
Epidemiologic Trends of Rabies in Domestic Animals in Southern Thailand, 1994-2008

� 2008 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 146A:861–872 (2008)
The Italian National Survey for Prader–Willi Syndrome:An Epidemiologic Study
Graziano Grugni,1* Antonino Crino,2 Laura Bosio,3 Andrea Corrias,4 Marina Cuttini,2
Teresa De Toni,5 Eliana Di Battista,5 Adriana Franzese,6 Luigi Gargantini,7 Nella Greggio,8
Lorenzo Iughetti,9 Chiara Livieri,10 Arturo Naselli,5 Claudio Pagano,8 Giovanni Pozzan,8
Letizia Ragusa,11 Alessandro Salvatoni,12 Giuliana Trifiro,13 Luciano Beccaria,14 Maria Bellizzi,15
Jaele Bellone,4 Amelia Brunani,1 Marco Cappa,2 Gabriella Caselli,9 Valeria Cerioni,3
Maurizio Delvecchio,16 Daniela Giardino,1 Francesco Iannı,17 Luigi Memo,18 Alba Pilotta,19
Cristoforo Pomara,20 Giorgio Radetti,21 Michele Sacco,22 Annarosa Sanzari,23 Alessandro Sartorio,1
Giorgio Tonini,24 Roberto Vettor,8 Federico Zaglia,25 and Giuseppe Chiumello3 on behalf of theGenetic Obesity Study Group of the Italian Society of Pediatric Endocrinology
and Diabetology (ISPED)1Italian Auxological Institute Foundation, Research Institute, Verbania, Italy2Bambino Gesu Children’s Hospital, Research Institute, Palidoro-Rome, Italy
3S. Raffaele Hospital, Research Institute, Milan, Italy4University of Turin, Turin, Italy
5University of Genoa, Genoa, Italy6University of Naples, Naples, Italy
7Civic Hospital of Treviglio (BG), Treviglio, Italy8University of Padua, Padua, Italy
9University of Modena and Reggio Emilia, Modena and Reggio Emilia, Italy10University of Pavia, Pavia, Italy
11Oasi Maria S.S., Research Institute, Troina (EN), Italy12University of Varese, Varese, Italy13S. Giuseppe Hospital, Milan, Italy14A. Manzoni Hospital, Lecco, Italy
15Civic Hospital of Trento, Trento, Italy16University of Bari, Bari, Italy
17Cervello Hospital, Palermo, Italy18Civic Hospital of Treviso, Treviso, Italy
19University of Brescia, Brescia, Italy20University of Foggia, Foggia, Italy
21Regional Hospital of Bolzano, Bolzano, Italy22Casa Sollievo della Sofferenza, Research Institute, S. Giovanni Rotondo (FG), Italy
23S. Anna Hospital, Como, Italy24Burlo Garofalo, Research Institute, Trieste, Italy
25Major Civic Hospital, Verona, Italy
Received 3 March 2007; Accepted 20 August 2007
Twenty-five medical centers and the Prader–Willi Syndrome(PWS) Association collaborated on a study which attemptedto identify all people with genetically confirmed diagnosisof PWS living in Italy. Investigators of the participatingcenters contacted PWS subjects and/or their family, filled in aspecially developed form with the required data andforwarded this information by email. The study identified425 subjects (209 males and 216 females, between the ages of
0.4–46.7). Two hundred thirty-eight patients had del15,104 had UPD15, 4 demonstrated a translocation affectingchromosome 15 and 79 showed a positive methylationtest. There were fewer subjects found over the age of 35,probably due to the low rate of identification of older PWSpatients as well as the high mortality rate. There were agreater number of male children and adolescents withPWS whilst, amongst adults, there were more females. As
*Correspondence to: Graziano Grugni, M.D., Ph.D., Division ofAuxology, S. Giuseppe Hospital, Research Institute, Italian AuxologicalInstitute Foundation, PO Box 1, 28921 Verbania, Italy.E-mail: [email protected]
DOI 10.1002/ajmg.a.32133

expected, the majority of subjects with PWS were obese,especially in adult life. Nevertheless, it is noteworthy that26% of patients aged between 6 and 17 were normal weight.A total of 212 subjects had received GH treatment, of which141 were still receiving therapy, while the remaining 71 hadstopped. In children and adolescents (233 cases), 89 subjectshad never undergone GH therapy. Eighteen PWS patientshad died in the past 20 years. Obesity-related cardiovascularand respiratory diseases were the cause of death, bothduring childhood and after 18 years of age. Three childrendied suddenly whilst undergoing GH therapy. Respiratory
infection and cardiac illness were the causes of death intwo cases. There was no definitive cause of death foundin the third case. Overall, there was no increase in numberof deaths during GH treatment, suggesting that GH adminis-tration in patients with PWS, as a group, does not increasethe risk of death. � 2008 Wiley-Liss, Inc.
Key words: Prader–Willi syndrome; obesity; GH therapy;death
How to cite this article: Grugni G, Crino A, Bosio L, Corrias A, Cuttini M, De Toni T, Di Battista E, Franzese A,Gargantini L, Greggio N, Iughetti L, Livieri C, Naselli A, Pagano C, Pozzan G, Ragusa L, Salvatoni A,Trifiro G, Beccaria L, Bellizzi M, Bellone J, Brunani A, Cappa M, Caselli G, Cerioni V, Delvecchio M,
Giardino D, Iannı F, Memo L, Pilotta A, Pomara C, Radetti G, Sacco M, Sanzari A, Sartorio A, Tonini G,Vettor R, Zaglia F, Chiumello G on behalf of the Genetic Obesity Study Group of the Italian Society of Pediatric
Endocrinology and Diabetology (ISPED). 2008. The Italian National Survey for Prader–Willisyndrome: An epidemiologic study. Am J Med Genet Part A 146A:861–872.
INTRODUCTION
Prader–Willi syndrome (PWS) is a complex multi-system disorder due to the absent expression of thepaternally active genes in the PWS critical region onchromosome 15 [Bittel and Butler, 2005]. In approx-imately 70–75% of affected individuals there is adeletion of the paternal chromosome 15q11-q13(del15) [Buiting and Horsthemke, 2006]. Most ofthe remaining subjects have a maternal uniparentaldisomy for chromosome 15 (UPD15). A small per-centage of patients may have abnormalities ofthe imprinting center or translocations involvingchromosome 15. Many features of PWS reflecthypothalamic dysfunction, including compulsiveappetite, body temperature instability, a high painthreshold, sleep disorders, hypogonadism andaltered GH secretion [Goldstone, 2004]. The syn-drome has two phases. Initially, PWS is characterizedby severe neonatal hypotonia, feeding problems anda failure to thrive. This is followed by hyperphagiaand weight gain between the ages of 1 and 6, leadingmost PWS patients to develop morbid obesity[Gunay-Aygun et al., 2001; McCandless and Cassidy,2006]. The complications associated with obesity arethe main risk factors for death in subjects withPWS [Einfeld et al., 2006]. This has prompted manyinvestigators to recommend strict control on calorieintake in order to improve life expectancy [Butler,2006].However, there is an increasing awareness thatpoor health outcomes for PWS subjects may notbe caused by obesity alone [Priano et al., 2006]. It iscurrently unclear whether risk of critical illnesses forPWS subjects is influenced by GH deficiency (GHD).A GH/IGF-I-mediated control of cardiac risk inPWS has recently been reported [Marzullo et al.,2005]. In this context, reducedGH secretion has beendocumented in both children and adults with PWS[Burman et al., 2001; Hoybye, 2004; Carrel et al.,
2006]. Therefore, GH therapy has been approved bythe member states of the European Union for thelong-term treatment of growth failure in childrenwith PWS. A large number of studies have shownthat GH treatment improves linear growth, finalheight, physical strength and agility and pulmonaryfunction in children with PWS [Allen and Carrel,2004]. Nevertheless, there have been fatal casesduring the first months of GH administration inyoung patients with PWS [Eiholzer, 2005; Craig et al.,2006]. Information regarding GH therapy in adultswith PWS is also beginning to emerge [Hoybye,2007], but further studies are needed to better clarifyits role in adulthood.
