainstatherosclerosis.HDLremovescholesteroloutoflipid-richtissuesandtransportsittotheliver,aprocesscommonlyreferredtoasreversecholesteroltransport(RCT).49,50RCTisacomplicated process involving various steps. First, cholesterol efflux from peripheral cells,includingfoamcellsinthearterialwall,isfacilitatedbytheATP-bindingcassette(ABC)transportersABCA1orABCG1,whichmediatetheeffluxofintracellularlipidstolipid-poorApoA1(nascentHDL)andmatureHDLinthecirculation,respectively.51,52UponuptakebyHDL,theeffluxedcholesterolisesterified,via lecithincholesterolacyltransferase (LCAT),andtransferredtothecoreofHDL,resulting intheremodelingandmaturationoftheHDLparticle.Next,thecholesterol inHDListransferredtothe liver,eitherviaselectiveuptakeofHDL-cholesterolbyscavengerreceptorBI(SR-BI),holoparticleuptakeviatheLDLr,orindirectly,viathetransferofcholesteroltoother,TG-rich,lipoproteins,throughcholesterylestertransferprotein(CETP)whicharesubsequentlyalsofluxedbacktotheliver.Here,thecholesteroltakenupbytheliverisexcretedintobileandfecesorusedassubstratefordenovocholesterolsynthesis.Importantly,miceandratsnaturallylackCETPactivity,andhenceCETP-inducedcholesteroltransferdoesnotoccurintheseanimals.53
SinceHDLcanmediatecholesteroleffluxfromlipid-richmacrophages,HDLhasbeenidentifiedasanimportantanti-atherogenicparticle.54Theanti-atherogenicpropertiesofHDL,however,extendbeyondtheremovalofexcess lipidfromthevascularwall.54,55Forexample,HDLcanalsoexertantioxidanteffects,asunderoxidizingconditions,thepresenceofHDLcansignificantlydecreaselipid peroxide concentrations within the LDL particle.56,57 In addition, HDL inhibits monocyteadhesiontothevesselwall,bysuppressingtheexpressionofendothelialadhesionandmigrationmolecules.58Finally,HDLalsoprotectsagainstdamageinflictedbyinflammatorymediatorstotheendothelium,andpreventsthrombosisbyupregulatingnitricoxide(NO)productioninendothelialcells.59-61
ConsideringthewidearrayofatheroprotectivefunctionsofHDL,highHDLconcentrationswerelong thought to be associatedwith a reduction in coronary artery disease (CAD) risk. Indeed,epidemiologicalstudieshaveindicatedthatlowHDLcholesterolisassociatedwithanincreasedrisk.However,pharmacological inductionofplasmaHDLcholesterol levelsdidnotreduceCADrisk.62 In line, increased macrophage cholesterol efflux capability of human serum is alsoindependent of the HDL cholesterol level.63,64 HDL cholesterol efflux capacity, however, didstrongly correlate with the concentration of lipid-poor ApoAI (pre-β HDL).65,66 To explore thedevelopmentofanovelHDLbased-atheroprotectivetherapyagainstitisthusimportanttofocus

Chapter1
14
onmodulatingnascentHDL (withhighcholesteroleffluxcapacity) rather thanHDLcholesterollevels.67
1.2 Generalpathogenesisunderlyingatheroscleroticlesiondevelopment
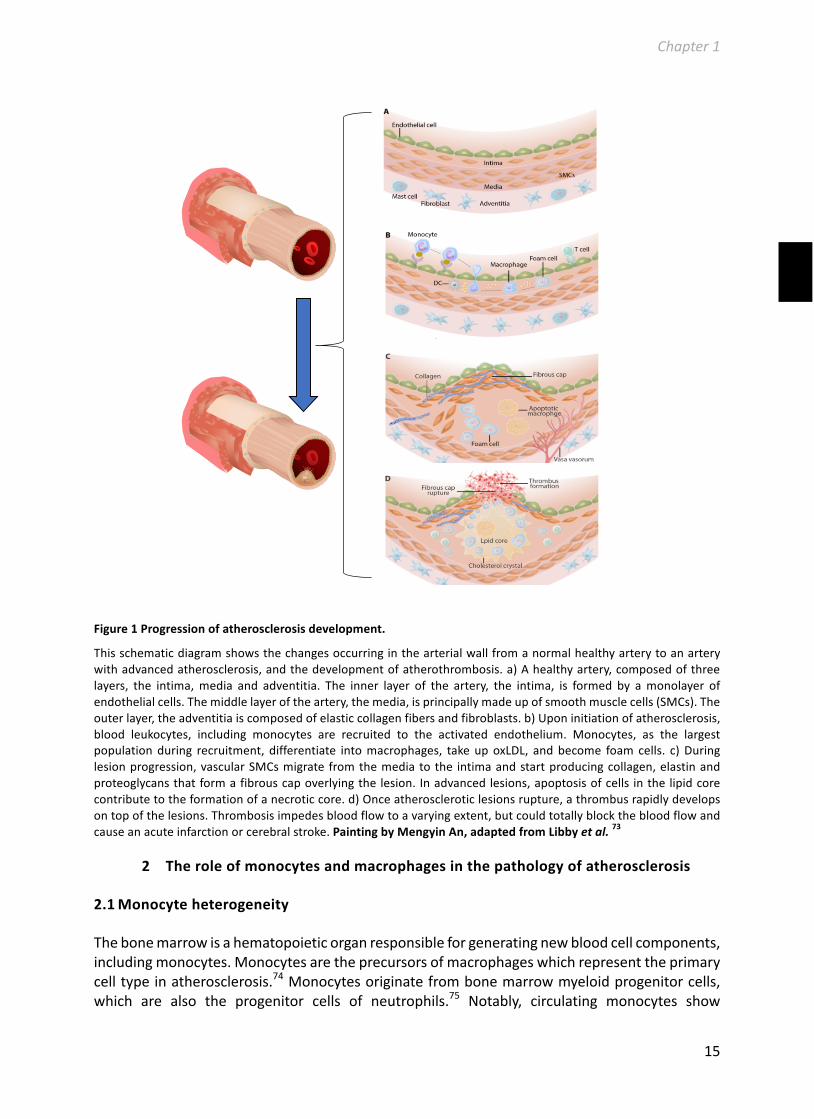
Thedevelopmentofatherosclerosisistheconsequenceofachronicinflammatoryreactionofthevascularwall, in response todyslipidemiaandendothelialdistress, involving the inflammatoryrecruitmentofleukocytesandtheactivationofresidentvascularcells.68Accordingtheresponse-to-injuryhypothesis,thedevelopmentofatherosclerosisisinitiatedbydysfunctionofthearterialendothelium.69Cardiovascularriskfactors,suchassmoking,hypertension,inflammation,age,andlipids (inparticularLDL),areknowntoaggravateendotheliumdysfunctionandactivation.Thiscauses the activated endothelial cells to start expressing surface factors that stimulate theinfiltrationofmonocytesfromthebloodstreamintotheintimaandsubintimalspace,wheretheydifferentiateintomacrophages.70LDLretainedinthearterialwall,mostlyafterextensiveoxidativemodification71,isphagocytizedbythemonocyte-derivedmacrophages,leadingtotheformationoflipid-richfoamcellsaswellthestartofachronicinflammatoryprocess.Monocytes,Tcellsandmastcellsallmigratetothesiteofactioninresponsetoinflammatorysignalsproducedatthesiteof theearly atherosclerotic lesion. These cells in turnwill contribute to the immune reaction,creating a progressive inflammatory environment in the developing plaque which furtheraccelerates atherosclerosis development. When the plaque macrophages are unable tosufficientlyeffluxtheirexcesscholesterol,theybecomeheavilylipid-ladenfoamcells.Ultimately,thesefoamcellsgrowinsizeanddie,therebyreleasingallcellularcholesterol intotheplaque,causing intraplaquecytotoxicityand furtheraggravationof the inflammatory response.At thisstage,theatheroscleroticplaqueconsistsofalipidcorewhichcontainscholesterol,cellulardebrisandinfiltratedimmunecells,coveredbyafibrouscap.Thiscollagen-richfibrouscapoverlyingthelipid-coreoftheplaqueistheconsequenceofvascularsmoothmusclecells(VSMC)proliferationandprovidesstabilitytotheplaque.However,thinningofthisfibrouscapbymediatorssecretedbyinflammatorycellscanultimatelyresultinplaquerupture.71Inthisadvancedstageoflesiondevelopment,plaquestabilityandcorrelatedsusceptibilitytoplaquerupture,isdeterminedbythebalancebetweenVSMCsthatprotecttheplaque,andcytotoxicfactorsreleasedbyimmunecells/endothelialcellsthatdamagethefibrouscap.72Astableplaqueisusuallyrichinextracellularmatrix and smooth muscle cells and in most cases does not cause acute clinical symptoms.Ruptureoftheatheroscleroticplaque,orerosionoftheendotheliallayerleadtotheformationofa thrombus on top of the atherosclerotic lesion, the culprit for the development of acutecardiovascularevents(Figure1).
Chapter1
15
Figure1Progressionofatherosclerosisdevelopment.
Thisschematicdiagramshowsthechangesoccurringinthearterialwallfromanormalhealthyarterytoanarterywithadvancedatherosclerosis,andthedevelopmentofatherothrombosis.a)Ahealthyartery,composedofthreelayers, the intima,media and adventitia. The inner layer of the artery, the intima, is formed by amonolayer ofendothelialcells.Themiddlelayeroftheartery,themedia,isprincipallymadeupofsmoothmusclecells(SMCs).Theouterlayer,theadventitiaiscomposedofelasticcollagenfibersandfibroblasts.b)Uponinitiationofatherosclerosis,blood leukocytes, including monocytes are recruited to the activated endothelium. Monocytes, as the largestpopulationduring recruitment, differentiate intomacrophages, takeupoxLDL, andbecome foam cells. c)Duringlesionprogression,vascularSMCsmigrate fromthemediatothe intimaandstartproducingcollagen,elastinandproteoglycansthatformafibrouscapoverlyingthelesion. Inadvancedlesions,apoptosisofcells inthelipidcorecontributetotheformationofanecroticcore.d)Onceatheroscleroticlesionsrupture,athrombusrapidlydevelopsontopofthelesions.Thrombosisimpedesbloodflowtoavaryingextent,butcouldtotallyblockthebloodflowandcauseanacuteinfarctionorcerebralstroke.PaintingbyMengyinAn,adaptedfromLibbyetal.73
2 Theroleofmonocytesandmacrophagesinthepathologyofatherosclerosis
2.1 Monocyteheterogeneity
Thebonemarrowisahematopoieticorganresponsibleforgeneratingnewbloodcellcomponents,includingmonocytes.Monocytesaretheprecursorsofmacrophageswhichrepresenttheprimarycelltypeinatherosclerosis.74Monocytesoriginatefrombonemarrowmyeloidprogenitorcells,which are also the progenitor cells of neutrophils.75 Notably, circulating monocytes show

Chapter1
16
morphologicalheterogeneity.Inhumans,monocytesareidentifiedbytheexpressionofsurfacereceptorsCD14andCD16.Basedontheexpressionlevelsofthesetwomarkers,monocytesaredividedintotwomainsubtypes.Thefirstsubtypeisaso-called‘classical’monocyte,expressinghighlevelsofCD14andverylowlevelsofCD16(CD14hiCD16-).Thisclassicalmonocytepopulationalsohasahighproducingcapacityforpro-inflammatorycytokines.Thesecondsubtypeisthe‘non-classical’monocyte,whichischaracterizedbylowexpressionlevelsofCD14andhighexpressionlevels of CD16 (CD14+CD16++). Thesemonocytes are also called patrollingmonocytes, as theyconstantlypatrolthevasculature,andareinvolvedintheearlyresponsestopathogensandtissuerepair.76,77
Inmice,monocytesareclassifiedintotwosubtypesbasedontheexpressionofLy6CandCCR2,withLy6ChiCCR2+monocytesrepresentingtheequivalentofhumanCD14hiCD16-monocytes,andLy6ClowCCR2-beingequivalenttohumanCD14+CD16++monocytes.78-80Afterinfiltrationintothearterial wall, the monocytes are exposed to specific environmental factors, triggering theirdifferentiation into different types of macrophages that differentially contribute to theprogressionofatherosclerosis.81
2.2 M1/M2macrophagesandatherosclerosis
Similarastheirmonocyteprecursors,multipletypesofmacrophagescanbedistinguished.82Sincedifferentmacrophageactivation formswereproposedbyMackness83 in1962andGordon84 in1992, for M1 and M2 macrophages, respectively, more and more researchers have startedinvestigatingtherelationshipbetweenmacrophagephenotypesandatherosclerosis.BothM1andM2macrophageshavebeenimplicatedinatherosclerosis.Invitro,theM1macrophagephenotypecanbeinducedbyincubationwithLPSorIFN-gamma.85UponLPSactivation,macrophagessecretehigh levels of pro-inflammatory cytokines, including interleukin 1β (IL-1β), IL-6, IL-12, tumornecrosisfactorα(TNF-α),butlowlevelsofanti-inflammatorycytokinessuchasIL-10.Assuch,M1macrophages actively contribute to the persistent inflammatory environment in theatherosclerotic plaque, and thereby accelerate atherosclerosis development. In contrast, M2macrophagesareknowntoprotectagainstatherosclerosis.M2macrophagescanbeinducedbyIL-4, IL-10 and IL-13 and, uponactivation, producehigh levels of anti-inflammatory cytokines,including IL-10,and lowlevelsofpro-inflammatorycytokines,suchas IL-12.Besidesproducinganti-inflammatorycytokines,M2macrophagesalsoenhancetheproductionofpro-fibroticfactors,includingcollagen,andtherebypromotetissuerepairandremodeling.86Hence,M2macrophagesprotectagainstatherosclerosisnotonlybydecreasingthelocalinflammatorystatusoftheplaquebutalsobyincreasingplaquestability.Inmice,M1macrophagesexpresshighlevelsofinduciblenitricoxidesynthase(iNOS),whichrendersiNOSamurineM1markergene.InadditiontoiNOS,high expression levels of the pro-inflammatory cytokines TNFα, IL-1β, and IL-12 are alsoconsideredasM1markers.Incontrast,murineM2macrophagesareknowntoexpresshighlevelsofarginase1(Arg1).Additionally,YM1andFIZZ1andscavengerreceptors(CD204,87CD16388)arealsoconsideredM2macrophagemarkers.81

Chapter1
17
2.3 Macrophagephenotypeandfoamcellsusceptibility
Besidesplayingaroleintheimmuneresponse,anotherimportantfunctionofmacrophagesistoengulfforeignagents,includingoxidizedLDL(oxLDL).89MacrophagestakeupoxLDLandbecomefoamcells,aprocesswhichisconsideredtobeoneofthehallmarksofatherosclerosis.SR-A90andCD36 (scavenger receptor class B member 3) are the main receptors involved in foam cellformation, being responsible for up to 90% of the oxLDL uptake by macrophages in vitro.91Interestingly, both SR-A92 and CD3693 are upregulated duringM2macrophage differentiation,suggestinganincreasedsusceptibilityofM2macrophagestobecomefoamcells.Indeed,vanTitsetal. found that compared toM1macrophages,M2macrophagesaremoreprone to takeupoxLDL and become foam cells.94 This suggests that, in contrast to their atheroprotective anti-inflammatoryrole,M2macrophagesarealsolikelytoplayapro-atherogenicrolebypromotingmacrophagefoamcellformation.
2.4 Macrophagephenotypeswitch
Theprocessofmacrophagepolarizationisdynamic,asmacrophagescanrapidlyswitchfromonephenotype to another in response to a changing microenvironment.95-97 In atherosclerosis,numerous factors affect the lesional microenvironment, including cholesterol oxidation,inflammation mediators, infiltrated immune cells, growth factors, dead cells and othersubstances.98 Therefore, the lesional microenvironment changes with the different stages ofatherosclerosisdevelopment,therebyfurtherinfluencingmacrophagepolarization.99DaSilvaandcolleaguesfoundthatcholesterol loadingofhumanmacrophages limitedtheircapabilitytobeprimed to M1 macrophages, but not to M2 macrophages, suggesting an anti-inflammatorypropertyoffoamcells.100Furthermore,inresponsetooxidizedphospholipids,aproductoflipidoxidation,macrophagesareprimedtoaso-calledMoxphenotype.101Finally,inresponsetohaemandhaemoglobinexposureafterintraplaquehemorrhage,macrophagescanbepolarizedtowardsan Mhem phenotype.102 Noteworthy, both Mox and Mhem macrophages display a reducedcapacitytoengulfoxLDL,andarethusconsideredlesspronetofoamcellformation.101,103AllM1,M2,Mox,andMhemmacrophagephenotypeshavebeendemonstratedinatheroscleroticlesions.However,M1andM2macrophagesaresuggestedtoactasthemainprecursorsformacrophagefoamcells.104
3 Experimentalmousemodelsandstrategiesforstudyingatherosclerosis
3.1 Miceandatherosclerosissusceptibility
Multipleanimalspecieshavebeenusedasexperimentalmodelstostudyatheroscleroticlesiondevelopment,includingpigs,rabbits,monkeys,non-humanprimatesandmice.105-110Amongthesenon-humanmodels,micenowareconsideredthebestchoiceforstudyingatherogenesis,duetotheirlowcost,highreproductionrateandshorttimeframefordiseasedevelopment.106AlthoughC57BL/6(hereafterreferredtoasWTmice)isthemousestrainmostsensitivetothedevelopmentofatherosclerosis,ascomparedtoothermurinestrains,C57Bl/6micearestillrelativelyresistanttodiet-inducedatherosclerosis.106Persistenthypercholesterolemia,reflectingplasmacholesterollevelsexceeding300mg/dL,isneededtoinduceatherosclerosisdevelopmentinmice.111Themainreason thatmice ingeneral are resistant toatherosclerosis is theirdistinctplasma lipoproteinprofile,ascomparedtohumans.112Thefactthatmicelackthecholesterolestertransferprotein(CETP)andexertalowabilitytoabsorbdietarycholesterolcausesacardiometaboliclipidprofile,

Chapter1
18
reflected by consistently high plasma levels of HDL cholesterol and low plasma levels of LDLcholesterol.112Therefore,exposingmicetohighconcentrationsofdietarycholesterolaloneisnotsufficienttoinduceatherosclerosisdevelopment.Toenhancetheatherosclerosissusceptibilityofmice, genetic modification is required for induction of a sufficiently high pro-atherogeniclipoproteinprofile.Sincethe1990s,thetechniqueofhomologousrecombinationinembryonicstemcellsmade it possible to selectively knockout genes involved in themetabolismofpro-atherogenic lipoproteins.113 Currently, the most frequently used mouse models to studyatherosclerosisareLDLreceptor(LDLr)knockout(KO)andApoEKOmice.112
3.2 ApoEKOmiceandLDLrKOmice
ApoEisaconstituentofnon-LDLlipoproteinsandservesasanessentialligandfortheuptakeoftheselipoproteinsbytheliver.114Therefore,micelackingApoEshowimpairedclearanceofplasmacholesterol, resulting in severe hypercholesterolemia. ApoE KOmice fed a regular chow dietdisplayplasmacholesterol levelsof>500mg/dL,whichcanmainlybeattributedto increasedlevelsofchylomicronsandVLDL,whereasplasmaHDL-cholesterolisdecreased.115ApoEKOmicedevelop extensive atherosclerotic lesions.116,117 Under normal chow conditions, spontaneousatheroscleroticlesiondevelopmentisobservedintheaorticsinuswithin3-4monthsofage.Intheolder mice, atherosclerotic lesions are visible throughout the aorta at locations of principalbranches.118,119 Importantly, this process can be accelerated by feeding ApoE KOmice a highfat/highcholesteroldiet.120
TheLDL-receptorregulatesplasmacholesterollevelsbyremovingIDLandLDLfromplasma.MicelackingtheLDLr,ascomparedtoWTmice,displaya2-foldhigherplasmacholesterollevel(~230mg/dL)when fed a regular chowdiet, as compared toWTmice. This increase canmainly beattributed to an increase in cholesterol within the IDL/LDL fraction.121,122 Furthermore, LDLrdeficiencyalso leads toa small increase inVLDL-cholesterol levels.Moreover,HDL-cholesterollevelsareincreased.122ThemildhypercholesterolemiainducedbyLDLrdeficiencyhowever,isnotsufficienttoeffectivelyinduceatherosclerosisdevelopmentinmiceonchow.Interestingly,plasmacholesterollevelsofLDLrKOmicearehighlyresponsivetodietaryinterventions,123,124andahigh-fat/high-cholesterol (Western-type) diet is known to induce severe hypercholesterolemia andrapid atherosclerosis development.122,125,126 Similar to ApoE KO mice, atherosclerotic lesiondevelopmentinLDLrKOmiceonWestern-typedietisinitiatedintheaorticroot.127Intermediateaortic lesiondevelopmentoccurswithin3monthsofWestern-typedietfeeding,andadvancedlesionsarepresentintheaortaafter5monthsofdietarychallenge.123,124,126Acommonlyusedapproachtoinvestigatethefunctionofaspecificgeneinatherosclerosis,iscrossbreedingofmicedeficientforthegeneofinterestwiththehypercholesterolemicLDLrKOorApoEKOmice.Sincegeneratingdoubleknockoutmiceisatimeconsumingandcostlyapproach,thereisanongoingsearchforalternativemethodstoinducehypercholesterolemiainmice.
3.3 Bonemarrowtransplantation
Hematopoieticstemcelltransplantationtoanestablishedmousemodelofatherosclerosis,suchastheapoEorLDLrKOmice,isaneffectivestrategytogeneratechimericmicewithtargetgenealterations in bonemarrow-derived cells of an atherosclerosis-pronebackground.Oneof thestrengthsofthismodel isthat intherecipientsspecificallythegenotypeofthebonemarrow-derived cells,which represent themajorplayers inatherosclerosis, is alteredand thusallowsanalysis of the specific contribution of a gene of interest in blood cells. Hematopoietic cell

Chapter1
19
transplantation not only helps to ease the time and money needed for the generation ofsophisticated cell type-specific knockoutmousemodels, but also allows a closermechanisticinsightintothecellularbiologyunderlyingatherosclerosisdevelopment.128
Beforebirth,bloodcellsarederivedfromthefetalliverandspleen,however,afterbirththebonemarrowbecomestheprimaryoriginforthegenerationofbloodcells.Therefore,bonemarrowisnormallythesourceofhematopoieticcellsfortransplantation,especiallyinmurinemodels.Inbonemarrowtransplantationstudies(Figure2),LDLrKOmicearenormallychosenoverApoEKOmiceastheatherosclerosis-pronerecipients.Thisisbecause1)thelipidprofileofLDLrKOmice,characterizedbyahighIDL/LDLcholesterolfraction,resemblesstronglytheplasmalipidprofilesofhumandyslipidemicpatients,1292)themorphologyoftheatheroscleroticlesionsinLDLrKOmiceresemblehumanatheroscleroticplaques,130,131and,mostimportantly,3)thepresenceoftheLDLrinthedonorbonemarrowdoesnotaffectthepro-atherogeniclipoproteinprofileandatherosclerosis susceptibility of the LDLr KO recipients.132-134 In contrast, several studies havedemonstrated that restoration of ApoE in bone marrow-derived cells normalizes serumcholesteroltoWTlevelsandreducesatherosclerosisdevelopmentinApoEKOrecipients.135-138
Figure2Schematicdiagramofabonemarrowtransplantation(BMT)procedureinmiceforstudyingatherosclerosis.
1)Originalbonemarrowofatherosclerosis-pronerecipientmice(oftenLDLrKOmice)isdestroyedbyalethaldoseofirradiation(9Gy).2)Therecipientsreceivefive-milliondonorbonemarrowcells,lackingoroverexpressingthegeneofinterest.3)Recipientsareallowedtorecoverfor8weeksonachowdiet.Oneweekbeforetheirradiation,andthroughoutthecompleterecoveryperiod,recipientmicereceiveantibioticsviadrinkingwater.4)Afterrecovery,therecipientsarechallengedwithanatherogenicWestern-typediettoinduceatherosclerosisdevelopment.
4 Signalingpathwaysandatherosclerosis
Asmentionedbefore,macrophagesare themain cell type in atherosclerotic lesionsand theirphenotypeandactivation status influence their exact role in thepathogenesisof thedisease.Importantly, macrophage phenotype and activation are highly dependent on the lesionalmicroenvironment and the intracellular signaling pathways that are activated within themacrophages.
Macrophageactivation isaverycomplicatedprocess.Fromreceivingan initial stimulus to thepointthateventuallythemacrophage’sphenotypicfunctionalproteinproductionisaltered,thisprocessentailsactivationofacomplexsetofsignalingpathways,andtranscriptionalandpost-transcriptional regulatory networks.96,139 In a simplified summary, as shown in figure 3 thefollowing key steps can be distinguished: 1�macrophage surface receptors recognizeenvironmental stimuli; 2) the signals are amplified and transmitted to the nucleus by protein
②①
③
④

Chapter1
20
kinase transducers; and 3) the nuclear transcriptional/post-transcriptional factors regulatemacrophage-specificgeneexpressionandthusdictatemacrophagepolarizationandfunctions.139This complicated activation process provides multiple possibilities to design novel strategies,addressing the different activation steps, to reprogram specific macrophage phenotypes fortherapeuticbenefit.Inthisthesis,fourgenesinvolvedinmacrophageactivationatdifferentlevels,wereinvestigated.MacrophagepolarizationtotheM1andM2phenotypehaslongservedasaparadigmforstudyingatherosclerosis.Inducingneweffectoractivitiesbyactivatedmacrophagesisconsideredasanattractivetherapeuticapproachforatherosclerosistreatment.139-143Toexplorepotential novel therapeutic targets, we first evaluated and discussed the role of the M2macrophage signature gene Arg1, a key player in the nitric oxide/L-arginine pathway, inatherosclerosis development. Protein kinases play essential role in the transcriptional andepigeneticregulationofmacrophagepolarization,139,144,145andarethemostintensivelystudiedproteintargetsinpharmacologyresearch.146-150InthisthesistheatheroscleroticroleofAkt2andMKP2, key members of the protein kinase B and mitogen-activated protein kinases familyrespectively, are addressed. Furthermore, transcription factors, critical regulators of geneexpression,havelongbeenproposedtoexecuteessentialregulatoryfunctionsinthepathogenesisofatherosclerosis.151Inaddition,inthisthesis,wefocusontheupstreamstimulatoryfactors(Usfs),recently identifiedlipid-relatedtranscriptionfactors,152thatareregulatorsofseveral importantcellularprocesses153andhenceareexpectedtoinfluenceatherosclerosisdevelopment.Belowthebackgroundof1)theNitricoxide/L-arginineandArg1,2)proteinkinasesandtheirinhibitorsAkt2andMKP2,and3)Usfsinatherosclerosisisdescribedinmoredetailbelow.
4.1 Nitricoxide/L-argininepathwayinatherosclerosis
Nitricoxide(NO)isanimportantsignalingmoleculethat influencesmanycellularprocesses.154Thecardiometabolicrelatedfunctionsofthismoleculeinclude:1)preventionofendothelialcellapoptosis,155,1562)reductionofoxidativestress,inducedbyreactiveoxygenspecies(ROS),1573)inhibitionofsmoothmusclecellproliferation,and4)inhibitionofvascularcelladhesionmolecule-1 (VCAM-1) expression and, hence, inhibition of monocyte recruitment.158 In line with theseathero-protective functions of NO, several animal studies have confirmed that decreasingNOproduction induces atherosclerosis,159 while increasing NO production attenuatesatherosclerosis.160,161
Important to note is that there are indications that the protective role of NO is both tissue-specific159,160,162-164anddose-dependent.160,162,165NOisaproductofnitricoxidesynthases(NOSs).TheNOSfamilyhasthreemembers,endothelialNOS(eNOS),neuronalNOS(nNOS),andinducibleNOS(iNOS).154eNOSandnNOSareconstitutivelyexpressed.166Asindicatedbythename,eNOSisprimarilyproducedbyendothelial cellsand,nNOSbyneurons.Theexpressionof iNOScanbeinducedbystimulators,especiallyinflammatorycytokines.167Moreimportantly,iNOSisexpressedbymacrophages, and highly upregulated in response to lipopolysaccharide and inflammatorycytokines,168leadingtoanenhancedproductionofNO.

Chapter1
21
Figure3RegulationinmacrophageactivationandtargetfactorsArg1,Akt2,MKP2andUsf1studiedinmoredetailinthisthesis.
1) Environmental stimuli activate cellular effector receptors. 2) The signals are amplified and transmitted to thenucleusbyproteinkinasetransducers, includingAkt2orMKP2.3)Transcriptionfactors,suchasUsf1,regulatethestimulus-specificgeneexpression,eventuallyleadingtophenotypicfunctionalproteinproduction,suchasArg1.
IncreasingevidenceindicatesthatNOproducedbynNOSandeNOSisatheroprotective,whileNOproduced by iNOS ismore likely pro-atherogenic.169 Diminished levels of bioavailable NO areassociatedwithendothelialdysfunction,170theindicatorofearlyatherosclerosis.DeletionofeNOSinApoEdeficientmiceleadstoincreasedatherosclerosisdevelopment,159andeNOSgenetransferimprovesatherosclerosis,160,161furtherevidencingtheathero-protectiveroleofNOderivedfromeNOS.ThebeneficialeffectsofnNOSinatherosclerosisdescribedbyKuhlencordtetal.,171weresurprising at first, since nNOS is primarily detected outside of cardiovascular system.172-174However, in their study, nNOS expression was demonstrated in the atherosclerotic lesion,predominantly in smooth muscle cells, and to a lesser extent in macrophages, but not inendothelial cells.175 nNOS-derivedhydrogenperoxide inducesendothelial dysfunction,but themechanismunderlyingtheprotectiveeffectsofnNOSinatherosclerosisisnotclearyet.169,176NOproducedbyiNOSoflesionalmacrophages,ispro-atherogenicinhumans.177Insupport,deletionof iNOS inmice reduces their susceptibility to the development of atherosclerosis.163,164,178 Inadditiontoitscelltype-specificeffectsonatherosclerosis,NOalsoinfluencesatherogenesisinadose-dependentmanner:lowconcentrationsofNOimprovesatherosclerosis,160,161butmassiveNOproductionworsensatherosclerosis.162

Chapter1
22
iNOS, a signaturemarker ofM1macrophages, uses the substrate L-arginine, to produce NO.Interestingly,thissamesubstrateisalsousedbyArginase-1(Arg1),animportantM2macrophagemarker,which catalyzes the conversion of L-arginine to urea and ornithine, the latter being aprecursorforcollagenproduction.DeletionofiNOSisknowntocauseadecreaseinNOproduction,resultinginanincreasedavailabilityofitssubstrateL-arginineforArg1mediated-convertionintoornithine.Hence, iNOS deletion can indirectly lead to an enhanced production of collagen,179thereby improving atherosclerotic plaque stability. Conversely, deletion of Arg1might lead toincreased substrate availability for the production of NO. As mentioned above, NO inatherosclerosisiswell-studied,however,theroleofArg1inatheroscleroticplaquedevelopmentiscurrentlystillunknown.
4.2 ProteinkinaseB(Akt)inatherosclerosis
Protein kinase B is a serine/threonine-specific protein kinase, ofwhich three isoforms can bedistinguished:Akt1,Akt2,andAkt3.180Aktplaysanimportantroleinmanycellularprocesses,suchas apoptosis,181 proliferation,182 migration,183 transcription,184 and insulin responsiveness.185Importantly,Aktsignalingisalsoknowntoinfluenceatheroscleroticlesiondevelopment.
Akt1iswidelyexpressedinalltissues,whereasAkt2expressionis limitedtometabolictissues,such as adipose tissue, liver, and skeletal muscle, and Akt3 is preferentially expressed inbrain.180,186,187Interestingly,macrophagesexpressallthreeAktisoforms.188RecentstudiesshowedthatAkt isoformsdifferentiallycontributetomacrophagepolarization.189,190Forexample,uponStaphylococcusaureusinfection,Akt1deficientmacrophagesshowedupregulatedexpressionoftheM1signaturegene iNOS.189Conversely,uponLPSstimulation,Akt2knockoutmacrophagesdisplay anM2-likephenotype,as evidencedby augmentedexpressionof theM2macrophagemarkersArg1,YM-1,andFIZZ-1.190,191Sofar,theroleofAkt3inmacrophagepolarizationremainsunknown.Inadditiontotheirrolesinmacrophagepolarization,Aktsalsodifferentiallyinfluencemacrophagefoamcellformation.PreviousstudieshaveshownthatAkt1doesnotaffectoxLDL-induced cholesterol accumulation in macrophages,192 whereas Akt2 promotes acLDL-inducedfoam cell formation,191 and Akt3 protects macrophages against acLDL-induced foam cellformation.193
In linewith the different roles of the Akt isoforms inmacrophage polarization and foam cellformation,Aktisoformsalsodistinctlycontributetoatherosclerosisdevelopment.Akt1hasbeenreportedtohaveanatheroprotectiverole.192However,thiseffectislikelyduetoAkt1ofvascularoriginandnotmacrophageAkt1.Indeed,bonemarrow-specificdeletionofAkt1didnotinfluenceatherosclerosis susceptibility.192Moreover, Fernandez-Hernanode and colleagues showed thatwholebodyAkt1deletioninhibitstheproliferationandmigrationofvascularsmoothmusclecells(VSMCs), leading to the development of vulnerable atherosclerotic plaques with increasednecrosis and a smaller collagen-rich fibrous cap.194 Akt3 is barely detectable in the healthyvasculature.195 However, in line with the importance of Akt3 to limit macrophage foam cellformation,anincreasedsusceptibilitytoatherosclerosisdevelopmentwasobservedinAkt3totalbodyandbonemarrow-specificknockoutmice.193
TheroleofAkt2inatherosclerosisismorecomplex.RensingandcolleaguesfoundthattotalbodydeletionofAkt2inducessmallerbutunstableatheroscleroticlesions,withamajorcausativeroleforVSMCderivedAkt2 in thedecreased lesional collagencontentand increasednecrotic coreformation.196TheunstablephenotypeinducedbyAkt2lossislikelyduetodisturbancesinVSMCs

Chapter1
23
migration, proliferation, andmetalloproteinase production.196 However, a study from anothergroup found that total bodyAkt2 deletion does not affect atherosclerosis development.191 Toexclude the contribution of smooth muscle cell Akt2, Babaev et al. used fetal liver celltransplantationandRotllanetal.usedbonemarrowtransplantationtospecificallydeleteAkt2inhematopoietic cells of LDLr KO mice.188,191 Both studies indicated that hematopoietic Akt2deficiency protects LDLr KO mice against diet-induced atherosclerosis. Akt2 deletion inmacrophages induced M2 macrophage polarization, decreased macrophage migration andinhibitedmacrophagefoamcellformation;processesthatarelikelyresponsiblefortheobserveddecreaseinatherosclerosissusceptibilityoftheLDLrKOrecipients.191,197Thesefindingsindicatethat both VSMC Akt2 and hematopoietic Akt2 play a role in atherogenesis. Importantly, theprotectiveeffectofhematopoieticAkt2deficiencywasindependentofAkt1andAkt3,becausebonemarrow-specificdeletionofAkt1andAkt3ledtounchangedor increasedatherosclerosisdevelopment,whilethephenotypeofthereducedsusceptibilitytoatherosclerosispersisteduponcombinedAkt2/Akt1orAkt2/Akt3deletioninbonemarrow.192,193,198-200
4.3 MAPKs/MKPspathwayinatherosclerosis
4.3.1 MAPKsandatherosclerosis
MAPKsareafamilyofproteinkinasesthatspecificallyphosphorylateserine/threonineresidues.Threemajorsubfamiliescanbedistinguished:extracellularregulatedproteinkinase(ERK),c-JunN-terminal kinase (JNK) and p38. All these subfamilies have been reported to participate inatherosclerosisdevelopment.
4.3.1.1 ERKandatherosclerosis
Invivo,ERKexpressionisincreasedinatheroscleroticlesionsofcholesterol-fedrabbits.201Invitro,ERKisrapidlyactivateduponoxLDLstimulationinmacrophages.202ThesefindingsindicatearoleforERKinfoamcellformation.Indeed,inhibitionofERK1/2,bytheirupstreamMEK1/2inhibitorU0126,significantlydecreasedfoamcellformationbothinvivoandinvitro,whichismostlikelythe consequence of upregulated cholesterol efflux transporters ABCA1 and ABCG1.203,204 Inagreement with the decreased susceptibility to foam cell formation, atherosclerotic lesiondevelopmentwasreducedinApoEknockoutmicetreatedwiththeinhibitorU0126.Thesefindingssuggestedananti-atheroscleroticroleforERK1/2inhibition.
ERK also influences macrophage polarization. Inhibition of ERK by the inhibitors U0126, orPD0325901 led toanM2-likemacrophagephenotype, reflectedby increasedM2markergeneexpression.205,206Furthermore,theERKinhibitor–dependentincreaseofM2macrophagemarkergeneexpressionislikelyindependentofthepre-existingpolarizationstateofthemacrophage,206asre-primingofLPS-polarizedM1macrophagesbyIL-4/IL-13stillinducedashifttowardstheM2phenotype.ThesefindingssuggestthatskewingofmacrophagestowardsanM2phenotypemightalsocontributetotheobservedathero-protectiveeffectofERKinhibition.
4.3.1.2 P38andatherosclerosis
P38,alsocalledmitogen-activatedproteinkinase11(MAPK11),has4isoforms:p38α,p38β,p38γ,and p38δ.207 p38α, the most well-studied isoform of p38, is rapidly phosphorylated inmacrophagesinresponsetoLPSandisresponsibleforthesubsequentinductionintheproductionof pro-inflammatory cytokines.208-211 Genetic deletion of p38α in macrophages results in an

Chapter1
24
impaired TLR4-mediated LPS-induced innate immune response, reflected by a decreasedproductionofthepro-inflammatorycytokinesTNF-αandIL-12.212Furthermore,p38activationisenhanced in IL-4-induced alternatively activatedmacrophages, and inactivation of p38 led todecreased IL-4-inducedM2marker expression.213 Collectively, these findings suggest that p38activationislikelyneededforbothM1andM2macrophagepolarizationandfunction.
Theroleofp38infoamcellformationisnotclearyet.Inactivationofpanp38byapharmaceuticalinhibitor prevents foam cell formation in vitro.214,215 However, genetic deficiency of p38α inmacrophages does not affect foam cell susceptibility,216 albeit it does enhance macrophageapoptosis.217Severalstudieshavebeenperformedtoaddresstheroleofp38inatherosclerosis.Systemic p38 inhibition, either via pharmaceutical inhibition or genetic deletion of the p38substrateMK2,protectsApoEKOmiceagainstatherosclerosis,218,219indicatingapro-atherogenicrole of p38. Interestingly, Seimon et al. showed that macrophage-specific deletion of p38αpromotes atherosclerosis development.217 In contrast, Kardakaris et al., using the sameexperimentalsetup,foundthatp38αdeficiencyhadnoeffectonatherosclerosisdevelopment.216Hence,theroleofmacrophagep38inatherosclerosisremainscontroversial.
4.3.1.3 JNKandatherosclerosis
c-JunN-terminalkinase(JNK),alsonamedmitogen-activatedproteinkinase8(MAPK8),ispresentin three isoforms: JNK1, JNK2, and JNK3.220 Of these three isoforms, JNK1 and JNK2 areubiquitouslyexpressed,whereasJNK3ismainlyexpressedinbrain,andtoalesserextendintheheartandtestes.220JNK1andJNK2arebothexpressedinmacrophages,221andtheiractivationisknowntoregulatevariousmacrophagefunctions,includingpolarization,foamcellformation,andprogramedcelldeath.
Inhibition of JNK activation leads to impaired macrophage development, proliferation, andsurvival,222suggestingabroadfunctionofJNKinmacrophages.Invitro,JNK1wasshowntoberesponsible for cytokine and NO production by LPS-stimulated M1 macrophages.223,224
Furthermore,invivo,deletionofJNK1reducedmacrophagemigrationandinfiltrationinamurinearthritismodel.225 On the other hand, JNK2was shown to stimulate oxLDL-induced foam cellformation.202Conversely,oxLDLtriggersJNK2activationinmacrophagesandfacilitatesscavenger-mediated foam cell formation in a CD36-JNK-SR-A loop manner.226 In accordance, enhancedactivationofJNK2isobservedinmacrophage-richatheroscleroticlesions.227,228
In line with the distinct effects of JNK isoforms on macrophage function, JNK isoforms alsocontributetoatherosclerosisdifferently.GeneticdeletionofJNK2protectsApoEKOmiceagainsthigh-fatdietinducedatherosclerosis,andthisislikelyattributedtothelackofJNK2-inducedfoamcell formation.227 Interestingly, hematopoietic JNK1 deficiency promotes atherosclerosisdevelopment in LDLr KO mice, likely caused by the lack of JNK1-mediated regulation ofmacrophagesurvival.221
4.3.1.4 InteractionsbetweenMAKPmembers
MAPK cascades can be activated by either intra- or extracellular stimulators and signalingmolecules. Depending on the cell type, stimulus signal strength and dynamics,MAPK can beactivated differently and serve distinct functions. Importantly, each member of MAPK familyclosely interacts with the othermembers, and inmost cases is subject to negative feedbackregulation.229

Chapter1
25
4.3.2 MAPkinasephosphatasesandatherosclerosis
TheactivityofMAPKsistightlyregulatedbyMAPkinasephosphatases(MKPs)thatbelongtothegroupofdual-specificityphosphatases(DUSPs),whichincludesatleast10members.230MKPsarenormally referred to as the typical DUSPs.231 Atypical DUSPs lack theMAPK-bindingmotif orkinase-interacting motif (MKB/KIM), which determines the dephosphorylation activity ofDUSPs.232,233Basedontheirsubcellularlocalization,MKPsaredividedintothreegroups.Thefirstgroup consists of MKP1 (DUSP1), PAC1 (DUSP2), MKP2 (DUSP4), and DUSP5, which are allinducibleMKPsthatarelocatedinthenucleus.ThesecondgroupisrepresentedbyMKP3(DUSP6),MKP-X(DUSP7)andMKP4(DUSP9),whicharealllocatedinthecytoplasm.Finally,thethirdgroupis comprised of DUSP8,MKP5 (DUSP10) andMKP-7 (DUSP16), which are located in both thenucleusandthecytoplasmofacell.230,234,235MKPsinactivateMAPKsbydephosphorylatingtheirphosphoserine/threonine and phosphotyrosine residues.236,237 Noteworthy, the expression ofgroup 1 members, being inducible MKPs,230,234,235 is induced in response to stimulation withLPS,238 a potent activator ofMAPKs. Therefore, this group ofMKPs is likely to act asMAPKsfeedbackloopregulator.230,239
All threemembersof theMAPK familyare involved inmacrophagepolarizationand foamcellformation,indicatingthatMKPsaremostlikelyalsoinvolvedintheregulationoftheseprocessesandtherebyinfluenceatherosclerosisdevelopment.Indeed,severalstudieshavedemonstratedthatvariousmacrophage functionsareaffectedbydepletionofMKPs,especially the inducibleMKPs. For instance, deletion ofMKP1 led to the upregulation of severalM1 signature genes,includingTNF-α,Il-6,IL-1β,andCCL2.240,241Inaddition,PAC1deficientmacrophagesdisplayedanincreasedproductionofpro-inflammatorycytokinesinresponsetoLPSstimulation.242ThesedatasuggestanimportantroleforMKP1andPAC1inregulatingLPS-inducedM1polarization.UnliketheclearrolesofMKP1andPAC1intheregulationofM1polarization,thecurrentfindingsontheroleofMKP2inmacrophagepolarizationremaincontradictory.Al-Mutairietal.foundthatuponLPS stimulation, genetic loss of MKP2 in bone marrow-derived macrophages (BMDM) led toenhancedproductionofpro-inflammatorycytokines,whiletheproductionofanti-inflammatorycytokineswasdecreased.243Incontrast,CornellandcolleaguesfoundthatuponLPSstimulation,MKP2deletioninBMDMsresultedindecreasedTNF-αandIL-10production.244DUSP5deficientmacrophages showed no altered cytokine or chemokine production in response to LPSstimulation.245However,despitesomeindicationsforaroleofMKPsinmacrophagepolarizationand function, so far only limited research has been done addressing the role of MKPs inatherosclerosis.246-248
4.4 Usftranscriptionfactorsandatherosclerosis
Upstreamstimulatoryfactors(Usfs)areDNA-bindingproteins,featuredasahelix-loop-helixmotifandleucinerepeat,thatserveastranscriptionfactors.249BybindingtotargetDNAasUsfhomo-andheterodimers,Usfsregulatetargetgeneexpression.139,250AccumulatingevidenceshowedthatdisturbedUsfsignalingnormallyleadstometabolicdisorders,especiallyinthecaseofUsf1.251-254Usf1isubiquitouslyexpressed,andhasabroadrangeoftargetgenes,ofwhichthegenesinvolvedinlipidandglucosemetabolismaremostwidelystudied.255Usf1isalsoknowntoregulategeneexpressioninresponsetostressors,suchasultraviolet(UV)irradiation,256insulin,257andgrowthfactor.258,259Moreover,inresponsetoPI3kinase/Aktsignaling,Usf1regulatesthetranscriptionofgenesimplicatedincellularapoptosisandcellcyclearrest.260

Chapter1
26
Usf1 is a direct target of p38 and AMPK, which both have been shown to be involved inmacrophage polarization.254,256,261-263 Furthermore, Usf1 is phosphorylated by protein kinaseCK2.264Proteinkinasesare important forregulationofvariouscellular functions.However, theexact roleofUsf1 incell function regulation is stillunclear. Importantly,mutations inUsf1arestrongly associatedwith familial combinedhyperlipidemia, a commonhereditary dyslipidemiawith a prevalence of 20% in CVD patients.265 In support of this association, a strong pro-atherogenic role for Usf1 is found in LDLr KOmice lacking Usf-1, which display a remarkabledecreasedintheirsusceptibilitytoatheroscleroticlesiondevelopment.152Themechanismbehindthiseffect,however,ispoorlydefined.
5 OutlineofthisThesis
Currently, therapeutic strategies to prevent atherosclerosis are primarily based on the use ofcholesterolloweringdrugs,e.g.statins.However,themorbidityandmortalityofcardiovasculardiseasecausedbyatherosclerosisremainshigh.Therefore,identificationofnovelpharmaceuticaltargetssuitableforthedevelopmentofnoveldrugsisstronglyneeded.Theaimofthisthesisistounraveltheroleofvariouscandidategenesinmacrophageactivationandtheirsubsequentroleinatherosclerosis.
InChapter2,wedeterminedtheroleofhematopoieticArg1,aclassicmarkerofM2macrophageactivation,inatherosclerosis.WeshowthatalthoughArg1deficiencypromotesmacrophagefoamcellformation,itdoesnotimpactonatherosclerosisdevelopment.
Akt2 is a key player in the PI3K/Akt transduction pathway that regulates M2 macrophagepolarization.190 InChapter3,weaddressedtheeffectsofbonemarrowAkt2-reconstitution, inAkt2/LDLrdKOmice.WeshowthathematopoieticAkt2promotesfoamcellformation,butdoesnotalteratherosclerosisdevelopmentinAkt2/LDLrdKOmice.
Next,wedetermined theeffectsof theupstreamregulatorsMKP2,of theMAPK transductionpathway,inatherosclerosis.MKP2wasfoundtoplayanimportantroleinregulatingmacrophagefunction, and in Chapter 4 we show that MKP2 deficiency skews macrophages to an M2phenotypeassociatedwithanenhancedsusceptibilitytofoamcellformation.Inline,deletionofMKP2inbonemarrow-derivedcellsleadstoincreasedatherosclerosisdevelopment.
Additionally,wefocusedonthebonemarrow-specificeffectsofUsf1,anupstreamtranscriptionstresssensorinatherosclerosis. InChapter5,weshowthatUsf1inbonemarrow-derivedcellsprotectsagainstatherosclerosis.
Finally,theoverallconclusionsandfutureperspectivesofthisthesisarediscussedinChapter6.
6 References
1. LibbyP,RidkerPM,MaseriA.Inflammationandatherosclerosis.Circulation.2002;105:1135-11432. DaviesMJ,WoolfN.Atherosclerosis:Whatisitandwhydoesitoccur?BrHeartJ.1993;69:S3-113. RossR.Thepathogenesisofatherosclerosis:Aperspectiveforthe1990s.Nature.1993;362:801-
8094. Townsend N, Nichols M, Scarborough P, Rayner M. Cardiovascular disease in europe--
epidemiologicalupdate2015.EurHeartJ.2015;36:2696-2705

Chapter1
27
5. Thompson RC, Allam AH, Lombardi GP,Wann LS, SutherlandML, Sutherland JD, SolimanMA,FrohlichB,MininbergDT,MongeJM,VallodolidCM,CoxSL,Abdel-MaksoudG,BadrI,MiyamotoMI, el-HalimNurel-DinA,Narula J, FinchCE, ThomasGS.Atherosclerosis across4000yearsofhumanhistory:Thehorusstudyoffourancientpopulations.Lancet.2013;381:1211-1222
6. CoiterV.Deaviumsceletisetpraecipiusmusculis.15757. KhanIA,MehtaNJ.Initialhistoricaldescriptionsoftheanginapectoris.JEmergMed.2002;22:295-
2988. Parry CH.An inquiry into the symptoms and causes of the syncope anginosa commonly called
angina pectoris; illustrated by dissections: By caleb hillier parry, mdmember of the college ofphysiciansoflondon,andoftheroyalmedicalsocietyofedinburgh;andoneofthephysiciansofthebathgeneralhospital.R.Cruttwell;andsoldbyCadellandDavies,Strand,London;1799.
9. LaRosaJC,PedersenTR.Foreword.EuropeanHeartJournalSupplements.2004;6:C1-C410. SlijkhuisW,MaliW,AppelmanY.A historical perspective towards a non-invasive treatment for
patientswithatherosclerosis.NethHeartJ.2009;17:140-14411. AscherE,HollierLH,StrandnessDE,TowneJB,HaimoviciH,CalligaroK,KentKC,MonetaGL,Pearce
WH,RicottaJJ.Haimovici'svascularsurgery.JohnWiley&Sons;2008.12. TedguiA,MallatZ.Cytokinesinatherosclerosis:Pathogenicandregulatorypathways.PhysiolRev.
2006;86:515-58113. Mehta NJ, Khan IA. Cardiology's 10 greatest discoveries of the 20th century. Tex Heart Inst J.
2002;29:164-17114. GottoAM,Jr.Evolvingconceptsofdyslipidemia,atherosclerosis,andcardiovasculardisease:The
louisf.Bishoplecture.JAmCollCardiol.2005;46:1219-122415. MayerlC,LukasserM,SedivyR,NiedereggerH,SeilerR,WickG.Atherosclerosisresearchfrompast
topresent--onthetrackoftwopathologistswithopposingviews,carlvonrokitanskyandrudolfvirchow.VirchowsArch.2006;449:96-103
16. RossR.Atherosclerosis--aninflammatorydisease.NEnglJMed.1999;340:115-12617. RossR,HarkerL.Hyperlipidemiaandatherosclerosis.Science.1976;193:1094-110018. GofmanJW,LindgrenFT,ElliottH.Ultracentrifugalstudiesoflipoproteinsofhumanserum.JBiol
Chem.1949;179:973-97919. KeysA.Coronaryheartdiseaseinsevencountries.Circulation.1970;41:186-19520. PrasslR,LaggnerP.Lipoproteinstructureanddynamics:Lowdensitylipoproteinviewedasahighly
dynamicandflexiblenanoparticle.INTECHOpenAccessPublisher;2012.21. NordestgaardBG,Tybjaerg-HansenA.Idl,vldl,chylomicronsandatherosclerosis.EurJEpidemiol.
1992;8Suppl1:92-9822. NikkilaE.Familiallipoproteinlipasedeficiencyandrelateddisordersofchylomicronmetabolism.
Metabolicbasisofinheriteddisease/[editedby]JohnB.Stanbury...[etal.].198323. NordestgaardBG,ZilversmitDB.Largelipoproteinsareexcludedfromthearterialwallindiabetic
cholesterol-fedrabbits.JLipidRes.1988;29:1491-150024. Proctor SD, Mamo JC. Intimal retention of cholesterol derived from apolipoprotein b100–and
apolipoprotein b48–containing lipoproteins in carotid arteries of watanabe heritablehyperlipidemicrabbits.Arteriosclerosis,thrombosis,andvascularbiology.2003;23:1595-1600
25. PalS,SemorineK,WattsGF,MamoJ. Identificationof lipoproteinsof intestinalorigininhumanatheroscleroticplaque.ClinChemLabMed.2003;41:792-795
26. GianturcoSH,RamprasadMP,LinAH,SongR,BradleyWA.Cellularbindingsiteandmembranebinding proteins for triglyceride-rich lipoproteins in humanmonocyte-macrophages and thp-1monocyticcells.JLipidRes.1994;35:1674-1687
27. GianturcoSH,BrownSA,ViaDP,BradleyWA.Thebeta-vldlreceptorpathwayofmurinep388d1macrophages.JLipidRes.1986;27:412-420
28. Tomkin GH, Owens D. The chylomicron: Relationship to atherosclerosis. Int J Vasc Med.2012;2012:784536
29. Bansal S, Buring JE, Rifai N, Mora S, Sacks FM, Ridker PM. Fasting compared with nonfastingtriglyceridesandriskofcardiovasculareventsinwomen.JAMA.2007;298:309-316

Chapter1
28
30. Al-Aubaidy HA, Jelinek HF. Oxidative stress and triglycerides as predictors of subclinicalatherosclerosisinprediabetes.RedoxReport.2014;19:87-91
31. RidkerPM.Fastingversusnonfastingtriglyceridesandthepredictionofcardiovascularrisk:Doweneedtorevisittheoraltriglyceridetolerancetest?Clinicalchemistry.2008;54:11-13
32. VanderLaanPA,ReardonCA,ThistedRA,GetzGS.VLDLbestpredictsaorticrootatherosclerosisinldlreceptordeficientmice.JLipidRes.2009;50:376-385
33. GibbonsGF,Wiggins D, BrownAM,Hebbachi AM. Synthesis and function of hepatic very-low-densitylipoprotein.BiochemSocTrans.2004;32:59-64
34. KugiyamaK,DoiH,MotoyamaT, SoejimaH,Misumi K, KawanoH,NakagawaO, YoshimuraM,Ogawa H, Matsumura T, Sugiyama S, Nakano T, Nakajima K, Yasue H. Association of remnantlipoprotein levels with impairment of endothelium-dependent vasomotor function in humancoronaryarteries.Circulation.1998;97:2519-2526
35. Krauss RM, Lindgren FT, Williams PT, Kelsey SF, Brensike J, Vranizan K, Detre KM, Levy RI.Intermediate-density lipoproteins and progression of coronary artery disease inhypercholesterolaemicmen.Lancet.1987;2:62-66
36. KugiyamaK,DoiH,TakazoeK,KawanoH,SoejimaH,MizunoY,TsunodaR,SakamotoT,NakanoT,NakajimaK,OgawaH,SugiyamaS,YoshimuraM,YasueH.Remnant lipoprotein levels infastingserumpredictcoronaryeventsinpatientswithcoronaryarterydisease.Circulation.1999;99:2858-2860
37. FukushimaH, Kugiyama K, Sugiyama S, HondaO, Koide S, Nakamura S, KawanoH, SoejimaH,MiyamotoS,YoshimuraM,SakamotoT,OgawaH.Comparisonofremnant-likelipoproteinparticlesinpostmenopausalwomenwithandwithoutcoronaryarterydiseaseandinmenwithcoronaryarterydisease.AmericanJournalofCardiology.2001;88:1370-1373
38. MasuokaH,KameiS,WagayamaH,OzakiM,KawasakiA,TanakaT,KitamuraM,KatohS,ShintaniU,MisakiM, SugawaM, ItoM,NakanoT.Associationof remnant-likeparticle cholesterolwithcoronaryarterydiseaseinpatientswithnormaltotalcholesterollevels.AmericanHeartJournal.2000;139:305-310
39. Wasan KM, Brocks DR, Lee SD, Sachs-Barrable K, Thornton SJ. Impact of lipoproteins on thebiologicalactivityanddispositionofhydrophobicdrugs:Implicationsfordrugdiscovery.NatRevDrugDiscov.2008;7:84-99
40. KraussRM,BurkeDJ. Identificationofmultiplesubclassesofplasma lowdensity lipoproteins innormalhumans.JLipidRes.1982;23:97-104
41. Coresh J, Kwiterovich PO, Jr. Small, dense low-density lipoprotein particles and coronary heartdiseaserisk:Aclearassociationwithuncertainimplications.JAMA.1996;276:914-915
42. RajmanI,EachoPI,ChowienczykPJ,RitterJM.Ldlparticlesize:Animportantdrugtarget?BrJClinPharmacol.1999;48:125-133
43. deGraaf J,Hak-LemmersHL,HectorsMP,Demacker PN,Hendriks JC, StalenhoefAF. Enhancedsusceptibility to in vitro oxidation of the dense low density lipoprotein subfraction in healthysubjects.ArteriosclerThromb.1991;11:298-306
44. Hurt-Camejo E, Olsson U, Wiklund O, Bondjers G, Camejo G. Cellular consequences of theassociation of apob lipoproteins with proteoglycans. Potential contribution to atherogenesis.ArteriosclerThrombVascBiol.1997;17:1011-1017
45. VakkilainenJ,MakimattilaS,Seppala-LindroosA,VehkavaaraS,LahdenperaS,GroopPH,TaskinenMR, Yki-Jarvinen H. Endothelial dysfunction in men with small ldl particles. Circulation.2000;102:716-721
46. AustinMA,KingMC,VranizanKM,KraussRM.Atherogenic lipoproteinphenotype.Aproposedgeneticmarkerforcoronaryheartdiseaserisk.Circulation.1990;82:495-506
47. BerneisK,RizzoM.Ldlsize:Doesitmatter.SwissMedWkly.2004;134:720-72448. Rajman I, EachoPI, Chowienczyk P, Ritter J. Ldl particle size: An important drug target?British
journalofclinicalpharmacology.1999;48:125-13349. SpadyDK.Reversecholesteroltransportandatherosclerosisregression.Circulation.1999;100:576-
578

