
THE COMPLEXES OF IRON(ll), COBALT(Il), AND NICKEL(ll)
273
THE COMPLEXES OF IRON(ll), COBALT(Il), AND NICKEL(ll) WITH LIGANDS CONTAINING THE CONJUGATED METHINE GROUP DISSERTATION Presented In Partial Fulfillment of the Requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University By ROBERT CARL STOUFER, B. A., B. S. The Ohio State University 1958 Approved by v Adviser Department of Chemistry
-
Upload
khangminh22 -
Category
Documents
-
view
6 -
download
0
Transcript of THE COMPLEXES OF IRON(ll), COBALT(Il), AND NICKEL(ll)

THE COMPLEXES OF IRON(ll), COBALT(Il), AND NICKEL(ll) WITH LIGANDS CONTAINING THE CONJUGATED
METHINE GROUP
DISSERTATIONPresented In Partial Fulfillment of the Requirements
for the Degree Doctor of Philosophy in the Graduate School of The Ohio State
University
ByROBERT CARL STOUFER, B. A., B. S.
The Ohio State University1958
Approved by
v Adviser Department of Chemistry

ACKNOWLEDGMENTS
The author gratefully acknowledges the help of all those persons who have made this work possible, particularly Professor Daryle H. Busch whose enthusiasm, encouragement, and helpful suggestions made the work contained in this dissertation a very rewarding experience. The Invaluable suggestions contributed by Professor WayneE. Hadley, with respect to the magnetic properties of compounds, are gratefully acknowledged. The author wishes to thank, publicly, Dr. Melvin M. Morris and Mr. DonaldC. Jicha for their help In running many of the infrared spectra included in this dissertation, and his wife, Virginia, for her help in many intangible ways and for her part in the preparation of the final manuscript. The author also wishes to extend his appreciation, for the wonderful cooperation he received In preparing this manuscript, to Mrs. Robert Holslnger, Jr., who prepared all of the diagrams and spectra for photographing, to Mrs. Leo Moore who typed a portion of the first draft, and, especially, to Mrs. Francis J. Bradley who typed the final manuscript.
11

CONTENTS
CHAPTER PageI . INTRODUCTION.............. 1
II. COMPLEXES PREPARED BY THE REACTION OP IRON(II), COBALT(II), AND NICKEL(ll)WITH BIACETYLDIHYDRAZONE, 2-PYRIDIN- ALHYDRAZONE, 2,6-PYRIDINDIALDIHYDRA- ZONE, AND 2-PYRIDINAL-P-TOLYLIMINE....... 34A. Introduction.......................... 34B. Experimental.......................... 4o
1. Preparations and Analytical Data.. 4o2. Conductivity Measurements ...... 5o3. Magnetic Measurements............. 574. Ultraviolet and Visible Spectra... oO5* Infrared Spectra.............. 6l6. Continuous Variations Study of the
System, Nickel(Il)-Biacetyldi- hydrazone......................... 6l
7. Resolution of Optical Isomers 62C. Results........... 64
1. Conductivity Measurements.......... 642. Magnetic Measurements....... 683 . Application of Magnetic Data to
Problems of Valency and Stereochemistry......... 76
4. Ultraviolet and Visible Spectra...1045 . Infrared Spectra... ..........1306. Continuous Variations Study of the
System, Nickel(ll)-Biacetyldi- hydrazone........................ 179
H i

lvCONTENTS (Contd.)
CHAPTER Page7- Resolution of Optical Isomers 183
D. Discussion............................. 186
III. THE COMPLEXES OF NICKEL(lI) AND DIMETHYLGLY- OXIME, BIACETYLOXIMEMETHOXIME, BIACETYLHYDRAZONEOXIME, ANDBIACETYLHYDRAZONEMETHOXIME................ 201A. Introduction........................... 201B. Experimental............................ 207
1. Preparations and Analytical Data.. 2072. Physical Measurements.............. 212
C. Results................................. 2121. Conductivity Measurements.......... 2122. Magnetic Measurements........... 2143 . Ultraviolet and Visible Spectra... 215
D. Discussion............................. 226IV. SUMMARY.................... ..................232
APPENDIX I Magnetic Susceptibility Measurements.. 237
APPENDIX II Magnetic Susceptibility Measurements.. 246APPENDIX III Temperature Dependence of Magnetic
Susceptibility........................ 248APPENDIX IV .................................. . 250AUTOBIOGRAPHY ........................................262

LIST OF TABLES
TABLE Page1. Hybridized Atomic Orbitals and Directional
Characteristics................... 72. Configurations and Bond Types of Transition
Metal Complexes..*........ 103* Energy of the d Orbital Levels in Crystal
Fields of Different Symmetries (in Terms of D£)...................................... 21
4. Molar Conductances of the Complexes Iron(ll),Cobalt(ll), and Nickel(ll) with BdH. PAH,PdAdH, and PAT. 10"3 Solutions, 25 C 66
5. Magnetic Moments Expected for Some Complexesof Iron(ll), Cobalt(Il), and Nickel(Il)... 80
6 . Molar Susceptibilities of Ligands and Anions 8l7. Molar Susceptibilities and Magnetic Moments
of Complexes............... 828 . Magnetic Susceptibilities of Some Spin-
Paired Iron(ll) Complexes as a Function of Temperature ..... 84
9 . Temperature Dependence of the MagneticMoment of [Co(PdAdH)2 ]l2 ................... 9°
10. Contributions of the Two Spin-states ofCobalt(II) to the Observed Magnetic Susceptibility of [Co(PdAdH)2 JI2 .......... 94
11. Contributions of the Two Spin-states ofCobalt(ll) to the Observed Magnetic Susceptibility of [Co(PdAdH)gJIg.......... 95
12. Temperature Dependence of the MagneticMoments of Normal-[Ni(PdAdH)p]lp and Iso-[Ni(PdAdH)2 ]l2 ..........1............. 101
v

viLIST OF TABLES (Contd.)
TABLE Page13* Ultraviolet and Visible Absorption Bands of
Biacetyldihydrazone; 2-Pyridinalhydrazone, and Their Complexes... .................... 109
14. Ultraviolet and Visible Absorption Bands ofPyridlndialdihydrazone, 2-Pyridinal-p- tolylimine and Their Complexes............ 110
15* Infrared Absorption Bands (cm-1) forBiacetyldlhydrazone (BdH) and Its Complexes................................... 131
16. Infrared Absorption Bands (cm-1) forPyridinalhydrazone and Its Complexes 133
17. Infrared Absorption Bands (cm-1) forPyridindlaldihydrazone and Its Complexes.. 135
18. Infrared Absorption Bands (cm-*) forPyridinal-p-tolylimine and Its Complexes.. 138
19. Continuous Variations Study of the Systems,Nickel(II) -Biacetyldlhydrazone............ l8l
20. Racemization of LFe(CitH_.J'J)1) ] [ (SbO)(c 4V 6)]2 .................. 184
21. Molar Conductances of the Nonionic Complexesof Nickel(ll) with H-BOM, H-BHO and BHM... 213
22. Molar Susceptibilities of Ligands andAnions................. 214
2 3 . Molar Susceptibilities of Complexes......... 21524. Ultraviolet and Visible Absorptions of
Ni(ll) Complexes and Ligands.............. 2212 5 . Calibration of Field with Water.............. 24l26. Calibration of Field with Ferrous Ammonium
Sulfate 6-Hydrate.......................... 242

LIST OF TABLES {Contd.)vii
TABLE Page27. Magnetic Susceptibility Data for Solid
Samples of Ligands...................... 24328. Magnetic Susceptibility Data for Solid
Samples of Complexes*...................... 24429. Calibration of Magnetic Field with Ferrous
Ammonium Sulfate 0 -Hydrate.............. 24630. Magnetic Susceptibility Data for Solid
Samples of Complexes. .................. . 24731. Temperature Dependence of the Molar
Susceptibilities of Some Spin-paired Iron(ll) Complexes.......*................. 248
32. Temperature Dependence of the MolarSusceptibility of [Co(PdAdH)2 3l2 .......... 249
33. Temperature Dependence of the Molar Susceptibilities of the Nickel(ll) Complexes of Pyridlndialdihydrazone....... 249
34. Infrared Absorption Bands (cm"l) for Dimethylglyoxlme and Its Nickel(ll) Complexes.................................. 256
35. Infrared Absorption Bands (cm~l) for 2,3- Butanedlone-2-0xime and Biacetylhydrazone- oxime....................................... 257
36. Infrared Absorption Bands (cm"3-) for [Nl(H-BH0)oClo3> [Ni(BH0)o ], andIn i (h -b h o ) |3lg........... .................. 258
37. Infrared Spectra of 2,3-Butanedione-2-Methoxime, Biacetylhydrazonemethoxime, and [Ni(H-BOMJ2Cl2 3............................ 259
38. • Infrared Spectra of Biacetylhydrazoneraethox- ime and lNi(BHM)2Cl2 3...................... 260

LIST OF FIGURES
FIGURE Page1. Electronic Configuration of Nickel(ll)....... 8
2. Diagrammatic Representations of the AngularParts of the 3d Orbitals.................... 14
3. Geometric Molecular Structure with Respectto the Cartesian C o o r d i n a t e s . . lo
4. Splitting the d Orbitals In Fields ofDifferent Symmetries........................ 17
5* Occupancy of <1 Orbitals........................ 18
6. Configuration of a dJ ion..................... 21
7* Correlation Diagram............................ 258. Brass Cryostat................................. 59
9* Splitting the d Orbitals of a d^ Ion inOctahedral and Tetrahedral Fields.......... 72
10. Splitting of the d Orbitals of a dJ Ion inPlanar and Tetragonal Fields.......... . 74
11. Temperature Dependence of the MagneticSusceptibilities of Some Spin-paired Iron(ll) Complexes.............. 86
12. Temperature Dependence of the MagneticSusceptibility of the Two Spin States of [Co(PdAdH)2 ]l2 ............ 97
13. Temperature Dependence of the MagneticSusceptibility of the Two Spin States of [Co(PdAdH)2 ]l2 ............................... 93
14. Temperature Dependence of the MagneticSusceptibility of the Two Isomeric Formsof [Ni(PdAdH)2 ]l2 ........................... 1°2
viii

ixLIST OP FIGURES (Contd.)
FIGURE Page15. Ultraviolet Spectra of Biacetyldlhydrazone
and Its Complexes.......................... 11216. Visible Spectra of the Complexes of
Biacetyldlhydrazone........................ 11417. Ultraviolet Spectra of Pyridinalhydrazone
and Its Complexes.......................... llo18. Visible Spectra of the Complex of
Pyridinalhydrazone......................... 11919* Ultraviolet Spectra of Pyridindialdihydra-
zone and Its Complexes......... 12020. Visible Spectra of the Complexes of
Pyridlndialdihydrazone.................... 12221. Ultraviolet Spectra of the Nlckel(ll)
Complexes of Pyridlndialdihydrazone...... 12522. Visible Spectra of the Nickel(ll) Complexes
of Pyridlndialdihydrazone................. 1262 3 . Ultraviolet Spectra of Pyridin-p-tolyliraine
and Its Complexes .......... 12724. Visible Spectra of the Complexes of
Pyridinal-p-tolylimine............ 1292 5 . Infrared Spectra of 2,3-Butanedione and
Biacetyldlhydrazone........................ 1432 6 * Cls and Trans-Configuration of
Biacetyldlhydrazone........................ 1462 7 . Diagrammatic Representation of the Inter
action in Biacetyldlhydrazone............. 14828. Infrared Spectrum of the Iron(Il) Complex
of Biacetyldlhydrazone.................... 1512 9 . Infrared Spectra of the Cobalt(ll) and
Nickel(ll) Complexes of Biacetyldlhydrazone ........................................ 152

XLIST OF FIGURES (Contd.)
FIGURE Page30. Infrared Spectra of 2 -PyridInaldehyde
and Pyridinalhydrazone. ............. 1^331* Infrared Spectrum of [Fe(PAH)^]l2 ............ lo232. Infrared Spectra of the Cobalt(II) and
Nlckel(Il) Complexes of Pyridinalhydrazone 163
33* Infrared Spectra of 2 ,6-Pyridindialdehydeand Pyridlndialdihydrazone................. 167
34. Infrared Spectra of the Iron(ll) andCobalt(ll) Complexes of Pyridlndialdihydra- zone...... 170
3 5 . Infrared Spectra of the Nickel(ll) Complexof P y r i d l n d i a l d i h y d r a z o n e . . 171
3 6 . Infrared Spectra of Pyridinal-p-tolylimineand the Iron(ll) Complex of Pyridinal-p- tolylimine......... 17*5
37* Infrared Spectra of the Cobalt(II) andNlckel(ll) Complexes of Pyrldinal-p- toly limine................................... 173
3 8 . Enhancement as a Function of Per centNickel(ll) Chloride in the System,Nickel(Il) -Biacetyldlhydrazone............ 182
39* Change In Observed, Rotation of [Fe(ChHioNh)o3[( S b O)(C^H^O^)with Respect to Time 185
40. Three Geometric Forms of Dimethylglyoxime.*. 203
41. Ultraviolet Spectra of Dimethylglyoxime andBiacetylhydrazoneoxime ..... 217
42. Ultraviolet Spectra of the Nickel(ll)Complexes of Dimethylglyoxime and Biacetylhydrazoneoxime. •••••..... 218
4 3 . Visible Spectra of the Nickel(ll) Complexesof Dimethylglyoxime and Biacetylhydrazoneoxime. ..... 219

xiLIST OF FIGURES (Contd.)
FIGURE Page44* Ultraviolet Spectra of Biacetyloxlmemth-
oxime, BiacetyIhydrazonemethoxime. [NI(BHM)2C12 ]J and [Ni(H-ROM) 2C 12 J........ 220
45. Visible Spectra of fNi(H-BOM)2C12 ] and[Ni(BHM)2Cl2 ]................................ 225
46. Magnetic Field as a Function of SampleLength.......... 239
47. Magnetic Field as a Function of SamplePlacement with Respect to the Center ofthe Pole Piece.... ................... 240
48. Infrared Spectra of Dimethylglyoxlme,rNifDMG) 2 J, and [Ni(H-DMG)2Cl2 3........... 251
49. Infrared Spectra of 2,3-butanedlone-2-oximeand Biacetylhydrazoneoxime................. 252
50. Infrared Spectra of the Nickel(ll) Complexesof Biacetylhydrazoneoxime .......... 253
51. Infrared Spectra of 2,3-butanedione-2-methoxlme. Biacetyloxtmemethoxime and rNi(H-BOM)2Cl2 3.............................. 254
52. Infrared Spectra of BiacetyIhydrazonemethoxime and [Ni(BHM)2Cl2 3..................... 255

I. INTRODUCTION
The tremendous strides which have been made in the field of inorganic chemistry during the past 60 years, particularly with respect to an adequate theory of valency and stereochemistry, were initiated, strangely enough, by an Alsatian-born organic chemist named Alfred Werner. Prior to the time of Werner, a large number of coordination compounds were known. They were recognized simply as addition compounds resulting from the union of two or more stable inorganic compounds; however, no satisfactory theory was available to account' for their existence and properties. Werner provided the theory.
Werner set forth the essential part of his theory of coordination chemistry, which was distinctly different from the concepts held by Berzelius, Bloomstrand, Jorgensen, and others, in a paper titled "Beitrag zur Kbnstltution anorgan- ischer Verbldungen.m1 His theory was developed in its
^A. Werner, Z. anorg. Chem., 2.* 2^7 (1893) •
entirety and recorded in "Neuere Anschaungen auf den Gebiete der anorganischen Chemle," published in 1905* Werner stated
2A. Werner, "Neuere Anschaungen auf den Oeblete der anorganischen Chemle," translated by Headley. Longmans, Green, and Company, New York, 1911*
1

that neutral molecules or oppositely charged ions were grouped or coordinated around a central ion in a "first sphere of attraction" in a spatially symmetrical arrangement. He called the number of groups which could be attached or coordinated to the central ion the "coordination number" and considered it to be a characteristic property of that ion. The suggested symmetrical arrangements of groups about a central ion included the square plane, the tetrahedron, and the octahedron. As a result of the stoichiometry of certain compounds which were known at that time (particularly those with a coordination number of six) the existence of stereoisomers was predicted by the coordination theory* In order to gain acceptance for his theory, it was necessary that Werner prove the existence of these Isomers. And, indeed, many of these missing compounds were prepared, the octahedral structure of the hexacoordinated compounds being established by the resolution of many complexes into their optically active forms. Modern x-ray studies have provided conclusive proof of Werner's ideas on stereochemistry.
Within the framework of this theory and as a result of the elegant classical experimental work of both Werner and Jorgensen, a suitable explanation was found for many perplexing problems involving physical and chemical properties of coordination compounds.

3Although Werner's theory provided a logical explana
tion for the stereochemistry and related properties of complex Inorganic compounds It contributed nothing to a clearer picture of bonding within the complex* Werner did propose that there were two types of valence, a "primary" valence, which corresponded to the classical electrovalence, and a "secondary" valence, which represented the "first sphere of attraction;" however, with the discovery that certain groups could simultaneously satisfy both valencies In certain compounds, the distinction between these two types of valence became somewhat nebulous* Complete acceptance of Werner's views did not come until the development of the electronic theory of valence by Lewis and his contemporaries, these concepts provided a rational explanation for directional properties In bonds while being completely consistent with Werner's views concerning stereochemistry*3
3j. c* Bailar, Jr., "Chemistry of Coordination Compounds," Reinhold, New York, 1936, Chapter 2*
About this same time Kbssel arrived at the conclusionthat complexes were held together primarily by electrostatic
aforces* Using such an electrostatic picture, he suggested
*W* Eossel, Z* Elektrochea.. 26, 314 (1920); Z* Phys* 1# 395 (1920); Naturwlssenschaften, 77 339, 360 (1913);“TT7 598 (1923); AnnTT h y s *. 43* 223 71916).

that the strongest bonding should result from a small central metal atom of high electrostatic charge. There was a considerable degree of success In the application of this theory to practical chemical problems. An extension of this theory was used by Xossel In the treatment of acid- base phenomena, which he considered to follow as a natural consequence of the coordination theory.
-\J. C. Bailar, Jr., o£. clt., Chapter 3*
6 7Garrick * carried out quantitative calculations in
fp. J. Oarriclc, Phil. Mag.. [7j 2. (1930); C 7 ]10 . 76 (1930);C7]U, 7'»TTT93T7V
•7F. Basolo and R. G. Pearson, "Mechanisms of Inorganic Reactions," John Wiley and Sons, Inc., London,1958, p. 46.
terms of an electrostatic model for various coordination numbers and stereochemistries, using the potential energy equations of classical electrostatics, and found the results to be in excellent agreement with experimentally determined bond energies In many cases. This excellent agreement was limited, however, to certain groups of compounds .
As a result of the development of quantum mechanics in the late nineteen twenties and early thirties, three modern concepts of bonding came Into being. They are (1)

5the valence bond theory,^ (2) the crystal field theory^*
®L. Pauling, "Nature of the Chemical Bond," Cornell University Press, Ithaca, New York, 1948.
^H. Bethe, Ann* Physik, 2> ^97 (1929)*10W. 0. Penney and R. Schlapp, Phys. Rev., 41, 194
(1932).n j. H. Van Vleck, Phys. Rev., 39, 103 (1932); 41,
208 (1932).
12 1^and (3) the molecular orbital theory. ' These three
12R. S. Mulliken, Phys. Rev., 41, 49 (1932); J. Chem. Phys., 2> 375 (1935).
x3j. H. Van Vleck, J. Chem. Phys., 2, 903, 807 (1935).
theories will be considered separately.The valence bond treatment has enjoyed the most wide
spread popularity among practicing inorganic chemists because it provided a simple, qualitative explanation of the stereochemistry and magnetic properties of many complex compounds. Quite early in the development of this theory It was realized that the orbitals on the central ion which were available for bond formation did not possess the symmetry properties required for the geometries which were known to exist for many types of compounds (assuming that the strongest covalent bond would be formed by the maximum overlap of the orbitals of the central atom with those of the coordinated group or ligands). In addition, equivalent

orbitals were required for the formation of equivalent bonds and, in many cases, the number of atomic orbitals of equivalent energy was not as great as the number of equivalent bonds which were known to be formed* The solu
1 btion to this problem-1 was found by assuming that the
^Pauling, o£. cit., pp. 82-86.
atomic orbitals utilized in bonding were not the orbitals known to exist in the free, gaseous ions, or atoms, but that the bonding atomic orbitals were derived from the original orbitals in such a manner that the number of equivalent bonding orbitals was the same as the number of equivalent bonds formed. These bonding atomic orbitals were called hybridized atomic orbitals. In addition, it was required that the orbitals be directed In space in the same manner as the groups attached to the central metal atom. A listing of the moBt commonly encountered geometries In complex inorganic compounds, together with the hybridized atomic orbitals which possess the proper directional characteristics, is found in Table 1. In this manner the stereochemistry of many complex compounds was explained.
Not only could known structures be rationalized within the framework of this theory, but, In certain instances (compounds involving four to eight d electrons), structures could be predicted from simple magnetic moment

TABLE 1HYBRIDIZED ATOMIC ORBITALS AND DIRECTIONAL CHARACTERISTICS
CoordinationNumber
HybridizedOrbitals
GeometricStructure
1 £ linear1 £ linear2 !£ linear3 s£_ trigonalk S£3 tetrahedralk
. 2 *»£ square planar5 dsp3, spd3 trigonal bipyraraidal5 d2sp2 spd3 square pyramidal6 d2s£3, S£3d2 octahedral6 dS£2-£d tetragonal bipyramidal*
* See Ref* 20.
measurements• Since the magnetic moment of a complex la frequently In accord with the apin-only formula, jx =Vn(n*2), (n la the number of unpaired electrons and la the magnetic moment expressed In Bohr magnetons) and since the total number of electrons in the system Is known, it follows that the electronic configuration la also known; therefore, the orbitals available for hybrid bond formation are known*From the stoichiometry of the complex and a knowledge of the

orbitals available for bond formation, the structure maybe postulated* Consider, for example, two hypothetical
XI XIcomplexes of nickel (II), Ni A^ and Ni where A and B are monodentate, coordinating groups* Compound NiA^ is found to have a magnetic moment of approximately 3 Bohr magnetons. From the spin-only formula it is determined that there must be two unpaired electrons. From Figure 1,
3d
uithil □ M i l
3d 4s
liduiifii □ M MFigure l
Ground state of Nl+ *
Excited state of N1+ +
,+*■ 8it is seen that this is the ground state of Ni T , a d configuration* Since nickel has formed four equivalent bonds and since there are three vacant *l£ orbitals and one vacant orbital, the bonding metal orbitals would be considered sp3 hybrids, from which it follows that the structure would be tetrahedral (Table 1)* Compound NIB^ is determined to be diamagnetic (no unpaired electrons); therefore, some combination of one 3d, one 4ji, and two orbitals is possible* For the formation of four, equivalentbonds, this theory predicts the use of four dsp hybrid

orbitals and, hence, a square planar configuration. The possible electronic configurations, as related to the number of d electrons and the bonding orbitals utilized in some first transltion-serles metal complexes, are listed in Table 2 with the calculated spin-only moments correspond ing to these configurations.
The valence bond treatment will be used, in part, throughout the remainder of this work because of the familiarity and convenience of the concepts in discussing, in a qualitative manner, the stereochemistry of complex compounds.
The valence bond theory suffers from severe limitations as any attempt to place it on a semiquantltatlve basis will reveal.*5 it does not explain the spectra of
15d. H. Busch, J. Chem. Jfl., 2 1 * 376, 498 (1956).
metal complexes, nor does it explain quantitative variations In magnetic moments which are in excess of the spin- only value. In addition, this theory does not predict which of several possible configurations will result from the formation of a complex compound with a given stoichiometry.
Although ligand field theory (crystal field theory) was originally applied to crystalline materials, It Is also applicable to any orderly arrangement of electrically

TABLE 2CONFIGURATIONS AND BOND TYPES OF TRANSITION METAL COMPLEXES
Ion or Compound Coordination Electronic Configuration Unpaired MagneticNumber 3d 4s. 4£ 4d Electrons Moment
(Calc.)
Ti(III)Gaseous Ion
Ti(lIl)Octahedral 3d^s4j>3 bonds
V(II)Gaseous Ion
V(ll)Octahedralbonds
Cr(ll)Gaseous Ion
Cr(lliOctahedral 4£4£^4d bonds
Cr(ll)Qctahedral 3dz4s7£3 bonds
mi i I Iii i t e m
a a ci m m
- ram 6 im n r r
r c n n m m
□ c m m
0 iniiiiu) mh h i <i i i □ c m m
BE rcrcnj
QE ciniiD
m
□ an mGD ZZZ S E
m
i
i
3
4
4
1.73 B.M.
ii it
3.88 "
, ii ii ♦
4.90 "
11 ti
2 .8 5 nMo
80

TABUS 2 (Contd.)CONFIGURATIONS AND BOND TYPES OF TRANSITION METAL COMPLEXES
Ion or Compound Coordination Number 3d
Electronic Configuration Unpaired Magnetic 4a 4p 4d Electrons Moment
(Calc.)
Mn(II)Gaseous Ion h n h i n n □ c m c n 5 5.92 B.M.
Mn(lllOctahedral4s4£:,4dz bonds e HHHhhl Eg X m E D H E E S !
t t 11
Mn( II)0ctahedral 3d4s4£3 bonds [HXEKDIE m CD 1*73 "
Fe(II)Gaseous Ion □ cm m 4 .9 0 "
Fe(lllOctahedral 4_s4£34d bonds EE It II
Fe(ll)Ocfcahedral 3dz4«4£5 bonds EE _ C D 0 0.00 "
Co(II)Gaseous Ion x x a a n □ cm m 3 3 .8 8 "
80

