
Isolation of Strong Antioxidants from Paeonia Officinalis Roots ...

Clinica Chimica Acta 436 (2014) 332–347
Contents lists available at ScienceDirect
Clinica Chimica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /c l inch im
Antioxidants and human diseases
Peramaiyan Rajendran a, Natarajan Nandakumar b, Thamaraiselvan Rengarajan a, Rajendran Palaniswami d,Edwinoliver Nesamony Gnanadhas e, Uppalapati Lakshminarasaiah f, Jacob Gopas b,c, Ikuo Nishigaki a,⁎a NPO-International Laboratory of Biochemistry, 1-166, Uchide, Nakagawa-ku, Nagoya 454-0926, Japanb Shraga Segal Department of Microbiology, Immunology and Genetics, Ben-Gurion University of the Negev, Israelc Oncology Department Soroka University Medical Center, Be'er-Sheva 84105, Israeld Department of Applied Zoology and Biotechnology, Vivekananda College (A Gurukula Institute of Life Training), Affiliated to Madurai Kamaraj University, ThiruvedakamWest, Madurai 625234, Indiae Avram and Stella Goldstein-Goren Department of Biotechnology Engineering, Ben-Gurion University of the Negev, Beer-Sheva 84105, Israelf Department of Clinical Biochemistry and Pharmacology, Soroka University Medical Center, Ben-Gurion University of the Negev, Be'er-Sheva 84105, Israel
⁎ Corresponding author.E-mail address: [email protected] (I. Nishigaki
http://dx.doi.org/10.1016/j.cca.2014.06.0040009-8981/© 2014 Elsevier B.V. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 4 April 2014Received in revised form 4 June 2014Accepted 5 June 2014Available online 13 June 2014
Keywords:AntioxidantOxidative stressDiabetesCancer
Oxidative stress plays a pivotal role in the development of human diseases. Reactive oxygen species (ROS) thatincludes hydrogen peroxide, hyphochlorus acid, superoxide anion, singlet oxygen, lipid peroxides, hypochloriteandhydroxyl radical are involved in growth, differentiation, progression and death of the cell. They can reactwithmembrane lipids, nucleic acids, proteins, enzymes and other small molecules. Low concentrations of ROS has anindispensable role in intracellular signalling and defence against pathogens, while, higher amounts of ROS play arole in number of human diseases, including arthritis, cancer, diabetes, atherosclerosis, ischemia, failures in im-munity and endocrine functions. Antioxidants presumably act as safeguard against the accumulation of ROSand their elimination from the system. The aim of this review is to highlight advances in understanding of theROS and also to summarize the detailed impact and involvement of antioxidants in selected human diseases.
© 2014 Elsevier B.V. All rights reserved.
Contents
1. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3332. Measuring oxidative stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333
2.1. Pro-oxidants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3333. Potential antioxidant biomarkers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334
3.1. Superoxide dismutase (SOD) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.2. Catalase (CAT) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.3. Glutathione peroxidase (GPx) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.4. Thiol index . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.5. Xanthine oxidase (XO) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.6. NADPH Oxidase . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3343.7. A biomarker of cardiovascular damage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 334
4. Mechanism of antioxidant action and nutrients . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3355. Antioxidants vs human diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 336
5.1. Antioxidant vs arthritis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3365.2. Anti oxidant vs cancer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3385.3. Antioxidant vs diabetes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3395.4. Antioxidant vs arthrosclerosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3415.5. Anti oxidant vs neurodegerative diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 342
6. Conclusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 342References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 343
).

333P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
1. Introduction
The beneficial effect of antioxidants on themaintenance of health inhuman has become an important subject that has engaged manyscientists across theworld over the last decade. In the last few years, an-tioxidants have become the indispensable nutrients of the nutritionalworld. Antioxidants are important in terms of their ability to protectagainst oxidative cell damage that can lead to conditions, such asAlzheimer's disease, cancer, heart disease and also linked with chronicinflammation [1]. It is defined as the substances which at low concen-tration significantly inhibits or delay the oxidative process, while oftenbeing oxidized themselves. Recent reports suggest that several endoge-nous and exogenous antioxidants are used to neutralize free radicalsand protect the body from free radicals by maintaining redox balance[2,3]. Singh et al. (2010) [4] quoted that antioxidants have gone from“Miracle Molecules” to “Marvellous Molecules” and finally to “Physio-logical Molecules” that they play a vital role in metabolic pathwaysand protect cells. However recent conflicting evidence has forced thescientists to dig deeper in order to explore the role of antioxidants andpro-oxidants, since free radical reactions have been implicated inevery human pathological condition which includes neurodegenerativedisorders like Alzheimer's disease, Parkinson's disease, multiplesclerosis, amyotrophic lateral sclerosis, memory loss, depression andcardiovascular diseases such as atherosclerosis, ischemic heart disease,cardiac hypertrophy, hypertension, shock and trauma. Further, it alsoimplicated in pulmonary disorders which include inflammatory lungdiseases such as asthma and chronic obstructive pulmonary diseaseand additionally diseases associated with premature infants such asbronchopulmonary dysplasia, periventricular leukomalacia, intraven-tricular hemorrhage, retinopathy of prematurity and necrotizing en-terocolitis and in some autoimmune diseases like rheumatoid arthritisand also in several renal disorders such as glomerulonephritis,tubulointerstitial nephritis, chronic renal failure, proteinuria, uremiaand finally gastrointestinal diseases like peptic ulcer, inflammatorybowel disease and colitis, diabetes, tumors and cancers [5].
2. Measuring oxidative stress
As oxidative stress is indicative of an inequity between oxidants andantioxidants, methods for quantifying oxidative stress mostly includestraight or indirect measurement of oxidants and antioxidants [6,7]. Inthe following segment, some principles and commonly used methodsfor the measurement of oxidative stress and damage will be brieflyoutlined.
2.1. Pro-oxidants
The most abundant free radicals in biological systems are theoxygen-centered free radicals and their metabolites, usually referredto as ROS [7]. ROS are formed continuously as normal by-products ofcellular metabolism; and, in low concentrations, they are essential forseveral physiological processes, including protein phosphorylation,transcription factor activation, cell differentiation, apoptosis, oocytematuration, steroidogenesis, cell immunity, and cellular defense againstmicroorganisms [7]. However, when produced in excess, ROS can dam-age cell functionality as they can harm cellular lipids, proteins, and DNA[7,8]. The plasma level of ROMs is considered an indicator of free-radicalproduction [7]. ROMs is a collective term that includes not only oxygen-centered free radicals such as the superoxide anion and hydroxylradical, but also some non-radical derivatives of oxygen, such as hydro-gen peroxide (H2O2) and hypochlorous acid [9]. A ROMs kit has beendeveloped to assess oxidant levels in plasma and other biological fluids;and the ROMs test has been validated by electron spin resonance [10],which is considered the “gold standard” for measuring total oxidativestatus. However, electron spin resonance is not suitable for routine anal-ysis, as themethod is complex and requires specific technical assistance
not available in most laboratories. The utility of the ROMs assay inmonitoring oxidative stress in goats [11], sheep [12], and dairy cows[13] has been reported.
The concentrations of individual oxidant components can bemeasured separately in the laboratory; but such measurements aretime-consuming, labor intensive, and costly. Free-radical analyticalsystem 4 technology has been shown to offer a quick, simple, precise,and reliable method for assessing the oxidative status in dairy cows[14] and in horses [15]. Such technology is particularly useful in thefield, where it is not always practical or possible to get samples to alaboratory immediately. The possibility of assessing oxidative stressdirectly in blood samples provides veterinarians with a simple andreliable method for measuring oxidative stress in clinical situationssuch as the monitoring of therapy and in the antioxidant supplementa-tion of domestic animals. However, given the lack of reference valuesfor ROMs in ruminants, it is difficult to establish if and when these ani-mals are actually experiencing oxidative stress. Therefore, it is importantto calculate the specific referral ranges; because a correct biochemicalevaluation of oxidative status is an essential premise to prevent andeventually to treat the effects of oxidative stress in ruminant medicine.Advanced oxidation protein products (AOPPs) are terminal products ofproteins exposed to free radicals and arise from the reaction betweenplasma proteins and chlorinated oxidantsmediated bymyeloperoxidase,a neutrophil enzyme [16,17]. In humans, AOPPs have been linked toseveral diseases, such as chronic renal failure [18], diabetes mellitus[19], diabetic nephropathy [20], coronary artery diseases [21], andobesity [22]. Chronic accumulation of AOPPs has been demonstrated topromote inflammatory processes in the diabetic kidney [20] and inchronic renal failure [18], indicating that these products might be a by-product of neutrophil activation during infections. Studies in ruminantshave shown higher levels of AOPPs than normal in lambs [23] anddairy cows [24] supplemented with Yerba Mate (Ilex paraguanensis).More information about the role of protein oxidation in ruminants'health and about oxidated proteins could be obtained by a comparisonof AOPPs with other indicators of protein oxidation, such as advancedglycation end products (AGE). However, a correlation between AGEand inflammatory parameters is usually not found or is only weak, andthe induction of proinflammatory activities caused by AOPPs seems tobe more intense [25,26], suggesting that oxidative stress is more closelylinked to inflammation and acute-phase reactions than to the advancedglycation process and its end products. AOPPs could thus better describeacute inflammation, whereas AGE might serve more as a marker ofchronic long-lasting damage [25]. These observations are highly relevant,as increased levels of AOPPs could indicate the presence of inflammatoryprocesses that can potentially compromise the correct embryonicdevelopment in dairy cows [14,27]. Lipids, in particular those that arepolyunsaturated, are prone to oxidation. Lipids are one of the mostsusceptible substrates to damage by free radicals, and biomarkers oflipid peroxidation are considered the best indicators of oxidative stress[28]. Malondialdehyde (MDA) is one of several low-molecular-weightend products formed during radical-induced decomposition of polyun-saturated fatty acids [29]. MDA readily reacts with thiobarbituric acid,producing a red pigment that can be easilymeasured by spectrophotom-etry in the form of thiobarbituric acid-reactive substances (TBARS, [29]).It is worth noting that the MDA assay has been criticized for its lowspecificity and for artifact formation, since only a fraction of the MDAmeasured is actually generated in vivo. Furthermore, the TBARS assay,a common method used to measure MDA, is considered inaccurate,and returns results that differ according to the assay conditions used[30]. For example, studies on dairy cows have yielded contrasting results,with some reports failing to show any significant changes in plasmaMDA concentrations during the peripartum period [31]; whereas inother studies MDA or TBARS concentrations were shown to increasearound calving [8,16,32]. This apparent discrepancy could also havebeenmainly due to the great variation in individual MDA concentrationsmeasured in the studies by Castillo et al. [31]. Similarly, studies on

334 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
transported cattle have failed to report a consistent change in their MDAconcentration. It seems that this discrepancy is partly due to the differentmethodologies employed to assess MDA levels. TBARS represent a widerange of lipid peroxidation products, and the thiobarbituric acid reactionis rather non-specific for MDA [33]. High-pressure liquid chromatogra-phywould be expected to be highly specific, and perhaps, more accuratethan the spectrophotometric procedures [34]. More recently, an ELISA-based method for the detection of isoprostane, which is considered tobe themost reliablemarker of lipid oxidation [35], has become commer-cially available and might be able to shed more light on the role of lipidperoxidation during the peripartum period in ruminants.
3. Potential antioxidant biomarkers
Epidemiologic literature that focuses on antioxidant status andchronic disease risk has in the past relied primarily upon biomarkersof exposure to antioxidant nutrients. However, this approach is inessence using exposure estimates to a select group of nutrients as a sur-rogate for estimating actual oxidative defense or oxidative stress status.Emphasis is now being placed on developing functional biomarkers ofoxidative stress status, that is, biomarkers that integrate the effect ofexposure to oxidants coupled with the full range of antioxidantprotectivemechanisms in vivo. Many of such biomarkers are beingstudied including various measures of oxidation products of lipid,DNA, and protein (Table 1). Some of these biomarkers are now beingapplied in research of pathologies related to oxidative stress.
3.1. Superoxide dismutase (SOD)
The term superoxide dismutase characterizes a family of enzymaticproteins differing in their structure and cofactors, among them beingMn-SOD and Cu–Zn-SOD. SOD activity enhances the spontaneousdismutation of superoxide radicals to H2O2 [52]. SOD can be measuredby utilizing the technique of Misra and Fridovich [43], which is basedon inhibition of the formation of nicotine amide adenine dinucleotide,phenazine methosulfate, and amino blue tetrazolium formazan.
3.2. Catalase (CAT)
The end product of the dismutation reaction, i.e., H2O2, can be re-moved by the activity of the enzyme catalase. CAT is an enzyme witha very high KM for its substrate and can remove H2O2 present in highconcentrations [52]. The activity of CAT can be measured through the
Table 1Biomarkers of oxidative stress.
Biomarkers Location Reference
Carbonyls Extracellular Mohanty et al. [36]Lipid peroxidation•Malondialdehyde Extracellular Ohkawa et al. [37]•F2-isoprostane Extracellular Collins et al. [38]•4-Hydroxynonenal Extracellular Halliwell and Gutteridge [39]
Ferric reducing ability of plasma Extracellular Benzie and Strain [40]Plasma vitamins•Vitamin C Extracellular Roe and Kuether [41]•Vitamin E Extracellular Teissier [42]
Antioxidant enzymes•Superoxide dismutase Intracellular Misra and Fridovich [43]•Catalase Intracellular Aebi and Bergmeyer [44]•Glutathione peroxidase Intracellular Floh'e andG¨unzler [45]•GSH/GSSG ratio in erythrocyte Intracellular Hissin and Hilf [46]
Prooxidant enzymes•Xanthine oxidase Intracellular Haining and Legan [47]•NADPH oxidase Intracellular Nauseef [48]
Others•Endothelial microparticles Extracellular Burger and Touyz [49]•Endothelial progenitor cells Extracellular Touyz and Schiffrin [50]•Ischemia modified albumin Extracellular Sinha et al. [51]
non spontaneous decomposition of hydrogen peroxide, by the methoddescribed by Aebi [44].
3.3. Glutathione peroxidase (GPx)
The biochemical function of glutathione peroxidase is to reduce lipidhydroperoxides to their corresponding alcohols and to reduce free H2O2
to water. In contrast to catalase, peroxidase possesses a high affinity forand can remove H2O2 even when it is present in a low concentration[52]. Its activity can be estimated by the method described by Floheand Gunzler [45].
3.4. Thiol index
The GSH/GSSG index is a parameter of the intracellular redox status.GSH is the major endogenous antioxidant produced by cells; and itparticipates directly in the neutralization of free radicals and reactive-oxygen compounds, as well as in maintaining exogenous antioxidantssuch as vitamins C and E in their reduced forms [53]. GSH and GSSGlevels can be measured in erythrocytes by the method described byHissin and Hilf [46].
3.5. Xanthine oxidase (XO)
XO is a key enzyme involved in the formation of reactive oxygenspecies, and it plays a major role in cell oxidative stress. This enzymecatalyzes the oxidation of hypoxanthine to xanthine and can furthercatalyze the oxidation of xanthine to uric acid [54]. Xanthine oxidasecan be estimated by the method of Haining and Legan [47].
3.6. NADPH Oxidase
TheNADPH-oxidase complex utilizes electrons to produce superoxideradicals from the oxygen molecule. NADPH oxidase may be measured bya chemiluminescence [55] or electrochemical method among others.Approaches to quantitate oxygen consumption, extracellular release ofO2∙ or H2O2, and intracellular O2
∙ production provide reliable assessmentof NADPH oxidase activity in a given population of cells [56].
3.7. A biomarker of cardiovascular damage
Endothelial microparticles (EMPs) are nuclear fragments of cellularmembrane shed from oxidatively stressed or damaged cells. Typicallydefined as having a diameter of 0.1 to 1.0mm, thesemicroparticles con-tain surface proteins and cytoplasmic material of the parental cells [57].
Many biomonitoring studies have investigated the role of antioxi-dants in reducing oxidatively generated DNA damage in urine andwhite blood cells. A collective interpretation is difficult because manystudies lack sufficient control and have unreasonably high baselinelevels of oxidatively damaged DNA. to investigating antioxidant effectshas been the use of biomarkers of oxidatively damaged DNA. This isbased on the mechanistic rationale that dietary antioxidants inhibitthe oxidation of DNA. With the possibility of detecting 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG), the first of many antioxidantsupplementation studies with focus on this lesion in WBC appeared inthe beginning of the 1990s. At the same time, reliable detection of uri-nary 8-oxodG excretion was achieved, and this was soon followed byantioxidant supplementation studies using urinary 8-oxodG excretionas key biomarker. However, by far the most popular method in antiox-idant intervention trials has been the comet assay that detects DNAstrand breaks (SB). An enzyme-modified version of the comet assayhas been developed to detect oxidatively altered nucleobases byincluding a DNA digestion step with DNA glycosylase or endonucleaseenzymes [58]. The characteristics of the intervention studies areoutlined in Table 2.