Chapter1
29
50. Favari E, Chroni A, Tietge UJ, Zanotti I, Escola-Gil JC, Bernini F. Cholesterol efflux and reversecholesteroltransport.HandbExpPharmacol.2015;224:181-206
51. RosensonRS,BrewerHB,Jr.,DavidsonWS,FayadZA,FusterV,GoldsteinJ,HellersteinM,JiangXC,PhillipsMC,RaderDJ,RemaleyAT,RothblatGH,TallAR, Yvan-Charvet L.Cholesterol effluxandatheroprotection: Advancing the concept of reverse cholesterol transport. Circulation.2012;125:1905-1919
52. DuffyD,RaderDJ.Updateonstrategies to increasehdlquantityand function.NatRevCardiol.2009;6:455-463
53. HogarthCA,RoyA,EbertDL.Genomicevidencefortheabsenceofafunctionalcholesterylestertransferproteingeneinmiceandrats.CompBiochemPhysiolBBiochemMolBiol.2003;135:219-229
54. BarterP.Theroleofhdl-cholesterolinpreventingatheroscleroticdisease.EuropeanheartjournalSupplements.2005;7:F4-F8
55. NoferJR,KehrelB,FobkerM,LevkauB,AssmannG,vonEckardsteinA.Hdlandarteriosclerosis:Beyondreversecholesteroltransport.Atherosclerosis.2002;161:1-16
56. MacknessMI,ArrolS,DurringtonPN.Paraoxonasepreventsaccumulationoflipoperoxidesinlow-densitylipoprotein.FEBSLett.1991;286:152-154
57. Mackness MI, Arrol S, Abbott C, Durrington PN. Protection of low-density lipoprotein againstoxidative modification by high-density lipoprotein associated paraoxonase. Atherosclerosis.1993;104:129-135
58. ClayMA,PyleDH,RyeKA,VadasMA,Gamble JR,BarterPJ.Timesequenceof the inhibitionofendothelial adhesion molecule expression by reconstituted high density lipoproteins.Atherosclerosis.2001;157:23-29
59. Yuhanna IS, Zhu Y, Cox BE, Hahner LD, Osborne-Lawrence S, Lu P, Marcel YL, Anderson RG,MendelsohnME,HobbsHH,ShaulPW.High-densitylipoproteinbindingtoscavengerreceptor-biactivatesendothelialnitricoxidesynthase.NatMed.2001;7:853-857
60. MineoC,YuhannaIS,QuonMJ,ShaulPW.Highdensitylipoprotein-inducedendothelialnitric-oxidesynthaseactivationismediatedbyaktandmapkinases.JBiolChem.2003;278:9142-9149
61. Riwanto M, Landmesser U. High density lipoproteins and endothelial functions: Mechanisticinsightsandalterationsincardiovasculardisease.JLipidRes.2013;54:3227-3243
62. KeeneD,PriceC,Shun-ShinMJ,FrancisDP.Effectoncardiovascularriskofhighdensitylipoproteintargeted drug treatments niacin, fibrates, and cetp inhibitors: Meta-analysis of randomisedcontrolledtrialsincluding117411patients.Bmj-BritMedJ.2014;349
63. KheraAV,CuchelM,de laLlera-MoyaM,RodriguesA,BurkeMF, JafriK,FrenchBC,Phillips JA,MucksavageML,WilenskyRL,MohlerER,RothblatGH,RaderDJ.Cholesteroleffluxcapacity,high-densitylipoproteinfunction,andatherosclerosis.NewEnglandJournalofMedicine.2011;364:127-135
64. RohatgiA,KheraA,BerryJD,GivensEG,AyersCR,WedinKE,NeelandIJ,YuhannaIS,RaderDR,deLemosJA,ShaulPW.Hdlcholesteroleffluxcapacityandincidentcardiovascularevents.NEnglJMed.2014;371:2383-2393
65. AnnemaW,Dikkers A, de Boer JF, vanGreevenbroekMMJ, van der Kallen CJH, Schalkwijk CG,StehouwerCDA,DullaartRPF,TietgeUJF.Impairedhdlcholesteroleffluxinmetabolicsyndromeisunrelatedtoglucosetolerancestatus:Thecodamstudy.SciRep-Uk.2016;6
66. SaleheenD,ScottR, JavadS,ZhaoW,RodriguesA,PicataggiA,LukmanovaD,MucksavageML,LubenR,BillheimerJ,KasteleinJJP,BoekholdtSM,KhawKT,WarehamN,RaderDJ.Associationofhdl cholesteroleffluxcapacitywith incidentcoronaryheartdiseaseevents:Aprospectivecase-controlstudy.LancetDiabetesEndo.2015;3:507-513
67. SiddiqiHK,KissD,RaderD.Hdl-cholesterolandcardiovasculardisease:Rethinkingourapproach.CurrOpinCardiol.2015;30:536-542
68. Weber C, Schober A, Zernecke A. Micrornas in arterial remodelling, inflammation andatherosclerosis.CurrentDrugTargets.2010;11:950-956

Chapter1
30
69. HanssonGK.Mechanismsofdisease-inflammation,atherosclerosis,andcoronaryarterydisease.NewEnglandJournalofMedicine.2005;352:1685-1695
70. LeyK,LaudannaC,CybulskyMI,NoursharghS.Gettingtothesiteofinflammation:Theleukocyteadhesioncascadeupdated.NatureReviewsImmunology.2007;7:678-689
71. SinghRB,MengiSA,XuYJ,ArnejaAS,DhallaNS.Pathogenesisofatherosclerosis:Amultifactorialprocess.ExpClinCardiol.2002;7:40-53
72. FishbeinMC.Thevulnerableandunstableatheroscleroticplaque.CardiovascPathol.2010;19:6-1173. Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of
atherosclerosis.Nature.2011;473:317-32574. Rubin R, Strayer DS, Rubin E. Rubin's pathology: Clinicopathologic foundations of medicine.
LippincottWilliams&Wilkins;2008.75. VolkmanA,GowansJL.Theoriginofmacrophagesfrombonemarrowintherat.BrJExpPathol.
1965;46:62-7076. PasslickB,FliegerD,Ziegler-HeitbrockHW.Identificationandcharacterizationofanovelmonocyte
subpopulationinhumanperipheralblood.Blood.1989;74:2527-253477. Ziegler-HeitbrockHW,FingerleG,StrobelM,SchrautW,StelterF,SchuttC,PasslickB,PforteA.The
novel subset of cd14+/cd16+ bloodmonocytes exhibits features of tissue macrophages. Eur JImmunol.1993;23:2053-2058
78. GeissmannF,JungS,LittmanDR.Bloodmonocytesconsistoftwoprincipalsubsetswithdistinctmigratoryproperties.Immunity.2003;19:71-82
79. PalframanRT,JungS,ChengCY,WeningerW,LuoY,DorfM,LittmanDR,RollinsBJ,ZweerinkH,RotA,vonAndrianUH.Inflammatorychemokinetransportandpresentationinhev:Aremotecontrolmechanismformonocyterecruitmenttolymphnodesininflamedtissues.JournalofExperimentalMedicine.2001;194:1361-1373
80. Yang J,ZhangL,YuC,YangX-F,WangH.Monocyteandmacrophagedifferentiation:Circulationinflammatorymonocyteasbiomarkerforinflammatorydiseases.Biomarkerresearch.2014;2:1
81. MantovaniA,GarlandaC,LocatiM.Macrophagediversityandpolarization inatherosclerosis:Aquestionofbalance.ArteriosclerThrombVascBiol.2009;29:1419-1423
82. ColinS,Chinetti-GbaguidiG,StaelsB.Macrophagephenotypesinatherosclerosis. ImmunolRev.2014;262:153-166
83. MackanessGB.Cellularresistancetoinfection.JournalofExperimentalMedicine.1962;116:381-&84. Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage
mannosereceptoractivity:Amarkerofalternativeimmunologicmacrophageactivation.JExpMed.1992;176:287-292
85. MartinezFO,SicaA,MantovaniA,LocatiM.Macrophageactivationandpolarization.FrontBiosci-Landmrk.2008;13:453-461
86. Chistiakov DA, Bobryshev YV, Nikiforov NG, Elizova NV, Sobenin IA, Orekhov AN. Macrophagephenotypic plasticity in atherosclerosis: The associated features and the peculiarities of theexpressionofinflammatorygenes.IntJCardiol.2015;184:436-445
87. Dewals BG, Marillier RG, Hoving JC, Leeto M, Schwegmann A, Brombacher F. Il-4ralpha-independentexpressionofmannose receptorandym1bymacrophagesdependson their il-10responsiveness.PLoSNeglTropDis.2010;4:e689
88. RoszerT.Understandingthemysteriousm2macrophagethroughactivationmarkersandeffectormechanisms.MediatInflamm.2015
89. Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol.1999;17:593-623
90. deWintherMPJ,vanDijkKW,HavekesLM,HofkerMH.Macrophagescavengerreceptorclassa-amultifunctionalreceptorinatherosclerosis.ArteriosclThromVas.2000;20:290-297
91. KunjathoorVV,FebbraioM,PodrezEA,MooreKJ,AnderssonL,KoehnS,RheeJS,SilversteinR,HoffHF,FreemanMW.Scavengerreceptorsclassai/iiandcd36aretheprincipalreceptorsresponsiblefortheuptakeofmodifiedlowdensitylipoproteinleadingtolipidloadinginmacrophages.JournalofBiologicalChemistry.2002;277:49982-49988

Chapter1
31
92. CantonJ,NeculaiD,GrinsteinS.Scavengerreceptorsinhomeostasisandimmunity.NatureReviewsImmunology.2013;13:621-634
93. HuangSCC,EvertsB,IvanovaY,O'SullivanD,NascimentoM,SmithAM,BeattyW,Love-GregoryL,LamWY,O'NeilCM,YanC,DuH,AbumradNA,UrbanJF,ArtyomovMN,PearceEL,PearceEJ.Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. NatureImmunology.2014;15:846-855
94. vanTitsLJ,StienstraR,vanLentPL,NeteaMG,JoostenLA,StalenhoefAF.Oxidizedldlenhancespro-inflammatoryresponsesofalternativelyactivatedm2macrophages:Acrucialroleforkruppel-likefactor2.Atherosclerosis.2011;214:345-349
95. ZhengXF,HongYX,FengGJ,ZhangGF,RogersH,LewisMAO,WilliamsDW,XiaZF,SongB,WeiXQ.Lipopolysaccharide-induced m2 to m1 macrophage transformation for il-12p70 production isblockedbycandidaalbicansmediatedup-regulationofebi3expression.PlosOne.2013;8
96. Wang N, Liang HW, Zen K. Molecular mechanisms that influence the macrophage m1-m2polarizationbalance.FrontImmunol.2014;5
97. ShireyKA,PletnevaLM,PucheAC,KeeganAD,PrinceGA,BlancoJCG,VogelSN.Controlof rsv-induced lung injury by alternatively activated macrophages is il-4r alpha-, tlr4-, and ifn-beta-dependent.MucosalImmunol.2010;3:291-300
98. YurdagulA,FinneyAC,WoolardMD,OrrAW.Thearterialmicroenvironment:Thewhereandwhyofatherosclerosis.BiochemicalJournal.2016;473:1281-1295
99. TabasI,BornfeldtKE.Macrophagephenotypeandfunctionindifferentstagesofatherosclerosis.CirculationResearch.2016;118:653-667
100. daSilvaRF,LappalainenJ,Lee-RueckertM,KovanenPT.Conversionofhumanm-csfmacrophagesinto foam cells reduces their proinflammatory responses to classical m1-polarizing activation.Atherosclerosis.2016;248:170-178
101. KadlA,MeherAK,SharmaPR,LeeMY,DoranAC,JohnstoneSR,ElliottMR,GruberF,HanJ,ChenWS, Kensler T, RavichandranKS, IsaksonBE,WamhoffBR, LeitingerN. Identificationof a novelmacrophage phenotype that develops in response to atherogenic phospholipids via nrf2.CirculationResearch.2010;107:737-U155
102. BoyleJJ,HarringtonHA,PiperE,ElderfieldK,StarkJ,LandisRC,HaskardDO.Coronaryintraplaquehemorrhage evokes a novel atheroprotective macrophage phenotype. American Journal ofPathology.2009;174:1097-1108
103. FinnAV,NakanoM,PolavarapuR,KarmaliV,SaeedO,ZhaoXQ,YazdaniS,OtsukaF,DavisT,HabibA,NarulaJ,KolodgieFD,VirmaniR.Hemoglobindirectsmacrophagedifferentiationandpreventsfoam cell formation in human atherosclerotic plaques. Journal of the American College ofCardiology.2012;59:166-177
104. LeitingerN,SchulmanIG.Phenotypicpolarizationofmacrophagesinatherosclerosis.ArteriosclerThrombVascBiol.2013;33:1120-1126
105. Freiman PC, Mitchell GG, Heistad DD, Armstrong ML, Harrison DG. Atherosclerosis impairsendothelium-dependentvascularrelaxationtoacetylcholineandthrombininprimates.CirculationResearch.1986;58:783-789
106. Getz GS, Reardon CA. Animal models of atherosclerosis. Arterioscler Thromb Vasc Biol.2012;32:1104-1115
107. LuginbühlH,PauliB,RatcliffeHL.Atherosclerosisinswineandswineasamodelforthestudyofatherosclerosis.ComparativePathologyoftheHeart.119-126
108. RussellJ.Ofmiceandmen,ratsandatherosclerosis.CardiovascularResearch.2003;59:810-811109. ShoreB,ShoreV.Rabbitsasamodelforthestudyofhyperlipoproteinemiaandatherosclerosis.
AdvancesinExperimentalMedicineandBiology.1976:123-141110. VesselinovitchD,GetzGS,HughesRH,WisslerRW.Atherosclerosisintherhesusmonkeyfedthree
foodfats.Atherosclerosis.1974;20:303-321111. GetzGS,ReardonCA.Dietandmurineatherosclerosis.Arteriosclerosis,thrombosis,andvascular
biology.2006;26:242-249

Chapter1
32
112. Kapourchali FR, SurendiranG,Chen L,Uitz E, BahadoriB,MoghadasianMH.Animalmodelsofatherosclerosis.WorldJClinCases.2014;2:126-132
113. WhitmanSC.Apracticalapproachtousingmice inatherosclerosis research.ClinicalBiochemistReviews.2004;25:81-93
114. KuipersF,vanReeJM,HofkerMH,WoltersH,In'tVeldG,HavingaR,VonkRJ,PrincenHM,HavekesLM.Alteredlipidmetabolisminapolipoproteine-deficientmicedoesnotaffectcholesterolbalanceacrosstheliver.Hepatology.1996;24:241-247
115. PlumpAS,ForteTM,EisenbergS,BreslowJL.Atherogenicbeta-vldlintheapoe-deficientmouse-composition,origin,andfate.Circulation.1993;88:2-2
116. Meir KS, Leitersdorf E. Atherosclerosis in the apolipoprotein-e-deficient mouse: A decade ofprogress.ArteriosclerThrombVascBiol.2004;24:1006-1014
117. BreslowJL.Mousemodelsofatherosclerosis.Science.1996;272:685-688118. Reddick RL, Zhang SH,MaedaN. Atherosclerosis inmice lacking apo-e - evaluation of lesional
developmentandprogression.ArteriosclerosisandThrombosis.1994;14:141-147119. NakashimaY,PlumpAS,RainesEW,BreslowJL,RossR.Apoe-deficientmicedeveloplesionsofall
phases of atherosclerosis throughout the arterial tree. Arteriosclerosis and Thrombosis.1994;14:133-140
120. IshibashiS,BrownMS,GoldsteinJL,GerardRD,HammerRE,HerzJ.Hypercholesterolemiainlowdensitylipoproteinreceptorknockoutmiceanditsreversalbyadenovirus-mediatedgenedelivery.JClinInvest.1993;92:883-893
121. Zadelaar S, Kleemann R, Verschuren L, de Vries-Van derWeij J, van der Hoorn J, Princen HM,KooistraT.Mousemodelsforatherosclerosisandpharmaceuticalmodifiers.ArteriosclerThrombVascBiol.2007;27:1706-1721
122. IshibashiS,GoldsteinJL,BrownMS,HerzJ,BurnsDK.Massivexanthomatosisandatherosclerosisincholesterol-fedlowdensitylipoproteinreceptor-negativemice.JClinInvest.1994;93:1885-1893
123. KowalaMC,RecceR,BeyerS,GuC,ValentineM.Characterizationofatherosclerosisinldlreceptorknockoutmice:Macrophageaccumulationcorrelateswithrapidandsustainedexpressionofaorticmcp-1/je.Atherosclerosis.2000;149:323-330
124. MaY,WangW, Zhang J, LuY,WuW,YanH,WangY.Hyperlipidemiaandatherosclerotic lesiondevelopmentinldlr-deficientmiceonalong-termhigh-fatdiet.PLoSOne.2012;7:e35835
125. TangiralaRK,RubinEM,PalinskiW.Quantitationofatherosclerosisinmurinemodels:Correlationbetweenlesionsintheaorticoriginandintheentireaorta,anddifferencesintheextentoflesionsbetween sexes in ldl receptor-deficient and apolipoprotein e-deficient mice. J Lipid Res.1995;36:2320-2328
126. Lichtman AH, Clinton SK, Iiyama K, Connelly PW, Libby P, Cybulsky MI. Hyperlipidemia andatherosclerotic lesiondevelopment in ldl receptor-deficientmice feddefinedsemipurifieddietswithandwithoutcholate.ArteriosclThromVas.1999;19:1938-1944
127. IshibashiS,GoldsteinJL,BrownMS,HerzJ,BurnsDK.Massivexanthomatosisandatherosclerosisincholesterol-fedlow-density-lipoproteinreceptor-negativemice.JournalofClinicalInvestigation.1994;93:1885-1893
128. Linton MF, Fazio S. Macrophages, lipoprotein metabolism, and atherosclerosis: Insights frommurinebonemarrowtransplantationstudies.CurrentOpinioninLipidology.1999;10:97-105
129. ChorroFJ,Such-BelenguerL,Lopez-MerinoV.Animalmodelsofcardiovasculardisease.RevEspCardiol.2009;62:69-84
130. MaYL,WangWY,ZhangJ,LuYL,WuWY,YanH,WangYP.Hyperlipidemiaandatheroscleroticlesiondevelopmentinldlr-deficientmiceonalong-termhigh-fatdiet.PlosOne.2012;7
131. RosenfeldME,PolinskyP,VirmaniR,KauserK,RubanyiG,SchwartzSM.Advancedatheroscleroticlesionsintheinnominatearteryoftheapoeknockoutmouse.ArteriosclThromVas.2000;20:2587-2592
132. Herijgers N, VanEck M, Groot PHE, Hoogerbrugge PM, VanBerkel TJC. Effect of bone marrowtransplantation on lipoprotein metabolism and atherosclerosis in ldl receptor knockout mice.ArteriosclThromVas.1997;17:1995-2003

Chapter1
33
133. Boisvert WA, Spangenberg J, Curtiss LK. Role of leukocyte-specific ldl receptors on plasmalipoproteincholesterolandatherosclerosisinmice.ArteriosclThromVas.1997;17:340-347
134. FazioS,HastyAH,CarterKJ,MurrayAB,Price JO,LintonMF.Leukocyte lowdensity lipoproteinreceptor(ldl-r)doesnotcontributetoldlclearanceinvivo:Bonemarrowtransplantationstudiesinthemouse.JLipidRes.1997;38:391-400
135. LintonMF,AtkinsonJB,FazioS.Preventionofatherosclerosisinapolipoproteine-deficientmicebybone-marrowtransplantation.Science.1995;267:1034-1037
136. FazioS,LintonMF.Murinebonemarrowtransplantationasanovelapproachtostudyingtheroleofmacrophagesinlipoproteinmetabolismandatherogenesis.TrendsCardiovasMed.1996;6:58-65
137. Boisvert WA, Spangenberg J, Curtiss LK. Treatment of severe hypercholesterolemia inapolipoproteine-deficientmicebybone-marrowtransplantation.JournalofClinicalInvestigation.1995;96:1118-1124
138. VanEckM, Herijgers N, Yates J, PearceNJ, Hoogerbrugge PM,Groot PHE, VanBerkel TJC. Bonemarrowtransplantationinapolipoproteinedeficientmice-effectofapoegenedosageonserumlipid concentrations, (beta)vldl catabolism, and atherosclerosis. Arterioscl Throm Vas.1997;17:3117-3126
139. LawrenceT,NatoliG.Transcriptionalregulationofmacrophagepolarization:Enablingdiversitywithidentity.NatRevImmunol.2011;11:750-761
140. Bekkering S, Joosten LA, van der Meer JW, Netea MG, Riksen NP. The epigenetic memory ofmonocytesandmacrophagesasanoveldrugtargetinatherosclerosis.ClinTher.2015;37:914-923
141. Sica A, Mantovani A. Macrophage plasticity and polarization: In vivo veritas. J Clin Invest.2012;122:787-795
142. Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, Curtiss LK. Transcriptionalrepressionofatherogenicinflammation:Modulationbyppardelta.Science.2003;302:453-457
143. Moore KJ, Sheedy FJ, Fisher EA.Macrophages in atherosclerosis: A dynamic balance.Nat RevImmunol.2013;13:709-721
144. WangX,CaoQ,YuL,ShiH,XueB,ShiH.EpigeneticregulationofmacrophagepolarizationandinflammationbyDNAmethylationinobesity.JCIInsight.2016;1:e87748
145. SaeedS,QuintinJ,KerstensHH,RaoNA,AghajanirefahA,MatareseF,ChengSC,RatterJ,BerentsenK,vanderEntMA,SharifiN,Janssen-MegensEM,TerHuurneM,MandoliA,vanSchaikT,NgA,BurdenF,DownesK,FrontiniM,KumarV,Giamarellos-BourboulisEJ,OuwehandWH,vanderMeerJW,JoostenLA,WijmengaC,MartensJH,XavierRJ,LogieC,NeteaMG,StunnenbergHG.Epigeneticprogrammingofmonocyte-to-macrophagedifferentiationandtrained innate immunity.Science.2014;345:1251086
146. Rask-AndersenM,ZhangJ,FabbroD,SchiothHB.Advancesinkinasetargeting:Currentclinicaluseandclinicaltrials.TrendsinPharmacologicalSciences.2014;35:60-76
147. LiuPX,ChengHL,RobertsTM,ZhaoJJ.Targetingthephosphoinositide3-kinasepathwayincancer.NatureReviewsDrugDiscovery.2009;8:627-644
148. HommesDW,PeppelenboschMP,vanDeventerSJ.Mitogenactivatedprotein(map)kinasesignaltransductionpathwaysandnovelanti-inflammatorytargets.Gut.2003;52:144-151
149. English JM, Cobb MH. Pharmacological inhibitors of mapk pathways. Trends Pharmacol Sci.2002;23:40-45
150. KaminskaB.Mapksignallingpathwaysasmoleculartargetsforanti-inflammatorytherapy-frommolecularmechanismstotherapeuticbenefits.Bba-ProteinsProteom.2005;1754:253-262
151. TugalD,LiaoX,JainMK.Transcriptionalcontrolofmacrophagepolarization.ArteriosclerThrombVascBiol.2013;33:1135-1144
152. LaurilaPP,SoronenJ,KooijmanS,ForsstromS,BoonMR,SurakkaI,KaiharjuE,CoomansCP,VanDen Berg SA, Autio A, Sarin AP, Kettunen J, Tikkanen E,Manninen T,Metso J, Silvennoinen R,MerikantoK,RuuthM,PerttilaJ,MakelaA,IsomiA,TuomainenAM,TikkaA,RamadanUA,SeppalaI,LehtimakiT,ErikssonJ,HavulinnaA,JulaA,KarhunenPJ,SalomaaV,PerolaM,EhnholmC,Lee-RueckertM,VanEckM,RoivainenA,TaskinenMR,PeltonenL,MervaalaE,JalankoA,HohtolaE,

Chapter1
34
OlkkonenVM,Ripatti S,KovanenPT,RensenPC,SuomalainenA, JauhiainenM.Usf1deficiencyactivates brown adipose tissue and improves cardiometabolic health. Sci Transl Med.2016;8:323ra313
153. HorbachT,GotzC,KietzmannT,DimovaEY.Proteinkinasesasswitchesforthefunctionofupstreamstimulatoryfactors:Implicationsfortissueinjuryandcancer.FrontPharmacol.2015;6
154. HouYC,JanczukA,WangPG.Currenttrendsinthedevelopmentofnitricoxidedonors.CurrPharmDes.1999;5:417-441
155. Forstermann U, Sessa WC. Nitric oxide synthases: Regulation and function. Eur Heart J.2012;33:829-837,837a-837d
156. XuF,SunYN,ChenYG,SunY,LiRJ,LiuCX,ZhangC,WangR,ZhangY.Endothelialcellapoptosisisresponsiblefortheformationofcoronarythromboticatheroscleroticplaques.TohokuJExpMed.2009;218:25-33
157. PieriniD,BryanNS.Nitricoxideavailabilityasamarkerofoxidativestress.AdvancedProtocolsinOxidativeStressIII.2015:63-71
158. CybulskyMI,IiyamaK,LiH,ZhuS,ChenM,IiyamaM,DavisV,Gutierrez-RamosJC,ConnellyPW,Milstone DS. A major role for vcam-1, but not icam-1, in early atherosclerosis. J Clin Invest.2001;107:1255-1262
159. KuhlencordtPJ,GyurkoR,HanF, Scherrer-CrosbieM,AretzTH,HajjarR,PicardMH,HuangPL.Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease inapolipoprotein e/endothelial nitric oxide synthase double-knockout mice. Circulation.2001;104:448-454
160. Hayashi T, Sumi D, Juliet PAR,Matsui-Hirai H, Asai-Tanaka Y, Kano H, Fukatsu A, Tsunekawa T,Miyazaki A, Iguchi A, Ignarro LJ. Gene transfer of endothelial no synthase, but not enos plusinduciblenos,regressedatherosclerosisinrabbits.CardiovascularResearch.2004;61:339-351
161. Mujynya-LudungeK,ViswambharanH,DriscollR,MingXF,vonSegesserLK,KappenbergerL,YangZH, Vassalli G. Endothelial nitric oxide synthase gene transfer restores endothelium-dependentrelaxationsandattenuateslesionformationincarotidarteriesinapolipoproteine-deficientmice.BasicResearchinCardiology.2005;100:102-111
162. OzakiM, Kawashima S, Yamashita T,Hirase T,NamikiM, InoueN,HirataK, YasuiH, SakuraiH,YoshidaY,MasadaM,YokoyamaM.Overexpressionofendothelialnitricoxidesynthaseacceleratesatherosclerotic lesion formation in apoe-deficient mice. Journal of Clinical Investigation.2002;110:331-340
163. DetmersPA,HernandezM,MudgettJ,HassingH,BurtonC,MundtS,ChunS,FletcherD,CardDJ,LisnockJ,WeikelR,BergstromJD,ShevellDE,Hermanowski-VosatkaA,SparrowCP,ChaoYS,RaderDJ, Wright SD, Pure E. Deficiency in inducible nitric oxide synthase results in reducedatherosclerosisinapolipoproteine-deficientmice.JournalofImmunology.2000;165:3430-3435
164. KuhlencordtPJ,Chen JQ,HanF,Astern J,HuangPL.Geneticdeficiencyof induciblenitricoxidesynthasereducesatherosclerosisandlowersplasmalipidperoxidesinapolipoproteine-knockoutmice.Circulation.2001;103:3099-3104
165. Mujynya-LudungeK,ViswambharanH,DriscollR,MingXF,vonSegesserLK,KappenbergerL,YangZ, Vassalli G. Endothelial nitric oxide synthase gene transfer restores endothelium-dependentrelaxationsandattenuateslesionformationincarotidarteriesinapolipoproteine-deficientmice.BasicResCardiol.2005;100:102-111
166. FishJE,MarsdenPA.Endothelialnitricoxidesynthase:Insightintocell-specificgeneregulationinthevascularendothelium.CellMolLifeSci.2006;63:144-162
167. GreenSJ,SchellerLF,MarlettaMA,SeguinMC,KlotzFW,SlayterM,NelsonBJ,NacyCA.Nitric-oxide - cytokine-regulation of nitric-oxide in host-resistance to intracellular pathogens.ImmunologyLetters.1994;43:87-94
168. ForstermannU,SessaWC.Nitricoxidesynthases:Regulationandfunction.EuropeanHeartJournal.2012;33:829-+
169. Lowenstein CJ. Beneficial effects of neuronal nitric oxide synthase in atherosclerosis.ArteriosclThromVas.2006;26:1417-1418

Chapter1
35
170. Verbeuren TJ, Coene MC, Jordaens FH, Vanhove CE, Zonnekeyn LL, Herman AG. Effect ofhypercholesterolemia on vascular reactivity in the rabbit .2. Influence of treatment withdipyridamole on endothelium-dependent and endothelium-independent responses in isolatedaortasofcontrolandhypercholesterolemicrabbits.CirculationResearch.1986;59:496-504
171. Kuhlencordt PJ, Hotten S, Schodel J, Rutzel S, Hu K, Widder J, Marx A, Huang PL, Ertl G.Atheroprotective effects of neuronal nitric oxide synthase in apolipoprotein e knockout mice.ArteriosclThromVas.2006;26:1539-1544
172. BredtDS,GlattCE,HwangPM,FotuhiM,DawsonTM,SnyderSH.Nitricoxidesynthaseproteinandmrnaarediscretelylocalizedinneuronalpopulationsofthemammaliancnstogetherwithnadphdiaphorase.Neuron.1991;7:615-624
173. MungrueIN,BredtDS.Nnosataglance:Implicationsforbrainandbrawn.JCellSci.2004;117:2627-2629
174. Papapetropoulos A, Rudic RD, Sessa WC. Molecular control of nitric oxide synthases in thecardiovascularsystem.CardiovascularResearch.1999;43:509-520
175. SchodelJ,PadmapriyaP,MarxA,HuangPL,ErtlG,KuhlencordtPJ.Expressionofneuronalnitricoxidesynthasesplicevariantsinatheroscleroticplaquesofapoeknockoutmice.Atherosclerosis.2009;206:383-389
176. CapettiniLSA,CortesSF,SilvaJF,Alvarez-LeiteJI,LemosVS.Decreasedproductionofneuronalnos-derived hydrogen peroxide contributes to endothelial dysfunction in atherosclerosis. Brit JPharmacol.2011;164:1738-1748
177. ButteryLD,SpringallDR,ChesterAH,EvansTJ,StandfieldEN,ParumsDV,YacoubMH,PolakJM.Induciblenitricoxidesynthaseispresentwithinhumanatheroscleroticlesionsandpromotestheformationandactivityofperoxynitrite.LabInvest.1996;75:77-85
178. Miyoshi T, Li YH, Shih DM,Wang XP, Laubach VE,Matsumoto AH, HelmGA, Lusis AJ, ShiWB.Deficiency of inducible no synthase reduces advanced but not early atherosclerosis inapolipoproteine-deficientmice.LifeSciences.2006;79:525-531
179. AlbinaJE,AbateJA,MastrofrancescoB.Roleofornithineasaprolineprecursorinhealingwounds.JSurgRes.1993;55:97-102
180. GonzalezE,McGrawTE.Theaktkinasesisoformspecificityinmetabolismandcancer.CellCycle.2009;8:2502-2508
181. Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. Pi3k/akt and apoptosis: Size matters.Oncogene.2003;22:8983-8998
182. LiangJ,SlingerlandJM.Multiplerolesofthepi3k/pkb(akt)pathwayincellcycleprogression.CellCycle.2003;2:339-345
183. Morales-RuizM,FultonD,SowaG,LanguinoLR,FujioY,WalshK,SessaWC.Vascularendothelialgrowthfactor-stimulatedactinreorganizationandmigrationofendothelialcellsisregulatedviatheserine/threoninekinaseakt.CirculationResearch.2000;86:892-896
184. BrunetA,BonniA,ZigmondMJ,LinMZ,JuoP,HuLS,AndersonMJ,ArdenKC,BlenisJ,GreenbergME.Aktpromotescellsurvivalbyphosphorylatingandinhibitingaforkheadtranscriptionfactor.Cell.1999;96:857-868
185. ZengGY,NystromFH,RavichandranLV,CongLN,KirbyM,MostowskiH,QuonMJ.Rolesforinsulinreceptor,pi3-kinase,andaktininsulin-signalingpathwaysrelatedtoproductionofnitricoxideinhumanvascularendothelialcells.Circulation.2000;101:1539-1545
186. YuHX,LittlewoodT,BennettM.Aktisoformsinvasculardisease.VascPharmacol.2015;71:57-64187. Fayard E, Tintignac LA, Baudry A, Hemmings BA. Protein kinase b/akt at a glance. J Cell Sci.
2005;118:5675-5678188. BabaevVR,HebronKE,WieseCB,TothCL,DingL,ZhangY,MayJM,FazioS,VickersKC,LintonMF.
Macrophagedeficiencyofakt2reducesatherosclerosisinldlrnullmice.JLipidRes.2014;55:2296-2308
189. XuF,KangY,ZhangH,PiaoZ,YinH,DiaoR,XiaJ,ShiL.Akt1-mediatedregulationofmacrophagepolarization in a murine model of staphylococcus aureus pulmonary infection. J Infect Dis.2013;208:528-538

Chapter1
36
190. Arranz A, Doxaki C, Vergadi E,Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E,AndroulidakiA,VenihakiM,MargiorisAN,StathopoulosEN,TsichlisPN,TsatsanisC.Akt1andakt2protein kinases differentially contribute tomacrophage polarization.ProcNatl Acad Sci U S A.2012;109:9517-9522
191. RotllanN,Chamorro-JorganesA,AraldiE,WanschelAC,AryalB,ArandaJF,GoedekeL,SalernoAG,RamirezCM,SessaWC,SuarezY,Fernandez-HernandoC.Hematopoieticakt2deficiencyattenuatestheprogressionofatherosclerosis.FasebJournal.2015;29:597-610
192. Fernandez-Hernando C, Ackah E, Yu J, Suarez Y, Murata T, Iwakiri Y, Prendergast J, Miao RQ,BirnbaumMJ,SessaWC.Lossofakt1leadstosevereatherosclerosisandocclusivecoronaryarterydisease.CellMetab.2007;6:446-457
193. DingL,BiswasS,MortonRE,SmithJD,HayN,ByzovaTV,FebbraioM,PodrezEA.Akt3deficiencyinmacrophagespromotesfoamcellformationandatherosclerosisinmice.CellMetab.2012;15:861-872
194. Fernandez-Hernando C, Jozsef L, Jenkins D, Di Lorenzo A, SessaWC. Absence of akt1 reducesvascularsmoothmusclecellmigrationandsurvivaland inducesfeaturesofplaquevulnerabilityandcardiacdysfunctionduringatherosclerosis.ArteriosclerThrombVascBiol.2009;29:2033-2040
195. YuH,LittlewoodT,BennettM.Aktisoformsinvasculardisease.VasculPharmacol.2015;71:57-64196. RensingKL,deJagerSCA,StroesES,VosM,TwicklerMTB,Dallinga-ThieGM,deVriesCJM,Kuiper
J,BotI,vonderThusenJH.Akt2/ldlrdoubleknockoutmicedisplayimpairedglucosetoleranceanddevelopmorecomplexatheroscleroticplaquesthanldlrknockoutmice.CardiovascularResearch.2014;101:277-287
197. BabaevVR,HebronKE,WieseCB,TothCL,DingL,ZhangYM,MayJM,FazioS,VickersKC,LintonMF. Macrophage deficiency of akt2 reduces atherosclerosis in ldlr null mice. Journal of LipidResearch.2014;55:2296-2308
198. PasterkampG,vanderLaanSW,HaitjemaS,ForoughiAslH,SiemelinkMA,BezemerT,vanSettenJ,DichgansM,MalikR,WorrallBB,SchunkertH,SamaniNJ,deKleijnDP,MarkusHS,HoeferIE,MichoelT,deJagerSC,BjorkegrenJL,denRuijterHM,AsselbergsFW.Humanvalidationofgenesassociatedwithamurineatheroscleroticphenotype.ArteriosclerThrombVascBiol.2016;36:1240-1246
199. Blackhart B, Yao Z,McCarthy B. An expression system for human apolipoprotein b100 in a rathepatomacellline.JournalofBiologicalChemistry.1990;265:8358-8360
200. Babaev VR, Ding L, Zhang Y, May JM, Linton MF. Loss of macrophage akt2/akt3 or akt1/akt2decreasescellsurvivalandsuppressesearlyatherosclerosisinldlrnullmice.2015
201. HuY,DietrichH,MetzlerB,WickG,XuQ.Hyperexpressionandactivationofextracellularsignal-regulatedkinases(erk1/2)inatheroscleroticlesionsofcholesterol-fedrabbits.ArteriosclerThrombVascBiol.2000;20:18-26
202. Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A cd36-dependentsignalingcascadeisnecessaryformacrophagefoamcellformation.CellMetabolism.2006;4:211-221
203. ZhangL,ChenY,YangX,YangJ,CaoX,LiX,LiL,MiaoQR,HajjarDP,DuanY,HanJ.Mek1/2inhibitorsactivate macrophage abcg1 expression and reverse cholesterol transport-an anti-atherogenicfunctionoferk1/2inhibition.BiochimBiophysActa.2016;1861:1180-1191
204. ChenY,DuanY,YangX,SunL,LiuM,WangQ,MaX,ZhangW,LiX,HuW,MiaoRQ,XiangR,HajjarDP,HanJ.Inhibitionoferk1/2andactivationoflxrsynergisticallyreduceatheroscleroticlesionsinapoe-deficientmice.ArteriosclerThrombVascBiol.2015;35:948-959
205. ParkSJ,LeeKP,KangS,LeeJ,SatoK,ChungHY,OkajimaF,ImDS.Sphingosine1-phosphateinducedanti-atherogenic and atheroprotective m2 macrophage polarization through il-4. Cell Signal.2014;26:2249-2258
206. LongME,EddyWE,GongKQ,Lovelace-MaconLL,McMahanRS,CharronJ,LilesWC,ManiconeAM.Mek1/2inhibitionpromotesmacrophagereparativeproperties.JImmunol.2017;198:862-872

Chapter1
37
207. YangY,KimSC,YuT,YiYS,RheeMH,SungGH,YooBC,ChoJY.Functionalrolesofp38mitogen-activatedproteinkinase inmacrophage-mediated inflammatory responses.Mediators Inflamm.2014;2014:352371
208. LeeJC,LaydonJT,McdonnellPC,GallagherTF,KumarS,GreenD,McnultyD,BlumenthalMJ,HeysJR,LandvatterSW,StricklerJE,MclaughlinMM,SiemensIR,FisherSM,LiviGP,WhiteJR,AdamsJL,Young PR. A protein-kinase involved in the regulation of inflammatory cytokine biosynthesis.Nature.1994;372:739-746
209. KimSH,KimJ,SharmaRP.Inhibitionofp38anderkmapkinasesblocksendotoxin-inducednitricoxide production and differentially modulates cytokine expression. Pharmacological Research.2004;49:433-439
210. ZhuW,DowneyJS,GuJ,DiPadovaF,GramH,HanJH.Regulationoftnfexpressionbymultiplemitogen-activatedproteinkinasepathways.JournalofImmunology.2000;164:6349-6358
211. PerregauxDG,DeanD,CronanM,ConnellyP,GabelCA.Inhibitionofinterleukin-1betaproductionbyskf86002:Evidenceoftwositesofinvitroactivityandofatimeandsystemdependence.MolPharmacol.1995;48:433-442
212. KangYJ,ChenJ,OtsukaM,MolsJ,RenS,WangY,HanJ.Macrophagedeletionofp38alphapartiallyimpairslipopolysaccharide-inducedcellularactivation.JImmunol.2008;180:5075-5082
213. Jimenez-Garcia L, Herranz S, Luque A, Hortelano S. Critical role of p38 mapk in il-4-inducedalternativeactivationofperitonealmacrophages.EuropeanJournalofImmunology.2015;45:273-286
214. ZhaoM,LiuYW,WangXF,NewLG,HanJH,BrunkUT.Activationofthep38mapkinasepathwayisrequiredforfoamcellformationfrommacrophagesexposedtooxidizedldl.Apmis.2002;110:458-468
215. YinR.Pparγphosphorylationmediatedbyjnkmapk:Apotentialroleinmac-rophage-derivedfoamcellformation.Actapharmacologicasinica.2006;27:1146-1152
216. Kardakaris R, Gareus R, Xanthoulea S, Pasparakis M. Endothelial and macrophage-specificdeficiencyofp38alphamapkdoesnotaffectthepathogenesisofatherosclerosisinapoe-/-mice.PLoSOne.2011;6:e21055
217. SeimonTA,WangY,HanS,SenokuchiT,SchrijversDM,KuriakoseG,TallAR,TabasIA.Macrophagedeficiencyofp38alphamapkpromotesapoptosisandplaquenecrosisinadvancedatheroscleroticlesionsinmice.JClinInvest.2009;119:886-898
218. SeegerFH,SeddingD,LangheinrichAC,HaendelerJ,ZeiherAM,DimmelerS.Inhibitionofthep38map kinase in vivo improves number and functional activity of vasculogenic cells and reducesatheroscleroticdiseaseprogression.BasicResCardiol.2010;105:389-397
219. JagaveluK,TietgeUJ,GaestelM,DrexlerH,SchiefferB,BavendiekU.Systemicdeficiencyofthemapkinase-activatedproteinkinase2reducesatherosclerosisinhypercholesterolemicmice.CircRes.2007;101:1104-1112
220. WaetzigV,HerdegenT.Context-specificinhibitionofjnks:Overcomingthedilemmaofprotectionanddamage.TrendsPharmacolSci.2005;26:455-461
221. BabaevVR,YeungM,ErbayE,DingL,ZhangY,MayJM,FazioS,HotamisligilGS,LintonMF.Jnk1deficiencyinhematopoieticcellssuppressesmacrophageapoptosisandincreasesatherosclerosisinlow-densitylipoproteinreceptornullmice.ArteriosclerThrombVascBiol.2016;36:1122-1131
222. Himes SR, Sester DP, Ravasi T, Cronau SL, Sasmono T, Hume DA. The jnk are important fordevelopmentandsurvivalofmacrophages.JImmunol.2006;176:2219-2228
223. Sanchez-TilloE,ComaladaM,XausJ,FarreraC,ValledorAF,CaellesC,LloberasJ,CeladaA.Jnk1isrequired for the induction of mkp1 expression in macrophages during proliferation andlipopolysaccharide-dependentactivation.JournalofBiologicalChemistry.2007;282:12566-12573
224. UtsugiM,DobashiK,OnoA,IshizukaT,HisadaT,KogaY,ShimizuY,KawataT,MatsuzakiS,AokiH,KamideY,MoriM.Jnk1andjnk2differentlyregulateil-12productioninthp-1macrophagecells.Cytokine.2010;51:127-131
225. Guma M, Ronacher LM, Firestein GS, Karin M, Corr M. Jnk-1 deficiency limits macrophage-mediatedantigen-inducedarthritis.ArthritisRheum.2011;63:1603-1612

Chapter1
38
226. Muslin AJ. Mapk signalling in cardiovascular health and disease: Molecular mechanisms andtherapeutictargets.ClinSci.2008;115:203-218
227. RicciR,SumaraG,SumaraI,RozenbergI,KurrerM,AkhmedovA,HersbergerM,ErikssonU,EberliFR, BecherB, Boren J, ChenM,CybulskyMI,MooreKJ, FreemanMW,Wagner EF,Matter CM,Luscher TF. Requirement of jnk2 for scavenger receptor a-mediated foam cell formation inatherogenesis.Science.2004;306:1558-1561
228. MetzlerB,HuYH,DietrichH,XuQB.Increasedexpressionandactivationofstress-activatedproteinkinases/c-junnh(2)-terminalproteinkinasesinatheroscleroticlesionscoincidewithp53.AmericanJournalofPathology.2000;156:1875-1886
229. FeyD,CroucherDR,KolchW,KholodenkoBN.Crosstalkandsignalingswitchesinmitogen-activatedproteinkinasecascades.FrontPhysiol.2012;3
230. CauntCJ,KeyseSM.Dual-specificitymapkinasephosphatases (mkps): Shaping theoutcomeofmapkinasesignalling.FEBSJ.2013;280:489-504
231. Salojin K, Oravecz T. Regulation of innate immunity by mapk dual-specificity phosphatases:Knockoutmodelsrevealnewtricksofoldgenes.JLeukocBiol.2007;81:860-869
232. HuangCY,TanTH.Dusps,tomapkinasesandbeyond.CellBiosci.2012;2233. PattersonKI,BrummerT,O'BrienPM,DalyRJ.Dual-specificityphosphatases:Critical regulators
withdiversecellulartargets.BiochemicalJournal.2009;418:475-489234. TheodosiouA,AshworthA.Mapkinasephosphatases.GenomeBiol.2002;3:REVIEWS3009235. CampsM,NicholsA,ArkinstallS.Dualspecificityphosphatases:Agenefamilyforcontrolofmap
kinasefunction.FASEBJ.2000;14:6-16236. Lang R, HammerM,Mages J. Duspmeet immunology: Dual specificitymapk phosphatases in
controloftheinflammatoryresponse.JournalofImmunology.2006;177:7497-7504237. Sim J, Yi K, Kim H, Ahn H, Chung Y, Rehman A, Jang SM, Lee KH, Jang K, Paik SS.
Immunohistochemical expression of dual-specificity protein phosphatase 4 in patients withcolorectaladenocarcinoma.GastroenterolResPract.2015;2015:283764
238. HamJE,OhEK,KimDH,ChoiSH.Differentialexpressionprofilesandrolesofinducibleduspsanderk1/2-specific constitutive dusp6 and dusp7 in microglia. Biochem Biophys Res Commun.2015;467:254-260
239. KondohK,NishidaE.Regulationofmapkinasesbymapkinasephosphatases.BiochimBiophysActa.2007;1773:1227-1237
240. Abraham SM, Clark AR. Dual-specificity phosphatase 1: A critical regulator of innate immuneresponses.BiochemSocT.2006;34:1018-1023
241. SmallieT,RossEA,AmmitAJ,CunliffeHE,TangT,RosnerDR,RidleyML,BuckleyCD,SaklatvalaJ,Dean JL, Clark AR. Dual-specificity phosphatase 1 and tristetraprolin cooperate to regulatemacrophageresponsestolipopolysaccharide.JImmunol.2015;195:277-288
242. JeffreyKL,BrummerT,RolphMS,LiuSM,CallejasNA,GrumontRJ,GillieronC,MackayF,GreyS,CampsM,RommelC,GerondakisSD,MackayCR.Positiveregulationofimmunecellfunctionandinflammatoryresponsesbyphosphatasepac-1.NatureImmunology.2006;7:274-283
243. Al-MutairiMS,CadalbertLC,McGachyHA,ShweashM,SchroederJ,KurnikM,SlossCM,BryantCE,AlexanderJ,PlevinR.Mapkinasephosphatase-2playsacriticalrole inresponseto infectionbyleishmaniamexicana.PlosPathog.2010;6
244. CornellTT,RodenhouseP,CaiQ,SunL,ShanleyTP.Mitogen-activatedproteinkinasephosphatase2regulatestheinflammatoryresponseinsepsis.InfectionandImmunity.2010;78:2868-2876
245. RushworthLK,KidgerAM,DelavaineL,StewartG,vanSchelvenS,DavidsonJ,BryantCJ,CaddyeE,East P, Caunt CJ, Keyse SM. Dual-specificity phosphatase 5 regulates nuclear erk activity andsuppressesskincancerbyinhibitingmutantharvey-ras(hrasq61l)-drivenserpinb2expression.ProcNatlAcadSciUSA.2014;111:18267-18272
246. LuoLJ,LiuF,WangXY,DaiTY,DaiYL,DongC,GeBX.Anessentialfunctionformkp5intheformationofoxidizedlowdensitylipid-inducedfoamcells.CellSignal.2012;24:1889-1898

Chapter1
39
247. ShenJ,ChandrasekharanUM,AshrafMZ,LongE,MortonRE,LiuY,SmithJD,DiCorletoPE.Lackofmitogen-activatedproteinkinasephosphatase-1protectsapoe-nullmiceagainstatherosclerosis.CircRes.2010;106:902-910
248. ImaizumiS,GrijalvaV,PricemanS,WuL,SuF,Farias-EisnerR,HamaS,NavabM,FogelmanAM,ReddyST.Mitogen-activatedproteinkinasephosphatase-1deficiencydecreasesatherosclerosisinapolipoprotein e nullmice by reducingmonocyte chemoattractant protein-1 levels.MolGenetMetab.2010;101:66-75
249. Gregor PD, Sawadogo M, Roeder RG. The adenovirus major late transcription factor usf is amemberofthehelix-loop-helixgroupofregulatoryproteinsandbindstoDNAasadimer.Genes&Development.1990;4:1730-1740
250. SiritoM, Lin Q,Maity T, SawadogoM. Ubiquitous expression of the 43- and 44-kda forms oftranscriptionfactorusfinmammaliancells.NucleicAcidsRes.1994;22:427-433
251. PajukantaP,LiljaHE,SinsheimerJS,CantorRM,LusisAJ,GentileM,DuanXJ,Soro-PaavonenA,Naukkarinen J, Saarela J, Laakso M, Ehnholm C, Taskinen MR, Peltonen L. Familial combinedhyperlipidemiaisassociatedwithupstreamtranscriptionfactor1(usf1).NatGenet.2004;36:371-376
252. NgMC,Miyake K, SoWY, Poon EW, Lam VK, Li JK, Cox NJ, Bell GI, Chan JC. The linkage andassociationofthegeneencodingupstreamstimulatoryfactor1withtype2diabetesandmetabolicsyndromeinthechinesepopulation.Diabetologia.2005;48:2018-2024
253. CoonH, Xin Y, Hopkins PN, Cawthon RM,Hasstedt SJ, Hunt SC.Upstream stimulatory factor 1associatedwithfamilialcombinedhyperlipidemia, ldlcholesterol,andtriglycerides.HumGenet.2005;117:444-451
254. Sanchez AP, Zhao JH, You Y, Decleves AE, Diamond-Stanic M, Sharma K. Role of the usf1transcriptionfactorindiabetickidneydisease.AmJPhysiol-Renal.2011;301:F271-F279
255. Naukkarinen J,GentileM,Soro-PaavonenA, Saarela J,KoistinenHA,PajukantaP,TaskinenMR,PeltonenL.Usf1anddyslipidemias:Convergingevidenceforafunctionalintronicvariant.HumanMolecularGenetics.2005;14:2595-2605
256. GalibertMD,CarreiraS,GodingCR.Theusf-1transcriptionfactorisanoveltargetforthestress-responsive p38 kinase and mediates uv-induced tyrosinase expression. Embo Journal.2001;20:5022-5031
257. Nowak M, Helleboid-Chapman A, Jakel H, Martin G, Duran-Sandoval D, Staels B, Rubin EM,PennacchioLA,TaskinenMR,Fruchart-NajibJ,FruchartJC. Insulin-mediateddown-regulationofapolipoprotein a5 gene expression through the phosphatidylinositol 3-kinase pathway: Role ofupstreamstimulatoryfactor.MolCellBiol.2005;25:1537-1548
258. ImagawaS,FujiiS,DongJ,FurumotoT,KanekoT,ZamanT,SatohY,TsutsuiH,SobelBE.Hepatocytegrowthfactorregulatesebox-dependentplasminogenactivatorinhibitortype1geneexpressioninhepg2livercells.ArteriosclThromVas.2006;26:2407-2413
259. ChandaD,LiTG,SongKH,KimYH,SimJG,LeeCH,ChiangJYL,ChoiHS.Hepatocytegrowthfactorfamilynegativelyregulateshepaticgluconeogenesisviainductionoforphannuclearreceptorsmallheterodimer partner in primary hepatocytes. Journal of Biological Chemistry. 2009;284:28510-28521
260. TerragniJ,NayakG,BanerjeeS,MedranoJL,GrahamJR,BrennanJF,SepulvedaS,CooperGM.Thee-boxbindingfactorsmax/mnt,mitf,andusf1actcoordinatelywithfoxotoregulateexpressionofproapoptoticandcellcyclecontrolgenesbyphosphatidylinositol3-kinase/akt/glycogensynthasekinase3signaling.JBiolChem.2011;286:36215-36227
261. CorreS,PrimotA,BaronY,LeSeyecJ,GodingC,GalibertMD.Targetgenespecificityofusf-1 isdirectedviap38-mediatedphosphorylation-dependentacetylation.JournalofBiologicalChemistry.2009;284:18851-18862
262. Corre S, Primot A, Sviderskaya E, Bennett DC, Vaulont S, Goding CR, GalibertMD. Uv-inducedexpressionofkeycomponentofthetanningprocess,thepomcandmc1rgenes,isdependentonthe p-38-activated upstream stimulating factor-1 (usf-1). Journal of Biological Chemistry.2004;279:51226-51233

Chapter1
40
263. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5'-monophosphate-activated protein kinasepromotes macrophage polarization to an anti-inflammatory functional phenotype. J Immunol.2008;181:8633-8641
264. LuppS,GotzC,KhadoumaS,HorbachT,DimovaEY,BohrerAM,KietzmannT,MontenarhM.Theupstreamstimulatoryfactorusf1isregulatedbyproteinkinaseck2phosphorylation.CellSignal.2014;26:2809-2817
265. Naukkarinen J, Ehnholm C, Peltonen L. Genetics of familial combined hyperlipidemia. CurrentOpinioninLipidology.2006;17:285-290

Chapter2
Chapter
HematopoieticArginase1deficiencyresultsindecreasedleukocytosisandincreasedfoamcellformationbutdoesnotaffectatherosclerosis
BaoyanRena*,ErikVanKampena*,TheoJ.C.VanBerkela,SheenaM.Cruickshankb,MirandaVanEcka
*BothAuthorscontributedequallytothiswork
aDivisionofBiopharmaceutics,ClusterBioTherapeutics,LeidenAcademicCentreforDrugResearch,LeidenUniversity,Leiden,TheNetherlands
bManchesterImmunologyGroup,FacultyofLifeSciences,TheUniversityofManchester,Manchester,UK
Atherosclerosis256(2017):35-46.

Chapter2
42
rginase1(Arg1),aM2macrophagemarker,playsacriticalroleinanumberofimmunologicalfunctions inmacrophages,whichare themain cell type facilitating atherosclerotic lesion
development. Arg1 uses the substrate L-arginine to create L-ornithine, a precursor moleculerequiredforcollagenformationandvascularsmoothmusclecelldifferentiation.ByreducingL-arginineavailability,Arg1limitstheproductionofNitricOxide(NO),apro-atherogenicfactorinmacrophages.Inendothelialcells,conversely,NOisstronglyanti-atherogenic.However,untilnow,the role of Arg1 in atherosclerosis is largely unknown. The aim of this study is to specificallyinvestigatetheeffectofArg1deletioninhematopoieticcellsonatherosclerosissusceptibility.
LDLrKOmiceweretransplantedwithArg1flox/flox;Tie2-Cre(Arg1KO)bonemarrow(BM)orwildtype(WT)BM.After8weeksrecoveryonchowdiet,recipientmicewerefedaWestern-TypeDiet(WTD)for10weekstoinduceatherosclerosis.After10weeksWTDchallenge,bloodleukocytecountsweredecreasedby25%(p<0.001),andspleenleukocytesweredecreasedby35%(p=0.05)inLDLrKO mice transplanted with Arg1 KO BM, compared to mice transplanted with WT BM. ThedecreaseinleukocyteswasduetolowerBlymphocytecounts.However,oxLDL-specificantibodieswereincreasedinplasmaofLDLrKOmicetransplantedwithArg1KOBMcomparedtoWTBMtransplanted controls, whereas oxLDL-specific IgM was not affected. On the other hand,peritonealfoamcellsinArg1KOBMrecipientswereincreased3-fold(p<0.001)comparedtoWTBMrecipients.Nochangeinbloodcholesterolwasfound.Despitechangesinleukocytecountsandmacrophagefoamcellformation,wedidnotobservedifferencesinatheroscleroticplaquesizeorplaquemacrophagecontentintheaorticroot.Surprisingly,therewasalsonodifferenceinplaquecollagencontent,indicatingthatabsenceofmacrophageArg1functiondoesnotreduceplaquestability.
DeletionofArg1 inhematopoieticcellsadverselyaffectsblood leukocytecountsand increasesfoamcell formation.However,noeffectsonatherosclerosiscouldbedemonstrated, indicatingthathematopoieticArg1functionisnotadecisivefactorinatheroscleroticplaqueformation.
Introduction
InhibitionoftheactivityoftheenzymeArg1isconsideredapromisingnoveltherapeuticstrategyforthetreatmentofcardiovasculardisease.1Inline,arginaseinhibitionbyN(omega)-hydroxy-nor-l-arginine(nor-NOHA)improvesendothelialfunctioninfamilialhypercholesterolemiapatientsandreduces atherosclerotic lesion development in carotid arteries of apolipoprotein E (ApoE)knockoutmiceexposedtolowshearstress.2-4Arg1influencesanumberofprocessesimplicatedin the pathogenesis of atherosclerosis.5-8 It is expressed in endothelial cells, vascular smoothmuscle cells (VSMCs) and macrophages, which are all important cellular components of theatheroscleroticplaque.1Dependingonthecelltypeitisexpressedin,Arg1functionisexpectedtoexertdifferenteffectsonatheroscleroticplaqueformation.
TheprimaryfunctionofArg1isproductionofureaandL-ornithinefromL-arginine.9L-arginine,however, is also used as a substrate by the enzymes inducible- and endothelial Nitric OxideSynthase(iNOSandeNOS)fortheproductionoftheendothelial-protectivesignallingmoleculenitricoxide(NO).2,10BycompetitionforthecommonsubstrateL-arginine,Arg1canthusindirectlyinhibitthesynthesisofNO.11,12Inline,endothelialArg1contributestoendothelialactivationandvascularstiffnessbyreducingtheL-argininepool, leadingtoeNOSuncouplingandreducedNOproduction.10,13Thisresultsinendothelialactivationandincreasedrecruitmentofimmunecellsto the plaque.10,13 However, atheroprotective effects have also been described for Arg1 in
A

Chapter2
43
macrophagesandVSMCs.ByproducingL-ornithine,Arg1contributestothesynthesisofL-prolinebytheenzymeOrnithineAminoTransferase(OAT),whichisaprecursorforcollagenbiosynthesis.Ornithinecanalsobemetabolisedintopolyamines,whichleadstoincreasedVSMCdifferentiationand decreased inflammation.14-16 In agreement, lentiviral-mediated upregulation of Arg1 in aballoon-injuryrabbitmodel inhibitedplaque inflammationandaugmentedVSMCproliferation.Plaquesizewas,however,notaffected.8
InmacrophagesArg1isfoundinthealternativelyactivatedM2cells,amacrophagesubtypewithan anti-inflammatory and wound healing function.6 Downregulation of Arg1 expression andinhibitionofArg1activity inRaw264.7macrophagesresultedinaugmentedLPS-inducedTNF-αand IL-6 secretion.8 On the other hand, Arg1 in macrophages suppresses Th2 dependentinflammationbydampeningtheproductionofanti-inflammatorycytokinesbyCD4+Tcellsandsuppressing T-cell proliferation in mice infected with the trematode Schistosoma mansoni.17Differentialgeneexpressionanalysisinmacrophagesofatherosclerosis-susceptibleand-resistantrabbitssuggestedthathighmacrophageArg1expressionwasassociatedwithlowatherosclerosissusceptibility.18 Furthermore,M2macrophages are foundpredominantly in carotid plaquesofasymptomatic patients that have more stable plaques,19 indicating the positive associationbetweenmacrophagesoftheM2phenotypeandatheroscleroticplaquestabilization.However,thefunctionalroleofmacrophageArg1inatheroscleroticplaquedevelopment iscurrentlystillunknown.
InthisstudywespecificallyassessedthecontributionofhematopoieticArg1tothedevelopmentof atherosclerosis, by transplanting bone marrow from Arg1flox/flox;Tie2Cre mice intoatherosclerosis-susceptibleLDLreceptorknockout(LDLrKO)mice.
MaterialandMethods
Animals
LDLrKOmiceandWTC57Bl/6wereobtainedfromtheJacksonLaboratoryandexpandedattheFacultyofScience,LeidenUniversity.Arg1flox/flox;Tie2Cre(Arg1KO)mice20werebredattheFacultyofLifeSciences,UniversityofManchester.AllanimalstudiesintheNetherlandswereapprovedbythe regulatory authority of Leiden University and carried out in compliance with the Dutchgovernmentguidelines.AllanimalworkintheUnitedKingdomwasperformedinaccordancewithHomeOfficeregulations.
mRNAExpressionAnalysisbyRealTimePCR
Thioglycollate-elicitedperitonealmacrophages(PMs)from12-weekoldmaleC57Bl/6micewereobtainedafterinjectionof1mLof3%thioglycollatesolution5dayspriortotheexperiment.Afteradherenceandwashing,themacrophageswereincubatedwith/without10µg/mLoxidizedlowdensitylipoprotein(oxLDL,preparedasdescribedpreviously21)for24hours.Afterthat,cellswerecollected for total RNA isolation.22 Subsequently, RevertAid M-MuLV enzyme (Fermentas,Burlington,Canada)wasusedtotranscribeRNAtocDNA.QuantitativePCR(qPCR,ABIPRISM7500system,FosterCity,CA)wasusedtoaccessthemRNAexpressionlevelsofgenesinterestedusingSYBRGreenreagents(AppliedBiosystems).RPL27and36B4wereusedashousekeepinggenes.

