
Sweat but no gain: Inhibiting proliferation of multidrug resistant cancer cells with “ersatzdroges”
Upload
khangminh22Category
view
1download
0
1
THE EXTENSIVE PROLIFERATION OF HUMAN
CANCER CELLS WITH EVER-SHORTER TELOMERES
Rebecca Dagg
A thesis submitted in fulfilment of the requirements for the degree of
Doctor of Philosophy
Faculty of Medicine
The University of Sydney
2017

i
Statement of Originality
This is to certify that to the best of my knowledge, the content of this thesis is my own work. This
thesis has not been submitted for any degree or other purposes.
I certify that the intellectual content of this thesis is the product of my own work and that all the
assistance received in preparing this thesis and sources have been acknowledged.
Rebecca Dagg

ii
Abstract
Cellular immortalisation is currently regarded as an essential step in malignant transformation and is
consequently considered a hallmark of cancer. Acquisition of replicative immortality is achieved by
activation of a telomere lengthening mechanism (TLM), either telomerase or the alternative
lengthening of telomeres (ALT), to counter normal telomere attrition. However, a substantial
proportion of glioblastomas, liposarcomas, retinoblastomas, and osteosarcomas are reported to be
TLM-negative. The lack of serial untreated malignant human tumour samples over time has made it
impossible to examine telomere length over time and therefore determine whether they are truly
TLM-deficient, or whether this is the result of false-negative assays. Here we describe a subset (11%)
of high-risk neuroblastomas (NB; MYCN-amplified or metastatic disease) that lack evidence of any
significant TLM activity despite a 51% 5-year mortality rate. NB cell lines (COG-N-291 and LA-N-6)
derived from such tumours proliferated for 500 population doublings (PDs) with ever-shorter
telomeres (EST). COG-N-291 and LA-N-6 cells had exceptionally long and heterogeneous telomere
lengths as measured by terminal restriction fragment (TRF) analysis (mean TRF of 31 and 37.8 kb,
respectively) and telomere fluorescence in situ hybridisation. Both cell lines were telomerase negative
during culturing and did not have elevated markers of ALT or associated gene mutations. The
telomeres of these cells shortened by 80 and 55 bases/PD, consistent with telomere attrition due to
normal cell division, but did not reach senescence after 500 PDs in culture. This is conclusive evidence
that cells from highly malignant, lethal tumours are able to undergo continuous proliferation in spite
of an EST phenotype. The EST phenotype was rescued by activation of telomerase (via transduction
with hTERT expression constructs) or ALT (spontaneous occurrence of a nonsense TP53 mutation,
followed by spontaneous activation of ALT after 100 PDs). We also found that NB EST cells are very
sensitive to topoisomerase I inhibitors indicating the potential to target the EST phenotype with
topoisomerase I inhibitors in high-risk NB.

iii
Acknowledgements
The first person that I need to thank is my supervisor Dr Loretta Lau. I cannot express how grateful I
am that you took a chance and employed me eight years ago. I am the scientist that I am today because
of you. Thank you for your guidance and support and putting up with short deadlines with my writing.
You have been a great teacher, friend and mentor and I couldn’t have asked for a better supervisor.
I would also like to thank my co-supervisor Prof. Roger Reddel. I am constantly astounded by
your replies to emails at all times of the day and I am so grateful for your guidance and insights. Thank
you for always having an open door, because of you I have learnt to be a better scientist and critical
thinker. The success of my PhD is in large part due to the support that I have received from both of
my supervisors.
To all of the members of the CCRU past and present you have made the long hours in the lab
far more enjoyable and become some of my dearest friends. I would especially like to thank Vanita for
always answering my questions (no matter how many times I had to ask about the same form), Belinda
for always offering me advice with a smile, and lunches and dinners spent laughing with Radikha, Jess,
Cuc, Kylie, Michael, Greg, Nick, Amanda, Oksana, Natalie, Namratha, Camilla, and Yu Wooi. Yuyan your
support has helped me more than I can express and I will miss being able to chat with you at the bench.
To Peta, Sarah, Naz, Kaitlin and Amy I can’t believe we decided to start Stress whilst doing our PhD but
I’m so proud of what we achieved! Ladies you have become some of my dearest friends and I can’t
imagine having survived the last few years without you. I also need to thank the past and present
members of the cancer research and telomere length regulation units at CMRI for all of the advice and
assistance I’ve received which have made this a better thesis.
During my PhD I have spent more time in the lab than anywhere else and I would like to thank
all of my friends and family for being so understanding about the events that I’ve missed, left early or
been late to, or had to reschedule around experiments. You have all been so understanding and I

iv
cannot say how much I love you and appreciate your support. I would especially like to thank Chris
and my grandparents for being an unending source of support, you have helped me become who I am
today and I love you more than I can say. To Mum and Katherine, I don’t think that there are words to
say what you mean to me, but to start with I love you. You have seen me at my best and worst during
the PhD and done everything you can to make sure that I’m still sane at the end of it. Thank you for
always listening, being there when I need you, and doing everything from eating cake with me to
getting posters printed when I was on the other side of the world. I never doubted that I would get to
the end of this because of your unending love, support and encouragement. In many ways, I think of
this as something that we have achieved together, so thank you for getting onto this rollercoaster with
me and helping me become the person that I am today.

v
Publications
Dagg RA, Pickett HA, Neumann AA, Napier CE, Henson JD, Teber ET, Arthur JW, Reynolds CP, Murray
J, Haber M, Lau L MS, Reddel RR (2017). Extensive proliferation of human cancer cells with ever-shorter
telomeres. Cell Reports. Vol 20;19(12): 2544-2556.
Hayward NK, Wilmott JS, Waddell N, Johansson PA, Field MA, Nones K, Patch A, Kakavand H,
Alexandrov LB, Burke H, Jakrot V, Kazakoff S, Holmes O, Leonard C, Sabarinathan R, Mularoni L, Wood
S, Xu Q, Waddell N, Tembe V, Pupo GM, Paoli-Iseppi RD, Vilain RE, Shang P, Lau L MS, Dagg RA,
Schramm S, Pritchard A, Dutton-Regester K, Newll F, Fitzgerald A, Shang CA, Grimmond SM, Pickett
HA, Jean Y. Yang JY, Stretch JR, Behren A, Kefford RF, Hersey P, Long GV, Cebon J, Shackleton M,
Spillane AJ, Saw R PM, Lopez-Bigas N, Pearson JV, Thompson JF, Scolyer RA, Mann GJ (2017). Whole
genome landscapes of major melanoma subtypes. Nature. Vol 11;545(7653): 175-180.
Bradbury P, Turner K, Mitchell C, Griffin K, Lau L, Dagg RA, Taran E, Cooper-White J, Fabry B, O’Neill GM
(2017). The focal adhesion targeting (FAT) domain of p130 Crk associated substrate (p130Cas) confers
mechanosensing function. Journal of Cell Science. Vol 1;130(7): 1263-1273.
Scarpa A, Chang DK, Nones k, Corbo V, Patch A, Bailey P, Lawlor RT, Johns AL, Miller DK, Mafficini A,
Rusev B, Scardoni M, Antonello D, Barbi S, Sikora KO, Cingarlini S, Vicentini C, Simpson S, Quinn MCJ,
Bruxner TJC, Christ AN, Harliwong I, Idrisoglu S, Manning S, Nourse C, Nourbakhsh E, Wilson PJ,
Anderson MJ, Fink JL, Newell F, Waddell N, Holmes O, Kazakoff SH, Leonard C, Wood S, Xu Q, Nagaraj
SH, Amato E, Dalai I, Bersani S, Cataldo I, Capelli P, Davì MV, Landoni L, Malpaga A, Miotto M, Whitehall
V, Leggett B, Khanna KK, Harris J, Jones MD, Humphris J, Chantrill LA, Chin V, Nagril A, Pajic M, Scarlett

vi
CJ, Pinho A, Rooman I, Toon C, Wu J, Pinese M, Cowley M, Giry-Laterriere M, Mawson A, Humphrey
ES, Colvin EK, Chou A, Lovell JA, Jamieson NB, Duthie F, Gingras M, Muzny D, Dagg RA, Lau LMS, Lee
M, Pickett HA, Reddel RR, Samra JS, Kench JG, Merrett ND, Epari K, Nguyen NQ, Zeps N, Falconi M,
Simbolo M, Butturini G, Fassan M, Australian Pancreatic Cancer Genome Initiative, Gill AJ, Wheeler
DA, Gibbs RA, Musgrove EA, Bassi C, Tortora G, Pederzoli P, Pearson JV, Waddell N, Biankin AV and
Grimmond SM (2017). Whole-genome landscape of pancreatic neuroendocrine tumours. Nature. Vol
543: 65–71.
Henson JD, Lau L MS, Koch S, Martin La Rotta N, Dagg RA, Reddel RR (2016). The C-circle assay for
Alternative-Lengthening-of-Telomeres activity. Methods. http://dx.doi.org/10.1016/j.ymeth.2016.
08.01
Farooqi AS, Dagg RA, Choi LM R, Shay JW, Reynolds P, Lau L MS (2014). Alternative Lengthening of
Telomeres in neuroblastoma cell lines is associated with a lack of MYCN genomic amplification and
with p53 pathway aberrations. Journal of Neuro-Oncology. Vol 119(1): DOI:10.1007/s11060-014-
1456-8.
Lee M, Hills M, Conomos D, Stutz M, Dagg RA, Lau L MS, Reddel RR, Pickett HA (2014). Telomere
extension by telomerase and ALT generates variant repeats by mechanistically distinct processes.
Nucleic Acids Research. Vol 42 (3): 1733-1746.
Lau L MS, Dagg RA, Henson J, Au A, Royds J, Reddel RR (2013). Detection of Alternative Lengthening
of Telomeres (ALT) by telomere quantitative PCR. Nucleic Acids Research. Vol 41 (2): e34.

vii
List of Abbreviations
ɸ29 phi 29 polymerase
β-actin beta-actin
32P phosphorus-32
123I-MIBG meta-iodobenzylguanidine
6-thio-dG 6-thio-2’-deoxyguanosine
36B4 acidic ribosomal phosphoprotein P0
53BP1 p53-binding protein 1
γ-H2AX H2AX phosphorylated at serine 139
ALT alternative lengthening of telomeres
ABDIL antibody dilution buffer
ACRF Australian Cancer Research Foundation
ALU Short stretch of DNA, transposable element
ANOVA analysis of variance
APB ALT‐associated PML body
ALK anaplastic lymphoma kinase
ASF1 anti‐silencing factor 1
ATM ataxia telangiectasia mutated
ATO arsenic trioxide
ATP adenosine triphosphate
ATR ataxia telangiectasia and Rad3‐related
ATRA all trans retinoic acid
ATRX alpha thalassemia/mental retardation syndrome X‐linked
AU arbitrary units
Alt-NHEJ alternative non-homologous end joining
BCA Bicinchoninic acid
BLM Bloom syndrome
bp base pair
BRCA1 breast cancer type 1 susceptibility protein
BSA bovine serum albumin
C Cytosine
CAF1 chromatin assembly factor 1

viii
CC/C-circle C‐rich telomere circle
cDNA complementary DNA
CDKN1B cyclin dependent kinase inhibitor 1B
cells/mL Cells/millilitre
ChIP chromatin immunoprecipitation
CHAPS 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulphonate
c-Myc V-Myc Avian Myelocytomatosis Viral Oncogene Homolog
CO2 carbon dioxide
COG Children’s Oncology Group
CpG 5’-C-phosphate-G-3’
CST CTC1-STN1-TEN1 complex
Ct cycle threshold
DABCO 1,4 diazabicyclo(2.2. 2)octane
DAPI 4',6‐diamidino‐2‐phenylindole dihydrochloride
dATP deoxyadenosine triphosphate
DAXX death domain‐associated protein 6
DC dyskeratosis congenital
DDR DNA damage response
dGTP deoxyguanosine triphosphate
DKC1 dyskerin gene
D‐loop displacement loop
DMEM Dulbecco’s modified Eagle’s medium
DMs double-minute chromatin bodies
DMSO Dimethylsulphoxide
DNA deoxyribonucleic acid
DNAse Deoxyribonuclease
DNMT DNA methyltransferase: 1, 3a or 3b
dNTP deoxyribonucleotide triphosphate
DOX Doxorubicin
dsDNA double-stranded DNA
DTT Dithiothreitol
dTTP deoxythymidine triphosphate
EDTA ethylenediaminetetraacetic acid
EGTA ethyleneglycoltetraacetic acid

ix
EFS event-free survival
EST ever-shorter telomeres
ExoI exonuclease I
F Forward
FANCA Fanconi anaemia group A
FAND2 Fanconi anaemia group D2
FBS fetal bovine serum
FEN1 flap endonuclease 1
FFPE formalin-fixed paraffin-embedded
FISH fluorescence in situ hybridisation
g Gravity
G Guanine
GAPDH glyceraldehyde‐3‐phosphate dehydrogenase
G‐circle G‐rich telomere circle
GD2 ganglioside 2
G-MCSF granulocyte macrophage colony-stimulating factor
G-quadruplex secondary structures formed in guanine rich sequences
GWAS genome-wide association study
G1 phase cell cycle phase gap 1
G2 phase cell cycle phase gap 2
H2AX Histone variant H2AX
H3 histone H3
H3.3 histone variant H3.3
H3F3A H3 Histone Family Member 3A
H3K9me3 H3 trimethylated at lysine 9
H4 histone H4
H4K20me3 H4 trimethylated at lysine 20
HBG Haemoglobin
HCl hydrochloric acid
HEPES 4‐(2‐hydroxyethyl)piperazine‐1‐ethanesulphonic acid
HIRA histone regulator A
HJ Holliday junction
HMTase histone methyltransferase
hnRNPs heterogeneous nuclear ribonucleoproteins

x
HP1 heterochromatin protein-1
HPV human papillomavirus
hr(s) hour(s)
HRP horseradish peroxidase
HSRs homogeneously staining regions
hTERT human telomerase reverse transcriptase
HSP70 heat shock protein 70
HSP90 heat shock protein 90
hTR human telomerase RNA
IARC International Agency for Research on Cancer
IC50 inhibitor concentration required for 50% growth inhibition
IF Immunofluorescence
IgG immunoglobulin G
IL-2 interleukin 2
IMDM Iscove's Modified Dulbecco's Media
Indel insertion or deletion
INPC International neuroblastoma pathology classification system
IP Immunoprecipitation
INRGSS International neuroblastoma risk group staging system
INSS International neuroblastoma staging system
ITS insulin-transferrin-selenium-sodium pyruvate
IVA Ingenuity Variant Analysis
kb Kilobase
KCl potassium chloride
kDa Kilodalton
kg Kilogram
KOH potassium hydroxide
LgT large T antigen
LHC-MM Laboratory of Human Carcinogenesis mesothelial medium
LOH Loss of heterozygosity
M Molar
M/MUT Mutant
MAPK mitogen-activated protein kinase
MEM minimum essential medium

xi
meta‐TIF metaphase‐TIF
MgCl2 magnesium chloride
min(s) minute(s)
mL Millilitre
mM Millimolar
MMS21 methyl methanesulphonate-sensitivity 21
MOPS 3-(N-morpholino)propanesulphonic acid
MRN MRE11/RAD50/NBS1
mRNA messenger RNA
MYCN V-Myc Avian Myelocytomatosis Viral Oncogene Neuroblastoma Derived
Homolog
n number of replicates
NaCl sodium chloride
NaF sodium fluoride
NaHCO3 sodium bicarbonate
Na2HPO4 disodium hydrogen phosphate
NaOH sodium hydroxide
Na3V04 sodium orthovanadate
NB Neuroblastoma
NBS1 Nijmegen breakage syndrome 1 (Nibrin) gene
NCBI National Center for Biotechnology Information
NEAA non-essential amino acids
ng Nanogram
ng/mL nanogram/millilitre
ng/µg nanogram/microgram
ng/µL nanogram/microliter
nM Nanomolar
nm Nanometre
NHEJ non-homologous end joining
NP-40 Nonidet P-40
NuRD nucleosome remodelling and histone deacetylation
ORC origin replication complex
p16INK4A cyclin-dependent kinase inhibitor 2A
p21 cyclin-dependent kinase inhibitor 1

xii
PAGE polyacrylamide gel electrophoresis
PanNET pancreatic neuroendocrine tumour
PBS phosphate-buffered saline
PCNA proliferating cell nuclear antigen
PCR polymerase chain reaction
PCT pressure cycling technology
PD(s) population doubling(s)
PFA Paraformaldehyde
pH potential of hydrogen
PML promyelocytic leukaemia
PML-NB promyelocytic leukaemia nuclear body
PMSF phenylmethanesulphonyl fluoride
PNA peptide nucleic acid
POT1 protection of telomeres protein 1
PPTP Pediatric Preclinical Testing Program
pRb retinoblastoma protein
PTEN phosphatase and tensin homolog
PVDF polyvinylidene fluoride
qPCR quantitative polymerase chain reaction
R Reverse
RAP1 repressor-activator protein 1
RFC replication factor C
RNA ribonucleic acid
RNase Ribonuclease
RNaseH1 ribonuclease H1
RPMI-1640 Roswell Park Memorial Institute 1640 Medium
RPA replication protein A
RTEL1 regulator of telomere elongation helicase 1
RT-PCR reverse transcriptase polymerase chain reaction
SCG single copy gene
SD standard deviation
SDS sodium dodecyl sulphate
sec Second
SIOPEN International Society of Paediatric Oncology Europe Neuroblastoma

xiii
shelterin protein complex that binds the telomere
SKP2 S-phase kinase-associated protein 2
SLX4 SLX4 Structure-Specific Endonuclease Subunit gene
SNP single nucleotide polymorphism
SNV single nucleotide variation
S-phase cell cycle phase- DNA synthesis
SSC saline-sodium citrate
ssDNA single-stranded DNA
STR short tandem repeat
SV structural variation
SV40 Simian virus 40
SWATH sequential window acquisition of theoretical fragment ion spectra
TBE tris-borate-EDTA
TBS tris-buffered saline
TC telomere content
T-circle telomere circle
TE tris-EDTA
TEP telomerase-associated protein 1
TERC telomerase RNA component gene
TERT telomerase reverse transcriptase gene
TERRA telomeric repeat containing RNA
TIF telomere dysfunction induced focus
TIN2 TRF1- and TRF2-interacting nuclear factor 2
T-loop telomere loop
TLM telomere lengthening mechanism
TOPO IIIα topoisomerase III alpha
TP53/p53 tumour protein 53
TPE telomere position effect
TPP1 Shelterin protein encoded by the ACD gene
TRAP telomere repeat amplification protocol
TRF terminal restriction fragment
TRF1 telomeric repeat-binding factor 1
TRF2 telomeric repeat-binding factor 2
Tris tris (hydroxymethyl) aminomethane

xiv
T-SCE telomere-sister chromatid exchange
TTBS Tween-20, tris buffered saline
TZAP zinc finger and BTB domain containing 48 (ZBTB48)
U/µg Unit/microgram
µg Microgram
µg/mL microgram/millilitre
µg/well microgram/well
µL Microliter
µm Micrometre
µM Micromolar
USA United States of America
UTR untranslated region
UV Ultraviolet
VAV2 Vav Guanine Nucleotide Exchange Factor 2
V/cm volts/centimetre
V/sec volts/second
v/v volume/volume
WGS whole genome sequence
WRN Werner syndrome RecQ like helicase
w/v weight/volume
W/WT wild type
XRCC3 X-Ray Repair Cross Complementing 3 gene
YFP yellow fluorescent protein
yr Year

xv
Table of Contents
Statement of Originality ........................................................................................................................... i
Abstract ................................................................................................................................................... ii
Acknowledgements ................................................................................................................................ iii
Publications ............................................................................................................................................. v
List of Abbreviations ............................................................................................................................. vii
Table of Contents .................................................................................................................................. xv
List of Figures ........................................................................................................................................ xxi
List of Tables ....................................................................................................................................... xxiii
Chapter 1: Introduction .......................................................................................................................... 2
1.1. Telomere biology ......................................................................................................................... 2
1.1.1. Telomeres and chromosome end-protection ....................................................................... 2
1.1.2. Telomeric chromatin ............................................................................................................. 5
1.1.3. Telomere replication ............................................................................................................. 7
1.1.4. Telomere transcription ......................................................................................................... 8
1.2. Telomeres and replicative aging .................................................................................................. 9
1.2.1. Senescence ............................................................................................................................ 9
1.2.2. Immortalisation ................................................................................................................... 11
1.3. Telomere length regulation ....................................................................................................... 14
1.4. Telomere lengthening mechanisms ........................................................................................... 14

xvi
1.4.1. Telomerase .......................................................................................................................... 14
1.4.2. Alternative Lengthening of Telomeres (ALT) ...................................................................... 19
1.4.3. Telomere lengthening maintenance negative samples ...................................................... 35
1.5. Neuroblastoma .......................................................................................................................... 36
1.5.1. Disease stages ..................................................................................................................... 36
1.5.2. Risk stratification................................................................................................................. 37
1.5.3. Treatment ........................................................................................................................... 41
1.5.4. Predisposition genes ........................................................................................................... 43
1.5.5. Genetic features of sporadic neuroblastoma ..................................................................... 43
1.5.6. Prognostic relevance of telomerase activity ....................................................................... 46
1.5.7. ALT in neuroblastoma ......................................................................................................... 48
1.6. Aims of the project..................................................................................................................... 49
Chapter 2: Materials and Methods ....................................................................................................... 52
2.1. Buffers and solutions ................................................................................................................. 52
2.2. Patient samples .......................................................................................................................... 52
2.3. Cell culture ................................................................................................................................. 53
2.4. Vectors and viral transfections .................................................................................................. 53
2.5. Cell proliferation assay ............................................................................................................... 55
2.6. Immunostaining and fluorescence in situ hybridisation (FISH) ................................................. 55
2.6.1. ATRX and DAXX immunofluorescence (IF) .......................................................................... 55
2.6.2. APB detection...................................................................................................................... 58
2.6.3. MetaTIF assay ..................................................................................................................... 59

xvii
2.6.4. Telomere FISH on metaphase spreads ............................................................................... 61
2.6.5. Telomere sister chromatid exchange (T-SCE) analysis ........................................................ 61
2.7. DNA extraction and quantitation ............................................................................................... 62
2.8. C-circle assay .............................................................................................................................. 62
2.8.1. Slot-blot detection .............................................................................................................. 62
2.8.2. Exonuclease treatment of DNA ........................................................................................... 63
2.8.3. Telomere quantitative polymerase chain reaction (qPCR) detection ................................ 64
2.9. Telomere length analysis ........................................................................................................... 65
2.9.1. Telomere restriction fragment (TRF) analysis ..................................................................... 65
2.9.2. Telomere qPCR .................................................................................................................... 65
2.10. T-circle detection ..................................................................................................................... 67
2.11. Telomere chromatin immunoprecipitation (ChIP)................................................................... 67
2.11.1. Cell lysis and chromatin isolation...................................................................................... 67
2.11.2. Immunoprecipitation (IP) .................................................................................................. 68
2.11.3. DNA isolation .................................................................................................................... 68
2.11.4. Quantitation ...................................................................................................................... 68
2.12. Labelling probes with 32P ......................................................................................................... 69
2.13. Whole genome sequencing (WGS) and bioinformatics ........................................................... 69
2.13.1. WGS and read alignment .................................................................................................. 69
2.13.2. Variant annotations .......................................................................................................... 71
2.14. Sanger sequencing ................................................................................................................... 72
2.15. RNA extraction ......................................................................................................................... 73

xviii
2.16. qPCR analysis ........................................................................................................................... 73
2.16.1. Reverse transcriptase qPCR (RT-qPCR) ............................................................................. 73
2.16.2. Genomic qPCR ................................................................................................................... 73
2.17. Telomeric repeat-containing transcript (TERRA) ..................................................................... 74
2.18. Telomerase activity .................................................................................................................. 74
2.18.1. Protein Lysis ...................................................................................................................... 74
2.18.2. Immunoprecipitation ........................................................................................................ 75
2.18.3. Telomeric repeat amplification protocol (TRAP) .............................................................. 75
2.19. Immunoblotting ....................................................................................................................... 75
2.20. Statistical analyses ................................................................................................................... 77
Chapter 3: A subgroup of high-risk MYCN non-amplified neuroblastomas lacks a telomere lengthening
mechanism ............................................................................................................................................ 79
3.1. Introduction ............................................................................................................................... 79
3.2. Results ........................................................................................................................................ 80
3.2.1. Unique subgroup of high-risk MYCN non-amplified neuroblastoma ................................. 80
3.2.2. ALT-negative and telomerase-negative cancer cell lines .................................................... 84
3.3. Discussion ................................................................................................................................... 97
Chapter 4: Long-term proliferation occurs in cancer cells with ever-shorter telomeres ................... 101
4.1. Introduction ............................................................................................................................. 101
4.2. Results ...................................................................................................................................... 102
4.2.1. Cells with ever-shorter telomeres are capable of long-term proliferation ...................... 102
4.2.2. Characteristics of EST cells ................................................................................................ 102

xix
4.2.3. Genetics of EST cells .......................................................................................................... 109
4.3. Discussion ................................................................................................................................. 111
Chapter 5: Activation of a telomere lengthening mechanism rescues the ever-shorter telomere
phenotype ........................................................................................................................................... 115
5.1. Introduction ............................................................................................................................. 115
5.2. Results ...................................................................................................................................... 115
5.2.1. Activation of telomerase rescues the EST phenotype ...................................................... 115
5.2.2. Activation of ALT rescues the EST phenotype .................................................................. 120
5.2.3. EST neuroblastoma cell lines are unable to generate t-circles ......................................... 124
5.3. Discussion ................................................................................................................................. 127
Chapter 6: Neuroblastoma EST cells are sensitive to topoisomerase inhibitors ................................ 131
6.1. Introduction ............................................................................................................................. 131
6.2. Results ...................................................................................................................................... 132
6.2.1. EST cells are sensitive to topoisomerase inhibitors .......................................................... 132
6.2.2. EST cells do not exhibit increased cell death with potential ALT inhibitors or after induction
of replication stress ..................................................................................................................... 136
6.2.3. EST cells have alterations to genes involved in DNA damage repair and replication ....... 137
6.3. Discussion ................................................................................................................................. 137
Chapter 7: Discussion .......................................................................................................................... 143
7.1. The ever-shorter telomeres phenotype in human cancer ....................................................... 143
7.2. Targeting telomere biology in the clinic .................................................................................. 145
7.3. Conclusion ................................................................................................................................ 146

xx
Chapter 8: References ......................................................................................................................... 149
Appendix I: Information Relating to Chapter 2 ................................................................................... 193
Appendix II: Data Pertaining to Chapter 4 .......................................................................................... 199

xxi
List of Figures
Figure 1.1. Structure of the human telomere. ........................................................................................ 4
Figure 1.2. Telomere length during cell division. .................................................................................. 13
Figure 1.3. Potential templates of homologous recombination mediated telomere lengthening. ..... 27
Figure 1.4. Prevalence of ALT in human cancer based on tumour tissue type. .................................... 33
Figure 2.1. Schematic of neuroblastoma (NB) tumours used in this study based on MYCN amplification
status. .................................................................................................................................................... 53
Figure 3.1. A subgroup of high-risk MYCN non-amplified neuroblastoma with long telomeres are ALT-
negative................................................................................................................................................. 81
Figure 3.2. A unique subgroup of high-risk ALT-negative/MYCN non-amplified/TC≥15 NBs have poor
survival. ................................................................................................................................................. 83
Figure 3.3. Two neuroblastoma cell lines (COG-N-291 and LA-N-6) have long and heterogeneous
telomere lengths similar to the ALT-negative/TC≥15 tumour group. .................................................. 86
Figure 3.4. The COG-N-291 and LA-N-6 cell lines are phenotypically similar to the ALT-negative/TC≥15
tumour group. ....................................................................................................................................... 89
Figure 3.5. The cell lines COG-N-291 and LA-N-6 lack characteristic of ALT. ........................................ 90
Figure 3.6. COG-N-291 and LA-N-6 lack DNA damage at the telomere and characteristics of ALT. .... 92
Figure 3.7. The level of telomere bound proteins does not differ between ALT neuroblastoma cell lines
and COG-N-291 and LA-N-6. ................................................................................................................. 93
Figure 3.8. Some melanoma tumours are ALT-negative with TC≥15. .................................................. 95
Figure 3.9. ALT-negative/long telomere melanoma cell lines are telomerase positive. ...................... 96
Figure 4.1. Two neuroblastoma cell lines (COG-N-291 and LA-N-6) are capable of long term
proliferation despite the ever-shorter telomere (EST) phenotype. ................................................... 103
Figure 4.2. EST cells are capable of proliferation for hundreds of population doublings. ................. 104

xxii
Figure 4.3. Characteristics of EST cells during continuous proliferation for 600 population doublings.
............................................................................................................................................................ 105
Figure 4.4. A subpopulation of COG-N-291 cells are responsible for sporadic increases in C-circles in
the first 300 PDs of culture. ................................................................................................................ 107
Figure 4.5. Periodically a population of COG-N-291 cells activate an ALT mechanism but a slower
proliferation rate prevents them outgrowing the EST cells. .............................................................. 108
Figure 4.6. Characteristics of COG-N-291 sublines during continuous long-term proliferation. ....... 110
Figure 5.1. Retroviral transduction of hTERT results in telomerase activity in LA-N-6 cells. .............. 116
Figure 5.2. Ectopic expression of telomerase rescues the EST phenotype. ....................................... 118
Figure 5.3. No change to ALT characteristics following ectopic expression of hTERT. ....................... 119
Figure 5.4. A LA-N-6 subline spontaneously acquired a premature stop codon in TP53. .................. 120
Figure 5.5. Activation of ALT results in rescue of the EST phenotype. ............................................... 122
Figure 5.6. Loss of p53 results in activation of ALT in an EST culture (LA-N-6). .................................. 123
Figure 5.7. LA-N-6 MUT p53 cells did not activate ALT due to aberrations to ATRX or DAXX. ........... 125
Figure 5.8. EST cells do not form t-circles after activation of ALT or extreme telomere lengthening by
telomerase activity. ............................................................................................................................ 127
Figure 5.9. EST cells express genes involved in t-circle formation...................................................... 128
Figure 6.1. EST cells are sensitive to topoisomerase inhibitors. ......................................................... 133
Figure 6.2. Telomere biology does not influence sensitivity of NB cells to common NB therapies. .. 135
Figure 6.3. Telomere biology does not predict the response of cell lines to inhibitors that cause
replication stress or reduce factors critical to the ALT mechanism. ................................................... 139

xxiii
List of Tables
Table 1.2. Neuroblastoma risk group classification for stages L1/L2, L1 and MS. ................................ 39
Table 1.3. Neuroblastoma risk group classification of stage L2............................................................ 39
Table 1.4. Neuroblastoma risk group classification of stage M. ........................................................... 39
Table 2.1. Composition of common buffers and solutions. .................................................................. 52
Table 2.2. Characteristics of NB cell lines ............................................................................................. 54
Table 2.3. Telomere lengthening mechanism of control cell lines ....................................................... 54
Table 2.4. Cell culture medium. ............................................................................................................ 56
Table 2.5. Seeding density of cell lines in the cell proliferation assay. ................................................. 58
Table 2.6. List of drugs and stock concentrations................................................................................. 60
Table 2.7. List of primers. ...................................................................................................................... 66
Table 2.8. Antibodies for telomere ChIP. .............................................................................................. 70
Table 2.8. TRAP PCR conditions. ........................................................................................................... 76
Table 2.9. Antibodies and conditions used for Western blot analysis. ................................................. 76
Table 3.1. Characteristics of 35 neuroblastoma cell lines. ................................................................... 85
Table 6.1. IC50 values for common NB chemotherapeutics. .............................................................. 136
Table 6.2. IC50 values for inhibitors that cause replication stress and inhibit factors associated with
ALT....................................................................................................................................................... 138
Table A1.1. Primers used to Sanger sequence ATRX. ......................................................................... 193
Table A1.2. Primers used to Sanger sequence DAXX. ......................................................................... 195
Table A1.3. PCR cycling conditions for program 1. ............................................................................. 196
Table A1.4. PCR cycling conditions for program 2. ............................................................................. 196
Table A1.5. PCR cycling conditions for program 3. ............................................................................. 197
Table A1.6. PCR cycling conditions for program 4. ............................................................................. 197
Table A1.7. PCR cycling conditions for program 5. ............................................................................. 198

xxiv
Table A1.8. PCR cycling conditions for program 6. ............................................................................. 198
Table A2.1. Characteristics of high-risk NB ALT tumours (n=36). ....................................................... 199
Table A2.2. Characteristics of high-risk MYCN amplified NB tumours (n=55). ................................... 201
Table A2.3. Characteristics of high-risk ALT-negative/long telomere NB tumours (n=17). ................ 204
Table A2.4. Characteristics of high-risk MYCN non-amplified/short telomere NB tumours (n=41). . 205
Table A2.5. Genes examined by whole genome sequencing. ............................................................ 208
Table A2.6. Genetic events identified by whole genome sequencing. ............................................... 249

1
Chapter 1:
Introduction

2
Chapter 1: Introduction
1.1. Telomere biology
1.1.1. Telomeres and chromosome end-protection
Chromosomes contain genetic material that must be conserved for correct cellular function. The linear
chromosomes of mammalian cells terminate with 3-12 kilobases (kb) of tandem arrays of TTAGGG
repeats (Moyzis et al., 1988), known as the telomere. The proximal 2 kb region of human telomeres
consist of non-random arrays of variant (TCAGGG, TGAGGG, and TTGGGG) and canonical (TTAGGG)
repeats (Allshire et al., 1989; Baird et al., 1995; Coleman et al., 1999). The telomere sequence is
predominantly double-stranded with the terminal 100-200 bases on the G-rich strand ending in a 3’
single-stranded overhang (Makarov et al., 1997) (Figure 1.1.A). To prevent recognition of the telomeric
3’ overhang as a double-stranded DNA (dsDNA) break which would lead to activation of a DNA damage
response (DDR) at chromosome ends or chromosome end-to-end fusion, the telomere forms a protein-
DNA complex that can induce a unique chromatin structure at the telomere known as the T-loop (Griffith
et al., 1999) (Figure 1.1.B). A T-loop is formed when the 3’ overhang invades the upstream duplex
telomeric DNA forming a Holliday junction (HJ) (Liu and West, 2004) before annealing to the
complementary C-rich strand inducing a displacement or D-loop (Figure 1.1.B). This sequesters the
single-stranded telomeric DNA and provides a protective cap that defines the end of the chromosome,
ultimately preventing it initiating a DDR (de Lange, 2004).
Telomeres provide specific binding sites for a number of proteins, of which the six-subunit
protein complex known as shelterin is the best characterised. The shelterin complex is involved in the
formation and stabilisation of the T-loop structure and is critical to preventing a DDR at the telomere
(Liu et al., 2004a; Ye et al., 2004c) (Figure 1.1.C, D). The shelterin complex binds to telomeric DNA
through three sequence-specific DNA binding proteins: telomeric repeat binding factor 1 (TRF1) and

3

4
Figure 1.1. Structure of the human telomere.
(A) The human telomere is a predominantly double-stranded sequence of (TTAGGG)n repeats that
ends in a 3’ single-stranded overhang of 100-200 bp on the G-rich strand. (B) The single-stranded 3’
overhang can invade the duplex sequence to form a T-loop. (C & D) The shelterin complex is able to
bind telomeric DNA through TRF1 and TRF2 interaction with the dsDNA of the telomere and through
POT1 binding to the ssDNA of the 3' overhang or the D-loop. TIN2 and TPP1 connect POT1 to TRF1
and TRF2 and RAP1 binds directly to TRF2.
telomeric repeat binding factor 2 (TRF2) which bind directly to double-stranded telomeric DNA
(Benarroch-Popivker et al., 2016; Bilaud et al., 1997; Broccoli et al., 1997; Chong et al., 1995; van
Steensel et al., 1998), and protection of telomeres 1 (POT1) which specifically binds to the single-
stranded telomeric DNA (Baumann and Cech, 2001; Loayza et al., 2004). TRF1 mediates T-loop formation
by bending the dsDNA, and this allows the single-stranded overhang to invade the duplex telomere
sequence and form a HJ, a process mediated by TRF2 (Griffith et al., 1999; Stansel et al., 2001).
Telomeres represent a challenge to the DNA replication machinery and cells require TRF1 to ensure
efficient telomere replication (Chastain et al., 2016; Martinez et al., 2009; Sfeir et al., 2009; Stewart et
al., 2012). TRF2 prevents an ataxia telangiectasia mutated (ATM) dependent DDR (Celli and de Lange,
2005; Denchi and de Lange, 2007; Karlseder et al., 2004) through two mechanisms: firstly the formation
and stabilisation of the T-loop structure (Doksani et al., 2013; Griffith et al., 1999; Poulet et al., 2009)
which prevents classical non-homologous end joining (NHEJ) at telomeres (Smogorzewska et al., 2002;
van Steensel et al., 1998) or cleavage of the structure by resolvases (Poulet et al., 2009) and secondly,
by direct inhibition of ATM (Okamoto et al., 2013). TRF2 recruits and binds the shelterin component
repressor-activator protein 1 (RAP1) to the telomere, which aids in suppressing illegitimate
recombination events at telomeres (Li et al., 2000; Martinez et al., 2010; Sfeir et al., 2010; Ye and de
Lange, 2004). The fifth component of the shelterin complex, TRF1- and TRF2-interacting nuclear factor
2 (TIN2), can bind both TRF1 and TRF2 and acts to stabilise the binding of these two proteins to the

5
telomere (Houghtaling et al., 2004; Kim et al., 2004; Kim et al., 1999; Liu et al., 2004a; Ye et al., 2004c).
TPP1, the final component of the shelterin complex, directly binds to POT1 and with TIN2 mediates POT1
interaction with TRF1 and TRF2 (Frescas and de Lange, 2014a; Frescas and de Lange, 2014b; Liu et al.,
2004b; Takai et al., 2011; Ye et al., 2004b). The TPP1/POT1 complex suppresses ataxia telangiectasia
and Rad3 related (ATR) dependent DNA repair (Denchi and de Lange, 2007; Wu et al., 2006), likely
through exclusion of the DDR protein replication protein A (RPA) from the single-stranded overhang
(Gong and de Lange, 2010).
In addition to the T-loop, telomeres can form G-quadruplex secondary structures. G-
quadruplexes are formed by G-rich DNA sequences which are able to associate with each other by
Hoogsteen base pairing (Gellert et al., 1962; Gilbert and Feigon, 1999). G-quadruplexes have been
detected in telomere sequence in vitro (Sen and Gilbert, 1988; Sundquist and Klug, 1989) and in vivo in
organisms including ciliates (Paeschke et al., 2008; Paeschke et al., 2005) and humans (Biffi et al., 2013).
Such structures can induce DNA damage at the telomere and hence must be unfolded by helicases
including Bloom syndrome helicase (BLM) for correct cellular functions including DNA replication (Wu
et al., 2015).
1.1.2. Telomeric chromatin
DNA is condensed into chromatin, a structure comprised of DNA bound to histone proteins, to allow all
genetic material to fit into the nucleus of a cell. Chromatin also provides a mechanism to regulate DNA
processes such as transcription. There are two types of chromatin; heterochromatin and euchromatin.
Heterochromatin is a compact structure that renders the DNA transcriptionally inactive and is
consequently associated with gene sparse regions of the chromosome (Pontecorvo, 1944). In contrast,
euchromatin is less condensed and therefore found in regions of the genome that are transcriptionally
active (Pontecorvo, 1944). The nucleosome is the basic subunit of chromatin and is comprised of 147
base pairs (bp) of DNA wrapped tightly around a disc-shaped histone octamer (Kornberg, 1974;
Kornberg and Thomas, 1974). A histone octamer contains two molecules each of H2A, H2B, H3 and H4

6
histones (Felsenfeld and Groudine, 2003; Kornberg, 1974; Kornberg and Thomas, 1974). Histones
sterically prevent factors such as RNA polymerase from binding to the DNA, and chromatin remodelling
is therefore required to allow transcription to occur. There are two methods of chromatin remodelling:
nucleosomes can slide or be temporarily disassembled by adenosine triphosphate (ATP) dependent
molecules (Cote et al., 1994; Lia et al., 2006; Saha et al., 2002; Saha et al., 2005), or the post-translational
modification of histones (acetylation or methylation) can modulate DNA-histone binding or recruit other
proteins to the nucleosome (Pogo et al., 1966; Schubeler et al., 2004).
Telomeric chromatin has features consistent with heterochromatin, including histone H3 tri-
methylation at lysine 9 (H3K9me3) and histone H4 tri-methylation at lysine 20 (H4K20me3) (Garcia-Cao
et al., 2004). The histone methyltransferase (HMTase) Suv39h mediates trimethylation of H3K9 (Peters
et al., 2001) which recruits heterochromatin protein-1 (HP1), necessary for chromatin compaction, to
the telomere and subtelomere regions (Cheutin et al., 2003). HP1 then recruits the HMTase Suv4-20h
resulting in trimethylation of H4K20 (Benetti et al., 2007b). Telomeric heterochromatin can also
influence the chromatin structure of reporter genes in the subtelomere resulting in gene silencing
through a process known as the telomere position effect (TPE) (Baur et al., 2001; Gottschling et al.,
1990). There is a low level of H3 and H4 acetylation at sub- and telomeric regions in mortal cells (Benetti
et al., 2007a). Mammalian telomeres lack CpG sequences and hence are not methylated, compared to
the sub-telomeric regions which may be highly methylated by the action of the DNA methyltransferases
DNMT1, DNMT3a and DNMT3b (Brock et al., 1999; Gonzalo et al., 2006). Nucleosomes have been
detected in human telomeres although they have an altered nucleosome structure compared to non-
telomeric regions of the genome (Ichikawa et al., 2014; Lejnine et al., 1995; Makarov et al., 1993;
Tommerup et al., 1994). Repetitive sequences are predicted to be a difficult substrate to maintain
packaged as a nucleosome, suggesting telomeres may also have rapid nucleosome turnover
(Schneiderman et al., 2009).

7
1.1.3. Telomere replication
Mammalian telomeres are replicated throughout S phase (Arnoult et al., 2010; Verdun and Karlseder,
2006; Wright et al., 1999) from replication start sites largely originating in the subtelomere (Drosopoulos
et al., 2012). However, there is emerging evidence that replication can also initiate in the telomere itself
(Drosopoulos et al., 2012; Kurth and Gautier, 2010; Sfeir et al., 2009). Telomeric structures such as the
T-loop, G-quadruplexes and heterochromatin present a barrier to replication forks (Paeschke et al.,
2011). Therefore, proteins that unwind structures within the telomere or restart stalled replication forks
associate with the telomere during S or G2 phase (Chawla et al., 2011; Crabbe et al., 2004; Gu et al.,
2012; Sfeir et al., 2009; Uringa et al., 2012; Vannier et al., 2013; Ye et al., 2010). To facilitate replication
of the telomere, telomeric structures must unfold either in response to the DNA polymerase or by active
unwinding by helicases (Vannier et al., 2012). The helicases Werner syndrome ATP-dependent helicase
(WRN), regulator of telomere elongation helicase 1 (RTEL1), and BLM are recruited to the mammalian
telomere by TRF1, TRF2 and POT1 which subsequently remove T-loop and G-quadruplex structures to
allow telomeric replication (Crabbe et al., 2004; Drosopoulos et al., 2015; Opresko et al., 2002; Sarek et
al., 2015; Sfeir et al., 2009; Vannier et al., 2012; Wu et al., 2015; Zaug et al., 2005). There is evidence
that during telomere replication single-stranded DNA (ssDNA) becomes exposed and induces the
formation of a structure similar to the stalled replication fork (Verdun and Karlseder, 2006). This is
detected by MRE11 and RPA which trigger an ATR/ATM-dependent response that leads to the
completion of replication (Verdun and Karlseder, 2006).
Upon completion of replication, there is a small 3’ overhang caused by the inability of the
replicating DNA to fill the terminal RNA primer during lagging strand synthesis (Levy et al., 1992). This
generates an ATM-dependent DNA damage response, indicated by the localisation of MRE11 and ATM
to the telomere during G2/M (Verdun and Karlseder, 2006). Consequently, the end of the telomere is
processed by Apollo, Exo1, the CST (CTC1-STN1-TEN1) complex and POT1 (Chow et al., 2012; Huang et
al., 2012; Lam et al., 2010; Stewart et al., 2012; Wu et al., 2012b; Wu et al., 2010) to generate a long 3’
overhang before the shelterin proteins and recombination proteins including RAD51, RAD52 and XRCC3

8
(X-Ray Repair Cross Complementing 3 protein) reform the T-loop at the end of the telomere (Amiard et
al., 2007; Doksani et al., 2013; Stansel et al., 2001; Verdun and Karlseder, 2006).
1.1.4. Telomere transcription
Telomeres are transcribed into long non-coding RNA termed TERRA (telomere repeat-containing RNA)
by RNA polymerase II from transcription initiation sites in the subtelomere (Azzalin et al., 2007;
Nergadze et al., 2009; Schoeftner and Blasco, 2008). TERRA transcripts consist of UUAGGG repeats
ranging in size from 100 bases to 9 kb (Azzalin et al., 2007) and about 7% of human TERRA transcripts
have a poly-A tail (Azzalin and Lingner, 2008; Porro et al., 2010). Initial reports suggested approximately
25% of telomeres in human cells transcribe TERRA from specific transcription start sites defined by CpG
DNA islands (Nergadze et al., 2009; Porro et al., 2010), although, controversially, it has recently been
proposed that only one or two subtelomeres are responsible for TERRA transcription (Lopez de Silanes
et al., 2014; Montero et al., 2016). Cohesin, involved in regulating sister-chromatid cohesion, and CTCF,
a chromatin organising factor, are components of human subtelomeres which are involved in the
regulation of TERRA expression (Deng et al., 2012). DNMT3B, a DNA methyltransferase which regulates
the methylation of repetitive sequences (Hansen et al., 1999), has also been implicated in the control of
TERRA transcription (Yehezkel et al., 2008). Telomere length is involved in the regulation of TERRA levels
via telomeric H3K9me3 density (Arnoult et al., 2012). Interaction between TRF1 and RNA polymerase II
is also involved in regulating TERRA levels (Schoeftner and Blasco, 2008). TERRA expression is associated
with the cell cycle, with levels peaking during early S phase before decreasing towards the end of S
phase/G2 (Arnoult et al., 2012; Flynn et al., 2011; Porro et al., 2010). The low TERRA levels in late S phase
facilitate DNA replication of the telomeres and as cells pass through mitosis and into the following G1
phase TERRA expression increases again (Porro et al., 2010).
TERRA transcripts are retained at telomeres post-transcription (Azzalin et al., 2007; Ho et al.,
2008; Schoeftner and Blasco, 2008) and can form DNA-RNA hybrids due to the GC-rich nature of the
sequences (Arora et al., 2014; Balk et al., 2013; Pfeiffer et al., 2013). Telomere DNA-RNA hybrids are

9
regulated by the RNA endonuclease RNaseH1 (Arora et al., 2014). TERRA is also able to form G-
quadruplex structures and can bind to telomere interacting proteins (Biffi et al., 2014; Xu et al., 2010).
Heterogeneous nuclear ribonucleoproteins (hnRNPs) bind to telomeric ssDNA (Lopez de Silanes et al.,
2010), an interaction regulated by TERRA (Yamada et al., 2016). TERRA assists in T-loop formation by
promoting POT1 binding to the telomeric ssDNA after DNA replication through its interaction with
hnRNPA1 which will lead to RPA displacement from the ssDNA to allow POT1 binding (Flynn et al., 2011).
Telomeric transcripts can bind to TRF1 and TRF2 (Azzalin et al., 2007; Deng et al., 2009; Schoeftner and
Blasco, 2008) and telomeric heterochromatin formation is in part mediated by the interaction of TRF2
and TERRA (Deng et al., 2009). This interaction also recruits the origin replication complex (ORC) to the
telomere for the initiation of replication and formation of heterochromatin (Deng et al., 2009).
1.2. Telomeres and replicative aging
1.2.1. Senescence
Normal somatic cells have a limited proliferative capacity in vitro (Hayflick, 1965; Hayflick and
Moorhead, 1961). As proliferation ceases the cells undergo numerous biochemical and morphological
changes as they enter a state of permanent growth arrest known as senescence. It was postulated by
Olovnikov that the end replication problem was responsible for the initiation of senescence (Olovnikov,
1971). The end replication problem refers to the inability of the DNA replication machinery to fully
replicate the end of linear DNA molecules. DNA polymerases are unidirectional and require a primer to
initiate synthesis therefore, the gap caused by the terminal RNA primer cannot be filled which ultimately
results in the loss of telomeric sequence during each round of cell division (Levy et al., 1992). However,
this process generates only a small overhang, which is converted to a 100-200 bp overhang by an
exonuclease that acts in the 5’ to 3’ direction, shortening the C-rich telomere strand and thereby
increasing the length of the G-rich single-stranded telomeric tail (Makarov et al., 1997). In fibroblasts
telomere lengths decrease by 31-85 bp/population doubling (PD) (Harley et al., 1990) and mean
telomere lengths decrease with age in the human population (Hastie et al., 1990; Lindsey et al., 1991).

10
Thus, the terminal telomere sequence is lost at the end of each cell division until the cell is prompted to
exit the cell cycle.
Cells require functional telomeres to form structures such as the T-loop to prevent chromosome
end fusions or recognition of the end of the chromosome as a dsDNA break. However, as normal cell
division erodes the telomere sequence the telomeres become critically short and dysfunctional.
Depletion of shelterin proteins results in a dissociation between telomere length and the induction of
senescence (Karlseder et al., 2002; van Steensel et al., 1998) implying that alterations to the telomere
structure, and not just shorter telomere lengths, result in activation of senescence due to the DDR that
forms at a subset of telomeres (d'Adda di Fagagna et al., 2003). This DDR is visualised by the localisation
of DDR components such as p53-binding protein 1 (53BP1) or phosphorylated histone H2AX (γ-H2AX) to
the telomere where they form a telomere dysfunction induced focus (TIF) (Takai et al., 2003).
Visualisation of telomeres and γ-H2AX in metaphase spreads allow an accurate quantification of TIFs
(Cesare et al., 2009; Thanasoula et al., 2010). In young primary human cells a very low frequency of TIFs
is observed however, they accumulate in presenescent cells until at least five foci are present in
senescent nuclei (Kaul et al., 2012). Consequently, it has been postulated that telomeres exist in three
states (Cesare et al., 2013). The first state is a ‘closed-state’ where telomeres form structures that
prevent a DDR at the end of the chromosome. If the structure fails to form, the cells have an
‘intermediate-state’ telomere which induces a DDR but is still able to prevent repair by end-joining; if a
sufficient number of intermediate-state telomeres is present, then senescence is induced, associated
with changes in the level of histones and consequently the chromatin architecture (O'Sullivan et al.,
2010). The ‘uncapped-state’ allows chromosome end fusions to occur when cells continue to proliferate
beyond the normal induction of senescence.
Dysfunctional telomeres are only one of the stimuli that can induce senescence. Indeed, chronic
activation of a DDR regardless of the genomic site will induce senescence (Nakamura et al., 2008).
Double-stranded DNA breaks, potent activators of senescence, can be the result of ionising radiation,

11
topoisomerase inhibitors, and other types of cytotoxic chemotherapies (Chang et al., 2002; Novakova
et al., 2010; Robles and Adami, 1998; Schmitt et al., 2002; Sedelnikova et al., 2004). Oxidative stress
results in DNA damage to a nucleotide or a ssDNA break which gets converted to a dsDNA break during
base excision repair or DNA replication (Kuzminov, 2001; Wilstermann and Osheroff, 2001). The
resulting dsDNA break can ultimately initiate senescence. Oxidative stress has also been implicated in
telomere shortening by inducing damage at the G-rich sites of the telomere sequence (Kawanishi and
Oikawa, 2004). Therefore, senescence is frequently induced as a result of directly or indirectly generated
dsDNA damage.
Senescence can also be induced by upregulated or unbalanced mitogenic and proliferation-
associated signals. Chronic activation of the mitogen-activated protein kinase (MAPK) signalling
pathway will induce senescence in normal cells (Lin et al., 1998). Mutations to the Ras pathway including
the H-RASV12 mutant (Serrano et al., 1997) and oncogenic mutations to BRAF (Michaloglou et al., 2005)
also result in senescence when the p53 and p16INK4A pathways are intact. Subsequent studies
determined that senescence is mediated by activation of the DNA damage signalling pathway in
response to oncogenic mutations (Bartkova et al., 2006; Di Micco et al., 2006; Mallette et al., 2007).
There is emerging evidence that genes frequently altered in cancer, including c-Myc (Wu et al., 2007),
CDKN1B (Lin et al., 2010), SKP-2 (Lin et al., 2010) and PTEN (Chen et al., 2005), are involved in mediating
senescence in normal cells.
1.2.2. Immortalisation
Senescence is primarily established or maintained by activation of the p53/p21 and p16INK4A/pRB
pathways (Beausejour et al., 2003). Therefore, the p53, pRb and p16INK4A proteins are considered tumour
suppressor proteins and inactivation of their normal function is associated with an increased
proliferative capacity in cells (Beausejour et al., 2003; Gire and Wynford-Thomas, 1998). The normal
function of p53 can be abrogated as the result of spontaneous loss of the wild type TP53 allele of Li-
Fraumeni cells (a syndrome where one TP53 allele contains a null-mutation) (Rogan et al., 1995),

12
transduction with mutated TP53 (Bond et al., 1994), or infection with human papillomavirus (HPV).
Regardless of the mechanism, loss of p53 function results in proliferation beyond the PDs that generally
induce senescence (Bond et al., 1999). Adenovirus, HPV and Simian virus 40 (SV40) are DNA viruses that
express proteins capable of binding to and inactivating p53 and pRB tumour suppressor proteins. When
the SV40 T-antigen (binds p53 and pRB), E1A/E1B (binds pRB/p53 respectively) adenovirus genes, or the
E6/E7 (binds p53/pRB respectively) HPV genes are artificially expressed in human fibroblasts the cells
have an increased proliferative capacity (Shay et al., 1991). Loss of up- or down-stream genes in the p53
or pRB pathways also result in the bypass of senescence (Brown et al., 1997; Dimri et al., 2000;
Huschtscha et al., 1998; O'Loghlen et al., 2015; Okamoto et al., 1994; Vogt et al., 1998). Therefore, a
critical step in the development of cells with indefinite replicative potential, or cellular immortality, is
inactivation of the p53 or pRB pathways (Duncan et al., 1993; Lehman et al., 1993; Rogan et al., 1995;
Shay et al., 1995).
With bypass of senescence by inactivation of the p53 or pRB pathways the telomere sequence
continues to be eroded during each round of cell division. The short, unprotected telomere ends are
able to undergo classic NHEJ or alternative end joining (alt-NHEJ) (Rai et al., 2010; Sfeir and de Lange,
2012) when there are <13 pure TTAGGG repeats distal to the final telomere variant repeat (Capper et
al., 2007). The resultant di-centric chromosomes lead to chromatin bridges between the daughter cells
during cell division and the resolution of these bridges causes genomic instability (Artandi et al., 2000;
Soler et al., 2005). The telomeres will eventually become critically short and induce the cells to enter a
period of crisis where most cells will undergo apoptosis (Girardi et al., 1965). In the case of human
fibroblasts, approximately 1 in 107 cells escape crisis (Huschtscha and Holliday, 1983) by activation of a
telomere lengthening mechanism (TLM) which stabilises the telomere lengths and allows unlimited
proliferation of the cells hence the immortal nature of post-crisis cells (Counter et al., 1992; Meyerson
et al., 1997; Rogan et al., 1995) (Figure 1.2). Cellular immortalisation is a hallmark of cancer (Hanahan
and Weinberg, 2000; Hanahan and Weinberg, 2011) and to date two TLMs have been described:

13
Figure 1.2. Telomere length during cell division.
Cells that have constitutively active telomerase activity such as embryonic stem cells may
completely maintain telomere length (blue line). Stem cells that express tightly regulated levels of
telomerase partially maintain telomere length (green line). In normal somatic cells telomere
erosion occurs as a result of normal cell division (grey line) until cells reach senescence. A
proportion of cells may acquire an abrogation to the p53 or pRb pathway that allows the cells to
overcome senescence. Cell division continues until cells enter crisis. A small proportion of cells may
escape crisis through activation of a telomere lengthening mechanism which allows continued cell
division without telomere length erosion (red line).
telomerase (Greider and Blackburn, 1989) and alternative lengthening of telomeres (ALT) (detailed in
section 1.4.).

14
1.3. Telomere length regulation
The telomere length of normal human cells is determined by a balance of lengthening and shortening
events (Gilson and Londono-Vallejo, 2007). Overlengthening of telomeres by activation of a TLM results
in telomere trimming and the generation of t-circles (Pickett et al., 2009). A t-circle refers to double-
stranded circular telomeric DNA that has a nick in both the C- and G-rich strands preventing the structure
from becoming supercoiled (Cesare and Griffith, 2004; Wang et al., 2004). They are generated when
telomere lengths are extended past a threshold length, likely due to resolution of the HJ formed by the
T-loop (Pickett et al., 2009). A number of genes are involved in the regulation of t-circles (XRCC3
(Compton et al., 2007; Wang et al., 2004), NBS1 (Compton et al., 2007; Wang et al., 2004), SLX4 (Sarkar
et al., 2015), ZBTB48 (encodes TZAP) (Li et al., 2017) and RTEL1 (Deng et al., 2013)) and their involvement
in homologous recombination and DNA replication supports this hypothesis. Recently, TZAP has been
implicated in the initiation of telomere trimming as long telomeres have a reduced concentration of
shelterin proteins which allows TZAP to bind to the telomere, and it is this switch from shelterin to TZAP
that is proposed to initiate telomere trimming (Li et al., 2017). T-circles are conserved in mammals
(Pickett et al., 2011; Rivera et al., 2017), yeast (Lustig, 2003), and plants (Watson and Shippen, 2007)
and observed in normal human and mouse cells (Pickett et al., 2011) indicating that telomere trimming
is a conserved process with a role in normal telomere biology. This process is tightly controlled and does
not result in activation of a DNA damage response or telomere fusions (Pickett et al., 2009).
1.4. Telomere lengthening mechanisms
1.4.1. Telomerase
1.4.1.1. Structure and function
In 1985 Greider and Blackburn discovered an enzyme in Tetrahymena capable of synthesising telomeric
sequence (Greider and Blackburn, 1985) by reverse transcribing the template region of its RNA subunit
(Greider and Blackburn, 1989) and in 1989 Morin described the human equivalent, telomerase (Morin,

15
1989). Telomerase, a ribonucleoprotein, is comprised of the catalytic reverse transcriptase domain
hTERT (human telomerase reverse transcriptase) (Nakamura et al., 1997), the human telomerase RNA
hTR (Feng et al., 1995), and the associated protein dyskerin (Cohen et al., 2007). In vitro studies indicate
that telomerase is a dimer comprised of two molecules each of hTERT, hTR and dyskerin (Cohen et al.,
2007; Sauerwald et al., 2013; Wenz et al., 2001) although there is debate about whether a higher order
structure is strictly necessary for telomerase function as several studies indicate that telomerase is
functional as a monomer (Alves et al., 2008; Errington et al., 2008). Experiments to isolate subunits of
the enzyme responsible for telomere elongation in yeast Saccharomyces cerevisiae utilised mutants
defective for telomere addition (Lundblad and Szostak, 1989). This led to a continual loss of telomere
length and ultimately senescence, a phenotype known as ever shorter telomeres (EST) with the gene
subsequently termed EST1. Further studies identified other EST genes including EST2 which was
subsequently identified as the catalytic component of telomerase in S.cerevisiae (Counter et al., 1997;
Lendvay et al., 1996).
The RNA subunit of telomerase permits de novo addition of six nucleotide repeats of telomere
sequence, 5’-GGTTAG-3’, to the 3’ single-stranded overhang of telomeres. The hTR transcript is
comprised of 451 nucleotides with an eleven nucleotide (5’-CUAACCCUAAC-3’) template region (Feng
et al., 1995), encoded from the TERC gene. The hTR RNA is composed of four domains; a pseudoknot
domain (CR2/CR3), a H/ACA box domain (CR6/CR8), CR4/CR5 domain, and C7 domain (Chen et al., 2000).
The H/ACA box domain is a characteristic of small nucleolar RNA and the region also contains a Cajal
body targeting sequence (CAB box) which facilitates localisation and retention of telomerase to Cajal
bodies (Ganot et al., 1997). Cajal bodies are subnuclear structures that contain several proteins such as
coilin and TCAB1. They provide a site for RNA processing and ribonucleoprotein assembly (Bauer and
Gall, 1997; Jady et al., 2003) and facilitate the recruitment of telomerase to telomeres.
The reverse transcriptase hTERT features: a reverse transcriptase motif located in the C-terminal
end of the protein, a telomerase specific region (T motif), dissociates activities of telomerase (DAT)

16
domain, the telomerase essential N-terminal (TEN) domain and the N-terminal end of the protein
(Armbruster et al., 2001; Moriarty et al., 2002; Nakamura et al., 1997; Xia et al., 2000). hTERT N-terminal
regions are essential for hTR binding (Moriarty et al., 2002; Xia et al., 2000), whilst the C-terminus of
hTERT and the TEN domain are responsible for the processivity of the telomerase enzyme (Akiyama et
al., 2015; Bryan et al., 2000; Hossain et al., 2002; Peng et al., 2001), and the DAT domain is essential for
correct telomerase localisation to the telomere (Armbruster et al., 2004).
Telomerase requires accessory proteins for in vivo synthesis, subcellular localisation and
function. The proteins dyskerin, NHP2, NOP10, GAR1, Nopp140, NAF1, pontin/reptin and TCAB1
associate with the hTR/hTERT complex (Venteicher et al., 2009; Venteicher et al., 2008). The H/ACA
ribonucleoproteins dyskerin, GAR1, NOP10 and NHP2 form a complex that can bind to H/ACA RNAs such
as hTR (Ganot et al., 1997; Henras et al., 1998; Watkins et al., 1998). This complex is required to stabilise
hTR and facilitate the synthesis of telomerase. Pontin and reptin act as a complex that mediates
telomerase synthesis through an interaction with hTERT and dyskerin (Venteicher et al., 2008). These
proteins are hypothesised to accumulate and stabilise hTR, potentially by pontin’s ATPase activity, and
are present at a pre-telomerase intermediate complex with hTERT. TCAB1 binds the CAB box region of
hTR, accumulates telomerase at Cajal bodies, and traffics it to the telomere (Chen et al., 2015; Stern et
al., 2012). Recently, additional proteins including ATR and ATM were identified as contributing to
telomerase recruitment to the telomere (Lee et al., 2015; Tong et al., 2015). Therefore, accurate
telomerase function in vivo requires an array of telomerase-associated proteins in addition to the
enzyme components hTR, hTERT and dyskerin.
1.4.1.2. Role in cancer and normal cells
Telomerase activity in normal human and cancer cells has been extensively characterised by telomerase
activity assays, most commonly the telomerase repeat amplification assay (TRAP) (Kim et al., 1994).
Telomerase activity is tightly controlled in normal human tissue, with activity detected during
embryogenesis in tissues such as the liver, skin, lung and muscle which is subsequently downregulated

17
prior to birth (Ulaner and Giudice, 1997; Ulaner et al., 2001; Wright et al., 1996). However, highly
proliferative tissues such as male germ cells, haematopoietic stem cells, epithelial cells including the
gastrointestinal tract and hair follicle, and activated lymphocytes require tightly regulated telomerase
activity to maintain their cell population (Bachor et al., 1999; Broccoli et al., 1995; Counter et al., 1995;
Harle-Bachor and Boukamp, 1996; Hiyama et al., 1996; Hiyama et al., 1995b; Ramirez et al., 1997;
Tanaka et al., 1998; Yashima et al., 1998; Yui et al., 1998).
Telomere dysfunction can result in short telomere syndromes. Dyskeratosis congenita was the
first short telomere syndrome identified and is generally caused by mutations in the genes for hTR
(Mitchell et al., 1999; Vulliamy et al., 2001a), hTERT (Armanios et al., 2005) or dyskerin (Heiss et al.,
1998) that lead to defective telomerase, excessively short telomeres and reduced proliferation of highly
regenerative cells such as the bone marrow and skin (Drachtman and Alter, 1992; Tummala et al., 2015).
Mutations in telomerase genes, RTEL1, TCAB1, TIN2, TPP1 and PARN (Calado et al., 2011; Guo et al.,
2014; Kocak et al., 2014; Stuart et al., 2015; Tummala et al., 2015) have been identified in aplastic
anaemia (Vulliamy et al., 2002), Hoyeraal-Hreidarsson syndrome (Knight et al., 1999; Marrone et al.,
2007), Coats plus syndrome (Anderson et al., 2012; Polvi et al., 2012), Revesz syndrome (Savage et al.,
2008), and idiopathic pulmonary fibrosis (Armanios et al., 2007; Tsang et al., 2012). Patients who have
germ-line mutations in a telomerase gene also have significantly shorter telomeres than healthy age
matched individuals (Alder et al., 2008; Marrone et al., 2007; Vulliamy et al., 2002; Vulliamy et al.,
2001b).
To ensure that telomerase activity only occurs in specific normal cell types there is tight
regulation over the expression of hTERT and hTR. hTERT is a limiting factor for telomerase expression as
it has either low expression or is absent in telomerase-negative cells and ectopic expression of hTERT in
these cells is sufficient to reactivate telomerase (Bodnar et al., 1998; Counter et al., 1998b; Hahn et al.,
1999; Meyerson et al., 1997; Wen et al., 1998). Expression of hTERT can be regulated at several levels;
including transcription, alternative splicing, and post-translational modifications (Cong et al., 1999; Cong

18
et al., 2002; Horikawa et al., 1999; Liu et al., 1999; Takakura et al., 1999). hTR expression is present at
limiting levels in all cell types and can become upregulated once hTERT is expressed (Avilion et al., 1996;
Cao et al., 2008a; Cao et al., 2008b; Feng et al., 1995). Immortalisation is considered a critical step to
oncogenesis, achieved when cells in crisis activate a TLM. Telomerase is responsible for the
immortalisation of up to 90% of human cancers (Kim et al., 1994; Shay and Bacchetti, 1997) by
preferentially extending the shortest telomeres in the cell (Britt-Compton et al., 2009; Hemann et al.,
2001; Teixeira et al., 2004). Initially two models were proposed to explain the presence of telomerase
activity in cancer cells; the expansion of a stem cell population that failed to undergo differentiation and
subsequently formed cancers (Greaves, 1996), or the dysregulation of endogenously expressed
telomerase to overcome crisis (Shay and Wright, 1996). Subsequent studies identified upregulation of
endogenously expressed hTERT or hTR as frequent events in cancer cells suggesting telomerase activity
is upregulated during tumourigenesis to bypass crisis (Bell et al., 2015; Cao et al., 2008b; Fredriksson et
al., 2014; Horn et al., 2013; Huang et al., 2013; Horikawa et al., 1999; Peifer et al., 2015; Wang et al.,
1998; Wu et al., 1999; Zhang et al., 2000). Endogenous expression of hTERT can be upregulated in cancer
cells through a number of mechanisms including abnormally high levels of Myc activity (Horikawa et al.,
1999; Wang et al., 1998; Wu et al., 1999), gene amplification (Cao et al., 2008b; Zhang et al., 2000),
C228T and C250T mutations in the promoter region leading to the introduction of transcription factor
binding sites (Bell et al., 2015; Fredriksson et al., 2014; Horn et al., 2013; Huang et al., 2013), and
structural rearrangements upstream of the promoter region that result in super-enhancer elements
(Peifer et al., 2015).
1.4.1.3. Telomerase inhibitors as anti-cancer therapeutics
Telomerase is an attractive target for anti-cancer therapies because its expression is crucial for the
majority of cancers and a minority of normal cells. There are a number of strategies for telomerase anti-
cancer therapeutics: the direct inhibition of telomerase, stabilisation of the telomere structure to
prevent telomerase binding, or inducing cell death specifically in telomerase expressing cells (Shay and
Keith, 2008). A number of potential anti-cancer therapies have progressed to clinical trials including the

19
antisense oligonucleotide GRN163L (targeting hTR) (Chanan-Khan et al., 2008; Roth et al., 2010; Ruden
and Puri, 2013), immunotherapy GV1001 (hTERT peptide based vaccine) (Bernhardt et al., 2006;
Brunsvig et al., 2011; Buanes et al., 2009; Kyte, 2009; Ruden and Puri, 2013; Shaw et al., 2010a; Shaw et
al., 2010b), and immunotherapy GRNVAC1 (dendritic cell vaccine to stimulate TERT-specific CD4+ &
CD8+ cells) (DiPersio et al., 2009; Khoury et al., 2010; Ruden and Puri, 2013). However, the results of
these trials are still pending and there is currently no data available suggesting that these drugs improve
patient outcome by inhibiting telomerase activity (Jafri et al., 2016). There are also concerns about the
length of time required for telomerase inhibitors to induce telomere shortening and hence cell death or
senescence. This has led to the development of novel drugs such as 6-thio-2'-deoxyguanosine (6-thio-
dG), a nucleoside analogue which can be incorporated into telomerase mediated de novo synthesised
telomeric DNA, resulting in telomere dysfunction which leads to rapid cell death (Mender et al., 2015).
There is also evidence that treating cells with a combination of PARP and telomerase inhibitors reduces
the time required to observe telomere shortening (Lu et al., 2014; Seimiya et al., 2005). So, whilst there
are currently no telomerase targeted therapies as a standard cancer treatment, telomerase remains a
potential target for the development of novel therapies.
1.4.2. Alternative Lengthening of Telomeres (ALT)
The most common TLM used by human cancers is telomerase, however, a subset of cancer cells and
immortalised cell lines utilise the alternative lengthening of telomeres (ALT) to confer immortalisation.
ALT is defined as any telomerase-independent method of telomere length maintenance, which generally
utilises a DNA template to synthesise de novo telomeric sequence (Dilley et al., 2016; Dunham et al.,
2000). The existence of ALT in human cells was first demonstrated by the observation that some
immortalised cell lines maintain their telomere length without any detectable telomerase activity (Bryan
et al., 1997; Bryan et al., 1995) and it has subsequently been estimated 10-15% of human cancers utilise
the ALT mechanism to achieve unlimited proliferation (Henson et al., 2002). The molecular details of
the ALT mechanism are not yet fully understood but studies have ascertained a number of
characteristics that are hallmarks of the ALT mechanism.