Table 2Administrations of dietary antioxidant with assessment of oxidatively DNA damage in white blood cells and urine.
Supplement per day Subjects Age(years)
Effect References
β-carotene (20 mg) for 14 weeks 122 M (S) 39 (NR) No effect on urinary excretion of 8-oxodG/24-h (HPLC) [59]β-carotene (30 mg) for 1 months 14 M (NS) 19–22 No effect on urinary excretion of 8-oxodG/24-h (HPLC) [60]Carotenoidsa for 3 weeks 32 MF(NS) 32 ± 11 No effect versus baseline, but decreased urinary excretion of 8-oxodG ELISA)
in active group post-supplementation[61]
VitaminC (500 mgas plain or slow release formulations)and vitamin E (182 mg) for 4 weeks
48 M (S) 39 ± 12 Decreased ENDOIII and FPG sites (Comet) in the group that ingested tabletswith the slowrelease vitamin C formulation
[62]
Multi-vitamin tablet (100 mg vitamin C, 280 mg vitaminE, and 25 mg b-carotene) for 20 weeks
100 M (50S) 50–59 Decreased ENDOIII sites (Comet) after 20 weeks in WBC [63]
Vegetable consumption (3.6 vs 12 servings) for2 weeks
64 F (NR) 23–81 Lower 8-oxodG in WBC (HPLC) among those subjects eating 12 servings per day [64]
Kiwi fruit (1–3 pieces) for 3 weeks 14 MF (NS) 26–54 Decreased ENDOIII and FPG (Comet) sites [65]Cranberry juice (750 ml) for 2 weeks 20 F (2S) 18–40 No effect on ENDOIII (Comet) in WBC [66]Blackcurrant juice or anthocyanine drink(475–1000 ml) for 3 weeks
57 MF (6S) 19–52 No effect on ENDOIII and FPG sites (Comet) in WBC [67]
Green tea or black tea (four cups) for 1–4 months 120 MF(S) 18–79 Decreased excretion of 8-oxodG (ELISA) in spot urine samples after 4 moin green tea group, but not before
[68,69]
Two interventions of green tea with 300 ml for 7 daysor 32 oz for 7 days
68MF (13S) 18–45 Decreased 12-h urinary excretion of 8-oxodG (HPLC) [70]
Green tea extract b for 3 weeks 16 M (8S) 20–31 No effect related to supplementation on 24-h urinary excretion of 8-oxodG(HPLC) but a decreased excretion during the study in all groups
[71]
Green tea (500 or 1000 mg) among high-risk subjectsof liver cancer for 3 months
124MF(NR)
NR Lower 8-oxodG/24-h (HPLC) after 3 months supplementation but not after1 month
[72]
Soya-hypocotyl tea (N1000 ml) for 1 months 38 F (NR) NR Decreased excretion of 8-oxodG (ELISA) in active group (statisticaltest not reported)
[73]
Soy milk, rice milk, or cow milk (1 l) for 4 weeks 10 M (NS) 20–50 Decreased ENDOIII sites (Comet) for soy milk [74]Polyphenol-rich olive oils (25 ml) for 40 days 12 M (NS) 20–22 Decreased excretion of 8-oxodG (HPLC) in spot urinary samples following
supplementation in a dose-dependent manner[75]
Olive oil (25 ml) with three different content of phenoliccompounds for 3 weeks
182 M (NS) 20–60 Decreased 8-oxodG/24-h urinary excretion, but no difference in urinary excretionof 8-oxoGua/24-h. No difference in the effect of the different olive oils
[76]
Antioxidant rich diet or tabletsc for 5 weeks 55 MF (NR) 71 ± 6 No effect versus baseline, but decreased 24-h urinary excretion of 8-oxodG(ELISA) in active group post-supplementation
[77]
Number of subjects indicated as males (M) and females (F). Smokers (S) and non-smokers (NS) are indicated in brackets. Lacking information is indicated as NR (not reported) Age isshown as range or mean ± standard deviation. aSupplement constitute b-carotene (6 mg), a-carotene (1.4 mg), lycopene (4.5 mg), bixin (11.7 mg), lutein (4.4 mg), and paprika carot-enoids (2.2 mg), b Consisted of 1000 mg extract/kg bodyweight inmeat patties (total phenolics were 23.5 mg/10 MJ), cConsisted of tablets with vitamin antioxidants (400 mg vitamin C,150 mg vitamin E, 4 mg b-carotene), capsules (90 mg vitamin C, 18 IU vitamin E, 2.4 mg b-carotene, and powder or extract of fruits and berries), or a carotenoid-rich diet.
335P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
4. Mechanism of antioxidant action and nutrients
It is reported that antioxidants can execute protective role againstfree radicals by a variety of different mechanism including the catalyticsystems to neutralize or divert ROS, binding or inactivation ofmetal ionsprevents generation of ROS by Haber–Weiss reaction, suicidal and chainbreaking antioxidants scavenge and destroy ROS or absorb energy,electron and quenching of ROS. In 21st century, demand for intake ofantioxidant food or dietary antioxidant increasing with the hope tokeep body healthy and free from diseases [78,79] and the potentialbeneficial effects of antioxidants in protecting against disease havebeen well established. It is increasingly thought that nutrition mayplay a vital role in helping to defend against oxidative stress and damage
Table 3Effects of the health associated to the intake of Antioxidants.
Antioxidants Effect of health
Vitamin C Protects against cancers, Protects from heart disease, Improvement of tcartilage, joints and skin, Maintaining a healthy immune system, Improantibody production, Increase in the absorption of nutrients, Increasesagainst H2O2-induced DNA strand breaks
Vitamin E Prevents coronary heart disease, Prevents the formation of blood clotsincidence of breast and prostate cancers, Brain protection, Reduces lonof dementia Decreases risk of Parkinson's disease
Polyphenol Inhibit oxidation of LDL, Inhibit platelet aggregation, Improve endothelLower risk of myocardial infarction, Effect anticarcinogenic Prevent neudiseases, Protect against neurotoxic drugs, treatment of diabetes, treatmosteoporosis, Inhibit non-heme iron absorption
Cu, Zn, Mn, Se Cofactors of antioxidant enzymes SOD-Cu/Zn, Mn-SOD and GSH-PoxOther carotenoids(lycopene)
Protection against oxidation of lipids, LDL, proteins and DNA. Abductioradical scavenging
induced by free radicals. Therefore, certain nutrients and dietary com-ponents with antioxidant properties are important for the protectionagainst oxidative stress injury of the body. Food consumption is amajor source of exogenous antioxidants and has been estimated that atypical diet provides more than 25,000 bioactive food constituents asnutrients and many of this may modify a multitude of processes thatare related to different diseases. Generally, antioxidants are abundantin vegetables and fruits and are also found in grain cereals, peas,legumes, nuts and other food products. At this juncture, a systematicsurvey has also identified more than 3100 antioxidants in foods, likebeverages, spices, herbs and supplements which are regularlyconsumed by different cultures [80]. It has been speculated that the de-crease in the intake of nutritional and antioxidants rich food may
References
he health ofvement in theprotection
Barry [85], Liu et al. [86], Wang et al. [87], Wintergerst et al. [88],Woo et al. [89], Thankachan et al. [90] and Riso et al. [91].
Decreasesg-term risk
Pryor [92], Traber et al. [93], Weinstein et al. [94], Muller et al. [95],Devore et al. [96] and Miyake et al. [97].
ial dysfunctionrodegenerativeent to prevent
Manach et al. [98], Russo et al. [99], Sch¨achinger et al. [100],Corder et al. [101], Yang et al. [102] Halliwell [103], Pan et al. [104],Zunino et al. [105], Atmaca et al. [106], Hurrell et al. [107].
Visioli F et al. [108]n and free Visioli F et al. [108]

336 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
increase the chances of oxidative stress whichmay lead to cell damage,therefore intake of such natural antioxidants may give protective effectagainst free radical induced diseases [80,81]. In view of the importanceof antioxidants, Suntres [82] reported that antioxidant liposomes willhold an important role in future research on antioxidants, and alsoreports that it facilitates the delivery of antioxidants to specific sites aswell as achieving prophylactic and therapeutic action. In addition,Bouayed and Bohn [83] have suggested that the balance between oxida-tion and anti-oxidation is critical in maintaining a healthy biologic sys-tem and low doses of antioxidants may be favorable to this system,but high quantities may disrupt the balance. It is true that antioxidantsare beneficial and display a useful role in the maintenance of the ho-meostasis in human ROS system, but so are pro-oxidants, thereforethe scientific community should search deeper into the kinetics andin vivo mechanisms of antioxidants to uncover the optimal concentra-tions or desired functions in order to push forward against cancer,neurodegenerative and cardiovascular diseases [84] (Table 3). Variousstudies related to free radicals, oxidative stress and antioxidant activityof food reveals the prominent beneficial role of antioxidant and its spe-cific role against different diseases individually. However the collectiveand concise data on the role of antioxidants in human diseases will bebetter and most appropriate one in order to know the exact role of an-tioxidants in all kinds of human diseases (Fig. 1). Hence, an attempthas been made in this review, to summarize the detailed role andimpact of antioxidants in certain selected dreadful human diseases.
Fig. 1. Antioxidants/oxidative s
5. Antioxidants vs human diseases
5.1. Antioxidant vs arthritis
Chronic autoimmune inflammation,which is commonly observed inrheumatoid arthritis (RA), disrupts the delicate balance between boneresorption and calcification causing the destruction of the bone andjoints. Multiple aetiologies are suspected to contribute to the formationof RA, including defective articular cartilage structure and biosynthesis,joint trauma, joint instability, congenital and developmental abnormal-ities, and inflammatory conditions [109,110]. Oxidative stress induceddamage to essential cell components caused by oxygen free radicals isa mechanism in the patho-biology of degenerative rheumatoid arthritis[109]. Hydroxyl radicals cause degradation of isolated proteoglycans,and hypochlorous acid (HOCl) fragments of collagen. Hydrogen perox-ide, which is highly diffusible, readily inhibits cartilage proteoglycansynthesis, by interfering with ATP synthesis, in part by inhibiting theglycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase inchondrocytes, aggravating the effects of proteolytic and free-radical-mediated cartilage degradation. Peroxynitrite and HOCl may facilitatecartilage damages by inactivating TIMPs. TIMP-1 inhibits stromelysins,collagenases and gelatinasesand this ability is lost after nitrusoxide (NO3
−) or HOCl treatment. HOCl can also activate latent forms ofneutrophil collagenases and gelatinase with obvious consequences.Hypochlorous acid, NO3
− andO2− reactwith ascorbate, which is essential
tress and health diseases.
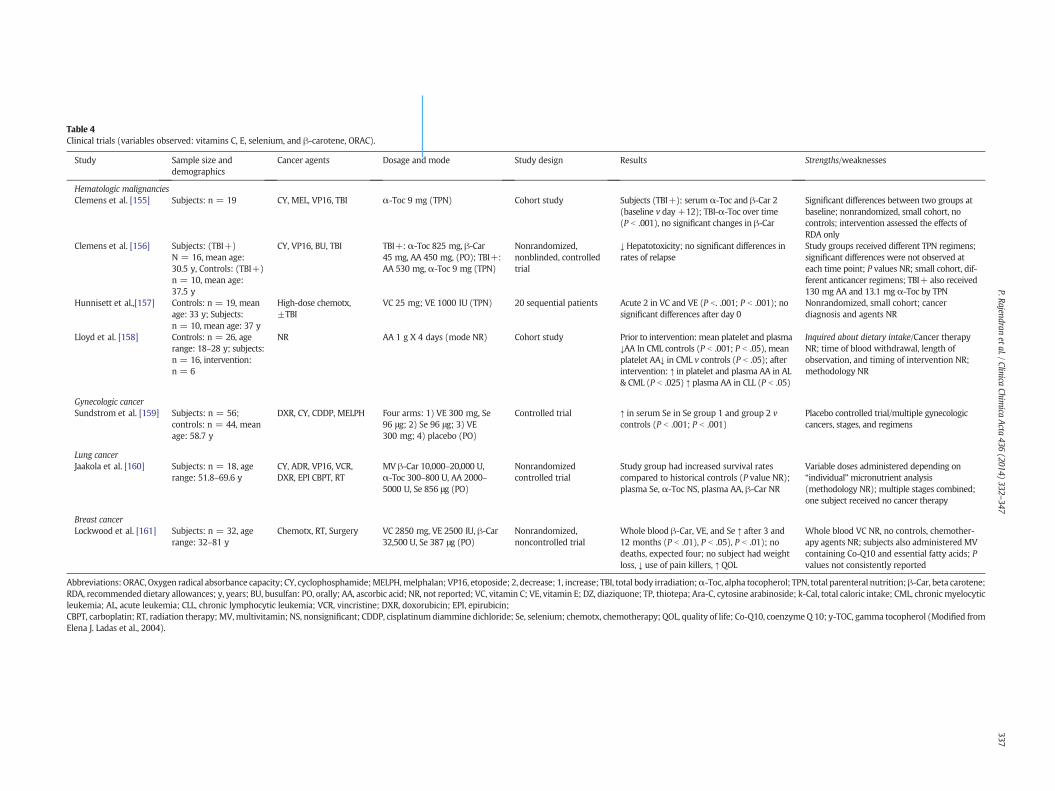
Table 4Clinical trials (variables observed: vitamins C, E, selenium, and β-carotene, ORAC).
Study Sample size anddemographics
Cancer agents Dosage and mode Study design Results Strengths/weaknesses
Hematologic malignanciesClemens et al. [155] Subjects: n = 19 CY, MEL, VP16, TBI α-Toc 9 mg (TPN) Cohort study Subjects (TBI+): serum α-Toc an -Car 2
(baseline v day +12); TBI-α-Toc er time(P b .001), no significant changes β-Car
Significant differences between two groups atbaseline; nonrandomized, small cohort, nocontrols; intervention assessed the effects ofRDA only
Clemens et al. [156] Subjects: (TBI+)N = 16, mean age:30.5 y, Controls: (TBI+)n = 10, mean age:37.5 y
CY, VP16, BU, TBI TBI+: α-Toc 825 mg, β-Car45 mg, AA 450 mg, (PO); TBI+:AA 530 mg, α-Toc 9 mg (TPN)
Nonrandomized,nonblinded, controlledtrial
↓ Hepatotoxicity; no significant di rences inrates of relapse
Study groups received different TPN regimens;significant differences were not observed ateach time point; P values NR; small cohort, dif-ferent anticancer regimens; TBI+ also received130 mg AA and 13.1 mg α-Toc by TPN
Hunnisett et al.,[157] Controls: n = 19, meanage: 33 y; Subjects:n = 10, mean age: 37 y
High-dose chemotx,±TBI
VC 25 mg; VE 1000 IU (TPN) 20 sequential patients Acute 2 in VC and VE (P b. .001; P .001); nosignificant differences after day 0
Nonrandomized, small cohort; cancerdiagnosis and agents NR
Lloyd et al. [158] Controls: n = 26, agerange: 18–28 y; subjects:n = 16, intervention:n = 6
NR AA 1 g X 4 days (mode NR) Cohort study Prior to intervention: mean platele and plasma↓AA In CML controls (P b .001; P 05), meanplatelet AA↓ in CML v controls (P .05); afterintervention: ↑ in platelet and pla a AA in AL& CML (P b .025) ↑ plasma AA in L (P b .05)
Inquired about dietary intake/Cancer therapyNR; time of blood withdrawal, length ofobservation, and timing of intervention NR;methodology NR
Gynecologic cancerSundstrom et al. [159] Subjects: n = 56;
controls: n = 44, meanage: 58.7 y
DXR, CY, CDDP, MELPH Four arms: 1) VE 300 mg, Se96 μg; 2) Se 96 μg; 3) VE300 mg; 4) placebo (PO)
Controlled trial ↑ in serum Se in Se group 1 and g up 2 vcontrols (P b .001; P b .001)
Placebo controlled trial/multiple gynecologiccancers, stages, and regimens
Lung cancerJaakola et al. [160] Subjects: n = 18, age
range: 51.8–69.6 yCY, ADR, VP16, VCR,DXR, EPI CBPT, RT
MV β-Car 10,000–20,000 U,α-Toc 300–800 U, AA 2000–5000 U, Se 856 μg (PO)
Nonrandomizedcontrolled trial
Study group had increased surviv l ratescompared to historical controls ( alue NR);plasma Se, α-Toc NS, plasma AA, -Car NR
Variable doses administered depending on“individual”micronutrient analysis(methodology NR); multiple stages combined;one subject received no cancer therapy
Breast cancerLockwood et al. [161] Subjects: n = 32, age
range: 32–81 yChemotx, RT, Surgery VC 2850 mg, VE 2500 IU, β-Car
32,500 U, Se 387 μg (PO)Nonrandomized,noncontrolled trial
Whole blood β-Car, VE, and Se ↑ ter 3 and12 months (P b .01), P b .05), P b 01); nodeaths, expected four; no subject ad weightloss, ↓ use of pain killers, ↑ QOL
Whole blood VC NR, no controls, chemother-apy agents NR; subjects also administered MVcontaining Co-Q10 and essential fatty acids; Pvalues not consistently reported
Abbreviations: ORAC, Oxygen radical absorbance capacity; CY, cyclophosphamide;MELPH,melphalan; VP16, etoposide; 2, decrease; 1, increase; TBI, total body irradiation;α-Toc, alpha copherol; TPN, total parenteral nutrition;β-Car, beta carotene;RDA, recommended dietary allowances; y, years; BU, busulfan: PO, orally; AA, ascorbic acid; NR, not reported; VC, vitamin C; VE, vitamin E; DZ, diaziquone; TP, thiotepa; Ara-C, cytosin rabinoside; k-Cal, total caloric intake; CML, chronic myelocyticleukemia; AL, acute leukemia; CLL, chronic lymphocytic leukemia; VCR, vincristine; DXR, doxorubicin; EPI, epirubicin;CBPT, carboplatin; RT, radiation therapy;MV,multivitamin; NS, nonsignificant; CDDP, cisplatinumdiammine dichloride; Se, selenium; chemotx, chemotherapy; QOL, quality of life; Co 10, coenzyme Q 10; y-TOC, gamma tocopherol (Modified fromElena J. Ladas et al., 2004).
337P.Rajendran
etal./ClinicaChim
icaActa
436(2014)
332–347
d βovin
ffe
b
tb .b
smCL
ro
aP vβ
af.h
toe a
-Q