Chapter2
44
Microarrayanalysis
Twelve-weekoldfemaleLDLrKOmicewerefirstfedWestern-typeDiet(WTD;SpecialDietServices)that contains 15% cacao butter and 0.25% cholesterol for a run-in period of 2 weeks beforebilateralperivascularcollarplacementinthecarotidarteries.ThenthemicewerechallengedwithWTDforanother2weekstoinduceearlyatheroscleroticlesiondevelopment.Thecarotidarterieswere isolated directly after the run-in WTD period (baseline group) or 2 weeks after collar-placement(atheroscleroticplaquegroup)formicroarrayanalysisaspreviouslydescribed.23
BoneMarrowTransplantation
Bonemarrow frommale C57Bl/6WT controls and Arg1 KOmice (around 12weeks old) waspreparedforbonemarrowtransplantation(BMT)to12weeksoldfemaleLDLrKOrecipientmice.Inbrief,lethallyirradiatedrecipientsreceived5x106bonemarrowcellsviatailveininjection.Themicewereallowedtorecoverfor8weeksonchowdiet(RM3;SpecialDietServices),afterwhichtheywerefedWTDtoinduceatherosclerosis.Afterthe10-weekWTDchallenge,themicewereanaesthetized by a lethal dose of anesthetic mixture that contained rompun, ketamine andatropine. Mice were bled and perfused with PBS, after which organs were isolated. ThehematologicchimerismwasconfirmedingenomicDNAofrecipientsbonemarrowusingthePCRmethod(SupplementaryFigure1A-B).
GenerationofBoneMarrow-DerivedMacrophages(BMDMs)
BonemarrowfromLDLrKOrecipientstransplantedwithWTBMorArg1KOBMwasisolatedatsacrifice for the in vitro experiments. Bone marrow-derived macrophages were obtained asdescribed previously.24 Macrophages were cultured for 24 hours with or without 100µg/mLacetylated-lowdensity lipoprotein (acLDL). Thepreparationof acLDL is describedpreviously.25Subsequentlythecellswereanalyzedbyanautomatedveterinaryhaematologyanalyzer(SysmexCorporation, XT-2000iV, Japan) for foamcell formation asdescribedpreviously.26,27 Briefly, theSysmex XT-2000iV analyzer applies a similar principle for cell differential analysis as patentedfluorescentflowcytometricanalysis.28Lasersidescatterandsidefluorescencelightswereusedfor separating cell clusters. Lipid-rich macrophages (foam cells) are larger and contain moreabundantgranulescompared to thenon-foamcells.26,27Thus inadifferential scattergram, thelipid-rich macrophage population shifts to a larger scale on the side scatter axis and sidefluorescentlightaxis,enablinggatingofaseparate,shiftedpopulationofmacrophagefoamcells.
FlowCytometryAnalysisandWBCDifferentialAnalysis
Blood samples, anti-coagulated with EDTA, as well as single splenic cell suspensions, wereobtainedusinga70µmcellstrainer(734-0003,VWR),andusedforFACSanalysis.Erythrocytelysisbuffer(0.15MNH4Cl,10mMNaHCO3,0.1mMEDTA,pH=7.3)wasusedtolyseredbloodcellsinthebloodandsplenocytepreparations.Consecutively,thecellswereanalyzedonaFACSCantoII(BD Biosciences, Mountain View, CA) using the relevant FACS antibodies (all obtained fromeBioscience).
AnautomatedHaematologyanalyzer(XT-2000iV,SysmexCorporation,Japan)wasusedtoanalyseleukocytecounts inspleenandbloodsamples.Furthermore,peritoneal leukocytescollectedatsacrifice from the bone marrow transplanted animals, were analyzed for quantification ofmacrophagefoamcellsformation.

Chapter2
45
SerumCholesterolLevelDetermination
Total and free cholesterol concentrations in serum were determined using an enzymaticcolorimetricmethodasdescribedpreviously.29Absorbancewasreadat490nm.
ELISAassayforanti-oxLDLantibodies
Copper-oxLDL was prepared as previously described.21 The mouse immunoglobulin isotypingELISAkitwasobtainedfromBDBiosciences(CatalogNo.550487).HRPlabelledpolyclonalrabbitanti-Ratimmunoglobulins(Ig)wereobtainedfromDAKO(ProductNo.P045001).TotalIgandIgMantibodiesagainstoxLDLweremeasuredbyamodifiedELISA. Inbrief,oxLDLwascoated inanELISAplate(Corning,NY,14831)inaconcentrationof10µg/mLovernightat4°Cincoatingbuffer(0.42%w/vNaHCO3,0.53%Na2CO3,pHof9.6).Afterwashingwith0.05%tween-20-PBS,wellswereblockedfor30minutesin1%BSAblockingbuffer.Afterwashing,4uLplasmawasaddedandplateswereincubatedatroomtemperatureforanhour,thenwashed.FordeterminationoftotalIg, theHRP-labelled rat anti-mouse Ig antibody included in theBDELISA kitwas added.Afterincubationfor1houratroomtemperatureandsubsequentwashing,theplateisreadyforcolourdevelopment. For the IgM determination, after incubation with IgM-specific rat anti-mouseantibody for1hour,wellswere incubatedwithHRP-labeledrabbitanti-rat Ig foranotherhourbeforecolourdevelopment,uponfollowingthemanufacturer'sinstruction.Opticaldensity(OD)wasobtainedby reading theplate inaplate reader (Biotek,poowerWave340)at450nmand570nm.Wavelengthcorrectionwasperformedbysubtractingthevaluesobtainedat570nmfromthevaluesat450nm.
HistologicalAnalysisoftheAorticRoot
After10-weekWTDfeeding,miceweresacrificed.Seven-µmserialsectionsoftheaorticrootwerecutusingaLeicacryostat.Oilred-Ostaining,MoMa2stainingandMasson’sTrichromekitwereusedforvisualizationofplaquearea,macrophagepositiveareaandcollagencontentrespectivelyasdescribedpreviously.29Quantificationwasperformedusing theLeica imageanalysis system(LeicaLtd,Cambridge,UK).29
StatisticalAnalysis
Student’sT-testortwo-wayANOVAwereusedtodeterminethestatisticallysignificantdifferences(GraphpadPrismsoftware).AWelchcorrectionwasappliedtotheT-testinthecaseofunequalvariancesinthedataset.Thestatisticalsignificancewassetat0.05.Resultsareshownasthemean±SEM.

Chapter2
46
Results
Arg1 expression is induced in macrophages by oxLDL loading and in carotid arteries uponinductionofcollar-inducedatherosclerosis
Theearlystagesofatheroscleroticlesiondevelopmentarecharacterizedbytheaccumulationoflipid-ladenmacrophages.Therefore, firsttheeffectsof incubationofmacrophageswithoxLDL,and inductionofcollar-inducedatherosclerosis in thecarotidarteryon theexpressionofArg1weredetermined.InagreementwithapreviousstudyfromGallardo-Soleretal.,30Arg1mRNAexpressionwasincreased4.3-foldinthioglycollate-elicitedperitonealmacrophagesafter24hoursoxLDL(10µg/mL)loading(p<0.001;figure1A)afteraqPCRanalysis.Oil-redOstainingconfirmedthat after oxLDL incubation, foam cells were successfully induced (Figure 1B). Moreover,microarray analysis showed thatArg1 expressionwas significantly increased in collar-inducedearlyatheroscleroticlesionsinthecarotidarteryofLDLrKOmice(4.5-fold,p<0.01;figure1C).
Figure 1 Arg1 expression was induced by oxLDL loading in wild-type (WT) thioglycollate-elicited peritonealmacrophages(PMs)orWestern-typediet(WTD)andcollar-inducedatherosclerosisinthecarotidartery.A)Thioglycollate-elicitedPMswereincubatedwith/without10µg/mLoxLDLfor24hours,andthenqPCRwasusedforanalysisofArg1geneexpression.(n=6).B)Oil-redOstainingofPMsafterincubationwith/without10µg/mLoxLDLfor 24 hours. C) LDLr KOmice were challenged withWTD 2 weeks before the collar placement (baseline). Theexperimentalgroupwas fedWTDforanother2weeksaftercollarplacement (atheroscleroticplaque).Thecollar-inducedatheroscleroticplaquesincarotidarteriesofLDLrKOmicewereusedformicroarrayanalysisofwhichtheeffects onArg1 expression are shown. (n=6). Results are expressed asmean± SEM, significancewas assessedbystudentT-test.**p<0.01;***p<0.001.
Arg1deletioninbonemarrow-derivedcellsofLDLrKOmiceincreasesfoamcellaccumulationintheperitoneuminabsenceofeffectsonserumtotalcholesterollevels
To generate a mouse model that specifically lacks Arg1 in bone marrow-derived cells, bonemarrow(BM)fromArg1flox/flox;Tie2Cre(Arg1KO)miceandWTcontrolswastransplantedintoLDLrKO recipients. InArg1flox/flox;Tie2CremiceArg1 has beendeleted in cells of the hematopoieticlineagesandinendothelialcells.19,31
Nieseetal.previouslyshowedthatArg1deficiencydoesnotaffectbonemarrowengraftment.32InagreementPCRanalysisongenomicDNAisolatedfrombonemarrowafter10weeksofWTDfeedingconfirmedsuccessfuldisruptionofArg1functionalityinthebonemarrowandperitonealcellsoftheLDLrKOrecipientmice(supplementaryfigureA-C).
DifferentialhaematologyanalysiswasperformedonperitonealcellsharvestedfromLDLrKOmice,transplantedwithArg1KOorWTbonemarrowafter10weeksofWTDfeeding.Nodifferenceintotalnumberofperitonealleukocytes(datanotshown)orthepercentageofmacrophageswithin
A
PBS oxLDL
Oil-redO
B C

Chapter2
47
the peritoneal leukocyte populationwere foundbetweenArg1 KOBM recipients andWTBMrecipients(p>0.05,figure2A).Interestingly,a3-foldlargerfractionoffoamcellsintheperitoneumofArg1KOBMrecipientswasfoundwhencomparedtotheWTBMrecipients(p<0.001,figure2B).Thisincreasedfoamcellformationcouldnotbeattributedtoincreasedserumcholesterollevels,asnodifferenceinserumfreecholesterol(datanotshown)ortotalcholesterolwasfound(p>0.05;figure2C).
Figure2IncreasedinvivoperitonealfoamcellformationinLDLrKOmicetransplantedwithArg1KOBMinabsenceofeffectsonserumcholesterolandaugmentedinvitrofoamcellformationinbonemarrow-derivedmacrophages(BMDM)lackingArg1.
Serumandperitonealleukocyteswerecollectedatsacrificeafter10weeksofhigh-fat,high-cholesterolWestern-typediet feeding. A)Macrophage percentage of total peritoneal leukocytes in BMT recipients (n=9-14). B) Foam cellpercentage of total peritoneal leukocytes in the BMT recipients (n=9-14). C) Total cholesterol level in the BMTrecipients (n=9-14). D) In vitro, BMDMs fromWTBMor Arg1 KOBM recipientswere incubated 24 hours in thepresenceorabsenceof100µg/mLacLDL.Foamcellpercentagewasdetectedusingahaematologyanalyser(n=6-10).Resultsareexpressedasmean±SEM,significancewasassessedbystudentT-testor2-wayANOVA.*p<0.05;**p<0.01;***p<0.001.
Arg1 KO BMDMs show increased foam cell formation and tend to differentiate to an M2macrophagephenotypeuponacLDLstimulation.
BMDMs fromWT andArg1 KOmicewere treatedwith 100µg/mL acLDL for 24 hours to gainmechanisticinsightintotheobservedincreasedfoamcellformationinvivo.AcLDL-inducedlipidloadingledtofoamcellformationinbothWTBMDMsandArg1KOBMDMs(4.7±0.3%to10.4±1.1% forWTBMDMs,p<0.01; and6.5 ± 0.66% to 15.1 ± 1.8% forArg1KOBMDMs,p<0.001,respectively; figure 2D). Interestingly, both before and after lipid loading Arg1 KO BMDMsdisplayeda50%increaseinfoamcellformationcomparedtoWTBMDMs(p<0.05,figure2D).ThemRNAexpressionofthegenesrelatedtofoamcellformationwasalsoassessedbyqPCRanalysisinBMDMsincubatedwith/without100µg/mLacetylatedLDLfor24hours.UponacLDLloading,the expression of SR-B1, a receptor for native and modified lipoproteins, was effectivelydownregulatedinWTBMDMs(p<0.05ascomparedtonon-loadedcells;figure3A),whereasArg1KOBMDMsfailedtodownregulateSR-B1uponacLDLloading(p>0.05,figure3A).Nodifferences
A B
C D

Chapter2
48
wereobservedinLDLrexpressionbetweenthe2genotypeseitherwithorwithoutacLDLloading(p>0.05;figure3B).Foamcellformationisdeterminedbythebalancebetweencholesteroluptakeandsynthesisontheonehandandcholesteroleffluxontheotherhand.AlthoughtheexpressionofABCA1,theprimarycholesteroleffluxtransporter,wasincreasedintheBMDMsloadedwithacLDL (WT, 15.7-fold, p<0.001; Arg1 KO, 3.7-fold, p<0.01; figure 3C), no difference in ABCA1expressionwasfounduponcomparisonoftheArg1KOandtheWTBMDMs(p>0.05,figure3C).Notably,expressionofSREBP1was2.45-foldhigherinArg1KOBMDMscomparetoWTBMDMSin response to acLDL loading, whereas no difference was found under control non-loadedconditions (Control, p>0.05; +acLDL, p<0.05; figure 3D). The difference in SREBP1 expressionbetweenacLDL loadedArg1KOandWTmacrophages isexplainedbya failureof theArg1KOmacrophagestodownregulateSREBP-1inresponsetoacLDLloading(WT,p<0.01;Arg1KO,p>0.05;figure 3D). Furthermore, theM1 andM2markers iNOS and FIZZ-1 were determined in bothgenotypesofBMDMsbeforeandafteracLDLloading.Undercontrolnon-loadedconditions,nodifferenceswerefoundintheexpressionofiNOSorFIZZ-1.However,inresponsetoacLDLloading,Arg1 deficiency in BMDMs led to an M2-like phenotype, as evidenced by significantlydownregulated iNOS expression (67%decrease,p<0.05; figure 3E) and extremely upregulatedFIZZ-1expression(19-fold,p<0.001;figure3F),whereasnosuchchangeswereobservedinWTBMDMs.
Furthermore,NOproductionwasdeterminedintheculturemediumofWTandArg1KOBMDMs,bothundercontrolconditionsandafteracLDLlipidloading.Two-wayANOVAshowedanincreasedNO production in Arg1 KO BMDMs compared to the WT BMDMs (p=<0.05, figure 3G). NOproductionwassignificantlyincreased(1.6-fold,p<0.05,figure3G)inArg1KOBMDMs,butnotinWT BMDMs in response to acLDL loading, leading to 2.2-fold (p<0.01, figure 3G) higher NOconcentrationsinthesupernatantofArg1KOBMDMsundertheseconditions.

Chapter2
49
Figure3GeneexpressionandnitricoxideproductioninWTandArg1KObonemarrow-derivedmacrophages(BMDMs)afterincubationwithorwithout100µg/mLacLDLfor24hours.
RelativemRNAexpressionofA)SR-B1,B)LDLr,C)ABCA1,D)SREBP1,E)iNOS,andF)FIZZ-1.(Controlgroup,n=9-12;+acLDL group, n=3-4). G) Nitric oxide concentrations in the supernatant of BMDMs incubated with or without100µg/mLacLDL24hours.(Controlgroup,n=3;+acLDLgroup,n=4).Resultsareexpressedasmean±SEM,significancewasassessedbystudentT-testor2-wayANOVA.*p<0.05;**p<0.01;***p<0.001.
BonemarrowArg1deficiencyaffectsneitheratheroscleroticplaquesizenorplaquecompositioninLDLrKOmice.
After10weeksWTDfeeding,theaorticrootwassectionedandstainedwithOilred-Otoanalyzeatherosclerotic lesion development. Despite the observed increase in macrophage foam cellformation upon deletion of Arg1, no difference in plaque size was found between the twoexperimentalgroups(647±29x103µm2forWTBMrecipientsvs.634±26x103µm2forArg1KOBMrecipients,figure4A).MacrophagesintheplaquewerevisualizedbyMOMA-2staining.Nodifferenceinplaquemacrophagecontentasafractionoftotalplaquesizewasobserved(0.171±0.011WTBMvs.0.169±0.016Arg1KOBM,figure4B).CollagenintheplaquewasstainedusingaMasson’sTrichromemethodandPicrosiriusRedstaining.Atrendtowardsareduction inthecollagencontentofplaquesofArg1KOBMrecipientswasfoundinMasson’sTrichrome-stainedsections(0.102±0.009WTBMvs.0.084±0.004Arg1KOBM,p=0.06,figure4C).However,analysisofPicrosiriusRedstainingdidnotindicateanydifferencebetweenthegroups(0.124±0.013WTBMvs.0.105±0.012Arg1KOBM,figure4D).
ASR-B1 BLDLr
CABCA1 DSREBP1
EiNOS FFIZZ GNitricoxide

Chapter2
50
Figure4DeletionofArg1inbonemarrow-derivedcellsdoesnotinfluenceatheroscleroticlesiondevelopmentafter10weeksWTDfeeding.
A)PlaqueswerestainedwithOil-RedOandplaqueareawasquantified.(p>0.5;WTBMT,n=16;Arg1KOBMT,n=15).B)PlaqueareastainedpositivelywithMOMA-2antibodywasmeasuredandnormalizedfortotallesionsize.(p>0.5;WTBMT,n=14;Arg1KOBMT,n=16).C)PlaquecollagenwasvisualizedusingMasson’sTrichrome(MTC)stainingandplaquecollagencontentwasanalysed.(p=0.064;WTBMT,n=15;Arg1KOBMT,n=15).D)ASiriusRedstainingwasperformedtoexamineplaquecollagencontentfurther.Collagencontentwasanalysed(p>0.5;WTBMT,n=11;Arg1KOBMT,n=12).E)Representativeimagesofthestainingsdescribedabove.Resultsareexpressedasmean±SEM,significancewasassessedbystudent’sT-test.
TransplantationofArg1KObonemarrowintoLDLrKOrecipientsresultsinreducedsplenocyteandbloodleukocytecounts
Flowcytometrywasusedtoassesswhether lossofArg1functionality inbonemarrow-derivedcellsinthetransplantedLDLrKOmiceaffectedleukocytenumbersinthecirculationorthespleen.Onchowdiet,nodifferenceintotalbloodleukocytenumberswasdetected(p>0.05,figure5A)betweentherecipientmicewithWTBMandthemicewithArg1KOBM.WTDfeedingfor10weeksincreasedtotalleukocytecountsfrominbloodofLDLrKOmicetransplantedwithWTBM(chowdiet:11410±708cells/µl;WTD:15672±689cells/µl;p<0.01;figure5A).MicetransplantedwithArg1KObonemarrow,however,failedtoshowanincreaseinbloodleukocytecountsuponWTD
Arg1KOBM
WTBM
OilRedO MoMa2 MTC SiriusRed
A
C
B
D
E

Chapter2
51
feeding(chowdiet:12092±906cells/µl;WTD:11573±491cells/µl;p>0.05;figure5A).Therefore,itledtolowertotalleukocytecountsinthebloodofArg1KOBMrecipientsascomparedWTBMrecipientsafter10weeksofWTDfeeding (p<0.001, figure5A).Comparisonof thesubtypesofWBC,showedthat,incontrasttoWTBMrecipientsinwhichlymphocyteswere1.6-foldincreases(p<0.001)after10weeksWTDfeeding,lymphocytesdidnotincreaseinLDLrKOmicetransplantedwith Arg1 KO BM (Figure 5B). No differences were found in neutrophils, monocytes, andeosinophils(datanotshown).Next,flowcytometricanalysiswasusedtodeterminetheeffectsonthe different cellular subsets in blood at sacrifice. No differenceswere found in the absoluteamountsofCD11b+cells,CD11b+/Ly6Chiinflammatory,CD11b+/Ly6Clow+medpatrollingmonocytesandCD11b+/Ly6G+neutrophils,northeamountsofCD4+Thelpercells,CD25+/CD4+activatedThelpercellsandCD8+cytotoxicT-cells (figure5C-H).Unexpectedly, thedecrease in totalbloodleukocytesappearedtobedrivenbya2-folddecreaseincirculatingCD19+Bcells(p<0.01,figure5I).Representativeflowcytometryplotsareshowninfigure5J.
Atthetimeofsacrifice,spleensweretakenandweighed.Organweightwasnormalizedfortotalbodyweight.Asmallbutsignificant10%decreaseinspleenweightwasfoundinLDLrKOmicetransplantedwithArg1KOBM(p<0.05,figure6A),whiletherewerenodifferencesintotalbodyweight(datanotshown).Correspondingly,spleensfromtheArg1KOBMrecipientscontained35%less splenocytes (p=0.052, figure 6B). Next, splenocyte composition was assessed by flowcytometry.Theabsolutenumbersof leukocytesubtypes, includingCD11b+cells,CD11b+/Ly6Chipro-inflammatory monocytes, CD11b+/Ly6Clow+med patrolling cells, CD11b+/Ly6G+ neutrophils,CD19+BcellsandCD8+TcellsinthespleenofArg1KOrecipientsshowedasignificantdecreaseor a trend towards a decrease, suggesting that the decrease in splenocyte number was notattributabletoonespecificcelltype(p<0.05,figure6C-D).However,fractionalanalysisshoweda23%increaseinCD4+andCD25+/CD4+TcellsinthespleenofLDLrKOmicethatreceivedArg1KOBMcomparedtotherecipientsthatreceivedWTBM(*p<0.05;$p<0.05,respectively.Figure6E).Representativeflowcytometryplotsareshowninfigure6F.
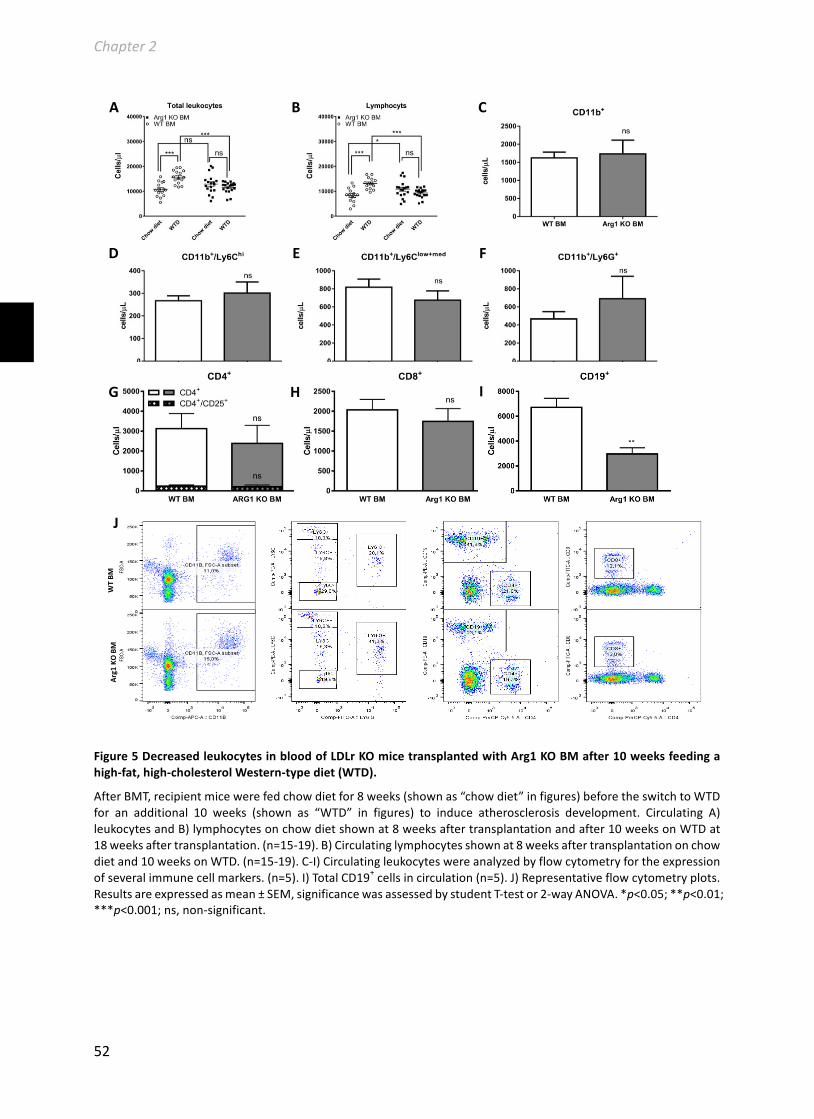
Chapter2
52
Figure5DecreasedleukocytesinbloodofLDLrKOmicetransplantedwithArg1KOBMafter10weeksfeedingahigh-fat,high-cholesterolWestern-typediet(WTD).
AfterBMT,recipientmicewerefedchowdietfor8weeks(shownas“chowdiet”infigures)beforetheswitchtoWTDfor an additional 10 weeks (shown as “WTD” in figures) to induce atherosclerosis development. Circulating A)leukocytesandB)lymphocytesonchowdietshownat8weeksaftertransplantationandafter10weeksonWTDat18weeksaftertransplantation.(n=15-19).B)Circulatinglymphocytesshownat8weeksaftertransplantationonchowdietand10weeksonWTD.(n=15-19).C-I)Circulatingleukocyteswereanalyzedbyflowcytometryfortheexpressionofseveralimmunecellmarkers.(n=5).I)TotalCD19+cellsincirculation(n=5).J)Representativeflowcytometryplots.Resultsareexpressedasmean±SEM,significancewasassessedbystudentT-testor2-wayANOVA.*p<0.05;**p<0.01;***p<0.001;ns,non-significant.
Arg1KOBM
WTBM
C A B
D E F
G H I
J
I
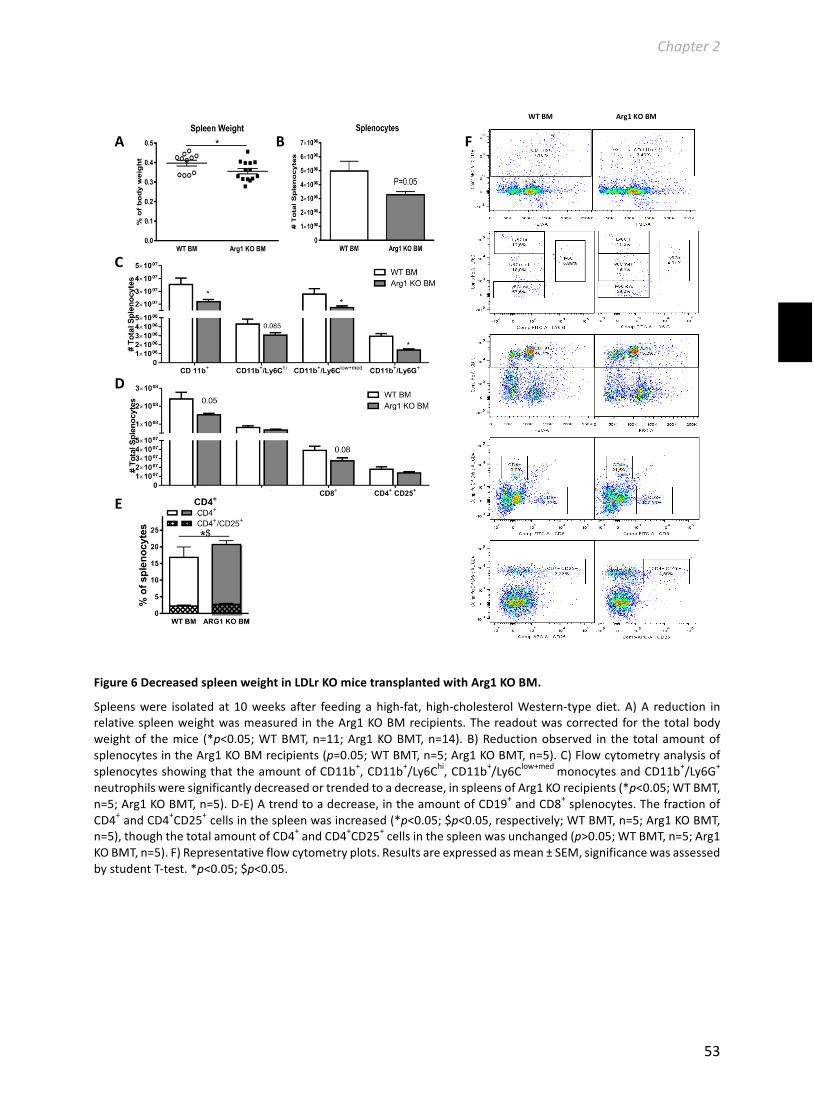
Chapter2
53
Figure6DecreasedspleenweightinLDLrKOmicetransplantedwithArg1KOBM.
Spleenswere isolatedat 10weeks after feedingahigh-fat, high-cholesterolWestern-typediet.A)A reduction inrelativespleenweightwasmeasured intheArg1KOBMrecipients.Thereadoutwascorrectedforthetotalbodyweightof themice (*p<0.05;WTBMT,n=11;Arg1KOBMT,n=14).B)Reductionobserved in the totalamountofsplenocytesintheArg1KOBMrecipients(p=0.05;WTBMT,n=5;Arg1KOBMT,n=5).C)FlowcytometryanalysisofsplenocytesshowingthattheamountofCD11b+,CD11b+/Ly6Chi,CD11b+/Ly6Clow+medmonocytesandCD11b+/Ly6G+neutrophilsweresignificantlydecreasedortrendedtoadecrease,inspleensofArg1KOrecipients(*p<0.05;WTBMT,n=5;Arg1KOBMT,n=5).D-E)Atrendtoadecrease,intheamountofCD19+andCD8+splenocytes.ThefractionofCD4+andCD4+CD25+cellsinthespleenwasincreased(*p<0.05;$p<0.05,respectively;WTBMT,n=5;Arg1KOBMT,n=5),thoughthetotalamountofCD4+andCD4+CD25+cellsinthespleenwasunchanged(p>0.05;WTBMT,n=5;Arg1KOBMT,n=5).F)Representativeflowcytometryplots.Resultsareexpressedasmean±SEM,significancewasassessedbystudentT-test.*p<0.05;$p<0.05.
WTBMArg1KOBM
A B
C
D
F
E

Chapter2
54
Arg1deficiencyinthebonemarrowofLDLrKOmiceinducescirculatingoxLDL-specificantibodylevels.
Thetotal levelofox-LDLspecific immunoglobulins (Ig)and its isotype IgMweredetermined inplasmaoftheLDLrKOmicetransplantedwithWTorArg1KOBMafter10weeksWTDchallenge.Asshowninfigure7,deletionofArg1inthebonemarrowofLDLrKOmiceledtoincreasedtotaloxLDL-specific Ig levels (1.2-fold,p<0.05 compared to theWTBM LDLr KO recipients after 10weeksWTDchallenge,whichwasnotdrivenbyIgM.
Figure7IncreasedoxLDL-specificantibodylevelsinLDLrKOmicetransplantedwithArg1KOBMafter10weeksWTDfeeding.
TotaloxLDL-specificantibodyandisotypeIgMlevelswereassessedusingELISA.Resultsareexpressedasmean±SEM,significancewasdeterminedbystudent’sT-test.*p<0.05ascomparedbetweenLDLrKOrecipientwithWTBMandrecipientswithArg1KOBMintheleveloftotaloxLDL-Specificantibody(Ig).ns,non-significant.
Discussion
InthecurrentstudywedeterminedforthefirsttimetheeffectsofArg1deletioninbonemarrow-derivedcellsonmacrophagefoamcellformationandatherosclerosissusceptibility.WeshowthatArg1expressionisincreasedinelicitedperitonealmacrophagesuponoxLDL-inducedlipidloadingand during the development of early murine atherosclerotic lesions in the carotid-artery. Inagreement, Gallardo-Soler, Alejandro, et al., previously demonstrated thatArg1 expression ishighly induced in bone marrow-derived macrophage foam cells upon oxLDL or acLDLstimulation.30InhibitionofPPAR-γ/δsuppressesthelipid-inducedincreaseinmacrophageArg1expression,30indicatingthatArg1expressionisincreasedduringmacrophagefoamcellformationprobablyduetoPPAR-γ/δactivation.Combined,theseresultsindicatethatArg1mightparticipateinmacrophagefoamcellformation,therebyinfluencingatherosclerosisdevelopment.However,so far there had been no reports on the effects of Arg1 function on macrophage foam cellformation. Interestingly, Arg1 KO BMDMs showed increased foam cell formation both undercontrol conditions and after acLDL-induced lipid-loading compared to theWTBMDMs.GenesinvolvedinfoamcellformationwereinvestigatedtogaininsightintothemechanismbehindtheenhancedfoamcellformationinabsenceofArg1.NodifferencewasfoundintheexpressionofABCA1, theprimarycholesterolexporttransporter,betweenWTandArg1KOmacrophages. Inresponse to acLDL loading, Arg1 deficient macrophages however did display an impairedsuppression of SREBP1 and SR-B1 compared to WT macrophages. SREBP1 is an importantregulatorofcellularlipidbiosynthesis,33whileSR-B1facilitatestheuptakeofnativeandmodifiedlipoproteins.34 Enhanced foam cell formation of macrophages lacking Arg1 might thus beexplained by defective suppression of cellular lipid biosynthesis and modified LDL uptake.Moreover,uponlipidloading,macrophageslackingArg1seemmorelikelytoskewtowardsthe

Chapter2
55
M2phenotype,asevidencedbythesignificantlydownregulatedM1marker iNOSandstronglyupregulatedM2marker FIZZ-1 expression in Arg1 KOmacrophages.M2macrophages exhibitincreased cholesterol loadingbyoxLDL compared toM1macrophages,providingonepossiblemechanism for the increased foamcell formation inourmodel.35,36Collectively, these findingssuggestalinkbetweenArg1expressionandmacrophagefoamcellformation.AsArg1andiNOScompete for thecommonsubstrateL-arginine,macrophageArg1deficiency indirectly leads toenhancedsynthesisofNOuponlipidloading.TheobserveddownregulationofiNOSintheArg1deficientmacrophagescouldthusbeduetonegativefeedbackbyNO.10,37-41Notably,inagreementwithourfindings,excessNOhasrecentlybeenshowntoinduceoxLDL-inducedmacrophagefoamcellformationbyinhibitionoftheLXR-ABCA1-cholesteroleffluxaxis.42
Nextweinvestigatedwhethertheobservedincreasedfoamcellformationinvitroalsotranslatedinto increased macrophage foam cell formation in vivo and augmented atherosclerosissusceptibility.Hereto,bonemarrowfromArg1flox/flox;Tie2CremicewastransplantedintoLDLrKOmice,a commonlyusedmodel to studyatherosclerosis.Wedecided touseArg1flox/flox;Tie2CremiceasdonorsastheTie2-credeleterhaspreviouslybeenshowntoleadtoacompleteablationofArg1activityinmacrophages,whileinArg1flox/flox;LysMCreonly~80%reductioninmacrophageArg1activitywasachieved.20InlinewiththeobservedincreaseinlipidaccumulationinArg1KOmacrophagesinvitro,foamcellformationintheperitonealcavityofLDLrKOmicetransplantedwithArg1KObonemarrowwasincreasedafter10weeksWTDfeeding.
Foam cell formation in the peritoneal cavity is a marker for atherosclerotic plaquedevelopment.43,44However, nodifferences inplaque sizeorplaquemacrophage contentweredetectedbetweentheLDLrKOmicetransplantedwithArg1KOorWTbonemarrow.Increasedexpression of Arg1 in balloon-injured rabbits results in augmented plaque stability as aconsequence of enhanced VSMC proliferation.8 Moreover, Arg1flox/flox;Tie2Cre (Arg1 KO) miceexhibitimpairedcutaneouswoundhealing,tosomeextentduetoimpairedcollagendeposition.43ThereducedcollagencontentinwoundsofArg1flox/flox;Tie2Cre(Arg1KO)micewasattributedtoincreasedcollagendegradationbyneutrophilsandmacrophageslackingArg1.45Inatherosclerosis,collagendepositionprovidesstabilitytotheatheroscleroticplaque.However,nosignificanteffectonthecollagen-contentofatheroscleroticplaquesofLDLrKOmicetransplantedwithArg1KObonemarrowwasfound.Intheaorticrootlesions,smoothmusclecells,themainproducersofcollagen deposited in atherosclerotic lesions,46were not affected by deletion of Arg1 in bonemarrow-derived cells.47 Conclusively, deletion of Arg1 in bonemarrow-derived cells does notaffectatherosclerosissusceptibilitydespiteaclearincreaseinmacrophagefoamcellformation,indicatingothercompensatorymechanisms.
Tie2isexpressedinallhematopoieticlineagecells.20Hence,inourbonemarrowtransplantationmodelusingtheArg1flox/flox;Tie2Cremiceasdonors,Arg1isnotonlydeletedinmacrophages,butinallhematopoieticcells.48ForlongithasbeenthoughtthatArg1isonlyexpressedinthemyeloidlineage and not in the lymphoid lineages.49 Indeed Arg1 is expressed primarily in anti-inflammatory alternatively activated macrophages, however it can also be detected inneutrophils,50,51andinnatelymphoidcellsII(ILC2).52
To investigate whether bone marrow Arg1 deficiency had any atheroprotective effects tocounteracttheobservedincreaseinfoamcellformation,theleukocytesinbloodandspleen,themajorhematopoieticorganandanimportantreservoirformonocytes,wereanalysedintheBMtransplantedmice.InagreementwithpreviousstudiesbyNieseetal.,32deletionofArg1inbone

Chapter2
56
marrow-derived cells did not affect circulating leukocytes on regular chow diet.Hypercholesterolemia is commonly known to induce leukocytosis in animal models.53,54 andhumans.55,56Uponchallengewithahighfat/highcholesterolWTD,LDLrKOmicetransplantedwithArg1KObonemarrowfailedtoincreaseleukocytecountsinthecirculation.Theinflammatoryandpro-atherogenic CD11b+/Ly6Chi subset of monocytes gives rise to classically activated M1macrophagesintheatheroscleroticplaque.57,58However,nodifferencewasfoundinthenumbersofCD11b+/Ly6Chimonocytesinblood,norintheamountofcirculatingneutrophilsbetweenthe2groupsofbonemarrowrecipientsafter10weeksWTDchallenge.Hypercholesterolemia-inducedmonocytosis andneutrophiliawas thusnot affected in thebloodbydeletionofArg1 inbonemarrow. In addition to myeloid cell mobilization from bone marrow, high fat diet-inducedinflammatoryconditionslikeatherosclerosiscouldalsoinducehematopoieticstemandprogenitorcells (HSPCs) settlement in the spleen and leading to local production of monocytes andneutrophils.59,60Spleen-derivedmonocytesandneutrophilseventuallyinfiltrateintothegrowingatherosclerotic lesion, giving rise to foam cell formation and pro-inflammatory cytokineproduction.60Notably,30%ofthetotalnumberofaorticmonocyteswerespleen-derivedLy-6Chimonocytes,whicharealsoreportedtocontributetofoamcell formation inthe lesions.60Arg1depletioninbonemarrowleadstoasignificantreductionoftheCD11b+cells,CD11b+/ly6Clow+medmonocytesandneutrophilsinthespleenofLDLrKOmiceandatendencytowardsadecreaseinpro-inflammatoryCD11b+/Ly6Chimonocytes.ThismightprovideanatheroprotectivemechanismcounteractingtheincreasedfoamcellinLDLrKOmicetransplantedwithArg1KOBM.
AsL-arginineisrequiredforCD4+T-cellfunctionandmaturation,Arg1-mediateddepletionofL-arginineby leukocytes results indecreasedTcellproliferation.17,61Tcell countsandactivationstatuswerethereforealsoinvestigated.However,therewasnodifferenceintheamountofCD4+cellsandCD4+/CD25+inbloodandspleen,althoughamodestfractionalincreaseinbothsubtypeswasfoundinthespleen.ThisindicatesthatleukocyteArg1isnotastrongregulatingfactorofT-cellproliferationinthespleen.Notably,strikinglyloweramountsofCD19+BcellswerefoundinboththecirculationandthespleenofArg1KOtransplantedLDLrKOmiceascomparedtoWTtransplanted animals after 10 weeks WTD challenge, explaining the decrease in total bloodleukocytecounts.L-arginineisanessentialaminoacidforBcellmaturationinthebonemarrowandarginase-mediated L-argininedepletion leads to reducedB cell emigration from thebonemarrowandreducedBcellnumbersinthespleenandlymphnodes.62Ifanything,leukocyteArg1deletionisthusanticipatedtoenhanceBcellemigrationfrombonemarrow,whichclearlycannotexplainthereducedBcellnumbersinblood.Bcells,astheantibodyproducingcellsoftheimmunesystem,playanimportantroleinatherosclerosis.Anti-oxLDLantibodies,especiallyIgManti-oxLDL,are inversely related toatheroscleroticplaquesize inexperimental studies.63Therefore,oxLDLantibody levelsweredetermined inplasmaobtained fromrecipientmiceafter10weeksWTDfeeding. To our surprise, despite the reduced numbers of B cells in the LDLr KO recipientstransplantedwithArg1KOBM,totaloxLDL-specificantibodylevelswereincreased.OxLDL-specificIgMthat issuggestedtobeatheroprotective,63however,wasnotchanged.Although inclinicalcardiovasculardiseasedivergingresultshavebeendescribedontheassociationbetweenoxLDL-specificantibodiesandatherosclerosis,animalstudiesconsistentlysuggestanatheroprotectiverole for oxLDL antibodies.64 Whatever the mechanism behind the reduction in B cells andincreasedcirculatinglevelsofoxLDL-specificantibodies,itmightcounteractthepro-atherogeniceffectsofenhancedfoamcellformationintheabsenceofArg1.

Chapter2
57
We conclude that despite leading to an increase in foam cell formation and a decrease incirculating B cells, Arg1 deficiency in bone marrow-derived cells does not significantly affectatheroscleroticplaquedevelopment.
Acknowledgments
Thisstudywassupportedby‘theNetherlandsCardioVascularResearchInitiative:theDutchHeartFoundation,DutchFederationofUniversityMedicalCenters, theNetherlandsOrganization forHealth Research and Development, and the Royal Netherlands Academy of Sciences’ for theGENIUSproject‘Generatingthebestevidence-basedpharmaceuticaltargetsforatherosclerosis’(CVON2011),theNetherlandsOrganizationforScientificResearch(VICIGrant91813603(M.V.E)),andtheMedicalResearchCouncil(grantG.14000449(SMC)).M.V.E.isanEstablishedInvestigatoroftheNetherlandsHeartFoundation(Grant2007T056).E.V.KwassupportedbyTheNetherlandsHeartFoundation(Grant2009B075andB.R.byagrantfromtheChinaScholarshipCouncil.
WethankMartineBotforsharingtheMicroarraydata.WethankRonaldvanderSluis,MarcovanderStoep,GijsvanPuijveldeandAmandaC.Foksfortheirexperthelpwithanimalsacrifice.

Chapter2
58
Supplementarymaterial
GenotypingofexpressionofTie2Creandthecorrectexcisionofexons7and8fromtheArg1gene.At18weeksaftertransplantationandafter10weeksofWTDfeedingtherecipientanimalsweresacrificed and genomicDNAwas isolated from thebonemarrowandperitoneal cavity of therecipientsandsubjectedtogenotypingPCRanalysis.A)GenotypingbyPCRshowspresenceofTie2CreinrepresentativemicehavingreceivedtheArg1flox/flox;Tie2CreBM.B-C)ThepresenceoftheArg1Δconstruct,i.e.thesuccessfulCre-mediatedexcisionofexons7and8fromtheArg1gene,wasalsodetectedusingasetof3primers.GenotypingbyPCRshowspositivebandsfortheArg1ΔproductinArg1flox/floxTie2Cretransplantedmice,indicatingsuccessfuldeletionofexons7and8inbonemarrowandperitonealcells.AfaintbandindicatingthepresenceofWTDNAcanstillbeseen,thisisinaccordancewithpreviousstudieswhereatransplantationefficiencyof95%wasdemonstrated24.
References
1. PernowJ,JungC.Arginaseasapotentialtargetinthetreatmentofcardiovasculardisease:Reversalofargininesteal?CardiovascRes.2013;98:334-343
2. GaoX,XuX,BelmadaniS,ParkY,TangZ,FeldmanAM,ChilianWM,ZhangC.Tnf-alphacontributesto endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury.ArteriosclerThrombVascBiol.2007;27:1269-1275
3. Shemyakin A, Kovamees O, Rafnsson A, Bohm F, Svenarud P, SettergrenM, Jung C, Pernow J.Arginaseinhibitionimprovesendothelialfunctioninpatientswithcoronaryarterydiseaseandtype2diabetesmellitus.Circulation.2012;126:2943-2950
4. OlivonVC,Fraga-SilvaRA,SegersD,DemougeotC,deOliveiraAM,SavergniniSS,BerthelotA,deCrom R, Krams R, Stergiopulos N. Arginase inhibition prevents the low shear stress-induceddevelopment of vulnerable atherosclerotic plaques in apoe−/− mice. Atherosclerosis.2013;227:236-243
WT BM Arg1 KO BM
Arg
1 Δ
WT BM Arg1 KO BM
Wt
Δ
Tie2
Cre
Wt
Cre
Arg
1 Δ
WT BM Arg1 KO BM
Wt
Δ
(C)
Peritonealcells
Bonemarrow
Bonemarrow
(A)
(B)

Chapter2
59
5. RyooS,GuptaG,BenjoA,LimHK,CamaraA,SikkaG,LimHK,SohiJ,SanthanamL,SoucyK,TudayE,BarabanE,IliesM,GerstenblithG,NyhanD,ShoukasA,ChristiansonDW,AlpNJ,ChampionHC,HusoD,BerkowitzDE.Endothelialarginaseii:Anoveltargetforthetreatmentofatherosclerosis.CircRes.2008;102:923-932
6. Khallou-LaschetJ,VarthamanA,FornasaG,CompainC,GastonAT,ClementM,DussiotM,LevillainO,Graff-DuboisS,NicolettiA,CaligiuriG.Macrophageplasticityinexperimentalatherosclerosis.PLoSOne.2010;5:e8852
7. SansonM,DistelE,FisherEA.Hdlinducestheexpressionofthem2macrophagemarkersarginase1andfizz-1inastat6-dependentprocess.PLoSOne.2013;8:e74676
8. WangXP,ZhangW,LiuXQ,WangWK,YanF,DongWQ,ZhangY,ZhangMX.Arginaseienhancesatheroscleroticplaquestabilizationbyinhibitinginflammationandpromotingsmoothmusclecellproliferation.EurHeartJ.2014;35:911-919
9. Durante W. Role of arginase in vessel wall remodeling. The role of arginase in endothelialdysfunction.2015;4:32
10. Berkowitz DE,White R, Li D,Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D,Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity andcontributestoendothelialdysfunctioninagingbloodvessels.Circulation.2003;108:2000-2006
11. SonokiT,NagasakiA,GotohT,TakiguchiM,TakeyaM,MatsuzakiH,MoriM.Coinductionofnitric-oxidesynthaseandarginase i incultured ratperitonealmacrophagesand rat tissues invivobylipopolysaccharide.JBiolChem.1997;272:3689-3693
12. Buga GM, Singh R, Pervin S, Rogers NE, Schmitz DA, Jenkinson CP, Cederbaum SD, Ignarro LJ.Arginase activity in endothelial cells: Inhibition by ng-hydroxy-l-arginine during high-output noproduction.American Journal of Physiology-Heart andCirculatory Physiology. 1996;271:H1988-H1998
13. WhiteAR,Ryoo S, LiDC, ChampionHC, Steppan J,WangDM,NyhanD, ShoukasAA,Hare JM,Berkowitz DE. Knockdown of arginase i restores no signaling in the vasculature of old rats.Hypertension.2006;47:245-251
14. Durante W, Liao L, Peyton KJ, Schafer AI. Thrombin stimulates vascular smooth muscle cellpolyaminesynthesisbyinducingcationicaminoacidtransporterandornithinedecarboxylasegeneexpression.CircRes.1998;83:217-223
15. DuranteW,LiaoL,ReynaSV,PeytonKJ,SchaferAI.Physiologicalcyclicstretchdirects l-argininetransportandmetabolismtocollagensynthesisinvascularsmoothmuscle.FASEBJ.2000;14:1775-1783
16. ZhangMH,CaragineT,WangHC,CohenPS,BotchkinaG,SodaK,BianchiM,UlrichP,CeramiA,SherryB,TraceyKJ.Spermineinhibitsproinflammatorycytokinesynthesisinhumanmononuclearcells:Acounterregulatorymechanismthatrestrainstheimmuneresponse.JournalofExperimentalMedicine.1997;185:1759-1768
17. PesceJT,RamalingamTR,Mentink-KaneMM,WilsonMS,ElKasmiKC,SmithAM,ThompsonRW,Cheever AW, Murray PJ, Wynn TA. Arginase-1-expressing macrophages suppress th2 cytokine-driveninflammationandfibrosis.PlosPathog.2009;5:e1000371
18. TeupserD,BurkhardtR,WilfertW,HaffnerI,NebendahlK,ThieryJ.Identificationofmacrophagearginaseiasanewcandidategeneofatherosclerosisresistance.Arteriosclerosis,thrombosis,andvascularbiology.2006;26:365-371
19. ChoKY,MiyoshiH,KurodaS,YasudaH,KamiyamaK,NakagawaraJ,TakigamiM,KondoT,AtsumiT.Thephenotypeofinfiltratingmacrophagesinfluencesarterioscleroticplaquevulnerabilityinthecarotidartery.JStrokeCerebrovascDis.2013;22:910-918
20. ElKasmiKC,QuallsJE,PesceJT,SmithAM,ThompsonRW,Henao-TamayoM,BasarabaRJ,KonigT,SchleicherU,KooMS,KaplanG,FitzgeraldKA,TuomanenEI,OrmeIM,KannegantiTD,BogdanC,Wynn TA, Murray PJ. Toll-like receptor-induced arginase 1 in macrophages thwarts effectiveimmunityagainstintracellularpathogens.NatImmunol.2008;9:1399-1406

Chapter2
60
21. Steinbrecher UP. Oxidation of human low density lipoprotein results in derivatization of lysineresidues of apolipoprotein b by lipid peroxide decomposition products. J Biol Chem.1987;262:3603-3608
22. Chomczynski P, SacchiN. Single-stepmethodof rna isolationby acid guanidinium thiocyanate-phenol-chloroformextraction.AnalBiochem.1987;162:156-159
23. MeursI.Abctransportersandscavengerreceptorbi:Importantmediatorsoflipidmetabolismandatherosclerosis. Division of Biopharmaceutics, Leiden/Amsterdam Center for Drug Research(LACDR),LeidenUniversity;2011.
24. VanEckM,BosIS,HildebrandRB,VanRijBT,VanBerkelTJ.Dualroleforscavengerreceptorclassb, type i on bone marrow-derived cells in atherosclerotic lesion development. Am J Pathol.2004;165:785-794
25. BasuSK,GoldsteinJL,AndersonGW,BrownMS.Degradationofcationizedlowdensitylipoproteinandregulationofcholesterolmetabolisminhomozygousfamilialhypercholesterolemiafibroblasts.ProcNatlAcadSciUSA.1976;73:3178-3182
26. ZhaoY,PenningsM,HildebrandRB,YeD,Calpe-BerdielL,OutR,KjerrulfM,Hurt-CamejoE,GroenAK,HoekstraM,JessupW,ChiminiG,VanBerkelTJ,VanEckM.Enhancedfoamcell formation,atheroscleroticlesiondevelopment,andinflammationbycombineddeletionofabca1andsr-biinbone marrow-derived cells in ldl receptor knockout mice on western-type diet. Circ Res.2010;107:e20-31
27. OutR, JessupW,LeGoffW,HoekstraM,Gelissen IC,ZhaoY,KritharidesL,ChiminiG,Kuiper J,ChapmanMJ,HubyT,VanBerkelTJ,VanEckM.Coexistenceoffoamcellsandhypocholesterolemiainmicelackingtheabctransportersa1andg1.CircRes.2008;102:113-120
28. MathersR,EvansG,BlebyJ,TornowT.Evaluationofthesysmexxt-2000ivhaematologyanalyserforrat,dogandmousewholebloodsamples.ComparativeClinicalPathology.2008;17:137-144
29. vanKampenE,BeaslasO,HildebrandRB,LammersB,VanBerkelTJ,OlkkonenVM,VanEckM.Orp8deficiencyinbonemarrow-derivedcellsreducesatheroscleroticlesionprogressioninldlreceptorknockoutmice.PLoSOne.2014;9:e109024
30. Gallardo-Soler A, Gomez-Nieto C, Campo ML, Marathe C, Tontonoz P, Castrillo A, Corraliza I.Arginaseiinductionbymodifiedlipoproteinsinmacrophages:Aperoxisomeproliferator-activatedreceptor-gamma/delta-mediatedeffectthatlinkslipidmetabolismandimmunity.MolEndocrinol.2008;22:1394-1402
31. WijnandsKAP,HoeksemaMA,MeestersDM,vandenAkkerNMS,MolinDGM,BriedeJJ,GhoshM,KohlerSE,vanZandvoortMAMJ,deWintherMPJ,BuurmanWA,LamersWH,PoezeM.Arginase-1deficiency regulates arginine concentrations and nos2-mediated no production duringendotoxemia.PlosOne.2014;9:e86135
32. Niese KA, Collier AR, Hajek AR, Cederbaum SD, O'Brien WE, Wills-Karp M, Rothenberg ME,ZimmermannN.Bonemarrowcellderivedarginaseiisthemajorsourceofallergen-inducedlungarginasebutisnotrequiredforairwayhyperresponsiveness,remodelingandlunginflammatoryresponsesinmice.BMCimmunology.2009;10:1
33. Bengoechea-Alonso MT, Ericsson J. SREBP in signal transduction: Cholesterol metabolism andbeyond.CurrOpinCellBiol.2007;19:215-222
34. PluddemannA,NeyenC,GordonS.Macrophage scavenger receptorsandhost-derived ligands.Methods.2007;43:207-217
35. Oh J, Riek AE,Weng S, PettyM, Kim D, ColonnaM, CellaM, Bernal-Mizrachi C. Endoplasmicreticulum stress controlsm2macrophagedifferentiation and foam cell formation. J Biol Chem.2012;287:11629-11641
36. Chu EM, Tai DC, Beer JL, Hill JS. Macrophage heterogeneity and cholesterol homeostasis:Classically-activatedmacrophagesareassociatedwithreducedcholesterolaccumulationfollowingtreatmentwithoxidizedldl.BiochimBiophysActa.2013;1831:378-386
37. ChangCI,LiaoJC,KuoL.Arginasemodulatesnitricoxideproductioninactivatedmacrophages.AmJPhysiol.1998;274:H342-348