20
1.4.2.1. Phenotypic characteristics of ALT
For over a decade since the discovery of ALT there was no biochemical assay to determine ALT activity
and the only definitive way to confirm cells utilise the ALT mechanism is to examine telomere length in
telomerase-negative cells over time. However, this requires serial untreated samples from the same
patient which are generally impossible to procure. Hence, a combination of ALT markers have
historically been used to determine ALT activity in human cells.
One of the most notable phenotypes of ALT is the presence of long and heterogeneous telomere
lengths within cells. Telomere lengths, in a mass population of cells, are observable by Southern blot
analysis of pulse field gel electrophoresed terminal restriction fragment (TRF) lengths, considered the
gold-standard technique to measure telomere lengths from a population of cells. From this technique,
the range of telomere lengths in ALT cells varies from <3 kb to >50 kb with an average length of 20 kb
(Bryan, 1997; Bryan et al., 1995; Grobelny et al., 2000; Murnane et al., 1994; Yeager et al., 1999)
compared to telomerase-positive cells which have relatively homogeneous telomere lengths with an
average length usually less than 10 kb (Bryan et al., 1995; Park et al., 1998). However, a large amount
of high quality genomic DNA (at least 2 g) is required to perform the analysis, often a limitation when
examining tumour samples. Telomere lengths within a single cell can be observed by telomere
fluorescent in situ hybridisation (FISH) on metaphase spreads. The telomere probe is visible at a range
of intensities within an individual ALT cell, with some chromosomes lacking a detectable signal
altogether, so called signal-free ends. Comparisons of FISH and TRF analysis confirm that TRFs reflect
the heterogeneous nature of telomere lengths within a single ALT cell (Lansdorp et al., 1998; Perrem et
al., 2001). Alternatively, telomere FISH can be performed on interphase cells allowing the examination
of telomere lengths in a range of sample types including formalin-fixed paraffin-embedded (FFPE)
tissues. Although long and heterogeneous telomere lengths are a marker of ALT in most situations,
ectopic overexpression of hTR and/or hTERT can result in extreme telomere lengthening and an average
length comparable to ALT cells (Cao et al., 2008b; Pickett et al., 2009). Therefore, telomere length alone
is not sufficient to indicate ALT activity. The ability to examine individual telomeres has also shown that

21
telomeres in ALT are very dynamic with rapid unsynchronised elongation and deletion events (Murnane
and Sabatier, 2004).
Promyelocytic leukaemia nuclear bodies (PML-NBs) are present in the nuclei of the majority of
mammalian cells (Bernardi and Pandolfi, 2007) and have been implicated in a number of cellular
processes including cell cycle regulation and DDR (Bernardi and Pandolfi, 2003; Dellaire and Bazett-
Jones, 2004; Dellaire et al., 2006), tumour suppression (He et al., 1997), induction of senescence and
apoptosis (Bernardi and Pandolfi, 2003; Ferbeyre et al., 2000; Jiang and Ringertz, 1997), transcriptional
regulation (Luciani et al., 2006; Zhong et al., 2000), protein refolding and degradation (Mattsson et al.,
2001), the immune system (Zheng et al., 1998), and viral replication (Chung and Tsai, 2009). PML-NBs
contain a number of proteins, including Sp100 and isoforms of PML, and in ALT cells a subset also contain
telomeric DNA and shelterin proteins, in addition to DNA repair, recombination and replication factors
(Henson et al., 2002; Jiang et al., 2005; Jiang et al., 2007; Yeager et al., 1999; Zhong et al., 2007). Hence,
such foci are termed ALT-associated promyelocytic leukemia nuclear bodies (APBs) (Yeager et al., 1999).
APBs associate with the termini of chromosomal telomeric DNA implying that this is a transient structure
and the PML-NBs can dissociate from the telomere to allow normal cellular processes to occur
(Molenaar et al., 2003). Methionine restriction in cells leads to an increase in the number of discernible
APBs and has shown that TRF1, TRF2, TIN2, RAP1, PML, HP1 and the MRN (comprised of MRE11, RAD50
and NBS1) complex are necessary for APB formation (Henson et al., 2002; Jiang et al., 2005; Jiang et al.,
2007; Zhong et al., 2007). APB formation also requires proteins of the SMC5/6 complex (Potts and Yu,
2007). It has been hypothesised that APBs are the site of ALT activity, however, to date there is no
conclusive proof that this is the case. Evidence in support of this hypothesis includes a reduction in the
number of APBs following inhibition of ALT (Jiang et al., 2005; Perrem et al., 2001) by sequestration of
the MRN complex (a complex involved in homologous recombination), and the increased frequency of
APBs when cells approach senescence (Fasching et al., 2007; Jiang et al., 2007; Jiang et al., 2009). There
is also evidence that the intensity of telomeric FISH signals increase in a subset of APBs compared to
other telomeres (Yeager et al., 1999). Additionally, APB levels increase during the G2 phase of the cell

22
cycle which is when homologous recombination occurs most frequently and is likely when the ALT
mechanism is active (Grobelny et al., 2000; Wu et al., 2000). The APB assay requires interphase cells and
can be performed on a range of fixed cells including FFPA tissues and has often been used, in conjunction
with telomere length, to determine the frequency of ALT in tumour cohorts.
ALT cells have a high abundance of extrachromosomal telomeric repeats which can be either
linear or circular in form (Cesare and Griffith, 2004; Nabetani and Ishikawa, 2009; Ogino et al., 1998;
Pickett et al., 2009; Tokutake et al., 1998; Wang et al., 2004). Extrachromosomal telomeric DNA likely
results from rapid deletion events, and is a feature of ALT cells. T-circles are the product of telomere
trimming (see section 1.3.) and are more abundant in ALT cells than normal cells or endogenously
expressing telomerase cells (Cesare and Griffith, 2004; Fasching et al., 2005; Henson et al., 2009; Wang
et al., 2004). This greater proportion of t-circles is likely due to resolution of the T-loop during telomere
trimming of over-lengthened telomeres as a result of the rapid elongation events caused by the ALT
mechanism (Pickett and Reddel, 2012). When telomeres are over-lengthened by telomerase, generally
via overexpression of hTERT and/or hTR, abundant t-circles are also generated at levels comparable to
ALT-positive cell lines indicating it is telomere length and not a specific aspect of the ALT mechanism
that results in t-circle formation (Pickett et al., 2009).
C-circles are a type of circular telomeric DNA that appear to be closely related to the ALT
mechanism. C-circles are a C-rich circular telomeric sequence that is only partially double-stranded and
therefore able to self-prime a rolling circle amplification assay (Henson et al., 2009). G-circles are also
specific to ALT cells however, they occur in far lower frequency than C-circles (Henson et al., 2009). C-
circles are a specific and quantitative marker of ALT activity, not found in mortal or telomerase-positive
cells but detectable in all ALT cell lines assayed to date (Henson et al., 2009). Most importantly,
treatment of ALT cells with high levels of YFP-Sp100 fusion protein to sequester the MRN complex and
inhibit ALT, produced a rapid decrease in C-circle levels (Henson et al., 2009). Patients with ALT-positive
osteosarcomas had detectable C-circles in their blood samples (Henson et al., 2009), signifying the

23
potential the C-circle assay has as a routine clinical test to diagnose ALT tumours. The discovery of C-
circles and a simple, rapid detection method for ALT is an important development for identifying TLMs
in cancer patients and facilitates the development of anti-cancer therapeutics to target TLMs.
Telomere variant repeats have also been examined in the context of ALT. Minisatellites are
tandem variant repeat sequences 6-100 bp in length (Vergnaud and Denoeud, 2000). Notably, there is
increased instability at the MS32 minisatellite locus in ALT cells although the frequency of mutation
varies between cell lines (Jeyapalan et al., 2005). Further structural differences have been noted
between the telomeres of ALT cells and non-ALT cells. In mortal or telomerase-positive cells, variant
repeats are confined to the 2 kb region proximal to the end of the telomere (Baird et al., 1995; Coleman
et al., 1999). However, homologous recombination in ALT cells has led to the dispersal of variant repeats
throughout their telomeres, most frequently the TCAGGG variant but TTCGGG and GTAGGG have also
been observed (Conomos et al., 2012; Varley et al., 2002). Orphan nuclear receptors have been
identified at ALT telomeres through binding to the variant telomeric sequence TCAGGG, which in turn
recruits the zinc finger protein ZNF827 to the telomere (Conomos et al., 2014; Conomos et al., 2012;
Dejardin and Kingston, 2009). Subsequently, the histone deacetylation and nucleosome remodelling
complex NuRD is recruited to the telomere through its binding with ZNF827 which leads to telomeric
chromatin remodelling and facilitates the telomere-telomere interaction required for homologous
recombination and ALT activity (Conomos et al., 2014).
The telomeric transcript TERRA can form DNA-RNA hybrids with telomeres and has been found
to localise to APBs (Arora et al., 2014). TERRA levels are higher in ALT-positive cells than telomerase-
positive cells and these levels are associated with telomere length (Arora et al., 2014; Episkopou et al.,
2014; Kreilmeier et al., 2016; Ng et al., 2009). The association between ALT activity and TERRA level may
be the result of reduced sub-telomeric methylation in ALT cells (Ng et al., 2009) or the reduced
chromatin compaction at telomeres which promotes not only the ALT mechanism but telomere
transcription (Episkopou et al., 2014). Similarly, depletion of the chromatin remodeller α-thalassemia

24
mental retardation X-linked (ATRX) protein, an event observed in a subset of ALT cells and tumours
(discussed in section 1.4.2.3) (Heaphy et al., 2011a; Lovejoy et al., 2012), results in decreased TERRA
expression in telomerase-positive cells due to increased telomeric chromatin compaction, reduced RNA
polymerase II recruitment, and decreased cohesion abundance at telomeres (Eid et al., 2015; Episkopou
et al., 2014). A study in S. cerevisiae suggests that TERRA expression is sufficient to induce recombination
(Yu et al., 2014). The evidence thus suggests that TERRA may be involved in telomere maintenance,
although we do not currently understand the role of TERRA in the human ALT mechanism.
Telomere-sister chromatid exchange (T-SCE) is a reflection of recombination at the telomere
and has been used as a marker for ALT (Bechter et al., 2004; Londono-Vallejo et al., 2004). Whilst some
studies reported that post-replicative telomere exchange events were detected at a greater frequency
in ALT-positive cells compared to telomerase-positive cells (Bechter et al., 2004; Londono-Vallejo et al.,
2004), there is also evidence that some telomerase-positive cell lines have comparable T-SCE levels to
ALT cells (Vera et al., 2008). T-SCEs may be more frequent in ALT cells because of the increased number
of short, more recombinogenic telomeres resulting from the ALT mechanism. However, T-SCEs can also
result from dsDNA break repair in the telomere and hence are not universally indicative of ALT activity
(Doksani and de Lange, 2016).
As identification of the ALT mechanism relies on the presence of phenotypic characteristics,
some more specific for ALT than others, it is important to confirm the classification via as many
techniques as possible. Given the specificity, and comparative ease of detection, the C-circle assay in
conjunction with telomere length testing can be rapidly used to identify ALT in tumour samples, even
with limited quantities of DNA (Lau et al., 2013).
1.4.2.2. Mechanistic details of ALT
The exact details of the ALT mechanism are currently unknown. The present model proposes that the 3’
single-stranded overhang of a telomere invades double-stranded telomeric DNA and anneals to the
complementary telomere strand which can act as a primer to synthesise new telomeric DNA, which is

25
then converted to dsDNA. As outlined below, there are a number of potential sources of template
telomeric DNA, including: sister chromatid exchange, replication within the t-loop structure,
homologous recombination from linear extrachromosomal DNA, and DNA rolling circle replication
(summarised in Figure 1.3.A, B, C, D).
Homologous recombination was first hypothesised to be involved in ALT telomere length
maintenance because of the dynamic nature of the telomeres observed in ALT cells. Homologous
recombination at the telomere can be detected in vivo in mammalian cells by inserting a DNA tag into
one telomere and demonstrating that it is copied to other non-homologous telomeres (Dunham et al.,
2000; Neumann et al., 2013; O'Sullivan et al., 2014) or duplicated in the original location (Muntoni et
al., 2009). This was observed in ALT but not telomerase-positive cells. These observations indicate that
ALT cells can use a telomere on a sister chromatid, another telomere, or even itself as a template to
copy telomeric sequence (Figure 1.3.A, B, C). This process appears to rely on break-induced replication
(Dilley et al., 2016; Roumelioti et al., 2016).
Break-induced replication is a process to repair DNA damage at the termini of the chromosome
(McEachern and Haber, 2006). A recent study indicates replication factor C (RFC) loads proliferating cell
nuclear antigen (PCNA) onto the exposed end of the chromosome, which is recognised as a site of DNA
damage, leading to polymerase δ recruitment to the telomere (Dilley et al., 2016). This leads to one end
of the break invading a template which is used for homology-directed telomere synthesis. In addition to
linear chromosomes, circular extrachromosomal telomeric DNA can act as a template for telomere
elongation via rolling circle amplification machinery (Henson et al., 2009; Natarajan and McEachern,
2002)(Figure 1.3.D). Introduction of exogenous t-circles to the telomerase negative Kluyveromyces lactis

26

27
Figure 1.3. Potential templates of homologous recombination mediated telomere lengthening.
Homologous recombination can use a number of templates to mediate telomere lengthening as
proposed in the ALT mechanism. These include (A) another telomere, (B) the T-loop of the same
telomere, and possibly (C) linear extrachromosomal DNA or (D) circular extrachromosomal DNA.
resulted in incorporation of the exogenous sequence into the chromosomal telomere and subsequent
spreading of the sequence by homologous recombination (Natarajan et al., 2003; Natarajan and
McEachern, 2002; Topcu et al., 2005).
The telomere templates used for homology-directed synthesis may be spatially close to the
termini of the telomere for example a sister chromatid, or spatially separated, such as a distant
telomere. To allow telomeres to find templates over long distances the cells require a mechanism to
move telomeres within the nuclei. Loss of the shelterin protein TRF2 results in a DDR at the telomere
and an increased mobility within the nuclei (Dimitrova et al., 2008). A spontaneous DDR is commonly
detected at the telomere of ALT cells (Cesare et al., 2009) and a subset of ALT telomeres have directional
movement which allows transient binding with each other and APBs (Molenaar et al., 2003). Of late,
evidence has emerged of a HOP2-MND1- and RAD51 dependent homology search mechanism that
drives rapid, directional, long-range movement of telomeres leading to telomere clustering, ultimately
initiating homology based telomere synthesis (Cho et al., 2014; Zhao and Sung, 2015). It is currently
unknown what proportion of the telomere ends utilise long-range movement to find template DNA for
the ALT mechanism.
1.4.2.3. Genetic changes and telomeric chromatin in ALT
It has been presumed that for the ALT mechanism to occur in cells, p53 would need to be inhibited to
allow recombination events to occur. There is some indirect evidence to suggest this is the case: in
glioblastoma, 78% of ALT tumours were p53 deficient but only 21% of telomerase-positive tumours

28
lacked p53 (Chen et al., 2006). However, this finding may be specific to glioblastoma, as ALT has also
been detected in other tumour types, such as primary neuroblastoma (NB) tumours which have a very
low incidence of p53 deficiency (Vogan et al., 1993; Kurihara et al., 2014; Lundberg et al., 2011; Onitake
et al., 2009; Valentijn et al., 2015). Of the ALT cell lines characterised to date only four have a wild type
TP53 allele and only one of these is known to have normal p53 protein levels (Henson and Reddel, 2010).
These data suggest that inhibition of p53 may be required in the majority of circumstances to permit
the ALT mechanism to activate, however, it is clearly not an absolute requirement for ALT to occur.
Genes that are involved in chromatin remodelling have recently been implicated in activation
of the ALT mechanism. An association between ALT-positive tumours and mutations to ATRX or Death-
Associated protein 6 (DAXX) was established in pancreatic neuroendocrine tumours (PanNETs) (Heaphy
et al., 2011a; Jiao et al., 2011; Scarpa et al., 2017). Studies in other tumour types including glioblastoma
(Schwartzentruber et al., 2012), liposarcoma (Lee et al., 2015) and NB (Kurihara et al., 2014) have
confirmed there is a correlation between ALT-positive tumours and ATRX mutations, although DAXX
mutations are rarely observed. Mutations in the histone variant H3.3, encoded by the gene H3F3A, have
also been associated with ALT (Schwartzentruber et al., 2012). ALT-positive cell lines frequently lack
ATRX expression (Bower et al., 2012; Lovejoy et al., 2012) although this is not necessarily due to loss-of-
function mutations in the gene (Lovejoy et al., 2012). A study of 22 ALT cell lines indicated that loss of
ATRX expression is not necessarily the result of somatic mutations, 19/22 cell lines had loss of ATRX
and/or DAXX protein but only 7/19 cell lines contained a loss-of-function mutation in ATRX and 0/19
had mutations in DAXX or H3F3A (Lovejoy et al., 2012). It is clear that loss of ATRX, DAXX and/or H3.3
expression is not solely responsible for initiation of the ALT mechanism as 14% of ALT cell lines exist
with normal levels of ATRX and DAXX and knockdown of either protein did not result in ALT
immortalisation of mortal cells (Lovejoy et al., 2012; Napier et al., 2015).
ATRX is a member of the ATP dependent SWI2/SNF2 family (Picketts et al., 1996) and mutations
to the ATRX gene cause alpha thalassemia in patients with a normal alpha globin gene which led to the

29
hypothesis that ATRX is involved in the regulation of gene expression (Gibbons et al., 1995). ATRX is
localised to PML bodies, pericentric heterochromatin, and sub-telomeric and telomeric DNA where the
protein is involved in regulating DNA methylation and the chromatin structure (De La Fuente et al., 2004;
Gibbons et al., 2000; Ritchie et al., 2008). To perform its chromatin remodelling function, ATRX forms a
complex with DAXX (Xue et al., 2003). DAXX can bind to H3.3 and the ATRX/DAXX complex can then
deposit H3.3 into ribosomal, telomeric, and pericentric repeat sequences (Drane et al., 2010; Goldberg
et al., 2010; Lewis et al., 2010). H3.3 is a histone variant associated with nucleosomes undergoing rapid
turnover at transcriptionally active sites (Ahmad and Henikoff, 2002). Therefore, H3.3 may be stabilising
the telomere chromatin structure as it does at other sites of rapid nucleosome turnover. Loss of either
ATRX or H3.3 in mouse embryonic stem cells resulted in an increased number of cells with five or more
TIFs (Wong et al., 2010) demonstrating that both proteins are important to maintaining telomere
function. ATRX and H3.3 are involved in maintaining the heterochromatin state across the genome and
mediate silencing of genetic regions including endogenous retroviral elements (Elsasser et al., 2015;
Sadic et al., 2015) and imprinted loci (Voon et al., 2015). Defective function of ATRX or H3.3 results in
hypo-methylation (Voon et al., 2015) and ALT cells have reduced compaction of telomeric chromatin
compared to telomerase-positive cells (Episkopou et al., 2014). However, knockdown of ATRX in isogenic
telomerase and ALT-positive cells led to increased telomeric chromatin compaction suggesting other
factors mediate the reduced telomeric chromatin compaction observed in ALT cells (Episkopou et al.,
2014). Furthermore, depletion of ASF1, a histone chaperone that interacts with histone regulator A
(HIRA) and the chromatin assembly factor 1 (CAF1) complex, led to the activation of ALT phenotypes in
a mortal and telomerase-positive cell line providing further evidence that normal chromatin structure
is critical in repressing ALT in normal cells (O'Sullivan et al., 2014).
1.4.2.4. ALT proteins
The hypothesis that ALT exploits normal homologous recombination pathways is supported by the
involvement of proteins utilised in DDR, homologous recombination, and DNA replication in the ALT
mechanism. Notably, a large proportion of these proteins are present in APBs lending support to the

30
hypothesis that they are the active site of the ALT mechanism (Henson and Reddel, 2010). Proteins
required for homologous recombination including RAD51, RAD52, RPA, NBS1, SLX4, BLM, MRN, BRCA1
and BRIT1, have been identified in the APBs of ALT-positive cells (Bhattacharyya et al., 2009; Cesare and
Reddel, 2010; Conomos et al., 2014; Wan et al., 2013; Wilson et al., 2013; Yeager et al., 1999). The
members of the MRN complex, involved in the early steps of homologous recombination (van den Bosch
et al., 2003), were some of the first proteins identified as critical for telomere lengthening by ALT. The
MRN complex recruits ATM to dsDNA breaks and facilitates resection of the chromosome end to create
the 3’ overhang for strand invasion and subsequent homologous recombination (Lee and Paull, 2007).
Therefore, the MRN complex may be involved in the initiation of ALT activity by recruiting ATM to the
telomere and creating a larger 3’ overhang at the telomere end to be used for strand invasion of
template telomeric DNA. Sp100 binds to and sequesters the MRN complex via its interaction with NBS1,
and overexpression of Sp100 led to the repression of ALT, where normal cell division based telomere
length attrition and a reduction in APBs was observed (Jiang et al., 2005). In support of the hypothesis
that the MRN complex is critical for the ALT mechanism, depletion of any individual component of the
complex resulted in loss of ALT mediated telomere maintenance (Zhong et al., 2007). The SMC5/6
recombination complex is comprised of 8 subunits, three of which (methy methanesulphonate-
sensitivity 21 (MMS21), SMC5, and SMC6) are required for ALT-mediated telomere maintenance (Potts
and Yu, 2007). The MMS21 SUMO ligase is required to sumoylate the telomere binding proteins TRF1,
TRF2, TIN2, and RAP1 (Potts and Yu, 2007; Wu et al., 2012a). Whilst the exact role of the SMC5/6
complex in ALT remains to be elucidated, the complex may recruit the telomere to APBs via sumoylation
of shelterin proteins.
Other components of dsDNA break repair machinery and DNA replication including Fanconi
anaemia group A (FANCA) (Fan et al., 2009), Fanconi anaemia group D2 (FANCD2) (Fan et al., 2009), flap
enonuclease 1 (FEN1) (Saharia and Stewart, 2009), and MUS81 endonuclease (Zeng et al., 2009), have
been implicated in the ALT mechanism although depletion of these proteins does not result in overall
loss of telomere length due to ALT inhibition. RecQ family DNA helicases are important for the resolution

31
of atypical DNA structures and mutations to the proteins result in chromosomal abnormalities and
disease in patients (Croteau et al., 2014). RecQ like proteins in humans include WRN, RECQL4, and BLM
(Ellis et al., 1995; Killoran and Keck, 2006; Yu et al., 1996), mutations in which are responsible for
Werner, Rothmund-Thomson, and Bloom syndrome, respectively. The binding partners of the BLM
protein, as identified by mass spectrometry, include heat shock protein 90 (HPSP90), telomerase-
associated protein 1 (TEP1), topoisomerase III alpha (TOPO IIIα), and the shelterin proteins TRF1 and
TRF2 (Bhattacharyya et al., 2009). Depletion of TOPO IIIα in ALT but not telomerase-positive cells
resulted in increased cell death and DNA damage at the telomere but a concurrent reduction in the level
of TRF2 protein could also account for these observations (Bhattacharyya et al., 2009; Temime-Smaali
et al., 2008). Interestingly, the WRN protein is not essential to the ALT mechanism (Laud et al., 2005)
despite its role in the separation of dsDNA, likely due to redundancy in the function of other RecQ
helicases. Indeed, recent studies have suggested that many DDR and homologous recombination
proteins maintain normal telomere function rather than, or perhaps in addition to, being required for
the ALT mechanism (Fan et al., 2009; Saharia and Stewart, 2009; Saharia et al., 2010; Verdun and
Karlseder, 2006; Zeng and Yang, 2009).
1.4.2.5. ALT in cancer and normal cells
The role of ALT has not been established in normal human cells and currently the best evidence for ALT
function in embryogenesis and normal somatic cells comes from murine models. Telomere length
analysis of oocytes and early embryos revealed that telomeres were short in oocytes but then
lengthened until blastocysts were formed (Liu et al., 2007). Telomerase activity remained consistently
low until blastocyst formation, demonstrating that telomerase activity alone was not responsible for the
increase in telomere length and indeed, the early cleavage embryos contained evidence of telomere
sister chromatid exchange and recombination suggestive of the ALT mechanism (Liu et al., 2007).
However, there is not complete concordance with these results, as a subsequent study incorporating a
tag into a single telomere only observed copying of the telomere via recombination in somatic cells and
not germ-line cells (Neumann et al., 2013). This discrepancy may be due to the different detection

32
methods used by each group and the inability of the telomere tag to detect intra-telomeric
recombination arising from the ALT mechanism (Neumann et al., 2013). The ALT mechanism has also
been detected in a range of eukaryotes including mosquitoes (Mason and Biessmann, 1995), the plant
Arabidopsis thaliana (Riha et al., 2001), the worm Caenorhabditis elegans (Cheng et al., 2012; Lackner
et al., 2012) and the yeasts C. cerevisiae (Lundblad and Blackburn, 1993) and K. lactis (McEachern and
Blackburn, 1996). These studies provide evidence that ALT activity is present in a range of cells and the
ALT mechanism observed in human cancers may be the result of a dysregulated normal TLM.
The prevalence of ALT in human cancers has not been as widely studied as telomerase due to
the difficulty in obtaining enough tumour material for assays and the need to perform multiple assays
for the identification of the ALT mechanism. However, this has changed with the recent development
of a telomere quantitative polymerase chain reaction (qPCR) based assay to detect C-circles which
allows ALT to be detected from as little as 30 ng of DNA (Lau et al., 2013). Lau et al. (2013) described a
cut-off for C-circle levels indicative of an active ALT mechanism, detected by qPCR, which gives 100%
concordance with APB assay results and the conventional C-circle assay detected by a P32-radiolabelled
telomere probe, validated in a cohort of 23 cell lines and 43 tumours including glioblastoma, melanoma,
and soft tissue sarcoma samples. The prevalence of ALT has been studied in cancers including
osteosarcoma, gastric carcinoma, soft tissue sarcoma, astrocytoma, mesothelioma, adrenocortical
carcinoma, ovarian carcinoma, melanoma, NB, medulloblastoma, breast carcinoma, colon carcinoma,
Ewings sarcoma, PanNETs and lung carcinoma (Figure 1.4. indicates prevalence of ALT according to
tumour tissue type) (Bryan et al., 1997; Costa et al., 2006; Else et al., 2008; Guilleret et al., 2002; Hakin-
Smith et al., 2003; Heaphy et al., 2011b; Henson et al., 2005; Jiao et al., 2011; Johnson et al., 2005;
Matsuo et al., 2009; Montgomery et al., 2004; Omori et al., 2009; Onitake et al., 2009; Sanders et al.,
2004; Scarpa et al., 2017; Subhawong et al., 2009; Ulaner et al., 2004; Ulaner et al., 2003; Villa et al.,
2008; Yan et al., 2002).

33
Figure 1.4. Prevalence of ALT in human cancer based on tumour tissue type.
The prevalence of ALT varies depending on the tumour tissue type with ALT yet to be identified in
any hematopoietic, pancreas or prostate tumours. Adapted from Dilley and Greenberg 2015.
The association between upregulation of ALT and prognosis is dependent on the tumour type.
ALT is utilised for immortalisation by approximately 50% of osteosarcomas but there is no difference in
survival between ALT-positive and ALT-negative patients (Henson et al., 2005). Interestingly, 29% of
liposarcomas are ALT-positive and these patients have a worse survival than telomerase-positive
patients (Pricolo et al., 1996). In contrast, glioblastoma patients with ALT tumours have a longer median
survival compared to non-ALT tumours (Hakin-Smith et al., 2003). The prevalence of ALT is also highly
variable in different tumour types, up to 60% of astrocytomas and sarcomas (neuroepithelial and
mesenchymal origin respectively) are ALT-positive compared to carcinomas (epithelial in origin) where
5-15% of tumours utilise ALT. This difference may be due to the regulation of ALT in specific cell types
(Henson and Reddel, 2010).

34
Normal cells appear to contain repressors of the ALT mechanism since the fusion of the ALT cell
line GM847 and normal fibroblasts resulted in a rapid loss of telomere sequence and onset of
senescence (Perrem et al., 1999). Hybridisation of ALT and telomerase-positive immortalised cell lines
created cells that utilised only one telomere maintenance mechanism (Bower et al., 2012; Ishii et al.,
1999; Perrem et al., 2001). This implies that telomerase-positive cells contain a repressor of ALT and
ALT-positive cells contain repressors of telomerase (Bower et al., 2012). Hence, the tumour types with
low or no ALT activity may contain repressors of ALT and tumour types with higher ALT prevalence may
more tightly regulate telomerase expression.
1.4.2.6. Anti-cancer therapeutics and ALT
To date, there are no ALT targeted anti-cancer therapeutics available, although there is some debate
regarding the potential to use ATR inhibitors as an anti-ALT therapy (Deeg et al., 2016; Flynn et al., 2015).
A better understanding of the mechanism and the prevalence of ALT is necessary to develop treatments
and determine patient groups eligible for targeted therapies. There is also evidence that cells within a
single tumour can utilise both telomerase and ALT (Bryan et al., 1997), although it is unclear if both exist
in a single cell or it is the result of heterogeneity within the tumour. It is possible to artificially generate
telomerase expression in ALT immortalised cell lines, and this has been used to demonstrate that both
mechanisms can co-exist in a single cell (Cerone et al., 2001; Grobelny et al., 2001; Perrem et al., 2001).
Consequently, anti-cancer therapies targeted to a TLM must take into consideration whether one or
both TLMs are present in the tumour.
Another concern with telomere maintenance mechanism targeted therapies is the possibility
that cells can change their active mechanism if one becomes non-functional. Inhibition of hTERT in a
telomerase positive cell line resulted in cells that could maintain telomere length and grow for over 200
PDs once telomerase became inactive, although the cells possessed no typical features of ALT such as
APBs or long and heterogeneous telomere lengths (Kumakura et al., 2005). This indicates, in principle,
that cells have the ability to activate alternative pathways for telomere length maintenance if their TLM

35
becomes compromised. Whilst, there is no evidence to suggest that human cancers treated with a
telomerase targeted anti-cancer therapeutic activate the ALT mechanism to continue telomere length
maintenance, telomerase inhibition has resulted in the emergence of ALT cells in mouse cells (Hu et al.,
2012). Therefore, targeting telomerase as an anti-cancer therapeutic has the potential to select for ALT-
positive cells which would be resistant to such therapies supporting the need to develop anti-cancer
therapeutics against both TLMs.
1.4.3. Telomere lengthening maintenance negative samples
Immortalisation via activation of a TLM is currently regarded as a hallmark of cancer (Hanahan and
Weinberg, 2000; Hanahan and Weinberg, 2011) although, it has been postulated that immortalisation
may not be required in cancers with a low level of tumour cell death or genetically simple cancers that
do not require extensive clonal evolution (Kipling, 1995; Parkinson, 1996; Reddel, 2000). A human
fibroblast isolated from a middle-aged person is capable of an additional 20-40 PDs (Hayflick, 1965). If
every cell survives, 40 PDs can generate 240 cells which would form a tumour mass of 1 kg. However, cell
death occurs in most tumours due to factors like an inadequate blood supply resulting in hypoxia and
necrosis, or due to non-viable cells created during the clonal evolution of the tumour. Consequently,
the majority of tumours activate a TLM to obtain the proliferative capacity to form a tumour mass. This
may not be required for tumours that have long telomere lengths in their progenitor cells, in cancers
such as leukaemias which do not undergo cell death due to limited blood supply, and in genetically
simple cancers. In keeping with the hypothesis that immortalisation is not an absolute requirement of
carcinogenesis, a substantial proportion of liposarcomas (Costa et al., 2006; Jeyapalan et al., 2008),
glioblastoma (Hakin-Smith et al., 2003), retinoblastomas (Gupta et al., 1996), and osteosarcomas
(Ulaner et al., 2003) have been reported as TLM-negative. While these tumours may be truly TLM-
negative, this could also be the result of false negative assays or an as-yet unidentified ALT mechanism
lacking the usual phenotypic characteristics. To date, it has been impossible to test which of these
scenarios is true, as the definitive test for telomere length maintenance is to examine telomere length
over time which would require serial samples of untreated human tumours, samples impossible to

36
obtain. It is increasingly important to define what is happening in this group of tumours as TLM-
inhibitors are moving toward clinical use and TLM-negative tumours would be resistant to such
treatment. Large-scale studies are also moving towards genetic markers such as telomere length and
hTERT and ATRX/DAXX mutations to define TLMs (Barthel et al., 2017; Pekmezci et al., 2017; Scarpa et
al., 2017; Valentijn et al., 2015) and the molecular details of TLM-negative tumours would ensure that
tumours are not misclassified.
1.5. Neuroblastoma
Neuroblastoma (NB) is an embryonal cancer that arises in the sympathetic nervous system. The disease
predominantly affects infants or young children, with an annual incidence of 29 patients/million infants
<1-year-old, which is reduced to one patient/million in children 10-14 years old (Lau and Irwin, 2017).
NB is the most common extracranial solid tumour in children, responsible for 7-10% of cancers in
children under 15 years, yet it accounts for 15% of deaths in paediatric oncology patients (Maris et al.,
2007).
1.5.1. Disease stages
The majority of NB patients (65%) present with a primary tumour in the abdomen, most commonly
adjacent to the adrenal gland. Other sites of primary disease include the pelvis, chest and neck (Maris
et al., 2007). Metastatic disease is present in nearly half of patients at diagnosis, either in lymph nodes
adjacent to the primary tumour, or at distant sites such as the bone marrow, cortical bones, the liver
and non-adjacent lymph nodes (Maris et al., 2007). Diagnosis can be made by the histology of the
tumour or the presence of tumour cells in the bone marrow combined with elevated catecholamine
levels in the urine. The tumour size and invasiveness and metastatic sites can be determined with CT or
MRI scan in conjunction with radiolabelled meta-iodobenzylguanidine (123I-MIBG).
Established in 1993, The International Neuroblastoma Staging System (INSS) (Brodeur et al.,
1993) is a classification system based on clinical and biological prognostic markers. However, a more

37
recent system of classification, the International Neuroblastoma Risk Group Staging System (INRGSS)
(Cohn et al., 2009), has been developed to ensure uniform risk stratification and comparison of clinical
trials across large international bodies such as the Children’s Oncology Group (COG) and the
International Society of Paediatric Oncology Europe Neuroblastoma (SIOPEN). The INRGSS classifies NB
into stages L1, L2, M, and MS. Stage L1 and L2 represent locoregional disease in the absence and
presence of image-defined risk factors, respectively. Stage M (equivalent to INSS stage 4) includes all
metastatic disease unless classified stage MS. Stage MS is similar to INSS Stage 4S with metastases
limited to the liver, skin and bone marrow, except that it includes patients up to age 18 months and
large primary tumours (L1 or L2) (Monclair et al., 2009). Stage MS, which will be referred to as stage 4S
in remainder of this thesis, is a unique group of NB that has the propensity to spontaneously regress in
the absence of treatment (D'Angio et al., 1971). The staging of NB reflects the heterogeneity of the
disease, ranging from tumours that can spontaneously regress, to metastatic disease that conveys a 2-
year event free survival of only 66% (Yu et al., 2010).
1.5.2. Risk stratification
There are a number of biological and clinical factors that need to be considered when stratifying NB
patients into risk groups, including: stage, age at diagnosis, histology of the tumour, differentiation
within the tumour, MYCN amplification, 11q aberration, and ploidy of the tumour. Based on these
features the INRGSS classifies NB patients into 16 pre-treatment risk categories (A to R) which are
further classified into risk subsets based on the 5-year event-free survival (EFS) of each category (Table
1.2); very low risk (5-year EFS >85%), low-risk (5-year EFS >75 to ≤85%), intermediate-risk (5-year EFS
≥50% to ≤75%) and high-risk (5-year EFS <50%) (Cohn et al., 2009; Monclair et al., 2009).
1.5.2.1. Histology and degree of differentiation
NB tumours are comprised of small, round blue cells with reduced cytoplasm and darkly staining nuclei.
A panel of antibodies is used to distinguish NB from other solid tumour types and the International
Neuroblastoma Pathology Classification System (INPC) uses a modified Shimada system to determine

38
the degree of Schwannian stroma (NB, stroma-poor; ganglioblastoma intermixed, stroma-rich;
ganglioneuroma, stroma-dominant; or ganglioneuroblastoma, composite rich/dominant and stroma-
poor), neuroblast differentiation (differentiating, poorly differentiated, or undifferentiated) and mitosis-
karyorrhexis index (high, intermediate or low) of the tumour (Shimada et al., 1999). The tumours are
classified into favourable or unfavourable histology groups based on the combination of these features
and age at diagnosis (Shimada et al., 2001).
1.5.2.2. Age at diagnosis
The median age at diagnosis of NB is 19 months, and 90% of patients are younger than 5 years of age at
diagnosis (London et al., 2005). Age at diagnosis correlates to the EFS of patients with Stage 4 MYCN
non-amplified NB, with children >24 months at diagnosis (6-year EFS of 31%) having a significantly
poorer prognosis than patients 12 to 18 months old at diagnosis (6-year EFS of 74%) (Schmidt et al.,
2005).
1.5.2.3. MYCN amplification
The proto-oncogene MYCN is located at chromosome 2p24 and amplification of the gene occurs in
approximately 22% of primary tumours and is associated with high-risk NB across all stages of disease
(Ambros et al., 2009; Bagatell et al., 2009; Brodeur et al., 1984; Cohn et al., 2009; Seeger et al., 1985).
Amplified MYCN DNA can be found in double-minute chromatin bodies (DMs) or homogeneously
staining regions (HSRs) which can be detected by MYCN FISH in the nuclei of NB cells (Corvi et al., 1994;
Schwab et al., 1984). MYCN amplification is defined as ≥10 copies of MYCN in a diploid cell or a >4-fold
increase in signal compared to chromosome 2. MYCN oncoprotein is a transcription factor that binds to
the promoter region of target genes involved in differentiation, cellular proliferation, and apoptosis,
through binding to E-box recognition sites (Murphy et al., 2009). Transgenic mouse models of MYCN
overexpression result in spontaneous NB, indicating MYCN expression has a role in NB oncogenesis
(Weiss et al., 1997), however, it remains to be elucidated by which mechanisms MYCN amplification
results in aggressive NB tumours. Currently, there is no successful therapeutic to target MYCN but as

39
Table 1.2. Neuroblastoma risk group classification for stages L1/L2, L1 and MS.
Stage L1/L2 L1 MS
Age (months) <18
Histology
Ganglioneuroma maturing;
ganglineuroblastoma intermixed
Any except ganglioneuroma
maturing or ganglineuroblastoma
intermixed
Differentiation
MYCN Non-
amplified Amplified Non-amplified Amplified
11q loss of heterozygosity
No Yes
Ploidy
INRG category A B K C Q R
Risk group Very low Very low High Very low High High
Table 1.3. Neuroblastoma risk group classification of stage L2.
Stage L2
Age (months) <18 ≥18
Histology
Any except ganglioneuroma maturing or ganglineuroblastoma
intermixed
Ganglineuroblastoma nodular or neuroblastoma
Differentiation Differentiating Poorly or un-differentiated
MYCN Non-amplified Non-amplified Non-amplified Amplified
11q loss of heterozygosity
No Yes No Yes
Ploidy
INRG category D G E H H N
Risk group Low Intermediate Low Intermediate Intermediate High
Table 1.4. Neuroblastoma risk group classification of stage M.
Stage M
Age (months) <18 <12 Dec-18 <18 ≥18
Histology
Differentiation
MYCN Non-amplified Non-amplified Non-amplified Amplified
11q loss of heterozygosity
Ploidy Hyperdiploid Diploid Diploid
INRG category F I J O P
Risk group Low Intermediate Intermediate High High

40
Aurora kinase A is required to stabilise MYCN protein, the inhibitor MLN8237 is currently in clinical trial
in combination with chemotherapy (Mosse et al., 2012) (clinical trial: NCT01601535). Inhibition of the
MYCN downstream target ornithine decarboxylase by difluoromethylorthinine is also in clinical trial
(clinical trial: NCT02030964).
1.5.2.4. Tumour ploidy and chromosome aberrations
Tumour ploidy is an independent prognostic factor, predominantly in patients less than 18 months of
age (George et al., 2005; Schleiermacher et al., 2010). The ploidy of a NB tumour refers to the DNA
content of the cells and is indicative of the type of genetic rearrangement that the tumour has
undergone. Hyperdiploid tumours have whole chromosome gains and therefore, little structural
rearrangement of chromosomes, which confers a more favourable outcome for patients (Kaneko et al.,
1987; Look et al., 1991). In contrast, diploid tumours are associated with advanced-stage disease and
poor prognostic markers including MYCN amplification and loss of 1p or 11q (Attiyeh et al., 2005; Cohn
et al., 2009; Kaneko et al., 1987; Look et al., 1991).
Loss of heterozygosity (LOH) of 11q has been identified in 34-45% of primary NBs (Attiyeh et al.,
2005; Guo et al., 1999; Plantaz et al., 1997; Spitz et al., 2003). 11q LOH does not correlate with MYCN
amplification but is associated with high-risk features such as unfavourable histology and poor survival.
Consequently, LOH of 11q has been identified as a prognostic factor in MYCN non-amplified tumours
and is used in the INRGSS to predict outcome in the stage L2 MYCN non-amplified tumours. The short
arm of chromosome 1 (1p) is deleted in 25-35% of NBs and correlates with MYCN amplification (Attiyeh
et al., 2005; Brodeur et al., 1981; Christiansen et al., 2000; Kaneko et al., 1987; Plantaz et al., 1997).
Allelic loss at 1p36 is associated with an increased risk of relapse, although it is only an independent
prognostic factor in low or intermediate risk tumours. Gain of additional copies of 17q, often occurs
through unbalanced translocation with chromosome 1 or 11, and is associated with aggressive disease
(Bown et al., 1999; Van Roy et al., 1997). It has been suggested that changes to ploidy are required for
the gain of tumour driver genes on 17q and a loss of tumour suppressor genes on 1p or 11q, however,

41
a recent study identified loss of 1p and 11q in relapse but not matched primary tumours (Eleveld et al.,
2015), suggesting these events are not tumour initiating but rather a step in aggressive tumour
evolution.
1.5.3. Treatment
NB treatment may vary depending on clinical trials group. The following therapies reflect COG protocols
for NB treatment. Low-risk NB has a good prognosis and a 5-year EFS of 75-85% (Monclair et al., 2009).
As such, minimal treatment is offered to patients who have tumours with favourable biology. If surgery
is performed, partial resection with residual disease is not a risk for relapse in these patients (Strother
et al., 2012). Chemotherapy (with or without surgery) consisting of low doses of carboplatin,
cyclophosphamide, doxorubicin and etoposide is given to patients who have life-threatening symptoms,
progressive disease after surgery, or unfavourable tumour biology in the resected tumour. In contrast,
stage 4S NB with no high-risk markers can be observed and given supportive care. Treatment with
chemotherapy is only given if the patient becomes symptomatic or the tumour has unfavourable biology
as most tumours spontaneously regress without any intervention (Nickerson et al., 2000).
Intermediate risk NB have a poorer survival (5-year EFS 50-75%) than low risk patients, and are
treated with chemotherapy and surgery (Baker et al., 2010). Chemotherapy can consist of 4 or 8 cycles
of carboplatin, cyclophosphamide, doxorubicin and etoposide. Four cycles are given for tumours with
favourable biology whilst 8 cycles are used when the biology is unfavourable.
The standard regimen for high-risk NB treatment consists of three phases: induction,
consolidation, and post-consolidation (Matthay et al., 1999; Park et al., 2011). The current COG trial
protocol for induction therapy involves 6 cycles of dose-intensive chemotherapy. The first two cycles
include a combination of topotecan and cyclophosphamide, then a combination of etoposide and
cisplatin for cycles 3 and 5, and for cycles 4 and 6 a combination of doxorubicin, cyclophosphamide and
vincristine (Park et al., 2011). The response to induction therapy is a prognostic indicator for high-risk
patients, and correlates with patient survival after the completion of therapy (Cheung et al., 1991).

42
Consequently, tumours are monitored with MIBG scoring systems at diagnosis and at the end of
induction therapy (Yanik et al., 2013). Local resection of the tumour is recommended after 4-6 cycles of
chemotherapy, although it is debatable that complete resection improves the outcome of patients
(Alonso et al., 2006; Castel et al., 2002; Du et al., 2014; Simon et al., 2013). Patients then receive
consolidation therapy consisting of myeloablative chemotherapy and autologous stem cell
transplantation. Myeloablative chemotherapy comprises of treatment with melphalan, followed by
carboplatin and etoposide. After completion of consolidation therapy, patients may also receive
radiation treatment. Though patients receive multi-modal therapy, relapse remains common in high-
risk NB, suggesting that minimal residual disease is an important factor in recurrence. To treat minimal
residual disease, it is standard to use non-cytotoxic differentiation therapy (treatment with 13-cis
retinoic acid), as post-consolidation therapy for high-risk NB (Matthay et al., 2009b) in addition to
immunotherapy with anti-ganglioside 2 (GD2) chimeric antibody (Simon et al., 2011) and cytokines
(interleukin-2 [IL-2] and granulocyte macrophage colony-stimulating factor [GMCSF]) (Yu et al., 2010).
Despite intensive treatment many high-risk patients have refractory disease or experience
recurrence, and there is no curative therapy for the majority of these patients (London et al., 2011).
Indeed, stage 4 patients with relapsed disease only have a 5-year overall survival of 8% (London et al.,
2011). Relapsed patients generally present with metastatic disease, that has acquired mutations as a
result of primary therapy and are consequently drug resistant (Carr-Wilkinson et al., 2010; Keshelava et
al., 2001; Tweddle et al., 2001). Therefore, patients that are treated with further chemotherapy, receive
agents with a mode of action distinct from therapies used in the initial treatment. Examples of relapse
therapy include temozolomide with irinotecan, topotecan in combination with etoposide or
cyclophosphamide, high dose 131I-MIBG therapy and a second autologous stem cell transplantation
(Kushner et al., 2006; Matthay et al., 2009a; Simon et al., 2007). Patients can also be enrolled in early
phase clinical trials.

43
1.5.4. Predisposition genes
Familial NB is rare, accounting for only 1-2% of NB cases, and is generally autosomal dominant with
incomplete penetrance (Kushner et al., 1986; Maris et al., 2002). Correlation between mutations and
cases of familial NB led to candidate predisposition regions being located at 16p, 12p and 2p23-26 (Maris
et al., 1997), ultimately resulting in the identification of the anaplastic lymphoma kinase (ALK) gene. A
proportion of the ALK mutations identified in patients are the result of germline missense mutations
(Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mosse et al., 2008). Patients with
NB associated with Hirschsprung disease or congenital central hypoventilation have germline PHOX2B
mutations (Mosse et al., 2004; Trochet et al., 2004). Consequently, the two most common
predisposition genes associated with NB tumour formation are ALK and PHOX2B. Single nucleotide
polymorphisms (SNPs) that have been reported to increase the risk of NB include HACE1 (Diskin et al.,
2012), LIN28B (Diskin et al., 2012), BARD1 (Capasso et al., 2009; Pugh et al., 2013), FGFR4 (Whittle et
al., 2016), CHEK2 (Pugh et al., 2013), PINK1 (Pugh et al., 2013), TP53 (Pugh et al., 2013) and LM01 (Wang
et al., 2011).
1.5.5. Genetic features of sporadic neuroblastoma
Despite the ability to stratify patients there is very little understood about how these factors drive the
NB tumour. Recently, studies utilising next generation sequencing techniques have been undertaken to
determine the genetic mutations that are responsible for driving NB pathogenesis in an effort to design
targeted therapies. Genome-wide association studies (GWAS) have identified several SNPs that are
associated with sporadic NB. Low-risk NB is associated with SNPs in DUSP12, HSD17B12, and
DDX4/IL31RA at chromosome 5q11.2 (Nguyen le et al., 2011), whereas high-risk tumours are associated
with SNPs within or upstream of 6p22 (Maris et al., 2008), HACE1 (Diskin et al., 2012), LIN28B (Diskin et
al., 2012), BARD1 (Capasso et al., 2009), FGFR4 (Whittle et al., 2016), and LM01 (Wang et al., 2011). A
common copy number variation in chromosome 1q21 that effects NBPF23 expression was also
identified in NB tumours (Diskin et al., 2009). In addition to familial NB, ALK mutations are present in

44
sporadic cases: 6-12% carry somatic mutations, 15-22% have gained copies of ALK and 3-4% have ALK
amplification (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mosse et al., 2008;
Pugh et al., 2013). Recent evidence suggests that there is epigenetic regulation of ALK expression in
samples with wild type ALK sequence (Gomez et al., 2015). Due to the prevalence of upregulated ALK
expression in unfavourable NB, ALK has become a target of novel NB therapies. Currently the ALK
inhibitor, crizotinib, is in clinical trial (Mosse et al., 2013) (clinical trial NCT01606878) and newer
inhibitors are under development.
With the increased ability to perform whole exome and whole genome sequencing several
genome wide studies (Cheung et al., 2012; Molenaar et al., 2012; Peifer et al., 2015; Pugh et al., 2013;
Sausen et al., 2013) have been conducted to determine the somatic alterations present in NB tumours.
As expected, genetic events previously linked to NB prognosis were identified including MYCN
amplification, 1p and 11q deletion and 17q gains. A very low level of recurrent gene mutations was
detected, only 12-14 amino acid affecting mutations were present per tumour (Molenaar et al., 2012;
Peifer et al., 2015; Pugh et al., 2013; Sausen et al., 2013). This is consistent with evidence that cancers
in paediatric patients carry fewer genetic alterations than adult cancers (Alexandrov et al., 2013; Jones
et al., 2008; Parsons et al., 2011; Vogelstein et al., 2013). The most frequently mutated genes in high-
risk NB are ALK (9.2%), ATRX (9.6%), PTPN11 (2.9%), MYCN (1.7%), and NRAS (0.83%) (Pugh et al., 2013).
Of interest, alterations to the ATRX gene were seen at rates comparable to ALK (Molenaar et al., 2012;
Peifer et al., 2015; Pugh et al., 2013) and predominantly in older patients without MYCN amplification
(Cheung et al., 2012; Peifer et al., 2015; Pugh et al., 2013). This suggests that ALT may be a significant
factor in NB pathogenesis, particularly for older patients. One study also identified the chromatin
remodelling genes ARID1A and ARID1B as altered in 11% of tumours and associated with poor survival
(Sausen et al., 2013). The Rac/Rho and neuritogenesis pathways were identified as frequently defective
in NB tumours in one study (Molenaar et al., 2012) however, this was not evident in the high-risk tumour
cohort of Pugh and colleagues (Pugh et al., 2013). Whilst mutations to TP53 are one of the most common

45
events in cancer overall, the gene is rarely mutated at diagnosis in NB (Vogan et al., 1993), although 15%
of recurrent tumours have acquired a TP53 mutation (Carr-Wilkinson et al., 2010).
Chromothripsis is a form of chromosomal rearrangement caused by the shattering of a
chromosomal region which results in the random reassembly of most of the fragments by DNA repair
mechanisms (Stephens et al., 2011). Analysis of structural rearrangements in NB identified
chromothripsis in 18% of high-stage tumours and absent in low-stage tumours (Molenaar et al., 2012).
Consequently, chromothripsis correlated with poor outcome although, subsequent studies found
chromothripsis in only 0-4% of NB (Peifer et al., 2015; Sausen et al., 2013). Another structural event that
has been identified in 24-31% of high-risk NB is a structural rearrangement 30-50 kb upstream of the
hTERT promoter on chromosome 5 (Peifer et al., 2015; Valentijn et al., 2015). This rearrangement results
in an upregulation of hTERT mRNA expression due to super-enhancer elements now adjacent to the
hTERT transcription start site (Peifer et al., 2015; Valentijn et al., 2015). The implication of these studies
is that NB is not a disease driven by a single mutation but rather by rare mutations, copy number
variation, structural rearrangements, and possibly epigenetic changes.
Mutations to the p53 pathway have been identified as events that contribute to drug resistance
and ultimately relapse in NB (Keshelava et al., 2001). Recently, next generation sequencing approaches
have been used to identify novel mutations acquired by relapse tumours. Relapse tumours have a 1.5-
2-fold increase in single nucleotide variations (SNVs) compared to primary tumours (Eleveld et al., 2015;
Schramm et al., 2015). Like primary tumours, relapse is not driven by one causative genetic aberration.
Schramm and colleagues identified genes involved in the epithelial-mesenchymal transition, a process
involved in cell migration, invasion and metastasis, as the most common pathway with aberrations in
relapse tumours (Schramm et al., 2015) however, this has yet to be validated in another tumour cohort.
The increased allelic frequency of CHD5, a putative tumour suppressor gene, from the primary to relapse
tumour suggests that clonal selection of the primary tumour occurs, likely due to therapy (Schramm et
al., 2015). A similar clonal evolution was observed in relapse tumours with activating ALK mutations in

46
which both increased allelic fraction of the ALK mutation, and an increase in the overall number of
tumours effected by ALK mutations (7-9% of primary tumours to 20-26% of relapse tumours) was
observed (Padovan-Merhar et al., 2016; Schleiermacher et al., 2014). Activating mutations to the
RAS/MAPK pathway have been identified in primary tumours and several studies have found this is
enriched in relapse tumours due to novel mutations and clonal selection (Eleveld et al., 2015; Padovan-
Merhar et al., 2016). Hence, the genome of NB tumours continually evolve under the selective pressure
of treatment and biopsies of relapse tumours are required for the use of targeted therapy in these
patients.
1.5.6. Prognostic relevance of telomerase activity
Telomerase expression in NB was first described over two decades ago. The initial studies utilised the
TRAP assay to determine telomerase activity. In 1995 Hiyama and colleagues detected telomerase
activity in 94% of tumours although, the level of activity ranged from high (20% of tumours) to low (76%
of tumours) (Hiyama et al., 1995a). Low telomerase activity was detected in the majority of stage 1 and
2 tumours whereas high telomerase activity was observed in stage 3 and 4 tumours. This correlated
with a poor patient outcome in 75% of tumours with high telomerase activity but only 3.3% of tumours
which exhibited low telomerase activity. Another study reported a similar number of tumours (96%) had
detectable telomerase activity and that the level of activity correlated with patient outcome (Hiyama et
al., 1997). It was therefore, concluded that telomerase activity could be a prognostic factor for NB.
Further studies confirmed that telomerase activity correlated with NB prognostic risk group
although, the total percentage of NB tumours reported as telomerase-positive decreased compared to
the initial two studies. These studies described 20% (Poremba et al., 1999), 24% (Krams et al., 2001),
29% (Poremba et al., 2000), 35% (Choi et al., 2000), and 37% (Onitake et al., 2009) of NBs as telomerase-
positive. The discrepancy between studies may be the result of false-positive TRAP results due to the
sensitivity of the PCR. Approximately 10% of localised NB tumours maintain telomere length via
telomerase compared to nearly 60% of metastatic disease (Poremba et al., 1999). Despite the varying

47
percentages of telomerase-positive NBs there is a correlation between high telomerase activity and
poor prognosis (30-45% 5-year overall survival depending on tumour cohort) (Ohali et al., 2006; Onitake
et al., 2009; Poremba et al., 2000).
Telomerase activity has also been identified in some, but not all, stage 4S tumours that exhibit
progressive disease (Brinkschmidt et al., 1998) and has been associated with fatal 4S tumours (Choi et
al., 2000). It has been proposed that the spontaneous regression of stage 4S tumours is the result of
telomere shortening and subsequent cellular senescence that occurs due to the low telomerase activity
in these tumours (Brinkschmidt et al., 1998). Indeed, examination of the telomere length of progressive
liver metastases biopsied from a single patient at different time points showed telomere shortening in
spite of low levels of telomerase (Hiyama et al., 1995a).
There are several ways that NB cells can upregulate telomerase activity. The amplification of
MYCN has been associated with upregulated telomerase activity, in one study 65% of tumours with high
telomerase activity were also MYCN amplified (Hiyama et al., 1997). This correlation is likely due to
MYCN driven telomerase activity as the hTERT promoter contains binding sequences for the MYCN
transcription factor (Cong et al., 1999; Mac et al., 2000; Wang et al., 1998). Expression of hTERT can also
be upregulated as a result of two mutations upstream of the promoter. However, this appears to be a
rare event in NB with 0/131 primary tumours harbouring either a C228T or a C250T mutation and only
3/19 cell lines (all derived from the same tumour) having a C228T mutation (Lindner et al., 2015).
Instead, up to 31% of NB tumours activate telomerase via chromosome 5 structural rearrangements
(Peifer et al., 2015; Valentijn et al., 2015). A breakpoint 30-50 kb upstream or 6-40 kb downstream of
the hTERT promoter results in the introduction of super-enhancer elements and the consequent
upregulation of hTERT mRNA expression. This event is mutually exclusive with MYCN amplification and
also with ATRX aberrations (Peifer et al., 2015; Valentijn et al., 2015). Telomerase activity has also been
identified in all but four NB cell lines screened to date (4/40 cell lines; Farooqi et al., 2014).

48
Given the correlation between telomerase activity and poor prognosis in NB, the mechanism
has become a target for the development of novel NB therapy. Long term treatment (4-6 weeks) with
the telomerase inhibitor telomestatin, at non-toxic concentrations in four NB cell lines, results in a
reduction in the growth rate of cells, a decrease in telomere length, and an increase in apoptosis (Binz
et al., 2005). This suggests that therapies targeting TLMs may have therapeutic relevance in NB.
1.5.7. ALT in neuroblastoma
A subgroup of NB tumours upregulates telomerase activity to maintain their telomere length however,
between 20 and 60% of high-risk tumours do not have detectable telomerase activity (Krams et al.,
2001; Onitake et al., 2009; Poremba et al., 2000; Poremba et al., 1999). The first evidence of ALT in NB
relied on telomere length to identify the mechanism. Long and heterogeneous telomere lengths, a
phenotypic characteristic of ALT, were observed in 5/9 telomerase-negative patients who had
protracted but fatal disease (Onitake et al., 2009). Another study utilising telomere length as a marker
of ALT indicated the mechanism may be used by up to 59% of NB tumours (Pezzolo et al., 2015). A
subsequent study that utilised telomere length and APBs to determine ALT activity confirmed that ALT
tumours have a poor outcome, approximately 45% 5-year overall survival, despite lacking poor
prognostic markers such as MYCN amplification (Lundberg et al., 2011).
Aberrations of the ATRX gene have been reported in NB tumours (Kurihara et al., 2014;
Lundberg et al., 2011; Onitake et al., 2009; Valentijn et al., 2015) and correlate with an upregulated ALT
mechanism in other tumour types (Chen et al., 2014; Heaphy et al., 2011a; Jiao et al., 2012; Jiao et al.,
2011). In NB, alterations to the ATRX gene associate with long telomeres, suggestive of ALT, and patients
with these tumours are older at diagnosis (ATRX mutations in 44% of patients aged ≥12 years, 17% of
patients aged 18 months to <12 years and 0% patients aged <18 months) (Cheung et al., 2012). A
retrospective examination of the tumours by Onitake and colleagues found all ALT tumours had an
aberration to ATRX or DAXX and that ATRX alterations led to a 5-year overall survival of approximately
40% (Kurihara et al., 2014). Mutations in ATRX or DAXX have been identified in 10-22% of high-risk NB

49
tumours (Cheung et al., 2012; Peifer et al., 2015; Valentijn et al., 2015). In most studies alterations to
the ATRX, hTERT, and MYCN genes are mutually exclusive (Cheung et al., 2012; Peifer et al., 2015;
Valentijn et al., 2015) and lead to a similarly poor overall survival rate of approximately 25% (Valentijn
et al., 2015).
A caveat of using large genome studies to identify ALT is that ATRX and DAXX mutations are not
a universal feature of the mechanism and relying on these genetic events would underestimate ALT in
NB. To date four telomerase-negative NB cell lines derived from metastatic disease have been identified
(Choi et al., 2000; Farooqi et al., 2014). Only one of these cell lines (CHLA-90) exhibited classical ALT
phenotypes including abundant C-circles and APBs, was MYCN non-amplified and ATRX and TP53
mutated, whilst a second cell line (SK-N-FI) retained wild type ATRX/DAXX expression but otherwise
exhibited classical features of ALT (Farooqi et al., 2014). Therefore, it appears that ALT is present and
clinically relevant in NB, although no study to date has utilised C-circles, the quantifiable ALT marker, to
identify ALT in NB tumours.
1.6. Aims of the project
High-risk NB confers a very poor prognosis and the identification of novel biomarkers is required for the
development of better therapy for these patients. Recent studies have identified mutations to genes
involved in TLMs as relatively frequent events in NB (Cheung et al., 2012; Peifer et al., 2015; Valentijn
et al., 2015). Telomerase activity is associated with poor outcome (Ohali et al., 2006; Onitake et al.,
2009; Poremba et al., 2000), whilst the identification of ALT has relied on genetic markers or telomere
length to identify the presence of this mechanism rather than C-circles, the active marker of ALT (Cheung
et al., 2012; Lundberg et al., 2011; Onitake et al., 2009; Peifer et al., 2015; Valentijn et al., 2015).
Consequently, the initial aim of this study was to determine the prognostic relevance of ALT in high-risk
NB utilising telomere length and abundant C-circles (Lau et al., 2013). Unexpectedly, we identified a
subgroup of high-risk tumours that lacked C-circles but had a telomere content indistinguishable from

50
ALT tumours and no telomerase activity. Therefore, the aim of this study became to investigate the
telomere biology of ALT-negative/long telomere NB. Specifically we sought to:
1. Determine if a subgroup of fatal malignant NB tumours can develop in the absence of a TLM
and the consequence of extensive proliferation on the telomere length of these cells.
2. Examine the effect of activation of a TLM on ALT-negative/long telomere NB cells.
3. Study the association between telomere biology and sensitivity to chemotherapeutics in NB.
4. Investigate similar phenotype in other tumour types.

