
Axonal transport defects: a common theme in neurodegenerative diseases
Upload
independentCategory
view
1download
0
1
The transition metals copper and iron in neurodegenerative diseases
Susana Rivera-Mancía1 , Iván Pérez-Neri1, Camilo Ríos1, Luis Tristán-López1, Liliana Rivera-
Espinosa2, Sergio Montes1*.
1. Neurochemistry Department, National Institute of Neurology and Neurosurgery
‘Manuel Velasco Suárez’, Mexico City, Mexico.
2. Pharmacology Department; National Institute of Pediatrics, Mexico City, Mexico
* Corresponding author.
National Institute of Neurology and Neurosurgery ‘Manuel Velasco Suárez’.
Insurgentes Sur 3877,
La Fama, Tlalpan,
Mexico City 14269
Mexico.
e-mail: [email protected]
Tel.: + 52 55 5606 3822 Ext. 2006; fax +52 55 5424 0808

2
ABSTRACT
Neurodegenerative diseases constitute a worldwide health problem. Metals like iron and
copper are essential for life, but they are also involved in several neurodegenerative
mechanisms such as protein aggregation, free radical generation and oxidative stress.
The role of Fe and Cu, their pathogenic mechanisms and possible therapeutic relevance
are discussed regarding four of the most common neurodegenerative diseases,
Alzheimer’s, Parkinson’s and Huntington’s diseases as well as amyotrophic lateral
sclerosis. Metal-mediated oxidation by Fenton chemistry is a common feature for all
those disorders and takes part of a self-amplifying damaging mechanism, leading to
neurodegeneration. The interaction between metals and proteins in the nervous system
seems to be a crucial factor for the development or absence of neurodegeneration. The
present review also deals with the therapeutic strategies tested, mainly using metal
chelating drugs. Metal accumulation within the nervous system observed in those
diseases could be the result of compensatory mechanisms to improve metal availability
for physiological processes.
Keywords: Alzheimer’s disease; Parkinson’s disease; Amyotrophic lateral sclerosis;
Huntington’s disease; Copper; Iron.
Abbreviations: AD, Alzheimer’s disease; PD, Parkinson’s disease; HD, Huntington’s
disease; ALS, Amyotrophic lateral sclerosis; ROS, Reactive oxygen species; ATP7A,
Copper-transporting P-type ATPase; TfR, Transferrin receptor; DMT1, Divalent metal
transporter 1; Abeta, Amyloid beta; APP, Amyloid precursor protein; MMP, Matrix
metalloprotease; CHO, Chinese Hamster Ovary; CSF, Cerebrospinal fluid; Cp,
Ceruloplasmin; SOD, Superoxide dismutase; EGCG, epigallocatechin-3-gallate; Akt,
Protein kinase B; GSK, Glycogen synthase kinase; CQ, Clioquinol; GTSM,
Glyoxalbis(N(4)-methyl-3-thiosemicarbazonato); SN, substantia nigra; SNpc,
Substantia nigra pars compacta; NMDA, N-methyl-D-aspartate; NO, nitric oxide; 6-
OHDA, 6-hydroxydopamine; Ireg-1, ferroportin; MPTP, 1-methyl-4-phenyl-1,2,3,6-
tetrahydropyridine; MPP+, 1-methyl-4-phenylpyridinium; FALS, Familial amyotrophic
lateral sclerosis; HFE, Hemochromatosis gene; MBR, Metal binding region; WTL,
Wild-type like; TTM, Ammonium tetrathiomolybdate; Htt, Huntingtin, LDH, Lactate
dehydrogenase; ESC, Embryonic stem cells.

3
1. Introduction
In recent years, neurodegenerative diseases have become an important worldwide health
issue. Those diseases affect the nervous system and share features such as selective
neuronal death, protein aggregation, oxidative stress, mitochondrial dysfunction,
transition metal accumulation and inflammation [1-5]. Neurodegenerative diseases are
associated with ageing but their exact etiology remains still unknown [1].
Almost all living organisms require Fe, Cu and other transition metals to correctly carry
out their most essential metabolic processes. Metals are involved in several important
functions in the nervous system: Fe is required to support the brain’s high respiratory
rate as well as for myelination, gene expression and neurotransmitters synthesis [6,7].
Cu is also required for mitochondrial respiration, neurotransmitter biosynthesis and as a
cofactor for antioxidant enzymes [8]
Although transition metals are important for life, it has been evidenced that they are also
involved in neuronal damage in many neurodegenerative disorders. Neurodegenerative
diseases associated with the disruption of brain metal homeostasis include Alzheimer’s
(AD), Parkinson’s (PD) and Huntington’s (HD) diseases as well as amyotrophic lateral
sclerosis (ALS) [5,9,10]. It has been observed that patients with neurodegenerative
diseases accumulate metals in their nervous system [11,12], suggesting a role of metals
in those disorders. Fe homeostasis is frequently altered in neurodegenerative
disorders[12,13]; under certain conditions, this metal is the most potent pro-oxidant due
to its high availability [14]. ]. The excessive production of reactive oxygen species
(ROS), oxidizes proteins, DNA and phospholipids leading to structural and functional
alterations [15]. Metal-binding proteins and DNA may therefore be vulnerable. Most of
the reactions involving Fe are related to Fenton chemistry, a series of reactions that
initiates with transition metals and hydrogen peroxide leading to the formation of highly
unstable radicals that affect biological macromolecules [5].
Proteins involved in metal transport and distribution in the nervous system, such as
copper transporter protein 1 and ATP7A (Copper-transporting P-type ATPase) for Cu
[2,16], transferrin and TfR (transferrin receptor) for Fe, and DMT1 (Divalent metal
transporter 1) for both Cu and Fe [17], could be involved in their altered status in the
brain of patients with neurodegenerative diseases.
This review focuses on the role of Fe, Cu and the proteins related to them, in the
underlying mechanisms of neurodegenerative diseases such as AD, PD, ALS and HD,
as well as some attempts that have been carried out to treat them.

4
2. Alzheimer’s disease
Several factors have been involved in the etiology of AD: aging, oxidative stress and
metal brain accumulation. This disorder is characterized by memory impairment,
progressive decline in cognitive function and dementia [18]. The increasing prevalence
of dementia [19] is a matter of public health and social concern since nowadays there is
no effective treatment or preventive management for this neurological disorder.
Several histopathological studies have documented that AD is characterized by the
presence of a) senile plaques, mainly formed by Amyloid beta (Abeta) peptides located
extracellularly and b) neurofibrillary tangles, an altered intracellular arrangement of the
Tau protein [20,21]. Evidence from genetic and biochemical studies support the
hypothesis that accumulation of insoluble aggregates composed of Abeta peptides
constitutes one of the main components in the pathogenesis and subsequent events of
AD [22,23]. Abeta peptides (39-43 amino acid polypeptides) are generated from the
Amyloid Precursor Protein (APP) [24], a membrane protein widely distributed in the
brain whose function is still unknown. The APP is cleaved by a group of protein-
processing complexes called secretases [23]. The β and secretases have been involved
in the generation of Abeta 1-40 and 1-42 peptides; the latter aggregates easily and is
frequently present in senile plaques and cerebrovascular amyloid deposits [25,26] (Fig
1).
Transition metals are necessary for the correct functioning of antioxidant systems in the
cell; however, an increasing body of evidence has been published regarding the role of
metals in AD [27-29], especially redox active metals; since it has been suggested that
these metals could be involved in most events underlying the pathogenesis and
progression of this neurological disease including oxidative stress and protein
aggregation.
_____________________________________________________________fig.1
2.1. Fe homeostasis in Alzheimer’s disease
Fe is involved in the pathophysiology of AD as suggested by its presence in senile
plaques and neurofibrillary tangles in post mortem brains from AD patients [29]. Fe has
been associated with free radical generation through the Fenton reaction, leading to the
formation of the highly-reactive hydroxyl radical. It is worth mentioning that the
hydroxyl radical not only affects cell molecules such as membrane lipids, proteins and
nucleic acids but also seems to contribute to Abeta aggregation by promoting covalent
binding between peptide monomers [30]. The association between Abeta and transition

5
metals such as Fe and Cu may also lead to the generation of hydrogen peroxide,
exacerbating the oxidative damage [31,32].
The role of Fe in this disorder is supported by the studies linking hereditary
hemochromatosis to AD [33] and the presence of an iron responsive element in the
untranslated region of the APP gene [34]. Additionally, ferritin is also present in senile
plaques in brains from patients with AD [35]. Supporting an important role for iron in
AD, a study carried out by Ding et al. [36] showed positive correlation between iron
levels in the hippocampus, measured by phase imaging, and the Mini-Mental State
Examination of AD patients.
An interesting strategy for AD treatment has been Fe chelation, since this metal is
involved in free radicals generation and thus, in oxidative stress. Reports concerning the
Fe chelating approach have been historically documented [37] and current attempts are
looking for molecules not only with metal-binding properties but also with the ability to
diminish oxidative stress, as will be discussed later.
2.2. Cu homeostasis in AD
Current evidence suggests that AD development involves an altered Cu homeostasis.
On the one hand, some studies support the idea that Cu participates in the development
of AD as a noxious metal. On the other hand, some other findings suggest that AD
could be the result of diminished availability of Cu in neurons. In regard of the former,
it should be mentioned that high content of Cu has been found in amyloid plaques from
AD brains [29]. In the triple transgenic murine model of AD; exposure to Cu in the
drinking water for three and nine months produced the exacerbation of both Abeta and
Tau pathologic consequences [38]. Such study suggested that Cu may be influencing
not only the senile plaque, but also the neurofibrillary tangles. It should be noted that
the Abetas involved in the pathogenesis of AD show high affinity for Cu [39]. Cu
binding possibly promotes Abeta toxicity through the formation of hydrogen peroxide
and the subsequent generation of free radicals through the Fenton reaction, as has been
extensively reported for Fe [40,41]; this effect may involve a one-electron Cu (II)
reduction by Abeta [31,40]. Consequently, a cascade of events related to oxidative
stress and subsequent neuronal death occurs (Fig. 1). It has been reported that the Abeta
inhibits the cytochrome c oxidase complex of the mitochondrial electron transport chain
[42] and other studies indicate that this inhibition may be further increased by the
presence of Cu (II) ions, requiring approximately 0.75 mole of metal per mole of

6
polypeptide to inhibit cytochrome c oxidase and to promote peptide aggregation
[43,44]. This effect may be due not only to the ability of the Abeta-Cu complex to
generate hydrogen peroxide but also to the formation of an intermediate reactive
product that interacts with cytochrome c oxidase [43], suggesting another cell damage
mechanism elicited by Cu. In glutamatergic neurotransmission Cu is co-released with
glutamate and substantial amounts of Cu ions, susceptible to bind Abeta (coming from
the action of secretases on APP) can be found in the synaptic space [45] this
extracellular binding also enhance Abeta oligomerization and precipitation and thus the
formation of senile plaques (Fig. 1).
The function of APP is still unknown; however, it has been suggested that APP-Abeta
may act as a Cu carrier system since APP knockout mice showed an increased Cu
content in the cortex as compared to wild type animals [46]; those findings are
complementary to human studies [47,48] and if true, it would explain why copper
deficiency down-regulates APP transcription [49] and the Abeta’s high affinity for
copper [39].
On the contrary, evidence supporting a deficiency of Cu in AD is based on findings now
discussed. Cu concentration in the CSF from AD patients was inversely correlated to
Abeta [50]. Experimentally, Cu has been associated with the upregulation of the Abeta
degrading metalloproteases MMP-2 and MMP-3 in the rodent lung [51,52]; other
studies suggest that this may also occur in brain [53]. Studies in APP-overexpressing
Chinese Hamster Ovary (CHO) cells showed that increasing Cu concentrations reduced
Abeta synthesis and thus reduced amyloidogenesis[54]. Those studies showed that Cu
acts at different levels: it participates in APP processing into non amyloidogenic
derivatives while its deficiency reduces Abeta degradation [55].
Cu deficiency may also influence the activity of Cu-binding proteins in AD. In this
regard, it has been observed a reduced Cu/Zn superoxide dismutase activity [56] in the
cerebrospinal fluid (CSF) from AD patients when compared to controls and the activity
of cytochrome c oxidase, another Cu dependent protein, is also reduced in AD [57].
Ceruloplasmin (Cp), a multicopper ferroxidase necessary for the oxidation of Fe2+ to
Fe3+ and subsequent binding of Fe to transferrin [58], could be an important factor in
AD because in this protein converge both Cu and Fe homeostasis. However, there are
conflicting reports in the literature; Cp content has been significantly increased in most
brain regions of AD patients compared to elderly controls [59]. Whereas, decreased
levels were found in the temporal cortex [60], probably due to methodological

7
differences. Regarding Cp ferroxidase activity, a tendency toward decrease was
observed in the CSF from AD patients [61]. In vitro, it has been observed, that chelation
of about 27% of total Cu in the neuroblastoma cell line SY5Y produces an appreciable
increase in the intracellular Fe level [55], due to the loss of Cp ferroxidase activity.
Serum levels of both Cu and Cp were significantly higher in a group of AD patients
versus controls, according to Squitti et al. [47]. The same authors, in a different study
[48], could not find an association between serum Cp bound-Cu and cognitive
impairment or the increased concentration of Cu in CSF. A recent report by our group
showed a trend towards an increased CSF free Cu concentration in AD patients,
accompanied by reduced Cu-Zn SOD and ferroxidase (Cp) activities [56]. The evidence
discused above suggests that altered homeostasis of Cu exists in AD and that such
alteration can lead to a redox dysequilibrium by altering the functioning of important
enzymes like Cu-Zn SOD and Cp. Therapies focused on metal chelation and recently,
on the transport of Cu into the central nervous system, have been tested.
2.3. Metal-related therapies for AD
A trial in AD patients with deferoxamine, a Fe chelator, showed a delayed loss of daily
living skills, compared to the group receiving placebo [37]; however, the reduced
crossing of deferoxamine through the blood-brain barrier, due to its molecular size and
the functional groups within its structure, have limited its clinical application.
Fe chelators with several other mechanisms of action have been assayed in vitro. In
neuroblastoma SH-SY5Y cells, the drug M-30 along with other congeners (VK-28 and
HLA-20), prevented serum deprivation-induced apoptosis; they also stimulated cell
differentiation and neurite growth. In CHO cells stably transfected with the APP gene,
M-30 reduced APP expression and increased the soluble forms of Abeta [61].
Some vegetal derivatives have also been tested in AD models, because of their
antioxidant, anti-inflammatory, metal-binding and membrane-crossing characteristics.
The green tea flavonoid, epigallocatechin-3-gallate (EGCG) reduced Abeta formation,
both in vitro and in vivo, by modifying APP metabolism leading to soluble non-
amyloidogenic products [62]. Those actions can be due to its Fe chelating properties
[63]. Curcumin, a polyphenolic derivative of turmeric, reduced Abeta aggregation and
the formation of senile plaques in mice expressing APP [64]. However, this derivative
did not lead to a significant effect in a double-blind clinical pilot study [65]. Blat et al.
[66] have recently used a modified octapeptide (NAPVSIPQ=Asn-Ala-Pro-Val-Ser-Ile-

8
Pro-Gln) that inhibited lipid peroxidation and decreased the hydroxyl radical formation
possibly through Fe chelation. Deferiprone, another iron chelator, produced a decrease
in iron signals as measured by MRI in the dentate nuclei of Friedreich ataxia patients
and neurologic improvement was also noted [67]. Deferiprone analogues have been
covalently attached to nanoparticles as a strategy to increase its crossing through the
blood-brain barrier. Those nanoparticle preparations were tested in fixed tissue from AD
patients and in cultured cells; they removed Fe and adsorbed Apolipoprotein E [68,69].
Their effect on animal models remains to be determined.
Regarding therapies aimed to restore Cu normal levels in the AD brain, it has been
observed that Cu chelators also increase the solubilisation of Abeta deposits from post-
mortem AD brain tissue in vitro [70]. Treatment to AD patients with D-penicillamine, a
Cu-chelating compound, reduced oxidative stress; however, this effect was not reflected
in their cognitive decay-rate [71]. The Cu chelator pyrrolidine dithiocarbamate
prevented the cognitive deficit, reduced Tau phosphorylation by interfering with the
Akt/GSK-3β pathway in transgenic mice. Interestingly, pyrrolidine dithiocarbamate
increased Cu content in the cortex as compared to wild type or APP/PS1 mice [72].
Then, it is possible that pyrrolidine dithiocarbamate not only chelates Cu, otherwise it
could move Cu from locations where Cu is in excess to locations where Cu is needed.
This fact could explain why the use of other Cu chelators resulted in no neurological
improvement [71]. Supporting this hypothesis, Clioquinol (CQ), a metal-binding
compound that crosses the blood-brain barrier, reduced the number of senile plaques in
the brain of transgenic mice [73], delayed cognitive impairment and decreased plasma
Abeta levels in AD patients [74]. A mechanistic study using APP-expressing CHO cells
showed that the complex of CQ with Cu(II) decreased the extracellular Abeta levels by
upregulating the matrix metalloproteases MMP-2 and MMP-3 that cleave Abeta. The
CQ-Cu complex transports the metal inside the cells [53]. Furthermore, in APP-
expressing mice, treatment with CQ alone enhanced mortality, whereas co-treatment
with Cu reduced it (versus CQ group) and also increased brain Cu levels [75]. CQ
would be a potential therapy for AD; however, its most important drawback is that it
was implicated in an epidemic of sub-acute myelo-optic neuropathy in the Japanese
population during the 1970s. Another strategy aimed to adequately transport Cu into the
cell is the use of the complex Glyoxalbis(N(4)-methyl-3-thiosemicarbazonato)-
copper(II) (Cu-GTSM), a metal bis(thiosemicarbazone). Treatment of APP-CHO cells
with Cu-GTSM showed that Cu(II) bioavailability significantly increased and the levels