Chapter2
61
38. GobertAP,DaulouedeS, LepoivreM,Boucher JL,BouteilleB,BuguetA,CespuglioR,VeyretB,VincendeauP.L-arginineavailabilitymodulateslocalnitricoxideproductionandparasitekillinginexperimentaltrypanosomiasis.InfectImmun.2000;68:4653-4657
39. Han Y-J, Kwon Y-G, Chung H-T, Lee S-K, Simmons RL, Billiar TR, Kim Y-M. Antioxidant enzymessuppressnitricoxideproductionthroughtheinhibitionofnf-κbactivation:Roleofh2o2andnitricoxideininduciblenitricoxidesynthaseexpressioninmacrophages.NitricOxide.2001;5:504-513
40. WooA,ShinW,CuongTD,MinB,LeeJH,JeonBH,RyooS.Arginaseinhibitionbypiceatannol-3'-o-β-d-glucopyranoside improves endothelial dysfunction via activation of endothelial nitric oxidesynthaseinapoe-nullmicefedahigh-cholesteroldiet.Internationaljournalofmolecularmedicine.2013;31:803-810
41. Kleinert H, Schwarz PM, ForstermannU. Regulation of the expression of inducible nitric oxidesynthase.BiolChem.2003;384:1343-1364
42. Zhao JF, Shyue SK, Lin SJ,Wei J, Lee TS. Excessnitric oxide impairs lxr(alpha)-abca1-dependentcholesteroleffluxinmacrophagefoamcells.JCellPhysiol.2014;229:117-125
43. LiAC,BinderCJ,GutierrezA,BrownKK,PlotkinCR,PattisonJW,ValledorAF,DavisRA,WillsonTM,WitztumJL.Differentialinhibitionofmacrophagefoam-cellformationandatherosclerosisinmicebypparα,β/δ,andγ.TheJournalofclinicalinvestigation.2004;114:1564-1576
44. Yvan-Charvet L, Ranalletta M, Wang N, Han S, Terasaka N, Li R, Welch C, Tall AR. Combineddeficiencyofabca1andabcg1promotesfoamcellaccumulationandacceleratesatherosclerosisinmice.JClinInvest.2007;117:3900-3908
45. CampbellL,SavilleCR,MurrayPJ,CruickshankSM,HardmanMJ.Localarginase1activityisrequiredforcutaneouswoundhealing.JInvestDermatol.2013;133:2461-2470
46. NadkarniSK,BoumaBE,deBoerJ,TearneyGJ.Evaluationofcollageninatheroscleroticplaques:Theuseoftwocoherentlaser-basedimagingmethods.LasersinMedicalScience.2009;24:439-445
47. Bentzon JF, Weile C, Sondergaard CS, Hindkjaer J, Kassem M, Falk E. Smooth muscle cells inatherosclerosis originate from the local vesselwall andnot circulating progenitor cells in apoeknockoutmice.Arteriosclerosis,thrombosis,andvascularbiology.2006;26:2696-2702
48. TangY,HarringtonA,YangX,FrieselRE,LiawL.Thecontributionofthetie2+lineagetoprimitiveanddefinitivehematopoieticcells.Genesis.2010;48:563-567
49. BronteV,ZanovelloP.Regulationofimmuneresponsesbyl-argininemetabolism.NatRevImmunol.2005;5:641-654
50. Luckner-Minden C, Fischer I, Langhans C-D, SchillerM, Kropf P,Müller I, Hohlfeld JM, Ho AD,MunderM.Humaneosinophilgranulocytesdonotexpresstheenzymearginase.JLeukocyteBiol.2010;87:1125-1132
51. MunderM,MollinedoF,CalafatJ,CanchadoJ,Gil-LamaignereC,FuentesJM,LucknerC,DoschkoG,SolerG,EichmannK,MullerFM,HoAD,GoernerM,ModolellM.Arginase i isconstitutivelyexpressed in human granulocytes and participates in fungicidal activity.Blood. 2005;105:2549-2556
52. BandoJK,NussbaumJC,LiangHE,LocksleyRM.Type2innatelymphoidcellsconstitutivelyexpressarginase-iinthenaiveandinflamedlung.JLeukocBiol.2013;94:877-884
53. Feldman DL, Mogelesky TC, Liptak BF, Gerrity RG. Leukocytosis in rabbits with diet-inducedatherosclerosis.ArteriosclerThromb.1991;11:985-994
54. MurphyAJ,AkhtariM,TolaniS,PaglerT,BijlN,KuoCL,WangM,SansonM,AbramowiczS,WelchC,BochemAE,KuivenhovenJA,Yvan-CharvetL,TallAR.Apoeregulateshematopoieticstemcellproliferation,monocytosis,andmonocyteaccumulation inatherosclerotic lesions inmice.JClinInvest.2011;121:4138-4149
55. DreselHA,ViaDP,StohrM,ElchnerU,GnassoA,PostiglioneA,BlinN,Augustin J, SchettlerG.Observations on leukocytes from patients with severe familial hypercholesterolemia.Arteriosclerosis.1986;6:259-264
56. TolaniS,PaglerTA,MurphyAJ,BochemAE,AbramowiczS,WelchC,NagareddyPR,HolleranS,Hovingh GK, Kuivenhoven JA, Tall AR. Hypercholesterolemia and reduced hdl-c promote

Chapter2
62
hematopoietic stem cell proliferation and monocytosis: Studies in mice and fh children.Atherosclerosis.2013;229:79-85
57. GeissmannF,JungS,LittmanDR.Bloodmonocytesconsistoftwoprincipalsubsetswithdistinctmigratoryproperties.Immunity.2003;19:71-82
58. SwirskiFK,LibbyP,AikawaE,AlcaideP,LuscinskasFW,WeisslederR,PittetMJ.Ly-6c(hi)monocytesdominate hypercholesterolemia-associated monocytosis and give rise to macrophages inatheromata.JournalofClinicalInvestigation.2007;117:195-205
59. Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity.2013;39:806-818
60. RobbinsCS,ChudnovskiyA,RauchPJ,FigueiredoJ-L,IwamotoY,GorbatovR,EtzrodtM,WeberGF,UenoT,vanRooijenN.Extramedullaryhematopoiesisgeneratesly-6chighmonocytesthatinfiltrateatheroscleroticlesions.Circulation.2012;125:364-374
61. ChoiBS,Martinez-FaleroIC,CorsetC,MunderM,ModolellM,MullerI,KropfP.Differentialimpactof l-argininedeprivationon the activation andeffector functionsof t cells andmacrophages. JLeukocBiol.2009;85:268-277
62. deJongeWJ,KwikkersKL,teVeldeAA,vanDeventerSJ,NolteMA,MebiusRE,RuijterJM,LamersMC, Lamers WH. Arginine deficiency affects early b cell maturation and lymphoid organdevelopmentintransgenicmice.JClinInvest.2002;110:1539-1548
63. HosseiniH,LiY,KanellakisP,TayC,CaoA,TippingP,BobikA,TohBH,KyawT.Phosphatidylserineliposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive igmproducingb1alymphocytes.CardiovascRes.2015;106:443-452
64. HultheJ.Antibodiestooxidizedldlinatherosclerosisdevelopment--clinicalandanimalstudies.ClinChimActa.2004;348:1-8

Chapter3
Chapter
HematopoieticAkt2restorationenhancesfoamcellformationbutdoesnotaffectatherosclerosisinAkt2/LDLreceptordoubleknockout
miceBaoyanRen,MennoHoekstra,RonaldJ.vanderSluis,MaraKröner, Janine G. Geerling, Ilze Bot, Miranda Van Eck
Leiden Academic Centre for Drug Research, Cluster BioTherapeutics, Division of Biopharmaceutics, Leiden, TheNetherlands
Manuscriptinpreparation

Chapter3
64
KTserine/threonineKinase2(Akt2)playsakeyroleininsulinsignalingandcardiovasculardisease. LDL receptor knockout (LDLr KO) mice lacking Akt2 are glucose intolerant, but
atherosclerosissusceptibilityisnotaffectedorevenslightlydecreased.DisruptionofAkt2functionspecificallyinbonemarrow-derivedcellsleadstoadramaticreductioninatheroscleroticlesiondevelopmentinLDLrKOmice,indicatingapotentpro-atherogeniceffectofhematopoieticAkt2.The contribution of Akt2 in bone marrow-derived cells to the effects on atherosclerosissusceptibilityinAkt2/LDLrdoubleKO(dKO)mice,however,iscurrentlyunclear.
Inthisstudy,werestoredbonemarrowAkt2inAkt2/LDLrdKOmicebytransplantingfunctionalAkt2containingLDLrKObonemarrowintoAkt2/LDLrdKOrecipients.Akt2/LDLrdKOrecipientstransplantedwithAkt2/LDLrdKObonemarrowservedascontrols.Inlinewithapro-atherogenicroleformacrophageAkt2,enhancedfoamcellformationwasobservedintheperitonealcavityofAkt2/LDLrdKOmicetransplantedwithLDLrKOBMaccumulation(3weeksWTD,+88.0%,P<0.05;5weeksWTD,+59.5%,P=0.07).Surprisingly,bonemarrowAkt2restorationinAkt2/LDLrdKOmicedidnotaffectglucosetoleranceoratherosclerosisdevelopment.Thenulleffectonatherosclerosiscanlikelybeexplainedbythefactthatthepro-atherogenicincreaseinfoamcellformationwascounteractedbyabeneficialchangeintheinflammatorystatus,sinceAkt2restorationsuppressedLPS-inducedM1macrophagepolarization(evidencedby-77.8%iNOS/Arg1(P<0.001)expression,-20.9%TNF-alpha(P<0.01)secretion,and+90%IL-10(P<0.001)secretion).
Inconclusion,restorationofAkt2inAkt2/LDLrdKOmacrophagessuppressesM1polarizationandincreasesmacrophagefoamcellformation,butdoesnotaffectatherosclerosissusceptibilityofinAkt2/LDLrdKOmicereconstitutedwithAkt2positivebonemarrow.
Introduction
ProteinkinaseB(PKBorAkt)isafamilyofserine/threonine-specificproteinkinases,discoveredbyStaalandcolleagues in1977.1Aktplayscritical roles invariouscellularprocesses, includingproliferation, apoptosis, migration, angiogenesis and glucose metabolism.2-6 Three highlyhomologousisoformshavebeenidentified:Akt1,Akt2,andAkt3.Eachisoformhasadistincttissuedistributionandfunction.7-10WhereasAkt1isubiquitouslyexpressedandplaysakeyroleinthemodulationofcellsurvival,8,9,11Akt3ispredominantlyexpressedinthebrainandisessentialforpostnatalbraindevelopment.10Inaddition,Akt2ishighlyexpressedininsulin-responsivetissuesandplaysacrucialroleintheregulationofinsulinsignaling.12-14Akt2deficiencyinmiceinducesinsulin resistance and a type 2 diabetic phenotype.13-15 Although type 2 diabetes is generallyassociatedwithanincreasedriskforthedevelopmentofcardiovasculardisease,16Akt2deficiencydoesnotaugmentatherosclerosisdevelopment in the LDL receptor knockoutbackground.15-21Thus, Akt2 is likely to exert an anti-atherosclerotic function that compensates for the pro-atherogeniceffectsoftheglucoseintolerantphenotype.
Macrophages play an essential role in all stages of atherosclerotic lesion development andatherosclerosissusceptibilityisinfluencedbythephenotypeoftheseimmunecells.Grossly,twodistincttypesofmacrophagescanbedistinguished:1)pro-inflammatoryM1macrophagesand2)anti-inflammatory, healing M2 macrophages. Importantly, Akt2 deficiency is known to skewmacrophagestoaphenotypehallmarkedby:1)attenuatedinflammatorycytokineproduction,222) impairedmigration capability,23 and3) reduced foamcell formation.17 In linewith thispro-atherogenicfunctionofAkt2,disruptionofAkt2functionspecificallyinbonemarrow-derivedcellsreducesatheroscleroticlesiondevelopmentinLDLrKOmice.17,23Thecontributionofmacrophage
A

Chapter3
65
Akt2 to atherosclerosis susceptibility in glucose intolerant Akt2/LDLr double KO (dKO) mice,however,iscurrentlyunclear.
Insulin resistanceandglucose intolerancebothaffectmacrophageactivationandpolarization.Whereas some studies have established that high glucose levels induce an M1 macrophagephenotype,Chenmingandcolleaguesreportedthatperitonealexudatemacrophagesandbonemarrow-derivedmacrophagesfromstreptozotocin-induceddiabeticmiceexhibitamoreM2-likemacrophagephenotype.24Thisphenotypecouldalsobeinducedinvitrobyexposingmacrophagestohighglucoselevelsinthemedium.Moreover,themacrophagesfromdiabeticmiceshowedanimpaired inflammatory response to LPS/IFN-γ stimulation,which could be reversed by insulintreatment. Importantly, similarobservationsweremadeafter treatmentwithanAkt inhibitor,underliningaroleforAktinmacrophagepolarization.Toelucidatewhethertheatheroprotectivepotential ofmacrophage Akt2 remains present under insulin resistant conditions, the currentstudy was aimed at investigating the effects of hematopoietic Akt2 on atherosclerotic lesiondevelopment inglucose intolerantAkt2/LDLrdKOmicebyreconstitutingthesemicewithAkt2positivebonemarrow.
MaterialandMethods
Animals
Low-densitylipoproteinreceptorknockout(LDLrKO)miceandAkt2/LDLrdoubleKO(Akt2/LDLrdKO)micewereobtainedfromJackson laboratory (BarHarbor,ME,USA).Allmiceused inthecurrentstudywerebredandmaintainedattheGorlaeusLaboratoriesinLeiden,theNetherlands.Thestudieswereapprovedby theDutchEthicsCommitteeand regulatoryauthorityat LeidenUniversity. The animal experiments were carried out in compliance with Dutch governmentguidelinesandtheDirective2010/63/EUoftheEuropeanParliamentontheprotectionofanimalsusedforscientificpurposes.
Bonemarrowtransplantation(BMT)
TheendogenousBMoffemaleAkt2/LDLrdKOrecipientmice(13-15weeksold)wasdestroyedbytotal-body X-ray irradiation (9-Gy) 1 day before the bone marrow transplantation (BMT).Subsequently,five-millionBMcells,freshlyisolatedfromLDLrKOmiceorAkt2/LDLrdKOmicewastransplanted into the recipient mice via tail vein injection. After BMT, recipient mice weremaintainedonantibioticwater(83mg/Lciprofloxacinand67mg/LpolymyxinBsulfateand2.5g/L sugar) and chowdiet (RM3; SpecialDiet Services) for 8weeks. Thereafter, themicewerechallengedwithWestern-typediet(WTD,SpecialDietServices,productcode824171)for3or5weeks to induceatherosclerosisdevelopment.Atsacrifice, themicewereanaesthetizedbyananaestheticmixturecontainingxylazine(12.5mg/kg),ketamine(100mg/kg)andatropine(125μg/kg)andsubjectedtototalbodyPBSperfusionbeforeorgancollection.

Chapter3
66
Oralglucosetolerancetest(OGTT)
Theoralglucosetolerancetestwasperformedintherecipientmiceat4weeksonWTDaftera16-hoursfastingperiod.GlucoselevelsoftailbloodweredeterminedbyusingamanualACCU-CHEKCompactglucosemonitor(RocheDiagnostics,Almere,theNetherlands).
Blood glucose levels before glucose administration were determined as baseline (t=0).Immediately,themicewereadministered2gr/kgglucosebyoralgavageusinga10%Beta-D(+)-glucose(Sigma-Aldrich,catalognumber:G8270)solutioninPBS.Additionalbloodsamplesweretakenat15,30,45,60,90and120minutesafterglucoseadministration.
SerumCholesterolassay
Plasmatotalcholesterolandfreecholesterollevelsweredeterminedusingastandardenzymaticcolorimetricassay(chemicalsfromSigmaAldrich,USAorRocheDiagnostics,Mannheim,Germany)aspreviouslydescribed.25
HaematologyAnalysis
Anautomatedhaematologyanalyzer(XT-2000iV,SysmexCorporation,Japan)wasusedtoanalyseleukocytesandquantifymacrophagefoamcellformationintheperitoneumoftheAkt2/LDLrKOBMTrecipientsasdescribedpreviously.26
HistologicalAnalysisofAtherosclerosis
SectionsoftheaorticrootweremadebyaLeicaCM3050Scryostat.Subsequently,thesectionswerestainedwithOilred-Oforneutrallipids,withMasson’sTrichrome(HT15-1,4,SigmaAldrich)for collagen and with MOMA-2 (Research Diagnostics Inc) for macrophages as describedpreviously.27TheatheroscleroticplaquesizeandcompositionweredeterminedusingLeicaQwinImagingsoftware(LeicaLtd,Cambridge,UK).
CellcultureBone marrow-derived macrophages (BMDMs) from LDLr KO and Akt2/LDLr dKO mice weregeneratedbyculturingbonemarrowcellswith20ng/mLM-CSF(eBioscience,catalognumber:14-8983-80)for7days.Toinducefoamcellformation,BMDMswereincubatedwith20μg/mLcopper-oxidized LDL (oxLDL) for 24 hours.28 To primeM1 orM2macrophages, BMDMs wereincubated for 24 hours with 100 ng/mL LPS (Escherichia coli0111:B4; Sigma Aldrich, catalognumber:L2630-10MG)or20ng/mLIL-4(Peprotech,catalog:214-14).BMDMsthatweretreatedwithPBSinsteadofoxLDL,LPSorIL-4servedascontrol.Macrophageculturesupernatantswerecollectedandstoredat-20°Cuntilfurtheranalysis.
AnalysisofgeneexpressionbyquantitativePCR(qPCR)
TotalRNAwasisolatedusingtheguanidiniumthiocyanatemethod.29cDNAwassynthesizedusingaRevertAidM-MuLVenzyme(Fermentas,Burlington,Canada).ThemRNAexpressionlevelsweremeasuredona7500FastReal-TimePCRsystem(ABIPRISM7500;AppliedBiosystems,FosterCity,CA)usingSYBRgreentechnology(AppliedBiosystems).Theaveragecyclethreshold(CT)of36B4(acidic ribosomal phosphoprotein P0) and RPL27 (Ribosomal Protein L27) were used ashousekeepinggeneexpressioncontrolvalues.

Chapter3
67
Cytokineanalysisandnitricoxideassay
ThesupernatantsoftheBMDMcellcultureswerethawedandthecontentofthecytokinesTNF-α, IL-6,and IL-10weredeterminedaccordingtothemanufacturer’s’ instructions (allELISAkitswerepurchasedfromBDBiosciences,UnitedStates).Thenitricoxide(NO)concentrationinthesupernatantswasdeterminedusingtheGriessmethodaccordingtothemanufacturer'sprotocol(Sigma-Aldrich).
Statisticalanalysis
Allthevaluesareexpressedasmeans±SEM.ThestatisticalsignificantdifferencesbetweenthegroupsweretestedusingtheunpairedStudent’st-testortwo-wayANOVAwithGraphPadPrismsoftware(GraphPadSoftwareInc.,SanDiego,California,USA).AWelchcorrectionwasappliedtothet-testincaseofunequalvariancesinthedataset.Atwo-sidedPvaluelowerthan0.05wasdefinedasstatisticallysignificant.
Results
BonemarrowAkt2restorationdoesnotaffectbloodglucose levelsandglucosetoleranceofAkt2/LDLrdKOmice
TotalbodyAkt2deficiencyinLDLrKOmiceleadstoimpairedglucosetolerance.18ToestablishthecontributionofbonemarrowAkt2totheregulationofglucosemetabolisminAkt2/LDLrKOmice,bloodglucoselevelsweredeterminedandaglucosetolerancetestwasperformedat12weeksafterBMT,whenmicewerefedaWTDfor4weeks.RestorationofbonemarrowAkt2didnotimpactonfastingbloodglucoselevelsaftereither2or4weeksofWTDfeeding(p>0.05;figure1A-B).UponoralglucoseadministrationbothAkt2/LDLrdKOmicetransplantedwithAkt2/LDLrdKOBMorLDLrKOBMshowedarapidincreaseinbloodglucoselevels.Inbothgroupsthepeakglucosevaluewasreachedat30minutesaftertheglucosebolusinjection(Akt/LDLrdKOBMvs.LDLrKOBM:18.3±0.7mMvs19.2±0.5mM,p<0.05comparedtobasalglucoselevels;figure1C).It isworthtonotethat,120minutesafteroralglucoseadministration,theplasmaglucoselevelsinbothgroupsfailedtogetbacktobasallevels(p<0.01comparedtobasalglucoselevels,figure 1C), supporting the glucose intolerant phenotype of the Akt2/LDLr dKO recipientmice.Conclusively, we observed no difference in glucose handling between the two experimentalgroups,indicatingthatrestorationofbonemarrowAkt2inAkt2/LDLrdKOmicedoesnotinfluencetheglucoseintolerantphenotypeoftherecipientmice.
BonemarrowAkt2restorationdoesnotaffectatherosclerosisdevelopmentinAkt2/LDLrdKOmice
To assess the effect of Akt2 restoration in bone marrow-derived cells on atherosclerosissusceptibility of Akt2/LDLr dKO mice, the atherosclerotic plaque size in the aortic root wasdeterminedafter3and5weeksofWTDfeeding.Atbothtimepointsthelesionswereprimarilycomposed of macrophage foam cells with no deposited collagen (Figure 2A). Surprisingly,Akt2/LDLrdKO recipients transplantedwith LDLrKOBMcontaining functionalAkt2developedatheroscleroticlesionsofasimilarsizeasAkt2/LDLrdKOtransplantedcontrols,bothafter3weeksWTDchallenge(Akt2/LDLrdKOBMvs.LDLrKOBM:119×103±14×103μm2vs.125×103±21×103
μm2,p>0.05;figure2Band2C)andafter5weeksWTDfeeding(Akt2/LDLrdKOBMvs.LDLrKOBM:154×103±14×103μm2vs.169×103±24×103μm2,p>0.05;figure2Band2C).Thus,incontrast
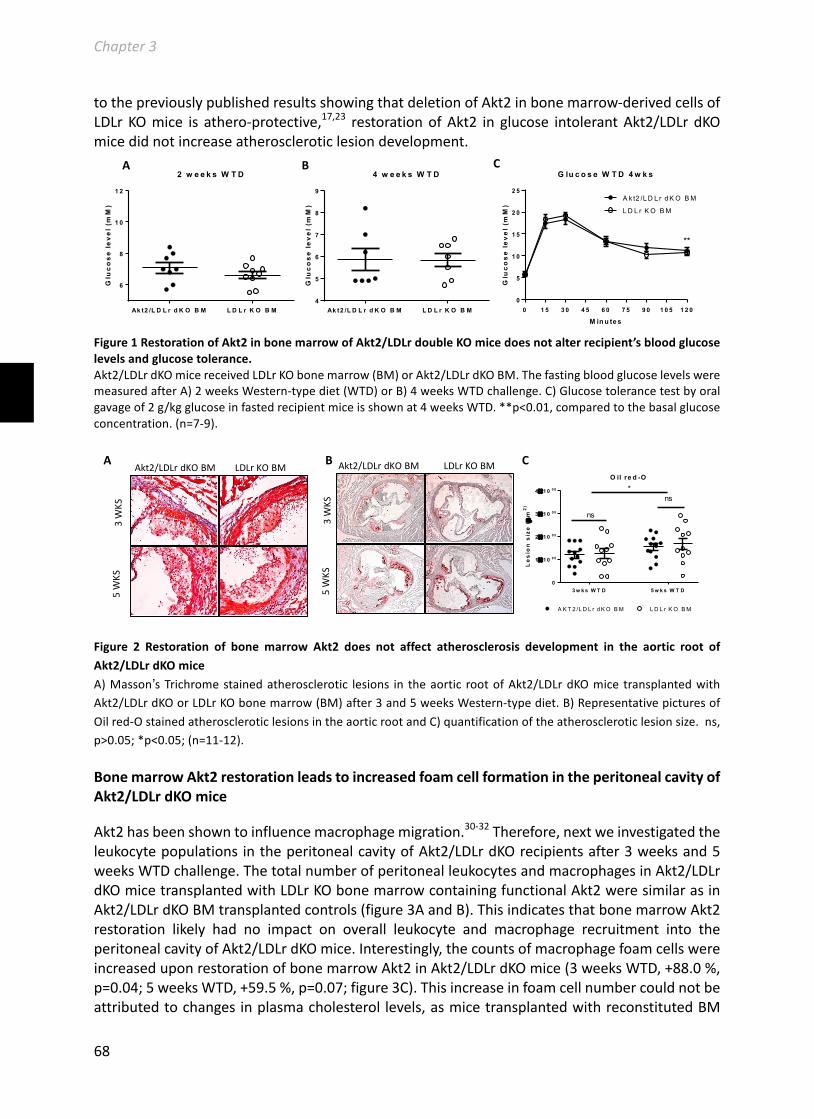
Chapter3
68
tothepreviouslypublishedresultsshowingthatdeletionofAkt2inbonemarrow-derivedcellsofLDLrKOmice is athero-protective,17,23 restorationofAkt2 in glucose intolerantAkt2/LDLrdKOmicedidnotincreaseatheroscleroticlesiondevelopment.
Figure1RestorationofAkt2inbonemarrowofAkt2/LDLrdoubleKOmicedoesnotalterrecipient’sbloodglucoselevelsandglucosetolerance.Akt2/LDLrdKOmicereceivedLDLrKObonemarrow(BM)orAkt2/LDLrdKOBM.ThefastingbloodglucoselevelsweremeasuredafterA)2weeksWestern-typediet(WTD)orB)4weeksWTDchallenge.C)Glucosetolerancetestbyoralgavageof2g/kgglucoseinfastedrecipientmiceisshownat4weeksWTD.**p<0.01,comparedtothebasalglucoseconcentration.(n=7-9).
Figure 2 Restoration of bonemarrow Akt2 does not affect atherosclerosis development in the aortic root ofAkt2/LDLrdKOmiceA)Masson’sTrichromestainedatherosclerotic lesions in theaortic rootofAkt2/LDLrdKOmice transplantedwithAkt2/LDLrdKOorLDLrKObonemarrow(BM)after3and5weeksWestern-typediet.B)RepresentativepicturesofOilred-OstainedatheroscleroticlesionsintheaorticrootandC)quantificationoftheatheroscleroticlesionsize.ns,p>0.05;*p<0.05;(n=11-12).
BonemarrowAkt2restorationleadstoincreasedfoamcellformationintheperitonealcavityofAkt2/LDLrdKOmice
Akt2hasbeenshowntoinfluencemacrophagemigration.30-32Therefore,nextweinvestigatedtheleukocytepopulationsintheperitonealcavityofAkt2/LDLrdKOrecipientsafter3weeksand5weeksWTDchallenge.ThetotalnumberofperitonealleukocytesandmacrophagesinAkt2/LDLrdKOmicetransplantedwithLDLrKObonemarrowcontainingfunctionalAkt2weresimilarasinAkt2/LDLrdKOBMtransplantedcontrols(figure3AandB).ThisindicatesthatbonemarrowAkt2restoration likely had no impact on overall leukocyte and macrophage recruitment into theperitonealcavityofAkt2/LDLrdKOmice.Interestingly,thecountsofmacrophagefoamcellswereincreaseduponrestorationofbonemarrowAkt2inAkt2/LDLrdKOmice(3weeksWTD,+88.0%,p=0.04;5weeksWTD,+59.5%,p=0.07;figure3C).Thisincreaseinfoamcellnumbercouldnotbeattributedtochangesinplasmacholesterol levels,asmicetransplantedwithreconstitutedBM
4 w e e k s W T D
Ak t2 /L D L r d K O B M L D L r K O B M4
5
6
7
8
9
Glu
co
se
le
ve
l (m
M)
0 1 5 3 0 4 5 6 0 7 5 9 0 1 0 5 1 2 00
5
1 0
1 5
2 0
2 5
G lu c o s e W T D 4 w k s
M in u te s
Glu
co
se
le
ve
l (m
M)
A k t2 /L D L r d K O B M
L D L r K O B M
**
2 w e e k s W T D
Ak t2 /L D L r d K O B M L D L r K O B M
6
8
1 0
1 2
Glu
co
se
le
ve
l (m
M)
A B C
3 w k s W T D 5 w k s W T D0
1´1 0 0 5
2´1 0 0 5
3´1 0 0 5
4´1 0 0 5
O il re d -O
Le
sio
n s
ize
(µ
m2
)
A K T 2 /L D L r d K O B M L D L r K O B M
ns
ns*
Akt2/LDLrdKOBM LDLrKOBM
3WKS
5WKS
Akt2/LDLrdKOBM LDLrKOBM
3WKS
5WKS
C A B

Chapter3
69
Akt2showedsimilarplasmacholesterollevelsascomparedtocontrolmice,bothonchowdietandafteraWTDchallenge(table1).Moreover,theplasmalipoproteindistributionprofilewasalsonotaltereduponrestorationofAkt2inbonemarrow-derivedcellsofAkt2/LDLrdKOmice(figure4).
Figure 3 Restoration of bone marrowAkt2 in Akt2/LDLr dKO mice inducesfoam cell formation in the peritonealcavity.
Peritoneal leukocytes were harvestedfrom Akt2/LDLr dKO recipientstransplanted with LDLr KO BM andAkt2/LDLr dKO BM after 3 weeks or 5weeksWestern-typediet(WTD)feeding.A) total leukocyte counts, B)macrophage counts and C) foam cellcounts were determined by using aSysmex hematology analyzer. D)Example picture of the hematologyanalysis (from the 3 weeks WTDchallengegroup).*p<0.05.n=11-12.
Ak t2 /L D L r d K O B M L D L r K O B M0
2´1 0 5
4´1 0 5
6´1 0 5
8´1 0 5
1´1 0 6
Cel
ls/m
ou
se
* P value 0.0368
Ak t2 /L D L r d K O B M L D L r K O B M0
2´1 0 5
4´1 0 5
6´1 0 5
8´1 0 5
1´1 0 6
Cel
ls/m
ou
se
P value 0.0718
Ak t2 /L D L r d K O B M L D L r K O B M0
5 .0´1 0 0 7
1 .0´1 0 0 8
1 .5´1 0 0 8
Cel
ls/m
ouse
P value 0.0797
Ak t2 /L D L r d K O B M L D L r K O B M0
5 .0´1 0 0 7
1 .0´1 0 0 8
1 .5´1 0 0 8
Cel
ls/m
ouse
P value 0.1724
3weeksWTD 5weeksWTD
Leukocytes
Ak t2 /L D L r d K O B M L D L r K O B M0
1´1 0 0 7
2´1 0 0 7
3´1 0 0 7
4´1 0 0 7
5´1 0 0 7
Cel
ls/m
ou
se
P value 0.3335
Ak t2 /L D L r d K O B M L D L r K O B M0
1´1 0 0 7
2´1 0 0 7
3´1 0 0 7
4´1 0 0 7
5´1 0 0 7
Cel
ls/m
ouse
P value 0.1018
3weeksWTD 5weeksWTD
Macrophages
3weeksWTD 5weeksWTD
Foamcells
A
B
C
D
LDLrKOBM Akt2/LDLrdKOBM

Chapter3
70
Table1RestorationofbonemarrowAkt2doesnotimpactplasmacholesterollevelsinAkt2/LDLrdKOmice.
Plasmawas collectedby tail bleedingofAkt2/LDLr dKO recipients that receivedLDLrKOBMandAkt2/LDLrdKOBMwhileonchowdietandatsacrificeafter3and5weeksWestern-typediet(WTD)feeding.Thefreecholesterolandtotalcholesterollevelsweredetermined.(n=6-12).
Figure 4 Restoration of bone marrowAkt2 does not impact on plasmacholesterol distribution among thedifferent types of lipoproteins inAkt2/LDLrdKOmice.
Plasmawascollectedbytail-bleedingofAkt2/LDLr dKO recipients that receivedLDLr KO BM or Akt2/LDLr dKO BM atsacrificeafter3weeksWestern-typediet(WTD) feeding. Cholesterol lipoproteindistribution was determined by FPLC.(n=6-12ofpooledplasma).
Akt2promotesfoamcellformationinLDLrdeficientmacrophagesbyregulatingtheexpressionofcholesterolinfluxandeffluxgenes
TogainfurtherinsightintothemechanismsunderlyingtheincreasedfoamcellformationofLDLrKOmacrophageswithfunctionalAkt2invivo,aninvitrofoamcellformationassaywasperformedusing LDLr KO and LDLr/Akt2 dKO BMDMs. After 24 hours of oxLDL incubation, LDLr KOmacrophages showed significantly more cellular lipid accumulation as evidenced by moreextensiveOil red-O staining compared toAkt2/LDLr dKOmacrophages (figure 5A). ThemRNAexpressionofSR-AandCD36,themainreceptorsforoxLDLuptake,33wassignificantlyhigherinLDLrKOmacrophagescomparedtoAkt2/LDLrdKOmacrophages(SR-A,+60.7%,p<0.001;figure5B;CD36, +24.8%, p<0.001, figure 5C) after oxLDL stimulation. The expression of low-densitylipoproteinreceptor-relatedprotein1(LRP1,1.16±0.05vs0.87±0.13,p>0.05,figure5D)wasnotaffectedandtheexpressionofVLDLr,responsiblefortheuptakeofunmodifiedapoE-containinglipoproteins,34,35wasdecreased(-59.1%,p<0.05,figure5E)inLDLrKOmacrophagescomparedtoAkt2/LDLrdKOmacrophages.Inaddition,theexpressionofthecholesteroleffluxgenesABCA1andABCG1wasdecreasedinLDLrKOmacrophagescomparedtoAkt2/LDLrdKOcontrols(ABCA1,-39.0%,p<0.05,figure5F;ABCG1,-40.5%,p<0.01,figure5G).Insummary,theseresultssuggestthatLDLrKOmacrophageswithfunctionalAkt2aremoresusceptibletooxLDL-inducedfoamcellformationduetoaugmenteduptakeofoxLDLanddecreasedcellularcholesterolefflux.
Chowdiet WTD
mg/mL FC TC FC TC
Akt2/LDLrdKOBM 0.75±0.05 2.39±0.12
3weeks 3.26±0.19 9.43±1.10
5weeks 2.65±0.16 7.35±0.68
LDLrKOBM0.77±0.03 2.45±0.103weeks 2.68±0.44 7.32±1.89
5weeks 2.51±0.24 7.23±0.60
0 2 4 6 8 10 12 14 16 18 20 22 240
500
1000
1500
2000
2500
Fractions
Cho
lest
erol
leve
l (µg
/µL)
Akt2/LDLr dKO BM
LDLr KO BM
VLDL
LDL
HDL
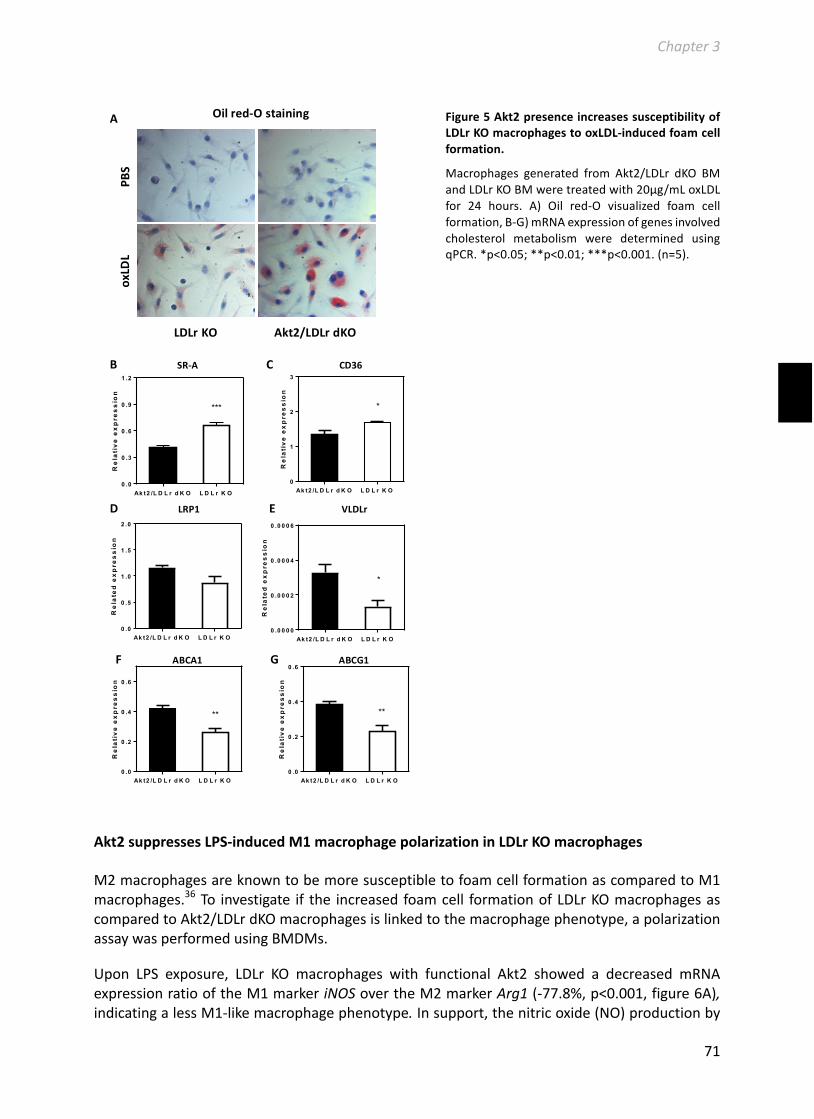
Chapter3
71
Figure5Akt2presenceincreasessusceptibilityofLDLrKOmacrophagestooxLDL-inducedfoamcellformation.
Macrophages generated from Akt2/LDLr dKO BMandLDLrKOBMweretreatedwith20μg/mLoxLDLfor 24 hours. A) Oil red-O visualized foam cellformation,B-G)mRNAexpressionofgenesinvolvedcholesterol metabolism were determined usingqPCR.*p<0.05;**p<0.01;***p<0.001.(n=5).
Akt2suppressesLPS-inducedM1macrophagepolarizationinLDLrKOmacrophages
M2macrophagesareknowntobemoresusceptibletofoamcellformationascomparedtoM1macrophages.36To investigate if the increasedfoamcell formationofLDLrKOmacrophagesascomparedtoAkt2/LDLrdKOmacrophagesislinkedtothemacrophagephenotype,apolarizationassaywasperformedusingBMDMs.
Upon LPS exposure, LDLr KO macrophages with functional Akt2 showed a decreased mRNAexpressionratiooftheM1markeriNOSovertheM2markerArg1(-77.8%,p<0.001,figure6A),indicatingalessM1-likemacrophagephenotype.Insupport,thenitricoxide(NO)productionby
A
Akt2/LDLrdKO
Oilred-Ostaining PB
S
LDLrKO
oxLD
L
Ak t2 /L D L r d K O L D L r K O0 .0
0 .2
0 .4
0 .6
A b c a 1
Re
lati
ve e
xp
res
sio
n
**
Ak t2 /L D L r d K O L D L r K O0 .0
0 .5
1 .0
1 .5
2 .0
L rp 1
Re
late
d e
xp
res
sio
n
Ak t2 /L D L r d K O L D L r K O0 .0
0 .2
0 .4
0 .6
A b c g 1
Re
lati
ve e
xp
res
sio
n
**
Ak t2 /L D L r d K O L D L r K O0 .0 0 0 0
0 .0 0 0 2
0 .0 0 0 4
0 .0 0 0 6
V ld lr
Re
late
d e
xp
res
sio
n
*
Ak t2 /L D L r d K O L D L r K O0
1
2
3
C d 3 6
Re
lati
ve e
xp
res
sio
n
*
Ak t2 /L D L r d K O L D L r K O0 .0
0 .3
0 .6
0 .9
1 .2
S r -a
Re
lati
ve
ex
pre
ss
ion
***
CCD36 BSR-A
EVLDLr DLRP1
GABCG1 FABCA1
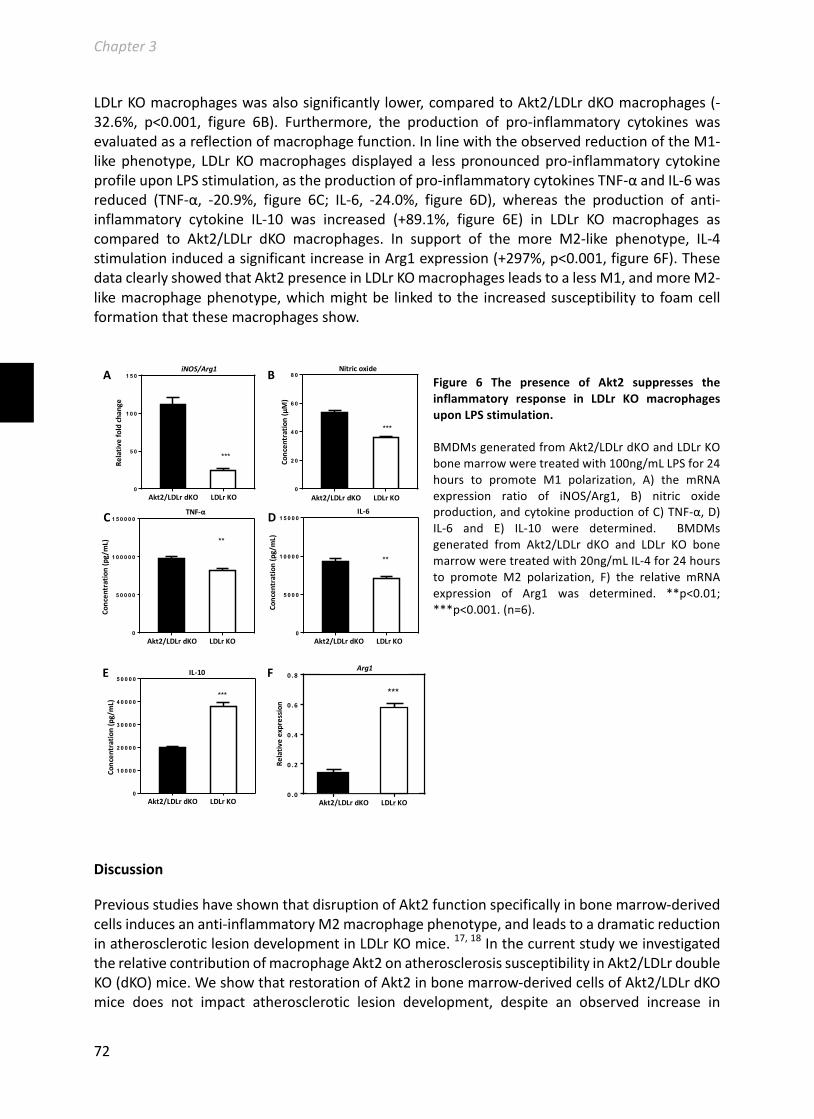
Chapter3
72
LDLrKOmacrophageswasalsosignificantlylower,comparedtoAkt2/LDLrdKOmacrophages(-32.6%, p<0.001, figure 6B). Furthermore, the production of pro-inflammatory cytokines wasevaluatedasareflectionofmacrophagefunction.InlinewiththeobservedreductionoftheM1-likephenotype, LDLrKOmacrophagesdisplayeda lesspronouncedpro-inflammatory cytokineprofileuponLPSstimulation,astheproductionofpro-inflammatorycytokinesTNF-αandIL-6wasreduced (TNF-α, -20.9%, figure 6C; IL-6, -24.0%, figure 6D), whereas the production of anti-inflammatory cytokine IL-10 was increased (+89.1%, figure 6E) in LDLr KO macrophages ascompared to Akt2/LDLr dKO macrophages. In support of the more M2-like phenotype, IL-4stimulationinducedasignificantincreaseinArg1expression(+297%,p<0.001,figure6F).ThesedataclearlyshowedthatAkt2presenceinLDLrKOmacrophagesleadstoalessM1,andmoreM2-likemacrophagephenotype,whichmightbe linkedtothe increasedsusceptibility to foamcellformationthatthesemacrophagesshow.
Figure 6 The presence of Akt2 suppresses theinflammatory response in LDLr KO macrophagesuponLPSstimulation.
BMDMsgeneratedfromAkt2/LDLrdKOandLDLrKObonemarrowweretreatedwith100ng/mLLPSfor24hours to promote M1 polarization, A) the mRNAexpression ratio of iNOS/Arg1, B) nitric oxideproduction,andcytokineproductionofC)TNF-α,D)IL-6 and E) IL-10 were determined. BMDMsgenerated from Akt2/LDLr dKO and LDLr KO bonemarrowweretreatedwith20ng/mLIL-4for24hoursto promote M2 polarization, F) the relative mRNAexpression of Arg1 was determined. **p<0.01;***p<0.001.(n=6).
Discussion
PreviousstudieshaveshownthatdisruptionofAkt2functionspecificallyinbonemarrow-derivedcellsinducesananti-inflammatoryM2macrophagephenotype,andleadstoadramaticreductioninatheroscleroticlesiondevelopmentinLDLrKOmice.17,18InthecurrentstudyweinvestigatedtherelativecontributionofmacrophageAkt2onatherosclerosissusceptibilityinAkt2/LDLrdoubleKO(dKO)mice.WeshowthatrestorationofAkt2inbonemarrow-derivedcellsofAkt2/LDLrdKOmice does not impact atherosclerotic lesion development, despite an observed increase in
A rg 1
A kt 2 / L D L r d KO L D L r KO0 .0
0 .2
0 .4
0 .6
0 .8
***
Relative
exp
ress
ion
Ak t2 /L D L r d K O L D L r K O0
5 0 0 0
1 0 0 0 0
1 5 0 0 0IL -6
Co
nc
en
tra
tio
n (
pg
/mL
)
**
Ak t2 /L D L r d K O L D L r K O0
1 0 0 0 0
2 0 0 0 0
3 0 0 0 0
4 0 0 0 0
5 0 0 0 0IL -1 0
Co
nc
en
tra
tio
n (
pg
/mL
) ***
E F
Ak t2 /L D L r d K O L D L r K O0
2 0
4 0
6 0
8 0N itr ic o x id e
Co
nc
en
tra
tio
n (µ
M)
***
Ak t2 /L D L r d K O L D L r K O0
5 0 0 0 0
1 0 0 0 0 0
1 5 0 0 0 0T N F -a
Co
nc
en
tra
tio
n (
pg
/mL
)
**
Ak t2 /L D L r d K O L D L r K O0
5 0
1 0 0
1 5 0iN O S /A rg 1
Re
lati
ve
fo
ld c
ha
ng
e
***
A
C
B
D
Relativ
efoldchange
Concen
tration(pg/mL)
Concen
tration(μM)
Concen
tration(pg/mL)
Concen
tration(pg/mL)
Relativ
eexpression
iNOS/Arg1 Nitricoxide
TNF-α IL-6
IL-10 Arg1
Akt2/LDLrdKOLDLrKO Akt2/LDLrdKOLDLrKO
Akt2/LDLrdKOLDLrKO
Akt2/LDLrdKOLDLrKO Akt2/LDLrdKOLDLrKO
Akt2/LDLrdKOLDLrKO

Chapter3
73
macrophagefoamcellformation.Thislackofeffectonlesiondevelopmentcanlikelybeexplainedby the observation that Akt2 restoration in macrophages also reduced the macrophagepolarization towards the pro-inflammatory M1 macrophage phenotype, and correspondinglyloweredtheproductionofpro-inflammatorycytokines.
TotalbodyAkt2lossisknowntoleadtoglucoseintoleranceinLDLrKOmice.17,18WehereshowthatrestorationofbonemarrowAkt2inAkt2/LDLrdKOmicedoesnotchangethebloodglucoselevelsnortheglucoseintolerantphenotypeofthesemice,suggestingthatmacrophageAkt2isnotinvolvedintheregulationofglucosehomeostasis.Inlinewiththis,Babaevetal.alsofoundthatdeletionofbonemarrowAkt2 inLDLrKOmicedidnotaffectglucose levels.23Thesedataindicate that bone marrow Akt2 is not a decisive factor for the observed glucose intolerantphenotypeofmicewithatotal-bodyAkt2deletion.Indeed,Akt2maintainsglucosehomeostasismainlybyregulatinghepaticglucoseproductionandskeletalmuscleinsulinresistance,celltypesthatarenotaffectedbybonemarrowtransplantation.37-39
PreviousstudieshavedemonstratedthatbonemarrowAkt2playsapro-atherogenicroleinnon-diabeticLDLrKOmice,asevidencedbyreducedatheroscleroticlesiondevelopmentuponbonemarrow-specificdeletionofAkt2.17,23Inourstudy,wedidnotobserveanyimpactofbonemarrowAkt2 restorationon atherosclerotic lesion size in glucose intolerantAkt2/LDLr dKOmice fed aWestern-typediet.Despitethelackofeffectonlesionsize,Akt2restorationinbonemarrowdidpromote macrophage foam cell formation in these mice. This increased susceptibility tomacrophage foam cell formation of mice with functional bone marrow Akt2 could not beattributedtochangesintheplasmacholesterollevelsorlipoproteindistribution.However,invitrostudiesshowedthattheincreaseinmacrophagefoamcellformationislikelytobecausedbyanincreasedinfluxofoxidizedLDLtogetherwithadecreasedcholesteroleffluxasevidencedbyanincreased mRNA expression of the LDL cholesterol uptake receptors SR-A1 and CD36 and adecreasedexpressionofcholesteroleffluxpumpsABCA1andABCG1.Inagreementwithourstudy,Rotllan et al. also found that macrophage Akt2 promotes foam cell formation on a wildtypebackground.17
Macrophagesusceptibilitytofoamcell formation is influencedbythemacrophagephenotype,with M2 macrophages being more prone to become foam cells as compared to M1macrophages.36Indeed,inlinewiththeincreasedfoamcellformation,LDLrKOmacrophageswithfunctionalAkt2showedasuppressedM1macrophagephenotypeinresponsetoLPSstimulationcompared to LDLrKOmacrophages lackingAkt2. This result is in contrast toprevious findingsshowing that Akt2 deficiency leads to a more M2-like macrophage phenotype after LPSstimulation,comparedtoWTmacrophages.22,23,40Severaldifferencesbetweenourcurrentstudyandthepreviousstudiescanbedistinguished,suchasthedurationoftheLPSstimulationperiod,theLPSconcentrationused,strainvariabilities(LDLrKOvsWT),41,42macrophageheterogeneities(bone marrow-derived macrophages vs peritoneal macrophages), and culture mediumcompositions.Hence,furtherresearchintothistopiciswarranted.
Itshouldbenotedthatboththepreviousstudiesonmacrophagepolarization22,23,40andthebonemarrowtransplantationexperimentsofRotllan17andBabaevetal.23appliedAkt2deficientcellswithafunctionalLDLr.Forbonemarrowtransplantation-basedatherosclerosisstudies,thisisacommonlyusedstrategyasitpreventstherequirementofextensivecross-breedingtogenerateLDLrKOdonorslackingthegeneofinterest.However,althoughpreviousstudieshaveshownthatthe presence or absence of the LDLr in bone marrow-derived cells only minimally affects

Chapter3
74
atherosclerosisperse,43,44itcannotbeexcludedthatuponinteractionwiththegeneofinterestabsence or presence of the LDLr does affectmacrophage function and atherosclerotic lesiondevelopment. Therefore, further analysis is warranted to confirm this possible interactionbetweenLDLrandAkt2.Understandingtheunderlyingmechanismsofapossiblegeneinteractionisbeyondthescopeofourcurrentstudy.However,thedifferenceintheLPS-inducedphenotypeofAkt2deficientmacrophagesintheLDLrKObackgroundinourcurrentstudyascomparedtothewildtypebackgroundinearlierstudies,mightproveamotivationforinitiatingfurtherresearch.
Taken together,our study showed that: 1)bonemarrowAkt2 isnot adecisive factor inAkt2-regulated glucose metabolism, and 2) bone marrow Akt2 promotes macrophage foam cellformationbutdoesnotaffectatherosclerosisdevelopmentinAkt2/LDLrdKOmice,whichmightbeduetothecounteractinganti-inflammatoryeffectsof theAkt2-inducedsuppressedM1-likemacrophagephenotype.
Acknowledgments
TheauthorsthankJoyaE.Nahon,OlgaS.C.Snip,HenrikeKerbstadt,LidewijR.deLeeuwandRickvanderGeestfortheexcellenttechnicalassistanceandhelpfuldiscussions.
Thisworkissupportedbythe‘NetherlandsCardioVascularResearchInitiative:theDutchHeartFoundation,DutchFederationofUniversityMedicalCenters, theNetherlandsOrganization forHealth Research and Development, and the Royal Netherlands Academy of Sciences for theGENIUSproject“Generatingthebestevidence-basedpharmaceuticaltargetsforatherosclerosis”(CVON2011-19)andtheNetherlandsOrganizationforScientificResearch(VICIGrant91813603(M.V.E). M.V.E. is an Established Investigator of the Netherlands Heart Foundation (Grant2007T056).B.RenwassupportedbytheChinaScholarshipCouncil.
References
1. StaalSP,HartleyJW,RoweWP.Isolationoftransformingmurineleukemiavirusesfrommicewithahighincidenceofspontaneouslymphoma.ProcNatlAcadSciUSA.1977;74:3065-3067
2. SongG,OuyangG,BaoS.Theactivationofakt/pkbsignalingpathwayandcellsurvival.JCellMolMed.2005;9:59-71
3. KandelES,SkeenJ,MajewskiN,DiCristofanoA,PandolfiPP,FelicianoCS,GartelA,HayN.Activationofakt/proteinkinasebovercomesag(2)/mcellcyclecheckpointinducedbyDNAdamage.MolCellBiol.2002;22:7831-7841
4. SardielloM,PalmieriM,diRonzaA,MedinaDL,ValenzaM,GennarinoVA,DiMaltaC,DonaudyF,EmbrioneV,PolishchukRS,BanfiS,ParentiG,CattaneoE,BallabioA.Agenenetworkregulatinglysosomalbiogenesisandfunction.Science.2009;325:473-477
5. PalmieriM,PalR,NelvagalHR,LotfiP,StinnettGR,SeymourML,ChaudhuryA,BajajL,BondarVV,BremnerL,SaleemU,TseDY,SanagasettiD,WuSM,NeilsonJR,PereiraFA,PautlerRG,RodneyGG,Cooper JD,SardielloM.Mtorc1-independent tfebactivationviaakt inhibitionpromotescellularclearanceinneurodegenerativestoragediseases.NatCommun.2017;8:14338
6. Chen J, Somanath PR, Razorenova O, ChenWS, Hay N, Bornstein P, Byzova TV. Akt1 regulatespathologicalangiogenesis,vascularmaturationandpermeabilityinvivo.NatMed.2005;11:1188-1196
7. YuH,LittlewoodT,BennettM.Aktisoformsinvasculardisease.VasculPharmacol.2015;71:57-648. ChenWS,XuPZ,GottlobK,ChenML,SokolK,ShiyanovaT,RoninsonI,WengW,SuzukiR,TobeK,
Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygousdisruptionoftheakt1gene.GenesDev.2001;15:2203-2208

Chapter3
75
9. ChoH,ThorvaldsenJL,ChuQW,FengF,BirnbaumMJ.Akt1/pkbalphaisrequiredfornormalgrowthbutdispensableformaintenanceofglucosehomeostasisinmice.JournalofBiologicalChemistry.2001;276:38349-38352
10. TschoppO,YangZZ,BrodbeckD,DummlerBA,Hemmings-MieszczakM,WatanabeT,MichaelisT,FrahmJ,HemmingsBA.Essentialroleofproteinkinasebgamma(pkbgamma/akt3)inpostnatalbraindevelopmentbutnotinglucosehomeostasis.Development.2005;132:2943-2954
11. GreenBD,JabbourAM,SandowJJ,RiffkinCD,MasourasD,DauntCP,SalmanidisM,BrumattiG,HemmingsBA,GuthridgeMA,PearsonRB,EkertPG.Akt1istheprincipalaktisoformregulatingapoptosisinlimitingcytokineconcentrations.CellDeathDiffer.2013;20:1341-1349
12. BoucherJ,KleinriddersA,KahnCR.Insulinreceptorsignalinginnormalandinsulin-resistantstates.CshPerspectBiol.2014;6
13. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR,WilliamsRM,AceriniCL,DungerDB,BarfordD,UmplebyAM,WarehamNJ,DaviesHA,SchaferAJ,StoffelM,O'Rahilly S, Barroso I. A familywith severe insulin resistance and diabetes due to amutationinakt2.Science.2004;304:1325-1328
14. FrankishH.Akt2-deficientmiceshowsymptomsoftype2diabetes.Lancet.2001;357:1771-177115. GarofaloRS,OrenaSJ,RafidiK,TorchiaAJ,StockJL,HildebrandtAL,CoskranT,BlackSC,BreesDJ,
WicksJR,McNeishJD,ColemanKG.Severediabetes,age-dependent lossofadiposetissue,andmild growth deficiency in mice lacking akt2/pkb beta. Journal of Clinical Investigation.2003;112:197-208
16. EmergingRisk FactorsC, SarwarN,GaoP, Seshasai SR,GobinR, KaptogeS,DiAngelantonioE,IngelssonE,LawlorDA,SelvinE,StampferM,StehouwerCD,LewingtonS,PennellsL,ThompsonA,SattarN,WhiteIR,RayKK,DaneshJ.Diabetesmellitus,fastingbloodglucoseconcentration,andrisk of vascular disease: A collaborative meta-analysis of 102 prospective studies. Lancet.2010;375:2215-2222
17. RotllanN,Chamorro-JorganesA,AraldiE,WanschelAC,AryalB,ArandaJF,GoedekeL,SalernoAG,RamirezCM,SessaWC,SuarezY,Fernandez-HernandoC.Hematopoieticakt2deficiencyattenuatestheprogressionofatherosclerosis.FasebJournal.2015;29:597-610
18. RensingKL,deJagerSC,StroesES,VosM,TwicklerMT,Dallinga-ThieGM,deVriesCJ,KuiperJ,BotI, von der Thusen JH. Akt2/ldlr double knockout mice display impaired glucose tolerance anddevelop more complex atherosclerotic plaques than ldlr knockout mice. Cardiovasc Res.2014;101:277-287
19. Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: Molecular mechanisms.CardiovascDiabetol.2002;1
20. ColwellJA,WinocourPD,LopesvirellaM,HalushkaPV.Newconceptsaboutthepathogenesisofatherosclerosisindiabetes-mellitus.AmericanJournalofMedicine.1983;75:67-80
21. KanterJE,KramerF,BarnhartS,AverillMM,Vivekanandan-GiriA,VickeryT,LiLO,BeckerL,YuanW,ChaitA,BraunKR,Potter-PerigoS,SandaS,WightTN,PennathurS,SerhanCN,HeineckeJW,Coleman RA, Bornfeldt KE. Diabetes promotes an inflammatory macrophage phenotype andatherosclerosisthroughacyl-coasynthetase1.PNatlAcadSciUSA.2012;109:E715-E724
22. Arranz A, Doxaki C, Vergadi E,Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E,AndroulidakiA,VenihakiM,MargiorisAN,StathopoulosEN,TsichlisPN,TsatsanisC.Akt1andakt2protein kinases differentially contribute tomacrophage polarization.ProcNatl Acad Sci U S A.2012;109:9517-9522
23. BabaevVR,HebronKE,WieseCB,TothCL,DingL,ZhangYM,MayJM,FazioS,VickersKC,LintonMF. Macrophage deficiency of akt2 reduces atherosclerosis in ldlr null mice. Journal of LipidResearch.2014;55:2296-2308
24. SunC,SunL,MaH,PengJ,ZhenY,DuanK,LiuG,DingW,ZhaoY.Thephenotypeandfunctionalalterations of macrophages in mice with hyperglycemia for long term. J Cell Physiol.2012;227:1670-1679