51
Chapter 2:
Materials and Methods

52
Chapter 2: Materials and Methods
2.1. Buffers and solutions
The buffers and solutions used throughout the methods are listed in Table 2.1.
Table 2.1. Composition of common buffers and solutions.
Solution Composition
6X DNA loading buffer 0.0001% (v/v) SDS 15% (w/v) Ficoll 400
0.25% (w/v) Bromophenol blue 6X TBE (Tris, borate and EDTA)
10X phosphate buffered saline (PBS) 1.37 M NaCl 0.1 M Na2HPO4
0.018 M KH2PO4
20X saline-sodium citrate (SSC) 3 M NaCl 0.3 M Tri-sodium citrate
Tris, borate and EDTA (TBE) 0.9 M Tris pH 8 0.9 M Boric acid
0.02 M ethylenediaminetetraacetic acid (EDTA)
Tris-EDTA (TE) 10 mM Tris pH 7.6 0.1 mM EDTA
2.2. Patient samples
The study cohort contained 149 high-risk NB samples: 19 frozen tumour specimens from The Children’s
Hospital at Westmead Tumour Bank (Australia), 46 DNA samples from the Children’s Cancer Institute
Tumour Bank (Australia), and 84 samples (35 frozen and 49 DNA) from the Children’s Oncology Group
Neuroblastoma Biobank (USA). High-risk NB was defined as tumours with MYCN amplification or
metastatic disease in patients aged ≥12 months. The samples were obtained at original diagnosis and
data on sex, age at diagnosis, tumour stage, MYCN status (Figure 2.1), and survival were retrieved from
the clinical database from the source of the sample. The Sydney Children’s Hospital Network and
Northern Hospital Network Human Research Ethics Committees provided ethics approval for this study.

53
Figure 2.1. Schematic of neuroblastoma (NB) tumours used in this study based on MYCN
amplification status.
One hundred and forty-nine high-risk NB tumours were examined in this study, 55 were MYCN
amplified and 94 were MYCN non-amplified tumours.
2.3. Cell culture
NB cell lines (characteristics of 6 cell lines predominantly used in the thesis in Table 2.2), and ALT and
telomerase lines used as controls (Table 2.3), were cultured with medium supplemented with fetal
bovine serum (FBS) and additional additives, as listed in Table 2.4. All cell lines were grown in a
humidified incubator at 37°C with atmospheric oxygen and 5% CO2, except COG-N-328h which was
grown under normoxic conditions (5% oxygen balanced with nitrogen). Cell lines were authenticated by
16-locus short tandem repeat (STR) profiling and were found to be free of Mycoplasma species by
CellBank Australia (Children’s Medical Research Institute, Westmead, NSW, Australia).
2.4. Vectors and viral transfections
Plasmid contructs pBABE-hTERT (Counter et al., 1998a), pBABE-hTR (Wong and Collins, 2006), pBABE
hTR/hTERT (Wong and Collins, 2006), and pBABE empty vector (Morgenstern and Land, 1990) were
obtained from Addgene (Plasmid #1771, 27666, 27665, 1764, respectively). Retrovirus was generated
by Lipofectamine 2000 (Life Technologies) transfection of Phoenix-AMPHO cells (ATCC CRL-3213) with
the plasmids, followed by harvest of the retrovirus 24 hours (hrs) later. LA-N-6 cells were retrovirally
transduced by two rounds of viral infection with 4 µg/mL polybrene (Sigma) and retrovirus. Cells that
stably expressed the plasmid were selected with culture medium containing 0.3 µg/mL puromycin (Life

54
Table 2.2. Characteristics of NB cell lines
Cell Line Patient Characteristics
Tissue sample of origin
Tumour Formation in
Mice
References
SH-SY5Y Stage 4, Female,
48 months old at diagnosis
Progressive disease, post
chemotherapy, bone marrow
Yes (Ross et al., 1983) (Nevo et al., 2008)
SK-N-BE2c Stage 4, Male,
24 months old at diagnosis
Progressive disease, post
chemotherapy, bone marrow
Yes (Biedler et al., 1978) (Ciccarone et al. 1989) (Reynolds et al., 1988)
SK-N-FI
Stage 4, Male,
132 months old at diagnosis
Progressive disease, post
chemotherapy, bone marrow
Yes (Reynolds et al., 1988) (Tsutsumimoto et al.,
2014) (Farooqi et al., 2014)
CHLA-90
Stage 4, Male,
102 months old at diagnosis
Progressive disease, bone
marrow transplant
(Keshelava et al., 1998)
LA-N-6
Stage 4, Male,
60 months old at diagnosis
Progressive disease, post
chemotherapy, bone marrow
Yes (Reynolds et al., 1988) (Wada et al., 1993)
COG-N-291 Progressive
disease, post chemotherapy
(Farooqi et al., 2014)
Table 2.3. Telomere lengthening mechanism of control cell lines
Cell Line Telomere Lengthening Mechanism
Reference
A2182 Telomerase + (Ng et al., 2009)
DOS16 ALT + (Henson et al., 2009)
G292 ALT + (Bryan et al., 1997)
GM639 Telomerase + (Bryan et al., 1995)
GM847 ALT + (Bryan et al., 1995)
HeLa 1.2.11 Telomerase + (Whitaker et al., 1995)
HFF5 Mortal (Bryan et al., 1995)
HT1080 Telomerase + (Whitaker et al., 1995)
HT1080 hTR Telomerase + (Pickett et al., 2009)
IIICF/c ALT + (Bryan et al., 1995)
MeT-4A ALT + (Bryan et al., 1995)
MG-63 Telomerase + (Bryan et al., 1997)
MRC-5 Mortal (Huschtscha and Holliday, 1983)
SAOS-2 ALT + (Bryan et al., 1997)
U-2 OS ALT + (Bryan et al., 1997)

55
Technologies) for 7 days. Sub-clones were isolated by plating cells, post puromycin selection, at a density
of 100 cells/mL with conditioning medium.
2.5. Cell proliferation assay
Cells were seeded in 96-well plates according to the density in Table 2.5 and treated with serial dilutions
of drugs (6 wells were treated with every drug concentration; Table 2.6) at 37°C with 5% CO2 for 6 PDs.
Drugs were replenished every 72 hrs and experiments were performed in triplicate. At the end of
treatment 10 µL of AlamarBlue (Thermo Fischer Scientific) was added to each well containing 100 µL of
medium/drug and incubated at 37°C with 5% CO2 for 4 hrs. AlamarBlue is a fluormetric indicator, where
the change in fluorescence correlates with the number of metabolically active cells. The fluorescence
was measured in a fluorescence plate reader (EnSpire, Perkin Elmer) with an excitation wavelength 570
nm and an emission wavelength at 585 nm. The results were expressed as a percentage of the untreated
control and GraphPad Prism6 was used to determine the IC50 dose (the dose that inhibited 50% of
untreated control cells growth).
2.6. Immunostaining and fluorescence in situ hybridisation (FISH)
2.6.1. ATRX and DAXX immunofluorescence (IF)
Cells grown in two-well chamber slides were washed once with 1X PBS and then fixed with 4% PFA for
10 minutes (mins) at room temperature. The cells were rinsed with 2X PBS and permeabilised in 0.1%
Triton-X for 10 mins, and then washed again with 2X PBS. Cells were blocked, at room temperature, with
5% goat serum/bovine serum albumin (BSA) for 20 mins before the cells were incubated for 1 hr at 37°C
with either anti-ATRX or DAXX rabbit polyclonal antibody (Sigma) diluted 1:500 in 3% BSA/4X SSC. To
remove excess antibody, the cells were washed three times with 0.1% Tween-20/4X SSC and were then
incubated with Alexa Fluor 594 goat anti-rabbit secondary antibody (Invitrogen) diluted 1:1000 in 3%
BSA/4X SSC. After incubation at room temperature for 1 hr, the cells were washed three times with 0.1%
Tween-20/4X SSC and the DNA was stained with 20 µg/mL 4, 6 diamidino-2-phenylindole (DAPI) in 1X

56
Table 2.4. Cell culture medium.
Cell Line Cell Type Medium FBS (%) Additives
A2182 Lung Carcinoma DMEM- with L-glutamine 10
CHLA-42 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-90 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-95 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-119 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-122 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-136 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-140 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-171 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-172 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHLA-225 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
CHP-134 Neuroblastoma RPMI-1640 10 1% L-glutamine
COG-N-291 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
COG-N-328h Neuroblastoma IMDM 20 2% L-glutamine
1X ITS
COG-N-347 Neuroblastoma IMDM 20 2% L-glutamine
1X ITS
DOS16 Leiomyosarcoma RPMI-1640 10
FU-NB-2006 Neuroblastoma IMDM 15 2% L-glutamine
1X ITS
G292 Osteosarcoma McCoy's 5a Medium
Modified 10
GM639 Fibroblast, skin DMEM- with L-glutamine 10
GM847 Fibroblast, skin DMEM- with L-glutamine 10
HeLa 1.2.11 Cervical carcinoma RPMI-1640 10 1% L-glutamine
HFF5 Fibroblast, foreskin DMEM- with L-glutamine 10
HT1080 Fibrosarcoma DMEM- with L-glutamine 10

57
Abbreviations: DMEM, Dulbecco’s Modified Eagle’s Medium; IMDM, Iscove's Modified Dulbecco's
Medium; LHC-MM, Laboratory of Human Carcinogenesis Mesothelial Medium; MEM, minimum
essential medium; ITS, insulin-transferrin-selenium-sodium pyruvate; NEAA, non-essential amino acids;
RPMI-1640, Roswell Park Memorial Institute 1640.
HT1080 hTR Fibrosarcoma DMEM- with L-glutamine 10
IIICF/c
Fibroblast, breast,
Li-Fraumeni
syndrome
DMEM- with L-glutamine 10
IMR32 Neuroblastoma DMEM- with L-glutamine 10
KELLY Neuroblastoma RPMI-1640 10 1% L-glutamine
LA-N-1 Neuroblastoma MEM-F12 with L-glutamine 10 1% NEAA
LA-N-2 Neuroblastoma MEM-F12 with L-glutamine 10 1% NEAA
LA-N-5 Neuroblastoma RPMI-1640 10 1% L-glutamine
LA-N-6 Neuroblastoma RPMI-1640 10 1% L-glutamine
MeT-4A Mesothelial LHC-MM 3
MG-63 Osteosarcoma DMEM- with L-glutamine 10
MRC-5 Fibroblast, lung DMEM- with L-glutamine 10
NB69 Neuroblastoma RPMI-1640 10 1% L-glutamine
SAOS-2 Osteosarcoma DMEM- with L-glutamine 10
SH-EP Neuroblastoma DMEM- with L-glutamine 10
SH-SY5Y Neuroblastoma DMEM- with L-glutamine 10 1% NEAA
SK-LU-1 Adenocarcinoma DMEM- with L-glutamine 10
SK-N-AS Neuroblastoma DMEM- with L-glutamine 10
SK-N-BE1 Neuroblastoma RPMI-1640 10 1% L-glutamine
SK-N-BE2c Neuroblastoma DMEM- with L-glutamine 10
SK-N-DZ Neuroblastoma DMEM- with L-glutamine 10
SK-N-FI Neuroblastoma RPMI-1640 10 1% L-glutamine
SK-N-SH Neuroblastoma DMEM- with L-glutamine 10
SMS-KANR Neuroblastoma RPMI-1640 10 1% L-glutamine
SMS-KCN Neuroblastoma RPMI-1640 10 1% L-glutamine
SMS-KCNR Neuroblastoma RPMI-1640 10 1% L-glutamine
SMS-LHN Neuroblastoma RPMI-1640 10 1% L-glutamine
SMS-SAN Neuroblastoma RPMI-1640 10 1% L-glutamine
U-2 OS Osteosarcoma DMEM- with L-glutamine 10

58
Table 2.5. Seeding density of cell lines in the cell proliferation assay.
Cell Line Seeding Density (X104 cells/well)
COG-N-291 1.6
CHLA-90 0.25
GM639 0.125
GM847 0.1
HeLa 1.2.11 0.38
HT1080 0.1
HT1080 hTR 0.2
LA-N-6 2.5
SK-N-BE2c 0.15
SK-N-FI 1
SH-SY5Y 0.375
U-2 OS 0.125
PBS for 1 min. The slides were rinsed with water and mounted in DABCO (2.33% antifade compound
1,4-Diazabicyclo[2.2.2]octane [Sigma], 90% [v/v] glycerol, 20 mM Tris-HCl pH 8.0). Cells were
considered ATRX or DAXX positive when foci were present in the nucleus.
2.6.2. APB detection
Cell cultures were harvested by trypsinisation and resuspended in buffered hypotonic solution (United
Biosciences). The cells (110,000 cells/mL) were cytocentrifuged onto SuperFrost Plus glass slides
(Menzel-Glaser) at 110 x g for 10 min in a Shandon Cytospin 4 with medium acceleration. Slides were
permeabilised at room temperature for 10 mins with KCM buffer (120 mM KCl, 20 mM NaCl, 10 mM Tris
pH 7.5, 0.1% Triton X-100), rinsed in 1X PBS, and fixed in 4% (v/v) formaldehyde in 1X PBS at room
temperature for 10 mins. After several rinses with 1X PBS, the slides were treated with 100 µg/mL
DNase-free RNase A (Sigma) in antibody dilution buffer (ABDIL; 20 mM Tris pH 7.5, 2% BSA, 0.2% fish
gelatin, 150 mM NaCl, 0.1% Triton X-100, 0.1% sodium azide) at 37°C for 30 mins. The slides were then
incubated at 37°C for 1 hr with mouse anti-PML antibody (PG-M3; Santa Cruz), diluted 1:250 in ABDIL.
Slides were washed three times with 0.1% Tween-20/1X PBS and then incubated with Alexa Fluor 594
goat anti-mouse secondary antibody (Invitrogen; 1:1000). Following the incubation at 37°C for 30 mins,
the cells were washed three times with 0.1% Tween-20/1X PBS, and proteins were cross-linked with 4%

59
PFA for 10 mins at room temperature. The cells were dehydrated with an ethanol series (70%, 90%, and
100%) and then telomeric DNA was detected using the 5’ labelled telomere-specific peptide nucleic acid
(PNA) probe, Alexa 488-(CCCTAA)3 PNA probe (PNA Bio) (Chen et al., 1999; Perrem et al., 2001). After
the probe was placed on the slide, the DNA was denatured at 80°C for 3 mins and the probe was
hybridised for 3 hrs at room temperature. Unhybridised probe was removed by washing with Buffer A
(70% formamide, 0.01 M Tris pH 7.5, 0.1% BSA) for 15 mins, and then the nuclei were stained with DAPI
(20 µg/mL) in wash Buffer B (0.1 M Tris pH 7.5, 0.15 M NaCl, 0.08% Tween-20) for 3 mins. The slides
were washed with 1X PBS and rinsed in water before mounting in DABCO antifade mounting medium.
A Zeiss Axio Imager Z2 with ApoTome was used to capture Z-stacked images (ACRF Telomere Analysis
Centre). Quantitation was completed on three independent experiments, on at least 100
cells/experiment, using Zeiss ZEN software. An APB was defined as the colocalisation of telomeric DNA
and PML protein of any size (Yeager et al., 1999).
2.6.3. MetaTIF assay
Cell cultures were treated with 50 µg/mL vinblastine for 1 hr at 37°C in 5% CO2 to obtain metaphase
chromosomes. Cells were harvested by trypsinisation and resuspended in buffered hypotonic solution
at 37°C. NB cultures were incubated for 20 mins whereas control cell lines were incubated for 5 mins.
To obtain metaphase spreads 110,000 cells/mL were cytocentrifuged onto SuperFrost Plus glass slides
at 450 x g (high acceleration) for 10 min in a Shandon Cytospin 4. Slides were then permeabilised with
KCM buffer for 10 mins, rinsed in 1X PBS three times, and fixed in 4% (v/v) formaldehyde in 1X PBS for
10 mins at room temperature. The slides were treated with 100 µg/mL RNase A in ABDIL at 37°C for 30
mins, and then incubated for 1 hr at 37°C with mouse anti-γH2AX antibody (Ser139 Clone JBW301;
Merck Millipore) diluted 1:300 with ABDIL. To remove excess antibody, the slides were washed three
times and then incubated with Alexa Fluor 594 anti-mouse secondary antibody (1:1000 in ABDIL) at 37°C
for 30 mins. Slides were washed three more times with 0.1% Tween-20/1X PBS, cross-linked with 4%
PFA for 10 mins, and dehydrated with increasing concentrations of ethanol (70%, 90% and 100%).
Telomeric DNA was detected using the Alexa 488-(CCCTAA)3 PNA probe, applied to the slide prior to

60
Table 2.6. List of drugs and stock concentrations.
Drug Company Target Diluent Highest Drug
Concentration
Dilution
Factor
for Serial
Dilution
All trans
retinoic acid AbCam Differentiation induction Ethanol 45 µM 3
Aphidicolin AbCam DNA replication inhibitor DMSO 30 µM 6
Arsenic Trioxide Sigma-
Aldrich
Complex mode of action,
causes apoptosis
1 M
NaOH 10 µM 3
Cisplatin Cayman
Chemicals Cross-links DNA
0.9%
Saline
(w/v)
30 µM 4
Doxorubicin Cayman
Chemicals
Interferes with enzymes
involved in DNA
replication
DMSO 1.5 µM 5
Etoposide Cayman
Chemicals
Topoisomerase II
inhibitor DMSO 400 µM 10
Hydroxyurea AbCam Ribonucleotide reductase
inhibitor DMSO 1600 µM 2
Irinotecan Cayman
Chemicals Topoisomerase I inhibitor DMSO 600 µM 6
KU-60019 Selleckchem ATM inhibitor DMSO 24 µM 2
Mitomycin C Selleckchem
Interferes with enzymes
involved in DNA
replication
Water 3 µg/mL 4
ML216 Sigma-
Aldrich Bloom helicase inhibitor DMSO 225 µM 3
Temozolomide Cayman
Chemicals
Alkylating agent, induce
DNA damage DMSO 400 µM 6
Topotecan Cayman
Chemicals Topoisomerase I inhibitor DMSO 90 µM 12
VE-822 Selleckchem ATR inhibitor DMSO 7 µM 5
Vincristine AbCam Mitotic inhibitor DMSO 0.05 µM 6
denature at 80°C for 3 mins. After hybridisation of the probe at room temperature for 3 hrs, the slides
were washed with Buffer A for 15 mins. The DNA was stained with 20 µg/mL DAPI/Buffer B for 3 mins
and rinsed with water several times before mounting in DABCO. Images were obtained on the Metafer

61
slide scanning system (ACRF Telomere Analysis Centre) and a meta-TIF was defined as the localisation
of γ-H2AX protein to the end of the chromosome (with or without the presence of telomere signal)
(Cesare et al., 2009). Quantitation was performed, in triplicate, on at least 1000 chromosome ends.
2.6.4. Telomere FISH on metaphase spreads
Metaphase chromosomes were obtained by treating cells with vinblastine (50 µg/mL) overnight for 16
hrs. Cell cultures were harvested by trypsinisation, resuspended in buffered hypotonic solution at 37°C
for 20 mins, fixed (3:1 methanol/glacial acetic mix) and dropped onto SuperFrost Plus glass slides. The
slides were baked at room temperature overnight, then treated with 100 µg/mL RNase A at 37°C for 30
mins, and then dehydrated with increasing concentrations of ethanol. FISH was performed with an Alexa
Fluor 488 telomere specific PNA probe applied to the slide prior to denaturing the DNA for 3 mins at
80°C. After a 3 hr hybridisation, the slides were washed for 15 mins with Buffer A to remove excess
probe. A second wash (with Buffer B) was performed for 5 mins at room temperature, and then the
slides were stained with DAPI (20 µg/mL in PBS) for 3 mins to visualise the DNA. The slides were washed
with 1X PBS, rinsed with water and mounted in DABCO. Fluorescence microscopy images were obtained
on the Metafer slide scanning system (ACRF Telomere Analysis Centre) and the intensity of the telomere
signal was quantitated using TFL-Telo software (Zijlmans et al., 1997) in fifteen metaphase spreads for
each sample.
2.6.5. Telomere sister chromatid exchange (T-SCE) analysis
T-SCE analysis was performed as previously described (Bailey et al., 2004). Briefly, cells were cultured in
medium containing a 3:1 ratio of BrdU:BrdC (Sigma) with vinblastine (50 µg/mL) added for the final 16
hrs. Metaphases were harvested and resuspended in buffered hypotonic solution at 37°C for 20 mins.
Chromosomes were then fixed with 3:1 ice-cold methanol:acetic acid and dropped onto SuperFrost Plus
glass slides. The slides were incubated at 37°C for 30 mins with 100 µg/mL DNase-free RNase A (Sigma)
in 2X SSC. Slides were postfixed with 4% formaldehyde in PBS for 10 mins and then dehydrated in an
ice-cold ethanol series (75, 85, 100%) for 2 mins each. Metaphase spreads were stained with 0.5 µg/mL

62
Hoechst 33258 at room temperature for 15 mins and exposed to long wave 365 nm UV light for 45 mins.
An exonuclease III (NEB) digest was then performed for 30 mins at 37°C. The chromosomes were
denatured for 5 mins at 80°C in 70% formamide/30% 2X SSC before the slides were dehydrated in an
ice-cold ethanol series as previously described. The slides were incubated overnight with 0.3 µg/ml
TAMRA–OO-(TTAGGG)3 PNA probe (PNA Bio) and washed with Buffer A for 15 mins followed by a wash
with Buffer B for 5 mins at room temperature. Slides were incubated with 0.3 µg/ml Alexa 488–OO-
(CCCTAA)3 PNA probe overnight and washed with buffer A and B as previously described prior to staining
with DAPI (20 µg/mL in PBS) for 3 mins to visualise the DNA. Lastly, the slides were washed with 1X PBS,
rinsed with water and mounted in DABCO. Imaging was performed on the Metafer slide scanning system
(ACRF Telomere Analysis Centre).
2.7. DNA extraction and quantitation
Genomic DNA was extracted by lysis at 37°C overnight with 2% sodium dodecyl sulphate (SDS) buffer
containing 50 mM Tris, 20 mM EDTA, and 200 µg/mL Pronase protease (Sigma). Lysis was followed by 2
hrs salt precipitation at 4°C with 1.4 M NaCl, followed by serial centrifugation (15 mins at 3900 x g) until
the white precipitant was removed. The DNA was precipitated at 4°C overnight with 100% ethanol (2.5
X volume of lysis buffer with NaCl), before centrifugation at 3900 x g for 30 mins. The DNA pellet was
washed with 70% ethanol, centrifuged for 15 mins, and then allowed to air-dry. The DNA was
resuspended in TE and quantitated with a Qubit Fluorometer (Invitrogen).
2.8. C-circle assay
2.8.1. Slot-blot detection
Rolling circle amplification of C-circles was performed on 10 ng of genomic DNA. The assay was
performed with and without Φ29 polymerase for each sample. The DNA (10 µL) was added to 10 µL of
the amplification mix, for a 20 µL reaction containing 4 µM DL-Dithiothreitol (DTT), 200 µg/mL BSA, 0.1%
Tween-20, 1X Φ29 buffer (New England BioLabs), 7.5 Units Φ29 polymerase (New England BioLabs), and

63
1 mM each of dATP, dGTP and dTTP (Roche), which was incubated at 30°C for 8 hrs and then at 65°C for
20 mins to heat inactivate the enzyme (Henson et al., 2009). The C-circle assay product was diluted with
100 µL of 2X SSC, and using slot-blot apparatus, blotted onto a Biodyne B 0.45 µM nylon membrane
(Pall). The membrane was dried and then the DNA was UV-cross-linked onto the membrane in a
Stratagene UV Stratalinker 1800. PerfectHyb Plus buffer (Sigma) was used to pre-hybridise the
membrane for 1 hr at 37°C, and a 32P-ATP-labelled (CCCTAA)3 telomeric probe was hybridised to the
DNA overnight, under native conditions. The membrane was washed four times at 37°C with 0.5X
SSC/0.1% SDS for 15 mins and exposed to a storage phosphor screen, which was scanned on a Typhoon
imager (GE Healthcare) the following day. Signals were quantitated using ImageQuant (GE Healthcare)
and the level of C-circles was calculated by subtracting the 32P-probe signal of the C-circle assay without
Φ29 from that of the C-circle assay with Φ29.
2.8.2. Exonuclease treatment of DNA
Genomic DNA was pretreated with 6 U/ug of the restriction enzymes HinfI and RsaI (New England
BioLabs) and 25 ng/µg RNase (Roche) at 37°C for 12 hrs, followed by heat inactivation of the enzymes
at 80°C for 20 mins. The digested DNA was ethanol precipitated (0.3 volume of 10 M ammonium acetate
and 3.3 volumes of 100% ethanol with 1 µL glycogen) and resuspended in TE, prior to quantitation by
the Qubit Fluorometer. To remove linear DNA from the C-circle reaction, 1 µg of pretreated DNA was
digested with lambda exonuclease (12.5 U/µg; New England BioLabs), exonuclease I (100 U/µg; New
England BioLabs), and 1X lambda buffer (New England BioLabs) at 37°C for 20 mins followed by 80°C for
20 mins (Henson et al., 2009). The DNA was quantitated, and additional rounds of exonuclease digestion
were performed, until there was <100 ng of DNA remaining. As a control, a mock reaction without
exonucleases was also carried out for each sample. The exonuclease digested DNA, and control sample,
were precipitated by ethanol precipitation as described above and resuspended in 20 µL Tris. To perform
the C-circle amplification reaction, 5 µL of the DNA was diluted with 5 µL of Tris and added to the
reaction. The rolling circle amplification product was quantitated by slot-blot analysis as described in
section 2.8.1.

64
2.8.3. Telomere quantitative polymerase chain reaction (qPCR) detection
Rolling circle amplification of C-circles was performed on 16 ng (in 5 µL) of genomic DNA, with and
without Φ29 polymerase for each sample. The total reaction volume was 10 µL (including DNA), and the
components and conditions were the same as section 2.8.1, with a modification to the amount of Φ29
polymerase (Lau et al., 2013), which was halved. The 10 µL C-circle assay product was diluted with 30
µL of TE, and 5 µL of the diluted product was used for each PCR. For every sample, the following PCRs
were performed in triplicate: telomere PCR of C-circle assay/Φ29+, telomere PCR of C-circle assay/
Φ29-, single copy gene (SCG) PCR of C-circle assay/Φ29+, and SCG PCR of C-circle assay/Φ29-. The SCG
for NB cell lines and tumours was VAV2 (Lau et al., 2013), whilst the SCG for oesophageal and PanNETs
was HBG, and the SCG for melanoma tumours and cell lines was 36B4 (Lau et al., 2013). Primer
sequences are listed in Table 2.7. A standard curve of DNA was included for each PCR, to allow for
subsequent analysis. Concentrations in the telomere PCR were 2.5, 1, 0.2, 0.04, 0.02, 0.01 ng/µL and
concentrations of the SCG genes were 2.5, 1.25, 0.625, 0.3125, 0.156, 0.078 ng/µL. Each qPCR reaction
consisted of 1X Rotor-Gene SYBR Green (Qiagen), 5 µL C-circle assay product or standard DNA, and the
primer sets. The final primer concentrations were, telomere: forward 500 nM and reverse 500 nM,
VAV2: forward 700 nM and reverse 400 nM, 36B4: forward 300 nM and 500 nM reverse, and HBG:
forward 500 nM and reverse 500 nM. PCR conditions were, telomere: 95°C for 15 mins, 30 cycles of 95°C
for 7 sec and 58°C for 10 sec, VAV2: 95°C for 5 mins, 43 cycles of 95°C for 15 sec, 61°C for 30 sec, and
72°C for 20 sec, and 36B4/HBG: 95°C for 5 mins, 40 cycles of 95°C for 15 sec and 58°C for 30 sec. All PCR
conditions concluded with a melt curve analysis. Analysis was conducted using the two standard curves
method (Cawthon, 2002), and the telomere product was corrected for loading with the SCG product. All
PCR results were expressed as the mean of triplicate reactions. The relative telomeric DNA content (TC)
of a sample was obtained from the mean normalised C-circle assay/Φ29- reaction, relative to the TC of
SK-N-FI or IIICF/c, ALT-positive cell lines with arbitrary TC values of 14.0 and 30.0, respectively. The
relative C-circle level of a sample was the normalised C-circle assay/Φ 29+ subtracted by the normalised
C-circle assay/Φ29-, relative to the C-circle level of SK-N-FI and IIICF/c, with arbitrary values of 196 and

65
100, respectively. Long telomeres were indicated by a relative TC≥15, and a sample was considered ALT+
when the relative C-circle level was ≥7.5 (Lau et al., 2013).
2.9. Telomere length analysis
2.9.1. Telomere restriction fragment (TRF) analysis
Genomic DNA was digested at 37°C for 12 hrs with 6 U/µg of the restriction enzymes HinfI and RsaI (New
England BioLabs) and 25 ng/µg RNase (Roche) (Perrem et al., 2001), followed by heat inactivation of the
enzymes at 80°C for 20 mins. Prior to gel electrophoresis, the DNA was ethanol precipitated (0.3 volume
of 10 M ammonium acetate and 3.3 volumes of 100% ethanol with 1 µL glycogen) and then resuspended
in TE. The digested DNA was quantitated by the Qubit Fluorometer and 1.5 µg/well was loaded on a 1%
(w/v) agarose gel in 0.5X TBE. To resolve bands in COG-N-291 and LA-N-6, the DNA was separated using
pulsed-field gel electrophoresis (BioRad) in 14°C recirculating 0.5X TBE buffer at 6 V/sec for 16 hrs, with
an initial switch time of 1 and final switch time of 6. Gels containing other samples were run for 14 hrs.
The gels were dried for 2 hrs at 65°C, denatured in 0.82 M NaOH/2.5 M NaCl for 1 hr, and neutralised in
0.7 M Tris pH 8/1.5 M NaCl for 1 hr at room temperature. Gels were pre-hybridised for 1 hr at 50°C in
Church buffer (0.5 M Na2HPO4 pH 7.2, 1 mM EDTA, 7% SDS, and 1% BSA) and then hybridised with a 32P-
ATP-labelled (CCCTAA)4 telomeric probe overnight. The gels were washed for 30 mins in 4X SSC, three
times at room temperature and once at 50°C, and then exposed to a storage phosphor screen. The
screen was scanned the following day with a Typhoon imager and the signals were quantitated using
Las4000-Multigauge software. The unweighted mean TRF length was calculated using Telorun software
(Baur et al., 2004), which utilised linear regression analysis of molecular weight marker positions to
calculate telomere length.
2.9.2. Telomere qPCR
The TC of cells, independent of C-circles, can be determined via the protocol described in section 2.8.3.
with modification to the input sample. Rather than rolling circle amplification product, 5 µL of 1 ng/µL

66
Table 2.7. List of primers.
Gene Primer sequence Methods section
Telomere F: 5’-CGGTTTGTTTGGGTTTGGGTTTGGGTTTGGGTTTGGGTT- 3’
R: 5’-GGCTTGCCTTACCCTTACCCTTACCCTTACCCTTACCCT- 3’ 2.8.3.
VAV2 F: 5’ -TGGGCATGACTGAAG ATGAC- 3’
R: 5’ -ATCTGCCCTCACCTTCTCAA- 3’ 2.8.3.
36B4 F: 5’ -CAGCAAGTGGGAAGGTGTAATCC- 3’
R: 5’ -CCCATTCTATCATCAACGGGTACAA- 3’ 2.8.3.
HBG F: 5’ -GCTTCTGACACAACTGTGTTCACTAGC- 3’
R: 5’ - CACCAACTTCATCCACGTTCACC- 3’ 2.8.3.
TP53 exon 2-3
F: 5’ -CCAGGTGACCCAGGGTTGGA- 3’
R: 5’ -AGCATCAAATCATCCATTGC- 3’ 2.14.
TP53 exon 4
F: 5’ -TGCTCTTTTCACCCATCTAC- 3’
R: 5’ -ATACGGCCAGGCATTGAAGT- 3’ 2.14.
TP53 exon 5-6
F: 5’ -TGTTCACTTGTGCCCTGACT- 3’
R: 5’ -TTAACCCCTCCTCCCAGAGA- 3’ 2.14.
TP53 exon 7
F: 5’ -CTTGCCACAGGTCTCCCCAA- 3’
R: 5’ -AGGGGTCAGCGGCAAGCAGA- 3’ 2.14.
TP53 exon 8-9
F: 5’ -TTGGGAGTAGATGGAGCCT- 3’
R: 5’ -AGTGTTAGACTGGAAACTTT- 3’ 2.14.
TP53 exon 10
F: 5’ -CAATTGTAACTTGAACCATC- 3’
R: 5’ -GGATGAGAATGGAATCCTAT- 3’ 2.14.
TP53 exon 11
F: 5’ -AGACCCTCTCACTCATGTGA- 3’
R: 5’ -TGACGCACACCTATTGCAAG- 3’ 2.14.
H3F3A exon 1
F: 5’ -GATTTTGGGTAGACGTAATCTTCA- 3’
R: 5’ -AACAAAAAGATGGATAACAGGAAA- 3’ 2.14.
H3F3A exon 2
F: 5’ -TTTCTTTGAAGCTGCCCACT- 3’
R: 5’ -TCCAAATTGTTGCATGTGCT- 3’ 2.14.
H3F3A exon 3
F: 5’ -AGCATCTTGCCCAGTCATTTT- 3’
R: 5’ -CAGGTATTGGCAGTTTTTCCA- 3’ 2.14.
TERT F: 5’-CGGAAGAGTGTCTGGAGCAA- 3’
R: 5’-TCGTAGTTGAGCACGCTGAACAG- 3’ 2.16.1.
TERC F: 5’-CTAACCCTAACTGAGAAGGGCGT- 3’
R: 5’-GGCGAACGGGCCAGCAGCTGAC- 3’ 2.16.1.
Dyskerin F: 5’-GCCGAAATACAACACGCTGAAG- 3’
R: 5’-CAGAGGATTTGAACCACATGCA- 3’ 2.16.1.
GAPDH F: 5‘-ACCCACTCCTCCACCTTTG- 3’
R: 5’-CTCTTGTGCTCTTGCTGGG- 3’ 2.16.1.
SLX4 F: 5’-CTGTGCCATCAAAGCAGAAA-3’
R: 5’-CGGAGATTTTTCTGGGAACA- 3’ 2.16.1.
XRCC3 F: 5’-CCATTCCGCTGTGAATTTG-3’
R: 5’-GCCTCTGTCACCTGGTTGAT- 3’ 2.16.1.
RTEL1 F: 5’-CACTCGCAACTCACACAGGT-3’
R: 5’-TTACGGCACAAGTGGATCTG- 3’ 2.16.1.
ZBTB48 F: 5‘-GGGTGAGGAAGTTTGAGTGC- 3’
R: 5’-ACCACCACCATCTTCTCGTC- 3’ 2.16.1.
NBS1 F: 5‘-CCCACCTCTTGATGAACCAT- 3’
R: 5’-CCCACCTCCAAAGACAACTG- 3’ 2.16.1.

67
MYCN F: 5’-GCCGGAAGAGACAGATAAGC-3’
R: 5’-TTCTCCACAGTGACCACGTC-3’ 2.16.2.
M2 5’-AAT CCG TCG AGC AGA GTT-3’ 2.18.3.
ACX 5’-GCGC GGC TTA CCC TTA CCC TTA CCC TAA CC-3’ 2.18.3.
Abbreviations: F, forward; R, reverse. Primer sequences for section 2.8.3. were described in Henson et al., 2017 & Lau et al., 2013. TP53 primer sequences were obtained from the International Agency for Research on Cancer (IARC) website (http://p53.iarc.fr/Download/TP53_DirectSequencing_IARC.pdf). H3F3A primers were obtained from (Bower et al., 2012) and RT-qPCR primers from section 1.15.1 were obtained from (Cao et al., 2008b). The M2 and ACX primers were described in (Kim and Wu, 1997; Piatyszek et al., 1995).
genomic DNA was added to the PCR. The relative TC of a sample was obtained from the mean of
triplicate telomere reactions normalised to the SCG reactions.
2.10. T-circle detection
T-circles were detected using two-dimensional TRF analysis. Genomic DNA was restriction digested with
HinfI and RsaI, using the TRF protocol. Digested DNA was separated in a 0.6% agarose gel at 1 V/cm for
13.5 hr, in the first dimension. Lanes were excised and run in a 1.1% agarose gel containing 300 ng/mL
ethidium bromide at 6 V/cm for 4 hrs, in the second dimension. Following the TRF protocol in section
2.9.1, gels were dried, denatured and hybridised overnight to a 32P-ATP-labelled (CCCTAA)4 telomeric
probe in Church buffer. Gels were washed, exposed to a storage phosphor screen and scanned on a
Typhoon imager.
2.11. Telomere chromatin immunoprecipitation (ChIP)
2.11.1. Cell lysis and chromatin isolation
Cells were harvested by trypsinisation, washed in 1X PBS and collected as 1X107 cell pellets. Pellets were
resuspended in 1 mL of cell lysis buffer (5 mM HEPES-KOH pH 8, 85 mM KCl, 0.4% [v/v] NP-40, and,
added fresh each time, 1X complete protease inhibitor (Roche), and 1 mM phenylmethylsulphonyl
fluoride [PMSF]) and incubated on ice for 10 mins. To pellet the nuclei, samples were centrifuged at
6000 x g for 15 sec at 4°C, and the supernatant discarded. The chromatin bound proteins were cross-
linked at room temperature with 1% (v/v) formaldehyde in cell lysis buffer, lacking NP-40, for 10 mins.

68
To end the fixation, 75 µL of 1.5 M glycine was incubated with the nuclei for 5 mins at room temperature.
The nuclei were centrifuged at 4°C for 15 sec at 6000 x g, and the supernatant was discarded. Nuclei
were lysed with 50 µL nuclei lysis buffer (50 mM Tris-HCl pH 8.1, 1 mM EDTA, 1% [v/v] SDS, added fresh
each time: 1X complete protease inhibitor, and 1 mM PMSF) on ice for 10 mins, and then diluted with
750 µL buffer A, containing 20 mM HEPES-KOH pH 8, 2 mM MgCl2, 300 mM KCl, 1 mM EDTA, 10% (v/v)
glycerol, and 0.1% (v/v) Triton X-100. The DNA was fragmented by sonication on a Branson sonifier in
an ice/ethanol bath. Sonication was performed with a duty cycle of 10, and 60 second pulse/pause cycles
at outputs 3, 4, and 5. Solubilised chromatin was isolated by retaining the supernatant after
centrifugation at 17,000 x g for 10 mins at 4°C.
2.11.2. Immunoprecipitation (IP)
IP was conducted in a buffer of 20 mM HEPES-KOH pH 7.9, 150 mM KCl, 1.5 mM EDTA, 1% (v/v) Triton
X-100, and, added fresh each time, 1X complete protease inhibitor, and 1 mM PMSF. The chromatin
(112 µL) was rotated at 4°C for 1 hr with 225 µL of the IP buffer, and the antibody as indicated in Table
2.8. A pre-blocked suspension of Protein G agarose beads (Roche), were bound to the antibody by
rotating 60 µL/sample at 4°C overnight. The next day, the chromatin/antibody/bead complexes were
collected by centrifugation at room temperature for 30 sec at 17,000 x g. Unbound chromatin and
antibodies were removed by washes: two washes with buffer A and once with TE, with the centrifuge
step repeated between each wash.
2.11.3. DNA isolation
Immunoprecipitated DNA was eluted from the beads by digestion of the antibody with 200 µg of
proteinase K (Sigma) in elution buffer (50 mM NaHCO3, 1% [v/v] SDS) at 45°C for 2 hrs. The DNA was
purified with the Qiagen PCR purification kit and eluted in 140 µL of TE.
2.11.4. Quantitation
The amount of telomeric chromatin immunopurified by each antibody was quantitated using slot-blot
analysis. The purified DNA was denatured with 980 µL of 0.46 M NaOH and heated at 95°C for 5 mins.

69
The cooled samples were then halved, and 560 µL loaded into a slot-blot well and blotted onto
Biodyne B 0.45 µM nylon membrane (Pall). The remainder of the sample was used to make a duplicate
membrane. For the purpose of quantitation, a standard was included for every sample; 1%, 5%, 10%
and 25% purified input (lacking IP) was also blotted. The membranes were air-dried, cross-linked in a
Stratagene UV Stratalinker 1800, and pre-hybridised with Church buffer at 50°C for 1 hr. One membrane
was hybridised overnight with a 32P-ATP-labelled (CCCTAA)3 telomere probe, and the other was
hybridised with 32P-dATP-labelled ALU sequence. The following day, both membranes were washed four
times: telomere membranes with 2X SSC at room temperature for 15 mins, and ALU membranes with
0.1% SDS/0.2X SSC at 37°C for 15 mins, and then exposed to a phosphor screen overnight. The phosphor
screen was imaged with a Typhoon imager and then signals were quantitated using ImageQuant
software. Standard curves were generated for each sample and used to normalise the telomeric DNA
immunoprecipitated. All experiments were performed on two biological replicates.
2.12. Labelling probes with 32P
Experiments used two types of C-rich telomere probes, [CCCTAA]3 or [CCCTAA]4. Both probes were end
labelled with ATP-γ32-P radiation (Perkin Elmer) by a T4 polynucleotide kinase (New England BioLabs) at
37°C for 30 mins. Unlabelled probe was removed using a Quick Spin Mini Oligo Column (Roche). The
same method was used to end-label the GAPDH probe in section 2.17. The ALU probe in section 2.11.4,
was labelled with α-32P-dATP (Perkin Elmer) using the DECAprime II Random Primed DNA Labeling Kit
(Ambion). Unlabelled DNA was removed using a Quick Spin Mini DNA Column (Roche).
2.13. Whole genome sequencing (WGS) and bioinformatics
2.13.1. WGS and read alignment
Whole genome sequencing was performed on the Illumina HiSeq X Ten platform, by Macrogen Inc.
(Korea) and the Kinghorn Centre for Clinical Genomics/Garvan Medical Research Institute, to generate
paired-end (2 × 150 bp) sequence reads. Subsequent analysis was performed by Dr Erdahl Teber. The

70
Table 2.8. Antibodies for telomere ChIP.
reads were trimmed, and poor quality reads were removed using Trimmomatic (v0.32; parameters:
ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:20 MINLEN:75). The
remaining paired-end reads were aligned to NCBI hg19 reference genome using BWA (v0.7.10; main
parameters: mem -M). Using Picard (v1.119), the generated BAM file was sorted; duplicates were
removed, and the reads placed into read groups and indexed. Picard (v1.1.19) modules,
CollectAlignmentSummaryMetrics and CollectMultipleMetrics, determined the mapping quality
metrics. Insertion or deletion (indel) realignment was carried out with RealignerTargetCreator and
IndelRealigner, and known indel sites were based on Mills_and_1000G_gold_standard.indels.hg19.vcf
and 1000G_phase1.indels.hg19.vcf (GATK v3.2.2 resource bundle). This was followed by
BaseRecalibrator (parameters for KnownSites are: Mills_ and_1000G_gold_standard.indels.hg19.vcf,
1000G_phase1.indels.hg19.vcf,dbsnp_138.hg19.vcf) and PrintReads (GATK v3.2.2). MuTect v1.1.4
(Cibulskis et al., 2013) (parameters: --dbsnp dbsnp_138.hg19.vcf --cosmic cosmic_v71.vcf) was used to
generate single nucleotide variants (SNVs), only a ‘PASS’ entry in the FILTER column was accepted. Indels
were generated using GATK v3.2.2 HaplotypeCaller (paremeters: --genotyping_mode DISCOVERY -
stand_call_conf 30 -stand_emit_conf 10), filtered by GATK VariantFiltration (parameters: --
filterExpression "QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0"), and were decomposed and left
Antibody Company Isolated from Dilution
TERF21P/ RAP1 Novus Biologicals
(NB100-292) Rabbit 1:40
TRF2 Novus Biologicals
(NB110-57130) Rabbit 1:70
Normal Mouse IgG Merck Millipore
(12-371) Mouse 1:30
Normal Rabbit IgG Cell Signaling Technology
(2729) Rabbit 1:30
Histone H3 Cell Signaling Technology
(4620) Rabbit 1:40
H3K9me3 Abcam
(ab8898) Rabbit 1:30
H3K4me2 Cell Signaling Technology
(9725) Rabbit 1:50

71
normalised (Vt normalisation reference, http://genome.sph.umich.edu/wiki/Vt) (Tan et al., 2015). Short
indel calls were generated by PINDEL v0.2.4w (Ye et al., 2009a) and were left normalised. For each cell
line, a final vcf file was generated, and used in all downstream analysis. This file contained the
intersecting variant calls between normalised Haplotypecaller and PINDEL, all unique HaplotypeCaller
indels sized 1 to 9 bp, unique PINDEL calls ≥100 bp, and all SNV ‘PASS’ Mutect calls.
Delly v0.6.7 (Rausch et al., 2012) identified structural variant (SV) calls to detect the four SV
types (deletion, DEL; duplication, DUP; inversion, INV; translocation, TRA). The results were
concatenated and sorted using vcftools (v0.1.13) and annotated into Class A and Class B categories,
whilst TxDb.Hsapiens.UCSC.hg19.knownGene (v3.2.2) R package was used to map the transcript and
gene structures. A Class A annotation indicated the gene(s) overlapping at a breakpoint, and the Class B
annotation, indicated the gene(s) between two breakpoints or within the SV.
2.13.2. Variant annotations
To determine potentially pathogenic genetic alterations Ingenuity Variant Analysis (IVA, v4.2.20160927)
was used to filter and annotate SNV and indel results. To remove variants present in a healthy
population, the analysis parameters included: a read depth of at least 10, variants had to be outside of
the top 1% most exonically variable genes in healthy genomes (1000 genomes), and variants observed
in at least 0.001% of the 1000 genomes project, ExAc and NHLBI ESP exomes were excluded. The next
step was to determine potentially deleterious events using the predicted deleterious filter. From the
settings used we obtained a gene list including mutations: pathogenic or likely pathogenic, established
in literature, gene fusion, inferred activating mutation by Ingenuity, predicted gain of function by BSIFT,
frameshift/in-frame indel or start/stop codon change, missense unless predicted tolerated by SIFT or
PolyPhen-2, splice site loss up to 2 bases into intron or as predicted by MaxEntScan, or a loss of function
structural variant.

72
2.14. Sanger sequencing
All ATRX and DAXX exons were amplified using the primers, PCR buffer and PCR conditions previously
reported (Jiao et al., 2011), and summarised in Appendix I. The protocol for TP53 sequencing was
described on the International Agency for Research on Cancer (IARC) website
(http://p53.iarc.fr/Download/TP53_DirectSequencing_IARC.pdf) and the primers are listed in Table 2.7.
Briefly, the Qiagen HotStar Taq (0.8 Units) was used to amplify the TP53 exons 2-3 and 7 in a reaction
mix containing 1X PCR buffer, 1X Q-solution, 0.2 mM each dNTP, 0.4 µM forward primer and 0.4 µM
reverse primer. The PCR conditions for exon 2-3 were: 94°C for 2 mins, followed by 50 cycles of 94°C for
30 sec, 61°C for 45 sec and 72°C for 45 sec, and the final cycle of 72°C for 10 mins, and for exon 7 were:
95°C for 15 mins, followed by 50 cycles of 94°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec, and the
final cycle of 72°C for 10 mins. Whereas, 0.8 Units of Taq Platinum (Invitrogen) were used to amplify
TP53 exons 4, 5-6, 8-9 and 11 in a reaction containing 1X PCR buffer, 1.5 mM MgCl2, 0.2 mM each dNTP,
0.4 µM forward primer and 0.4 µM reverse primer. Exons 4, 5-6, 8-9, and 11 were amplified with the
conditions: 94°C for 2 mins, followed by 20 cycles of 94°C for 30 sec, 63°C for 45 sec (-0.5°C every 3
cycles) and 72°C for 60 sec, then 30 cycles of 94°C for 30 sec, 60°C for 45 sec and 72°C for 60 sec, and
the final cycle of 72°C for 10 mins. Finally, TP53 exon 10 was amplified with the PCR conditions: 94°C for
2 mins, followed by 20 cycles of 94°C for 30 sec, 58.5°C for 45 sec (-0.5°C every 3 cycles) and 72°C for 60
sec, then 30 cycles of 94°C for 30 sec, 55°C for 45 sec and 72°C for 60 sec, and the final cycle of 72°C for
10 mins. The gene H3F3A was sequenced using primers previously reported to amplify exons 1, 2 and 3
of the gene (Bower et al., 2012). Invitrogen’s Taq Platinum was used to amplify the exons, using the
primer sequences listed in Table 2.7, with the PCR conditions: 95°C for 5 mins, followed by 30 cycles of
94°C for 30 sec, 60°C for 1 min and 72°C for 2 mins, and the final cycle of 72°C for 10 mins. Prior to
sequencing, all PCR products were purified with the QIAquick PCR Purification Kit (Qiagen), and
subsequent Sanger sequencing was conducted at the Australian Genome Research Facility.

73
2.15. RNA extraction
RNA was extracted from cell pellets using the RNeasy Mini Kit (Qiagen) with on column DNA digestion
(Qiagen DNase). The RNA was resuspended in RNase-free water and quantitated using a NanoDrop
spectrophotometer.
2.16. qPCR analysis
2.16.1. Reverse transcriptase qPCR (RT-qPCR)
Complementary DNA (cDNA) was synthesised from 1 µg of RNA using the Superscript III First Strand
Synthesis Kit (Invitrogen) with random primers. Transcripts of TERT, TERC, DKC1, and GAPDH were
detected as previously described (Cao et al., 2008b), using the primers in Table 2.7. Expression of NBS1,
XRCC3, RTEL1, ZBTB48, and SLX4 mRNA was detected using the primers described in Table 2.7. PCR
reactions contained 1X QuantiFAST SYBR Green PCR buffer (Qiagen), and 0.4 µM forward and reverse
primers for TERC, DKC1, NBS1, XRCC3, RTEL1, ZBTB48, SLX4 and GAPDH transcripts or 0.5 µM forward
and reverse primers for TERT transcripts. The cDNA reaction was diluted 1:2.5 with TE, and 2 µL was
loaded into each PCR reaction. The PCR was performed in triplicate with the following conditions: 1 cycle
at 95°C for 15 mins, followed by 40 cycles of 95°C for 15 sec and 60°C for 60 sec for TERT, TERC, DKC1,
and GAPDH or 40 cycles of 95°C for 15 sec, 60°C for 20 sec and 72°C for 30 sec for NBS1, XRCC3, RTEL1,
and SLX4 or 40 cycles of 95°C for 15 sec and 60°C for 60 sec for ZBTB48 and then melt curve analysis.
The level of expression was calculated using a delta delta cycle threshold (Ct) method, with GAPDH as a
reference gene, and expressed as RNA level relative to the appropriate control sample.
2.16.2. Genomic qPCR
MYCN copy number was determined by qPCR of 5 ng of genomic DNA. PCR reactions were performed
in triplicate using 1X QuantiFAST SYBR Green PCR buffer (Qiagen) containing 0.5 µM of forward and
reverse primers (Table 2.7) with the PCR conditions: 95°C for 5 mins then 43 cycles of 95°C for 15 sec,
61°C for 30 sec and 72°C for 20 sec, and melt curve analysis. Samples were quantitated using a standard

74
curve method, MYCN was normalised to the VAV2 gene, and copy number was expressed relative to the
normal DNA control (Applied Biosystems).
2.17. Telomeric repeat-containing transcript (TERRA)
The quantitation of TERRA was performed by slot-blot analysis of 1 µg of DNase treated RNA (Ng et al.,
2009). In addition to a DNase treated RNA sample for each cell line, 1 µg of RNA was also treated with
25 ng/µg RNase A (Sigma) to confirm that there was no DNA contamination. All samples were denatured
at 65°C for 15 mins in a solution of 66% (v/v) formamide, 2.6 M formaldehyde, and 13% (v/v) 3-(N-
morpholino)propanesulphonic acid (MOPS) solution (0.5 M sodium acetate, 0.01 M EDTA, 0.2 M MOPS).
The denatured RNA was diluted with 100 µL of 2X SSC and slot-blotted onto a Biodyne B nylon
membrane. Duplicates of all membranes were prepared. Membranes were dried and the RNA was
crossed-linked to the membrane using the Stratagene UV Stratalinker 1800. After UV-cross-linking, both
membranes were pre-hybridised with Church buffer at 55°C for 1 hr, then one membrane was
hybridized with a 32P-ATP-labelled (CCCTAA)3 telomeric probe and the other a 32P-labelled GAPDH probe
(ACCCACTCCTCCACCTTTG), overnight at 55°C. The following day, the membranes were washed three
times for 15 mins at room temperature with 2X SSC/0.1% SDS and then at 50°C with 0.2X SSC/0.1% SDS
for 15 mins. Membranes were exposed to a storage phosphor screen and scanned on a Typhoon imager.
Images were quantitated with ImageQuant software and the amount of TERRA was corrected for loading
by dividing by the intensity of the corresponding GAPDH transcript. Three biological replicates were
analysed.
2.18. Telomerase activity
2.18.1. Protein Lysis
Cells were trypsinised, washed with 1X PBS and pelleted. An equal number of cells were lysed at 4°C for
30 mins with 3-([3-cholamidopropyl] dimethylammonio)-1-propanesulphonate (CHAPS) lysis buffer (10
mM Tris-HCl pH 7.5, 1 mM MgCl2, 1 mM ethylene glycol-bis[2-aminoethylether]-N,N,N',N'-tetraacetic

75
acid [EGTA], 10% glycerol, 10% CHAPS), supplemented with 1mM PMSF and 5 mM 2-mercaptoethanol.
Following lysis, the samples were centrifuged at 15,500 x g for 20 mins at 4°C, and then the supernatant
was transferred to a new tube to remove cellular debris.
2.18.2. Immunoprecipitation
To purify the lysate and remove potential PCR inhibitors, anti-hTERT antibody (10 µg; obtained from Dr
Scott Cohen) was bound to telomerase by rotating samples at 4°C for 30 mins (Cohen and Reddel, 2008).
A 50% (v/v) suspension of Protein G beads (Roche), pre-blocked in BSA (New England BioLabs), was then
bound to the antibody at 4°C for 1 hr. The telomerase/antibody/bead complex was collected in an
Illustra micro-spin column (GE Healthcare) with vacuum suction, and the supernatant discarded. The
beads were washed with ice cold lysis buffer to remove any unbound proteins. To release the
telomerase enzyme from the bead/antibody complex, 1.5 mM antigenic peptide solution was combined
with the complex in the column and incubated at room temperature for 1 hr before collection of the
supernatant.
2.18.3. Telomeric repeat amplification protocol (TRAP)
Immunoprecipitated telomerase was added to 0.09 µg of the M2 primer (Table 2.7) and allowed to
extend via telomerase mediated base addition in a buffer containing 45 mM Tris pH 8.8, 11 mM
ammonium sulphate, 4.5 mM MgCl2, 10 mM EDTA pH 8, 76 mM 2-mercaptoethanol, 113 µg/mL BSA
(Ambion), 1 mM each of dNTPs (Roche), 0.045 µg of the reverse primer ACX (Table 2.7), and 2 Units Taq
polymerase (Roche). The TRAP reaction was performed under the conditions in Table 2.8. PCR products
were separated on a 10% denaturing polyacrylamide gel in 0.5X TBE and detected with SYBR Green I
(Molecular Probes), visualised on a Typhoon imager.
2.19. Immunoblotting
Cells were harvested, pelleted and resuspended in cold lysis buffer supplemented with complete
protease inhibitor (Roche), 100 mM NaF, and 200 µM Na3VO4 immediately before use. For analysis of

76
Table 2.8. TRAP PCR conditions.
Number of cycles Step Temperature Time
1 cycle Telomerase extension of
primer 25°C 30 mins
1 cycle Taq Hot start 95°C 2 mins
35 cycles Denaturation 95°C 10 sec
Annealing 50°C 25 sec
Extension 72°C 30 sec
1 cycle Denaturation 95°C 15 sec
Annealing 50°C 25 sec
Extension 72°C 60 sec
Table 2.9. Antibodies and conditions used for Western blot analysis.
Name Manufacturer Isolated
from Blocking agent Diluent Dilution Incubation
conditions
anti-ATRX Abcam
(ab72124) Rabbit 5% (w/v) skim milk 1X TBS 1:500
Room temperature,
2.5 hrs
anti-DAXX Sigma
(HPA008736-100UL)
Rabbit 5% (w/v) skim milk 1X TBS 1:250 Room
temperature, 2 hrs
anti-p53 (DO-1)
Merck Millipore (OP43L)
Mouse 5% (w/v) skim milk/1X TTBS
Same as blocking
agent 1:1000
Room temperature,
1 hr
anti-p21 (Waf1/Cip1)
Cell Signalling (2947)
Rabbit 5% (w/v) BSA/1X
TTBS
Same as blocking
agent 1:1000
Room temperature,
1 hr
anti-GAPDH ThermoFisher
Scientific (MA5-15738)
Mouse 5% (w/v) skim milk/1X TTBS
Same as blocking
agent 1:5000
Overnight, 40C
anti-β-actin Sigma
(A2228-100UL) Mouse 5% (w/v) skim milk 1X TBS 1:3000
Overnight, 40C
anti-HSP70 Sigma
(H5147-100UL)
Mouse 5% (w/v) skim milk 1X TBS 1:3000 Overnight,
40C
ATRX and DAXX proteins the cells were lysed in EBC buffer (50 mM Tris pH 8, 120 mM NaCl, and 0.5%
(v/v) NP-40), and for p53 and p21 immunoblots the cells were lysed in RIPA buffer (50 mM Tris pH 8,
150 mM NaCl, 0.1% (v/v) SDS, 1% (v/v) NP-40, and 0.5% (v/v) sodium deoxycholate). Cells were lysed
for 30 mins at 4°C and centrifuged at 15,500 x g for 20 mins to remove cell debris. The protein
concentration of the whole cell extract was determined against known concentrations of BSA standards,
using the bicinchoninic acid (BCA) protein assay kit.

77
Equal concentrations of protein lysates were mixed with NuPAGE LDS Sample Buffer (Life
Technologies) and denatured by boiling at 95°C for 10 mins. Proteins were resolved on a 3–8% NuPAGE
Tris-acetate polyacrylamide SDS gel (Life Technologies) (running buffer: 2.5 mM tricine, 2.5 mM Tris,
0.005% (v/v) SDS, pH 8.24) for ATRX, on a 10% Tris-glycine SDS polyacrylamide gel for DAXX, and on a
12% Tris-glycine SDS polyacrylamide gel for p53 and p21 (running buffer: 0.02 M Tris, 0.2 M glycine,
0.1% SDS (v/v), pH 8.3). Separated proteins were transferred to a methanol-activated 0.45 mm PVDF
membrane (Millipore), in chilled transfer buffer (22 mM Tris, 171 mM glycine, 0.01% (v/v) SDS, 20% (v/v)
methanol), and blocked for 1 hr at room temperature and then probed according to the conditions in
Table 2.9. The membranes were washed with a solution of 0.1% Tween-20, 50 mM Tris, and 150 mM
NaCl (TTBS) three times for 15 mins each, then probed at room temperature for 1 hr with HRP-
conjugated secondary antibodies (DakoCytomation) diluted 1:5000 in TTBS, and followed by another
wash series with TTBS. Chemiluminescence detection was performed using SuperSignal West Pico
chemiluminescent substrate (ThermoFisher Scientific) at room temperature for 5 mins, exposed to X-
ray film (Fujifilm), and developed on an X-ray developer (Konica).
2.20. Statistical analyses
GraphPad Prism 6 was used to generate graphs and perform statistical analysis. The Kaplan-Meier
product-limit method was used to estimate survival, comparisons of survival curves were performed by
the log-rank test, and medians and proportions were compared by the Mann-Whitney test and Chi-
square test, respectively. Two data sets were compared with unpaired Student’s two-tailed t tests or
two-tailed Mann-Whitney tests, and multiple data sets were compared using one-way ANOVA.