9
of secreted Abeta were reduced in a dose-dependent manner [76]. Further studies
showed that Cu-GTSM increased the copper bioavailability in cultured cells in about
400%. This compound also enhanced the phosphorylation and further inhibition of
GSK-3β, the kinase involved in the modification of Tau protein and the formation of
neurofibrillary tangles. Transgenic APP/PS1 animals treated with this compound
showed an enhanced performance in the Y maze as compared to those animals without
treatment, furthermore, levels of trimeric Abeta were also reduced in those animals
treated with the copper complex [77].
Additionally, a series of Cu-complexed compounds, have also been tested in APP-
expressing CHO cells. Those complexes were able to boost the metal content inside the
cells but only those that released the metal were able to increase the degradation of
Abeta by activating the PI3K phosphorylation cascade and the expression of matrix
metalloproteases responsible of the degradation of Abeta [76].
Recent innovations regarding drug design have led to the synthesis of drugs with
acetylcholinesterase activity and Cu/Fe chelating properties; those compounds also
inhibited Abeta aggregation. Theoretically, the new synthesized molecules may
constitute potential therapeutic tools, only after completing in vivo and safety studies
[78].
2.4. AD: Concluding remarks
Cu has shown dual properties in AD. Abeta binds Cu with high affinity and this union is
involved in the generation of free radicals and inhibition of mitochondrial function. Cu
ions (coming from disrupted Cu transport or from physiological release during
glutamatergic transmission) are also involved with the formation of senile plaques by
precipitating Abeta oligomers. On the other hand, substantial evidence suggests that low
intracellular Cu: a) is involved with the biosynthesis of Abeta from APP, b) limited
functioning of copper dependent antioxidant enzymes i.e. SOD and c) diminished
copper dependent ferroxidase (ceruloplasmin) activity that in the long term would lead
to Fe cell deposition thus increasing oxidative stress. It remains to be determined if
current experimental therapies in AD that include the transport of Cu into cells are in
fact a real alternative. It is worth mentioning that drugs binding Cu could be
redistributing the metal from rich-Cu compartments to areas where the metal is needed.
3. Parkinson’s disease

10
PD is the second most prevalent neurodegenerative disorder worldwide [79]. It is
mainly characterized by motor disturbances such as tremor, rigidity and bradykinesia
[80,81], although cognitive and behavioural abnormalities have also been reported
[82,83]. PD occurs following dopaminergic neuronal death within the substantia nigra
pars compacta (SNpc) [79-81] but the etiology of this selective neurodegeneration is
still unknown [84,85], although several genetic and environmental factors have been
implicated [80,85]. Among those factors, occupational exposure to transition metals,
especially Fe and Cu, has been proposed as a risk factor for the development of PD
[86,87]. Several studies support the role of transition metals brain accumulation in the
pathophysiology of PD that may be independent on the environmental exposure,
suggesting that metal homeostasis in PD is altered. The neurotoxic potential of Fe has
been consistently reported while in the case of Cu the evidence is not completely
conclusive as will be discussed later.
3.1. Fe homeostasis in Parkinson’s disease
Although Fe is very important for physiological processes in several organs including
the brain, its role in the pathophysiology of PD has been extensively studied and a wide
body of evidence showing its neurotoxic effects has been reported [88-90], especially
on tyrosine hydroxylase-immunopositive neurons as demonstrated by Fe microinjection
to the SN [90].
Total Fe levels have been found to be increased in the SNpc but not in the cerebellum,
caudate nucleus, putamen or cerebral cortex from post mortem PD brains [91-94],
suggesting that the underlying mechanisms for Fe accumulation may be specific for the
SNpc. In contrast, levels of this metal were reduced in the globus pallidus compared to
control values [92]. It should be noted that non-significant difference in Fe content has
been found in brain tissue showing moderate neurodegeneration [94]; thus, it could be
considered that Fe accumulation in PD might be the consequence of the underlying
mechanisms of neuronal death since otherwise its levels should be expected to be
increased since the early stages of the disorder; thus, some mechanisms may initiate
neuronal death at the early stages of the disorder and lead to Fe accumulation that, in
turn, may potentiate oxidative damage.
Interestingly, ferritin-reactive microglia has been found surrounding degenerating
neurons [91]. Since ferritin is the main Fe-binding protein, this result suggests that Fe
accumulates within microglial cells and several hypotheses may be suggested in this

11
regard. On the one hand, it is possible that microglia release Fe that could be toxic to the
surrounding neurons; on the other hand, Fe may accumulate in both neurons and
microglia but the former may be more sensitive to the toxic effect of this metal and thus
surviving ferritin-positive microglia may be found in post mortem brain tissue
surrounding degenerating neurons. The role of microglia in brain Fe accumulation
deserves further investigation.
Whatever the cell type responsible for Fe accumulation, the content of this metal is
increased in the SNpc in PD and it has been detected in living patients by neuroimaging
methods. Transcranial sonography has revealed that the SN in PD patient is
hyperechogenic, most likely due to metal deposits in this region [91,95,96]. This
hyperechogenicity may be present in up to 90% of PD patients [91] and may also be
found in healthy subjects, but even in this case it reflects nigrostriatal dysfunction to
some extent, since it is associated with decreased 18Fdopa uptake [91,95,97]. Also,
hyperechogenicity in healthy elderly subjects is associated with motor alterations such
as hypokinesia [91]. SN hyperechogenicity is most likely due to Fe accumulation since
the echogenicity of post mortem human brain tissue correlates well with Fe content, but
not with that of Cu, magnesium, zinc or calcium [91,97]. Other imaging findings further
support Fe accumulation in the SNpc in PD [98,99] and its role on nigrostriatal
dysfunction in PD.
Brain Fe content increases during normal ageing and it is associated with a reduced
motor performance [100]. Fe content in PD is higher than expected by normal ageing
and the underlying mechanism for such accumulation remains to be completely
elucidated [11] but several studies have shed some light in this regard. Excitotoxicity
might be involved in PD and may lead to Fe accumulation. Fe uptake is enhanced by
NMDA-receptor activation through nitric oxide (NO) signalling [89]. Both glutamate
and NO are involved in excitotoxic death which is suggested to occur in PD (Fig. 2);
thus, Fe accumulation in this disorder may be associated with neuronal death through
glutamate receptors [89]. Further studies support this neurotoxic pathway. Fe chelation
reduces NMDA-induced excitotoxicity [89].
NO increases Fe uptake through a transferrin-independent mechanism most likely
mediated by divalent metal transporter (DMT1) [89]. Furthermore, high NO
concentrations are able to displace Fe from Fe-sulfur centers in some proteins such as
mitochondrial complex II, forming dinitrosyl-Fe complexes [101] thus increasing the
potentially neurotoxic free-Fe pool. Fe may potentiate excitotoxic cell death since

12
glutamate release after ischemia/reperfusion is higher in animals fed with a Fe-
supplemented diet [88].
It is well known that Cu proteins Cp and hephaestin oxidize Fe+2 to Fe+3 in order to
facilitate iron removal as it has been demonstrated by iron overload and
neurodegeneration in the double knockout mice lacking both Cp and hephaestin [102].
The possibility of a role for these two proteins in PD has been explored [103,104]. In a
model of PD using 6-hydroxydopamine (6-OHDA) a decreased expression of
hephaestin was found [103], while mutations of the Cp gene have been associated to PD
[104]. Therefore, it is possible that iron overload could be in part a consequence of
altered oxidation of Fe, preventing its extrusion.
Recently, upregulation of DMT1 in the SNpc of PD patients and in the SNpc of mice
exposed to MPTP, a neurotoxin known to induce several features of PD, has been
demonstrated [105]. These results were confirmed by treating the dopaminergic cell line
MES23.5 with MPP+, the active metabolite of MPTP; upregulation of DMT1 was also
found [106]. The up-regulation of DMT1 was correlated to Fe accumulation.
Additionally, rodents carrying a mutation that impairs DMT1 Fe transport were partially
protected from injury caused by both MPTP and 6-OHDA. These evidences point
towards a direct involvement of DMT1 in Fe accumulation and consequently, in the
pathophysiology of PD.
Once Fe is accumulated in PD it could enhance neuronal death through oxidative stress.
Fe induces lipid peroxidation [107,108] and increases ROS production by 6-OHDA
auto-oxidation [109]. Fe also stimulates the formation of intracellular aggregates of -
synuclein and favours oxidative damage [110]. It is known that autosomal dominant PD
is related to mutations in -synuclein that enhance aggregation of the protein [111]
therefore, those individuals with mutations in -synuclein could be more susceptible to
oxidative damage by Fe.
Although Fe accumulation in PD is not explained by ageing itself, ageing modulates Fe
neurotoxicity. Adult mice (12-24 months old) fed with Fe during the neonatal period
showed reduced striatal dopamine content while their young counterparts (2 months
old) have unchanged dopamine levels following the same treatment [80,112]. However,
although brain Fe accumulation in young animals is not likely to produce neuronal
death by itself, early postnatal Fe administration potentiates MPTP-induced dopamine

13
depletion during adulthood [113], suggesting that Fe accumulation in young animals
may lead to neurotoxicity.
As mentioned before, Fe (II) may lead to neuronal damage due to oxidative stress
through the Fenton reaction with hydrogen peroxide (H2O2) [114,115]. The H2O2 supply
for this reaction may arise from monoamine oxidase activity and also from Fe-induced
oxidation of dopamine [79] among other possible sources, leading to the enhancement
of this damaging mechanism. Increased glutathione biosynthesis is observed in cell
survival after Fe overload [114] suggesting that oxidative stress is directly involved in
Fe neurotoxicity.
_____________________________________________________________Fig.2.
3.2. Cu homeostasis in Parkinson’s disease
Several studies have shown that Cu may lead to both toxic and protective effects under
certain experimental conditions. Toxic effects have been reported for peripheral tissues
while its protective effects have been found against certain paradigms of neuronal
damage. Since brain Cu content has been reported to be decreased in PD [92,116], the
toxic effects that occur at high concentrations are not likely to be involved in the
pathophysiology of this disorder while its protective effects are relevant in the case of a
possible Cu-deficiency in PD.
Multivariate analyses have shown that occupational co-exposure to both Pb and Cu for
20 years or more significantly increases the risk for PD (odds ratio 5.0); however, the
association of exposure to Cu only did not reach statistical significance in some studies
[86]. Thus, it remains to be determined if Cu exposure itself is associated with PD or if
it depends on the simultaneous exposure to other risk factors.
Cu has been implicated in the pathophysiology of PD since its concentration has been
found altered in the brain and CSF from patients with this disorder [56,117]. In a post
mortem study, Cu content was shown to be significantly higher in the reticular
formation in PD cases [94]. In contrast, several studies suggest that brain Cu levels are
deficient in PD. Total Cu content is reduced in the SNpc [92] and the caudate nucleus
[59], but not in the cerebellum, globus pallidus, putamen or the dorsolateral prefrontal
cortex [94] from PD brains. Regarding Cu content in CSF, while total Cu concentration
is not changed in PD patients compared to controls [56,118] free Cu is increased and
positively correlated to both disease duration and Unified Parkinson’s Disease Rating
Scale motor scores [56]. It is possible that free Cu levels might lead to oxidative stress

14
and neuronal death in PD through the Fenton reaction; also, free Cu could be increased
due to the uncoupling from its binding sites in antioxidant proteins (such as Cp and
SOD) leading to oxidative stress.
Cu (II), as well as other metals, binds to -synuclein with dissociation constants in the
micromolar range (40-500 M) and induces oligomerization of this protein when
incubated either alone [119,120] or in combination with H2O2 [121,122]. Also, Cp-
bound Cu leads to ROS-mediated -synuclein aggregation when incubated with H2O2
[123]. However, this effect has been studied in vitro and is dependent on the
experimental conditions tested; in this regard, some studies did not find any effect of Cu
(II) (at a Cu:protein ratio of 10:1) on -synuclein oligomerization and, in contrast,
suggest that this cation might inhibit the spontaneous aggregation of the protein [124].
Thus, the effect of Cu on -synuclein oligomerization in vivo remains to be elucidated.
Cu (II) (0.2-1.0 M) although is inactive by itself, enhances cysteine autoxidation-
induced neurotoxicity [125]. However, Cu deficiency could also lead to nigrostriatal
dysfunction since rats fed with Cu-deficient diets during gestation and lactation show
reduced striatal dopamine content [126]. Furthermore, both acute and chronic Cu (II)
administration has shown neuroprotective effects against both MPP+ and quinolinic acid
neurotoxicities [127-129]. Moreover, in contrast to Fe, Cu chelation is not protective
against MPTP injury [130] and even, as in the case of diethyldithiocarbamate, can
enhance neurotoxicity [131].
The neuroprotective effect of Cu may involve the modulation of Fe transport. Cu
reduces Fe uptake possibly through neuronal DMT1 [115]. Decreased DMT1
expression is associated with neuronal survival following Fe overload [132]; thus, an
inhibitory effect of Cu on Fe uptake is also expected to be neuroprotective. Both Oral
and intracerebroventricular Cu administration are neuroprotective against dopaminergic
degeneration; oral Cu administration may lead to this effect by decreasing Fe levels
since Cu competes with Fe for intestinal absorption [115], thus decreasing Fe uptake,
and consequently, the brain content of this metal.
Fe transport modulation through ferroportin (Ireg-1) may also be involved in the
neuroprotective effect of Cu since this protein mediates Fe efflux (Fig. 2) in neurons
and astrocytes [115,132]. Increased ferroportin expression is associated with neuronal
survival after Fe overload [115]. Cu-deficient diets reduce ferroportin expression in the
rat liver [133] leading possibly to Fe accumulation; in patients with non-alcoholic fatty

15
liver disease, low hepatic Cu content is associated with a decreased ferroportin
expression, thus contributing to Fe accumulation in those patients [133]. According to
those studies, Fe accumulation may be the consequence of Cu deficiency. As a matter of
fact, Fe accumulates in several tissues during Cu deficiency [115], supporting this
hypothesis.
Cu-deficient diets lead to a reduced Cp activity [134]. Cu (II) (20 nM) induces Cp
expression in cultured liver cells [135] and chelation of this ion leads to the opposite
effect [136]. Cu chelation may lead to intracellular Fe accumulation by decreasing
ferroportin expression [136]. The underlying mechanism for this effect most likely
involves Cu-mediated Cp activity, since ferroportin targeting to the astrocyte plasma
membrane is absent in Cp knockout animals [136]. This interaction is due to the
ferroxidase activity of Cp since this activity at the plasma membrane reduces the
extracellular Fe (II) concentration leading to an increased expression of ferroportin to
compensate for the Fe (II) depletion by increasing its efflux [136].
As an altered metal homeostasis seems to exist also in PD, some attempts aiming to
regulate metal levels have been made in order to treat this disorder.
3.3 Metal-related therapies for Parkinson’s disease
As discussed in section 3.1, several studies have consistently shown that Fe is
accumulated in the SNpc of PD patients. Also, a wide body of evidence supporting the
neurotoxic potential of Fe overload has been reported [88-90]; however, no therapeutic
approach targeting Fe accumulation in PD has been performed to date. Fe chelation is
neuroprotective in animal models [89,137,138] but may not be convenient in the clinical
practice, not only due to the interference with the physiological role of this metal but
also to the lack of a specific Fe binding and the potential adverse effects of Fe chelators
[1]. Thus, beyond Fe chelation, different attempts focusing upstream (Fe intake) or
downstream (antioxidant effects) events have been performed. High Fe intake in the diet
is a risk factor for PD [139] possibly by increasing brain Fe concentration. This suggests
that dietary Fe restriction may be beneficial in PD as has been found in experimental
models [140]. It is possible that a single mechanism of action is not sufficient to slow
the progression of a complex disorder such as PD. Since not only Fe overload
[80,90,112], but also Fe restriction [140], leads to nigrostriatal dysfunction it is possible
that metal homeostasis, rather than excess or deficiency, needs to be achieved in PD but
this issue awaits further investigation.