Chapter3
76
25. vanKampenE,BeaslasO,HildebrandRB,LammersB,VanBerkelTJ,OlkkonenVM,VanEckM.Orp8deficiencyinbonemarrow-derivedcellsreducesatheroscleroticlesionprogressioninldlreceptorknockoutmice.PLoSOne.2014;9:e109024
26. Ren BY, Van Kampen E, Van Berkel TJC, Cruickshank SM, Van EckM.Hematopoietic arginase 1deficiencyresultsindecreasedleukocytosisandincreasedfoamcellformationbutdoesnotaffectatherosclerosis.Atherosclerosis.2017;256:35-46
27. LammersB,ZhaoY,FoksAC,HildebrandRB,KuiperJ,VanBerkelTJC,VanEckM.Leukocyteabca1remainsatheroprotectiveinsplenectomizedldlreceptorknockoutmice.PlosOne.2012;7
28. Steinbrecher UP. Oxidation of human low-density-lipoprotein results in derivatization of lysineresidues of apolipoprotein-b by lipid peroxide decomposition products. Journal of BiologicalChemistry.1987;262:3603-3608
29. ChomczynskiP,SacchiN.Thesingle-stepmethodofrnaisolationbyacidguanidiniumthiocyanate-phenol-chloroformextraction:Twenty-somethingyearson.NatureProtocols.2006;1:581-585
30. ZhouGL,TuckerDF,BaeSS,BhathejaK,BirnbaumMJ,FieldJ.Opposingrolesforakt1andakt2inrac/paksignalingandcellmigration.JBiolChem.2006;281:36443-36453
31. Al-Jarallah A, Chen X, Gonzalez L, Trigatti BL. High density lipoprotein stimulatedmigration ofmacrophagesdependsonthescavengerreceptorclassb,typei,pdzk1andakt1andisblockedbysphingosine1phosphatereceptorantagonists.PLoSOne.2014;9:e106487
32. Zhang BG,Ma YJ, Guo H, Sun BC, Niu RF, Ying GG, ZhangN. Akt2 is required formacrophagechemotaxis.EuropeanJournalofImmunology.2009;39:894-901
33. KunjathoorVV,FebbraioM,PodrezEA,MooreKJ,AnderssonL,KoehnS,RheeJS,SilversteinR,HoffHF,FreemanMW.Scavengerreceptorsclassa-i/iiandcd36aretheprincipalreceptorsresponsiblefortheuptakeofmodifiedlowdensitylipoproteinleadingtolipidloadinginmacrophages.JBiolChem.2002;277:49982-49988
34. Krieger M, Herz J. Structures and functions of multiligand lipoprotein receptors: Macrophagescavengerreceptorsandldlreceptor-relatedprotein(lrp).AnnuRevBiochem.1994;63:601-637
35. Takahashi S, Kawarabayasi Y, Nakai T, Sakai J, Yamamoto T. Rabbit very low-density-lipoproteinreceptor - a low-density-lipoprotein receptor-like proteinwith distinct ligand specificity.PNatlAcadSciUSA.1992;89:9252-9256
36. van Tits LJH, Stienstra R, van Lent PL, Netea MG, Joosten LAB, Stalenhoef AFH. Oxidized ldlenhancespro-inflammatoryresponsesofalternativelyactivatedm2macrophages:Acrucialroleforkruppel-likefactor2.Atherosclerosis.2011;214:345-349
37. Cho H,Mu J, Kim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, 3rd, Kaestner KH, BartolomeiMS,ShulmanGI,BirnbaumMJ.Insulinresistanceandadiabetesmellitus-likesyndromeinmicelackingtheproteinkinaseakt2(pkbbeta).Science.2001;292:1728-1731
38. StrombergA,JanssonM,FischerH,RullmanE,HagglundH,GustafssonT.Bonemarrowderivedcellsinadultskeletalmuscletissueinhumans.SkeletMuscle.2013;3
39. MalletVO,MitchellC,MezeyE,FabreM,GuidottiJE,ReniaL,CoulombelL,KahnA,GilgenkrantzH.Bonemarrow transplantation inmice leads to aminor population, of hepatocytes that canbeselectivelyamplifiedinvivo.Hepatology.2002;35:799-804
40. VergadiE,VaporidiK,TheodorakisEE,DoxakiC,LagoudakiE,IeronymakiE,AlexakiVI,HelmsM,Kondili E, Soennichsen B, Stathopoulos EN, Margioris AN, Georgopoulos D, Tsatsanis C. Akt2deficiency protects from acute lung injury via alternativemacrophage activation andmir-146ainductioninmice.JImmunol.2014;192:394-406
41. Reeves PP, Wang L. Genomic organization of lps-specific loci. Curr Top Microbiol Immunol.2002;264:109-135
42. Patil PB, Sonti RV. Variation suggestive of horizontal gene transfer at a lipopolysaccharide (lps)biosynthetic locus inxanthomonasoryzaepv.Oryzae, thebacterial leafblightpathogenof rice.BmcMicrobiol.2004;4
43. Herijgers N, Van Eck M, Groot PH, Hoogerbrugge PM, Van Berkel TJ. Effect of bone marrowtransplantation on lipoprotein metabolism and atherosclerosis in ldl receptor-knockout mice.ArteriosclerThrombVascBiol.1997;17:1995-2003

Chapter3
77
44. Herijgers N, Van Eck M, Groot PH, Hoogerbrugge PM, Van Berkel TJ. Low density lipoproteinreceptorofmacrophagesfacilitatesatheroscleroticlesionformationinc57bl/6mice.ArteriosclerThrombVascBiol.2000;20:1961-1967


Chapter4
Chapter
MacrophageMKP2deficiencyisassociatedwithanM2-drivenfoamcellphenotypeandincreasesatherosclerosissusceptibilityofLDLreceptorknockoutmice
BaoyanRen1,MennoHoekstra1,JanineJ.Geerling1,PeterVanSantbrink1,RobinPlevin2,MirandaVanEck1
1Divisionof Biopharmaceutics, Cluster BioTherapeutics, LeidenAcademicCentre forDrugResearch, Leiden, TheNetherlands
2StrathclydeInstituteforPharmacyandBiomedicalSciences,UniversityofStrathclyde,Scotland,UnitedKingdom
Submittedforpublication

Chapter4
80
everal studies have established a role for mitogen-activated protein kinases (MAPK) inatherogenesis. The exact role ofMAPK phosphatase 2 (MKP2/DUSP4), aMAPK upstream
regulator,inthisprocessis,however,stillunknown.ThisstudythereforeaimedatinvestigatingtheroleofMKP2inatherosclerosisdevelopment.
MKP2deficiencyinculturedmacrophages isassociatedwith increasedJNKactivation(1.5-fold,p<0.05) and a shift towards an M2-like macrophage phenotype as compared to wild-typemacrophages.ThisisreflectedbydecreasedmRNAexpressionofiNOS(-83.7%,p<0.001)andTNF-α(-41.8%,p<0.001)andincreasedArg1(2.7-fold,p<0.001)andYM-1(2.6-fold,p<0.05)expression.MacrophageslackingMKP2exhibitincreasedexpressionlevelsofthescavengerreceptorsSR-A(2.6-fold,p<0.01)andCD36(2.2-fold,p<0.001),leadingtoanenhancedpredispositiontobecomefoamcells.TransplantationofMKP2knockoutbonemarrowintolethallyirradiatedhyperlipidemicLDLreceptorknockoutmiceconfirmedtheatheroprotectiveeffectofmacrophageMKP2.A1.3-foldincrease(p<0.05)inatheroscleroticlesionsizewasobservedinmicereconstitutedwithMKP2knockoutbonemarrowascomparedtowild-typebonemarrowrecipients.Theincreaseinlesionsizecoincidedwitha30%decrease(p<0.01)inlesionalcollagencontent,suggestingthatlossofmacrophageMKP2isassociatedwithlargerlesionswitharelativelyunstableplaquephenotype.
Inconclusion,wehaveshownthatMKP2deficiency(1)skewsculturedmacrophagestoanM2phenotype, resulting in an enhanced susceptibility to become foam cells, and (2) increasesatherosclerosissusceptibilityinvivo.
Introduction
Atherosclerosis is ahyperlipidemia-inducedchronic inflammatorydiseasecharacterizedby thedeposition of macrophage foam cells in the arterial wall.1,2 Inside atherosclerotic lesionsmacrophagepopulationswithadifferentinflammatoryphenotypecanbedistinguished,includingM1andM2macrophages.3Ingeneral,pro-inflammatoryM1macrophagesareregardedaspro-atherogenic,whileanti-inflammatoryM2macrophagesareconsideredtobeatheroprotective.4Interestingly,M2ascomparedtoM1macrophagesaremorepronetooxLDL-inducedfoamcellformation,5,6highlightingadynamicatheroscleroticroleformacrophagesubpopulations.
MAPKsasserine/threonine-specificproteinkinasesactivatevariouscellularsignalingtransductionpathwaysbyphosphorylatingdownstreamtargetgenes.7ThethreemainsubfamiliesofMAPKs,i.e.extracellularsignal-regulatedkinase(ERK),stress-activatedproteinkinase(p38)8,9andc-JunN-terminalkinase(JNK),10,11play importantrolesduringatherosclerosisdevelopment.12MAPKsactivity iscontrolledbyafamilycalledMAPKphosphatases(MKPs).MKPsinactivateMAPKsbydephosphorylating theirphosphoserine/threonineandphosphotyrosine residues.13-15TheMKPfamilycontainsatleast10well-characterizedmembers,dividedover2sub-familiesdependingontheir subcellular distribution and immediate-early or late gene regulation.16 Two membersbelonging to thesub-familyof immediate-earlygene regulatorswithanuclear localizationareMKP1andMKP2.16TheexpressionofMKP1andMKP2isinducedbyMAPKactivation,17whiletheyin turn inactivate MAPKs. Hence, they are considered the most potent regulators of theMAPK/MKPfeedbackloop.16,17
MKP2hasamolecularweightof42.9-kDaandisubiquitouslyexpressedinvarioustissues.18,19Itsexpressionisinducedinresponsetogrowthfactors,20hormones,21oxidativestress,22,23UVlight19andlipopolysaccharides(LPS).24MKP2expressionisalsohighlyresponsivetocholesterol-richdiet
S

Chapter4
81
andfattyacids.25ArecentstudyshowedthatMKP2expressionisstronglyregulatedinactivatedmacrophages.26 However, so far many contradictory findings on the effect of MKP2 onmacrophage function have been described. In response to LPS, bone marrow-derivedmacrophages lackingMKP2acquireanM2-likemacrophagephenotype,reflectedbyenhancedArg1anddecreasediNOSactivities.27Thisphenotypewasnotalwayssupportedbythecytokineproductionprofile;Al-Mutairietal.foundapotentiatedpro-inflammatorycytokineproduction,whileCornelletal.foundattenuatedpro-inflammatorycytokineproductionbyMKP2knockoutmacrophages in response to LPS27,28 Additionally, overexpression of MKP2 in macrophagessignificantlydecreasedJNKactivationandtheexpressionofinflammatorymediators.29
Interestingly,apro-atherogenicfunctionofMKP1wasrecentlyestablishedinmice.30However,therole of MKP2 in the pathogenesis of atherosclerosis is still unclear. In the current study, wethereforeinvestigatedtheroleofMKP2inmacrophagepolarizationandfoamcellformation invitroandevaluatedtheimpactofmacrophageMKP2deficiencyonatherosclerosissusceptibilityinvivo.
Materialsandmethods
Animals
Breeding pairs of C57BL/6 wild-type (WT) mice and MKP2 knockout (MKP2 KO) mice wereobtainedfromthePhysiology&PharmacologylaboratoriesinGlasgow,UnitedKingdom.27Low-density lipoproteinreceptorKO(LDLrKO)micewereboughtfromTheJacksonLaboratory(BarHarbor,ME,USA)andexpandedlocallyattheGorlaeusLaboratories,Leiden,TheNetherlands.Allanimalworkwasapprovedby theDutchEthicsCommitteeand regulatoryauthorityat LeidenUniversityandwascarriedoutincompliancewithDutchgovernmentguidelinesandtheDirective2010/63/EUoftheEuropeanParliamentontheprotectionofanimalsusedforscientificpurposes.
Isolationofthioglycollate-elicitedperitonealmacrophages
WTandMKP2KOmicewereintraperitoneallyinjectedwith3%Brewer’smodifiedthioglycollatemedium(Becton,DickinsonandcompanySparks,MD,USA;Productnumber:211716) toelicitmacrophageinfiltrationintotheperitonealcavity.Fivedayslater,peritonealmacrophageswereharvestedthroughperitoneallavagewithPBS.CellswereculturedovernightincompleteDMEMmedium(LonzaWalkersville,USA,catalognumber:BE12-708F)containing10%fetalcalfserum(HyClone™ Calf Serum (U.S.), catalog number: SH30073.03). Non-adherent cellswerewashedawaytoacquiretheperitonealmacrophageculturesusedforfurtherresearch.
PhosphorylationlevelsofERK,JNK,p38MAPKdeterminationbyELISAassay
A total of 30.000 peritoneal macrophages were plated per well in a 96-wells culture plate.PhosphorylationlevelsofMAPK(P38,ERK,JNK)wereanalysedafter24hoursusingacell-basedELISA kit (RayBiotech, Norcross, GA, USA. catalog number: CBEL-ERK-SK) according to themanufacturer’sinstructions.Absorbanceswerereadat450nmand570nmusingaplatereader(modelPowerWave340,Biotek,USA).

Chapter4
82
mRNAexpressiondeterminationusingReal-timePCR
TotalRNA isolationandcDNAsynthesiswasperformedasdescribedpreviously.31,32ThemRNAexpressionofgenesofinterestwasdeterminedusinga7500FastReal-TimePCRsystem(AppliedBiosystems,CA)usingSYBRgreen(GCBiotech,catalognumber:QT625-20supplier)technology.Theaveragecyclethreshold(CT)ofRPL27and36B4wasusedashousekeepingcontrol.
Lipidassays
Plasma triglycerides, total cholesterol and free cholesterol were determined using standardenzymaticcolorimetricassays.ThetriglyceridescolorimetricassaykitwasobtainedfromRocheDiagnostics(catalognumber:11488872216).Thecholesterolassaywasperformedasdescribedpreviously.33 Precipath control serum (Roche, catalog number:11285874122) was used asstandardfortheassays.Thedistributionofcholesteroloverthedifferentlipoproteinclasseswasassessed by fast protein liquid chromatography using a Superose 6 column (GE Healthcare,Uppsala,Sweden).
Cytokineproteinmeasurements
TNF-α, IL-6, IL-10, IL-12p40, and MCP-1 protein levels were measured by enzyme-linkedimmunosorbent assays (BD Biosciences Pharmingen, San Diego, CA) according to themanufacturer’sprotocols.
HematologyanalysisandFlowCytometryAnalysis
Total leukocytesand leukocytesubtypeswereanalyzedusinganautomatedSysmexXT-2000iVVeterinaryHaematologyanalyzer(SysmexCorporation).Fluorescence-activatedcellsorting(FACS)analysis was performed on a FACS Canto II machine (BD Biosciences, CA, USA) using FACSantibodies(eBioscience).
BoneMarrowTransplantation
LDLrKOrecipientmicewereexposedto9GyX-rayirradiation34todestroytheendogenousbonemarrow.Onedayafter, five-millionbonemarrowcells, freshly isolated fromWTandMKP2KOdonormice,weretransplantedintothelethallyirradiatedLDLrKOrecipientsviatailveininjection.Bonemarrowrecipientmicewereallowedtorecoverfor8weeksonaregularchowdiet,afterwhichtheywereswitchedtoaWestern-typediet(WTD)for9weekstoinducethedevelopmentofatheroscleroticlesions.
HistologicalAnalysisoftheAorticRoot
Atsacrifice,miceweresubjectedtowholebodyperfusionwithPBS.Subsequently,heartswereisolatedandfixedin4%Formal-Fixxbuffer(ThermoScientific™Shandon™)for24hours,beforeembedding in Tissue-Tek O. C. T. compound (Sakura Finetek, USA) overnight. Ten-microncryosectionsoftheaorticrootwerecutusingaLeicaCM3050scryostat.Atherosclerotic lesionarea(inµm2)andlesionalcollagenandmacrophagecontentweredeterminedbyrespectivelyOilredOstaining,Masson’sTrichrome(MTC)stainingandMOMA-2immunohistochemicalstaining,respectively (dilution1:50,ResearchDiagnostics Inc).Allquantificationanalysisof thesectionswasperformedbyablindedoperatorusingtheLeicaimageanalysissystem(LeicaLtd,Cambridge,UK).

Chapter4
83
StatisticalAnalysis
Dataareexpressedasmeans±SEM.Statisticalsignificantdifferencesbetweenthegroupsweredetermined by two-tailed unpaired Student’s t-test or 2-way ANOVA using GraphPad Prismsoftware(GraphPadSoftwareInc.,SanDiego,California,USA).Welchcorrectionwasappliedtothe t-test in the case of unequal variances in the dataset. A p value of <0.05was consideredstatisticallysignificant.
Results
LossofMKP2inmacrophagesleadstoenhancedJNKactivation
TostudytheroleofMKP2inmacrophageMAPKactivation,thephosphorylationstatusofJNK,p38and ERK1/2 (hereafter referred to as ERK) were determined. Loss of MKP2 in macrophagesresultedinanenhancedactivationofJNK(1.5-fold,p<0.05;Figure1A),whereasp38activationwasdecreased inMKP2KOmacrophages as compared toWTmacrophages (-25.5%,p<0.001;Figure1B).MacrophageERKactivationwasnotaffectedbyMKP2deficiency(p>0.05;Figure1C).
Figure1MKP2deficiencyalterstherelativephosphorylationstatusofMAP-kinasesinperitonealmacrophages
PhosphorylatedandtotalJNK,p38,andERKproteinlevelsweremeasuredbyELISAinthioglycollate-elicitedperitonealmacrophagesfromWTandMKP2KOmice.Dataareexpressedasopticaldensity(OD)ratiosofA)phosphorylatedJNK/total JNK, B) phosphorylated P38/total P38 and C) phosphorylated ERK/total ERK. *p<0.05, ***p<0.001, nsp>0.05.
MKP2deficiencyskewsmacrophagestoanM2-likephenotype
JNKandp38areinvolvedinmacrophagepolarization.35-37AsMKP2deficiencyaffectedbothJNKand p38 activation,we next investigatedwhetherMKP2 deficiency also affectedmacrophagepolarization.RelativemRNAexpressionlevelsoftheM1markersiNOS(-83.7%,p<0.001)andTNF-α(-41.8%,p<0.001)weresignificantlydecreasedinMKP2KOmacrophagesascomparedtoWTmacrophages(Figure2A-B).Incontrast,mRNAexpressionoftheM2markersArg1andYM-1wassignificantly increased inMKP2KOmacrophagesascomparedtoWTmacrophages,by2.7-fold(p<0.001;Figure2C)and2.5-fold(p<0.05;Figure2D)respectively.Collectively,thesedatasuggestthatlossofMKP2polarizesmacrophagestowardsanM2phenotype.
A C B
W T M K P 2 K O0 .0
0 .1
0 .2
0 .3
0 .4
E R K
OD
p-E
RK
/OD
ER
K
ns
W T M K P 2 K O0 .0
0 .2
0 .4
0 .6
p 3 8
OD
p-p
38
/OD
p3
8 ***
W T M K P 2 K O0
1
2
3
J N K
OD
p-J
NK
/OD
JN
K
*

Chapter4
84
Figure 2 Thioglycollate-elicited macrophageslacking MKP2 are skewed towards an M2-likephenotype.Relative mRNA expression levels of the M1macrophagemarkersA)iNOSandB)TNF-αandtheM2 macrophage markers C) Arg1 and D) YM-1were measured by quantitative PCR. *p<0.05,**P<0.01,***p<0.001.
MKP2 deletion upregulates the expression of lipid uptake genes and enhancesmacrophage
foamcellformation.
ExpressionofSR-AandCD36,themainreceptorsforuptakeofmodifiedLDL,areupregulatedinM2macrophages.6,38Asaresult,M2macrophagesaremoresusceptibletofoamcellformation.39Therefore, we hypothesized that MKP2 deletion in macrophages, via stimulation of M2polarization,islikelytopredisposemacrophagestofoamcellformation.InagreementwiththemoreM2-likemacrophagephenotype,SR-A(2.6-fold;p<0.01)andCD36(2.2-fold;p<0.001)mRNAexpressionwashigherinMKP2KOmacrophages,ascomparedtoWTmacrophages(Figure3A-B).ThemRNAexpressionofABCA1,ABCG1andHMG-CoAreductaseweresimilarbetweenthetwogenotypes(p>0.05;Figures3C-E),suggestingthatcholesteroleffluxandendogenouscholesterolsynthesiswerelikelynotaffectedbythelossofMKP2.Probably,asaresultoftheaugmentedSR-AandCD36expression,MKP2KOmacrophagesalreadyformedfoamcellswhenmaintainedin10%fetal calf serum-containing culture medium for 24 hours; an effect barely found in WTmacrophagesunderthesamecultureconditions(Figure3G).ThissupportsthehypothesisthatMKP2lossinmacrophagespredisposesthecellstobecomefoamcells.Correspondingly,wealsoobservedmoreextensivefoamcellformationinMKP2KOmacrophages,ascomparedtoMKP2KOmacrophagesafteroxLDLtreatment(Figure3G).
W T M K P 2 K O0 .0 0 0 0
0 .0 0 0 1
0 .0 0 0 2
0 .0 0 0 3
0 .0 0 0 4
iN O SR
ela
tiv
e e
xp
res
sio
n
***
W T M K P 2 K O0 .0 0 0
0 .0 0 4
0 .0 0 8
0 .0 1 2
T N F -a
Re
lati
ve
ex
pre
ss
ion
***
W T M K P 2 K O0 .0 0 0
0 .0 0 2
0 .0 0 4
0 .0 0 6
0 .0 0 8
A rg 1
Re
lati
ve
ex
pre
ss
ion **
W T M K P 2 K O0 .0 0 0
0 .0 0 1
0 .0 0 2
0 .0 0 3
0 .0 0 4
Y M -1R
ela
tiv
e e
xp
res
sio
n
*
A B
C D

Chapter4
85
Figure3MKP2deficiencypredisposesthioglycollate-elicitedmacrophagestofoamcellformation.
Cellswere incubatedinmediumwithorwithout10µg/mLoxLDLfor24hours.RelativemRNAexpressionlevelsofthecholesterol uptake genes A) SR-A and B) CD36, thecholesterol efflux genes C) ABCA1 and D) ABCG1, and E)HMG-Coa reductase (HMGCR) that mediates de novocholesterol synthesis, weremeasured by quantitative PCRundernon-lipidloadingcondition.G)LipidaccumulationwasvisualizedbyOilredOstaining.*p<0.05,nsp>0.05.
HematopoieticMKP2deficiency in LDLrKOmicedoesnotalterplasma lipidprofileorwhitebloodcellcounts
Toverifythepotentialpro-atherogeniceffectofmacrophageMKP2deficiencyonatherosclerosisdevelopment invivo, lethally irradiatedLDLrKOmice,transplantedwithWTorMKP2KObonemarrow,werechallengedwithWTDfor9weeksto inducethedevelopmentofatheroscleroticlesions. HematopoieticMKP2 deficiency did not impact on plasma free cholesterol and totalcholesterol levels (p>0.05, Figure 4A) or to the distribution of cholesterol over the differentlipoproteinclasses(Figure4B).Thetotalbloodleukocytecountwasalsonotdifferentbetweenthetwogroupsofbonemarrowrecipients (p>0.05,Figure4C).Wefurtheranalysedthebloodleukocytecompositionprofileusingflowcytometry.Nodifferencewasfoundinthepercentageofneutrophils,totalmonocytes,Ly6ClowpatrollingmonocytesorLy6Chipro-inflammatorymonocytes(p>0.05,Figure4D).Representativeflowcytometricplotsofcirculatingleukocytesareshowninfigure4E.
PBS oxLDL
WT
MKP
2KO
G
W T M K P 2 K O0 .0 0
0 .0 1
0 .0 2
0 .0 3
0 .0 4
0 .0 5
A B C A 1
Re
lati
ve
ex
pre
ss
ion p = 0 .0 9
W T M K P 2 K O0 .0 0
0 .0 1
0 .0 2
0 .0 3
0 .0 4
A B C G 1
Re
lati
ve
ex
pre
ss
ion
ns
W T M K P 2 K O0 .0 0 0
0 .0 0 5
0 .0 1 0
0 .0 1 5
H M G -C o A
Re
lati
ve
ex
pre
ss
ion
ns
C D E
W T M K P 2 K O0 .0
0 .5
1 .0
1 .5
2 .0
C D 3 6
Re
lati
ve
ex
pre
ss
ion *
W T M K P 2 K O0 .0
0 .1
0 .2
0 .3
0 .4
0 .5
S R -A
Re
lati
ve
ex
pre
ss
ion *
A B
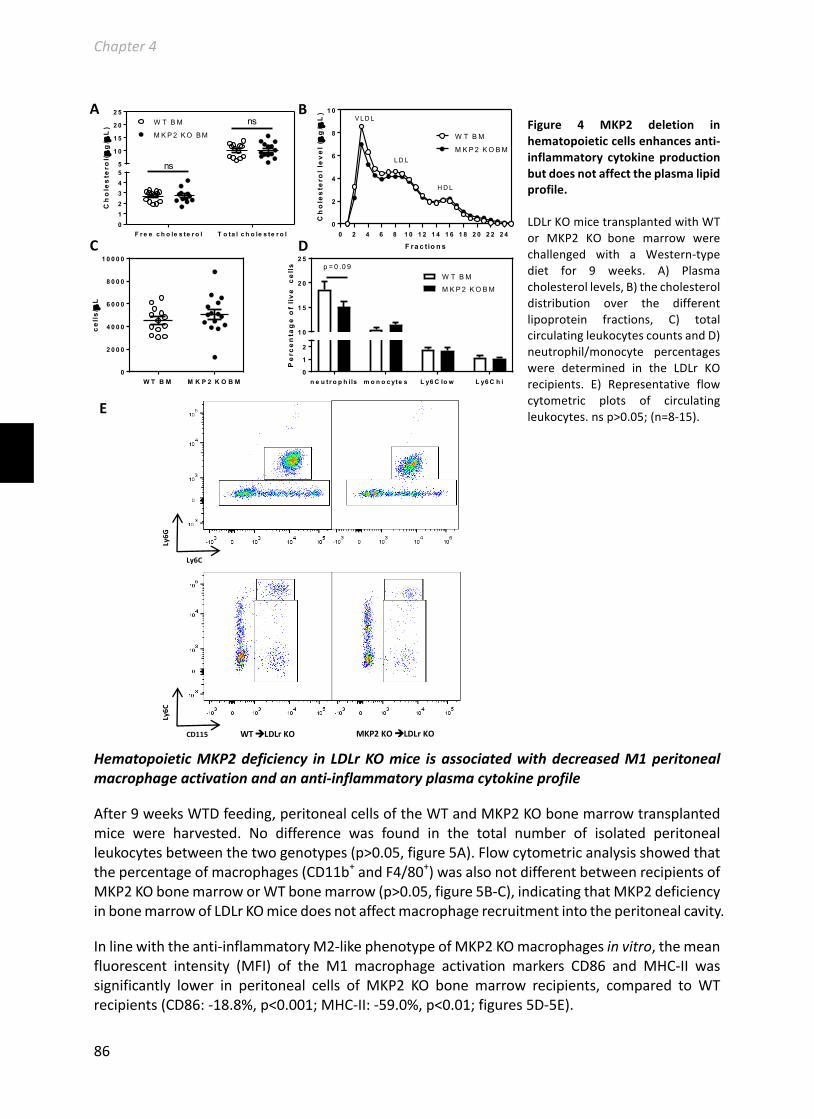
Chapter4
86
Figure 4 MKP2 deletion inhematopoieticcellsenhancesanti-inflammatory cytokineproductionbutdoesnotaffecttheplasmalipidprofile.
LDLrKOmicetransplantedwithWTor MKP2 KO
bone marrow were
challenged with a Western-typediet for 9 weeks. A) Plasmacholesterollevels,B)thecholesteroldistribution over the differentlipoprotein fractions, C) totalcirculatingleukocytescountsandD)neutrophil/monocyte percentageswere determined in the LDLr KOrecipients. E) Representative flowcytometric plots of circulatingleukocytes.nsp>0.05;(n=8-15).
HematopoieticMKP2deficiency in LDLrKOmice isassociatedwithdecreasedM1peritonealmacrophageactivationandananti-inflammatoryplasmacytokineprofile
After9weeksWTDfeeding,peritonealcellsoftheWTandMKP2KObonemarrowtransplantedmice were harvested. No difference was found in the total number of isolated peritonealleukocytesbetweenthetwogenotypes(p>0.05,figure5A).Flowcytometricanalysisshowedthatthepercentageofmacrophages(CD11b+andF4/80+)wasalsonotdifferentbetweenrecipientsofMKP2KObonemarroworWTbonemarrow(p>0.05,figure5B-C),indicatingthatMKP2deficiencyinbonemarrowofLDLrKOmicedoesnotaffectmacrophagerecruitmentintotheperitonealcavity.
Inlinewiththeanti-inflammatoryM2-likephenotypeofMKP2KOmacrophagesinvitro,themeanfluorescent intensity (MFI) of the M1 macrophage activation markers CD86 and MHC-II wassignificantly lower in peritoneal cells of MKP2 KO bone marrow recipients, compared toWTrecipients(CD86:-18.8%,p<0.001;MHC-II:-59.0%,p<0.01;figures5D-5E).
n e u tr o p h ils m o n o c yte s L y6 C lo w L y6 C h i0
1
2
1 0
1 5
2 0
2 5
Pe
rce
nta
ge
of
liv
e
ce
lls
W T B M
M K P 2 K O B M
p = 0 .0 9
F r e e c h o le s te r o l T o ta l c h o le s te r o l0123455
1 0
1 5
2 0
2 5C
ho
les
tero
l(µ
g/µ
L) W T B M
M K P 2 K O B M
ns
ns
H D L
L D L
V L D L
0 2 4 6 8 1 0 1 2 1 4 1 6 1 8 2 0 2 2 2 40
2
4
6
8
1 0
F ra c tio n s
Ch
ole
ste
rol
lev
el
(µg
/µL
)
W T B M
M K P 2 K O B M
A B
W T B M M K P 2 K O B M0
2 0 0 0
4 0 0 0
6 0 0 0
8 0 0 0
1 0 0 0 0
cell
s/µ
L
C D
E
Ly6C
Ly6G
WTàLDLrKO MKP2KOàLDLrKO CD115
Ly6C

Chapter4
87
Wenextinvestigatedplasmacytokinelevelsinthetwogroupsofbonemarrowtransplantedmiceafter9weeksWTDfeeding.LDLrKOmicewithMKP2KObonemarrowshowedastriking6.9-foldhigher plasma level of the anti-inflammatory cytokine IL-10 as compared to LDLr KO micereconstitutedwithWTbonemarrow(817.3±106.1pg/mLforMKP2KOversus118.4±35.68pg/mLforWT;p<0.001;Figure5F).Infurthersupportofamoreanti-inflammatoryphenotype,MKP2KObone marrow transplanted mice exhibited decreased plasma levels of the pro-inflammatorycytokineIL-12p40ascomparedtotheirWTbonemarrowrecipientcontrolmice(8.5±1.6pg/mLforMKP2KOversus30.3±5.3pg/mLforWT;p<0.01;Figure5G).
Figure5HematopoieticMKP2deficiencyinLDLrKOmiceisassociatedwithdecreasedM1macrophageactivationintheperitonealcavity. LDLrKOmicereconstitutedwithWTorMKP2KObonemarrowwerechallengedwithaWestern-typedietfor9weeks.Peritoneal leukocyte populationswere analyzedby flow cytometry. A) Total peritoneal leukocytes. B) Peritonealmacrophages as percentage of total leukocytes. C) Representative flow cytometric plots of CD11b+ / F4/80+
macrophagesandgatingstrategyusedintheanalysis.D)MedianfluorescenceintensityofCD86andE)MHC-IIinWT(open bars) andMKP2 KO peritonealmacrophages (closed bars). Plasma IL-10 (F) and IL-12p40 (G) levels weredeterminedbyELISA.**p<0.01,***p<0.001,nsp>0.05;(n=8-15).
A B D
MKP2KOBM WTBM
Comp-Pacific Blue-A :: CD11Bx
Comp-PE-Cy7-A :: CD19
FSC-Ax
Comp-APC-Cy7-A :: LIVE_DEAD
FSC-Ax
FSC-H
Timex
FSC-A
0 103
104
105
106
Comp-Pacific Blue-A :: CD11B
0
-103
103
104
105
Com
p-AP
C-A
:: F
4_80
Gatingstrategy
W T B M M K P 2 K O B M0
2 0 0 0
4 0 0 0
6 0 0 0
8 0 0 0
C D 8 6
MF
I
***
W T B M M K P 2 K O B M0
2 0 0 0
4 0 0 0
6 0 0 0
8 0 0 0
M H C -IIM
FI
**
E C
W T B M M K P 2 K O B M0
5 0 0
1 0 0 0
1 5 0 0
2 0 0 0
2 5 0 0IL 1 0
Co
nc
en
tra
tio
n (
pg
/ml)
***
W T B M M K P 2 K O B M0
2 0
4 0
6 0
8 0
1 0 0IL 1 2 p 4 0
Co
nc
en
tra
tio
n (
pg
/ml)
**
F G
W T B M M K P 2 K O B M0
2 0
4 0
6 0
8 0
F 4 /8 0
Pe
rce
nta
ge
of
liv
e
ce
lls ns
W T B M M K P 2 K O B M0
2´1 0 0 6
4´1 0 0 6
6´1 0 0 6
8´1 0 0 6
P e r ito n e a l le u k o c y te s
Ce
lls
pe
r m
ou
se
ns
CD11b
F4-80

Chapter4
88
HematopoieticMKP2deficiency in LDLrKOmice isassociatedwith increasedatheroscleroticlesiondevelopment
Atherosclerotic lesionsizewasanalysed inOil red-Ostainedsectionsof theaortic rootafter9weeksWTD feeding.Bonemarrow-specificMKP2deficiencywasassociatedwithan increasedatheroscleroticlesionsize(2.1±0.2×105µm2forMKP2KOrecipientsversus1.6±0.1×105µm2forWTrecipients;p<0.05;Figure6A).Nodifference in lesionalmacrophagecontentwasobservedbetweenbothgroups(Figure6B).However,comparedtoWTbonemarrowrecipientmice,MKP2KObonemarrowtransplantedmicedidshowalowerlesionalcollagencontent(27±2%forMKP2KOversus38±3%forWT;p<0.01;Figure6C),suggestingthatlossofMKP2inbonemarrow-derivedcellsisassociatedwithlargerlesionswithalessstablephenotype.
Figure6HematopoieticMKP2deletioninLDLrKOmicepromotesatherosclerosis. LDLrKOmicereconstitutedwithWTorMKP2KObonemarrowwerechallengedwithaWestern-typedietfor9weeks.A)OilredO-positiveatheroscleroticlesionareawithintheaorticroot.B)Lesionalmacrophagecontent.C)Plaquecollagencontent.*p<0.05,**p<0.01,nsp>0.05.
A
B
C
OilredO
MKP2KOBM WTBM
W T B M M K P 2 K O B M0
2 0
4 0
6 0
8 0
1 0 0M a c r o p h a g e a r e a %
Mac
rop
hag
e (%
lesi
on
are
a)
ns
W T B M M K P 2 K O B M0
2 0
4 0
6 0
8 0
1 0 0P la q u e c o lla g e n %
Co
llag
en (
% le
sio
n a
rea)
**
L e s io n a r e a
Le
sio
n a
rea
(´
105µ
m2
)W T B M M K P 2 K O B M
0
1
2
3
4
5
*
MoMa-2
MKP2KOBM WTBM
MTC
MKP2KOBM WTBM

Chapter4
89
PeritonealmacrophageM1markersarenegativelycorrelatedtoaortic lesionsizeinLDLrKO
recipients
TogaininsightinthepossiblemechanismunderlyingtheincreasedatherosclerosissusceptibilityofLDLrKOmicetransplantedwithMKP2KObonemarrow,atheroscleroticlesionsizewasplottedagainstthecytokineconcentrationsinplasma,ortheexpressionoftheM1macrophagemarkersonperitonealcells.Theconcentrationofanti-inflammatorycytokinesIL-10andIL-12intheplasmaoftherecipientmicedidnotcorrelatewiththeatheroscleroticlesionsizeintheaorticroot(p>0.05,figure7Aand7B).However,theMFIoftheM1macrophagemarkersCD86andMHCIIwerefoundtonegativelycorrelatewithatheroscleroticlesionsizeintheLDLrKOrecipients(r=-0.584,p<0.05forCD86;r=-0.6963,p<0.01forMHCII).
Figure7ExpressionofperitonealmacrophageM1markersarenegativelycorrelatedtoaorticlesionsizeinLDLrKOrecipients CorrelationanalysisontheplasmaconcentrationofIL-10(A)andIL-12(B)andperitonealmacrophageexpressionofCD86(C)andMHC-II(D)withaorticrootatheroscleroticlesionsize.WhitedotsrepresentindividualWTbonemarrowtransplantedmice,whileblackdotsrepresentindividualMKP2KObonemarrowrecipientmice.Solidlinesrepresentregressionlines.Dottedlinesrepresentthe95%confidenceintervalforindividualpredictions.(n=16-25).
Discussion
In the current study,we investigated the role ofMKP2 inmacrophage polarization, foam cellformation and atherosclerosis. Deletion of MKP2 in macrophages is associated withhyperactivation of the MAPK family member JNK, enhanced polarization towards an M2-likephenotypeandaggravatedfoamcellformation invitro.Correspondingly,bonemarrow-specificMKP2deficientmiceshowedacceleratedatheroscleroticlesiondevelopmentinvivo.
MKP2 is a member of the subclass of nuclear inducible MKPs, which also contains MKP1.HematopoieticdeletionofMKP1inducesatherosclerosissusceptibilityofLDLrKOmicetoasimilar
r 0.2639 P value summary ns
0 5 0 0 1 0 0 0 1 5 0 0 2 0 0 00
1´1 0 0 5
2´1 0 0 5
3´1 0 0 5
4´1 0 0 5
IL 1 0 (p g /m L )
Le
sio
n s
ize
(µ
m2
)
W T B M M K P 2 K O B M
0 2 0 4 0 6 00
1´1 0 0 5
2´1 0 0 5
3´1 0 0 5
4´1 0 0 5
IL 1 2 (p g /m L )
Le
sio
n s
ize
(µ
m2
)
W T B M M K P 2 K O B M
P value summary ns r -0.2932
P value .018
4 0 0 0 5 0 0 0 6 0 0 0 7 0 0 0 8 0 0 0 9 0 0 0-1´1 0 0 5
0
1´1 0 0 5
2´1 0 0 5
3´1 0 0 5
C D 8 6 (M F I)
Le
sio
n s
ize
(µ
m2
)
W T B M M K P 2 K O B M
r -0.584 P value 0.0039
0 2 0 0 0 4 0 0 0 6 0 0 0 8 0 0 0 1 0 0 0 0-1´1 0 0 5
0
1´1 0 0 5
2´1 0 0 5
3´1 0 0 5
M H C -II (M F I)
Le
sio
n s
ize
(µ
m2
)
W T B M M K P 2 K O B M
r -0.6963
A B
D C

Chapter4
90
extendaswhatweshowforhematopoieticMKP2deletion.40TheeffectsofMKP1deletionwereprimarilyattributedtoeffectsonmonocytemigration,inresponsetoMCP1.Inthecurrentstudy,we showed that hematopoietic MKP2 deletion did not affect blood monocyte counts andphenotypenormacrophagemigration, indicatingthathematopoieticMKP1andMKP2,despitetheirsimilarMAPKinactivatingfunction,differentiallyinfluenceatherosclerosis.
MKP2ishighlyhomologouswithMKP1attheC-terminalcatalyticdomain,41buttheirN-terminaldomains are less closely related.19 The unique sequence in the N-terminal domain of MKPsdeterminetheirMAPKsubstratepreference.42MKP1isabletodephosphorylateallthreeMAPKs,butitseffectsonatherogenesisarelikelythroughdeactivationofJNKandp38.43,44MKP2appearstohaveonlyERK2andJNKaspreferredsubstratesbutnotp38.19Notably,ourcurrentfindings,togetherwithpreviouslypublishedstudies,indicatethatthesubstratepreferenceofMKP2iscell-typedependent.
Here we show that deletion of MKP2 in thioglycollate-elicited macrophages stimulated JNKactivation.Inline,arecentlypublishedstudybyMashaeletal.showedthat,uponLPSstimulation,JNK activation was largely increased in bone marrow-derived macrophages lacking MKP2.45However, the JNK activation status was not changed under basal conditions in this type ofmacrophages.45Thisisprobablyduetoaphenotypicdifference,asnaïvebonemarrow-derivedmacrophages are monocyte-like, while peritoneal macrophages are activated in response tothioglycollate.46 In agreement with the Mashael et al. study,27 ERK activation in peritonealmacrophageswasnotaffectedintheabsenceofMKP2.Wedidalsoobserveadecreaseinp38phosphorylation on the MKP2 KO macrophages. This is, however, most likely not a directconsequenceoftheMPK2loss,sinceMKPsdeactivateMAPKs.JNKandp38negativelyregulateeachother’sactivationinmanycelltypes,47,48includingthioglycollate-elicitedmacrophages.49Assuch,thedecreasedphosphorylationofp38inMKP2KOmacrophagescanperhapsbeattributedto the enhanced JNK activation. Taken together, our study suggests that JNK is the preferredsubstrateofMKP2inmurinemacrophages.
Macrophage polarization is extensively regulated by phosphorylation and subsequentdephosphorylationofproteinsinvolvedincellsignaltransductionpathways.Associatedwiththeaugmented JNK activation in peritoneal macrophages lacking MKP2, the expression of M1signaturegeneswasdownregulatedandM2signaturegenesupregulated,suggestinganM2-likephenotype.Similartoourstudy,Stuartetal.,alsofoundenhancedexpressionandactivityoftheM2markersArg1innaiveMKP2deficientbonemarrow-derivedmacrophages.However,otherinvitro studies showed that JNK activation is enhanced and prolonged during LPS-induced M1macrophage polarization50,51 Correspondingly, JNK deficient macrophages display a lowerexpression of M1-associated pro-inflammatory cytokines and chemokines upon LPSstimulation.29,35Theseresultsareincontrasttoourfindings,probablybecauseinthelatterstudiesthe macrophages were stimulated with LPS, an extremely potent stimulator, that skews M2macrophagestowardsanM1phenotype.52LPSbindingtotoll-likereceptor2stimulatestheJNKactivation-induced production of pro-inflammatory cytokines.53 In agreement, Mashael et al.showedapro-inflammatory cytokineproductionprofile in the “M2-like”MKP2deficientbonemarrow-derivedmacrophagesuponstimulationwithLPS.27ThisindicatesthatlossofMKP2mightleadtoamacrophagephenotypethat ishyper-responsivetoLPSstimulation.Moreresearch isneededtoconfirmthishypothesisandtoaddresstheexactmechanismofhowMKP2regulatesmacrophagepolarization.

Chapter4
91
Macrophagephenotype isan important factordeterminingatherosclerosis susceptibility.54M2macrophages were considered as promising therapeutic targets for the treatment ofatherosclerosisduetotheiranti-inflammatoryproperties.55However,recentevidencesuggeststhatM2macrophagesaremoresusceptibletofoamcellformation.39,56Inthecurrentstudy,forthefirsttimeastrikingeffectofMKP2deficiencyonmacrophagefoamcellformationwasshown.M2 macrophages are more susceptible to oxLDL-induced foam cell formation57 due to anincreased expression of the scavenger receptors CD36 and SR-A.38,58 SR-A and CD36 areresponsibleforupto90%oftheoxLDLuptakebymacrophagesinvitro.59InlinewiththeM2-likephenotypeofMKP2KOmacrophages,theexpressionofCD36andSR-A,aswellastheassociatedfoamcellformation,wereinducedintheabsenceofmacrophageMKP2.JNKactivationstimulatesoxLDL-induced foam cell formation by inducing the CD36 / JNK / SR-A pathway.60 Hence, wespeculatethattheMKP2deficiency-inducedmacrophagefoamcellformationisJNK-dependentandactslikelythroughtheCD36/JNK/SR-Apathway(Figure8,leftpanel).
InlinewiththeobservationthatMKP2KOmacrophagesweremorepronetodevelopintofoamcells,bonemarrow-specificdeletionofMKP2increasedatheroscleroticlesiondevelopmentintheaorticrootofLDLrKOmice.Interestingly,althoughtheatheroscleroticlesionswerelargeruponMKP2 deletion in bone marrow-derived cells, the collagen content was lower. This can beexplained by a previous finding showing that M2 macrophages are responsible for collagendegradation.61 In linewith theanti-inflammatorypropertiesofM2macrophages,plasma IL-10levelswerehighlyelevated,whileplasmaIL-12p40levelsweredecreasedintheMKP2KObonemarrow recipients as compared to WT bone marrow recipients. IL-10 is a prototypic anti-inflammatory cytokine, andalthough it hasbeen shown to stimulateoxLDL-induced foamcellformation,itisingeneralconsideredtobeathero-protective.62,63Therefore,theincreasedIL-10production in the current study is unlikely to have contributed to the increased lesion sizesobserved in MKP2 KO bone marrow transplanted mice. This is supported by the correlationanalysis,whichshowedthattheM1macrophagemarkerexpression,ratherthanplasmaIL-10/12cytokinelevels,issignificantlyassociatedwithatheroscleroticlesionsizeintheaorticroot.Assuch,weanticipatethattheM2polarizationandenhancedfoamcellformation,ratherthanthereducedinflammation,underliestheaugmentedatherosclerosisdevelopmentduetoMKP2loss(Figure8,rightpanel).
Inadditiontomacrophages,MKP2isalsoexpressedinotherbonemarrow-derivedcells,includingBcells,Tcells,anddendriticcells.64-66AlthoughinourbonemarrowtransplantationmodelMKP2deficiencydidnotinfluenceBcellandTcellcounts,wecannotruleoutapotentialcontributionofMKP2inthesecellstotheprotectionagainstatherosclerosis.
Inconclusion,wehaveshownthat(1)MKP2deficiencypredisposesinvitroculturedmacrophagestoacquireanM2phenotype,resultinginanenhancedsusceptibilitytobecomefoamcells,and,(2)MKP2deficiencyinbonemarrow-derivedcellsenhancesthesusceptibilitytoatheroscleroticlesiondevelopmentinvivo(Figure8).

Chapter4
92
Figure8DiagramofMKP2effectsdeletiononmacrophageactivationandatherosclerosis. MKP2 deficiency in macrophages stimulates JNK activation, leading to M2 polarization. CD36 expression isupregulatedinM2macrophageswhichinturnstimulatesJNKphosphorylationandsubsequentSR-Aupregulation.UnderoxLDLconditions,stimulationoftheCD36/JNK/SR-Apathwaypromotesfoamcellformation.MKP2inducedM2 macrophage and foam cell formation overweigh the athero-protective role of elevated anti-inflammatorycytokineproduction,promotingatherosclerosisdevelopmentinvivo.
Acknowledgments
TheauthorsthankFrankSchaftenaar,GijsvanPuijvelde,RonaldvanderSluis,AmberOuweneel,HiddeDouna,MaraKröner,RickvanderGeest,LidewijdeLeeuw,JoyaNahon,OlgaSnip,RenataMartinsCardosoandHenrikeKerbstadtforthetechnicalassistanceandhelpfuldiscussions.
Sourcesoffunding
Thisworkissupportedbythe‘NetherlandsCardioVascularResearchInitiative:theDutchHeartFoundation,DutchFederationofUniversityMedicalCenters, theNetherlandsOrganization forHealth Research and Development, and the Royal Netherlands Academy of Sciences for theGENIUSproject“Generatingthebestevidence-basedpharmaceuticaltargetsforatherosclerosis”(CVON2011-19)andtheNetherlandsOrganizationforScientificResearch(VICIGrant91813603(M.V.E). M.V.E. is an Established Investigator of the Netherlands Heart Foundation (Grant2007T056).B.RenwassupportedbytheChinaScholarshipCouncil.
Disclosures
Noconflictsofinterest,financialorotherwise,aredeclaredbytheauthors.

Chapter4
93
References
1. LibbyP.Inflammationinatherosclerosis.ArteriosclerThrombVascBiol.2012;32:2045-20512. Bobryshev YV. Monocyte recruitment and foam cell formation in atherosclerosis. Micron.
2006;37:208-2223. MooreKJ,TabasI.Macrophagesinthepathogenesisofatherosclerosis.Cell.2011;145:341-3554. JohnsonJL,NewbyAC.Macrophageheterogeneity inatheroscleroticplaques.CurrOpinLipidol.
2009;20:370-3785. vanTitsLJ,StienstraR,vanLentPL,NeteaMG,JoostenLA,StalenhoefAF.Oxidizedldlenhances
pro-inflammatoryresponsesofalternativelyactivatedm2macrophages:Acrucialroleforkruppel-likefactor2.Atherosclerosis.2011;214:345-349
6. Canton J, Neculai D, Grinstein S. Scavenger receptors in homeostasis and immunity. Nat RevImmunol.2013;13:621-634
7. ZhangW,LiuHT.Mapksignalpathwaysintheregulationofcellproliferationinmammaliancells.CellRes.2002;12:9-18
8. CobbMH.Mapkinasepathways.ProgBiophysMolBiol.1999;71:479-5009. Kyriakis JM, Avruch J. Sounding the alarm: Protein kinase cascades activated by stress and
inflammation.JBiolChem.1996;271:24313-2431610. Ip YT,Davis RJ. Signal transductionby the c-junn-terminal kinase (jnk) - from inflammation to
development.CurrentOpinioninCellBiology.1998;10:205-21911. Force T, Bonventre JV. Growth factors and mitogen-activated protein kinases. Hypertension.
1998;31:152-16112. Muslin AJ. Mapk signalling in cardiovascular health and disease: Molecular mechanisms and
therapeutictargets.ClinSci.2008;115:203-21813. Muslin AJ. Mapk signalling in cardiovascular health and disease: Molecular mechanisms and
therapeutictargets.ClinSci(Lond).2008;115:203-21814. Salojin K, Oravecz T. Regulation of innate immunity by mapk dual-specificity phosphatases:
Knockoutmodelsrevealnewtricksofoldgenes.JLeukocBiol.2007;81:860-86915. Sim J, Yi K, Kim H, Ahn H, Chung Y, Rehman A, Jang SM, Lee KH, Jang K, Paik SS.
Immunohistochemical expression of dual-specificity protein phosphatase 4 in patients withcolorectaladenocarcinoma.GastroenterolResPract.2015;2015:283764
16. CauntCJ,KeyseSM.Dual-specificitymapkinasephosphatases (mkps): Shaping theoutcomeofmapkinasesignalling.FEBSJ.2013;280:489-504
17. KondohK,NishidaE.Regulationofmapkinasesbymapkinasephosphatases.BiochimBiophysActa.2007;1773:1227-1237
18. ChuY,SolskiPA,Khosravi-FarR,DerCJ,KellyK.Themitogen-activatedproteinkinasephosphatasespac1,mkp-1,andmkp-2haveuniquesubstratespecificitiesandreducedactivityinvivotowardtheerk2sevenmakermutation.JBiolChem.1996;271:6497-6501
19. ChuYF,SolskiPA,KhosraviFarR,DerCJ,KellyK.Themitogen-activatedproteinkinasephosphatasespac1,mkp-1,andmkp-2haveuniquesubstratespecificitiesandreducedactivityinvivotowardtheerk2sevenmakermutation.JournalofBiologicalChemistry.1996;271:6497-6501
20. HirschDD, StorkPJS.Mitogen-activatedprotein kinasephosphatases inactivate stress-activatedproteinkinasepathwaysinvivo.JournalofBiologicalChemistry.1997;272:4568-4575
21. GomezNV, Gorostizaga AB, GarciaMMMS, Brion L, Acquier A, Gonzalez-Calvar SI,Mendez CF,PodestaEJ,PazC.Mapkphosphatase-2(mkp-2)isinducedbyhcgandplaysaroleintheregulationofcyp11a1expressioninma-10leydigcells.Endocrinology.2013;154:1488-1500
22. Lee JS, Ellis BE. Arabidopsis mapk phosphatase 2 (mkp2) positively regulates oxidative stresstolerance and inactivates the mpk3 and mpk6 mapks. Journal of Biological Chemistry.2007;282:25020-25029
23. ShenWH,WangJL,WuJJ,ZhurkinVB,YinYX.Mitogen-activatedproteinkinasephosphatase2:Anoveltranscriptiontargetofp53inapoptosis.CancerResearch.2006;66:6033-6039

Chapter4
94
24. Crowell S,Wancket LM, Shakibi Y, XuPP,Xue JJ, Samavati L,Nelin LD, LiuYS.Post-translationalregulationofmitogen-activatedproteinkinasephosphatase(mkp)-1andmkp-2inmacrophagesfollowing lipopolysaccharide stimulation the role of the c termini of the phosphatases indeterminingtheirstability.JournalofBiologicalChemistry.2014;289:28753-28764
25. JiaoH,TangP,ZhangY.Mapkinasephosphatase2regulatesmacrophage-adipocyteinteraction.PLoSOne.2015;10:e0120755
26. JeffreyKL,BrummerT,RolphMS,LiuSM,CallejasNA,GrumontRJ,GillieronC,MackayF,GreyS,CampsM,RommelC,GerondakisSD,MackayCR.Positiveregulationofimmunecellfunctionandinflammatoryresponsesbyphosphatasepac-1.NatureImmunology.2006;7:274-283
27. Al-MutairiMS,CadalbertLC,McGachyHA,ShweashM,SchroederJ,KurnikM,SlossCM,BryantCE,AlexanderJ,PlevinR.Mapkinasephosphatase-2playsacriticalrole inresponseto infectionbyleishmaniamexicana.PlosPathog.2010;6
28. CornellTT,RodenhouseP,CaiQ,SunL,ShanleyTP.Mitogen-activatedproteinkinasephosphatase2regulatestheinflammatoryresponseinsepsis.InfectionandImmunity.2010;78:2868-2876
29. JiaoHP,TangP,ZhangYL.Mapkinasephosphatase2regulatesmacrophage-adipocyteinteraction.PlosOne.2015;10
30. ImaizumiS,GrijalvaV,PricemanS,WuL,SuF,Farias-EisnerR,HamaS,NavabM,FogelmanAM,ReddyST.Mitogen-activatedproteinkinasephosphatase-1deficiencydecreasesatherosclerosisinapolipoprotein e nullmice by reducingmonocyte chemoattractant protein-1 levels.MolGenetMetab.2010;101:66-75
31. ChomczynskiP,SacchiN.Thesingle-stepmethodofrnaisolationbyacidguanidiniumthiocyanate-phenol-chloroformextraction:Twenty-somethingyearson.NatProtoc.2006;1:581-585
32. Ren BY, Van Kampen E, Van Berkel TJC, Cruickshank SM, Van EckM.Hematopoietic arginase 1deficiencyresultsindecreasedleukocytosisandincreasedfoamcellformationbutdoesnotaffectatherosclerosis.Atherosclerosis.2017;256:35-46
33. vanKampenE,BeaslasO,HildebrandRB,LammersB,VanBerkelTJC,OlkkonenVM,VanEckM.Orp8 deficiency in bonemarrow-derived cells reduces atherosclerotic lesion progression in ldlreceptorknockoutmice.PlosOne.2014;9
34. VanEckM,BosIST,HildebrandRB,VanRijBT,VanBerkelTJC.Dualroleforscavengerreceptorclassb,typeionbonemarrow-derivedcellsinatheroscleroticlesiondevelopment.AmericanJournalofPathology.2004;165:785-794
35. HanMS,JungDY,MorelC,LakhaniSA,KimJK,FlavellRA,DavisRJ.Jnkexpressionbymacrophagespromotesobesity-inducedinsulinresistanceandinflammation.Science.2013;339:218-222
36. KangYJ,ChenJ,OtsukaM,MolsJ,RenS,WangY,HanJ.Macrophagedeletionofp38αpartiallyimpairs lipopolysaccharide-induced cellular activation. The Journal of Immunology.2008;180:5075-5082
37. Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol.2013;13:679-692
38. HuangSCC,EvertsB,IvanovaY,O'SullivanD,NascimentoM,SmithAM,BeattyW,Love-GregoryL,LamWY,O'NeilCM,YanC,DuH,AbumradNA,UrbanJF,ArtyomovMN,PearceEL,PearceEJ.Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. NatureImmunology.2014;15:846-855
39. DaSilvaF,LappalainenJ,Lee-RueckertMD,KovanenPT.Conversionofhumanm-csfmacrophagesinto foam cells reduces their proinflammatory responses to classical m1-polarizing activation.CardiovascularResearch.2016;111:S59-S59
40. KimHS,TavakoliS,PieferLA,NguyenHN,AsmisR.Monocyticmkp-1isasensorofthemetabolicenvironment and regulates function andphenotypic fate ofmonocyte-derivedmacrophages inatherosclerosis.SciRep-Uk.2016;6
41. Hutter D, Chen PL, Barnes J, Liu YS. The carboxyl-terminal domains ofmkp-1 andmkp-2 haveinhibitory effects on their phosphatase activity. Molecular and Cellular Biochemistry.2002;233:107-117