Based on these data, the main objectives of thisstudy were: (1) to identify patients with PWS in Italy,(2) to measure the prevalence of overweight andobesity, (3) to assess the use of GH treatment, (4) todetect the number and the causes of death, and (5) toexplore the possible relationships between deathand GH therapy.
PATIENTS AND METHODS
In collaboration with the Italian PWS Associationand in a national collaborative study, all known casesof genetically confirmed PWS were collected. Datawere obtained by 25 medical centers for pediatricand adult patients of the Italian Network for RareDiseases (Italian National Survey for PWS: INSUP),including hospital inpatient services, endocrineclinics, genetic centers and intensive care units. Foreach center, information was collected by retro-spectively reviewing all medical charts of subjectsdiagnosed with PWS and followed between January1986 and June 2006 inclusive. Data were comparedto that obtained through a retrospective review of allrecords of patients registered at the Italian PWS
862 GRUGNI ET AL.
American Journal of Medical Genetics Part A

Association. During the active period of thestudy, between January and June 2006, investigatorsfrom the participating centers re-contacted allpatients and/or their family. For the purpose of thestudy, a form was developed and made availablethrough e-mail. Clinicians had to report initials,gender, and date of birth of patients, in order topreserve anonymity as well as to avoid doublecounting. Required data included a test confirming agenetic alteration on the 15q11-13 region, length/height, weight and body mass index [BMI¼weight (kg)/height2 (m)], presence or absence ofGH therapy, dosage and duration of GH treatment,vital status (alive or dead) at June 30, 2006 and, fordeceased patients, age and cause of death. Variousgenetic tests for PWS have been performed in16 participating centers: (a) chromosome studieswere performed on peripheral blood lympho-cytes using banding techniques to investigatestructural alterations; (b) fluorescence in situ hybrid-ization (FISH) was used to detect microdeletions,with commercially available probes coveringthe 15q11-13 PWS critical region, according tothe manufacturers protocols; (c) the methylationpattern analysis of the PWS region was carried out todetect deletions, UPD and IC defects, according tostandard diagnostic protocols for Southern blotand bisulphite methylation polymerase chain reac-tion (PCR) [Chotai and Payne, 1998; Clayton-Smithand Laan, 2003]; (d) microsatellite analysis wasapplied to detect UPD, with markers mapped tosegment 15q11-14. Supine length or standingheight was determined by a Harpenden Stadiometer(Holtain Limited, Crymych, Dyfed, UK). Body weightwas measured to the nearest 0.1 kg, by usingstandardized equipment. The Centers for DiseaseControl and Prevention Growth Charts were used tocalculate weight-for-length centiles in infants frombirth to 24 months [Kuczmarski et al., 2002]. Over-weight and obesity were established at weight-for-length >85th centile and >95th centile, respec-tively. The published international standards forsex-and age-specific BMI centiles were used forsubjects aged 2–18 years [Cole et al., 2000]. Wedefined as overweight and obese those patientswith a BMI more than the centile curves, that at age18 passed through the cut-off point of 25 and30 for adult overweight and obesity, respectively.In addition, BMI Standard Deviation Score (BMI SDS)of subjects younger than 18 years was derivedfrom population standards [Cole et al., 1995]. Finally,the BMI cut-off points of 25–30 to define over-weight and >30 to define obesity were used forindividuals >18 years of age. Data on causes ofdeath were obtained from the medical files.The study protocol was approved by the ad hocEthical Committee of the Italian Auxological InstituteFoundation.
Statistical Analysis
Data are expressed as mean� SE. The Student’s t-test or analysis of variance was performed fornormally distributed values. Otherwise, a non-para-metric test was used. Significance was set at P< 0.05.
Proportions were computed for categorical varia-bles, and means and medians for continuous ones,together with appropriate variability measures and95% confidence intervals (CI). The probability ofsurvival and 95% confidence interval (CI) aroundthe survival curve were estimated according to theKaplan–Meier method [Kaplan and Meier, 1958].Survival was measured from birth to the end ofdata collection on the 30th June 2006, or to whendeath occurred. All deaths were counted as failures,whether or not they were disease-related. Compar-ison of survival among different sub-groups ofpatients was made using the log-rank test. Multi-variate Cox proportional hazards analysis [Coxand Oakes, 1984] was used to explore the factorspotentially associated with survival. Variables in-cluded in the model were gender, year of birth,genetic subtype, presence of overweight andobesity, GH treatment and duration.
Statistical analysis was carried out using theStata software, version 9.0 (StataCorp. 2005, StataStatistical Software: Release 9.0. StataCorp., CollegeStation, TX).
RESULTS
Population Data
Four hundred twenty-five subjects with PWS,209 males and 216 females, were identified.Four hundred eight individuals were registered inthe Italian PWS Association, while 17 families didnot enter the patient support society. Of the total of425 patients, 177 were cared for in 10 centers innorthwest Italy, 72 in seven centers in northeastItaly and 176 in 8 centers in the center and south. Allbut nine patients were Caucasian. Five subjectsoriginated from North Africa, two from India, onefrom Taiwan and one from the Philippines. Therewasonepair of identical twins (males). Four hundredseven subjects were still alive as of June 30, 2006.
Genetic mechanisms are reported in Table I.A positive methylation test was found in 79 patients,but the underlying genetic defect was not identified.Of the 346 subjects who performed FISH and/ormicrosatellite analysis, 68.8% had a del15, 30.05%had a UPD15 and 1.15% had a de novo transloca-tion involving chromosome 15. Of the latter, twocases with a nonreciprocal translocation leadingto 15pter-q13 monosomy have previously beendescribed [Rivera et al., 1990]. The remainingtwo patients showed a 45,XY,der(9)t(9;15)(q34,q12),-15 and a 45,XX,der(15)t(15;20)(q12;q13.3),
ITALIAN NATIONAL SURVEY FOR PRADER–WILLI SYNDROME 863
American Journal of Medical Genetics Part A
-15 karyotype, respectively. A slightly higherproportion of UPD15 was detected in children under6 years of age (21 cases, 36.8%). There were no casesof imprinting defect found.
In our population, ages ranged from 0.4 to46.7 years (mean� SE: 17.2� 0.53 years). Table Ishows the distribution of subjects, grouped in6-year intervals. There were 233 patients (54.8%)younger than 18 years whereas there were 192 adults(45.2%). The median age was 13.7 years (range0.5–45.4 years) for males, and 19.0 years (range0.4–46.7 years) for females. One hundred thirty(62.2%) of the males and 103 (47.7%) of thefemales were below 18 completed years of age. Onthe contrary, data from the older age groupshows a prevalence of female subjects (males/females: 79/113). The difference of age distributionbetween males and females was statistically signi-ficant (P¼ 0.03). In terms of genetic mechanisms,age was similar in subjects with del15 and inindividuals with UPD15 (18.5� 0.7 years vs. 16.6�1.0 years, respectively).