78
Chapter 3:
A subgroup of high-risk MYCN non-
amplified neuroblastomas lacks a
telomere lengthening mechanism

79
Chapter 3: A subgroup of high‐risk MYCN non‐amplified
neuroblastomas lacks a telomere lengthening
mechanism
3.1. Introduction
The acquisition of replicative immortality is a hallmark of human cancer, achieved by activation of a TLM
to counter the normal telomere attrition that accompanies cellular proliferation (Hanahan and
Weinberg, 2000; Hanahan and Weinberg, 2011). However, a substantial proportion of glioblastomas
(Hakin-Smith et al., 2003), liposarcomas (Costa et al., 2006; Jeyapalan et al., 2008), osteosarcomas
(Ulaner et al., 2003), and retinoblastomas (Gupta et al., 1996) have been reported as telomerase-
negative with no evidence of ALT activity. The consensus view in the telomere research field has been
that these results are due to false-negative assays; however, there can be other plausible explanations.
Firstly, telomerase-negative tumours may be able to maintain their telomere length by a different ALT
mechanism which lacks the known phenotypic characteristics of ALT. A second potential explanation is
that it is theoretically possible some tumours can achieve long term proliferation without an active TLM
(Reddel, 2000). To date, it has been impossible to demonstrate the latter scenario as serial sampling of
human tumours which are required to show continuous telomere shortening over extended periods of
time cannot usually be obtained. Therefore, suitable cell line models are required to determine if an
active TLM is a requirement for the long-term proliferation of malignant cells.
NB is an embryonal tumour of the sympathetic nervous system (Maris, 2010). Telomerase
activity has been identified as a poor prognostic factor in NB (Ohali et al., 2006; Onitake et al., 2009;
Poremba et al., 2000; Poremba et al., 1999) although it is absent in 20-60% of metastatic NB (Krams et
al., 2001; Onitake et al., 2009; Poremba et al., 2000; Poremba et al., 1999), supporting the presence of

80
ALT in NB. Thus far, ALT has either been diagnosed in NB tumours by long telomeres (a feature of ALT)
in the absence of telomerase (Onitake et al., 2009) or by the presence of mutation/deletion in ATRX
(Cheung et al., 2012; Kurihara et al., 2014; Molenaar et al., 2012; Peifer et al., 2015; Valentijn et al.,
2015), a chromatin remodelling factor where loss of function is associated with ALT activity (Bower et
al., 2012; Chen et al., 2014; Heaphy et al., 2011a; Lovejoy et al., 2012; Schwartzentruber et al., 2012).
However, C-circles, a quantifiable marker of ALT activity (Henson et al., 2009), have not been used to
detect ALT in any of these studies. Relying solely on long telomeres and the absence of telomerase can
potentially misclassify tumours as ALT-positive, especially in embryonal tumours that tend to have
longer telomeres, while using only ATRX mutations will likely underestimate the prevalence of ALT as
ATRX mutations are not a universal feature of ALT (Lovejoy et al., 2012).
We hypothesised that (1) a proportion of high-risk NB with long telomeres were ALT-negative
and (2) a subset of ALT-positive high-risk NB tumours were ATRX wild-type. Utilising quantitative PCR to
measure telomere content (TC) and C-circle levels (Lau et al., 2013) in a cohort of high-risk NB, we
detected ALT in a significantly higher frequency than previously reported on the basis of ATRX mutation
frequency, supporting the hypothesis that some ALT-positive NB are ATRX wild-type. Furthermore, we
found that a subset of MYCN non-amplified tumours with long telomeres lacked ALT activity. We also
went on to screen a large panel of NB cell lines and identified two NB cultures with exceptionally long
telomeres which were negative for both telomerase and C-circles.
3.2. Results
3.2.1. Unique subgroup of high-risk MYCN non-amplified neuroblastoma
Here we describe a cohort of 149 high-risk NB screened for C-circles and TC using telomere quantitative
PCR where ALT-positive tumours were defined as those with a relative C-circle level of ≥7.5 compared
to the ALT+ IIICF/c cell line with an arbitrary value of 100 (Lau et al., 2013) (Appendix II Table A2.1-4).
Twenty-four percent (36/149) of tumours were found to be C-circle/ALT-positive. None (0/36) of the
ALT-positive tumours had MYCN amplification, consistent with other studies reporting MYCN

81
Figure 3.1. A subgroup of high-risk MYCN non-amplified neuroblastoma with long telomeres are
ALT-negative.
(A) Relative telomere content (TC) in arbitrary units (AU), measured by telomere qPCR for individual
patient tumours (n=149), are plotted according to ALT status (ALT+ n=36, ALT- n=113). ALT+ was
defined by a relative C-circle (CC) level ≥7.5 compared to the ALT+ IIICF/c cell line with an arbitrary
value of 100. (B) Age at diagnosis for individual patient tumours is plotted according to ALT status
(n=149). (C) Relative TC of MYCN non-amplified tumours (n=94) is plotted according to ALT status
(ALT+ n=36, ALT- n=58). A subgroup of ALT- tumours (n=17) had long telomeres (TC≥15, indicated
by the bracket). (D) Age at diagnosis for individual MYCN non-amplified ALT-negative patient
tumours (n=58) is plotted according to TC.
Data points represent the average of triplicate TC measurements by qPCR. Horizontal red bar
indicates group median. * P<0.01

82
amplification and ATRX aberrations are mutually exclusive (Cheung et al., 2012; Kurihara et al., 2014;
Peifer et al., 2015; Pugh et al., 2013; Valentijn et al., 2015). This is contrast with ALT-negative tumours
where 49% (55/113) had MYCN amplification. The relative TC of ALT-positive tumours was significantly
higher than that of ALT-negative tumours (median 19.7 vs. 6.2; P<0.0001; Figure 3.1.A); this was not due
to younger patients in the ALT-positive group as patients with ALT NB were significantly older at
diagnosis than patients with ALT-negative NB (median age 4.4 vs. 2.3 years; P<0.0001; Figure 3.1.B). Of
note, within the MYCN non-amplified group, 29% (17/58) of tumours with long telomeres (TC≥15) were
ALT-negative (Figure 3.1.C). In addition, patients with ALT-negative/TC≥15 tumours were older at
diagnosis than patients with ALT-negative/TC<15 tumours (median age 4.67 vs. 2.72 years; P<0.01;
Figure 3.1.D). There was sufficient DNA to assess telomere length by Southern blot terminal restriction
fragment (TRF) analysis for 45 samples, including four of the 17 ALT-negative/TC≥15 tumours. The TRF
telomere length profile of the four ALT-negative/TC≥15 tumours was long and heterogeneous (relative
TC≥15 corresponds to mean TRF length 20 kb; Figure 3.2.A), with a mean length indistinguishable from
ALT-positive tumours (P>0.05). Frozen tumour samples were available for the four ALT-negative/TC≥15
tumours which allowed us to perform TRAP assays; all four tumours were telomerase-negative (Figure
3.2.B). This suggests that a subgroup of metastatic NB with long telomeres may not require a TLM to
allow long term proliferation as a result of their extensive telomere reserves.
The molecular details of ALT upregulation are not well defined but there is a correlation
between ALT activity and the loss of expression of ATRX or its binding partner DAXX (Bower et al., 2012;
Heaphy et al., 2011a; Lovejoy et al., 2012). Inactivation of p53 is also strongly correlated with ALT in cell
lines although this is less apparent in tumours (Henson and Reddel, 2010). Next-generation sequencing
results were available for 32 tumour samples; 13 ALT-positive and 19 ALT-negative. This includes
previously reported whole exome sequence data for 26 samples (Pugh et al., 2013) and whole genome
sequence data for nine tumours obtained as part of this study (of which 3 had existing whole exome
data from Pugh et al., 2013). Of the 13 ALT-positive tumours, eight had an ATRX mutation, one had a

83
Figure 3.2. A unique subgroup of high-risk ALT-negative/MYCN non-amplified/TC≥15 NBs have
poor survival.
(A) Terminal restriction fragment (TRF) analysis of 13 selected tumour samples. Telomere content
(TC) and C-circle (CC) status measured by telomere qPCR is indicated at the bottom with mean TRF
lengths. Black boxes represent the four ALT-negative/TC≥15 samples, green boxes represent the
ALT-positive samples, and ALT-negative/TC<15 are indicated by white boxes. (B) Telomere repeat
amplification protocol (TRAP) to detect telomerase activity in six selected tumour samples. Cell
lysates were immunopurified with anti-hTERT antibody to remove potential inhibitors. The ALT
sample U-2 OS was negative for telomerase and SH-SY5Y was included as a telomerase positive
control. (C, D) Event-free and overall survival of ALT-negative/TC≥15 (ALT-/ TC≥15) (n=17), ALT-
positive (n=36), and MYCN amplified (amp) (n=55) tumours.
TRF and IP-TRAP were performed by Dr Loretta Lau.

84
DAXX mutation, and one had a TP53 mutation. In contrast, abnormalities of ATRX, DAXX or TP53 were
observed in 0/19 ALT-negative tumours.
It is well established that MYCN amplification results in a worse outcome for patients than MYCN
non-amplified NB tumours (Ambros et al., 2009; Seeger et al., 1985). In our cohort, the outcome for
patients with ALT NB was as poor as the outcome for MYCN amplified patients (5-year event-free
survival 28% vs. 24%; 5-year overall survival 36% vs 28%; Figure 3.2.C, D), despite all ALT-positive
tumours being MYCN non-amplified. Like ALT NB, ALT-negative/TC≥15 NB also had later events or
deaths and a poor survival that was not significantly different from the ALT tumours, although this may
be due to the number of cases in each subgroup (5-year overall survival 49% vs. 36%; P=0.1908; Figure
3.2.C, D), ultimately leading to death in 51% of patients with this subgroup of disease.
3.2.2. ALT-negative and telomerase-negative cancer cell lines
3.2.2.1. Neuroblastoma
To determine whether ALT-negative/TC≥15 NB cells lack a TLM, or have an as-yet unidentified
telomerase-independent mechanism, the telomere length of these cells must be examined over time.
To perform this study, we screened 35 cell lines derived from metastatic NB for a phenotype
corresponding to the ALT-negative/TC≥15 tumours (TC≥15 and C-circle negative via quantitative PCR
and MYCN non-amplified). We identified three cell lines that have a TC≥15 (CHLA-90, COG-N-291, LA-N-
6) and one cell line with a TC of 14 (SK-N-FI); two cell lines (SK-N-FI and CHLA-90) were C-circle positive,
as previously reported (Farooqi et al., 2014; Lau et al., 2013), and two were C-circle negative (COG-N-
291 and LA-N-6; Table 3.1) and also had the highest TC of the 35 cell lines (25 and 38, respectively). TRF
analysis confirmed long and heterogeneous telomere lengths in COG-N-291 and LA-N-6 cells (mean 31.0
and 37.8 kb, respectively), comparable to U-2 OS, an ALT cell line with very long telomeres (mean TRF
38.8 kb; Figure 3.3.A). The TRF blots of LA-N-6 and COG-N-291 showed a banding pattern that has only
been observed when ALT activity was suppressed in ALT-positive cells by overexpression of Sp100 a
component of PML bodies (Jiang et al., 2005), and when telomerase-mediated telomere extension was

85
Table 3.1. Characteristics of 35 neuroblastoma cell lines.
Telomere content
SD C-
circle SD hTERT SD hTR SD Dyskerin SD MYCN SD
LA-N-6 38.0 0.047 -0.4 0.036 0.0 0.01 1.1 0.07 0.5 0.05 3.9 0.4
COG-N-291 25.0 0.006 -3.3 0.022 0.0 0.00 0.0 0.00 0.9 0.13 2.7 0.2
CHLA-90 15.6 0.005 139 0.285 0.1 0.06 2.8 0.44 0.8 0.04 18.9 0.8
SK-N-FI 14.0 0.013 196 0.695 0.0 0.00 0.3 0.04 1.4 0.24 4.2 0.5
CHLA-119 2.0 0.002 2.1 0.014 0.1 0.02 0.7 0.10 0.9 0.14 262 16.0
CHLA-122 0.8 0.001 -0.1 0.000 3.0 0.40 4.2 0.29 1.2 0.08 154 14.5
CHLA-136 0.8 0.003 -0.1 0.000 0.5 0.07 1.7 0.07 1.3 0.13 141 24.0
CHLA-140 1.2 0.009 -0.2 0.001 4.6 0.56 2.2 0.34 0.3 0.01 6.9 0.7
CHLA-171 1.3 0.001 0.3 0.001 3.3 0.28 1.1 0.07 0.6 0.11 2.7 0.1
CHLA-225 2.5 0.001 0.0 0.006 2.2 0.29 2.6 0.29 1.5 0.18 127 14.2
CHLA-42 0.4 0.001 0.3 0.001 0.6 0.09 2.5 0.08 1.0 0.13 4.8 0.6
CHLA-95 1.0 0.001 0.1 0.002 0.6 0.04 0.7 0.07 0.8 0.10 495 41.9
CHP-134 1.3 0.002 -0.1 0.001 2.1 0.07 1.0 0.14 1.6 0.07 485 60.5
COG-N-328h 0.5 0.001 -0.1 0.000 0.9 0.13 1.9 0.15 2.0 0.22 252 26.7
COG-N-347 0.4 0.002 -0.1 0.000 0.6 0.08 1.5 0.11 0.3 0.02 1066 77.0
FU-NB-2006 0.6 0.000 0.3 0.004 1.0 0.25 5.5 0.53 1.2 0.18 5.7 0.7
IMR32 0.5 0.002 0.0 0.002 1.0 0.10 1.0 0.11 1.0 0.10 120 25.4
KELLY 0.6 0.001 0.0 0.002 2.5 0.26 0.8 0.08 0.7 0.07 676 56.5
LA-N-1 1.3 0.001 0.0 0.002 0.3 0.05 0.8 0.13 1.0 0.09 392 85.5
LA-N-5 0.5 0.000 0.0 0.001 2.7 0.25 1.2 0.14 1.4 0.17 256 10.7
NB69 1.1 0.000 0.1 0.002 2.7 0.36 0.7 0.07 0.6 0.09 4.5 0.7
SH-EP 2.5 0.002 0.0 0.004 8.1 0.50 1.1 0.12 1.3 0.11 1.0 0.1
SH-SY5Y 1.2 0.003 -0.2 0.001 2.9 0.31 1.2 0.06 0.8 0.05 3.7 0.3
SK-N-AS 1.8 0.004 0.2 0.001 2.3 0.29 0.3 0.02 1.0 0.14 2.8 0.7
SK-N-BE1 1.8 0.005 0.3 0.005 1.0 0.14 1.6 0.14 0.4 0.05 475 41.6
SK-N-BE2c 0.6 0.001 0.0 0.002 0.4 0.05 0.9 0.11 1.0 0.14 115 8.4
SK-N-DZ 2.1 0.036 -0.3 0.001 0.8 0.11 0.9 0.08 1.0 0.08 220 36.3
SK-N-SH 1.2 0.001 -0.1 0.002 1.8 0.19 0.9 0.15 0.6 0.07 3.1 0.3
SMS-KANR 0.4 0.000 0.0 0.002 0.8 0.11 1.6 0.08 1.1 0.12 164 17.7
SMS-KCN 0.7 0.001 0.1 0.000 0.4 0.06 0.7 0.07 0.5 0.04 909 55.7
SMS-KCNR 0.4 0.000 0.0 0.002 0.5 0.07 1.2 0.09 0.6 0.02 537 32.5
SMS-LHN 1.3 0.002 -0.3 0.001 0.2 0.02 2.3 0.33 1.3 0.17 6.2 0.7
SMS-SAN 0.5 0.001 0.0 0.001 0.2 0.03 1.8 0.10 0.9 0.10 835 64.1
Note: Relative hTERT, hTR, and dyskerin mRNA levels (samples normalised to IMR32 set to 1) and MYCN copy number were measured by qPCR and expressed as the mean of triplicates.

86
Figure 3.3. Two neuroblastoma cell lines (COG-N-291 and LA-N-6) have long and heterogeneous
telomere lengths similar to the ALT-negative/TC≥15 tumour group.
(A) TRF analysis of a panel of seven cell lines. ALT-positive (ALT+) and telomerase-positive (TEL+) cell
lines are indicated. The HT1080 hTR line is a clone of HT1080 with telomeres over-lengthened due to
overexpression of hTR and upregulation of telomerase. (B) Representative images of telomere
fluorescent in situ hybridisation (FISH) including the ALT cell line U-2 OS and telomerase-positive cell
line SH-SY5Y. Telomeres are represented by green signals at the end of the chromosomes. (C)
Quantitation of telomere FISH in a panel of eleven cell lines. Dots in graph represent individual
telomere signals with mean telomere fluorescence indicated by red horizontal bar. 15 metaphases
were analysed per cell line. (D) Percentage of chromosome ends without telomere signal (images
from Figure 3.3.B). 15 metaphase spreads were analysed per cell line.

87
inhibited by deletion of the essential DNA helicase RTEL1 (Uringa et al., 2012). Telomere fluorescence
in situ hybridisation (FISH) analysis showed COG-N-291 and LA-N-6 have a greater range of fluorescent
intensities and fewer short telomeres (signal-free ends) than ALT cell lines (Figure 3.3.B, C, D).
Although COG-N-291 and LA-N-6 had very long telomeres, they did not have elevated markers
of ALT or telomerase activity. Both cell lines were telomerase-negative by TRAP (Figure 3.4.A). To
confirm this was not the result of PCR inhibitors present in the sample lysate resulting in a false-negative
TRAP result, the samples were spiked with a telomerase-positive lysate (Figure 3.4.B). The presence of
telomerase activity in the spiked samples confirmed these two cell lines were telomerase-negative. The
cells also lacked detectable mRNA expression for hTERT, the catalytic subunit of telomerase (Table 3.1,
Figure 3.4.C). Although a low level of APBs was identified in COG-N-291 and LA-N-6, the level was similar
to telomerase-positive samples and significantly lower than ALT-positive cell lines (P<0.001; Figure
3.4.D). C-circle levels were measured using a second method where a 32P-labelled telomeric probe was
used to detect products of the C-circle rolling circle amplification catalysed by Φ29 polymerase (Henson
et al., 2009). This detection method identified very low levels of signal above the no Φ29 polymerase
control (Figure 3.4.E). To determine whether this low level of signal was due to extension of genomic
linear telomeric DNA or amplification of C-circles, exonuclease digestion of linear telomeric DNA was
performed prior to the C-circle assay. The presence of radioactive signals following exonuclease
digestion confirmed that the cells contained very low levels of C-circles (Figure 3.4.F).
Both COG-N-291 and LA-N-6 lacked genetic variations associated with ALT activation, and like
the ALT-negative/TC≥15 tumours were MYCN non-amplified (Table 3.1). Whole genome sequencing
indicated both cell lines were wild-type for H3F3A, TP53, ATRX and DAXX genes (Appendix II Table A2.5,
6) and was confirmed by Sanger sequencing, although it has been previously reported that LA-N-6
possesses a homozygous deletion of CDKN2A (Garcia et al., 2010) and COG-N-291 possesses a TP53
polymorphism (Farooqi et al., 2014). Consistent with wild type TP53, the downstream target p21 was
induced when cells were treated with the DNA damaging agent doxorubicin (Figure 3.5.A). Full- length

88

89
Figure 3.4. The COG-N-291 and LA-N-6 cell lines are phenotypically similar to the ALT-
negative/TC≥15 tumour group.
(A) TRAP assay to detect telomerase activity in a panel of NB cell lines. Cell lysates were
immunopurified (IP) with anti-hTERT antibody to remove potential PCR inhibitors. (B) Telomerase
activity measured by TRAP. All telomerase-negative samples were spiked with lysate from the
telomerase-positive sample, SH-SY5Y, to confirm that no PCR inhibitors were present. (C) Melt curves
from SYBR Green hTERT RT-qPCR. Arrows indicate that primer dimers formed when no template was
present in CHLA-90 (ALT+) and LA-N-6. Second peak represents product formed by qPCR
amplification of template in telomerase-positive samples (SK-N-BE2c and SH-SY5Y). (D) ALT-
associated promyelocytic leukaemia bodies (APBs) in a panel of cell lines (co-localisation of telomeric
foci (green) and PML nuclear body (red), indicated by arrows). ALT-positive (ALT+) and telomerase-
positive (TEL+) cell lines are indicated. Data represent mean (+SD) of 3 independent experiments.
Scale bar represents 3 µM. (E) Representative C-circle (CC) assay with and without Φ29 polymerase,
followed by detection using a 32P labelled telomeric probe in slot-blot analysis. CC level was
calculated relative to that of the ALT-positive U-2 OS cell line (designated to be 100 AU). ALT-positive
(ALT+) and telomerase-positive (TEL+) cell lines are indicated. (F) Confirmation of C-circle assay
products in COG-N-291 and LA-N-6 at three different population doublings (PD). To remove linear
telomeric DNA prior to the C-circle assay, restriction enzyme-treated DNA was subjected to
exonuclease digestion. MeT-4A and DOS16 are ALT-positive cell lines with low CC levels (Henson et
al., 2009). SK-N-BE2c, a telomerase-positive cell line, was used as negative control.
ATRX and DAXX proteins were also expressed in both cell lines (Figure 3.5.B) and localised to the nucleus
(Figure 3.5.C). As expected for TP53 wild-type cultures (Cesare et al., 2009), the cells do not accumulate
DNA damage at the telomere (Figure 3.6.A). The level of the telomeric transcript TERRA has been
associated with telomere length and accordingly, levels are higher in ALT cells than telomerase-positive
cells (Arora et al., 2014; Episkopou et al., 2014; Ng et al., 2009). However, TERRA levels in COG-N-291
and LA-N-6 were well below those of any other sample with long telomeres (Figure 3.6.B). Taken

90
Figure 3.5. The cell lines COG-N-291 and LA-N-6 lack characteristic of ALT.
(A) p53 and p21 immunoblot with GAPDH loading control. Cultures were treated for 72 hrs with a
doxorubicin (DOX) dose equivalent to their IC50. p53 status is indicated for each cell line (W, wild
type; M, mutant). (B) ATRX and DAXX immunoblot with HSP70 loading control. CHLA-90 and U-2 OS
(ATRX-negative cell line with partial deletion of ATRX gene) were used as negative controls. (C)
Representative images of ATRX and DAXX foci (red) in the nuclei (blue) of NB cell lines.

91
together these data suggest that COG-N-291 and LA-N-6 cells lack the genetic and/or epigenetic
(Episkopou et al., 2014) changes that allow upregulation of ALT in some cancer cells.
To determine whether differences in the chromatin structure of COG-N-291 and LA-N-6 were
responsible for preventing upregulation of ALT in these cells, we performed telomere chromatin
immunoprecipitation (ChIP) for shelterin proteins and histone 3 (H3) marks. There was no difference in
the level of TRF2 or RAP1 shelterin proteins bound to the telomeres of ALT or COG-N-291 and LA-N-6
cells (Figure 3.7.A). Similarly, there was no increase in repressive histone marks at H3 compared to ALT
NB cell lines (Figure 3.7.B).
From the work described in this section, we conclude that the cell line screen has identified two
cell lines (COG-N-291 and LA-N-6) that are phenotypically and genetically similar to the ALT-
negative/TC≥15 tumours.

92
Figure 3.6. COG-N-291 and LA-N-6 lack DNA damage at the telomere and characteristics of ALT.
(A) Representative images of meta-TIF assay (telomere foci green and γ-H2AX red, arrows indicate
damage at telomere). Bottom panel is percentage of chromosome ends with or without telomere
signal that have DNA damage. p53 status is indicated below each cell line (WT wild type; M, mutant;
LgT, SV40 large T antigen). Mean +SD from 3 independent experiments. (B) Telomeric repeat-
containing RNA (TERRA) was quantitated from DNase treated RNA, followed by detection using a 32P
labelled telomeric or GAPDH (loading control) probe in slot-blot analysis. The TERRA levels of ALT
samples (CHLA-90, SK-N-FI, U-2 OS, SK-LU-1, SAOS-2 and G292), telomerase-positive cells (SH-SY5Y,
SK-N-BE2c, A2182 and MG63) and mortal cell lines (HFF5 and MRC-5) were compared to COG-N-291
and LA-N-6. Bar graph shows mean (+SD) TERRA levels from 3 independent experiments.

93
Figure 3.7. The level of telomere bound proteins does not differ between ALT neuroblastoma cell
lines and COG-N-291 and LA-N-6.
(A) Telomere-ChIP against the shelterin complex components RAP1 and TRF2. The telomerase-
positive cell lines SK-N-BE2c and SH-SY5Y, and ALT-positive cell lines SK-N-FI and CHLA-90 are
included for comparison. Quantitation of the percentage of telomeric pulled down is shown in the
graph. Mean +SD from 2 independent experiments. (B) Telomere-ChIP against histone H3 and
histone marks. The telomerase-positive cell line SH-SY5Y and ALT-positive cell lines SK-N-FI and
CHLA-90 are included for comparison. Quantitation of the percentage of telomeric pulled down
and normalised to H3 is shown in the panel below. Mean +SD from 2 independent experiments.

94
3.2.2.2. Other tumour types
In addition to NB, we screened a cohort of 94 PanNET tumours, 48 oesophageal tumours, and 2 cohorts
of melanoma tumours (n=82 and n=62) for the ALT-negative/TC≥15 phenotype. Four melanoma
tumours met the criteria, C-circle negative and TC≥15, whilst one oesophageal tumour had a TC just
below the cut-off for long telomeres (Figure 3.8.A, B, C, D). As with the NB tumours, to determine if
these samples are utilising an as-yet unidentified ALT mechanism, or if they proliferate despite lack of a
TLM, we require cell line models to examine telomere length over time. Subsequently, 146 melanoma
cell lines were examined for C-circle level and TC. There were four melanoma cell lines with longer
telomere lengths (TC>10; Figure 3.9.A) and no C-circles, however, they had detectable mRNA expression
for hTERT and hTR RNA (Figure 3.9.B), components of telomerase, and had telomerase activity (Figure
3.9.C). Therefore, the ALT-negative/TC≥15 phenotype is present in melanoma tumours but the current
study did not identify any corresponding cell line model.

95
Figure 3.8. Some melanoma tumours are ALT-negative with TC≥15.
(A) Relative TC in arbitrary units (AU) of pancreatic neuroendocrine tumours (PanNET), measured by
telomere qPCR for individual patient tumours (n=94), plotted according to ALT status (ALT+ n=33,
ALT- n=61). ALT+ was defined by a relative C-circle level ≥7.5 compared to the ALT+ IIICF/c cell line
with an arbitrary value of 100. (B) Relative TC of oesophageal tumours (n=48) is plotted according to
ALT status (ALT+ n=0, ALT- n=48). (C) Relative TC of melanoma tumours cohort 1 (n=82) is plotted
according to ALT status (ALT+ n=12, ALT- n=70). ALT-negative samples with TC≥15 compared to ALT+
IIICF/c cell line with arbitrary value of 30 are indicated by the bracket. (D) Relative TC of melanoma
tumours cohort 2 (n=62) is plotted according to ALT status (ALT+ n=1, ALT- n=61), ALT-negative
samples with TC≥15 compared to ALT+ IIICF/c cell line with arbitrary value of 30 are indicated by the
bracket.
Data points represent the average of triplicate measurements by qPCR. Horizontal red bar indicates
mean.

96
Figure 3.9. ALT-negative/long telomere melanoma cell lines are telomerase positive.
(A) Relative telomere content, in arbitrary units (AU), of ALT-negative cell lines. Data points represent
the average of triplicate TC measurements by qPCR. A cohort of 146 melanoma cell lines are
represented. The box indicates 4 melanoma cell lines with long telomeres (TC>10). (B) mRNA
expression of the telomerase components hTR and hTERT, measured by RT-qPCR, for the TC>10
melanoma cell lines in Figure 3.8.A. (C) TRAP to detect telomerase activity in the long telomere TC>10
melanoma cell lines from Figure 3.8.A, B. The cell lines LA-N-6 and U-2 OS (ALT) are included as
negative controls whilst SH-SY5Y is a telomerase positive NB cell line.

97
3.3. Discussion
In the past two decades, numerous reports have identified telomerase activity as a poor prognostic
factor in NB (Ohali et al., 2006; Onitake et al., 2009; Poremba et al., 2000; Poremba et al., 1999).
However, reports also identified a subgroup of metastatic disease that does not utilise telomerase for
continued proliferation (Choi et al., 2000; Krams et al., 2001; Poremba et al., 2000; Poremba et al., 1999;
Reynolds et al., 1997). Studies of ALT in NB tumours thus far have relied on telomere length (Onitake et
al., 2009), or ATRX/DAXX mutations (Cheung et al., 2012; Kurihara et al., 2014; Molenaar et al., 2012;
Peifer et al., 2015; Valentijn et al., 2015) to identify the mechanism. This is the first study of ALT in NB
tumours to utilise telomere length and C-circles, a quantifiable marker of ALT activity, to determine the
prevalence of ALT in metastatic NB.
We found 24% of high-risk NB were ALT-positive and that MYCN amplification and ALT were
mutually exclusive, in agreement with previous studies (Cheung et al., 2012; Farooqi et al., 2014;
Molenaar et al., 2012; Peifer et al., 2015; Pugh et al., 2013). Interestingly, ALT-positive patients had
more late events than MYCN amplified patients. The presence of ALT correlated with poor outcome in
high-risk NB as previously reported in ATRX mutated NB tumours (Kurihara et al., 2014; Lundberg et al.,
2011; Onitake et al., 2009; Valentijn et al., 2015), liposarcoma, and malignant fibrous histiocytoma
tumours (Henson and Reddel, 2010). Thirteen ALT tumours had whole exome or whole genome
sequence data available and four of these tumours were wild type for ATRX and DAXX. Mutations in
ATRX/DAXX, and consequently the prevalence of the ALT mechanism, have been reported in 10-22% of
high-risk NB tumours (Cheung et al., 2012; Peifer et al., 2015; Valentijn et al., 2015). The broad range of
ALT prevalence reported via ATRX/DAXX sequencing indicates inter-study variables impact significantly
on the final prevalence estimates obtained from sequencing. These studies under-report the prevalence
of ALT by 2-12% compared to ALT in high-risk NB detected by the presence of C-circles. Therefore,
relying on ATRX and DAXX mutations to identify ALT-positive NB tumours would underestimate the
prevalence of this mechanism.

98
Of note, we identified a subgroup of high-risk NB that had telomere lengths similar to the length
of ALT cells but with very low levels of C-circles and no telomerase activity. In addition, 51% of the
patients in this subgroup died from the disease. This is distinct from Stage 4S NB, a group of unique
tumours that generally spontaneously regress and has a survival of 70-90% (Nickerson et al., 2000; van
Noesel et al., 1997). Like ALT-negative/telomerase-negative tumours reported in liposarcoma,
glioblastoma, osteosarcoma, and retinoblastoma (Costa et al., 2006; Gupta et al., 1996; Hakin-Smith et
al., 2003; Jeyapalan et al., 2008; Ulaner et al., 2003), serial tumour samples are required to determine if
these tumours represent an unidentified ALT mechanism lacking phenotypic characteristics of ALT, or if
these tumours do not require a TLM for long term proliferation. The only definitive way to determine
which hypothesis is correct, is to determine what happens to telomere length over time. In the first
scenario telomere lengths would be maintained, whilst in the second telomeres would shorten over
time. To determine if telomere length shortened during tumour growth we had to identify
corresponding cell line models, as it is not possible to obtain serial tumours samples from the same
patient over an extended period of time when the high-risk NB is left untreated. Two NB cell lines, COG-
N-291 and LA-N-6, derived from patients who died of metastatic disease, were identified with
characteristics that correspond to the ALT-negative/TC≥15 tumours. Interestingly, these cells have a
very low level of C-circle activity (at least 2 times lower than MeT-4A which has one of the lowest C-
circle levels reported in ALT-positive cells (Henson et al., 2009)) which indicates a very low level of ALT
activity in these cells. The only definitive way to determine if this is sufficient for telomere maintenance
is to examine telomere length over time. Subsequent chapters of this thesis describe the consequences
of continued proliferation on telomere length in these cells.
We were unable to identify potential TLM-negative cell lines in other tumour types. The criteria
used to identify ALT-negative/TC≥15 NB may not reflect the phenotype of ALT-negative/telomerase-
negative cells in other tumour types. NB is a childhood cancer and consequently the tumour initiating
cells have not undergone as many cycles of replication induced telomere shortening as in adult cancers.
The NB ALT-negative/TC≥15 cells have extreme telomere lengths, hence cell lines derived from these

99
tumours still have very long telomeres despite the PDs, and consequent telomere shortening,
undergone by the cells during the establishment of the cell line. Whilst adult tumour types, such as
melanoma, have ALT-negative/TC≥15 tumours, they do not have the extreme telomere lengths seen in
NB. Hence cell lines derived from these tumours may not have lengths above the normal range after the
PDs required to derive a cell line from the tumour. Indeed, this hypothesis is supported by the
observation, by the Decottignies laboratory, that melanoma cells with telomeres which are at the upper
end of the normal length do not have a TLM (Viceconte et al., 2017). Consequently, when screening
adult cancer types for ALT-negative/telomerase-negative cell lines the telomere length cut-off may need
to be adjusted.
In conclusion, telomere biology plays an important role in NB pathogenesis as activation of
either TLM confers poor outcome, although the disease course differs between ALT and telomerase-
positive tumours. Currently, TLM inhibitors are being developed as anti-cancer therapies and may be
effective in improving the outcome in high-risk NB with TLM. However, we require a greater
understanding of the ALT-negative/TC≥15 phenotype to determine whether such inhibitors would be
effective in these tumours. Success of targeted therapies requires appropriate biomarkers to predict
response. Establishing the TLM status of a tumour is therefore required prior to treatment with TLM
inhibitors or other TLM-targeted therapies. Care must be taken to correctly define ALT tumours, because
neither high telomere content nor ATRX/DAXX mutation is sufficient to classify ALT tumours, and are
best complemented with C-circles as an ALT activity assay.

100
Chapter 4: Long-term proliferation
occurs in cancer cells with ever-shorter
telomeres

101
Chapter 4: Long‐term proliferation occurs in cancer cells
with ever‐shorter telomeres
4.1. Introduction
A subgroup of high-risk NB tumours lack telomerase activity and upregulate the ALT mechanism to
maintain telomere lengths; however, previous identification of ALT in NB has been limited to
examination of telomere length (Onitake et al., 2009) or ATRX/DAXX mutations (Cheung et al., 2012;
Kurihara et al., 2014; Molenaar et al., 2012; Peifer et al., 2015; Valentijn et al., 2015). In the previous
chapter we examined ALT in high-risk NB by simultaneous detection of long telomeres and abundant C-
circles. This method identified ALT in 24% of high-risk NB, as well as a subgroup of TLM-negative tumours
that had extremely long telomeres. In previous studies, TLM-negative tumours were assumed to result
from false-negative assays. Yet, it is possible that the cells utilise a yet to be identified mechanism to
lengthen telomeres. It has also been hypothesised that extreme telomere length in tumour progenitor
cells would be sufficient for the tumour to achieve long term proliferation, without the need to activate
a TLM (Reddel, 2000). The aim of this chapter was to determine the consequence of continued
proliferation on the telomere length of two NB cell lines, COG-N-291 and LA-N-6. These cells are
characteristic of ALT-negative/TC≥15 tumours when screened by qPCR. However, detection by 32P
labelled telomere probe revealed the cells have a very low level of C-circles (compared to MeT-4A, an
ALT cell line with low C-circle levels (Henson et al., 2009)) indicative of low levels of ALT activity. To
determine if this was sufficient to maintain telomeres in these cells, telomere length was examined
during four years of continuous cell culture. We found that the COG-N-291 and LA-N-6 telomeres
undergo continuous shortening during continuous proliferation for 500 PDs, a phenotype we termed
ever-shorter telomeres (EST) in accordance with terminology from S. cerevisiae mutants (Lundblad and
Szostak, 1989).

102
4.2. Results
4.2.1. Cells with ever-shorter telomeres are capable of long-term proliferation
To determine whether the NB cell lines COG-N-291 and LA-N-6 utilise an alternative non-telomerase
method to maintain telomeres, or if they are truly TLM-negative, we examined the telomere length of
the cells during continual proliferation for 500 PDs. If the first scenario was true, telomere lengths would
be maintained, whilst truly TLM-negative cells would have continuous telomere length shortening.
Southern blot TRF analysis showed the telomeres of both cell lines shortened continuously during
culture (Figure 4.1.A, B), at a rate of 80 and 55 bp/PD for COG-N-291 and LA-N-6, respectively (Figure
4.2.A). Normal somatic cell division results in a 31-85 bp/PD loss of telomere sequence (Harley et al.,
1990). Hence, the shortening exhibited by COG-N-291 and LA-N-6 is the result of cell division related
telomere erosion, indicating the cells lack a TLM. A reduction in telomere content by qPCR confirmed
the telomere shortening demonstrated by TRF analysis (Figure 4.2.B). Despite shortening telomeres and
a very slow proliferation rate (3.4 days/PD for COG-N-291 and 3 days/PD for LA-N-6), the cells have yet
to reach senescence after 500 PDs and over four years in culture (Figure 4.2.C). Therefore, NB cells
derived from lethal high-risk tumours continue to proliferate long-term in spite of their EST phenotype.
4.2.2. Characteristics of EST cells
Telomerase activity and levels of ALT characteristics were monitored during continuous proliferation of
the EST cells. The cells remained telomerase negative throughout the culture period (Figure 4.3.A) and
the APB levels did not change (Figure 4.3.B). The C-circle levels were compared to the ALT cell line MeT-
4A set at 10 arbitrary units. This ALT cell line has one of the lowest levels of C-circles previously detected
(Henson et al., 2009). The C-circles in COG-N-291 and LA-N-6 persisted at very low levels with sporadic
increases for the first 300 PDs in culture, but for the next 200 PDs of culture there was a stabilisation of
C-circle levels, at an amount similar to MeT-4A (Figure 4.3.C). ATRX and DAXX were expressed across
the period of culture (Figure 4.3.D).

103
Figure 4.1. Two neuroblastoma cell lines (COG-N-291 and LA-N-6) are capable of long term
proliferation despite the ever-shorter telomere (EST) phenotype.
(A, B) TRF analysis of COG-N-291 and LA-N-6 telomere lengths at progressive population doublings
(PD). The white gaps between lanes and different well heights indicate individual TRF analysis
performed at progressive time points throughout the culture period. Individual blots are aligned
with each other by molecular weight marker.
Initial half of TRFs were completed by Dr Loretta Lau.

104
Figure 4.2. EST cells are capable of proliferation for hundreds of population doublings.
(A) The average rate of telomere shortening was calculated by plotting the average TRF length of
four individual TRF bands (indicated by asterisks in Figure 4.1) against the number of population
doublings (PD). (B) Telomere content measured by telomere qPCR. Data represent mean + SD, n=3.
(C) Growth curves of COG-N-291 and LA-N-6 demonstrate long-term proliferation.
Calculations in panel (A) completed by Dr Loretta Lau.

105
Figure 4.3. Characteristics of EST cells during continuous proliferation for 600 population
doublings.
(A) Telomerase activity was monitored by TRAP at the specified PDs in COG-N-291 and LA-N-6 cells.
SH-SY5Y and SK-N-BE2c were used as positive controls, and SK-N-FI and CHLA-90 (ALT+) as negative
controls. (B) Frequency of APBs in EST cells at early and late population doublings (PD). Data

106
represent mean +SD of 3 independent experiments. (C) C-circle levels detected using 32P labelled
telomeric DNA probe on slot blot. Levels were calculated relative to those of the ALT+ MeT-4A cell
line (designated to be 10 AU). SK-N-BE2c (telomerase-positive) was included as a negative control.
(D) ATRX and DAXX immunoblot at specified population doublings (PD) with β-actin as loading
control.
IP-TRAP was, in part, performed by Dr Christine Napier.
4.2.2.1. ALT and the EST phenotype
C-circles were present at low levels in early populations of the EST cell lines. The cells undergo
continuous telomere shortening during this time, indicating that a small amount of ALT activity may be
present in these cells but at levels insufficient to confer telomere maintenance. This is consistent with
the observation that telomere shortening can occur in telomerase-positive normal cells (Broccoli et al.,
1995; Marion et al., 2009), and that ALT activity has been detected in somatic cells of mice (Neumann
et al., 2013). However, within the first 300 PDs of culture the cells had sporadic increases in C-circle
levels despite continuous telomere shortening ultimately resulting in a level of C-circles similar to the
ALT cell line MeT-4A after PD400 and telomere length stabilisation. To determine if a subpopulation of
the EST mass culture was responsible for the sporadic increases in C-circle levels at early PDs, COG-N-
291 cells at PD247 (the first timepoint where COG-N-291 had increased C-circle levels, Figure 4.3.C) were
sub-cloned by plating cells at very low density until colonies from a single cell arose. The TRF analysis of
subclones indicated clone 4 has long and heterogeneous telomere lengths, typical of ALT cells, whilst
the remaining cell lines have the banding pattern seen in the EST mass culture (Figure 4.4.A). The level
of C-circles in clone 4 was at least five-fold higher than the other COG-N-291 subclones (Figure 4.4.B),
which indicates that a subpopulation of cells is responsible for the increased C-circle level in the mass
culture.

107
Figure 4.4. A subpopulation of COG-N-291 cells are responsible for sporadic increases in C-circles
in the first 300 PDs of culture.
(A) TRF of the telomere length of COG-N-291 clones isolated at PD247. (B) C-circle slot blot detected
with 32P-labelled telomeric DNA probe. SK-N-FI was used as an ALT-positive, C-circle positive control
and SK-N-BE2c a telomerase-positive, C-circle negative control.
The TRF pattern and C-circle level in COG-N-291 clone 4 suggests that these cells have
upregulated the ALT mechanism whilst the other subclones isolated were EST cells. Examination of
telomere length over time by both TRF and qPCR analysis showed telomere maintenance in clone 4,
whereas clones 3 and 11 exhibited the EST phenotype (Figure 4.5.A, B). As expected, none of the
subclones exhibited telomerase activity during 100 PDs of culture (Figure 4.5.C), confirming that, by
definition, the clone 4 cells must be utilising some form of an ALT mechanism to maintain telomere

108
Figure 4.5. Periodically a population of COG-N-291 cells activate an ALT mechanism but a slower
proliferation rate prevents them outgrowing the EST cells.
(A) TRF analysis of the COG-N-291 subclones telomere lengths at progressive population doublings
(PD). (B) Telomere content measured by telomere qPCR in arbitrary units (AU). Data represent
mean + SD, n=3. (C) Telomerase activity was monitored by TRAP at the specified PDs. SH-SY5Y was

109
included as a positive control and U-2 OS as a negative control. (D) Growth curves of COG-N-291
subclones over 50 population doublings (PD).
lengths. The EST subclones had faster growth rates than clone 4; 3.3 days/PD for clone 3, 2 days/PD for
clone 11 vs. 3.8 days/PD for clone 4 but this did not result in senescence in the cultures after 100 PDs of
continuous proliferation (Figure 4.5.E). The slower growth rate of clone 4 implies that activation of the
ALT mechanism has not provided a survival advantage for these cells, and consequently they do not
become the predominant population in a mixed culture with the EST cells in the first 300 PDs of growth
(Figure 4.3C). Presumably, further molecular changes, caused by long term culture, were required for
these ALT-like cells to become the predominant population of cells observed after PD400 leading to the
increased C-circle levels (Figure 4.3C).
Subclone 4 was able to upregulate the ALT mechanism to a sufficient level to confer telomere
length maintenance; however, these cells do not possess any of the other characteristic features of ALT.
As expected, clone 4 cells maintained a high level of C-circles during culture whilst the EST subclones
had very low levels although, like the mass culture, clone 3 had a sporadic increase in C-circles (Figure
4.6.A). There was no increase in the level of APBs observed in clone 4 compared to the EST controls
(Figure 4.6.B). Gene mutations associated with ALT, such as alterations to ATRX, DAXX and TP53, were
absent in the clone 4 culture and ATRX and DAXX full length proteins were expressed (Figure 4.6.C).
There was also no increase in TERRA levels with activation of ALT (Figure 4.6.D) and as expected for TP53
wild type cells meta-TIFs did not accumulate (Figure 4.6.E).
4.2.3. Genetics of EST cells
To determine whether EST cells had genetic events that prevented sufficient upregulation of a TLM to
counteract telomere attrition, we used whole genome sequencing to examine 656 genes related to
telomere biology, ALT, and processes relating to RNA or DNA replication, repair and damage response

110
Figure 4.6. Characteristics of COG-N-291 sublines during continuous long-term proliferation.
(A) C-circle levels of three sub-clones of COG-N-291 (clone 4, 3, 11) detected using 32P-labelled
telomeric DNA probe on slot blot. Levels were calculated (as arbitrary units [AU]) relative to those
of the ALT cell line MeT-4A set to 10 AU. SK-N-BE2c (telomerase-positive) was included as a
negative control. (B) Frequency of APBs in COG-N-291 sublines compared to the COG-N-291 mass
culture. Graph shows the mean +SD of three independent experiments. (C) ATRX and DAXX
immunoblot with GAPDH as loading control. (D) Graph of meta-TIF assay results, percentage of
chromosome ends with or without telomere signal that have DNA damage. Mean +SD from 3

111
independent experiments. (E) TERRA detected by slot-blot analysis with GAPDH loading control.
Quantitation in bottom panel from 3 independent experiments (mean +SD).
(Appendix II Table A2.5). To represent each of the three telomere-biology groups (EST, ALT and
telomerase-positive), a total of 6 cell lines and 9 tumours were analysed (two NB cell lines and three
tumours per group). As matched normal DNA was not available for the samples, we filtered the data
using Ingenuity Variant Analysis to exclude genetic events previously reported in publicly available data
sets of normal genomes. Relevant alterations were those predicted to have unknown or deleterious
consequences and were not present in the ALT or telomerase-positive samples. There were no specific
genetic events identified for the EST phenotype (Appendix II Table A2.6).
4.3. Discussion
Cellular immortalisation, via activation of a TLM to counteract normal telomere attrition, is considered
to be an essential step in malignant transformation and therefore a hallmark of cancer (Hanahan and
Weinberg, 2000; Hanahan and Weinberg, 2011). To form a mass of approximately 1 kg, a single cell
needs to go through 40 cell divisions (240 cells), which many types of normal cells are able to achieve
(Parkinson, 1996; Reddel, 2000). However, this would require all cells to survive, which is not possible
in a tumour environment where there is an inadequate supply of oxygen and nutrients. Oncogenesis
also requires multistep oncogenic mutation and associated clonal evolution, resulting in an increase in
the number of cell divisions required for a tumour mass to form, and consequently the cells may require
immortalisation in order to form a clinically significant tumour (Parkinson, 1996; Reddel, 2000).
However, it has been postulated that immortalisation is not required if the progenitor cells have long
telomeres, the tumours are genetically simple such as paediatric cancers (Alexandrov et al., 2013; Jones
et al., 2008; Parsons et al., 2011; Vogelstein et al., 2013) and don’t require multiple rounds of clonal
evolution, or the tumour environment supports a high surviving fraction such as leukaemias (Reddel,
2000). The presence of the EST phenotype in malignant NB cells provides direct evidence to support the

112
hypothesis that immortalisation is not an absolute requirement for oncogenic development. The NB EST
cells have very long telomeres that can act as a substitute for an active TLM, permitting the cell divisions
required for additional oncogenic changes which are essential to the formation of malignant tumours.
The discovery of malignant and lethal TLM-negative cancer cells has implications for the
development of TLM inhibitors for clinical use as anti-cancer therapies. Tumours that rely on extreme
telomere length, and not a TLM, for continual proliferation will be resistant to these inhibitors. The
existence of EST cells illustrates the need to assay for TLMs prior to therapy with such drugs. The
similarity of the telomere lengths in EST and ALT cells indicate that it is not appropriate to define ALT by
telomere length alone: measurement by telomere qPCR, TRF or telomere FISH is not sufficient to
differentiate the two phenotypes. Furthermore, care must be taken when utilising C-circles to define
ALT; presence of low levels of C-circles does not indicate sufficient ALT activity to counter the EST
phenotype. This is further complicated by the evidence that EST cultures have periodic increases in C-
circles. Although misclassification of such a phenotype as ALT could result in the use of an ALT inhibitor
that targets a subpopulation of the tumour cells, the bulk of the tumour would be resistant to such
treatment.
The majority of ALT cell lines report inactivation of p53 function, either by viral oncoproteins or
TP53 mutations (Henson and Reddel, 2010). This implies that p53 has a role in supressing ALT, likely due
to p53 mediated senescence or apoptosis which may be triggered by the high levels of chromosomal
instability (Montgomery et al., 2004; Scheel et al., 2001; Ulaner et al., 2004) and telomeric DNA damage
signals present in ALT cells (Cesare et al., 2009; Stagno d'Alcontres et al., 2007). COG-N-291 subclone 4
is an example of a cell line that can maintain telomeres by a telomerase-independent mechanism while
retaining functional p53. As expected, these cells do not accumulate DNA damage signals at the
telomere, although they have reduced proliferation rates compared to the EST cells. In a mixed culture
of ALT and EST COG-N-291 cells, clone 4 is unable to outgrow the EST cells due to this reduced rate of
proliferation, resulting in their removal from the population and the fluctuating C-circle levels we

113
observed. Previous studies indicate ATRX can supress the ALT mechanism (Clynes et al., 2015; Napier et
al., 2015). Clone 4 cells lack abundant APBs but have wild type ATRX and DAXX, which may act with p53
to suppress some of the characteristics of ALT. However, a small proportion of ALT cell lines retain ATRX
and DAXX protein, whilst exhibiting classic ALT phenotypes (Lovejoy et al., 2012), indicating that
retention of p53, ATRX and DAXX protein is not soley responsible for the repressed ALT phenotype in
COG-N-291 clone 4 cells.
In conclusion, this chapter has provided evidence that lethal, malignant human cancers may, in
some cases, contain cells that can proliferate long term with continuously shortening telomeres. The
extreme telomere length which was presumably present in the progenitor cells gave rise to EST cells
that do not require immortalisation for proliferation in excess of 500 PDs. The overlap in some features
of the EST and ALT phenotypes indicates that appropriate assaying for TLMs is required before the use
of TLM inhibitors as anti-cancer therapeutics.

114
Chapter 5: Activation of a telomere
lengthening mechanism rescues the
ever-shorter telomere phenotype

115
Chapter 5: Activation of a telomere lengthening
mechanism rescues the ever‐shorter telomere
phenotype
5.1. Introduction
The previous chapter describes two NB EST cell lines that were capable of extensive proliferation in spite
of their continually shortening telomeres. Examination of C-circle levels in these cells identified a
subpopulation of cells that were able to activate a telomerase-independent TLM, although these cells
did not possess many of the characteristics of ALT. In principle, these cells show that activation of a TLM
would rescue the EST phenotype. The aim of this chapter is to provide evidence that ectopic expression
of hTERT in EST cells results in telomerase activity, and that TP53 mutated EST cells activate a typical
ALT mechanism, and ultimately, that activation of either TLM rescues the EST phenotype. In addition to
proving that activation of a TLM abolishes the EST phenotype, we determined that the EST lines were t-
circle deficient, suggesting that the EST phenotype may arise due to a failure of t-circle formation and
telomere trimming.
5.2. Results
5.2.1. Activation of telomerase rescues the EST phenotype
To confirm that activation of a TLM would abolish the EST phenotype we activated telomerase in LA-N-
6 cells at PD83. As established in Chapter 3, LA-N-6 cells lack expression of the telomerase catalytic
subunit, hTERT. Retroviral transduction of hTERT, or of hTERT plus hTR (telomerase RNA subunit; Figure
5.1.A), resulted in telomerase activity (Figure 5.1.B). To confirm that the telomerase activity detected

116
Figure 5.1. Retroviral transduction of hTERT results in telomerase activity in LA-N-6 cells.
(A) mRNA expression of the three telomerase components (hTR, hTERT and dyskerin; measured by
RT-qPCR) in LA-N-6 infected with plasmids for either hTR (clones, hTR C3 & C4), hTERT (clones,
hTERT C4 & C23) or hTR/hTERT (clones, hTR/hTERT C19 & C23). (B) Telomerase activity detected
by TRAP. U-2 OS and SH-SY5Y cells are negative and positive controls, respectively, for telomerase
activity. (C) TRAP assay performed on heat-inactivated lysates (55°C for 20 mins) from Figure 5.1.B
shows that there is no PCR product contamination. (D) Growth curves of LA-N-6 derivatives.
*P<0.0003

117

118
Figure 5.2. Ectopic expression of telomerase rescues the EST phenotype.
(A) TRF analysis of the telomere length of LA-N-6 clones at the indicated PDs. (B) Verification of TRF
results by telomere qPCR at the indicated PDs. qPCR data represent mean of triplicates +SD. (C)
Relative telomere fluorescent signals measured from telomere FISH (15 metaphases analysed per
cell line). Dots represent individual telomere signals and red horizontal bar indicates mean. (D) Graph
indicates the percentage of chromosome ends without telomere signal in telomere FISH from Figure
5.2.C.
was not due to PCR contamination a proportion of all samples had the telomerase enzyme heat
inactivated prior to the TRAP assay. These samples were negative on a TRAP gel (Figure 5.1.C) confirming
that the banding pattern in Figure 5.1.B was due to the presence of the telomerase enzyme in these
cells. As expected, expression of hTR alone did not result in telomerase activity. Activation of telomerase
led to a significant increase in the growth rate of these cells compared to LA-N-6 transduced with hTR
alone (Figure 5.1.D). Telomere length was examined by both TRF and qPCR, which showed that the EST
phenotype was abolished in all clones expressing telomerase (Figure 5.2.A, B). Two clones expressing
hTERT (C4 and C23) and one clone expressing hTR/hTERT (C19) maintained telomere length, whereas
one hTR/hTERT expressing clone (C23) underwent telomere lengthening over the 50 PDs of culture
(Figure 5.2.A, B). In the telomerase-positive cells, the shortest telomeres were preferentially elongated
(Figure 5.2.C, D). In contrast, two independent clones with overexpression of hTR did not abolish the
EST phenotype (Figure 5.2.A, B). The activation of telomerase resulted in elongated telomere lengths in
LA-N-6 but as expected this did not result in any changes to ALT characteristics. The level of APBs, C-
circles and TERRA did not differ between the hTR, hTERT, and hTR/hTERT clones (Figure 5.3.A, B, C, D).
Consequently, we have demonstrated that activation of telomerase in an EST cell line rescued the EST
phenotype.

119
Figure 5.3. No change to ALT characteristics following ectopic expression of hTERT.
(A) APB (localisation of telomere FISH signal to PML body) analysis of LA-N-6 derived cell lines. Mean
+SD from 3 independent experiments. (B) C-circle levels were detected by slot blot analysis. MeT-4A
and SK-N-BE2c were included as the ALT-positive control and the telomerase-positive negative
control, respectively. (C) Quantitation of C-circle results in Figure 5.3.B. (D) TERRA detected by slot-
blot analysis. Quantitation in bottom panel from 3 independent experiments (mean +SD).

120
Figure 5.4. A LA-N-6 subline spontaneously acquired a premature stop codon in TP53.
(A) Chromatograph from Sanger sequencing of LA-N-6 TP53 wild-type cells (LA-N-6 WT p53) and
the LA-N-6 subline that spontaneously acquired a stop codon (Y236*) (LA-N-6 MUT p53). (B) p53
immunoblot of LA-N-6 cells with wild-type and mutant TP53. GAPDH was used as loading control.
Western blot performed by Dr Loretta Lau.
5.2.2. Activation of ALT rescues the EST phenotype
Activation of telomerase in telomerase-negative cells only requires ectopic expression of the telomerase
components hTR and hTERT (Weinrich et al., 1997). By comparison activating ALT is far more
challenging. Currently, no one gene has been identified that is responsible for activating the ALT
mechanism. In the previous chapter, we provided evidence that COG-N-291 cells can spontaneously
activate ALT. In contrast to COG-N-291, LA-N-6 cells have a single copy of TP53 and consequently there
is a high likelihood that these cells may develop a spontaneous TP53 mutation during long-term culture.
Therefore, to determine if activation of ALT can rescue the EST phenotype, we developed a strategy to
identify a spontaneously activated ALT mechanism in LA-N-6 TP53 mutated cells.

121

122
Figure 5.5. Activation of ALT results in rescue of the EST phenotype.
(A) TRF analysis of the LA-N-6 culture from acquisition of the p53 mutation (PD90) to PD654.
(B) Telomere content of LA-N-6 MUT p53 cells measured by telomere qPCR. Mean +SD, n=3.
(C) Percentage of chromosome ends free of telomere signal (obtained from telomere FISH) in LA-
N-6 parental (wild-type TP53) and LA-N-6 MUT p53 cells. Quantitation from 15 metaphase spreads.
(D) Growth curves of LA-N-6 parental (wild-type TP53) and LA-N-6 MUT p53 culture. (E) Telomerase
activity was monitored by TRAP at the specified PDs in LA-N-6 MUT p53 cells. Lysates were
immunopurified (IP) with anti-hTERT antibody to remove potential inhibitors which could lead to
false negative results. SH-SY5Y and SK-N-BE2c were used as positive controls.
We first identified a subculture of LA-N-6 that spontaneously acquired a TP53 mutation at PD90. The
subclone had spontaneously acquired a base change in exon 7 of TP53 (Figure 5.4.A), resulting in a
premature stop codon (Y236*) and loss of p53 protein expression (hereafter referred to as LA-N-6 MUT
p53; Figure 5.4.B). As expected, the loss-of-function mutation was insufficient to activate ALT, and the
telomeres of this LA-N-6 MUT p53 subculture continued to shorten until PD209, i.e., for another 119 PD
(Figure 5.5.A). By PD222, the telomere length profile of the cells had changed to have the typical
appearance of ALT (Figure 5.5.A). Over the next 99 PD, the mean telomere length fluctuated slightly,
but after PD240 the continuous telomere shortening previously exhibited by these cells had ceased,
demonstrating that a TLM had been activated (Figure 5.5.A). Telomere qPCR confirmed telomere length
maintenance in LA-N-6 MUT p53 cells after PD209 (Figure 5.5.B). From PD222 onwards, the shortest
telomeres in the TRF were elongated, an observation supported by a decrease in the percentage of
chromosomes with telomere signal-free ends (Figure 5.5.C). The growth rate of the culture was
unchanged by the loss of p53 at PD90 and by activation of a TLM at PD209 (Figure 5.5.D). The LA-N-6
MUT p53 cells exhibited telomere maintenance in the absence of telomerase (Figure 5.5.E), therefore
the TLM that was activated was, by definition, an ALT mechanism. Consistent with this conclusion, we
observed a 2 to 4-fold increase in C-circle levels from PD222 until PD503, after which a further 2 to

123
Figure 5.6. Loss of p53 results in activation of ALT in an EST culture (LA-N-6).
(A) C-circle assay product was detected using slot-blot analysis. Levels were calculated relative to
the ALT-positive MeT-4A cell line with an arbitrary unit of 10 and graphed on the right. SK-N-BE2c
was included as a negative control. (B) Percentage of APB positive cells in TP53 wild type LA-N-6 at

124
PD95 compared to LA-N-6 mutant cells at PD704 (mean +SD, n=3 independent experiments). (C)
TERRA level in the ALT-positive LA-N-6 MUT p53 PD618 compared to TP53 wild-type LA-N-6 PD93
cells. Quantitation shows mean +SD (n=3 independent experiments). (D) Telomere sister chromatid
exchange (T-SCE) events indicated by colocalisation of red and green FISH labelled telomeres
(shown by arrows) and quantitated in the graph (mean +SD, n=2 independent experiments).
4-fold increase was observed (Figure 5.6.A), there was a 4-fold increase in APBs (Figure 5.6.B), a 2-fold
increase in TERRA (Figure 5.6.C), and a 4-fold increase in T-SCE events (Figure 5.6.D). The mechanism of
ALT activation in LA-N-6 MUT p53 cells is currently unknown; although down-regulated, ATRX and DAXX
expression was not abolished, and the proteins continued to localise to the nucleus (Figure 5.7.A, B).
Although loss of p53 removes a barrier to the accumulation of DNA damage responses at the telomere
(Cesare et al., 2009), the number of meta-TIFs remained unchanged (Figure 5.7.C). Irrespective of the
mechanism utilised to activate ALT, our results demonstrate that ALT is able to rescue the EST
phenotype.
5.2.3. EST neuroblastoma cell lines are unable to generate t-circles
During the investigation of spontaneous ALT activation in COG-N-291 clone 4 and LA-N-6 MUT p53 cells,
we examined the level of t-circles, a type of double-stranded circular telomeric DNA. T-circles are
abundant in ALT cells (Cesare and Griffith, 2004), and it has been proposed that this is the result of
telomere trimming following telomere over-lengthening by ALT (Pickett and Reddel, 2012). T-circles
have also been shown to form as a result of telomere trimming in cells that have had telomeres over-
lengthened by telomerase (Pickett et al., 2009). The two EST cell lines lack t-circles, and as expected, t-
circles are present in the two ALT NB cell lines (Figure 5.8.A). Contrary to what was expected, no t-circles
were generated in the LA-N-6 clones after activation of telomerase (Figure 5.8.B), even though all hTERT
or hTERT/hTR expressing clones had extremely long telomeres after activation of telomerase. The
LA-N-6 MUT p53 culture also failed to generate t-circles, even after hundreds of PDs utilising the ALT
mechanism, and perhaps less surprisingly the COG-N-291 clone 4 cells also lacked t-circles (Figure 5.8.C).

125
Figure 5.7. LA-N-6 MUT p53 cells did not activate ALT due to aberrations to ATRX or DAXX.
(A) ATRX and DAXX immunoblot with HSP70 loading control. (B) Immunofluorescence indicating
the presence of ATRX or DAXX foci (red) in the nucleus (blue). CHLA-90 is included as the ATRX
negative control and SH-SY5Y as the positive control for ATRX and DAXX expression. (C) Graph of
meta-TIF assay results demonstrating percentage of chromosome ends which have both DNA
damage and telomere signal (-H2AX/TTAGGG+) and chromosome ends which have DNA damage
in the absence of telomere signal (-H2AX/TTAGGG-) in LA-N-6 TP53 wild type PD96 and LA-N-6
MUT p53 PD707. Mean +SD from 3 independent experiments.
These results suggest that both of the EST cell lines are unable to undergo telomere trimming and
generate t-circles. A number of proteins are known to be involved in t-circle formation including: XRCC3
(Compton et al., 2007; Wang et al., 2004), NBS1 (Wang et al., 2004), SLX4 (Sarkar et al., 2015), TZAP (Li
et al., 2017), and RTEL1 (Deng et al., 2013), but whole genome sequencing of the EST cells did not reveal
any mutations in these genes (Appendix II Table A2.6) and all NB cell lines have detectable transcript for
the five genes involved in t-circle formation (Figure 5.9).

126

127
Figure 5.8. EST cells do not form t-circles after activation of ALT or extreme telomere lengthening
by telomerase activity.
(A) Two-dimensional gel electrophoresis of restriction-digested genomic DNA. Denatured gels were
hybridised with a 32P-labelled C-rich telomeric DNA probe. T-circle arcs are indicated by the arrow.
(B) hTR and hTERT expressing LA-N-6 clones do not have t-circles at early or later PDs. (C) T-circles
(indicated by arrow) are present in the ALT control but not in LA-N-6 MUT p53 or COG-N-291
clone 4.
The investigation of t-circles was conducted by A/Prof Hilda Pickett.
5.3. Discussion
This chapter describes the features of EST cultures that have activated a TLM, and thereby abolished
the EST phenotype. Spontaneous activation of a telomerase independent TLM, which is by definition
ALT, in a p53-mutant LA-N-6 clone (presented in this chapter) resulted in a different phenotype than a
COG-N-291 clone (described in Chapter 4). The COG-N-291 clone had a reduced proliferation rate with
no increase in APBs and TERRA, compared to COG-N-291 EST cells. In contrast, the LA-N-6 clone had a
similar proliferation rate, and increased APB and TERRA levels compared to LA-N-6 EST cells.
Interestingly, both cultures have a typical ALT TRF profile, an increase in C-circles, and are wild type for
ATRX and DAXX. The most notable difference between the ALT COG-N-291 and LA-N-6 cultures relates
to TP53 status. The COG-N-291 clone (clone 4) is the first cell line reported to retain expression of wild
type TP53 and full length ATRX protein whilst utilising a telomerase-independent mechanism to prevent
telomere shortening (Henson and Reddel, 2010; Lovejoy et al., 2012). The presence of functional p53
may account for the reduced proliferation rate in the COG-N-291 ALT clone that was not observed in
LA-N-6. Currently, we do not understand the mechanism that these cells have used to activate telomere
lengthening, but as we know that a proportion of ALT NB tumours are wild type for both TP53 and ATRX
(Chapter 3), this cell line will provide a valuable model for understanding this disease.

128
Figure 5.9. EST cells express genes involved in t-circle formation.
(A) Transcript levels of NBS1, TZAP, RTEL1, SLX4, and XRCC3, genes involved in t-circle formation.
The cell lines GM847, SK-N-FI and CHLA-90 are ALT cell lines that form t-circles.
The EST cells that we have identified indicate that a TLM may not be required for the formation
of metastatic and lethal tumours, presumably when the progenitor cells have telomeres long enough to
sustain extensive proliferation, though we do not know how this initial lengthening occurred in EST NB
cells. Telomerase is active during human embryogenesis, prior to downregulation during the late stages
of gestation, leaving somatic cells telomerase-negative (Ulaner and Giudice, 1997; Wright et al., 1996).
Telomere trimming, a process that generates t-circles, regulates telomere length in cells that have been
over-lengthened by telomerase (Pickett et al., 2009). We propose that a failure of telomere trimming,
and subsequently lack of t-circle formation, in EST NB cells resulted in in excessive telomere lengths
during embryogenesis. The EST NB cells lacked detectable t-circles, even in circumstances where their
long telomeres were lengthened further by telomerase. Abundant t-circles are also a feature of
lengthening by ALT (Cesare and Griffith, 2004), but t-circles were not detected in either of the COG-N-

129
291 or LA-N-6 clones that spontaneously activated an ALT mechanism. A lack of telomere trimming may
cause inhibition to the full expression of the ALT phenotype as observed in the ALT COG-N-291 or LA-N-
6 clones. Presumably, the EST cells downregulated telomerase activity as normal during embryogenesis,
leaving excessively lengthened telomeres, which have allowed very extensive proliferation to occur
without requiring significant upregulation of a TLM. As with tumours that activate a TLM, additional
oncogenic changes would need to have occurred to result in the development of malignant EST tumours,
and the excessively long telomeres presumably provide sufficient reserves of telomere content to
obviate the need for an activated TLM.
In summary, we have shown that activation of a TLM is sufficient to rescue the EST phenotype.
EST cells lack t-circles implying a failure of telomere trimming, regardless of the mechanism (either
activation of telomerase or ALT) used to overlengthen the telomeres. This is the first example of a
disease that may result from a failure of telomere trimming. Despite treatment in these patients, EST
tumours are lethal in approximately 50% of patients and the telomere trimming mechanism may
provide a unique way to target therapies to these cells.

130
Chapter 6: Neuroblastoma EST cells are
sensitive to topoisomerase inhibitors

131
Chapter 6: Neuroblastoma EST cells are sensitive to
topoisomerase inhibitors
6.1. Introduction
The aggressive behaviour of high-risk NB requires extensive cellular proliferation made possible via
activation of a TLM, either telomerase or ALT, or by an EST phenotype which is characterised by
extremely long telomeres that allow proliferation without significant upregulation of a TLM. In Chapter
3 we show that the prognosis of each of these groups of tumours is poor: the 5-year overall survival is
28% for patients with MYCN amplified tumours (which are generally telomerase positive (Hiyama et al.,
1997)), 36% for ALT-positive patients, and 49% for EST NB. In addition, telomere biology has an impact
on disease course, with ALT and EST NB patients having late events compared to MYCN amplified
tumours. Recently, several large scale genomic studies have been conducted to elucidate driver
mutations in NB (Molenaar et al., 2012; Pugh et al., 2013). Whilst these studies were unable to identify
a driver mutation, approximately 10% of tumours had mutations to the ATRX gene which is associated
with ALT activity (Cheung et al., 2012; Kurihara et al., 2014; Molenaar et al., 2012; Pugh et al., 2013), or
rearrangements upstream of hTERT which results in reactivation of telomerase (Peifer et al., 2015;
Valentijn et al., 2015). Therefore, telomere biology has become an attractive target for the development
of novel NB therapies.
Over the last two decades telomerase inhibitors have gone through various stages of
development, resulting in a number of clinical trials in diseases such as breast and lung cancer,
glioblastoma, and lympho-proliferative disorders (Arndt and MacKenzie, 2016). In contrast, ALT
inhibitors have yet to enter clinical trials and are still being developed in preclinical laboratory models
(Deeg et al., 2016; Flynn et al., 2015).