Chapter4
95
42. JeffreyKL,CampsM,RommelC,MackayCR.Targetingdual-specificityphosphatases:Manipulatingmapkinasesignallingandimmuneresponses.NatRevDrugDiscov.2007;6:391-403
43. AminiN,BoyleJJ,MoersB,WarboysCM,MalikTH,ZakkarM,FrancisSE,MasonJC,HaskardDO,Evans PC. Requirement of jnk1 for endothelial cell injury in atherogenesis. Atherosclerosis.2014;235:613-618
44. ZakkarM,ChaudhuryH,SandvikG,EnesaK,LuongLA,CuhlmannS,MasonJC,KramsR,ClarkAR,Haskard DO, Evans PC. Increased endothelial mitogen-activated protein kinase phosphatase-1expression suppresses proinflammatory activation at sites that are resistant to atherosclerosis.CirculationResearch.2008;103:726-732
45. Al-MutairiMS,CadalbertLC,McGachyHA,ShweashM,SchroederJ,KurnikM,SlossCM,BryantCE,AlexanderJ,PlevinR.Mapkinasephosphatase-2playsacriticalrole inresponseto infectionbyleishmaniamexicana.PLoSPathog.2010;6:e1001192
46. WangC,YuX,CaoQ,WangY,ZhengG,TanTK,ZhaoH,ZhaoY,WangY,HarrisD.Characterizationofmurinemacrophagesfrombonemarrow,spleenandperitoneum.BMCImmunol.2013;14:6
47. PengT,ZhangT,LuX,FengQ.Jnk1/c-fosinhibitscardiomyocytetnf-alphaexpressionviaanegativecrosstalkwitherkandp38mapkinendotoxaemia.CardiovascRes.2009;81:733-741
48. StepniakE,RicciR,EferlR,SumaraG,SumaraI,RathM,HuiL,WagnerEF.C-jun/ap-1controlsliverregenerationbyrepressingp53/p21andp38mapkactivity.GenesDev.2006;20:2306-2314
49. HallJP,DavisRJ.Inhibitionofthep38pathwayupregulatesmacrophagejnkanderkactivities,andtheerk,jnk,andp38mapkinasepathwaysarereprogrammedduringdifferentiationofthemurinemyeloidm1cellline.JCellBiochem.2002;86:1-11
50. MatsuguchiT,MusikacharoenT,JohnsonTR,KraftAS,YoshikaiY.Anovelmitogen-activatedproteinkinasephosphatase is an important negative regulator of lipopolysaccharide-mediated c-junn-terminal kinase activation in mouse macrophage cell lines. Molecular and Cellular Biology.2001;21:6999-7009
51. HambletonJ,WeinsteinSL,LemL,DeFrancoAL.Activationofc-junn-terminalkinaseinbacteriallipopolysaccharide-stimulatedmacrophages.PNatlAcadSciUSA.1996;93:2774-2778
52. ZhengXF,HongYX,FengGJ,ZhangGF,RogersH,LewisMAO,WilliamsDW,XiaZF,SongB,WeiXQ.Lipopolysaccharide-induced m2 to m1 macrophage transformation for il-12p70 production isblockedbycandidaalbicansmediatedup-regulationofebi3expression.PlosOne.2013;8
53. MatsuguchiT,MusikacharoenT,OgawaT,YoshikaiY.Geneexpressionsoftoll-likereceptor2,butnot toll-like receptor4, is inducedby lpsand inflammatorycytokines inmousemacrophages. JImmunol.2000;165:5767-5772
54. Moore KJ, Sheedy FJ, Fisher EA.Macrophages in atherosclerosis: A dynamic balance.Nat RevImmunol.2013;13:709-721
55. LeeWJ,TateyaS,ChengAM,Rizzo-DeLeonN,WangNF,HandaP,WilsonCL,ClowesAW,SweetIR,BomsztykK,SchwartzMW,KimF.M2macrophagepolarizationmediatesanti-inflammatoryeffectsofendothelialnitricoxidesignaling.Diabetes.2015;64:2836-2846
56. Wolfs IMJ, Donners MMPC, de Winther MPJ. Differentiation factors and cytokines in theatherosclerotic plaque micro-environment as a trigger for macrophage polarisation. ThrombHaemostasis.2011;106:763-771
57. van Tits LJH, Stienstra R, van Lent PL, Netea MG, Joosten LAB, Stalenhoef AFH. Oxidized ldlenhancespro-inflammatoryresponsesofalternativelyactivatedm2macrophages:Acrucialroleforkruppel-likefactor2.Atherosclerosis.2011;214:345-349
58. CantonJ,NeculaiD,GrinsteinS.Scavengerreceptorsinhomeostasisandimmunity.NatureReviewsImmunology.2013;13:621-634
59. KunjathoorVV,FebbraioM,PodrezEA,MooreKJ,AnderssonL,KoehnS,RheeJS,SilversteinR,HoffHF,FreemanMW.Scavengerreceptorsclassa-i/iiandcd36aretheprincipalreceptorsresponsiblefortheuptakeofmodifiedlowdensitylipoproteinleadingtolipidloadinginmacrophages.JBiolChem.2002;277:49982-49988

Chapter4
96
60. Rahaman SO, Lennon DJ, Febbraio M, Podrez EA, Hazen SL, Silverstein RL. A cd36-dependentsignalingcascadeisnecessaryformacrophagefoamcellformation.CellMetabolism.2006;4:211-221
61. MadsenDH,LeonardD,MasedunskasA,MoyerA,JurgensenHJ,PetersDE,AmornphimolthamP,SelvarjA,YamadaSS,BrennerDA,BurgdorfS,EngelholmLH,BehrendtN,HolmbeckK,WeigertR,Bugge TH.M2-like macrophages are responsible for collagen degradation through a mannosereceptor-mediatedpathway.JournalofCellBiology.2013;202:951-966
62. Han X, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multipleatherogenicmacrophagefunction.ThrombHaemost.2015;113:505-512
63. HanX,BoisvertWA.Theroleofil-10inatherosclerosis.INTECHOpenAccessPublisher;2012.64. BarbourM, Plevin R, Jiang HR. Map kinase phosphatase 2 deficient mice develop attenuated
experimentalautoimmuneencephalomyelitisthroughregulatingdendriticcellsandtcells.SciRep-Uk.2016;6
65. HuangCY,LinYC,HsiaoWY,LiaoFH,HuangPY,TanTH.Dusp4deficiencyenhancescd25expressionand cd4(+) t-cell proliferation without impeding t-cell development. European Journal ofImmunology.2012;42:476-488
66. Schmid CA, Robinson MD, Scheifinger NA, Muller S, Cogliatti S, Tzankov A, Muller A. Dusp4deficiency causedbypromoterhypermethylationdrives jnk signalingand tumor cell survival indiffuselargebcelllymphoma.JournalofExperimentalMedicine.2015;212:775-792

Chapter5
Chapter
EnhancedatheroscleroticlesiondevelopmentinLDLreceptorknockoutmicelackingUpstreamStimulatingFactor1(Usf1)inbone
marrow-derivedcells
Baoyan Ren1, Pirkka-Pekka Laurila2,3,9, Reeni B. Hildebrand1, Jarkko Soronen3,4, Vanessa Frodermann1, Zhuang Li5,Mariëtte R. Boon5,6, Janine J. Geerling1, Patrick C.N. Rensen5,6, Christian Ehnholm3, Petri T. Kovanen7, MattiJauhiainen3,8,MennoHoekstra1,MirandaVanEck1
1 Division of Biopharmaceutics, Cluster BioTherapeutics, Leiden Academic Centre for Drug Research, GorlaeusLaboratories,Leiden,TheNetherlands
2DepartmentofMedicalGenetics,UniversityofHelsinki,Helsinki,Finland3GenomicsandBiomarkersUnit,NationalInstituteforHealthandWelfare,Biomedicum1,Helsinki,Finland4PharmaceuticalsDivisionBayerOyBOF-PH-MRA-MA,MedicalAffairsPO,Espoo,Finland5DepartmentofMedicine,DivisionofEndocrinology,LeidenUniversityMedicalCenter,Leiden,TheNetherlands6 Einthoven Laboratory for Experimental Vascular Medicine, Leiden University Medical Center, Leiden, The
Netherlands7WihuriResearchInstitute,Biomedicum,Helsinki,Finland8MinervaFoundationInstituteforMedicalResearch,Biomedicum,Helsinki,Finland9InstituteforMolecularMedicineFinland,FIMM,Helsinki,Finland
Submittedforpublication

Chapter5
98
pstreamStimulatoryFactor1(Usf1),aubiquitoustranscriptionfactorassociatedwithfamilialcombinedhyperlipidemia,regulatestheexpressionofgenes involvedin lipidmetabolism.
PreviousstudiesshowedthatmicelackingUsf1developedabeneficialcardiometabolicprofile.Inthisstudy,weinvestigatedtheatherogeniceffectofhematopoieticUsf1inlow-densitylipoproteinreceptor(LDLr)knockout(KO)mice.
BonemarrowfromUsf1KOmiceandwild-typemicewastransplantedintomaleLDLrKOmice.After8weeksrecoveryonaregularchowdiet,themicewerechallengedwithapro-atherogenicWestern-typedietfor20weeks.SpecificdeletionofhematopoieticUsf1alsoprotectedagainstdiet-inducedobesity.However,opposedtototal-bodyUsf1deficiency,deletionofhematopoieticUsf1inLDLrKOmiceledtoasignificantincreaseinatheroscleroticlesionsize(130%,p<0.05).Theincreased atherosclerosis susceptibility coincided with increased neutrophil counts in thecirculation(200%,p<0.01)andelevatedVLDLcholesterollevels(162%,p<0.05).Interestingly,thephenotype inducedbyhematopoieticUsf1deficiency inLDLrKOmice is likelyattributedtoanimpairedclearanceofVLDLbywhiteadiposetissue(WAT).ThemRNAexpressionofPeroxisomeproliferator activated receptor gamma, Lipoprotein lipase and VLDL receptor, key players inregulationofVLDLclearance,weredownregulatedinWATofUsf1KObonemarrowrecipients(-41.7%, p<0.05; -30.1%, p<0.05; and -49.4%, p<0.005; respectively) and associated with adecreasedlipidcontentinWAT.
Takentogether,theseresultssuggestthathematopoieticablationofUsf1doesnotaccountforthebeneficialeffectsofglobalUsf1deletion.
Introduction
UpstreamStimulatoryFactor1(Usf1)isaubiquitouslyexpressedtranscriptionfactorthatplaysanimportantroleinlipidmetabolism.1Usf1regulatestheexpressionofmanygenesinvolvedinlipidmetabolism,includingapolipoproteinA2(apoA2),2apoA5,3apoC3,4apoE,5hepaticlipase(HL),6ATP-bindingcassettetransporter1(ABCA1),7,8andfattyacidsynthase(FASN).9-11TwoformsoftheUsfproteinhavebeenidentified,whicharereferredtoasUsf1andUsf2,respectively.12TheUsfproteinsformhetero-and(lesscommon)homo-dimersandbindtotheE-boxmotif.AlthoughtheUsfgenesareubiquitouslyexpressedinmammaliancells,therelativeabundanceoftheUsf1andUsf2geneproductsvariesamongcelltypes.13Importantly,thefunctionofUsfsismodulatedinacell-specific manner.14 Variants of Usf1 have been associated with familial combinedhyperlipidemia (FCHL), characterized by increased serum total cholesterol, triglycerides orboth.11,15Whole-bodyorliver-specificover-expressionofhumanUsf1significantlydecreasedtotalplasma cholesterol levels in C57BL/6Jmice, while triglycerides tended to be slightly higher.16Conversely,Laurilaetal.showedthatdeletionofUsf1inC57Bl/6Jmiceledtoelevatedplasmatotal cholesterol, primarily inhighdensity lipoprotein (HDL)particles, anddecreasedvery lowdensitylipoprotein(VLDL)triglycerides.17
Inadditiontoitsroleinlipidmetabolism,thereareindicationsthatUsf1mightalsomodulatetheimmuneresponse.AcorrelationwasfoundbetweenUsf1andIL-6ontranscriptionallevelintheliveroftransgenicmiceoverexpressinghumanUsf1.13,16Moreover,downregulationofUsf1intheRAW264.7macrophagecell lineupregulatesmitochondrialuncouplingprotein2(UCP2)whichsuppresses the production of pro-inflammatorymitochondria-derived reactive oxygen species(mtROS).18 In agreement, global Usf1 deficiency also led to lower circulating inflammatorycytokinesinmice.17Collectively,thesedataimplythatUsf1mightplayanimportantroleinlipid
U

Chapter5
99
metabolismandtheimmuneresponse.However,onlylimitedresearchonthelinkbetweenUsf1andatherosclerosishasbeendescribed.17,19-21RecentstudiesbyLaurilaandcolleaguesshowedthatlow-densitylipoprotein(LDL)receptor(r)knockout(KO)micelackingUsf1displayremarkablydecreased susceptibility to atherosclerotic lesion development.17 However, the mechanismsunderlying thereductionofatherosclerosis inducedbyUsf1deficiencyarepoorlydefined.Forinstance, it is not known whether Usf1 merely affects blood lipid levels or whether it alsomodulates atherosclerosis susceptibility by impacting the immune system. Bone marrow-transplantation(BMT)allowstospecificallydeleteUsf1inBM-derivedleukocytes.Theaimofthecurrent study was, therefore, to specifically assess the role of Usf1 in immune cells and theconsequencesforatherosclerosisdevelopment.Hereto,BMfromUsf1KOmicewastransplantedinto male LDLr KO mice and atherosclerosis susceptibility was determined after 20 weekschallengewithapro-atherogenicWestern-TypeDiet(WTD).
Materialandmethods
Animalsandbonemarrowtransplantation
LDLrKOmice(C57Bl/6Jbackground)purchasedfromtheJacksonLaboratories,weremaintainedand bred under standard laboratory conditions at the Gorlaeus Laboratories in Leiden, theNetherlands. All animal work was approved by the Dutch Ethics Committee and regulatoryauthority at Leiden University and was carried out in compliance with Dutch governmentguidelinesandtheDirective2010/63/EUoftheEuropeanParliamentontheprotectionofanimalsusedforscientificpurposes.
Usf1KOandwild-type(WT)littermates(bothC57BL/6Jbackground)werebredattheNationalInstitute for Health and Welfare, and University of Helsinki, in compliance with the Finnishgovernmentguidelines.ExperimentswereconductedinconformitywiththeFinnishregulationsand theEuropeanparliamentDirective2010/63/EU.Otherdetails regardingUsf1KOmiceareavailableinarecentreport.17
Bones were harvested from Usf1 KO and WT mice and transported to Leiden in Dulbecco'smodifiedEaglemedium(DMEM).Within36hoursofcollectionofthebones,bonemarrow(BM)wasisolated.LDLrKOmicerecipients(male,approx.12weeksold)weretransplantedwitheitherUsf1KOBMorWTBM.BMTwasperformedbyintravenoustailveininjectionof5×106cellsintotherecipients,onedayafter lethal irradiation (Röntgen,8Gy).Therecipientswereallowedtorecoverfor8weeksonachowdiet(RM3;SpecialDietServices).Subsequently,themicewerefedapro-atherogenicWTD,containing0.25%cholesterol,15%cocoabutterand1%cornoil (SDS,Sussex,UK).After20weeksofWTDfeeding,themiceweresacrificed. Inshort,themicewereanaesthetizedusingamixofxylazine,ketamineandatropine.Bloodwascollectedbyretro-orbitalbleeding(forflowcytometricanalysisandtestingonaveterinaryhaematologyanalyzer(Sysmex))orbytailcut(forlipidanalysis).Subsequently,theanimalswereperfusedwithPBS,andtheheartandotherorganswerecollectedforfurtherresearch.Erythrocytesinthebloodwerelysedwitherythrocyte lysisbuffer (0.15mol/LNH4Cl,10mmol/LNaHCO3,0.1mmol/LEDTA,pH7.3)andsubsequentlythewhitebloodcellswereusedforflowcytometricanalysis.

Chapter5
100
Plasmalipiddetermination
After4hoursfasting,bloodwascollectedviatailsamplinginpotassium-EDTAmicrovetteCB300tubes(Sarstedt,Nümbrecht,Germany),andcentrifugedwith2,000rpmat4°Cfor5minutestoseparateouttheplasma.Freecholesterolandtotalcholesterollevelsweredeterminedinplasmaaspreviouslydescribed.22Furthermore,plasmawasusedforlipoproteinprofileanalysisusingfastprotein liquid chromatography (FPLC) using a high-resolution size-exclusion chromatographySuperose6HRcolumn(3.2×30mm;Smart-System,Pharmacia,Uppsala,Sweden).
Hepaticlipidextraction
TotallipidswereextractedfromliversamplesusingtheBligh&Dyermethodthatwasdescribedpreviously,23 anddissolved in 2%Triton X-100. The cholesterol and triglyceride content in thehomogenate weremeasured and divided by the protein content as determined using a BCAassay,24andexpressedas“µglipid/mgprotein”.
Adiposetissuelipidcontentquantification
Paraffinembeddedsections(5µm)fromgonadalwhiteadiposetissue(WAT)andinterscapularbrownadiposetissue(BAT)ofthetransplantedLDLrKOrecipientswerepreparedandstainedbyhematoxylinandeosin.Thelipid–droplet-positiveareaaspercentageofthetotalWATandBATareawasqualifiedusingImageJsoftware(version1.47).25
Sysmexhaematologyanalyserandflowcytometry
Blood leukocyte counts were analysed using an automated Sysmex XT-2000iV VeterinaryHaematologyanalyser(SysmexCorporation).Fluorescentactivatedcellsorting(FACS)analysiswasperformedonaFACSCantoIIapparatus(BDBiosciences,MountainView,CA)todetectcellsurfacemarkers on blood cells. The antibodies anti-Ly6C, anti-Ly6G and anti-CD11b were all fromeBioscience,Ltd.Nilered(Sigma-Aldrich,USA)andusedtodetectlipid-richleukocytes.DatawereanalysedusingFlowJoSoftwarev10(TreeStarInc).
Atheroscleroticlesionanalysisinaorticroot
Heartswerefixedin4%ShandonZincFormal-Fixx(ThermoFisherScientific,9990245)for24hours,andsubsequentlyembeddedinTissue-TekO.C.T.compound(SakuraFinetek,USA)until furtherprocessing. Cryosections (7 μm) at the level of the aortic sinus were obtained using a LeicaCM3050scryostat.Lipid-richatheroscleroticplaqueswerestainedwithOilRedO.Plaquearea(inµm2)quantitationwasperformedusingaLeicaimageanalysissystem(LeicaLtd,Cambridge,UK).
mRNAexpressionanalysisbyrealtimePCR
TotalRNAwasisolatedfromliver,gonadalwhiteadiposetissueandinterscapularbrownadiposetissuesamplesobtainedatsacrificeafter20weeksWTDchallenge.cDNAweresynthesizedusingRevertAidM-MuLVreversetranscriptase(ThermoScientific,USA)accordingtothemanufacturer’sprotocol (ThermoScientific,USA).Quantitativegeneexpressionwasmeasuredona7500FastReal-TimePCRsystem(AppliedBiosystems,CA)usingSensiMixSYBRgreen(GCbiotechB.V.,TheNetherlands)technology.Theaverageexpressionofthehousekeepinggenesβ-actin,RPL27and

Chapter5
101
36B4wasusedasareferenceforcalculationoftherelativeexpressionofthegenesofinterest.TheprimersequencesareshowninTable1.
Table1qPCRprimersequences.
Primer sequences used for qPCR Gene Orientation of primers Sequences
36B4 Forward primer CTGAGTACACCTTCCCACTTACTGA Reverse primer CGACTCTTCCTTTGCTTCAGCTTT
ABCA1 Forward primer AGAGCAAAAAGCGACTCCACATAGAA Reverse primer CGGCCACATCCACAACTGTCT
ApoA Forward primer ATGTGTCCCAGTTTGAATCCTCCT Reverse primer TTTCTCCAGGTTATCCCAGAAGTCC
β-actin Forward primer AACCGTGAAAAGATGACCCAGAT Reverse primer CACAGCCTGGATGGCTACGTA
CD68 Forward primer TGCCTGACAAGGGACACTTCGGG Reverse primer GCGGGTGATGCAGAAGGCGATG
FASN Forward primer GGCGGCACCTATGGCGAGG Reverse primer CTCCAGCAGTGTGCGGTGGTC
HMG-coA Forward primer CGAGCCACGACCTAATGAAGAATG Reverse primer TGCATCACTAAGGAACTTTGCACC
LPL Forward primer CCCCTAGACAACGTCCACCTC Reverse primer TGGGGGCTTCTGCATACTCAAA
LRP Forward primer CTTCTGGTGGCTGGCGTGGTG Reverse primer CATCCGCTGGTGCTGGAAGCC
MTTP Forward primer TCTCACAGTACCCGTTCTTGGT Reverse primer GAGAGACATATCCCCTGCCTGT
PPARg Forward primer AAGCCCTTTGGTGACTTTATGGAGCC Reverse primer TGCAGCAGGTTGTCTTGGATGTCC
RPL27 Forward primer CGCCAAGCGATCCAAGATCAAGTCC Reverse primer AGCTGGGTCCCTGAACACATCCTTG
SCD1 Forward primer TACTACAAGCCCGGCCTCC Reverse primer CAGCAGTACCAGGGCACCA
SR-B1 Forward primer AAACAGGGAAGATCGAGCCAGTAG Reverse primer CGTAGTGAAGAACCTGGGGCAT
UCP1 Forward primer CCAAGCTGTGCGATGTCCATGTACA Reverse primer AAACATGATGACGTTCCAGGACCCG
Usf1 Forward primer AGTTGGGAGATACAAAGTCCTCCG Reverse primer TGCACTGTTCCCTCTTCGGTT
VLDLr Forward primer TGGAGATGAAGACTGTGCGG Reverse primer CGAAGTCAGACTCAGCACACG
Statisticalanalysis
All values are expressed as means ± SEM. Differences between the groups were statisticallyanalysedwithanunpairedStudent’sT-testortwo-wayANOVAusingGraphPadPrismsoftware(GraphPadSoftware Inc., San Diego, California, USA).Welch correctionwas applied in case ofunequal variances in the dataset. A two-sided P value lower than 0.05 was considered asstatisticallysignificant.

Chapter5
102
Results
HematopoieticUsf1deficiencylowersbodyweightgaininLDLrKOmice
TotalbodydeletionofUsf1leadstoabeneficialmetabolicprofileinC57BL/6miceassociatedwithleanness,increasedlipolysisandimprovedinsulinsensitivitycomparedtotheirWTlittermates.17ToinvestigatewhetherhematopoieticUsf1deficiencyinfluencesbodyweightgainofthemice,changesinbodyweightweremonitoredthroughoutthestudy.MicelackingUsf1inbonemarrow-derivedcellsgainedlessweightcomparedtomicethatreceivedWTBM(Figure1A).The2groupsstartedtodivergeatweek18afterBMT(10thweekonWTD)(Figure1B).Atsacrifice(20thweekonWTD),LDLrKOmicewithUsf1KOBMwere2.5g(8.5%)lighterinweightcomparedtomicetransplantedwithWTBM(Figure1C).
Interestingly, Usf1 mRNA expression in the gonadal white adipose tissue of LDLr KO micereconstitutedwithUsf1KOBMwas44%lowercomparedtothemicethatreceivedWTBM(p<0.05;Figure1D).ThedecreasewaslikelynottheresultofareducedmacrophagecontentoftheadiposetissueasnosignificantdifferencewasfoundintheexpressionofthemacrophagemarkerCD68betweenthe2groups(p=0.11;figure1E).ThereducedUsf1expressioninwhiteadiposetissuewasassociatedwith30%lowerexpressionoflipoproteinlipase(LPL,p<0.05;Figure1F)and48%lower very-low density lipoprotein receptor (VLDLr, p<0.05; Figure 1G) expression, genesresponsible for VLDL-TG-derived fatty acid uptake, and 42% lower Peroxisome proliferator-activatedreceptor-γ(PPARg,p<0.05;Figure1H)expression,whichisanimportanttranscriptionalfactorregulatingLPLandVLDLrexpression.Theexpressionofintracellularlipolysisrelatedgenes,including hormone-sensitive lipase (Hsl), adipose triglyceride lipase (Atgl), and the glucosetransportertype4(Glut4)andlipiddroplet-associatedprotein(Plin)werenotchanged(datanotshown).Collectively,thesedataindicatedecreaseduptakeofVLDL-TG-derivedfattyacidsbywhiteadiposetissueupondeletionofUsf1 inbonemarrow-derivedcells.Therefore, lipidcontentofWATwasmeasuredinparaffin-embeddedgonadalWAT.Inlinewiththebodyweightandgeneexpressiondata,adecreasedWATlipidcontentwasobservedintheUsf1BMTmicecomparedtotheWTmice,(-7%,p<0.05;Figure1I).Morphologicalexaminationalsoconfirmedasmalldecreaseinadipocytecellsize(Figure1J).
PlasmacholesteroliselevatedinLDLrKOmicereconstitutedwithUsf1KObonemarrow
DeletionofUsf1inbonemarrow-derivedcellsofLDLrKOmiceresultedinasignificantincreaseintotalcholesterolandfreecholesterollevels(+36%,p<0.01and+26%,p<0.05respectively;Figure2A-B)after20weeksonWTD,whilethiseffectwasnotobservedonchowdiet(datanotshown).Determination of the lipoprotein distribution pattern showed that the increase in plasmacholesterollevelsonWTDcouldbeattributedtoincreasedVLDLcholesterollevels(+60%,p<0.05;Figure2CandD).Moreover,atrendtowardshigherplasmatriglycerideswasobservedinLDLrKOmicewithUsf1KOBMonWTD(+17%,p=0.12;Figure2E).NoeffectofBMUsf1deletionwasfoundonplasmaglucoselevelsintheLDLrKOrecipients(datanotshown).

Chapter5
103
Figure1DeletionofhematopoieticUsf1inLDLrKOmiceattenuatesbodyweightgainandadiposity.
LDLrKOmicereceived5×106bonemarrowcellsintravenouslyfromeitherWTorUsf1KOmiceafter8GyX-raylethal
irradiation.Themicewereallowedtorecoverfor8weeksonchowdiet,andwerethenfedWTDdietfor20weekstoinduceatherosclerosis.A)BodyweightgainofLDLrKOrecipientsreconstitutedwithWTbonemarrow(opencircles“○”)orUsf1KObonemarrow(closedcircles“●”)from0-28weeksafterBMT.2-wayANOVAwasusedtoanalyzethestatisticalsignificantdifferenceintime.B)RelativebodyweightofLDLrKOrecipientswithUsf1KOBMcorrectedbythebodyweightofrecipientswithWT,thedistinctivebodyweightdifferencestartsonweek15(7weeksWTDfeeding).C)BodyweightofLDLrKOrecipientsatsacrifice.D-H)RelativemRNAexpressionofUsf1,CD68,LPL,VLDLrandPPARgininguinaladiposetissueofLDLrKOmicewithWTbonemarrow(openbar)orUsf1KObonemarrow(closedbar).I)LipidcontentofgonadalWATinLDLrKOrecipientswithWTbonemarrow(openbar)orUsf1KObonemarrow(closedbar). J) Representative histology photographs of hematoxylin/eosin stained paraffin sections of gonadal whiteadiposetissue(originalmagnification10X).*P<0.05**P<0.01ascomparedtoLDLrKOmicereconstitutedwithWTbonemarrow(n=7-14).
Bodyweight Usf1KOBMTrecipients
Bodyweight
LPL VLDLr PPARg

Chapter5
104
Figure 2 Deletion of hematopoietic Usf1 inLDLr KO mice induces plasma cholesterollevels.
After20weeksWTDfeeding,plasmafromLDLrKOmicereconstitutedwithWTbonemarroworUsf1 KO bone marrow was collectedsubsequentlyto4hoursfasting.A)Totalplasmacholesterol and B) free plasma cholesterollevelsofLDLrKOrecipientsofWTbonemarrow(open bars) or Usf1 KO bonemarrow (closedbars). C) Plasma samples were fractionatedwith FPLC size-exclusion chromatography andcholesterol levels were measured from thesefractions. Fractions 2-6 represent VLDL,fractions 7-14 LDL, fractions 15-19HDL.Opencircles(○)representLDLrKOrecipientswithWTbonemarrow,closedcircles(●)representLDLrKO recipients with Usf1 KO bonemarrow. D)AreaunderthecurveofcholesterolcontentinVLDL particles from recipients with WT bonemarrow (open bar) or with Usf1 KO bonemarrow (closed bar). E) Plasma triglycerideslevels on of recipients. *P<0.05, **P<0.01 ascomparedtomicewithWTbonemarrow(n=4-5).
DeletionofhematopoieticUs1inLDLrKOmiceincreasedhepaticcholesterylesteraccumulation,butkeptunaffectedthemRNAexpressionofgenesinvolvedinhepaticlipidmetabolism
In order to investigate if the increased plasma cholesterol in BM-specific Usf1 KO mice wasassociatedwithanalteredhepaticlipidmetabolism,thelipidcontentandmRNAexpressionlevelsof genes involved in lipid homeostasis were determined. BM-specific Usf1 deletion led toincreasedOilredOstainingforneutrallipids(Figure3A)intheliverafter20weeksWTDfeeding,accompaniedbyincreasedcholesterylesteraccumulationevidencedbyquantitativeanalysisafterBlighandDyerextraction(+72%,p<0.05;Figure3B),whilefreecholesterolandtriglycerideswerenotaffected(p>0.05;figure3C).ThisisincontrasttothetotalbodyUsf1knockoutmicewhichwere protected against hepatic steatosis, in line with their overall beneficial metabolicphenotype.17
DespitetheobservedincreaseincholesterylesteraccumulationintheliverofLDLrKOmicelackingUsf1inbonemarrow-derivedcells,theliverexpressionofgenesinvolvedinlipidsynthesis,i.e.3-hydroxy-3-methylglutaryl-coenzymeA reductase (HMG-coA reductase),microsomal triglyceridetransferprotein(MTTP),stearoyl-CoAdesaturase-1(SCD1),FASNandthecholesterolesterificationenzymeacyl-CoA:cholesterolacyltransferase(ACAT)werenotaffected(Supplementaryfigure1A).Furthermore,whiletotalbodyUsf1deletioninmiceleadstoincreasedplasmaHDLlevels,17BM-specific Usf1 deletion did not affect HDL cholesterol. Not surprisingly, genes involved in HDL
F re e c h o le s te ro l
W T B M T U s f1 K O B M T0
2
4
6
Ch
ole
ste
rol
lev
el
(mg
/mL
)
*
W T B M T U s f1 K O B M T0
2 0 0 0
4 0 0 0
6 0 0 0
8 0 0 0
P la s m a T G
Ch
ole
ste
rol
lev
el
(µg
/mL
) P =0 .1 2
W T B M T U s f1 K O B M T0
2
4
6
8
1 0
V L D L
AU
C
*
T o ta l c h o le s te ro l
W T B M T U s f1 K O B M T0
5
1 0
1 5
Ch
ole
ste
rol
lev
el
(mg
/mL
)
**
T o ta l c h o le s te ro l
0 2 4 6 8 1 0 1 2 1 4 1 6 1 8 2 0 2 2 2 40
1
2
3
4
5
W T B M T
U s f1 K O B M T
*
F ra c tio n s
Ch
ole
ste
rol
lev
el
(mg
/mL
)
V L D L
L D LH D L
C
B A
D E

Chapter5
105
metabolism, i.e.ApoA1 andABCA1 were not affected in the liver of Usf1 KO BMT recipientscomparedtoWTBMTrecipients(Supplementaryfigure1B).Alsonoeffectswereobservedontheexpressionofgenesinvolvedincholesterolclearancei.e.low-densitylipoproteinreceptor-relatedprotein1(LRP1)andscavengerreceptorclassBtypeI(SR-BI)(Supplementaryfigure1C).Moreover,thehepaticexpressionofUsf1wasunchanged(p>0.05;Supplementaryfigure1D).However,a2.7-foldincreaseinexpressionwasfoundoftheKupffercellmarkerCD68 inliversofLDLrKOmicereconstitutedwithUsf1KOBMascomparedtocontrols(p<0.001;Figure3D),suggestingincreasedhepaticinflammationinducedbytheaugmentedcholesterylesteraccumulationintheliver.
Figure3EffectofhematopoieticUsf1deficiencyonhepaticlipidaccumulationafter20weeksWTDfeeding.
A) RepresentativeimagesofOilredOandH&Estainedsections(originalmagnification10x).LipidwasextractedfromliversandthecontentofB)cholesterylester,C)freecholesterolandtriglycerideweremeasuredandnormalizedforproteinlevel.D)RelativemRNAexpressionofCD68inliver.OpenbarsrepresenttherecipientsofWTbonemarrow,closedbarsrepresentstherecipientsofUsf1KObonemarrow.*P<0.05;***P<0.001ascomparedtoWTBMTlivers(n=8).
HematopoieticUsf1doesnotaffectbrownadiposetissueofLDLrKOrecipients
Activatedbrownadiposetissue(BAT)efficientlytakesupfattyacidsreleasedfromtriglyceride-richlipoproteins(TRL)suchaschylomicronsandVLDLuponlipolysisoftheircoretriglyceridesleadingtorapidclearanceofthegeneratedchylomicronandVLDLremnantsbytheliver.26,27Micewithtotal-bodyUsf1deletiondisplayelevateduptakeofTRL-derivedfattyacidsbyBAT,andreducedBATlipidcontentaswellassmallerbrownadipocytesizeduetoenhancedBATthermogenesis.17ThiseffectwasindependentofUCP1expression,thespecificuncouplingproteinofBAT,whichwasnotchangedduetotheglobalUsf1deletion.17Inthecurrentstudy,mRNAexpressionofUCP1inBATofLDLrKOmicewasalsonotalteredupondeletionofUsf1 inbonemarrow-derivedcells.(p>0.05;Supplementaryfigure2A).Moreimportantly,incontrasttothetotalbodyUsf1KOmice,thelipidcontentofBATwasnotaffectedbyBMUsf1deletion(p>0.05,Supplementaryfigure2B),suggestingasmallercontributionofBAT,ifany,tothereducedbodyweightoftheLDLrKOmicelackinghematopoieticUsf1.
W T B M T U s f1 K O B M T0
5
1 0
1 5
2 0
2 5
H e p a t ic c h o le s te ro l e s te r
To
tal
ch
ole
ste
rol
*
B C D 6 8
W T B M T U s f1 K O B M T0 .0
0 .2
0 .4
0 .6
Re
lati
ve
ex
pre
ss
ion
***
C
F r e e l c h o le s te r o l T r ig lyc e r id e s0
2 0
4 0
6 0
8 0
1 0 0
H e p a t ic lip id c o n te n t
Lip
id m
g/
mg
pro
tein
W T B M T
U s f1 K O B M T
ns
nsD
A WTàLDLrKO Usf1KOàLDLrKO

Chapter5
106
Circulating neutrophil and monocyte counts are increased in LDLr KO mice lackinghematopoieticUsf1
Highfat,highcholesterolfeedingnotonlyleadstohepaticinflammation,butalsotoaugmentedsystemicinflammatorymarkersinmice.28-30GlobalUsf1deficiencypreviouslyshowedprotectionagainstlow-gradesystemicinflammation,aconditionassociatedwithmetabolicdisturbances.17To explorewhether deletion of Usf1 in bonemarrow-derived cells also affected the systemicinflammatorystatusunderWTDfeedingconditions,thecirculatingleukocyteprofilewasassessedusing flowcytometryandhaematologicalanalysis (Sysmex).After20weeksofWTDfeeding,atrendtowardshighertotalwhitebloodcell(WBC)countswasobserved(+19%,p=0.09;Figure4A).TheobservedtrendtoincreasedWBCcountswasattributedtosignificantlyhigheramountsofcirculatingneutrophils(+63%,p<0.01;Figure4B),andmonocytes(+45%,p<0.05;Figure4B),butnot lymphocytes (data not shown). The results were confirmed by flow cytometric analysis.ComparedtotheWTcontrols,Usf1KOtransplantedmiceshowedanincreaseinthetotalamountof circulating CD11b+ cells (+68%, p<0.05; Figure 5A-B); increased CD11b+Ly6G+ neutrophils(+100%, p<0.01; Figure 5C-D); and a small trend towards an increase in CD11b+Ly6Chi pro-inflammatory monocytes (+23%, p=0.11; Figure 5E-F), but no difference in CD11b+Ly6Clowpatrollingmonocytes(p>0.05,Figure5E-F).
Figure4EffectofhematopoieticUsf1deficiencyoncirculatingleukocytesinLDLrKOmice.After20weeksofWTDfeeding, thecirculating leukocyteprofilewasassessedusingahematologicalanalyzer.A)Totalleukocytecounts,B)Neutrophilandmonocytecounts.OpenbarsrepresentLDLrKOmicereconstitutedwithWTbonemarrow,closedbarsrepresentmicewithUsf1KObonemarrow.*P<0.05ascomparedtoWTBMTmice(n=11-16).
T o ta l le u k o c y te s
Ce
lls
*1
03
/µL
W T B M T U s f1 K O B M T0
2
4
6
8 0.0882A B
N e u tr o p h ils M o n o c yte s0
1
2
3
B lo o d le u k o c y te s
Ce
lls
*1
03
/µL
W T B M T
U s f1 K O B M T
*
*

Chapter5
107
Figure5FlowcytometricanalysisofbloodcellsinLDLrKOmicewithhematopoieticUsf1deficiency.
Circulating leukocytes in LDLrKO recipientswereanalysedby flowcytometry after 20ofweeksWTD feeding.A)AbsolutenumbersofCD11b+cells,C)CD11b+/LY6G+neutrophilsandE)CD11b+/Ly6Clowanti-inflammatorymonocytesandCD11b+/Ly6Chighpro-inflammatorymonocyteswereanalyzedintheLDLrKOrecipientmicewithWTbonemarrow(open bars) or Usf1 KO bone marrow (closed bars). B, D, F) Representative flow cytometric plots. *P<0.05; ascomparedtoWTBMTmice(n=6).
HematopoieticUsf1deficiency inLDLrKOmiceaggravates intracellular lipidaccumulation inbloodleukocytes
Nileredwasusedtoquantifythecellularneutrallipidcontentofcirculatingbloodcellsusingflowcytometry.31,32InagreementwiththeelevatedVLDL-cholesterollevels,anincreasedpercentageoffoamyleukocyteswithahighnileredintensitywereobservedinUsf1KOBMTmicecomparedtoWTBMTcontrolsonWTD(Figure6A),indicatinganinductionoftheamountoflipid-ladencellsinthebloodstream.

Chapter5
108
Figure6LDLrKOmicewithhematopoieticUsf1deficiencydisplayaggravatedintracellularlipidaccumulationinblood.
After20weeksWTDfeeding,A)Percentageofnileredpositivecirculatingleukocytes.OpenbarsrepresentrecipientsofWTbonemarrow,andclosedbarsrecipientsofUsf1KObonemarrow.**P<0.01(n=6)B)Representative flowcytometricplotsofnileredstainingofcirculatingleukocytes.(n=6).
Usf1deficiencyinbonemarrow-derivedcellsofLDLrKOmiceleadstoincreasedatherosclerosissusceptibility
SinceUsf1deletioninbonemarrow-derivedcellsofLDLrKOmicewasassociatedwithincreasedVLDLcholesterolandtheappearanceofmorelipid-richinflammatorycellsinthecirculation,wenextassessedtheeffectofhematopoieticUsf1deficiencyonatherosclerosissusceptibilityintheLDLrKOrecipients.Asexpected,after20weeksWTDfeeding,largeraorticrootatheroscleroticlesionswerefoundintheUsf1KOBMTmicecomparedtotheWTBMTcontrols(+31%,p<0.05;Figure7A-B).Totalcholesterollevelscorrelatedwelltoatheroscleroticlesionsizes(p<0.05;figure7C),indicatingtheelevatedVLDLcholesterolislikelyresponsiblefortheincreasedsusceptibilitytoatherosclerosisofthehematopoieticUsf1KOmice.
Figure7BonemarrowUsf1deficiencyincreasesatherosclerosissusceptibilityofLDLrKOmice.
After20weeksofWTDfeeding,atheroscleroticlesiondevelopmentwasassessed.A)AtheroscleroticlesionsizeintheaorticrootofLDLrKOmicereconstitutedwitheitherWTbonemarrow(openbars)orUsf1KObonemarrow(closedbars). B) Representative aortic root atherosclerotic lesion stained for neutral lipids with Oil red O. *P<0.05 ascomparedtoWTBMTmice(n=16-18).
Discussion
Inthecurrentstudy,weshowthatdeletionofUsf1inhematopoieticcellsprotectsLDLrKOmiceagainst diet-induced obesity, but leads to increased levels of cholesterol in circulating VLDLparticlesandelevatedcountsoflipid-ladeninflammatoryleukocytesinthecirculationculminatingintoanincreasedsusceptibilitytoatherosclerosis.
A Usf1 KOà LDLr KO WT à LDLr KO B
W T B M T U s f1 K O B M T0
5 .0´1 0 0 5
1 .0´1 0 0 6
1 .5´1 0 0 6
A o r t ic ro o t le s io n s iz e
Ao
rtic
ro
ot
les
ion
are
a (
µm
2)
*

Chapter5
109
ArecentlypublishedstudybyLaurilaetal.showedthattotalbodyUsf1KOmicearealsoprotectedagainstdiet-inducedobesity.17However,incontrasttoourfindingsuponselectivedeletionofUsf1inbonemarrow-derivedcells,totalbodyUsf1KOmicedisplayedabeneficialcardiometaboliclipidprofile with decreased VLDL-triglycerides, and elevated HDL-cholesterol and were protectedagainstatheroscleroticlesiondevelopment.Inthiscontext,itisimportanttonotethattheactivityandfunctionofUsfsiscell-typedependent.14ThemostpronouncedtissueeffectintheglobalKOmicewas ascribed to BAT, although the contribution of other tissues could not be ruled out.SpecificdeletionofUsf1inbonemarrow-derivedcells,asexpected,onlyminimallyaffectedtheexpressionofUsf1inliversoftheUsf1KOBMTmice,whileexpressioninBATwasnotaffected.
GlobalUsf1deletionprotectsmiceagainstthedevelopmentofdiet-inducedobesitybyincreasingtheactivationofBAT.17Interestingly,inthecurrentstudywefoundthatspecificdeletionofUsf1inbonemarrow-derivedcellsalsoprotectedLDLrKOmicefromdiet-inducedobesity,suggestingthatalsobone-marrowdependentmechanismscouldaccountforthereducedbodyweightoftheglobalknockouts.17Incontrasttototal-bodyUsf1KOmice,inourBM-specificUsf1KOmodel,BATlipidcontentwasnotchangednorwastheexpressionofUCP1altered,suggestingthatbrownfatactivationisnotacausativefactorintheleanphenotypeobservedintheUsf1KOBMTmice.TofullyexcludeBATactivityasacausativefactorinhematopoieticUsf1deficiency-inducedprotectionagainstweightgain,morecomprehensivemeasurementsofBATactivity,includingmeasurementsofBAToxygenconsumptionanduptakeoflipidsandglucose,wouldhavetobeperformed.
Adipose tissue mass can grow as a result of the expansion of the number of adipocytes(hyperplasia)orgrowthofthesizeofexistingcells(hyperthrophy).KnockdownofUsf1in3T3-L1cells, a murine adipocyte model, by small interfering RNA (siRNA) represses adipogenesis,10indicatingthatUsf1alsohasdirecteffectsonadipogenesis.Notably,inourstudy,afterchallengewithWTDfor20weeks,Usf1mRNAexpressionwaslargelydecreasedinadiposetissueofUsf1KOBMTmice as compared toWT BMT controls. By performing a BMT with GFP-expressing BMYuyamaetal.previouslyshowedthatupto16.7%oftheadipocytesinmicechallengedwithahighfat diet for 7 weeks were derived from BM progenitors.10 The decreasedUsf1 expression inadiposetissueofUsf1KOtransplantedLDLrKOmiceisthuslikelynotonlytheconsequenceofdeletionofUsf1inadiposetissuemacrophages(ATMs),butalsoinadipocytesfromBM-origin.ThedecreasedUsf1expressioninadiposetissuecoincidedwith lowerVLDLrexpression.VLDLrdeficiency protects against obesity by lowering adipose tissuemass,which is associatedwithsmalleradipocytesizeduetoareductioninintracellularlipiddropletdeposition.33AnalysisofthelipidcontentofWATinmicewithBMUsf1deletionshowedasmalldecrease(-7%)inlipidcontent,butthis isprobablynotsufficienttoexplaintheoverall8.5%lowerbodyweight.Theleanbodyweightphenotypeisthuslikelyalsotheconsequenceofanimpairedexpansionofthenumberofadipocytes,whichisinlinewiththepreviouslypublishedroleofUsf1inadipogenesis.10
Incontrasttototal-bodyUsf1KOmicethatdisplayabeneficialcardiometabolicprofile,17inthecurrentstudyweshowedthatselectivedeletionofUsf1inbonemarrow-derivedcellsinLDLrKOmice led to increasedplasma levelsofpro-atherogenicVLDL-cholesterolwhileHDL-cholesterolwasnotaffected.NoeffectswereobservedonthehepaticexpressionofgenesinvolvedinVLDLclearance(LRP1andSR-BI)orVLDLsynthesis (HMG-coAreductase,MTTP), suggestingthattheobservedaugmentedVLDLcholesterollevelsareunlikelytheresultofanalteredhepaticuptake.
Besidestheliver,whiteadiposetissueisanimportantorganforcholesterolstorage.34Adipocytesin rodentsareasignificantsite forcholesterolsynthesisandstorage,35and, inobesesubjects

Chapter5
110
adiposetissuestoresupto50%ofthetotalbodycholesterol.34,36Conversely,LDLrKOmicelackingadipose tissue display severe hyperlipidemia due to impaired plasma cholesterol clearance.37Similarly, our mice lacking Usf1 in bone marrow-derived cells remained lean and displayedincreased plasma VLDL-cholesterol levels upon WTD feeding. Notably, WAT of LDLr KO micereconstituted with Usf1 KO BM showed a decreased lipid content compared WAT of micetransplanted with WT BM, indicating reduced adipose tissue lipid deposition. Therefore, wehypothesizedthatanimpairedVLDL-TG-derivedfattyacidclearancecapacityoftheadiposetissueupon hematopoietic Usf1 deletion might be a causative factor in the observed elevation ofcirculatingcholesterollevels,likelybyimpairinghepaticVLDLremnantclearance.LPLandVLDLraretwokeyproteinsinvolvedinVLDLclearancebywhiteadiposetissue.38AdipocytescanremoveVLDLparticlesdirectlyfromthecirculationviatheVLDLr.39,40orafterhydrolysisoftheparticles’coretriglyceridestofreefattyacidsbyLPL,generatingaVLDLremnantparticle.41TheexpressionoftheVLDLrandLPL isregulatedbyPPAR-γactivation.42Yuyamaetal.previouslyshowedthatknockdownofUsf1intheadipocytecellline3T3-L1downregulatedPPARgexpressioninthiscelltype.10Notably,PPARgexpressionwasdecreasedinadiposetissueofLDLrKOmicetransplantedwithUsf1KOBM. In supportof thedecreasedactivityofPPAR-γ inadipose tissueof LDLrKOtransplantedwithUsf1KOBM,boththeexpressionoftheVLDLrandLPLweredecreased.WespeculatethatdeletionofUsf1inbonemarrow-derivedcellsmightleadtoelevatedplasmaVLDLlevelsthroughinhibitionofVLDLclearancebyadiposetissuethroughimpairmentofthePPAR-γ-VLDLr/LPLaxis.
TheexcessiveamountsofVLDLintheplasmaduetoimpairedVLDLclearancebyadiposetissue,couldcyclebacktotheliver,43leadingtoincreasedhepaticcholesterylesteraccumulation.Indeed,cholesteroldepositioninliversofLDLrKOrecipientsreconstitutedwithUsf1KOBMwasincreased.Interestinglyand inagreementwithourfindings,JonesandcolleaguespreviouslyshowedthatconditionaldeletionofPPAR-γinadiposetissueprotectsmicefromhighfatdiet-inducedobesity,andstimulateslipidaccumulationintheliver.44TheaugmentedcholesteroldepositioninliversofLDLrKOmicetransplantedwithUsf1KOBMcoincidedwithadramaticincreaseinhepaticCD68expression compared to WT transplanted controls, suggesting augmented hepaticinflammation.45,46Moreover,augmentedsystemicinflammationwasfoundasevidencedbytheobserved increase in neutrophil counts and the trend towards increased pro-inflammatorymonocytecountsinthecirculationuponhematopoieticUsf1deletion.
Besidesinducinglipidaccumulationintheliver,highlevelsofVLDLandVLDLremnantcholesterolalsorapidlygiveriseto lipiddropletformation inmonocytes,both inhumansand inmice.47,48
Consistently, BM-specific deficiencyofUsf1 in LDLr KOmice led to increased countsof foamymonocytes in the circulation. Lipid-rich monocytes in blood are predictive markers for thedevelopmentofatherosclerosis49andVLDLandremnantswerereportedtobethebestpredictorof aortic root atherosclerosis in the LDLr KO model.50 In agreement, we observed largeratheroscleroticlesionsintheaorticsinusoftheUsf1KOBMTmiceafter20weeksWTDfeeding,correlatingwiththeaugmentedserumcholesterolinthecirculationoftheseanimals.Interestingly,in contrast to the observed 1.3-fold increase in atherosclerotic plaque area in LDLr KO micetransplantedwithUsf1KOBM,Laurilaetal.17recentlyreportedthattotalbodyUsf1/LDLrdoubleKOmiceexhibita4-folddecreaseinatheroscleroticplaquesizeafter20weeksofWTDfeedinginenfaceaorticsections,whichisinlinewiththeimprovedcardiometaboliclipidprofileintheseanimals.Importantly,thisobservationwassupportedbya45%reductioninatheroscleroticplaquearea in humans being homozygous for an allele which induces 18 % decrease in usf1expression.17,51Thus,thebeneficialmetaboliceffectscausedbyglobalUsf1deficiencyinmiceand

Chapter5
111
humansareabletoovercomethedetrimentaleffectsofUsf1 inbonemarrow-derivedcellsasshowninthecurrentstudy(Figure8).
In conclusion, our study revealed a potential role of hematopoieticUsf1 in VLDLmetabolism,obesityandatherosclerosisdevelopmentandhighlightstheimportanceofstudyingtissue-specificeffectsofgenemodifyingstrategiesinanimalmodels.
Figure8EffectofhematopoieticUsf1onVLDLmetabolism,adiposity,andatherosclerosissusceptibility.
1)BonemarrowtransplantationdeletesUsf1inallbonemarrow-derivedcellsofLDLrKOrecipients.2)WTDfeedinginducesthegenerationofnoveladipocytesofwhichafractionisderivedfrombonemarrowprogenitorslackingUsf1,contributingtodecreasedadiposeUsf1expressionintheUsf1KOBMTmice.3)AdiposeUsf1positivelyregulatestheadiposetissueabilitytotakeupplasmaVLDL-TG-derivedfattyacidsthroughtranscriptionalregulationofVLDLrandLPLexpressionviaPPAR-γ.4)ClearanceofVLDLremnantisimpairedduetoreducedUsf1expressioninadiposetissue,leading to elevated plasma VLDL-cholesterol 5) Increased cycling of VLDL-cholesterol to the liver contributes toincreased hepatic lipid accumulation in Usf1 BMT mice. 6) Increased plasma VLDL-cholesterol promotesatherosclerosissusceptibilityinLDLrKOrecipients.7)Reducedlipiddepositionandimpairedadipogenesisco-leadtothedecreasedbodyweightgainandadiposity.Theblack linesandarrowsindicatethenormalpathways.TheredcrossmeansknockoutandredarrowsindicatetheeffectsofhematopoieticUsf1deficiency.

Chapter5
112
Acknowledgments
Thisstudywassupportedby‘theNetherlandsCardioVascularResearchInitiative:theDutchHeartFoundation,DutchFederationofUniversityMedicalCenters, theNetherlandsOrganisation forHealth Research and Development, and the Royal Netherlands Academy of Sciences’ for theGENIUSproject‘Generatingthebestevidence-basedpharmaceuticaltargetsforatherosclerosis’(CVON2011),TheNetherlandsOrganizationforScientificResearch(VICIGrant91813603(M.V.E)),theFinnishFoundationforCardiovascularResearch(M.J.,P.-P.L.,andJ.S.),JennyandAnttiWihuriFoundation(M.J.andJ.S.),PaavoNurmiFoundation(M.J.,P.-P.L.,andJ.S.),AcademyofFinland(grant#257545toM.J.,grants#283045),FinskaLäkaresällskapet(P.-P.L.andJ.S.),AarneKoskeloFoundation(P.-P.L.),EmilAaltonenFoundation(P.-P.L.),BiomedicumHelsinkiFoundation,(P.-P.L.and J.S.), Foundation forDiabetesResearch (P.-P.L. and J.S.),Orion-FarmosFoundation (P.-P.L.),MagnusEhrnroothFoundation (P.-P.L.), LivochHälsa (P.-P.L), JaneandAatosErkkoFoundation(M.J.), Jalmari andRauhaAhokas Foundation (J.S.), Sigrid Juselius Foundation (P-P.L), and TheFinnishMedicalFoundation(P.-P.L.).WihuriResearchInstituteismaintainedbyJennyandAnttiWihuriFoundation (P.T.K.).M.V.E.andP.C.N.R.areEstablished Investigatorsof theDutchHeartFoundation(grants2007T056and2009T038,respectively).B.R.wassupportedbyagrantfromtheChinaScholarshipCouncil(CSC).