TABLE 2 (Contd.)CONFIGURATIONS AND BOND TYPES OF TRANSITION METAL COMPLEXES
Ion or Compound CordinationNumber 3d
Electronic Configuration Unpaired Magnetic 4** 4£ 4d Electrons Moment
(Calc.)
Co(IllTetrahedral *£*£ bonds
Co(II)Octahedral 4s_4|>34d2 bonds
Co(II)Planar b°nda
D! 0 1 9 3 0 1 03 I M Q ] 3 3-88 B.M.
m ii ii
Ni(II)Gaseous Ion
Nl(IllC4«4o3ilOctahedral ^ bonds
H 0 K D 9 1
ami□ HD m 2 a-85 ”
irMtrir.Ti•i n
N1(II)P1I3d4s4p
Lanar 3d4s4£ bonds
4 m m 0 hd m ° 0.00

13interacting atomic or polyatomic groups such as may be found in complex compounds.1^ This theory is primarily
^ F. Basolo and R. G. Pearson, loc. clt., Chapter 2.
concerned with repulsions and, as a consequence, is not specifically concerned with bonding. The basic concepts may be rather easily understood in a qualitative manner through the use of a physical model. In Figure 2 the five d orbitals of the penultimate electron shell are shown with reference to a Cartesian set of coordinates. The orbitals are drawn singly in order to clearly show their individual characteristics. The effect of an octahedral field on these orbitals may be seen as follows. If six negative ions, or dipolar molecules, are brought up to the central atom along the Cartesian axes, it is Immediately seen from a qualitative electrostatic picture that electrons occupying the d orbitals which are closer to the ligands (those orbitals which are directed along the Cartesian coordinatesi.e., d 2 2 and d 2) are repelled to a greater degree than—y zthose In the remaining orbitals. Consequently the dx2 ^2 and dg2 orbitals will be associated with a higher energy than is true of the dyZ, and dxz orbitals which are not directed along the axes. The magnitude of the difference in the energies of the d orbitals is a function of the strength of the electrostatic field which gives rise

14DIAGRAMMATIC REPRESENTATIONS OF THE ANGULAR
PARTS OF THE 3d O R B I T A L S

to the "splitting" of the energy levels. Consequently,the splitting of the energy levels depends on the natureof the ligands used. Figure 3 serves as a guide in theapplication of this concept to other geometries in additionto the octahedral case. Figure 4 indicates the types ofenergy separations which are expected from these geometries.If, in the octahedral case, the ligands are identical, thesplitting of the d orbitals will result in a lower tripletof energy levels (tgg orbitals), which includes the dxy,dL , and d orbitals, and an upper doublet (e orbitals),—yz' —xz —gwhich is comprised of the d 2 2 and the d 2 orbitals.“A "j *"ZFigure 5 depicts the occupancy of the d orbitals in octahedral ions with one to ten d electrons. The reference energy of the degenerate d orbitals may be represented as that of the same orbitals in a spherically symmetrical field, equal in magnitude to that of the field which Is under consideration. The total energy separation between the e and the t^ orbitals is defined as IQDq. The spllt--g “2g ---ting of these energy levels is such that the center of gravity of the system is unchanged from the spherical field case, consequently, the energy of each electron in the upper doublet is + 6Pg while the energy of each electron in the lower triplet is -4Dq. The value of may be determined experimentally from spectral data. One other quantity must be defined In order to make this theory

O R I E N T A T I O N OF G E O ME T R I C M O L E C U L A R S T R U C T U R E WI TH R E S P E C T TO THE C A R T E S I A N C O O R D I N A T E S
O c t a h e d r o n
Square Plane Tetrohedron
Figure 3

Oi'trfhnlr*! lof
T e t r a h e d r a l I o n S p l i t t i n g ‘.d t ' l v r - f o l d D r j i r i n ' r a t * - d ' L * -v f l in « nt l i - v l r u » U t u ' F l r i d u( O c u . i f d r a l S y m m e t r y
S p l i t t i n g of F i v e - f o l d D e g e n e r a t e d - L e v e l in an E l e c t r o s t a t i c F i e l d o f T e t r a h e d r a l S y m m e t r y
y*S plitting of F iv e -fo ld D egen erate d -L e v e l in an E le c tr o s ta t ic F ie ld of Square P lanar Sym m etry
(L igands in ay P lane)
S p litt in g o f F iv e - fo ld D eg en e r a te d - L e v e l ia an E le c t r o s ta t ic F ie ld o f T e tr a g o n a l S ym m etry
(E lon ga ted s - a x is )
Figure 4

18
I L
£ ___ o r ______ ______
r t L _ii_ _r t
O c c u p a n c y of d - O r b i ta l s in th e O c ta h e d r a l lo g s h av ing E le c t r o n ic S t r u c tu r e s d 1 th ro u g h d
_tt tit L.10
_iL JL _ILO ccupancy of d-O rbitals in the O ctahedral Ions having
E lectron ic Structures d ^ through d10
Figure 5

19operational. This quantity is the crystal field stabilization energy (CFSE) and is defined as follows for the octahedral case:
CFSE - (-ADq*n + 6Dq*m) n = no. of electrons in the orbitals m = no. of electrons in the eg orbitals
In other words, as long as there are more electrons in theto- orbitals than in the e orbitals, there is a decrease in the energy of the system (as compared to a spherical field case) which results from the splitting of the d levels. As a consequence of the value of -4Dq for the lower triplet and + 6Dq for the upper doublet, the CFSE must be zero if all five d orbitals are equally occupied. Applying these concepts to the octahedral case, it is seenthat, as the d orbitals are occupied, the first threeelectrons will go into the lower triplet with parallel spins in accordance with Hund's rule, since these orbitals are of lower energy than those of the upper doublet. The next two electrons may either occupy the lower triplet or the upper doublet depending upon the energy separation between the t2g and the e^ orbitals. If the energy separation is greater than the energy required to pair these electrons with those which are already present in the lower triplet, pairing will occur and the resulting magnetic

20moment will be smaller than the maximum possible. However, if the splitting is smaller than the energy required to pair the electrons, they will occupy the upper doublet and the maximum magnetic moment will result. The electronic configurations which may occur for an octahedral ion of the first series are shown in Figure 5*
In order to determine which stereochemistry is favored for a given ion, the relative energy levels for the d orbitals in fields of different symmetries must be known. Table 3 lists this information for a number of different geometric structures. Using these values, the crystal field stabilization energies may then be calculated in the manner indicated previously.
An additional consideration which has been found tobe of considerable utility is the predictive manipulation
17of ligand field theory is the JahnTeller effect. This
*7jahn &nd Teller, Proc. Roy. Soc., London, Al6l, 108 (1937).
concept requires that, If orbital degeneracy exists within a molecule, the molecule \e distorted in such a manner that the degeneracy be removed. As a consequence of this effect, many complexes which might otherwise be considered to be octahedral are, in actuality, tetragonal In structure.Such is the effect in a "spin-paired" d* configuration, for example (see Figure 6).

TABLE 3ENERGY OP THE d ORBITAL LEVELS IN CRYSTAL FIEIDS OF
DIFFERENT SYMMETRIES (IN TERMS OF Dc[)*
C.N. Structure d 2 2 - x - y d 2 —z ixy -xz d-yz
1 t -3*14 5.14 -3*14 0.57 0.572 linear- -6.28 10.28 -6.28 1.14 1.143 trigonal^ 5.46 -3.21 5.46 -3.86 -3.864 tetrahedral -2.67 -2.67 1.78 1.78 1.784 square planar^ 12.28 -4.28 2.28 -5.14 -5*145 trigonal
blpyramld -0.82 7.07 -0.82 -2.72 -2.725 square pyramid0 9.14 0.86 -0.86 -4.57 -4.576 octahedron 6.00 6.00 -4.00 -4.00 -4.007 pentagonal
blpyramld0 2.82 4.93 2.82 -5.28 -5.28
* Values taken from reference 16. ^ Bonds lie along z axis, t Bonds In the x£ plane.0 Pyramid base In xy. plane.
1d 2 _p d p-x -y* -z2
n u 11d d d—xy -yz —xz
Figure 6
V - y 2 ]
* Z S
1 [
-xyiiiid__ d-yz —xz

22Several excellent references are available which
discuss this theory (ligand field theory) and some of Its applications In considerable detail
F. Basolo and R. 0. Pearson, loc» cit., Chapter 2.19L. E. Orgel and J. S. Griffith, Quart. Revs., 11,
381 (1957).20R. S. Nyholm, "Complex Compounds of the Transition
Metals," Report to the Xth Solvay Council, Brussels, May, 1956.
Another modern treatment which has received considerable attention Is the molecular orbital theory. Whereas the valence bond theory considers only bonding between atoms and crystal field theory considers only electrostatic repulsions, the molecular orbital theory considers both repulsions and attractions since this theory Is concerned with molecular orbitals in which the energy of the electron Is quantized with respect to the molecule as a whole. That both attractions and repulsions arise naturally in the course of a molecular orbital treatment gives promise that such a theory will encompass the parts of the problem covered separately by crystal field theory and valence bond theory. According to the usual approximate approach to a complex problem In terms of molecular orbital theory, the molecular orbitals are derived from atomic orbitals of the appropriate symmetry and energy. The relative energies of the molecular orbitals with respect to the atomic orbitals from which

they are derived thus provide a basis for determining whether a given molecular orbital contributes toward attraction or repulsion between atoms* The formation of molecular orbitals from two atomic orbitals, according to the method of linear combination of atomic orbitals (LCAO approach), results in the derivation of a number of molecular orbitals equal to the number of atomic orbitals utilized* In general, half of the molecular orbitals will be of lower energy than the atomic orbitals (bonding) and half will be of a higher energy than the atomic orbitals (antibonding)• Those atomic orbitals which are unchanged In energy may be considered non-bonding* The molecular orbitals which are formed In an octahedral complex may
piserve as an illustration* A pair of molecular orbitals
metal atom by combining with an orbital of each ligand containing a pair of electrons on the x, -x, y, and -y axes (this ligand orbital might be some combination of the s. and £ orbitals which projects In the bond direction)* The metal d^2 orbital would overlap orbitals on all six ligands* Four additional pairs of bonding and antibonding molecular
central metal atom upon proper combination with the orbitals
21F. Basolo and R. 0* Pearson, loc• cit., p. 41
may be constructed from the d 2 2 orbital of the central
orbitals result from orbitals of the

24of the ligands. Three of the d. orbitals are not involvedin the formation of molecular orbitals of this particularclass (d , d , and d orbitals). These orbitals are-xy —yz —xznon-bonding. The molecular orbitals which have Just been constructed are called sigma orbitals; i.e., they are symmetrical about the molecular axis. In the same manner, gi molecular orbitals, e.g., molecular orbitals which are notsymmetrical about the molecular axis, may be constructed
22 23from the remaining atomic orbitals of the central atom. '
Eyring, J. Walter and G. E. Kimball, "QuantumChemistry," John Wiley and Sons, New York, 1944, pp. 227-231.
23K. W. H. Stevens, Proc. Roy. Soc., London, A219,5^2 (1953) •
Neglecting gi bonding, for the present, this combination may be represented, schematically in the form of a correlation diagram, Figure 7* It is apparent that this picture accounts for the magnetic properties of complexes in much the same way as does crystal field theory. If, for example, the correlation diagram in Figure 7 Is that of a complex in which the central atom has electrons which singly occupy the non-bonding orbitals and the antl-bondlng orbitals, the magnetic moment of this complex would approximate the expected moment of the free ion. However, if another ligand were used to form a complex of this metal and if the energies of the orbitals of this ligand were such that the

25
Metal Orbitals Molecular O rbitals Ligand Orbitols
(2)A nti-bond ing O rbitals
BondingOrbitalsJJu (6)
oio (2)
C o rre la tion Diagram
a * singly degenerate e * doubly d eg en e ra te t • triply degenerate
Figure 7

energy separation between the nonbonding and next higher antibonding molecular orbital is larger than the energy required to pair the electrons with the nonbonding orbitals, the electrons would be paired and the magnetic moment would be a minimum* It should be emphasized that the same molecular orbitals are involved in bonding regardless of whether the electrons are paired to the maximum possible extent*In contrast, the valence bond theory would demand that the
3 2spin-free complex use sp d hybridized atomic orbitals and2 ^that the spin-paired complex use d sp hybridized atomic
orbitals*It is also possible to use this representation to
account for the ultraviolet and visible spectra of these complexes, the absorptions arising from electronic transitions between the highest filled orbitals and the lowest
oil 2*5 26unfilled ones* ' ' Using the spectra obtained2kL. E* Orgel, J. Cheat. Phys., 2£, 1819 (1955)*25J* J* Griffith, J* Inorg. Nuclear Cheat*, 2. 1,229 (1956). “ ~26J* Owen, Proc* Roy* Soc*, London, A227, 183 (1955)*
experimentally, it is then possible to calculate the energy separations between these levels* The actual energy separations between these levels may be regarded as a function of the nature of the central metal atom and of the ligands and thus of the bond strengths*

Since the valence bond theory and the ligand field theory are complementary, a combination of the two provides a powerful set of concepts by which the magnetic properties, stereochemistries and spectra of coordination compounds may be explained in a qualitative manner. Furthermore, these properties may be predicted in a rough, quantitative manner. Although pi bonding arises naturally in the molecular orbital treatment, this aspect of valency presents an additional complexity in the case of the combined valence bond and crystal field points of view. Nonetheless, some compounds which do not at first appear to be properly described by these theories are found to confom if pi bonding is considered.
Pi bonding may be of two general types. The central metal atom may possess paired electrons which are sharedwith ligand molecules through available £ orbJtals or d
27orbitals on the ligand or the ligand molecules may have
27H. Eyring, J. Valter and G. £• Kimball, loc. cit.
pairs of electrons in £ orbitals which are not used in sigmabond formation and which are of the proper symmetry to over-
Ofi oqlap the t_ orbitals of the metal atom. * 7 In either“2gpO
L. E. Orgel and J. S. Griffith, loc. cit.29L. B. Orgel, "Some Applications of Crystal-field
Theory to Problems in Transition-Metal Chemistry," Report to the Xth Solvay Council, Brussels. May, 1956.

28case the double bonding, which results from the formation of both a sigma and a pi bond, gives rlBe to the formation of bonding (t2g and antibonding t ) orbitals from the originally nonbonding t set. In the first case, the electrons responsible for pi bonding are the metal d electrons which originally occupied the nonbonding t2g level. Since the bonding t^ level lies below the nonbonding tgg level (from which It is derived), and since the antibonding e level is unaffected by gl bond formation, It follows that the separation between these levels must increase In
Q Q
this case (lQDq is increased). On this basis, Orgel has
3°Ibld.
suggested that complexes In which double bonding of this type occurs are more likely to be spin-paired than complexes In which no such bonding Is present.
The modern theories which have Just been discussed are used, both quantitatively and qualitatively, to Interpret the properties of many transltlon-metal complexes.In this respect, It Is probably true that the complexes of Iron (II), cobalt (II), and nickel (II), have received more attention than the complexes of any other trio of metal Ions. The reason for this tremendous Interest may be associated with the large number of complexes formed by these Ions (with ligands of many different types) and the

large variation In properties of these compounds. Whereas, many metals tend to exhibit properties which are consistent throughout all of their complexes or compounds, Iron (II), cobalt (II), and nickel (II) form a variety of complexes, these compounds may be spin-paired or spin-free and the distribution of ions or groups about the central metal ion may be octahedral, square planar, tetrahedral, square pyramidal, or tetragonal blpyramidal.
Among the ligands forming complexes with these metals which have received considerable attention are those which contain the conjugated dimethine g r o u p s . Typical
J W. W. Brandt, P. P. Dwyer, and E. C. Gyarfas, Chem. Hevs., £4, 970 (195^)•
- -
C ------- C\ X X /N N
Iexamples of this type of ligand are 2, 2 1-bipyridine (dlpy, structure II), o-phenanthrollne (o-phen, structure III), dimethylglyoxlme (H-DMG, structure IV), and blacetyblsme- thylimine (BbM, structure V)• Although the unsaturated

30nitrogen atoms are the electron donors in all the structures shown above, these nitrogen atoms may be roughly divided into three distinct classes. There are the aromatic, heterocyclic diamines (structures II and III), the dioximes (structure IV), and the alkyl diimines (structure V)• In general, it may be stated that the bidentate chelating agents form complexes which are more stable than their unidentate counterparts. This enhancement of stability as more and more of these donor atoms are physically connected, i.e., as the polydentate character of the ligand increases, is called the chelate effect. There are two contributing factors to this effect. One is the decrease in entropy of reaction which accompanies the increase In coordinatingcapacity of the ligand (this effect is approximately the
32same for all metals). The second effect Involves an
32J. C* Bailar, Jr., loc. cit., Chapter 5*
Increase in enthalpy of bonding. As a consequence of the bidentate nature of these ligands and the rather good basicproperties of the unsaturated nitrogen atom, these ligands
33in many cases, form complexes of unusual stability.
33- Vf. Vf. Brandt, F. P. Dwyer, and E. C. Gyarfas, loc.cit*

31Ligands of nixed character have been prepared, e.g.,
34pyridinalmethylimine. This ligand contains both a cyclic
34D. H. Busch and J. C. Bailar, Jr., J. Am* Chen* Soc., J 8 , 1137 (1936). ”
amine nitrogen atom and an alkyl imine nitrogen atom* The iron (IX) complex formed with this ligand is quite similarto the analogous complex formed by iron (II) with 2, 2'-
35bipyridine and o-phenanthroline. It is significant that
35Ibid.
pyridinalmethylimine contains the same conjugated methlne structural unit found in Structures II-V*
VI
Despite the fact that these ligands all contain the same conjugated dimethine structural linkage, divergent properties of the complexes which they form have been n o t e d . 37 In general^ the o< -dioximes form complexes
W. Brandt, P. P. Dwyer, and E. C* Qyarfas, loc*cit.
37R. C. Stoufer and D* H. Busch, J* Am. Chera. Soc., J8, 6016 (1956). “
which are quite distinct from those complexes formed with

32the aromatic, heterocyclic diamines, the alkyl diimines, and ligands of mixed type, e.g., pyridinalmethylimine.
As a consequence of these apparent anomalies, a study of the complexes formed by these metals with ligands which are mixed in type, or of Intermediate character (to the °(-dioximes on one hand and the aromatic, heterocyclic diamines and alkyl diinines on the other), might lead to a better understanding of the systems already studied and the relationships which exist between them.
The work presented in this dissertation is concerned with the synthesis and properties of the complexes formed by iron(ll), cobalt(ll), and nickel(ll), with ligands of intermediate character to the Q^-dioximes and the aromatic heterocyclic diamines and ligands of mixed type. The ligands used in this investigation include biacetyldihydra- zone (BdH), pyrdinalhydrazone (PAH), pyridindialdihydra- zone (PdAdH), pyridinal-p-tolylimine (PAT), biacetyl- hydrazoneoxime (H-BHO), biacetylhydrazonemethoxime (BHM), and biacetyloxlmemethoxime (H-BOM). Various physical methods have been used to characterize these complexes. These include determination of molar conductance, molar susceptibility, ultraviolet, visible, and infrared spectra, and resolution studies. The magnetic studies indicate the effect of the electrostatic field (ligand field) produced by the coordinating group on the electronic configuration

of the central metal atom whereas the spectral studies reflect the effect of the metal atom on the electronic structure of the coordinating groups and the type of interaction between the metal atom and the ligand molecules. The conductance studies reveal the number and type of ions which are dissociated in a given solvent medium by a formula weight of the complex under consideration. In many instances the conductance data aid in the determination of the number and type of coordinating groups within a given complex. Resolution studies indicate the degree of stability of complexes and, when considered in, conjunction with analytical data, the structures.

II. COMPLEXES PREPARED BY THE REACTION OF IRON(ll), OOBALT(II), AND NICKEL(ll) WITH BIACETYLDIHYDRAZONE,
2-PYRIDINAIHYDRA2DNE, 2,6-PYRIDINDIALDIHYDRAZONE,AND 2-PYRIDINAL-p-TOLYLIMINE
A. Introduction
The aromatic heterocyclic diamines and triamines(such as 2,2‘-bipyridine and o-phenanthroline) and theoC -dioximes (such as dimethylglyoxime) are among thebest known complexing agents for iron(ll), cobalt(ll),and nickel(II). As a matter of convenience, the formergroups of ligands will be called the dipyridyl group andthe latter, the dimethylglyoxime group.
The octahedral complexes formed by iron(ll) withthe ligands of the dipyridyl series are invariably spin-paired. In general, these complexes are quite stable aswitnessed by the fact that some of them have been resolved
1 2 3into optical isomers. ' The rather high stabilities
1F. P. Dwyer, J. Proc. Roy. Soc., N. S. Wales, 85. 135 (1952).
o 0. T. Morgan and F. H. Burstall, J. Chem. Soc., 1931, 2213. “
3A. Werner, Ber., 4£, 433 (1912).
and the intense absorptions in the visible spectra of these iron(Il) complexes have been explained by a number of
34

35investigators on the basis of multiple bonding between
4 5 6 7 8metal atom and ligand. * * Certainly, the concept of
4L. E. Orgel, J. Chem. Phys., 2£, 1819 (1955)-^L. E. Orgel, "Some Applications of Crystal Field
Theory to Problems in Transition-Metal Chemistry,M Report to the Xth Solvay Council, Brussels, May, 1956.
°W. W. Brandt, F. P. Dwyer, and E. C. Gyarfas, Chem. Revs., £4, 970 (1954).
^P. Krumholz, J. Am. Chem. Soc., 75* 2163 (1953)•®F. H. Burstall and R. S. Nyholm, J. Chem. Soc.,
1952, 3570.
multiple bonding between a metal atom and donor atoms is9 10 11 12neither new or unusual* ' ' According to modern
^L. Pauling, "Nature of the Chemical Bond," Cornell University Press, Ithaca, New Yorkj 1948.
^W. W. Brandt, F. P. Dwyer, and E. C. Gyarfas, loc.cit.
*■4). P. Craig, A. Maccoll, R. S. Nyholm, L. E. Orgel, and L. E. Sutton, J. Chem. Soc., 1954, 332.
12R. S. Nyholm, J. Chem. Soc., 1951, 3245.
theory, octahedral complexes may form a maximum of three pi bonds with the ligand molecules.1^ The iron(Il) atom
Eyring, J. Walter, and G. E. Kimball, "Quantum Chemistry," John Wiley and Sons, New York, 1944, pp. 227- 231.
(d^ configuration) possesses three pairs of electrons in the nonbonding tgg orbitals which can be donated to the

36ligands through vacant £2 orbitals. This type of interaction was considered earlier in the discussion of £i bonding.
In addition to the complexes formed by iron(ll)with 2,2 '-bipyrldine and o-phenanthroline with a ligand-metal ratio of three to one, complexes in which the ratio
14 1*5 16is two-to-one and one-to-one are known to exist. *
l4P. Krumholz, J. Chem. Soc., Jl, 3654 (19^0).15*\J. H. Baxendale and P. George, Trans. Faraday
Soc., 46, 55 (1950).■^F. Basolo and F. P. Dwyer, J. Am. Chem. Soc.,
l£, 1454 (1954).
The two-to-one complexes, which are violet in color, may be prepared by the thermal decomposition of the three-to- one complex. These two-to-one complexes are unstable in aqueous solution with respect to disproportionation, yielding the red three-to-one and the yellow one-to-one complexes.^'^ It has been pointed out1^ that the relative
17Ibid.l8W. W. Brandt, F. P. Dwyer, and E. C. Gyarfas, loc.
cit.Irving and R. S. P. Williams, J. Chem. Soc.,
1953, 3192.
stabilities of these iron(ll) complexes (the three-to-one complex is more stable than the one-to-one, which is, in

37turn, more stable than the two-to-one complex) are notthose usually observed (one-to-one complex more stablethan the two-to-one which Is, In turn, more stable than
20the three-to-one complex). Irving and Williams have
2QIbld.
suggested that this apparent anomaly arises from orbital stabilization of the three-to-one complex (spin-pairing).
The cobalt(ll) complexes of these ligands which have been reported are generally six coordinate and spin- free. These complexes have not been resolved into optical isomers. This is to be expected since a rather high correlation exists between complexes which are spin-paired
21and those which have been resolved into optical isomers.
21J. C. Bailar, Jr., "Chemistry of the Coordination Compounds," Reinhold, New York, 1956, Chapter 4.
The fact that spin-pairing does not occur in the•7
case of a d f configuration, such as that of cobalt(ll), while it does occur in the case of a d configuration, may be explained from several points of view. It should be recalled, according to ligand field theory, that an electrostatic field of cubic symmetry splits the electronic d levels Into a low-lying triplet (tgg orbitals) and an upper doublet (e^ orbitals). For the d^ configuration, this splitting corresponds to a crystal field stabilization

energy of 24 Da in the case of a spin-paired complex and 4 D£ in the case of a spin-free complex (see Table 3)•The difference between these two is 20 D£. This difference is called the extra crystal field stabilization energy. This value, minus the pairing energy, is an indication of the stability of the spin-paired state relative to that of the spin-free state. By the same type of reasoning, co- balt(ll) (d^) has an extra crystal field stabilization
s’energy of 10 D£, only one half as much as that of the db ion. The pairing energies of the d° and d ions should be comparable; therefore, less is gained through spin- pairing in the case of cobalt(ll) than in the case of lron(ll), i.e., the total energy of the system is not lowered to as great an extent in the former case.
A molecular orbital picture reveals that the spin- paired complex of iron(ll) has no antibonding electrons in the e orbitals while the spin-paired cobalt(ll) complex has one such electron (Figure 7)• From this, it is con- eluded that the spin-paired d configuration would be expected to be more stable than the spin-paired d^ configuration. This approach leads to the same conclusions as does ligand field theory*
The valence bond theory predicts the same resultsin the following way. A spin-paired, octahedral complex
2 ^of cobalt(ll) would use d sp-* hybridized bonds, requiring

39that one electron be promoted to the 5s. level. This promotion Is energetically expensive. In the case of a spin- paired iron(ll) complex, no such promotion is involved.
By an argument similar to that used in the case of the octahedral cobalt(ll) complexes, it would be predicted that the octahedral nickel(Il) complexes formed with the dipyridyl type of ligands also would be spin-free. Experimentally, this is found to be true; however, a behavior is noted, in this instance, which is not predicted. The octahedral complexes of nickel(ll) with 2,2'-bipyrldineand o-phenanthroline resemble those of iron(ll) in that
22 2^ 24they have been resolved into their antipodal forms. *
p. Dwyer, loc♦ cit.^ A . Werner, loc. cit.24F. P. Dwyer and E. C. Gyarfas, J. Proc. Roy. Soc.,
N. S. Wales, 8^, 232 (1950).
The tris-(o-phenthroline)-nickel(ll) complex is not completely racemized after fifty hours while the analogous 2,2'-bipyridine complex is estimated to have a half-life of fifteen minutes at 1'J°C This behavior constitutes
25f . P. Dwyer, loc. cit.26W. W. Brandt, F. P. Dwyer, and E. C. Gyarfas, loc.
cit.
a significant deviation from the usual correlation found