16
Therapeutic strategies regarding copper have been also tested. Rojas et al. [141]
administered EGb761, a well-defined mixture of active compounds extracted from
Ginkgo biloba, to mice treated with MPP+. EGb761 pretreatment resulted in the
prevention of changes in copper levels observed in mice only treated with MPP+. The
fact that copper homeostasis is returned to normality may contribute to the protective
effects of EGb761 in this model. Pretreatment with CuSO4 to rats treated with MPP+
prevented protein nitration, tyrosine hydroxylase inactivation as well as the dopamine-
depleting effect of MPP+ [128]. Probably, strategies aimed to restore Cu brain levels can
be helpful in PD treatment.
3.4. PD: Concluding remarks
A central role for Fe accumulation in mesencephalic tissue from patients is observed in
PD, the reason for this effect is still unknown; however, Fe transporters DMT1 and
ferroportin seem to be involved. Fe burden in dopaminergic brain areas takes a major
importance since the catabolic route of dopamine produces hydrogen peroxide that in
presence of Fe favors Fenton reactions and excessive oxidative stress. The ferroxidase
activity of ceruloplasmin could play an important role, since isoform variations and low
activity of this enzyme (separately) have been linked to increased nigral ecography that
in turn, is related to Fe accumulation. Cu may also influence the content of Fe in
neurons, not only because of the effect elicited by ceruloplasmin, but also on iron
transporters DMT-1 and ferroportin. More studies are necessary to explore the
relationship between Cu and Fe and to propose related-strategies in PD.
4. Amyotrophic lateral sclerosis
Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease of unknown
etiology clinically manifested by weakness and wasting of the affected muscles with
pyramidal signs. ALS is characterized by the progressive loss of motor neurons of the
anterior horns in the spinal cord, bulb and cortex [142].
ALS seems to be sporadic in 90% of all cases, while familial amyotrophic lateral
sclerosis (FALS) showing dominant autosomal inheritance, represents 10% of the cases
of the disease [143]. Over 110 FALS-linked mutations throughout the SOD1 gene are
related to approximately 20% of the FALS cases [144,145]. In FALS, SOD1 acquires a
toxic function as demonstrated by transgenic models showing that mice overexpressing

17
the human mutant enzyme G93A exhibit features of ALS [146] while those lacking the
enzyme do not develop the disease [147].
Cytoplasmic aggregates have been found in motor neurons from sporadic and familial
ALS patients and from transgenic mice models of ALS. Those aggregates include
Bunina bodies [148], skein-like inclusions [149] and Lewy body-like inclusions [150].
Interestingly, Lewy body-like inclusions are immunoreactive for SOD1 [151] and the
presence of this protein in several enzyme mutant aggregates correlates with disease
onset and progression [152].
Superoxide dismutases are the major antioxidant enzymes involved in free radical
scavenging. SOD1, SOD2 and SOD3, catalyze the dismutation of superoxide anions
yielding H2O2 and O2, preventing intracellular damage [153]. Human SOD1 is a 32 kDa
homodimeric metalloenzyme containing one Cu and one Zn ion per subunit [145]. The
Cu ion bound to the SOD1 active site has a catalytic function, while the Zn ion
maintains the enzyme structure [154]. The association between SOD1 mutations and
FALS suggests that oxidative injury is involved in this disorder [155].
4.1 Fe homeostasis in amyotrophic lateral sclerosis
Increased spinal cord Fe levels reported in ALS [156,157] are possibly involved in
oxidative damage through the Fenton reaction. It has been suggested that Fe
accumulation may be due to increased uptake of this metal [11] since lactoferrin is
increased in ALS affected motoneurons [158]. Ferritin is upregulated in SOD1-G93A
mice just prior to end-stage disease, suggesting an increased Fe deposition [159].
Moreover, in ALS patients, CSF ferric reducing ability is decreased, while the content
of oxidized proteins is increased both in CSF and plasma [142]. SOD activity modulates
the levels of TfRs, ferritin and Ireg-1 [153].
The expression of proteins associated with iron homeostasis, DMT1, TfR1, the iron
exporter Fpn and Cp has been studied in a transgenic mice model of ALS; a caudal-to-
rostral gradient in the mRNA levels of these proteins, with the highest levels rostrally in
the cervical region, were found [160]. Such a distribution correlates with the caudal-to-
rostral progression of the disease in SOD1-G37R transgenic mice.
Interestingly, Mizuno and co-workers [161] found that transferrin colocalizes with
Bunina bodies in the spinal cord of ALS patients; therefore, transferrin possibly
interacts with cystatin C since they are the only known proteins in Bunina bodies [162].

18
Another evidence supporting the involvement of Fe in this disorder is that the
prevalence of HFE (hemochromatosis gene) mutation in ALS patients is the second
most frequent in this disease [163]. HFE interacts with the TfR to sense Fe levels [164];
its polymorphisms have been associated with hereditary hemochromatosis [165], a
genetic disorder resulting in free Fe accumulation in parenchymal tissues. Moreover,
HFE mutations are associated with a decreased expression of SOD1, -tubulin and -
actin [163]. Therefore, it is possible that HFE polymorphisms in ALS are associated
with an altered Fe homeostasis and, consequently, to oxidative damage in this disease
[166].
4.2. Cu homeostasis in Amyotrophic lateral sclerosis
As SOD1 contains Cu and Zn, altered levels of those metals have been associated with
ALS pathology. In patients with this disorder, Cu levels have been reported only in CSF
and serum, and they vary from low [167] to unchanged when compared to controls [56].
However, in transgenic models of FALS, Cu levels are increased in the spinal cord of
rats [168] and mice [169-171].
Changes in spinal cord Cu content could be explained, at least in part, by the
downregulation of atp7b gene (encoding a Cu-transporting ATPase) in SOD1 transgenic
mice [159]; however, Jonsson and co-workers [172] suggested that the deficient Cu-
coupling to SOD1 is not due to a general decrease in tissue Cu uptake, but to an altered
process in the folding of proteins.
Altered Zn and Cu levels could be the consequence of structural changes in SOD1. The
FALS SOD1 proteins can be divided into two groups according to their metal content
[173] and the position of the specific mutation. Metal content in wild-type-like (WTL)
mutant SOD is nearly identical to that found in the wild type protein, whereas mutations
at the metal-binding region (MBR) or at the electrostatic and Zn loop elements [174]
lead to a deficiency in Zn and Cu content [173]. WTL-SOD1 mutants show high
reactivity with hydrogen peroxide and produce site-specific oxidative damage to the
MBR, compromising metal binding, while MBR mutants appear to aggregate with no
further modification [174,175]. Hence, both types of mutants may aggregate involving
metal uncoupling.
An increasing body of evidence suggests that SOD1 stability is dependent on its metal-
binding state. Some hypotheses hold that the balance between normal and toxic SOD1
functioning depends on Zn binding at the active site of the enzyme [176] (Fig. 3);

19
experimental models have shown that in the absence of Zn the catalytic reaction of
SOD1 runs backwards, producing reactive oxygen species [177]. SOD1 proteins that
have been oxidatively inactivated by reaction with hydrogen peroxide lose their affinity
for Cu and consequently they are more likely to aggregate than the undamaged protein
[178]. It has been observed that even wild-type human SOD1, in its metal-free state,
may form large, stable, soluble, amyloid-like protein oligomers under relatively mild
conditions, although the intrasubunit disulfide bond remains intact, suggesting that the
gain of a toxic SOD1 function in ALS may be related to the inability of this protein to
achieve or to maintain the metallated state [145,179]. The same holds true for ALS
mutants, that are completely unfolded in the metal-free state [180]. In fact, it has been
observed that Zn binding in ALS mutants can lead to a complete SOD1 folding,
reducing oligomeric fractions [180,181]. Metal-free WT and ALS-associated SOD1
mutants form disulfide-linked oligomers only when both Cys6 and Cys111 are present
[145,182]. It is possible that the lack of metal ions distorts SOD1 structure, exposing the
Cys residues and promoting protein aggregation [154].
_________________________________________________________________Fig.3.
The loss of Cu and Zn from SOD1 also facilitates the reduction of the intrasubunit
disulfide bond between Cys57 at the Zn loop and Cys146 at the β-barrel, thus leading to
the dissociation of SOD1 subunits, a fact that greatly increases the formation of
insoluble aggregates [183,184].
The loss of Zn(II) in turn alters Cu(II) coordination through a shared histidine ligand
[177]. In vitro experiments have shown that the Cu(II) ion at the active site can react
with hydrogen peroxide, leading to the oxidation of the Cu(II)-coordinating histidine
residues and the inactivation of SOD1, thus promoting enzyme unstability [185]. Zn
uncoupling alters human SOD structure even more than any ALS mutation that has been
crystallographically characterized, producing the opening of the 4 Å wide channel that
normally avoids small molecules to access the catalytic Cu [186]. It has been
hypothesized that when the Zn(II) ion is unbound to SOD1, a Cu ion undergoes a one-
electron reaction with molecular oxygen to form superoxide anion, that further reacts
with NO forming peroxynitrite [177]. This pro-oxidant function promotes inactivation
of the mutant enzymes, which may also lead to Cu ion release from the inactivated
protein [187]. It has also been suggested that Zn loss from wild-type SOD could be
involved in 98% of ALS patients without SOD mutations [186].

20
It has been reported that a FALS-linked SOD1 mutant, H46R, abnormally binds Cu at a
cysteine residue (Cys111) outside the active site [188]; that residue is important to
maintain protein stability [189]. As mentioned above, mutant SOD1 exhibits a
decreased affinity for Zn(II) and an increased affinity for Cu(II), the last one probably
mediated by the Cys111 residue [189]; this incorrectly coordinated Cu can be highly
redox-active and therefore potentially toxic. This could lead to a similar effect as that of
Cp, which becomes pro-oxidant when Cu is abnormally bound outside its active site
[190].
Another study supporting the role for Cu in ALS is that performed by Kiaei et al. [191];
Cu deficiency induced by the Mobr allele (inhibiting the activity of an ATPase that
transports Cu(II) across the intestinal lumen, as it occurs in Menkes disease) in a mice
model of ALS. In that study, a slight increase in the life span of the double transgenic
mice was found compared with that of the mice carrying only the SOD1-G86R mutation
and treated orally with the Cu chelator D-penicillamine. It is noteworthy that the double
mutant animals for the two pathogenic mutations (Cu depletion plus mutant SOD1)
lived significantly longer than the single SOD1 mutant mice. Then, despite mutant SOD
can strongly bind Cu, its depletion could be beneficial in ALS.
Cu has been also implicated in apoptosis. Either Zn-deficient wild-type or mutated SOD
initiate apoptosis in cultured motor neurons even in the presence of brain-derived
neurotrophic factor; NO-dependent mechanisms are involved [177]. Interestingly, it has
been proposed that Cu activates the Fas apoptotic pathway [2]. Cu accumulation
induces conformational changes in the X-linked inhibitor of apoptosis protein that, in
turn, plays an important role in intracellular Cu homeostasis [192], leading to its
degradation and decreasing its ability to inhibit caspase activity [193]. Accordingly, Cu
liberated from Zn deficient SOD could potentially initiate apoptosis (Fig, 3).
SOD1 localization has been related to the enzyme binding metals, while partially or
metal-free SOD1 is inserted into the mitochondria, the holoenzyme is not. Interestingly,
in ALS patients and transgenic mice, the mutant protein is encountered in mitochondria
[194,195], this effect could be related to the metallated state of the enzyme since
Okado-Matsumoto and Fridovich [196] reported that in mouse neuroblastoma N2A
cells, the entry of both wild-type and mutant SOD1 into the mitochondria depends on its
metal-coupling state.
4.3 Metal-related therapies for amyotrophic lateral sclerosis

21
Therapeutic agents and strategies that reduce the transgenic ALS mice pathology extend
their survival for only a few days. Antibiotics of the β-lactam type have been proposed
as a treatment since they are also metal chelators [197]. Administration of the Cu
chelators DP-109, DP-460 [198], penicillamine [199], N-acetylcysteine [200] and
trientine [201-203] have been effective in delaying the disease onset, improving motor
performance and slowing the disease progression, in ALS mouse models. Treatment
with the Cu chelator diethyldithiocarbamate reduced hydroxyl radical production [204]
and increased cell survival in in vitro models of FALS [205]. Recently, in a transgenic
model of ALS Tokuda et al. [171] found that ammonium tetrathiomolybdate (TTM), a
Cu chelator used for the treatment of Wilson’s disease, led to delayed disease onset,
longer survival and slower progression than that of other agents tested before, besides
restoring Cu levels. Tokuda et al. [171] suggested that the removal of the Cu ion bound
to Cys111 in mutant SOD1 may underlie the effect of TTM. An advantage of TTM is
that it chelates both intracellular and extracellular Cu ions, whereas other agents like D-
penicillamine and trientine remove only extracellular free Cu [206]. This fact suggests
that it is necessary that chelating therapies for ALS should be aimed to remove
intracellular Cu deposits.
Regarding iron chelation, the treatment with salicylaldehyde isonicotinoyl hydrazone, a
lipophilic iron chelator in transgenic SOD1-G37R mice resulted in the increase animal
life span by 5 weeks. This drug also helped to preserve neurons and diminished the
number of iron containing cells without signs of anemia [160].
4.4. ALS: Concluding remarks.
ALS is characterized by the malfunction of Cu-Zn SOD; misfolding of the protein, as
well as metal binding alterations are implicated in this effect. The diminished
antioxidant capacity of motor cell is further aggravated by the SOD1 gain of toxic
function, in which Cu bound to the protein plays a central role. Free Cu ions are also
suspected to participate in the cascade of events that end with cell death. Additionally,
involving of SOD1 in regulating iron proteins plays an important role in Fe
accumulation, complicating oxidative stress. The strategy of combining antioxidants
and metal chelation may have some potential, especially Cu chelators to withdraw
misplaced Cu in the enzyme as well as intra and extra cellular free ions.
5. Huntington’s disease

22
Huntington’s disease (HD) is an autosomal dominantly inherited neurodegenerative
disorder characterized by progressive motor, cognitive and psychiatric deterioration
[207]. It is caused by the expansion of an unstable CAG trinucleotide repeat within the
first exon of the IT-15 gene encoding huntingtin (Htt) protein [208,209]. The function
of Htt protein has not been completely elucidated; however, it is possibly involved in
endocytosis, vesicular trafficking [210], embryonic development [211] and
transcriptional regulation [212]. The CAG repeat in Htt shows between 10 to 29 copies
in healthy subjects and it is expanded to 36-121 in HD [213]. The CAG repeat yields a
polyglutamine stretch within the protein [214]. The mutant Htt is widely distributed in
most brain regions as well as in peripheral tissues [215] and acquires an unusual
conformation, which is hypothesized to produce cell toxicity. Additionally, altered
metal homeostasis it has been implicated in HD pathology [12,208,216].
Neuronal loss and brain atrophy in HD patients occur mainly in the caudate and
putamen [59], although they may also occur in other regions such as the cerebral cortex,
thalamus, globus pallidus, cerebellum as well as in white matter tracts [217,218].
5.1. Fe homeostasis in Huntington’s disease
Fe accumulation has been reported in the basal ganglia of HD patients [12,216]. Dexter
et al. [12] measured post-mortem metal levels in brain tissue from HD patients and
found that total Fe was increased in the putamen and caudate nuclei (44% and 56% over
controls respectively), the same brain areas also showed extensive pathological
disturbances, as a consequence of the disease. In the same study, ferritin levels were
unchanged between HD patients and control subjects in all of the brain regions
examined. However, other authors have found increased ferritin levels in HD brains
[13,219], those discrepancies may be due to experimental issues. Also, increased Cp
levels in HD brains [59] and reduced CSF Cp ferroxidase activity [56] have been
reported. Those findings suggest generalized disruption of Fe homeostasis that may be
due, at least in part, to functional changes in Htt, a phenomenon involved in the
regulation of the Fe pathway. In turn, Htt expression may be influenced by Fe, as
suggested by the studies showing its upregulation in the presence of the Fe chelator
deferoxamine [211]. In this regard, the loss of wild-type Htt [220,221] and its altered
function as observed in mutant Htt, may increase brain free Fe levels [13]; such an
effect, may be toxic through the Fenton reaction, leading to free radical production, lipid
peroxidation [56,222], DNA and protein damage and finally, cell death [13].