Weight
Weight-for-length, height, weight, and BMI datawere available for 342 subjects (Table I). Onehundred seventy-one patients were younger than18 years of age (96 males). In this group 36 (21.1%,19 males) and 69 (40.3%, 40 males) subjects wereoverweight and obese, respectively. Sixty-twopatients were normal weight (36.3%, 35 males) and4 individuals <2 years of age were underweight
(¼weight-for-length <3rd centile; 2.3%, 2 males).BMI SDS ranged from �3.4 to 15.1 (mean� SE:3.65� 0.32). No differences were detected betweenmales and females (3.60� 0.43 vs. 3.69� 0.48:P¼ 0.8). BMI SDS was similar in subjects with del15and UPD15 (del15: 3.55� 0.42, UPD15: 3.37� 0.63;P¼ 0.8).
BMI data were available for 171 adults with PWS(71 males). One hundred forty-five subjects wereobese (84.8%, 56 males), whereas 22 individuals wereoverweight (12.9%, 13males).Only fouradult patientshad normal BMI (2.3%, 2 males). Mean BMI was41.2� 0.95 (range 20.7–72.2). BMI was similar in thetwo sexes (males: 39.8� 1.44, females: 42.3� 1.25;P¼ 0.2), as well as in patients with del15 and UPD15(del15: 40.6� 1.16, UPD15: 42.4� 1.94; P¼ 0.4).
Overall, the proportion of obesity was 62.6% (95%CI 57.2–67.7%). The difference between males(57.5%; 95%CI 49.6–65.1%) and females (67.4%,95% CI 59.9–74.3%) was only marginally significantand disappeared after adjusting for age. The pro-portion of overweight was 19.2% (95% CI 13.5–26.0)and 14.9% (95% CI 9.9–21.0%) in males and females,respectively. In addition, the proportions of obesityand overweight were similar in PWS patients withdifferent genotypes (data not shown).
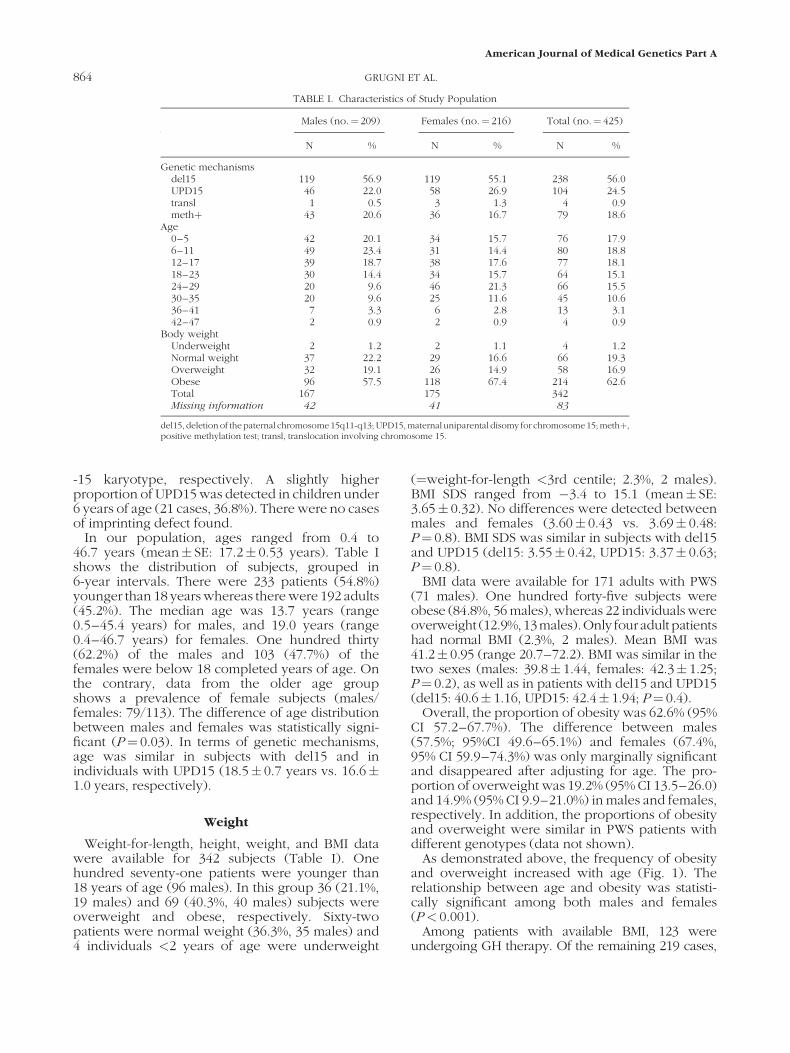
As demonstrated above, the frequency of obesityand overweight increased with age (Fig. 1). Therelationship between age and obesity was statisti-cally significant among both males and females(P< 0.001).
Among patients with available BMI, 123 wereundergoing GH therapy. Of the remaining 219 cases,
TABLE I. Characteristics of Study Population
Males (no.¼ 209) Females (no.¼ 216) Total (no.¼ 425)
N % N % N %
Genetic mechanismsdel15 119 56.9 119 55.1 238 56.0UPD15 46 22.0 58 26.9 104 24.5transl 1 0.5 3 1.3 4 0.9methþ 43 20.6 36 16.7 79 18.6
Age0–5 42 20.1 34 15.7 76 17.96–11 49 23.4 31 14.4 80 18.812–17 39 18.7 38 17.6 77 18.118–23 30 14.4 34 15.7 64 15.124–29 20 9.6 46 21.3 66 15.530–35 20 9.6 25 11.6 45 10.636–41 7 3.3 6 2.8 13 3.142–47 2 0.9 2 0.9 4 0.9
Body weightUnderweight 2 1.2 2 1.1 4 1.2Normal weight 37 22.2 29 16.6 66 19.3Overweight 32 19.1 26 14.9 58 16.9Obese 96 57.5 118 67.4 214 62.6Total 167 175 342Missing information 42 41 83
del15, deletion of thepaternal chromosome15q11-q13; UPD15,maternal uniparental disomy for chromosome15; methþ,positive methylation test; transl, translocation involving chromosome 15.