40between spin-paired complexes and complexes which have been resolved into optical isomers. It is even more unexpected to note that the three-to-one o-phenanthroline complex of iron(Il) racemizes more rapidly than does the analogous nickel(II) c o m p l e x . I t should be pointed out,
2^J. C. Bailar, Jr., loc. clt., Table 8.8.
however, that there appears to be a difference in the mechanism of raceraizatlon (the rate of racemization of the nickel(Il) complex corresponds to a dissociative processwhile the racemization of the Iron(ll) complex must in-
28volve an intramolecular rearrangement)•
28Ibid.
In addition to the three-to-one complexes formed by nickel(II) with these ligands, a number of two-to-one complexes have been p r e p a r e d . 2 ^ 30 These are of the general
2^W. W. Brandt, P. P. Dwyer, and £. C. Gyarfas, loc»clt.
30A. E. Martell and M. Calvin, Chemistry of the Netal Chelate Compounds," Prentice Hall, Inc., New York, 1952, Table 6.3.
form NltJUOg^'nKgO, where AA Is a bldentate ligand and X represents a uninegative group or ion such as a chloride ion. n Is found to have any value from 0-3. The anhydrous two-to-one complexes may be prepared in a manner completely

41analogous to that used to prepare the two-to-one complexes of Iron(II). These complexes are bright green In color while the hydrates are blue-3^ Again, as In the case of
Basolo and F. P. Dwyer, loc. clt.
the analogous iron(ll) complexes, these substances are unstable with respect to dlsproportlonatlon In aqueous solution.
The properties of the complexes foraied by iron(Il), cobalt(ll), and nickel(ll) with ligands of the dimethyl- glyoxime type are, In some Instances, quite different from those with ligands of the dipyrldyl type discussed abovej however, It will be observed, In the characterization of these dimethylglyoxime complexes that similarities do exist.
It has been demonstrated that dimethylglyoxime andO Oiron(ll) coordinate In a ratio of two-to-one. This
3 % . Sone, Bull. Chem. Soc. Japan, 25, 1 (1952) •
reaction produces a dark red solution which looses itscolor rather rapidly unless stabilized by some base such
33 34as ammonia, pyridine, hydrazine, or hydroxylamine.
33ibld.Tschugaeff, 2. anorg. Chem. 46, 160 (1950; 41, 1678 (1908) . ~

42Whereas the best characterized complexes of cobalt
(II) with ligands of the dipyridyl type are octahedral and spin-free, cobalt(ll) reacts with dimethylglyoxime in a ratio of one-to-two, forming a brown, nonionic, spin- paired complex.
Tschugaeff, Z. anorg. Chem., 46, 144 (1905)*
In contrast to the very stable three-to-one complexes formed by nickel(Il) with ligands of the dipyridyl group, no three-to-one complexes of nickel(ll) with the ligands of the dimethylglyoxime group have been prepared.In addition to the well-known bis-(dimethylglyoxime) nickel(ll), a second two-to-one complex of nickel(Il) with dimethylglyoxime has been prepared.36>37 ^he
3 % , Paneth and E. Thilo, Z. anorg* Chem*, 147, 196(1925).
37a . g . Sharpe and D. B. Wakefield, J. Chem. Soc., 1957, 496.
composition of this second complex corresponds to the general formula ftfi (AA^X^. It should be recalled that a number of complexes with this general formula were reported for the dipyridyl group of ligands. The complexes of this latter group of ligands disproportionate in aqueous solution yielding a three-to-one complex and a one-to-one complex, whereas the analogous complex of nlckel(II) with

dimethylglyoxime yields the familiar red, water insoluble, nonionic complex, bis-(dimethylglyoxime)-nickel(II) in water. The properties of the -dioxime complexes of nickel(ll) will be discussed more fully in the Introduction to Part III of this dissertation.
Krumholz-5 demonstrated that the °< -diimines, e.g.,
3®P. Krumholz, loc. clt.
biacetylbismethylimine (structure V), form complexes with iron(ll) which are quite similar to those formed by the cyclic diamines. More recently,39*40 the preparation of
W. Brandt, F. P. Dwyer, and E. C. Gyarfas, loc.clt.
40D. H. Busch and J. C. Bailar, Jr., J. Am. Chem. Soc., J8, 1137 (1956).
ligands involving mixed functional groups (of both the cyclic amine type and of the imine type) have been reported, e.g., pyridinalmethylimine (structure VI). The six coordinate iron(ll) complexes with ligands of mixed type are quite similar to those formed by iron(II) with ligands of the dipyridyl group.
Busch and Bailar^ reported the preparation of an
^Ibid.
iron(ll) complex with biacetyldihydrazone (BdH, structure VII)• The analytical data and magnetic moment (^eff a 5*32

44B.M.) Indicate the formula [Fe(BdH)^JFeCl^, in which the
hocation is spin-paired and the anion Is spin-free* Nyholm
42 iiR. S. Nyholm, Complex Compounds of the TransitionMetals," Report to the Xth Solvay Council, Brussels, May,1956*
has stated that there are no known examples of tetrahedral complexes of ions with a d*3 configuration. It is interesting to note that this complex may provide an example of such an ion, FeCl^*. The analytical data preclude the possibility of an ion such as [FeCl^(HgO)2 ]= and a square planar configuration appears to be quite Improbable. It Is possible that the anion consists of Infinite chains of edge shared FeCl^ octahedra, or octahedra formed by coordinating both with the chloride ions and with the functional groups of the BdH which are unused in coordinationto the cation. In connection with the latter suggestion,
43it is significant that Stratton has presented evidence
j. Stratton, dissertation, The Ohio State University, 1958*
for similar intermolecular bonding in a bis-(salicylalhydra- zone)-nlckel(Il)•
It appears that the ligand field produced by the BdH may be considerably larger than that produced by the chloride ions. This is not unexpected Blnce BdH contains

45the same conjugated methine linkage characteristic of both the <*v-dioxlme and the dipyridyl class of ligands (compare structures II, IV, and VII). This is entirely consistent with the molecular orbital and ligand field theory approach, i.e., a ligand which is capable of jdI bonding pro-
lili ItRduces a relatively great ligand field. '
^ J . S. Griffith, L. E. Orgel, Quart. Revs., 11,381 (1957). ------------ -----
45L. E. Orgel, J. Chem. Phys., 2£, 1819 (1955)- 46L. E. Orgel, "Some Applications of Crystal Field
Theory to Problems in Transitlon-Metal Chemistry," Report to the Xth Solvay Council, Brussels, May, 1956.
The work presented In Part II of this dissertation is concerned with the preparation and characterization of the iron(Il), cobalt(ll), and nickel(ll) complexes of biacetyldihydrazone, pyridinalhydrazone (structure VIII), and pyridindialdihydrazone (PdAdH, structure IX). The synthesis and properties of pyridinal-p-tolylimine (PAT, structure X) complexes are also studied. Several of the latter group of complexes were first prepared by Bahr and Thamlitz.^7
*7 G. Bahr and H. Thamlitz, A. anorg. allgem. Chem. m , 3 (1955). ------- ---------------
555 .OH,\ / *VII
o H^ NH,
N ‘VIII

46
Blacetyldihydrazone might be expected to resemble both the dipyridyl group and the dimethylglyoxime group of ligands. Certainly the electronegativity and electron rich character of the NHg group would appear to resemble the OH group of the C-dioximes. Therefore, the study of the complexes formed by iron(ll), cobalt(Il), and nickel (II) with BdH and with PAH and PdAdH (which are intermediate in structure between BdH and 2,2'-bipyridine), may provide a better understanding of the factors which contribute to the divergence of properties observed in the complexes of the dipyridyl group and the dimethylglyoxime group of ligands.
B. Experimental
1. Preparations and Analytical DataBlacetyldihydrazone.- Blacetyldihydrazone was pre
pared by the method of Busch and Bailar,^® m.p., 158°.
J. Busch and J. C. Ballar, Jr., loc. cit.

Anal. Calcd. for C^H N^: C, 42.08; H, 8.83; N, 49*09.Found: C, 42.26, 42.22; H, 8 .8 9, 8 .97; N, 48.95, 49.03*
Trls-(blacetyldihydrazone)-lron(ll) Iodide Hydrate.- Two and twelve hundredths grams of Iron(ll) chloride 4-hydrate (0 .0107 rnole) was dissolved in 10 ml. of water. The solution was filtered, warmed and added to a second solution containing 4.0 g. (O.O35 mole) of biace- tyldihydrazone dissolved in a minimum amount of hot water. (Alternatively, dry ligand was dissolved In a warm solution of the metal salt.) The resulting solution was dark red. Approximately 1 g. of potassium iodide per ml. of solution was added and the solution cooled in an Icebath.A dark rust-colored product crystallized. The crystals were filtered with suction and washed with a small amount of cold water. The product was recrystallized after dissolution in warm water (60°) by adding potassium iodide, as described above. The purified product was washed with small amounts of cold water, absolute ethanol and anhydrous ether, In that order, and dried in vacuo over f2°5 at room temperature; yield 82 per cent. Anal. Calcd. for [Fe(C4Hl0N ^ l C 2 '3H20 : C, 20.43; H, 5*14; N, 2 3.8 3.Found: C, 20.42; H, 4.92; N, 24.04.
Bis-(blacetyldihydrazone)-cobalt(II) Chloride.- Two grams of cobalt(ll) chloride 6 -hydrate (0.0084‘ mole) was dissolved in 30 ml* of ethanol. This solution was

48wanned and added to a solution prepared by dissolving 2 .0
g. of blacetyldihydrazone (0.0175 mole) in 150 ml* of boiling ethanol. The mixing resulted in a brown-red colored solution from which a maroon-colored crystalline product was isolated by filtering with suction. The crystals were washed several times with small portions of cold absolute ethanol and anhydrous ether, and dried in vacuo at room temperature over P20^, yield 99 per cent. Anal. Calcd. fort Co(C^H10N^)2 ] Cl2 : C, 26.80; H, 5*61; N,31.30. Pound: C, 26.75, 26.49; H, 5*81, 5*64; N, 3 0 .5 8,30.71.
Trls-(blacetyldihydrazone)-cobalt(IX) Iodide*- Two grams of cobalt(ll) chloride 6 -hydrate (0.0084 mole) was dissolved in a minimum amount of cold water. This was added to a solution which was prepared by dissolving 2 .9 6
g. (0 .0 2 6 mole) of blacetyldihydrazone In a minimum amount of hot water. The mixing resulted in a dark red-brown solution. The solution was cooled in an ice-bath and approximately 1 g. of potassium iodide per ml. of solution was added. A dark amber, crystalline product separated. The crystals were filtered with suction and washed with cold water and absolute ethanol. The product was redls- solved in a small amount of warm water, filtered and again crystallized by the addition of potassium Iodide* The purified product was filtered with suction, washed with

49cold water, absolute ethanol and anhydrous ether, In that order, and dried in vacuo at room temperature; yield 73*5
per cent. Anal. Calcd. for [Co(C^H^N^)3 ^ 2 : C* 2 1 *99;H, 4.62; N, 2 5 .6 5 . Found: C, 21.91, 21.83; H, 4.64, 4.70;N, 25.59, 25.42.
Bis-(blacetyldihydrazone)-nickel(II) Chloride.- This substance was prepared as a light blue powder according to the method reported for the similar cobalt compound. Anal. Calcd. for tNi(C^Hl0N^)2 )Cl2 : C, 26.85; H, 5*64;N, 31*40; Ni, 16.40; Cl, 19*80. Found: C, 26.81, 2 6 .76;H. 5 -8 6, 5 .6 9 ; N, 3 1.11, 31.19; Ni, 16.55, 16.63; Cl, 19.59, 19.74.
Tris-(blacetyldihydrazone)-nlckel(ll) Chloride 2-
Hydrate.- Two grams of nickel(ll) chloride 6 -hydrate (0 .0 0 8 6 mole) was dissolved in 50 ml. of ethanol and added with stirring to a solution of blacetyldihydrazone prepared by dissolving 3*1 g- of blacetyldihydrazone (0 .0 2 7
mole) in250 ml. of boiling ethanol. The color of the resultant solution was dark pink. The solution was cooled in an lce-bath which resulted in the separation of a red-brown, needle-like crystalline product which was filtered with suction, washed with absolute ethanol and anhydrous ether, and dried in vacuo at room temperature; yield 6 8 .7 per cent. Anal. Calcd. for [Ni(C^H10N^)^Clgi C, 28.40; H, 6.70, N, 33.15. Found: C, 28.37, 28.48; H, 6 .6 7, 6 .8 5;N, 33.12, 33.20.

Pyrldinalhydrazone.- Fifty grams of freshly distilled 2 -pyridinaldehyde (0.49 mole) was added, drop-wise (with extreme caution), and with stirring to 16 g. of anhydrous hydrazine (0 .5 0 mole). The temperature of the reaction mixture was maintained near 0° C . by means of an ice-bath. As the addition of aldehyde proceeded, the reaction mixture assumed a light straw color and became rather v I b c o u s . After the addition of aldehyde was complete, the mixture was transferred to a distillation flask and heated slowly, on an oil bath, to 130°C. at a pressure 5 mm. Only the water formed by the reaction and the excess hydrazine were removed. The high boiling component which remained in the distillation flask was analyzed; yeild, 81.1 per cent. Anal. Calcd. for CgH^N^: C, 59.48H, 5-83; N, 34.69* Found: C, 59*61; H, 6.00; N, 34-68.
Trls-(pyrldinalhydrazone)-iron(Xl) Iodide.- Six and six-tenths grams of 2-pyridinaldehyde (0.061 mole) was added dropwlse and with stirring to an aqueous solution containing 1*95 g* (0.061 mole) of 95 per cent hydrazine. To this was added a second solution prepared by dissolving 4 g. of iron(Il) chloride 4-hydrate (0.020 mole) in 20 ml. of water. The resulting blood-red solution was filtered and allowed to stand for five minutes. Approximately 0.5 g. of potassium iodide for each ml. of solution was added, whereupon a red-black oil-like material separated. Upon

51prolonged and vigorous stirring, the oil became more viscous and finally began to form small red-colored particles. After complete conversion from the oil to the solid, the product was filtered with suction and washed with water.The red-black product was redissolved in 70 ml. of water by gently warming to 40°. Again, upon the addition of potassium iodide an oil-like product began to deposit on the sides of the beaker. At this point the solution was stirred vigorously and maintained at a constant temperature
i oof 40 until small maroon-colored crystals began to form. The oil on the sides of the beaker slowly disappeared as the volume of the crystalline product increased. The solution was cooled in an lce-bath and filtered with suction. The crystals were washed thoroughly with cold water, ethanol and absolute ether and dried in vacuo over P205 ; yield 93*5 per cent. Anal. Calcd. for [Fe(CgH^N^)^ ] I2 : C, 31.27; H, 3*35; N, 18.24. Found: C, 31*10,31.23; H, 3.67, 3-77; N, 17-90, 17-76.
Trls-(pyridlnalhydrazone)-coba1t(II) Iodide and Tris-(pyridlnalhydrazone)-nickel(II) Iodide.- These complexes were prepared in a manner analogous to that of the iron compound as described above (however, no difficulty was encountered in obtaining the product in a crystalline form); yield (cobalt complex), 9^*0 per cent; (nickle complex), 8 7 . 7 per cent. Anal. Calcd. fort CotCgH^N^)3 ^ 2 *

52C, 31*97; H, 3*13; N, 18.64. Found: C, 32.00, 31*87;H, 3*31, 3*32; N, 18.29, 1 8.3 2 . Anal. Calcd. for[Ni(C6H N ) ]l : C, 31*98; H, 3*13; N, 1 8.6 5 . Found:7 3 3 2c, 3 1 .6 9 , 31.77; H, 3 .3 2, 3 .3 8; N, 1 8.5 1, 1 8.4 5 .
2 ,6-Pyrldlndlaldlhydrazone.- Four grams (0.0296 mole) of 2,6-pyridindialdehyde was dissolved in 70 ml. of warm absolute ethanol. This solution was added drop-wise to a second solution which had been prepared by adding 2 g. of 95 per cent hydrazine (O.O594 mole) to 20 ml. of absolute ethanol. A crystalline product, light tan in color, was separated upon cooling the solution in an ice-bath.This product was filtered with suction and washed with cold absolute ethanol. The crystalline product was redissolved in 70 ml. of warm absolute ethanol and the product again separated by cooling. The crystals were filtered with suction, washed with cold absolute ethanol and anhydrous ether and dried in vacuo over P2°5* y ield 80.0 per cent. Anal. Calcd. for CyH^N^: C, 51*52, H, 5*56,N, 42.92. Found: C, 51*50, 51*41; H, 5*54, 5*71; N, 42.67, 42.67*
Bls-(2,6-pyrldlndialdlhydrazone)-lron(ll) Iodide.- Four grams of iron(ll) chloride 4-hydrate (0.0201 mole) was dissolved in 20 ml. of water and filtered. This was added to a second solution which was prepared by dissolving 6 .5 6 g. of 2,6-pyridindialdihydrazone (0.0402 mole) in

53warm water. The black-red solution was filtered and to the filtrate was added approximately 20 g. of potassium Iodide whereupon the dark maroon, crystalline product separated. The product was filtered with suction and redissolved In 300 ml. of warm water. After filtration the solution was cooled and a black needle-like product crystallized. The crystals were filtered with suction, washed with cold water, absolute alcohol and anhydrous ether and dried in vacuo over *2^5 * yield 8 9 .6 per cent. Anal.Calcd. for [ FefC^HgN^] Igi C, 26.43; H, 2.85; N, 22.02. Found: C, 26.44, 2 6 .5 6; H, 3*01, 3.02; N, 21.88, 21.94.
Bis-(2,6-pyrldlndlaldihydrazone)-cobalt(XI) Iodide and No rmal-Bis-(2,6-pyridindlaldihydra zone)-nlcke1(II) Iodide.- These complexes were prepared in a manner analogous to that of the bis-(2,6-pyridindialdihydrazone)- lron(ll) iodide. The cobalt complex was Isolated in the form of black needle-like crystals which became brown upon crushing; yield 86.3 per cent. The nickel complex was separated as a tan-colored powder; yield 79*6 per cent. Anal. Calcd. for [C o f C ^ N ^ ] ^ : C, 26.31; H, 2.84; N,21.92. Found: C, 26.21, 26.43; H, 2.79, 2.88; N, 21.98,21.83. Anal. Calcd. for [ N i C C j H ^ ) ^ ^ : C, 26.32; H,2.84; N, 21.93* Found: C, 26.20, 26.47; H, 3*09, 2.99;N, 21.86, 21.75*

54Iso-Bis-(Pyrldindlaldlhydrazone)-nickel(II) Iodide.-
Pour and thirty-six hundredths grains of pyridindialdihydra- zone (0.0268 mole) was dissolved In 250 ml. of absolute alcohol. The resulting solution was boiled until the volume had been reduced to approximately 100 ml. To this boiling solution was added, with stirring, a hot alcoholic solution of nlckel(ll) chloride 6-hydrate which had been prepared by dissolving 3*16 g. of nickel(Il) chloride 6- hydrate (0.0132 mole) in 30 ml. of absolute ethanol. Immediately, a tan-colored product began to separate; however, the color of the solid changed rapidly to an olive green color. The green product was filtered from the mixture and dissolved in 30 ml. of warm water. The resulting solution was filtered four times to remove a small amount of a black, water insoluble material. The product was crystallized as an olive green solid, by adding approximately 1 g. of sodium iodide per ml. of solution. This product was filtered with suction, washed with several small portions of cold water and absolute ethanol, and dried in vacuo over Yield 87*4 per cent. Anal.Calcd. for [Hi(C^H^N^)^Jlg: C, 26.32; H, 2.84; N, 21.93;I, 39*73* Pound: C, 26.06; H, 2.90; N, 21.75; I, 39.61.
2-Pyridlnal-p-tolylimlne.- This product was prepared essentially in the manner given by Q. Bahr and H;

55• • 49Thamlltz except that the product was not fractionally
Bahr and H. Thamlltz, loc. clt.
distilled but merely crystallized from ligroin; m.p. c i t e d , 58*5°; found, 57-58°; yield 63.0 per cent. Anal.
50ibld.
Calcd. for C13Hl2N2 : C, 79*56; H, 6.12; N, 14.28. Pound: C, 79.67, 79,45; H, 6.19, 6.23; N, 14.35, 14.18.
Trls-(2-pyr idina1-p-10lylimine)-cobalt(II) Iodide3-Hydrate.- Six and six-tenths grams of 2-pyridlnal-p- tolylimine (O.O336 mole) was added to a solution prepared by dissolving 2 .6 7 g. of cobalt(II) chloride b-hydrate (0.0H2 mole) in 20 ml. of water. The resultant solution, which was dark red in color, was filtered to remove excess ligand. This filtrate was added with vigorous stirring to a saturated potassium iodide solution (50 ml.) (mixing of the solution in this order prevents the formation of a putty-like mass of product which Is handled with difficulty)• The product separated as small orange particles which were filtered with suction. The product was washed thoroughly with water and permitted to dry on the filter. It was then dried in vacuo over p2°5* 72.1per cent. Anal. Calcd. for [CofC^H^NgJ^Ig'SHgO:49*02; H, 4.41; N, 8.80. Pound: C, 48.15, 48.28; H,4.20, 4.12; N, 8.72, 8.5O.

Trl3-(2 -pyridlnal-p-tolyllmlne)-nickel(ll) Iodide.-This complex was prepared In the same manner as the cobaltcomplex. The product was separated as a fine, orangecolored solid (slightly different color from that of thecobalt); yield 90-6 per cent. Anal. Calcd. for[N1 (C13H 12N2 )3 ]I2 *2H20 : C, 49-97; H, 4.30; N, 8 .9 8.Pound: C, 49-50, 49-30; H, 3 .9 2, 4.15; N, 8 .9 1, 8.86.
Bls-(2-pyrldinal-p-tolylimlne)-lron(ll) Chloride,Tris-(2-pyridlnal-p-tolylimine)-lron(ll) Iodide and Bis-(2-pyrldinal-p-tolylimine)-nlckel(ll) Chloride.- Thesecomplexes were prepared after the manner of BShr and ♦. 61T h a m l l t z , a n d their compositions were confirmed by
51Ibid.
analysis.All analyses were by the Galbraith Microanalytlcal
Laboratories, Knoxville, Tennessee.2. Conductivity Measurements
Conductances were measured using an Industrial Instruments Inc. Model RC-16B conductivity bridge and a cell with a cell constant of 1.398 cm"*. All measurements were made at 25° C. using 10"^ M solutions (based on formula weight), and a bridge frequency of lOOOc.p.s. The absolute methanol used for these measurements was protected from the atmosphere and had a specific conductance of less than 10-? ohm"*.

573* Magnetic Measurements
All of the magnetic measurements were made by the Gouy method using ferrous ammonium sulfate 6 -hydrate and water as standards. The field strength was approximately 7000 gauss. The sample tubes (approximately 5 mm. inside diameter) used for the powdered samples were calibrated with water delivered from a calibrated T.D. plpet. Each sample tube was fitted with a standard taper, ground glass stopper and suspended from the balance by a fine aluminum wire, such that the bottom of the sample tube was centered between the two inch pole pieces of the magnet.
The first series of measurements was made using a Consolidated Engineering Corporation magnet (equipped with two inch, tapered pole-pieces) and power supply. A Chris- tian-Becker analytical balance of 0.1 mg. sensitivity was used to determine the apparent change of weight in the applied field.
The second series of measurements was made using the magnet described above; however, the pole pieces were separated by a distance which was somewhat greater than that in the first series of measurements. This increase In gap-width was attended by a small decrease in field strength for a given value of the current through the colls. An Ainsworth semi-micro balance of 0.003 mg. sensitivity was used to determine the apparent change in

58weight of the sample In the field. Under operating conditions, the sensitivity of this balance Is estimated to be0 .0 3 mg.
The data arising from a study of the temperature dependence of the molar susceptibilities of several complexes are found in Appendix III. The magnet and balance used were those described above for the second series of determinations. The brass cryostat is shown in Figure 8. The susceptibilities of several complexes were determined at five different temperatures (from 80°K-373°K). Approximately thirty minutes were allotted for the sample to attain thermal equilibrium at each temperature(with the exception of the determination at room temperature). Steamwas passed through the Jacket of the cryostat for the
odeterminations which were made at 373 K. Liquid freon was used to obtain temperatures between 232°K and l83°K (thevapor pressure - temperature relationship has been deter-
52mined). The pressure above the liquid freon in the
^2E. Neilson, master's thesis, The Ohio State University, 1957 •
reservoir of the cryostat was controlled by means of a barostat. Liquid nitrogen was used for the determinations at 80°Kj however, some difficulty was experienced in maintaining this temperature. Due to the rather large heat leak of the cryostat, and its small capacity (approximately

Filling tu b e (g lo ss )
Exhausti ■7
V acuum , jacket ^ sp ace
Constructed of | j brass stock
k - ' HB rass C ryosta t
To evacuate vacuum jacket
Figure 8

603/4 liter), a single filling was not sufficient to maintain the temperature at 80°K during the course of a determination. Therefore, nitrogen was drawn into the cryostat in a continuous stream from an external reservoir by pumping on the system. The pressure was maintained at 760 mm, - 20 mm.
For the weighings which were made at temperatures below 3 0 0°K, two determinations of the sample weight without the magnetic field were required because of the condensation of atmospheric moisture on the sample tube- One weighing was made before the weight determination in the field, the other, at an equivalent time interval, after the weight determination in the field. Assuming that the rate of condensation remained constant during this period of time, the two weighings made without the magnetic field were averaged. This average was used in calculating the apparent change of weight of the sample in the magnetic field (^uJ s uJp - uJe where uJp - the weight of the sample in the field and U)0 ■ the average of the two weighings without the field).
4. Ultraviolet and Visible SpectraThe ultraviolet and visible spectra were obtained
using a Model 10 Cary Recording Spectrophotometer and matched cells with fused quartz windows. The ultraviolet region was considered to include the range 2000A to 3000*1,

61o othe visible, 3000 A to 7000 A. The concentrations used
in the determination of the ultraviolet spectra were ap- _5proximately 30 M, while the concentrations used for the
visible region were 10“3 _ io-^ M.5* Infrared Spectra
The infrared spectra were obtained by means of a Model 21 Perkin-Elraer Infrared Recording Spectrophotometer equipped with sodium chloride optics. The potassium bromide pellet method was used for solid samples and sodium chloride capillary cells were used for liquid samples.
Deuteration of selected samples was accomplished by dissolution of the compound in the minimum amount of D20.The DgO was pumped off and the samples were dried in vacuo over PgO^.
6. Continuous Variations Study of the Nlckel(ll) Blacetyldihydrazone SystemThe Continuous Variations Study was made using
53w. C. Vosburgh and G. R. Cooper, J. Am. Chem. Soc., £2, 437 (1941). ~
the Model 10, Cary Recording Spectophotometer and matched cells with fused quartz windows. The stock solutions of blacetyldihydrazone and nickel(II) chloride were prepared
j)(2 x 10" M) using an analyzed sample of biacetyldihydra- zone and reagent grade Ni C12 *6h 20. These solutions were then mixed in various proportions, permitted to stand for

62thirty minutes, and the visible spectrum of each solution determined. A total of sixteen solutions was utilized (including 100 per cent blacetyldihydrazone and 100 per cent NiC^) • The enhancement of absorption (absorbance of the solution at a given wavelength minus the absorbance of the solution of NICI2 at a concentration equal to the concentration of Ni++in the mixture, assuming Beer's Law) was plotted against the per cent of nickel in the solution for two selected wavelengths in order to determine the ratio of ligand to metal.
Resolution of Optical IsomersThe tris-(biacetyldihydrazone)-iron(ll) iodide 3-
hydrate was prepared in the manner described earlier. The resolving agent, silver d-antimonyl tartrate was prepared by dissolving 30 g. of K(SbO)(C^H^Og)(0.09 mole) in 100 ml. of hot water. To this was added, with stirring, 15 ml. of a solution containing 15*29 6 * of AgNO^ (0 .0 9 mole). Immediately, a white, microcrystalline product began to separate. The solution was permitted to cool and the white product filtered with suction, washed with several portions of water, and dried in vacuo over P2°5 * 95per cent.
The d-antimonyl tartrate salt of [Fe(BdH)g)++ was prepared in the following manner. Three and one-half grams of [Fe(BdH)33l2 *3H20 (0.00495 mole) was dissolved in 35 ml.