Chapter5
113
Supplementarymaterial
SupplementaryFigure1HepaticlipidmetabolismwasnotaffectedinLDLrKOmicelackingUsf1inbonemarrow-derivedcells.
After20weeksWTDfeeding,liversfromLDLrKOmicereconstitutedWTbonemarroworUsf1KObonemarrowwerecollectedformRNAexpressionanalysis.A)RelativemRNAexpressionofgenesinvolvedinVLDLmetabolism,B)genesinvolvedinHDLmetabolism,C)genesinvolvedincholesterolclearance,andD)hepaticUsf1wereanalyzed.OpenbarsrepresentliversfrommicereconstitutedwithWTbonemarrow(n=8),closedbarsrepresentsmicetransplantedwithUsf1KObonemarrow(n=8).
LRP1SR-BI
HMG-coAMTTPSCD1FASNACAT1ACAT2 ApoA1ABCA1
WTBMTUsf1KOBMT

Chapter5
114
SupplementaryFigure2Brownadiposetissue(BAT)morphologyinLDLrKOmicereconstitutedwitheitherWTorUsf1KObonemarrowafter20weeksWestern-typedietfeeding.
A)mRNAexpressionofUCP1,B)lipidcontentinBATwasanalyzed(n=9-16);nsindicatesnon-significantdifference.C)Representativehistologyphotographsofhematoxylin-stainedparaffinsectionsofintercapularadiposetissue(originalmagnification10×).
References
1. BrunzellJD,PorteD,Jr.,BiermanEL.Reversibleabnormalitiesinpostheparinlipolyticactivityduringthe late phase of release in diabetes mellitus (postheparin lipolytic activity in diabetes).Metabolism.1975;24:1123-1137
2. RibeiroA,PastierD,KardassisD,ChambazJ,CardotP.Cooperativebindingofupstreamstimulatoryfactorandhepaticnuclearfactor4drivesthetranscriptionofthehumanapolipoproteina-iigene.JBiolChem.1999;274:1216-1225
3. Nowak M, Helleboid-Chapman A, Jakel H, Martin G, Duran-Sandoval D, Staels B, Rubin EM,PennacchioLA,TaskinenMR,Fruchart-NajibJ,FruchartJC. Insulin-mediateddown-regulationofapolipoprotein a5 gene expression through the phosphatidylinositol 3-kinase pathway: Role ofupstreamstimulatoryfactor.MolCellBiol.2005;25:1537-1548
4. PastierD,LacorteJM,ChambazJ,CardotP,RibeiroA.Twoinitiator-likeelementsarerequiredforthe combined activation of the human apolipoprotein c-iii promoter by upstream stimulatoryfactorandhepaticnuclearfactor-4.JBiolChem.2002;277:15199-15206
5. SaleroE,GimenezC,ZafraF.Identificationofanon-canonicale-boxmotifasaregulatoryelementintheproximalpromoterregionoftheapolipoproteinegene.BiochemJ.2003;370:979-986
6. vanDeursenD,JansenH,VerhoevenAJ.Glucoseincreaseshepaticlipaseexpressioninhepg2livercellsthroughupregulationofupstreamstimulatoryfactors1and2.Diabetologia.2008;51:2078-2087
7. YangXP,FreemanLA,KnapperCL,AmarMJ,RemaleyA,BrewerHB,Jr.,Santamarina-FojoS.Thee-boxmotifintheproximalabca1promotermediatestranscriptionalrepressionoftheabca1gene.JLipidRes.2002;43:297-306
8. PanX,JiangXC,HussainMM.Impairedcholesterolmetabolismandenhancedatherosclerosisinclockmutantmice.Circulation.2013;128:1758-1769

Chapter5
115
9. CasadoM,ValletVS,KahnA,VaulontS.Essentialroleinvivoofupstreamstimulatoryfactorsforanormaldietaryresponseofthefattyacidsynthasegeneintheliver.JBiolChem.1999;274:2009-2013
10. YuyamaM,FujimoriK.Suppressionofadipogenesisbyvalproicacidthroughrepressionofusf1-activatedfattyacidsynthesisinadipocytes.BiochemJ.2014;459:489-503
11. PajukantaP,LiljaHE,SinsheimerJS,CantorRM,LusisAJ,GentileM,DuanXJ,Soro-PaavonenA,Naukkarinen J, Saarela J, Laakso M, Ehnholm C, Taskinen MR, Peltonen L. Familial combinedhyperlipidemiaisassociatedwithupstreamtranscriptionfactor1(usf1).NatGenet.2004;36:371-376
12. SawadogoM.Multipleformsofthehumangene-specifictranscriptionfactorusf.Ii.DNAbindingpropertiesandtranscriptionalactivityofthepurifiedhelausf.JBiolChem.1988;263:11994-12001
13. SiritoM, Lin Q,Maity T, SawadogoM. Ubiquitous expression of the 43- and 44-kda forms oftranscriptionfactorusfinmammaliancells.NucleicAcidsRes.1994;22:427-433
14. QyangY,LuoX,LuT,IsmailPM,KrylovD,VinsonC,SawadogoM.Cell-type-dependentactivityoftheubiquitoustranscriptionfactorusfincellularproliferationandtranscriptionalactivation.MolCellBiol.1999;19:1508-1517
15. CoonH, Xin Y, Hopkins PN, Cawthon RM,Hasstedt SJ, Hunt SC.Upstream stimulatory factor 1associatedwithfamilialcombinedhyperlipidemia, ldlcholesterol,andtriglycerides.HumGenet.2005;117:444-451
16. WuS,Mar-HeymingR,DugumEZ,KolaitisNA,QiH,PajukantaP,CastellaniLW,LusisAJ,DrakeTA.Upstream transcription factor 1 influences plasma lipid andmetabolic traits inmice.HumMolGenet.2010;19:597-608
17. LaurilaPP,SoronenJ,KooijmanS,ForsstromS,BoonMR,SurakkaI,KaiharjuE,CoomansCP,VanDen Berg SA, Autio A, Sarin AP, Kettunen J, Tikkanen E,Manninen T,Metso J, Silvennoinen R,MerikantoK,RuuthM,PerttilaJ,MakelaA,IsomiA,TuomainenAM,TikkaA,RamadanUA,SeppalaI,LehtimakiT,ErikssonJ,HavulinnaA,JulaA,KarhunenPJ,SalomaaV,PerolaM,EhnholmC,Lee-RueckertM,VanEckM,RoivainenA,TaskinenMR,PeltonenL,MervaalaE,JalankoA,HohtolaE,OlkkonenVM,Ripatti S,KovanenPT,RensenPC,SuomalainenA, JauhiainenM.Usf1deficiencyactivates brown adipose tissue and improves cardiometabolic health. Sci Transl Med.2016;8:323ra313
18. BallWB,MukherjeeM,SrivastavS,DasPK.Leishmaniadonovaniactivatesuncouplingprotein2transcriptiontosuppressmitochondrialoxidativeburstthroughdifferentialmodulationofsrebp2,sp1andusf1transcriptionfactors.IntJBiochemCellBiol.2014;48:66-76
19. KristianssonK,IlveskoskiE,LehtimakiT,PeltonenL,PerolaM,KarhunenPJ.Associationanalysisofallelicvariantsofusf1incoronaryatherosclerosis.ArteriosclerThrombVascBiol.2008;28:983-989
20. FanYM,HernesniemiJ,OksalaN,LevulaM,RaitoharjuE,CollingsA,Hutri-KahonenN,JuonalaM,MarniemiJ,LyytikainenLP,SeppalaI,MennanderA,TarkkaM,KangasAJ,SoininenP,SaleniusJP,KloppN,IlligT,LaitinenT,Ala-KorpelaM,LaaksonenR,ViikariJ,KahonenM,RaitakariOT,LehtimakiT.Upstreamtranscriptionfactor1(usf1)allelicvariantsregulatelipoproteinmetabolisminwomenandusf1expressioninatheroscleroticplaque.SciRep.2014;4:4650
21. Collings A, Hoyssa S, FanM, KahonenM, Hutri-Kahonen N,Marniemi J, JuonalaM, Viikari JS,RaitakariOT,LehtimakiTJ.Allelicvariantsofupstreamtranscriptionfactor1associatewithcarotidarteryintima-mediathickness:Thecardiovascularriskinyoungfinnsstudy.CircJ.2008;72:1158-1164
22. VanEckM,DeWintherMP,HerijgersN,HavekesLM,HofkerMH,GrootPH,VanBerkelTJ.Effectofhuman scavenger receptor class a overexpression in bonemarrow-derived cells on cholesterollevels and atherosclerosis in apoe-deficientmice.Arterioscler ThrombVascBiol. 2000;20:2600-2606
23. BlighEG,DyerWJ.Arapidmethodoftotallipidextractionandpurification.CanJBiochemPhysiol.1959;37:911-917

Chapter5
116
24. SmithPK,KrohnRI,HermansonGT,MalliaAK,GartnerFH,ProvenzanoMD,FujimotoEK,GoekeNM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem.1985;150:76-85
25. BerbeeJF,BoonMR,KhedoePP,BarteltA,SchleinC,WorthmannA,KooijmanS,HoekeG,MolIM,JohnC,JungC,VazirpanahN,BrouwersLP,GordtsPL,EskoJD,HiemstraPS,HavekesLM,SchejaL,Heeren J, Rensen PC. Brown fat activation reduces hypercholesterolaemia and protects fromatherosclerosisdevelopment.NatCommun.2015;6:6356
26. KhedoePP,HoekeG,KooijmanS,DijkW,BuijsJT,KerstenS,HavekesLM,HiemstraPS,BerbeeJF,BoonMR,RensenPC.Brownadiposetissuetakesupplasmatriglyceridesmostlyafterlipolysis.JLipidRes.2015;56:51-59
27. HoekeG,KooijmanS,BoonMR,RensenPC,BerbeeJF.Roleofbrownfatinlipoproteinmetabolismandatherosclerosis.CircRes.2016;118:173-182
28. AssiniJM,MulvihillEE,SutherlandBG,TelfordDE,SawyezCG,FelderSL,ChhokerS,EdwardsJY,Gros R, Huff MW. Naringenin prevents cholesterol-induced systemic inflammation, metabolicdysregulation,andatherosclerosisinldlr(-)/(-)mice.JLipidRes.2013;54:711-724
29. FunkeA,SchreursM,Aparicio-VergaraM,SheedfarF,GrubenN,KloosterhuisNJ,Shiri-SverdlovR,GroenAK,vandeSluisB,HofkerMH,KoonenDP.Cholesterol-inducedhepaticinflammationdoesnot contribute to the development of insulin resistance in male ldl receptor knockout mice.Atherosclerosis.2014;232:390-396
30. WoutersK,vanGorpPJ,BieghsV,GijbelsMJ,DuimelH,LutjohannD,KerksiekA,vanKruchtenR,MaedaN,StaelsB,vanBilsenM,Shiri-SverdlovR,HofkerMH.Dietarycholesterol,ratherthanliversteatosis, leads to hepatic inflammation in hyperlipidemic mouse models of nonalcoholicsteatohepatitis.Hepatology.2008;48:474-486
31. Greenspan P,Mayer EP, Fowler SD. Nile red: A selective fluorescent stain for intracellular lipiddroplets.JCellBiol.1985;100:965-973
32. Naukkarinen J, Nilsson E, Koistinen HA, Soderlund S, Lyssenko V, Vaag A, Poulsen P, Groop L,TaskinenMR,PeltonenL.Functionalvariantdisruptsinsulininductionofusf1:Mechanismforusf1-associateddyslipidemias.CircCardiovascGenet.2009;2:522-529
33. GoudriaanJR,TackenPJ,DahlmansVE,GijbelsMJ,vanDijkKW,HavekesLM,JongMC.Protectionfromobesityinmicelackingthevldlreceptor.ArteriosclerThrombVascBiol.2001;21:1488-1493
34. KrauseBR,HartmanAD.Adiposetissueandcholesterolmetabolism.JLipidRes.1984;25:97-11035. KovanenPT,NikkilaEA,MiettinenTA.Regulationofcholesterolsynthesisandstorageinfatcells.J
LipidRes.1975;16:211-22336. GrantRW,DixitVD.Adiposetissueasanimmunologicalorgan.Obesity(SilverSpring).2015;23:512-
51837. WangM,GaoM,LiaoJ,QiY,DuX,WangY,LiL,LiuG,YangH.Adiposetissuedeficiencyresultsin
severehyperlipidemiaandatherosclerosisinthelow-densitylipoproteinreceptorknockoutmice.BiochimBiophysActa.2016;1861:410-418
38. RobertsCK,BarnardRJ,LiangKH,VaziriND.Effectofdietonadiposetissueandskeletalmusclevldlreceptorandlpl:Implicationsforobesityandhyperlipidemia.Atherosclerosis.2002;161:133-141
39. VaziriND,NorrisK.Lipiddisordersandtheirrelevancetooutcomesinchronickidneydisease.BloodPurif.2011;31:189-196
40. TakahashiS,SuzukiJ,KohnoM,OidaK,TamaiT,MiyaboS,YamamotoT,NakaiT.Enhancementofthe binding of triglyceride-rich lipoproteins to the very low density lipoprotein receptor byapolipoproteineandlipoproteinlipase.JBiolChem.1995;270:15747-15754
41. Goudriaan JR, Espirito Santo SM,Voshol PJ, TeusinkB, vanDijk KW, vanVlijmenBJ, Romijn JA,Havekes LM, Rensen PC. The vldl receptor plays a major role in chylomicron metabolism byenhancinglpl-mediatedtriglyceridehydrolysis.JLipidRes.2004;45:1475-1481
42. TaoH,AakulaS,AbumradNN,HajriT.Peroxisomeproliferator-activatedreceptor-gammaregulatesthe expression and function of very-low-density lipoprotein receptor. Am J Physiol EndocrinolMetab.2010;298:E68-79

Chapter5
117
43. PattersonBW,MittendorferB,EliasN,SatyanarayanaR,KleinS.Useofstableisotopicallylabeledtracerstomeasureverylowdensitylipoprotein-triglycerideturnover.JLipidRes.2002;43:223-233
44. JonesJR,BarrickC,KimKA,LindnerJ,BlondeauB,FujimotoY,ShiotaM,KestersonRA,KahnBB,MagnusonMA.Deletionofppargammainadiposetissuesofmiceprotectsagainsthighfatdiet-inducedobesityandinsulinresistance.ProcNatlAcadSciUSA.2005;102:6207-6212
45. Dixon LJ, Barnes M, Tang H, Pritchard MT, Nagy LE. Kupffer cells in the liver. ComprehensivePhysiology.2013
46. DuarteN,CoelhoIC,PatarraoRS,AlmeidaJI,Penha-GoncalvesC,MacedoMP.Howinflammationimpingesonnafld:Aroleforkupffercells.BiomedResInt.2015;2015:984578
47. denHartighLJ,Connolly-RohrbachJE,ForeS,HuserTR,RutledgeJC.Fattyacids fromvery low-density lipoprotein lipolysis products induce lipiddroplet accumulation inhumanmonocytes. JImmunol.2010;184:3927-3936
48. JacksonWD,WeinrichTW,WoollardKJ.Very-lowandlow-densitylipoproteinsinduceneutrallipidaccumulationandimpairmigrationinmonocytesubsets.SciRep.2016;6:20038
49. XuL,DaiPerrardX,PerrardJL,YangD,XiaoX,TengBB,SimonSI,BallantyneCM,WuH.Foamymonocytesformearlyandcontributetonascentatherosclerosisinmicewithhypercholesterolemia.ArteriosclerThrombVascBiol.2015;35:1787-1797
50. VanderLaanPA,ReardonCA,ThistedRA,GetzGS.VLDLbestpredictsaorticrootatherosclerosisinldlreceptordeficientmice.JLipidRes.2009;50:376-385
51. Auro K, Kristiansson K, Zethelius B, Berne C, Lannfelt L, TaskinenMR, JauhiainenM, PerolaM,PeltonenL,SyvanenAC.Usf1genevariantscontributetometabolictraitsinmeninalongitudinal32-yearfollow-upstudy.Diabetologia.2008;51:464-472


Chapter6
119
Chapter
Summaryandperspectives

Chapter6
120
Tableofcontents
6.1 Englishsummary..................................................................................................120
6.1.1 Introduction.........................................................................................................1216.1.2 TherapeutictargetingofM2macrophagesforatherosclerosistreatment........1216.1.3 Theimportanceofmacrophagephenotypemarkersinatherogenesis..............1226.1.4 Theimportanceofproteinkinasepathwaysinmacrophagephenotypepolarizationandatherogenesis..........................................................................................................1236.1.5 Theimportanceofupstreamstimulatoryfactor1inatherogenesis..................1256.1.6 Concludingremarksandfutureperspectives......................................................126
6.2 Nederlandsesamenvatting...................................................................................129
6.2.1 Inleiding................................................................................................................1296.2.2M2macrofagenalstherapeutischdoelwitvoordebehandelingvanatherosclerose 129..........................................................................................................................1296.2.3 Hetbelangvanmacrofaagfenotype-merkersindeontwikkelingvanatherosclerose 130...........................................................................................................................1296.2.4 Hetbelangvaneiwitkinase-signaleringsroutesinmacrofaagpolarisatieenatheroscleroseontwikkeling...........................................................................................1326.2.5 Hetbelangvanupstreamstimulatoryfactor1inatheroscleroseontwikkeling.1346.2.6 Concluderendeopmerkingenentoekomstperspectieven..................................134

Chapter6
121
English summary
6.1.1 Introduction
ardiovasculardisease(CVD)isthenumberonecauseofdeathintheWesternworld.In2012,theWorldHealthOrganizationreportedthatmorethan30%oftotaldeathswascausedby
CVD,whichoftenresultsfromatherosclerosis.1Atherosclerosisisabloodvesselnarrowingandhardeningdiseasecharacterizedbydepositionofcholesterollocallyinthearterialwallandleadingtoalow-gradechronicinflammation.2Strikingly,basedon2011-2014data,11%ofUnitedStates’adultshavetotalserumcholesterollevelsthatexceedthethresholdof“highbloodcholesterol”(>240mg/dL)for increasedCVDrisk.3Currenttherapeuticstrategiestopreventatherosclerosisaremainlyaimedatloweringserumlow-densitylipoprotein(LDL)cholesterol,e.g.byusingstatinsto inhibit de novo cholesterol synthesis.4 Although an approximate 25% to 35% death ratereductionisachievedbytheuseofcholesterolloweringdrugsinthelastseveraltensofyears,5-7the remaining high incidence of CVD indicates a clear necessity for exploring alternativetherapeutictargetsforreducingtheresidualriskofatherosclerosis.
6.1.2 TherapeutictargetingofM2macrophagesforatherosclerosistreatment
Thedevelopmentofatherosclerosisisacomplexmulti-factorialprocess.8,9Depositionofmodifiedlipoproteins,richincholesterol,inthearterialwallcausesmonocytestobeattractedtotheintimalarea, where they differentiate into macrophages that ingest the modified lipoproteins. Thisprocess, leading to the formation of macrophage foam cells, hallmarks the development ofatherosclerosis.Thus,beingthemajorcelltypeoftheearlyatheroscleroticlesion,macrophagessignificantlycontributetothelesionalmicroenvironmentandstronglyinfluenceatherosclerosisprogression.
Increasingevidenceindicatesthattheheterogeneityofmacrophagesplaysanimportantroleinshaping thesize, composition,andclinical consequencesofatherosclerotic lesions.Twomajortypesofmacrophagescanbedistinguished in lesions:pro-inflammatoryM1macrophagesandanti- or less inflammatory M2 macrophages.10 The distribution of the different types ofmacrophages in the lesion are both regional and stage-specific. In the early stage ofatherosclerosis, the lesion ismainly comprisedofM2macrophages,whereas in the advancedstage, M1 macrophages represent the leading macrophage phenotype.11 Furthermore, M1macrophages dominate the rupture-prone shoulder regions of the atherosclerotic lesion,10whereasM2macrophages aremore often found in stable locations like peripheral regions inasymptomaticplaques.12Thesefindingsimplythatthemacrophagephenotypedistributionisanimportantdeterminingfactorinthedevelopmentofatherosclerosis.
M1macrophagesexpresshighlevelsiNOSwhichfacilitatestheproductionofNO.HighlevelsofNO produced by lesional macrophages, thus indicating an enhanced production of oxidativespecies,wereshowntobepro-atherogenic.13-15Moreover,M1macrophagesproducemanypro-inflammatory cytokines, which aggravate atherosclerosis progression. Besidesmodulating theinflammatoryresponse,M1macrophagesarealsonegativelycorrelatedwithcapthicknessofthelesion,16 an important determinant of the susceptibility to plaque rupture. Plaque rupture inadvancedatherosclerosiscanleadtothrombusformationandtheblockadeofthearteriallumen,which represent the underlying cause of acute cardiovascular symptoms such as myocardialinfarction and stroke. In contrast, M2 macrophages have long been regarded to beatheroprotective.17M2macrophagesproducehighlevelsofanti-inflammatorycytokinessuchas
C

Chapter6
122
IL-10andTGF-β,whichinhibitatherosclerosisdevelopment.18,19Furthermore,M2macrophagesexpresshighlevelsofArg1,amajorplayerinL-argininemetabolism.Byregulatingthesynthesisofproline,themajorprecursorofcollagen,M2macrophagescancontributetoplaquestabilitybyincreasing collagen production.20 In addition, Arg1 reducesNOproduction by competingwithiNOS for the common substrate L-arginine. Therefore, M2 macrophages protect againstatherosclerosis,by inhibiting lesionprogressionandby increasingplaquestability.Collectively,inducingtherepolarizationofM1toM2macrophagethusseemsapromisingtherapeuticstrategyforthetreatmentofatherosclerosis.
We therefore first studied the role of the M2 marker Arg1 in atherosclerosis development.Subsequently the importance of Akt2 andMKP2, kinases that both have been implied inM2macrophage polarization, in atherosclerotic lesion development was investigated. Evidence isaccumulating showing that identification of disease-associated genemutations is a successfulstrategy for the identification of novel drug targets.Therefore, finally, the contribution ofhematopoieticUsf1,ageneassociatedwithfamilialcombinedhyperlipidaemia,toatheroscleroticlesionformationwascharacterized.
6.1.3 Theimportanceofmacrophagephenotypemarkersinatherogenesis
Arg1andiNOS,classicmacrophagephenotypemarkergenes,sharethesamesubstrateL-arginineand hence negatively regulate each other’s activity.21 The role of iNOS which facilitates theproductionofNO,apotentsignalingmolecule,inatherosclerosis,hasbeenwell-studied.17,18,22-27However,abouttheroleofArg1inatheroscleroticlesiondevelopmentstilllittleisknown.Hence,inChapter2,weaimedtounraveltheroleofmacrophageArg1inatherosclerosis.LDLrKOmicereceivedatransplantationofArg1KOorwild-typebonemarrow,inordertospecificallyinvestigatetheroleofhematopoieticArg1.Wefoundthat,deletionofArg1inmacrophagesinducesamoreM2-likephenotypeinvitro.M2macrophageshavepreviouslybeenreportedtobemorepronetofoamcell formation.28 In line, lossofArg1inmacrophages ledtoanincreasedsusceptibilitytofoamcellformationinresponsetoacetylatedLDL(acLDL)loadinginvitro.Inagreementwiththesefindings,bonemarrowArg1deficiencyalsoresultedinenhancedfoamcellformationinvivo,inabsenceofeffectsoncirculatingcholesterollevelsinLDLrKOrecipientsfedaWestern-typediet.However, the enhanced macrophage foam cell formation did not alter the susceptibility toatheroscleroticlesiondevelopment,assimilarlesionsizeswerefoundinLDLrKOrecipientsthatreceivedeitherArg1KOorWTbonemarrow.Furthermore,thelesionalcollagencontentwasalsonotaffectedbymacrophage-specificArg1loss.Thismightbeexplainedbythefactthatsmoothmuscle cells, the main producers of lesional collagen, are not affected by the bone marrowtransplantation procedure. To further clarify the compensatory mechanisms that mightcounteract the enhanced foam cell formation, we investigated the leukocyte profile in thecirculationandspleenoftheArg1KObonemarrowtransplantedmice.DeletionofArg1inbonemarrowledtoadecreaseinBcells.Bcellscanprotectagainstatherosclerosisbymediatingtheproduction of antibodies against oxLDL.29However, despite the observed reduction in B cells,oxLDL-specificantibodieswereincreasedinLDLrKOmicetransplantedwithArg1KObonemarrow.DeletionofArg1inbonemarrow-derivedcellsalsoledtosignificantlyloweramountsofCD11b+cells,CD11b+/ly6Clow+medmonocytesandneutrophilsinthespleenofLDLrKOmiceandatendencytowardsdecreasedinpro-inflammatoryCD11b+/ly6Chighcells.Consideringthat30%ofthetotalnumberofmonocytesinatheroscleroticlesionsarederivedfromthespleen,30thismightprovideanatheroprotectivemechanism,counteractingtheincreasedfoamcellformationintheLDLrKOmicetransplantedwithArg1KObonemarrow.Inconclusion,Arg1inbonemarrow-derivedcells

Chapter6
123
affectsfoamcellformationandcirculatingBcells,butdoesnotaffectatherosclerosisdevelopmentin LDLr KO mice, suggesting that hematopoietic Arg1 is unlikely a therapeutic target foratherosclerosistreatment.
Repolarizationofpro-inflammatoryM1macrophages to less inflammatoryM2macrophages isconsideredapromisingstrategytoreduceatherosclerosis.However,simplychangingoneofthemarkersusedtodiscriminatebetweentheM1andM2macrophagephenotype,namelyArg1,istoo simplistic and one shouldmerely consider amore holistic approach tometabolically andimmunologicallyreprogrammacrophages.
Macrophages represent a continuous spectrum of cells with different markers. The currentM1/M2classificationofthemacrophagesystemisnotsufficientforillustratingthefullspectrumofmacrophagefunctionsandactivationpathways.Invitro,macrophagescanbeskewedtoanM1phenotypebyincubationwithLPS/IFN-γandtoanM2phenotypebyIL-4/IL-13.Invivo,howeverwithin the atherosclerotic lesion, however, awide varietyof factors arepresent thatmaynotinducesuchaclear-cutM1orM2phenotypeas invitro.31,32This indeedresults inabunchofmacrophagephenotypesthatlackorexpresstheclassicM1/M2macrophagephenotypemarkersonlytoalimitedextend.33Thislargelylimitsthepotentialvalueofknownmacrophagephenotypemarkergenesasdiagnosticindicatorsandtherapeutictargets.
M1andM2macrophagesusedifferentsignalingpathwaystofueltheireffectorfunctions.34 Foramoreholisticapproachtoinfluencemacrophagemetabolicandinflammatoryfunctions,targetingmicroRNAs, have been suggested as a successful strategy, as they are importantposttranscriptionalfinetunersofmanybiologicalandmetabolicprograms,havebeensuggestedas a successful strategy.Notably, recently iswas shown that inhibition ofmicroRNA-33 skewsmacrophages toanM2phenotypeand reducesatherosclerosis.34 Interestingly, thisapparentlyworks via the protein kinase AMPK. Protein kinases are pivotal for stimulus-triggered cellularprocesses.35 Except AMPK, macrophage activation is also tightly regulated by thephosphoinositide-3-kinase/protein B (PI3K/Akt) pathway andmitogen-activated protein kinase(MAPK)pathway.36-38Inthecontextofthisthesis,westudiedtheroleofAkt2,amemberofthePI3K/Akt pathway and MKP2, a phosphatase involved in the de-activation of MAPK inatherogenesis.
6.1.4 The importance of protein kinase pathways in macrophage phenotypepolarizationandatherogenesis
Akt2 is a potent macrophage polarization regulator.39-41 Macrophage Akt2 deficiency skewsmacrophagestowardsanM2phenotypeandprotectsagainstdiet-inducedatherosclerosisinnon-diabeticLDLrKOmice.42,43Inadditiontoregulatingmacrophagepolarization,Akt2isalsoamajorregulatoroftheinsulinpathway.TotalbodyAkt2deficiencyleadstoimpairedglucosetoleranceinmice,however,ithaslittletonoeffectonatherosclerosisdevelopment.43,44Tobetterunderstandthe role of macrophage Akt2 in atherosclerosis under glucose intolerant conditions, glucoseintolerantAkt2/LDLrdKOmicewere transplantedwitheither LDLrKOorAkt2/LDLrdKObonemarrow,therebyenablinginvestigationofmacrophage-specificrestorationofAkt2expression.InChapter 3 data are presented that indicating that macrophage Akt2 does not significantlycontributetothetype2diabeticphenotypeandatherosclerosisdevelopmentinAkt2/LDLrdKOmice.PreviousstudiesshowedthatmacrophageAkt2deficiencyisassociatedwithadecreasedsusceptibilitytofoamcellformation.43Inagreement,wefoundthatAkt2restorationincreased

Chapter6
124
foamcellformation.Inlinewiththeaugmentedfoamcellformation,43LDLrKOmacrophageswithfunctionalAkt2signalingexhibitedanM2macrophagephenotype.Interestingly,thisfindingisincontrast to previous studies showing that Akt2 deficiency promotes M2 macrophagepolarization.42,45,46Inthisview,itisworthtonotethatinourstudy,theeffectsofAkt2werestudiedin LDLr KO macrophages, whereas previous studies investigated Akt2 function in wild-typemacrophages. Overall, the results described in chapter 3 indicate that macrophage Akt2restoration leads to more pronounced foam cell formation, but a less inflammatory M2macrophage phenotype; ultimately, leading to unaltered atherosclerosis development in thetransplantedAkt2/LDLrdKOmice.Inadditiontothecounteractingeffectsofenhancedfoamcellformation and reduced inflammation upon Akt2 restoration, likely also the glucose intolerantbackgroundcontributes to theobservedunchangedatherosclerosis susceptibilityofAkt2/LDLrdKOmice transplanted with Akt2 positive/LDLr KO bonemarrow. It is worth noting that themacrophagesinvestigatedinthecurrentstudywereLDLrdeficient,whichisdifferentascomparedtothepreviouslypublishedstudies.42,45,46Macrophageactivationistheresultofacollaborativeinteractionofseveralgeneproductsinresponsetoanextracellularsignalingstimulus.47Geneticvariation could possibly disturb the cellular signaling pathways, thereby affectingmacrophageactivationandultimately leadtoa totallydifferentphenotype.48Hence,wespeculatethat thepresenceoftheLDLronmacrophagesmighthaveimportantconsequencesforM2macrophagepolarizationinducedbyAkt2deficiency.Toconfirmthisfurtherdedicatedresearchisneeded.Ourstudy in this thesis expanded the knowledge of Akt2 in macrophage polarization andatherosclerosis. It underlined that theatherosclerotic roleofAkt2mightbe influencedby thegeneticandmetabolicbackgroundoftheatherosclerosis-pronemousemodelused.
MAPKphosphatases(MKP)representafamilyofphosphatasesthataretightlyrelatedtomitogen-activatedproteinkinases(MAPKs).49TheMKPfamilyconsistsofatleasttendifferentmembers,each MKP family member being able to bind and de-activate their substrate MAPKs bydephosphorylatingtheirphosphoserine/threonineandphosphotyrosineresidues.50,51ThespecificfunctionsofMKPsaredifferentforeachfamilymember,whichisatleastpartlyduetotheirdistinctMAPKsubstratepreference.49Interestingly,MPK2isinvolvedinmacrophagepolarization52,andthus represents a potential target for the treatment of atherosclerosis. Chapter 4 thereforefocuseson theeffectofMKP2onmacrophage functionandatherosclerosisdevelopment.Wedemonstrated that MKP2 deficiency enhanced the activation of the MAPK member JNK inthioglycollate-elicitedmacrophages.Inaddition,macrophageslackingMKP2expressanM2-likephenotype,andinline,displayanenhancedsusceptibilitytofoamcellformationinresponsetooxLDL.Next,MKP2deficientbonemarrowwastransplantedintoLDLrKOmice.After9weeksWTDfeeding,thelipoproteincholesteroldistributionprofileoftherecipientmicewasnotaffectedbybonemarrowMKP2loss.InagreementwiththeM2macrophagepolarizationinvitro,peritonealmacrophagesfromMKP2KObonemarrowrecipientsshowedadecreasedexpressionoftheM1markersCD86andMHCII.Furthermore,MKP2KObonemarrowrecipientsdisplayed increasedproduction of the anti-inflammatory cytokine IL-10, whereas the production of the pro-inflammatory cytokine IL-12 was reduced. Strikingly, despite the observed decrease in pro-atherogeniccytokines,an increase inatherosclerotic lesionsizewasobservedafter9weeksofWTD diet feeding. This result indicated that the anti-inflammatory effect induced by MKP2deletionintheendwaslikelyoverruledbytheaugmentedfoamcellsusceptibilityoftheMKP2knockoutmacrophages.Importantly,thecollagencontentinthelesionsoftheMKP2bonemarrowrecipientswaslowerthaninthelesionsoftheWTbonemarrowrecipients,indicatingthatbonemarrowMKP2alsoleadstoanunstableatherosclerosisphenotype.Invitro,theMKP2deficiency-

Chapter6
125
inducedM2macrophagephenotypehadonlyalimitedeffectonfoamcellformationinabsenceofoxLDLstimulation.Therefore,itislikelythathighlevelsofcirculatingcholesterolmagnifytheeffect ofMKP2 deficiency on foam cell formation and ultimately overrule the beneficial anti-inflammatoryeffects in theLDLrKO recipients. Fromthis view, itwouldbevery interesting toexpandthecurrentstudyandinvestigatetheroleofMKP2inatherosclerosisdevelopmentundercholesterol-loweringconditions.
Theproteinkinasesuperfamilymembersandtheproteinsregulatingtheiractivityarethemostintensively studiedproteindrug targets in currentpharmacological research.Basedona2014report, there weremore than 3000 approved and experimental kinase-based drugs in activeclinicaltrials.53Themajorityofthetrialswithkinase-targetingagentsfocusonthetreatmentofcancer,but the field isnowexpanding to the treatmentof inflammatorydiseases.53 Emergingevidence indicates thatmodulation of the kinase pathway could effectively altermacrophagepolarizationinabeneficialway.54-59Therefore,targetingkinasesisconsideredapotentstrategyforthedevelopmentofnovelpharmaceuticaltherapiesforthetreatmentofatherosclerosis.Inthisthesis,itisindeedshownthattargetingproteinkinaseactivity,i.e.byrestorationofkinaseAKT2ordisruptionofkinase-inactivatingMKP2,isaneffectivewaytostimulatemacrophageM2polarizationandinduceanti-inflammatoryeffects.Inbothstudies,however,theM2polarizationalsoledtoaugmentedfoamcellformationandeventuallyhadeithernoeffectonatheroscleroticlesiondevelopmentorevenledtolargerlesions.
6.1.5 Theimportanceofupstreamstimulatoryfactor1inatherogenesis
The hallmark of atherosclerosis is foam cell formation, a pathological process forwhich bothmacrophagesand lipidsare indispensable. In the firstpartof this thesis,wefocusedongenesinvolvedinM2macrophagepolarization.Inthesecondpartofthesis,wefocusedonUsf1,ageneinvolved in lipidmetabolism. Patients suffering from familial combined hyperlipidemia (FCHL)displayahighrisktodevelopprematurecoronaryarterydisease.60,61Usf1 isageneassociatedwithfamilialcombinedhyperlipidemia(FCHL),characterizedby increasedLDLandtriglyceridesconcentrations, an effect often accompanied by decreased HDL levels.60 A previous studysuggestedanimportantroleoftotalbodyUsf1inlipidmetabolismandatherogenesis.62Inchapter5,weinvestigatedtheroleofhematopoieticUsf1inatherosclerosis.Inthisstudy,wefoundthatbone marrow restricted Usf1 deficiency induced a different phenotype in LDLr KO mice ascomparedtototalbodyUsf1deficiency.Forinstance,totalbodyUsf1deletionledtoabeneficialcardiometabolic lipid profile, marked by decreased athero-promoting VLDL cholesterol andtriglyceride levels,and increased levelsofathero-protectiveHDLcholesterol. Incontrast,bonemarrow Usf1 deletion led to elevated VLDL cholesterol levels and increased susceptibility toatherosclerosis. This effect can likelybeattributed toan impaired clearanceofVLDLbywhiteadipose tissue. Overall, both literature and our study demonstrated that Usf1 alterationsignificantlyaffectatherosclerosisdevelopment.62Interestingly,incontrasttothepro-atherogenicrole of total body Usf1,62 hematopoietic-specific Usf1 is athero-protective. Our study thushighlightedatissuespecificroleofUsf1inatherosclerosisdevelopment.ThisshouldbetakenintoaccountwhendevelopingnovelpharmaceuticsaimedatinhibitingUsf1activityordownregulatingitsexpressiontocombatatherosclerosis.

Chapter6
126
6.1.6 Concludingremarksandfutureperspectives
Using knockoutmice and the bonemarrow transplantation tool, we evaluated the effects ofgenetic modulation involved in macrophage polarization and lipid metabolism on thepathogenesisof atherosclerosis. First a classicM2marker geneArg1was studied (Chapter2),followedbykeyregulatorsofM2macrophageactivation,Akt2(Chapter3)andMKP2(Chapter4).IntheendwediscussedtheroleofUsf1(Chapter5),ageneassociatedwithfamilialcombinedhyperlipidaemiaassociatedgene, inatherosclerosis.Besides theseselectedgenes,manyothermolecules,thatarepossiblyalsoinvolvedinmacrophageactivationandlipidmetabolism,arestillopenforevaluation.
Macrophages are essential for themaintenance of tissue homeostatic functions by regulatinggene expression in response to the local tissue environment.The plasticity of macrophagephenotypes is highly manipulable, which is interesting as the different types of macrophagephenotypescontributetoatherogenesisinadistinctway.Thesefeaturesprovideapossibilitytodevelop cell- or gene-based therapies for treating atherosclerosis by modifying macrophagephenotypes.Themostsuccessfultherapytargetingmacrophagefunctiontodate, ispulmonarymacrophagetransplantationtotreathereditarypulmonaryalveolarproteinosis.63,64Theideaoftherapeutic targeting of macrophage phenotype was developed first for the treatment oftumors.65,66However,allclinicaltrialsfaileduntilnow,67-74whichislargelyduetotherapidlossoftheanti-tumorM1phenotypewhenthepre-educatedmacrophagesarere-exposedtothetumormicroenvironment.75-77 Therefore, establishing how to “lock” the macrophage polarizationphenotypeinvivo,andconsequentlymodulatethemicroenvironment,ratherthanbeexposedtomodulation by the local microenvironment is the key point for improving the efficiency ofmacrophage-basedtherapies.
Targeting macrophage genes at the molecular level to modify macrophage polarization asdescribed in this thesis, is an alternative. By modifying the expression of genes involved inmacrophagepolarization,thephenotypecanbefavorablyalteredandmaylockpolarizationofthemacrophageintoaspecificphenotype.Forexample,MKP2lossskewsmacrophagesfromanM1toamoreM2-likephenotype.Furthermore,afterbonemarrowtransplantation, LDLrKOmicetransplantedwithMKP2deficientbonemarrowshowedananti-inflammatoryplasmacytokineprofile.Thisfindingsuggeststhatgeneticmodulationofthemacrophagephenotypeand“locking”ofthefavorablephenotypeisfeasibleinmice.Therapeuticstrategiesaimedatalteringproteinkinase activity, as shown in this thesis, are an effective way to stimulate macrophage M2polarizationandinduceanti-inflammatoryeffects.
Another interesting strategy is to interfere in epigenetic regulation ofmonocyte/macrophagegeneexpression.Epigeneticmodifications, suchasDNAmethylation,histonemodificationandregulation via non-coding RNAs, alter DNA accessibility and chromatin structure, therebyregulatingthepatternsofgeneexpression.78-83Emergingliteraturesuggestsanimportantroleforepigeneticregulationinmacrophagepolarizationandthedevelopmentofatherosclerosis.84-88Forinstance,Jmjd3,ahistone3Lys27(H3K27)demethylase,facilitatesexpressionofIRF4,akeyM2-promotingtranscriptionfactor,andregulatestheM2macrophagepolarizationwithoutaffectingM1 macrophages.89,90 Deleting histone deacetylase 3 (Hdac3) skews macrophage to the M2phenotype, improves lipid handling and increases atherosclerosis plaque stability.91Moreover,treatmentwith5-aza-2ʹ-deoxycytidine,aninhibitorofDNAmethylation,decreasesmacrophageinflammation and reduces atherosclerosis development in LDLr KO mice.92,93 Besides, the

Chapter6
127
epigenetic changes seem also transplantable. Van Kampen and colleagues found that bonemarrowfrommiceexposedtolong-termWestern-typedietchallengeshowshypomethylationofCpGregionsinthegenesencodingPu.1andIRF8andthattransplantationofthesebonemarrowcells to LDLr KO mice on a low-fat, no cholesterol chow diet leads to increasedmonocyte/macrophageproliferationanddifferentiation,andaortic rootatherosclerosisplaqueformation.94 Further research to increase the understanding of epigenetic changes inmacrophagesduringatheroscleroticlesiondevelopmentandtheepigeneticpathwayscontrollingtheirinflammatoryrepertoirewouldgreatlyhelpthefuturedevelopmentofnoveltherapiesinthecombatagainstatherosclerosis.78
From an inflammation point of view, the anti-inflammatory M2 macrophage phenotype isbeneficial for slowing down the progression of atherosclerosis. However, M2 macrophagesacceleratefoamcellformationwhichlargelyreducesthetherapeuticvalueofM2macrophagesunderhighcholesterolconditions.Theenhancedfoamcellformationresultsinpro-atherogeniceffectsandmaycounteracttheanti-inflammatoryatheroprotectiveeffectsoftheM2macrophage(Chapter3and4).Therefore,itisimportantthatnoveltherapeuticstrategiesaimedatskewingmacrophages to an anti-inflammatory M2 phenotype are combined with plasma cholesterolloweringdrugs.
Massive lowering of plasma cholesterol is a valuable strategy to induce regression ofatheroscleroticlesionsinmousemodelsofatherosclerosis.Interestingly,inregressingplaquesaconsistentincreaseinM2overM1macrophagesisobserved.95-97Averyrecentstudyindicatedthatinfluxofmacrophagesisimportanttoinducelesionregression,likelytoremovedebrisfromregressingplaques.98UpontransplantationofapoEKOaorta’swithplaquestoaCCR2KOmouse,norecruitmentofnewmacrophageswasseenintotheexistinglesionswasseenandnoregressionof atherosclerosis could be induced. CCR2 is a chemokine receptor crucial for monocyterecruitmenttoatheroscleroticlesionsandamarkerofclassical,pro-inflammatorymonocytes.99Ithas been speculated that the classical Ly6Chigh/CCR2+ monocytes are the precursors for M1macrophages, but this has not been firmly established. Considering that the majority of thepatientsentertheclinicwithestablishedatheroscleroticlesions,itwillbeimportanttoinvestigateif induction of M2 macrophages on top of cholesterol lowering, can promote or impedeatherosclerosisregression.
As described in this thesis, bonemarrow transplantation is an effective way to modify geneexpressioninmacrophages.Bonemarrowtransplantationisanexcellenttooltouncoverif,andviawhichmechanisms,ageneofinterestinfluencesatherosclerosisdevelopment,andtoprovideessential information on macrophage gene candidates as potential pharmacological targets.However, with respect to clinical application, bone marrow transplantation requiresmyeloablation,andhence isnot suitableasadirect therapy foratherosclerosis treatmentnotsuitable. To modulate macrophage gene expression, several strategies can be pursued. Forinstance,smallinterferingRNA(siRNA),shorthairpinRNA(shRNA),andmicroRNA(miRNA)canbe used for gene-silencing,100-104 while lentiviral vectors and plasmid vectors are suitable foroverexpressionofageneofinterest.105,106Besidesusinggeneticengineeringmethodstodirectlytargetgeneexpression,pharmacologicalinhibitorsandagonist/antagonistsofageneofinterestcanalsobespecificallydeliveredtomacrophagesusingmacrophage-receptor-basedor ligand-anchoredmicro-/nano-carriers.107-109 In fact, nanoparticles loadedwith pioglitazone, a potentactivatorofPPAR-γ,significantlyskewmonocyte/macrophagetoananti-inflammatoryphenotypein ApoE KOmice.110 Furthermore, nanoparticles containing contrast agents used in computed

Chapter6
128
tomography (CT) andmagnetic resonance imaging (MRI), are also apotent tool to imageandcharacterize theplaque in vivo, providingbasis for the further clinical treatment strategies.109Overall,thesestrategiesofferpossibilitiestospecificallytargetgenesandsignalingpathwaysthatskewmacrophagefunctionsandmightproveinterestinginfurtherfutureclinicaltreatment.
Macrophage heterogeneity is not only pivotal for atherosclerosis, but also for various otherdiseaseswithextensiveinflammationcomponents,suchascancer,Parkinson’sdisease,obesity,diabetes, and arthritis.111-117 Therefore, targeting macrophages would not only providepossibilitiesforthetreatmentofcardiovasculardisease,butcanalsobebeneficialformultipleotherinflammation-basedpathologies.
Inconclusion,modulationofmacrophagepolarizationcombinedwithcholesterolloweringagents,mightprovideapromisingstrategytotreatatherosclerosis. Itwouldbe interestingtoevaluatewhetherselectivetargetingofmacrophagepolarizationis indeedaneffectiveapproachforthetreatmentofatherosclerosisandforthereductionofcardiovascularrisk.

Chapter6
129
6.2 Nederlandsesamenvatting6.2.1 Inleiding
art-envaatziekten(HVZ)isdenummerééndoodsoorzaakindeWestersewereld.In2012rapporteerdedeinternationaleWorldHealthOrganizationthatmeerdan30%vanhettotaal
aantal overlijdens in de wereld waren veroorzaakt door HVZ, dat vaak het gevolg is vanslagaderverkalking ofwel atherosclerose.1 Atherosclerose, een aandoening die leidt totvernauwing en verstijving van bloedvaten, wordt gekarakteriseerd door een afzetting vancholesterollokaalindevaatwandmeteenchronischeontstekingvandebloedvatwandalsgevolg.2Een belangrijke oorzaak voor deze cholesterolafzettingen is onder andere een hoog totaalcholesterolniveau inhetbloed. Inditkader ishetbelangrijk tevermeldendatgegevensuitdejaren 2011-2014 laten zien dat een opmerkelijke hoeveelheid van 11% van de Amerikaansevolwasseneneentotaalserum-cholesterolniveauheeftdatdedrempelwaardevoordedefinitievan‘hoogcholesterol’(>240mg/dL)alsrisicofactorvoorHVZoverschrijdt.3Dehuidigetherapieënvooratherosclerosezijnervooralopgerichtomdehogecholesterolniveausgetransporteerddoorhetzogehetenlage-dichtheidslipoproteïne(LDL)inhetbloedteverlagen,bijvoorbeelddoorhetgebruikvanstatinesomdenovocholesterolsyntheseteremmen.4Indeafgelopendecenniaheeftmen dankzij deze cholesterolverlagende therapieën een afname van het risico op HVZ vanongeveer25-35%kunnenbewerkstellingen.5-7Echter,deresterendehogeincidentievanHVZgeeftaandatereenduidelijkenoodzaakisvooralternatievetherapeutischedoelwittenteneindehetrestrisicovanatheroscleroseteverlagen.
6.2.2 M2 macrofagen als therapeutisch doelwit voor de behandeling vanatherosclerose
Deontwikkelingvanatheroscleroseiseencomplexenmultifactorieelproces.8,9Deafzettingvangemodificeerdecholesterolrijkelipoproteïnenindeintimavandearteriëlevaatwandzorgtervoordatmonocytennaar dit gebied vande vaatwandworden getrokken,waar zij differentiëren inmacrofagen die de gemodificeerde lipoproteïnen opnemen. Dit proces, dat resulteert in devorming van zogeheten macrofaag-schuimcellen, is het belangrijkste kenmerk vanatheroscleroseontwikkeling.Omdatmacrofagenhetbelangrijksteceltypevertegenwoordigendataanwezigisindevroegeatherosclerotischeplaque,leverenzijeensignificantebijdrageaanhetmicromilieu in de plaque en hebben zij een sterke invloed op de vordering van hetatherosclerotischziekteproces.
Intoenemendematewordtbewezendatdeheterogeniteitvanmacrofageneenbelangrijkerolspeelt in het bepalen van de grootte, compositie en klinische consequenties vanatherosclerotische plaques. In deze plaques kunnen twee macrofaag-subtypen gedefinieerdworden:pro-inflammatoireM1macrofagenenanti-,ofminder,inflammatoireM2macrofagen.10Deverdelingvandeverschillendetypemacrofagenindeplaqueiszowelregio-alsfasespecifiek.IndevroegefasevanatheroscleroseatheroscleroseontwikkelingbestaatdeplaquevooraluitM2macrofagen,terwijlindemeergevorderdefasejuistM1macrofagenhetvoornaamstefenotypevertegenwoordigen.11DaarnaastzijnM1macrofagendominantaanwezig indeschouderregio’svandeatherosclerotischeplaque,eendeelvandeplaquedaterggevoeligisvooreenruptuur.M2macrofagenworden inaandeanderekant juistvakeraangetroffen in locatieswaardeplaquemeer stabiel is, zoals de perifere regio’s in asymptomatische plaques.12 Deze bevindingenimplicerendatdeverdelingvanhetmacrofaagfenotypeeenbelangrijkebepalendefactorisvoorhetontwikkelenvanatherosclerose.
H

Chapter6
130
M1macrofagenbrengengrotehoeveelhedeniNOStotexpressie,eenenzymdatdeproductievanNOfaciliteert.Eerderestudieshebbenlatenziendathetatherosclerotischziekteproceswordtgestimuleerd door juist die macrofagen in de atherosclerotische plaque met een hoge NO-productie.13-15M1macrofagenproducerendaarnaastveelpro-inflammatoirecytokinen,diedeontwikkelingvanatheroscleroseversnellen.NaastdemodulatievandeontstekingsreactiezijndehoeveelhedenM1macrofagenindeplaqueooknegatiefgecorreleerdmetdediktevanhetkapseldatdeplaqueafdekt.16Eenafnameindediktevanditkapseliseenbelangrijkerisicofactorvoorhetscheurenvandeplaque,eencomplicatiewaardoorereenbloedpropkanontstaandiehetbloedvatlokaalofverderopindebloedstroomkanafsluitenendaarmeeacutecardiovasculairesymptomen zoals een hart- of herseninfarct kan veroorzaken. In tegenstelling tot de M1macrofagen,wordenM2macrofagen juistveelalbeschouwdalsbeschermendtenaanzienvanatheroscleroseontwikkeling.17M2macrofagenproducerenhogeniveausvananti-inflammatoirecytokinen zoals IL-10 en TGF-β die erom bekend staan de ontwikkeling van atherosclerose teremmen.18, 19Daarnaast brengenM2macrofagen grote hoeveelhedenArg1 tot expressie, eenenzymdateensleutelrolspeelinhetmetabolismvanL-arginine.Arg1kanmetiNOScompeterenvoor hun gedeelde substraat L-arginine. Op deze manier verlaagt de Arg1 expressie in M2macrofagendeNOproductiedoordezecellen,waardoorM2macrofagendeontwikkelingvanatherosclerose kunnen tegengaan. Daarnaast kunnen M2 macrofagen de stabiliteit van eenplaque vergroten door de synthese van proline, de belangrijkste voorloper van collageen, teverhogen.20M2macrofagenbeschermendustegenatherosclerosedoorenerzijdsdegroeivandeplaque te remmen en anderzijds de stabiliteit van de plaque te verhogen.Op basis van dezebevindingenkandeverschuivingvandemacrofaagpolarisatievaneenM1naareenM2fenotypebeschouwd worden als een veelbelovende therapeutische strategie voor de behandeling vanatherosclerose.
Omdezehypothesetetesten,hebbenweeerstonderzochtwelkeroldeM2markerArg1speeltindeontwikkelingvanatherosclerose.DaarnahebbenwederelevantievanAkt2enMKP2,beidekinaseswaarvangesuggereerdwordtdatzebetrokkenzijnbijdepolarisatievanmacrofagennaarhet M2 fenotype, in de ontwikkeling van atherosclerose bestudeerd. Tot slot, omdat er intoenemende mate bewijs wordt gevonden dat de identificatie van ziekte-geassocieerdegenmutaties een succesvolle strategie is voor het ontdekken van nieuwe therapeutischedoelwitten,hebbenweonderzochtwelkerolUSf1,eengendatgeassocieerdwordtmetfamiliarehyperlipidemie,speeltinhetontstaanvanatherosclerose.
6.2.3 Het belang van macrofaagfenotype-merkers in de ontwikkeling vanatherosclerose
Arg1eniNOS,klassiekemerkersvoorrespectievelijkhetM2enM1macrofaagfenotype,delenhetgezamenlijkesubstraatL-arginineenremmenhiermeeelkaarsactiviteit.21DerolvaniNOS,datdeproductie van het potente signaleringsmolecuul NO faciliteert, is reeds uitgebreidbestudeerd.17,18,22-27Echter,derolvanArg1indeontwikkelingvanatheroscleroseisnogonbekend.In hoofdstuk 2 hebben we daarom getracht om de rol van macrofaag-specifiek Arg1 in deontwikkeling van atherosclerose te ontrafelen. Hiertoe werd in LDLr KO muizen het eigenbeenmergvervangendoorbeenmergvanofwelArg1KOofwildtypedonoren.Naherstelvandebeenmergtransplantatiewerden de dieren eenWesters dieet gevoed omde ontwikkeling vanatherosclerosetestimuleren.AllereerstvondenwedatuitschakelingvanArg1inmacrofageninvitroeenmeerM2-gelijkendfenotypeveroorzaakte.ZowarenArg1KOmacrofagenmeergevoeligvoorhetontwikkelenvanschuimcellennablootstellingaangeacetyleerdLDL(acLDL)invitro.Dit

Chapter6
131
is inovereenstemmingmetde literatuur,waarineerder is latenziendatM2macrofagenmeergevoeligzijnvoorschuimcelvorming.28Inovereenstemmingmetdezeinvitrobevindingen,leiddebeenmerg-specifieke deficiëntie van Arg1 in LDLr KO muizen ook tot een verhoogdeschuimcelvorming,zonderdatereeneffectopdebloedcholesterolspiegelswaargenomenwerd.Echter, deze verhoogde schuimcelvorming had geen effect op de gevoeligheid vooratheroscleroseontwikkeling, aangezien de plaquegroottes in de Arg1 KObeenmerggetransplanteerde LDLr KO muizen vergelijkbaar waren aan die van WTbeenmerggetransplanteerdeLDLrKOmuizen.Daarnaastwasookdehoeveelheidcollageenindeplaquesnietverschillendtussendetweegroepen,watverklaardzoukunnenwordendoorhetfeitdatdegladdespiercellen,debelangrijksteproducentenvancollageen indeatherosclerotischeplaque,nietaangedaanwordendoordebeenmergtransplantatieprocedure.Desalnietteminiser,ondanksdeverhoogdeschuimcelvormingalsgevolgvanbeenmergspecifiekeArg1-deletieinLDLrKO muizen, geen sprake van een verhoogde atheroscleroseontwikkeling. Dit duidt op demogelijkheiddatereencompenserendmechanismeinwerkingisgetredendatdeeffectenvandeverhoogdeschuimcelvormingtegengaat.Omdezemechanismentekunnenverklarenhebbenwedeleukocytprofieleninzowelhetbloedalsindemiltvandemuizenonderzocht.Hieruitbleekdat beenmergspecifieke Arg1-deletie leidde tot een significante verlaging van het aantalmonocyten en neutrofiele granulocyten in de milt. Aangezien 30% van het totaal aantalmonocytenindeatherosclerotischeplaquevanuitdemiltkomen,30kandezegevondenverlagingvan het aantal monocyten in de milt mogelijk een verklaring vormen voor het feit dat deontwikkelingvanatherosclerosenietisverhoogdinLDLrKOmuizenmetArg1deficiëntbeenmergondanksdeverhoogdeschuimcelvorming.SamenvattendbeïnvloedtArg1inbeenmergafgeleidecellendeschuimcelvormingdoormacrofagen,maarheefthetgeeneffectopdeontwikkelingvanatheroscleroseinLDLrKOmuizen.Arg1inbeenmergafgeleidecellenisdaaromwaarschijnlijkgeengoedtherapeutischdoelwitvoordebehandelingvanatherosclerose.
De repolarisatie van pro-inflammatoire M1 macrofagen naar minder inflammatoire M2macrofagenwordtbeschouwdalseenveelbelovendestrategieomatheroscleroseteverminderen.Echter,hetsimpelwegveranderenvaneenvandespecifiekeM1-markergenen,zoalsverlagingvanArg1, is een te simplistische benadering voor deze strategie. Men zal daarom op een meerholistische wijze het metabolisme van en het afweermechanisme in macrofagen moetenherprogrammerenomdaadwerkelijkeeneffectopatherosclerosetekunnenbewerkstelligen.
Macrofagenvertegenwoordigeneengrootspectrumvancellendieelkgekenmerktwordendoorverschillendemerkers.DehuidigeM1/M2classificatievanmacrofagen isonvoldoendeomhetvolledige spectrum van macrofaagfunctie en –activering weer te kunnen geven. MacrofagenkunneninvitrodoorincubatiemetLPS/IFN-γgestimuleerdwordentotveranderinginmacrofagenmet een M1 fenotype, terwijl incubatie met IL-4/IL-13 er juist voor zorgt dat macrofagenveranderingincellenmeteenM2fenotype.Indeatherosclerotischeplaqueinvivoiserechtereen grote variëteit aan cytokinen en andere signaleringsmoleculen aanwezig, waardoor deaanwezige macrofagen mogelijk ook minder duidelijke fenotypen kunnen aannemen.31,32 Inatherosclerotische plaques worden inderdaad veel macrofaagfenotypen gevonden waarin deexpressievandeklassiekeM1/M2markersslechtsbeperktofzelfsgeheelnietaanwezigis.33Ditzorgtervoordathetgebruikvanmacrofaagfenotype-merkergenenalsdiagnostischhulpmiddeloftherapeutischedoelwitvoordebehandelingvanatheroscleroselastigis.
M1enM2macrofagenmakengebruikvanverschillendesignaleringspadenomhuneffectorfunctietotuitingtekunnenbrengen.34Omopeenmeerholistischewijzehetmetabolismevanenhet

Chapter6
132
afweermechanismeinmacrofagentereprogrammeren,kanhetlonenomdestrategieterichtenopmicroRNA’saangeziendezeveelbiologischeenmetaboleprogramma’sopposttranscriptioneelniveau reguleren. In dit opzicht is het belangrijk om temelden dat recent is aangetoond datremmingvanmicroRNA-33macrofagennaareenM2fenotypekandrijvenenatherosclerosekanverminderen.34DitproceslijktgemedieerdtewordendoordeeiwitkinaseAMPK.Eiwitkinaseszijnessentieelvoorstimulus-geïnduceerdecellulaireprocessen.35Macrofaagactivatiewordt,naastdeAMPK-signaleringsroute,ookgereguleerddoorde fosfoinositide-3-kinase/eiwitB (KI3K/Akt)enmitogen-geactiveerde eiwitkinase (MAPK) signaleringsroutes.36-38 Binnen de context van ditproefschrifthebbenwij zowelde rolvanAkt2als lidvandeKI3K/Akt-route,alsde rolvanhetfosfataseMKP2diedeMAPK-routedeactiveert,indeontwikkelingvanatherosclerosebestudeerd.Dezesignaleringsrouteswordenhieronderinmeerdetailbeschreven.
6.2.4 Het belang van eiwitkinase-signaleringsroutes in macrofaagpolarisatie enatheroscleroseontwikkeling
Akt1iseensterkeregulatorvanmacrofaagpolarisatie.39-41Akt2-deficiëntieinmacrofagenleidttoteensterkerM2macrofaagfenotypeenbeschermdtegendieet-geïnduceerdeatherosclerose inniet-diabetischeLDLrKOmuizen.42,43Naastheteffectopmacrofaagpolarisatie isAkt2ookeenbelangrijke speler in de signaleringsroute van insuline. Wanneer Akt2 in het gehele lichaamafwezigis,resulteertditineenverlaagdeglucosetolerantieinmuizenterwijlerweinigtotgeeneffect zichtbaar was op de ontwikkeling van atherosclerose.43, 44 Om de rol van macrofaag-specifiekAkt2inatheroscleroseonderglucoseintoleranteconditiesbetertebegrijpen,hebbenwijin hoofdstuk 3 glucoseintolerante Akt2/LDLr dKO muizen getransplanteerd met LDLr KO ofAkt2/LDLr dKO beenmerg, om zodoende het effect vanmacrofaag-specifiek herstel van Akt2-expressietekunnenbestuderen.DeresultatenlatenziendatAkt2inmacrofagengeensignificantebijdragelevertaanhettype2diabetesfenotype,nochaandeontwikkelingvanatheroscleroseinAkt2/LDLr dKO muizen. Eerdere studies hebben aangetoond dat macrofaag-specifieke Akt2-deficiëntiegeassocieerdismeteenverlaagdevatbaarheidvoormacrofaag-schuimcelvorming.43Onzestudieondersteuntdezebevindingen,omdatinonzeopzethetherstelvanAkt2expressieinmacrofagendeschuimcelvormingvandezecellenjuistverhoogde.IntegenstellingtotdeeerderebevindingendatAkt2-deficientieleidttoteenM2macrofaagfenotype,latenonzestudiesindeLDLrKOachtergrondziendatAkt2expressieinmacrofagenleiddetoteenM2macrofaagfenotype.Samengevat laten wij in hoofdstuk 3 zien dat het herstel van Akt2 specifiek in macrofagenresulteerdeineensterkeschuimcelvorming,maarhetM2macrofaagfenotypeversterktwaardoornaarverwachtingdemacrofagenminderinflammatoirzijn.Gezamenlijkleiddedituiteindelijktoteenonveranderdeatheroscleroseontwikkeling indegetransplanteerdeAkt2/LDLrdKOmuizen.Naast de tegenhangende effecten van een verhoogde schuimcelvorming en verlaagdeinflammatoirereponsnaAkt2-herstel,speeltdeglucoseintoleranteachtergrondvandemuizenwaarschijnlijkookeenrolindeonveranderdegevoeligheidvoordeontwikkelingatheroscleroseinonzemuizen.InditkaderishetbelangrijkomtevermeldendatdemacrofagendiewijinonsonderzoekhebbenbestudeerdeenLDLr-deficiënteachtergrondhadden,terwijleerderestudiesvaakopeenwildtypeachtergrondzijnuitgevoerd.42,45,46Macrofaagactiveringishetgevolgvaneen samenwerkende interactie tussen verschillende genproducten, in reactie op eenextracellulaire signaleringsstimulus.47 Genetische variatie, zoals een LDLr KO versus wildtypeachtergrond, kan mogelijk de cellulaire signaleringsroutes beïnvloeden. De activatie vanmacrofagen inverschillendegenetischeachtergrondenzoudaarmeeuiteindelijkkunnen leidentotheelverschillendeeffcetenopmacrofaagfenotypeenfunctie.48Wespeculerendaaromdatdeaanwezigheid van de LDLr op macrofagen mogelijk belangrijke consequenties heeft voor de

Chapter6
133
polarisatievanmacrofagennaareenM2fenotypeonderAkt2-deficiënteomstandigheden.Omdezehypothese te kunnenbevestigen is echter verderonderzoeknodig.Desalnietteminheeftonzestudie,zoalsbeschreveninhoofdstuk3vanditproefschrift,dekennisoverderolvanAkt2in macrofaagpolarisatie en atherosclerose vergroot, en onderstreept deze de notie dat degenetischeenmetaboleachtergrondvanhetmuismodeldatwordtgebruiktderolvanAkt2 inatheroscleroseontwikkelingkanbeïnvloeden.
MAPK fosfatases (MKP’s) vertegenwoordigen een familie van minstens tien verschillendefosfatases die elk sterk gerelateerd zijn aandeMAPK’s.49MKP’s hebbenelk een verschillendevoorkeur voor specifieke MAPK’s, die ze kunnen deactiveren via binding en daaropvolgendedefosforylatievandefosfoserine/treonine-enfosfotyrosineresiduen.50,51ElkMKP-familielidheefteenanderefunctie,watdeelsveroorzaaktwordtdoordiensspecifiekevoorkeurvoorhetMAPK-substraat.49 In dit kader is het interessant om te melden dat MKP2 betrokken is bijmacrofaagpolarisatie52, en daarmee dus een potentieel therapeutisch doelwit is voor debehandeling van atherosclerose. Hoofdstuk 4 richt zich daarom op het effect van MKP2 opmacrofaagfunctieenatheroscleroseontwikkeling.WehebbenlatenziendatMKP2-deficiëntiedeactivatie van het MAPK-familielid JNK verhoogt in thioglycollaat-opgewekte macrofagen.Daarnaast laten we zien dat macrofagen die geen MKP2 tot expressie brengen een M2-macrofaagfenotypelatenzienen, inlijndaarmee,meervatbaarzijnvoorschuimcelvormingnablootstellingaangeoxideerdLDL.OmderolvanMKP2inatheroscleroseontwikkelingverderteontrafelen,hebbenweMKP2-deficiëntbeenmerggetransplanteerdnaarLDLrKOmuizen.Dezemuizen lieten, na 9 weken een Westers dieet gevoerd te zijn, geen verandering zien in hetdistributieprofielvancholesteroloverdelipoproteïne-fractiesinhetbloed.Inlijnmetdeinvitroaangetoonde versterkte M2-macrofaagpolarisatie, observeerden we minder M1 markers opperitoneale cellen in demacrofaag-specifiekeMKP2-deficiëntemuizen, en zagenwe dat dezemuizen meer anti-inflammatoire en juist minder pro-inflammatoire cytokinen produceerden.Echter,ondanksdeverlaginginpro-atherogenecytokineproductie,zagenwedattransplantatiemetMKP2-deficiëntbeenmergleiddetoteenverhoogdeatheroscleroseontwikkelingna9wekenvanWestersedieetvoeding.Belangrijkomtevermelden isdatdehoeveelheidcollageen indeplaquesvandierendiemetMKP2-deficiëntbeenmerggetransplanteerdwarenlagerwasdaninde plaques van de controledieren. Samengevat laten deze resultaten zien dat de anti-inflammatoire effecten van MKP2-deficiëntie uiteindelijk tenietgedaan werden door deverhoogde vatbaarheid voor schuimcelvorming van de MKP2-deficiënte macrofagen. Het isdenkbaar dat de hoge cholesterolniveaus in het bloed van de macrofaagspecifieke MKP2-deficiënte LDLr KO muizen op een Westers dieet het effect van MKP2-deficiëntie opschuimcelvormingdusdanigvergrootheeft,datdegunstigeanti-inflammatoireeffectenvanMKP2overstemdwerden.Inditkaderzouhetinteressantzijnomdehuidigestudieverderuittebreidenen de rol van MKP2 in atheroscleroseontwikkeling te bestuderen onder condities waarin decholesterol-bloedspiegelsactiefverlaagdworden.
Deeiwitkinase-superfamilie,alsmedederegulatorenvanhunactiviteit,zijndemeest intensiefonderzochtegeneesmiddel-doelwitten inhethuidigefarmacologischeonderzoek.Opbasisvaneenrapportuit2014,bestaanermeerdan3000goedgekeurdeenexperimentelegeneesmiddeleninklinischetrialsdiegebaseerdzijnopkinases.53Hoeweldemeerderheidvandeklinischetrialsdiegebruikmakenvankinase-gerichtegeneesmiddelenzichrichtenopdebehandelingvankanker,breidditveldzichinmiddelsookuitnaardebehandelingvanontstekingsziekten.53Erkomtsteedsmeerbewijsdatlaatziendatdemodulatievandekinase-signaleringsrouteopeeneffectieveengunstigemaniermacrofaagpolarisatiekanbeïnvloeden.54-59Vanuitditoogpuntwordthetgebruik

Chapter6
134
vankinase-gerichtemiddelenbeschouwdalseenpotentestrategieomnieuwetherapieënvoordebehandelingvanatheroscleroseteontwikkelen.Inditproefschriftlatenwijinderdaadziendathetmodulerenvaneiwitkinase-activiteit,bijvoorbeelddoorhetherstellenvandeexpressievandekinaseAkt2(hoofdstuk3)ofhetverstorenvanexpressievandekinase-inactiverendefosfataseMKP2(hoofdstuk4),eeneffectievemanierisommacrofaagpolarisatierichtinghetM2-fenotypetedrijvenenomanti-inflammatoireeffectentebewerkstellingen.BeidestudieslatenechterziendatdeverhoogdeM2-polarisatieookleiddetoteenverhoogdeschuimcelvorming,waardoordeatheroscleroseontwikkelinguiteindelijknietaangedaanofzelfsversterktwerd.
6.2.5 Hetbelangvanupstreamstimulatoryfactor1inatheroscleroseontwikkeling
Het belangrijkste kenmerk van atherosclerose is schuimcelvorming, een pathologisch proceswaarvoor zowelmacrofagen als lipiden onmisbaar zijn. In het eerste deel van dit proefschrifthebbenweonsgerichtopgenendiebetrokkenzijnbijdepolarisatievanmacrofagenrichtingeenM2fenotype.Inhettweededeelvanditproefschriftrichtenweonsopeengendatbetrokkenisbij het lipidenmetabolisme. Patiënten die lijden aan zogeheten familiaire gecombineerdehyperlipidemie (FCHL) hebben een hoog risico op het ontwikkelen van vroegtijdige coronairevaatziekten.60,61FCHLwordtgekarakteriseerddoorverhoogdeLDL-entriglycerideniveausdievaakgepaardgaanmeteenverlaginginHDL-niveaus.60Usf1iseengendatgeassocieerdwordtmetFCHL,60endateenbelangrijkerollijkttespeleninhetlipidenmetabolismeendeontwikkelingvanatherosclerose.62 Inhoofdstuk 5, hebbenwe daaromdemacrofaag-specifieke rol vanUsf1 inatheroscleroseonderzocht.Indezestudiehebbenwelatenziendatbeenmerg-specifiekeUsf1-deficiëntieeenanderfenotypelietzieninLDLrKOmuizendanUsf1wordtuitgeschakeldinalleweefselsvanhetlichaam.WanneerUsf1inhetgehelelichaamafwezigwas,leiddedittoteengunstigcardiometaboollipidenprofiel,watgekenmerktwerddooreenverlagingindeniveausvan‘slecht’VLDL-cholesterolentriglyceriden,terwijlhet‘goede’HDL-cholesterolniveaujuistverlaagdwas. Echter, beenmerg-specifieke Usf1-deficiëntie leidde tot een verhoging in VLDL-cholesterolniveauseneenverhoogdegevoeligheidvoordeontwikkelingvanatherosclerose.Diteffectkanwaarschijnlijk toegeschrevenwordenaaneenverlaagdeVLDL-klaringdoorhetwittevetweefsel. Op basis van onze eigen studies en eerdere studies uit de literatuur kan in hetalgemeengesteldwordtdatmodulatievanUsf1deontwikkelingvanatherosclerosebeïnvloed.62Eeninteressantebevindinginonzestudiesisdat,integenstellingtotdepro-atherogenerolvanalgeheleUSf1expressie62,beenmergspecifiekeUsf1-expressiejuisteenbeschermenderolheeftin het kader van atheroscleroseontwikkeling. Onze studie onderstreept dus eenweefselafhankelijkerolvanUsf1inatherosclerose,waarmeerekeninggehoudendienttewordenbijhetontwikkelenvannieuwegeneesmiddelengerichtophetremmenvanUsf1-activiteitofdedownregulatievandiensexpressieomatherosclerosetegentegaan.
6.2.6 Concluderendeopmerkingenentoekomstperspectieven
In dit proefschrift is, middels het gebruik van knockout-muizen en debeenmergtransplantatietechniek,onderzochtwelkeffectdemodulatievangenenbetrokkenbijmacrofaagpolarisatie (hoofdstuk 2-4) en het lipidenmetabolisme (hoofdstuk 5) heeft op depathogenese van atherosclerose. Naast de in dit proefschrift bestudeerde genen, zijn er veleandere moleculen die een rol spelen in macrofaagpolarisatie en het lipidenmetabolisme, enderhalveeenmogelijkinteressantdoelwitzijnvoordebehandelingvanatherosclerose.