864 GRUGNI ET AL.
American Journal of Medical Genetics Part A
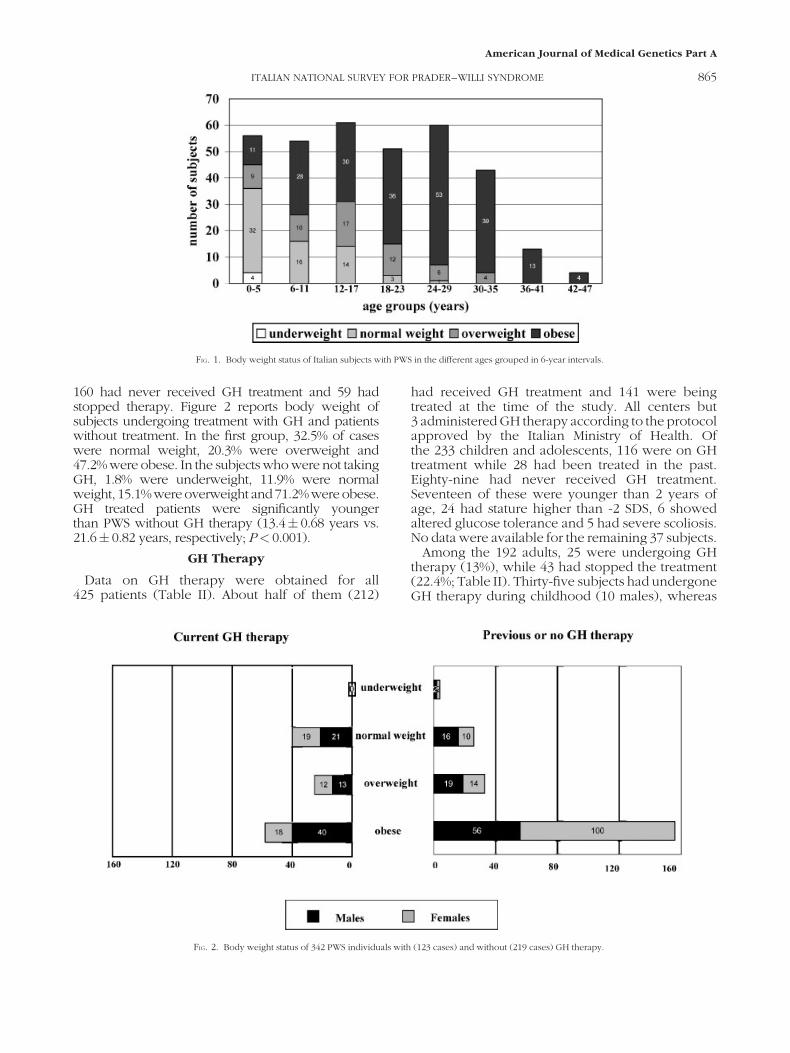
160 had never received GH treatment and 59 hadstopped therapy. Figure 2 reports body weight ofsubjects undergoing treatment with GH and patientswithout treatment. In the first group, 32.5% of caseswere normal weight, 20.3% were overweight and47.2% were obese. In the subjects who were not takingGH, 1.8% were underweight, 11.9% were normalweight, 15.1%wereoverweight and71.2%wereobese.GH treated patients were significantly youngerthan PWS without GH therapy (13.4� 0.68 years vs.21.6� 0.82 years, respectively; P< 0.001).
GH Therapy
Data on GH therapy were obtained for all425 patients (Table II). About half of them (212)
had received GH treatment and 141 were beingtreated at the time of the study. All centers but3 administered GH therapy according to the protocolapproved by the Italian Ministry of Health. Ofthe 233 children and adolescents, 116 were on GHtreatment while 28 had been treated in the past.Eighty-nine had never received GH treatment.Seventeen of these were younger than 2 years ofage, 24 had stature higher than -2 SDS, 6 showedaltered glucose tolerance and 5 had severe scoliosis.No data were available for the remaining 37 subjects.
Among the 192 adults, 25 were undergoing GHtherapy (13%), while 43 had stopped the treatment(22.4%; Table II). Thirty-five subjects had undergoneGH therapy during childhood (10 males), whereas
FIG. 1. Body weight status of Italian subjects with PWS in the different ages grouped in 6-year intervals.
FIG. 2. Body weight status of 342 PWS individuals with (123 cases) and without (219 cases) GH therapy.
ITALIAN NATIONAL SURVEY FOR PRADER–WILLI SYNDROME 865
American Journal of Medical Genetics Part A

17 (12 males) were treated both during childhoodand after 18 years of age. Furthermore, 16 cases (7males) received GH treatment only in adulthood. All68 adults treated with GH had reduced GH responsesto the standard stimulation tests.
One hundred twenty-five out of the 238 patientswith del15 had undergone GH therapy (52.5%).A very similar percentage was observed in subjectswith UPD15 (52.9%).
The duration of treatment ranged between 2 and169 months. Among currently treated patients, themean duration was 52� 2 months in subjectsyounger than 18 years (range 2–129 months) and41� 4 months in adults (range 6–169 months). Forpatients who stopped therapy, GH administrationwas interrupted after 34� 3 months in children and
adolescents (range 6–84 months) and 24� 6 monthsin adults (range 6–103 months).
The median dose of GH was 0.196 mg/kg/week(range 0.11–0.43 mg/kg/week) in children andadolescents, and 0.044 mg/kg/week (range 0.011–0.079 mg/kg/week) in adults.
Deaths
Eighteen patients died in the 20-year periodcovered by the study. Deaths occurred in 12 out ofthe 25 participating centers. Table III shows clinicalcharacteristics and causes of death. Eleven subjectshad a del15, 5 had a UPD15 and 2 showed a positivemethylation test. Among the 6 cases under theage of 18 (4 males, range 0.5–17.1 years), 3 children(2 males) died while receiving GH treatment[Grugni et al., 2005; Sacco and Di Giorgio, 2005]. Allthese patients were obese and died during thefirst months of therapy. Bronchopneumonia wassuspected in one case, while the 3.8-year-old girlwas well the night before her death. A postmortemwas performed on the remaining subject (Patient 3)and the cause of death was most likely cardiac[Pomara et al., 2005]. From the other 3 cases under18 years of age, who had never undergone GHtherapy, two males were severely obese. One diedfrom acute cardiorespiratory failure and the otherfrom bacterial bronchopneumonia. Following anepisodeof febrile gastroenteritis, one girl of 6monthsunexpectedly died with severe metabolic acidosis,acute massive hemolysis and intractable hyper-kalemia [Zaglia et al., 2005]. However, followingautopsy a well defined cause of death could not beestablished.
TABLE II. GH Therapy in the Study Population
Males Females Total
N % N % N %
PWS< 18 years 130 103 233Previous 14 10.8 14 13.6 28 12.0Current 63 48.4 53 51.5 116 49.8Total 77 59.2 67 65.1 144 61.8Never 53 40.8 36 34.9 89 38.2
PWS� 18 years 79 113 192Previous 12 15.2 31 27.4 43 22.4Current 17 21.5 8 7.1 25 13.0Total 29 36.7 39 34.5 68 35.4Never 50 63.3 74 65.5 124 64.6
Total PWS 209 216 425Previous 26 12.4 45 20.8 71 16.7Current 80 38.3 61 28.3 141 33.2Total 106 50.7 106 49.1 212 49.9Never 103 49.3 110 50.9 213 50.1
TABLE III. Clinical Findings and Causes of Death of PWS Patients
Patients SexGenetic
mechanisms BMIAge at death
(years) GH therapy Cause of death
1 F del15 NO 0.5 No Refractory metabolic acidosis and massive hemolysis2 F del15 22.9 3.8 Yes Unexplained3 M UPD 28.0 3.9 Yes Myocardial contraction band necrosis linked with ventricular fibrillation4 M UPD 22.6 6.3 Yes (?) Bronchopneumonia5 M methþ 46.2 16.0 No Bronchopneumonia6 M del15 52.4 17.1 No Cardiorespiratory failure7 M del15 52.4 18.5 No Cardiac failure8 M UPD 28.2 21.9 No Unexplained9 M del15 47.5 22.3 No Bronchopneumonia10 M del15 72.2 23.8 No Cardiorespiratory failure11 M del15 36.2 26.1 prev Cerebral vascular accident12 F UPD 60.3 28.5 No Gastric perforation following intragastric balloon insertion13 M methþ 50.7 29.6 No Cardiac failure14 F del15 71.9 32.1 No Cardiac arrest15 F UPD 52.8 34.0 No Cardiorespiratory failure16 M del15 36.8 34.4 No Cardiorespiratory failure17 F del15 54.6 36.1 No Myocardial infarction18 M del15 49.9 39.3 No Choked on food
F, female; M, male; del15, deletion of the paternal chromosome 15q11-q13; UPD15, maternal uniparental disomy for chromosome 15; methþ, positive methylation test;BMI, body mass index (kg/m2); NO, not obese; prev, patient previously treated with GH.