63of water. To this was added 3*94 g* of Ag(SbO)(C^H^Og).The Agl formed by the reaction and the excess Ag(SbO)(Cj^H^O^) were removed by filtration. The filtrate was
oevaporated in a rotary evaporator (at 5 C), to 20 ml.The solid product which had separated during the evaporation was filtered with suction, washed with several small portions of cold water, and dried in vacuo over ^2^5 *Yield (based on separation of one isomer), J6 per cent.This product was recrystallized twice by dissolving the solid product in the minimum amount of warm water and quickly cooling in an ice-bath. Yield, 18 . 8 per cent.Anal. Calcd. for [ Fe(CifH 10N lt) ][ (SbO) (C^H^Og)] g : C,24.76; H, 3*95; N, 17.33. Found: C, 24.89, 24.72; H,4.05, 3*87; N, 17.10, 17.29. The specific rotation obtained is reported in the Results Section, p. 186.
An attempt was made to resolve [Fe(PAH)_]I0 and [FetPAT)^]^ according to the procedure described above; however, the d-antimonyl tartrate salts of these complex cations could not be crystallized from water as a result of the formation of either a tarry-like mass or a glass when the volume of the solution was reduced to the maximum possible extent. Variations of temperature over a considerable range (90° - 0° C.) did not appear to facilitate crystallization.
All determinations of rotation were made with a photoelectric Rudolph Model 80 high precision polarlmeter.

64C . Results
1. Conductivity MeasurementsConductance data have been used for a number of
54years as an aid to structure determination. Werner used
54 nJ A. Werner, Neuere Auschaungen auf den Gebiete der anorganischen Chemie,11 translated by Headley* Longmans, Green, and Company, New York, 1911.
conductance data to determine the number of ions within the "inner sphere of coordination" in compounds such as [Ni(NH0)£ ]C1 and [Co(NH ) ltCl ]C1. There is the possibil-3 D 3 3 4 2ity, however, that such data may be misleading for systemsin which the solvent molecules replace one or more of the coordinating groups. In the case oftCotNH^)^ Cl^],
55Ibid.
Werner observed that the initial conductance was quite low(which was in agreement with the formulation given above),i.e., all of the chloride ions were coordinated to the cobalt; however, he also observed that the conductance increased with time. This, he attributed to the replacement of the chloride Ions with water molecules from the solvent.
In this study, the conductances of eighteen complexes have been determined. Absolute methanol was used as the solvent for two reasons. (1) All of the complexes are soluble in methanol and (2) the complexes which contain

6 5a ligand to metal ratio of two-to-one do not appear to disproportionate in methanol as they do in water ([Ni(RdH)2 Clg ] for example). The conductance data are reported in Table 4. The anion conductances were calculated from data
r y
given in Maclnnes and in Glasstone using Walden's rule
56d . A. Maclnnes, "The Principles of Electrochemistry," Rinhold Publishing Corporation, New York, 1939, Chapter 19*
57S . Glasstone, "An Introduction to Electrochemistry," D. Van Nostrand Company, Inc., New York, 1942, Chapter II•
which associates the limiting equivalent conductance of an electrolyte, A , with the viscosity, ) , of the solvent In which it is dissolved. The values derived In this
o Omanner for anion conductances in 10-o m solutions at 25
are as follows: Cl”, 50*9 ohm "S I”, 61.3 ohm C10^“,7 1 .0 ohm-1.
The molar conductances of the complexes In which bidentate or tridentate ligands occupy all six coordination positions are In agreement with the formulation of these compounds as bi-univalent electrolytes. The cation conductances of these complexes lie between 43.5 ohm”1 and 54.4 ohm”1. The molar conductances of the complexes of the general formula [ M(AA)2X2 3 (where AA is a bidentate ligand and X is an unldentate ligand with a single negative charge) pose somewhat of a problem. These complexes have

TABLE 4MOLAR CONDUCTANCES OP THE COMPLEXES OP IRON(ll),
COBALT(ll), AND NICKEL(II) WITH BdH, PAH, PdAdH, AND PAT. 10“3 M SOLUTIONS, 25°C
Compound (ohm-*) cation (ohm
[Fe(BdH)3 ]l2 1 6 6 . 8 44.2[Co(BdH)2Cl2 ] 100.9 (5 0 .0 )*[Co(BdH)3 Jl2 166.4 43*8LNi(BdH)2Cl2 ] 1 1 6 . 0 (65*1)*[Ni(BdHJ3 JCl2 145*3 43*5[Nl(BdH)2 (ClO^)2 ] 152*9[Fe(PAH)3 ]l2 174.9 5 2 . 2
[Co (p a h )3 3i 2 172.0 49.4[n i (p a h )3 ]i 2 174.8 52.2[Fe(PdAdH)2 3l2 176.3 53*7[Co(PdAdH)2 ]I2 173*0 50.4Normal-[Ni(PdAdH)2 Jl2 177*0 54.4I s o - t N i f P d A d H ^ U C l O ^ g 183*9 41.9[Fe(PAT)2Cl2 ]*2H20 137*1 •?
*
[Fe(PAT)3 3l2 *2H20 164.5 41.9[c o (p a t )3 3i 2 *3H2o 163*9 41.3[N1(PAT)2 (C104)2 ] 170.5 ?[N1(PAT)3 ]I2 *2H20 170.5 47*9
* This value la calculated by assuming that one chloride Ion has been replaced by methanol.

67been formulated as six coordinates from which one would predict very small conductance values; however, the conductances listed In Table 4 are more nearly equal to those expected of a uni-univalent electrolyte in methanol.^8*59
58Ibld.noD. A. Maclnnes, loc. clt.
Those conductance values which approximate a unl-univalent—1 60electrolyte in methanol ( A NaCl r 96.9 ohm ) probably
6oIbld.
result from a reaction between the complex and the solvent molecules in which an average of slightly more than one unidentate chloride ion or perchlorate ion per molecule is replaced by methanol. Support is found for this explanation in the observation that these complexes (of the form [M(AA^X2]) dissolve very slowly in methanol, the dissolution being accompanied by a definite change in color in the case of tNi(BdH)2C121, (blue to pink). Although the color change, indicated above, is observed when this type of complex dlsproportionates in aqueous solution, the measured molar conductance is too small to support a similar disproportions! ion in methanol. It is significant that all of the complexes of this general type, with the exception of [Pe(PAT)2Cl2 )-2H20 are obtained as a consequence of their very low solubility In ethanol.

68In the case of L Fe (P A T ) 1 2 3'2H20 , one might sug
gest that a formulation such as [Pe(PAT)2C1H20]C1*H20-i
would better account for the conductance of (137*1 ohm- ); however; partial substitution of the remaining chloride ion would be necessary in order to account for the rather large conductance value. Alternatively, it would be necessary to explain an exceptionally high cation conductance (86.2 ohm-^)• One other formulation for this compound is possible, [Fe(PAT)2 (H20)2 ]C12 ; however, this requires a cation conductance of 35*3 ohm-1, which is rather small.
In summary, it may be stated that all the complexes, in which bidentate or tridentate ligands occupy the six coordination positions of the metal ion, have conductances which are consistent with their existence as bi-univalent electrolytes. The data obtained for the remaining complexes, although not detailed, preclude the existance of tetrahedral complexes and indicate that, the complexes are six coordinate in the solid state. The molar conductance values which are much larger than those expected for a non-electrolyte may be explained by the following equations:
Cm Ca a ) ^ ] + c h 3o h = [m (a a )2 (c h 3o h ) X]+ + X “[M(AA)2(CH3OH)X]+ + CH3OH = [M(AA)2 (CH3OH)2 3++ + x"
2• Magnetic MeasurementsMagnetic data form a very important part of the
physical characterization of coordination compounds. Their

use Is three-fold- These data aid in the determination of the oxidation state of the central metal ion, they reflect the type of bonding between the metal atom and the groups coordinated about it, and, under ideal circumstances, they give an indication of the stereochemistry of the molecules. Before considering these three uses in more detail, It is well to consider the effects of Intramolecular or intra- ionic electric fields on magnetic moments-
Type I- Weak Field-- Ideal magnetic behavior Is observed when the electrons responsible for the paramagnetism of the Ion are well shielded from external fields,i.e., the effective electric fields are not strong enough to greatly perturb either or L. In addition, in the case of Immediate Interest, one J value lies much lower in energy (with respect to kT) than all other jJ values- This type of behavior Is found among the rare earth ions, for example, In which the electrons responsible for the paramagnetism occur in the 4f orbitals of the ions- In this case, the magnetic moment is usually in agreement with the equation
where £, the Lande splitting factor is given by the expression
6 • -L (L+1)2J(J+l)
and J ■ | Lts | • In general, the magnetic moments of the

70rare earth type of ion are found to be independent of the stereochemical environment. However, if all of the J
1P. W. Selwood, "Magnetochemistry," Second Edition, Interscience Publishers, Inc., New York, 1956, Chapter VIII.
values (in general, J takes the values | L-S /, L-S +1,.... L+S-l, L+S) are separated by an energy much smaller than kT, the magnetic moment is found to correspond to either the equation given above or to the equation
JU~ l/^S(S+l)+L(L+l)The extent of spin-orbit coupling determines which equation Is applicable to the determination of the magnetic moment.041 In actuality, it appears that few compounds have
H. Van Vleck, "Theory of Electric and Magnetic Susceptibilities, Oxford University Press, London, 1932, pp. 229-237.
magnetic moments which are in accord with this latter equation. (Some spin-free cobalt(II) complexes appear to be in agreement, having magnetic moments as large as 5*2 B.M. These will be discussed in more detail later.)
^ R . S. Nyholm, loc. cit.6*R. S. Nyholm, Quart. Revs., I, 377 (1953).
Type II. Intermediate Field.- In general, the magnetic moments of the complexes formed by the metals of

71the first transition series are found to correspond more
orbital contribution to the magnetic moment having been quenched. Quenching occurs when an electric field removes the orbital degeneracy of an atom. It is observed, however, that this formula applies more precisely to the first half of the transition series than to the last half, in which the orbital quenching Is not complete,^5*66
the applied electric (ligand) field only partially removes the orbital degeneracy. As a consequence, a new value of L, smaller than the L value characteristic of the ground state of the given gaseous Ion must be assumed in order to account for the magnetic moment of the bonded ion. In this case, one of the first two equations may apply, using the new L value*
suiting from Incomplete quenching Is dependent upon the stereochemical environment of the ion. This may be illustrated, in a qualitative manner from a consideration of the effect of ligand fields of octahedral symmetry and of tetrahedral symmetry on the splitting of the d orbital levels of a d^ ion.
closely to the spin-only formula, l), the
65Ibid.fZfi
R. S. Nyholm, loc. cit.
In many cases, the small orbital contribution re

Splitting of Five-fold Splitting of Five-foldDegenerate d.-Level in Degenerate d-Level inan Octahedral Field a Tetrahedral Field
Figure 9
From the splitting produced by a ligand field of octahedral symmetry, it is apparent that there is an orbital degeneracy in the lower energy t2 g orbitals, which should give rise to an appreciable orbital contribution to the moment. From the apparent degeneracy of three, L equals one and the magnetic moment, with orbital contribution is 4.1 Bohr Magnetons. If the separation of the _tggand e orbitals is relatively small, additional orbital ~“Sdegeneracy will result and the magnetic moment will increase accordingly.
The splitting pattern Is Inverted In the case of a ligand field of tetrahedral symmetry. In this case, no orbital degeneracy exists in either the lower eg orbitals or the upper levels. This indicates that no orbital degeneracy is expected unless the splitting between eg and tgg is small enough that some interaction can occur, giving rise to a small orbital contribution. It Is not

unreasonable to expect this type of Interaction to occur since the splitting between the two energy levels Is much less In the tetrahedral case than In the octahedral case.*
67J. S. Griffith and L. E. Orgel, loc. clt.
As a result of the orbital degeneracy In the lower level for the octahedral case, and since no degeneracy of this type exists In the tetrahedral case, one would expectthe magnetic moment of a spin-free, octahedral complex of
;he 68
7this d ion to have the greater positive deviation fromthe spin-only moment.
roR. S. Nyholm, "Complex Compounds of the Transition Metals," Report to the Xth Solvay Council, Brussels, May, 1956.
Type III. Strong Field.- In the event that the ligand field of an ion exceeds a certain critical value, the spin degeneracy is lifted. According to ligand field theory, the spin degeneracy is lifted in those instances In which 10 D£ is greater than the energy required to pair electrons.
For an odd number of electrons, spin-pairing can not result in a diamagnetic ion. The paramagnetic moments of such spin-paired complexes are sensitive to the stereochemical environments of the ions. In the square planar complexes of cobalt(II), a large, positive deviation from

74the moment expected for one unpaired electron is observed
/T Q
( j X e ff = 2.1-2.9 B.M.). In contrast, the magnetic
69Ibid.
moments of six coordinate cobalt(ll) 70 71‘ complexes should
7°Ibld.71r. S. Nyholm, Quart. Revs., 7, 377 (1953)•
more nearly agree with the expected value of 1.73 B.M.(see below). However, the John-Teller principal indicatesthat no regular octahedral structures should occur in the
7case of spin-paired d ions. A tetragonal distortion is expected. The magnetic moments may be explained, in a qualitative way, on the basis of the splitting of the d levels in ligand fields of planar and tetragonal symmetries, In both splitting patterns, it appears that there will be
d e 2 — xe-y^ - y -
d~xy
t i -Z
XL£cy
A L .” X Z
Splitting of Five-fold Degerate d-Level in a Square Tlanar Field
JjL—xz
Splitting of Five-fold Degenerate d-Level in a TetragonaT Field
Figure 10

75
no orbital degeneracy; however, if the separation between the two upper levels is small, an effective degeneracy may exist, giving rise to an orbital contribution to the magnetic moment. One might expect the contribution to be larger for the tetragonal case than for the planar, since the two upper levels and have a smaller energyseparation in the first case. However, it can be shown that no orbital contribution can arise from an orbital multiplicity involving only the d_2 „2 and d o orbitals.— A —y - 2 c -
72W . B. Hadley, private communication.
This case appears to be analogous to the "non-magneticn73doublet which Bethe J describes in the case of octahedral
7 % . Bethe, Ann. Physlk., 3# 113 (1929); A. Physlk., 60, 218 (1930)•
copper(ll), in order to account for the small orbital contribution to the magnetic moment. Consequently, the tetragonal case should be associated with a magnetic moment comparable to the spin only value.
In contrast, the planar, spin-paired configuration involves, in its orbital degeneracy, the dx2 -y 2 and dorbitals. A substantial orbital contribution may be as-
74soclated with this structure. Nyholm' states that the
S. Nyholm, "Complex Compounds of the Transition Metals." Report to the Xth Solvay Council, Brussels, May, !956.

76available experimental data indicate that the moment of a
7planar d ion occurs between 2 .1 and 2.9 B.M. As will be pointed out in the discussion section, the upper portion of this range of values may be associated with a considerable population of a quartet state (three unpaired electrons) .
3• Application of Magnetic Data to Problems of Valency and StereochemistryIn many instances, the oxidation state of the cen
tral metal atom of a complex may be determined, in a straightforward manner, using the spin-only formula to calculate the number of unpaired electrons. It is required, however, that each of the possible oxidation states be associated with a different number of unpaired electrons.
75Pauling'^ made the first avowed attempt to correlate
^ L . Pauling, J. Am. Chem. Soc.. 53j 167 (1931)*
magnetic data and bond type in the enunciation of a "Magnetic Criterion of Bond Type." Effectively, he stated that if no change of magnetic moment accompanies complex formation, the complex is "essentially ionic" (spin-free). On the other hand, a change (decrease) in the moment accompanying complex formation indicates that the complex is "essentially covalent" (spin-paired). In order to avoid

77use of these misleading terms, the statement may be modified to indicate that the bonding involved in a spin- paired complex of a given ion is usually more covalent than that in a spin-free complex of the same ion.^
S. Griffith and L. E. Orgel, loc. cit.
An application of this concept may be made, for example, to the six coordinate complexes of metals containing between four and eight d electrons. These complexes may be either spin-free or spin-paired, the spin-state being determined by the strength of the ligand field. In the majority of cases, a differentiation can be made between these two states by calculating the number of unpaired electrons (spin-only formula) from the experimentally determined moment.
The increase in covalent character associated with spin pairing may be accounted for on the basis of a shortened metal-ligand bond distance In passing from a spin- free to a spin-paired complex of a given ion. In terms of ligand field theory, this shortened bond distance arises from a decrease In the repulsive energy between ligand and metal atom as the electrons, originally in the e orbitals (closer to the ligand atoms), are removed and paired with those in the tg orbitals (farther from the ligand atoms)• This shortened bond distance results in

78an increased interaction between the metal atoms and ligand molecules.
77L. E. Orgel, J. Chem. Phys., 2£, 1819 (1955)*
The same conclusions are drawn from a molecular orbital picture in which there is a decrease in the number of antibonding electrons as the complex passes from the spin-free to spin-paired state.7^ This has been discussed
7®J. S. Griffith and L. E. Orgel, loc. cijt.
previously in conjunction with the d^ configuration.Application of magnetic data to structure determin
ation is successful only for those systems in which each configuration is characterized by a unique magnetic moment. In addition, it Is necessary that the moment, characteristic of each configuration, either can be predicted from theoretical considerations or that it be expected to lie within a limited range of values which is indicated by previous experience with related complexes.
There are two general types of effects which give rise to a difference in moment for two possible structures of the same ion. They are a difference in spin degeneracy and a difference in effective orbital degeneracy of the metal ion.

79In the first ease, application of the spin-only
formula to the experimentally determined magnetic moment may give an indication of the structure. For example, two configurations are possible for a four coordinate complex of cobalt(ll). These are square planar and tetrahedral. The moment expected of a tetrahedral ion should fall in the range 4.4-4 .8 B.M. while that associated with the spin-paired planar structure is 2.1-2.9 B.M.
The application of changes in orbital degeneracy to the determination of structure is a more recent development. As indicated earlier, It is a simple matter, in many caBes, to predict, qualitatively, the relative magnitudes of orbital contributions to the magnetic moments of two different structures, using the splitting patterns of the d orbitals.
The values of the magnetic moments which are expected for the tetrahedral, square planar, octahedral, and tetragonal ions of iron(ll), cobalt(ll) and nickel(Il) are listed in Table 5*
The molar susceptibilities of BdH, PAH, PdAdH,PAT, and eighteen of their complexes have been determined.These susceptibilities are tabulated in Tables 6 and 7*
All of the lron(ll) complexes which are coordlnatelysaturated by the polydentate ligands have negative susceptibilities indicating quite conclusively that the complexes

80
TABLE 5MAGNETIC MOMENTS EXPECTED FOR SOME COMPLEXES OF
IRON(II), COBALT(ll), AND NICKEL(ll)*
Ion Tetrahedral Square Planar Octahedral Tetragonal
Iron(ll) 4*9 B*M.a 5*0 B.M.a '+Cobalt(ll) 4.4-4*7a 2 .1-2 .9b 4.8b -5*2 l-9bNlckel(ll) 3*Ba (b) 2*83-3*2a '+ (b)
* All values are taken from references 7^ and 79 unless otherwise stated*
a Spin-free*b Spin-paired*+ Based on data Included In references 3° and 6 l*
79b * N. Figgis and R* S. Nyholm, J* Chem. Soc«,12Si- 12.

8 1
TABLE 6
MOLAR SUSCEPTIBILITIES OP LIGANDS AND ANIONS
Ligand or AnionNumber of
Determinations * 106
Biacetyldihydrazone (4) - 70.6 ~ 23
2-Pyrldinalhydrazonea - 98 t 192,6-Pyridindialdihydrazone (2) -1 1 3 .8 - 192-Pyridinal-p-tolylimine (2) - 61.3 ~ 13Iodide ion** - 5 0 .6 - 1 .6
Chloride ion1* - 2 3 .4 - 1 .3
Perchlorate ionb - 3 0.2 (derived)Water - 13
a Value calculated from that of 2,6-pyridindialdihydra zone and Pascal*b atonic susceptibilities as given by reference cited below, p. 5 1*
^ Values from P. tf. Selwood, "Magnetochemlstry," Interscience Publishers, Inc., New York, N.Y.,19^3, P . 36 .

MOLAR SUSCEPTIBILITIESTABLE 7 AND MAGNETIC MOMENTS OF
82
COMPLEXES
CompoundNumber of
Determinations ^ M * X° 6 (complex)A e f f
(metal Ion)
[Fe{BdH)3 ]l2 (3) -227 0.45[Co(BdH)2Cl2 ] (2) 9774 4.91[Co(BdH)3 ]l2 (2) 6923 4.17[Nl(BdH)2Cl2 ] (3) 3848 3 .1 2
tNi(BdH)3 ]Cl2 (2) 3679 3.10[Fe(PAH)3 3l2 (3) -111 O .83
[co(pah)3 ]i2 ' (2) 8429 4.63[ni(pah)3 Ji2 (2) 3730 3.15[Fe(PdAdH)2 ]l2 (3) - 62 0 .8l[Co(PdAdH)2 3l2 (3) 3280 2 .9 6
Normal-[Ni(PdAdH)2 3l2 (3) 4134 3 .2 8
Iso-[Nl(PdAdH)2 ]l2 (2) 3842 3 .1 8
[Fe(PAT)2Cl2 ]*2H20 (3) 11577 5-34[Fe(PAT)3 ]l2 -2H20 (3) -358 0
[co(pat)3 )i2 *3H2o (2) 10225 5.06[Nl(PAT)2C 12 J *2H20 (2) 4413 3.32[ni(pat)2 (cio4)2 ]*h2o (2) 4094 3.24[N1(PAT)3 ]I2 #2H20 (2 ) 4028 3.24

83are spin-paired. It may be significant that after corrections have been made for the diamagnetic contributions of the ligand molecules and anions, small, paramagnetic moments (0.if5 - O .83 B.M.) may be attributed to the Iron atoms. The only exception Is [Fe(PAT)3 )1 2 *2^0 which remains diamagnetic after the diamagnetic corrections have been applied.
There are several possible factors which may contribute to the small, paramagnetic moment which is ob-
floserved. It has been pointed out by Orgel that, on the
®°J. S. Griffith and L. E. Orgel, loc. clt.
basis of crystal field theory, spin-pairing Is not expected of the Iron(ll) ion In complexes with the ligands of the dipyridyl class (specifically, o-phenanthroline)•The fact that these complexes are spin-paired Indicates that £i bonding Is a very important factor. He concludes from this that these complexes must lie very close to the cross over point between spin-paired and spin-free states (the energy separation '-'kT) • As a consequence of such a relatively small energy separation, it would be expected that the two spln-states might coexist. The relative population of the states would be determined by a Boltz- man distribution and would be a function of the temperature.