23
Other possible mechanism involving Fe homeostasis is the mutant Htt-mediated
stimulation of the lysosomal autophagy and proteosome systems that, under normal
conditions, quickly degrade ferritin following its Fe-mediated oxidation [13,223,224].
Ferritin plays an important role in Fe homeostasis by sequestering this metal; in turn, Fe
levels regulate ferritin expression, which increases with Fe accumulation [225,226].
Simmons et al. [13] analyzed the specific localization of ferritin in the brain from
transgenic R6/2 mice and HD patients; they found that ferritin was predominantly
increased in microglia; those cells appeared dystrophic, suggesting that they may be
dysfunctional and contribute to HD progression. The early increase in microglial ferritin
in the R6/2 mice carrying the Htt mutation occurs when nuclear inclusions first appear
[13,227-229], possibly implying a direct link between ferritin and nuclear inclusions.
Oligodendroglia is also possibly involved in HD pathophysiology, as myelination
impairment (reviewed in [215]) and increased oligodendroglial density have been found
in the brain of HD patients [230]. Differentiation and proliferation of those cells is
dependent on Fe stores [231]. It has been hypothesized that elevated oligodendrocyte
ferritin levels could be an attempt to accumulate Fe to support myelination [215].
It is also possible that increased total Fe content involves an altered
compartmentalization. In this regard, Lumsden et al. [232], using zebrafish embryos,
found that Htt knockdown led to cellular Fe deficiency despite of the availability of this
metal. Increased levels of TfR1 transcripts were observed in Htt-deficient zebrafish. Htt
appears to act downstream of the TfR-mediated Fe endocytosis, thus implicating Htt in
Fe release from endocytic compartments into the cytosol [232].
On the other hand, the activities of many Fe-dependent enzymes are decreased in HD
patients; those include aconitase and mitochondrial complexes I-IV [233,234], which
are important for energy metabolism. The most consistent finding in HD is a decreased
activity of mitochondrial complexes II, III and IV [233,234]. It has been reported that
aconitase, a Fe-sulphur (Fe-S) containing enzyme important for the tricarboxylic acid
cycle and Fe homeostasis [235,236], as well as complexes II and III, are susceptible to
inhibition by reactive oxygen species [237,238]. Decreased activity of those enzymes,
as observed in HD, could lead to a self-amplifying cycle of respiratory chain inhibition
and free radical generation [239]. Then, high Fe levels in HD brain could be indirectly
disrupting the energetic metabolism by free radicals generation. It has been proposed
that free radicals damage [4Fe-4S] centers, inactivating several enzymes, releasing their
catalytic Fe ions and increasing oxidative injury through the Fenton reaction [240]. It

24
should be noted that altered activities of complexes II and III have been associated with
basal ganglia degeneration [233], as occurs in HD.
Compromised function of the electron transport chain leads to reduced ATP levels and
consequently, to the failure of several ATP-dependent ion pumps. Thus, membrane
repolarization will be affected, releasing the voltage-gated Mg+2 block of the NMDA
channel and allowing its activation, even at basal glutamate levels [241]. As caudate and
putamen nuclei are Fe-rich areas and receive excitatory inputs, a synergic toxic effect
between Fe and glutamate may be suggested to occur [11].
Consistent with the damage to mitochondrial respiratory complexes, increased lactate
concentrations have been reported in the basal ganglia and occipital cortex [242,243].
Although it seems that damage to oxidative metabolism involves Fe dysregulation to a
great extent, it may also involve other mechanisms since reduced lactate dehydrogenase
activity has also been reported in the brain of R6/2 mice following Cu accumulation
[208]. Increased lactate concentration may decrease pH contributing to Fe release from
ferritin stores [244].
5.2. Cu homeostasis in Huntington’s disease
Both increased [12] and decreased [59] brain Cu levels have been found in HD patients,
compared to controls. Recently, our group found that CSF free Cu concentration is
associated with the clinical stage and the time after onset in HD patients [56].
Consistent with the interaction between Cu and the Abeta [40], Fox et al. [208] found
that this metal promotes Htt aggregation (Fig. 4) and interacts with histidine (His)
residues in the N-terminus of the protein. Moreover, they proposed that reduced lactate
dehydrogenase activity in HD is due, at least in part, to Cu-mediated enzymatic
inhibition possibly leading to neurodegeneration. More studies are needed in relation to
Cu role in HD.
_________________________________________________________________Fig.4.
5.3. Metal-related therapies for Huntington’s disease
Even though several attempts have been made, there is no effective treatment for HD. A
possible alternative could be focused on metals. Since Fe accumulation leads to
oxidative stress it has been suggested that Fe chelators could be beneficial in this
disorder. Firdaus et al. [245] reported that pre-treatment with deferoxamine to COS-7
cells transiently transfected with a Htt mutant vector showed decreased inclusions body

25
size, suggesting a role for Fe in the formation of those aggregates. However, Htt is up-
regulated in embryonic stem cells (ESC) following Fe chelation with deferoxamine,
leading to nuclear and perinuclear abnormalities in both ESC and STHdh+/Hdh+ striatal
cells [211].
CQ reduced polyglutamine expanded levels in vitro and reduced the pathology and
behavioural abnormalities of R6/2 transgenic mice, but it is not known if those effects
were due to metal chelation or to other mechanisms [246], although it could be
suggested that the effect of CQ may involve Cu (II) binding in a 1:2 metal:ligand
stoichiometry [247]. Furthermore, it is possible as in AD [53], that CQ moves Cu from
sites where it accumulates to other sites where it is needed.
EGCG chelates Cu and modulates early events in Htt misfolding. It reduced toxicity in a
Drosophila model of HD, probably by scavenging free radicals or chelating metal ions
[248].
5.4. HD: Concluding remarks
Htt mutant expression in HD has been linked with events that produce Fe accumulation
in basal ganglia, thus this metal is presumably involved in the development of HD. In
fact, mutant Htt and Fe are engaged in mechanisms that suggest that one favours the
accumulation of the other causing neurodegeneration. As in the others
neurodegenerative diseases the disruption in Fe homeostasis, the free radicals as well as
protein precipitation are involved. Cu role for this disease is still less clear than that of
Fe and deserves future investigation. Iron chelation could be an interesting approach in
transgenic rodent models of the disease.
________________________________________________________________Table 1
6. Conclusions
All of the evidence discussed above suggests that disturbances of metal functioning,
regulation and distribution are likely to occur. Mechanisms of damage elicited by Cu
and Fe common to AD, PD, ALS and HD include: a) Free radical production, b) Protein
aggregation and c) Metal transport alteration. Although there are some studies regarding
the role of metal transporters (Table 1) in neurodegenenerative diseases, this is a
growing study field, having the possibility of studying novel therapeutic strategies,
since metal transporters are involved in metal brain distribution, intracellular
localization as well as disposal from brain. It is possible that the final pictures of metal

26
status in neurodegenerative diseases obeys to altered compartamentalization of metals
and thus we observe the sum of disturbances caused by the disease and by tissue
compensative actions.
Detailed studies on the links between altered metal transport and neurodegeneration will
be helpful to facilitate the search for effective therapeutic strategies to avoid damage
caused by metal dyshomeostasis.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Acknowledgements
S Montes, I Pérez-Neri and C Ríos receive grants from CONACyT (51541, 83521 and
47425 respectively). Rivera-Mancía S and Tristán-López L receive fellowships from
CONACyT (203330 and 207021 respectively)

27
References
[1] A. Gaeta, R. C. Hider, The crucial role of metal ions in neurodegeneration: the basis for a promising therapeutic strategy, Br. J. Pharmacol. 146 (2005) 1041-1059.
[2] C. W. Levenson, Trace metal regulation of neuronal apoptosis: from genes to behavior, Physiol. Behav. 86 (2005) 399-406.
[3] D. L. Price, New order from neurological disorders, Nature 399 (1999) A3-A5.
[4] C. A. Rottkamp, A. Nunomura, K. Hirai, L. M. Sayre, G. Perry, M. A. Smith, Will antioxidants fulfill their expectations for the treatment of Alzheimer disease?, Mech. Ageing Dev. 116 (2000) 169-179.
[5] M. B. Youdim, M. Fridkin, H. Zheng, Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases, Mech. Ageing Dev. 126 (2005) 317-326.
[6] J. L. Beard, J. R. Connor, Iron status and neural functioning, Annu. Rev. Nutr. 23 (2003) 41-58.
[7] P. Ponka, Cellular iron metabolism, Kidney Int. Suppl. 69 (1999) S2-11.
[8] M. M. Peña, J. Lee, D. J. Thiele, A delicate balance: homeostatic control of copper uptake and distribution, J. Nutr. 129 (1999) 1251-1260.
[9] J. R. Connor, S. A. Benkovic, Iron regulation in the brain: histochemical, biochemical, and molecular considerations, Ann. Neurol. 32 Suppl. (1992) S51-S61.
[10] T. A. Rouault, Systemic iron metabolism: a review and implications for brain iron metabolism, Pediatr. Neurol. 25 (2001) 130-137.
[11] D. Berg, M. B. Youdim, Role of iron in neurodegenerative disorders, Top. Magn. Reson. Imaging 17 (2006) 5-17.
[12] D. T. Dexter, P. Jenner, A. H. Schapira, C. D. Marsden, Alterations in levels of iron, ferritin, and other trace metals in neurodegenerative diseases affecting the basal ganglia. The Royal Kings and Queens Parkinson's Disease Research Group, Ann. Neurol. 32 Suppl. (1992) S94-100.
[13] D. A. Simmons, M. Casale, B. Alcon, N. Pham, N. Narayan, G. Lynch, Ferritin accumulation in dystrophic microglia is an early event in the development of Huntington's disease, Glia 55 (2007) 1074-1084.
[14] O. I. Aruoma, H. Kaur, B. Halliwell, Oxygen free radicals and human diseases, J. R. Soc. Health 111 (1991) 172-177.

28
[15] J. Agar, H. Durham, Relevance of oxidative injury in the pathogenesis of motor neuron diseases, Amyotroph. Lateral Scler. Other Motor Neuron Disord. 4 (2003) 232-242.
[16] E. Madsen, J. D. Gitlin, Copper and iron disorders of the brain, Annu. Rev. Neurosci. 30 (2007) 317-337.
[17] H. Gunshin, B. Mackenzie, U. V. Berger, Y. Gunshin, M. F. Romero, W. F. Boron, S. Nussberger, J. L. Gollan, M. A. Hediger, Cloning and characterization of a mammalian protein-coupled metal-ion transporter, Nature 388 (1997) 482-488.
[18] B. J. Kelley, R. C. Petersen, Alzheimer's disease and mild cognitive impairment, Neurol. Clin. 25 (2007) 577-609, v.
[19] C. P. Ferri, M. Prince, C. Brayne, H. Brodaty, L. Fratiglioni, M. Ganguli, K. Hall, K. Hasegawa, H. Hendrie, Y. Huang, A. Jorm, C. Mathers, P. R. Menezes, E. Rimmer, M. Scazufca, Alzheimer's Disease International, Global prevalence of dementia: a Delphi consensus study, Lancet 366 (2005) 2112-2117.
[20] K. A. Jellinger, C. Bancher, Neuropathology of Alzheimer's disease: a critical update, J. Neural. Transm. Suppl. 54 (1998) 77-95.
[21] C. L. Masters, G. Multhaup, G. Simms, J. Pottgiesser, R. N. Martins, K. Beyreuther, Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels, EMBO J. 4 (1985) 2757-2763.
[22] C. B. Eckman, E. A. Eckman, An update on the amyloid hypothesis, Neurol. Clin. 25 (2007) 669-682.
[23] D. J. Selkoe, Alzheimer's disease: genes, proteins, and therapy, Physiol. Rev. 81 (2001) 741-766.
[24] R. E. Tanzi, J. F. Gusella, P. C. Watkins, G. A. Bruns, P. St George-Hyslop, M. L. Van Keuren, D. Patterson, S. Pagan, D. M. Kurnit, R. L. Neve, Amyloid beta protein gene: cDNA, mRNA distribution, and genetic linkage near the Alzheimer locus, Science 235 (1987) 880-884.
[25] J. T. Jarrett, E. P. Berger, P. T. Lansbury, Jr., The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer's disease, Biochemistry 32 (1993) 4693-4697.
[26] F. Prelli, E. M. Castano, S. G. van Duinen, G. T. Bots, W. Luyendijk, B. Frangione, Different processing of Alzheimer's beta-protein precursor in the vessel wall of patients with hereditary cerebral hemorrhage with amyloidosis-Dutch type, Biochem. Biophys. Res. Commun. 151 (1988) 1150-1155.
[27] P. J. Crouch, A. R. White, A. I. Bush, The modulation of metal bio-availability as a therapeutic strategy for the treatment of Alzheimer's disease, FEBS J. 274 (2007) 3775-3783.

29
[28] G. Liu, W. Huang, R. D. Moir, C. R. Vanderburg, B. Lai, Z. Peng, R. E. Tanzi, J. T. Rogers, X. Huang, Metal exposure and Alzheimer's pathogenesis, J. Struct. Biol. 155 (2006) 45-51.
[29] M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell, W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci. 158 (1998) 47-52.
[30] K. J. Barnham, F. Haeffner, G. D. Ciccotosto, C. C. Curtain, D. Tew, C. Mavros, K. Beyreuther, D. Carrington, C. L. Masters, R. A. Cherny, R. Cappai, A. I. Bush, Tyrosine gated electron transfer is key to the toxic mechanism of Alzheimer's disease beta-amyloid, FASEB J. 18 (2004) 1427-1429.
[31] X. Huang, C. S. Atwood, M. A. Hartshorn, G. Multhaup, L. E. Goldstein, R. C. Scarpa, M. P. Cuajungco, D. N. Gray, J. Lim, R. D. Moir, R. E. Tanzi, A. I. Bush, The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction, Biochemistry 38 (1999) 7609-7616.
[32] D. Schubert, M. Chevion, The role of iron in beta amyloid toxicity, Biochem. Biophys. Res. Commun. 216 (1995) 702-707.
[33] S. Moalem, M. E. Percy, D. F. Andrews, T. P. Kruck, S. Wong, A. J. Dalton, P. Mehta, B. Fedor, A. C. Warren, Are hereditary hemochromatosis mutations involved in Alzheimer disease?, Am. J. Med. Genet. 93 (2000) 58-66.
[34] J. T. Rogers, J. D. Randall, C. M. Cahill, P. S. Eder, X. Huang, H. Gunshin, L. Leiter, J. McPhee, S. S. Sarang, T. Utsuki, N. H. Greig, D. K. Lahiri, R. E. Tanzi, A. I. Bush, T. Giordano, S. R. Gullans, An iron-responsive element type II in the 5'-untranslated region of the Alzheimer's amyloid precursor protein transcript, J. Biol. Chem. 277 (2002) 45518-45528.
[35] S. R. Robinson, D. F. Noon, J. Kril, G. Halliday, Most amyloid plaques contain ferritin rich cells, Alz. Res. 1 (1995) 191-196.
[36] B. Ding, K. M. Chen, H. W. Ling, F. Sun, X. Li, T. Wan, W. M. Chai, H. Zhang, Y. Zhan, Y. J. Guan, Correlation of iron in the hippocampus with MMSE in patients with Alzheimer's disease, J. Magn. Reson. Imaging 29 (2009) 793-798.
[37] D. R. Crapper McLachlan, A. J. Dalton, T. P. Kruck, M. Y. Bell, W. L. Smith, W. Kalow, D. F. Andrews, Intramuscular desferrioxamine in patients with Alzheimer's disease, Lancet 337 (1991) 1304-1308.
[38] M. Kitazawa, D. Cheng, F. M. LaFerla, Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD, J. Neurochem. 108 (2009) 1550-1560.
[39] C. S. Atwood, R. C. Scarpa, X. Huang, R. D. Moir, W. D. Jones, D. P. Fairlie, R. E. Tanzi, A. I. Bush, Characterization of copper interactions with alzheimer amyloid beta peptides: identification of an attomolar-affinity copper binding site on amyloid beta1-42, J. Neurochem. 75 (2000) 1219-1233.