866 GRUGNI ET AL.
American Journal of Medical Genetics Part A

Twelve subjects died after reaching the age of18 (8 males, range 18.5–39.3 years), 11 of whom hadnever received GH therapy. One male had under-gone GH treatment during childhood but had beenwithdrawn 8 years before his death. All patients hadmorbid obesity with the exception of one male whowas overweight. Autopsy was not carried out on anyof the adults with PWS. However, in the majorityof the patients (9/12 cases) death was associatedwith complications conventionally related toobesity,such as cardiopulmonary disorders and arterialhypertension. A very obese female died from theconsequences of intragastric balloon insertion, whilea 39.3-year-old male choked after ingestion of rawmeat. Finally, in the case of one overweight patient,cause of death was not identified.
Risk of Death
Overall survival of PWS patients and 95% CI isshown in Figure 3a. The frequency of death duringGH therapy was 1.41% (3/212). Fifteen out of213 PWS patients who were not on GH treatmentdied (7.04%). Taking into consideration only thosepatients under 18 years of age, 3 out of 144 subjectsdied during GH therapy (2.08%) and there were3 deaths among the 89 subjects who had neverreceived GH treatment (3.37%). Survival withrespect to GH treatment (any versus never) isreported in Figure 3b. Patients who had neverreceived treatment tended to be older, whichexplains their longer curve. However, no differenceof survival according to GH therapy was found(log-rank test 0.67, P¼ 0.413).
The frequency of death in males and femaleswas 5.74% (12/209) and 2.77% (6/216), respectively.As far as genetic mechanisms are concerned, theoccurrence of death was very similar for bothgenotypes (del15: 11/238¼ 4.62%; UPD15: 5/104¼4.80%).
When the effect of GH therapy, obesity, geneticdefect and gender were considered together in amultivariate Cox model, only gender had a margin-ally significant relationship with survival (P< 0.05),with a decreased risk of death for females comparedto men (OR 0.38, 95% CI 0.14–1.02).
DISCUSSION
Prader–Willi syndrome is the single most com-monly known genetic cause of obesity. It has beenestimated to have a population prevalence of about1:10,000 to 1:52,000 [Whittington et al., 2001]. Morerecently, a prevalence varying from 1:49,911 up to1:91,802 was reported [Vogels et al., 2004]. Takinginto consideration the Italian population (approxi-mately 57 million people), our identification of425 subjects with PWS leads us to believe that thereare a significant number of undiagnosed PWS cases,
rather than a lower minimum prevalence (1:134,117). This potential source of error is likely related toolder patients, born before the syndrome was wellcharacterized and in whom the diagnosis was neversuspected. In addition, the high rate of mortalityabove age 30 [Whittington et al., 2001] might havecontributed to the low identification rate for olderPWS subjects. This would explain why, in ourpopulation, there were few subjects over the age of35 (17 cases, 4%).
In terms of genetic mechanisms, the percentages ofdifferent genetic abnormalities found in this reportare in accordance with the literature [Gunay-Aygunet al., 2001]. On the other hand, a greater proportionof patients with UPD15 has recently been reportedin young children with PWS, likely due to theincreasing maternal age at conception [Whittingtonet al., 2007]. In this respect, the percentage of UPD15was higher than that which would have beennormally expected in our patients aged 0–5 years(36.8%), but data about maternal age was notincluded in our database.
This study would seem to confirm that PWSaffects males and females equally [Cassidy, 1997;Wattendorf and Muenke, 2005]. Nevertheless, therewas a higher number of male patients under 18 yearsof age whereas the majority of adults were female.These findings may indicate random variation insmall populations. However, the possibility cannotbe excluded that with PWS, as in the generalpopulation, women outlive men. On the other hand,such variations may also suggest an increasedmortality rate in male patients with PWS. Thiscorroborates previously reported evidence on27 deaths related to subjects with PWS [Schrander-Stumpel et al., 2004], and which raised the questionof a gender-related mortality risk being present inPWS. Nevertheless, interpretation of the resultsshouldbe cautious, and the collection andevaluationof further cases are needed in order to betterunderstand the role of gender in sudden death inPWS.
As expected, the INSUP study documented ahigh prevalence of obesity, especially in adult life.The low rate of obesity in the first 5 years wasin accordance with the natural course of PWS,characterized by a more evident tendency towardsexcessiveweight gain after 3 years of age [Holm et al.,1993; Zipf, 2004]. Furthermore, it is important topoint out that about 1/4 of patients aged 6–17 yearswere normal weight (26%, 30 cases), in contrast withprevious reports [Wollmann et al., 1998; Butler et al.,2006]. The relevance of these findings, however,remains to be established. In fact, the indexes ofweight excess adopted in our study are widely usedas a simple summary measure of overweight inboth children and adults. Nevertheless, it is knownthat weight-for-height, as well as BMI, are not anexact measure of adiposity in PWS, because they
ITALIAN NATIONAL SURVEY FOR PRADER–WILLI SYNDROME 867
American Journal of Medical Genetics Part A

underestimate percentage of body fat [Goldstoneet al., 2002; Kennedy et al., 2006]. This is consistentwith a study reporting increased percentage of fatmass in young underweight PWS children [Eiholzeret al., 2004]. PWS subjects have a higher fat mass thanthose with simple obesity, under the same degreeof weight excess, both in children and in adults
with PWS [Brambilla et al., 1997; Theodoro et al.,2006]. In light of this, dual-energy X-ray photonabsorptiometry (DXA) seems to be the best availabletechnique to evaluate body composition. Never-theless, its use is impracticable for epidemiologicalstudies because of the high costs involved. As far asour normal weight subjects are concerned, it is of
FIG. 3. Survival of PWS patients: overall (a) and by GH treatment (b). GHT, GH therapy. [Color figure can be viewed in the online issue, which is available atwww.interscience.wiley.com.]
868 GRUGNI ET AL.
American Journal of Medical Genetics Part A

note that 40 out of 66 patients (21 males) wereundergoing GH therapy from 31� 2 months (range:6–58 months), while 3 subjects (2 males) haddiscontinued GH treatment. In addition, 18 subjects(12males) not undergoingGH therapywereyoungerthan 3 years. The effect ofGHadministrationonbodycomposition in children and adolescents with PWShas been evaluated in several studies [Burman et al.,2001]. During the first year of GH therapy there is asignificant improvement in body composition byincreasing lean body mass and reducing fat mass[Carrel et al., 1999; Carrel et al., 2004]. After 24monthsof therapy, PWS patients have experienced sustaineddecreases in fat mass and increases in fat-free mass[Myers et al., 2000]. Based on these findings, we mayhypothesize that body fat percentage of our 40 PWSsubjects with normal BMI, who were undergoingGH therapy, is lower than expected. However, DXAanalysis is required to confirm this particular aspect.On the other hand, GH therapy alone is ineffectivein controlling body weight in individuals with PWS[Eiholzer and Whitman, 2004; Hoybye, 2004]. In thiscontext, it is of note that a higher percentage ofobesity was found in our patients not undergoingGH therapy, compared with those undergoing GHtreatment. Nevertheless, this difference could beexplained by the increasing frequency of obesitywith age, since the first group was significantly olderthan the GH-treated patients. Weight managementneeds a multidisciplinary medical approach, includ-ing dietary restrictions, hormones replacement,appropriate treatment of behavioral and psycholog-ical problems, as well as occupational and physicaltherapy [Eiholzer andWhitman, 2004]. In this respect,weight control seems to improve in PWS patientswho have had the benefit of early diagnosis andtreatment [Vogels and Fryns, 2004]. Thus, it is notsurprising that the great majority of our adult subjectswith PWS are obese or overweight (97.7%). In fact,older subjects, born before the syndrome waswell established, have most likely been diagnosedlater than younger patients, as previously reported[Vogels et al., 2004], and have not received anti-cipatory care and appropriate treatment for weightexcess.