In an attempt to establish or disprove the postulated equilibrium between spin-free and spin-paired states of iron(ll) complexes, the magnetic susceptibilities of four iron(ll) complexes, including the diamagnetic PAT complex, were determined as a function of temperature (Table 8 and Appendix III).
TABLE 8
MAGNETIC SUSCEPTIBILITIES OP SOME SPIN-PAIRED IRON(Il) COMPLEXES AS A FUNCTION OF TEMPERATURE
Compound T. °K. M eff
[Fe(BdH)oil^ 230080
0 . If2 0 .2 0
[Fe(PAH)3 ]l2 300232206183
80
0.910.850.830.790 .1 8
[Fe(PdAdH)g ]l2 300232206183
80
0 .6 60 .6 10 .6 10 .6 10 .3 9
[Fe(PAT)3 3l2 *2H20 300232186
80
0 .0 00 .0 00 .3 00 .2 2
These data are inconclusive* Although a definite trend is observed in each series of determinations (A w becomes smaller as the temperature decreases) the magnitude

85of the changes is quite small (Figure 11). For example, the difference in £> w for [FefBdH)^)^ 300°K and at 80°K is only 8 x 10"-* g. This trend may only reflect the increasing experimental uncertainty associated with the lower temperature measurements (discussed in experimental section)•
Q1Orgel states that difficulty would be anticipated
^ L . E. Orgel, "Some Applications of Crystal Field Theory to Problems in Transition Metal Chemistry," Report To the Xth Solvay Council, Brussels, May, 1956.
in determinations of this type unless the two forms be present in comparable amounts and 3ugge3ts that optical measurements might be more suitable. It might be conceded that such measurements (optical) enjoy their maximum advantage to systems in which there is a very small amount of spin-paired complex relative to the amount of spin- free. It would appear that paramagnetic resonance techniques should prove very useful in the case under consideration, since the concentration of spin-paired complex is relatively great compared to that of the spin-free complex (ensuring that the spin-free complex be magnetically dilute).
There are several additional factors which may, in part, account for the residual paramagnetism of these iron (II) complexes. The calculation of an effective moment for the metal is based on the assumption that the magnetic

86
♦ 3 0 0
O [Fe(PAT)3 ] I 2 ■ 3H Z0 A [Fe (BdH )3] I 2 x [Fe( PAH)3] I 2 □ [Fe ( PdAdH)2 ] I 2
♦200
+ 100
-100
-200
- 3 0 0
3 0 0100 200 Tem perature, *K
Figure 11* Temperature Dependence of the Magnetic Susceptibilities of Some Spin-paired Iron(ll) Complexes*

87properties of the constituents of the molecule (metal ion, ligand, and anion) are additive. There is no doubt that
Or,this assumption is not true; however, the magnitude and
82p, w. Selwood, loc. clt., Chapter VI.
direction of this inaccuracy is somewhat less certain. For example, certain aliphatic organic compounds experience a change in molecular diamagnetism when cyclized, and polymerization also brings about changes. Secondly, the precision with which the molar susceptibilities of the ligand molecules may be determined Is not good (Table 6). It may well be that a very precise determination of the ligand susceptibilities would result in lower values, thus constituting a smaller diamagnetic correction to the molar susceptibility of the complex. Finally, the apparent change in weight of these spin-paired samples, when placed In a magnetic field, is a small number resulting from the difference of two, relatively very large numbers. One or two-tenths of a milligram difference affects the molar susceptibility substantially.
In contrast to the spin-paired nature of the complexes Just discussed, the only iron(Il) complex in which two of the six coordination positions are occupied by '’strange" groups (either chloride Ion or water) Is strongly paramagnetic (J -eff * 5*3*1 B.M.). This observation is

88entirely consistent with the properties of other lron(Il) complexes which are coordinated to ligand molecules through four unsaturated nitrogen atoms and to two different groups such as HgO or X. The existence of [PefPATjgCl^]*2H20 as
^ L . E . Orgel, loc. cit.
a tetrahedral complex is precluded by the large magneticmoment. It will be recalled that tetrahedral iron(ll)complexes should have magnetic moments which are very closeto the spin-only value, the orbital degeneracy being in the
84"non magnetic" doublet (d^2 2 and — 2 orbitals)* There
B. Hadley, loc. cit.
appears to be no experimental evidence available to substantiate this value, however. From this point of view, It appears, also, that the existence of discrete FeCl^= Ions in the compound formulated as [Fe(BdH) [FeCl^] is very questionable ( JU - 5*32 B.M.)*
The magnetic moments of the cobalt(II) complexes, which are listed in Table 7, appear, at first glance, to
Qc 0^offer a challenge to Nyholm*s * correlation of structure
85r . s . Nyholm, "Complex Compounds of the Transition Metals," Report to the Xth Solvay Council, Brussels, May, 1956.
86B* N. Figgis and R. S. Nyholm, loc. cit.

with magnetic susceptibility. Of the five cobalt(ll) complexes included in this listing, only two have moments between 4.9 and 5*6 B.M. They are [Co(BcLH)gClgJ and [Co(PAT)3 Jig*3H20* In the first case, a tetrahedral structure, formulated as [Co(BdH)2 )0 1 2, appears quite improbable if one is to place any confidence in the effects of configuration on orbital multiplicity. It is, therefore, concluded that thiB complex is octahedral. The tris-(pyridinal-p-tolylimine) complex fits into the pattern very well, exhibiting a high moment as expected of an octahedral ion. The most striking result is observed In the case of [Co(P d A d H ) has a mon,ent of 2 .9 6
B.M. The analytical data preclude the possibility of an oxidation state other than two for the cobalt atom and it seems unlikely that the coordination number is less than six, although the moment could be explained more easily on the basis of a planar, spin-paired, four coordinate complex (usual range o fM e f f = 2 *I~2 *9 B.M*)*
87C a l v i n 1 has studied the temperature dependence of a
87M. Calvin and C. H. Berklew, J. Am. Chem. Soc., 6 8 , 2267 (1946).
number of planar cobalt(II) complexes which appear to exhibit a similar pattern of behavior.
An alternate explanation Involves a spin-paired tetragonal structure. As indicated above, a tetragonal

90complex should have a magnetic moment which is considerably smaller (usual range of M e = 1-8-1.9 3.M.) f than the experimental value, 2 .9 6 B.M. In an attempt to resolve this problem, the magnetic susceptibility of the complex was determined at six different temperatures (373°* 300°, 232°, 206°, 133°, and 80° K). The data are listed in Table 9 and Appendix III.
TABLE 9TEMPERATURE DEPENDENCE OF THE MAGNETIC MOMENT OF
[Co(PdAdH)2 ]l2
T. °K. ^ e f f
373 3.69*300 3.12*300 3.04232 2.49206 2.33183 2 .2 0
80 1.90
* Measurement made after repacking sample tube.
The moment of this complex is observed to vary from 1 .9 0 B.M. (at 80° K.) to 3*69 B.M. (at 373° £*)• The value of 1.90 B.M. is entirely consistent with a tetragonal configuration. The abnormally high value of the moment at

91room temperature and above may result from an equilibrium mixture of the spin-paired and the spin-free states according to the Boltzman distribution factor,
Z1EKeq = Ae
If an equilibrium mixture does exist for this co- balt(ll) complex, it follows that the simplest model, in terms of which the observed moment may be expressed, may be represented as the sum of the individual contributions of the two spin-states, i.e.,
_ N *V + N Y M " 1 ^ M x 2 M 2
where is the mole fraction and*Xj^, the molar susceptibility of the spin-free complex while N2 and ~X hold the same significance for the spin-paired complex. M represents the experimental value of the magnetic susceptibility. The above equation may be solved for the mole fraction of the spin-free complex.
Nx = (Xm- X m 2) Xm1-Xm2
The equilibrium constant is given by the expression Keq 3
Prom the Boltzman distribution function it Is apparent that log K should be a linear function of l/T. Appropriate values must be assigned t o % M ^ and order to calculate Keq.

92As pointed out earlier, the value of should2
correspond very closely to the calculated spin-only value for one unpaired electron, JJL = 1*73 B.M. E x p e r i mentally, it has been determined that the moment usually
Q Q
lies very d o s e to 1.90 B.M. The choice of a magnetic
S. Nyholm, l o c . c l t .
moment is somewhat more difficult in the case of the spin- free component. As Indicated earlier, the majority of spin-free octahedral complexes have moments between 4.8 B.M. and 5*2 B.M. These values are very close to the magnetic moment calculated using the equation =V 4S(S+l) +L(L+l) where L = 3 and S a 3/2. The value,L m 3 > classifies this as a weak field case since no orbital degeneracy has been removed by the ligand field, i.e., the splitting between the tg and the e^ orbitals (10 Dq) Is not sufficient to remove any orbital degeneracy. However, in the example of Immediate interest, the field would appear to be better classified as strong, or nearly strong, since 10 Is sufficiently large to cause spin- pairing in a fraction of the complex. On this basis, it appears reasonable to expect that the orbital degeneracy will be determined by the orbital levels. This d e generacy corresponds to L - 1, or 2L + 1 = 3 * Substitution of the values L - 1 and S = 3/2 into the formula,

_____________ 93yM-eff = \ A s (S+1)+L(L+1), results In a magnetic moment of 4.1 B.M.
The equilibrium constant values for the complex, bis-(pyridindialdihydrazone)-cobalt(ll) iodide, were calculated using, for the spin-free-state, both the 4.1 B.M. and the 5.2 B.M. values, with the spin-paired value of 1.9 B.M. The data are reported in Tables 10 and 11.From the Boltzman distribution function, the following equation may be obtained which relates the equilibrium constant with the temperature.
-AERT A S
K eq = Ae where log A - + R .
A plot of log Keq as a function of l/T should give a straight line (providing A H and A s remain relatively constant over the temperature range under consideration) with a slope equal to - A h /2.3R and an intercept equal toA s . Figures 12 and 13 reveal that the 5*2-1 *9 B.M. values for the spin-free and spin-paired states, respectively result in a straight line which passes through five of the six points (the point at the lower extremity of the temperature range, 80° K., showing a relatively large deviation) while the 4.1-1.9 B.M. values do not result in a straight line. The deviation of the point at 80° from the straight line which passes through the remaining points is not too significant since a small adjustment of the effective

TABLE 10CONTRIBUTIONS OP THE TWO SPIN-STATES OP COBALT(ll) TO THE OBSERVED
MAGNETIC SUSCEPTIBILITY OF [Co(PdAdH32I2*
T N1 n2 106 10° KeqlogKeq
373 0.693 5574 0.307 1206 4234 0.44 0.356300 0 .3 8 8 6926 0 .6 1 2 1499 3o06 1.58 0.198232 0.149 8965 0.851 1940 299O 5.71 0.757206 0.095 10088 0.905 2183 2936 1 0 .0 0 1 .0 0 0
183 0.054 11357 0.946 2457 2934 1 7 .5 2 1.24480 O.03O 26000 0.970 5625 5245 3 2 .3 3 1 .5 0 9
* 4.1 B.M.; y&g, 1.9.

TABLE 11CONTRIBUTIONS OF THE TWO SPIN-STATES OF COBALT(ll) TO
THE OBSERVED MAGNETIC SUSCEPTIBILITY OF [Co(PdAdH)2 3l2*
T N1 n2 7 ^ M 2x 10° X Mx 106 KeqlogKeq
373 O .390 8978 O.blO 1206 *23* 1.56 0.193300 0 .2 1 8 11156 0 .7 8 2 1*99 360b 3.59 0.555232 0 .08* 14*39 O .916 19*0 2990 IO.9O I.O38
206 0.05* 162*8 0 .9*6 2183 2936 17.52 1 .2**183 0 .0 3 0 18291 O.97O 2*57 293* 32.33 1.50980 0 .0 1 0 *1875 O.99O 5625 52*5 99.00 1.996
* 5*2 B . M . j ^ g , 1.9 B.M.
VOui

moment assumed In the spin-paired case (1*9 B.M.) would place this point on the line while not affecting the remaining points significantly. This is easily seen from the fact the effective moment which Is calculated from the 80° point is equal to 1*9 B.M., the value which was chosen a standard for the spin-paired isomer.
As a consequence of the straight line which resulted from the substitution of 1.9 and 5*2 B.M., for the magnetic moment of the spin-paired and spin-free complexes respectively, subsequent calculations will be based on these values. On this basis, A H, the heat of conversion from one spin-forra to the other, was calculated to be -2.14 IcCal (spin-freespin-paired). This value appears to be entirely reasonable for an equilibrium In which the two species are present in approximately equal amounts at room temperature.
The value of ^ S was calculated to be 0.439 e.u. This change in entropy includes the change In spin-multi- plicity In going from the spin-free to the spin-paired state. The change in entropy which is attributable to a change In spin-multiplicity may be calculated from the expression 4 s / R r In 1/2. Prom this, it is seen that, if only the change in spin-multiplicity were involved, the change in entropy, A S , would be -1*38 e.u. Obviously, there are other factors Involved which contribute

97
0.0
04
02
- 0.2
i x I03 VFigure 12. Temperature Dependence of the MagneticSusceptibility of the Two Spin-3tates
of [Co(PdAdH)?]I2.

2 0
.9 B.M.
*
0 8
0 6
0 4
0.2
* 10s, *K-1
Figure 13. Temperature Dependence of the MagneticSusceptibility of the Two Spin-statesof [Co(PdAdH)23l2.

99to the observed change in entropy; however, these can not be evaluated at the present time from the available data.
The magnetic moments of the tris-(biacetyldihydra- zone) and tris-(pyridinalhydrazone) complexes of cobalt(ll), which are somewhat below the values expected for octahedral, spin-free complexes, may reflect the same type of equilibrium as appears to exist in the case of the pyri- dindialdihydrazone complex, with this exception. Instead of the equilibrium involving comparable amounts of the spin-free and spin-paired states, the BdH and PAH complexes are primarily spin-free at room temperature. Certainly, the temperature dependence of these moments
8 Qshould be determined. Figgins ^ has measured the
piggins, master*s thesis, The Ohio State University, 1958.
susceptibility of the cobalt(ll) complex of pyridindial- bismethylimine and reported a value of 2.31 B.M. These data indicate that a series of cobalt(II) complexes exists In which the energy separation between spin-free and spin- paired states Is comparable to kT. A detailed study of such a series of complexes might be expected to reveal a number of significant relationships.
The magnetic moments of the nlckel(ll) complexes of these ligands (3*10-3*32 B.M.) lie In the range of

100values which is expected for spin-free octahedral complexes. This is in agreement with the formulation of the complexes containing mixed ligands as [Ni(AA)2^2 ]•One point of particular interest arises, however. The two isomeric nickel(ll) complexes of PdAdH are difficult to explain on the basis of geometrical isomerism. Alternatively, it was anticipated that the isomers might be associated with different electronic configurations (spin- free and spin-paired); however, the magnetic moments, as determined at room temperature, are almost identical.In order to pursue the matter further, the moments of these complexes were determined over a range of temperatures (80° K - 300°K)• The data are given In Table 12 and Appendix III. A plot of the molar susceptibilities,^ as a function of l/T (Figure 14) shows that the behavior of the two complexes Is virtually identical and that the magnetic moments of both deviate from the Curie law ( /Cm l/T) at low temperatures. It is Interesting to speculate concerning the cause of this apparent deviation. It may be associated with a change in spin-multi- plicity such as was observed in the case of bis-(pyridin- dialdihydrazone)-cobalt(II) iodide. If this is the real cause of the deviation, the susceptibility should continue to fall rapidly as the temperature is decreased. This postulate is consistent with the data which are available.

1 0 1
TABLE 12TEMPERATURE DEPENDENCE OP THE MAGNETIC MOMENTS OP
NORMAL-[Ni(PdAdH)2 ]I2 AND ISO-[Ni(PdAdH)2 ]l2
Compound T. °K. ^ e f f
Norma1-[N1(PdAdH)Q ]I9 300 3.38232 3*27206 3*21191 3*1580 2.34
Iso-[Ni(PdAdH)P ]l0 300 3*10232 3*01206 2*93199 2*9880 2*19
The three-to-one complex which nickel(ll) forms with2 ,2 1-bipyridine and o-phenanthroline have been resolvedinto optical isomers, indicating an unusually high degreeof covalent character (this point was discussed in a pre-vlous section)• The complex:, Ni(diarsine) 903 is spin-
9®r , S. Nyholm, loc* cit.
paired and six coordinate, presumably a tetragonal configuration* As pointed out earlier, the ligand, pyridindial- dlhydrazone, has some of the same structural characteristics found in the ligands of the dlpyridyl class and, to a lesser extent, in dlarslne, l*e., all of these ligand molecules have orbitals of £i symmetry which are available for £l bond formation with the metal, using d electron

102
oX
X
O Norrool - [Ni(PdAdH)2] I*
A iso - [N ifP d A d H fe ] I 2
■r X 10*. •K“l
Figure 14. Temperature Dependence of the MagneticSusceptibility of the Tiro IsomericForms of [Kl(PdAdH)2]l2*

103pairs of the metal atom. In fact, the high degree of covalent character observed In the complexes formed by these ligands is attributed to strong £l bond formation. Therefore, it might be anticipated that pyridindialdehydra- zone would interact with the nickel(ll) atom in a similar manner. Nyholm^1 states that all of the spin-paired six
91R. S. Nyholm, loc. clt.
coordinate complexes of nickel(ll) contain ligands which "have a marked capacity for double bond formation using d. electron pairs of the metal atom.” As a consequence of the possibility of interaction of this type in bis- (pyridindialdihydrazone)-nickel(ll) iodide and since the analogous cobalt(ll) complex does appear to be a mixture of spin-paired and spin-free complex (approximately 2/3 of the complex is in the spin-paired state at room temperature) the postulate of spin-pairing at low temperatures is given added support.
In summary, the iron(ll) complexes, In which six nitrogen atoms of the methine type are coordinated to the metal atom, are spin-paired. The only iron(Il) complex in which two of the b I x positions are occupied by halide and/or water is spin-free. The cobalt(II) complexes are octahedral and spin-free with the exception of [Co(FdAdH)2 ) Ig which appears to be an equilibrium mixture of spin-free

104and spin-paired complex. The nickel(ll) complexes are all spin-free and octahedral.
4. Ultraviolet and Visible SpectraThe ultraviolet spectra of BdH, PAH, PdAdH, and
PAT have been determined together with the ultravioletand visible spectra of eighteen of their complexes. Atthe time these spectra were determined, it was intendedthat they be used to characterize the ligands and theircomplexes on the basis of the shifts of absorption maxima,characteristic of the ligand molecules, which occur as aconsequence of the differences in degree and of type ofinteraction between these ligands and iron(ll), cobalt(XI), and nickel(ll)• In addition, there are certainother high intensity bands which appear to be related tothe ease of oxidation or reduction of the metal atom and
92ligand group. Recently, however, it has come to the
attention of this writer that a large volume of literature exists which treats, quantitatively, the spectra of Inorganic complexes. One region which is of particular inter est lies in the near infrared. With regard to these recent development^ the spectra reported herein are rather limited in their usefulness (the spectral range Is
c. Bailar, Jr., loc. clt., Chapter 18
since data were not obtained In the

105near infrared region. However, these spectra will be discussed from the more modern point of view whenever possible.
Before the time of Werner, it was suggested that the colors of compounds were characteristic of certain groups contained in the molecule or, of particular structures.^ Then followed a period in which a large amount
93Ibid.
of qualitative work associated the presence of characteristic bands with specific groups or combinations of groups
04in the complexes. However, a quantitative treatment of
QiLSee, for example, R. Tsuchida, Bull. Chem. Soc.Japan, (5) 13, 388 (1938); (5) 13, 393 (T53H) ;"TT7 (1936); 11, 721 (1936); (6) 12, 437 (1938)..
78T
ultraviolet and visible spectra was not realized until the development of molecular orbital and crystal field theory.93
93L. E. Orgel, Quart. Revs. 8, 442 (1954).
For the purposes of this brief discussion, the absorptions of Interest may be separated into three general groups, (l) those which are characteristic of the ligand molecules, (2) those which are associated with a transition of electrons from orbitals which are localized on

106the metal atom, and (3) those absorptions which result from electronic transitions within the central metal atom (d-d transitions). The intensity of these absorptions Is a function of the probability of the transition
96(according to the spectroscopic selection rules).
96Ibid,
The absorptions, which are characteristic of the ligands, may be charge transfer (an example Is found in the case of halogen donors)^ or they may be associated
9?L. E. Orgel, "Some Applications of Crystal Field Theory to Problems In Transition Metal Chemisty," Report to the Xth Solvay Council, Brussels, May, 1956*
with electronic transitions within the £i electron system of the ligand molecule (the absorptions characteristic of 2,2'-bipyridine which occur at 238 myU.. and 280 m JJ.») •
Alteration of these absorptions upon complexing might be expected to reflect the degree of interaction between the metal atom and the ligand molecules.
The name, charge transfer, is descriptive of the type of transition presumed to occur in the second group of absorptions. An example or two will serve to illustrate the postulated nature of these transitions. One type of charge transfer involves a transition from the highest occupied d orbital of the metal atom (these orbitals are localized

107on the metal atom) to the lowest, empty t*u ormolecular orbital which Is part ligand jdI and sigma, andpart metal In character (primarily associated withthe ligand) (See correlation diagram, Figure 7*) Thistype of transition may be described as 3d. - in charac-
98ter. These transitions occur in the complexes of metal
98Ibld.
ions which are easily oxidized (for example, [Fe(dipyridyl)3 ] | |
). Another type of transition is from the bonding a and molecular orbitals (primarily ligand) to an emptyd orbital (primarily localized on the metal atom). The absorptions ariBing from this type of transition are frequently observed to lie near 200 mjU. Charge transfer bands occur in both the ultraviolet and visible regions of the spectra and are usually very intense ( C -10^).The intensity of such bands indicates that the transitions are permitted, in terms of the selection rule. The very intense colors associated with compounds in which a metal exists in more than one oxidation state have been attributed
OQto charge transfer between the metal atoms." The first
V. Sldgwlck, "Chemical Elements and Their Compounds,” Oxford Press, London, 1950.
two classes of absorptions await detailed explanation on the basis of molecular orbital theory*

108Whereas the charge transfer bands arise as a conse
quence of transitions to or from orbitals which are associated, primarily, with the ligand to those of the metal atom, the d-d transitions, which correspond to very low absorptions, ( 10)100 are restricted to the metal
100L. E. Orgel, loc. cit.
orbitals* The low intensity of these bands is a result of the low probability of £ - £ transitions (orbitally forbidden). The relative positions of these absorptions may, in some cases, be predicted from crystal field theory and, accordingly, the value of 10 Dq may be determined from the position of one or more of these bands.101 One
101See, for example, (a) C. K. Jorgensen, Acta. Chem. Scand. 9, 180 (1955); 2 * 116 (1955)* (b) C. J.Ballhausen ana C. K. Jorgensen, Acta. Chem. Scand.,397 (1955)* (c) C. J. Ballhausen and £• K. Jorgensen,Kgl. Danske. Vldenskab. Matt, fys - Medd*, (l*) 2£ (1955)•
or two of these bands frequently occur in the visible portion of the spectrum, the remaining bands lying in the near Infrared region of the spectrum.
The absorptions which are characteristic of the spectra of the compounds of immediate interest are listed In Tables 13 and 14.
There is one absorption which is common to the majority of the spectra of the metallic complexes of this

TABLE 13ULTRAVIOLET AND VISIBLE ABSORPTION BANDS OF BIACETYUDIHYDRAZONE,
2 -PYRIDINAIHYDRAZONE AND THEIR COMPLEXES
Compound A max ^max A max ^max Amax <^"max A max ^max
Blac etyldlhydrazone - - 263 (b) 28000 - - - -[Fe(BdH)33l2 225 58000 289 (b) 17000 - - 443 6200[Co(BdH)33l2 228 48000 262 (b) 45000 - - - -[Nl(BdH)3)Cl2 - - 264 (b) 23000 - - 490 222-Pyrldinalhydrazone - - 259 (sh) 6000 286 7800 - -[Fe(PAH)3]l2 225 50000 258 19000 294 34000 480 7400[Co(pah)33i2 226 40000 262 26000 289 30000 -[ni(pah)33i2 225 46000 253 (sh) 26000 305 28000 - -
(b) A broad absorption.(Bh) A shoulder.

m u
m m o ® i® visible ABS«raoii kids op 2-pmDM-p-wifflffi ad i m p conpihes
Compounds Amax fmax Aiax pm Aaax fm Aiax fm A m fm \m fm
2,6-Pyridlndlaldihydrazone 230(«h) lWOO 265 20000 ppp m m m 307 13000 ppp ppp ppp ppp
W h m j U l ■ PP ■■■ 280 33000 ■ PP MVP PPP PPP ppp ppp PPM ppp
2 « 229 ♦5000 258 43000 ««• ppp 334 13000 419 8300 521 7000[Co(PMdH)2)l2 226 55000 276 38000 ■ PM PPM 3»5 13000 ♦85 1100 PPP ppp
Nomil-[«l(PMOfi)2 ]l2 225 46000 208 51000 PPM PPM 364 11000 ppp ppp PPP ppp
Iw-[»1( P M ) 2](C10 ,)2 ppp PPP 288 51000 PPM ppp 365 10000 ■ PP pp« PPP ppp
2-Pyridinal-p-tolylimine PPP 239 11000 PPM ppp PPP ppp ■ PP pp« PPP ppp
«•» ppp 247 12000 290 10000 331 8000 ppp ppp PPP ppp
(PefPAlJjCljI^O ppp ppp 239 23000 284 11000 331 7800 555 860 580 900[Pa(PAT)3 Jl2 -2HgO 223 32000 PPP PPP 286 10000 325 7000 540 2400 580 3100[Co(PAT)3)I2-3I20 223 25000 241 16000 286 12000 331 9600 PPP PPP PPP ppp
Ih M c k ^ I «pp ppp 241 23000 289 11000 331 7700 PPP PPP PPP ppp
[Pl(PAl)j)l2'2I20 ? ? 241 14000 294 9000 331 9600 PPP PPP PPP ppp
lb) Broad ibiorptloRi (ah) ShouId«r<

Illseries which Is observed to lie between 223 nyu and 229 mp. This absorption Is attributed to the characteristic charge transfer of the iodide ion, which is reported to occur at 230 mp.^02 It may be seen from Tables 13 and 14 that this
l02L. E. Orgel, Quart. Revs., 8, ^22 (1954).
band is present in the spectrum of every iodide salt and that it is absent from the spectra of the chlorides and perchlorates (which are reported to exhibit corresponding bands below 200 mp)* Additional confidence may be placed in this assignment since the observed molar extinction co- efficients, approximately 10 , are in agreement with those which have been observed in other systems. This band will not be considered further except in those cases in which it appears to obscure another band.
The ultraviolet spectrum of biacetyldihydrazone, Figure 15# is characterized by a single broad band which occurs at 263 nip. This absorption is probably associated with an electronic transition within the £i system of the molecule. The relatively large molar extinction (28000) indicates that this transition is probably £ - u in charac ter.
The spectra of the paramagnetic three-to-one complexes of cobalt(ll) and nlckel(ll) with BdH reveal that this band is not shifted upon complexing, but that it 1b

Tra
nsm
itta
nce
112B ia c e ty td ih y d r a io n e
[C o (B d H )s ] lI c m cell,
[N t (B dH )3] l
20
4 0
6 0
8 0
2 2 9
Wave Length in M il lim icrons
Figure 15* Ultraviolet Spectra of Biacetyldlhydrazone and Its Complexes.