30
[40] X. Huang, M. P. Cuajungco, C. S. Atwood, M. A. Hartshorn, J. D. Tyndall, G. R. Hanson, K. C. Stokes, M. Leopold, G. Multhaup, L. E. Goldstein, R. C. Scarpa, A. J. Saunders, J. Lim, R. D. Moir, C. Glabe, E. F. Bowden, C. L. Masters, D. P. Fairlie, R. E. Tanzi, A. I. Bush, Cu(II) potentiation of alzheimer abeta neurotoxicity. Correlation with cell-free hydrogen peroxide production and metal reduction, J. Biol. Chem. 274 (1999) 37111-37116.
[41] C. Opazo, X. Huang, R. A. Cherny, R. D. Moir, A. E. Roher, A. R. White, R. Cappai, C. L. Masters, R. E. Tanzi, N. C. Inestrosa, A. I. Bush, Metalloenzyme-like activity of Alzheimer's disease beta-amyloid. Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2), J. Biol. Chem. 277 (2002) 40302-40308.
[42] S. J. Kish, C. Bergeron, A. Rajput, S. Dozic, F. Mastrogiacomo, L. J. Chang, J. M. Wilson, L. M. DiStefano, J. N. Nobrega, Brain cytochrome oxidase in Alzheimer's disease, J. Neurochem. 59 (1992) 776-779.
[43] P. J. Crouch, K. J. Barnham, J. A. Duce, R. E. Blake, C. L. Masters, I. A. Trounce, Copper-dependent inhibition of cytochrome c oxidase by Abeta(1-42) requires reduced methionine at residue 35 of the Abeta peptide, J. Neurochem. 99 (2006) 226-236.
[44] P. J. Crouch, R. Blake, J. A. Duce, G. D. Ciccotosto, Q. X. Li, K. J. Barnham, C. C. Curtain, R. A. Cherny, R. Cappai, T. Dyrks, C. L. Masters, I. A. Trounce, Copper-dependent inhibition of human cytochrome c oxidase by a dimeric conformer of amyloid-beta1-42, J. Neurosci. 25 (2005) 672-679.
[45] M. L. Schlief, A. M. Craig, J. D. Gitlin, NMDA receptor activation mediates copper homeostasis in hippocampal neurons, J. Neurosci. 25 (2005) 239-246.
[46] A. R. White, R. Reyes, J. F. Mercer, J. Camakaris, H. Zheng, A. I. Bush, G. Multhaup, K. Beyreuther, C. L. Masters, R. Cappai, Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice, Brain Res. 842 (1999) 439-444.
[47] R. Squitti, P. Pasqualetti, F. G. Dal, F. Moffa, E. Cassetta, D. Lupoi, F. Vernieri, L. Rossi, M. Baldassini, P. M. Rossini, Excess of serum copper not related to ceruloplasmin in Alzheimer disease, Neurology 64 (2005) 1040-1046.
[48] R. Squitti, G. Barbati, L. Rossi, M. Ventriglia, F. G. Dal, S. Cesaretti, F. Moffa, I. Caridi, E. Cassetta, P. Pasqualetti, L. Calabrese, D. Lupoi, P. M. Rossini, Excess of nonceruloplasmin serum copper in AD correlates with MMSE, CSF [beta]-amyloid, and h-tau, Neurology 67 (2006) 76-82.
[49] S. A. Bellingham, D. K. Lahiri, B. Maloney, F. S. La, G. Multhaup, J. Camakaris, Copper depletion down-regulates expression of the Alzheimer's disease amyloid-beta precursor protein gene, J. Biol. Chem. 279 (2004) 20378-20386.
[50] D. Strozyk, L. J. Launer, P. A. Adlard, R. A. Cherny, A. Tsatsanis, I. Volitakis, K. Blennow, H. Petrovitch, L. R. White, A. I. Bush, Zinc and copper modulate

31
Alzheimer Abeta levels in human cerebrospinal fluid, Neurobiol. Aging 30 (2009) 1069-1077.
[51] I. Y. Adamson, R. Vincent, J. Bakowska, Differential production of metalloproteinases after instilling various urban air particle samples to rat lung, Exp. Lung Res. 29 (2003) 375-388.
[52] W. Wu, J. M. Samet, R. Silbajoris, L. A. Dailey, D. Sheppard, P. A. Bromberg, L. M. Graves, Heparin-binding epidermal growth factor cleavage mediates zinc-induced epidermal growth factor receptor phosphorylation, Am. J. Respir. Cell Mol. Biol. 30 (2004) 540-547.
[53] A. R. White, T. Du, K. M. Laughton, I. Volitakis, R. A. Sharples, M. E. Xilinas, D. E. Hoke, R. M. Holsinger, G. Evin, R. A. Cherny, A. F. Hill, K. J. Barnham, Q. X. Li, A. I. Bush, C. L. Masters, Degradation of the Alzheimer disease amyloid beta-peptide by metal-dependent up-regulation of metalloprotease activity, J. Biol. Chem. 281 (2006) 17670-17680.
[54] T. Borchardt, J. Camakaris, R. Cappai, C. L. Masters, K. Beyreuther, G. Multhaup, Copper inhibits beta-amyloid production and stimulates the non-amyloidogenic pathway of amyloid-precursor-protein secretion, Biochem. J. 344 Pt 2 (1999) 461-467.
[55] M. A. Cater, K. T. McInnes, Q. X. Li, I. Volitakis, F. S. La, J. F. Mercer, A. I. Bush, Intracellular copper deficiency increases amyloid-beta secretion by diverse mechanisms, Biochem. J. 412 (2008) 141-152.
[56] M. C. Boll, M. Alcaraz-Zubeldia, S. Montes, C. Rios, Free copper, ferroxidase and SOD1 activities, lipid peroxidation and NO(x) content in the CSF. A different marker profile in four neurodegenerative diseases, Neurochem. Res. 33 (2008) 1717-1723.
[57] I. Maurer, S. Zierz, H. J. Moller, A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients, Neurobiol. Aging 21 (2000) 455-462.
[58] S. J. Texel, X. Xu, Z. L. Harris, Ceruloplasmin in neurodegenerative diseases, Biochem. Soc. Trans. 36 (2008) 1277-1281.
[59] D. A. Loeffler, P. A. LeWitt, P. L. Juneau, A. A. Sima, H. U. Nguyen, A. J. DeMaggio, C. M. Brickman, G. J. Brewer, R. D. Dick, M. D. Troyer, L. Kanaley, Increased regional brain concentrations of ceruloplasmin in neurodegenerative disorders, Brain Res. 738 (1996) 265-274.
[60] J. R. Connor, P. Tucker, M. Johnson, B. Snyder, Ceruloplasmin levels in the human superior temporal gyrus in aging and Alzheimer's disease, Neurosci. Lett. 159 (1993) 88-90.
[61] Y. Avramovich-Tirosh, T. Amit, O. Bar-Am, H. Zheng, M. Fridkin, M. B. Youdim, Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer's disease, J. Neurochem. 100 (2007) 490-502.

32
[62] K. Rezai-Zadeh, D. Shytle, N. Sun, T. Mori, H. Hou, D. Jeanniton, J. Ehrhart, K. Townsend, J. Zeng, D. Morgan, J. Hardy, T. Town, J. Tan, Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice, J. Neurosci. 25 (2005) 8807-8814.
[63] L. Reznichenko, T. Amit, H. Zheng, Y. vramovich-Tirosh, M. B. Youdim, O. Weinreb, S. Mandel, Reduction of iron-regulated amyloid precursor protein and beta-amyloid peptide by (-)-epigallocatechin-3-gallate in cell cultures: implications for iron chelation in Alzheimer's disease, J. Neurochem. 97 (2006) 527-536.
[64] F. Yang, G. P. Lim, A. N. Begum, O. J. Ubeda, M. R. Simmons, S. S. Ambegaokar, P. P. Chen, R. Kayed, C. G. Glabe, S. A. Frautschy, G. M. Cole, Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo, J. Biol. Chem. 280 (2005) 5892-5901.
[65] L. Baum, C. W. Lam, S. K. Cheung, T. Kwok, V. Lui, J. Tsoh, L. Lam, V. Leung, E. Hui, C. Ng, J. Woo, H. F. Chiu, W. B. Goggins, B. C. Zee, K. F. Cheng, C. Y. Fong, A. Wong, H. Mok, M. S. Chow, P. C. Ho, S. P. Ip, C. S. Ho, X. W. Yu, C. Y. Lai, M. H. Chan, S. Szeto, I. H. Chan, V. Mok, Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease, J. Clin. Psychopharmacol. 28 (2008) 110-113.
[66] D. Blat, L. Weiner, M. B. Youdim, M. Fridkin, A Novel iron-chelating derivative of the neuroprotective peptide NAPVSIPQ shows superior antioxidant and antineurodegenerative capabilities, J. Med. Chem. 51 (2008) 126-134.
[67] N. Boddaert, K. H. Le Quang Sang, A. Rötig, A. Leroy-Willig, S. Gallet, F. Brunelle, D. Sidi, J. C. Thalabard, A. Munnich, Z. I. Cabantchik, Selective iron chelation in Friedreich ataxia: biologic and clinical implications, Blood 110 (2007) 401-408.
[68] G. Liu, P. Men, P. L. Harris, R. K. Rolston, G. Perry, M. A. Smith, Nanoparticle iron chelators: a new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalance, Neurosci. Lett. 406 (2006) 189-193.
[69] G. Liu, P. Men, W. Kudo, G. Perry, M. A. Smith, Nanoparticle–chelator conjugates as inhibitors of amyloid- aggregation and neurotoxicity: A novel therapeutic approach for Alzheimer disease, Neurosci. Lett. 455 (2009) 187-190.
[70] R. A. Cherny, J. T. Legg, C. A. McLean, D. P. Fairlie, X. Huang, C. S. Atwood, K. Beyreuther, R. E. Tanzi, C. L. Masters, A. I. Bush, Aqueous dissolution of Alzheimer's disease Abeta amyloid deposits by biometal depletion, J. Biol. Chem. 274 (1999) 23223-23228.
[71] R. Squitti, P. M. Rossini, E. Cassetta, F. Moffa, P. Pasqualetti, M. Cortesi, A. Colloca, L. Rossi, A. Finazzi-Agro, d-penicillamine reduces serum oxidative stress in Alzheimer's disease patients, Eur. J. Clin. Invest. 32 (2002) 51-59.

33
[72] T. M. Malm, H. Iivonen, G. Goldsteins, V. Keksa-Goldsteine, T. Ahtoniemi, K. Kanninen, A. Salminen, S. Auriola, G. T. Van, H. Tanila, J. Koistinaho, Pyrrolidine dithiocarbamate activates Akt and improves spatial learning in APP/PS1 mice without affecting beta-amyloid burden, J. Neurosci. 27 (2007) 3712-3721.
[73] R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. McLean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters, A. I. Bush, Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice, Neuron 30 (2001) 665-676.
[74] C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi, C. L. Masters, Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial, Arch. Neurol. 60 (2003) 1685-1691.
[75] S. Schäfer, F. G. Pajonk, G. Multhaup, T. A. Bayer, Copper and clioquinol treatment in young APP transgenic and wild-type mice: effects on life expectancy, body weight, and metal-ion levels, J. Mol. Med. 85 (2007) 405-413.
[76] P. S. Donnelly, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, R. A. Cherny, R. A. Sharples, A. F. Hill, Q. X. Li, C. L. Masters, K. J. Barnham, A. R. White, Selective intracellular release of copper and zinc ions from bis(thiosemicarbazonato) complexes reduces levels of Alzheimer disease amyloid-beta peptide, J. Biol. Chem. 283 (2008) 4568-4577.
[77] P. J. Crouch, L. W. Hung, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, A. Nurjono, A. Caragounis, T. Du, K. M. Laughton, I. Volitakis, A. I. Bush, Q. X. Li, C. L. Masters, R. Cappai, R. A. Cherny, P. S. Donnelly, A. R. White, K. J.Barnham, Increasing Cu bioavailability inhibits A oligomers and tau phosphorylation, Proc. Natl. Acad. Sci. U. S. A. 106 (2009) 381-386.
[78] M. L. Bolognesi, A. Cavalli, L. Valgimigli, M. Bartolini, M. Rosini, V. Andrisano, M. Recanatini, C. Melchiorre, Multi-target-directed drug design strategy: from a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer's disease, J. Med. Chem. 50 (2007) 6446-6449.
[79] S. Bharath, M. Hsu, D. Kaur, S. Rajagopalan, J. K. Andersen, Glutathione, iron and Parkinson's disease, Biochem. Pharmacol. 64 (2002) 1037-1048.
[80] B. K. Barlow, D. A. Cory-Slechta, E. K. Richfield, M. Thiruchelvam, The gestational environment and Parkinson's disease: evidence for neurodevelopmental origins of a neurodegenerative disorder, Reprod. Toxicol. 23 (2007) 457-470.
[81] D. Kaur, J. Andersen, Does cellular iron dysregulation play a causative role in Parkinson's disease?, Ageing Res. Rev. 3 (2004) 327-343.

34
[82] D. J. Burn, A. I. Tröster, Neuropsychiatric complications of medical and surgical therapies for Parkinson's disease, J Geriatr. Psychiatry Neurol. 17 (2004) 172-180.
[83] P. M. Rees, C. J. Fowler, C. P. Maas, Sexual function in men and women with neurological disorders, Lancet 369 (2007) 512-525.
[84] R. Graumann, I. Paris, P. Martinez-Alvarado, P. Rumanque, C. Perez-Pastene, S. P. Cardenas, P. Marin, F. az-Grez, R. Caviedes, P. Caviedes, J. Segura-Aguilar, Oxidation of dopamine to aminochrome as a mechanism for neurodegeneration of dopaminergic systems in Parkinson's disease. Possible neuroprotective role of DT-diaphorase, Pol. J. Pharmacol. 54 (2002) 573-579.
[85] J. Jankovic, Searching for a relationship between manganese and welding and Parkinson's disease, Neurology 64 (2005) 2021-2028.
[86] J. M. Gorell, E. L. Peterson, B. A. Rybicki, C. C. Johnson, Multiple risk factors for Parkinson's disease, J. Neurol. Sci. 217 (2004) 169-174.
[87] M. E. Götz, K. Double, M. Gerlach, M. B. Youdim, P. Riederer, The relevance of iron in the pathogenesis of Parkinson's disease, Ann. N. Y. Acad. Sci. 1012 (2004) 193-208.
[88] M. Castellanos, N. Puig, T. Carbonell, J. Castillo, J. Martinez, R. Rama, A. Dávalos, Iron intake increases infarct volume after permanent middle cerebral artery occlusion in rats, Brain Res. 952 (2002) 1-6.
[89] J. H. Cheah, S. F. Kim, L. D. Hester, K. W. Clancy, S. E. Patterson, III, V. Papadopoulos, S. H. Snyder, NMDA receptor-nitric oxide transmission mediates neuronal iron homeostasis via the GTPase Dexras1, Neuron 51 (2006) 431-440.
[90] K. L. Double, G. M. Halliday, J. Henderson, F. M. Griffiths, T. Heinemann, P. Riederer, M. Gerlach, The dopamine receptor agonist lisuride attenuates iron-mediated dopaminergic neurodegeneration, Exp. Neurol. 184 (2003) 530-535.
[91] S. Behnke, D. Berg, G. Becker, Does ultrasound disclose a vulnerability factor for Parkinson's disease?, J. Neurol. 250 Suppl. 1 (2003) I24-I27.
[92] D. T. Dexter, F. R. Wells, A. J. Lees, F. Agid, Y. Agid, P. Jenner, C. D. Marsden, Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson's disease, J. Neurochem. 52 (1989) 1830-1836.
[93] A. E. Oakley, J. F. Collingwood, J. Dobson, G. Love, H. R. Perrott, J. A. Edwardson, M. Elstner, C. M. Morris, Individual dopaminergic neurons show raised iron levels in Parkinson disease, Neurology 68 (2007) 1820-1825.
[94] P. Riederer, E. Sofic, W. D. Rausch, B. Schmidt, G. P. Reynolds, K. Jellinger, M. B. Youdim, Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains, J. Neurochem. 52 (1989) 515-520.
[95] G. Becker, D. Berg, Neuroimaging in basal ganglia disorders: perspectives for transcranial ultrasound, Mov. Disord. 16 (2001) 23-32.

35
[96] R. Martínez-Hernández, S. Montes, J. Higuera-Calleja, P. Yescas, M. C. Boll, C. Rios, Plasma ceruloplasmin ferroxidase activity correlates with substantia nigra sonographic area of Parkinson's disease patients, J. Neurol. Neurosurg. Psychiatry Submitted Manuscript (2009).
[97] D. Berg, W. Roggendorf, U. Schroder, R. Klein, T. Tatschner, P. Benz, O. Tucha, M. Preier, K. W. Lange, K. Reiners, M. Gerlach, G. Becker, Echogenicity of the substantia nigra: association with increased iron content and marker for susceptibility to nigrostriatal injury, Arch. Neurol. 59 (2002) 999-1005.
[98] J. Vymazal, A. Righini, R. A. Brooks, M. Canesi, C. Mariani, M. Leonardi, G. Pezzoli, T1 and T2 in the brain of healthy subjects, patients with Parkinson disease, and patients with multiple system atrophy: relation to iron content, Radiology 211 (1999) 489-495.
[99] S. Brar, D. Henderson, J. Schenck, E. A. Zimmerman, Iron accumulation in the substantia nigra of patients with Alzheimer disease and parkinsonism, Arch. Neurol. 66 (2009) 371-374.
[100] W. A. Cass, R. Grondin, A. H. Andersen, Z. Zhang, P. A. Hardy, L. K. Hussey-Andersen, W. S. Rayens, G. A. Gerhardt, D. M. Gash, Iron accumulation in the striatum predicts aging-related decline in motor function in rhesus monkeys, Neurobiol. Aging 28 (2007) 258-271.
[101] G. C. Brown, V. Borutaite, Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols, Biochim. Biophys. Acta 1658 (2004) 44-49.
[102] M. Hadziahmetovic, T. Dentchev, Y. Song, N. Haddad, X. He, P. Hahn, D. Pratico, R. Wen, Z. L. Harris, J. D. Lambris, J. Beard, J. L. Dunaief, Ceruloplasmin/hephaestin knockout mice model morphologic and molecular features of AMD, Invest. Ophthalmol. Vis. Sci. 49 (2008) 2728-2736.
[103] J. Wang, H. Jiang, J. X. Xie, Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats, Eur. J. Neurosci. 25 (2007) 2766-2772.
[104] H. Hochstrasser, P. Bauer, U. Walter, S. Behnke, J. Spiegel, I. Csoti, B. Zeiler, A. Bornemann, J. Pahnke, G. Becker, O. Riess, D. Berg, Ceruloplasmin gene variations and substantia nigra hyperechogenicity in Parkinson disease, Neurology 63 (2004) 1912-1917.
[105] J. Salazar, N. Mena, S. Hunot, A. Prigent, D. Alvarez-Fischer, M. Arredondo, C. Duyckaerts, V. Sazdovitch, L. Zhao, L. M. Garrick, M. T. Nuñez, M. D. Garrick, R. Raisman-Vozari, E. C. Hirsch, Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease, Proc. Natl. Acad. Sci. U. S. A. 105 (2008) 18578-18583.
[106] S. Zhang, J. Wang, N. Song, J. Xie, H. Jiang, Up-regulation of divalent metal transporter 1 is involved in 1-methyl-4-phenylpyridinium (MPP+)-induced apoptosis in MES23.5 cells, Neurobiol. Aging 30 (2009) 1466-1476.