132
The EST NB cell lines have very low levels of C-circles which likely represent a very low level of
ALT activity that is insufficient to confer telomere maintenance in these cells. In yeast, ALT acts
preferentially on the shortest telomeres (Fu et al., 2014) although, Murnane and colleagues suggest that
this is not necessarily the case in human cells (Murnane et al., 1994). In the experiments described in
this chapter, we tested the hypothesis that the low level of ALT in EST cells may preferentially lengthen
the shortest telomeres as critically short telomeres will eventually trigger p53-mediated senescence or
apoptosis. If this was the case, even though ALT activity is clearly insufficient to prevent telomere
shortening, EST cells might rely on low levels of ALT to survive, and consequently inhibition of ALT would
result in increased cell death. The ALT mechanism relies on homologous recombination. APBs potentially
play a role in this process as proteins associated with homologous recombination are found at APBs
(Pickett and Reddel, 2015). One such protein, RPA, is associated with replication stress via its interaction
with ATR (Zou and Elledge, 2003). It has been suggested that ALT cells are acutely sensitive to ATR
inhibitors (Flynn et al., 2015), implying that replication stress has a role in ALT activity that can be
targeted by anti-cancer therapeutics. Therefore, the aim of this chapter was to determine the sensitivity
of EST cells to common NB chemotherapeutics, and to determine if treatment with drugs that target
the ALT mechanism led to an increased rate of cell death.
6.2. Results
6.2.1. EST cells are sensitive to topoisomerase inhibitors
EST cells represent a novel prognostic group of NB, with an approximately 50% 5-year mortality in high-
risk patients. To develop better treatments for these patients we first need to understand the role
telomere biology plays in the response of high-risk NB to chemotherapy. We asked whether cells of
different telomere biology groups (ALT-positive, telomerase-positive, or EST) had different sensitivity to
common NB chemotherapeutics. Using AlamarBlue, a redox indicator that provides a quantitative
measure of cell viability, we determined the effect of chemotherapeutics on the proliferation of six
telomerase-positive (SH-SY5Y and SK-N-BE2c NB cell lines; HT1080, GM639, HT1080 hTR and

133
Figure 6.1. EST cells are sensitive to topoisomerase inhibitors.
Cell lines were treated with increasing concentrations of (A) doxorubicin, (B) etoposide, (C, D)
irinotecan, and (E, F) topotecan for 6 PDs before proliferation was quantified using AlamarBlue. U-
2 OS and GM847 ALT cell lines, and HT1080, GM639, HT1080 hTR and HeLa 1.2.11 telomerase-
positive cell lines were included as non-NB controls. Results in panel (A), (B), (C), (E) are expressed
as percentages of growth compared to untreated controls and as means of triplicate independent
experiments +SD. Panel (D) displays the percentage of cell growth, compared to control cells, when

134
cells were treated with 2.8 µM irinotecan. COG-N-291 had significantly reduced growth compared
to all cell lines except SH-SY5Y and HT1080 hTR whilst LA-N-6 had significantly reduced growth
compared to U-2 OS, SK-N-BE2c and HeLa 1.2.11 cells (when significant P<0.05). Panel (F) displays
the percentage of cell growth, compared to control cells, when cells were treated with 0.0004 µM
topotecan. COG-N-291 and LA-N-6 cells had significantly reduced growth compared to all other cell
lines (P<0.001).
HeLa 1.2.11 non-NB cell line), four ALT-positive (SK-N-FI and CHLA-90 NB cell lines; U-2 OS and GM847
non-NB cell lines), and the two EST (COG-N-291 and LA-N-6 NB cells) cell lines. As the cell lines in this
panel had a range of growth rates, of which COG-N-291 and LA-N-6 were the slowest, it was not possible
to treat for a fixed length of time, instead cells were treated for a duration of 6 PDs. The DNA damaging
agent doxorubicin reduced proliferation of all cell lines, although there was no differential response to
doxorubicin based on telomere biology (Figure 6.1.A; Table 6.1). Treatment with etoposide, a
topoisomerase II inhibitor, resulted in the most significant growth inhibition of COG-N-291 cells (IC50
0.08 µM; the dose to inhibit 50% of control growth) (Figure 6.1.B), although sensitivity to topoisomerase
II inhibitors is not a general feature of EST cells as LA-N-6 had an IC50 of 0.23 µM, similar to telomerase-
positive cell lines (0.17 to 0.56 µM). In contrast, EST cells show a striking sensitivity to the topoisomerase
I inhibitors, irinotecan and topotecan (Figure 6.1.C, D, E, F). At a dose of 2.8 µM irinotecan COG-N-291
had significantly reduced cell growth compared to all cell lines except SH-SY5Y and HT1080 hTR whilst
LA-N-6 cells had a similar trend of sensitivity with significantly reduced growth compared to U-2 OS, SK-
N-BE2c and HeLa.1.2.11 cells (where significant P<0.05, Figure 6.1D). The EST cells were particularly
sensitive to topotecan treatment, 0.0004 µM topotecan led to significantly reduced EST cell growth
compared to all other cell lines tested (P<0.001, Figure 6.1F). The IC50 of irinotecan and topotecan for
non-EST cell lines with long telomeres (U-2 OS, HT1080 hTR, and HeLa 1.2.11), was generally higher than
the EST cells (Figure 6.1.C, D; Table 6.1), indicating that the telomere length of EST cells is not entirely

135
Figure 6.2. Telomere biology does not influence sensitivity of NB cells to common NB therapies.
Cell lines were treated with increasing concentrations of (A) vincristine, (B) cisplatin, (C)
temozolomide, and (D) all-trans retinoic acid (ATRA) for 6 PDs before proliferation was quantified
using AlamarBlue. U-2 OS and GM847 ALT cell lines, and HT1080, GM639, HT1080 hTR and HeLa
1.2.11 telomerase-positive cell lines were included as controls. Results are expressed as
percentages of growth compared to untreated controls and as means of triplicate independent
experiments +SD.
responsible for their sensitivity to topoisomerase I inhibitors. The response of cell lines to vincristine,
cisplatin and temozolomide (Figure 6.2.A, B, C) varied depending on the cell line and was not associated
with telomere length maintenance phenotype. All-trans retinoic acid (ATRA), a differentiating agent, did
not reduce the proliferation of cells in a treatment period of 6 PDs (Figure 6.2.D).

136
Table 6.1. IC50 values for common NB chemotherapeutics.
IC50 (µM)
Doxorubicin Etoposide Cisplatin Irinotecan Topotecan Temozolomide Vincristineb
All Trans
Retinoic Acid
LA-N-6 0.40 0.23 4.23 1.04 <0.0004 324.33 8.07 >45
COG-N-291 0.01 0.08 0.77 0.31 <0.0004 58.87 12.64 >45
SK-N-FI 0.69 3.65 2.62 5.64 0.07 >400 >25 >45
CHLA-90 0.20 8.11 6.60 30.44 0.42 >400 2.21 >45
GM847 0.01 0.41 1.57 6.14 0.02 191.54 0.59 >45
U-2 OS 0.02 0.74 5.93 11.64 0.16 >400 6.02 >45
SK-N-BE2c 0.03 0.29 16.19 8.59 0.09 >400 1.61 >45
SH-SY5Y 0.03 0.56 2.20 2.50 0.01 98.96 19.35 >45
HT1080 0.01 0.19 1.88 5.00 0.01 389.18 0.18 >45
GM639 0.01 0.17 1.75 3.59 0.01 126.93 1.02 >45
HT1080 hTRa 0.12 0.66 8.39 2.08 0.11 365.30 2.79
HeLa 1.2.11a 0.12 0.51 0.98 17.21 <0.0004 >400 22.67 aLong telomeres/telomerase-positive bIC50 (nM)
6.2.2. EST cells do not exhibit increased cell death with potential ALT inhibitors or after
induction of replication stress
The EST cells have a low level of ALT activity which we hypothesise is preferentially lengthening the
shortest telomeres, consequently these cells could be sensitive to agents that target the ALT
mechanism. Arsenic trioxide (ATO; results in PML degradation) and ML216 (BLM helicase inhibitor) have
been shown to downregulate C-circles in ALT cells (unpublished data). However, neither ALT nor EST
cells showed greater sensitivity for these agents than telomerase-positive cells (Figure 6.3.A, B; Table
6.2). Previous work by Flynn and colleagues found that ALT cells are hypersensitive to ATR inhibition
(Flynn et al., 2015). In this panel of cell lines, the IC50 values for the ATR inhibitor VE-822 ranged from
0.02-0.53 µM for telomerase-positive cells, 0.02-6.22 µM for ALT cells, and 0.39-3.52 µM for EST cells
(Figure 6.3.C; Table 6.2), which is consistent with a later report that shows ALT cells are not more
sensitive to ATR inhibition than telomerase-positive cells (Deeg et al., 2016). There was a slight increase
in the IC50 doses for EST and ALT cells in response to the ATM inhibitor Ku60019 (Figure 6.3.D; Table
6.2). Agents that induce replication stress, including mitomycin C, hydroxyurea, and aphidicolin reduce

137
proliferation in all cells; however, the extent of reduction was not related to the telomere biology of the
cells (Figure 6.3.E, F, G; Table 6.2). Whilst COG-N-291 cells were most sensitive to all three inhibitors,
the IC50s for LA-N-6 were similar to ALT and telomerase-positive cells, and we found no evidence that
ALT cells are more sensitive to replication stress than other cell types (Table 6.2). In summary, the
current study failed to identify any drug that specifically inhibits either ALT or telomerase-positive cells.
However, we were able to identify the significant sensitivity of EST cells to topoisomerase I inhibitors.
6.2.3. EST cells have alterations to genes involved in DNA damage repair and replication
We have previously shown (Chapter 3) that the EST NB cell lines have functional p53 capable of
activating downstream targets such as p21 and triggering p53-mediated apoptosis in response to DNA
damage. Mutations to pathways involved in DNA damage repair and replication would prevent the EST
cells from repairing damage caused by chemotherapeutics leading to apoptosis through the p53
pathway. Whole genome sequencing data revealed heterozygous alterations of a number of genes
involved in DNA damage repair and replication in the EST cell lines (Appendix II A2.6). The COG-N-291
cells have mutations in GEN1 a resolvase that cleaves Holliday junctions (Ip et al., 2008), HLTF which is
involved in DNA damage repair and replication fork reversal (Achar et al., 2011; Blastyak et al., 2010;
Kile et al., 2015), and MCM4, a gene involved in the initiation and progression of replication forks (Sheu
et al., 2014; Sheu et al., 2016). Moreover, LA-N-6 cells have altered EME2 which is involved in the
cleavage and restart of replication forks (Pepe and West, 2014), and POLM, a polymerase used for gap
filling in DNA damage repair by NHEJ (Pryor et al., 2015). The effect of heterozygous mutations of these
genes on DNA damage repair and replication remains to be elucidated.
6.3. Discussion
The poor survival rates of NB patients with high-risk disease indicate the need to develop better
therapies for these patients. Our results, in conjunction with a number of genomic studies (Cheung et
al., 2012; Kurihara et al., 2014; Molenaar et al., 2012; Peifer et al., 2015; Pugh et al., 2013; Valentijn et

138
Table 6.2. IC50 values for inhibitors that cause replication stress and inhibit factors associated with ALT.
IC50 (µM)
ML216 Arsenic Trioxide VE-822 Ku60019 Mitomycin Ca Hydroxyurea Aphidicolin
LA-N-6 >225 3.24 3.52 >24 0.94 429.24 0.62
COG-N-291 >225 3.51 0.39 >24 0.05 84.33 0.08
SK-N-FI >225 2.55 6.22 >24 >3 1199.17 2.38
CHLA-90 5.04 7.00 0.33 20.91 1.22 257.94 4.45
GM847 110.53 0.77 0.02 4.97 0.18 193.23 0.51
U-2 OS 95.98 2.19 0.09 13.37 1.32 731.70 16.13
SK-N-BE2c >225 4.88 0.07 12.91 0.51 348.27 0.19
SH-SY5Y >225 3.94 0.53 10.77 0.43 >1600 0.50
HT1080 26.98 1.26 0.04 3.52 0.52 229.45 0.97
GM639 95.11 1.26 0.02 11.79 0.49 262.79 0.57
aIC50 (µg/mL)
al., 2015), suggest that telomere biology may be a target for novel NB therapies. The EST cells are highly
sensitive to the topoisomerase I inhibitors topotecan and irinotecan. Topotecan and irinotecan are
derivatives of camptothecin, a topoisomerase I poison that stabilises the topoisomerase I-DNA cleavage
complex during DNA replication (Hsiang et al., 1985). This results in a dsDNA break when the advancing
DNA replication fork collides with the topoisomerase I-DNA complex leading to cell cycle arrest and
apoptosis (Hsiang et al., 1989). The differential response of EST cells to this form of DNA damage
compared with ALT or telomerase-positive cells was not due to therapy-acquired resistance in the ALT
or telomerase-positive NB cell lines as neither topotecan nor irinotecan were used to treat NB patients
from which these cells were derived more than two decades ago (Biedler et al., 1973; Keshelava et al.,
1998; Wada et al., 1993). Whilst COG-N-291 and LA-N-6 have obvious sensitivity to topoisomerase I
inhibitors the analysis is limited by the number of EST cell lines. The Pediatric Preclinical Testing Program
(PPTP) identified NB-1643 as acutely sensitive to topotecan in their cell line panel with an IC50 of 0.71
nM (Carol et al., 2010) and the Reynold’s lab has identified a further four cell lines that are telomerase-

139
Figure 6.3. Telomere biology does not predict the response of cell lines to inhibitors that cause
replication stress or reduce factors critical to the ALT mechanism.
Cell lines were treated with increasing concentrations of (A) arsenic trioxide (ATO) which degrades
PML, (B) the BLM inhibitor ML216, (C) the ATR inhibitor VE-822, (D) the ATM inhibitor KU60019, (E)

140
mitomycin C, (F) hydroxyurea, and (G) aphidicolin for 6 PDs before proliferation was quantified
using AlamarBlue. U-2 OS and GM847 ALT cell lines, and HT1080 and GM639 telomerase-positive
cell lines were included as controls. Results are expressed as percentages of growth compared to
untreated controls and as means of triplicate independent experiments +SD.
negative and lack C-circles (unpublished data). Therefore, further testing is required to establish if any
of these cell lines have the EST phenotype, with the potential to expand the EST NB cohort for further
drug screening. Genz-644282 is a recently developed new generation of topoisomerase I inhibitor, not
derived from camptothecin (Li et al., 2003). According to the PPTP screen, NB cells are generally more
sensitive to Genz-644282 than topotecan (Houghton et al., 2012), therefore, establishing the acute
sensitivity of EST cells to Genz-644282 will confirm EST cells are particularly sensitive to topoisomerase
I inhibitors.
One possible explanation for the EST cell sensitivity to topoisomerase inhibitors are the very
long telomeres of these cells. Telomeres are known to form G-quadruplex structures (Sen and Gilbert,
1988; Sundquist and Klug, 1989) which need to be removed prior to telomere replication in S. cerevisiae
(Paeschke et al., 2011). Long telomeres have a greater potential to form such structures which could
reduce the efficiency of telomeric replication and make complete DNA replication more difficult.
Furthermore, the replication fork must travel a further distance to fully replicate long telomeres. In this
study, we found no evidence that telomere length as a single factor was responsible for the EST
sensitivity to topoisomerase I inhibitors. The cell line U-2 OS has wild type TP53 which can induce p21
(Bensaad et al., 2006; Cesare et al., 2009) and has similar telomere lengths to EST cell lines however, it
is not sensitive to treatment with topotecan or irinotecan. Whole genome sequencing identified
aberrations in DNA damage repair and replication genes in the EST cell lines but further studies are
required to determine the effect of these heterozygous mutations. The EST cells may be particularly
sensitive to DNA damage by topoisomerase I inhibitors due to a combination of very long telomeres,

141
wild-type TP53 and impaired DNA repair/replication machinery, which means that the additional DNA
damage caused by topoisomerase I inhibition during replication, cannot be repaired by the cells leading
to p53-mediated apoptosis.
Topoisomerase levels may predict the response of cells to topoisomerase inhibitors (Burgess et
al., 2008). We are currently using pressure cycling technology (PCT) and sequential window acquisition
of all theoretical fragment ion spectra (SWATH) mass spectrometry (Guo et al., 2015) to measure the
level of proteins in EST, telomerase and ALT NB which will allow us to determine if the level of
topoisomerase I and II correlates with sensitivity to topoisomerase inhibitors. This study will also allow
us to determine the effect of heterozygous mutations on the protein expression of GEN1, HLTF, MCM4,
EME2, and POLM in the EST cells. We will also use the mass spectrometry results to determine a
proteomic profile of the EST cells compared to ALT and telomerase-positive NB to aid the development
of targeted therapies based on the telomere biology of NB cells.
The use of topoisomerase I inhibitors is a recent development in NB treatment. Topotecan has
only recently been incorporated into COG clinical trials as a part of induction therapy (Park et al., 2011)
or to treat relapsed/refractory disease (Garaventa et al., 2003) whilst irinotecan is only used to treat
relapsed or refractory NB (Kushner et al., 2006). Our results indicate that 11% of high-risk NB, i.e. the
EST tumours, could be acutely sensitive to upfront treatment with topotecan and irinotecan. Further
preclinical studies are required to confirm whether topoisomerase I inhibition results in better outcome
for EST tumours but we have shown that targeting telomere biology may lead to novel treatment
regimens for high-risk NB.

142
Chapter 7: Discussion

143
Chapter 7: Discussion
The acquisition of cellular immortality, via activation of a TLM, is currently considered a hallmark of
cancer (Hanahan and Weinberg, 2000; Hanahan and Weinberg, 2011). However, studies of some
cancers including glioblastoma (Hakin-Smith et al., 2003), liposarcoma (Costa et al., 2006; Jeyapalan et
al., 2008), osteosarcoma (Ulaner et al., 2003), and retinoblastoma (Gupta et al., 1996) have reported
that a substantial proportion of tumours are TLM-negative, and the widely-held explanation for this has
been that these are false negatives caused by deficiencies in the standard TLM assays. Our study has
identified a subset of NB tumours that lack telomerase activity, have very long telomeres, and have such
low levels of ALT that they are unable to prevent telomere shortening. Cells derived from such tumours
are capable of long-term proliferation despite ever-shorter telomeres. Subsequent investigations
showed NB EST cells were acutely sensitive to topoisomerase I inhibitors, indicating TLMs are potential
targets for novel NB therapies.
7.1. The ever-shorter telomeres phenotype in human cancer
Telomerase activity was established as a poor prognostic factor in NB over two decades ago (Poremba
et al., 2000; Poremba et al., 1999) and recent studies have utilised telomere length (Onitake et al., 2009),
or ATRX/DAXX mutations (Cheung et al., 2012; Kurihara et al., 2014; Molenaar et al., 2012; Peifer et al.,
2015; Valentijn et al., 2015) to identify ALT tumours. Ours is the first study to utilise C-circles as a
quantifiable marker of ALT activity to determine the prevalence of ALT in metastatic NB. Using this
method, we identified 11% of high-risk NB tumours as ALT-negative/TC≥15. These tumours lacked
telomerase activity, indicating that a subgroup of high-risk NBs lack evidence of a TLM, consistent with
previous studies (Choi et al., 2000; Krams et al., 2001; Poremba et al., 2000; Poremba et al., 1999;
Reynolds et al., 1997). These studies were unable to distinguish among the possibilities that the
apparent lack of significant ALT or telomerase activity was due to: (1) false-negative assays, (2) an as-
yet unidentified ALT mechanism lacking the common phenotypic characteristics of the "classic" ALT

144
mechanism, or (3) a true lack of significant TLM activity in the cells. The only conclusive test to determine
if cells are TLM-negative is to observe telomere length over time, which cannot be done ethically in
human tumour samples as it would require serial samples from the same patient over time in the
absence of treatment. Consequently, cell line models were required to determine which of these
scenarios was true. We present evidence that human cell lines derived from fatal metastatic disease are
capable of proliferating for 500 PDs, despite ever-shorter telomeres.
The observation of the EST phenotype in high-risk NB provides direct evidence that, as
previously postulated (Reddel, 2000), immortalisation is not an absolute requirement of oncogenesis.
Telomerase activity is detectable during the early stages of human embryogenesis however, it is
downregulated in late embryogenesis resulting in telomerase-negative somatic cells (Ulaner and
Giudice, 1997; Wright et al., 1996). NB is an embryonal cancer, and we propose that excessive
telomerase activity during embryogenesis resulted in very long telomeres in the EST progenitor cells
before telomerase activity was downregulated at the end of embryogenesis. The EST progenitor cells
would not require significant upregulation of a TLM to allow extensive proliferation to occur in these
cells although further oncogenic events would be required to form a malignant tumour. Telomere
trimming is a process that regulates telomere length in cells that have been over-lengthened by
telomerase (Pickett et al., 2009). The lack of t-circles, a product of telomere trimming, in LA-N-6 cells
with over-lengthened telomeres due to ectopic telomerase expression suggests that EST cells have a
failure of telomere trimming. T-circles are also detected in cells that have upregulated ALT (Cesare and
Griffith, 2004), however we were unable to detect t-circles either in COG-N-291 clone 4, or in the LA-N-
6 MUT p53 clone that spontaneously activated ALT, further confirming that these cells have a failure of
telomere trimming. This is the first report of a disease that may result from a failure of telomere
trimming, although the mechanism leading to failure of telomere trimming remains to be elucidated.
Whole genome sequencing did not identify genetic events unique to the EST cells and further studies
are required to examine their epigenetic and proteomic profile compared to ALT and telomerase-
positive cells and consequently identify events responsible for the failure of telomere trimming. The

145
failure of telomere trimming in EST cells may be responsible for their sensitivity to topoisomerase I
inhibitors. If this is the case, knockdown of genes associated with telomere trimming would lead to the
increased sensitivity of non-EST cells to topoisomerase I inhibitors, providing further evidence that the
EST cells are a disease resulting from the failure of telomere trimming.
Our study did not identify potential EST cell lines in other tumour types although this is likely
due to the criteria used to identify EST cell lines. The criteria for NB EST may not reflect the phenotype
of EST cells in other tumour types, as NB is a childhood cancer and consequently the tumour progenitor
cells have not undergone many cycles of replication induced telomere shortening. In contrast, the
progenitor cells of adult cancers may have undergone many rounds of cell division, and associated
telomere shortening, before oncogenesis occurs. Cell lines derived from these tumours may not have
very long telomere lengths after the PDs required to derive a cell line from the tumour. Indeed, data
from the Decottignies laboratory, found melanoma cells lacking evidence of a TLM had telomere lengths
at the upper end of the normal range (Viceconte et al., 2017). Another explanation for the lack of EST
cell lines in other tumour types may be the difficulty in deriving cell lines from such tumours. The NB
EST cell lines grow very slowly compared to cell lines with an upregulated TLM and consequently other
EST cell lines may never have been characterised or grown long term in laboratories due to the bias
towards with faster growing cell lines for in vitro experiments. Therefore, it is likely that cell line studies
will underestimate the occurrence of the EST phenotype.
7.2. Targeting telomere biology in the clinic
Since the discovery of telomerase, inhibitors have been developed to target the enzyme translating to
clinical trials in cancers including breast and lung cancer, glioblastoma, and lympho-proliferative
disorders, although to date there is no evidence of their effectiveness in treating malignancies (Arndt
and MacKenzie, 2016). In contrast, ALT inhibitors have yet to enter clinical trials and are currently being
developed in preclinical laboratory models (Deeg et al., 2016; Flynn et al., 2015). High-risk NB patients
have very poor survival rates and novel targets for therapy need to be identified. Our study, in addition

146
to a number of large scale genomic studies (Cheung et al., 2012; Kurihara et al., 2014; Molenaar et al.,
2012; Peifer et al., 2015; Pugh et al., 2013; Valentijn et al., 2015), indicate that telomere biology plays a
significant role in NB pathogenesis and may be a target for novel NB therapies. We have shown that NB
EST cells are highly sensitive to the topoisomerase I inhibitors topotecan and irinotecan and therefore
11% of high-risk NB could be acutely sensitive to upfront treatment with these agents. The identification
of further EST NB cell lines, currently underway through expansion of the cell line screen, will aid in the
identification of other agents that uniquely target these cells. The identification of EST cell lines in other
cancer types will facilitate further studies to determine if sensitivity to topoisomerase I inhibition is a
universal feature of EST cells.
EST cells provide the first evidence of a disease that may result from a deficiency in telomere
trimming. This provides a novel area for drug development as there is currently no therapy to target the
telomere trimming mechanism. The molecular details of the trimming mechanism are still under
investigation which may lead to the identification of novel therapeutic targets.
The development of drugs which target features of telomere biology require appropriate
biomarkers to predict response to therapy. The discovery of malignant and lethal EST cancer cells
illustrates the need to assay for TLMs prior to treatment with drugs that target these mechanisms. The
phenotypic similarity of EST and ALT cells illustrates that care must be taken to correctly define ALT
tumours: ATRX/DAXX mutations are not a universal feature of ALT cells (Lovejoy et al., 2012), telomere
length (as measured by telomere qPCR, TRF analysis or telomere FISH) is insufficient to differentiate ALT
and EST cells, and the presence of low levels of C-circles does not necessarily indicate sufficient ALT
activity to counter the EST phenotype. Therefore, studies that define a molecular profile for the EST cells
will be critical in developing accurate clinical assays to differentiate between EST and ALT cells.
7.3. Conclusion
In conclusion, this study has provided evidence of lethal, malignant human cancers that can proliferate
continuously for hundreds of PDs with ever-shorter telomeres. This indicates that the upregulation of a

147
TLM to confer immortalisation is not an absolute requirement for oncogenesis. Patients with high-risk
NB have a poor survival rate and we need to develop novel therapies to improve the outcome of these
patients. Our results, indicate that telomere biology is a relevant target for novel NB therapies and EST
tumours are potentially very susceptible to treatment with topoisomerase I inhibitors. The similarity of
the EST and ALT phenotypes indicates that rapid and specific assays for TLMs are required to facilitate
the use of TLM targeting inhibitors as anti-cancer therapeutics.

148
Chapter 8: References

149
Chapter 8: References
Achar, Y. J., Balogh, D., and Haracska, L. (2011). Coordinated protein and DNA remodeling by human HLTF on stalled replication fork. Proc Natl Acad Sci U.S.A. 108, 14073-14078. Ahmad, K., and Henikoff, S. (2002). Histone H3 variants specify modes of chromatin assembly. Proc Natl Acad Sci U.S.A. 99, 16477-16484. Akiyama, B. M., Parks, J. W., and Stone, M. D. (2015). The telomerase essential N-terminal domain promotes DNA synthesis by stabilizing short RNA-DNA hybrids. Nucleic Acids Res 43, 5537-5549. Alder, J. K., Chen, J. J. L., Lancaster, L., Danoff, S., Su, S. C., Cogan, J. D., Vulto, I., Xie, M. Y., Qi, X. D., Tuder, R. M., et al. (2008). Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U.S.A. 105, 13051-13056. Alexandrov, L. B., Nik-Zainal, S., Wedge, D. C., Aparicio, S. A., Behjati, S., Biankin, A. V., Bignell, G. R., Bolli, N., Borg, A., Borresen-Dale, A. L., et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415-421. Allshire, R. C., Dempster, M., and Hastie, N. D. (1989). Human telomeres contain at least three types of G-rich repeat distributed non-randomly. Nucleic Acids Res 17, 4611-4627. Alonso, M. M., Fueyo, J., Yung, W. K., and Gomez-Manzano, C. (2006). E2F1 and telomerase: alliance in the dark side. Cell Cycle 5, 930-935. Alves, D., Li, H. T., Codrington, R., Orte, A., Ren, X. J., Klenerman, D., and Balasubramanian, S. (2008). Single-molecule analysis of human telomerase monomer. Nat Chem Biol 4, 287-289. Ambros, P. F., Ambros, I. M., Brodeur, G. M., Haber, M., Khan, J., Nakagawara, A., Schleiermacher, G., Speleman, F., Spitz, R., London, W. B., et al. (2009). International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. Br J Cancer 100, 1471-1482. Amiard, S., Doudeau, M., Pinte, S., Poulet, A., Lenain, C., Faivre-Moskalenko, C., Angelov, D., Hug, N., Vindigni, A., Bouvet, P., et al. (2007). A topological mechanism for TRF2-enhanced strand invasion. Nat Struct Mol Biol 14, 147-154. Anderson, B. H., Kasher, P. R., Mayer, J., Szynkiewicz, M., Jenkinson, E. M., Bhaskar, S. S., Urquhart, J. E., Daly, S. B., Dickerson, J. E., O'Sullivan, J., et al. (2012). Mutations in CTC1, encoding conserved telomere maintenance component 1, cause Coats plus. Nat Genet 44, 338-342. Armanios, M., Chen, J. L., Chang, Y. P. C., Brodsky, R. A., Hawkins, A., Griffin, C. A., Eshleman, J. R., Cohen, A. R., Chakravarti, A., Hamosh, A., and Greider, C. W. (2005). Haploinsufficiency of telomerase reverse transcriptase leads to anticipation in autosomal dominant dyskeratosis congenita. Proc Natl Acad Sci U.S.A. 102, 15960-15964.

150
Armanios, M. Y., Chen, J. J. L., Cogan, J. D., Alder, J. K., Ingersoll, R. G., Markin, C., Lawson, W. E., Xie, M. Y., Vulto, I., Phillips, J. A., et al. (2007). Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med 356, 1317-1326. Armbruster, B. N., Banik, S. S. R., Guo, C. H., Smith, A. C., and Counter, C. M. (2001). N-terminal domains of the human telomerase catalytic subunit required for enzyme activity in vivo. Mol Cell Biol 21, 7775-7786. Armbruster, B. N., Linardic, C. M., Veldman, T., Bansal, N. P., Downie, D. L., and Counter, C. M. (2004). Rescue of an hTERT mutant defective in telomere elongation by fusion with hPot1. Mol Cell Biol 24, 3552-3561. Arndt, G. M., and MacKenzie, K. L. (2016). New prospects for targeting telomerase beyond the telomere. Nat Rev Cancer 16, 508-532. Arnoult, N., Schluth-Bolard, C., Letessier, A., Drascovic, I., Bouarich-Bourimi, R., Campisi, J., Kim, S. H., Boussouar, A., Ottaviani, A., Magdinier, F., et al. (2010). Replication timing of human telomeres is chromosome arm-specific, influenced by subtelomeric structures and connected to nuclear localization. PLoS Genet 6, e1000920. Arnoult, N., van Beneden, A., and Decottignies, A. (2012). Telomere length regulates TERRA levels through increased trimethylation of telomeric H3K9 and HP1a. Nat Struct Mol Biol 19, 948-956. Arora, R., Lee, Y., Wischnewski, H., Brun, C. M., Schwarz, T., and Azzalin, C. M. (2014). RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 5, 5220-31. Artandi, S. E., Chang, S., Lee, S. L., Alson, S., Gottlieb, G. J., Chin, L., and DePinho, R. A. (2000). Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature 406, 641-645. Attiyeh, E. F., London, W. B., Mosse, Y. P., Wang, Q., Winter, C., Khazi, D., McGrady, P. W., Seeger, R. C., Look, A. T., Shimada, H., et al. (2005). Chromosome 1p and 11q deletions and outcome in neuroblastoma. N Engl J Med 353, 2243-2253. Avilion, A. A., Piatyszek, M. A., Gupta, J., Shay, J. W., Bacchetti, S., and Greider, C. W. (1996). Human telomerase RNA and telomerase activity in immortal cell lines and tumor tissues. Cancer Res 56, 645-650. Azzalin, C. M., and Lingner, J. (2008). Telomeres: the silence is broken. Cell Cycle 7, 1161-1165. Azzalin, C. M., Reichenbach, P., Khoriauli, L., Giulotto, E., and Lingner, J. (2007). Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science 318, 798-801. Bachor, C., Bachor, O. A., and Boukamp, P. (1999). Telomerase is active in normal gastrointestinal mucosa and not up-regulated in precancerous lesions. J Cancer Res Clin Oncol 125, 453-460. Bagatell, R., Beck-Popovic, M., London, W. B., Zhang, Y., Pearson, A. D., Matthay, K. K., Monclair, T., Ambros, P. F., and Cohn, S. L. (2009). Significance of MYCN amplification in international neuroblastoma staging system stage 1 and 2 neuroblastoma: a report from the International Neuroblastoma Risk Group database. J Clin Oncol 27, 365-370. Bailey, S. M., Brenneman, M. A., and Goodwin, E. H. (2004). Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res 32, 3743-3751.

151
Baird, D. M., Jeffreys, A. J., and Royle, N. J. (1995). Mechanisms underlying telomere repeat turnover, revealed by hypervariable variant repeat distribution patterns in the human Xp/Yp telomere. EMBO J 14, 5433-5443. Baker, D. L., Schmidt, M. L., Cohn, S. L., Maris, J. M., London, W. B., Buxton, A., Stram, D., Castleberry, R. P., Shimada, H., Sandler, A., et al. (2010). Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N Engl J Med 363, 1313-1323. Balk, B., Maicher, A., Dees, M., Klermund, J., Luke-Glaser, S., Bender, K., and Luke, B. (2013). Telomeric RNA-DNA hybrids affect telomere-length dynamics and senescence. Nat Struct Mol Biol 20, 1199-1205. Barthel, F. P., Wei, W., Tang, M., Martinez-Ledesma, E., Hu, X., Amin, S. B., Akdemir, K. C., Seth, S., Song, X., Wang, Q., et al. (2017). Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet 49, 349-357. Bartkova, J., Rezaei, N., Liontos, M., Karakaidos, P., Kletsas, D., Issaeva, N., Vassiliou, L. V., Kolettas, E., Niforou, K., Zoumpourlis, V. C., et al. (2006). Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633-637. Bauer, D. W., and Gall, J. G. (1997). Coiled bodies without coilin. Mol Biol Cell 8, 73-82. Baumann, P., and Cech, T. R. (2001). Pot1, the putative telomere end-binding protein in fission yeast and humans. Science 292, 1171-1175. Baur, J. A., Wright, W. E., and Shay, J. W. (2004). Analysis of mammalian telomere position effect. Meth Mol Biol 287, 121-136. Baur, J. A., Zou, Y., Shay, J. W., and Wright, W. E. (2001). Telomere position effect in human cells. Science 292, 2075-2077. Beausejour, C. M., Krtolica, A., Galimi, F., Narita, M., Lowe, S. W., Yaswen, P., and Campisi, J. (2003). Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 22, 4212-4222. Bechter, O. E., Zou, Y., Walker, W., Wright, W. E., and Shay, J. W. (2004). Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res 64, 3444-3451. Bell, R. J., Rube, H. T., Kreig, A., Mancini, A., Fouse, S. D., Nagarajan, R. P., Choi, S., Hong, C., He, D., Pekmezci, M., et al. (2015). Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 348, 1036-1039. Benarroch-Popivker, D., Pisano, S., Mendez-Bermudez, A., Lototska, L., Kaur, P., Bauwens, S., Djerbi, N., Latrick, C. M., Fraisier, V., Pei, B., et al. (2016). TRF2-mediated control of telomere DNA topology as a mechanism for chromosome-end protection. Mol Cell 61, 274-286. Benetti, R., Garcia-Cao, M., and Blasco, M. A. (2007a). Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet 39, 243-250.

152
Benetti, R., Gonzalo, S., Jaco, I., Schotta, G., Klatt, P., Jenuwein, T., and Blasco, M. A. (2007b). Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol 178, 925-936. Bensaad, K., Tsuruta, A., Selak, M. A., Vidal, M. N., Nakano, K., Bartrons, R., Gottlieb, E., and Vousden, K. H. (2006). TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107-120. Bernardi, R., and Pandolfi, P. P. (2003). Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22, 9048-9057. Bernardi, R., and Pandolfi, P. P. (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat Rev Mol Cell Biol 8, 1006-1016. Bernhardt, S. L., Gjertsen, M. K., Trachsel, S., Moller, M., Eriksen, J. A., Meo, M., Buanes, T., and Gaudernack, G. (2006). Telomerase peptide vaccination of patients with non-resectable pancreatic cancer: a dose escalating phase I/II study. Br J Cancer 95, 1474-1482. Bhattacharyya, S., Keirsey, J., Russell, B., Kavecansky, J., Lillard-Wetherell, K., Tahmaseb, K., Turchi, J. J., and Groden, J. (2009). Telomerase associated protein 1, HSP90 and topoisomerase IIa associate directly with the BLM helicase in immortalized cells using ALT and modulate its helicase activity using telomeric DNA substrates. J Biol Chem 284, 14966-14977. Biedler, J. L., Helson, L., and Spengler, B. A. (1973). Morphology and growth, tumorigenicity, and cytogenetics of human neuroblastoma cells in continuous culture. Cancer Res 33, 2643-2652.
Biedler, J. L., Roffler-Tarlov, S., Schachner, M., Freedman, L. S. (1978). Multiple neurotransmitter synthesis by human neuroblastoma cell lines and clones. Cancer Res. 38, 3751-3757.
Biffi, G., Di Antonio, M., Tannahill, D., and Balasubramanian, S. (2014). Visualization and selective chemical targeting of RNA G-quadruplex structures in the cytoplasm of human cells. Nat Chem 6, 75-80. Biffi, G., Tannahill, D., McCafferty, J., and Balasubramanian, S. (2013). Quantitative visualization of DNA G-quadruplex structures in human cells. Nat Chem 5, 182-186.
Bilaud, T., Brun, C., Ancelin, K., Koering, C. E., Laroche, T., and Gilson, E. (1997). Telomeric localization of TRF2, a novel human telobox protein. Nat Genet 17, 236-239. Binz, N., Shalaby, T., Rivera, P., Shin-Ya, K., and Grotzer, M. A. (2005). Telomerase inhibition, telomere shortening, cell growth suppression and induction of apoptosis by telomestatin in childhood neuroblastoma cells. Eur J Cancer 41, 2873-2881. Blastyak, A., Hajdu, I., Unk, I., and Haracska, L. (2010). Role of double-stranded DNA translocase activity of human HLTF in replication of damaged DNA. Mol Cell Biol 30, 684-693. Bodnar, A. G., Ouellette, M., Frolkis, M., Holt, S. E., Chiu, C. P., Morin, G. B., Harley, C. B., Shay, J. W., Lichtsteiner, S., and Wright, W. E. (1998). Extension of life-span by introduction of telomerase into normal human cells. Science 279, 349-352. Bond, J. A., Haughton, M. F., Rowson, J. M., Smith, P. J., Gire, V., Wynford-Thomas, D., and Wyllie, F. S. (1999). Control of replicative life span in human cells: barriers to clonal expansion intermediate between M1 senescence and M2 crisis. Mol Cell Biol 19, 3103-3114.

153
Bond, J. A., Wyllie, F. S., and Wynford-Thomas, D. (1994). Escape from senescence in human diploid fibroblasts induced directly by mutant p53. Oncogene 9, 1885-1889. Bower, K., Napier, C. E., Cole, S. L., Dagg, R. A., Lau, L. M., Duncan, E. L., Moy, E. L., and Reddel, R. R. (2012). Loss of wild-type ATRX expression in somatic cell hybrids segregates with activation of Alternative Lengthening of Telomeres. PLoS ONE 7, e50062. Bown, N., Cotterill, S., Lastowska, M., O’Neill, S., Pearson, A. D. J., Plantaz, D., Meddeb, M., Danglot, G., Brinkschmidt, C., Christiansen, H., et al. (1999). Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N Engl J Med 340, 1954-1961. Brinkschmidt, C., Poremba, C., Christiansen, H., Simon, R., Schafer, K. L., Terpe, H. J., Lampert, F., Boecker, W., and Dockhorn-Dworniczak, B. (1998). Comparative genomic hybridization and telomerase activity analysis identify two biologically different groups of 4s neuroblastomas. Br J Cancer 77, 2223-2229. Britt-Compton, B., Capper, R., Rowson, J., and Baird, D. M. (2009). Short telomeres are preferentially elongated by telomerase in human cells. FEBS Lett 583, 3076-3080. Broccoli, D., Smogorzewska, A., Chong, L., and de Lange, T. (1997). Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet 17, 231-235. Broccoli, D., Young, J. W., and de Lange, T. (1995a). Telomerase activity in normal and malignant hematopoietic cells. Proc Natl Acad Sci USA 92, 9082-9086. Brock, G. J., Charlton, J., and Bird, A. (1999). Densely methylated sequences that are preferentially localized at telomere-proximal regions of human chromosomes. Gene 240, 269-277. Brodeur, G. M., Green, A. A., Hayes, F. A., Williams, K. J., Williams, D. L., and Tsiatis, A. A. (1981). Cytogenetic features of human neuroblastomas and cell lines. Cancer Res 41, 4678-4686. Brodeur, G. M., Pritchard, J., Berthold, F., Carlsen, N. L. T., Castel, V., Castleberry, R. P., Debernardi, B., Evans, A. E., Favrot, M., Hedborg, F., et al. (1993). Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J Clin Oncol 11, 1466-1477. Brodeur, G. M., Seeger, R. C., Schwab, M., Varmus, H. E., and Bishop, J. M. (1984). Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 224, 1121-1124. Brown, J. P., Wei, W., and Sedivy, J. M. (1997). Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 277, 831-834. Brunsvig, P. F., Kyte, J. A., Kersten, C., Sundstrum, S., Muller, M., Nyakas, M., Hansen, G. L., Gaudernack, G., and Aamdal, S. (2011). Telomerase peptide vaccination in NSCLC: a phase II trial in stage III patients vaccinated after chemoradiotherapy and an 8-year update on a phase I/II trial. Clin Cancer Res 17, 6847-6857. Bryan, T. M., Englezou, A., DallaPozza, L., Dunham, M. A., and Reddel, R. R. (1997). Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat Med 3, 1271-1274.

154
Bryan, T. M., Englezou, A., Gupta, J., Bacchetti, S., and Reddel, R. R. (1995). Telomere elongation in immortal human cells without detectable telomerase activity. EMBO J 14, 4240-4248. Bryan, T. M., Goodrich, K. J., and Cech, T. R. (2000). A mutant of Tetrahymena telomerase reverse transcriptase with increased processivity. J Biol Chem 275, 24199-24207. Buanes, T., Maurel, J., Liauw, W., Hebbar, M., and Nemunaitis, J. (2009). A randomized phase III study of gemcitabine (G) versus GV1001 in sequential combination with G in patients with unresectable and metastatic pancreatic cancer (PC). J Clin Oncol 27, 4601-4601. Burgess, D. J., Doles, J., Zender, L., Xue, W., Ma, B., McCombie, W. R., Hannon, G. J., Lowe, S. W., and Hemann, M. T. (2008). Topoisomerase levels determine chemotherapy response in vitro and in vivo. Proc Nat Acad Sci USA 105, 9053-9058. Calado, R. T., Brudno, J., Mehta, P., Kovacs, J. J., Wu, C., Zago, M. A., Chanock, S. J., Boyer, T. D., and Young, N. S. (2011). Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 53, 1600-1607. Cao, Y., Bryan, T. M., and Reddel, R. R. (2008a). Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci 99, 1092-1099. Cao, Y., Huschtscha, L. I., Nouwens, A. S., Pickett, H. A., Neumann, A. A., Chang, A. C., Toouli, C. D., Bryan, T. M., and Reddel, R. R. (2008b). Amplification of telomerase reverse transcriptase gene in human mammary epithelial cells with limiting telomerase RNA expression levels. Cancer Res 68, 3115-3123. Capasso, M., Devoto, M., Hou, C., Asgharzadeh, S., Glessner, J. T., Attiyeh, E. F., Mosse, Y. P., Kim, C., Diskin, S. J., Cole, K. A., et al. (2009). Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat Genet 41, 718-723. Capper, R., Britt-Compton, B., Tankimanova, M., Rowson, J., Letsolo, B., Man, S., Haughton, M., and Baird, D. M. (2007). The nature of telomere fusion and a definition of the critical telomere length in human cells. Genes Dev 21, 2495-2508. Carol, H., Houghton, P. J., Morton, C. L., Kolb, E. A., Gorlick, R., Reynolds, C. P., Kang, M. H., Maris, J. M., Keir, S. T., Watkins, A., et al. (2010). Initial testing of topotecan by the pediatric preclinical testing program. Pediatr Blood Cancer 54, 707-715. Carr-Wilkinson, J., O’Toole, K., Wood, K. M., Challen, C. C., Baker, A. G., Board, J. R., Evans, L., Cole, M., Cheung, N. K., Boos, J., et al. (2010). High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Cancer Res 16, 1108-1118. Castel, V., Tovar, J. A., Costa, E., Cuadros, J., Ruiz, A., Rollan, V., Ruiz-Jimenez, J. I., Perez-Hernandez, R., and Canete, A. (2002). The role of surgery in stage IV neuroblastoma. J Pediatr Surg 37, 1574-1578. Cawthon, R. M. (2002). Telomere measurement by quantitative PCR. Nucleic Acids Res 30, e47. Celli, G. B., and de Lange, T. (2005). DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol 7, 712-718. Cerone, M. A., Londono-Vallejo, J. A., and Bacchetti, S. (2001). Telomere maintenance by telomerase and by recombination can coexist in human cells. Hum Mol Gen 10, 1945-1952.

155
Cesare, A. J., and Griffith, J. D. (2004). Telomeric DNA in ALT cells is characterized by free telomeric circles and heterogeneous t-loops. Mol Cell Biol 24, 9948-9957. Cesare, A. J., Hayashi, M. T., Crabbe, L., and Karlseder, J. (2013). The telomere deprotection response is functionally distinct from the genomic DNA damage response. Mol Cell 51, 141-155. Cesare, A. J., Kaul, Z., Cohen, S. B., Napier, C. E., Pickett, H. A., Neumann, A. A., and Reddel, R. R. (2009). Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol 16, 1244-1251. Cesare, A. J., and Reddel, R. R. (2010). Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet 11, 319-330. Chanan-Khan, A. A., Munshi, N. C., Hussein, M. A., Elias, L., Benedetti, F., Smith, J., Khor, S. P., and Huff, C. A. (2008). Results of a phase I study of GRN163L, a direct inhibitor of telomerase, in patients with relapsed and refractory multiple myeloma (MM). Blood 112, 1263-1263. Chang, B. D., Swift, M. E., Shen, M., Fang, J., Broude, E. V., and Roninson, I. B. (2002). Molecular determinants of terminal growth arrest induced in tumor cells by a chemotherapeutic agent. Proc Natl Acad Sci USA 99, 389-394. Chastain, M., Zhou, Q., Shiva, O., Whitmore, L., Jia, P., Dai, X., Huang, C., Fadri-Moskwik, M., Ye, P., and Chai, W. (2016). Human CST facilitates genome-wide RAD51 recruitment to GC-Rich repetitive sequences in response to replication stress. Cell Rep 16, 1300-1314. Chawla, R., Redon, S., Raftopoulou, C., Wischnewski, H., Gagos, S., and Azzalin, C. M. (2011). Human UPF1 interacts with TPP1 and telomerase and sustains telomere leading-strand replication. EMBO J 30, 4047-4058. Chen, C., Hong, Y. K., Ontiveros, S. D., Egholm, M., and Strauss, W. M. (1999). Single base discrimination of CENP-B repeats on mouse and human Chromosomes with PNA-FISH. Mamm Genome 10, 13-18. Chen, J. L., Blasco, M. A., and Greider, C. W. (2000). Secondary structure of vertebrate telomerase RNA. Cell 100, 503-514. Chen, X., Bahrami, A., Pappo, A., Easton, J., Dalton, J., Hedlund, E., Ellison, D., Shurtleff, S., Wu, G., Wei, L., et al. (2014). Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7, 104-112. Chen, Y., Deng, Z., Jiang, S., Hu, Q., Liu, H., Songyang, Z., Ma, W., Chen, S., and Zhao, Y. (2015). Human cells lacking coilin and Cajal bodies are proficient in telomerase assembly, trafficking and telomere maintenance. Nucleic Acids Res 43, 385-395. Chen, Y., Takita, J., Choi, Y. L., Kato, M., Ohira, M., Sanada, M., Wang, L., Soda, M., Kikuchi, A., Igarashi, T., et al. (2008). Oncogenic mutations of ALK kinase in neuroblastoma. Nature 455, 971-974. Chen, Y. J., Hakin-Smith, V., Teo, M., Xinarianos, G. E., Jellinek, D. A., Carroll, T., McDowell, D., MacFarlane, M. R., Boet, R., Baguley, B. C., et al. (2006). Association of mutant TP53 with alternative lengthening of telomeres and favorable prognosis in glioma. Cancer Res 66, 6473-6476.

156
Chen, Z., Trotman, L. C., Shaffer, D., Lin, H. K., Dotan, Z. A., Niki, M., Koutcher, J. A., Scher, H. I., Ludwig, T., Gerald, W., et al. (2005). Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 436, 725-730. Cheng, C., Shtessel, L., Brady, M. M., and Ahmed, S. (2012). Caenorhabditis elegans POT-2 telomere protein represses a mode of alternative lengthening of telomeres with normal telomere lengths. Proc Natl Acad Sci USA 109, 7805-7810. Cheung, N. K., Heller, G., Kushner, B. H., Burch, L., and O’Reilly, R. J. (1991). Stage IV neuroblastoma more than 1 year of age at diagnosis: major response to chemotherapy and survival durations correlated strongly with dose intensity. Prog Clin Biol Res 366, 567-573. Cheung, N. K., Zhang, J., Lu, C., Parker, M., Bahrami, A., Tickoo, S. K., Heguy, A., Pappo, A. S., Federico, S., Dalton, J., et al. (2012). Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 307, 1062-1071. Cheutin, T., McNairn, A. J., Jenuwein, T., Gilbert, D. M., Singh, P. B., and Misteli, T. (2003). Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science 299, 721-725. Cho, N. W., Dilley, R. L., Lampson, M. A., and Greenberg, R. A. (2014). Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell 159, 108-121. Choi, L. M., Kim, N. W., Zuo, J. J., Gerbing, R., Stram, D., Lukens, J. N., Matthay, K. K., Seeger, R. C., and Reynolds, C. P. (2000). Telomerase activity by TRAP assay and telomerase RNA (hTR) expression are predictive of outcome in neuroblastoma. Med Pediatr Oncol 35, 647-650. Chong, L., van Steensel, B., Broccoli, D., Erdjument-Bromage, H., Hanish, J., Tempst, P., and de Lange, T. (1995). A human telomeric protein. Science 270, 1663-1667. Chow, T. T., Zhao, Y., Mak, S. S., Shay, J. W., and Wright, W. E. (2012). Early and late steps in telomere overhang processing in normal human cells: the position of the final RNA primer drives telomere shortening. Genes Dev 26, 1167-1178. Christiansen, M., Stevnsner, T., Bohr, V. A., Clark, B. F., and Rattan, S. I. (2000). Gene-specific DNA repair of pyrimidine dimers does not decline during cellular aging in vitro. Exp Cell Res 256, 308-314. Chung, Y. L., and Tsai, T. Y. (2009). Promyelocytic leukemia nuclear bodies link the DNA damage repair pathway with hepatitis B virus replication: implications for hepatitis B virus exacerbation during chemotherapy and radiotherapy. Mol Cancer Res 7, 1672-1685. Cibulskis, K., Lawrence, M. S., Carter, S. L., Sivachenko, A., Jaffe, D., Sougnez, C., Gabriel, S., Meyerson, M., Lander, E. S., and Getz, G. (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotech 31, 213-219. Ciccarone, V. Spengler, B. A., Meyers, M. B., Biedler, J. L., Ross, R. A. (1989). Phenotypic diversification in human neuroblastoma cells: expression of distinct neural crest lineages. Cancer Res 49, 219-225. Clynes, D., Jelinska, C., Xella, B., Ayyub, H., Scott, C., Mitson, M., Taylor, S., Higgs, D. R., and Gibbons, R. J. (2015). Suppression of the alternative lengthening of telomere pathway by the chromatin remodelling factor ATRX. Nat Commun 6, 7538-49.

157
Cohen, S. B., Graham, M. E., Lovrecz, G. O., Bache, N., Robinson, P. J., and Reddel, R. R. (2007). Protein composition of catalytically active human telomerase from immortal cells. Science 315, 1850-1853. Cohen, S. B., and Reddel, R. R. (2008). A sensitive direct human telomerase activity assay. Nat Methods 5, 355-360. Cohn, S. L., Pearson, A. D., London, W. B., Monclair, T., Ambros, P. F., Brodeur, G. M., Faldum, A., Hero, B., Iehara, T., Machin, D., et al. (2009). The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J Clin Oncol 27, 289-297. Coleman, J., Baird, D. M., and Royle, N. J. (1999). The plasticity of human telomeres demonstrated by a hypervariable telomere repeat array that is located on some copies of 16p and 16q. Hum Mol Genet 8, 1637-1646. Compton, S. A., Choi, J. H., Cesare, A. J., Ozgur, S., and Griffith, J. D. (2007). Xrcc3 and Nbs1 are required for the production of extrachromosomal telomeric circles in human alternative lengthening of telomere cells. Cancer Res 67, 1513-1519. Cong, Y. S., Wen, J., and Bacchetti, S. (1999). The human telomerase catalytic subunit hTERT: organization of the gene and characterization of the promoter. Hum Mol Genet 8, 137-142. Cong, Y. S., Wright, W. E., and Shay, J. W. (2002). Human telomerase and its regulation. Microbiol Mol Biol Rev 66, 407-425.
Conomos, D., Reddel, R. R., and Pickett, H. A. (2014). NuRD-ZNF827 recruitment to telomeres creates a molecular scaffold for homologous recombination. Nat Struct Mol Biol 21, 760-770.
Conomos, D., Stutz, M. D., Hills, M., Neumann, A. A., Bryan, T. M., Reddel, R. R., and Pickett, H. A. (2012). Variant repeats are interspersed throughout the telomeres and recruit nuclear receptors in ALT cells. J Cell Biol 199, 893-906. Corvi, R., Amler, L. C., Savelyeva, L., Gehring, M., and Schwab, M. (1994). MYCN is retained in single copy at chromosome 2 band p23-24 during amplification in human neuroblastoma cells. Proc Nat Acad Sci USA 91, 5523-5527. Costa, A., Daidone, M. G., Daprai, L., Villa, R., Cantu, S., Pilotti, S., Mariani, L., Gronchi, A., Henson, J. D., Reddel, R. R., and Zaffaroni, N. (2006). Telomere maintenance mechanisms in liposarcomas: association with histologic subtypes and disease progression. Cancer Res 66, 8918-8924. Cote, J., Quinn, J., Workman, J. L., and Peterson, C. L. (1994). Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI.SNF complex. Science 265, 53-60. Counter, C. M., Avilion, A. A., LeFeuvre, C. E., Stewart, N. G., Greider, C. W., Harley, C. B., and Bacchetti, S. (1992). Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J 11, 1921-1929. Counter, C. M., Gupta, J., Harley, C. B., Leber, B., and Bacchetti, S. (1995). Telomerase activity in normal leukocytes and in hematologic malignancies. Blood 85, 2315-2320.

158
Counter, C. M., Hahn, W. C., Wei, W., Dickinson Caddle, S., Beijersbergen, R. L., Lansdorp, P. M., Sedivy, J. M., and Weinberg, R. A. (1998a). Dissociation among in vitro telomerase activity, telomere maintenance, and cellular immortalization. Proc Natl Acad Sci USA 95, 14723-14728. Counter, C. M., Meyerson, M., Eaton, E. N., Ellisen, L. W., Caddle, S. D., Haber, D. A., and Weinberg, R. A. (1998b). Telomerase activity is restored in human cells by ectopic expression of hTERT (hEST2), the catalytic subunit of telomerase. Oncogene 16, 1217-1222. Counter, C. M., Meyerson, M., Eaton, E. N., and Weinberg, R. A. (1997). The catalytic subunit of yeast telomerase. Proc Natl Acad Sci USA 94, 9202-9207. Cox, K. E., Marechal, A., and Flynn, R. L. (2016). SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep 14, 1032-1040. Crabbe, L., Verdun, R. E., Haggblom, C. I., and Karlseder, J. (2004). Defective telomere lagging strand synthesis in cells lacking WRN helicase activity. Science 306, 1951-1953. Croteau, D. L., Popuri, V., Opresko, P. L., and Bohr, V. A. (2014). Human RecQ helicases in DNA repair, recombination, and replication. Annu Rev Biochem 83, 519-552. D’Adda di Fagagna, F., Reaper, P. M., Clay-Farrace, L., Fiegler, H., Carr, P., von Zglinicki, T., Saretzki, G., Carter, N. P., and Jackson, S. P. (2003). A DNA damage checkpoint response in telomere-initiated senescence. Nature 426, 194-198. D’Angio, G., Evans, A., and Koop, C. E. (1971). Special pattern of widespread neuroblastoma with a favourable prognosis. Lancet 297, 1046-1049. De La Fuente, R., Viveiros, M. M., Wigglesworth, K., and Eppig, J. J. (2004). ATRX, a member of the SNF2 family of helicase/ATPases, is required for chromosome alignment and meiotic spindle organization in metaphase II stage mouse oocytes. Dev Biol 272, 1-14. De Lange, T. (2004). T-loops and the origin of telomeres. Nat Rev Mol Cell Biol 5, 323-329. Deeg, K. I., Chung, I., Bauer, C., and Rippe, K. (2016). Cancer cells with alternative lengthening of telomeres do not display a general hypersensitivity to ATR inhibition. Front Oncol 6, doi: 10.3389/fonc.2016.00186. Dejardin, J., and Kingston, R. E. (2009). Purification of proteins associated with specific genomic loci. Cell 136, 175-186. Dellaire, G., and Bazett-Jones, D. P. (2004). PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bio Essays 26, 963-977. Dellaire, G., Ching, R. W., Ahmed, K., Jalali, F., Tse, K. C., Bristow, R. G., and Bazett-Jones, D. P. (2006). Promyelocytic leukemia nuclear bodies behave as DNA damage sensors whose response to DNA double-strand breaks is regulated by NBS1 and the kinases ATM, Chk2, and ATR. J Cell Biol 175, 55-66. Denchi, E. L., and de Lange, T. (2007). Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 448, 1068-1071.

159
Deng, Z., Glousker, G., Molczan, A., Fox, A. J., Lamm, N., Dheekollu, J., Weizman, O. E., Schertzer, M., Wang, Z., Vladimirova, O., et al. (2013). Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal-Hreidarsson syndrome. Proc Natl Acad Sci USA 110, E3408-E3416. Deng, Z., Norseen, J., Wiedmer, A., Riethman, H., and Lieberman, P. M. (2009). TERRA RNA binding to TRF2 facilitates heterochromatin formation and ORC recruitment at telomeres. Mol Cell 35, 403-413. Deng, Z., Wang, Z., Stong, N., Plasschaert, R., Moczan, A., Chen, H. S., Hu, S., Wikramasinghe, P., Davuluri, R. V., Bartolomei, M. S., et al. (2012). A role for CTCF and cohesin in subtelomere chromatin organization, TERRA transcription, and telomere end protection. EMBO J 31, 4165-4178. Di Micco, R., Fumagalli, M., Cicalese, A., Piccinin, S., Gasparini, P., Luise, C., Schurra, C., Garre’, M., Nuciforo, P. G., Bensimon, A., et al. (2006). Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638-642. Dilley, R. L. and Greenberg, R. A. (2015). Alternative telomere maintenance and cancer. Trends Cancer 1, 145-156. Dilley, R. L., Verma, P., Cho, N. W., Winters, H. D., Wondisford, A. R., and Greenberg, R. A. (2016). Break-induced telomere synthesis underlies alternative telomere maintenance. Nature 539, 54-58. Dimitrova, N., Chen, Y. C., Spector, D. L., and de Lange, T. (2008). 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature 456, 524-528. Dimri, G. P., Itahana, K., Acosta, M., and Campisi, J. (2000). Regulation of a senescence checkpoint response by the E2F1 transcription factor and p14ARF tumor suppressor. Mol Cell Biol 20, 273-285. DiPersio, J. F., Collins, R. H., Blum, W., Devetten, M. P., Stiff, P., Elias, L., Reddy, A., Smith, J. A., and Khoury, H. J. (2009). Immune responses in AML patients following vaccination with GRNVAC1, autologous RNA transfected dendritic cells expressing telomerase catalytic subunit hTERT. Blood 114, 262-262. Diskin, S. J., Capasso, M., Schnepp, R. W., Cole, K. A., Attiyeh, E. F., Hou, C., Diamond, M., Carpenter, E. L., Winter, C., Lee, H., et al. (2012). Common variation at 6q16 within HACE1 and LIN28B influences susceptibility to neuroblastoma. Nat Genet 44, 1126-1130. Diskin, S. J., Hou, C., Glessner, J. T., Attiyeh, E. F., Laudenslager, M., Bosse, K., Cole, K., Mosse, Y. P., Wood, A., Lynch, J. E., et al. (2009). Copy number variation at 1q21.1 associated with neuroblastoma. Nature 459, 987-991. Doksani, Y., and de Lange, T. (2016). Telomere-internal double-strand breaks are repaired by homologous recombination and PARP1/Lig3-dependent end-joining. Cell Rep 17, 1646-1656. Doksani, Y., Wu, J. Y., de Lange, T., and Zhuang, X. (2013). Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 155, 345-356. Drachtman, R. A., and Alter, B. P. (1992). Dyskeritosis-congenita- clinical and genetic-heterogeneity- report of a new case and review of the literature. Am J Pediatr Hematol Oncol 14, 297-304.

160
Drane, P., Ouararhni, K., Depaux, A., Shuaib, M., and Hamiche, A. (2010). The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev 24, 1253-1265. Drosopoulos, W. C., Kosiyatrakul, S. T., and Schildkraut, C. L. (2015). BLM helicase facilitates telomere replication during leading strand synthesis of telomeres. J Cell Biol 210, 191-208. Drosopoulos, W. C., Kosiyatrakul, S. T., Yan, Z., Calderano, S. G., and Schildkraut, C. L. (2012). Human telomeres replicate using chromosome-specific, rather than universal, replication programs. J Cell Biol 197, 253-266. Du, L., Liu, L., Zhang, C., Cai, W., Wu, Y., Wang, J., and Lv, F. (2014). Role of surgery in the treatment of patients with high-risk neuroblastoma who have a poor response to induction chemotherapy. J Pediatr Surg 49, 528-533. Duncan, E. L., Whitaker, N. J., Moy, E. L., and Reddel, R. R. (1993). Assignment of SV40-immortalized cells to more than one complementation group for immortalization. Exp Cell Res 205, 337-344. Dunham, M. A., Neumann, A. A., Fasching, C. L., and Reddel, R. R. (2000). Telomere maintenance by recombination in human cells. Nat Genet 26, 447-450. Eleveld, T. F., Oldridge, D. A., Bernard, V., Koster, J., Daage, L. C., Diskin, S. J., Schild, L., Bentahar, N. B., Bellini, A., Chicard, M., et al. (2015). Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet 47, 864-871. Eid, R., Demattei, M. V., Episkopou, H., Auge-Gouillou, C., Decottignies, A., Grandin, N., and Charbonneau, M. (2015). Genetic inactivation of ATRX leads to a decrease in the amount of telomeric cohesin and of telomere transcription in human glioma cells. Mol Cell Biol 35, 2818-2830. Ellis, N. A., Groden, J., Ye, T. Z., Straughen, J., Lennon, D. J., Ciocci, S., Proytcheva, M., and German, J. (1995). The blooms-syndrome gene-product is homologous to REQ helicases. Cell 83, 655-666. Elsasser, S. J., Noh, K. M., Diaz, N., Allis, C. D., and Banaszynski, L. A. (2015). Histone H3.3 is required for endogenous retroviral element silencing in embryonic stem cells. Nature 522, 240-244. Else, T., Giordano, T. J., and Hammer, G. D. (2008). Evaluation of telomere length maintenance mechanisms in adrenocortical carcinoma. J Clin Endocrinol Metab 93, 1442-1449. Episkopou, H., Draskovic, I., van Beneden, A., Tilman, G., Mattiussi, M., Gobin, M., Arnoult, N., Londono-Vallejo, A., and Decottignies, A. (2014). Alternative Lengthening of Telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res 42, 4391-4405. Errington, T. M., Fu, D., Wong, J. M. Y., and Collins, K. (2008). Disease-associated human telomerase RNA variants show loss of function for telomere synthesis without dominant-negative interference. Mol Cell Biol 28, 6510-6520. Fan, Q., Zhang, F., Barrett, B., Ren, K., and Andreassen, P. R. (2009). A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res 37, 1740-1754.

161
Farooqi, A. S., Dagg, R. A., Choi, L. M., Shay, J. W., Reynolds, C. P., and Lau, L. M. (2014). Alternative lengthening of telomeres in neuroblastoma cell lines is associated with a lack of MYCN genomic amplification and with p53 pathway aberrations. J Neurooncol 119, 17-26. Fasching, C. L., Bower, K., and Reddel, R. R. (2005). Telomerase-independent telomere length maintenance in the absence of alternative lengthening of telomeres-associated promyelocytic leukemia bodies. Cancer Res 65, 2722-2729. Fasching, C. L., Neumann, A. A., Muntoni, A., Yeager, T. R., and Reddel, R. R. (2007). DNA damage induces alternative lengthening of telomeres (ALT)-associated promyelocytic leukemia bodies that preferentially associate with linear telomeric DNA. Cancer Res 67, 7072-7077. Felsenfeld, G., and Groudine, M. (2003). Controlling the double helix. Nature 421, 448-453. Feng, J., Funk, W. D., Wang, S. S., Weinrich, S. L., Avilion, A. A., Chiu, C. P., Adams, R. R., Chang, E., Allsopp, R. C., Yu, J. H., et al. (1995). The RNA component of human telomerase. Science 269, 1236-1241. Ferbeyre, G., De Stanchina, E., Querido, E., Baptiste, N., Prives, C., and Lowe, S. W. (2000). PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 14, 2015-2027. Flynn, R. L., Centore, R. C., O’Sullivan, R. J., Rai, R., Tse, A., Songyang, Z., Chang, S., Karlseder, J., and Zou, L. (2011). TERRA and hnRNPA1 orchestrate an RPA-to-POT1 switch on telomeric single-stranded DNA. Nature 471, 532-536. Flynn, R. L., Cox, K. E., Jeitany, M., Wakimoto, H., Bryll, A. R., Ganem, N. J., Bersani, F., Pineda, J. R., Suva, M. L., Benes, C. H., et al. (2015). Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 347, 273-277. Fredriksson, N. J., Ny, L., Nilsson, J. A., and Larsson, E. (2014). Systematic analysis of noncoding somatic mutations and gene expression alterations across 14 tumor types. Nat Genet 46, 1258-1263. Frescas, D., and de Lange, T. (2014a). Binding of TPP1 to TIN2 is required for POT1a,b-mediated telomere protection. J Biol Chem 289, 24180-24187. Frescas, D., and de Lange, T. (2014b). TRF2-tethered TIN2 can mediate telomere protection by TPP1/POT1. Mol Cell Biol 34, 1349-1362. Fu, X. H., Duan, Y. M., Liu, Y. T., Cai, C., Meng, F. L., and Zhou, J. Q. (2014). Telomere recombination preferentially occurs at short telomeres in telomerase-null type II survivors. PloS ONE 9, e90644. Ganot, P., CaizerguesFerrer, M., and Kiss, T. (1997). The family of box ACA small nucleolar RNAs is defined by an evolutionarily conserved secondary structure and ubiquitous sequence elements essential for RNA accumulation. Genes Dev 11, 941-956. Garaventa, A., Luksch, R., Biasotti, S., Severi, G., Pizzitola, M. R., Viscardi, E., Prete, A., Mastrangelo, S., Podda, M., Haupt, R., and De Bernardi, B. (2003). A phase II study of topotecan with vincristine and doxorubicin in children with recurrent/refractory neuroblastoma. Cancer 98, 2488-2494. Garcia, I., Mayol, G., Rodríguez, E., Suñol, M., Gershon, T. R., Ríos, J., Cheung, N. K. V., Kieran, M. W., George, R. E., Perez-Atayde, A. R., et al. (2010). Expression of the neuron-specific protein CHD5 is an independent marker of outcome in neuroblastoma. Mol Cancer, 9, 277-291.