113somewhat less broad than in the spectrum of the free ligand. In contrast, the band is shifted to lower frequencies in the spectrum of the spin-paired iron(ll) complex (289 mp), indicating that the chelate ring may involve increased conjugation.
The visible spectra of these complexes Indicate that, again, the Iron(ll) complex is unique in that it has an intense band at 443 mji (Figure lo). This band would appear to correspond, in type, to the 522 mji band of trls-(dipyridyl)-lron(ll), even though this band Is at 422 mu and is somewhat more intense In the spectrum of the dipyridyl complex. There absorptions are attributed to acharge transfer which involves a transition from the high-
* *est, occupied d orbital into the lowest empty t, or t*— lu 2u103orbitals. The £ - u character of the transition
l°3ibld.
accounts for the rather large molar extinction coefficient (approximately 10^)• It is quite significant that the co- balt(ll) and nickel(Il) complexes do not exhibit such a band.
The nlckel(ll) complex has, in its spectrum, a band of low intensity (22) which is located at 490 m|i. The position and low Intensity of this band Indicate that it

Tron
sm
itto
nce
114
[Ni ( B d H ) 3 ] Cli c m c e l l , ;
[Co ( B d H ) 3 ] I 220
4 0
6 0
8 0
3 6 0 4 2 0 4 8 0 5 4 0 600 660
Wove L e n g t n m Mil l imicrons
Figure 16. Visible Spectra of the Complexes ofBlacetyldthydrazone.

115104Is a d-d transition. The spectrum shown for the
104C. J. Ballhausen, Kgl. Danske Videnskab.Selskab. Matt-fya Medd., (3)7^9 (19$5)•
analogous cobalt(ll) complex In Figure 15 has very high
transmittance In the visible region due to the low con
centration of the solution used to determine the spectrum.
A more concentrated solution indicated the existence of a weak band near the ultraviolet extreme of the visible spectrum; however, the resolution of the band did not permit the assignment of an absorption maximum.
The ultraviolet spectrum of pyridinalhydrazone reveals two band3, one at 259 mp which appears as a shoulder on the stronger 28b mp band (Figure 17) • Pyridine has three bands in this region, the strongest of which is at 25b mp. The other two bands occur as shoulder on this stronger, central band.^ 5 Bls(pyridinal)ethylinedlmlne
a . Friedel and M. Orchin, "Ultraviolet Spectra of Aromatic Compounds," John Wiley and Sons, New York,1951-
is observed to absorb at 260 nyi and at 280 nyu. On this basis, the higher frequency band is assigned to the pyridine nucleus (involving the £i system of the ring) and the lower absorption is assigned to the alkyl side chain C*N.
The 259 nip band of the free ligand, which is assigned to the aromatic nucleus, is relatively unchanged

Tro
nsm
itta
nce
1:62 - Pyridinalhydrozone
----------------- [ C o ( P A H ) 3 ] 120
4 0
6 0
8 0
2772 2 9 253 301 3 2 5 3 4 9 37 3Wave Length in Millimicrons
Figure 17- Ultraviolet Spectra of Pyridinalhydrazoneand Its Complexes.

117In the spectrum of the spin-paired iron(ll) complex; how
ever, It is shifted to higher frequencies in the spectra
of the cobalt(ll) and nickel(ll) complexes. This observa
tion may be explained on the basis of compensating effects
in the case of iron(ll) complex one of which is absent,
or at least not as pronounced in the complexes of cobalt
(Il) and nickel(ll). These effects will be discussed in
greater detail in the next section.
The 23o mp band of PAH is shifted to lower fre
quencies in all of the complexes, the shift being most
pronounced in the spectrum of the nickel(ll) complex (19 mp.). This shift amounts to 3 mp and 8 mp in the 3pectra of the cobalt(ll) and iron(ll) complexes, respectively.
It is not possible to correlate, directly, the relative
order of these shifts with the other physical properties
of these complexes; however, it might be associated with
a change in both the ground state and excited state of
the molecule resulting from multiple bonding between metal
ion and ligands, i.e., although, it might be predicted,
on a qualitative basis, that the iron(ll) ion would inter
act most strongly with the ligand molecules thereby giv
ing rise to a low frequency shift, the excited state might
not be affected by this increased interaction to the same
extent as the ground state. The net result of this inter
action might be a higher energy transition in the iron(ll)

118complexes than in the analogous cobalt(Il) and nickel(ll)
complexes, in which interaction is not so profound.
The visible spectra of these complexes (Figure 18)
are quite similar to those of the RdH series. The trans
mittance of the spin-free cobalt(ll) and nlckel(ll) com
plexes Is very high; therefore, no bands are assigned
even though one would expect to find at least one of the
low probability, d-d transition bands in the visible por
tion of these spectra. The charge transfer band of the
iron(ll) complex lies at 480 myA, somewhat lower in fre
quency and higher in intensity than in the spectrum of
the analogous RdH complex.
There are three absorptions in the ultraviolet
spectrum of pyridindialdihydrazone (Figure 19), a strong,
central absorption at 2o5 and shoulders on both the
low frequency (attributed-to the acyclic C=N) and the
high frequency side (307 n a n d 230 mjA- respectively). The entire region of absorption is quite broad and dif
fuse. The position of the main band is in agreement with
the position and intensity of the strong absorption in
the ultraviolet spectrum of 2,6-dimethylpyridine.The spectra of the complexes indicate that the
high frequency band at 230 myA is obscured by the charge transfer band attributed to the iodide ion (Figure 19)*
The 265 m ^ absorption of the free ligand is relatively

Tro
nsm
itta
nce
119
[C o (P A H ),J I -420[ Ni ( P A H ) , ] I -4
4 0
6 0
6 0
1007206 6 06 0 05 4 03 6 0 4 0 0
Wove Length m Millimicrons
4 2 0
Figure 18. Visible Spectra of the Complexes ofPyr id 1 na lhyd ra z one.

Tra
nsm
itta
nce
120
20
4 0
6 0
------------- 2 ,6 - P y r id in d ia ld i h y d ro z o n eI cm c e l l , 6 * lO- 5 M
------------ f e ( P d A d H ) 2] I 2I cm. cell, 2 x I 0 " 5 M
------------- [C o (P d 4 d H )2 ] I 2i cm . cell, 2 x I 0 - 5 M
..................[ N i ( P d A d H ) 2] l 2
8 0
3 4 93 2 52 7 7 3012 5 32 2 9W ave L eng th in M illim icrons
Figure 19. Ultraviolet Spectra of Pyridindialdihydrazoneand Its Complexes.

121unchanged In the spectra of the nickel(ll) complexes. In
contrast, the spectrum of the cobalt(ll) complex reveals
that this band has been shifted to lower frequencies (27o mji) , while this absorption is doubled in the spectrum of
the Iron(ll) complex, the stronger band occuring at 253
mya and a shoulder at 280 mjx. The 307 vcijx absorption is shifted toward the visible region of the spectra, the
shift being most marked in the spectrum of the nickel(ll)
complex (57 myi) . The band Is shifted 27 mya in the spectrum
of the Iron(ll) complex while that of the cobalt(ll) com
plex is Intermediate in position. This is In agreement
with the magnetic data which indicate that the cobalt(ll)
complex of PdAdH should be intermediate in nature, more
closely resembling the analogous Iron(ll) complex than the
nickel(ll) comples (Figure 20).
From the visible spectra of these complexes, It Is
immediately obvious that the transmittance of the cobalt
(II) complex is much less than in the spectra of the com
plexes which have been discussed previously. The absorp
tion of the cobalt(ll) complex which occurs at 485 nyu is
attributed to a charge transfer process. This assignment
is consistent with the trends noted in the ultraviolet
spectra and with the magnetic data discussed previously.
It Is interesting to note, at this point, that In all the
spectra, which have been considered, only the spectra of

Perc
ent
Tra
nsm
itta
nce
122
20
r*
4 0
60
8 0
10042 0 4 8 0 540 i
Wove Length in Millimicrons6 0 0 6 6 0 7203 6 0
Figure 20. Visible Spectra of the Complexes ofPyridindialdihydrazone.

123the spin-paired complexes (of iron(ll)) have contained
charge transfer bands even though the cobalt(II) ion
should be more easily oxidized and stabilized by complex
formation than the Iron(ll) Ion. The spectrum of the
Iron(Il) complex, rather than exhibiting the usual broad,
Intense charge transfer absorption, has two, strong, well-
resolved absorptions at 419 mji and $21 mji* The rather
broad features of this charge transfer band in the
spectra, which were discussed previously, may have served
to obscure the doublet nature of this band. The charge
transfer band in the spectrum of tris-(dipyridyl)-iron(ll)
iodide is a partially resolved d o u b l e t . O r g e l * ^ states
^°D. H. Busch and J. C. Bailar, Jr., loc. cit.
E. Orgel, "Some Applications of Crystal Field Theory to Problems in Transition Metal Chemistry," Report to the Xth Solvay Council, Brussels, May, 195o.
that there should be another absorption band in this same
region of the spectrum which arises from a transition from
the lowest occupied d orbital to the molecular or
bital; however, this band should be very much weaker than
the band In question since the transition is £ - £ in
character and, therefore, spectroscopically forbidden. It
can only be assumed, In this case, that the Interaction
between ligand and metal ion Is of such a nature that two
charge transfer processes of unequal energy may occur.

124It is to be noted that the lower frequency band of the
iron(ll) complex is nearly identical, in position, to the10 charge transfer band in the dipyridyl complex. c
1C8D. H. Busch and J. C. Eallar, Jr., loc. c i 1..
Figures 21 and 22 indicate that the only major dif
ference in the spectra of the isomers of [Ni(FdAdH)2 is found In the 225 mp region. This difference Is easily
explained on the basis that the complex which contains
the 225 mp band is an iodide salt while the other complex is a perchlorate salt. It is entirely possible that the
d_ - d transitions which may lie In the visible portion of
the spectra, are different, thus accounting for the dif
ference In color of the two samples.
The ultraviolet spectrum of pyridlnal-p-tolyllmine
has four bands which are rather poorly defined (Figure 23)• The 247 mp band has a shoulder on the high frequency side
at 239 mp. The third band lies In the central portion of
the spectrum at 290 mp while the last band is well toward the visible region at 331 m p. Specific assignment of
these bands Is rather difficult. It appears that the ab
sorption, which is characteristic of the tolyl group, is
coincident with one of the two higher bands of the spectrum,
which were assigned, previously, to the pyridine nucleus
and the side chain C = N.

Tra
nsm
itta
nce
125
20
40
60
N o r m o l — [ N i ( B t f A d H ) * ] I 2
6 0 ISO - [M i ( P d A d H )2] ( C I04)
100277
W o v e L e n g th in M il l im icro n s
253229 301 325 3 4 9
Figure 21. Ultraviolet Spectra of the Nlckel(Il)Complexes of PyrIdIndiaIdlhydrazone*

Tra
nsm
itta
nce
126
N o r m a I - [N i ( P d A d H ) 2 ] I2 I cm. c e l l , I x 1 0 " 4 M
I s o - [ Ni ( P d A d H ) 2 (Cl04)2]20
4 0
6 0
8 0
100 6 6 03 6 0 4 2 0 4 8 0
Wave Lenafh in Millimicrons
5 4 0 6 0 0
Figure 22. VlBlble Spectra of the Nlckel(ll)Complexes of Pyridlndialdihydrazone.

Tro
nsm
itto
nce
127
2-Pyrid ino l -p-to lilim m e ) cm. cell, 4 x 10"5 M
----------- [FetPATJJCIj 2HjjOi cm. cell , 2 x IO'9 M
-------------- [F e{P A T )j] I2 ■ 2h 8o "l c m c e l l , I x K)-9 M
x [Co(PAT)3 ] Is 3 H ,0 I cm. cell, 2 x K)‘ * M
[N i(P A T )£ (C l04 )e l I c m ce l l , 2 x IO-5 M
[ N i ( P A T ) , ] I 2 2^ l cm. cell , 2 x 10"* M
229 2 5 3 277 301 3 2 5
Wove Length in Millimicrons
Figure 23* Ultraviolet Spectra of Pyridlnal-p-tolylimineand Its Complexes*

The^ultraviolet spectra of the complexes of PAT
are not as well-resolved as that of the free ligand; how
ever, a few general shifts are noted. The two bands which
occur In the spectrum of the free ligand at 239 mp and 247 mp are merged in the spectra of the complexes, a single
absorption occuring at 241 mp. The 290 nji band is shifted to slightly higher frequencies upon complexing (approxi
mately 4 mp) except in the spectrum of the three-to-one
nickel(ll) complex in which it is lowered approximately 4
mp. The 331 mp band, although it is repressed consider
ably in the spectra of the complexes, is relatively
stationary.The spectra of the two-to-one and three-to-one com
plexes of iron(ll) are, again, the only ones which are characterized by absorptions in the visible region at the concentrations used (Figure 24)• The spin-paired complex has an absorption at 580 mp with a shoulder on the low frequency side at 5^0 mp* The molar extinction coefficients of these bands are much lower than those observed previously in the spectra of spin-paired iron(ll) complexes ( < 1C>3)* Hie Bpectrum of the spin-free iron(ll) complex reveals three absorptions, one at 580 mp with a shoulder on the high frequency side at 555 np and one on the low frequency side at 640 up. Although there have been no other examples of two-to-one iron(ll) complexes

Tro
nsm
itta
nce
129
[Fe (PAT), ] I2 2HaOI cm cell, 5 * IO'5
[Fe (PATfe] Cl2 2HaO20
[C o (P A T ) ,] I 2 3 h 20 i cm cell, 5 x 1 0 '9 *
[N i (PAT)2 (Cl0«)2]
• [Ni (P A T ) , ] I t 2Ht 04 0
6 0
8 0
1007206 6 06 0 05404 8 0
Wove Length in Millimicrons4 2 03 6 0
Fiftt** 2 k * Visible Spectra of the Complexes of Pyrldina 1-p - to lyllmlne.

130considered in this series, it is a little unexpected to find that this two-to-one complex absorbs in the visible region in a manner quite similar to that of the three-to- one. This absorption may be explained on the basis of a dissociation of the two-to-one complex which accompanies dissolution, resulting in the formation of the three-to- one complex. Alternatively, the two-to-one complex may lie very near the cross over point between a spin-paired and a spin-free state. As a result, there might be a certain amount of increased interaction, which is possible in this intermediate state, which gives rise to aweak charge transfer absorption.
5 . Infrared SpectraThe infrared spectra of biacetyldihydrazone (BdH),
pyridinalhydrazone (PAH), pyridindialdihydrazone (PdAdH), pyridinal-p-tolylimine (PAT), and nineteen complexes of these molecules with iron(ll), cobalt(ll), and nickel(ll) have been determined. Assignments have been proposed in the case of twenty-eight different absorptions which occur in these spectra (Tables 15, 16, 17, and 18). These assignments are in accord with the usual frequency ranges reported for the groups in question.1°9,110
1 0 9H. M. Randall, R. G. Fowler, J. Fuson and J. R. Dangl, "infrared Determinations of Organic Structures,"D. Van Nostrand Company, Inc., New York, 19^9•
H°L. J. Bellamy, "The Infrared Spectra of Complex Molecules," John Wiley and Sons, Inc., New York, 195^*

TABLE 15
m b e d j u r a bands (cb'1) pob bucetyibihydrazohe (sij m us corns
Assignment BUc° BdH If* [Pe(M)jjl2 [FelBdHlJlj* [Co(MI)2C1j] [Co{BaH)3Jla [ll(BJH)jClj] i m M j J c :
H20 ■ ■■■ 3 1 8 1m 3460 1 3413! 3356 i 3390 ! 3»oii 3401 ! 3378 !st“S NH2 336? vs (•■Ha 3269 V! ■ ■■a 3279 ! 3311! 32681 3279 !st«s NHg 3215 vs «■■■ 3185 V! mmmm 3215! 32151 3226 ! 3 1 6 5 1
st-a NP2 mmmm 2500 a ■ ■■■ 2513i ■ a a a a a a a a a a a ■ a a a
st-s f 2 mmmm 2342! * • ■ ■ 2342 w a a a a a a a a a a a a a a a a
st CH £994i 2933 •
3040 w 2939i
3 0 1 2» 2950 1
2907v« 2924 VI ■ • ■ •
2907 wa a a a
2 9 0 7 1a a a a
2 9 4 1»mmmm
2933 (A)a a a a
it OO 1724 vil6Sl(sh) rnmrnm
■ ■■■
I I M
■ «■■
■ ■ •■
■ a a a
*■■■
a a a a
a a a a
■ a a a
a a a p
mmmm
mmmm
a a a a
■ a a a
d E • • • « lWji ■ ■■■ 1621 (lit) ■ a a a 1633 M 1637 ! 16J4! 1639 (16)d 1 2 • • • • mmmm 12211 ■ ■■■ 1225 a a a a a a a a ■ a a a a a a a a
st C=K ■ ■■■ 15% B 1577 1 1595 1 15951 1623 : 16131 16131 1 6 1 3 1
1-a C-Cfij 1422 oa a a a
11641 1441 n H 5 6 1
1437 *l445 i
■ ■■■1 4 3 9 1
■ ■■a
I44lia a a a
14511()a a a a
1449 1a a a a
l44lia a a a

M ! 15 (CojM.
iNlprt Blic8 U Bd/ [Pe(Bffi)j)I2 IColMH) ! ICijM)! [liM^I [U(N
J-i MHj 1553« 1J70 a 13*8 j 1371a 1372« 13831 13811 13^3*
d ,r i j 7*1 7*9 •■■■ 790 725 739 CT
55* •••■ 582 . . . . J03 532
($2
0 liquid sample.
d e u M e d sample.

HjO
it-l ig it-iig St-8 NO, st-i NO, it CH 3030 »2817 a
2686 k
it c:0 1712 vj1"
TABLE 16D M ® ABSORPTION B »S ( a ' 1) FOR F fflO R IilM JM E m IIS CONFIDES
M M + [Pe(M)j)lg (Pe(PJB)j]lg+ [Co(fffl),]l, [NI(ffl),ll, Oll(Fffl)3 “ 2
3*25 S 33011 3*13 8 3 1 2 5,3367 s3175 s
239< blur)2»9» b(nr)
2923
1631 (shj — 1206
3125 s23932303
'3 2
3367 s
3125 s
1631 (shj 1631M
3*36
2299 s

TABLE lo (ContdJINFRARED ABSORPTION BANDS (cm’1) FOE mMMWtiM AND ITS COMPLEXES
iiiljownt Pi0 m pm* [Pe(PlH)j]lj [Pe(ME]jJlj+ ICofPIH)^ IhKpAH] Jl IlilMHjjJlj*
Band I (ring) 1582 s I0O5 (ah) 1 5 8 5! 1587 *
mmmm
1608 smmmm
I0O8 VSm m m m
I0O8 vsm m m m
1608 vs l6l0 V0
Band II (ring) 1570 (ah) 1570 (sh) 1567 m 15801 1577 » 1570 (64) 1576 (8b) 1578 (sh)
at C=N (acyclic)
mmmm 15*11 15*1 8 1558 m 1555 1 1555 s
Band III (ring)
1468 hi I473 i 1477 s 1477 s 1475 s 14810 14816 1479 s
Band IV (ring) 1435« 1433 • 1*35« 1445 i 1443 « 14*7» 1**7» l*43i
? m m m m mmmm 1302 * 1299 * (6) 1 3 0 7» 1305 * 1302 m? m m m m m m m m 1271* 1276 k 1282 m 1280 1 1272 a? m m m m mm m m 12*71 1239 « 1244 a 1242 a 1242 a?
d CE or ring vlb.
mm m m
11481mmmm
11(8 > 1151a?
1153 m12141 (b) 1152 m
1214 (0b) 1153 m (5)
1212 (04) 1151 * (*)
12121 (b) 11521 (4)
d CH d CH
7 6 3 ™m m m m
7 7 5 1
744 •777 a % 1
763 vs (b) 764 0 (b) 753 m
768 0 (b) 7*8 m
768 0 (4) 7*5»
769 s 7*9a
o Liquid sample*+ Deuterated Sample*

TABIE 17iroilffl M O T H B U S (cm'1) FOB P M M J i m i f f l B » IB CflUffi
Pi PdAdH Pdidl+ [ f « ( p m ) j ]i} lPi(em^Ii* [co(m)±
V>
at-a
a t-a ]©A
a t CS
a t C=0
3080 vw3022 w 2874 vw2724 va1695 w
* ■ « » mm m m
3215 III3265 HI ••••
*••• 2463 a
23018
2929 VW 2959 Ww
■ m m m m
m m m m m m m m
m m m m mmmm
M *
3 $ (sh) 3 $ 1
3?5?»
312511
33001
31*5 1
2 l5 1 i
m i
3«S» 3W81 (1)3 3H *
3155«
mmmm
mm m m
m m m m
2W 1 »2309 VI
1222 i

HJMJffl M O T M B ® (cm'1) FOR P m t M M B W f f l l E Jill) IIS COMES
Assignment PA FdAdH PaAdH+ [Fi(PdAdH)2]l2 lFe(PMi)2 ]l2+ [ColfdAdHljjlj [»l(Wi)2ll2 IM(PiM)2]I2
Band I (ring) 1587 b 1585 n 1582 vs 1608 vs 1005 s lol3 V! 1616 V8 1618 vs
Band II (ring) 1577 (ah) 1570 (sh) 1567 vs 1800 (sh) 1590 (sh) I6O3 (sh) lbOl (sh) 1597 M
at C»N (acyclic) »*#•
1603 vs■ •■a
1597 vsaaaa
1538 vs 1531s 1538 s 15(8 (sh)
1553 VB 15(8 vsm m m m
1 aaaa 1511b 1513 s m m m mm m m m
Band III (ring) U68 vs 1(6( vs 1179 b 1(77* 1(8( a l*79s 1*751
Band IV (ring)
m m m m 1(2$ a 1(12 • ? )* 1(25 a 1(37* 1(38 * (o)
? m m m m m m m m 1305 w 1 1305 (sh) 131(i 1
? m m m m 1271k 1277 s* 128( a (b) 128* 1 (*) 1290 a? m m m m M m 12(1» 12(71 12(7»(*) 12501
? m m m m 1227 a 1217 a (b) 12171 (*) 1221 (sh)? 120$ i 1208 m (b) 1206 s (b) •■•a
m m m m 12061

BFRAR1 ABSORPTION BAHDS (a'1) FOR PTRDINDIAimRAZONE AND ITS COMPLEXES
Assignment PA PdAdH PdAdH+ [FelPiiAdHljllj [Fe(PdAdH)j]lj+ [ColPiAdH)Jl [H(PdAdl)2ll2 iRlfPlAfflljllj*
d CH or 1166 w U 63 m II6311 llol 1 (8) 1168 ■ (8) 11701 (8) 1 1 671 (8) 1188 1 (b)d CH 807 a 8M vs 805 V8 W 1 79*« 8011 806 1 805 Bd CH 788 s 738 m 7 6 8 1 ?A6i 7W 1 7*7« 787* 746 m
* Dfuterite! Saople.

H M D ABSORPTION BAUDS (cl'1) FOR FYFIDDIAI-P-TOLraHIHE MD IIS C0HPIEU5
Assignment PAT [JeMjClj] (Pe(PAT)Jlj (Co(PAT)Jlj [HtPAUjClj] [|(l(PAl)j)lj
h2o 3460 ■ (b) 3*80 s 3484 a 3*361 (b) 3**8 vi (b) 3**3 vi (b)st CH n
2907 m 3021 1 2915v» 3030 * (dl)
2950 K (d2)3021 * 2915 m
30*0 » 2933 *
30J0 » 292* w
8t C=H(Acyclic)
M i 1 8 2 8 1 1585 i 1631 n 1 6 3 7 0 1 6 3 * 1
Band I (py ring)
1582 s M i l608 i 1597 1 1600 1 1600 1
Band II (py ring) 15*3 (ib) 15*8 (ib) 15*6 v 15*8 m 15*6 m 15*6 m
15811 1563 » 1575 (ib) 1583 « 1 5 6 7 1 1 5 6 5 1
binnnt(ring)
1506 i 1*97 vi UO6 1 150*i 1 5 0 8 1 1508 i
Bind III (py ring)
1(821 1*711 1*711 l*79i 1*81 ■ l*77i
Bind IT (py ring) l*35i l**7i 1443 a 1445 n 1*49 n 1449 a
it M (tolyl-Jf)
13*81 1 3 6 6« 1353 » (b) 1382 a 1366 V 1388 i

BBIE 18 (Contd.)
Assignment PAT [Pe(PAT)2Cl2J (Pe(PAT) Jl2 [Co(fAl),]l [ltl(FM)2Clj]
? 1292 vw 1307 » 1297 « (b) 1302 m 130b 1 130b 1
? 1289 VW 1271 If 1250 H 1271 w 1269 » 1272 *d CH or py ring vib. 1233 w 1212 k 1239 B 123fi» 1239 m 1 2 3 8»
p-substitutlon(tolyl)
1214 w 1215 w 1217 » 121b * 1215 w 1217 *
? 1198 m 1 2 0 3 1 1200 1 1200 s 12011 1202 ap-substitution(tolyl)
1088 vw 1112i 11151 11081 (b) 1112* 1110 a (b)
d CH or py ring vlb.1018 w 10l8« 1018 * (b) 1019 a 1021i 1021a
? • iaa W 1 (b) 912»(b) 912i 91b 1 913"d CH (tolyl-ring)
823 va (b) 820 vs (b) 823 a (b) 819 a (b) 819«(b) 819 a (b)
? ms 775 a (b, or) 771 a (b, nr) 773 a (b, nr) 7 8 1 » (b, nr) 772 a (b,nr]d CH (py ring)
ms 775 « (b, »r) 771 a (b, nr) 773 a (b, nr) 7 8 1 1 (b, nr) 772 a (b,nrj
d CH (py ring)
737 8 718 • 7b8 1 (b) 7*5»(b) 7*9 * 748 1
d CH (tolyl-ring)70b a (b) 711b 718« 711»(b) 713* 7b6 ■

140The magnetic data have Indicated that, in general,
the complexes which Iron(ll) forms with these ligands are spin-paired, whereas the analogous cobalt(ll) and nickel(ll) complexes are spin-free (with the exception of LCo(PdAdH)2 3 Ig). From the visible spectral data it is seen that only the iron(ll) complexes are all highly colored. It has been suggested by other investigators that the properties of the iron(ll) complexes which distinguish them from those of cobalt and nickel are a result of pi-bonding between the conjugated ligand and the tgg (non-bonding) electrons of the iron atom . * ^ ' ^ 2 The purpose of this study is to
***P. Krumholz, loc, clt.112D. H. Busch and J* C. Bailar, Jr., loc. cit.
correlate the change in spectral properties with apparent change In bond type between metal and ligand. Particular attention will be given to the double bond region of the spectra (175°-1^00 cra-^)• Differences in the Interaction between the metal atomB and ligands should be reflected by changes in the position or Intensity of the acyclic C*N stretching frequency and the stretching frequencies associated with the aromatic pyridine nucleus. Such effects have been considered by other Investigators**^ in systems
**3lbid.
involving iron(ll) and the conjugated dimethine group.