In our PWS population, about half of the subjectshave performed GH therapy, and one third wereundergoing treatment. Up to 2000, in order to receivetreatment, diagnosis of GH deficiency by standardstimulation tests was required, both in children andadult PWS patients. Subsequently, GH administra-tion was approved for children and adolescentswith genetically confirmed PWS without GH/IGFaxis testing before therapy. In this light, we haveevaluated how many patients under 18 years werereceiving GH therapy. One hundred and sixteen of233 children and adolescents with PWS were under-going GH treatment (49.8%), whereas a significantpercentage of cases had never received GH therapy
(38.2%). In the context of the wide spectrum ofproblems affecting PWS children, GH administrationwere not recommended to improve linear growthas standard practice, unless short stature becomesevident [Paterson and Donaldson, 2003]. In thisrespect, the majority of our patients not undergoingGH treatment, apart from those with metabolicand orthopedic contraindications, were very youngsubjects or had normal stature. However, theimportance of ‘‘non-growth’’ effects of GH appearmore consequential for many children and adoles-cents with PWS than changes in height [Allen andCarrel, 2004]. Beside the positive effects on lineargrowth andfinal height, it has beenwell documentedthat GH replacement therapy improves body com-position, muscle strength and behavior [Carrel et al.1999; Haqq et al., 2003]. Moreover, greater benefitson anthropometric parameters, body composition,and psychomotor development were seen in theearly years with the result that early implementationof GH therapy was recently recommended forPWS children [Eiholzer et al., 2004; Myers et al.,2007]. Therefore, we would recommend GH therapyfor a greater percentage of Italian PWS patients andat an earlier age, taking into consideration bothgrowthpattern andbody composition abnormalities.Nevertheless, considering the limited evidence onGH treatment in younger children with PWS, theeffects of GH administration should be closelymonitored to avoid significant adverse effects [Crinoet al., 2007] and sudden deaths. In our study, 12% ofpatients under 18 years of age discontinued GHadministration. Unfortunately, the events which ledto withdrawal were not included in our database.Further investigation is required in this area beforefinal conclusions can be drawn.
Among adults with PWS, 52 out of 192 subjectswere treated during childhood, whereas a significantnumber of cases (25 patients, 13%).were undergoingGH therapy with the lower dosages commonly usedin adulthood. In contrast to the patients under18 years of age, GH deficiency was demonstratedin all of the patients by appropriate standardstimulation tests. Currently, the role of GH treatmentin older patients with PWS is unclear. Preliminarystudies have demonstrated that GH deficiency maybe present in a significant percentage of PWS adults[Partsch et al., 2000; Grugni et al., 2006]. This findingseems to be supported by the beneficial effects of GHtherapy on PWS patients recently reported [Hoybyeet al., 2003; Bertella et al., 2007; Hoybye, 2007]. Itsrelevance, however, needs to be fully investigated inlarge cohorts of subjects with appropriate surveil-lance. The presence of a significant number of adultswith PWS treated with GH in our population alsoseems to suggest this approach.
A mortality rate of 3% a year across all ages andabout 7%ayear in thoseover 30 years of agehas beenreported in people with PWS [Whittington et al.,
ITALIAN NATIONAL SURVEY FOR PRADER–WILLI SYNDROME 869
American Journal of Medical Genetics Part A

2001]. This observation seems not to be supported byour data. In total, we detected 18 deaths in 425 PWSpatients between January 1986 and June 2006. Ourfindings lead us to believe that the mortality rateof PWS is lower than that previously observed[Smith et al., 2003; Vogels et al., 2004]. The cause ofthese discrepancies may be related to the elevatednumber of our PWS patients, compared with otherstudies. Nevertheless, it is possible that there maybe a significant number of deaths related toundiagnosed patients with PWS, particularly olderindividuals, not reported in our study. However,the reason for these differences remains to beestablished, and systematic prospective studiesshould be carried out.
In terms of genetic mechanisms, it has beenreported that UPD15 seems to be an independentrisk factor for death in adult patients with PWS [Smithet al., 2003]. In our study, we found no correlationbetween death and UPD15. In this respect, our resultshave demonstrated a very similar frequency of deathin PWS subjects with UPD15 when compared to PWSpatientswithdel15. It is ofnote thatour study took intoconsideration a greater number of patients than theprevious report, as well as a wider range of ages.
Complications associated with obesity are recog-nized as the main risk factors for death during thelife-span of patients with PWS [Zipf, 2004], whileinfectious disease seems to be the major causeof sudden death in children below the age of5 [Schrander-Stumpel et al., 2004; Stevenson et al.,2004]. The great majority of our PWS subjects thatdied were very obese, both during childhoodand after 18 years of age. Unfortunately, an autopsywas performed in very few cases. According toprevious reports [Smith et al., 2003; Vogels et al.,2004], obesity-related cardiovascular and respiratorydiseases were the cause of death in most of ourolder patients, and one severely obese patient diedbecause of complications of bariatric surgery. Inaddition, death due to choking was reported in one39.3-year-old male, confirming its role as a risk factorin older PWS subjects [Stevenson et al., 2007].
As far as younger patients are concerned, deathwas related to cardiac and pulmonary illnesses in themajority of our obese PWS subjects under the age of18 years.
Overall, these findings suggest that improvementin weight control remains the most importantcomponent of any PWS treatment program and willreduce mortality dramatically. Nevertheless, two ofour 18 PWS patients who died were not obese(11.1%). One of them was a 6-month-old girl whodied after a short period of fever and gastroenteritis,as previously reported [Schrander-Stumpel et al.,2004]. In addition, no definitive cause of death wasfound in the other patient, who was slightly over-weight. On the whole, these observations suggestthat patients with PWS may be susceptible to
additional diseases unrelated to obesity, which couldcompromise their health further. In this regard, therole ofGH/IGF-I axis dysfunction and its treatment inthe poor health outcomes of PWS adults remainsto be fully established, whereas the benefits of GHtherapy were well demonstrated, as mentionedabove, both in children and in adolescents withPWS. In spite of these beneficial effects, fatal eventsafter the start of GH treatment have been reportedin pediatric patients with PWS [Eiholzer, 2005;Craig et al., 2006], including three children in ourstudy [Grugni et al., 2005; Sacco and Di Giorgio,2005], raising the possibility that a causal link maybe present. Nevertheless, because of the lack ofreliable epidemiological studies on death rates inpatients with PWS worldwide, current data areunable to establish a cause–effect relationshipbetween mortality and GH therapy [Zipf, 2004]. Inaddition, the same causes of death reported in thecases treated with GH therapy were frequently alsoobserved in PWS children not undergoing GHtherapy [Nagai et al., 2005]. In this context, our studyhas not shown an increased number of deaths duringGH treatment, both in children and in adults withPWS. On the basis of these results, we suggest thatGH therapy in patients with PWS, as a group, doesnot seem to increase risk of death.
In conclusion, the INSUP study is the firstpopulation survey that was undertaken in order toestablish all cases of genetically confirmed diagnosisof PWS country-wide; previous epidemiologicalstudies have consisted of smaller populations andlimited age groups [Burd et al., 1990; Akefeldtet al., 1991; Ehara et al., 1995; Whittington et al.,2001; Vogels et al., 2004]. The present survey waslarge enough to look at prevalence of obesity in ourPWS population as well as to analyze the impact ofGH administration and to evaluate the causes ofdeath with or without GH treatment. From our dataGH therapy seems to be safe and does not affect lifeexpectancy.