162
Garcia-Cao, M., O’Sullivan, R., Peters, A., Jenuwein, T., and Blasco, M. A. (2004). Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nature Genetics 36, 94-99. Gellert, M., Lipsett, M. N., and Davies, D. R. (1962). Helix formation by guanylic acid. Proc Natl Acad Sci USA 48, 2013-2018. George, R. E., London, W. B., Cohn, S. L., Maris, J. M., Kretschmar, C., Diller, L., Brodeur, G. M., Castleberry, R. P., and Look, A. T. (2005). Hyperdiploidy plus nonamplified MYCN confers a favorable prognosis in children 12 to 18 months old with disseminated neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 23, 6466-6473. George, R. E., Sanda, T., Hanna, M., Frohling, S., Luther, W., Zhang, J., Ahn, Y., Zhou, W., London, W. B., McGrady, P., et al. (2008). Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 455, 975-978. Gibbons, R. J., McDowell, T. L., Raman, S., O’Rourke, D. M., Garrick, D., Ayyub, H., and Higgs, D. R. (2000). Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nature Genetics 24, 368-371. Gibbons, R. J., Picketts, D. J., Villard, L., and Higgs, D. R. (1995). Mutations in a putative global transcriptional regulator cause X-linked mental-retardation with alpha-thalassemia (ATR-X syndrome). Cell 80, 837-845. Gilbert, D. E., and Feigon, J. (1999). Multistranded DNA structures. Curr Opin Struct Biol 9, 305-314. Gilson, E., and Londono-Vallejo, A. (2007). Telomere length profiles in humans: all ends are not equal. Cell Cycle 6, 2486-2494. Girardi, A. J., Jensen, F. C., and Koprowsk.H (1965). SV40-induced transformation of human diploid cells- crisis and recovery. J Cell Comp Physiol 65, 69-84. Gire, V., and Wynford-Thomas, D. (1998). Reinitiation of DNA synthesis and cell division in senescent human fibroblasts by microinjection of anti-p53 antibodies. Mol Cell Biol 18, 1611-1621. Goldberg, A. D., Banaszynski, L. A., Noh, K. M., Lewis, P. W., Elsaesser, S. J., Stadler, S., Dewell, S., Law, M., Guo, X. Y., Li, X., et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678-691. Gomez, S., Castellano, G., Mayol, G., Sunol, M., Queiros, A., Bibikova, M., Nazor, K. L., Loring, J. F., Lemos, I., Rodriguez, E., et al. (2015). DNA methylation fingerprint of neuroblastoma reveals new biological and clinical insights. Epigenomics 7, 1137-1153. Gong, Y., and de Lange, T. (2010). A Shld1-controlled POT1a provides support for repression of ATR signaling at telomeres through RPA exclusion. Mol Cell 40, 377-387. Gonzalo, S., Jaco, I., Fraga, M. F., Chen, T. P., Li, E., Esteller, M., and Blasco, M. A. (2006). DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol 8, 416-U466.

163
Gottschling, D. E., Aparicio, O. M., Billington, B. L., and Zakian, V. A. (1990). Position effect at S. cerevisiae telomeres: reversible repression of Pol II transcription. Cell 63, 751-762. Greaves, M. (1996). Is telomerase activity in cancer due to selection of stem cells and differentiation arrest? Trends Genet 12, 127-128. Greider, C. W., and Blackburn, E. H. (1985). Identification of a specific telomere terminal transferase-activity in tetrahymena extracts. Cell 43, 405-413. Greider, C. W., and Blackburn, E. H. (1989). A telomeric sequence in the RNA of Tetrahymena telomerase required for telomere repeat synthesis. Nature 337, 331-337. Griffith, J. D., Comeau, L., Rosenfield, S., Stansel, R. M., Bianchi, A., Moss, H., and de Lange, T. (1999). Mammalian telomeres end in a large duplex loop. Cell 97, 503-514. Grobelny, J. V., Godwin, A. K., and Broccoli, D. (2000). ALT-associated PML bodies are present in viable cells and are enriched in cells in the G(2)/M phase of the cell cycle. J Cell Sci 113, 4577-4585. Grobelny, J. V., Kulp-McEliece, M., and Broccoli, D. (2001). Effects of reconstitution of telomerase activity on telomere maintenance by the alternative lengthening of telomeres (ALT) pathway. Hum Mol Gen 10, 1953-1961. Gu, P., Min, J. N., Wang, Y., Huang, C., Peng, T., Chai, W., and Chang, S. (2012). CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J 31, 2309-2321. Guilleret, I., Yan, P., Guillou, L., Braunschweig, R., Coindre, J. M., and Benhattar, J. (2002). The human telomerase RNA gene (hTERC) is regulated during carcinogenesis but is not dependent on DNA methylation. Carcinogenesis 23, 2025-2030. Guo, C., White, P. S., Weiss, M. J., Hogarty, M. D., Thompson, P. M., Stram, D. O., Gerbing, R., Matthay, K. K., Seeger, R. C., Brodeur, G. M., and Maris, J. M. (1999). Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene 18, 4948-4957. Guo, T., Kouvonen, P., Koh, C. C., Gillet, L. C., Wolski, W. E., Rost, H. L., Rosenberger, G., Collins, B. C., Blum, L. C., Gillessen, S., et al. (2015). Rapid mass spectrometric conversion of tissue biopsy samples into permanent quantitative digital proteome maps. Nat Med 21, 407-413. Guo, Y., Kartawinata, M., Li, J., Pickett, H. A., Teo, J., Kilo, T., Barbaro, P. M., Keating, B., Chen, Y., Tian, L., et al. (2014). Inherited bone marrow failure associated with germline mutation of ACD, the gene encoding telomere protein TPP1. Blood 124, 2767-2774. Gupta, J., Han, L. P., Wang, P., Gallie, B. L., and Bacchetti, S. (1996). Development of retinoblastoma in the absence of telomerase activity. J Natl Cancer Inst 88, 1152-1157. Hahn, W. C., Counter, C. M., Lundberg, A. S., Beijersbergen, R. L., Brooks, M. W., and Weinberg, R. A. (1999). Creation of human tumour cells with defined genetic elements. Nature 400, 464-468. Hakin-Smith, V., Jellinek, D. A., Levy, D., Carroll, T., Teo, M., Timperley, W. R., McKay, M. J., Reddel, R. R., and Royds, J. A. (2003). Alternative lengthening of telomeres and survival in patients with glioblastoma multiforme. Lancet 361, 836-838.

164
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100, 57-70. Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646-674. Hansen, R. S., Wijmenga, C., Luo, P., Stanek, A. M., Canfield, T. K., Weemaes, C. M., and Gartler, S. M. (1999). The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Nat Acad Sci USA 96, 14412-14417. HarleBachor, C., and Boukamp, P. (1996). Telomerase activity in the regenerative basal layer of the epidermis in human skin and in immortal and carcinoma-derived skin keratinocytes. Proc Nat Acad Sci USA 93, 6476-6481. Harley, C. B., Futcher, A. B., and Greider, C. W. (1990). Telomeres shorten during ageing of human fibroblasts. Nature 345, 458-460. Hastie, N. D., Dempster, M., Dunlop, M. G., Thompson, A. M., Green, D. K., and Allshire, R. C. (1990). Telomere reduction in human colorectal carcinoma and with ageing. Nature 346, 866-868. Hayflick, L. (1965). The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37, 614-636. Hayflick, L., and Moorhead, P. S. (1961). The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585-621. He, D. L., Mu, Z. M., Le, X. F., Hsieh, J. T., Pong, R. C., Chung, L. W., and Chang, K. S. (1997). Adenovirus-mediated expression of PML suppresses growth and tumorigenicity of prostate cancer cells. Cancer Res 57, 1868-1872. Heaphy, C. M., de Wilde, R. F., Jiao, Y., Klein, A. P., Edil, B. H., Shi, C., Bettegowda, C., Rodriguez, F. J., Eberhart, C. G., Hebbar, S., et al. (2011a). Altered telomeres in tumors with ATRX and DAXX mutations. Science 333, 425. Heaphy, C. M., Subhawong, A. P., Hong, S. M., Goggins, M. G., Montgomery, E. A., Gabrielson, E., Netto, G. J., Epstein, J. I., Lotan, T. L., Westra, W. H., et al. (2011b). Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am J Path 179, 1608-1615. Heiss, N. S., Knight, S. W., Vulliamy, T. J., Klauck, S. M., Wiemann, S., Mason, P. J., Poustka, A., and Dokal, I. (1998). X-linked dyskeratosis congenita is caused by mutations in a highly conserved gene with putative nucleolar functions. Nat Genet 19, 32-38. Hemann, M. T., Strong, M. A., Hao, L. Y., and Greider, C. W. (2001). The shortest telomere, not average telomere length, is critical for cell viability and chromosome stability. Cell 107, 67-77. Henras, A., Henry, Y., Bousquet-Antonelli, C., Noaillac-Depeyre, J., Gelugne, J. P., and Caizergues-Ferrer, M. (1998). Nhp2p and Nop10p are essential for the function of H/ACA snoRNPs. EMBO J 17, 7078-7090. Henson, J. D., Cao, Y., Huschtscha, L. I., Chang, A. C., Au, A. Y., Pickett, H. A., and Reddel, R. R. (2009). DNA C-circles are specific and quantifiable markers of alternative-lengthening-of-telomeres activity. Nat Biotechnol 27, 1181-1185.

165
Henson, J. D., Hannay, J. A., McCarthy, S. W., Royds, J. A., Yeager, T. R., Robinson, R. A., Wharton, S. B., Jellinek, D. A., Arbuckle, S. M., Yoo, J. Y., et al. (2005). A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin Cancer Res 11, 217-225. Henson, J. D., Lau, L. M., Koch, S., Martin La Rotta, N., Dagg, R. A., and Reddel, R. R. (2017). The C-Circle Assay for alternative-lengthening-of-telomeres activity. Methods 114, 74-84. Henson, J. D., Neumann, A. A., Yeager, T. R., and Reddel, R. R. (2002). Alternative lengthening of telomeres in mammalian cells. Oncogene 21, 598-610. Henson, J. D., and Reddel, R. R. (2010). Assaying and investigating Alternative Lengthening of Telomeres activity in human cells and cancers. FEBS Lett 584, 3800-3811. Hiyama, E., Hiyama, K., Ohtsu, K., Yamaoka, H., Ichikawa, T., Shay, J. W., and Yokoyama, T. (1997). Telomerase activity in neuroblastoma: is it a prognostic indicator of clinical behaviour? Eur J Cancer 33, 1932-1936. Hiyama, E., Hiyama, K., Yokoyama, T., Matsuura, Y., Piatyszek, M. A., and Shay, J. W. (1995a). Correlating telomerase activity levels with human neuroblastoma outcomes. Nat Med 1, 249-255. Hiyama, E., Tatsumoto, N., Kodama, T., Hiyama, K., Shay, J. W., and Yokoyama, T. (1996). Telomerase activity in human intestine. Int J Oncol 9, 453-458. Hiyama, K., Hirai, Y., Kyoizumi, S., Akiyama, M., Hiyama, E., Piatyszek, M. A., Shay, J. W., Ishioka, S., and Yamakido, M. (1995b). Activation of telomerase in human-lymphocytes and hematopoietic progenitor cells. J Immunol 155, 3711-3715. Ho, C. Y., Murnane, J. P., Yeung, A. K., Ng, H. K., and Lo, A. W. (2008). Telomeres acquire distinct heterochromatin characteristics during siRNA-induced RNA interference in mouse cells. Curr Biol 18, 183-187. Horikawa, I., Cable, P. L., Afshari, C., and Barrett, J. C. (1999a). Cloning and characterization of the promoter region of human telomerase reverse transcriptase gene. Cancer Res 59, 826-830. Horn, S., Figl, A., Rachakonda, P. S., Fischer, C., Sucker, A., Gast, A., Kadel, S., Moll, I., Nagore, E., Hemminki, K., et al. (2013). TERT promoter mutations in familial and sporadic melanoma. Science 339, 959-961. Hossain, S., Singh, S., and Lue, N. F. (2002). Functional analysis of the C-terminal extension of telomerase reverse transcriptase — A putative “Thumb” domain. J Biol Chem 277, 36174-36180. Houghtaling, B. R., Cuttonaro, L., Chang, W., and Smith, S. (2004). A dynamic molecular link between the telomere length regulator TRF1 and the chromosome end protector TRF2. Curr Biol 14, 1621-1631. Houghton, P. J., Lock, R., Carol, H., Morton, C. L., Gorlick, R., Anders Kolb, E., Keir, S. T., Reynolds, C. P., Kang, M. H., Maris, J. M., et al. (2012). Testing of the topoisomerase 1 inhibitor Genz-644282 by the pediatric preclinical testing program. Pediatr Blood Cancer 58, 200-209. Hsiang, Y. H., Hertzberg, R., Hecht, S., and Liu, L. F. (1985). Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260, 14873-14878.

166
Hsiang, Y. H., Lihou, M. G., and Liu, L. F. (1989). Arrest of replication forks by drug-stabilized topoisomerase I-DNA cleavable complexes as a mechanism of cell killing by camptothecin. Cancer Res 49, 5077-5082. Hu, J., Hwang, S. S., Liesa, M., Gan, B., Sahin, E., Jaskelioff, M., Ding, Z., Ying, H., Boutin, A. T., Zhang, H., et al. (2012). Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell 148, 651-663. Huang, C., Dai, X., and Chai, W. (2012). Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res 22, 1681-1695. Huang, F. W., Hodis, E., Xu, M. J., Kryukov, G. V., Chin, L., and Garraway, L. A. (2013). Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957-959. Huschtscha, L. I., and Holliday, R. (1983). Limited and unlimited growth of SV40-transformed cells from human diploid MRC-5 fibroblasts. J Cell Sci 63, 77-99. Huschtscha, L. I., Noble, J. R., Neumann, A. A., Moy, E. L., Barry, P., Melki, J. R., Clark, S. J., and Reddel, R. R. (1998). Loss of p16INK4 expression by methylation is associated with lifespan extension of human mammary epithelial cells. Cancer Res 58, 3508-3512. Ichikawa, Y., Morohashi, N., Nishimura, Y., Kurumizaka, H., and Shimizu, M. (2014). Telomeric repeats act as nucleosome-disfavouring sequences in vivo. Nucleic Acids Res 42, 1541-1552. Ip, S. C., Rass, U., Blanco, M. G., Flynn, H. R., Skehel, J. M., and West, S. C. (2008). Identification of Holliday junction resolvases from humans and yeast. Nature 456, 357-361. Ishii, Y., Tsuyama, N., Maeda, S., Tahara, H., and Ide, T. (1999). Telomerase activity in hybrids between telomerase-negative and telomerase-positive immortal human cells is repressed in the different complementation groups but not in the same complementation group of immortality. Mech Ageing Dev 110, 175-193. Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461, 1071-1078. Jady, B. E., Darzacq, X., Tucker, K. E., Matera, A. G., Bertrand, E., and Kiss, T. (2003). Modification of Sm small nuclear RNAs occurs in the nucleoplasmic Cajal body following import from the cytoplasm. EMBO J 22, 1878-1888. Jafri, M. A., Ansari, S. A., Alqahtani, M. H., and Shay, J. W. (2016). Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med 8, doi: 10.1186/s13073-016-0324-x. Janoueix-Lerosey, I., Lequin, D., Brugieres, L., Ribeiro, A., de Pontual, L., Combaret, V., Raynal, V., Puisieux, A., Schleiermacher, G., Pierron, G., et al. (2008). Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 455, 967-970. Jeyapalan, J. N., Mendez-Bermudez, A., Zaffaroni, N., Dubrova, Y. E., and Royle, N. J. (2008). Evidence for alternative lengthening of telomeres in liposarcomas in the absence of ALT-associated PML bodies. Int J Cancer 122, 2414-2421.

167
Jeyapalan, J. N., Varley, H., Foxon, J. L., Pollock, R. E., Jeffreys, A. J., Henson, J. D., Reddel, R. R., and Royle, N. J. (2005). Activation of the ALT pathway for telomere maintenance can affect other sequences in the human genome. Hum Mol Gen 14, 1785-1794. Jiang, W. Q., and Ringertz, N. (1997). Altered distribution of the promyelocytic leukemia-associated protein is associated with cellular senescence. Cell Growth Differ 8, 513-522. Jiang, W. Q., Zhong, Z. H., Henson, J. D., Neumann, A. A., Chang, A. C., and Reddel, R. R. (2005). Suppression of alternative lengthening of telomeres by Sp100-mediated sequestration of MRE11/RAD50/NBS1 complex. Mol Cell Biol 25, 2708-2721. Jiang, W. Q., Zhong, Z. H., Henson, J. D., and Reddel, R. R. (2007). Identification of candidate alternative lengthening of telomeres genes by methionine restriction and RNA interference. Oncogene 26, 4635-4647. Jiang, W. Q., Zhong, Z. H., Nguyen, A., Henson, J. D., Toouli, C. D., Braithwaite, A. W., and Reddel, R. R. (2009). Induction of alternative lengthening of telomeres-associated PML bodies by p53/p21 requires HP1 proteins. J Cell Biol 185, 797-810. Jiao, Y., Killela, P. J., Reitman, Z. J., Rasheed, A. B., Heaphy, C. M., de Wilde, R. F., Rodriguez, F. J., Rosemberg, S., Oba-Shinjo, S. M., Marie, S. K., et al. (2012). Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3, 709-722. Jiao, Y. C., Shi, C. J., Edil, B. H., de Wilde, R. F., Klimstra, D. S., Maitra, A., Schulick, R. D., Tang, L. H., Wolfgang, C. L., Choti, M. A., et al. (2011). DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199-1203. Johnson, J. E., Varkonyi, R. J., Schwalm, J., Cragle, R., Klein-Szanto, A., Patchefsky, A., Cukierman, E., von Mehren, M., and Broccoli, D. (2005). Multiple mechanisms of telomere maintenance exist in liposarcomas. Clin Cancer Res 11, 5347-5355. Jones, S., Zhang, X., Parsons, D. W., Lin, J. C., Leary, R. J., Angenendt, P., Mankoo, P., Carter, H., Kamiyama, H., Jimeno, A., et al. (2008). Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 321, 1801-1806. Kaneko, Y., Kanda, N., Maseki, N., Sakurai, M., Tsuchida, Y., Takeda, T., Okabe, I., and Sakurai, M. (1987). Different karyotypic patterns in early and advanced stage neuroblastomas. Cancer Res 47, 311-318. Karlseder, J., Hoke, K., Mirzoeva, O. K., Bakkenist, C., Kastan, M. B., Petrini, J. H., and de Lange, T. (2004). The telomeric protein TRF2 binds the ATM kinase and can inhibit the ATM-dependent DNA damage response. PloS Biol 2, E240. Karlseder, J., Smogorzewska, A., and de Lange, T. (2002). Senescence induced by altered telomere state, not telomere loss. Science 295, 2446-2449. Kaul, Z., Cesare, A. J., Huschtscha, L. I., Neumann, A. A., and Reddel, R. R. (2012). Five dysfunctional telomeres predict onset of senescence in human cells. EMBO Rep 13, 52-59. Kawanishi, S., and Oikawa, S. (2004). Mechanism of telomere shortening by oxidative stress. Ann NY Acad Sci 1019, 278-284.

168
Keshelava, N., Seeger, R. C., Groshen, S., and Reynolds, C. P. (1998). Drug resistance patterns of human neuroblastoma cell lines derived from patients at different phases of therapy. Cancer Res 58, 5396-5405. Keshelava, N., Zuo, J. J., Chen, P., Waidyaratne, S. N., Luna, M. C., Gomer, C. J., Triche, T. J., and Reynolds, C. P. (2001). Loss of p53 function confers high-level multidrug resistance in neuroblastoma cell lines. Cancer Res 61, 6185-6193. Khoury, H. J., Collins, R. H., Blum, W., Maness, L., Stiff, P., Kelsey, S. M., Reddy, A., Smith, J. A., and DiPersio, J. F. (2010). Prolonged administration of the telomerase vaccine GRNVAC1 is well tolerated and appears to be associated with, favorable outcomes In high-risk acute myeloid leukemia (AML). Blood 116, 904-904. Kile, A. C., Chavez, D. A., Bacal, J., Eldirany, S., Korzhnev, D. M., Bezsonova, I., Eichman, B. F., and Cimprich, K. A. (2015). HLTF’s Ancient HIRAN Domain Binds 3’ DNA Ends to Drive Replication Fork Reversal. Mol Cell 58, 1090-1100. Killoran, M. P., and Keck, J. L. (2006). Sit down, relax and unwind: structural insights into RecQ helicase mechanisms. Nucleic Acids Res 34, 4098-4105. Kim, N. W., Piatyszek, M. A., Prowse, K. R., Harley, C. B., West, M. D., Ho, P. L., Coviello, G. M., Wright, W. E., Weinrich, S. L., and Shay, J. W. (1994). Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011-2015. Kim, N. W., and Wu, F. (1997). Advances in quantification and characterization of telomerase activity by the telomeric repeat amplification protocol (TRAP). Nucleic Acids Res 25, 2595-2597. Kim, S. H., Beausejour, C., Davalos, A. R., Kaminker, P., Heo, S. J., and Campisi, J. (2004). TIN2 mediates functions of TRF2 at human telomeres. J Biol Chem 279, 43799-43804. Kim, S. H., Kaminker, P., and Campisi, J. (1999). TIN2, a new regulator of telomere length in human cells. Nat Genet 23, 405-412. Kipling, D. (1995). Telomerase: immortality enzyme or oncogene? Nat Genet 9, 104-106. Knight, S. W., Heiss, N. S., Vulliamy, T. J., Aalfs, C. M., McMahon, C., Richmond, P., Jones, A., Hennekam, R. C. M., Poustka, A., Mason, P. J., and Dokal, I. (1999). Unexplained aplastic anaemia, immunodeficiency, and cerebellar hypoplasia (Hoyeraal-Hreidarsson syndrome) due to mutations in the dyskeratosis congenita gene, DKC1. Br J Haematol 107, 335-339. Kocak, H., Ballew, B. J., Bisht, K., Eggebeen, R., Hicks, B. D., Suman, S., O’Neil, A., Giri, N., Maillard, I., Alter, B. P., et al. (2014). Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev 28, 2090-2102. Kornberg, R. D. (1974). Chromatin structure- repeating units of histones and DNA. Science 184, 868-871. Kornberg, R. D., and Thomas, J. O. (1974). Chromatin structure- oligomers of histones. Science 184, 865-868. Krams, M., Claviez, A., Heidorn, K., Krupp, G., Parwaresch, R., Harms, D., and Rudolph, P. (2001). Regulation of telomerase activity by alternate splicing of human telomerase reverse transcriptase mRNA in a subset of neuroblastomas. Am J Pathol 159, 1925-1932.

169
Kreilmeier, T., Mejri, D., Hauck, M., Kleiter, M., and Holzmann, K. (2016). Telomere Transcripts Target Telomerase in Human Cancer Cells. Genes 7, e46. Kumakura, S., Tsutsui, T. W., Yagisawa, J., Barrett, J. C., and Tsutsui, T. (2005). Reversible conversion of immortal human cells from telomerase-positive to telomerase-negative cells. Cancer Res 65, 2778-2786. Kurihara, S., Hiyama, E., Onitake, Y., Yamaoka, E., and Hiyama, K. (2014). Clinical features of ATRX and DAXX mutated neuroblastoma. J Pediatr Surg 49, 1835-1838. Kurth, I., and Gautier, J. (2010). Origin-dependent initiation of DNA replication within telomeric sequences. Nucleic Acids Res 38, 467-476. Kushner, B. H., Gilbert, F., and Helson, L. (1986). Familial neuroblastoma. Case reports, literature review, and etiologic considerations. Cancer 57, 1887-1893. Kushner, B. H., Kramer, K., Modak, S., and Cheung, N. K. (2006). Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. J Clin Oncol 24, 5271-5276. Kuzminov, A. (2001). Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA 98, 8241-8246. Kyte, J. A. (2009). Cancer vaccination with telomerase peptide GV1001. Expert Opin Investig Drugs 18, 687-694. Lackner, D. H., Raices, M., Maruyama, H., Haggblom, C., and Karlseder, J. (2012). Organismal propagation in the absence of a functional telomerase pathway in Caenorhabditis elegans. EMBO J 31, 2024-2033. Lam, Y. C., Akhter, S., Gu, P., Ye, J., Poulet, A., Giraud-Panis, M. J., Bailey, S. M., Gilson, E., Legerski, R. J., and Chang, S. (2010). SNMIB/Apollo protects leading-strand telomeres against NHEJ-mediated repair. EMBO J 29, 2230-2241. Lansdorp, P. M., Poon, S., Chavez, E., Dragowska, V., Zijlmans, M., Bryan, T., Reddel, R., Egholm, M., Bacchetti, S., and Martens, U. (1998). Telomeres in the haemopoietic system. In Telomeres and Telomerase, D.J. Chadwick, and G. Cardew, eds. (Chichester: John Wiley & Sons Ltd), pp. 209-218. Lau, L. M., Dagg, R. A., Henson, J. D., Au, A. Y., Royds, J. A., and Reddel, R. R. (2013). Detection of alternative lengthening of telomeres by telomere quantitative PCR. Nucleic Acids Res 41, e34. Lau, L. M. S., and Irwin, M. S. (2017). Neuroblastoma. eLS, 1-8. Laud, P. R., Multani, A. S., Bailey, S. M., Wu, L., Ma, J., Kingsley, C., Lebel, M., Pathak, S., DePinho, R. A., and Chang, S. (2005). Elevated telomere-telomere recombination in WRN-deficient, telomere dysfunctional cells promotes escape from senescence and engagement of the ALT pathway. Genes Dev 19, 2560-2570. Lee, S. S., Bohrson, C., Pike, A. M., Wheelan, S. J., and Greider, C. W. (2015). ATM kinase is required for telomere elongation in mouse and human cells. Cell Rep 13, 1623-1632.

170
Lee, J. C., Jeng, Y. M., Liau, J. Y., Tsai, J. H., Hsu, H. H., and Yang, C. Y. (2015). Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod Pathol 28, 1064-1073. Lee, J. H., and Paull, T. T. (2007). Activation and regulation of ATM kinase activity in response to DNA double-strand breaks. Oncogene 26, 7741-7748. Lehman, T. A., Modali, R., Boukamp, P., Stanek, J., Bennett, W. P., Welsh, J. A., Metcalf, R. A., Stampfer, M. R., Fusenig, N., Rogan, E. M., and Harris, C. C. (1993). P53 mutations in human immortalized epithelial cell lines. Carcinogenesis 14, 833-839. Lejnine, S., Makarov, V. L., and Langmore, J. P. (1995). Conserved nucleoprotein structure at the ends of vertebrate and invertebrate chromosomes. Proc Natl Acad Sci USA 92, 2393-2397. Lendvay, T. S., Morris, D. K., Sah, J., Balasubramanian, B., and Lundblad, V. (1996). Senescence mutants of Saccharomyces cerevisiae with a defect in telomere replication identify three additional EST genes. Genetics 144, 1399-1412. Levy, M. Z., Allsopp, R. C., Futcher, A. B., Greider, C. W., and Harley, C. B. (1992). Telomere end-replication problem and cell aging. J Mol Biol 225, 951-960. Lewis, P. W., Elsaesser, S. J., Noh, K. M., Stadler, S. C., and Allis, C. D. (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci USA 107, 14075-14080. Li, B., Oestreich, S., and de Lange, T. (2000). Identification of human Rap1: implications for telomere evolution. Cell 101, 471-483. Li, J. S., Miralles Fuste, J., Simavorian, T., Bartocci, C., Tsai, J., Karlseder, J., and Lazzerini Denchi, E. (2017). TZAP: A telomere-associated protein involved in telomere length control. Science 355, 638-641. Li, T. K., Houghton, P. J., Desai, S. D., Daroui, P., Liu, A. A., Hars, E. S., Ruchelman, A. L., LaVoie, E. J., and Liu, L. F. (2003). Characterization of ARC-111 as a novel topoisomerase I-targeting anticancer drug. Cancer Res 63, 8400-8407. Lia, G., Praly, E., Ferreira, H., Stockdale, C., Tse-Dinh, Y. C., Dunlap, D., Croquette, V., Bensimon, D., and Owen-Hughes, T. (2006). Direct observation of DNA distortion by the RSC complex. Mol Cell 21, 417-425. Lin, A. W., Barradas, M., Stone, J. C., van Aelst, L., Serrano, M., and Lowe, S. W. (1998). Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev 12, 3008-3019. Lin, H. K., Chen, Z., Wang, G., Nardella, C., Lee, S. W., Chan, C. H., Yang, W. L., Wang, J., Egia, A., Nakayama, K. I., et al. (2010). Skp2 targeting suppresses tumorigenesis by Arf-p53-independent cellular senescence. Nature 464, 374-379. Lindner, S., Bachmann, H. S., Odersky, A., Schaefers, S., Klein-Hitpass, L., Hero, B., Fischer, M., Eggert, A., Schramm, A., and Schulte, J. H. (2015). Absence of telomerase reverse transcriptase promoter mutations in neuroblastoma. Biomed Rep 3, 443-446.

171
Lindsey, J., McGill, N. I., Lindsey, L. A., Green, D. K., and Cooke, H. J. (1991). In vivo loss of telomeric repeats with age in humans. Mutat Res 256, 45-48. Liu, D., O’Connor, M. S., Qin, J., and Songyang, Z. (2004a). Telosome, a mammalian telomere-associated complex formed by multiple telomeric proteins. J Biol Chem 279, 51338-51342. Liu, D., Safari, A., O’Connor, M. S., Chan, D. W., Laegeler, A., Qin, J., and Songyang, Z. (2004b). PTOP interacts with POT1 and regulates its localization to telomeres. Nat Cell Biol 6, 673-680. Liu, K., Schoonmaker, M. M., Levine, B. L., June, C. H., Hodes, R. J., and Weng, N. P. (1999). Constitutive and regulated expression of telomerase reverse transcriptase (hTERT) in human lymphocytes. FASEB J 13, A619-A619. Liu, L., Bailey, S. M., Okuka, M., Munoz, P., Li, C., Zhou, L. J., Wu, C., Czerwiec, E., Sandler, L., Seyfang, A., et al. (2007). Telomere lengthening early in development. Nat Cell Biol 9, 1436-41. Liu, Y., and West, S. C. (2004). Happy Hollidays: 40th anniversary of the Holliday junction. Nat Rev Mol Cell Biol 5, 937-944. Loayza, D., Parsons, H., Donigian, J., Hoke, K., and de Lange, T. (2004). DNA binding features of human POT1: a nonamer 5’-TAGGGTTAG-3’ minimal binding site, sequence specificity, and internal binding to multimeric sites. J Biol Chem 279, 13241-13248. London, W. B., Castel, V., Monclair, T., Ambros, P. F., Pearson, A. D., Cohn, S. L., Berthold, F., Nakagawara, A., Ladenstein, R. L., Iehara, T., and Matthay, K. K. (2011). Clinical and biologic features predictive of survival after relapse of neuroblastoma: a report from the International Neuroblastoma Risk Group project. J Clin Oncol 29, 3286-3292. London, W. B., Castleberry, R. P., Matthay, K. K., Look, A. T., Seeger, R. C., Shimada, H., Thorner, P., Brodeur, G., Maris, J. M., Reynolds, C. P., and Cohn, S. L. (2005). Evidence for an age cutoff greater than 365 days for neuroblastoma risk group stratification in the Children’s Oncology Group. J Clin Oncol 23, 6459-6465. Londono-Vallejo, J. A., Der-Sarkissian, H., Cazes, L., Bacchetti, S., and Reddel, R. R. (2004). Alternative lengthening of telomeres is characterized by high rates of telomeric exchange. Cancer Res 64, 2324-2327. Look, A. T., Hayes, F. A., Shuster, J. J., Douglass, E. C., Castleberry, R. P., Bowman, L. C., Smith, E. I., and Brodeur, G. M. (1991). Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: a Pediatric Oncology Group study. J Clin Oncol 9, 581-591. Lopez de Silanes, I., Grana, O., Luigia De Bonis, M. L., Dominguez, O., Pisano, D. G., and Blasco, M. A. (2014). Identification of TERRA locus unveils a telomere protection role through association to nearly all chromosomes. Nat Commun 5, 4723. Lopez de Silanes, I., Stagno d’Alcontres, M., and Blasco, M. A. (2010). TERRA transcripts are bound by a complex array of RNA-binding proteins. Nat Commun 1, 33. Lovejoy, C. A., Li, W. D., Reisenweber, S., Thongthip, S., Bruno, J., de Lange, T., De, S., Petrini, J. H. J., Sung, P. A., Jasin, M., et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PloS Genetics 8, 16.

172
Lu, R., Pal, J., Buon, L., Nanjappa, P., Shi, J., Fulciniti, M., Tai, Y.T., Guo, L., Yu, M., Gryaznov, S., et al. (2014). Targeting homologous recombination and telomerase in Barrett’s adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene 33, 1495-1505. Luciani, J. J., Depetris, D., Usson, Y., Metzler-Guillemain, C., Mignon-Ravix, C., Mitchell, M. J., Megarbane, A., Sarda, P., Sirma, H., Moncla, A., et al. (2006). PML nuclear bodies are highly organised DNA-protein structures with a function in heterochromatin remodelling at the G2 phase. J Cell Sci 119, 2518-2531. Lundberg, G., Sehic, D., Lansberg, J. K., Ora, I., Frigyesi, A., Castel, V., Navarro, S., Piqueras, M., Martinsson, T., Noguera, R., and Gisselsson, D. (2011). Alternative lengthening of telomeres—an enhanced chromosomal instability in aggressive non-MYCN amplified and telomere elongated neuroblastomas. Genes Chromosomes Cancer 50, 250-262. Lundblad, V., and Blackburn, E. H. (1993). An alternative pathway for yeast telomere maintenance rescues est1- senescence. Cell 73, 347-360. Lundblad, V., and Szostak, J. W. (1989). A mutant with a defect in telomere elongation leads to senescence in yeast. Cell 57, 633-643. Lustig, A. J. (2003). Clues to catastrophic telomere loss in mammals from yeast telomere rapid deletion. Nat Rev Genet 4, 916-923. Mac, S. M., D’Cunha, C. A., and Farnham, P. J. (2000). Direct recruitment of N-myc to target gene promoters. Mol Carcinog 29, 76-86. Makarov, V. L., Hirose, Y., and Langmore, J. P. (1997). Long G tails at both ends of human chromosomes suggest a C strand degradation mechanism for telomere shortening. Cell 88, 657-666. Makarov, V. L., Lejnine, S., Bedoyan, J., and Langmore, J. P. (1993). Nucleosomal organization of telomere-specific chromatin in rat. Cell 73, 775-787. Mallette, F. A., Gaumont-Leclerc, M. F., and Ferbeyre, G. (2007). The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev 21, 43-48. Marion, R. M., Strati, K., Li, H., Tejera, A., Schoeftner, S., Ortega, S., Serrano, M., and Blasco, M. A. (2009). Telomeres acquire embryonic stem cell characteristics in induced pluripotent stem cells. Cell Stem Cell 4, 141-154. Maris, J. M. (2010). Recent advances in neuroblastoma. N Engl J Med 362, 2202-2211. Maris, J. M., Hogarty, M. D., Bagatell, R., and Cohn, S. L. (2007). Neuroblastoma. Lancet 369, 2106-2120. Maris, J. M., Kyemba, S. M., Rebbeck, T. R., White, P. S., Sulman, E. P., Jensen, S. J., Allen, C., Biegel, J. A., and Brodeur, G. M. (1997). Molecular genetic analysis of familial neuroblastoma. Eur J Cancer 33, 1923-1928. Maris, J. M., Mosse, Y. P., Bradfield, J. P., Hou, C., Monni, S., Scott, R. H., Asgharzadeh, S., Attiyeh, E. F., Diskin, S. J., Laudenslager, M., et al. (2008). Chromosome 6p22 locus associated with clinically aggressive neuroblastoma. N Engl J Med 358, 2585-2593.

173
Maris, J. M., Weiss, M. J., Mosse, Y., Hii, G., Guo, C., White, P. S., Hogarty, M. D., Mirensky, T., Brodeur, G. M., Rebbeck, T. R., et al. (2002). Evidence for a hereditary neuroblastoma predisposition locus at chromosome 16p12-13. Cancer Res 62, 6651-6658. Marrone, A., Walne, A., Tamary, H., Masunari, Y., Kirwan, M., Beswick, R., Vulliamy, T., and Dokal, I. (2007). Telomerase reverse-transcriptase homozygous mutations in autosomal recessive dyskeratosis congenita and Hoyeraal-Hreidarsson syndrome. Blood 110, 4198-4205. Martinez, P., Thanasoula, M., Carlos, A. R., Gomez-Lopez, G., Tejera, A. M., Schoeftner, S., Dominguez, O., Pisano, D. G., Tarsounas, M., and Blasco, M. A. (2010). Mammalian Rap1 controls telomere function and gene expression through binding to telomeric and extratelomeric sites. Nat Cell Biol 12, 768-780. Martinez, P., Thanasoula, M., Munoz, P., Liao, C., Tejera, A., McNees, C., Flores, J. M., Fernandez-Capetillo, O., Tarsounas, M., and Blasco, M. A. (2009). Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev 23, 2060-2075. Mason, J. M., and Biessmann, H. (1995). The unusual telomeres of Drosophila. Trends Genet 11, 58-62. Matsuo, T., Shay, J. W., Wright, W. E., Hiyama, E., Shimose, S., Kubo, T., Sugita, T., Yasunaga, Y., and Ochi, M. (2009). Telomere-maintenance mechanisms in soft-tissue malignant fibrous histiocytomas. J Bone Joint Surg Am 91, 928-937. Matthay, K. K., Quach, A., Huberty, J., Franc, B. L., Hawkins, R. A., Jackson, H., Groshen, S., Shusterman, S., Yanik, G., Veatch, J., et al. (2009a). Iodine-131—metaiodobenzylguanidine double infusion with autologous stem-cell rescue for neuroblastoma: a new approaches to neuroblastoma therapy phase I study. J Clin Oncol 27, 1020-1025. Matthay, K. K., Reynolds, C. P., Seeger, R. C., Shimada, H., Adkins, E. S., Haas-Kogan, D., Gerbing, R. B., London, W. B., and Villablanca, J. G. (2009b). Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. J Clin Oncol 27, 1007-1013. Matthay , K. K., Villablanca , J. G., Seeger , R. C., Stram , D. O., Harris , R. E., Ramsay , N. K., Swift , P., Shimada , H., Black , C. T., Brodeur , G. M., et al. (1999). Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-Retinoic Acid. N Engl J Med 341, 1165-1173. Mattsson, K., Pokrovskaja, K., Kiss, C., Klein, G., and Szekely, L. (2001). Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc Natl Acad Sci USA 98, 1012-1017. McEachern, M. J., and Blackburn, E. H. (1996). Cap-prevented recombination between terminal telomeric repeat arrays (telomere CPR) maintains telomeres in Kluyveromyces lactis lacking telomerase. Genes Dev 10, 1822-1834. McEachern, M. J., and Haber, J. E. (2006). Break-induced replication and recombinational telomere elongation in yeast. Annu Rev Biochem 75, 111-135.

174
Mender, I., Gryaznov, S., and Shay, J. W. (2015). A novel telomerase substrate precursor rapidly induces telomere dysfunction in telomerase positive cancer cells but not telomerase silent normal cells. Oncoscience 2, 693-695. Meyerson, M., Counter, C. M., Eaton, E. N., Ellisen, L. W., Steiner, P., Dickinson Caddle, S., Ziaugra, L., Beijersbergen, R. L., Davidoff, M. J., Liu, Q., et al. (1997). hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90, 785-795. Michaloglou, C., Vredeveld, L. C., Soengas, M. S., Denoyelle, C., Kuilman, T., van der Horst, C. M., Majoor, D. M., Shay, J. W., Mooi, W. J., and Peeper, D. S. (2005). BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 436, 720-724. Mitchell, J. R., Wood, E., and Collins, K. (1999). A telomerase component is defective in the human disease dyskeratosis congenita. Nature 402, 551-555. Molenaar, C., Wiesmeijer, K., Verwoerd, N. P., Khazen, S., Eils, R., Tanke, H. J., and Dirks, R. W. (2003). Visualizing telomere dynamics in living mammalian cells using PNA probes. EMBO J 22, 6631-6641. Molenaar, J. J., Koster, J., Zwijnenburg, D. A., van Sluis, P., Valentijn, L. J., van der Ploeg, I., Hamdi, M., van Nes, J., Westerman, B. A., van Arkel, J., et al. (2012). Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589-593. Monclair, T., Brodeur, G. M., Ambros, P. F., Brisse, H. J., Cecchetto, G., Holmes, K., Kaneko, M., London, W. B., Matthay, K. K., Nuchtern, J. G., et al. (2009). The International Neuroblastoma Risk Group (INRG) staging system: an INRG Task Force report. J Clin Oncol 27, 298-303. Montero, J. J., Lopez de Silanes, I., Grana, O., and Blasco, M. A. (2016). Telomeric RNAs are essential to maintain telomeres. Nat Commun 7, 12534. Montgomery, E., Argani, P., Hicks, J. L., DeMarzo, A. M., and Meeker, A. K. (2004). Telomere lengths of translocation-associated and nontranslocation-associated sarcomas differ dramatically. Am J Pathol 164, 1523-1529. Morgenstern, J. P., and Land, H. (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res 18, 3587-3596. Moriarty, T. J., Huard, S., Dupuis, S., and Autexier, C. (2002). Functional multimerization of human telomerase requires an RNA interaction domain in the N terminus of the catalytic subunit. Mol Cell Biol 22, 1253-1265. Morin, G. B. (1989). The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell 59, 521-529. Mosse, Y. P., Laudenslager, M., Khazi, D., Carlisle, A. J., Winter, C. L., Rappaport, E., and Maris, J. M. (2004). Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet 75, 727-730. Mosse, Y. P., Laudenslager, M., Longo, L., Cole, K. A., Wood, A., Attiyeh, E. F., Laquaglia, M. J., Sennett, R., Lynch, J. E., Perri, P., et al. (2008). Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 455, 930-935.

175
Mosse, Y. P., Lim, M. S., Voss, S. D., Wilner, K., Ruffner, K., Laliberte, J., Rolland, D., Balis, F. M., Maris, J. M., Weigel, B. J., et al. (2013). Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. Lancet Oncol 14, 472-480. Mosse, Y. P., Lipsitz, E., Fox, E., Teachey, D. T., Maris, J. M., Weigel, B., Adamson, P. C., Ingle, M. A., Ahern, C. H., and Blaney, S. M. (2012). Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clin Cancer Res 18, 6058-6064. Moyzis, R. K., Buckingham, J. M., Cram, L. S., Dani, M., Deaven, L. L., Jones, M. D., Meyne, J., Ratliff, R. L., and Wu, J. R. (1988). A highly conserved repetitive DNA-sequence, (TTAGGG)N, present at the telomeres of human-chromosomes. Proc Natl Acad Sci USA 85, 6622-6626. Muntoni, A., Neumann, A. A., Hills, M., and Reddel, R. R. (2009). Telomere elongation involves intra-molecular DNA replication in cells utilizing alternative lengthening of telomeres. Hum Mol Gen 18, 1017-1027. Murnane, J. P., and Sabatier, L. (2004). Chromosome rearrangements resulting from telomere dysfunction and their role in cancer. Bio Essays 26, 1164-1174. Murnane, J. P., Sabatier, L., Marder, B. A., and Morgan, W. F. (1994). Telomere dynamics in an immortal human cell line. EMBO J 13, 4953-4962. Murphy, D. M., Buckley, P. G., Bryan, K., Das, S., Alcock, L., Foley, N. H., Prenter, S., Bray, I., Watters, K. M., Higgins, D., and Stallings, R. L. (2009). Global MYCN transcription factor binding analysis in neuroblastoma reveals association with distinct E-box motifs and regions of DNA hypermethylation. PloS One 4, e8154. Nabetani, A., and Ishikawa, F. (2009). Unusual telomeric DNAs in human telomerase-negative immortalized cells. Mol Cell Biol 29, 703-713. Nakamura, A. J., Chiang, Y. J., Hathcock, K. S., Horikawa, I., Sedelnikova, O. A., Hodes, R. J., and Bonner, W. M. (2008). Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin 1, 6. Nakamura, T. M., Morin, G. B., Chapman, K. B., Weinrich, S. L., Andrews, W. H., Lingner, J., Harley, C. B., and Cech, T. R. (1997). Telomerase catalytic subunit homologs from fission yeast and human. Science 277, 955-959. Napier, C. E., Huschtscha, L. I., Harvey, A., Bower, K., Noble, J. R., Hendrickson, E. A., and Reddel, R. R. (2015). ATRX represses alternative lengthening of telomeres. Oncotarget 6, 16543-16558. Natarajan, S., Groff-Vindman, C., and McEachern, M. J. (2003). Factors influencing the recombinational expansion and spread of telomeric tandem arrays in Kluyveromyces lactis. Eukaryotic Cell 2, 1115-1127. Natarajan, S., and McEachern, M. J. (2002). Recombinational telomere elongation promoted by DNA circles. Mol Cell Biol 22, 4512-4521. Nergadze, S. G., Farnung, B. O., Wischnewski, H., Khoriauli, L., Vitelli, V., Chawla, R., Giulotto, E., and Azzalin, C. M. (2009). CpG-island promoters drive transcription of human telomeres. RNA 15, 2186-2194.

176
Neumann, A. A., Watson, C. M., Noble, J. R., Pickett, H. A., Tam, P. P., and Reddel, R. R. (2013). Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev 27, 18-23. Nevo, I., Sagi-Assif, O., Botzer, L. E., Amar, D., Maman, S., Kariv, N., Leider-Trejo L. E., Savelyeve L., Schwab M., Yron, I., and Witz, I. P. (2008). Generation and characterisation of novel local and metastatic human neuroblastoma variants. Neoplasia 10, 817-827. Ng, L. J., Cropley, J. E., Pickett, H. A., Reddel, R. R., and Suter, C. M. (2009). Telomerase activity is associated with an increase in DNA methylation at the proximal subtelomere and a reduction in telomeric transcription. Nucleic Acids Res 37, 1152-1159. Nguyen le, B., Diskin, S. J., Capasso, M., Wang, K., Diamond, M. A., Glessner, J., Kim, C., Attiyeh, E. F., Mosse, Y. P., Cole, K., et al. (2011). Phenotype restricted genome-wide association study using a gene-centric approach identifies three low-risk neuroblastoma susceptibility Loci. PloS Genet 7, e1002026. Nickerson, H. J., Matthay, K. K., Seeger, R. C., Brodeur, G. M., Shimada, H., Perez, C., Atkinson, J. B., Selch, M., Gerbing, R. B., Stram, D. O., and Lukens, J. (2000). Favorable biology and outcome of stage IV-S neuroblastoma with supportive care or minimal therapy: a Children’s Cancer Group study. J Clin Oncol 18, 477-486. Novakova, Z., Hubackova, S., Kosar, M., Janderova-Rossmeislova, L., Dobrovolna, J., Vasicova, P., Vancurova, M., Horejsi, Z., Hozak, P., Bartek, J., and Hodny, Z. (2010). Cytokine expression and signaling in drug-induced cellular senescence. Oncogene 29, 273-284. O’Loghlen, A., Martin, N., Krusche, B., Pemberton, H., Alonso, M. M., Chandler, H., Brookes, S., Parrinello, S., Peters, G., and Gil, J. (2015). The nuclear receptor NR2E1/TLX controls senescence. Oncogene 34, 4069-4077. O’Sullivan, R. J., Arnoult, N., Lackner, D. H., Oganesian, L., Haggblom, C., Corpet, A., Almouzni, G., and Karlseder, J. (2014). Rapid induction of alternative lengthening of telomeres by depletion of the histone chaperone ASF1. Nat Struct Mol Biol 21, 167-174. O’Sullivan, R. J., Kubicek, S., Schreiber, S. L., and Karlseder, J. (2010). Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17, 1218-1225. Ogino, H., Nakabayashi, K., Suzuki, M., Takahashi, E., Fujii, M., Suzuki, T., and Ayusawa, D. (1998). Release of telomeric DNA from chromosomes in immortal human cells lacking telomerase activity. Biochemical and Biophysical Research Communications 248, 223-227. Ohali, A., Avigad, S., Ash, S., Goshen, Y., Luria, D., Feinmesser, M., Zaizov, R., and Yaniv, I. (2006). Telomere length is a prognostic factor in neuroblastoma. Cancer 107, 1391-1399. Okamoto, A., Demetrick, D. J., Spillare, E. A., Hagiwara, K., Hussain, S. P., Bennett, W. P., Forrester, K., Gerwin, B., Serrano, M., Beach, D. H., and Harris, C. C. (1994). Mutations and altered expression of p16INK4 in human cancer. Proc Natl Acad Sci USA 91, 11045-11049. Okamoto, K., Bartocci, C., Ouzounov, I., Diedrich, J. K., Yates, J. R., III, and Denchi, E. L. (2013). A two-step mechanism for TRF2-mediated chromosome-end protection. Nature 494, 502-505.

177
Olovnikov, A. M. (1971). Principle of marginotomy in template synthesis of polynucleotides. Dokl Akad Nauk SSSR 201, 1496-1499. Omori, Y., Nakayama, F., Li, D., Kanemitsu, K., Semba, S., Ito, A., and Yokozaki, H. (2009). Alternative lengthening of telomeres frequently occurs in mismatch repair system-deficient gastric carcinoma. Cancer Sci 100, 413-418. Onitake, Y., Hiyama, E., Kamei, N., Yamaoka, H., Sueda, T., and Hiyama, K. (2009). Telomere biology in neuroblastoma: telomere binding proteins and alternative strengthening of telomeres. J Pediatr Surg 44, 2258-2266. Opresko, P. L., von Kobbe, C., Laine, J. P., Harrigan, J., Hickson, I. D., and Bohr, V. A. (2002). Telomere binding protein TRF2 binds to and stimulates the Werner and Bloom syndrome helicases. J Biol Chem 277, 41110-41119. Padovan-Merhar, O. M., Raman, P., Ostrovnaya, I., Kalletla, K., Rubnitz, K. R., Sanford, E. M., Ali, S. M., Miller, V. A., Mosse, Y. P., Granger, M. P., et al. (2016). Enrichment of Targetable Mutations in the Relapsed Neuroblastoma Genome. PloS Genet 12, e1006501. Paeschke, K., Capra, J. A., and Zakian, V. A. (2011). DNA replication through G-quadruplex motifs is promoted by the Saccharomyces cerevisiae Pif1 DNA helicase. Cell 145, 678-691. Paeschke, K., Juranek, S., Simonsson, T., Hempel, A., Rhodes, D., and Lipps, H. J. (2008). Telomerase recruitment by the telomere end binding protein-b facilitates G-quadruplex DNA unfolding in ciliates. Nat Struct Mol Biol 15, 598-604. Paeschke, K., Simonsson, T., Postberg, J., Rhodes, D., and Lipps, H. J. (2005). Telomere end-binding proteins control the formation of G-quadruplex DNA structures in vivo. Nat Struct Mol Biol 12, 847-854. Park, J. R., Scott, J. R., Stewart, C. F., London, W. B., Naranjo, A., Santana, V. M., Shaw, P. J., Cohn, S. L., and Matthay, K. K. (2011). Pilot Induction Regimen Incorporating Pharmacokinetically Guided Topotecan for Treatment of Newly Diagnosed High-Risk Neuroblastoma: A Children’s Oncology Group Study. J Clin Oncol 29, 4351-4357. Park, K. H., Rha, S. Y., Kim, C. H., Kim, T. S., Yoo, N. C., Kim, J. H., Roh, J. K., Noh, S. H., Min, J. S., Lee, K. S., et al. (1998). Telomerase activity and telomere lengths in various cell lines: Changes of telomerase activity can be another method for chemosensitivity evaluation. International Journal of Oncology 13, 489-495. Parkinson, E. K. (1996). Do telomerase antagonists represent a novel anti-cancer strategy? BrJCancer 73, 1-4.
Parsons, D. W., Li, M., Zhang, X., Jones, S., Leary, R. J., Lin, J. C., Boca, S. M., Carter, H., Samayoa, J., Bettegowda, C., et al. (2011). The genetic landscape of the childhood cancer medulloblastoma. Science 331, 435-439. Peifer, M., Hertwig, F., Roels, F., Dreidax, D., Gartlgruber, M., Menon, R., Kramer, A., Roncaioli, J. L., Sand, F., Heuckmann, J. M., et al. (2015). Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 526, 700-704.

178
Pekmezci, M., Rice, T., Molinaro, A. M., Walsh, K. M., Decker, P. A., Hansen, H., Sicotte, H., Kollmeyer, T. M., McCoy, L. S., Sarkar, G., et al. (2017). Adult infiltrating gliomas with WHO 2016 integrated diagnosis: additional prognostic roles of ATRX and TERT. Acta Neuropathol. DOI: 10.1007/s00401-017-1690-1 Peng, Y., Mian, I. S., and Lue, N. F. (2001). Analysis of telomerase processivity: Mechanistic similarity to HIV-1 reverse transcriptase and role in telomere maintenance. Molecular Cell 7, 1201-1211. Pepe, A., and West, S. C. (2014). MUS81-EME2 promotes replication fork restart. Cell Rep 7, 1048-1055. Perrem, K., Bryan, T. M., Englezou, A., Hackl, T., Moy, E. L., and Reddel, R. R. (1999). Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene 18, 3383-3390. Perrem, K., Colgin, L. M., Neumann, A. A., Yeager, T. R., and Reddel, R. R. (2001). Coexistence of alternative lengthening of telomeres and telomerase in hTERT-transfected GM847 cells. Mol Cell Biol 21, 3862-3875. Peters, A. H., O’Carroll, D., Scherthan, H., Mechtler, K., Sauer, S., Schofer, C., Weipoltshammer, K., Pagani, M., Lachner, M., Kohlmaier, A., et al. (2001). Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107, 323-337. Pezzolo, A., Pistorio, A., Gambini, C., Haupt, R., Ferraro, M., Erminio, G., De Bernardi, B., Garaventa, A., and Pistoia, V. (2015). Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 6, 7493-7503. Pfeiffer, V., Crittin, J., Grolimund, L., and Lingner, J. (2013). The THO complex component Thp2 counteracts telomeric R-loops and telomere shortening. EMBO J 32, 2861-2871. Piatyszek, M. A., Kim, N. W., Weinrich, S. L., Hiyama, K., Hiyama, E., Wright, W. E., and Shay, J. W. (1995). Detection of telomerase activity in human cells and tumors by a telomeric repeat amplification protocol (TRAP). Methods Cell Sci 17, 1-15. Pickett, H. A., Cesare, A. J., Johnstone, R. L., Neumann, A. A., and Reddel, R. R. (2009). Control of telomere length by a trimming mechanism that involves generation of t-circles. EMBO J 28, 799-809. Pickett, H. A., Henson, J. D., Au, A. Y., Neumann, A. A., and Reddel, R. R. (2011). Normal mammalian cells negatively regulate telomere length by telomere trimming. Hum Mol Genet 20, 4684-4692. Pickett, H. A., and Reddel, R. R. (2012). The role of telomere trimming in normal telomere length dynamics. Cell Cycle 11, 1309-1315. Pickett, H. A., and Reddel, R. R. (2015). Molecular mechanisms of activity and derepression of alternative lengthening of telomeres. Nat Struct Mol Biol 22, 875-880. Picketts, D. J., Higgs, D. R., Bachoo, S., Blake, D. J., Quarrell, O. W. J., and Gibbons, R. J. (1996). ATRX encodes a novel member of the SNF2 family of proteins: Mutations point to a common mechanism underlying the ATR-X syndrome. Hum Mol Gen 5, 1899-1907. Plantaz, D., Mohapatra, G., Matthay, K. K., Pellarin, M., Seeger, R. C., and Feuerstein, B. G. (1997). Gain of chromosome 17 is the most frequent abnormality detected in neuroblastoma by comparative genomic hybridization. Am J Pathol 150, 81-89.

179
Pogo, B. G. T., Allfrey, V. G., and Mirsky, A. E. (1966). RNA Synthesis and histone acetylation during course of gene activation in lymphocytes. Proc Natl Acad Sci USA 55, 805-12. Polvi, A., Linnankivi, T., Kivela, T., Herva, R., Keating, J. P., Makitie, O., Pareyson, D., Vainionpaa, L., Lahtinen, J., Hovatta, I., et al. (2012). Mutations in CTC1, encoding the CTS telomere maintenance complex component 1, cause cerebroretinal microangiopathy with calcifications and cysts. Am J Hum Genet 90, 540-549. Pontecorvo, G. (1944). Structure of heterochromatin. Nature 153, 365-367. Poremba, C., Scheel, C., Hero, B., Christiansen, H., Schaefer, K. L., Nakayama, J., Berthold, F., Juergens, H., Boecker, W., and Dockhorn-Dworniczak, B. (2000). Telomerase activity and telomerase subunits gene expression patterns in neuroblastoma: a molecular and immunohistochemical study establishing prognostic tools for fresh-frozen and paraffin-embedded tissues. J Clin Oncol 18, 2582-2592. Poremba, C., Willenbring, H., Hero, B., Christiansen, H., Schafer, K. L., Brinkschmidt, C., Jurgens, H., Bocker, W., and Dockhorn-Dworniczak, B. (1999). Telomerase activity distinguishes between neuroblastomas with good and poor prognosis. Ann Oncol 10, 715-721. Porro, A., Feuerhahn, S., Reichenbach, P., and Lingner, J. (2010). Molecular dissection of telomeric repeat-containing RNA biogenesis unveils the presence of distinct and multiple regulatory pathways. Mol Cell Biol 30, 4808-4817. Potts, P. R., and Yu, H. T. (2007). The SMC5/6 complex maintains telomere length in ALT cancer cells through SUMOylation of telomere-binding proteins. Nat Struct Mol Biol 14, 581-590. Poulet, A., Buisson, R., Faivre-Moskalenko, C., Koelblen, M., Amiard, S., Montel, F., Cuesta-Lopez, S., Bornet, O., Guerlesquin, F., Godet, T., et al. (2009). TRF2 promotes, remodels and protects telomeric Holliday junctions. EMBO J 28, 641-651. Pricolo, V. E., Finkelstein, S. D., Wu, T. T., Keller, G., Bakker, A., Swalsky, P. A., and Bland, K. I. (1996). Prognostic value of TP53 and k-ras-2 mutational analysis in stage III carcinoma of the colon. Am J Surg 171, 41-46. Pryor, J. M., Waters, C. A., Aza, A., Asagoshi, K., Strom, C., Mieczkowski, P. A., Blanco, L., and Ramsden, D. A. (2015). Essential role for polymerase specialization in cellular nonhomologous end joining. Proc Natl Acad Sci USA 112, E4537-4545. Pugh, T. J., Morozova, O., Attiyeh, E. F., Asgharzadeh, S., Wei, J. S., Auclair, D., Carter, S. L., Cibulskis, K., Hanna, M., Kiezun, A., et al. (2013). The genetic landscape of high-risk neuroblastoma. Nat Genet 45, 279-284. Rai, R., Zheng, H., He, H., Luo, Y., Multani, A., Carpenter, P. B., and Chang, S. (2010). The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J 29, 2598-2610. Ramirez, R. D., Wright, W. E., Shay, J. W., and Taylor, R. S. (1997). Telomerase activity concentrates in the mitotically active segments of human hair follicles. J Invest Dermatol 108, 113-117. Rausch, T., Zichner, T., Schlattl, A., Stutz, A. M., Benes, V., and Korbel, J. O. (2012). DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333-i339.

180
Reddel, R. R. (2000). The role of senescence and immortalization in carcinogenesis. Carcinogenesis 21, 477-484.
Reynolds, C. P., Brodeur, G. M., Tomayko, M. M., Donner, L., Helson, L., Seeger, R. C., Triche, T. .J. (1988). Biological classification of cell lines derived from human extra-cranial neural tumors. Prog Clin Biol Res. 271, 291-306.
Reynolds, C. P., Zuo, J. J., Kim, N. W., Wang, H., Lukens, J. N., Matthay, K. K., and Seeger, R. C. (1997). Telomerase expression in primary neuroblastomas. Eur J Cancer 33, 1929-1931. Riha, K., McKnight, T. D., Griffing, L. R., and Shippen, D. E. (2001). Living with genome instability: plant responses to telomere dysfunction. Science 291, 1797-1800. Ritchie, K., Seah, C., Moulin, J., Isaac, C., Dick, F., and Berube, N. G. (2008). Loss of ATRX leads to chromosome cohesion and congression defects. J Cell Biol 180, 315-324. Rivera, T., Haggblom, C., Cosconati, S., and Karlseder, J. (2017). A balance between elongation and trimming regulates telomere stability in stem cells. Nat Struct Mol Biol 24, 30-39. Robles, S. J., and Adami, G. R. (1998). Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibrolasts. Oncogene 16, 1113-1123. Rogan, E. M., Bryan, T. M., Hukku, B., Maclean, K., Chang, A. C., Moy, E. L., Englezou, A., Warneford, S. G., Dalla-Pozza, L., and Reddel, R. R. (1995). Alterations in p53 and p16INK4 expression and telomere length during spontaneous immortalization of Li-Fraumeni syndrome fibroblasts. Mol Cell Biol 15, 4745-4753. Roos, R. A., Spengler, B. A., Biedler, J. L. (1983). Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J Natl Cancer Inst 71, 741-747. Roth, A., Harley, C. B., and Baerlocher, G. M. (2010). Imetelstat (GRN163L)--telomerase-based cancer therapy. Recent Results Cancer Res 184, 221-234. Roumelioti, F. M., Sotiriou, S. K., Katsini, V., Chiourea, M., Halazonetis, T. D., and Gagos, S. (2016). Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep 17, 1731-1737. Ruden, M., and Puri, N. (2013). Novel anticancer therapeutics targeting telomerase. Cancer Treat Rev 39, 444-456. Sadic, D., Schmidt, K., Groh, S., Kondofersky, I., Ellwart, J., Fuchs, C., Theis, F. J., and Schotta, G. (2015). Atrx promotes heterochromatin formation at retrotransposons. EMBO Rep 16, 836-850. Saha, A., Wittmeyer, J., and Cairns, B. R. (2002). Chromatin remodeling by RSC involves ATP-dependent DNA translocation. Genes Dev 16, 2120-2134. Saha, A., Wittmeyer, J., and Cairns, B. R. (2005). Chromatin remodeling through directional DNA translocation from an internal nucleosomal site. Nat Struct Mol Biol 12, 747-755.