36
[107] D. S. Maharaj, H. Maharaj, S. Daya, B. D. Glass, Melatonin and 6-hydroxymelatonin protect against iron-induced neurotoxicity, J. Neurochem. 96 (2006) 78-81.
[108] A. C. Muller, A. Dairam, J. L. Limson, S. Daya, Mechanisms by which acyclovir reduces the oxidative neurotoxicity and biosynthesis of quinolinic acid, Life Sci. 80 (2007) 918-925.
[109] E. Mendez-Alvarez, R. Soto-Otero, A. Hermida-Ameijeiras, M. E. Lopez-Martin, J. L. Labandeira-Garcia, Effect of iron and manganese on hydroxyl radical production by 6-hydroxydopamine: mediation of antioxidants, Free Radic. Biol. Med. 31 (2001) 986-998.
[110] N. Ostrerova-Golts, L. Petrucelli, J. Hardy, J. M. Lee, M. Farer, B. Wolozin, The A53 T alpha-synuclein mutation increases iron-dependent aggregation and toxicity, J. Neurosci. 20 (2000) 6048-6054.
[111] A. Gupta, V. L. Dawson, T. M. Dawson, What causes cell death in Parkinson's disease?, Ann. Neurol. 64 (Suppl.) (2008) S3-S15.
[112] D. Kaur, J. Peng, S. J. Chinta, S. Rajagopalan, D. A. Di Monte, R. A. Cherny, J. K. Andersen, Increased murine neonatal iron intake results in Parkinson-like neurodegeneration with age, Neurobiol. Aging 28 (2007) 907-913.
[113] A. Fredriksson, T. Archer, Effect of postnatal iron administration on MPTP-induced behavioral deficits and neurotoxicity: behavioral enhancement by L-Dopa-MK-801 co-administration, Behav. Brain Res. 139 (2003) 31-46.
[114] P. Aguirre, P. Valdés, P. Aracena-Parks, V. Tapia, M. T. Núñez, Upregulation of -glutamate-cysteine ligase as part of the long-term adaptation process to iron accumulation in neuronal, Am. J. Physiol. Cell Physiol. 292 (2007) C2197-C2203.
[115] M. Arredondo, M. T. Núñez, Iron and copper metabolism, Mol. Aspects Med. 26 (2005) 313-327.
[116] B. F. Popescu, M. J. George, U. Bergmann, A. V. Garachtchenko, M. E. Kelly, R. P. E. McCrea, K. Lüning, R. M. Devon, G. N. George, A. D. Hanson, S. M. Harder, L. D. Chapman, I. J. Pickering, H. Nichol, Mapping metals in Parkinson's and normal brain using rapid-scanning x-ray fluorescence, Phys. Med. Biol. 54 (2009) 651-653.
[117] D. Beshgetoor, M. Hambidge, Clinical conditions altering copper metabolism in humans, Am. J Clin. Nutr. 67 (1998) 1017S-1021S.
[118] M. C. Boll, J. Sotelo, E. Otero, M. caraz-Zubeldia, C. Rios, Reduced ferroxidase activity in the cerebrospinal fluid from patients with Parkinson's disease, Neurosci. Lett. 265 (1999) 155-158.
[119] R. M. Rasia, C. W. Bertoncini, D. Marsh, W. Hoyer, D. Cherny, M. Zweckstetter, C. Griesinger, T. M. Jovin, C. O. Fernandez, Structural characterization of copper(II) binding to alpha-synuclein: Insights into the

37
bioinorganic chemistry of Parkinson's disease, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 4294-4299.
[120] V. N. Uversky, J. Li, A. L. Fink, Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson's disease and heavy metal exposure, J. Biol. Chem. 276 (2001) 44284-44296.
[121] E. N. Lee, S. Y. Lee, D. Lee, J. Kim, S. R. Paik, Lipid interaction of alpha-synuclein during the metal-catalyzed oxidation in the presence of Cu2+ and H2O2, J. Neurochem. 84 (2003) 1128-1142.
[122] S. R. Paik, H. J. Shin, J. H. Lee, Metal-catalyzed oxidation of alpha-synuclein in the presence of Copper(II) and hydrogen peroxide, Arch. Biochem. Biophys. 378 (2000) 269-277.
[123] K. S. Kim, S. Y. Choi, H. Y. Kwon, M. H. Won, T. C. Kang, J. H. Kang, The ceruloplasmin and hydrogen peroxide system induces alpha-synuclein aggregation in vitro, Biochimie 84 (2002) 625-631.
[124] R. Lowe, D. L. Pountney, P. H. Jensen, W. P. Gai, N. H. Voelcker, Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain, Protein. Sci. 13 (2004) 3245-3252.
[125] X. F. Wang, M. S. Cynader, Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity, J. Neurosci. 21 (2001) 3322-3331.
[126] S. H. Sun, B. L. O'Dell, Low copper status of rats affects polyunsaturated fatty acid composition of brain phospholipids, unrelated to neuropathology, J. Nutr. 122 (1992) 65-73.
[127] M. Alcaraz-Zubeldia, P. Rojas, C. Boll, C. Rios, Neuroprotective effect of acute and chronic administration of copper (II) sulfate against MPP+ neurotoxicity in mice, Neurochem. Res. 26 (2001) 59-64.
[128] M. Rubio-Osornio, S. Montes, F. Perez-Severiano, P. Aguilera, E. Floriano-Sanchez, A. Monroy-Noyola, C. Rubio, C. Rios, Copper reduces striatal protein nitration and tyrosine hydroxylase inactivation induced by MPP+ in rats, Neurochem. Int. 54 (2009) 447-451.
[129] A. Santamaria, A. Flores-Escartin, J. C. Martinez, L. Osorio, S. Galvan-Arzate, J. Pedraza-Chaverri, P. D. Maldonado, O. N. Medina-Campos, M. E. Jimenez-Capdeville, J. Manjarrez, C. Rios, Copper blocks quinolinic acid neurotoxicity in rats: contribution of antioxidant systems, Free Radic. Biol. Med. 35 (2003) 418-427.
[130] M. B. Youdim, E. Grunblatt, S. Mandel, The copper chelator, D-penicillamine, does not attenuate MPTP induced dopamine depletion in mice, J. Neural Transm. 114 (2007) 205-209.
[131] C. Rios, R. Alvarez-Vega, P. Rojas, Depletion of copper and manganese in brain after MPTP treatment of mice, Pharmacol. Toxicol. 76 (1995) 348-352.

38
[132] P. Aguirre, N. Mena, V. Tapia, M. Arredondo, M. T. Núñez, Iron homeostasis in neuronal cells: a role for IREG1, BMC Neurosci. 6 (2005) 3.
[133] E. Aigner, I. Theurl, H. Haufe, M. Seifert, F. Hohla, L. Scharinger, F. Stickel, F. Mourlane, G. Weiss, C. Datz, Copper availability contributes to iron perturbations in human nonalcoholic fatty liver disease, Gastroenterology 135 (2008) 680-688.
[134] S. Auclair, C. Feillet-Coudray, C. Coudray, S. Schneider, M. U. Muckenthaler, A. Mazur, Mild copper deficiency alters gene expression of proteins involved in iron metabolism, Blood Cells Mol. Dis. 36 (2006) 15-20.
[135] D. Das, N. Tapryal, S. K. Goswami, P. L. Fox, C. K. Mukhopadhyay, Regulation of ceruloplasmin in human hepatic cells by redox active copper: identification of a novel AP-1 site in the ceruloplasmin gene, Biochem. J. 402 (2007) 135-141.
[136] I. De Domenico, D. M. Ward, M. C. di Patti, S. Y. Jeong, S. David, G. Musci, J. Kaplan, Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin, EMBO J. 26 (2007) 2823-2831.
[137] D. Kaur, F. Yantiri, S. Rajagopalan, J. Kumar, J. Q. Mo, R. Boonplueang, V. Viswanath, R. Jacobs, L. Yang, M. F. Beal, D. DiMonte, I. Volitaskis, L. Ellerby, R. A. Cherny, A. I. Bush, J. K. Andersen, Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease, Neuron 37 (2003) 899-909.
[138] T. Shang, S. Kotamraju, S. V. Kalivendi, C. J. Hillard, B. Kalyanaraman, 1-Methyl-4-phenylpyridinium-induced apoptosis in cerebellar granule neurons is mediated by transferrin receptor iron-dependent depletion of tetrahydrobiopterin and neuronal nitric-oxide synthase-derived superoxide, J. Biol. Chem. 279 (2004) 19099-19112.
[139] C. C. Johnson, J. M. Gorell, B. A. Rybicki, K. Sanders, E. L. Peterson, Adult nutrient intake as a risk factor for Parkinson's disease, Int. J. Epidemiol. 28 (1999) 1102-1109.
[140] C. W. Levenson, R. G. Cutler, B. Ladenheim, J. L. Cadet, J. Hare, M. P. Mattson, Role of dietary iron restriction in a mouse model of Parkinson's disease, Exp. Neurol. 190 (2004) 506-514.
[141] P. Rojas, S. Montes, N. Serrano-García, J. Rojas-Castañeda, Effect of EGb761 supplementation on the content of copper in mouse brain in an animal model of Parkinson's disease, Nutrition 25 (2009) 482-485.
[142] G. Siciliano, S. Piazza, C. Carlesi, C. A. Del, M. Franzini, A. Pompella, G. Malvaldi, M. Mancuso, A. Paolicchi, L. Murri, Antioxidant capacity and protein oxidation in cerebrospinal fluid of amyotrophic lateral sclerosis, J. Neurol. 254 (2007) 575-580.

39
[143] M. Orth, A. H. Schapira, Mitochondria and degenerative disorders, Am. J. Med. Genet 106 (2001) 27-36.
[144] P. M. Andersen, Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene, Curr. Neurol. Neurosci. Rep. 6 (2006) 37-46.
[145] L. Banci, I. Bertini, A. Durazo, S. Girotto, E. B. Gralla, M. Martinelli, J. S. Valentine, M. Vieru, J. P. Whitelegge, Metal-free superoxide dismutase forms soluble oligomers under physiological conditions: a possible general mechanism for familial ALS, Proc. Natl. Acad. Sci. U. S. A. 104 (2007) 11263-11267.
[146] M. E. Gurney, H. Pu, A. Y. Chiu, M. C. Dal Canto, C. Y. Polchow, D. D. Alexander, J. Caliendo, A. Hentati, Y. W. Kwon, H. X. Deng, ., Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation, Science 264 (1994) 1772-1775.
[147] A. G. Reaume, J. L. Elliott, E. K. Hoffman, N. W. Kowall, R. J. Ferrante, D. F. Siwek, H. M. Wilcox, D. G. Flood, M. F. Beal, R. H. Brown, Jr., R. W. Scott, W. D. Snider, Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury, Nat. Genet. 13 (1996) 43-47.
[148] K. Okamoto, S. Hirai, M. Amari, M. Watanabe, A. Sakurai, Bunina bodies in amyotrophic lateral sclerosis immunostained with rabbit anti-cystatin C serum, Neurosci. Lett. 162 (1993) 125-128.
[149] P. N. Leigh, B. H. Anderton, A. Dodson, J. M. Gallo, M. Swash, D. M. Power, Ubiquitin deposits in anterior horn cells in motor neurone disease, Neurosci. Lett. 93 (1988) 197-203.
[150] T. Kato, T. Katagiri, A. Hirano, T. Kawanami, H. Sasaki, Lewy body-like hyaline inclusions in sporadic motor neuron disease are ubiquitinated, Acta Neuropathol. 77 (1989) 391-396.
[151] S. Matsumoto, H. Kusaka, H. Ito, N. Shibata, T. Asayama, T. Imai, Sporadic amyotrophic lateral sclerosis with dementia and Cu/Zn superoxide dismutase-positive Lewy body-like inclusions, Clin. Neuropathol. 15 (1996) 41-46.
[152] H. X. Deng, Y. Shi, Y. Furukawa, H. Zhai, R. Fu, E. Liu, G. H. Gorrie, M. S. Khan, W. Y. Hung, E. H. Bigio, T. Lukas, M. C. Dal Canto, T. V. O'Halloran, T. Siddique, Conversion to the amyotrophic lateral sclerosis phenotype is associated with intermolecular linked insoluble aggregates of SOD1 in mitochondria, Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 7142-7147.
[153] R. Danzeisen, T. Achsel, U. Bederke, M. Cozzolino, C. Crosio, A. Ferri, M. Frenzel, E. B. Gralla, L. Huber, A. Ludolph, M. Nencini, G. Rotilio, J. S. Valentine, M. T. Carrì, Superoxide dismutase 1 modulates expression of transferrin receptor, J. Biol. Inorg. Chem. 11 (2006) 489-498.
[154] L. Banci, I. Bertini, M. Boca, S. Girotto, M. Martinelli, J. S. Valentine, M. Vieru, SOD1 and amyotrophic lateral sclerosis: mutations and oligomerization, PLoS ONE 3 (2008) e1677.

40
[155] M. F. Beal, Mitochondrial dysfunction in neurodegenerative diseases, Biochim. Biophys. Acta 1366 (1998) 211-223.
[156] P. G. Ince, P. J. Shaw, J. M. Candy, D. Mantle, L. Tandon, W. D. Ehmann, W. R. Markesbery, Iron, selenium and glutathione peroxidase activity are elevated in sporadic motor neuron disease, Neurosci. Lett. 182 (1994) 87-90.
[157] E. J. Kasarskis, L. Tandon, M. A. Lovell, W. D. Ehmann, Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study, J. Neurol. Sci. 130 (1995) 203-208.
[158] B. Leveugle, G. Spik, D. P. Perl, C. Bouras, H. M. Fillit, P. R. Hof, The iron-binding protein lactotransferrin is present in pathologic lesions in a variety of neurodegenerative disorders: a comparative immunohistochemical analysis, Brain Res. 650 (1994) 20-31.
[159] M. K. Olsen, S. L. Roberds, B. R. Ellerbrock, T. J. Fleck, D. K. McKinley, M. E. Gurney, Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord, Ann. Neurol. 50 (2001) 730-740.
[160] S. Y. Jeong, K. I. Rathore, K. Schulz, P. Ponka, P. Arosio, S. David, Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis, J. Neurosci. 29 (2009) 610-619.
[161] Y. Mizuno, M. Amari, M. Takatama, H. Aizawa, B. Mihara, K. Okamoto, Transferrin localizes in Bunina bodies in amyotrophic lateral sclerosis, Acta Neuropathol. 112 (2006) 597-603.
[162] K. Okamoto, Y. Mizuno, Y. Fujita, Bunina bodies in amyotrophic lateral sclerosis, Neuropathology 28 (2008) 109-115.
[163] X. S. Wang, S. Lee, Z. Simmons, P. Boyer, K. Scott, W. Liu, J. Connor, Increased incidence of the Hfe mutation in amyotrophic lateral sclerosis and related cellular consequences, J. Neurol. Sci. 227 (2004) 27-33.
[164] R. E. Fleming, W. S. Sly, Mechanisms of iron accumulation in hereditary hemochromatosis, Annu. Rev. Physiol. 64 (2002) 663-680.
[165] R. Stendel, T. A. Pietila, K. Lehmann, R. Kurth, O. Suess, M. Brock, Ruptured intracranial dermoid cysts, Surg. Neurol. 57 (2002) 391-398.
[166] K. J. Thompson, S. Shoham, J. R. Connor, Iron and neurodegenerative disorders, Brain Res. Bull. 55 (2001) 155-164.
[167] E. Kapaki, C. Zournas, G. Kanias, T. Zambelis, A. Kakami, C. Papageorgiou, Essential trace element alterations in amyotrophic lateral sclerosis, J. Neurol. Sci. 147 (1997) 171-175.
[168] T. Ahtoniemi, G. Goldsteins, V. Keksa-Goldsteine, T. Malm, K. Kanninen, A. Salminen, J. Koistinaho, Pyrrolidine dithiocarbamate inhibits induction of