338 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
for cartilage function, leading to low levels of ascorbate in synovial fluid.Low concentrations of hydrogen peroxide (H2O2), O2
.− or both, acceler-ate bone resorption by osteoclasts, whereas nitric oxide (NO.) inhibits itand promotes chondrocyte apoptosis, thereby inhibits proteoglycansynthesis and activates latent metalloproteinases and cyclooxygenase.ROS, produced by activated phagocytes, could alter the antigenic behav-ior of immunoglobulin G, producing fluorescent protein aggregates thatcan further activate phagocytic cells. Radical-exposed IgG is able to bindrheumatoid factor and results in the generation of C3a. This reactionmay be self-perpetuating within the rheumatoid joint, suggesting thatfree radicals play a role in the chronicity of the inflammatory reactionwhich is a key question regarding to which extent free radicals contrib-ute to the consequences of inflammation, such as the cartilage andbone destruction. Reactive oxygen intermediates can also function assignaling messengers to activate transcription factors, like NF-κBChange all to NF-κBand activator protein 1(AP-1), and induce geneexpression [111]. All this knowledge might serve to apply a rationalselection of antioxidants for possible therapeutic purposes, of theinflammatory joint disease.
Many raw foods contains natural antioxidants, including enzymessuch as superoxide dismutase, glutathione peroxidase and catalase,which are usually inactivated during food processing, and non-enzymic antioxidants such as carotenoids (e.g. canthaxanthin andastaxanthin in some farmed fish), β-carotene, lutein, lycopene, tocoph-erols (in oils) and other compounds in plants. The latter include othercarotenoids and ascorbate (vitamin C). The plasma concentrations ofthese are largely determined by dietary intake and it is possible thattwo or more antioxidants can act together synergistically [112]. Inview of the high antioxidant content of the French diet which is richin fruit, vegetables and red wine [113,114], it is intriguing to note therelatively low incidence of arthritis [115]. More work is required on tis-sue distributions and bioavailability of antioxidant molecules withinjoints since lipophilic antioxidant molecules, such as vitamin E orβ-carotene, may not have the same access to tissues as hydrophilic an-tioxidants, such as vitamin C. Perhaps, the differences in their effects indisease processes may depend on the hydrophilicity of the antioxidantmolecules concerned and the resulting pattern of tissue distribution indifferent tissue areas. One major problem is that there is no assaycurrently available to measure oxidant activity within joints and possi-bly the activity could occur from an alternative mechanism.
5.2. Anti oxidant vs cancer
Reactive Oxygen Species is a hallmark of human cancer. ROS andtheir functions with respect to the cancer initiation and signalling incancer cells is a prime concern of cancer research. Tobacco smoke alsoplays a very important role in increasing the risk for inflammation andcancer due to its high carcinogenic potential and the synergistic effectswith other respirable particulate to generate ROS and catalyse redox re-actions in the cells of humans, leading to oxidative stress and increasedproduction ofmediators of inflammation [116–118]. Inflammatory cellsare particularly effective in generating most of the reactive oxygenspecies. The activation of the redox metabolism of the inflammatorycells generates a highly oxidative environment within an organ of aero-bic organisms. Much of the oxygen biochemistry, through the activationof plasma membrane NADPH oxidase, of macrophages and neutrophilsis directed towards the release of superoxide anion, hydrogen peroxideand hydroxyl radicals [119–121]. Inflammation acts through the forma-tion of ROS and reactive nitro species (RNS) which cause oxidativedamage to cellular components. Many pro-inflammatory mediators,especially cytokines, chemokines and prostagladins, turn on the angio-genesis switches mainly controlled by vascular endothelial growthfactors [122]. Cancer-associated inflammation is also linked withimmune-suppression that allows cancer cells to evade detection bythe immune system. Inflammation is a critical component of tumor pro-gression. Many cancers arise from sites of infection, chronic irritation
and inflammation. It is now becoming clear that the tumor microenvi-ronment, which is largely orchestrated by inflammatory cells, is anindispensable participant in the neoplastic process, fostering prolifera-tion, survival and migration. In addition, tumor cells have co-optedsome of the signalling molecules of the innate immune system such asselectins, chemokines and their receptors for invasion, migration andmetastasis. These insights are fostering new anti-inflammatory thera-peutic approaches to cancer development [123]. Pathological angiogen-esis is a hallmark of cancer and various ischaemic and inflammatorydiseases. Chronic inflammation is associated with angiogenesis, aprocess that helps cancer cells to grow. Angiogenesis is necessary forexpansion of tumor mass and macrophage, platelets, fibroblasts andtumor cells are a major source of angiogenic factors (vascular endothe-lial growth factors, chemokines, NO, etc.) [124,125]. The inflammationin the tumor microenvironment is characterized by leukocyte infiltra-tion, ranging in size, distribution and composition, as: tumor-associated macrophages (TAM), mast cells, dendritic cells, natural killer(NK) cells, neutrophils, eosinophils and lymphocytes. These cells producea variety of cytotoxic mediators such as ROS and RNS, serine and cysteineproteases, membrane perforating agents, matrix metalloproteinase(MMP), tumor necrosis factor α (TNFα), interleukins (IL-1, IL-6, IL-8),interferons (IFNs) and enzymes, likecyclooxygenase-2 (COX-2),lipooxygenase-5 (LOX-5) and phospholipase A2 (PLA2). Other re-searchers discovered that tumor-associated macrophages are key regula-tors of the link between inflammation and carcinogenesis [126] andexperimental studies showed that, tobacco smoke produce ROS thathave been associated with activation of mitogen-activated protein kinase(MAPK) family members and activation of transcription factors such asNF-κβ and AP-1. These signaling pathways have been implicated in pro-cesses of inflammation, apoptosis, proliferation, transformation and dif-ferentiation [127]. The activation protein-1 (AP-1) contributes to basalgene expression in biological systems. ROS can activate AP-1 throughseveral mechanisms and the effect of AP-1 activation is an increased cellproliferation due to increased expression of growth stimulatory genessuch as cyclin D1 and suppression of the protein p21waf. Studies showedthat AP-1 and NF-kB, inducible by tumor promoters of oxidative stimuli,show differential protein levels or activation in response to tumor pro-moters in JB6 cells. Researchers suggest that as long as oxidative eventsregulate AP-1 and NF-kβ transactivation, these oxidative events can beimportant molecular targets for cancer prevention [128,129]. NF-kB isexpressed and participates in a wide range of biological processes, suchas cell survival, differentiation, inflammation and growth [130]. NF-kB ac-tivation has been linked to a wide spectrum of extracellular stimuli andoxidants and subsequent involvement in the carcinogenic processthrough promotion of angiogenesis and tumor cell invasion andmetasta-sis [131,132].
Dietary factors like chlorogenic acid have also been found to protectagainst environmental carcinogen induced carcinogenesis by their up-regulation of phase II conjugating enzymes and suppression of ROS-mediated correct, AP-1, and MAPK activation [133]. Cellular defencesystem is comprised of several Phase II detoxification enzymes such asglutathione-S-transferases (GSTs), NADP (H) quinoneoxidoreductase(NQO1), Glutathione peroxidases (GPx), Catalase, superoxidedismutases (SODs), epoxide hydrolase, hemeoxygenase (HO-1), UDP-glucuronosyltransferases (UGTs), gammaglutamylcysteinesynthetaseandmanyothers. Expression of these proteins protects cells fromoxida-tive damage and can prevent mutagenesis and cancer [134–137]. Tocounterbalance the oxidative damage from ROS, aerobic organismshave created a variety of antioxidant mechanisms to maintain theirgenomic stability. These mechanisms include phase II detoxificationand other anti-oxidation enzymes that act in cellular defence such ascatalase and SOD, GPx, GST, other constitutive and inducible antioxi-dants, DNA repair enzymes, and other cellular mechanisms of genomicsurveillance, such as cell cycle checkpoint control systems [133]. More-over, several growth factors such as serum, insulin like growth factor I,or fibroblast growth factor 2 generates ROS in MIA PaCa-2 and PANC-1