181
Saharia, A., and Stewart, S. A. (2009). FEN1 contributes to telomere stability in ALT-positive tumor cells. Oncogene 28, 1162-1167. Saharia, A., Teasley, D. C., Duxin, J. P., Dao, B., Chiappinelli, K. B., and Stewart, S. A. (2010). FEN1 ensures telomere stability by facilitating replication fork re-initiation. J Biol Chem 285, 27057-27066. Sanders, R. P., Drissi, R., Billups, C. A., Daw, N. C., Valentine, M. B., and Dome, J. S. (2004). Telomerase expression predicts unfavorable outcome in osteosarcoma. J Clin Oncol 22, 3790-3797. Sarek, G., Vannier, J. B., Panier, S., Petrini, J. H., and Boulton, S. J. (2015). TRF2 recruits RTEL1 to telomeres in S phase to promote T-loop unwinding. Mol Cell 57, 1-14. Sarkar, J., Wan, B., Yin, J., Vallabhaneni, H., Horvath, K., Kulikowicz, T., Bohr, V. A., Zhang, Y., Lei, M., and Liu, Y. (2015). SLX4 contributes to telomere preservation and regulated processing of telomeric joint molecule intermediates. Nucleic Acids Res 43, 5912-5923. Sauerwald, A., Sandin, S., Cristofari, G., Scheres, S. H. W., Lingner, J., and Rhodes, D. (2013). Structure of active dimeric human telomerase. Nat Struct Mol Biol 20, 454-460. Sausen, M., Leary, R. J., Jones, S., Wu, J., Reynolds, C. P., Liu, X., Blackford, A., Parmigiani, G., Diaz, L. A., Jr., Papadopoulos, N., et al. (2013). Integrated genomic analyses identify ARID1A and ARID1B alterations in the childhood cancer neuroblastoma. Nat Genet 45, 12-17. Savage, S. A., Giri, N., Baerlocher, G. M., Orr, N., Lansdorp, P. M., and Alter, B. P. (2008). TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet 82, 501-509. Scarpa, A., Chang, D. K., Nones, K., Corbo, V., Patch, A. M., Bailey, P., Lawlor, R. T., Johns, A. L., Miller, D. K., Mafficini, A., et al. (2017). Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 543, 65-71. Scheel, C., Schaefer, K. L., Jauch, A., Keller, M., Wai, D., Brinkschmidt, C., van Valen, F., Boecker, W., Dockhorn-Dworniczak, B., and Poremba, C. (2001). Alternative lengthening of telomeres is associated with chromosomal instability in osteosarcomas. Oncogene 20, 3835-3844. Schleiermacher, G., Janoueix-Lerosey, I., Ribeiro, A., Klijanienko, J., Couturier, J., Pierron, G., Mosseri, V., Valent, A., Auger, N., Plantaz, D., et al. (2010). Accumulation of segmental alterations determines progression in neuroblastoma. J Clin Oncol 28, 3122-3130. Schleiermacher, G., Javanmardi, N., Bernard, V., Leroy, Q., Cappo, J., Rio Frio, T., Pierron, G., Lapouble, E., Combaret, V., Speleman, F., et al. (2014). Emergence of new ALK mutations at relapse of neuroblastoma. J Clin Oncol 32, 2727-2734. Schmidt, M. L., Lal, A., Seeger, R. C., Maris, J. M., Shimada, H., O'Leary, M., Gerbing, R. B., and Matthay, K. K. (2005). Favorable prognosis for patients 12 to 18 months of age with stage 4 nonamplified MYCN neuroblastoma: a Children's Cancer Group Study. J Clin Oncol 23, 6474-6480. Schmitt, C. A., Fridman, J. S., Yang, M., Lee, S., Baranov, E., Hoffman, R. M., and Lowe, S. W. (2002). A senescence program controlled by p53 and p16INK4A contributes to the outcome of cancer therapy. Cell 109, 335-346.

182
Schneiderman, J. I., Sakai, A., Goldstein, S., and Ahmad, K. (2009). The XNP remodeler targets dynamic chromatin in Drosophila. Proc Natl Acad Sci USA 106, 14472-14477. Schoeftner, S., and Blasco, M. A. (2008). Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol 10, 228-236. Schramm, A., Koster, J., Assenov, Y., Althoff, K., Peifer, M., Mahlow, E., Odersky, A., Beisser, D., Ernst, C., Henssen, A. G., et al. (2015). Mutational dynamics between primary and relapse neuroblastomas. Nat Genet 47, 872-877. Schubeler, D., MacAlpine, D. M., Scalzo, D., Wirbelauer, C., Kooperberg, C., van Leeuwen, F., Gottschling, D. E., O'Neill, L. P., Turner, B. M., Delrow, J., et al. (2004). The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev 18, 1263-1271. Schwab, M., Varmus, H. E., Bishop, J. M., Grzeschik, K. H., Naylor, S. L., Sakaguchi, A. Y., Brodeur, G., and Trent, J. (1984). Chromosome localization in normal human cells and neuroblastomas of a gene related to c-myc. Nature 308, 288-291. Schwartzentruber, J., Korshunov, A., Liu, X. Y., Jones, D. T., Pfaff, E., Jacob, K., Sturm, D., Fontebasso, A. M., Quang, D. A., Tonjes, M., et al. (2012). Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482, 226-231. Sedelnikova, O. A., Horikawa, I., Zimonjic, D. B., Popescu, N. C., Bonner, W. M., and Barrett, J. C. (2004). Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6, 168-170. Seeger, R. C., Brodeur, G. M., Sather, H., Dalton, A., Siegel, S. E., Wong, K. Y., and Hammond, D. (1985). Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N Engl J Med 313, 1111-1116. Seimiya, H., Muramatsu, Y., Ohishi, T., and Tsuruo, T. (2005). Tankyrase 1 as a target for telomere-directed molecular cancer therapeutics. Cancer Cell 7, 25-37. Sen, D., and Gilbert, W. (1988). Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334, 364-366. Serrano, M., Lin, A. W., McCurrach, M. E., Beach, D., and Lowe, S. W. (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593-602. Sfeir, A., and de Lange, T. (2012). Removal of shelterin reveals the telomere end-protection problem. Science 336, 593-597. Sfeir, A., Kabir, S., van Overbeek, M., Celli, G. B., and de Lange, T. (2010). Loss of Rap1 induces telomere recombination in the absence of NHEJ or a DNA damage signal. Science 327, 1657-1661. Sfeir, A., Kosiyatrakul, S. T., Hockemeyer, D., MacRae, S. L., Karlseder, J., Schildkraut, C. L., and de Lange, T. (2009). Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell 138, 90-103.

183
Shaw, V., Naisbitt, D., Costello, E., Farrell, J., Park, B. K., Neoptolemos, J., and Middleton, G. (2010a). TeloVac trial translational studies: investigation of the immune response to GV1001. Immunology 131, 164-164. Shaw, V. E., Naisbitt, D. J., Costello, E., Greenhalf, W., Park, B. K., Neoptolemos, J. P., and Middleton, G. W. (2010b). Current status of GV1001 and other telomerase vaccination strategies in the treatment of cancer. Expert Rev Vaccines 9, 1007-1016. Shay, J. W., and Bacchetti, S. (1997). A survey of telomerase activity in human cancer. Eur J Cancer 33, 787-791. Shay, J. W., and Keith, W. N. (2008). Targeting telomerase for cancer therapeutics. Br J Cancer 98, 677-683. Shay, J. W., Pereirasmith, O. M., and Wright, W. E. (1991). A role for both Rband p53 in the regulation of human cellular senescence. Exp Cell Res 196, 33-39. Shay, J. W., Tomlinson, G., Piatyszek, M. A., and Gollahon, L. S. (1995). Spontaneous in vitro immortalization of breast epithelial cells from a patient with Li-Fraumeni syndrome. Mol Cell Biol 15, 425-432. Shay, J. W., and Wright, W. E. (1996). The reactivation of telomerase activity in cancer progression. Trends Genet 12, 129-131. Sheu, Y. J., Kinney, J. B., Lengronne, A., Pasero, P., and Stillman, B. (2014). Domain within the helicase subunit Mcm4 integrates multiple kinase signals to control DNA replication initiation and fork progression. Proc Natl Acad Sci USA 111, E1899-1908. Sheu, Y. J., Kinney, J. B., and Stillman, B. (2016). Concerted activities of Mcm4, Sld3, and Dbf4 in control of origin activation and DNA replication fork progression. Genome Res 26, 315-330. Shimada, H., Ambros, I. M., Dehner, L. P., Hata, J., Joshi, V. V., Roald, B., Stram, D. O., Gerbing, R. B., Lukens, J. N., Matthay, K. K., and Castleberry, R. P. (1999). The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 86, 364-372. Shimada, H., Umehara, S., Monobe, Y., Hachitanda, Y., Nakagawa, A., Goto, S., Gerbing, R. B., Stram, D. O., Lukens, J. N., and Matthay, K. K. (2001). International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: a report from the Children's Cancer Group. Cancer 92, 2451-2461. Simon, T., Haberle, B., Hero, B., von Schweinitz, D., and Berthold, F. (2013). Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J Clin Oncol 31, 752-758. Simon, T., Hero, B., Faldum, A., Handgretinger, R., Schrappe, M., Klingebiel, T., and Berthold, F. (2011). Long term outcome of high-risk neuroblastoma patients after immunotherapy with antibody ch14.18 or oral metronomic chemotherapy. BMC Cancer 11, 21. Simon, T., Langler, A., Berthold, F., Klingebiel, T., and Hero, B. (2007). Topotecan and etoposide in the treatment of relapsed high-risk neuroblastoma: results of a phase 2 trial. J Pediatr Hematol Oncol 29, 101-106.

184
Smogorzewska, A., Karlseder, J., Holtgreve-Grez, H., Jauch, A., and de Lange, T. (2002). DNA ligase IV-dependent NHEJ of deprotected mammalian telomeres in G1 and G2. Curr Biol 12, 1635-1644. Soler, D., Genesca, A., Arnedo, G., Egozcue, J., and Tusell, L. (2005). Telomere dysfunction drives chromosomal instability in human mammary epithelial cells. Genes Chromosomes Cancer 44, 339-350. Spitz, R., Hero, B., Ernestus, K., and Berthold, F. (2003). Deletions in chromosome arms 3p and 11q are new prognostic markers in localized and 4s neuroblastoma. Clin Cancer Res 9, 52-58. Stagno d'Alcontres, M., Mendez-Bermudez, A., Foxon, J. L., Royle, N. J., and Salomoni, P. (2007). Lack of TRF2 in ALT cells causes PML-dependent p53 activation and loss of telomeric DNA. J Cell Biol 179, 855-867. Stansel, R. M., de Lange, T., and Griffith, J. D. (2001). T-loop assembly in vitro involves binding of TRF2 near the 3' telomeric overhang. EMBO J 20, 5532-5540. Stephens, P. J., Greenman, C. D., Fu, B., Yang, F., Bignell, G. R., Mudie, L. J., Pleasance, E. D., Lau, K. W., Beare, D., Stebbings, L. A., et al. (2011). Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell 144, 27-40. Stern, J. L., Zyner, K. G., Pickett, H. A., Cohen, S. B., and Bryan, T. M. (2012). Telomerase Recruitment Requires both TCAB1 and Cajal Bodies Independently. Mol Cell Biol 32, 2384-2395. Stuart, B. D., Choi, J., Zaidi, S., Xing, C., Holohan, B., Chen, R., Choi, M., Dharwadkar, P., Torres, F., Girod, C. E., et al. (2015). Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet 47, 512-517. Stewart, J. A., Wang, F., Chaiken, M. F., Kasbek, C., Chastain, P. D., Wright, W. E., and Price, C. M. (2012). Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J 31, 3537-3549. Strother, D. R., London, W. B., Schmidt, M. L., Brodeur, G. M., Shimada, H., Thorner, P., Collins, M. H., Tagge, E., Adkins, S., Reynolds, C. P., et al. (2012). Outcome after surgery alone or with restricted use of chemotherapy for patients with low-risk neuroblastoma: results of Children's Oncology Group study P9641. J Clin Oncol 30, 1842-1848. Subhawong, A. P., Heaphy, C. M., Argani, P., Konishi, Y., Kouprina, N., Nassar, H., Vang, R., and Meeker, A. K. (2009). The alternative lengthening of telomeres phenotype in breast carcinoma is associated with HER-2 overexpression. Mod Pathol 22, 1423-1431. Sundquist, W. I., and Klug, A. (1989). Telomeric DNA dimerizes by formation of guanine tetrads between hairpin loops. Nature 342, 825-829. Takai, H., Smogorzewska, A., and de Lange, T. (2003). DNA damage foci at dysfunctional telomeres. Curr Biol 13, 1549-1556. Takai, K. K., Kibe, T., Donigian, J. R., Frescas, D., and de Lange, T. (2011). Telomere protection by TPP1/POT1 requires tethering to TIN2. Mol Cell 44, 647-659.

185
Takakura, M., Kyo, S., Kanaya, T., Hirano, H., Takeda, J., Yutsudo, M., and Inoue, M. (1999). Cloning of human telomerase catalytic subunit (hTERT) gene promoter and identification of proximal core promoter sequences essential for transcriptional activation in immortalized and cancer cells. Cancer Res 59, 551-557. Tan, A., Abecasis, G. R., and Kang, H. M. (2015). Unified representation of genetic variants. Bioinformatics 31, 2202-2204. Tanaka, M., Kyo, S., Takakura, M., Kanaya, T., Sagawa, T., Yamashita, K., Okada, Y., Hiyama, E., and Inoue, M. (1998). Expression of telomerase activity in human endometrium is localized to epithelial glandular cells and regulated in a menstrual phase-dependent manner correlated with cell proliferation. Am J Pathol 153, 1985-1991. Teixeira, M. T., Arneric, M., Sperisen, P., and Lingner, J. (2004). Telomere length homeostasis is achieved via a switch between telomerase- extendible and -nonextendible states. Cell 117, 323-335. Temime-Smaali, N., Guittat, L., Wenner, T., Bayart, E., Douarre, C., Gomez, D., Giraud-Panis, M. J., Londono-Vallejo, A., Gilson, E., Amor-Gueret, M., and Riou, J. F. (2008). Topoisomerase IIIa is required for normal proliferation and telomere stability in alternative lengthening of telomeres. EMBO J 27, 1513-1524. Thanasoula, M., Escandell, J. M., Martinez, P., Badie, S., Munoz, P., Blasco, M. A., and Tarsounas, M. (2010). p53 prevents entry into mitosis with uncapped telomeres. Curr Biol 20, 521-526. Tokutake, Y., Matsumoto, T., Watanabe, T., Maeda, S., Tahara, H., Sakamoto, S., Niida, H., Sugimoto, M., Ide, T., and Furuichi, Y. (1998). Extra-chromosome telomere repeat DNA in telomerase-negative immortalized cell lines. Biochem Biophys Res Commun 247, 765-772. Tommerup, H., Dousmanis, A., and Delange, T. (1994). Unusual chromatin in human telomeres. Mol Cell Biol 14, 5777-5785. Tong, A. S., Stern, J. L., Sfeir, A., Kartawinata, M., de Lange, T., Zhu, X. D., and Bryan, T. M. (2015). ATM and ATR signalling regulate the recruitment of human telomerase to telomeres. Cell Rep 13, 1633-1646. Topcu, Z., Nickles, K., Davis, C., and McEachern, M. J. (2005). Abrupt disruption of capping and a single source for recombinationally elongated telomeres in Kluyveromyces lactis. Proc Natl Acad Sci USA 102, 3348-3353. Trochet, D., Bourdeaut, F., Janoueix-Lerosey, I., Deville, A., de Pontual, L., Schleiermacher, G., Coze, C., Philip, N., Frebourg, T., Munnich, A., et al. (2004). Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet 74, 761-764. Tsang, A. R., Wyatt, H. D. M., Ting, N. S. Y., and Beattie, T. L. (2012). hTERT mutations associated with idiopathic pulmonary fibrosis affect telomerase activity, telomere length, and cell growth by distinct mechanisms. Aging Cell 11, 482-490. Tsutsumimoto T., Williams, P., Yoneda, T. (2014). The SK-N-AS human neuroblastoma cell line develops osteolytic bone metastases with increased angiogenesis and COX-2 expression. J Bone Onc 3, 67-76.

186
Tummala, H., Walne, A., Collopy, L., Cardoso, S., de la Fuente, J., Lawson, S., Powell, J., Cooper, N., Foster, A., Mohammed, S., et al. (2015). Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest 125, 2151-2160.
Tweddle, D. A., Malcolm, A. J., Bown, N., Pearson, A. D., and Lunec, J. (2001). Evidence for the development of p53 mutations after cytotoxic therapy in a neuroblastoma cell line. Cancer Res 61, 8-13. Ulaner, G. A., and Giudice, L. C. (1997). Developmental regulation of telomerase activity in human fetal tissues during gestation. Mol Hum Reprod 3, 769-773. Ulaner, G. A., Hoffman, A. R., Otero, J., Huang, H. Y., Zhao, Z., Mazumdar, M., Gorlick, R., Meyers, P., Healey, J. H., and Ladanyi, M. (2004). Divergent patterns of telomere maintenance mechanisms among human sarcomas: sharply contrasting prevalence of the alternative lengthening of telomeres mechanism in Ewing's sarcomas and osteosarcomas. Genes Chromosomes Cancer 41, 155-162. Ulaner, G. A., Hu, J. F., Vu, T. H., Giudice, L. C., and Hoffman, A. R. (2001). Tissue-specific alternate splicing of human telomerase reverse transcriptase (hTERT) influences telomere lengths during human development. Int J Cancer 91, 644-649. Ulaner, G. A., Huang, H. Y., Otero, J., Zhao, Z., Ben-Porat, L., Satagopan, J. M., Gorlick, R., Meyers, P., Healey, J. H., Huvos, A. G., et al. (2003a). Absence of a telomere maintenance mechanism as a favorable prognostic factor in patients with osteosarcoma. Cancer Res 63, 1759-1763. Uringa, E. J., Lisaingo, K., Pickett, H. A., Brind'amour, J., Rohde, J. H., Zelensky, A., Essers, J., and Lansdorp, P. M. (2012). RTEL1 contributes to DNA replication, repair and telomere maintenance. Mol Biol Cell 23, 2782-2792. Valentijn, L. J., Koster, J., Zwijnenburg, D. A., Hasselt, N. E., van Sluis, P., Volckmann, R., van Noesel, M. M., George, R. E., Tytgat, G. A., Molenaar, J. J., and Versteeg, R. (2015). TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat Genet 47, 1411-1414. van den Bosch, M., Bree, R. T., and Lowndes, N. F. (2003). The MRN complex: coordinating and mediating the response to broken chromosomes. EMBO Rep 4, 844-849. van Noesel, M. M., Hahlen, K., Hakvoort-Cammel, F. G., and Egeler, R. M. (1997). Neuroblastoma 4S: a heterogeneous disease with variable risk factors and treatment strategies. Cancer 80, 834-843. Van Roy, N., Laureys, G., Van Gele, M., Opdenakker, G., Miura, R., van der Drift, P., Chan, A., Versteeg, R., and Speleman, F. (1997). Analysis of 1;17 translocation breakpoints in neuroblastoma: implications for mapping of neuroblastoma genes. E J Cancer 33, 1974-1978. van Steensel, B., Smogorzewska, A., and de Lange, T. (1998). TRF2 protects human telomeres from end-to-end fusions. Cell 92, 401-413. Vannier, J. B., Pavicic-Kaltenbrunner, V., Petalcorin, M. I., Ding, H., and Boulton, S. J. (2012). RTEL1 dismantles T loops and counteracts telomeric G4-DNA to maintain telomere integrity. Cell 149, 795-806. Vannier, J. B., Sandhu, S., Petalcorin, M. I., Wu, X., Nabi, Z., Ding, H., and Boulton, S. J. (2013). RTEL1 is a replisome-associated helicase that promotes telomere and genome-wide replication. Science 342, 239-242.

187
Varley, H., Pickett, H. A., Foxon, J. L., Reddel, R. R., and Royle, N. J. (2002). Molecular characterization of inter-telomere and intra-telomere mutations in human ALT cells. Nat Genet 30, 301-305. Venteicher, A. S., Abreu, E. B., Meng, Z. J., McCann, K. E., Terns, R. M., Veenstra, T. D., Terns, M. P., and Artandi, S. E. (2009). A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644-648. Venteicher, A. S., Meng, Z. J., Mason, P. J., Veenstra, T. D., and Artandi, S. E. (2008). Identification of ATPases pontin and reptin as telomerase components essential for holoenzyme assembly. Cell 132, 945-957. Vera, E., Canela, A., Fraga, M. F., Esteller, M., and Blasco, M. A. (2008). Epigenetic regulation of telomeres in human cancer. Oncogene 27, 6817-6833. Verdun, R. E., and Karlseder, J. (2006). The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell 127, 709-720. Vergnaud, G., and Denoeud, F. (2000). Minisatellites: mutability and genome architecture. Genome Res 10, 899-907. Viceconte, N., Dheur, M.S., Majerova, E., Pierreux, C.E., Baurain, J.F., van Baren, N., and Decottignies, A. (2017). Highly aggressive metastatic melanoma cells unable to maintain telomere length. Cell Reports 19, In press. Villa, R., Daidone, M. G., Motta, R., Venturini, L., De Marco, C., Vannelli, A., Kusamura, S., Baratti, D., Deraco, M., Costa, A., et al. (2008). Multiple mechanisms of telomere maintenance exist and differentially affect clinical outcome in diffuse malignant peritoneal mesothelioma. Clin Cancer Res 14, 4134-4140. Vogan, K., Bernstein, M., Leclerc, J. M., Brisson, L., Brossard, J., Brodeur, G. M., Pelletier, J., and Gros, P. (1993). Absence of p53 gene mutations in primary neuroblastomas. Cancer Res 53, 5269-5273. Vogelstein, B., Papadopoulos, N., Velculescu, V. E., Zhou, S., Diaz, L. A., Jr., and Kinzler, K. W. (2013). Cancer genome landscapes. Science 339, 1546-1558. Vogt, M., Haggblom, C., Yeargin, J., Christiansen-Weber, T., and Haas, M. (1998). Independent induction of senescence by p16INK4a and p21CIP1 in spontaneously immortalized human fibroblasts. Cell Growth Differ 9, 139-146. Voon, H. P., Hughes, J. R., Rode, C., De La Rosa-Velazquez, I. A., Jenuwein, T., Feil, R., Higgs, D. R., and Gibbons, R. J. (2015). ATRX plays a key role in maintaining silencing at interstitial heterochromatic loci and imprinted genes. Cell Rep 11, 405-418. Vulliamy, T., Marrone, A., Dokal, I., and Mason, P. J. (2002). Association between aplastic anaemia and mutations in telomerase RNA. Lancet 359, 2168-2170. Vulliamy, T., Marrone, A., Goldman, F., Dearlove, A., Bessler, M., Mason, P. J., and Dokal, I. (2001a). The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413, 432-435.

188
Vulliamy, T. J., Knight, S. W., Mason, P. J., and Dokal, I. (2001b). Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol Dis 27, 353-357. Wada, R. K., Seeger, R. C., Brodeur, G. M., Einhorn, P. A., Rayner, S. A., Tomayko, M. M., and Reynolds, C. P. (1993). Human neuroblastoma cell lines that express N-myc without gene amplification. Cancer 72, 3346-3354. Wan, B., Yin, J., Horvath, K., Sarkar, J., Chen, Y., Wu, J., Wan, K., Lu, J., Gu, P., Yu, E. Y., et al. (2013). SLX4 assembles a telomere maintenance toolkit by bridging multiple endonucleases with telomeres. Cell Rep 4, 861-869. Wang, J., Xie, L. Y., Allan, S., Beach, D., and Hannon, G. J. (1998). Myc activates telomerase. Genes Dev 12, 1769-1774. Wang, K., Diskin, S. J., Zhang, H., Attiyeh, E. F., Winter, C., Hou, C., Schnepp, R. W., Diamond, M., Bosse, K., Mayes, P. A., et al. (2011). Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature 469, 216-220. Wang, R. C., Smogorzewska, A., and de Lange, T. (2004). Homologous recombination generates T-loop-sized deletions at human telomeres. Cell 119, 355-368. Watkins, N. J., Gottschalk, A., Neubauer, G., Kastner, B., Fabrizio, P., Mann, M., and Luhrmann, R. (1998). Cbf5p, a potential pseudouridine synthase, and Nhp2p, a putative RNA-binding protein, are present together with Gar1p in all H BOX/ACA-motif snoRNPs and constitute a common bipartite structure. RNA 4, 1549-1568. Watson, J. M., and Shippen, D. E. (2007). Telomere rapid deletion regulates telomere length in Arabidopsis thaliana. Mol Cell Biol 27, 1706-1715. Weinrich, S. L., Pruzan, R., Ma, L., Ouellette, M., Tesmer, V. M., Holt, S. E., Bodnar, A. G., Lichtsteiner, S., Kim, N. W., Trager, J. B., et al. (1997). Reconstitution of human telomerase with the template RNA component hTR and the catalytic protein subunit hTRT. Nat Genet 17, 498-502. Weiss, W. A., Aldape, K., Mohapatra, G., Feuerstein, B. G., and Bishop, J. M. (1997). Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J 16, 2985-2995. Wen, J. P., Cong, Y. S., and Bacchetti, S. (1998). Reconstitution of wild-type or mutant telomerase activity in telomerase-negative immortal human cells. Hum Mol Gen 7, 1137-1141. Wenz, C., Enenkel, B., Amacker, M., Kelleher, C., Damm, K., and Lingner, J. (2001). Human telomerase contains two cooperating telomerase RNA molecules. EMBO J 20, 3526-3534. Whitaker, N. J., Bryan, T. M., Bonnefin, P., Chang, A. C., Musgrove, E. A., Braithwaite, A. W., and Reddel, R. R. (1995). Involvement of RB-1, p53, p16INK4 and telomerase in immortalisation of human cells. Oncogene 11, 971-976. Whittle, S. B., Reyes, S., Du, M., Gireud, M., Zhang, L., Woodfield, S. E., Ittmann, M., Scheurer, M. E., Bean, A. J., and Zage, P. E. (2016). A Polymorphism in the FGFR4 Gene Is Associated With Risk of Neuroblastoma and Altered Receptor Degradation. J Pediatr Hematol Oncol 38, 131-138.

189
Wilson, J. S., Tejera, A. M., Castor, D., Toth, R., Blasco, M. A., and Rouse, J. (2013). Localization-dependent and -independent roles of SLX4 in regulating telomeres. Cell Rep 4, 853-860. Wilstermann, A. M., and Osheroff, N. (2001). Base excision repair intermediates as topoisomerase II poisons. J Biol Chem 276, 46290-46296. Wong, J. M., and Collins, K. (2006). Telomerase RNA level limits telomere maintenance in X-linked dyskeratosis congenita. Genes Dev 20, 2848-2858. Wong, L. H., McGhie, J. D., Sim, M., Anderson, M. A., Ahn, S., Hannan, R. D., George, A. J., Morgan, K. A., Mann, J. R., and Choo, K. H. A. (2010). ATRX interacts with H3.3 in maintaining telomere structural integrity in pluripotent embryonic stem cells. Genome Res 20, 351-360. Wright, W. E., Piatyszek, M. A., Rainey, W. E., Byrd, W., and Shay, J. W. (1996a). Telomerase activity in human germline and embryonic tissues and cells. Dev Genet 18, 173-179. Wright, W. E., Tesmer, V. M., Liao, M. L., and Shay, J. W. (1999). Normal human telomeres are not late replicating. Exp Cell Res 251, 492-499. Wu, C. H., van Riggelen, J., Yetil, A., Fan, A. C., Bachireddy, P., and Felsher, D. W. (2007). Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci USA 104, 13028-13033. Wu, G. K., Lee, W. H., and Chen, P. L. (2000). NBS1 and TRF1 colocalize at promyelocytic leukemia bodies during late S/G(2) phases in immortalized telomerase-negative cells - Implication of NBS1 in alternative lengthening of telomeres. J Biol Chem 275, 30618-30622. Wu, K. J., Grandori, C., Amacker, M., Simon-Vermot, N., Polack, A., Lingner, J., and Dalla-Favera, R. (1999). Direct activation of TERT transcription by c-MYC. Nat Genet 21, 220-224. Wu, L., Multani, A. S., He, H., Cosme-Blanco, W., Deng, Y., Deng, J. M., Bachilo, O., Pathak, S., Tahara, H., Bailey, S. M., et al. (2006). Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell 126, 49-62. Wu, N., Kong, X. D., Ji, Z. J., Zeng, W. H., Potts, P. R., Yokomori, K., and Yu, H. T. (2012a). Scc1 sumoylation by Mms21 promotes sister chromatid recombination through counteracting Wapl. Genes Dev 26, 1473-1485. Wu, P., Takai, H., and de Lange, T. (2012b). Telomeric 3' overhangs derive from resection by Exo1 and Apollo and fill-in by POT1b-associated CST. Cell 150, 39-52. Wu, P., van Overbeek, M., Rooney, S., and de Lange, T. (2010). Apollo contributes to G overhang maintenance and protects leading-end telomeres. Mol Cell 39, 606-617. Wu, W. Q., Hou, X. M., Li, M., Dou, S. X., and Xi, X. G. (2015). BLM unfolds G-quadruplexes in different structural environments through different mechanisms. Nucleic Acids Res 43, 4614-4626. Xia, J. Q., Peng, Y., Mian, I. S., and Lue, N. F. (2000). Identification of functionally important domains in the N-terminal region of telomerase reverse transcriptase. Mol Cell Biol 20, 5196-5207.

190
Xu, Y., Suzuki, Y., Ito, K., and Komiyama, M. (2010). Telomeric repeat-containing RNA structure in living cells. Proc Natl Acad Sci USA 107, 14579-14584. Xue, Y., Gibbons, R., Yan, Z., Yang, D., McDowell, T. L., Sechi, S., Qin, J., Zhou, S., Higgs, D., and Wang, W. (2003). The ATRX syndrome protein forms a chromatin-remodeling complex with Daxx and localizes in promyelocytic leukemia nuclear bodies. Proc Natl Acad Sci USA 100, 10635-10640. Yamada, T., Yoshimura, H., Shimada, R., Hattori, M., Eguchi, M., Fujiwara, T. K., Kusumi, A., and Ozawa, T. (2016). Spatiotemporal analysis with a genetically encoded fluorescent RNA probe reveals TERRA function around telomeres. Sci Rep 6, 38910. Yan, P., Benhattar, J., Coindre, J. M., and Guillou, L. (2002). Telomerase activity and hTERT mRNA expression can be heterogeneous and does not correlate with telomere length in soft tissue sarcomas. I J Cancer 98, 851-856. Yanik, G. A., Parisi, M. T., Shulkin, B. L., Naranjo, A., Kreissman, S. G., London, W. B., Villablanca, J. G., Maris, J. M., Park, J. R., Cohn, S. L., et al. (2013). Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children's oncology group. J Nucl Med 54, 541-548. Yashima, K., Maitra, A., Rogers, B. B., Timmons, C. F., Rathi, A., Pinar, H., Wright, W. E., Shay, J. W., and Gazdar, A. F. (1998). Expression of the RNA component of telomerase during human development and differentiation. Cell Growth Differ 9, 805-813. Ye, J., Lenain, C., Bauwens, S., Rizzo, A., Saint-Leger, A., Poulet, A., Benarroch, D., Magdinier, F., Morere, J., Amiard, S., et al. (2010). TRF2 and apollo cooperate with topoisomerase 2a to protect human telomeres from replicative damage. Cell 142, 230-242. Ye, J. Z., and de Lange, T. (2004). TIN2 is a tankyrase 1 PARP modulator in the TRF1 telomere length control complex. Nat Genet 36, 618-623. Ye, J. Z., Hockemeyer, D., Krutchinsky, A. N., Loayza, D., Hooper, S. M., Chait, B. T., and de Lange, T. (2004a). POT1-interacting protein PIP1: a telomere length regulator that recruits POT1 to the TIN2/TRF1 complex. Genes Dev 18, 1649-1654. Ye, J. Z. S., Donigian, J. R., van Overbeek, M., Loayza, D., Luo, Y., Krutchinsky, A. N., Chait, B. T., and de Lange, T. (2004b). TIN2 binds TRF1 and TRF2 simultaneously and stabilizes the TRF2 complex on telomeres. J Biol Chem 279, 47264-47271. Ye, K., Schulz, M. H., Long, Q., Apweiler, R., and Ning, Z. (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865-2871. Yeager, T. R., Neumann, A. A., Englezou, A., Huschtscha, L. I., Noble, J. R., and Reddel, R. R. (1999). Telomerase-negative immortalized human cells contain a novel type of promyelocytic leukemia (PML) body. Cancer Res 59, 4175-4179. Yehezkel, S., Segev, Y., Viegas-Pequignot, E., Skorecki, K., and Selig, S. (2008). Hypomethylation of subtelomeric regions in ICF syndrome is associated with abnormally short telomeres and enhanced transcription from telomeric regions. Hum Mol Genet 17, 2776-2789.

191
Yu, A. L., Gilman, A. L., Ozkaynak, M. F., London, W. B., Kreissman, S. G., Chen, H. X., Smith, M., Anderson, B., Villablanca, J. G., Matthay, K. K., et al. (2010). Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med 363, 1324-1334. Yu, C. E., Oshima, J., Fu, Y. H., Wijsman, E. M., Hisama, F., Alisch, R., Matthews, S., Nakura, J., Miki, T., Ouais, S., et al. (1996). Positional cloning of the Warner's syndrome gene. Science 272, 258-262. Yu, T. Y., Kao, Y. W., and Lin, J. J. (2014). Telomeric transcripts stimulate telomere recombination to suppress senescence in cells lacking telomerase. Proc Natl Acad Sci USA 111, 3377-3382. Yui, J., Chiu, C. P., and Lansdorp, P. M. (1998). Telomerase activity in candidate stem cells from fetal liver and adult bone marrow. Blood 91, 3255-3262. Zaug, A. J., Podell, E. R., and Cech, T. R. (2005). Human POT1 disrupts telomeric G-quadruplexes allowing telomerase extension in vitro. Proc Natl Acad Sci USA 102, 10864-10869. Zeng, S., Xiang, T., Pandita, T. K., Gonzalez-Suarez, I., Gonzalo, S., Harris, C. C., and Yang, Q. (2009). Telomere recombination requires the MUS81 endonuclease. Nat Cell Biol 11, 616-623. Zeng, S., and Yang, Q. (2009). The MUS81 endonuclease is essential for telomerase negative cell proliferation. Cell Cycle 8, 2157-2160. Zhang, A., Zheng, C., Lindvall, C., Hou, M., Ekedahl, J., Lewensohn, R., Yan, Z., Yang, X., Henriksson, M., Blennow, E., et al. (2000). Frequent amplification of the telomerase reverse transcriptase gene in human tumors. Cancer Res 60, 6230-6235. Zhao, W., and Sung, P. (2015). Significance of ligand interactions involving Hop2-Mnd1 and the RAD51 and DMC1 recombinases in homologous DNA repair and XX ovarian dysgenesis. Nucleic Acids Res 43, 4055-4066. Zheng, P., Guo, Y., Niu, Q., Levy, D. E., Dyck, J. A., Lu, S., Sheiman, L. A., and Liu, Y. (1998). Proto-oncogene PML controls genes devoted to MHC class I antigen presentation. Nature 396, 373-376. Zhong, S., Salomoni, P., Ronchetti, S., Guo, A., Ruggero, D., and Pandolfi, P. P. (2000). Promyelocytic leukemia protein (PML) and Daxx participate in a novel nuclear pathway for apoptosis. J Exp Med 191, 631-640. Zhong, Z. H., Jiang, W. Q., Cesare, A. J., Neumann, A. A., Wadhwa, R., and Reddel, R. R. (2007). Disruption of telomere maintenance by depletion of the MRE11/RAD50/NBS1 complex in cells that use alternative lengthening of telomeres. J Biol Chem 282, 29314-29322. Zijlmans, J. M., Martens, U. M., Poon, S. S., Raap, A. K., Tanke, H. J., Ward, R. K., and Lansdorp, P. M. (1997). Telomeres in the mouse have large inter-chromosomal variations in the number of T2AG3 repeats. Proc Natl Acad Sci USA 94, 7423-7428.
Zou, L., and Elledge, S. J. (2003). Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 300, 1542-1548.

192
Appendices

193
Appendix I: Information Relating to Chapter 2
ATRX and DAXX sequencing was performed with the primers listed in Table A1.1. and A1.2., respectively.
The PCR conditions are listed in the subsequent tables.
Table A1.1. Primers used to Sanger sequence ATRX.
Exon PCR
product size
M13 primer Other primer PCR
Program Taq used in
PCR
35 552 ATGGTCCTGTGAATGCCATC GGCATTTAAGGGGACCAAAC 1 Taq Platinum (Invitrogen)
34 339 ACCTTGGGAAATCCCGAATA CAATGACTATCCATCCCTCCA 1 Taq Platinum (Invitrogen)
33 237 GTTGGCAAATGGAAGGATTC AGTAGGGGGTGGAGGGTACA 2 Taq Platinum (Invitrogen)
32 353 GGGTGAAAAGGGTGTTTTGT
T GCATAGGGAACCCTCAACAA 3
HotStar Taq (Qiagen)
31 372 GGTTTTAGTTTCTAGTACAGT
TGACCA GGGGAATGTGTTCCTAAAACC 1
Taq Platinum (Invitrogen)
30 500 GCAAAATTGCTGATGAGTTTT
T CATTTTATTATCCTTGAAAAAT
TCTGA 1
Taq Platinum (Invitrogen)
29 405 AAGAAATGAATTCTCTGAACT
CTTGA CCAACTTTGTTTCCCTCTCTG 1
Taq Platinum (Invitrogen)
28 329 TGATGAGCAAGGTGGAAAAT
C TGACACTGTTTTGCAACCTGA 1
Taq Platinum (Invitrogen)
27 313 AAATCCTGCTGGGATTTTTG GGGTAGTTTTGTTTCTTTTGTT
GC 1
Taq Platinum (Invitrogen)
26 392 CCCCATGGGTAGGTCTTTTT TTGCTTGTATTGGCCTAGCA 1 Taq Platinum (Invitrogen)
24-25 554 TTCCTCAGTCCTTCCTCAGC CCCCAAATACCAGAGAGCAC 3 HotStar Taq
(Qiagen)
23 289 GCTTCTCTACACTGCCAAAAG
TG TTCTGCTTCCAATAGATGCTTT 1
Taq Platinum (Invitrogen)
22 387 TTGTGGGTTTAGAAAGGGTA
AA TGCAAAACTGAAAAAGAACA
ACA 1
Taq Platinum (Invitrogen)
21 367 TGAGCATTTCATTGGGGAAT TGAAAGAGCGGGAAAGAAAA 1 Taq Platinum (Invitrogen)
20 449 TTAACCAAATACGGGAGCAG
A TTTCACAGCAGACTAAGATGA
ACC 1
Taq Platinum (Invitrogen)
19 576 GGCAAGAGGGATTAAAAGAT
GA TGGCGACATTAAGGGTGATT 1
Taq Platinum (Invitrogen)
18 387 TTGGAAATTCTGGCCGTTTA TTCCCACTGAAATATGCATCA
C 1
Taq Platinum (Invitrogen)
17 418 TCTTCAGCCCCTACGACTGT GAAGGAAAGTCCCCCTGTTC 1 Taq Platinum (Invitrogen)
16 379 CAATTGGATTTGTGGTGTGG CCACCCCACTCACCAATTTA 1 Taq Platinum (Invitrogen)

194
15 503 TCTTCCACCTTTTCCTGCTG AGGCTGGGTGTGTTGACTC 3 HotStar Taq
(Qiagen)
14 398 TGGAACAGAGAGGTAACAGC
A AACAAACCTCCCCTCAGGAT 1
Taq Platinum (Invitrogen)
13 245 TGCTCTGTTTTAATGTCGAGT
CA TGAAGGCATGGTCATTCAGA 1
Taq Platinum (Invitrogen)
12 314 CAGCTTCCCAAAGTGCTAGG CGAGGCATTTTAAAGGCTGA 3 HotStar Taq
(Qiagen)
11 391 TCTATTGGCACATTTATTTCT TTGGCCTCCCAAAGTCCTGAG
ATT 1
Taq Platinum (Invitrogen)
10 343 TTTGGAGTCCAGAGTTTAGA
CC AACTTGCAGGAAGACTGTGA
GCGA 1
Taq Platinum (Invitrogen)
9(g) 493 CATCTGATGCTGAGGAAAGT
TCTG GAGATCCCTGATACTGAATAC
TAGC 1
Taq Platinum (Invitrogen)
9(f) 649 TGAATCTTCATCTGATGGCAC
TGA TTTCTGTTCATCGCTGCTTCCC
TC 2
Taq Platinum (Invitrogen)
9(e) 640 AGGAATGGATAATCAAGGGC
ACA TCCTTTCCCTGTTGACTTCTCA
GC 1
Taq Platinum (Invitrogen)
9(d) 655 GGATAAGCGTAATTCTTCTGA
CAGTGC AGCACTTGCTTGCTGCTTCTTA
GG 1
Taq Platinum (Invitrogen)
9(c) 777 CCGGTGGTGAACATAAGAAA
TCTG AACTGTGACTCATCCTGCTCA
CCT 4
Taq Platinum (Invitrogen)
9(b) 663 CTTGTTCAGTTCCACTGCTGC
CAT CCTGTTCTGGCTCTGTAACCT
ACT 1
Taq Platinum (Invitrogen)
9(a) 742 CCAATGCAAGATGAGCCTTC GAGTAAGCAGATGACCTAAA
TTACCAC 1
Taq Platinum (Invitrogen)
8 226 CACACCAGTGTCCTGGAGAT
TT AGGAAACACTGAATGTTAGCT
CATCT 1
Taq Platinum (Invitrogen)
7 415 TGCCAAGGTTGTCATGTGCTT
AG GAAGTCTTCCAAGGGCAGAT
ACCA 1
Taq Platinum (Invitrogen)
6 276 CCAGCAATGTTGGCTTTATCT
GAACTG AAGCACATCCGATTTTCCAA 1
Taq Platinum (Invitrogen)
5 348 TTCCTTGTTGAGACCCACTGC
TCA GCCATGTTTGGTCGTTTGTAC
ATAGT 1
Taq Platinum (Invitrogen)
4 219 GCTAATTGTAGGGATGCCGT
TTCG CTCAGAATAGTGGTTGACATG
AGTTCAG 1
Taq Platinum (Invitrogen)
3 259 AGTGTGAGAATGGGTTTGTG
GAGT TGGGTATCAGTAGCCTTCGAC
ACA 1
HotStar Taq (Qiagen)
2 424 GGGCTTCTATAAAGCTTGCTA
ATCTGTC ACACCCACAACTGTAACATTT
CCC 1
Taq Platinum (Invitrogen)
1 418 TGTGCTTTGGAGGAGGTAGC
CAAT TAAGCAACACACAGGCCTAAC
CCA 1
Taq Platinum (Invitrogen)
M13 primer: refers to the addition of the M13 sequence (5’ -GTAAAACGACGGCCAGT- 3’) at the 5’ end of the
listed primer.

195
Table A1.2. Primers used to Sanger sequence DAXX.
Exon PCR
product size
M13 primers Other primers PCR
Program PCR Master Mix
8 409 GTGCCACATCCTGTCTCTTCC GGGACAGCTAATGCCAATCTG 5 Taq Platinum (Invitrogen)
7 404 AAGAGACAGGATGTGGCACG GTCTGCTGGGAGAGACTGGAC 2 Taq Platinum (Invitrogen)
6(b) 466 TCTCCCAGCAGACTCAGTTCC CAAAGGACGCATAGTGTCACC 2 Taq Platinum (Invitrogen)
6(a) 534 CAAGGGAACATTCTCCTCACC CGCCTCCATTGAAGGAAGTAG 1 Taq Platinum (Invitrogen)
5 383 GAGTCCAGGTTGACTGATGGG GAAAGGTTTCAAACAGGTGGC 2 Taq Platinum (Invitrogen)
4 397 ATGTCAGGTATGAGGCGGATG CATCAGTCAACCTGGACTCCC 2 Taq Platinum (Invitrogen)
3(d) 576 GTCAGAGCACTCAGCCCTTG GTAAGCTGATCCGCCTCTTTG 2 Taq Platinum (Invitrogen)
3(c) 543 TAACCCTCCCACACACCTCTC TGGCAGCCAAAGTTGTAGATG 1 Taq Platinum (Invitrogen)
3(b) 553 CGCCTGTTAACCTCTGGGTAG CATTCCTCTATAACCGGCAGC 1 Taq Platinum (Invitrogen)
3(a) 502 CAGAGGAAGCAGTAGTTCGGG AGGTGTGTGGGAGGGTTATTC 6 Taq Platinum (Invitrogen)
2 443 GGGTTAGTGGGAAAGAAAGG
AC GGGCTGGATGTTACTGAAACC 6
Taq Platinum (Invitrogen)

196
Table A1.3. PCR cycling conditions for program 1.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
3 cycles Denaturation 94°C 30 sec
Annealing 64°C 45 sec
Extension 70°C 1 min
41 cycles Denaturation 94°C 30 sec
Annealing 57°C 45 sec
Extension 70°C 1 min
1 cycle Extension 72°C 10 mins
Table A1.4. PCR cycling conditions for program 2.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
3 cycles Denaturation 94°C 30 sec
Annealing 65°C 45 sec
Extension 70°C 1 min
41 cycles Denaturation 94°C 30 sec
Annealing 59°C 45 sec
Extension 70°C 1 min
1 cycle Extension 72°C 10 mins

197
Table A1.5. PCR cycling conditions for program 3.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
20 cycles Denaturation 94°C 30 sec
Annealing
58.5°C
-0.5°C from
annealing temp
every 3 cycles
45 sec
Extension 72°C 1 min
30 cycles Denaturation 94°C 30 sec
Annealing 55°C 45 sec
Extension 72°C 1 min
1 cycle Extension 72°C 10 mins
Table A1.6. PCR cycling conditions for program 4.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
3 cycles Denaturation 94°C 30 sec
Annealing 67°C 45 sec
Extension 70°C 1 min
41 cycles Denaturation 94°C 30 sec
Annealing 60°C 45 sec
Extension 70°C 1 min
1 cycle Extension 72°C 10 mins

198
Table A1.7. PCR cycling conditions for program 5.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
3 cycles Denaturation 94°C 30 sec
Annealing 63°C 45 sec
Extension 70°C 1 min
41 cycles Denaturation 94°C 30 sec
Annealing 55°C 45 sec
Extension 70°C 1 min
1 cycle Extension 72°C 10 mins
Table A1.8. PCR cycling conditions for program 6.
Number of cycles Step Temperature Time
1 cycle Taq Hot start 94°C 2 mins
3 cycles Denaturation 94°C 30 sec
Annealing 62°C 45 sec
Extension 70°C 1 min
41 cycles Denaturation 94°C 30 sec
Annealing 55°C 45 sec
Extension 70°C 1 min
1 cycle Extension 72°C 10 mins

199
Appendix II: Data Pertaining to Chapter 4
Table A2.1. Characteristics of high-risk NB ALT tumours (n=36).
Sample
ID
Gen
der
MYCN
Age
Dx
(Yr)
Outcome
(Yr)
Telomere
Content
(Arbitrary
Unit)
Telomere
Content
Group
C-circle
Assay
Product
(Arbitrary
Unit)
C-circle
Assay
Classification
TRF
(kb)
Telomerase
Activity
CHW 21 F NA 4.49
DOD
(2.96yr) 68.7 long 106 + ND -
CHW 27 F NA 4.04
DOD
(2.1yr) 19.1 long 43.0 + ND +
CHW 3 F NA 3.65
DOD
(2.62yr) 16.7 long 41.0 + 28.3 ND
NB 157 M NA 4.24
DOD
(3.6yr) 72.1 long 134 + ND ND
NB 31 F NA 2.78
DOD
(4.24yr) 60.5 long 1311 + ND ND
NB 71 M NA 4.08
DOD
(1.32yr) 43.5 long 38.7 + ND ND
NB 74 F NA 3.44
OS
(2.77yr) 27.9 long 666 + ND ND
NB 8 F NA 5.41
DOD
(1.63yr) 20.2 long 249 + ND ND
NB 92 F NA 4.49
DOD
(2.96yr) 75.7 long 54.7 + ND ND
PAHSVL M NA 6.7
DOD
(7.62yr) 16.02 long 111 + 11.0 -
PAIHWT F NA 4.2
DOD
(4.27yr) 28.27 long 129 + 11.1 +
PAISZV M NA 5.1
OS
(5.35yr) 65.89 long 125 + 22.0 -
PAIUTE M NA 5.5
DOD
(4.01yr) 75.32 long 14.8 + 22.0 +
PAPKXS M NA 6.98
OS
(5.4yr) 17.57 long 15.1 + 18.4 +
PAPNHR M NA 3.6
OS
(5.08yr) 15.92 long 36.0 + 1.9 -
PAPTAN F NA 4.0
OS
(5.61yr) 26.45 long 141 + 19.9 -
PAPUWY
M
NA
5.4
DOD
(2.98yr)
23.85
long
16.0
+
19.3
+
PARACS F NA 3.2
OS
(3.61yr) 15.03 long 341 + ND ND

200
PARASL M NA 4.4
DOD
(0.53yr) 34.04 long 25.0 + 24.1 +
PARBGT F NA 2.6
DOD
(2.74yr) 29.58 long 14.0 + ND ND
PARKNP M NA 8.8
DOD
(1.75yr) 20.87 long 1149 + 20.2 -
PARYM
N M NA 4.4
DOD
(2.95yr) 42.07 long 462 + ND ND
PASAAN F NA 4.2
OS
(3.76yr) 16.39 long 348 + 24.8 +
PASJRH F NA 7.04
DOD
(3.33yr) 21.82 long 26.3 + 23.1 +
PASPIJ F NA 8.2
OS
(2.81yr) 18.86 long 11.2 + 29.2 -
PASRTR F NA 4.4
OS
(2.6yr) 18.08 long 7.6 + ND ND
PASVYG M NA 5.1
OS
(2.3yr) 23.94 long 45.8 + ND ND
CHW 24 F NA 2.69
DOD
(5.03yr) 14.1 short 249 + ND +
CHW 25 F NA 3.59
OS
(11.72yr) 12.7 short 111 + ND -
PALRGK M NA 5.4
DOD
(2.52yr) 13.84 short 104 + 17.0 +
PAMHM
K M NA 5.7
DOD
(3.18yr) 13.61 short 47.5 + 18.3 -
PANLET M NA 5.4
DOD
(3.32yr) 8.07 short 29.5 + 9.0 +
PANRVJ F NA 13.1
OS
(6.19yr) 5.30 short 41.7 + 9.8 -
PANUIF M NA 6.1
OS
(6.22yr) 7.21 short 14.5 + ND ND
PARVLK M NA 16.5
DOD
(2.36yr) 7.91 short 219 + 12.7 -
PASWLY F NA 2.7
OS
(2.72yr) 12.17 short 166 + 14.6 +
F = female NA = Not Amplified DOD = Died of Disease
M = male Yr = year OS = Overall survival
A= Amplified
ND = not determined
DX = Diagnosis

201
Table A2.2. Characteristics of high-risk MYCN amplified NB tumours (n=55).
Sample
ID Gen
der
MYCN
Age
Dx
(yr)
Outcome
(yr)
Telomere
Content
(Arbitrary
Unit)
Telomere
Content
Group
C-circle
Assay
Product
(Arbitrary
Unit)
C-circle
Assay
Classification
TRF
(kb)
Telomerase
Activity
CHW 7 F A 1.71
OS
(7.33yr) 18.2 long 1.4 - ND ND
NB 114 M A 1.73
DOD
(1.28yr) 18.1 long 0.0 - ND ND
NB 195 M A 2.27
DOD
(1.28yr) 23.6 long 0.0 - ND ND
NB 45 F A 3.75
DOD
(1.56yr) 20.4 long 0.0 - ND ND
PAKPHB M A 0.4
OS
(4.25yr) 35.25 long 0.0 - ND ND
PAPWFY F A 2.1
DOD
(2.75yr) 25.55 long 0.0 - ND ND
PARYXW F A 6.0
DOD
(1.17yr) 43.20 long 1.5 - ND ND
CHW 12 M A 1.78
DOD
(1.64yr) 10.8 short 0.0 - 14.9 ND
CHW 13 M A 0.78
DOD
(0.44yr) 6.5 short 0.0 - 10.4 ND
CHW 15 F A 1.96
DOD
(0.87yr) 4.7 short 0.0 - 8.6 ND
CHW 16 F A 2.19
OS
(5.54yr) 7.3 short 0.1 - 11.4 ND
CHW 2 F A 1.59
OS
(7.41yr) 9.1 short 1.3 - 11.9 ND
CHW 20 F A 2.51
OS
(4.98yr) 11.1 short 0.0 - ND ND
CHW 8 F A 1.32
OS
(5.62yr) 3.4 short 0.0 - 11.7 ND
NB 149 F A 1.11
OS
(8.33yr) 2.5 short 0.6 - ND ND
NB 166 F A 5.43
DOD
(1.09yr) 13.4 short 0.0 - ND ND
NB 169 F A 0.44
DOD
(0.66yr) 13.2 short 0.0 - ND ND
NB 171 M A 2.93
DOD
(0.61yr) 7.6 short 0.0 - ND ND
NB 172 F A 2.39
DOD
(0.65yr) 3.4 short 0.0 - ND ND
NB 23
F
A
2.01
OS
(9.33yr)
7.3
short
0.3
-
ND
ND

202
NB 40 F A 0.64
DOD
(0.05yr) 2.9 short 1.6 - ND ND
NB 43 F A 5.81
DOD
(0.86yr) 12.3 short 0.5 - ND ND
NB 44 F A 1.55
OS
(6.76yr) 3.3 short 0.0 - ND ND
NB 48 F A 0.73
DOD
(0.69yr) 9.7 short 0.0 - ND ND
NB 51 M A 1.22
DOD
(0.95yr) 2.5 short 0.0 - ND ND
NB 53 F A 1.51
DOD
(2.15yr) 3.0 short 0.3 - ND ND
NB 56 M A 1.38
DOD
(1.26yr) 8.0 short 0.0 - ND ND
NB 6 F A 1.41
DOD
(0.5yr) 14.0 short 0.0 - ND ND
NB 98 M A 0.91
DOD
(0.63yr) 10.0 short 0.0 - ND ND
PAKNYZ F A 1.7
DOD
(0.41yr) 4.44 short 0.0 - ND ND
PAKPXS M A 3.0
DOD
(0.07yr) 2.53 short 0.12 - ND ND
PALHHT F A 1.6
OS
(9.97yr) 2.09 short 0.27 - ND ND
PALUVT F A 2.4
DOD
(3.18yr) 4.17 short 0.72 - ND ND
PALZMB F A 0.8
DOD
(0.53yr) 2.29 short 0.18 - ND ND
PAMRAA F A 0.4
DOD
(0.5yr) 5.04 short 0.0 - ND ND
PANHRZ 1 A 4.1
OS
(6.86yr) 1.82 short 0.42 - ND ND
PANLTR F A 1.2
DOD
(0.41yr) 9.62 short 1.5 - ND ND
PAPRHX F A 10.1
DOD
(1.01yr) 5.30 short 0.82 - ND ND
PAPRPR M A 1.7
DOD
(4.32yr) 2.31 short 0.19 - ND ND
PAPRXW F A 2.4
OS
(5.62yr) 2.03 short 0.0 - ND ND
PARELJ F A 0.3
OS
(4.3yr) 6.75 short 0.26 - ND ND
PARETE
F
A
1.9
DOD
(0.7yr)
4.47
short
0.02
-
ND
ND

203
F = female NA = Not Amplified DOD = Died of Disease
M = male Yr = year OS = Overall survival
A= Amplified
ND = not determined
DX = Diagnosis
PARHDE F A 2.3
DOD
(1.3yr) 5.79 short 0.41 - ND ND
PARSCY M A 1.4
DOD
(0.64yr) 9.62 short 1.1 - ND ND
PARSUF F A 1.3
DOD
(1.02yr) 1.51 short 0.07 - ND ND
PARVVM F A 0.9
DOD
(0.83yr) 4.89 short 0.0 - ND ND
PARXBV M A 3.6
DOD
(1.32yr) 8.25 short 0.0 - ND ND
PARYRE M A 3.2
OS
(4.11yr) 4.38 short 0.29 - ND ND
PASBTL F A 1.3
DOD
(0.61yr) 3.80 short 0.42 - ND ND
PASFKX F A 0.7
OS
(3.01yr) 2.10 short 0.23 - ND ND
PASLXS M A 2.4
DOD
(0.33yr) 2.38 short 0.24 - ND ND
PASTGD F A 2.5
DOD
(1.5yr) 4.12 short 0.43 - ND ND
PASWFB M A 2.0
DOD
(0.2yr) 2.10 short 0.54 - ND ND
PASYIP F A 2.3
DOD
(0.6yr) 3.07 short 0.86 - ND ND
PASZPI F A 1.8
OS
(2.3yr) 9.61 short 0.32 - ND ND

204
Table A2.3. Characteristics of high-risk ALT-negative/long telomere NB tumours (n=17).
Sample
ID Gen
der
MYCN
Age
Dx
(yr)
Outcome
(yr)
Telomere
Content
(Arbitrary
Unit)
Telomere
Content
Group
C-circle
Assay
Product
(Arbitrary
Unit)
C-circle
Assay
Classification
TRF
(kb)
Telomerase
Activity
CHW 11 F NA 3.81
OS
(14.54yr) 22.3 long 3.8 - 24.3 -
CHW 9 F NA 4.67
DOD
(2.78yr) 21.1 long 2.3 - 36.3 -
NB 128 F NA 4.45
DOD
(1.74yr) 27.3 long 0.0 - ND ND
NB 19 F NA 3.29
OS
(11.98yr) 16.1 long 0.0 - ND ND
NB 3 F NA 3.05
DOD
(1.27yr) 19.4 long 0.0 - ND ND
NB 36 F NA 5.65
OS
(9.49yr) 15.2 long 0.0 - ND ND
NB 4 F NA 3.83
OS
(10.63yr) 73.5 long 0.0 - ND ND
NB 60 F NA 1.26
OS
(7.38yr) 18.3 long 0.5 - ND ND
NB 70 F NA 2.43
DOD
(3.71yr) 36.8 long 0.0 - ND ND
NB 95 M NA 5.30
OS
(12.5yr) 17.8 long 1.4 - ND ND
PAIDDG F NA 9.4
DOD
(2.5yr) 31.64 long 5.7 - ND ND
PAIFXV M NA 5.5
DOD
(3.04yr) 20.68 long 1.5 - 18.7 -
PALPYT F NA 5.8
OS
(9.75yr) 50.67 long 0.0 - ND ND
PAMFUX M NA 3.6
DOD
(4.52yr) 24.22 long 2.7 - ND ND
PAMTPS F NA 11.2
DOD
(5.23yr) 18.08 long 4.0 - ND ND
PARXFT F NA 5.1
OS
(2.05yr) 69.95 long 6.6 - 24.5 -
PATHVK M NA 5.8
OS
(2.0yr) 52.55 long 0.0 - ND ND
F = female NA = Not Amplified DOD = Died of Disease
M = male Yr = year OS = Overall survival
A= Amplified
ND = not determined
DX = Diagnosis

205
Table A2.4. Characteristics of high-risk MYCN non-amplified/short telomere NB tumours (n=41).
Sample
ID Gen
der
MYCN
Age
Dx
(yr)
Outcome
(yr)
Telomere
Content
(Arbitrary
Unit)
Telomere
Content
Group
C-circle
Assay
Product
(Arbitrary
Unit)
C-circle
Assay
Classification
TRF
(kb)
Telomerase
Activity
CHW 22 M NA 2.00
OS
(15.93yr) 2.9 short 1.5 - ND +
CHW 23 F NA 1.47
OS
(11.06yr) 2.5 short 0.2 - ND +
CHW 26 F NA 1.25
OS
(9.7yr) 4.2 short 0.0 - ND +
CHW 4 F NA 3.69
DOD
(2.47yr) 7.1 short 1.0 - 13.2 ND
NB 112 M NA 1.99
OS
(5.48yr) 7.4 short 5.0 - ND ND
NB 12 F NA 2.12
OS
(3.67yr) 9.4 short 0.5 - ND ND
NB 140 F NA 3.46
DOD
(0.78yr) 6.6 short 1.4 - ND ND
NB 17 M NA 3.59
DOD
(0.6yr) 5.7 short 1.4 - ND ND
NB 174 F NA 3.44
DOD
(1.07yr) 13.8 short 0.8 - ND ND
NB 198 F NA 4.18
DOD
(0.37yr) 2.7 short 0.0 - ND ND
NB 216 F NA 1.97
OS
(4.65yr) 6.1 short 0.0 - ND ND
NB 217 F NA 2.88
DOD
(0.97yr) 9.4 short 0.0 - ND ND
NB 226 M NA 3.99
DOD
(3.2yr) 5.9 short 1.3 - ND ND
NB 24 M NA 2.41
DOD
(1.68yr) 9.1 short 1.5 - ND ND
NB 25 M NA 6.63
DOD
(0.67yr) 5.4 short 0.0 - ND ND
NB 65 M NA 2.72
DOD
(5.71yr) 6.2 short 0.8 - ND ND
NB 77 F NA 1.55
DOD
(2.42yr) 6.3 short 1.8 - ND ND
NB 78 F NA 1.78
OS
(3.17yr) 4.6 short 0.0 - ND ND
PAHVCS M NA 1.2
OS
(5.52yr) 7.17 short 1.1 - 8.5 +

206
PAHXUD
M
NA
1.4
DOD
(3.24yr) 5.10 short 0.0 - 6.6 +
PAILRI M NA 1.2
OS
(8.25yr) 3.70 short 0.0 - 9.5 +
PALKNC M NA 1.0
OS
(1.77yr) 3.26 short 0.0 - 9.1 +
PALSRF F NA 1.0
OS
(8.96yr) 3.44 short 0.0 - 10.2 -
PAMHFI F NA 1.1
OS
(5.91yr) 6.20 short 0.0 - 8.7 -
PAMPXA F NA 1.9
OS
(7.39yr) 3.64 short 0.20 - ND ND
PAMRTG F NA 1.4
OS
(7.65yr) 8.86 short 1.5 - ND ND
PAMYBA F NA 12.6
DOD
(1.48yr) 3.63 short 0.34 - ND ND
PANHVF F NA 6.6
DOD
(3.56yr) 0.93 short 0.0 - 4.1 +
PAPFSL M NA 6.0
DOD
(0.61yr) 2.87 short 0.0 - 9.4 +
PAPMUT F NA 2.7
OS
(5.25yr) 5.30 short 1.1 - ND ND
PAPRGH M NA 1.2
OS
(5.14yr) 4.60 short 0.0 - 6.5 -
PAPZFW F NA 3.7
OS
(4.61yr) 2.13 short 0.80 - ND ND
PARADX F NA 4.4
DOD
(0.5yr) 5.39 short 0.32 - ND ND
PARGIW F NA 3.2
OS
(4.69yr) 2.37 short 0.56 - 9.1 +
PARNNG F NA 3.7
OS
(4.65yr) 3.69 short 0.0 - ND ND
PARRLH M NA 3.6
DOD
(1.48yr) 4.27 short 0.06 - ND ND
PASHVB M NA 5.1
OS
(3.17yr) 7.28 short 0.0 - 12.1 +
PASJNY M NA 3.4
DOD
(1.22yr) 5.41 short 0.0 - 7.9 +
PASLUP M NA 4.5
DOD
(2.53yr) 2.28 short 0.01 - 5.5 +
PASRII M NA 2.3
DOD
(1.8yr) 5.04 short 0.59 - ND ND
PASUCB F NA 2.8
OS
(2.45yr) 2.98 short 0.0 - 13.8 -

207
F = female NA = Not Amplified DOD = Died of Disease
M = male Yr = year OS = Overall survival
A= Amplified
ND = not determined
DX = Diagnosis

208
Table A2.5. Genes examined by whole genome sequencing.
Name Entrez ID Summary Synonym
ABL1 25
Protein tyrosine kinase involved in a
variety of cellular processes, including
cell division, DNA damage response,
adhesion, differentiation, and response
to stress, SWI/SNF complex JTK7, P150
ACD 65057 Shelterin protein TINT1, TPP1
ACTL6A 86
Required for SMARCA4/BRG1/BAF190A
containing remodelling complex BAF
with chromatin/nuclear matrix. BAF53A, INO80K
ACTL6B 51412 Role in remodelling mononucleosomes BAF53B
AIM2 9447
Recognises cytosolic dsDNA, putative
role in tumorigenic reversion and may
control cell proliferation. PYHIN4
AKT1 207
Regulates many processes including
metabolism, proliferation, cell survival,
growth and angiogenesis.
ALKBH 8846 DNA repair gene
ALKBH2 121642 DNA repair gene
APEX1 328
Role in the cellular response to
oxidative stress.
APITD1 378708
Involved in DNA damage and repair
(involved in Fanconi anaemia)
APTX 54840 Editing and processing nucleases aprataxin
ARID1A 8289 SWI/SNF complex SMARCF, C1orf4
ARID1B 57492 Involved in chromatin remodelling
ARID3A 1820 Transcription factor

209
ASF1A 25842 Histone chaperone HAsf1, HCIA, HSPC146
ASF1B 55723
Member of the H3/H4 family of histone
chaperone proteins HAsf1b
ASH1L 55870 Histone methyltransferase
ASH1, KMT2H,
KIAA1420
ASH2L 9070
Component of the Set1/Ash2 histone
methyltransferase (HMT) complex ASH2L1, Bre2
ASXL1 171023
Transcription coactivator activity,
chromatin modifier KIAA0978, MDS, BOPS
ATM 472 DNA damage response ATA, ATDC, ATC, ATD
ATP5C1 509 Subunit of mitochondrial ATP synthase. ATP5CL1, ATP5C
ATR 545 Conserved DNA damage response gene MEC1, FRP1
ATRIP 84126 Conserved DNA damage response gene AGS1
ATRX 546 Chromatin Modifier/epigenetic
remodeller RAD54, JMS, MRX52
BARD1 580
DNA damage repair, ubiquitination and
transcriptional regulation
BAZ1A 11177
Chromatin Modifier/epigenetic
remodeller WCRF18, HACF10
BAZ1B 9031
Chromatin Modifier/epigenetic
remodeller
WSTF, WBSCR10,
WBSCR9
BCCIP 56647 DNA repair and cell cycle TOK1
BCL2 596 Apoptosis Bcl-2, PPP1R50
BLM 641 DNA helicase RECQL3, RECQ2
BMI1 648
Chromatin Modifier/epigenetic
remodeller PCGF4, RNF51