ACKNOWLEDGMENTS
We are grateful to the patients with PWS and theirfamilies for their willingness to participate in thisresearch. Our special thanks to the Italian Prader–Willi Syndrome Association for its collaboration. Inparticular, we would specifically like to thank MariaAntonietta Ricci, Gilberto Michielli, Silvia Brumani,Franca Di Pierro and Donatella Garitta. We wouldalso like to thank Dr. M.L. Giovannucci Uzielli fromthe Department of Paediatrics of the University ofFlorence for her collaboration.
REFERENCES
Akefeldt A, Gillberg C, Larsson C. 1991. Prader–Willi syndrome ina Swedish rural county: Epidemiological aspects. Dev MedChild Neurol 33:715–721.
870 GRUGNI ET AL.
American Journal of Medical Genetics Part A

Allen DB, Carrel AL. 2004. Growth Hormone therapy for Prader–Willi syndrome: A critical appraisal. J Ped Endocrinol Metab17:1297–1306.
Bertella L, Mori I, Grugni G, Pignatti R, Ceriani F, Molinari E,Ceccarelli A, Sartorio A, Vettor R, Semenza C. 2007. Quality oflife and psychological well-being in GH-treated, adult PWSpatients: A longitudinal study. J Int Disab Res 51:302–311.
Bittel DC, Butler MG. 2005. Prader–Willi syndrome. Clinicalgenetics, cytogenetics and molecular biology. Expert Rev MolMed 7:1–20.
Brambilla P, Bosio L, Manzoni P, Pietrobelli A, Beccaria L,Chiumello G. 1997. Peculiar body composition in patientswith Prader–Labhart–Willi syndrome. Am J Clin Nutr 65:1369–1374.
Buiting K, Horsthemke B. 2006. Molecular genetic findings inPrader–Willi syndrome. In: Butler MG, Lee PDK, WhitmanBY, editors. Management of Prader–Willi syndrome. 3rdedition. New York: Springer. p 58–73.
Burd L, Vesely B, Martsolf J, Kerbeshian J. 1990. Prevalence studyof Prader–Willi syndrome in North Dakota. Am J Med Genet37:9–99.
Burman P, Ritzen EM, Lindgren AC. 2001. Endocrine dysfunctionin Prader–Willi syndrome: A review with special reference toGH. Endocr Rev 22:787–799.
Butler MG. 2006. Management of obesity in Prader–Willisyndrome. Nat Clin Pract Endocrinol Metab 2:592–593.
Butler MG, Hanchett JM, Thompson T. 2006. Clinical findings andnatural history of Prader–Willi syndrome. In: Butler MG, LeePDK, Whitman BY, editors. Management of Prader–Willisyndrome. 3rd edition. New York: Springer. p 3–48.
Carrel AL, Myers SE, Whitman BY, Allen DB. 1999. Growthhormone improves body composition, fat utilization, physicalstrength and agility, and growth in Prader–Willi syndrome: Acontrolled study. J Pediatr 134:215–221.
Carrel AL, Moerchen V, Myers SE, Bekx MT, Whitman BY, AllenDB. 2004. Growth hormone improves mobility and bodycomposition in infants and toddlers with Prader–Willisyndrome. J Pediatr 145:744–749.
Carrel AL, Lee PDK, Mogul HR. 2006. Growth Hormone inPrader–Willi syndrome. In: Butler MG, Lee PDK, WhitmanBY, editors. Management of Prader–Willi syndrome. 3rdedition. New York: Springer. p 201–241.
Cassidy SB. 1997. Prader–Willi syndrome. J Med Genet 34:917–923.
Chotai KA, Payne SJ. 1998. A rapid, PCR based test for differentialmolecular diagnosis of Prader–Willi and Angelman syn-dromes. J Med Genet 35:472–475.
Clayton-Smith J, Laan L. 2003. Angelman syndrome: A review ofthe clinical and genetic aspects. J Med Genet 40:87–95.
Cole TJ, Freeman JV, PreeceMA. 1995.Bodymass index referencecurves for the UK, 1990. Arch Dis Child 73:25–29.
Cole TJ, Bellizzi MC, Flegal KM, Dietz WH. 2000. Establishing astandard definition for child overweight and obesity world-wide: International survey. BMJ 7244:1240–1243.
Cox DR, Oakes D. 1984. Analysis of survival data. London:Chapman & Hall.
Craig ME, Cowell CT, Larsson P, Zipf WB, Reiter EO, Albertsson-Wikland K, Ranke MB, Price AD. 2006. Growth hormonetreatment and adverse events in Prader–Willi syndrome: Datafrom KIGS (the Pfizer International Growth Database). ClinEndocrinol 65:178–185.
Crino A, Di Giorgio G, Manco M, Grugni G, Maggioni A. 2007.Effects of growth hormone therapy on glucose metabolismand insulin sensitivity indices in prepubertal children withPrader–Willi syndrome. Horm Res 68:83–90.
Ehara H, Ohno H, Takeshita K. 1995. Frequency of Prader–Willisyndrome in the San-in district, Japan. Brain Dev 17:324–326.
Eiholzer U. 2005. Deaths in children with Prader–Willi syndrome:A contribution to the debate about the safety of growthhormone treatment in children with PWS. Horm Res 63:33–39.
Eiholzer U, Whitman B. 2004. A comprehensive teamapproach tothe management of patients with Prader–Willi syndrome. JPediatr Endocrinol Metab 17:1153–1175.
Eiholzer U, l’Allemand D, Schlumpf M, Rousson V, Gasser T,Fusch C. 2004. Growth hormone and body composition inchildren younger than 2 years with Prader–Willi syndrome.J Pediatr 144:753–758.
Einfeld SL, Kavanagh SJ, Smith A, Evans EJ, Tonge BJ, Taffe J.2006. Mortality in Prader–Willi syndrome. Am J Ment Retard111:193–198.
Goldstone AP. 2004. Prader–Willi syndrome: Advances ingenetics, pathophysiology and treatment. Trends EndocrinolMetab 15:12–20.
Goldstone AP, Brynes AE, Thomas EL, Bell JD, Frost G, Holland A,Ghatei M, Bloom SR. 2002. Resting metabolic rate, plasmaleptin concentrations, leptin receptor expression, and adiposetissue measured by whole-body magnetic resonance imagingin women with Prader–Willi syndrome. Am J Clin Nutr 75:468–475.
Grugni G, Livieri C, Corrias A, Sartorio A, Crino A. 2005. Deathduring GH therapy in children with Prader–Willi sindrome:Descriptionof twonewcases. J Endocrinol Invest 28:554–557.
Grugni G, Marzullo P, Ragusa L, Sartorio A, Trifiro G, Liuzzi A,Crino A. 2006. Impairment of GH responsiveness to combinedGH-releasing hormone and arginine administration in adultpatients with Prader–Willi syndrome. Clin Endocrinol 65:492–499.
Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, CassidySB. 2001. The changing purpose of Prader–Willi syndromeclinical diagnostic criteria and proposed revised criteria.Pediatrics 108:e92.
Haqq AM, Stadler DD, Jackson RH, Rosenfeld RG, Purnell JQ,LaFranchi SH. 2003. Effects of growth hormone on pulmonaryfunction, sleep quality, behavior, cognition, growth velocity,body composition, and resting energy expenditure in Prader–Willi syndrome. J Clin Endocrinol Metab 88:2206–2212.