41
immunoproteasome and decreases survival in a rat model of amyotrophic lateral sclerosis, Mol. Pharmacol. 71 (2007) 30-37.
[169] Q. X. Li, S. S. Mok, K. M. Laughton, C. A. McLean, I. Volitakis, R. A. Cherny, N. S. Cheung, A. R. White, C. L. Masters, Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model, Aging Cell 5 (2006) 153-165.
[170] E. Tokuda, S. Ono, K. Ishige, A. Naganuma, Y. Ito, T. Suzuki, Metallothionein proteins expression, copper and zinc concentrations, and lipid peroxidation level in a rodent model for amyotrophic lateral sclerosis, Toxicology 229 (2007) 33-41.
[171] E. Tokuda, S. Ono, K. Ishige, S. Watanabe, E. Okawa, Y. Ito, T. Suzuki, Ammonium tetrathiomolybdate delays onset, prolongs survival, and slows progression of disease in a mouse model for amyotrophic lateral sclerosis, Exp. Neurol. 213 (2008) 122-128.
[172] P. A. Jonsson, K. S. Graffmo, P. M. Andersen, T. Brännström, M. Lindberg, M. Oliveberg, S. L. Marklund, Disulphide-reduced superoxide dismutase-1 in CNS of transgenic amyotrophic lateral sclerosis models, Brain 129 (2006) 451-464.
[173] L. J. Hayward, J. A. Rodriguez, J. W. Kim, A. Tiwari, J. J. Goto, D. E. Cabelli, J. S. Valentine, R. H. Brown, Jr., Decreased metallation and activity in subsets of mutant superoxide dismutases associated with familial amyotrophic lateral sclerosis, J. Biol. Chem. 277 (2002) 15923-15931.
[174] J. S. Valentine, P. J. Hart, Misfolded CuZnSOD and amyotrophic lateral sclerosis, Proc. Natl. Acad. Sci. U. S. A. 100 (2003) 3617-3622.
[175] P. J. Hart, Pathogenic superoxide dismutase structure, folding, aggregation and turnover, Curr. Opin. Chem. Biol. 10 (2006) 131-138.
[176] D. V. Martyshkin, S. B. Mirov, Y. X. Zhuang, J. P. Crow, V. Ermilov, J. S. Beckman, Fluorescence assay for monitoring Zn-deficient superoxide dismutase in vitro, Spectrochim. Acta A Mol. Biomol. Spectrosc. 59 (2003) 3165-3175.
[177] A. G. Estévez, J. P. Crow, J. B. Sampson, C. Reiter, Y. Zhuang, G. J. Richardson, M. M. Tarpey, L. Barbeito, J. S. Beckman, Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase, Science 286 (1999) 2498-2500.
[178] J. J. Goto, E. B. Gralla, J. S. Valentine, D. E. Cabelli, Reactions of hydrogen peroxide with familial amyotrophic lateral sclerosis mutant human copper-zinc superoxide dismutases studied by pulse radiolysis, J. Biol. Chem. 273 (1998) 30104-30109.
[179] Z. A. Oztug Durer, J. A. Cohlberg, P. Dinh, S. Padua, K. Ehrenclou, S. Downes, J. K. Tan, Y. Nakano, C. J. Bowman, J. L. Hoskins, C. Kwon, A. Z. Mason, J. A. Rodriguez, P. A. Doucette, B. F. Shaw, J. S. Valentine, Loss of metal ions,

42
disulfide reduction and mutations related to familial ALS promote formation of amyloid-like aggregates from superoxide dismutase, PLoS ONE 4 (2009) e5004.
[180] Y. Furukawa, T. V. O'Halloran, Amyotrophic lateral sclerosis mutations have the greatest destabilizing effect on the apo- and reduced form of SOD1, leading to unfolding and oxidative aggregation, J. Biol. Chem. 280 (2005) 17266-17274.
[181] F. Arnesano, L. Banci, I. Bertini, M. Martinelli, Y. Furukawa, T. V. O'Halloran, The unusually stable quaternary structure of human Cu,Zn-superoxide dismutase 1 is controlled by both metal occupancy and disulfide status, J. Biol. Chem. 279 (2004) 47998-48003.
[182] J. Niwa, S. Yamada, S. Ishigaki, J. Sone, M. Takahashi, M. Katsuno, F. Tanaka, M. Doyu, G. Sobue, Disulfide bond mediates aggregation, toxicity, and ubiquitylation of familial amyotrophic lateral sclerosis-linked mutant SOD1, J. Biol. Chem. 282 (2007) 28087-28095.
[183] P. A. Doucette, L. J. Whitson, X. Cao, V. Schirf, B. Demeler, J. S. Valentine, J. C. Hansen, P. J. Hart, Dissociation of human copper-zinc superoxide dismutase dimers using chaotrope and reductant. Insights into the molecular basis for dimer stability, J. Biol. Chem. 279 (2004) 54558-54566.
[184] M. J. Lindberg, J. Normark, A. Holmgren, M. Oliveberg, Folding of human superoxide dismutase: disulfide reduction prevents dimerization and produces marginally stable monomers, Proc. Natl. Acad. Sci. U. S. A. 101 (2004) 15893-15898.
[185] T. Kurahashi, A. Miyazaki, S. Suwan, M. Isobe, Extensive investigations on oxidized amino acid residues in H(2)O(2)-treated Cu,Zn-SOd protein with LC-ESI-Q-TOF-MS, MS/MS for the determination of the copper-binding site, J. Am. Chem. Soc. 123 (2001) 9268-9278.
[186] B. R. Roberts, J. A. Tainer, E. D. Getzoff, D. A. Malencik, S. R. Anderson, V. C. Bomben, K. R. Meyers, P. A. Karplus, J. S. Beckman, Structural characterization of zinc-deficient human superoxide dismutase and implications for ALS, J. Mol. Biol. 373 (2007) 877-890.
[187] M. B. Yim, J. H. Kang, H. S. Yim, H. S. Kwak, P. B. Chock, E. R. Stadtman, A gain-of-function of an amyotrophic lateral sclerosis-associated Cu,Zn-superoxide dismutase mutant: An enhancement of free radical formation due to a decrease in Km for hydrogen peroxide, Proc. Natl. Acad. Sci. U. S. A. 93 (1996) 5709-5714.
[188] H. Liu, H. Zhu, D. K. Eggers, A. M. Nersissian, K. F. Faull, J. J. Goto, J. Ai, J. Sanders-Loehr, E. B. Gralla, J. S. Valentine, Copper(2+) binding to the surface residue cysteine 111 of His46Arg human copper-zinc superoxide dismutase, a familial amyotrophic lateral sclerosis mutant, Biochemistry 39 (2000) 8125-8132.
[189] S. Watanabe, S. Nagano, J. Duce, M. Kiaei, Q. X. Li, S. M. Tucker, A. Tiwari, R. H. Brown, Jr., M. F. Beal, L. J. Hayward, V. C. Culotta, S. Yoshihara, S. Sakoda, A. I. Bush, Increased affinity for copper mediated by cysteine 111 in

43
forms of mutant superoxide dismutase 1 linked to amyotrophic lateral sclerosis, Free Radic. Biol. Med. 42 (2007) 1534-1542.
[190] C. K. Mukhopadhyay, B. Mazumder, P. F. Lindley, P. L. Fox, Identification of the prooxidant site of human ceruloplasmin: a model for oxidative damage by copper bound to protein surfaces, Proc. Natl. Acad. Sci. U. S. A. 94 (1997) 11546-11551.
[191] M. Kiaei, A. I. Bush, B. M. Morrison, J. H. Morrison, R. A. Cherny, I. Volitakis, M. F. Beal, J. W. Gordon, Genetically decreased spinal cord copper concentration prolongs life in a transgenic mouse model of amyotrophic lateral sclerosis, J. Neurosci. 24 (2004) 7945-7950.
[192] E. Burstein, L. Ganesh, R. D. Dick, S. B. van De, J. C. Wilkinson, L. W. Klomp, C. Wijmenga, G. J. Brewer, G. J. Nabel, C. S. Duckett, A novel role for XIAP in copper homeostasis through regulation of MURR1, EMBO J. 23 (2004) 244-254.
[193] A. R. Mufti, E. Burstein, R. A. Csomos, P. C. Graf, J. C. Wilkinson, R. D. Dick, M. Challa, J. K. Son, S. B. Bratton, G. L. Su, G. J. Brewer, U. Jakob, C. S. Duckett, XIAP Is a copper binding protein deregulated in Wilson's disease and other copper toxicosis disorders, Mol. Cell 21 (2006) 775-785.
[194] P. Pasinelli, M. E. Belford, N. Lennon, B. J. Bacskai, B. T. Hyman, D. Trotti, R. H. Brown, Jr., Amyotrophic lateral sclerosis-associated SOD1 mutant proteins bind and aggregate with Bcl-2 in spinal cord mitochondria, Neuron 43 (2004) 19-30.
[195] C. M. Higgins, C. Jung, H. Ding, Z. Xu, Mutant Cu, Zn superoxide dismutase that causes motoneuron degeneration is present in mitochondria in the CNS, J. Neurosci. 22 (2002) RC215.
[196] A. Okado-Matsumoto, I. Fridovich, Amyotrophic lateral sclerosis: a proposed mechanism, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 9010-9014.
[197] H. F. Ji, L. Shen, H. Y. Zhang, Beta-lactam antibiotics are multipotent agents to combat neurological diseases, Biochem. Biophys. Res. Commun. 333 (2005) 661-663.
[198] S. Petri, N. Y. Calingasan, O. A. Alsaied, E. Wille, M. Kiaei, J. E. Friedman, O. Baranova, J. C. Chavez, M. F. Beal, The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis, J. Neurochem. 102 (2007) 991-1000.
[199] A. F. Hottinger, E. G. Fine, M. E. Gurney, A. D. Zurn, P. Aebischer, The copper chelator d-penicillamine delays onset of disease and extends survival in a transgenic mouse model of familial amyotrophic lateral sclerosis, Eur. J. Neurosci. 9 (1997) 1548-1551.
[200] O. A. Andreassen, A. Dedeoglu, P. Klivenyi, M. F. Beal, A. I. Bush, N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis, Neuroreport 11 (2000) 2491-2493.

44
[201] O. A. Andreassen, A. Dedeoglu, A. Friedlich, K. L. Ferrante, D. Hughes, C. Szabo, M. F. Beal, Effects of an inhibitor of poly(ADP-ribose) polymerase, desmethylselegiline, trientine, and lipoic acid in transgenic ALS mice, Exp. Neurol. 168 (2001) 419-424.
[202] S. Nagano, Y. Ogawa, T. Yanagihara, S. Sakoda, Benefit of a combined treatment with trientine and ascorbate in familial amyotrophic lateral sclerosis model mice, Neurosci. Lett. 265 (1999) 159-162.
[203] S. Nagano, Y. Fujii, T. Yamamoto, M. Taniyama, K. Fukada, T. Yanagihara, S. Sakoda, The efficacy of trientine or ascorbate alone compared to that of the combined treatment with these two agents in familial amyotrophic lateral sclerosis model mice, Exp. Neurol. 179 (2003) 176-180.
[204] M. Wiedau-Pazos, J. J. Goto, S. Rabizadeh, E. B. Gralla, J. A. Roe, M. K. Lee, J. S. Valentine, D. E. Bredesen, Altered reactivity of superoxide dismutase in familial amyotrophic lateral sclerosis, Science 271 (1996) 515-518.
[205] M. Azzouz, P. Poindron, S. Guettier, N. Leclerc, C. Andres, J. M. Warter, J. Borg, Prevention of mutant SOD1 motoneuron degeneration by copper chelators in vitro, J. Neurobiol. 42 (2000) 49-55.
[206] Y. Ogra, K. T. Suzuki, Targeting of tetrathiomolybdate on the copper accumulating in the liver of LEC rats, J. Inorg. Biochem. 70 (1998) 49-55.
[207] E. J. Wild, S. J. Tabrizi, Huntington's disease phenocopy syndromes, Curr. Opin. Neurol. 20 (2007) 681-687.
[208] J. H. Fox, J. A. Kama, G. Lieberman, R. Chopra, K. Dorsey, V. Chopra, I. Volitakis, R. A. Cherny, A. I. Bush, S. Hersch, Mechanisms of copper ion mediated Huntington's disease progression, PLoS ONE 2 (2007) e334.
[209] The Huntington's Disease Collaborative Research Group, A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes., Cell 72 (1993) 971-983.
[210] J. Velier, M. Kim, C. Schwarz, T. W. Kim, E. Sapp, K. Chase, N. Aronin, M. DiFiglia, Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways, Exp. Neurol. 152 (1998) 34-40.
[211] P. Hilditch-Maguire, F. Trettel, L. A. Passani, A. Auerbach, F. Persichetti, M. E. MacDonald, Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles, Hum. Mol. Genet. 9 (2000) 2789-2797.
[212] M. Garcia, D. Charvin, J. Caboche, Expanded huntingtin activates the c-Jun terminal kinase/c-Jun pathway prior to aggregate formation in striatal neurons in culture, Neuroscience 127 (2004) 859-870.
[213] E. J. Wild, S. J. Tabrizi, The differential diagnosis of chorea, Pract. Neurol. 7 (2007) 360-373.

45
[214] A. Schapira, R. Lodi, Assessment of in vitro and in vivo mitochondrial function in Friedreich's ataxia and Huntington's disease, Methods Mol. Biol. 277 (2004) 293-307.
[215] G. Bartzokis, P. H. Lu, T. A. Tishler, S. M. Fong, B. Oluwadara, J. P. Finn, D. Huang, Y. Bordelon, J. Mintz, S. Perlman, Myelin breakdown and iron changes in Huntington's disease: pathogenesis and treatment implications, Neurochem. Res. 32 (2007) 1655-1664.
[216] G. Bartzokis, J. Cummings, S. Perlman, D. B. Hance, J. Mintz, Increased basal ganglia iron levels in Huntington disease, Arch. Neurol. 56 (1999) 569-574.
[217] S. M. de la Monte, J. P. Vonsattel, E. P. Richardson, Jr., Morphometric demonstration of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington's disease, J. Neuropathol. Exp. Neurol. 47 (1988) 516-525.
[218] J. P. Vonsattel, R. H. Myers, T. J. Stevens, R. J. Ferrante, E. D. Bird, E. P. Richardson, Jr., Neuropathological classification of Huntington's disease, J. Neuropathol. Exp. Neurol. 44 (1985) 559-577.
[219] J. C. Chen, P. A. Hardy, W. Kucharczyk, M. Clauberg, J. G. Joshi, A. Vourlas, M. Dhar, R. M. Henkelman, MR of human postmortem brain tissue: correlative study between T2 and assays of iron and ferritin in Parkinson and Huntington disease, AJNR Am. J. Neuroradiol. 14 (1993) 275-281.
[220] E. Cattaneo, C. Zuccato, M. Tartari, Normal huntingtin function: an alternative approach to Huntington's disease, Nat. Rev. Neurosci. 6 (2005) 919-930.
[221] Y. Zhang, M. Li, M. Drozda, M. Chen, S. Ren, R. O. Mejia Sanchez, B. R. Leavitt, E. Cattaneo, R. J. Ferrante, M. R. Hayden, R. M. Friedlander, Depletion of wild-type huntingtin in mouse models of neurologic diseases, J. Neurochem. 87 (2003) 101-106.
[222] F. Pérez-Severiano, C. Rios, J. Segovia, Striatal oxidative damage parallels the expression of a neurological phenotype in mice transgenic for the mutation of Huntington's disease, Brain Res. 862 (2000) 234-237.
[223] K. E. Hoffman, K. Yanelli, K. R. Bridges, Ascorbic acid and iron metabolism: alterations in lysosomal function, Am. J. Clin. Nutr. 54 (1991) 1188S-1192S.
[224] J. Mehlhase, G. Sandig, K. Pantopoulos, T. Grune, Oxidation-induced ferritin turnover in microglial cells: role of proteasome, Free Radic. Biol. Med. 38 (2005) 276-285.
[225] N. Aziz, H. N. Munro, Iron regulates ferritin mRNA translation through a segment of its 5' untranslated region, Proc. Natl. Acad. Sci. U. S. A 84 (1987) 8478-8482.
[226] T. M. Hansen, H. Nielsen, N. Bernth, T. Moos, Expression of ferritin protein and subunit mRNAs in normal and iron deficient rat brain, Brain Res. Mol. Brain Res. 65 (1999) 186-197.

46
[227] S. W. Davies, M. Turmaine, B. A. Cozens, M. DiFiglia, A. H. Sharp, C. A. Ross, E. Scherzinger, E. E. Wanker, L. Mangiarini, G. P. Bates, Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation, Cell 90 (1997) 537-548.
[228] L. Mangiarini, K. Sathasivam, M. Seller, B. Cozens, A. Harper, C. Hetherington, M. Lawton, Y. Trottier, H. Lehrach, S. W. Davies, G. P. Bates, Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice, Cell 87 (1996) 493-506.
[229] C. A. Meade, Y. P. Deng, F. R. Fusco, M. N. Del, S. Hersch, D. Goldowitz, A. Reiner, Cellular localization and development of neuronal intranuclear inclusions in striatal and cortical neurons in R6/2 transgenic mice, J. Comp. Neurol. 449 (2002) 241-269.
[230] R. H. Myers, J. P. Vonsattel, P. A. Paskevich, D. K. Kiely, T. J. Stevens, L. A. Cupples, E. P. Richardson, Jr., E. D. Bird, Decreased neuronal and increased oligodendroglial densities in Huntington's disease caudate nucleus, J. Neuropathol. Exp. Neurol. 50 (1991) 729-742.
[231] G. Bartzokis, Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer's disease, Neurobiol. Aging 25 (2004) 5-18.
[232] A. L. Lumsden, T. L. Henshall, S. Dayan, M. T. Lardelli, R. I. Richards, Huntingtin-deficient zebrafish exhibit defects in iron utilization and development, Hum. Mol. Genet. 16 (2007) 1905-1920.
[233] S. E. Browne, A. C. Bowling, U. MacGarvey, M. J. Baik, S. C. Berger, M. M. Muqit, E. D. Bird, M. F. Beal, Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia, Ann. Neurol. 41 (1997) 646-653.
[234] M. Gu, M. T. Gash, V. M. Mann, F. Javoy-Agid, J. M. Cooper, A. H. Schapira, Mitochondrial defect in Huntington's disease caudate nucleus, Ann. Neurol. 39 (1996) 385-389.
[235] M. I. Lind, F. Missirlis, O. Melefors, H. Uhrigshardt, K. Kirby, J. P. Phillips, K. Söderhäll, T. A. Rouault, Of two cytosolic aconitases expressed in Drosophila, only one functions as an iron-regulatory protein, J. Biol. Chem. 281 (2006) 18707-18714.
[236] R. J. Ward, L. C. Kuhn, P. Kaldy, A. Florence, T. J. Peters, R. R. Crichton, Control of cellular iron homeostasis by iron-responsive elements in vivo, Eur. J. Biochem. 220 (1994) 927-931.
[237] A. Hausladen, I. Fridovich, Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not, J. Biol. Chem. 269 (1994) 29405-29408.
[238] S. J. Tabrizi, M. W. Cleeter, J. Xuereb, J. W. Taanman, J. M. Cooper, A. H. Schapira, Biochemical abnormalities and excitotoxicity in Huntington's disease brain, Ann. Neurol. 45 (1999) 25-32.