339P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
cells, which are human pancreatic adenocarcinoma cells that promotescell survival. Inhibiting ROS generation with the antioxidants or NADPHoxidase (Nox4) antisense, or MnSODover expression would stimulateapoptosis in PaCa-2 and PANC-1 cells. This mechanism might play animportant role in pancreatic cancer resistance for treatment and thusrepresent a novel therapeutic target. In gastrointestinal and coloncancer, oxidative pentose pathway (OPP) and the glutathione (GSH) an-tioxidant defense system play an important role in the regulation of cellgrowth and apoptosis. The OPPmodulate intracellular redox status andprovides NADPH for the synthesis of GSH, which is responsible for theinactivation of intracellular ROS that induce apoptosis and cell injury.Depletion of GSH increases the sensitivity of cells to ROS. Therapeuticinhibition of the OPP and/or the GSH defense system might increasethe sensitivity of gastric and colon cancer cells to anti-cancer therapy[138]. Recent studies have shown that the enzymatic product of thymi-dine phosphorylase (TP) generated ROS within cancer cells that help inmaintaining the growth of colon cancer cells thus may provide im-proved therapeutic results as well as a preventative effect on carcino-genesis of the colorectum [124]. Emerging evidence also suggests thatsupranutritional doses of selenite could induce typical apoptosis incolorectal cancer cells in vitro and in xenografttumors by inducingROS and it indicates that antioxidants can activate the apoptoticmachinery through redox-dependent activation and could be useful incancer therapy [139].
The MAPK signalling cascade is activated by a wide variety of recep-tors involved in growth and differentiation including receptor tyrosinekinases (RTKs), integrins and ion channels. This pathway has been re-ported to be activated in over 50% of acute myelogenous leukemia,acute lymphocytic leukemia and are frequently activated in breast,prostate cancers and other cancer types [140,141]. Under optimalgrowth conditions, transiently elevated ROS levels confer a growth ad-vantage to tumor cells. However, exposure of these cells to anticanceragents induces a prolonged increase in ROS levels resulting in potentiat-ing of apoptosis. Thus ROS modulates the ability of stress kinases tostimulate cell growth or cell death and it depends on signal intensityand signal duration [142]. This idea has been nurtured to explain thecomplex nature of ERK signaling in cell cycle regulation and in stress ac-tivated protein kinase signalling (SAPK). Transient, reduced activity ofSAPK promotes cell proliferation, whereas persistent, increased activityresults in cell killing [143]. Conversely, in drug-resistant tumors, phase IIenzymes or other antioxidant defences are often up regulated, shieldingcells from apoptosis. ASK1, an upstream regulator of SAPKs, is inhibitedin normal cells through its association with thioredoxin, which is anantioxidant capable of metabolizing ROS. Increased levels of ROS leadto the dissociation of this complex and thereby facilitate the activationof ASK1 and subsequently SAPKs [144]. A similar redox control hasbeen identified for JNKwhere increased level of ROS triggers thedetach-ment of JNK associated glutathione-S-transferase-π (GSTp) and therebyfacilitating JNK activation. Moreover, ROS-dependent activation of JNKmay also lead to knock down of a JNK phosphatase via an unknownmechanism linking ROS and stress induced kinases an important playerin cancer cell growth [145]. Growth inhibition of pancreatic cells is de-pendent on the efficiency of scavenging ROS as well as effective inhibi-tion of ERK1/2 signaling pathway activation as demonstrated by usingthe free radical scavenger N-Acetyl Cysteine (NAC) or the MEK/ERK1/2 inhibitor (PD98059). Moreover, ERK1/2 induction is dependent onROS production as demonstrated by a complete removal of ERK1/2phosphorylation by NAC. Thus ROS act as pro-survival, anti-apoptoticfactor in pancreatic cancer cells [52]. Moreover, treatment of SW620(colon cancer cells) with berberine, which is a major constituents ofCoptidisthizoma, resulted in activation of apoptosis by phosphorylationof JNK and p38 MAPK, as well as generation of the ROS [146]. Further-more, proline oxidase (POX), that often considered as a ‘housekeepingenzyme’, induce apoptosis through both intrinsic and extrinsicpathways and is involved in nuclear factor of activated T cells (NFAT)signaling and regulation of the MEK/ERK pathway in colon cancer
cells. It has been suggested that, as a nutrition factor, POXmaymodulateapoptosis signals induced by p53 or other anti-cancer agents andenhance apoptosis in stress situations [147].
Components of the cell signaling network, especially those whichconverge on the ubiquitous eukaryotic redox-sensitive transcriptionfactor nuclear factor-kappa B (NF-κB), have been implicated in patho-genesis of many inflammation-associated disorders. Under normalphysiologic conditions, NF-κB is sequestered in the cytoplasm by bind-ing to the inhibitory protein called IκBa. Phosphorylation and subse-quent ubiquitination results in degradation of IκBa by proteasomes.Phosphorylation of IκBa is mainly mediated by the IκB kinase (IKK)complex. Phosphorylation of specific serine residues of p65 subunit ofNF-κB has been considered to facilitate the translocation of NF-κB tonucleus and interaction with the coactivator CBP/p300. Induction ofκphase-2 detoxifying or antioxidant genes represents an important cel-lular defense in response to oxidative and electrophilic insults. Nucleartranscription factor erythroid 2p45 (NF-E2)-related factor 2 (Nrf2)plays a crucial role in regulating phase-2 detoxifying/antioxidant geneinduction. Like NF-κB, Nrf2 is present in the cytoplasm as an inactivecomplexwith the inhibitory protein subunit, in this case Keap1. Dissoci-ation of Nrf2 from Keap1 is prerequisite for nuclear translocation andsubsequent transactivational activity. Once translocated into nucleus,Nrf2 interacts with a small Maf protein, forming a heterodimer thatbinds to antioxidant responsive elements (ARE) or electrophile respon-sive elements (EpRE) present in the promoter/enhancer regions ofgenes encodingmany antioxidant and detoxifying enzymes. Keap1 con-tains several cysteine residues that function as sensors of redox changes.Oxidation or covalent modification of these critical cysteine thiolsdiminishes the affinity of Nrf2 for Keap1, releasing Nrf2 for nucleartranslocation. Dissociation of the Nrf2-Keap1 complex is also assumedto be stimulated through the phosphorylation of Nrf2 by distinct up-stream kinases such as MAPKs, PKC, PI3K, etc. As in the case of NF-κB,phosphorylation of Nrf2 is also considered to facilitate the interactionof this redox-sensitive transcription factor with CBP/p300.NRF2 isa known regulator of the antioxidant response [148,149]. NRF2-regulated phase II enzymes protect against the development of cancerby catalyzing reactions that convert highly reactive, carcinogenicchemicals to less reactive products [150,151]. Singh et al. (2012) [152]recently demonstrated that vitamin C and BHA provide protectionagainst E2-mediated oxidative DNA damage but the mechanism is notwell understood.Many antioxidants derived fromdietary andmedicinalplants have been found to modulate Nrf2-Keap1 signaling, therebypotentiating cellular antioxidant capacity or facilitating detoxificationof carcinogens and other toxicants [153,154]. Table 4 describes 7 obser-vational studies reporting changes in serum, plasma, or whole bloodlevels of vitamins Cand E, selenium,β-carotene, and total radical antiox-idant parameter (TRAP), which represents total body antioxidant status,in patients undergoing anticancer therapy. Information is grouped bymalignancy and includes subject demographics, cancer data, cancertreatment, end points evaluated, results, and comments. No specifictreatment was consistently associated with changes in the individualantioxidants (Table 4) [162].
5.3. Antioxidant vs diabetes
Glucose overload may damage the cells through oxidative stress.This is currently the basis of the “unifying hypothesis” thathyperglycemia-induced oxidative stress may account for the pathogen-esis of all diabetic complications [163,164]. Increasing evidence in bothexperimental and clinical studies suggests that oxidative stress plays amajor role in the pathogenesis of both types of diabetes mellitus. Freeradicals are formed disproportionately in diabetes by glucose oxidation,non-enzymaticglycation of proteins, and the subsequent oxidativedegradation of glycatedproteins [165]. This increased superoxideproduction causes the activation of 5 major pathways involved inthe pathogenesis of complications: polyol pathway flux, increased

Fig. 2. Shows the involvement of cellular antioxidant enzymes in cardiovascular diseases [216–227].
340 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
formation of AGEs (advanced glycation end products), increased ex-pression of the receptor for AGEs and its activating ligands, activationof protein kinase C isoforms, and overactivity of the hexosamine path-way. It also directly inactivates 2 critical anti-atherosclerotic enzymes,endothelial nitric oxide synthase and prostacyclin synthase. Throughthese pathways, increased intracellular reactive oxygen species causedefective angiogenesis in response to ischemia, activate a number ofpro-inflammatory pathways, and cause long-lasting epigenetic changesthat drive persistent expression of proinflammatory genes after glyce-mia is normalized (“hyperglycemic memory”). Atherosclerosis and car-diomyopathy in diabetes are caused in part by pathway-selectiveinsulin resistance, which increases mitochondrial ROS productionfrom free fatty acids and by inactivation of anti-atherosclerosis enzymesby ROS. Over expression of antioxidant enzymes in diabetic patientsprevents diabetic retinopathy, nephropathy, and cardiomyopathy[166]. Antioxidants have proven to play a minimal if any role in thetreatment of diabetic complications in humans [167]. Decreases in ex-pression, and in some instances the activity of antioxidant enzymes,has been previously reported in diabetic microvascular disease [168].Indeed, the over expression of Cu+Zn2+ superoxide dismutase (SOD)protects against end organ damage in models of type II diabetic ne-phropathy [169]. Other studies inmicewith genetic deletions of variousantioxidant enzymeshave also provided insight into the specific relativecontributions of Mn2+SOD [170] to the development of diabetes com-plications. Mn2+SODmimetics such asMnTBAP have also shown effica-cy in preventing ROS-induced injury in vitro, although the utility in vivoof such drugs may be limited [171,172]. Further strengthening a poten-tial role for the antioxidant Mn2+SOD, specific polymorphisms of theMn2+SOD gene are associated with the development of diabetic com-plications [173]. Interestingly, GPx-1-deficient mice have no increased
risk for microvascular disease, in particular diabetic complications[174], most likely because of redundancy with respect to GPx isoforms.Over-expression of catalase in experimental models of type 2 diabeticappears to be protective the diabetic complications [175]. In contrastto Mn2+SOD, however, studies in humans have indicated no relation-ship between catalase gene polymorphisms that interfere with itscellular expression and the incidence of type 2 diabetic patients [176].
Kunisaki et al. [177] investigated the effects of treatments with clas-sical antioxidants such as vitamin E,vitamin C and lipoic acid. Specifical-ly, vitamin E normalizes retinal blood flow and protein kinase C (PKC)activity in the vascular tissue of diabetic rats. One short-term experi-mental study showed that high doses of vitamin C can improve some as-pects of endothelial dysfunction in diabetes. Another experimentalstudy has demonstrated that intra-peritoneal administration of α-lipoic acid to streptozotocin (STZ) diabetic Wistar rats normalisesTBARS levels in plasma, and the retina, liver, and pancreas [178]. Fur-thermore, it has been reported that α-lipoic acid leads to a decrease inthe severity of diabetic neuropathy by maintaining GSH levels and/orby its direct antioxidant properties [179]. However, lipoic acid adminis-tration improved endothelial function in subjects with metabolic syn-drome. In another study to determine the effects of vitamin E onoxidative stress and cell membrane fluidity in the brains of diabetes-induced experimental rats, Hong et al. [180] reported that vitamin Ewas found to be effective for strengthening the antioxidant defense sys-tem. They noted a reduction of the accumulation of ROS such as super-oxide radicals, a decrease in the generation of oxidative damagingsubstances such as the carbonyl value, a significantly improved lipidcomposition, and maintenance of membrane fluidity in the brains ofthe rats. Coleman suggested that triple antioxidant therapy (vitamin E,lipoic acid and vitamin C) in diabetic volunteers attenuates the

341P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
experimental oxidative stress ofmet-hemoglobin formation in vitro andreduces haemoglobin glycation in vivo [181]. Panjwani et al. [182]suggested that vitamin C administered alone or in combination withvitamin E reduced the fall in ulnar nerve conduction velocity. Sivanand Reece [183] investigated whether dietary supplementation withvitamin E would reduce the incidence of diabetic embryopathy in anin vivo rat model. Fifty pregnant rats were designated as the controlgroup and received a normal diet while the diabetic group received vita-min E supplements (400 mg/day). This experimental study found thatsupplementation with vitamin E reduces the incidence of neural tubedefects bymore than 75% [184]. These findings suggest that vitamin E re-duces this oxidative load, confers a protective effect against diabeticembryopathy, and thus may potentially serve as a dietary prophylaxis inthe future. These results are in line with the reports of Chang et al. [184]which showed that diabetic embryopathy of rat or mouse embryos isprevented by vitamin C, vitamin E, SOD, N-acetyl-cysteine, or glutathioneethyl ester. Another study by Otero et al. [185] showed the effects ofsupplementation with vitamin E to diabetic mice. The beneficial effectof vitamin E observed in this model was shown to retard coronaryatherosclerosis accelerated by DM, and was demonstrated to be due to areduction in oxidative stress, and not secondary to a decrease in plasmaglucose or cholesterol since their respective plasma concentrationsremained unchanged in the diabetic mice supplemented with vitamin E[186]. Furthermore, it has been recently reported that macrophagesfrom diabetic mice are under excess oxidative stress, and that the antiox-idant vitamin E can attenuatemacrophage oxidative stresswhich exists inDM and leads to accelerated atherosclerosis development. In a placebo-controlled, randomized trial of diabetic patients, Reaven investigatedthe effect of 1600 IU of RRR-α-tocopherol supplementation daily for10 weeks on hyperglycemia-induced LDL modifications. The result ofthe study pointed to a reduction of approximately 60% of plasma LDLoxidation in diabetic patients, which was statistically significant whencompared to healthy controls [187]. These results are supported and con-firmed by other similar clinical data. Salonen et al. [188] found that dosesequal to or higher than 450 IU are sufficient to significantly amelioratethe susceptibility of LDL to oxidation, indicating that relativelyhigh doses of RRR-α-tocopherol for supplementation are needed.Furthermore, Bellomo and others have shown that the effects of RRR-α-tocopherolsupplementation on LDL oxidation are accompanied by aconcomitant reduction in autoantibody levels againsthyperglycemia-induced LDL modifications. Moreover, clinical studies have shownan inhibitory effect of RRR-α-tocopherol supplementation onthehyperglycemia-induced LDL modifications inT1DM and T2DM dia-betic patients. Based on nutrition recommendations and interventionsfor diabetes, there is no clear evidence of benefits that can be derivedfrom vitamin or mineral supplementation in people with diabetes.Routine supplementation with antioxidants, such as vitamins E and Cand carotene, is not advised because of lack of evidence of efficacy, andconcern related to long term safety, and therefore cannot be recom-mended [189]. The majority of studies included in this review supporta possible role of antioxidant supplementation in reducing the risk ofdiabetic complications
5.4. Antioxidant vs arthrosclerosis
The mechanisms of oxidative stress and antioxidants are in patientswith atherosclerosis have been evaluated in extensive animal experi-ments and clinical research. There is a relationship between atheroscle-rotic risk factors (ARF) and increased vascular production of ROS; themost important ARFs are heredity, age, hypertension, dyslipidemia,diabetes and smoking [190]. It has been reported that in in-vitro studiesan increased vascular production of ROS, particularly superoxide causesdeleterious effects in cell death [191,192]. ROS and reactive nitrogenspecies (RNS) are closely linked to the disease process. Incomplete scav-enging of ROS and RNS influences the mitochondrial lipid cardiolipin,stimulates the release of mitochondrial cytochrome c and finally
activates the intrinsic death pathway [193]. Local generation of RNSmay contribute to vascular tissue injury. Therefore, ROS and RNS partic-ipate as signalling molecules that regulate diverse pathophysiologicalsignallingpathways. High levels of ROS are potent inducers of the intrin-sic apoptotic pathway and tissue injury in pathophysiological condi-tions as an integral part of atherosclerotic plaque stabilization. Themost important sources of ROS production associated with CVD pathol-ogy are the mitochondrial respiratory chain, nicotinamide adeninedinucleotide phosphate oxidases (NADP oxidase). The pathogenesis ofatherosclerosis is further related to inflammation, immune responseand the proliferative process. Endothelial denuding injury leads toplatelet aggregation and releases platelet-derived growth factor,which triggers the proliferation of smooth muscle cells forming thenidus of the atherosclerotic plaque in the arterial intima, implicating in-flammatory changes in the development of the disease [194,195].Supplementing treatment with antioxidants might be helpful to pa-tients. Vitamin E reduces the consequences of lipid peroxide contentin mouse peritoneal macrophages (MPMs) [196]. E-selectin has beenproposed as an important factor in the development of the inflammato-ry process underlying atherothrombosis. The expression of E-selectin isdecreased by vitamin E in the diabetic rats [197]. Vitamin C has alsobeen investigated in studies on the prevention of atherosclerosis. Thefollowing proposals have been put forward regarding the mechanismsof vitamin C in preventing atherosclerosis: First, vitamin C has beenshown to prevent apoptosis caused by cytokines in cultured endothelialcells [198]. It also decreases the release of micro-particles derived fromendothelial cells and suppresses pro-apoptotic activity in congestiveheart failure patients in vivo [199]. Second, vitamin C stimulates alltypes of collagen synthesis by specific hydroxylase enzymes [200].Endothelial cell proliferation is, in part, associated with the synthesisof type IV collagen [201]. Thus, the lack of vitamin C prevents thegeneration of type IV collagen in cultured endothelial cells [202].Third, vitamin C protects the vascular endothelium by enhancing endo-thelial NO synthase. Endothelial NO synthase activity is inhibited byROSthat oxidize and deplete the co-factor tetrahydrobiopterin [203]. There-fore, vitamin C prevents the loss of NO synthase activity bymaintainingtetrahydrobiopterin [204]. Glutathione monoethyl ester, but not ascor-bic acid, exerted protective effects against ischemia–reperfusion injury.Interestingly, the protective effects of glutathione monoethyl ester areenhanced by co-administration with vitamin C in rat hearts subjectedto ischemia and reperfusion [205]. The oxidized LDL leads to increasedplatelet-endothelial cell adhesion, which can be prevented by superox-ide dismutase (SOD) and catalase [206]. In fact, the vascular extracellu-lar expression of SOD is stimulated by NO [207]. Narang et al. [208]reported that dietary palmolein oil, which is rich in monounsaturatedfatty acid and antioxidant vitamins, “protected rat heart from oxidativestress associated with ischemiareperfusion injury”. Das et al. [209]found “a role for c-Src [a family of proto-oncogenic tyrosine kinases]in post ischemic cardiac injury and dysfunction and demonstrate directcardio protective effects of [the tocotrienol-rich fraction of palm oil(TRF)]. The cardio protectiveproperties of TRF appear to be due toinhibition of c-Src activation and proteasome stabilization”. Carlsonet al. [210] using a rat model, reported: “Antioxidant vitamin therapy[vitamin C, vitamin E, vitamin A and zinc] abrogatedmyocardial inflam-matory cytokine signaling and attenuated sepsis-related contractiledysfunction, suggesting that antioxidant vitamin therapy may be apotential approach to treat injury and disease states characterized bymyocardial dysfunction”. Hypertension is directly regulated by thekidneys and cardiovascular system and adversely affects these organs.Tian et al. [211] found that in salt sensitive rat model of hypertension,vitamin C and vitamin E treatments “decreased renal inflammatorycytokines and chemokines, renal immune cells, NF-κB, and arterial pres-sure and improved renal function and damage”. As noted above, chroniczidovudine administration promotes cardiovascular damage, and vita-min C has been found to combat this effect in rats [212]. Diabetesmellitus may initiate increased myocardial vulnerability to ischemia–