Holm VA, Cassidy SB, Butler MG, Hanchett JM, Greenswag LR,Whitman BY, Greenswag F. 1993. Prader–Willi syndrome:Consensus diagnostic criteria. Pediatrics 91:398–402.
Hoybye C. 2004. Endocrine and metabolic aspects of adultPrader–Willi syndrome with special emphasis on the effect ofgrowth hormone treatment. Growth Horm IGF Res 14:1–15.
Hoybye C. 2007. Five-years growth hormone (GH) treatment inadults with Prader–Willi syndrome. Acta Paediatr 96:410–413.
Hoybye C, Hilding A, Jacobbson H, Thoren M. 2003. Growthhormone treatment improves body composition in adultswithPrader–Willi syndrome. Clin Endocrinol 58:653–661.
Kaplan EL, Meier P. 1958. Nonparametric estimation fromincomplete observations. J Am Stat Assoc 53:457–481.
Kennedy L, Bittel DC, Kibiryeva N, Kalra SP, Torto R, Butler MG.2006. Circulating adiponectin levels, body composition andobesity-related variables in Prader–Willi syndrome: Compar-ison with obese subjects. Int J Obes 30:382–387.
Kuczmarski RJ, Ogden CL, Guo SS, Grummer-Strawn LM, FlegalKM,Mei Z,Wei R, Curtin LR, RocheAF, Johnson CL. 2002. 2000CDC Growth Charts for the United States: Methods anddevelopment. Vital Health Stat 11:1–190.
Marzullo P, Marcassa C, Campini R, Eleuteri E, Minocci A, PrianoL, Temporelli P, Sartorio A, Vettor R, Liuzzi A, Grugni G. 2005.The impact of growth hormone/insulin-like growth factor-1and nocturnal breathing disorders on cardiovascular featuresof adult patients with Prader–Willi syndrome. J Clin Endo-crinol Metab 90:5639–5646.
McCandless SE, Cassidy SB. 2006. Diagnostic criteria for Prader–Willi syndrome. In: Butler MG, Lee PDK,Whitman BY, editors.Management of Prader–Willi syndrome. 3rd edition. NewYork: Springer. p 49–57.
Myers SE, Carrel AL, Whitman BY, Allen DB. 2000. Sustainedbenefit after 2 years of growth hormone on body composition,
ITALIAN NATIONAL SURVEY FOR PRADER–WILLI SYNDROME 871
American Journal of Medical Genetics Part A

fat utilization, physical strength and agility, and growth inPrader–Willi syndrome. J Pediatr 137:42–49.
Myers SE,WhitmanBY,Carrel AL,MoerchenV,BekxMT,AllenDB.2007. Two years of growth hormone therapy in young childrenwithPrader–Willi syndrome:Physicalandneurodevelopmentalbenefits. Am J Med Genet Part A 143A:443–448.
Nagai T, Obata K, Tonoki H, Temma S, Murakami N, Katada Y,Yoshino A, Sakazume S, Takahashi E, Sakuta R, Niikawa N.2005. Cause of sudden, unexpected death of Prader–Willisyndrome patients with or without growth hormone treat-ment. Am J Med Genet Part A 136A:45–48.
Partsch C-J, Lammer C, Gillessen-Kaesbach G, Pankau R. 2000.Adult patients with Prader–Willi syndrome: Clinical charac-teristics, life circumstances and growth hormone secretion.Growth Horm IGF Res 10:S81–S85.
Paterson WF, Donaldson MDC. 2003. Growth hormone therapyin the Prader–Willi syndrome. Arch Dis Child 88:283–285.
Pomara C, D’Errico S, Riezzo I, de Cillis GP, Fineschi V. 2005.Sudden cardiac death in a child affected by Prader–Willisyndrome. Int J Legal Med 119:153–157.
Priano L, Grugni G, Miscio G, Guastamacchia G, Toffolet L,Sartorio A, Mauro A. 2006. Sleep cycling alternating pattern(CAP) expression is associated with hypersomnia andGH secretory pattern in Prader–Willi syndrome. Sleep Med7:627–633.
Rivera H, Zuffardi O, Gargantini L. 1990. Nonreciprocal andjumping translocations of 15q1->qter in Prader–Willi syn-drome. Am J Med Genet 37:311–317.
Sacco M, Di Giorgio G. 2005. Sudden death in Prader–Willisyndrome during GH therapy. Horm Res 63:29–32.
Schrander-Stumpel CTRM,Curfs LMG, Sastrowijoto P, Cassidy SB,Schrander JJP, Fryns J-P. 2004. Prader–Willi syndrome:Causesof death in an international series of 27 cases. Am J Med GenetPart A 124A:333–338.
Smith A, Loughnan G, Steinbeck K. 2003. Death in adults withPrader–Willi syndrome may be correlated with maternaluniparental disomy. J Med Genet 40:e63.
Stevenson DA, Anaya TM, Clayton-Simth J, Hall BD, Van AllenMI, Zori RT, Zackai EH, Frank G, Clericuzio CL. 2004.Unexpected death and critical illness in Prader–Willisyndrome: Report of ten individuals. Am J Med Genet Part A124A:158–164.
Stevenson DA, Heinemann J, Angulo M, Butler MG, Loker J, RupeN, Kendell P, Clericuzio CL, Scheimann AO. 2007. Deaths dueto choking in Prader–Willi syndrome. Am J Med Genet Part A143A:484–487.
Theodoro MF, Talebizadeh Z, Butler MG. 2006. Body composi-tion and fatness patterns in Prader–Willi syndrome: Compar-ison with simple obesity. Obesity 14:1685–1690.
Vogels A, Fryns JP. 2004. Age at diagnosis, body mass index andphysical morbidity in children and adults with the Prader–Willi syndrome. Genet Couns 15:397–404.
Vogels A, Van Den Ende J, Keymolen K, Mortier G, Devriendt K,Legius E, Fryns JP. 2004. Minimum prevalence, birth incidenceand cause of death for Prader–Willi syndrome in Flanders. EurJ Med Genet 12:238–240.
Wattendorf DJ, Muenke M. 2005. Prader–Willi syndrome. AmFam Physician 72:827–830.
Whittington JE, Holland AJ, Webb T, Butler J, Clarke D, Boer H.2001. Population prevalence and estimated birth incidenceand mortality rate for people with Prader–Willi syndrome inone UK Health Region. J Med Genet 38:792–798.
Whittington JE, Butler JV, Holland AJ. 2007. Changing rate ofgenetic subtypes of Prader–Willi syndrome in the UK. EurJ Hum Genet 15:127–130.
Wollmann HA, Schultz U, Grauer ML, Ranke MB. 1998. Referencevalues for height and weight in Prader–Willi syndrome basedon 315 patients. Eur J Pediatr 157:634–642.
Zaglia F, Biban P, Zaffanello M. 2005. Unexpected death due torefractory metabolic acidosis and massive hemolysis in younginfant with Prader–Willi syndrome. Am J Med Genet Part A132A:219–221.
ZipfWB. 2004. Prader–Willi syndrome: The care and treatment ofinfants, children and adults. Adv Pediatr 51:409–434.
872 GRUGNI ET AL.
American Journal of Medical Genetics Part A
Copyright © 2022 FDOKUMEN











![[Reducing the incidence of ventilator-associated pneumonia following heart surgery: 13-year experience of epidemiologic surveillance in a teaching hospital]](https://static.fdokumen.com/doc/165x107/633733ca9c13609c6c0ee513/reducing-the-incidence-of-ventilator-associated-pneumonia-following-heart-surgery.jpg)