141In almost every spectrum reported here which was
determined by the KBr disk method, there Is a band attributable to water in the 350° cm“l region (Tables 15#16 , 17, and 18). This absorption will not be considered further except In those instances in which It may Interfere with the asymmetric NHg stretching vibrations charac teristlc of amines.
The following abbreviations are used in the first column of the table of assignments:^^
M. Randall, R. G. Fowler, J. Fuson and J. R. Dangl, "Infrared Determinations of Organic Structures,"D. Van Nostrand Company, Inc., New York, 1949*
st, bond stretching vibration d, deformation vibration r, rocking vibration
-s, symmetric vibration -a, asymmetric vibration ?, assignment uncertain
The following abbreviations are used in the body of the table of assignments to indicate the relative in tensities of the absorptions
U S l b U .
vs, very strong s, strong m, medium strong w, weak vw, very weak

142The following symbols In parentheses are used to
describe the nature of the absorption:
ll6Ibid.
fb), broad absorption(d), partly resolved doublet (average value)
(dl), short frequency member of a resolveddoublet
(d2), long frequency member of a resolveddoublet
(nr), not resolved(sh), shoulder on a band of higher intensity(d), unresolved doublet, frequency given is
average•
2,3-Butanedione»- The Infrared spectrum of biacetyldihydrazone(Figure 25) has been characterized previously) I*? however, two points should be emphasized. The
117H. M. Randall, R. G. Powler, N. Puson, and J. R. Dangl, loc * cit., Table 5 and p. 1 6 7.
position and character of the carbonyl absorption, when considered in conjunction with Raman spectra data, indicate that the interaction between the two "conjugated" C=0 bonds is very s m a l l . F u r t h e r m o r e , electron
1i8h. m. Randall, R. G. Powler, J. Puson, and J. R. Dangl, loc, cit., pp. 17,18.
H Q7L. J. Bellamy, loc. cit., p. 122.
diffraction data indicate that the molecule exists in a

Tren
t m
itt o
nce
143
100
00
60
40
202 , 3 - § u t a n « 4 t o M
!7009001900200090004000 3000
W on te v n b tn (cm "1)t O O r
90"*
60
co£40
B iac tfy M ih y d fao n *20
B O Oooo 30019002000 T O O4000 3000M m num b** , c m '1
(00
80
60
20
4000 3000 2000 000 TOO1000 900 800Wow nuinfewy, em '1
Figure 25* Infrared Spectra of 2,3-Butanedione andBlacetyldlhydrason*•

144120trans configuration. LuValle and Schomaker have
E. LuValle and V. Schomaker, J. Am. ChemSoc., 62, 988 (1940).
calculated that the trans configuration of biacetyl is1 .7 Kcal per mole more stable than the cis configuration
Biacetyl
Biacetyldlhydrazone (BdH).- The two strong bands of approximately equal intensity which occur at 3367 cm -1
and symmetric NHg stretching frequencies. The positions of these bands, which are somewhat lower than those usually associated with these vibrations (3 50 0 -3 3 0 0 cm"1), may occur as a result of interaction between the nitrogen atom of the NH2 group and the imlne nitrogen atom, both of which have electron pairs with pi-symmetry. Alternatively, the positions of these bands may be associated with the fact that this spectrum was obtained using a solid sample. Upon deuteration, (Figure 2 5) two new bands occur at 2500 cm"1 and 2342 cm”*. These bands are similar In shape and Intensity to the NH2 stretching bands.
Trans-configuration Cis-configuration
and 3215 cm -1 (Figure 2 5) are attributed to the asymmetric

Therefore, these two new bands are assigned to the two NI>2 stretching vibrations.
The spectral region of greatest interest is that associated with the C^N stretching frequency. The O N vibration is reported to occur in the 1690-1640 cm-1
region in the case of an isolated C=N bond. In the
l21L. J. Bellamy, loc. cit., p. 226.
spectrum of biacetyldihydrazone there are two absorption bands of similar intensity near loOO cm”1 (Table 15)*These two bands occur at 1645 cm-1 and 1582 cm-1. The NHg internal deformation band is reported to occur between I65O cm-1 and 1590 cm”1 .^ 22
J. Bellamy, loc. cit«, p. 218.
On the basis of the usual positions of these two bands one might expect the C=N stretching mode to be associated with the higher of the two bands in the 1600 cm”l region; however, deuteration of the sample results in a spectrum in which the 1645 cm"^ absorption is virtually eliminated while the 1582 cm”1 absorption is unchanged.As a consequence of this change upon deuteration, the 1645 cm”1 band is assigned to the NH2 internal deformation mode while the 1582 cm-1 band is assigned to the C=N stretching vibration.

3A6There are several possible factors which might con
tribute to the relatively low frequency associated with the C=N stretch. Considering the evidence for the existence of biacetyl in a trans planar configuration, it is not altogether unreasonable to expect a trans planar configuration for the corresponding hydrazone. It may be seen, in a qualitative manner, from the representations of both the cis and trans planar configurations that steric hindrance would be greater in the cis configuration than in the trans configuration. This steric hindrance (which arises from repulsions between the two methyl
KH
H
C
\ — c
H/H
H
I IITrans-Configuration Cis-Configuration
Figure 26

147groups and the two amine groups with themselves and with each other) would tend to distort the planarity of the molecule thus reducing the effective interaction of the £ 1 electrons.
In biacetyldihydrazone, one electron may be assigned to the orbital on each of the two imine nitrogen atoms and the two methine carbon atoms. In the planar configuration these orbitals lie perpendicular to the plane of the molecule and, according to valence bond theory, are positioned in a way which should give rise to the maximum possible overlap or interaction between these pi orbitals. Electronically, the system is analogous to butadiene. This interaction would serve to lower the O N stretching frequency with respect to that expected in the case of a single, Isolated C=N bond. It has been indicated from other investigations that the lowering In frequency is proportional to the degree of c o n j u g a t i o n . I n t e r -
123r. g. r. Bacon and W. S. Lindsay, J. Chem. Soc., 1958, 1382.
action between the amine nitrogen atom and the lmlne nitrogen atom to which It is bonded also appear to be of significance* This view Is supported by the fact that the O N stretching frequency Is found at higher values In the spectra of molecules such as benzaldazlne and pyrldlnal-p- tolylimine (discussed later In this Bectlon) In which

148conjugation of two adjacent C«N groups occurs but Is not complicated by the presence of an NH2 group. In order for the NH2 group to exhibit a conjugative effect, the structure of the group would have to be distorted toward the limiting trigonal planar array of sigma bonds in order to bring the unshared electron pair of the NHg group into pi symmetry with the £i electron system of the remainder of the molecule. It will be recalled that the NH2 stretching modes occur at lower frequencies than is commonly observed. This is in agreement with a decrease in electron density on the amine nitrogen atom. The interaction which is suggested is represented below, diagra- matically.
Figure 27

149The relative Intensities and positions of the
asymmetric and symmetric C-CH^ deformation which occur at 1464-1441 cm-* and 1370 cra" In the spectrum of biacetyldihydrazone are quite similar to those of biacetyl, the symmetrical deformation frequency giving rise to a much stronger band than the asymmetric mode. There is the difference, however, that the asymmetric mode is doubled in the spectrum of biacetyldihydrazone (see Figure 25)* This is an occurrence not uncommon in the solid state. ^
l2^L. J. Bellamy, loc. cit., p. 21.
This asymmetric C-CH^ deformation doublet is well resolved Into two bands occuring at 1664 cm~l and 1441 cm-*. The splitting of this band is also observed in the spectrum of the deuterated sample of biacetyldihydrazone.
Very little appears to be known concerning the lowfrequency, asymmetric and symmetric deformations associated with the two amine groups other than the fact that they are quite susceptible to changes occuring as a result of hydrogen bonding and other modes of interaction. It may be seen In Figure 25 that this region of the spectrum is substantially altered upon deuteration. Bellamy states that these vibrations should occur In the 9OO-65O cm“l region.*2^ Randall12^ reports that methyl amine has a
125l . J. Bellamy, loc. cit., pp. 218-219*126H. M. Randall, R. G* Fowler, N. Fuson and J. R.
Dangl, loc. cit., Table 5 and p. 6l.

150band at 774 cm-1 which he attributes to an NH2 deformation. On this basis, the strong, broad absorptions at 74l and 664 cm”1 appear to be associated with the asymmetric and symmetric deformations of the NHg group respectively. These bands are absent in the spectrum of the deuterated sample.
Complexes of Blacetyldlhydrazone.- A number of regular and significant changes are apparent in comparing the spectrum of biacetyldihydrazone with the spectra of the complexes of this ligand with iron(Il), cobalt(Il), and nickel(ll)• (Figures 28 and 2 9 .) The bands of these complexes which are attributed to the NH2 asymmetric stretching frequency appear at a position somewhat lower than in the spectrum of the free ligand. They occur in the range 3268-3311 cm”1 (Table 15)* In addition, these absorptions are somewhat less intense in the spectra of the complexes than the symmetrical stretching mode while, as stated above, these two stretching frequencies are approximately equal in intensity in the spectrum of the free ligand. On the other hand, the positions of the symmetrical NH2 stretching vibrations remain virtually unchanged from that found in the spectrum of the free ligand. Also, in the spectrum of a given complex, the relative intensity of this band, with respect to the other bands in the spectrum is quite similar to the corresponding relationship

151
too
eo
GO
I40
20
20004000 3000 1500 7001000 900 800< M numbm, cm '
DO
80
60 -
e•" 4 0 -
20DwtoroM [F tlB aH ),] J,
4000 3000 1900 noo 9002000 800-4W on
Figure 28. Infrared Spectra of the Iron(ll) Co#plexof Biacetyldihydrazone.

Tro
nsm
iito
nc*
152
100
80
60
40(
7008009001000150020009000 4000 3000Wove N tfnb tn (cm .'1)
100 f
*
EM
20
700800100060020003000 4000 3000Wove Number* (cm "1)
100 r
80 >
w
a
201[ N i l B d H M C I C U l i ]
T O O900 800noo5000 15004000 2000Wovt numbers, c m *1
Figure 29* Infrared Spectra of the Cobalt(ll) andNlckel(ll) Complexes of Biacetyldihydrazone.

153observed In the spectrum of the free ligand. It should be pointed out, however, that the observed change in position of the NH2 stretching vibrations is not conclusive because of the low dispersion of sodium chloride optics in this region.
In contrast to the lower positions of the asymmetric stretching frequencies in the spectra of the complexes, the NH2 deformation is shifted to slightly higher frequencies, the shift being most marked in the case of [PetBdHj^llg (19 cm"-1-) . This corresponds to approximately twice the shift observed for the cobalt(ll) and nickel(ll) complexes (Table 15)• Assignment of this band in the spectra of the cobalt and nickel complexes is rendered somewhat difficult because it appears merely as a shoulder on the stronger O N absorption.
Very significant shifts are apparent for the O N
stretching frequency throughout the series of five metal complexes (Table 15)• Whereas this band is at 1582 cm“l in the spectrum of the free ligand, it occurs at increasingly higher frequencies for the complexes in the following order: [Pe(BdH)3 3l2 < [Ni(BdH)2Cl2 3 = [Ni(BdH)3 3C12 s[Co(BdH)3 3l2 < [Co(BdH)2C12 3• The difference in the position of this band in the two-to-one cobalt complex and that in the spectrum of the free ligand is 41 cm"1. If one considers the O N stretching frequency to be sensitive to the strength of bonding or degree of interaction

15^between the metal atom and the donor atom, the relative positions of this vibration for the various complexes is entirely consistent with the types of complexes involved. The iron(ll) complex has an effective moment corresponding to no unpaired electrons. As a result of this spin- pairing, or concurrent with it, the nitrogen-metal distance should be somewhat shortened with respect to that found in the spin-free complexes . 7 This shortened metal-nitrogen
12?L. E. Orgel, Quart. Revs., 11, 381 (1957).
distance, also, would facilitate the formation of E l bonds between the metal and the ligand. As a result of multiple bonding between the metal atom and the ligand, the C*N stretching vibration would be expected to occur at lower frequencies in the spectrum of the spin-paired iron(ll) complex than in the spectra of the analogous, spin-free complexes of cobalt(ll) and nickel(ll).
An apparent anomaly arises from the relative positions of the C-N stretching frequencies of the spin-paired iron(ll) complex and of the free ligand. As a result of increased interaction between the metal atom and the biacetyldihydrazone, this stretching frequency would be expected to occur somewhat lower in the spectrum of the iron(II) complex than in the spectrum of the free ligand. The opposite is true (compare 1582 cm"1 and 1595 cm"1) .

155This seeming anomaly may be explained in terms of the interactions occuring in the biacetyldihydrazone molecule, as mentioned earlier. It was suggested that a large contributing factor to the low position of the C=N band in biacetyldihydrazone is the planar configuration of the molecule. Furthermore, it was suggested that the planar configuration was associated with a tran3-structure of the molecule, which would facilitate conjugation of the NHg group. However, upon formation of a chelate ring, the ligand must assume a cis-configuration. Consideration of a physical model suggested that, in the case of a cls- configuration, not only do the two adjacent methyl groups interfere with each other but the NHg group press back upon the molecule. These two factors then suggest a tendency toward distortion of the planar configuration upon coraplexing. As a result, the Interaction between the conjugated dimethine groups should be diminished.
Furthermore, the formation of the cis structure should very greatly limit the interaction of the electron pair on the NH2 group with the £i electron system of the molecule. Consequently, the C=N band would occur at a position somewhat higher in frequency than in the case of the free ligand. It is seen, then, that two opposed effects must be considered. The increased interaction of the metal with the ligand molecule tending to lower the CaN band and the decrease In Interaction between the

conjugated dimethine groups occurring as a result of the distortion of the planar molecule. The positions of the C=N stretching vibrations listed in Table 15 shows that the C-N band of the spin-paired iron(ll) complex occurs 15 cm*-*- above that of the free ligand indicating that the distortion of the ligand molecule is the dominant factor in determining the direction of the shift; however, the spectra of the tris-biacetyldihydrasone complexes of co- balt(ll) and nick:el(ll) reveal that the C=N band is approximately 33 cm-1 higher than in the spectrum of the free ligand. From this relationship, it appears that the conjugation between ligand and iron(ll) is substantially greater than in the analogous cobalt(ll) and nickel(ll) complexes.
The C-CH^ deformation vibrations experience a rather irregular shift from spectrum to spectrum, Table 1 5. A systematic alteration in relative intensities is observed* Whereas the symmetrical deformation is more in tense than the asymmetric deformation in the spectrum of the free ligand and of biacetyl, the intensity of the symmetrical mode is reduced, in the spectra of the complexes, to an intensity only slightly greater than that of the assymmetric deformation. The latter remains relatively unchanged in intensity.
The NH2 deformation which occurs near 7^1 cm"^ in the spectrum of the free ligand experiences an irregular

157shift among the spectra of the complexes, while the second band at 66^ cm-1 is shifted to approximately 682 cm' 1 in the spectra of the complexes (remaining unchanged from spectrum to spectrum).
The spectra which follow are somewhat more complex than those of biacetyldihydrazone and its complexes. The pyridine nucleus, which is common to all the compounds which are discussed below, gives rise to this increased complexity, and, in the case of pyrldinal-p-tolylimine, there is still more complexity as a result of the addition of the toly-group to the system. Even though there are a large number of peaks in each spectrum, consideration will be given, for the most part, only to those absorptions associated with the functional groups of the molecule which may be sensitive to changes in the type of complex. Some additional bands, which are easily assigned or which tend to characterize the spectra as a consequence of the regularity with which they occur from spectrum to spectrum (even though some are not assigned) are listed in Tables 16, 17# and 18.
2-Pyrldinaldehyde {PA.)* — The strongest absorption in the spectrum of pyrldinaldehyde is that associated with the C*0 stretching vibration of the aldehyde group (1712 cm-1)• On the low frequency side of this very intense absorption is a band of medium intensity (1664 cm-1) which is partially merged with that of the carbonyl (Figure 30)•
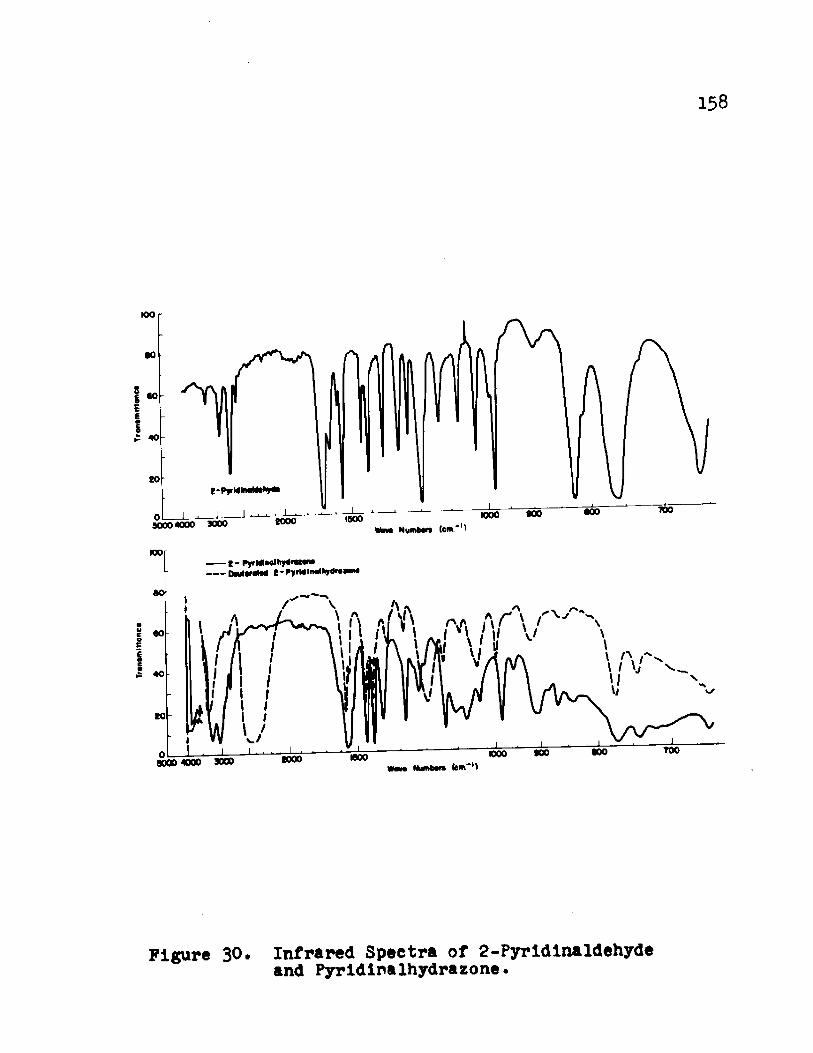
Tron
tmitt
OA
M
Tro
wni
itlo
nci
158
KK)
*0
eono• 0 0«oo10001900800030004000 3000
t-Py'MMiM'*''"* D w ta n M «-P»rl4 liw lh)r*4»**
1 \ / V vI 1 : llA /'I'A
9000 4000 3000 • m Hunbwm ten'1)
Figure 30. Infrared Spectra of 2-Pyridinaldehydeand Pyrldinalhydrazone.

159Although this peak remains unassigned It appears to be associated with the carbonyl peak since a similar shoulder is found in the spectra of biacetyl and 2,6-pyridindialde- hyde. This band or shoulder is not apparent in any of the spectra except those containing carbonyl groups.
Pour absorptions in the range from 1600-1400 cm-1 are particularly useful in comparing this spectrum with those of the hydrazone derivative and the hydrazone complexes. These bands appear to be most sensitive to the type of complex formed. They appear at 1582 cm-1, 1570 cm"1, 1468 cm-1, and 1435 cm"1 (Table 16) in the spectrum of PA and are assigned to the bond stretching vibrations within the pyridine ring. Pyridine exhibits characteristic absorptions at 1582 cm"1, 1570 cm”1, 1484 cm"1, and 1437 cm"1,12^*12^ which is good agreement with these
128H. M. Randall, R. 0. Fowler, N. Fuson and J. R. Dangl, loc. cit., p. 225 and Table 5*
129L . J. Bellamy, loc. cit., Chapter 16.
noted for the carboxaldehyde derivative. Since it is impossible to assign these peaks to individual stretching vibrations, Band I will be used to Indicate the 1532 cm"1 peak or high band of pyridine, Band IX, the 1570 cm'1 peak, Band III, the 1484 cm”1 peak, and Band IV, the 1437 cm"1 peak. A very strong peak which distinguishes this spectrum of PA from those of the corresponding

loOhydrazone and derivatives occurs at 1209 cm"1 (nearly as strong as the carbonyl absorption) (see Figure 30)•
2 -Fyrldlnalhydrazone (PAH) T h e asymmetric and symmetric NH2 and ND2 stretching frequencies of the pyri- dinalhydrazone lie very close to those of BdH (Tables 15 and lo); however, the NH2 deformation of PAH which occurs at 1631 cm-1 lies at a frequency somewhat lower than that of BdH which is at 1645 cm"1* This decrease in frequency of the NH2 deformation in the spectrum of PAH is accompanied by a similar, relative decrease in the position of the MDg deformation, BdH absorbing at 1221 cm"1 while PAH absorbs at 1206 cm"1. Again, deuteration is necessary to confirm the assignment of the NH2 deformation. The positions of the four ring vibrations of PAH are in agreement with those of the aldehyde. There is one stretching vibration in this spectrum, however, which should appear but which cannot be assigned. The missing band is associated with the acyclic O N stretching vibration which should also fall in the 1600 cm"1 region. There are two obvious possibilities. The band may coincide in frequency with one of the ring stretching vibrations of the pyridine nucleus or the Interaction between this bond and the pyridine ring is so great that the O N vibration is shifted out of this portion of the spectrum. The data which follow will indicate that the former possibility is most reasonable.

16 XComplexes of 2-ftrrldlnalhydrazone.- In general,
It may be stated that the NH2 stretching frequencies shift to somewhat lower frequencies upon complexing; however, it is not possible to discuss the quantitative aspects of this shift for two reasons. The first Is associated with the resolution of sodium chloride optics which was considered earlier. The second reason is that the water band present in the spectra of the complexes lies so close to the asymmetric NH2 band that the latter Is obscured In several Instances (see Figures 31 and 32)• Comparison of the ND2 stretching bands Is no more rewarding since the two vibrations are not resolved in the spectrum of the ligand.
The NHg deformation occuring near 1600 cm"l Is somewhat more difficult to assign in the spectrum of the complexes since this mode is not consistently resolved but tends to overlap the high band of pyridine which lies at somewhat higher frequencies in the spectra of the complexes than in the spectrum of the free hydrazone. A small shoulder, which disappears from the spectrum of the nlckel(ll) complex upon deuteration, indicates that the position is relatively unchanged from that found in the spectrum of the free ligand (Figure 32).
In contrast to the behavior noted above, several significant changes occur in the pyridine ring vibrations

162
lOOr
•0
60
40
TOO8009001000190020004000 3000i, cm'
DO)
60
60
40
20
D nM roM [F«(P4M)J I ,
4ooo— ste DOO
Figure 31. Infrared Spectre of [Fe(PAH)^]Ig.