342 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
reperfusion injury and pro/antioxidant imbalance. Resistance toischemia-induced ventricular arrhythmias and levels of endogenous an-tioxidants (α-tocopherol) has been found to be increased in diabetic ratmyocardium [213]. The currently available evidence needs to be con-firmed in clinical trials. Consumption of fruit and vegetable diets richin antioxidant nutrients can be recommended for all individuals, andthere is some indication that such a diet can be beneficial in the effectsof cardiovascular events. Fig. 2 shows the involvement of cellular anti-oxidant enzymes in cardiovascular diseases.
5.5. Anti oxidant vs neurodegerative diseases
Neurodegenerative disorders are a heterogeneous group of diseasesof the nervous system, including the brain, spinal cord, and peripheralnerves that have much different aetiology. ROS are particularly activein the brain and neuronal tissue as the excitatory amino acids andneurotransmitters, whose metabolism is factory of ROS, which areunique to the brain and serve as sources of oxidative stress. ROS attackglial cells and neurons, which are post-mitotic cells and therefore,they are particularly sensitive to free radicals, leading to neuronal dam-age [226]. It has been reported that deleterious effects of ROS on humancells may end in oxidative injury leading to programmed cell death i.e.apoptosis [227]. A multitude of genes involved in redox status, anti-inflammation and detoxification are transcribed by Nrf2–ARE pathwayactivation. These genes are known to be involved in cytoprotectionfrom various oxidative insult and cellular injuries in numerous differenttissues and organs including brain. Intracellular peroxidases are clearedby a group of enzymes that are transcribed by Nrf2–ARE pathway. Theperoxisomal CAT catalyses the conversion of H2O2 to water and molec-ular oxygen. However, the specific activity of CAT ismuch lower in brainthan peripheral tissue [228]. Glutathione-s-peroxidase (Gpx) is anotherenzyme that metabolizes H2O2 and depends on reduced glutathione(GSH). The oxidized GSH (GS-SG) is recycled to GSH by glutathionereductase (GR). Glutathione (GSH), a tripeptide γ-glutamyl-cysteinyl-glycine, is the most abundant low molecular weight thiol expressedubiquitously. It is widely recognized as an endogenous non-enzymaticantioxidant and an oxyradical scavenger, and is thereby critical tomain-taining a reducing environment in the cell and protect against oxidativedamage by ROS [229–233]. GSH has been implicated in a wide range ofmetabolic processes, including cell division, DNA repair, regulationof enzyme activity, and activation of transcription factors, modulationof anion and cation homeostasis, and protection against oxidativedamage [234].
GSTs are key detoxification enzymes that catalyze the conjugation ofvarious electrophiles, reactive alkenals, and numerous other xenobioticto GSH. These GSH-S-conjugates are removed from cells by the multi-drug resistant protein-1 (MRP-1), an ATP binding cassette (ABC) familyprotein [235,236]. MRP-1 is an integral plasma membrane protein thatexports glutathione-S conjugates out of the cell in an ATP-dependentmanner [237,238]. Studies have shown reduced GST activity in brainand ventricular fluids in AD [239]. Increased expression of GST leadsto increased resistance towards oxidative stress in neuroblastoma cellsand provides protection against HNE-mediated toxicity in neuronalcell culture [239]. Growing evidence demonstrates that the AD brain isunder tremendous oxidative stress. A significantly increased HO-1 ex-pression was reported in post-mortem AD temporal cortex and hippo-campus compared to aged-matched control [240]. Additionally, anincreased Nqo1 activity and expression was found in astrocytes andneurons of AD brain [229] and Nrf2 was predominantly localized incytoplasm in AD hippocampal neurons [241]. Furthermore, there is in-creased protein oxidation [242,243] and lipid peroxidation [244–246]in AD brain when compared to aged matched controls. Recent studiesin aged APP/PS1 AD mouse models showed reduced Nrf2, Nqo1, GCLcatalytic subunit (GCLC) and GCL modifier subunit (GCLM) mRNA andNrf2 protein levels [247]. Additionally, in a triple transgenic ADmouse, the GSH/GSSG ratio was reported to be reduced [248].
The hallmark of PD is a severe reduction of dopamine in all compo-nents of the basal ganglia. Dopamine and its metabolites are depletedin the caudate nucleus, putamen, globuspallidus, nucleus accumbens,the ventral tegmental area, and the substantianigra pars compacta andreticulata. Moderate losses of dopamine are found in the lateral hypo-thalamus, medial olfactory region and amygdaloid nucleus [249]. Inearly parkinsonism, there appears to be a compensatory increase indopamine receptors to accommodate the initial loss of dopamineneurons [250]. As the disease progresses, the number of dopamine re-ceptors are decreases, apparently due to the concomitant degenerationof dopamine target sites on striatal neurons. In the remainingneurons inpatients with PD, dopamine turnover seems greatly increased, judgingfrom the concentrations of homovannilic acid [HVA] in the nerve termi-nals in the striatum and the cell bodies and dendrites in the substantianigra [251] and the ROS production may very well increase in conse-quence. This hypothesis is strengthened by a study showing that theconcentrations of GSH decrease when dopamine turnover increaseafter reserpine treatment in rats, indicating increased activity of theperoxide scavenging enzyme GSH-Px [252]. If the increase in ROSproduction due to increased dopamine turnover is not buffered by thescavenging enzymes (SOD, catalase, and GSH-Px), the compensatoryhyperactivity of the dopaminergic neurons may become self-destructive. Chronic administration of L-DOPA would then only exacer-bate the production of destructive ROS [253]. The administration ofL-DOPA itself has been postulated to enhance the accumulation of ROS[254]. Another index of oxidative stress in PD might be the evidenceof a robust increase of NF-κB in the nuclei of dopaminergic neurons inthe substantia nigra of PD patients [255]. This clinical finding is consis-tent with in vitro data showing that oxidative stress induced by C2-ceramide treatment causes nuclear translocation of NF-κB in culturedmesencephalic neurons [256]. More recently, it has been shown thatthe neurotoxin 6-OHDA activates NF-κB in PC12 cells by enhancing in-tracellular ROS levels [257]. Interestingly, in this experimental model,NF-κB seems to sustain cell survival by stimulating the expression ofthe anti-apoptotic proteins Bcl-2 and Bfl-1144. Moreover, as alreadymentioned, the potent green tea polyphenol antioxidant EGCG exertsa neuroprotective effect in a MPTP mouse model of PD [258]. However,there are still few and controversial epidemiological data on this impor-tant point, which might be partly due to the intrinsic difficulties inperforming epidemiological surveys regarding the dietary habits oflarge populations. Nevertheless, it is desirable that future studiesaimed at investigating the relationship between dietary antioxidantintake and the relative risk for neurodegenerative disorders such asAD, PD, and ALS will throw more light on this very important aspect ofpublic health.
6. Conclusion
In summary the reactive oxygen species and oxidative stress play animportant role in the aetiology and progression of major human degen-erative diseases has triggered enormous and worldwide interest inendogenous and exogenous antioxidants. There is now abundant evi-dence that substances in fruit and vegetables are potent preventives ofvarious diseases, especially arthritis, cancer, heart disease, diabetesand neurodegenerative diseases. With these recent developments inscientific knowledge which firmly establish the links between foodfactors and the prevention of disease. The present review reveals avast number of relevant studies related to oxidative stress and selectedhuman diseases. The research field actually presents a variety ofapproaches that have been produced suggestive evidence that antioxi-dants might have an impact against many diseases. However, moresystematic research in this line is imperative in order to reveal the rela-tionship and involvement of antioxidants with other diseases. The largediversity in the molecular mechanisms and the methodology of antiox-idants discussed here are factors probably responsible for the consistentfindings of their prevention of diseases. This shows that it will be very

343P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
useful in controlling many oxidative stress mediated diseases asdiscussed above. In comparison with the effects of the antioxidantsanalysed in the previous study we can expect that many plant derivedcompounds which have excellent antioxidant property and were suc-cessfully investigated here and validates or corroborates the antioxidanthypothesis of many diseases [165,259–263]. Current review highlightsthe perspectives and possibilities of unrevealing the uncapped poten-tials of antioxidants in herbal plants for disease cure and warrants theneed for systematic research in the field to clear the insights in thisvery complex matter. However, recent research work with antioxidanttherapy suggests another promising area i.e., combining potential anti-oxidants and using them during the early onset of the disease as aprophylactic measure to prevent the disease that may eventuallyprove to be more effective in treating several devastating diseases.
References
[1] Dinstel RR, Cascio J, Koukel S. The antioxidant level of Alaska's wild berries: high,higher and highest. Int J Circumpolar Health 2013;72:796–802.
[2] Pham-Huy LA, He H, Pham-Huy C. Free radicals, antioxidants in disease and health.Int J Biomed Sci 2008;4:89–96.
[3] Valko M, Leibfritz D, Moncol J, Cronin MTD, Mazur M, Telser J. Free radicals andantioxidants in normal physiological functions and human disease. Int J BiochemCell Biol 2007;39:44–84.
[4] Singh PP, Chandra A, Mahdi F, Roy A, Sharma P. Reconvene and reconnect theantioxidant hypothesis in human health and disease. Indian J Clin Biochem2010;25:225–43.
[5] Sen S, Chakraborty R, Sridhar C, Reddy YSR, De B. Free radicals, antioxidants,diseases and phytomedicines: current status and future prospect. Int J Pharm SciRev Res 2010;3:91–100.
[6] Lykkesfeldt J, Svendsen O. Oxidants and antioxidants in disease: oxidative stress infarm animals. Vet J 2007;173:502–11.
[7] Miller JK, Brzezinska-Slebodzinska E, Madsen FC. Oxidative stress, antioxidants,and animal function. J Dairy Sci 1993;76:2812–23.
[8] Sugino N. Roles of reactive oxygen species in the corpus luteum. Anim Sci J2006;77:556–65.
[9] Reilly PM, Schiller HJ, Bulkley GB. Pharmacologic approach to tissue injurymediated by free radicals and other reactive oxygen metabolites. Am J Surg1991;161:488–503.
[10] Alberti A, Bolognini L, Macciantelli D, Caratelli M. The radical cation of N, N-Diethyl-para-phenylendiamine: a possible indicator of oxidative stress in biological samples.Res Chem Intermed 2000;26:253–67.
[11] Di Trana A, Celi P, Claps S, Fedele V, Rubino R. The effect of hot season and nutritionon the oxidative status and metabolic profile in dairy goats during mid lactation.Anim Sci 2006;82:717–22.
[12] Rizzo A, Mutinati M, Spedicato M, et al. First demonstration of an increased serumlevel of reactive oxygen species during the peripartal period in the ewes.Immunopharmacol Immunotoxicol 2008;30:741–6.
[13] Bernabucci U, Ronchi B, Lacetera N, Nardone A. Influence of body condition scoreon relationships between metabolic status and oxidative stress in periparturientdairy cows. J Dairy Sci 2005;88:2017–26.
[14] Merlo M, Celi P, Barbato O, Gabai G. Relationships between oxidative status andpregnancy outcome in dairy cows. Anim Prod Aust 2008;27:84.
[15] Celi P, Sullivan M, Evans D. The stability of the reactive oxygen metabolites(d-ROMs) and biological antioxidant potential (BAP) tests on stored horse blood.Vet J 2010;183:217–8.
[16] Fialová L, Malbohan I, Kalousová M, et al. Oxidative stress and inflammation inpregnancy. Scand J Clin Lab Invest 2006;66:121–7.
[17] Noyan T, Güler A, Sekeroglu MR, Kamaci M. Serum advanced oxidation proteinproducts, myeloperoxidase and ascorbic acid in pre-eclampsia and eclampsia.Aust N Z J Obstet Gynaecol 2006;46:486–91.
[18] Witko-Sarsat V, Friedlander M, Nguyen Khoa T, et al. Advanced oxidation proteinproducts as novel mediators of inflammation and monocyte activation in chronicrenal failure. J Immunol 1998;161:2524–32.
[19] Kalousová M, Skrha J, Zima T. Advanced glycation end-products and advancedoxidation protein products in patients with diabetes mellitus. Physiol Res2002;51:597–604.
[20] Shi XY, Hou FF, Niu HX, et al. Advanced oxidation protein products promote inflam-mation in diabetic kidney through activation of renal nicotinamide adeninedinucleotide phosphate oxidase. Endocrinology 2008;149:1829–39.
[21] Kaneda H, Taguchi J, Ogasawara K, Aizawa T, Ohno M. Increased level of advancedoxidation protein products in patients with coronary artery disease. Atherosclero-sis 2002;162:221–5.
[22] Atabek ME, Keskin M, Yazici C, et al. Protein oxidation in obesity and insulinresistance. Eur J Pediatr 2006;165:753–6.
[23] Celi P, Raadsma HW. The effects of Yerba Mate (Ilex paraguarensis) supplementa-tion on the productive performance of lambs. Ruminant physiology digestion,metabolism, and effects of nutrition on reproduction and welfare. , Proceeding ofthe XIth International Symposium in Ruminant PhysiologyFrance: Clermont-Ferrand; 2009. p. 804–5.
[24] Celi P, Raadsma HW. Effects of Yerba Mate (Ilex paraguariensis) supplementationon the productive performance of dairy cows during mid-lactation. Anim ProdSci 2010;50:339–44.
[25] Kalousová M, Zima T, Tesar V, Dusilová-Sulková S, Skrha J. Advanced glycoxidationend products in chronic diseasesclinical chemistry and genetic background. MutatRes 2005;579:37–46.
[26] Witko-Sarsat V, Nguyen-Khoa T, Jungers P, Drüeke TB, Descamps-Latscha B.Advanced oxidation protein products as a novel molecular basis of oxidative stressin uraemia. Nephrol Dial Transplant 1999;14(1):76–8.
[27] Merlo M, Barbato O, Stefani A, Celi P, Da Dalt L, Gabai G. Embryonic mortality andplasma advanced oxidation protein products (AOPP) increase in dairy cows. Cur-rent Topics in Dairy Production, Proceedings of the Dairy Research FoundationSymposium. SciELO Brazil; 2008. p. 49–53 [Camden, Australia].
[28] Georgieva NV. Oxidative stress as a factor of disrupted ecological oxidative balancein biological systems—a review. Bulg J Vet Med 2005;8:1–11.
[29] Janero DR. Malondialdehyde and thiobarbituric acid-reactivity as diagnostic indicesof lipid peroxidation and peroxidative tissue injury. Free Radic Biol Med1990;9:515–40.
[30] Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement, andsignificance. Am J Clin Nutr 1993;57:715S–24S.
[31] Castillo C, Hernández J, Valverde I, et al. Plasma malonaldehyde (MDA) andtotal antioxidant status (TAS) during lactation in dairy cows. Res Vet Sci2006;80:133–9.
[32] Bouwstra RJ, Goselink RM, Dobbelaar P, Nielen M, Newbold JR, van Werven T. Therelationship between oxidative damage and vitamin E concentration in blood,milk,and liver tissue from vitamin E supplemented and nonsupplementedperiparturient heifers. J Dairy Sci 2008;91:977–87.
[33] GriffithsHR,Moller L, BartoszG, et al. Biomarkers.Mol AspectsMed2002;23:101–208.[34] Lykkesfeldt J. Determination of malondialdehyde as dithiobarbituric acid adduct in
biological samples by HPLC with fluorescence detection: comparison withultraviolet–visible spectrophotometry. Clin Chem 2001;47:1725–7.
[35] Milne GL, Musiek ES, Morrow JD. F2-Isoprostanes as markers of oxidative stressin vivo: an overview. Biomarkers 2005;10:10–23.
[36] Mohanty JG, Bhamidipaty S, EvansMK, Rifkind JM. Afluorimetric semi-microplate for-mat assay of protein carbonyls in blood plasma. Anal Biochem 2010;400(2):289–94.
[37] Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues bythiobarbituric acid reaction. Anal Biochem 1979;95(2):351–8.
[38] Collins CE, Quaggiotto P, Wood L, O'Loughlin EV, Henry RL, Garg ML. Elevatedplasma levels of F2α isoprostane in cystic fibrosis. Lipids 1999;6(34):551–6.
[39] Halliwell B, Gutteridge JMC. Free radical in biology andmedicine. 4th ed. New York,NY, USA: Oxford University Press; 2007.
[40] Benzie IFF, Strain JJ. The ferric reducing ability of plasma (FRAP) as a measure of“antioxidant power”: the FRAP assay. Anal Biochem 1996;239(1):70–6.
[41] Roe HJ, Kuether CA. Detection of ascorbic acid in whole blood and urine throughthe 2, 4-dinitrophenyl-hydrazine derivative of dehydro ascorbic acid. J Biol Chem1943;147:399–407.
[42] Teissier E, Walters-Laporte E, Duhem C, Luc G, Fruchart JC, Duriez P. Rapid quanti-fication of alpha-tocopherol in plasma and low- and high-density lipoproteins. ClinChem 1996;42(1):430–5.
[43] Misra HP, Fridovich I. Therole of superoxide anion in the autoxidation of epinephrineand a simple assay for superoxide dismutase. J Biol Chem 1972;247(10):3170–5.
[44] Aebi M. Catalase. In: Bergmeyer HU, editor. Methods of enzymatic analysis, vol. 2.NY, USA: Verlag Chemie-Academic Press; 1974.
[45] Flohe L, GunzlerWA.Assays of glutathioneperoxidise.Met Enzymol 1984;105:114–20.[46] Hissin PJ, Hilf R. A fluorometric method for determination of oxidized and reduced
glutathione in tissues. Anal Biochem 1976;74(1):214–26.[47] Haining JL, Legan JS. Fluorometric assay for xanthine oxidase. Anal Biochem
1967;21(3):337–43.[48] Nauseef WM. Biological roles for the NOX family NADPH oxidases. J Biol Chem
2008;283(25):16961–5.[49] Burger D, Touyz RM. Cellular biomarkers of endothelial health: microparticles, en-
dothelial progenitor cells, and circulating endothelial cells. J Am Soc Hypertens2012;6(2):85–99.
[50] Touyz RM, Schiffrin E. Arterial hypertension. In: Hill J, editor. Muscle fundamentalbiology and mechanisms of disease, vol. 2. San Diego, Calif, USA: Elsevier; 2012.p. 1311–9.
[51] Sinha MK, Gaze DC, Tippins JR, Collinson PO, Kaski JC. Ischemiamodified albumin isa sensitive marker of myocardial ischemia after percutaneous coronary interven-tion. Circulation 2003;107(19):2403–5.
[52] Kohen R, Nyska A. Oxidation of biological systems: oxidative stress phenomena,antioxidants, redox reactions, and methods for their quantification. Toxicol Pathol2002;30(6):620–50.
[53] Scholz RW, Graham KS, Gumpricht E, Reddy CC. Mechanism of interaction ofvitamin E and glutathione in the protection against membrane lipid peroxidation.Ann N Y Acad Sci 1989;570:514–7.
[54] Harrison R. Structure and function of xanthine oxidoreductase: where are we now?Free Radic Biol Med 2002;33(6):774–97.
[55] Yamazaki T, Kawai C, Yamauchi A, Kuribayashi F. A highly sensitive chemilumines-cence assay for superoxide detection and chronic granulomatous disease diagnosis.Trop Med Health 2011;39(2):41–5.
[56] Nauseff WM. Detection of superoxide anion and hydrogen peroxide production bycellular NADPH oxidases. Biochim Biophys Acta 2014;1840(2):757–67.
[57] Moller P, Loft S. Dietary antioxidants and beneficial effect on oxidatively damagedDNA. Free Radic Biol Med 2006;41:388–415.
[58] Moller P. The alkaline comet assay: towards validation in biomonitoring of DNAdamaging exposures. Basic Clin Pharmacol Toxicol 2006;98:336–45.