210
BRCA1 672
Role in maintaining genomic stability,
and it also acts as a tumour suppressor. FANCS
BRCA2 675
Involved in double-strand break repair
and/or homologous recombination. FANCD1
BRD2 6046
Chromatin Modifier, transcriptional
regulator KIAA9001, RING3
BRD4 23476 Chromatin/epigenetic remodeller HUNKI, MCAP
BRINP1 1620
Involved in cell cycle and cell death
pathways
DBC1, DBCCR1,
FAM5A
BRIP1 83990 DNA-dependent ATPase FANCJ
BTAF1 9044 Regulates transcription MOT1
CABIN1 23523
Involved in replication-independent
chromatin assembly. PPP3IN, KIAA0330
CARM1 10498 Chromatin regulation PRMT4
CBX1 10951 Component of heterochromatin. CBX, M31, HP1Hsbeta
CBX2 84733 Involved in chromatin regulation CDCA6, SRXY5
CBX3 11335
Binds DNA and is a component of
heterochromatin.
CBX5 23468 Component of heterochromatin HP1A
CCAR2 23468
Protein is enriched in the
heterochromatin and associated with
centromeres. HEL25
CCNH 902 Transcription P34, P37, CycH, CAK
CCNO 10309 Cell cycle CCNU, UDG2, CILD29
CCNT1 904 Transcription HIVE1, CCNT, CYCT1
CDC25A 993 Cell cycle CDC25A2

211
CDC25B 994 Cell cycle CDC25HU2
CDK2 1017 Cell cycle P33(CDK2)
CDK7 1022
Cell cycle, transcription initiating, DNA
repair gene CAK, STK1
CDKN1A 1026 Cell cycle, DNA repair gene
CDKN1, P21, WAF1,
MDA-6, CAP20
CENPA 1058 Chromatin CENP-A
CENPT 80152 Chromatin C16orf56, ICEN22
CEP164 22897
Involved in microtubule organization,
DNA damage response, and
chromosome segregation.
Cep164,
NPHP15, KIAA1052
CHAF1A 10036
Involved in DNA replication and DNA
repair. CAF1
CHD1 1105
Chromatin Modifier/epigenetic
remodeller CHD-1
CHD2 1106
Chromatin Modifier/epigenetic
remodeller CHD-2, EEOC
CHD3 1107
NuRD (nucleosome remodelling and
histone deacetylation)
Mi2-ALPHA, ZFH, CHD-
3
CHD4 1108
NuRD (nucleosome remodelling and
histone deacetylation) Mi2-BETA, CHD-4
CHD7 55636
Chromatin regulator, transcription
regulator CRG, KAL5
CHD8 57680 Chromatin remodeller HELSNF1, AUTS18
CHEK1 1111 Conserved DNA damage response gene CHK1
CHEK2 11200 Conserved DNA damage response gene RAD53, CHK2
CHFR 55743 Cell Cycle RNF116, RNF196

212
CHTF18 63922 Cell replication
C16orf41, RUVBL,
CHL12
CLK2 1196 Transcription
CLOCK 9575 Transcription KAT13D, KIAA0334
CLSPN 63967 Cell cycle, DNA damage
CREBBP 1387
Chromatin regulator, transcription
factor RSTS, KAT3A
CTBP1 1487 Transcription regulator BARS
CTCF 10664 Chromatin regulator MRD21
CTCFL 140690 DNA binding protein CT27, HMGB1L1
CTDP1 9150 Involved in initiation of translation CCFDN, FCP1
CTR9 9646 Chromatin regulation SH2BP1, TSBP, P150
DAXX 1616 Chromatin remodeller DAP6, EAP1, BING2
DBF4B 80174 Roll in DNA replication and proliferation
ASKL1, DRF1, CHIFB,
ZDBF1B
DCLRE1A 9937
Required for DNA inter-strand cross-link
repair and cell cycle arrest
SNM1, PSO2, HSNM1,
HSNM1A, KIAA0086
DCLRE1B 64858
Involved in t-loop formation, telomere
replication and DNA damage
SNM1B, APOLLO,
HSNM1B
DCLRE1C 64421 Involved in DNA damage repair
SCIDA, ARTEMIS,
HSNM1C, DCLREC1C
DDB1 1642 Required for DNA repair
DDBA, XPCE, XAP1,
UV-DDB1
DDB2 1643 Required for DNA repair DDBB, XPE, UV-DDB2
DHX29 54505
RNA helicase involved in translation
initiation. DDX29

213
DHX36 170506
Role in transcriptional regulation and
mRNA stability.
DDX36, MLEL1, G4R1,
RHAU, KIAA1488
DHX58 79132 Recognises ssRNA, dsRNA LGP2, RLR
DDX60 55601 Binds ssRNA, dsRNA and dsDNA
DHX9 1660 Functions as a transcriptional activator. DDX9, LKP, RHA, NDH2
DKC1 1736 Component of telomerase
DKC, NOLA4, CBF5,
NAP57, Dyskerin
DNMT1 1786
Chromatin regulator, methylates CpG
residues.
DNMT, MCMT, AIM,
ADCADN, HSN1E
DNMT3A 1788
DNA methyltransferase, NuRD
associated TBRS
DNMT3B 1789
DNA methyltransferase, NuRD
associated M.HsaIIIB, ICF1
DNTT 1791 DNA polymerase TDT
DR1 1810 Chromatin regulator NC2B
E2F1 1869 Transcription activator RBBP3, RBAP1, E2F-1
E2F2 1870 Transcription factor, cell cycle
E2F3 1871
Transcription factor, also involved in cell
cycle and DNA repair KIAA0075
E2F4 1874
Transcription factor, also involved in cell
cycle and DNA repair
P107/P130-Binding
Protein
ECT2 1894 Involved in cell cycle ARHGEF31
EGF 1950 Involved in cell growth and proliferation HOMG4, URG
EHMT2 10919
Methyltransferase that methylates
lysine residues of histone H3. C6orf30, BAT8
EID3 493861
Involved in repair of DNA double-strand
breaks by homologous recombination NSE4B

214
ELP3 55140 Chromatin remodelling/regulation HELP3, KAT9
EME1 146956
Homologous recombination, interacts
with MUS81 to form a DNA structure-
specific endonuclease SLX2A, MMS4
EME2 197342
Homologous recombination, interacts
with MUS81 to form a DNA structure-
specific endonuclease SLX2B, Gs125
ENDOV 284131 Editing and processing RNAs
EP300 2033 histone acetyltransferase, remodeller P300, RSTS2, KAT3B
ERCC1 2067 Involved in DNA repair RAD10, UV20, COFS4
ERCC2 2068
Transcription factor also involved in
DNA repair
XPD, TTD1, COFS2,
EM9
ERCC3 2071
DNA helicase and transcription factor
also involved in DNA repair
RAD25, GTF2H, BTF2,
TTD2, XPB
ERCC4 2072 Involved in DNA repair XPF, RAD1, FANCQ,
XFEPS, XPF, ERCC11
ERCC5 2073 Involved in DNA repair
ERCM2, XPGC, COFS3,
UVDR, XPG
ERCC6 2074 Involved in DNA repair
CKN2, RAD26, ARMD5,
UVSS1, COFS
ERCC8 1161 Involved in DNA repair CKN1, UVSS2
ERI1 90459
RNA exonuclease, involved in histone
RNA degradation THEX1, HEXO
EWSR1 2130 Transcription factor- repressor
EXO1 9156 DNA exonuclease HEX1, EXOI
EZH2 2146
Chromatin remodeller and transcription
repressor

215
FAAP20 199990 Involved in DNA damage repair C1orf86, FP7162
FAAP24 91442 Involved in DNA damage repair C19orf40
FAAP100 80233 Involved in Fanconi anaemia DNA
damage response C17orf70
FAN1 22909 Editing and processing nucleases MTMR15
FANCA 2175 Involved in DNA repair FACA, FANCH
FANCB 2187 Involved in DNA repair
FANCC 2176 Involved in DNA repair FACC
FANCD2 2177 Involved in DNA repair FACD, FANCD
FANCE 2178 Involved in DNA repair FACE
FANCF 2188 Involved in DNA repair
FANCG 2189 Involved in DNA repair XRCC9
FANCI 55215 Involved in DNA repair KIAA1794
FANCL 55120 Involved in DNA repair PHF9, POG
FANCM 57697 Involved in DNA repair KIAA1596, FAAP250
FBL 2091
Methyltransferase that has the ability to
methylate both RNAs and proteins. RNU3IP1
FBXO18 84893 Involved in homologous recombination HFBH1
FEN1 2237 Involved in DNA repair DNase IV, RAD2
Foxp1 27086 NuRD associated, transcription
HSPC215,
QRF1, 12CC4, HFKH1B
Foxp2 93986 NuRD associated, transcription
TNRC10,
SPCH1, CAGH44
Foxp4 116113
NuRD associated, transcriptional
repressor HFKHLA, FKHLA

216
FSBP 100861412 Transcriptional repressor
FUS 2521 Chromatin regulator
ALS6, ETM4, FUS1,
POMP75, TLS
GAR1 54433 Involved in telomerase NOLA1
GEN1 48654
Homologous recombination,
Endonuclease which resolves Holliday
junctions
GET4 51608 Post translational trafficking C7orf20
GMNN 51053 Involved in cell cycle regulation MGORS6
GNL3 26354 Involved in cell proliferation
E2IG3, NNP47, NS,
C77032
GTF2B 2959 Transcription factor S300-II, TFIIB, TF2B
GTF2F1 2962
Transcription initiation factor that binds
to RNA polymerase II
RAP74, TF2F1, TFIIF,
BTF4
GTF2H1 2965 Transcription factor BTF2, TFIIH, TFB1, P62
GTF2H2 2966 Transcription factor
T-BTF2P44, TFIIH,
BTF2, P44
GTF2H3 2967 Transcription factor TFB4, TFIIH, BTF2, P34
GTF2H4 2968 Transcription factor TFB2, TFIIH, P52
H2AFX 3014 Chromatin Structure H2AX
H2AFY 9555 Chromatin Structure H2AF12M, MH2A1
H2AFZ 3015 Chromatin Structure H2AZ
HAT1 8520 Involved in nucleosome assembly KAT1
HDAC1 3065
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones RPD3L1, GON-10, HD1

217
HDAC10 83933
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HDAC2 3066
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
YAF1, HD2, RPD3, YY1-
Associated Factor 1
HDAC3 8841
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HDAC4 9759
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HDAC5 10014
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HD5, KIAA0600, NY-
CO-9
HDAC6 10013
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HDAC7 51564
Responsible for the deacetylation of
lysine residues on the N-terminal part
of the core histones
HELLS 3070 Chromatin regulation LSH, ICF4
HELQ 113510
DNA-dependent ATPase and 5 to 3 DNA
helicase. HEL308
HELZ 9931
Helicase that plays a role in RNA
metabolism
KIAA0054,
DHRC, HUMORF5
HES1 3280 Transcriptional repressor HRY, HL, HES-1

218
HEXIM1 10614
Transcriptional regulator which
functions as a general RNA polymerase
II transcription inhibitor. HIS1, EDG1, CLP1
HIC1 3090 Transcriptional repressor ZNF901, ZBTB29
HIRA 7290
Chromatin Modifier/epigenetic
remodeller TUPLE1, DGCR1, HIR
HIST1H1A 3024 Replication Dependent Histone H1F1, H1.1, H1a
HIST1H1B 3009
Replication Dependent Histone H1F5, H1.5, H1b, H1s-
3
HIST1H1C 3006 Replication Dependent Histone H1F2, H1.2, H1s-1, H1c
HIST1H1D 3007
Replication Dependent Histone H1F3, H1.3, H1d, H1s-
2
HIST1H1E 3008
Replication Dependent Histone H1F4, H1.4, H1e, H1s-
4
HIST1H1PS1 387325 Replication Dependent Histone DJ34B20.16, H1F6P
HIST1H1PS2 10338 Replication Dependent Histone H3FEP, PH3/e
HIST1H1T 3010 Replication Dependent Histone H1FT, H1t
HIST1H2AA 221613
Replication Dependent Histone BA317E16.2, H2AFR,
H2AA, TH2A
HIST1H2AB 8335 Replication Dependent Histone H2AFM, H2A/m
HIST1H2AC 8334 Replication Dependent Histone H2AFL
HIST1H2AD 3013 Replication Dependent Histone H2AFG, H2A/g, H2A.3
HIST1H2AE 3012 Replication Dependent Histone H2AFA, H2A/a, H2A.1
HIST1H2AG 8969
Replication Dependent Histone H2AFP, pH2A/f,
H2A/p, H2A.1b
HIST1H2AH 85235
Replication Dependent Histone H2AFALii, dJ86C11.1,
H2A/S

219
HIST1H2AI 8329 Replication Dependent Histone H2AFC, H2A/c
HIST1H2AJ 8331 Replication Dependent Histone H2AFE, H2A/E
HIST1H2AK 8330 Replication Dependent Histone H2AFD, H2A/d
HIST1H2AL 8332
Replication Dependent Histone H2AFI, H2A/I,
dJ193B12.9
HIST1H2AM 8336 Replication Dependent Histone H2AFN, H2A/n, H2A.1
HIST1H2BA 255626
Replication Dependent Histone bA317E16.3, STBP,
TSH2B, H2BFU
HIST1H2BB 3018 Replication Dependent Histone H2BFF, H2B/f
HIST1H2BC 8347 Replication Dependent Histone H2BFL, H2B/l, H2B.1
HIST1H2BD 8347 Replication Dependent Histone H2BFB, H2B/b
HIST1H2BE 8344 Replication Dependent Histone H2BFH, H2B/h, H2B.h
HIST1H2BF 8343 Replication Dependent Histone H2BFG, H2B/g
HIST1H2BG 8339 Replication Dependent Histone H2BFA, H2B/a, H2B.1A
HIST1H2BH 8345 Replication Dependent Histone H2BFJ, H2B/j
HIST1H2BI 8346 Replication Dependent Histone H2BFK, H2B/k
HIST1H2BJ 8970 Replication Dependent Histone H2BFR, H2B/r
HIST1H2BK 85236 Replication Dependent Histone H2BFT, H2BFAiii
HIST1H2BL 8340
Replication Dependent Histone H2BFC, H2B/c,
dJ97D16.4
HIST1H2BM 8342
Replication Dependent Histone H2BFE, H2B/e,
dJ160A22.3
HIST1H2BN 8341 Replication Dependent Histone H2BFD, H2B/d
HIST1H2BO 8348 Replication Dependent Histone H2BFN, H2B/n, H2B.2
HIST1H3A 8350 Replication Dependent Histone H3FA, H3/A

220
HIST1H3B 8358 Replication Dependent Histone H3FL, H3/l
HIST1H3C 8352 Replication Dependent Histone H3FC, H3/c, H3.1
HIST1H3D 8351 Replication Dependent Histone H3FB, H3/b,
HIST1H3E 8353 Replication Dependent Histone H3FD, H3/d, H3.1
HIST1H3F 8968 Replication Dependent Histone H3FI, H3/i
HIST1H3G 8355 Replication Dependent Histone H3FH, H3/h
HIST1H3H 8357 Replication Dependent Histone H3FK, H3/k, H3F1K
HIST1H3I 8354 Replication Dependent Histone H3FF, H3/f, H3.f
HIST1H3J 8356 Replication Dependent Histone H3FJ, H3/j
HIST1H4A 8359 Replication Dependent Histone H4FA
HIST1H4B 8366 Replication Dependent Histone H4FI, H4/I
HIST1H4C 8364
Replication Dependent Histone H4FG, H4/g,
dJ221C16.1
HIST1H4D 8360 Replication Dependent Histone H4FB, H4/b
HIST1H4E 8367 Replication Dependent Histone H4FJ, H4/j
HIST1H4F 8361 Replication Dependent Histone H4FC, H4/c, H4
HIST1H4G 8369 Replication Dependent Histone H4FL, H4/l
HIST1H4H 8365 Replication Dependent Histone H4FH, H4/h
HIST1H4I 8294 Replication Dependent Histone H4FM, H4/m
HIST1H4J 8363 Replication Dependent Histone H4FE, H4/e, H4F2iv
HIST1H4K 8362
Replication Dependent Histone H4FD, H4/d, H4F2iii,
dJ160A22.1
HIST1H4L 8368 Replication Dependent Histone H4FK, H4.k, H4/k
HIST2H2AA3 8337
Replication Dependent Histone H2AFO, HIST2H2AA,
H2A.2, H2A/q

221
HIST2H2AA4 723790
Replication Dependent Histone H2A/r, H2BFQ, H2B/q,
H2B.1
HIST2H2AB 317772 Replication Dependent Histone H2AB
HIST2H2AC 8338 Replication Dependent Histone H2A, H2AFQ, H2A/q
HIST2H2BA 337875 Replication Dependent Histone
HIST2H2BB 338391 Replication Dependent Histone
HIST2H2BC 337873 Replication Dependent Histone H2B/t, HIST2H2BD
HIST2H2BD 337874 Replication Dependent Histone H2BFO, H2B/s, H2B/o
HIST2H2BE 8349
Replication Dependent Histone H2B, H2BQ, H2BFQ,
H2BGL105
HIST2H2BF 440689 Replication Dependent Histone
HIST2H3A 333932 Replication Dependent Histone H3/N, H3/O
HIST2H3C 126961 Replication Dependent Histone H3/M, H3, H3.2
HIST2H3D 653604 Replication Dependent Histone
HIST2H4A 8370
Replication Dependent Histone H4F2, H4FN, HIST2H4,
H4/n
HIST2H4B 554313 Replication Dependent Histone H4/o
HIST3H2A 92815 Replication Dependent Histone MGC3165
HIST3H2BA 337872 Replication Dependent Histone
HIST3H2BB 128312 Replication Dependent Histone
HIST3H3 8290 Replication Dependent Histone H3FT, H3t, H3/g, H3.4
HIST4H4 121504 Replication Dependent Histone MGC24116
HLTF 6596
SWI/SNF family, helicase and ubiquitin
ligase functions SMARCA3

222
HMGA1 3159 Transcription
HMGIY, HMGA1A,
HMG-R
HMGA2 8091 Transcription
HMGIC, LIPO, BABL,
STQTL9
HMGB1 3146
Involved in transcription and chromatin
remodelling HMG1, SBP-1, HMG1
HMGB2 3148
Involved in transcription and chromatin
remodelling HMG2
HMGN1 3150
Involved in transcription and chromatin
remodelling HMG14
HMGN2 3151
Involved in transcription and chromatin
remodelling HMG17
HNRNPA0 10949 RNA processing HNRPA0
HNRNPA1 3178 RNA processing
HNRPA1, IBMPFD3,
ALS19, ALS20, UP 1
HNRNPA2B1 3181 RNA processing
HNRPA2B1, IBMPFD2,
HNRPA2, HNRPB1,
RNPA2
HNRNPD 3184
Involved in RNA processing and DNA
replication
AUF1, HNRPD, P37,
HnRNPD0, HNRPD
HSP90AA1 3320
Involved in cell cycle and signal
transduction
HSPC1, HSPCA, LAP2,
EL52, Hsp90
HSPA1L 3305 Protein stabilization HSP70T
HUS1 3364 Role in DNA repair
IDH1 3417 Chromatin regulation
IDPC, IDCD, HEL-216,
PICD
IDH2 3418 Chromatin regulation
IDPM, IDHM, D2HGA2,
IDH

223
IFIH1 64135 RNA helicase
MDA5, IDDM19,
IDDM19, RH116, AGS7
IGF1 3479 Growth factor and replication IGFI, IBP1, IGF-I, MGF
IKBKAP 8518
Involved in transcription and chromatin
remodelling
DYS, ELP1, IKAP, P150,
IKI3, DYS, FD
INO80 54617
DNA helicase and transcription factor
also involved in DNA repair INOC1, KIAA1259
INO80C 125476
Involved in transcriptional regulation,
DNA replication and DNA repair. C18orf37
INO80E 283899
Involved in transcriptional regulation,
DNA replication and DNA repair. CCDC95
ISG20 3669
Exoribonuclease that acts on single-
stranded RNA CD25, HEM45
KAT2A 2648
Histone acetyltransferase that
promotes transcriptional activation. GCN5L2, STAF97
KAT2B 8850
Histone acetyltransferase that
promotes transcriptional activation.
PCAF, CAF, Lysine
Acetyltransferase 2B
KAT5 10524 Transcriptional activation
HTATIP, TIP60,
HTATIP, ESA1, PLIP,
ZC2HC5
KDM1A 23028 Histone demethylase, NuRD associated
lysine (K)-specific
demethylase
1A (LSD1)
KDM3B 51780 Demethylates Lys-9 of histone H3
C5orf7, JMJD1B,
KIAA1082, NET22
KDM4A 9682
Demethylates Lys-9 and Lys-36 residues
of histone H3
JMJD2A, JHDM3A,
KIAA0677, TDRD14A

224
KDM4B 23030 Demethylates Lys-9 of histone H3
JMJD2B, KIAA0876,
TDRD14B, JHDM3B
KDM4C 23081
Demethylates Lys-9 and Lys-36 residues
of histone H3
JMJD2C, TDRD14C,
KIAA0780, GASC1,
JHDM3C
KDM5A 5927 Demethylates Lys-4 of histone H3
RBBP2, JARID1A,
RBBP2
KDM5B 10765
Demethylates Lys-4 of histone H3
JARID1B, RBP2-H1,
PLU-1, CT31, PUT1,
RBBP2H1A, PPP1R98
KDM5C 8242 Demethylates Lys-4 of histone H3
SMCX, JARID1C,
MRX13, MRXJ,
MRXSCJ, XE169,
DXS1272E
KDM6B 23135
Histone demethylase, demethylates
Lys-27 of histone H3
KMT2B
9757
Methylates Lys-4 of histone H3
MLL4, MLL1B, CXXC10,
KIAA0304, TRX2,
WBP7
KMT2D 8085 Methylates Lys-4 of histone H3
(H3K4me).
MLL2, TNRC21,
AAD10, KABUK1,
CAGL114, ALR
KMT2E 55904
Mono- and di-methylates Lys-4 of
histone H3 (H3K4me1 and H3K4me2)
MLL5, NKp44L,
HDCMC04P
KMT5A 387893
Monomethylates both histones and
non-histone proteins, involved in
mitosis
SETD8, H4-K20-
Specific Histone
Methyltransferase

225
KMT5B 51111 Trimethylates Lys-20 of histone H4.
SUV420H1, Lysine-
Specific
Methyltransferase 5B
KTM5C 84787 Histone methyltransferase
SUV420H2, Lysine (K)-
Specific
Methyltransferase 5C
KRAS 3845 Cell proliferation
KRIT1 889 Cell proliferation CCM1, CaM
LIG1 3978 DNA repair
LIG3 3980 DNA repair LIG2
LIG4 3981 DNA repair LIG4S
MAD2L2 10459 Cell cycle and DNA repair REV7, POLZ2, MAD2B
MAVS 57506 Detect intracellular dsRNA
VISA, IPS1, KIAA1271,
CARDIF
MBD1 4152
Transcriptional corepressor and
coactivator, Chromatin
Modifier/epigenetic remodeller
MBD2 8932
Transcriptional corepressor and
coactivator, NuRD complex
DMTase, NY-CO-41,
demethylase
MBD3 53615
Transcriptional corepressor and
coactivator, NuRD complex
MBD4 8930
Transcriptional corepressor and
coactivator
MCM2 4171 DNA replication
CCNL1, CDCL1,
MITOTIN, D3S3194,
KIAA0030
MCM3 4172 DNA replication
RLFB, HCC5, P1-
MCM3, P102

226
MCM4 4173 Initiation of DNA replication CDC21
MCM5 4174 Cell cycle CDC46
MCM6 4175 Initiation of DNA replication
MCM7 4176 DNA replication
MCM2, CDC47,
P1CDC47, PPP1R104,
PNAS146, P85MCM
MCPH1 79648 DNA damage response BRIT1, MCT
MCRS1 10445 Transcription regulator
MSP58, P78, ICP22BP,
INO80Q
MDC1 9656 Cell cycle and DNA damage response NFBD1, KIAA0170
MECP2 4204 Mediates transcriptional expression
RTT, MRX16, MRX79,
AUTSX3, RTS, MRXSL
MED12 9968 Transcription
TNRC11, FGS1,
ARC240, CAGH45,
HOPA, MED12S
MEN1 4221 Transcriptional regulator SCG2, Menin
MGMT 4255
Protein catalyses transfer of methyl
groups from O(6)-alkylguanine and
other methylated moieties of the DNA
to its own molecule, which repairs the
toxic lesions.
O6-Methylguanine-
DNA
Methyltransferase
MKI67 4288 Cell proliferation MIB-1, KIA, PPP1R105
MLH1 4292 DNA repair
FCC2, HMLH1, HNPCC,
COCA2
MLLT1 4298 Transcription regulator
YEATS1, LTG19, ENL,
CTC-503J8.6
MNAT1 4331 Transcription regulator
CAP35, RNF66, MAT1,
P36, TFB3

227
MPG 4350 DNA repair
ADPG, AAG, MDG,
APNG, PIG16, PIG11,
CRA36.1
MPLKIP 136647 Cell cycle TTDN1, C7orf11
MRE11A 4361
Component of the MRN complex, role
in double-strand break repair, DNA
recombination, maintenance of
telomere integrity and meiosis. MRE11
MSH2 4436
Post-replicative DNA mismatch repair
system
MSH3 4437 DNA repair
MRP1, DUP, DUC1,
DUG
MSH4 4438 Involved in meiotic recombination.
MSH5 4439 Involved in meiotic recombination. MUTSH5, NG23, G7
MSH6 2956 DNA repair
GTBP, HNPCC5,
HMSH6, HSAP, P160,
GTMBP
MTA1 9112
Transcriptional corepressor and
coactivator, NuRD complex
p70, MTA1L1, PID,
KIAA1264
MTA2 9219
Transcriptional corepressor and
coactivator, NuRD complex
p70, MTA1L1, PID,
KIAA1265
MTA3 57504
Transcriptional corepressor and
coactivator, NuRD complex
p70, MTA1L1, PID,
KIAA1266
MTF2 22823 Transcriptional activator
M96, TDRD19A,
DJ976O13.2, PCL2
MUS81 80198
DNA repair and homologous
recombination SLX3
MUTYH 4595 DNA repair HMYH

228
MYC 4609 Transcription factor
C-Myc, MYCC, MRTL,
BHLHe39
NAA10 8260
Catalytic subunit of the N-terminal
acetyltransferase A (NatA) complex
ARD1, ARD1A, TE2,
NATD, OGDNS,
DXS707, MCOPS1
NABP1 64859 DNA repair and cell cycle
SSB2, OBFC2A, SOSS-
B2
NABP2 79035 DNA repair and cell cycle
SSB1, OBFC2B,
LP3587, SOSS-B1
NBS1 4683
Component of MRN complex, DNA
repair NBS, NBN, ATV, P95
NCAPG 64151 Involved in meiosis
YCG1, CHCG, HCAP-G,
NYMEL3, NY-MEL-3,
CAPG
NCL 4691
Chromatin decondensation by binding
H1
NCOA1 8648 Involved in transcription
F-SRC-1, BHLHe42,
KAT13A, NCoA-1, SRC-
1, RIP160
NCOA3 8202 Chromatin Modifier
NCOR1 9611 Chromatin Modifier
NCOR2 9612 transcriptional regulator, promoting
chromatin condensation
Ndc80 10403 Involved in cell cycle
KNTC2, HEC, KNTC2,
TID3
NELFCD 51497 Transcription TH1L, HSPC130, NELFD
NFRKB 4798
Involved in transcriptional regulation
and DNA repair INO80G

229
NHEJ1 79840 DNA repair XLF, XRCC4-Like Factor
NHP2 55651 Involved in trafficking hTR NOLA2, DKCB2, NHP2P
NOP10 55505 Involved in trafficking hTR NOLA3, DKCB1
NSMCE2 286053
Component of the SMC5-SMC6
complex, a complex involved in DNA
double-strand break repair by
homologous recombination. C8orf36
NSMCE3 56160 DNA repair
NDNL2, MAGEG1,
HCA4, NSE3
NSMCE4A 54780 DNA repair C10orf86, NSE4A
NTHL1 4913 DNA repair NTH1, FAP3, OCTS3
NUTM1 256646 Chromatin regulation
C15orf55, FAM22H,
NUT
OBFC1 79991 DNA replication RPA-32, STN1, AAF44
OGG1 4968 DNA repair genes
MMH, HOGG1, OGH1,
MUTM
ORC1 4998 DNA replication
ORC1L, PARC1,
HSORC1
ORC2 4999 DNA replication ORC2L
ORC3 23595 DNA replication ORC3L, LATHEO, LAT
ORC4 5000 DNA replication ORC4L
ORC5 5001 DNA replication
ORC5L, ORC5P,
ORC5T, PPP1R117
ORC6 23594 DNA replication ORC6L
PABPC1 26986 RNA processing
PAB1, PABPC2,
PABPL1
PAF1 54623 Transcription PD2

230
PALB2 79728 DNA repair FANCN, PNCA3
PAPOLG 64895
Editing and processing nucleases, 3'
mRNA polyadenylation PAP2, PAPG
PARP1 142 DNA damage response
ADPRT1, PARP, PPOL,
ARTD1
PARP2 10038 DNA damage response
ADPRTL2, ADPRTL3,
ARTD2, PARP-2,
PADPRT-2
PARP4 143 DNA damage response
ADPRTL1, KIAA0177,
VAULT3, VWA5C,
P193, PH5P, PARP-4
PAX8 7849 Transcription factor
PAXIP1 22976
Involved in DNA damage response and
transcriptional regulation
PAXIP1L, TNRC2,
PACIP1, CAGF29,
CAGF28, PAXIP1L
PCNA 5111 DNA polymerase
PER1 5187 Transcriptional repressor PER, KIAA0482, RIGUI
PES1 23481
Involved in cell proliferation and RNA
processing PES
PGBD3 267004 Transcription
PHF1 5252 Transcriptional repressor
HPCl1, PCL1,
TDRD19C, MTF2L2
PHF2 5253 Lysine demethylase KIAA0662, JHDM1E
PHF20 51230 Transcriptional regulator
C20orf104, GLEA2,
HCA58, NZF, TZP
PHF20L1 51105 Involved in chromatin regulation
URLC1, CGI-72,
TDRD20B

231
PIF1 80119 DNA helicase C15orf20, PIF
PINX1 54984
Involved in chromosome segregation
and TRF1 and TERT accumulation in
nucleus
LPTL, LPTL,
PIN2/TERF1
Interacting,
Telomerase Inhibitor 1
PKMYT1 9088 Cell cycle PPP1R126, MyT1
PLK1 5347 Cell cycle PLK, STPK13, EC 2.7.11
PML 5371
Involved in tumour suppression,
transcriptional regulation, apoptosis,
senescence, DNA damage response, and
viral defence mechanisms.
PP8675, TRIM19,
RNF71, MYL
PMS1 5378 DNA repair
PMSL1, HNPCC3,
HPMS1, MLH2
PMS2 5395 DNA repair
PMSL2, HNPCC4,
PMS2CL, MLH4
POLA1 5422 DNA polymerase POLA, NSX, P180
POLA2 23649 DNA replication
POLB 5423 DNA polymerase
POLD1 5424 DNA polymerase
POLD, CDC2, CRCS10,
MDPL
POLD2 5425 Subunit of DNA polymerase
POLD3 10714 Subunit of DNA polymerase
p66, p68, KIAA0039,
PPP1R128
POLD4 57804 Subunit of DNA polymerase P12, POLDS
POLE 5426 Subunit of DNA polymerase POLE1, FILS, CRCS12
POLE2 5427 Subunit of DNA polymerase DPE2

232
POLE3 54107
Role in allowing polymerase epsilon to
carry out its replication and/or repair
function- chromatin remodelling P17, YBL1, CHARAC17
POLE4 56655
Role in allowing polymerase epsilon to
carry out its replication and/or repair
function- chromatin remodelling P12, YHHQ1
POLG 5428 Mitochondrial DNA polymerase
POLG1, POLGA, MDP1,
SANDO, MIRAS, SCAE,
PEO
POLH 5429 DNA polymerase RAD30A, XPV
POLI 11201 DNA polymerase RAD30B, RAD3OB
POLK 51426 DNA polymerase
DINB1, POLQ, DINB1,
DINP
POLL 27343 DNA polymerase BETAN, POLKAPPA
POLM 27434
DNA polymerase, involved in repair of
DNA double-strand breaks by non-
homologous end joining
POLN 353497 DNA polymerase POL4P
POLQ 10721
DNA polymerase that promotes
microhomology-mediated end-joining
POLR2A 5430 RNA polymerase II
POLR2B 5431 RNA polymerase subunit
POL2RB, HRPB140,
RPB2
POLR2C 5432 RNA polymerase subunit RPB3, HRPB33
POLR2D 5433 RNA polymerase subunit HSRBP4, RPB16, RBP4
POLR2F 5435 RNA polymerase subunit
POLRF, RPABC2, RPB6,
RPC15

233
POLR2G 5436 RNA polymerase subunit
RPB19, RPB7, HRPB19,
HsRPB7
POLR2H 5437 RNA polymerase subunit RPB17, RPABC3
POLR2I 5438 RNA polymerase subunit RPB9, RPB14.5
POLR2J 5439 RNA polymerase subunit
RPB11, HRPB14,
POLR2J1
POLR2K 5440 Subunit of RNA polymerase II RPB12, RPABC4
POLR2L 5441 RNA polymerase subunit RPB10, RPABC5
POLR3A 11128
Catalytic component of RNA
polymerase III
RPC1, HLD7, RPC155,
ADDH
POT1 25913 Shelterin complex GLM9, CMM10
PPARG 5468 Transcription factor NR1C3, GLM1, CIMT1
PPP1CA 5499 Involved in cell division PPP1A
PPP1CB 5500 Involved in cell division PP1B
PPP1CC 5501 Involved in cell division PP1C
PPP1R10 5514
Involved in cell cycle progression, DNA
repair and apoptosis
CAT53, PP1R10,
PNUTS, FB19, P99
PPP2R1A 5518
Involved in the negative control of cell
growth and division. MRD36, PR65A
PPP2R1B 5519
Involved in the negative control of cell
growth and division. PR65B
PPP2R2A 5520 Cell division and growth
PRDM16 63976
Transcriptional regulator
PFM13, MEL1,
KIAA1675, CMD1LL,
LVNC8

234
PRDM2 7799
Histone methyltransferase that
specifically methylates Lys-9 of histone
H3
PRIM1 5557
Polymerase that synthesizes small RNA
primers for the Okazaki fragments
made during discontinuous DNA
replication.
P49, DNA Primase
Subunit 48
PRIM2 5558
Polymerase that synthesizes small RNA
primers for the Okazaki fragments
made during discontinuous DNA
replication. PRIM2A, P58
PRKCA 5578
Involved in cell cycle checkpoint and
transcription regulation PKCA, AAG6
PRKCB 5579
Involved in cell cycle checkpoint and
transcription regulation
PRKCB2, PKCB,
PRKCB1
PRKDC 5591
DNA-dependent protein kinase involved
in DNA repair
HYRC, HYRC1, XRCC7,
DNAPK
PRMT1 3276 Arginine methyltransferase
HRMT1L2, IR1B4,
ANM1, HCP1
PRMT2 3275 Methylates H4 HRMT1L1
PRMT3 10196
Methylates (mono and asymmetric
dimethylation) the guanidino nitrogens
of arginyl residues in some proteins. HRMT1L3
PRMT5 10419 Chromatin regulator SKB1, HRMT1L5
PRMT6 55170 Methyltransferase activity HRMT1L6
PRPF19 27339
Involved in pre-mRNA splicing and DNA
repair.
PSO4, PRP19,
NMP200, UBOX4
PSIP1 11168 Transcriptional coactivator
PSIP2, DFS70, LEDGF,
P75, P52

235
PTGES3 10728 Involved in transcriptional regulation CPGES, TEBP, P23
PURA 5813 Transcriptional activator
MRD31, PUR1, PUR-
ALPHA
PYGO1 26108 Involved in signal transduction through
the Wnt pathway.
RAD1 5810 Involved in DNA damage response REC1, HRAD1
RAD17 5884
Involved in cell growth, maintenance of
chromosomal stability, and ATR-
dependent checkpoint activation upon
DNA damage. RAD24, CCYC, R24L
RAD18 56852
Involved in ubiquitination and DNA
damage repair RNF73
RAD21 5885
Involved in cell cycle, DNA repair and
apoptosis.
CDLS4, SCC1, MCD,
NXP1 1
RAD23B 5887 DNA repair gene P58
RAD50 10111
Component of the MRN complex, which
plays a central role in double-strand
break (DSB) repair, DNA recombination,
maintenance of telomere integrity and
meiosis. NBSLD
RAD51 5888
Homologous recombination DNA
damage response
RAD51A, RECA, BRCC5,
FANCR, HsT16930,
MRMV2
RAD51AP1 10635 Involved in DNA damage response PIR51
RAD51B 5890
Involved in DNA damage repair via
homologous recombination RAD51L1, REC2
RAD51C 5889
Involved in DNA damage repair via
homologous recombination FANCO, BROVCA3

236
RAD51D 5892
Involved in DNA damage repair via
homologous recombination
RAD51L3, BROVCA4,
TRAD, R51H3
RAD52 5893
Involved in double-stranded break
repair.
RAD54B 25788
Involved in DNA repair and mitotic
recombination. RDH54
RAD54L 8438
Involved in DNA repair and mitotic
recombination. RAD54A, HR54
RAD9A 5883 Involved in DNA damage response RAD9
RAP1A 5906
Regulates signalling pathways that
affect cell proliferation and adhesion RAP1
RAPGEF1 2889
Involved in signalling cascade that
induces apoptosis GRF2, C3G
RASSF1 11186
Involved in cell cycle and cell death
pathways
RDA32,123F2,
NORE2A, REH3P21
RB1 5925
Involved in chromatin regulation, cell
cycle and is a tumour suppressor gene OSRC
RBBP4 5928
Involved in histone acetylation and
chromatin assembly. Component NuRD RbAp48
RBBP5 5929
Involved in regulating gene induction
and H3 Lys-4 methylation RBQ3, SWD
RBBP7 5931
Core histone-binding subunit, NuRD
complex RbAp46
RBBP8 5932 DNA damage repair CtIP, SCKL2
RBL1 5933
Involved in cell division and
heterochromatin formation
RBL2 5934
Involved in cell division and
heterochromatin formation

237
RDM1 201299
May confer resistance to the antitumor
agent cisplatin. Binds to DNA and RNA. RAD52B
RECQL 5965 DNA helicase RECQ1
RECQL4 9401 DNA-dependent ATPase
RECQL5 9400
DNA helicase, role in DNA replication,
transcription and repair.
RELA 5970 Chromatin remodeller NFKB3
REV1 51455
Deoxycytidyl transferase involved in
DNA repair. REV1L
REV3L 5980
Interacts with MAD2L2 to form the
error prone DNA polymerase zeta
involved in translesion DNA synthesis. POLZ
RFC1 5981
Component of a complex that possesses
DNA-dependent ATPase activity
RFC2 5982 Component of a complex that possesses
DNA-dependent ATPase activity
RFC3 5983
Component of a complex that possesses
DNA-dependent ATPase activity
RFC4 5984
Component of a complex that possesses
DNA-dependent ATPase activity
RFC5 5985
Component of a complex that possesses
DNA-dependent ATPase activity
RIF1 55183 Conserved DNA damage response gene
RARRES3 5920 Tumour suppressor or growth regulator. RIG1, TIG3
RING1 6015 Involved in chromatin regulation
RMI1 80010
Involved in the dissolution of double
Holliday junctions

238
RMI2 116028 Involved in homologous recombination C16orf75
RNF168 165918
Involved in ubiquitination and DNA
damage repair
RNF2 6045 Chromatin modifier
RNF20 56254 Chromatin modifier
RNF4 6047
Chromatin modifier, regulates
degradation of PML
RNF40 9810 Chromatin modifier
RNF8 9025 Involved in DNA damage response
RPA1 6117
Involved in DNA replication and damage
response
RPA2 6118
Involved in DNA replication and damage
response
RPA3 6119
Involved in DNA replication and damage
response
RPA4 29935
Binds ssDNA and probably plays a role
in DNA repair.
RPAIN 84268 Participates in DNA metabolism RIP, HRIP
RRP8 23378 Chromatin regulator KIAA0409, NML
RTEL1 51750
TP-dependent DNA helicase implicated
in telomere-length regulation, DNA
repair and the maintenance of genomic
stability.
C20orf41, NHL,
DKCA4, DKCB5,
PFBMFT3
RUVBL1 8607
DNA-dependent ATPase and DNA
helicase activities, involved in chromatin
modification
TIP49, PONTIN, ECP54,
TIH1

239
RUVBL2 10856
DNA-dependent ATPase and DNA
helicase activities, involved in chromatin
modification
TIP48, REPTIN, ECP51,
TIH2
SART1 9092 Role in mRNA splicing
HOMS1, Snu66,
SNRNP110, Ara1
SATB1 6304 Chromatin remodeller
SETD7 80854
Histone methyltransferase that
specifically monomethylates Lys-4 of
histone H3 SET9, SET7, KMT7
SETDB1 9869
Regulates histone methylation, gene
silencing, and transcriptional
repression-trimethylates Lys-9 of
histone H3
SETMAR 6419 DNA repair METNASE, Mar1
SFR1 119392
Required for double-strand break repair
via homologous recombination C10orf78, MEIR5
SGO1 151648
Role in chromosome cohesion during
mitosis
SGOL1, CAID, NY-BR-
85, SGO
SHFM1 7979 Involved in DNA damage response
SHFD1, DSS1, SHSF1,
SEM1, ECD
SHPRH 257218
E3 ubiquitin-protein ligase involved in
DNA repair.
SIN3A 25942 Acts as a transcriptional repressor.
SIN3B 23309
Acts as a transcriptional repressor,
NuRD associated
SIRT1 23411
Transcriptional regulator involved in cell
cycle, response to DNA damage,
metabolism, apoptosis and autophagy. Sirtuin 1
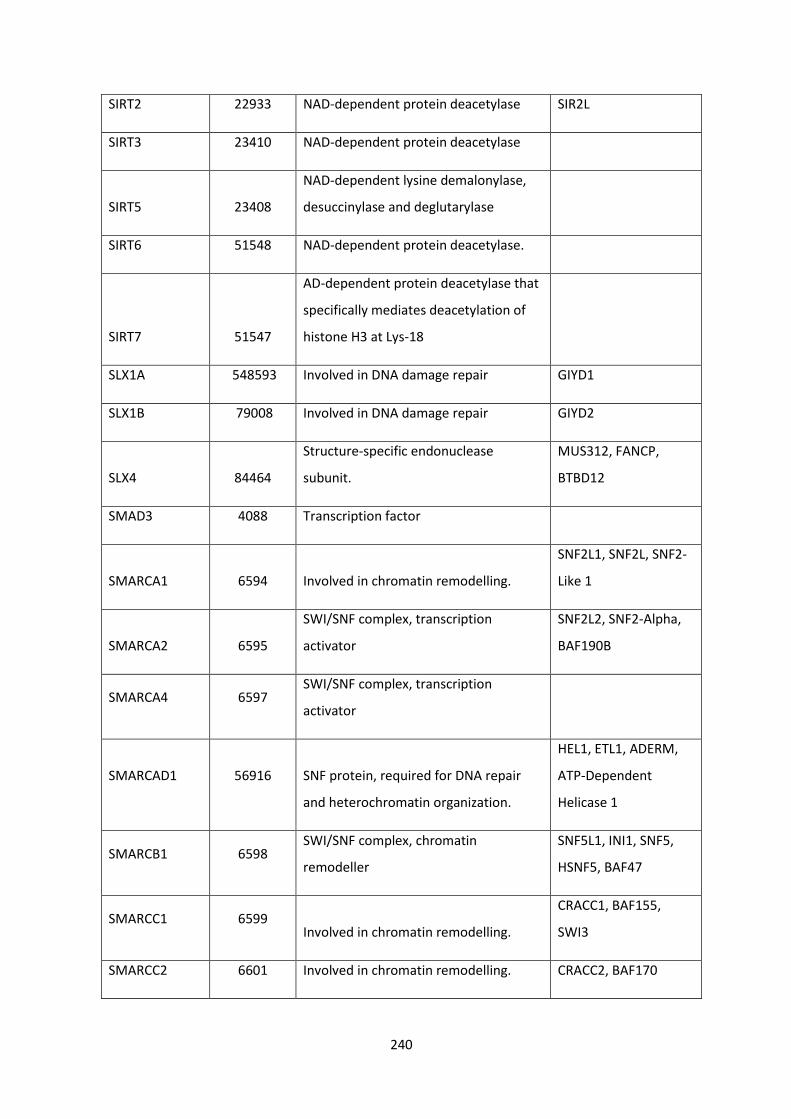
240
SIRT2 22933 NAD-dependent protein deacetylase SIR2L
SIRT3 23410 NAD-dependent protein deacetylase
SIRT5 23408
NAD-dependent lysine demalonylase,
desuccinylase and deglutarylase
SIRT6 51548 NAD-dependent protein deacetylase.
SIRT7 51547
AD-dependent protein deacetylase that
specifically mediates deacetylation of
histone H3 at Lys-18
SLX1A 548593 Involved in DNA damage repair GIYD1
SLX1B 79008 Involved in DNA damage repair GIYD2
SLX4 84464
Structure-specific endonuclease
subunit.
MUS312, FANCP,
BTBD12
SMAD3 4088 Transcription factor
SMARCA1 6594 Involved in chromatin remodelling.
SNF2L1, SNF2L, SNF2-
Like 1
SMARCA2 6595
SWI/SNF complex, transcription
activator
SNF2L2, SNF2-Alpha,
BAF190B
SMARCA4 6597 SWI/SNF complex, transcription
activator
SMARCAD1 56916 SNF protein, required for DNA repair
and heterochromatin organization.
HEL1, ETL1, ADERM,
ATP-Dependent
Helicase 1
SMARCB1 6598 SWI/SNF complex, chromatin
remodeller
SNF5L1, INI1, SNF5,
HSNF5, BAF47
SMARCC1 6599 Involved in chromatin remodelling.
CRACC1, BAF155,
SWI3
SMARCC2 6601 Involved in chromatin remodelling. CRACC2, BAF170

241
SMARCD1 6602 Involved in chromatin remodelling. CRACD1, BAF60A
SMARCD2 6603 Involved in chromatin remodelling. CRACD2, BAF60B
SMARCD3 6604 Involved in chromatin remodelling. CRACD3, BAF60C
SMC1A 8243
Involved in chromosome cohesion
during cell cycle and in DNA repair.
SMC1L1, CDLS2,
DXS423E
SMC2 10592
Cell cycle, DNA replication and DNA
repair SMC2L1, CAP-E
SMC3 9126
Cell cycle, DNA replication and DNA
repair CSPG6
SMC4 10051
Cell cycle, DNA replication and DNA
repair SMC4L1, CAP-C
SMC5 23137
Involved in DNA double-strand breaks
by homologous recombination. SMC5L1
SMC6 79677
Involved in DNA double-strand breaks
by homologous recombination. SMC6L1
SMG1 23049
RNA degradation and DNA damage
response
SMG5 23381
Involved in nonsense-mediated mRNA
decay, telomerase associated protein Est1p-Like Protein B
SMG6 23293
Involved in nonsense-mediated mRNA
decay
C17orf31, EST1-Like
Protein A
SMG7 9887
Plays a role in nonsense-mediated
mRNA decay.
C1orf16, EST1-Like
Protein C
SMUG1 23583 DNA repair gene FDG
SMYD2 56950
Methylates both histones and non-
histone proteins, including p53/TP53
and RB1.
KMT3C, ZMYND14,
HSKM-B

242
SP1 6667
Chromatin regulator, DNA damage
response
SP100 6672 Component of PML body
SP110 3431 Transcription factor IFI41, IFI75, VODI
SPO11 23626 Required for meiotic recombination
SPATA43, TOPVIA,
CT35
SPRTN 83932 DNA repair
c1orf124, PRO4323,
DVC1
SRCAP 10847 Chromatin regulator
SSB 6741 Involved in RNA metabolism LARP3
SSBP1 6742
involved in mitochondrial DNA
replication, binds ssDNA SSBP, SOSS-B1, MtSSB
SSRP1 6749 Chromatin remodeller
FACT, T160, SSRP1,
T160
SSU72 29101 Role in RNA processing and termination. HSPC182, PNAS-120
STAG1 10274 DNA replication SCC3A, SA1
STAG2 10735
Chromatin Modifier/epigenetic
remodeller SCC3B, SA2
STRA13 201254
Involved in DNA damage repair and
genome maintenance. CENPX, MHF2, MHF2
SUN1 23353
Involved in telomere attachment to
nuclear envelope and gametogenesis. UNC84A, KIAA0810
SUPT16H 11198 Chromatin remodeller
CDC68, SPT16, SPT16,
FACTP140
SUPT5H 6829
Involved in mRNA processing and
transcription elongation SPT5H

243
SUV39H1 6839 Chromatin remodeller
SUV39H, HTDG,
KMT1A, MG44,
Histone H3-K9
Methyltransferase 1
SUV39H2 79723 Chromatin remodeller
KMT1B, Histone H3-K9
Methyltransferase 2
SUZ12 23512 Chromatin remodeller CHET9, JJAZ1
SWI5 375757 Involved in homologous recombination C9orf119, SAE3
SYMPK 8189
Involved in histone mRNA 3-end
processing. SYM, SPK
TAF1 6872 Transcription and phosphorylation
DYT3, CCGS, CCG1,
BA2R, TAF2A
TBK1 29110 Ubiquitination NAK, T2K
TBP 6908 Transcription TF2D, GTF2D, SCA17
TCEA1 6917
Assembly of RNA Polymerase-II
Initiation Complex TFIIS, GTF2S
TCEB3 6924 Transcription TCEB3A
TDG 6996 Chromatin Structure and Modification
TDP1 55775 DNA repair gene
TDRD1 56165 piRNA metabolic process
TDRD10 126668
Nucleic acid binding and nucleotide
binding.
TDRD12 91646 ATP-dependent RNA helicase activity. ECAT8
TDRD3 81550
poly(A) RNA binding and chromatin
binding.
TDRD5 163589 Involved in DNA integrity TUDOR3

244
TDRD6 221400
Involved in proper precursor and
mature miRNA expression
SPATA36, TDR2,
BA446F17.4
TDRD7 23424 Post-transcriptional regulation CATC4, TRAP
TDRD9 122402
Nucleic acid binding and helicase
activity. C14orf75, NET54
TDRKH 11022 Nucleic acid binding and RNA binding. TDRD2
TELO2 9894 Regulator of the DNA damage response TEL2, CLK2, HCLK2
TEP1 7011 Telomerase TP1, TLP1, P240
TERC 7012 Telomerase TRC3, DKCA1, TR
TERF1 7013 Shelterin complex TRF1, TRBF1, PIN2
TERF2 7014 Shelterin complex TRF2, TRBF2
TERF2IP 54386
Shelterin complex and as a transcription
regulator.
RAP1, TERF2
Interacting Protein
TERT 7015 Telomerase
TCS1, EST2, TP2,
DKCA2, TRT
TET1 80312 Chromatin Modifier/epigenetic
remodeller, DNA demethylation CXXC6
TET2 54790 Chromatin Modifier/epigenetic
remodeller, DNA demethylation KIAA1546
TFAM 7019
Mitochondrial transcription regulation,
replication and repair. TCF6, TCF6L2
TGFB1 7040
Controls proliferation, differentiation,
adhesion, migration and other functions TGFB, DPD1
TIAL1 7073
RNA binding protein involved in
translational control, splicing and
apoptosis. TIAR, TCBP

245
TICAM1 148022
Protein kinase binding and signal
transducer activity. TRIF
TINF2 26277 Shelterin complex
TIN2, DKCA3, TERF1
(TRF1)-Interacting
Nuclear Factor 2
TMEM173 340061
Sensor of cytosolic DNA from bacteria
and viruses
STING, ERIS, HMITA,
MITA, SAVI, NET23
TMEM201 199953 Mitosis SAMP1, NET5
TMPO 7112
Nuclear organisation and DNA
replication LAP2
TNKS 8658
Wnt signalling pathway, telomerase and
vesicle trafficking. TIN1, TANK1, PARP5A
TNKS2 80351
Wnt signalling pathway, telomerase and
vesicle trafficking.
TNKS-2, TANK2,
PARP5B
TOP1 7150 Involved in DNA transcription TOPI
TOP2A 7153
Involved in DNA replication and
transcription TOP2
TOP2B 7155
Involved in DNA replication and
transcription Topo II Beta
TOP3A 7156 Involved in DNA replication Topo III-Alpha
TOP3B 8940
Involved in DNA replication and
transcription TOP3B1
TOPBP1 11073 Conserved DNA damage response gene TOP2BP1
TP53 7157
Conserved DNA damage response gene,
induces growth arrest or apoptosis
TP53BP1 7158 Conserved DNA damage response gene 53BP1

246
TP53I13 90313 Tumour suppressor
TP53 INDUCED
PROTEIN 13
TP53I3 9540 Member of TP53 signalling pathway
TP53 INDUCED
PROTEIN
TREX1 11277 Editing and processing nucleases DNase III
TREX2 11219 Editing and processing nucleases
TRIM28 10155
Chromatin Modifier/epigenetic
remodeller RNF96, KAP1, TIF1B
TRIM32 22954 E3 ubiquitin ligase activity. HT2A, LGMD2H
TRIM33 51592 E3 ubiquitin-protein ligase. TIF1G, RFG7
TRIM56 81844
poly(A) RNA binding and ubiquitin-
protein transferase activity. RNF109
TRIP13 9319
Chromatin Modifier/epigenetic
remodeller
TRRAP 8295
Chromatin Modifier/epigenetic
remodeller TRA1, STAF40, PAF400
TTI1 9675 Regulator of the DNA damage response.
SMG10, KIAA0406,
TELO2 Interacting
Protein 1
TTI2 80185 Regulator of the DNA damage response.
C8orf41, MRT39,
TELO2 Interacting
Protein 2
TUBA1B 10376 Chromatin, cell cycle, apoptosis Tubulin
TYMP 1890
Chromatin Modifier/epigenetic
remodeller
UBA52 7311
Ubiquitin gene, Ubiquitin A-52 Residue
Ribosomal Protein Fusion Product 1 RPL40

247
UBB 7314 Ubiquitin gene, ubiquitin B HEL-S-50
UBC 7316 Ubiquitin gene, ubiquitin C HMG20
UBE2A 7319 Ubiquitination and modification RAD6A
UBE2B 7320 Ubiquitination and modification RAD6B
UBE2N 7334 Ubiquitination and modification UBC13
UBE2T 29089
DNA damage repair and Fanconi
anaemia FANCT
UBE2V2 7336 Ubiquitination and modification MMS2
UBR5 51366 Transcriptional regulation
UHRF1 29128
Chromatin Modifier/epigenetic
remodeller
UIMC1 51720 DNA repair
RAP80, X2HRIP110,
RXRIP110
UPF1 5976
RNA-dependent helicase, involved in
decay of mRNA containing premature
stop codons RENT1, NORF1
USP1 7398
Negative regulator of DNA damage
repair HUBP
VCP 7415
Related to cell cycle and DNA damage
repair
WDR48 57599
Regulator of deubiquitinating
complexes. UAF1, P80
WDR77 79084
Chromatin Modifier/epigenetic
remodeller MEP50, P44
WEE1 7465 Cell cycle
WRAP53 55135 Telomerase and Cajal bodies TCAB1, DKCB3
WRN 7486 DNA helicase RECQL2, RECQ3

248
WRNIP1 56897 DNA synthesis
XAB2 56949 DNA repair gene HCNP, SYF1
XPA 7507 DNA repair gene XP1, XPAC
XPC 7508 DNA repair gene
XPCC, P125, RAD4,
XP3
XRCC1 7515 DNA repair gene RCC
XRCC2 7516 Homologous recombination RAD51-Like
XRCC3 7517 Homologous recombination RAD51-Like, CMM6
XRCC4 7518 DNA repair gene
XRCC5 7520 DNA repair gene
KU80, KUB2, Ku86,
NFIV, KARP1
XRCC6 2547
Single-stranded DNA-dependent ATP-
dependent helicase, involved in DNA
non-homologous end joining. G22P1, KU70
XRCC6P2 389901 DNA repair gene
ZBTB32 27033
Transcription regulation and DNA
binding (involved in Fanconi anaemia) ZNF538
ZBTB44 29068 Transcription regulation ZNF851
ZBTB48 3104
Nucleic acid binding and transcription
factor activity HKR3, ZNF855, TZAP
ZCCHC7 84186 RNA degradation
ZMYM2 7750 Transcription factor ZNF198
ZNHIT4 83444
Involved in transcriptional regulation,
DNA replication and probably DNA
repair.
INO80B
ZSWIM7 125150 Involved in homologous recombination SWS1

249
Table A2.6. Genetic events identified by whole genome sequencing.
Sample ID
Gene Symbol C
hro
mo
som
e
Variation Type
Gene Region Start site End site
Refer-ence Allele
Alter-native Allele
Allelic Fracti-
on Protein Variant
LA-N-6 AIM2 1 SNV:DEL Exon chr1:
159032486 chr1:
159032486 65.38
CHLA-90 ATRX X SV:DEL
chrx: 76935119
chrx: 76959029
PARKNP ATRX X SV:DEL
chrx: 76924445
chrx: 77028694
PAISZV ATRX X SV:DEL
chrx: 76937155
chrx: 76983145
PAPTAN BARD1 2 SNV:DEL Exon chr2:
215645502 chr2:
215645522 42.86
PARXFT BARD1 2 SNV:DEL Exon chr2:
215645502 chr2:
215645522 45.45
PARGIW BRIP1 17 SV:TRA Intron chr17:
59796638 chr2:
14401098
SK-N-BE2c CARM1 19 SNV Exon chr19:
11031414 chr19:
11031414 C A 46.67 P472T
PAIFXV CCNT1 12 SV:DEL
chr2: 25957192
chr12: 104379378
SH-SY5Y CHEK2 22 SNV:DEL Exon chr22:
29091856 chr22:
29091856 75
CHLA-90 CREBBP 16 SV:DUP Intron chr16:
3885945 chr16:
3919341
COG-N-291 DDX60 7 SV:TRA Intron chr7:
116809191
COG-N-291 E2F2 1 SV:DEL
chr1: 3801505
chr1: 30723742
LA-N-6 EME2 16 SNV:DEL Exon chr16:
1823389 chr16:
1823389 43.48
PAPTAN FOXP1 3 SNV
Intron/base before
exon chr3:
71408396 chr3:
71408396 C T 46.67
COG-N-291 GEN1 2 SV:DEL
chr2: 1485200
chr2: 27254414
PARGIW HIST1H1B 6 SNV:DEL Exon chr6:
27834678 chr6:
27834692 38.46
PAISZV HIST2H2BC 1 SV:DEL
chr1: 110246334
chr1: 181216287
COG-N-291 HLTF 3 SNV Exon chr3:
148789089 chr3:
148789089 C A 61.54 D282Y
PARXFT IDH1 2 SNV:DEL Promoter chr2:
209120302 chr2:
209120302 35
PARGIW INO80C 18 SNV:DEL Promoter;
5'UTR chr18:
33077852 chr18:
33077859 55.56
PARXFT KDM6B 17 SNV:DEL Exon chr17:
7751858 chr17:
7751863 54.55
SH-SY5Y KRAS 12 SNV Exon chr12:
25398284 chr12:
25398284 C A 45.45 G12V
COG-N-291 MCM4 8 SNV Exon chr8:
48875559 chr8:
48875559 T G 45.83 F218V

250
CHW_11 MCM6 2 SNV Exon chr2:
136624255 chr2:
136624255 A T 52 I220N
SH-SY5Y MRE11A 11 SNV Exon chr11:
94226952 chr11:
94226952 C G 56.25
PARKNP MSH2 2 SNV:DEL Exon chr2:
47693814 chr2:
47693815 69.23
SK-N-FI NCL 2 SNV:DEL Exon chr2:
232325414 chr2:
232325416 57.89
COG-N-291 NCL 2 SNV:DEL Exon chr2:
232325414 chr2:
232325414 48.15
PAIFXV NCL 2 SNV:DEL Exon chr2:
232325414 chr2:
232325414 100
PAIFXV NCL 2 SNV:DEL Exon chr2:
232326655 chr2:
232326655 39.47
PAIFXV NCOA3 20 SNV:DEL Exon chr20:
46279814 chr20:
46279822 44.44
PARGIW NCOA3 20 SNV:DEL Exon chr20:
46279863 chr20:
46279865 100
PAISZV NCOR1 17 SV:DEL
chr17: 16095144
chr17: 16095626
PAPTAN NCOR2 12 SNV:DEL Exon chr12:
124887058 chr12:
124887069 63.64
PAPFSL NSMCE2 8 SNV:DEL Exon chr8:
126114595 chr8:
126114595 66.67
LA-N-6 POLM 7 SV:DEL
chr7: 31199193
chr7: 64005420
PAIFXV POLQ 3 SNV:DEL Exon chr3:
121208703 chr3:
121208705 50
PAIFXV POLR2A 17 SNV Exon chr17:
7405325 chr17:
7405325 G A 22.22 S819N
SK-N-FI PPP2R2A 8 SNV:DEL Intron chr8:
26150637 chr8:
26150637 60
SK-N-FI PRDM2 1 SNV:DEL Intron chr1:
14074949 chr1:
14074950 57.14
SK-N-BE2c PRMT2 21 SNV Intron chr21:
48056789 chr21:
48056789 C A 40
SK-N-BE2c PRMT5 14 SNV Intron chr14:
23393931 chr14:
23393931 G T 14.81
PARKNP RAD17 5 SNV:DEL Intron chr5:
68667048 chr5:
68667050 46.67
PAPFSL RAD23B 9 SNV:DEL Intron chr9:
110046745 chr9:
110046747 60
SH-SY5Y RAD23B 9 SNV:DEL Intron chr9:
110046745 chr9:
110046747 100
SH-SY5Y RAD51B 14 SV:INV Intron chr14:
68907987 chr14:
69297996
PARGIW RB1 13 SNV:DEL Exon chr13:
48878084 chr13:
48878092 40
PARGIW RBBP8 18 SNV:DEL Intron chr18:
20513542 chr18:
20513549 64.71
PARGIW RBL1 20 SV:TRA Intron chr20:
35627929
PAPFSL RECQL4 8 SNV Exon chr8:
145738660 chr8:
145738660 C T 30.77 V802M
SH-SY5Y RECQL5 17 SNV Exon chr17: chr17: G A 80 R175C

251
73658807 73658807
PARGIW RIF1 2 SNV:DEL Exon chr2:
152320880 chr8:
152320882 23.08
PAISZV SETDB1 1 SNV Exon chr1:
150934631 chr1:
150934631 C A 29.03 S1052*
PAPTAN SMAD3 15 SV:DEL Intron chr15:
67404674 chr15:
67405604
PARKNP SMARCA2 9 SV:DEL Intron chr9:
2147643 chr9:
2151529
SH-SY5Y SMARCA4 19 SNV Exon chr19:
11134251 chr19:
11134251 C T 44.44 R973W
SK-N-FI SMARCA4 19 SNV Exon chr19:
11170813 chr19:
11170813 C T 100
R1587*; R1590*; R1653*; R1621*; R1588*; R1591*
SH-SY5Y SMC3 10 SNV:DEL Splice Site;
Intron chr10:
112360304 chr10:
112360305 60
SK-N-BE2c SMG1 16 SNV Exon chr16:
18827823 chr16:
18827823 C A 36.36 S3368I
LA-N-6 SP1 12 SNV Exon chr12:
53777044 chr12:
53777044 T C 46.15
L431P; L438P; L390P
SK-N-BE2c SRCAP 19 SV:TRA Intron chr19:
2599286
CHLA-90 STAG1 3 SV:DEL
chr3: 135906165
chr3: 136313178
COG-N-291 TCEB3 1 SV:DEL
chr1: 30723742
PARKNP TDRD12 19 SNV Exon chr19:
33222697 chr19:
33222697 G A 45.83 D31N
SH-SY5Y TERT 5 SNV Promoter chr5:
1295228 chr5:
1295228 G A 60
PAPTAN TMEM201 1 SNV Exon chr1:
9671877 chr1:
9671877 C G 80 S611C
CHLA-90 TP53 17 SNV Exon chr17:
7577082 chr17:
7577082 C T 100
E127K; E247K; E154K; E286K
SK-N-FI TP53 17 SNV Exon chr17:
7577544 chr17:
7577544 A C 100
M246R; M114R; M207R; M87R
SK-N-BE2c TP53 17 SNV Exon chr17:
7578526 chr17:
7578526 C A 100
C96F; C3F;
C135F
CHLA-90 TP53 17 SNV:DEL Intron chr17:
7588679 chr17:
7588679 100
SH-SY5Y TRRAP 7 SNV Exon chr7:
98581778 chr7:
98581778 G T 28.57 G3004W; G3033W
SH-SY5Y TTI2 8 SNV Exon chr8:
33364825 chr8:
33364825 C A 52.63 L283F
PAPTAN UIMC1 5 SV:TRA Intron chr5:
176387600

252
SK-N-BE2c VCP 9 SNV Exon chr9:
35064279 chr9:
35064279 C A 37.5 E194*
PAPFSL WRNIP1 6 SNV:DEL Exon chr6:
2766363 chr6:
2766374 37.5
CHW_11 ZMYM2 13 SNV Exon chr13:
20580560 chr13:
20580560 G T 32 S449I
SNV, single nucleotide variation; SNV:DEL, deletion of 1-20 bp; SV:DEL, structural variation: deletion; SV:TRA, structural variation: translocation; SV:INV, structural variation: inversion.
Copyright © 2022 FDOKUMEN




