47
[239] A. H. Schapira, Mitochondrial involvement in Parkinson's disease, Huntington's disease, hereditary spastic paraplegia and Friedreich's ataxia, Biochim. Biophys. Acta 1410 (1999) 159-170.
[240] S. I. Liochev, I. Fridovich, The role of O2.- in the production of HO.: in vitro and in vivo, Free Radic. Biol. Med. 16 (1994) 29-33.
[241] T. Grünewald, M. F. Beal, Bioenergetics in Huntington's disease, Ann. N. Y. Acad. Sci. 893 (1999) 203-213.
[242] B. G. Jenkins, W. J. Koroshetz, M. F. Beal, B. R. Rosen, Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy, Neurology 43 (1993) 2689-2695.
[243] B. G. Jenkins, H. D. Rosas, Y. C. Chen, T. Makabe, R. Myers, M. MacDonald, B. R. Rosen, M. F. Beal, W. J. Koroshetz, 1H NMR spectroscopy studies of Huntington's disease: correlations with CAG repeat numbers, Neurology 50 (1998) 1357-1365.
[244] J. G. Joshi, Ferritin: intracellular regulator of metal availability, in: J. R. Connor (Ed.), Metals and oxidative damage in neurological disorders, Plenum Press, New York, 1997, pp. 131-143.
[245] W. J. Firdaus, A. Wyttenbach, P. Giuliano, C. Kretz-Remy, R. W. Currie, A. P. Arrigo, Huntingtin inclusion bodies are iron-dependent centers of oxidative events, FEBS J. 273 (2006) 5428-5441.
[246] T. Nguyen, A. Hamby, S. M. Massa, Clioquinol down-regulates mutant huntingtin expression in vitro and mitigates pathology in a Huntington's disease mouse model, Proc. Natl. Acad. Sci. U. S. A. 102 (2005) 11840-11845.
[247] E. Ferrada, V. Arancibia, B. Loeb, E. Norambuena, C. Olea-Azar, J. P. Huidobro-Toro, Stoichiometry and conditional stability constants of Cu(II) or Zn(II) clioquinol complexes; implications for Alzheimer's and Huntington's disease therapy, Neurotoxicology 28 (2007) 445-449.
[248] D. E. Ehrnhoefer, M. Duennwald, P. Markovic, J. L. Wacker, S. Engemann, M. Roark, J. Legleiter, J. L. Marsh, L. M. Thompson, S. Lindquist, P. J. Muchowski, E. E. Wanker, Green tea (-)-epigallocatechin-gallate modulates early events in huntingtin misfolding and reduces toxicity in Huntington's disease models, Hum. Mol. Genet. 15 (2006) 2743-2751.
[249] W. Zheng, N. Xin, Z. H. Chi, B. L. Zhao, J. Zhang, J. Y. Li, Z. Y. Wang, Divalent metal transporter 1 is involved in amyloid precursor protein processing and Ageneration, FASEB J. 23 (2009) 4207-4217.
[250] R. N. Kalaria, S. M. Sromek, I. Grahovac, S. I. Harik, Transferrin receptors of rat and human brain and cerebral microvessels and their status in Alzheimer's disease, Brain Res. 585 (1992) 87-93.

48
[251] H. Xu, H. Jiang, J. Wang, J. Xie, Rg1 protects the MPP+ -treated MES23.5 cells via attenuating DMT1 up-regulation and cellular iron uptake, Neuropharmacology 58 (2010) 488-494.
[252] N. Song, H. Jiang, J. Wang, J. X. Xie, Divalent metal transporter 1 up-regulation is involved in the 6-hydroxydopamine-induced ferrous iron influx, J. Neurosci. Res
S. V. Kalivendi, S. Cunningham, S. Kotamraju, J. Joseph, C. J. Hillard, B. Kalyanaraman, Synuclein up-regulation and aggregation during MPP+-induced apoptosis in neuroblastoma cells: Intermediacy of transferrin receptor iron and hydrogen peroxide, J. Biol. Chem. 279 (2004) 15240-15247.

49
Figure captions
Fig. 1. Copper (Cu) and iron (Fe) in Alzheimer’s disease. (1) Cu ions in the
extracellular synaptic space are co-released with glutamate. Abeta, produced from APP
by secretases, possesses high affinity for Cu. Abeta and Cu ions coincide in synaptic
space, inducing the precipitation of Abeta. (2) Cu is suspected to consolidate the
formation of senile plaques by catalyzing new covalent bonds among polypeptides. Cu
is found in high quantities in amyloid plaques. (3) Intracellular Cu is involved with the
expression of matrix metalloproteases responsible for the cleavage of Abeta. (4) High
content of iron is involved with increased expression of APP through an Iron Response
Element (IRE), whereas depletion of copper decreases the expression of this protein. (5)
The binding of Abeta with Cu increases the production of free radicals by itself and by
inhibition of mitochondrial respiratory chain. APP, Amyloid beta protein precursor;
BACE, Beta secretase; ROS, Reactive Oxygen Species; NMDAR, ionotropic Glutamate
receptor NMDA type; PI3K, phosphoinositol-3-kinase; AKT, protein kinase B; GSK-3,
glycogen synthase kinase 3; MAPK, mitogen activated protein kinase; MMP, matrix
metalloproteases.
Fig. 2. The reciprocal modulation of Fe and Cu and its further association with
excitotoxicity in PD. Glutamate increases Fe uptake through a NO-dependent
mechanism involving DMT1 (1). Fe, in turn, may increase glutamate release (2).
Excessive glutamate induces excitotoxicity through a NMDA mediated mechanism (3).
Cu can protect neurons against Fe overload by competing for transport through DMT1
(4) and favoring Fe efflux through IREG1 (5). DMT1, divalent metal transporter 1;
IREG1, ferroportin; NMDAR, glutamate NMDA receptor ; NO, nitric oxide; NOS,
nitric oxide synthase.
Fig. 3. The role of SOD metal uncoupling in ALS. SOD mutations can be both MBR-
mutations (1) or WTL-mutations (2). MBR-mutant SOD has an altered metal content,
while WTL-mutant SOD generally preserves its normal metal content. WTL-mutant
SOD is susceptible to oxidative damage compromising metal binding (3). Therefore,
both types of mutations can conduce to alterations in SOD structure (4), involving
protein aggregation (5), gain of toxic function, oxidative damage and apoptosis. The
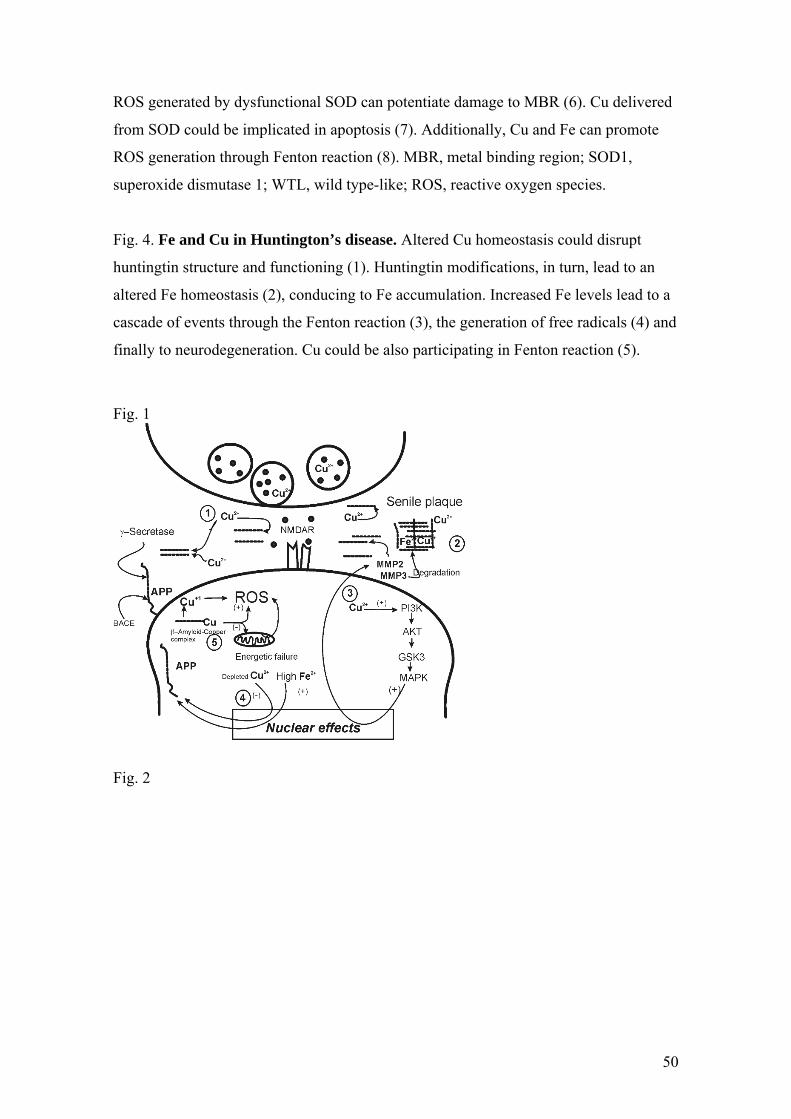
50
ROS generated by dysfunctional SOD can potentiate damage to MBR (6). Cu delivered
from SOD could be implicated in apoptosis (7). Additionally, Cu and Fe can promote
ROS generation through Fenton reaction (8). MBR, metal binding region; SOD1,
superoxide dismutase 1; WTL, wild type-like; ROS, reactive oxygen species.
Fig. 4. Fe and Cu in Huntington’s disease. Altered Cu homeostasis could disrupt
huntingtin structure and functioning (1). Huntingtin modifications, in turn, lead to an
altered Fe homeostasis (2), conducing to Fe accumulation. Increased Fe levels lead to a
cascade of events through the Fenton reaction (3), the generation of free radicals (4) and
finally to neurodegeneration. Cu could be also participating in Fenton reaction (5).
Fig. 1
Fig. 2
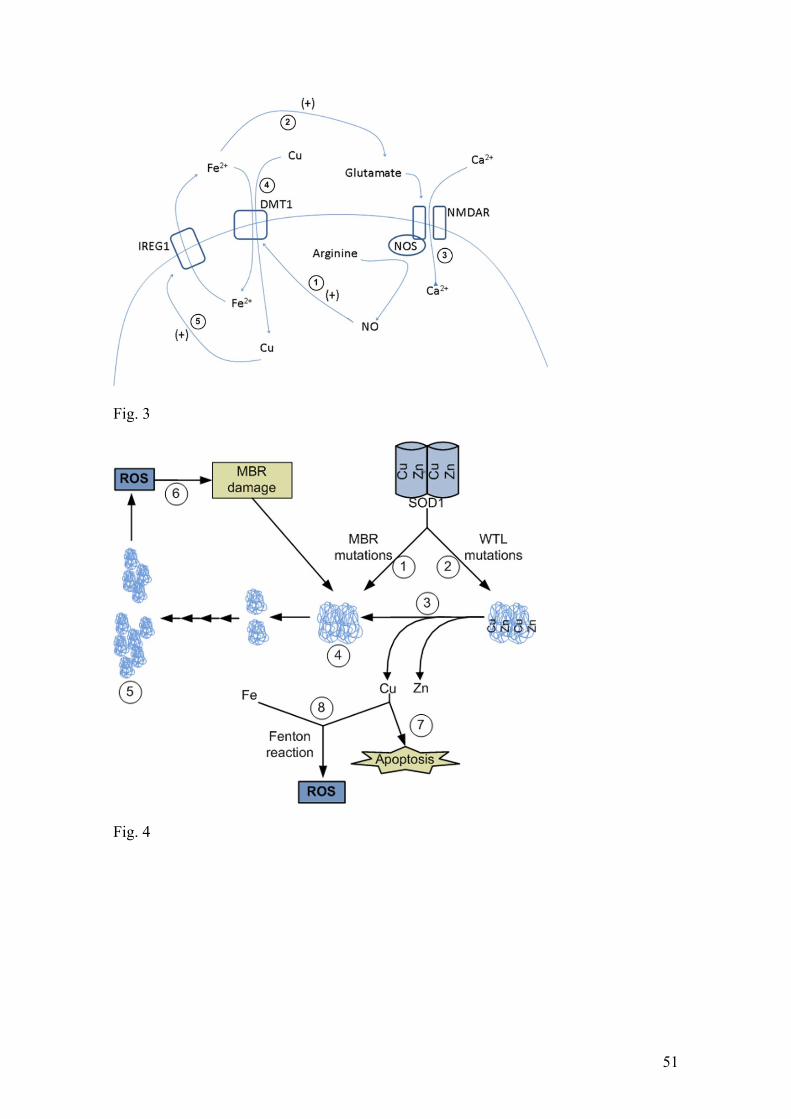
51
Fig. 3
Fig. 4
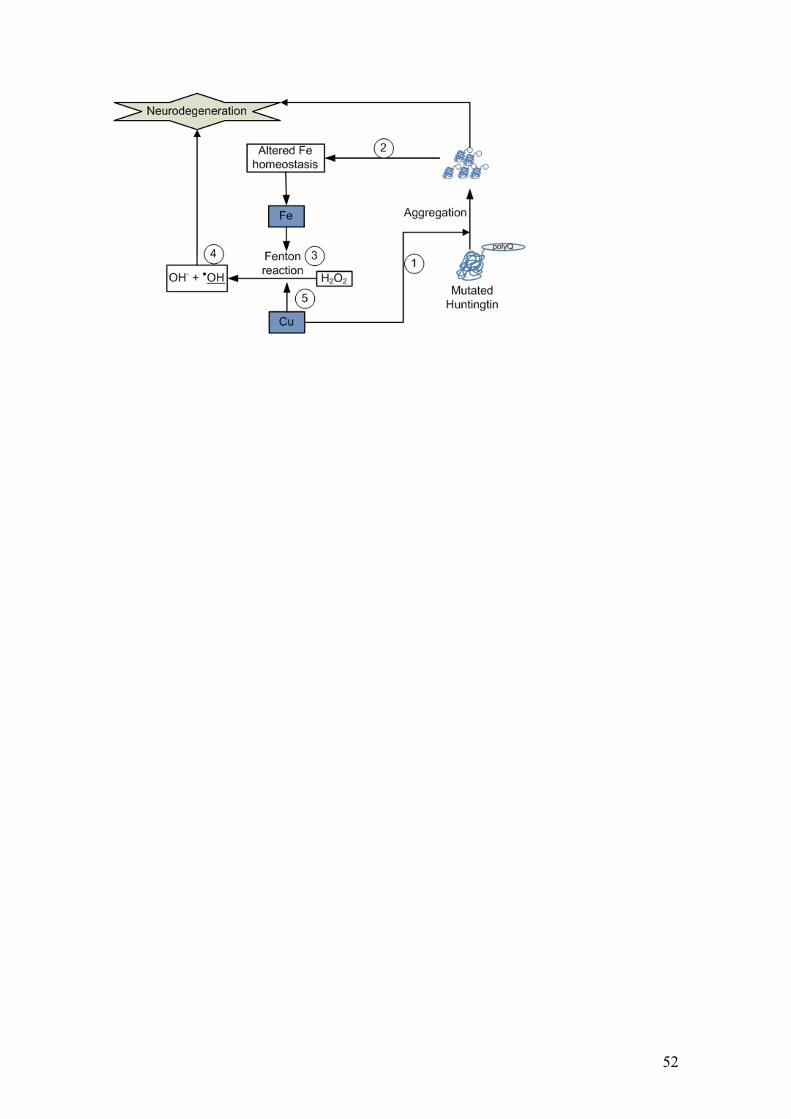
52
Copyright © 2022 FDOKUMEN




