344 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
[59] Boyle SP, Dobson VL, Duthie SJ, Kyle JAM, Collins AR. Absorption and protectiveeffects of flavonoid glycosides from an onion meal. Eur J Nutr 2000;39:213–23.
[60] Moller P, Loft S. Oxidative DNA damage in white blood cells of humans in dietaryantioxidant intervention studies. Am J Clin Nutr 2002;76:303–10.
[61] Sacheck JM, Milbury PE, Cannon JG, Roubenoff R, Blumberg JB. Effect of vitamin Eand eccentric exercise on selected biomarkers of oxidative stress in young andelderly men. Free Radic Biol Med 2003;34:1575–88.
[62] Collins AR, Cadet J, Moller L, Poulsen HE, Vina J. Are we sure we know how to mea-sure 8-oxo-7,8-dihydroguanine in DNA from human cells? Arch Biochem Biophys2004;423:57–65.
[63] Arendt BM, Ellinger S, Kekic K, et al. Single and repeated moderate consumption ofnative or dealcoholized red wine show different effects on antioxidant parametersin blood and DNA strand breaks in peripheral leukocytes in healthy volunteers: arandomized controlled trial (ISRCTN68505294). Nutr J 2005;4:33.
[64] Collins AR, Duthie SJ, Dobson VL. Direct enzymic detection of endogenous oxidativebase damage in human lymphocyte DNA. Carcinogenesis 1993;14:1733–5.
[65] Foksinski M, Rozalski R, Guz J, et al. Urinary excretion of DNA repair productscorrelates with metabolic rates as well as with maximum life spans of differentmammalian species. Free Radic Biol Med 2004;37:1449–54.
[66] Collins AR. Assays for oxidative stress and antioxidant status: applications toresearch into the biological effectiveness of polyphenols. Am J Clin Nutr2005;81:261S–7S.
[67] Bowen P, Chen L, Stacewicz-Sapuntzakis M, et al. Tomato sauce supplementationand prostate cancer: lycopene accumulation and modulation of biomarkers ofcarcinogenesis. Exp Biol Med 2002;227:886–93.
[68] Collins AR, Gedik CM, Olmedilla B, Southon S, Bellizzi M. Oxidative DNA damagemea-sured in human lymphocytes: large differences between sexes andbetween countries,and correlations with heart disease mortality rates. FASEB J 1998;12:1397–400.
[69] Moller P, Loft S. Interventions with antioxidants and nutrients in relation tooxidative DNA damage and repair. Mutat Res 2004;551:79–89.
[70] Choi SW, Benzie IF, Collins AR, Hannigan BM, Strain JJ. Vitamins C and E: acuteinteractive effects on biomarkers of antioxidant defence and oxidative stress.Mutat Res 2004;551:109–17.
[71] Tsilimigaki S, Messini-Nikolaki N, Kanariou M, Piperakis SM. A study on the effectsof seasonal solar radiation on exposed populations. Mutagenesis 2003;18:139–43.
[72] Panayiotidis M, Collins AR. Ex vivo assessment of lymphocyte antioxidant statususing the comet assay. Free Radic Res 1997;27:533–7.
[73] Moller P, Vogel U, Pedersen A, Dragsted LO, Sandstrom B, Loft S. No effect of 600 gfruit and vegetables per day on oxidative DNA damage and repair in healthyhuman non-smokers. Cancer Epidemiol Biomarkers Prev 2003;12:1016–22.
[74] Moller P, Viscovich M, Lykkesfeldt J, Loft S, Jensen A, Poulsen HE. Vitamin C supple-mentation decreases oxidative DNA damage in mononuclear blood cells ofsmokers. Eur J Nutr 2004;43:267–74.
[75] Giovannelli L, Saieva C, Masala G, et al. Nutritional and lifestyle determinants ofDNA oxidative damage: a study in a Mediterranean population. Carcinogenesis2002;23:1483–9.
[76] Gedik CM, Collins A. ESCODD (European Standards Committee on Oxidative DNADamage). Establishing the background level of base oxidation in human lympho-cyte DNA: results of an inter-laboratory validation study. FASEB J 2005;19:82–4.
[77] Françoise DG, Chantal MB. The many faces of endothelial microparticles. ArterThrom Vasc Bio 2011;31:27–33.
[78] Eguchi M, Monden K, Miwa N. Role of MAPK phosphorylation in cytoprotection bypro-vitamin C against oxidative stress-induced injuries in cultured cardiomyoblastsand perfused rat heart. J Cell Biochem 2003;90:219–26.
[79] Nguyen HT, HsiehMH, Gaborro A, Tinloy B, Phillips C, Adam RM. JNK/SAPK and p38SAPK-2 mediate mechanical stretch-induced apoptosis via caspase-3 and-9 inNRK-52E renal epithelial cells. Nephron Exp Nephrol 2006;102:49–61.
[80] Matsuzawa A, Ichijo H. Redox control of cell fate by MAP kinase: physiological rolesof ASK1-MAP kinase pathway in stress signaling. Biochim Biophys Acta Gen Subj2008;1780:1325–36.
[81] Torres M. Mitogen-activated protein kinase pathways in redox signaling. FrontBiosci 2003;8:D369–91.
[82] Donadelli M, DallaPozza E, Costanzo C, Scupoli MT, Piacentini P, Scarpa A. Increasedstability of P21(WAF1/CIP1) mRNA is required for ROS/ERK-dependent pancreaticadenocarcinoma cell growth inhibition by pyrrolidinedithiocarbamate. BiochimBiophys Acta 2006;1763:917–26.
[83] HsuWH, Hsieh YS, Kuo HC, et al. Berberine induces apoptosis in SW620 human co-lonic carcinoma cells through generation of reactive oxygen species and activationof JNK/p38 MAPK and FasL. Arch Toxicol 2007;81:719–28.
[84] Liu Y, Borchert GL, Surazynski A, Hu CA, Phang JM. Proline oxidase activates bothintrinsic and extrinsic pathways for apoptosis: the role of ROS/superoxides, NFATand MEK/ERK signaling. Oncogene 2006;25:5640–7.
[85] Barry I. Vitamin C: friends or foe? Nat Rev Cancer 2008;8:830.[86] Liu L, Zhao SP, Gao M, Zhou QCZ, Li YL, Xia B. Vitamin C preserves endothelial func-
tion in patients with coronary heart disease after a high-fat meal. Clin Cardiol2002;25:219–24.
[87] Wang Y, Hodge AM, Wluka AE, et al. Effect of antioxidante on knee cartilage andbone in healthy, middle-aged subjects: a cross-sectional study. Arthritis Res Ther2007;9:1–9.
[88] Wintergerst ES, Maggini S, Homig DH. Immune-enhancing role of vitamin C andzinc and effect on clinical conditions. Ann Nutr Metab 2006;50:85–94.
[89] Woo A, Kim JH, Jeong YJ, et al. Vitamin C acts indirectly to modulate isotypeswitching in mouse B cells. Anat Cell Biol 2010;43:25–35.
[90] Thankachan P, Walczyk T, Muthayya S, Kurpad AV, Hurrell RF. Iron absorption inyoung Indian women: the interaction of iron status with the influence of tea andascorbic acid. Am J Clin Nutr 2008;87:881–6.
[91] Riso P, Martini D, Moller P, et al. DNA damage and repair activity after broccoliintake in young healthy smokers. Mutagenesis 2010:1–8.
[92] Pryor WL. Vitamin E and heart disease: basic science to clinical intervention trials.Free Radic Biol Med 2000;28:141–61.
[93] Traber MG, Frei B, Beckman JS. Vitamin E revisited: do new data validate for chron-ic disease prevention? Curr Opin Lipidol 2008;19:30–8.
[94] Weinstein SJ, Wright ME, Lawson KA, et al. Serum and dietary vitamin E inrelation to prostate cancer risk. Cancer Epidemiol Biomarkers Prev2007;16:1253–8.
[95] Muller DPR. Vitamin E and neurological functions. Mol Nutr Food Res 2010;54:1–9.[96] Devore EE, Grodstein F, van Rooij FJ, et al. Dietary antioxidant and lonterm risk of
dementia. Arch Neurol 2010;67:819–25.[97] Miyake Y, FukushimaW, Tanaka K, et al. Dietary intake of antioxidant vitamins and
risk of Parkinson's disease: a case–control study in Japan. Eur J Neurol 2010. http://dx.doi.org/10.1111/j.1468-1331.2010. 03088.
[98] Manach C, Mazur A, Scalbert A. Polyphenols and prevention of cardiovascular dis-eases. Curr Opin Lipidol 2005;16:77–84.
[99] Russo P, Tedesco I, Russo M, Russo GL, Venezia A, Cicala C. Effects of de-alcoholatedred wine and its phenolic fractions on platelet aggregation. Nutr Metab CardiovascDis 2001;11:25–9.
[100] Schachinger V, Britten MB, Zeiher AM. Prognostic impact of coronary vasodilatordysfunction on adverse long-term outcome of coronary heart disease. Circulation2000;101:1899–906.
[101] Corder R, Mullen W, Khan NQ, et al. Oenology: red wine procyanidins and vascularhealth. Nature 2006;444:566.
[102] Yang CS, Landau JM, Huang MT, Newmark HL. Inhibition of carcinogenesis bydietary polyphenolic compounds. Ann Rev Nutr 2001;21:381–406.
[103] Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeuticimplications for antioxidant treatment. Drugs Aging 2001;18:685–716.
[104] Pan T, Jankovic J, Le W. Potential therapeutic properties of green tea polyphenols inParkinson's disease. Drugs Aging 2003;20:711–21.
[105] Zunino SJ, Storms DH, Stephensen CB. Diets rich in polyphenols and vitamin Ainhibit the development of Type I autoimmune diabetes in nonobese diabeticmice. J Nutr 2007;137:1216–21.
[106] Atmaca A, Kleerekoper M, Bayraktar M, Kucuk O. Soy isoflavones in the manage-ment of postmenopausal osteoporosis. Menopause 2008;15:748–57.
[107] Hurrell RF, Reddy M, Cook JD. Inhibition of non-haem iron absorption in man bypolyphenolic-containing beverages. Br J Nutr 1999;81:289–95.
[108] Visioli F, Grande S, Bogani P, Galli C. The role of antioxidants in the mediterraneandiets: focus on cancer. Eur J Cancer Prev 2004;13:337–43.
[109] de Oliveira El-Warrak A, Rouma M, Amoroso A, Boysen SR, Chorfi Y. Measurementof vitamin A, vitamin E, selenium, and L-lactate in dogs with and without osteoar-thritis secondary to ruptured cranial cruciate ligament. Can Vet J 2012;53:1285–8[La revue veterinairecanadienne].
[110] Roush JK, McLaughlin RM, Radlinsky MAG. Understanding the pathophysiology ofosteoarthritis. Vet Med 2002;97:108–17.
[111] Hadjigogos K. The role of free radicals in the pathogenesis of rheumatoid arthritis.Panminerva Med 2003;45:7–13.
[112] Liu C, Russell RM, Wang XD. Alpha-Tocopherol and ascorbic acid decrease theproduction of beta-apo-carotenals and increase the formation of retinoids frombeta-carotene in the lung tissues of cigarette smoke-exposed ferrets in vitro. JNutr 2004;134:426–30.
[113] Ortiz J,MarinR,NoriegaD,NavarroM,Arozarena I. Color, phenolics, andantioxidant ac-tivity of blackberry (RubusglaucusBenth.), blueberry (VacciniumfloribundumKunth.),and apple wines from Ecuador. J Food Sci 2013;78:C985–93.
[114] Pastor-Valero M. Fruit and vegetable intake and vitamins C and E are associatedwith a reduced prevalence of cataract in a Spanish Mediterranean population.BMC Ophthalmol 2013;13:52.
[115] Elmali N, Baysal O, Harma A, Esenkaya I, Mizrak B. Effects of resveratrol in inflam-matory arthritis. Inflammation 2007;30:1–6.
[116] Gardi C, Valacchi G. Cigarette smoke and ozone effect on murine inflammatory re-sponses. In: Valacchi G, editor. Environmental Stressors in Biology and Medicine;2012. p. 104–11.
[117] Sangani RG, Ghio AJ. Lung injury after cigarette smoking is particle related. Int JChron Obstruct Pulmon Dis 2011;6:191–8.
[118] Moller P, Folkmann JK, Forchhammer L, et al. Air pollution, oxidative damage toDNA, and carcinogenesis. Cancer Lett 2008;266:84–97.
[119] Lazennec G, Richmond A. Chemokines and chemokine receptors: new insights intocancer-related inflammation. Trends Mol Med 2010;16:133–44.
[120] Rodriguez-Vita J, Lawrence T. The resolution of inflammation and cancer. CytokineGrowth Factor Rev 2010;21:61–5.
[121] Porta C, Larghi P, Rimoldi M, et al. Cellular and molecular pathways linkinginflammation and cancer. Immunobiology 2009;214:761–77.
[122] Costa C, Incio J, Soares R. Angiogenesis and chronic inflammation: cause orconsequence? Angiogenesis 2007;10:149–66.
[123] Valavanidis A, Vlachogianni T, Fiotakis K, Loridas S. Pulmonary oxidative stress, in-flammation and cancer: respirable particulate matter, fibrous dusts and ozone asmajor causes of lung carcinogenesis through reactive oxygen species mechanisms.Int J Environ Res Public Health 2013;10:3886–907.
[124] Allavena P, Sica A, Solinas G, Porta C, Mantovani A. The inflammatory micro-environment in tumor progression: the role of tumor-associated macrophages.Crit Rev Oncol Hematol 2008;66:1–9.
[125] Benelli R, Lorusso G, Albini A, Noonan DM. Cytokines and chemokines as regulatorsof angiogenesis in health and disease. Curr Pharm Des 2006;12:3101–15.
[126] Vendramini-Costa DB, Carvalho JE. Molecular linkmechanisms between inflamma-tion and cancer. Curr Pharm Des 2012;18:3831–52.