Chapter6
135
Macrofagen en de plasticiteit van het fenotype van de macrofagen zijn essentieel voor hetonderhoud van homeostatische functies van weefsels. De doen ze door hun genexpressiepatronenteverandereninreactieopdelokaleomgevingvanhetweefsel.Omdatdeverschillendetypen macrofagen elk op een verschillende manier bijdragen aan de ontwikkeling vanatherosclerose, ishetmogelijkomcel-of gengebaseerde therapieën teontwikkelenwaarmeemacrofaagfenotypengemoduleerdkunnenwordenalsbehandelingtegenatherosclerose.Totnutoe is demeeste bekendemacrofaag-gerichte therapie demacrofaagtransplantatie die wordtgebruiktomeenerfelijk longaandoening,pulmonairealveolaireproteinose,tebehandelen.63,64Het idee om therapieën te ontwikkelen die specifiek gericht zijn op het moduleren vanmacrofaagfenotype, stamt uit het kankeronderzoeksveld, waar het ingezet werd voor debehandelingvantumoren.65,66Echter,totnutoezijnalleklinischetrialsopditgebiedgefaald,67-74voornamelijk omdatde voorbehandeldemacrofagennaherhaaldeblootstelling aandemicro-omgevingvandetumorhunanti-tumorM1-fenotypesnelverliezen.75-77Hetisdaaromvangrootbelangomuittezoekenhoehetmacrofaagfenotype‘vastgezet’kanwordeninvivozodatzijdemicro-omgeving van de tumor kunnen moduleren, in plaats van dat deze de macrofagenmoduleert.
Eenalternatiefhiervoorzijntherapieëndiezichrichtenophetmodulerenvanmacrofagenophetmoleculaire niveau, zoals beschreven in dit proefschrift. Door de expressie van genen diebetrokken zijnbijmacrofaagpolarisatie temoduleren, kanhet fenotypevandemacrofaag tengunstewordenveranderden‘vastgezet’worden.ZowordenmacrofagendooruitschakelingvanMKP2veranderdvanmacrofagenmeteenM1-fenotypeincellenmeteenmeerM2-fenotype.Inaanvulling hierop laten LDLr KOmuizen dieMKP2-deficiënt beenmerg hebben ontvangen eenmeer anti-inflammatoir cytokinenprofiel in hun bloed zien. Deze bevinding suggereert datgenetischemodulatievanhetmacrofaagfenotypeenhet‘vastzetten’vanditfenotypehaalbaarisinmuizen.Daarnaasthebbenwijinditproefschriftooklatenziendattherapeutischestrategieëngericht op het moduleren van eiwitkinase-activiteit ook een effectieve manier is om M2-macrofaagpolarisatietebewerkstelligenenanti-inflammatoireeffectenteinduceren.
Eenandereinteressantestrategieomgenexpressieinmonocytenen/ofmacrofagentemodulerenisdoorteinterfererenindeepigenetischeregulatievandezegenen.Epigenetischemodificatieskunnenwordenbewerkstelligd doorDNA-methylatie, histonmodificatie, en door regulatie vanniet-coderendeRNA’s.DezemodificatieszorgenervoordatdetoegankelijkheidvanhetDNAendechromatinestructuurverandert,waardoorgenexpressiepatronengewijzigdworden.78-83Indeliteratuurzijnersteedsmeerstudiesdielatenziendatepigenetischeregulatieeenbelangrijkerolspeeltinzowelmacrofaagpolarisatiealsatheroscleroseontwikkeling.84-88ZofaciliteertJmjd3,eenhiston 3 Lys27 (H3K27) demethylase, de expressie van IRF4, een belangrijkeM2-stimulerendetranscriptiefactor dieM2-macrofaagpolarisatie reguleert zonder een effect te hebben opM1-macrofagen.89,90 Daarnaast hebben eerdere studies laten zien dat uitschakeling van histondeacetylase3(Hdac3)macrofagenrichtingeenM2-fenotypedrijft,eengunstigeffectheeftopdeverwerking van lipiden, en de stabiliteit van atherosclerotische plaques vergroot.91 Tot slotverlaagt behandeling met een DNA-methylatieremmer, 5-aza-2’-deoxcycytidine, macrofaagontstekingsprocessenenverlaagthetdeontwikkelingvanatheroscleroseinLDLrKOmuizen.92,93Interessantgenoeglijkenepigenetischeveranderingenooktransplanteerbaartezijn.VanKampenen collega’s vonden dat beenmerg vanmuizen die gedurende lange tijd gevoed zijnmet eenWestersdieeteenhypomethylatievanCpG-regionenlietenzienindegenendiecoderenvoorPu.1enIRF8.94TransplantatievandezebeenmergcellennaarLDLrKOmuizendiegevoedwerdenmeteen controledieet dat laag in vet en cholesterol is, leidde tot een verhoging vandemonocyt-

Chapter6
136
/macrofaagproliferatieendifferentiatieeneenverhogingvandeatheroscleroseontwikkeling.Hetontrafelenvanepigenetischeveranderingeninmacrofagentijdensatheroscleroseontwikkeling,envan de epigenetische signaleringsroutes die de inflammatoire respons van macrofagenbeïnvloeden, zal de toekomstige ontwikkeling van nieuwe therapieën in de strijd tegenatherosclerosesterkkunnenondersteunen.78
Vanuithetoogpuntvandeontstekingsreactie,lijkthetdrijvenvanmacrofaagpolarisatierichtinghet anti-inflammatoire M2-fenotype een gunstige strategie voor het afremmen van deatheroscleroseontwikkeling.Echter,M2-macrofagenversnellendeschuimcelvorming,waardoorde therapeutischewaarde vanM2-macrofagen sterkt afneemtwanneer er sprake is vanhogecholesterolniveaus in de circulatie. De versterkte schuimcelvorming heeft een pro-atherogenewerkingenkanzelfshetanti-inflammatoire,beschermendeeffectvanM2-macrofagentenietdoen(hoofdstuk3en4).Hetisdaaromvangrootbelangdatnieuwetherapeutischestrategieëngerichtop het stimuleren van een M2-macrofaag fenotype gecombineerd worden metcholesterolverlagendemedicijnen.
Sterke verlaging van het cholesterolniveau in het bloed is eenwaardevolle strategie voor hetinducerenvanregressievanatherosclerotischeplaquesinmuismodellen.InditkaderisineerderestudieseenconstanteverhogingvandeM2-over-M1-ratiovanmacrofagenindeplaquesonderregressieconditiesaangetoond.95-97Hetisbelangrijktevermeldendateenrecentestudieheeftlatenziendatdeinfluxvangezondemacrofagenindeatheroslerotischeplaquebelangrijkisomuiteindelijkregressievandeplaquetekunneninduceren,waarschijnlijkomdatmacrofagennodigzijnvoorhetopruimenvandedodecellenenhetnecrotischeweefsel indekleinerwordendeplaques.98 Deze studie liet zien datwanneer plaque-bevattende apoE KO aorta’s naarmuizengetransplanteerd werden die deficiënt waren voor de chemokinereceptor CCR2, er geenrekruteringvannieuwemacrofagenplaatsvindt inde reedsbestaandeplaquesendatergeenregressie van atherosclerose bewerkstelligd kon worden. CCR2 is een chemokinereceptor dieessentieelisvoorderekruteringvanmonocytennaaratherosclerotischeplaques.99Gezienhetfeitdatdemeerderheidvandepatiëntendie zichmeldenmetHVZklachten reedsvergevorderdeatherosclerotische laesies heeft, is het belangrijk om te onderzoeken of de inductie vanM2-macrofaagpolarisatiebovenopcholesterolverlagingregressievanatheroscleroseontwikkelingkanversterkenofdatregressiehierdoorjuisttegengegaanwordt.
Beenmergtransplantatieiseeneffectievemanieromgenexpressieinmacrofagentemoduleren,zoalsook inditproefschriftbeschreven is.Omdezereden isbeenmergtransplantatieeenzeergeschiktmiddelomteontrafelenofenzoja,viawelkemechanismen,eengenvaninteressedeontwikkeling van atherosclerose kan beïnvloeden. Op deze manier biedt debeenmergtransplantatietechniekookessentiëleinformatieoverpotentiële‘drugable’macrofaag-kandidaatgenen. In de kliniek is de techniek echter niet bruikbaar, omdat voorbeenmergtransplantatieeenmyeloablatie,devernietigingvanendogeenbeenmerg,benodigdis.Ominmensenalsnogdeexpressievanmacrofaaggenentekunnenmoduleren,kunnenmeerderestrategieëntoegepastworden.ZokansmallinterferingRNA(siRNA),shorthairpinRNA(shRNA)enmicroRNA(miRNA)gebruiktwordenomdegenexpressieteniettedoen,100-104terwijllentiviraleenplasmavectorenjuistgebruiktkunnenwordenomeengenvaninteressetotoverexpressietebrengen.105,106Naasthetgebruikvangenetischemethodenomopdirectewijzedeexpressievangenen te moduleren, kan men ook farmacologische remmers en (ant)agonisten van het genspecifiekafleverenaanmacrofagenmiddelshetgebruikvanmacrofaagreceptor-gebaseerdeofligand-geankerde micro- of nanodeeltjes.107-109 Zo heeft een eerdere studie laten zien dat

Chapter6
137
behandeling van ApoE KO muizen met nanodeeltjes geladen met pioglitazon, een potenteactivator van PPAR-γ, monocyt-/macrofaagpolarisatie succesvol naar een anti-inflammatoirfenotypekandrijven.110Daarnaastishetgebruikvannanodeeltjesgeladenmetcontrastvloeistof,incombinatiemetdevisualisatietechniekenCTenMRI,eensuccesvollemethodegeblekenomatherosclerotischelaesies invivotevisualiserenentekarakteriseren,wateenbelangrijkebasisbiedt voor verdere klinische behandelstrategieën.109 Samengevat bieden deze verschillendetechnieken de mogelijkheid om te interfereren in specifieke genexpressiepatronen ensignaleringsroutes enmacrofaagfuncties te moduleren, en bieden als zodanig een basis voortoekomstigeklinischebehandelstrategieën.
Totslot,macrofaagheterogeniteitisnietalleenessentieelvooratherosclerose,maarookvoorveelandere ziekten die een sterke ontstekings component kennen, zoals kanker, de ziekte vanParkinson, obesitas, diabetes en arthritis.111-117 De ontwikkeling van macrofaag-gerichtetherapieën biedt dus niet alleen nieuwe behandelmogelijkheden op het gebied van hart- envaatziekten, maar zal ten gunste komen aan een grote variëteit van ontstekings-gebaseerdeaandoeningen.
Concluderendkangesteldwordendatmodulatie vanmacrofaagpolarisatie, in combinatiemetcholesterolverlaging, een veelbelovende strategie is voor de behandeling van atherosclerose.Vervolgonderzoekdientaantewijzenofmacrofaagpolarisatie-gerichtetherapieëninderdaadeeneffectievebenaderingvoordebehandelingvanatherosclerosezijn,enofzijdebeoogdeverlagingincardiovasculairrisicokunnenbewerkstelligen.

Chapter6
138
References
1. OrganizationWH.Cardiovasculardiseases(cvds).2013.FactSheet.20162. LibbyP,RidkerPM,MaseriA.Inflammationandatherosclerosis.Circulation.2002;105:1135-11433. BenjaminEJ,BlahaMJ,ChiuveSE,CushmanM,DasSR,DeoR,deFerrantiSD,FloydJ,FornageM,
GillespieC, IsasiCR,JimenezMC,JordanLC,JuddSE,LacklandD,LichtmanJH,LisabethL,LiuS,LongeneckerCT,MackeyRH,MatsushitaK,MozaffarianD,MussolinoME,NasirK,NeumarRW,Palaniappan L, Pandey DK, Thiagarajan RR, Reeves MJ, Ritchey M, Rodriguez CJ, Roth GA,RosamondWD,SassonC,TowfighiA,TsaoCW,TurnerMB,ViraniSS,VoeksJH,WilleyJZ,WilkinsJT,WuJH,AlgerHM,WongSS,MuntnerP,AmericanHeartAssociationStatisticsC,StrokeStatisticsS.Heartdiseaseand stroke statistics-2017update:A report from theamericanheart association.Circulation.2017;135:e146-e603
4. KonesR.Ispreventionafantasy,orthefutureofmedicine?Apanoramicviewofrecentdata,status,anddirectionincardiovascularprevention.TherAdvCardiovascDis.2011;5:61-81
5. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: Thescandinaviansimvastatinsurvivalstudy(4s).Lancet.1994;344:1383-1389
6. Shepherd J, Cobbe SM, Ford I, Isles CG, Lorimer AR,MacFarlane PW,McKillop JH, Packard CJ.Preventionofcoronaryheartdiseasewithpravastatininmenwithhypercholesterolemia.Westofscotlandcoronarypreventionstudygroup.NEnglJMed.1995;333:1301-1307
7. RidkerPM,CookN.Clinicalusefulnessofveryhighandverylowlevelsofc-reactiveproteinacrossthefullrangeofframinghamriskscores.Circulation.2004;109:1955-1959
8. LusisMJ,MarR,PajukantaI.Geneticsofatherosclerosis.AnnuRevGenomHumG.2004;5:189-2189. SinghRB,MengiSA,XuYJ,ArnejaAS,DhallaNS.Pathogenesisofatherosclerosis:Amultifactorial
process.ExpClinCardiol.2002;7:40-5310. Stoger JL, GijbelsMJ, van der Velden S,MancaM, van der Loos CM, Biessen EA, DaemenMJ,
Lutgens E, de Winther MP. Distribution of macrophage polarization markers in humanatherosclerosis.Atherosclerosis.2012;225:461-468
11. Khallou-LaschetJ,VarthamanA,FornasaG,CompainC,GastonAT,ClementM,DussiotM,LevillainO,Graff-DuboisS,NicolettiA,CaligiuriG.Macrophageplasticityinexperimentalatherosclerosis.PLoSOne.2010;5:e8852
12. deGaetanoM,CreanD,BarryM,BeltonO.M1-andm2-typemacrophageresponsesarepredictiveofadverseoutcomesinhumanatherosclerosis.FrontImmunol.2016;7:275
13. Radi R, Rodriguez M, Castro L, Telleri R. Inhibition of mitochondrial electron transport byperoxynitrite.ArchBiochemBiophys.1994;308:89-95
14. ButteryLDK,SpringallDR,ChesterAH,EvansTJ,StandfieldN,ParumsDV,YacoubMH,PolakJM.Induciblenitricoxidesynthaseispresentwithinhumanatheroscleroticlesionsandpromotestheformationandactivityofperoxynitrite.LaboratoryInvestigation.1996;75:77-85
15. LowensteinCJ.Beneficialeffectsofneuronalnitricoxidesynthaseinatherosclerosis.ArteriosclerThrombVascBiol.2006;26:1417-1418
16. Medbury HJ, James V, Ngo J, Hitos K, Wang Y, Harris DC, Fletcher JP. Differing association ofmacrophagesubsetswithatheroscleroticplaquestability.IntAngiol.2013;32:74-84
17. Peled M, Fisher EA. Dynamic aspects of macrophage polarization during atherosclerosisprogressionandregression.FrontImmunol.2014;5:579
18. MallatZ,BesnardS,DuriezM,DeleuzeV,EmmanuelF,BureauMF,SoubrierF,EspositoB,DuezH,Fievet C, Staels B, Duverger N, Scherman D, Tedgui A. Protective role of interleukin-10 inatherosclerosis.CircRes.1999;85:e17-24
19. MallatZ,GojovaA,Marchiol-FournigaultC,EspositoB,KamateC,MervalR,FradeliziD,TedguiA.Inhibitionoftransforminggrowthfactor-betasignalingacceleratesatherosclerosisandinducesanunstableplaquephenotypeinmice.CircRes.2001;89:930-934
20. PeterkofskyB,UdenfriendS.Conversionofprolinetocollagenhydroxyprolineinacell-freesystemfromchickembryo.JBiolChem.1963;238:3966-3977

Chapter6
139
21. MillsCD,KincaidK,AltJM,HeilmanMJ,HillAM.M-1/m-2macrophagesandtheth1/th2paradigm.JImmunol.2000;164:6166-6173
22. KuhlencordtPJ,GyurkoR,HanF, Scherrer-CrosbieM,AretzTH,HajjarR,PicardMH,HuangPL.Accelerated atherosclerosis, aortic aneurysm formation, and ischemic heart disease inapolipoprotein e/endothelial nitric oxide synthase double-knockout mice. Circulation.2001;104:448-454
23. Hayashi T, Sumi D, Juliet PAR,Matsui-Hirai H, Asai-Tanaka Y, Kano H, Fukatsu A, Tsunekawa T,Miyazaki A, Iguchi A, Ignarro LJ. Gene transfer of endothelial no synthase, but not enos plusinduciblenos,regressedatherosclerosisinrabbits.CardiovascularResearch.2004;61:339-351
24. Mujynya-LudungeK,ViswambharanH,DriscollR,MingXF,vonSegesserLK,KappenbergerL,YangZH, Vassalli G. Endothelial nitric oxide synthase gene transfer restores endothelium-dependentrelaxationsandattenuateslesionformationincarotidarteriesinapolipoproteine-deficientmice.BasicResearchinCardiology.2005;100:102-111
25. OzakiM, Kawashima S, Yamashita T,Hirase T,NamikiM, InoueN,HirataK, YasuiH, SakuraiH,YoshidaY,MasadaM,YokoyamaM.Overexpressionofendothelialnitricoxidesynthaseacceleratesatherosclerotic lesion formation in apoe-deficient mice. Journal of Clinical Investigation.2002;110:331-340
26. DetmersPA,HernandezM,MudgettJ,HassingH,BurtonC,MundtS,ChunS,FletcherD,CardDJ,LisnockJ,WeikelR,BergstromJD,ShevellDE,Hermanowski-VosatkaA,SparrowCP,ChaoYS,RaderDJ, Wright SD, Pure E. Deficiency in inducible nitric oxide synthase results in reducedatherosclerosisinapolipoproteine-deficientmice.JournalofImmunology.2000;165:3430-3435
27. KuhlencordtPJ,Chen JQ,HanF,Astern J,HuangPL.Geneticdeficiencyof induciblenitricoxidesynthasereducesatherosclerosisandlowersplasmalipidperoxidesinapolipoproteine-knockoutmice.Circulation.2001;103:3099-3104
28. vanTitsLJ,StienstraR,vanLentPL,NeteaMG,JoostenLA,StalenhoefAF.Oxidizedldlenhancespro-inflammatoryresponsesofalternativelyactivatedm2macrophages:Acrucialroleforkruppel-likefactor2.Atherosclerosis.2011;214:345-349
29. CaligiuriG,NicolettiA,PoirierB,HanssonGK.Protectiveimmunityagainstatherosclerosiscarriedbybcellsofhypercholesterolemicmice.JClinInvest.2002;109:745-753
30. RobbinsCS,ChudnovskiyA,RauchPJ,FigueiredoJL,IwamotoY,GorbatovR,EtzrodtM,WeberGF,UenoT,vanRooijenN,Mulligan-KehoeMJ,LibbyP,NahrendorfM,PittetMJ,WeisslederR,SwirskiFK.Extramedullaryhematopoiesisgeneratesly-6c(high)monocytesthatinfiltrateatheroscleroticlesions.Circulation.2012;125:364-U415
31. MartinezFO,GordonS.Them1andm2paradigmofmacrophageactivation:Timeforreassessment.F1000PrimeRep.2014;6:13
32. MosserDM,EdwardsJP.Exploringthefullspectrumofmacrophageactivation.NatRevImmunol.2008;8:958-969
33. JablonskiKA,AmiciSA,WebbLM,Ruiz-RosadoJdeD,PopovichPG,Partida-SanchezS,Guerau-de-Arellano M. Novel markers to delineate murine m1 and m2 macrophages. PLoS One.2015;10:e0145342
34. OuimetM,EdiriweeraHN,GundraUM,SheedyFJ,RamkhelawonB,HutchisonSB,RineholdK,vanSolingenC,FullertonMD,CecchiniK,RaynerKJ,SteinbergGR,ZamorePD,FisherEA,LokeP,MooreKJ.Microrna-33-dependentregulationofmacrophagemetabolismdirectsimmunecellpolarizationinatherosclerosis.JClinInvest.2015;125:4334-4348
35. SchultzeSM,HemmingsBA,NiessenM,TschoppO.Pi3k/akt,mapkandampksignalling:Proteinkinasesinglucosehomeostasis.ExpertRevMolMed.2012;14:e1
36. RaoKM.Mapkinaseactivationinmacrophages.JLeukocBiol.2001;69:3-1037. VergadiE, IeronymakiE, LyroniK,VaporidiK,TsatsanisC.Aktsignalingpathway inmacrophage
activationandm1/m2polarization.JImmunol.2017;198:1006-101438. LuyendykJP,SchabbauerGA,TencatiM,HolscherT,PawlinskiR,MackmanN.Geneticanalysisof
the roleof thepi3k-aktpathway in lipopolysaccharide-induced cytokineand tissue factor geneexpressioninmonocytes/macrophages.JImmunol.2008;180:4218-4226

Chapter6
140
39. George S, Rochford JJ, Wolfrum C, Gray SL, Schinner S, Wilson JC, Soos MA, Murgatroyd PR,WilliamsRM,AceriniCL,DungerDB,BarfordD,UmplebyAM,WarehamNJ,DaviesHA,SchaferAJ,StoffelM,O'Rahilly S, Barroso I. A familywith severe insulin resistance and diabetes due to amutationinakt2.Science.2004;304:1325-1328
40. FrankishH.Akt2-deficientmiceshowsymptomsoftype2diabetes.Lancet.2001;357:1771-177141. GarofaloRS,OrenaSJ,RafidiK,TorchiaAJ,StockJL,HildebrandtAL,CoskranT,BlackSC,BreesDJ,
WicksJR,McNeishJD,ColemanKG.Severediabetes,age-dependent lossofadiposetissue,andmild growth deficiency in mice lacking akt2/pkb beta. Journal of Clinical Investigation.2003;112:197-208
42. BabaevVR,HebronKE,WieseCB,TothCL,DingL,ZhangYM,MayJM,FazioS,VickersKC,LintonMF. Macrophage deficiency of akt2 reduces atherosclerosis in ldlr null mice. Journal of LipidResearch.2014;55:2296-2308
43. RotllanN,Chamorro-JorganesA,AraldiE,WanschelAC,AryalB,ArandaJF,GoedekeL,SalernoAG,RamirezCM,SessaWC,SuarezY,Fernandez-HernandoC.Hematopoieticakt2deficiencyattenuatestheprogressionofatherosclerosis.FasebJournal.2015;29:597-610
44. RensingKL,deJagerSC,StroesES,VosM,TwicklerMT,Dallinga-ThieGM,deVriesCJ,KuiperJ,BotI, von der Thusen JH. Akt2/ldlr double knockout mice display impaired glucose tolerance anddevelop more complex atherosclerotic plaques than ldlr knockout mice. Cardiovasc Res.2014;101:277-287
45. VergadiE,VaporidiK,TheodorakisEE,DoxakiC,LagoudakiE,IeronymakiE,AlexakiVI,HelmsM,Kondili E, Soennichsen B, Stathopoulos EN, Margioris AN, Georgopoulos D, Tsatsanis C. Akt2deficiency protects from acute lung injury via alternativemacrophage activation andmir-146ainductioninmice.JImmunol.2014;192:394-406
46. Arranz A, Doxaki C, Vergadi E,Martinez de la Torre Y, Vaporidi K, Lagoudaki ED, Ieronymaki E,AndroulidakiA,VenihakiM,MargiorisAN,StathopoulosEN,TsichlisPN,TsatsanisC.Akt1andakt2protein kinases differentially contribute tomacrophage polarization.ProcNatl Acad Sci U S A.2012;109:9517-9522
47. RomanoskiCE,LinkVM,HeinzS,GlassCK.Exploitinggenomicsandnaturalgeneticvariationtodecodemacrophageenhancers.TrendsImmunol.2015;36:507-518
48. KammengaJE.Thebackgroundpuzzle:Howidenticalmutationsinthesamegeneleadtodifferentdiseasesymptoms.TheFEBSJournal.2017
49. CauntCJ,KeyseSM.Dual-specificitymapkinasephosphatases (mkps): Shaping theoutcomeofmapkinasesignalling.FEBSJ.2013;280:489-504
50. Lang R, HammerM,Mages J. Duspmeet immunology: Dual specificitymapk phosphatases incontroloftheinflammatoryresponse.JournalofImmunology.2006;177:7497-7504
51. Sim J, Yi K, Kim H, Ahn H, Chung Y, Rehman A, Jang SM, Lee KH, Jang K, Paik SS.Immunohistochemical expression of dual-specificity protein phosphatase 4 in patients withcolorectaladenocarcinoma.GastroenterolResPract.2015;2015:283764
52. SaeedS,QuintinJ,KerstensHH,RaoNA,AghajanirefahA,MatareseF,ChengSC,RatterJ,BerentsenK,vanderEntMA,SharifiN,Janssen-MegensEM,TerHuurneM,MandoliA,vanSchaikT,NgA,BurdenF,DownesK,FrontiniM,KumarV,Giamarellos-BourboulisEJ,OuwehandWH,vanderMeerJW,JoostenLA,WijmengaC,MartensJH,XavierRJ,LogieC,NeteaMG,StunnenbergHG.Epigeneticprogrammingofmonocyte-to-macrophagedifferentiationandtrained innate immunity.Science.2014;345:1251086
53. Rask-AndersenM,ZhangJ,FabbroD,SchiothHB.Advancesinkinasetargeting:Currentclinicaluseandclinicaltrials.TrendsinPharmacologicalSciences.2014;35:60-76
54. Sag D, Carling D, Stout RD, Suttles J. Adenosine 5 '-monophosphate-activated protein kinasepromotes macrophage polarization to an anti-inflammatory functional phenotype. Journal ofImmunology.2008;181:8633-8641
55. Li C, Ding XY, Xiang DM, Xu J, Huang XL, Hou FF, Zhou QG. Enhanced m1 and impaired m2macrophagepolarization and reducedmitochondrial biogenesis via inhibitionof amp kinase inchronickidneydisease.CellPhysiolBiochem.2015;36:358-372

Chapter6
141
56. GuhaM,Mackman N. The phosphatidylinositol 3-kinase-akt pathway limits lipopolysaccharideactivationofsignalingpathwaysandexpressionofinflammatorymediatorsinhumanmonocyticcells.JBiolChem.2002;277:32124-32132
57. Jimenez-Garcia L, Herranz S, Luque A, Hortelano S. Critical role of p38 mapk in il-4-inducedalternativeactivationofperitonealmacrophages.EurJImmunol.2015;45:273-286
58. DingLX,GuHJ,GaoXM,XiongSD,ZhengB.Aurorakinasearegulatesm1macrophagepolarizationandplaysaroleinexperimentalautoimmuneencephalomyelitis.Inflammation.2015;38:800-811
59. CudejkoC,WoutersK,FuentesL,HannouSA,PaquetC,BantubungiK,BouchaertE,VanhoutteJ,FleuryS,RemyP,TailleuxA,Chinetti-GbaguidiG,DombrowiczD,StaelsB,PaumelleR.P16(ink4a)deficiency promotes il-4-induced polarization and inhibits proinflammatory signaling inmacrophages.Blood.2011;118:2556-2566
60. PajukantaP,LiljaHE,SinsheimerJS,CantorRM,LusisAJ,GentileM,DuanXJ,Soro-PaavonenA,Naukkarinen J, Saarela J, Laakso M, Ehnholm C, Taskinen MR, Peltonen L. Familial combinedhyperlipidemiaisassociatedwithupstreamtranscriptionfactor1(usf1).NatGenet.2004;36:371-376
61. CoonH, Xin Y, Hopkins PN, Cawthon RM,Hasstedt SJ, Hunt SC.Upstream stimulatory factor 1associatedwithfamilialcombinedhyperlipidemia, ldlcholesterol,andtriglycerides.HumGenet.2005;117:444-451
62. LaurilaPP,SoronenJ,KooijmanS,ForsstromS,BoonMR,SurakkaI,KaiharjuE,CoomansCP,VanDen Berg SA, Autio A, Sarin AP, Kettunen J, Tikkanen E,Manninen T,Metso J, Silvennoinen R,MerikantoK,RuuthM,PerttilaJ,MakelaA,IsomiA,TuomainenAM,TikkaA,RamadanUA,SeppalaI,LehtimakiT,ErikssonJ,HavulinnaA,JulaA,KarhunenPJ,SalomaaV,PerolaM,EhnholmC,Lee-RueckertM,VanEckM,RoivainenA,TaskinenMR,PeltonenL,MervaalaE,JalankoA,HohtolaE,OlkkonenVM,Ripatti S,KovanenPT,RensenPC,SuomalainenA, JauhiainenM.Usf1deficiencyactivates brown adipose tissue and improves cardiometabolic health. Sci Transl Med.2016;8:323ra313
63. HappleC,LachmannN,SkuljecJ,WetzkeM,AckermannM,BrennigS,MucciA,JirmoAC,GroosS,Mirenska A, Hennig C, Rodt T, Bankstahl JP, Schwerk N, Moritz T, Hansen G. Pulmonarytransplantation ofmacrophage progenitors as effective and long-lasting therapy for hereditarypulmonaryalveolarproteinosis.ScienceTranslationalMedicine.2014;6
64. SuzukiT,ArumugamP,SakagamiT, LachmannN,ChalkC, SalleseA,AbeS,TrapnellC,CareyB,MoritzT,MalikP,LutzkoC,WoodRE,TrapnellBC.Pulmonarymacrophagetransplantationtherapy.Nature.2014;514:450-+
65. LeeS,KivimaeS,DolorA,SzokaFC.Macrophage-basedcelltherapies:Thelongandwindingroad.JControlRelease.2016;240:527-540
66. Fidler IJ. Inhibition of pulmonary metastasis by intravenous injection of specifically activatedmacrophages.CancerRes.1974;34:1074-1078
67. AndreesenR,ScheibenbogenC,BruggerW,KrauseS,MeerpohlHG,LeserHG,EnglerH,LohrGW.Adoptive transfer of tumor cytotoxic macrophages generated in vitro from circulating bloodmonocytes:Anewapproachtocancerimmunotherapy.CancerRes.1990;50:7450-7456
68. Hennemann B, Scheibenbogen C, Schumichen C, Andreesen R. Intrahepatic adoptiveimmunotherapy with autologous tumorcytotoxic macrophages in patients with cancer. JImmunotherEmphasisTumorImmunol.1995;18:19-27
69. FaradjiA,BohbotA,FrostH,Schmitt-GoguelM,SiffertJC,DufourP,EberM,LallotC,WieselML,BergeratJP,etal.Phaseistudyofliposomalmtp-pe-activatedautologousmonocytesadministeredintraperitoneallytopatientswithperitonealcarcinomatosis.JClinOncol.1991;9:1251-1260
70. BurgerM,ThiounnN,DenzingerS,KondasJ,BenoitG,ChapadoMS,Jimenz-CruzFJ,KisbenedekL,SzaboZ,ZsoltD,GrimmMO,RomicsI,ThuroffJW,KissT,TombalB,WirthM,MunsellM,MillsB,KohT,ShermanJ.Theapplicationofadjuvantautologousantravesicalmacrophagecelltherapyvs.Bcginnon-muscleinvasivebladdercancer:Amulticenter,randomizedtrial.JTranslMed.2010;8:54
71. Eymard JC, LopezM, Cattan A, BoucheO, Adjizian JC, Bernard J. Phase i/ii trial of autologousactivatedmacrophagesinadvancedcolorectalcancer.EurJCancer.1996;32A:1905-1911

Chapter6
142
72. Hennemann B, Beckmann G, Eichelmann A, Rehm A, Andreesen R. Phase i trial of adoptiveimmunotherapyofcancerpatientsusingmonocyte-derivedmacrophagesactivatedwithinterferongammaandlipopolysaccharide.CancerImmunolImmun.1998;45:250-256
73. LesimpleT,MoisanA,GuilleF,LeberreC,AudranR,DrenouB,ToujasL.Treatmentofmetastaticrenalcellcarcinomawithactivatedautologousmacrophagesandgranulocyte-macrophagecolony-stimulatingfactor.JImmunother.2000;23:675-679
74. Baron-BodoW,DoceurP,LefebvreML,LabroquereK,DefayeC,CambourisC,PrigentD,SalcedoM,BoyerA,NardinA.Anti-tumorpropertiesofhuman-activatedmacrophagesproducedinlargescaleforclinicalapplication.Immunobiology.2005;210:267-277
75. Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumormicroenvironment.TrendsImmunol.2012;33:119-126
76. SquadritoML,DePalmaM.Macrophageregulationoftumorangiogenesis:Implicationsforcancertherapy.MolAspectsMed.2011;32:123-145
77. MosserDM,Edwards JP.Exploring the full spectrumofmacrophageactivation.NatureReviewsImmunology.2008;8:958-969
78. HoeksemaMA, deWintherMP. Epigenetic regulation of monocyte andmacrophage function.AntioxidRedoxSignal.2016;25:758-774
79. ErnstJ,KheradpourP,MikkelsenTS,ShoreshN,WardLD,EpsteinCB,ZhangX,WangL,IssnerR,Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. Mapping and analysis of chromatin statedynamicsinninehumancelltypes.Nature.2011;473:43-49
80. MargueronR,ReinbergD.Chromatinstructureandtheinheritanceofepigeneticinformation.NatRevGenet.2010;11:285-296
81. MattickJS.Rnadrivingtheepigeneticbus.EMBOJ.2012;31:515-51682. MattickJS,TaftRJ,FaulknerGJ.Aglobalviewofgenomicinformation--movingbeyondthegene
andthemasterregulator.TrendsGenet.2010;26:21-2883. ZhouVW,GorenA,BernsteinBE.Chartinghistonemodificationsandthefunctionalorganizationof
mammaliangenomes.NatRevGenet.2011;12:7-1884. TammenSA,FrisoS,ChoiSW.Epigenetics:Thelinkbetweennatureandnurture.MolAspectsMed.
2013;34:753-76485. MukaT,KoromaniF,PortillaE,O'ConnorA,BramerWM,TroupJ,ChowdhuryR,DehghanA,Franco
OH.Theroleofepigeneticmodificationsincardiovasculardisease:Asystematicreview.IntJCardiol.2016;212:174-183
86. GrimaldiV,VietriMT,SchianoC,PicasciaA,DePascaleMR,FioritoC,CasamassimiA,NapoliC.Epigeneticreprogramminginatherosclerosis.CurrAtherosclerRep.2015;17:476
87. HaiZ,ZuoW.AberrantDNAmethylationinthepathogenesisofatherosclerosis.ClinChimActa.2016;456:69-74
88. TakeuchO,AkiraS.Epigeneticcontrolofmacrophagepolarization.EurJImmunol.2011;41:2490-2493
89. SatohT,TakeuchiO,VandenbonA,YasudaK,TanakaY,KumagaiY,MiyakeT,MatsushitaK,OkazakiT,SaitohT,HonmaK,MatsuyamaT,YuiK,TsujimuraT,StandleyDM,NakanishiK,NakaiK,AkiraS.The jmjd3-irf4axis regulatesm2macrophagepolarizationandhost responsesagainsthelminthinfection.NatImmunol.2010;11:936-944
90. Ivashkiv LB. Epigenetic regulation of macrophage polarization and function. Trends Immunol.2013;34:216-223
91. HoeksemaMA,GijbelsMJJ,VandenBosscheJ,vanderVeldenS,SijmA,NeeleAE,SeijkensT,StogerJL, Meiler S, Boshuizen MCS, Dallinga-Thie GM, Levels JHM, Boon L, Mullican SE, Spann NJ,CleutjensJP,GlassCK,LazarMA,deVriesCJM,BiessenEAL,DaemenMJAP,LutgensE,deWintherMPJ.Targetingmacrophagehistonedeacetylase3stabilizesatheroscleroticlesions.EmboMolMed.2014;6:1124-1132
92. VandenBosscheJ,NeeleAE,HoeksemaMA,deHeijF,BoshuizenMCS,vanderVeldenS,deBoerVC, Reedquist KA, de Winther MPJ. Inhibiting epigenetic enzymes to improve atherogenicmacrophagefunctions.BiochemBiophResCo.2014;455:396-402

Chapter6
143
93. HaradaT,OhguchiH,GrondinY,KikuchiS,SagawaM,TaiYT,MazitschekR,HideshimaT,AndersonKC.Hdac3regulatesdnmt1expressioninmultiplemyeloma:Therapeuticimplications.Leukemia.2017
94. vanKampenE, JaminonA,vanBerkelTJ,VanEckM.Diet-induced(epigenetic)changes inbonemarrowaugmentatherosclerosis.JLeukocBiol.2014;96:833-841
95. FeigJE,ParathathS,RongJX,MickSL,VengrenyukY,GrauerL,YoungSG,FisherEA.Reversalofhyperlipidemia with a genetic switch favorably affects the content and inflammatory state ofmacrophagesinatheroscleroticplaques.Circulation.2011;123:989-U141
96. Feig JE,RongJX,ShamirR,SansonM,VengrenyukY,Liu JH,RaynerK,MooreK,GarabedianM,Fisher EA. Hdl promotes rapid atherosclerosis regression in mice and alters inflammatorypropertiesofplaquemonocyte-derivedcells.PNatlAcadSciUSA.2011;108:7166-7171
97. TroganE,FeigJE,DoganS,RothblatGH,AngeliV,TackeF,RandolphGJ,FisherEA.Geneexpressionchangesinfoamcellsandtheroleofchemokinereceptorccr7duringatherosclerosisregressioninapoe-deficientmice.PNatlAcadSciUSA.2006;103:3781-3786
98. K.RahmanYVeaJip.JCIinpress.201799. GuoJ,VanEckM,TwiskJ,MaedaN,BensonGM,GrootPHE,VanBerkelTJC.Transplantationof
monocyte cc-chemokine receptor 2-deficient bone marrow into apoe3-leiden mice inhibitsatherogenesis.ArteriosclThromVas.2003;23:447-453
100. Graff JW, Dickson AM, Clay G, McCaffrey AP, Wilson ME. Identifying functional micrornas inmacrophageswithpolarizedphenotypes.JBiolChem.2012;287:21816-21825
101. KimSS,YeCT,KumarP,ChiuI,SubramanyaS,WuHQ,ShankarP,ManjunathN.Targeteddeliveryofsirnatomacrophagesforanti-inflammatorytreatment.MolecularTherapy.2010;18:993-1001
102. AouadiM, Tesz GJ, Nicoloro SM,WangM, ChouinardM, Soto E, Ostroff GR, CzechMP. Orallydelivered sirna targeting macrophage map4k4 suppresses systemic inflammation. Nature.2009;458:1180-1184
103. PushparajPN,AarthiJJ,ManikandanJ,KumarSD.Sirna,mirna,andshrna:Invivoapplications.JDentRes.2008;87:992-1003
104. TroegelerA,LastrucciC,DuvalC,TanneA,CougouleC,Maridonneau-PariniI,NeyrollesO,Lugo-VillarinoG.Anefficientsirna-mediatedgenesilencinginprimaryhumanmonocytes,dendriticcellsandmacrophages.ImmunolCellBiol.2014;92:699-708
105. ZhaoW,LeiT,LiH,SunD,MoX,WangZ,ZhangK,OuH.Macrophage-specificoverexpressionofinterleukin-5attenuatesatherosclerosisinldlreceptor-deficientmice.GeneTher.2015;22:645-652
106. TranTH,RastogiR,ShelkeJ,AmijiMM.Modulationofmacrophagefunctionalpolaritytowardsanti-inflammatory phenotype with plasmid DNA delivery in cd44 targeting hyaluronic acidnanoparticles.SciRep.2015;5:16632
107. TiwariS,ChaturvediAP,TripathiYB,MishraB.Macrophage-specifictargetingofisoniazidthroughmannosylatedgelatinmicrospheres.AapsPharmscitech.2011;12:900-908
108. BrandhonneurN,ChevanneF,VieV,FrischB,PrimaultR,LePotierMF,LeCorreP.Specificandnon-specific phagocytosis of ligand-grafted plga microspheres by macrophages. Eur J Pharm Sci.2009;36:474-485
109. MatobaT,EgashiraK.Nanoparticle-mediateddrugdeliverysystemforcardiovasculardisease.IntHeartJ.2014;55:281-286
110. MatobaT,KogaJI,NakanoK,EgashiraK,TsutsuiH.Nanoparticle-mediateddrugdeliverysystemforatheroscleroticcardiovasculardisease.JCardiol.2017
111. KraakmanMJ,MurphyAJ, Jandeleit-DahmK,KammounHL.Macrophagepolarization inobesityandtype2diabetes:Weighingdownourunderstandingofmacrophagefunction?FrontImmunol.2014;5
112. PanniRZ,LinehanDC,DeNardoDG.Targetingtumor-infiltratingmacrophagestocombatcancer.Immunotherapy-Uk.2013;5:1075-1087
113. UdalovaIA,MantovaniA,FeldmannM.Macrophageheterogeneityinthecontextofrheumatoidarthritis.NatRevRheumatol.2016;12:472-485

Chapter6
144
114. Moreira AP, Hogaboam CM. Macrophages in allergic asthma: Fine-tuning their pro- and anti-inflammatoryactionsfordiseaseresolution.JInterfCytokRes.2011;31:485-491
115. VogelDYS,VereykenEJF,GlimJE,HeijnenPDAM,MoetonM,vanderValkP,AmorS,TeunissenCE,van Horssen J, Dijkstra CD. Macrophages in inflammatory multiple sclerosis lesions have anintermediateactivationstatus.JNeuroinflamm.2013;10
116. Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissuemacrophagepolarization.JournalofClinicalInvestigation.2007;117:175-184
117. Brynskikh AM, Zhao YL,Mosley RL, Li S, BoskaMD, KlyachkoNL, Kabanov AV, GendelmanHE,BatrakovaEV.Macrophagedeliveryoftherapeuticnanozymes inamurinemodelofparkinson'sdisease.Nanomedicine-Uk.2010;5:379-396
�

Abbreviations
145
AbbreviationsABC ATP-bindingcassetteacLDL acetylated-lowdensitylipoproteinAkt proteinkinaseBApo apolipoproteinArg1 arginase1Atgl adiposetriglyceridelipaseBAT brownadiposetissueBMDMs Bonemarrow-derivedmacrophagesBMT bonemarrowtransplantationCAD coronaryarterydiseaseCETP cholesterylestertransferproteindKO doubleknockoutDUSPs dual-specificityphosphatasesERK extracellularregulatedproteinkinaseFACS fluorescentactivatedcellsortingFASN fattyacidsynthaseFCHL familialcombinedhyperlipidemiaFPLC fastproteinliquidchromatographyGlut4 glucosetransportertype4HDL high-densitylipoproteinHL hepaticlipaseHSL hormone-sensitivelipaseIDL intermediate-densitylipoproteinIL-1β interleukin1βiNOS induciblenitricoxidesynthaseJNK c-JunN-terminalkinaseLDL low-densitylipoproteinLDLrKO LDLreceptorknockoutLPS lipopolysaccharidesLRP1 low-densitylipoproteinreceptor-relatedprotein1M1 classicallyactivatedmacrophagesM2 alternativelyactivatedmacrophagesMAPK mitogen-activatedproteinkinasesMKPs MAPkinasephosphatasesMTC Masson’sTrichromemtROS mitochondria-derivedreactiveoxygenspeciesMTTP microsomaltriglyceridetransferproteinnNOS endothelialNOS(eNOS),neuronalNOSNO nitricoxideOAT ornithineamino-transferaseOGTT oralglucosetolerancetestoxLDL oxidizedLDL

Abbreviations
146
p38 stress-activatedproteinkinasePlin lipiddroplet-associatedproteinRCT reversecholesteroltransportROS reactiveoxygenspeciesSMCs smoothmusclecellsSR-BI scavengerreceptorclassBtypeITG triglyceridesTNF-α tumornecrosisfactorαTRL triglyceride-richlipoproteinsUCP2 uncouplingprotein2Usfs upstreamstimulatoryfactorsVCAM-1 vascularcelladhesionmolecule-1VLDL very-low-densitylipoproteinVSMC vascularsmoothmusclecellsWAT whiteadiposetissueWBC whitebloodcellWT wild-typeWTD Western-TypeDiet

Listofpublications
147
Listofpublications
BaoyanRen, ErikVanKampen,Theo JCVanBerkel, SheenaM.Cruickshank,MirandaVanEck.Hematopoieticarginase1deficiency results indecreased leukocytosisand increased foamcellformationbutdoesnotaffectatherosclerosis.Atherosclerosis256(2017):35-46.
Baoyan Ren, Pirkka-Pekka Laurila, Reeni B. Hildebrand, Jarkko Soronen, Vanessa Frodermann,ZhuangLi,MariëtteR.Boon,JanineJ.Geerling,PatrickC.N.Rensen,ChristianEhnholm,PetriT.Kovanen,MattiJauhiainen,MennoHoekstra,MirandaVanEck.EnhancedatheroscleroticlesiondevelopmentinLDLreceptorknockoutmicelackingUpstreamStimulatingFactor1(Usf1)inbonemarrow-derivedcells.(submittedforpublication).
BaoyanRen,MennoHoekstra,JanineJ.Geerling,PeterVanSantbrink,RobinPlevin,MirandaVanEck.MacrophageMKP2 deficiency is associated with anM2-driven foam cell phenotype andincreasesatherosclerosissusceptibilityofLDLreceptorknockoutmice.(submittedforpublication).
BaoyanRen,MennoHoekstra,RonaldvanderSluis,MaraKröner, JanineG.Geerling, IlzeBot,MirandaVan Eck.HematopoieticAkt2 restoration enhances foam cell formationbut does notaffectatherosclerosisinAkt2/LDLreceptordoubleknockoutmice.(Manuscriptinpreparation).
OlgaS.CSnip,BoayanRen,D.HuyenTran,JanineJ.Geerling,MirandaVanEck.LeukocyteABCA1ImpedesProgressionofEstablishedAtheroscleroticLesionsafterDietaryCholesterolLoweringinLDLr-/-Mice.(Manuscriptinpreparation)
RickvanderGeest,JanineJ.Geerling,MennoHoekstra,BaoyanRen,LidewijR.deLeeuw,RichardVerbeek,JohannesM.vanNoort,MirandaVanEck.HeatshockproteinalphaB-crystallinpromotesmacrophagefoamcell formationandaggravatesearlyatherosclerosis inLDLreceptor-deficientmice.(Manuscriptinpreparation)
Jianhua Li, Qianzhong Han, Pengtao Gong, Tuo Yang, Baoyan Ren, Shijie Li, Xichen Zhang.Toxoplasma gondii rhomboid protein 1 (TgROM1) is a potential vaccine candidate againsttoxoplasmosis.Veterinaryparasitology184,2(2012):154-160.
GuilianYang, JianhuaLi,XichenZhang,QuanZhao,PengtaoGong,BaoyanRen,GuocaiZhang.Eimeria tenella: Cloning and characterization of telomerase reverse transcriptasegene.Experimentalparasitology124,4(2010):380-385.


CurriculumVitae
149
CurriculumVitae
Baoyan Renwas born on February 3rd 1985 in Dingzhou, P. R. China. She grew up there andgraduated fromExperimentalHigh School ofDingzhou in 2004. Afterwards, shewent to JilinUniversityandreceivedfive-yearseducation inVeterinaryMedicine. In2009,sheobtainedherbachelor degree and continued her three-years postgraduate study in the key laboratory ofZoonosisResearchunderthesupervisionofProf.Dr.XichenZhanginthesameuniversitywithafull scholarship funded by JilinUniversity. During hermaster studies, she focused on parasiteantigens as possible targets for cancer immunotherapy. She finished her master studies andreceivedhermasterdegreein2012.InLeiden,theNetherlands,shestartedherPhDprogramwithafullscholarshipfundedbyChinaScholarshipCouncil(CSC)inthatsameyear.TheresheworkedatthedivisionofBiopharmaceuticsoftheLeidenAcademicCentreforDrugResearch(LACDR)atLeidenUniversityunder supervisionofProf.Dr.Miranda vanEck.DuringherPhD studies, herresearchfocusedonbonemarrowtransplantationinmiceasatooltoinvestigateM2macrophageactivationpathwaysinatherogenesis,asdescribedinthisthesis.


PhDPortfolio
151
PhDPortfolio
CoursesandWorkshops2015 Effectivecommunication2015 CardiovascularPhD-trainingcourse2014 Timemanagement,self-management2014 Communicationinscience2014 Introductiontoteaching&supervisionforLACDRPhDstudents2014 LACDRPhDIntroductoryCourseonDrugResearch2014 Onbeingascientist2014 HealthPhysicsexpertlevel5B2013 ULLASummerschool2013 Proefdierkunde(LaboratoryAnimalScienceCourse)2012 LACDRcourseonAtherosclerosis
(Inter)NationalPosterPresentations2016 LACDRSpringSymposium,Leiden,TheNetherlands
2015 17thInternationalSymposiumonAtherosclerosis(ISA2015),Amsterdam,TheNetherlands,
2015 LACDRSpringSymposium,Leiden,TheNetherlands2015 CardiovascularPhD-trainingcourse,Arnhem,TheNetherlands2014 LACDRSpringSymposium,Leiden,TheNetherlands2014 20thAnnualScandinavianAtherosclerosisConference,Humlebæk,Denmark2014 The5thRembrandtSymposium,Noordwijkerhout,TheNetherlands2013 ULLAsummerschool,London,UK2013 The4thRembrandtSymposium,Noordwijkerhout,TheNetherlands2013 LACDRSpringSymposium,Leiden,TheNetherlands
Teaching
2016 9-monthresearchprojectMScstudentBio-PharmaceuticalSciences(BPS)2015 InternationalBPSSummerSchool2015 TherapeuticModulationofAtherosclerosisBScBPSLaboratorycourse2014 DrugAdministrationandDistributionBScBPSLaboratorycourse2014 10-weekresearchprojectsBScstudentsBPS
2014 4-monthresearchprojectErasmusMScstudentChemistryandPharmaceuticalTechnology(Parma,Italy)
2013 TherapeuticModulationofAtherosclerosisBScBPSLaboratorycourse2013 PharmaceuticaladministrationanddistributionBScBPSLaboratorycourse2013 10-weekresearchprojectsBScstudentsBPS


��: thesis final 20171117 for macbook version.docx ���: /Users/olive_ren/Library/Containers/com.microsoft.Word/Data/Documents ��: /Users/olive_ren/Library/Group Containers/UBF8T346G9.Office/User
Content.localized/Templates.localized/Normal.dotm � : � : ��: Office 365 ���: ��: ��� : 2017/10/30 PM2:56:00 ���: 172 ����� : 2017/11/18 AM12:27:00 �����: Office 365 �����: 29 �� �����: 2017/11/18 AM12:28:00 � ��� �: 152 �: 102,291 (�) ��: 583,061 (�)



![PhD Thesis [Corrected Version] - Minjie Cai](https://static.fdokumen.com/doc/165x107/6327dcb3e491bcb36c0b7db0/phd-thesis-corrected-version-minjie-cai.jpg)
