345P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
[127] Knaapen AM, Shi TM, Borm PJA, Schins RPF. Soluble metals as well as the insolubleparticle fraction are involved in cellular DNA damage induced by particulate mat-ter. Mol Cell Biochem 2002;234:317–26.
[128] Hsu TC, Young MR, Cmarik J, Colburn NH. Activator protein 1 (AP-1)- and nuclearfactor kappa B (NF-kappa B)-dependent transcriptional events in carcinogenesis.Free Radic Biol Med 2000;28:1338–48.
[129] Dhar A, Young MR, Colburn NH. The role of AP-1, NF-kappa B and ROS/NOS in skincarcinogenesis: the JB6 model is predictive. Mol Cell Biochem 2002;234:185–93.
[130] Sethi G, Sung B, Aggarwal BB. Nuclear factor-kB activation: from bench to bedside.Exp Biol Med 2008;233:21–31.
[131] Lee CH, Jeon YT, Kim SH, Song YS. NF-kappa B as a potential molecular target forcancer therapy. BioFactors 2007;29:19–35.
[132] Wang H, Cho CH. Effect of NF-kappa B signaling on apoptosis in chronicinflammation-associated carcinogenesis. Curr Cancer Drug Targets 2010;10:593–9.
[133] Kaefer CM, Milner JA. The role of herbs and spices in cancer prevention. J NutrBiochem 2008;19:347–61.
[134] Pool-Zobel B, Veeriah S, Bohmer FD. Modulation of xenobiotic metabolising en-zymes by anticarcinogens—focus on glutathione S-transferases and their role astargets of dietary chemoprevention in colorectal carcinogenesis. Mutat ResFundam Mol Mech Mutagen 2005;591:74–92.
[135] Barve A, Khor TO, Nair S, et al. gamma-Tocopherol-enriched mixed tocopherol dietinhibits prostate carcinogenesis in TRAMP mice. Int J Cancer 2009;124:1693–9.
[136] Kumaraguruparan R, Seshagiri PB, Hara Y, Nagini S. Chemoprevention of ratmammary carcinogenesis by black tea polyphenols: modulation of xenobiotic-metabolizing enzymes, oxidative stress, cell proliferation, apoptosis, and angiogen-esis. Mol Carcinog 2007;46:797–806.
[137] Lee SB, Kim CY, Lee HJ, Yun JH, Nho CW. Induction of the phase II detoxification en-zymeNQO1 in hepatocarcinoma cells by lignans from the fruit of Schisandrachinensisthrough nuclear accumulation of Nrf2. Planta Med 2009;75:1314–8.
[138] Acharya A, Das I, Chandhok D, Saha T. Redox regulation in cancer a double-edgedsword with therapeutic potential. Oxidative Med Cell Longev 2010;3:23–34.
[139] Huang F, Nie C, Yang Y, et al. Selenite induces redox-dependent Bax activation andapoptosis in colorectal cancer cells. Free Radic Biol Med 2009;46:1186–96.
[140] Mebratu Y, Tesfaigzi Y. How ERK1/2 activation controls cell proliferation and celldeath is subcellular localization the answer? Cell Cycle 2009;8:1168–75.
[141] McCubrey JA, Steelman LS, Abrams SL, et al. Targeting survival cascades induced byactivation of Ras/Raf/MEK/ERK, PI3K/PTEN/Akt/mTOR and Jak/STAT pathways foreffective leukemia therapy. Leukemia 2008;22:708–22.
[142] Tsanko S, Gechev S, Frank VB, Julie MS, Iliya D, Christophe L. Reactive oxygen spe-cies as signals that modulate plant stress responses and programmed cell death.Bio Essays 2006;28:1091–101.
[143] Moran B, David E, Alexander L. ROS, stress-activated kinases and stress signaling incancer. EMBO Rep 2002;3(5):420–5.
[144] Asha A, Ila D, Des C, Tapas S. Redox regulation in cancer, A double-edged swordwith therapeutic potential. Oxid Med Cell Longev 2010;3(1):23–34.
[145] McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activationof the MAP kinase signaling pathways. Antioxid Redox Signal 2006;8(9–10):1775–89.
[146] Christodoulou MI, Kontos CK, Halabalaki M, Skaltsounis AL, Scorilas A. Naturepromises new anticancer agents: interplay with the apoptosis-related BCL2 genefamily. Anticancer Agents Med Chem 2014;14(3):375–99.
[147] Young JS. NF-κB and Nrf2 as potential chemopreventive targets of someanti-inflammatory and antioxidative phytonutrients with anti-inflammatoryand antioxidative activities. Asia Pac J Clin Nutr 2008;17(S1):269–72.
[148] Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signalingpathway and its activation by oxidative stress. J Biol Chem 2009;284:13291–5.
[149] Dhakshinamoorthy S, Jaiswal AK. Small Maf (MafG and MafK) proteinsnegatively regulate antioxidant response element-mediated expression andantioxidant induction of the NAD(P)H : quinone oxidoreductase1 gene. J BiolChem 2000;275:40134–41.
[150] Li W, Kong AN. Molecularmechanisms of Nrf2-mediated antioxidant response.MolCarcinog 2009;48:91–104.
[151] Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcino-genesis 2010;31:90–9.
[152] Singh B, Bhat NK, Bhat HK. Induction of NAD(P)H-quinoneoxidoreductase 1 by an-tioxidants in female ACI rats is associated with decrease in oxidative DNA damageand inhibition of estrogen-induced breast cancer. Carcinogenesis 2012;33:156–63.
[153] Lee JS, Surh YJ. Nrf2 as a novel molecular target for chemoprevention. Cancer Lett2005;224:171–84.
[154] Yu X, Kensler T. Nrf2 as a target for cancer chemoprevention. Mutat Res FundamMol Mech Mutagen 2005;591:93–102.
[155] Clemens MR, Ladner C, Schmidt H, et al. Decreased essential antioxidants and in-creased lipid hydroperoxides following high-dose radiochemotherapy. Free RadicRes Commun 1989;7:227–32.
[156] Clemens MR, Waladkhani AR, Bublitz K, Ehninger G, Gey KF. Supplementation withantioxidants prior to bone marrow transplantation. Wien Klin Wochenschr1997;109:771–6.
[157] Hunnisett A, Davies S, McLaren-Howard J, Gravett P, Finn M, Gueret-Wardle D.Lipoperoxides as an index of free radical activity in bone marrow transplantrecipients. Preliminary observations. Biol Trace Elem Res 1995;47:125–32.
[158] Lloyd JV, Davis PS, Emery H, Lander H. Platelet ascorbic acid levels in normalsubjects and in disease. J Clin Pathol 1972;25:478–83.
[159] Sundström H, Korpela H, Viinikka L, Kauppila A. Serum selenium and glutathioneperoxidase, and plasma lipid peroxides in uterine, ovarian or vulvar cancer, andtheir responses to antioxidants in patients with ovarian cancer. Cancer Lett1984;24:1–10.
[160] Jaakkola K, Lähteenmäki P, Laakso J, Harju E, Tykkä H, Mahlberg K. Treatment withantioxidant and other nutrients in combination with chemotherapy and irradiationin patients with small-cell lung cancer. Anticancer Res 1992;12:599–606.
[161] Lockwood K, Moesgaard S, Hanioka T, Folkers K. Apparent partial remission ofbreast cancer in ‘high risk’ patients supplemented with nutritional antioxidants,essential fatty acids and coenzyme Q10. Mol Aspects Med 1994;15:s231–40.
[162] Ladas Elena J, Jacobson Judith S, Kennedy Deborah D, Teel Katherine, FleischauerAaron, Kelly Kara M. Antioxidants and cancer therapy: a systematic review. J ClinOncol 2004;22:517–28.
[163] Brownlee M. Biochemistry and molecular cell biology of diabetic complications.Nature 2001;414:813–20.
[164] Maritim AC, Sanders RA, Watkins JB. Diabetes, oxidative stress, and antioxidants: areview. J Biochem Mol Toxicol 2003;17:24–38.
[165] Nishigaki I, Rajendran P, Venugopal R, Ekambaram G, Sakthisekaran D, Nishigaki Y.Cytoprotective role of astaxanthin against glycated protein/iron chelate-inducedtoxicity in human umbilical vein endothelial cells. Phytother Res 2010;24:54–9.
[166] Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res2010;107:1058–70.
[167] Avignon A, Hokayem M, Bisbal C, Lambert K. Dietary antioxidants: do they have arole to play in the ongoing fight against abnormal glucose metabolism? Nutrition2012;28:715–21.
[168] Ceriello A, Morocutti A, Mercuri F, et al. Defective intracellular antioxidantenzyme production in type 1 diabetic patients with nephropathy. Diabetes2000;49:2170–7.
[169] DeRubertis FR, Craven PA, Melhem MF. Acceleration of diabetic renal injury in thesuperoxide dismutase knockout mouse: effects of tempol. Metab Clin Exp2007;56:1256–64.
[170] Hinerfeld D, Traini MD, Weinberger RP, et al. Endogenous mitochondrial oxidativestress: neurodegeneration, proteomic analysis, specific respiratory chain defects,and efficacious antioxidant therapy in superoxide dismutase 2 null mice. JNeurochem 2004;88:657–67.
[171] Nishikawa T, Edelstein D, Du XL, et al. Normalizing mitochondrial superoxide produc-tion blocks three pathways of hyperglycaemic damage. Nature 2000;404:787–90.
[172] Asaba K, Tojo A, Onozato ML, Goto A, Fujita T. Double-edged action of SODmimeticin diabetic nephropathy. J Cardiovasc Pharmacol 2007;49:13–9.
[173] Mollsten A, Marklund SL, Wessman M, et al. A functional polymorphism in themanganese superoxide dismutase gene and diabetic nephropathy. Diabetes2007;56:265–9.
[174] de Haan JB, Stefanovic N, Nikolic-Paterson D, et al. Kidney expression of glutathioneperoxidase-1 is not protective against streptozotocin-induced diabetic nephropa-thy. Am J Physiol Ren Physiol 2005;289:F544–51.
[175] Brezniceanu ML, Liu F, Wei CC, et al. Catalase overexpression attenuatesangiotensinogen expression and apoptosis in diabetic mice. Kidney Int2007;71:912–23.
[176] dos Santos KG, Canani LH, Gross JL, Tschiedel B, PiresSouto KE, Roisenberg I. Thecatalase-262C/T promoter polymorphism and diabetic complications in Caucasianswith type 2 diabetes. Dis Markers 2006;22:355–9.
[177] Kunisaki M, Bursell SE, Clermont AC, et al. Vitamin-E prevents diabetes-induced ab-normal retinal blood-flow via the diacylglycerol-protein kinase-C pathway. Am JPhysiol Endocrinol Metab 1995;269:E239–46.
[178] Mohora M, Greabu M, Muscurel C, Duţa C, Totan A. The sources and the targets ofoxidative stress in the etiology of diabetic complications. Rom J Biophys2007;17:63–84.
[179] Obrosova IG, Fathallah L, Greene DA. Early changes in lipid peroxidation andantioxidativedefense in diabetic rat retina: effect of DL-alpha-lipoic acid. Eur JPharmacol 2000;398:139–46.
[180] Hong JH, KimMJ, Park MR, et al. Effects on vitamin E on oxidative stress and mem-brane fluidity brain of streptozotocin-induced diabetic rats. Clin Chim Acta2004;340:107–15.
[181] Coleman MD, Fernandes S, Khanderia L. A preliminary evaluation of a novel meth-od to monitor a triple antioxidant combination (vitamins E, C and alpha-lipoicacid) in diabetic volunteers using in vitro methaemoglobin formation. EnvironToxicol Pharmacol 2003;14:69–75.
[182] Panjwani U, Yadav DK, Kumar A, Singh SB, Selvamurthy W. Effect of vitamin C andE supplementation in modulating the peripheral nerve conduction following coldexposure in humans. Int J Biometeorol 2003;48:103–7.
[183] Sivan E, Reece EA, Wu YK, Homko CJ, Polansky M, Borenstein M. Dietary vitamin Eprophylaxis and diabetic embryopathy: morphologic and biochemical analysis. AmJ Obstet Gynecol 1996;175:793–9.
[184] Chang TI, Horal M, Jain SK, Wang F, Patel R, Loeken MR. Oxidant regulation of geneexpression and neural tube development: insights gained from diabetic pregnancyon molecular causes of neural tube defects. Diabetologia 2003;46:538–45.
[185] Otero P, Bonet B, Herrera E, Rabano A. Development of atherosclerosis in the dia-betic BALB/c mice—prevention with Vitamin E administration. Atherosclerosis2005;182:259–65.
[186] Hayek T, Kaplan M, Kerry R, AviramM.Macrophage NADPH oxidase activation, im-paired cholesterol fluxes, and increased cholesterol biosynthesis in diabetic mice: astimulatory role for D-glucose. Atherosclerosis 2007;195:277–86.
[187] Reaven PD, Herold DA, Barnett J, Edelman S. Effects of vitamin-E on susceptibility oflow-density-lipoprotein and low-density lipoprotein subfractions to oxidation andon protein glycation in niddm. Diabetes Care 1995;18:807–16.
[188] Salonen JT, Ylaherttuala S, Yamamoto R, et al. Autoantibody against oxidized LDLand progression of carotid atherosclerosis. Lancet 1992;339:883–7.
[189] Bantle JP, Wylie-Rosett J, Albright AL, et al. Nutrition recommendations and inter-ventions for diabetes: a position statement of the American Diabetes Association.Diabetes Care 2008;31(1):S61–78.

346 P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
[190] Landmesser U, Drexler H. General concepts about oxidative stress. Dev CardiovascMed 2006;258:1–15.
[191] Ohara Y, Peterson TE, Harrison DG. Hypercholesterolemia increases endothelial su-peroxide anion production. J Clin Invest 1993;91:2546–51.
[192] Hink U, Li HG, Mollnau H, et al. Mechanisms underlying endothelial dysfunction indiabetes mellitus. Circ Res 2001;88:E14–22.
[193] Victor VM, Rocha M, Sola E, Banuls C, Garcia-Malpartida K, Hernandez-Mijares A.Oxidative stress, endothelial dysfunction and atherosclerosis. Curr Pharm Des2009;15:2988–3002.
[194] Sugamura K, Keaney Jr JF. Reactive oxygen species in cardiovascular disease. FreeRadic Biol Med 2011;51:978–92.
[195] Libby P, Ridker PM, Hansson GK. Leducq Transatlantic Network A. Inflammation inatherosclerosis from pathophysiology to practice. J Am Coll Cardiol 2009;54:2129–38.
[196] Fuhrman B, Volkova N, Aviram M. Oxidative stress increases the expression of theCD36 scavenger receptor and the cellular uptake of oxidized low-density lipopro-tein in macrophages from atherosclerotic mice: protective role of antioxidantsand of paraoxonase. Atherosclerosis 2002;161:307–16.
[197] Shirpoor A, Ansari M-HK, Heshmatian B, et al. Decreased blood pressure with acorresponding decrease in adhesive molecules in diabetic rats caused by vitaminE administration. J Diabetes 2012;4:362–8.
[198] Saeed RW, Peng T, Metz CN. Ascorbic acid blocks the growth inhibitory effect oftumor necrosis factor-alpha on endothelial cells. Exp Biol Med 2003;228:855–65.
[199] Rossig L, Hoffmann J, Hugel B, et al. Vitamin C inhibits endothelial cell apoptosis incongestive heart failure. Circulation 2001;104:2182–7.
[200] Libby P, Aikawa M. Vitamin C, collagen, and cracks in the plaque. Circulation2002;105:1396–8.
[201] Axelrad TW, Deo DD, Ottino P, et al. Platelet-activating factor (PAF) induces activa-tion of matrix metalloproteinase 2 activity and vascular endothelial cell invasionand migration. FASEB J 2004;18:568–70.
[202] Yoshikawa K, Takahashi S, Imamura Y, Sado Y, Hayashi T. Secretion of non-helicalcollagenous polypeptides of alpha 1(IV) and alpha 2(IV) chains upon depletion ofascorbate by cultured human cells. J Biochem 2001;129:929–36.
[203] Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase andatherosclerosis. Arterioscler Thromb Vasc Biol 2004;24:998–1005.
[204] Heller R, Unbehaun A, Schellenberg B, Mayer B, Werner-Felmayer G, Werner ER. L-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabiliza-tion of tetrahydrobiopterin. J Biol Chem 2001;276:40–7.
[205] Gao F, Yao CL, Gao EH, et al. Enhancement of glutathione cardioprotection by ascor-bic acid in myocardial reperfusion injury. J Pharmacol Exp Ther 2002;301:543–50.
[206] Vink H, Constantinescu AA, Spaan JAE. Oxidized lipoproteins degrade the endothe-lial surface layer—implications for platelet-endothelial cell adhesion. Circulation2000;101:1500–2.
[207] Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation ofthe vascular extracellular superoxide dismutase by nitric oxide and exercise train-ing. J Clin Invest 2000;105:1631–9.
[208] Narang D, Sood S, ThomasMK, Dinda AK, Maulik SK. Effect of dietary palm olein oilon oxidative stress associated with ischemic-reperfusion injury in isolated ratheart. BMC Pharmacol 2004;4:29.
[209] Das S, Powell SR,Wang P, et al. Cardioprotectionwith palm tocotrienol: antioxidantactivity of tocotrienol is linked with its ability to stabilize proteasomes. Am JPhysiol Heart Circ Physiol 2005;289:H361–7.
[210] Carlson D,Maass DL,White DJ, Tan J, Horton JW. Antioxidant vitamin therapy alterssepsis-related apoptotic myocardial activity and inflammatory responses. Am JPhysiol Heart Circ Physiol 2006;291:H2779–89.
[211] Tian N, Moore RS, Braddy S, et al. Interactions between oxidative stress and inflam-mation in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol 2007;293:H3388–95.
[212] Papparella I, Ceolotto G, Berto L, et al. Vitamin C prevents zidovudine-inducedNAD(P)H oxidase activation and hypertension in the rat. Cardiovasc Res2007;73:432–8.
[213] Matejikova J, Kucharska J, Pancza D, Ravingerova T. The effect of antioxidant treat-ment and NOS inhibition on the incidence of ischemia-induced arrhythmias in thediabetic rat heart. Physiol Res/Acad Sci Bohem 2008;57:S55–60.
[214] Chu Y, Iida S, Lund DD, et al. Gene transfer of extracellular superoxide dismutasereduces arterial pressure in spontaneously hypertensive rats: role of heparin-binding domain. Circ Res 2003;92:461–8.
[215] Sentman ML, Brannstrom T, Westerlund S, et al. Extracellular superoxidedismutase deficiency and atherosclerosis in mice. Arterioscler Thromb VascBiol 2001;21:1477–82.
[216] Yang H, Roberts LJ, Shi MJ, et al. Retardation of atherosclerosis by overexpression ofcatalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipo-protein E. Circ Res 2004;95:1075–81.
[217] van Empel VP, Bertrand AT, van Oort RJ, et al. EUK-8, a superoxide dismutase andcatalase mimetic, reduces cardiac oxidative stress and ameliorates pressureoverload-induced heart failure in the harlequin mouse mutant. J Am Coll Cardiol2006;48:824–32.
[218] Blankenberg S, Rupprecht HJ, Bickel C, et al. Glutathione peroxidase 1 activity andcardiovascular events in patients with coronary artery disease. N Engl J Med2003;349:1605–13.
[219] Forgione MA, Weiss N, Heydrick S, et al. Cellular glutathione peroxidase defi-ciency and endothelial dysfunction. Am J Physiol Heart Circ Physiol 2002;282:H1255–61.
[220] Kisucka J, Chauhan AK, Patten IS, et al. Peroxiredoxin1 prevents excessiveendothelial activation and early atherosclerosis. Circ Res 2008;103:598–605.
[221] Choi MH, Lee IK, Kim GW, et al. Regulation of PDGF signalling and vascular remod-elling by peroxiredoxin II. Nature 2005;435:347–53.
[222] Park JG, Yoo JY, Jeong SJ, et al. Peroxiredoxin 2 deficiency exacerbates atherosclero-sis in apolipoprotein E-deficient mice. Circ Res 2011;109:739–49.
[223] Guo X, Yamada S, Tanimoto A, et al. Overexpression of peroxiredoxin 4 attenuatesatherosclerosis in apolipoprotein E knockout mice. Antioxid Redox Signal2012;17:1362–75.
[224] Wei Y, Liu XM, Peyton KJ, et al. Hypochlorous acid-induced heme oxygenase-1gene expression promotes human endothelial cell survival. Am J Physiol CellPhysiol 2009;297:C907–15.
[225] Li C, Hossieny P,Wu BJ, Qawasmeh A, Beck K, Stocker R. Pharmacologic induction ofheme oxygenase-1. Antioxid Redox Signal 2007;9:2227–39.
[226] Gilgun-Sherki Y, Melamed E, Offen D. Oxidative stress induced-neurodegenerativediseases: the need for antioxidants that penetrate the blood brain barrier.Neuropharmacology 2001;40:959–75.
[227] Salganik RI. The benefits and hazards of antioxidants: controlling apoptosis andother protective mechanisms in cancer patients and the human population. J AmColl Nutr 2001;20:464S–72S.
[228] Ho YS, Xiong Y, MaWC, Spector A, Ho DS. Mice lacking catalase develop normally butshow differential sensitivity to oxidant tissue injury. J Biol Chem 2004;279:32804–12.
[229] Meister A. Mitochondrial changes associated with glutathione deficiency. BiochimBiophys Acta Mol Basis Dis 1995;1271:35–42.
[230] DarleyUsmar V, Halliwell B. Blood radicals—reactive nitrogen species, reactiveoxygen species, transition metal ions, and the vascular system. Pharm Res1996;13:649–62.
[231] Hansen JM, Go YM, Jones DP. Nuclear and mitochondrial compartmentation ofoxidative stress and redox signaling. Annu Rev Pharmacol Toxicol 2006:215–34.
[232] Schulz JB, Lindenau J, Seyfried J, Dichgans J. Glutathione, oxidative stress andneurodegeneration. Eur J Biochem 2000;267:4904–11.
[233] Sies H. Glutathione and its role in cellular functions. Free Radic Biol Med1999;27:916–21.
[234] Meister A, Anderson ME. Glutathione. Annu Rev Biochem 1983;52:711–60.[235] Sultana R, Butterfield DA. Oxidatively modified GST and MRP1 in Alzheimer's dis-
ease brain: implications for accumulation of reactive lipid peroxidation products.Neurochem Res 2004;29:2215–20.
[236] Renes J, De Vries EEG, Hooiveld G, Krikken I, Jansen PLM, Muller M. Multidrug re-sistance protein MRP1 protects against the toxicity of the major lipid peroxidationproduct 4-hydroxynonenal. Biochem J 2000;350:555–61.
[237] Nies AT, Jedlitschky G, Konig J, et al. Expression and immunolocalization ofthe multidrug resistance proteins, Mrp1–Mrp6 (ABCC1–ABCC6), in human brain.Neuroscience 2004;129:349–60.
[238] Conseil G, Deeley RG, Cole SPC. Polymorphisms of MRP1 (ABCC1) and related ATP-dependent drug transporters. Pharmacogenet Genomics 2005;15:523–33.
[239] Lovell MA, Xie C, Markesbery WR. Decreased glutathione transferase activity inbrain and ventricular fluid in Alzheimer's disease. Neurology 1998;51:1562–6.
[240] Schipper HM, Bennett DA, Liberman A, et al. Glial heme oxygenase-1 expression inAlzheimer disease and mild cognitive impairment. Neurobiol Aging 2006;27:252–61.
[241] Raina AK, Templeton DJ, Deak JC, Perry G, Smith MA. Quinone reductase (NQO1), asensitive redox indicator, is increased in Alzheimer's disease. Redox Rep 1999;4:23–7.
[242] Ramsey CP, Glass CA, Montgomery MB, et al. Expression of Nrf2 in neurodegener-ative diseases. J Neuropathol Exp Neurol 2007;66:75–85.
[243] Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage inAlzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med2001;7:548–54.
[244] MarkesberyWR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic BiolMed 1997;23:134–47.
[245] Lovell MA, Xie C, Markesbery WR. Acrolein is increased in Alzheimer's diseasebrain and is toxic to primary hippocampal cultures. Neurobiol Aging2001;22:187–94.
[246] Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation,is increased in the brain in Alzheimer's disease. Neurobiol Aging 1998;19:33–6.
[247] Lauderback CM, Hackett JM, Huang FF, et al. The glial glutamate transporter, GLT-1,is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain:the role of A beta 1-42. J Neurochem 2001;78:413–6.
[248] Kanninen K, Malm TM, Jyrkkanen H-K, et al. Nuclear factor erythroid 2-related fac-tor 2 protects against beta amyloid. Mol Cell Neurosci 2008;39:302–13.
[249] Resende R, Moreira PI, Proenca T, et al. Brain oxidative stress in a triple-transgenicmouse model of Alzheimer disease. Free Radic Biol Med 2008;44:2051–7.
[250] Martinez RCR, Hamani C, de Carvalho MC, et al. Intraoperative dopamine releaseduring globuspallidusinternus stimulation in Parkinson's disease. Mov Disord2013;28:2027–31.
[251] Passamonti L, Salsone M, Toschi N, et al. Dopamine-transporter levels drive striatalresponses to apomorphine in Parkinson's disease. Brain Behav 2013;3:249–62.
[252] Agid Y, Ruberg M, Javoy-Agid F, et al. Are dopaminergic neurons selectively vulner-able to Parkinson's disease? In: Narabayashi H, Nagatsu T, Yanagisawa N, MizunoY, editors. Advances in Neurology; Parkinson's Disease: From basic research totreatment; 1993. p. 148–64.
[253] SmeyneM, Smeyne RJ. Glutathione metabolism and Parkinson's disease. Free RadicBiol Med 2013;62:13–25.
[254] Smith TS, Parker WD, Bennett JP. L-dopa increases nigral production of hydroxylradicals in-vivo—potential l-dopa toxicity. Neuroreport 1994;5:1009–11.
[255] Wick MM, Byers L, Frei E. L-dopa—selective toxicity for melanoma cells invitro.Science 1977;197:468–9.
[256] Hunot S, Brugg B, Ricard D, et al. Nuclear translocation of NF-kappa B is increased indopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A1997;94:7531–6.
[257] Blum D, Torch S, Nissou MF, Verna JM. 6-Hydroxydopamine-induced nuclearfactor-kappaB activation in PC12 cells. Biochem Pharmacol 2001;62:473–81.

347P. Rajendran et al. / Clinica Chimica Acta 436 (2014) 332–347
[258] Levites Y, Youdim MBH, Maor G, Mandel S. Attenuation of 6-hydroxydopamine(6-OHDA)-induced nuclear factor-kappaB (NF-kappa B) activation and cell deathby tea extracts in neuronal cultures. Biochem Pharmacol 2002;63:21–9.
[259] Rajendran P, Ekambaram G, Sakthisekaran D. Cytoprotective effect of mangiferinon benzo(a)pyrene-induced lung carcinogenesis in Swiss albino mice. Basic ClinPharmacol Toxicol 2008;103:137–42.
[260] Ganapathy E, Peramaiyan R, Rajasekaran D, VenkataramanM, Dhanapal S. Modula-tory effect of naringenin on N-methyl-N′-nitro-N-nitrosoguanidine- and saturatedsodium chloride-induced gastric carcinogenesis in male Wistar rats. Clin ExpPharmacol Physiol 2008;35:1190–6.
[261] Rajendran P, Li F, Manu KA, et al. Gamma-Tocotrienol is a novel inhibitor of consti-tutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma:
potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. BrJ Pharmacol 2011;163:283–98.
[262] Rengarajan T, Rajendran P, Nandakumar N, Balasubramanian MP, Nishigaki I.Cancer preventive efficacy of marine carotenoid fucoxanthin: cell cycle arrestand apoptosis. Nutrients 2013;5:4978–89.
[263] Nandakumar N, Rengarajan T, Balamurugan A, Balasubramanian MP. Modulatingeffects of hesperidin on key carbohydrate-metabolizing enzymes, lipid profile,and mbrane-bound adenosine triphosphatases against 7,12-dimethylbenz(a)an-thracene-induced breast carcinogenesis. Hum Exp Toxicol 2013. http://dx.doi.org/10.1177/0960327113485252.
Copyright © 2022 FDOKUMEN




