X Tf
Ofli
mitl
ence
X
rron
ynTt
unc«
T
rons
nMtto
ncc
163
KX)
6 0
40>
20
0 _ ..S O O O 4 0 0 0 3 0 0 0 2 6 0 0 7 0 0COO 9 0 0 6 0 0B O O2000
Wove N um tun (cm H1
iO O
BO'
TOO1500 1000 900 BOO20004000 3000num ters, c m -'
?00
60T
60j
40
20O u M f M d [Ml (PAH),] I,
4000 3000 2000 1500 1000 900 800 TO
Figure 32. Infrared Spectra of the Cobalt(ll) andNlclcel(ll) Complexes of Pyrldlnalhydraxone.

of PAH upon complexing. The 1585 cm-1 band of PAH,Band 1, remains the strongest band of the spectra, but Is shifted to higher frequencies (1608 cm-1), and the relative intensity with respect to the other absorptions in the double bond region is enhanced (Figures 3 0, 3 1, and 32). The high frequency band of pyridine and the portion of the spectrum extending to approximately 20 cm"1 below it appears to be better resolved in the spectrum of the iron (II) complex than in the spectrum of the free ligand.This observation may be attributed to the fact that Band I is shifted more greatly toward higher frequencies than is Band II. As a result, Band II is revealed as a separate absorption rather than a shoulder on Band I. In the spectra of the cobalt(ll) and nickel(ll) complexes, however, Band II Is not resolved from Band I.
An additional band (15^1-1560 cm"1) appears in the double bond region of the spectra of the complexes (see Figures 30, 31, and 32)• This may be associated with the acyclic C*N stretching frequency, which has been shifted to lower frequencies upon complexing. This shift toward lower frequencies may be accounted for by the Increased resonance of the acyclic O N bond made possible by complexing with a metal atom, an effect anticipated if multiple bonding is Involved In the metal-llgand link. The assignment of the acyclic O N vibration to the band at 15*1-1 5 6 0 cm"1, is dependent on simultaneous assignment

of the band at about 1608 cm"1 to an aromatic ring vibra tion (py)* This assignment appears to be reasonable because in this system, the C=N band behaves in a manner analogous to the 15^1-1560 cm"1 band of the PAH complexes; i.e., this frequency occurs substantially lower in the spectrum of the spin-paired iron(ll) complex than is observed for the spin-free cobalt(II) and nickel(ll) complexes. The spin-pairing of iron(ll) with ligands containing the conjugated dimethine group indicates a more profound type of interaction than that found in the case of the spin-free cobalt(Xl) and nickel(Il). This is manifest in the 15^1 cm"1 peak which is assigned to the acyclic O N band in the spectrum of rFe(PAH) 1 and the higher 1555 cm-* peak assigned to this vibration for the analogous cobalt(ll) and nlckel(ll) complexes.By way of summary, it may be said that the C=N band was assigned on the basis of prior Identification of all other bands in the immediate region, on the basis of relative intensities, and of the analogy between the behavior of this band and the acyclic O N band of BdH and its complexes.
In the spectra of both the free ligand and the complexes, there is a band near 1150 cm -1 which remains relatively unchanged throughout the series. This band may be attributed to either a ring vibration or a CH

1661^0bending mode of the pyridine ring. J Two CH deformations
*3°H. M. Randall, R. G. Fowler, N. Fuson and J. R. Dangl, loc. cit., Table 5*
may be assigned with more certainty as occurring near 770 cm"* and 745 cm"** No assignment of an external NHg deformation has been made for this series of compounds because all of the bands in the deformation region which show any regularity have been assigned.
Four additional bands characterize the spectra of the complexes of PAH (Table 16)* These bands do not appear to have any counterpart in the spectrum of the free ligand. They occur at approximately 1302 cm“*, 1271 cm"*, 1247 cm"* and 1214 cm"*.
2 ,6 -Pyrldindlaldehyde (PdA).- In contrast to the well-defined double bond region of the 2 -pyridinaldehyde spectrum, the double bond region of the spectrum of 2 , 6 -pyridindialdehyde is very poorly defined (with the exception of the carbonyl band)* Definition in other portions of the two spectra Is comparable (see Figures 2 5,30, and 33)• Only one of the four ring vibrations, 1587
cm"*, (Band I) noted in the spectrum of PA is present in the spectrum of the dlcarboaldehyde of pyridine*
The carbonyl band of PdA lies at 1724 cm"* which is identical with that of blacetyl and somewhat higher than that of PA. Three strong bands (1351 cm"*, 1261 cm"*

Trantmittone*
Zfim PjrridmdKlJdthydt
O J - J J , . , ■ 1 , ^ ^ x ,_„ .________._ _______ 18000 4000 3000 8000 BOO WOO 900 90 0 TOOWom Numbcn tom.-1)too
80
60
4 0
202,6 - PyfidwKMolifthpdrator*
i.4000 3000 000 WOO 900 600 TOO
WM rxmtarv, an'*100
60
J 601s-«
20M utiroM I2,6 -PyrMMtaiUltiyffrQBni
2000 1900 ooo 900 TOO000W n number «, cm '1
Figure 33* Infrared Spectra of 2,6 -Pyrid indialdehyde and Pyridindialdlhydrazone*

168and 1209 cm-1) are characteristic of the middle frequency region of this spectrum. These peaks may be associated with the carbonyl groups since they do not occur In any of the derivatives of the dialdehyde.
The CH deformations fall within the expected region for a dl-substituted pyridine.
2 ,6 -Pyridindialdlhydrazone (PdAdH).- The spectrum of 2 ,6 -pyridindialdlhydrazone, which Is better defined than that of the dialdehyde, resembles the spectrum of PAH.The NHg stretching frequencies are quite similar, suffering the same limitations discussed earlier in connection with PAH. The NH^ deformation band occurs at a slightly higher frequency in the spectrum of PdAdH than in the spectrum of PAH. The assignment of this band is again confirmed by the spectrum of the deuterated sample. There is one additional peak in the 1600 cm"1 region which occurs on the high side of Band I. This peak may correspond to the absorption associated with the acyclic C»N stretching vibration. This acyclic CaN would appear to lie at a position somewhat higher than in the spectrum of PAH.This is in agreement with the assignments given the analogous methyl imlne derivatives, i.e., the acyclic C=N stretching modes are assigned at 163^ cm"1 and I06O cm"1 in pyridinalmethylimine and pyridindlalbismethyllmine respectively.

169As In the spectrum of PAH, no unequivocal assign
ment of an NH^ deformation mode could be made. The CH deformations are those expected for a 2 :6 -di-substituted pyridine.
The Complexes of 2,6 -Pyridindialdlhydrazone.- The spectra of normal- and iso-[Ni(PdAdH)2 Jl2 are identical; therefore, only one spectrum has been included in the figures (Figure 3^ and 35)•
As was true in the case of the previously discussed hydrazones, very little may be derived from considerations of the NH2 stretching frequencies. In addition, the external NH2 deformation frequency appears to be obscured by other bands In the double bond region of these spectra as a consequence of the shift of the 1587 cm-* band of pyridine toward higher frequencies.
In the spectra of these complexes, the 1585 cm-1
peak of pyridine (Band I) is shifted to higher frequencies and Is the strongest of the absorptions resulting from the double bonds of the pyridine ring and their interactions. Band II appears as a relatively weak shoulder in most instances on the low side of Band I, near 1595 cm”*. This is in agreement with the observations made in the spectra of pyrldinalmethylimine, pyridlndialbismethylimlne and their complexes. J As in the spectra of the complexes
131P. Figgis, loc. cit.

Tron
smrtt
Qnc
e T r
ans
rrntlo
r>c*
170
*X>
80
40
20
2000 15004000 3000 1000 900 800M m num bers. cm
100
80
60-
Oeuterotcd [F*(P0A0H)|] If
0i . - - . l . , . . .4000 3000 2000 1500 1000 900 800 700
Wave num bers, cm"*
too
80
£ 80 co
»-
5000 4000 3000 19002000 900 SCO TOOWav* Numban 1cm'1}
Figure 3^« Infrared Spectra of the Iron(ll) and Cobalt(ll)Complexes of Pyridindialdlhydrazone.

Tron
imift
onc*
T
ron»
mif
fonc
*
171
too
SO
60
40
4000 30^0 70080090020C0 1900 1000Wgvt nitmbtn, cm ' 1
lOOr
80 -
60
40
20 J Owt«r«l«d [N i(PdM H )|] 1,
0 scbs— sdso— L J—l—I I 1_1_I__1__ I___1___ I J i I i'700“2000 1900
4tavi flumbws, cm "1000 900 800
Figure 35* Infrared Spectra of the Nickel(II) Complexof Pyridindialdlhydrazone.

172of PAH, the band which lies in the range from 1531-135 3 cm"1 is attributed to the acyclic C=N stretching frequency. This assignment is based on the same reasoning as that used previously (Table 17); however, there is one difference noted with respect to the relative positions of this band in the spectra of the cobalt(ll) complexes of PAH and PdAdH. Whereas the spectrum of the PAH complex of cobalt indicated that the C=N stretching vibration was nearly identical to that of the analogous nickel complex, the spectrum of the PdAdH complex of cobalt indicated that the O N band is more nearly identical with that of the iron(ll) complex. The band occurs at a higher frequency in the spectrum of the analogous nickel(ll) complex. This observation serves as an additional Justification for the assignment of this band to the acyclic O N stretching frequency. Prom considerations of the visible spectra of [Co(PdAdH)2 3l2 and of the magnetic behavior of this complex, which indicate that the substance is essentially spin-paired, the infrared absorptions would be expected to resemble that of the iron(ll) complex. There is still one singularity in the spectrum of the iron(ll) complex, however. There is an additional band at 1511 cm"1 which appears as though it might have occurred as the result of a splitting of the 1531 cm"1
band. This might be brought about as the result of great

173Interaction between the acyclic C-N and the pyridine
ring* A parallel behavior is found In the splitting ofa CaC band when it Is conjugated with another aliphatic
1^2double bond. This effect is reported to be less
*^2L. J. Bellamy, loc. clt., p. 3 5.
pronounced in conjugation with aromatic nuclei.The two lower frequency vibrations of the pyridine
ring are raised to higher frequencies to some extent by complexing, but remain relatively unchanged throughout the series of complexes. This shift may be explained on the basis that any interaction within the chelate ring detracts from the resonance characteristic of the aromatic ring.
The CH deformations are those expected of a 2,6 di-substituted pyridine.
Once again, as in the spectra of the complexes of PAH four bands are apparent which appear to be characteristic only of the spectra of the complexes. These occur near 13°5 cm"*, 1280 cm"*, 1245 cm"* and 1220 cm"*.
2-Pyrldinal-p-tolyllmlne (PAT) .- In contrast to the spectrum of PAH in which the highest band in the double bond region was attributed to the pyridine ring, but in agreement with the spectrum of PdAdH, the highest frequency band In the spectrum of PAT is attributed to the

17 4acyclic C=N stretching frequency* This hand occurs at 1634 cm”1, i.e., slightly below the usual range for such groups. As proposed in the case of BdH, a contributing factor to the low position of the acyclic C»N band is the NH2 group. In this ligand, a phenyl group is attached to the Inline nitrogen in place of the NH2 group of BdH. This argument appears to be supported by the position of the acyclic C=N stretching mode in the spectra of pyridinal- methylimlne (1634 cm-1), pyridindialbismethylimine (1560
cm-1), benzaldazine (1620 cm-1) and pyridinaldazine (1620
133y. j. Stratton, loc. clt.
The high ring stretching frequency again occurs near 1582 cm”1, the other bands occuring at 1567 cm-1,1462 cm"1, and 1435 cm"l. These values are in agreement with the assignments made in the spectra of the hydrazone complexes discussed above.
Benzene Is reported to absorb near 1600 cm”1 and 1500 cm"1; however, external conjugation with benzene frequently gives rise to a low intensity band near 1575 cm”1.1^ The very weak absorption in the spectrum of PAT
J. Bellamy, loc. clt., pp. 6l, 62.
near 15^5 cm"1 may be associated with this external

175conjugation (Figure 36). The strong peak occuring at 1506 cm"1 is, without doubt, the second or low band reported for the phenyl group.
The peak which occurs at 1348 cm"l may be considered to arise from the C-N stretching vibration on the assumption that the nitrogen atom attached to the tolyl- group may be considered to behave, spectrally, in a manner similar to a tertiary, aromatic amine nitrogen. Tertiary, aromatic amines undergo a C-N stretching vibration between 1360 cm"1 and 1312 cm"1.135
1 3 5 l . j . Bellamy, loc. clt., p. 213*
No definite assignment can be made for the expected1600 cm"* band of the toly1-group. Since the intensityof this band is subject to considerable variation, it is entirely possible that it is obscured by one of the bands of pyridine. It certainly can not be assigned to the 1624 cm"* band on the basis of the shifts observed In thespectra of the complexes of PAT.
It has been pointed out that pyridine and a number of pyridine derivatives have bands near 1200 cm"1 and near 1100 cm"1 .*3^ 2-Methylpyridine has absorptions at
*36l . J. Bellamy, loc. clt., p. 235*
1240 cm"1 and 1043 cm"1, the latter being the stronger.

176100 -
BO |-
J 6 0 -
20-
2 - Pyrid inol - p - td il inun*
O .____200040 00 9000 1500 1000 900 800 T O O
cm'
100
60
60
20[F.(PAT), ] Cl,
90004000 2000 B O O 1000 900 BOO TOOWove numban, cm
100
8 0 -
60
20
01___L_.4000
_I_BOO
J—X _L2000
x— i L i900
x X9000
x X-J —TOOOOO BOOWav* numbM, cm
Figure 36. Infrared Spectra of Pyrldinal-p-tolylimine andthe Iron(Il) Complex of Pyrldinal-p-tolylimine.

177On this basis and due to the occurrence in position of these bands in the derivatives of PAT, the 1233 cm“* band and the 10i8 cm”* band of PAT are assigned as either a ring vibration or a CH deformation.
The remaining CH deformations of the toly-group occur at 823 cm-1 and 704 cm”* (two adjacent H atoms,1:4 substitution).*-^ In addition, para substitution is
*^L. J* Bellamy, loc. cit., p. 55*
reported to give rise to two weak bands near 1200 cm”* and 1100 cm”*. Therefore, the two bands in the spectrum of PAT occurring at 1214 cm"* and 1088 cm”* appear to be characteristic of the p-tolyl-group (Figures 36 and 37)* There are several additional bands listed in Table 18 which are not assigned but are Included because they are found to be characteristic, not only of the free ligand, but of the complexes as well.
The Complexes of 2-Pyrldinal-p-tolylimine.- Again, the region of most interest in characterizing the complexes of PAT is the double bond region. The trends which were observed In the spectra of the hydrazone complexes also occur in this case. The spin-paired, iron(ll) complex has its alkyl C»N stretching frequency at a considerably lower value than the spin-free complex of iron(II), [FefPATjgClg], or analogous cobalt(Il) and nickel(Il)

[C(«T>,] I,
4000 S000
“Y~"
[MtoTfcCld0 I J i 1________ i__ I-----i. ._ !___ I - l - j j 1 - - 1 L ■ -L - > j ) 1_________ -L--
« o o . 3oao 2000 t t o woo Soo ooo Tw
•Of-
t a
— 1 J tbKa 1 J l~J-- i J -1-Bb--J--«4x-80OD 1900 1000 900 100 TOO
Figure 37* Infrared Spectra of the Cobalt(ll) andNickel(ll) Complexes of Pyridinal-p-tolylimine.

179complexes. In fact, in the spectrum of [Fe(PAT)3)12 * the acyclic C*N band appears to cross below the high frequency band of the pyridine ring (assignment based on shape and relative intensity of the band, see Figure 36)* Tentatively, the O N band may be assigned at 1585 cm"l in the spectrum of this iron(ll) complex while it occurs slightly above 1630 cm"* in the spectra of the analogous cobalt(ll) and nlckel(ll) complexes.
There are several additional unassigned bands which appear to be characteristic of this series of spectra In general. These occur near 1300 cm--*-, 1270 cm-*, 1200 cm"*, 910 cm'*, and 775 cm"1 (Table 18).
6 • Continuous Variations Study of the System, Nickel(II)-BlacetyldlhydrazoneIt was observed that dissolution of the blue-colored
complex, dichloro bis-(biacelydihydrazone)- nickel(ll), In water resulted in a rose-colored solution. Subsequently, only the amber-colored complex, tris-(blacetyldlhydrazone) - nickel(ll) chloride, could be crystallized from the solution. In order to determine, if possible, the species present in solution and, in particular, to establish the existence or absence of the two-to-one complex In water (it will be recalled, from the preparations section, that this two-to-one complex was prepared in absolute alcohol), a Continuous Variations Study was made. During the course

180of this investigation, four distinct absorption maxima were observed. Two of these bands are attributable to the hydrated nickel(ll) Ion (658 mJX and 725 ^JX )• One band was observed in the spectrum of a solution containing equal amounts of ligand and nickel(ll) chloride (^ 5^2 m jX ) . The third band, at 490 vajU. , is characteristic of the three-to-one complex (Figure lo). For those solutions containing intermediate ratios of ligand to metal, the positions of these bands are shifted and the intensity of each band is less than the intensity observed under the conditions stated above.
Two frequencies were chosen which reflected absorptions characteristic of single species. The enhancements of absorption at each of these frequencies, corrected according to the method of Vosburgh and Cooper*^ are
C. Vosburgh and G. R. Cooper, loc. clt.
contained in Table 19* A plot of these values as a function of the mole per cent of nickel(ll) ion in solution (with respect to the total amount of metal salt and ligand present) was made (Figure 33)•
It is indicated, from this plot, that one-to-one and three-to-one complexes of nlckel(ll) with biacetylidi- hydrazone predominate in solution of the appropriate mole ratios of ligand to metal ion. No evidence was found for a two-to-one complex by this method.
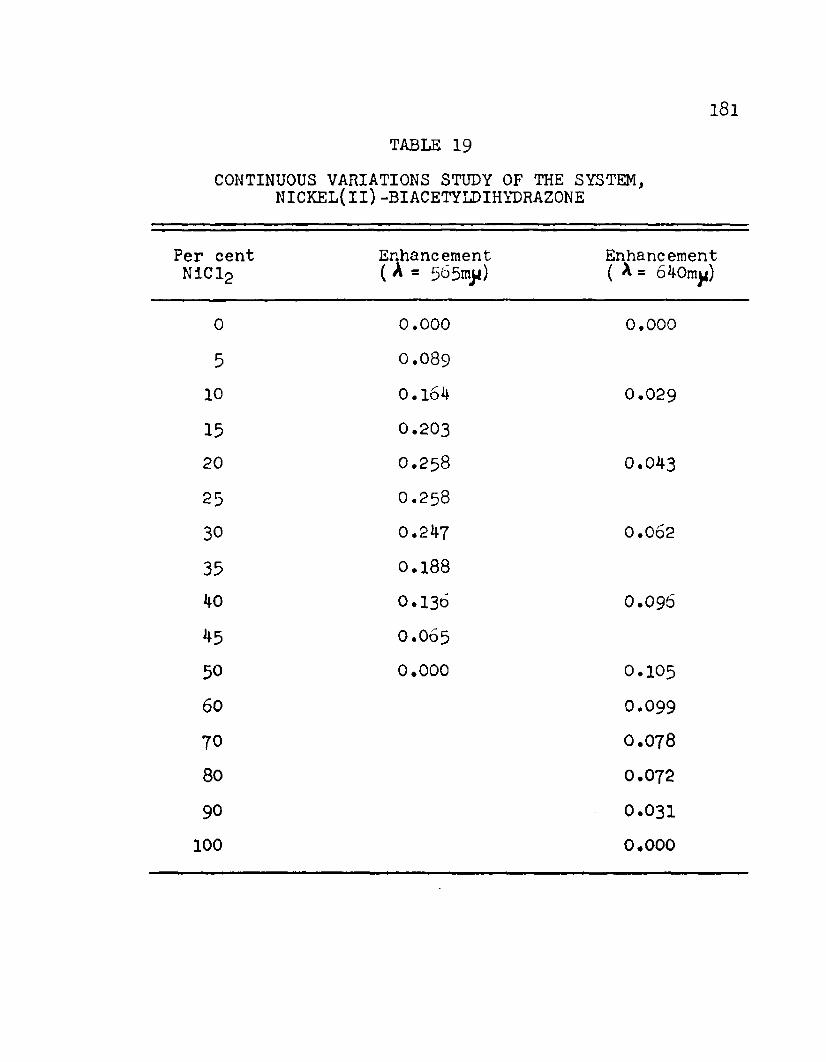
18iTABLE 19
CONTINUOUS VARIATIONS STUDY OF THE SYSTEM, NICKEL(II)-BIACETYLDIHYDRAZONE
Per cent NIC12
Enhancement( A = 565mjj)
Enhancement ( A = 640mj|)
0 0 .0 0 0 0 .0 0 0
5 0.08910 0.164 0 .0 2 9
15 0 .2 0 3
20 0 .2 5 8 0.04325 O .258
30 0.247 0 .0 6 2
35 O .188
40 0 .13b 0 .0 9 6
45 O.O65
50 0 .0 0 0 0 .1 0 5
60 0.09970 0 .0 7 8
80 0 .0 7 2
90 0.031100 0 .0 0 0

Enh
ance
men
t (p
erce
nt)
40
X* 565 m u3 5
30
2 5
20
X * 6 4 0 mp.
20 30 4 0 50 6 0 70
Percent nickel chloride
1008 0 9 0
Figure 38* Enhancement as a Function of Per cent Nickel (II) Chloride in the System, Nickel(ll)- Biacetyldihydrazone.

183The results of this study indicate that the re
action of dichloro-bis-(biacetyldlhydrazone)-nickel(ll) with water may be expressed by the equation,2[Ni(BdH)2Cl2 ] + 5H20 [Ni(BdH)3 ]++ + [Ni(BdH)(H20)^]+++2Cl
7. Resolution of Optical IsomersThe resolution of the trl3-(biacetyldihydrazone)-
iron(ll) Ion into optically active antipodes was attempted in order to obtain an indication of the covalent character of the hydrazone complex of iron(ll) and to establish, by this classical method, the octahedral configuration of this spin-paired complex.
Following the procedure outlined in the Experimental section, a dextro-rotatory diastereoisomer of this complex ion with d-antimonyl tartrate was isolated and re- crystallized. In an attempt to determine the rotation of this species, it was observed that the rotation changed with time, consequently, the rotation was measured as a function of time and the initial rotation (zero time) was determined graphically (0.452°), The observed rotations were then corrected for the rotation of the d-antimonyl tartrate (0.047° in a 0.05 per cent solution of diastereoisomer) • These data are reported in Table 20. From the rate expression for a racemization process, the expression shown below is obtained, where
- / « = kt

184TABLE 20
RACEMIZATION OF [Fe(ClfHl0N4) 3 ] L (SbO) (Cj+Hj+06) ]2*
Time (in Min.)CorrectedRotation -log
0 0.405a
*•5 O .385 0.951 0 .0 2 2
13.25 0.350 0.864 0.06418.25 0 .3 2 2 O .795 0 .1 0 0
23.5 0.304 0.751 0.12428.75 0.289 0.714 0.14637.75 0.257 0.635 0.19746.0 0.230 0 .5 6 8 0.24653.25 0.197 0.486 0.313
* 0.05 per cent solution (by weight) at 25°C 1 1°.a Determined Graphically.
Jt is the time, k is the rate constant, is the rotationat time zero, and is the rotation at time _t.
A A plot of -log as a function of time results inthe straight line shown in Figure 39* From the slope ofthis line, the value of the rate constant, k, is deter-
-1mined to be O .3 1 9 hr. . From this, the half life of this reaction is determined to be 2 .1 7 hourB.
- U

- Lo
g
185
032
0.28
024
0.20-
0 16
0.12
006
004
20 30Time (in minj
40 50 60
Figure 39. Change in Observed Rotation of tFe(C4Hi0H4)3 ]l(Sb0 )(C4H406) } ?with Respect to Time.

186pcO
The specific rotation, J of the diastereo-isomer is 90^°* Prom this it is calculated that the specific rotation attributable to the complex cation is 1976°. No attempt was made to obtain the 3imple iodide salt of this cation; however, from these data, a specific rotation of 1205° is predicted.
It is significant that rate of this racemization process is slower than the rate of racemization of either the analogous 2,21-bipyridine complex or the o-phenanthro- llne complex.^ 9 Certainly, this appears to indicate that
■39jj# r, Davies and P . P. Dwyer, Trans. Faraday Soc., 180 (1953)•
there Is a high degree of covalent character In the spin- paired hydrazone complex of iron(ll).
D. Discussion
The iron(ll) complexes which have been prepared with the ligands BdH, PAH, PdAdH, and PAT are of two general types, (l) [Fe(AA)2 ll2 or [FefAAAjgllg# an<2 (2) [Fe(AA)2X2 ]* A complex, corresponding to the latter formulation, was prepared with PAT, (Fe(PAT)2C12 3*2H20 . An attempt to prepare the analogous complex of BdH (by the same synthetic method) resulted in a complex of the formula [FetBdH^HFeCl^]. No attempt was made to prepare these two-to-one complexes by thermal degradation, a

187method which has been applied successfully to the analogous complexes of iron(ll) with 2,2'-bipyridine and o-
140phenanthroline• The conductance data indicate that
Basolo and F. P. Dwyer, loc. cit.
all of these complexes are octahedral despite the fact that some solvation of the nonionlc two-to-one complex occurs, resulting in a conductance which is larger than that expected for a non-electrolyte. In fact, the molar conductance (100 ohm "*) indicates that an average of more than one chloride ion is replaced per formula weight of complex. Irrefutable evidence for the octahedral structure of tris-(biacetyldihydrazone)-iron(ll) iodide was obtained from the resolution of this complex into its optically active forms. It is significant that the rate of racemization [Fe(BdH)^Slg'Ck = 0,319 hr. *) is substantially slower than the rate of racemization of either[Fetdipyridyl^Jlg or tFe(o-phenanthroline)^3Ig (k - 2 . 2 hr.
and k = 2.4 hr.”1 respectively).1**1' 1]*3' 1**1*
l4ljp. P. Dwyer, loc. clt.1**2G. T. Morgan and F. H. Burstall, loc. clt.l2*3A. Werner, Ber., 4£, 443 (1912).
C. Bailar, Jr., loc. cit., Table 8.8.
One possible explanation of this phenomenon will be

188presented In the discussion of the nickel(ll) complexes. Since all of the ligands used in this investigation contain the same conjugated methine structural unit, it maybe concluded, by analogy, that the remaining three-to- one complexes are octahedral and are capable of being resolved into their optically active forms. An attempt was made to resolve the three-to-one complexes of iron(ll) with PAH and PAT using the same method applied to the BdH complex (this method has also been applied successfullyto the resolution of the dipyridyl and o-phenanthroline
/ \\ 14^ 146 147complexes of iron(II)); ■ however, the very
p* Dwyer, loc. clt.T* Morgan and P. H. Burstall, loc. clt.
Werner, loc. cit.
large solubility of the diastereoisomer made it impossible to fractionally crystallize them from water. The same problem was encountered to a lesser extent in the dipyridyl and o-phenanthroline complexes.
The only two-to-one complex which was prepared, [Pe(PAT)2Cl2 3*2^0, was strongly paramagnetic. The complexes, in which all six positions were occupied by unsaturated nitrogen atoms, were spin-paired, and, essentially diamagnetic. This diamagnetism, in addition to the resolution of the spin-paired BdH complex into optically active antipodes, indicates a large amount of covalent

189character* In fact, it is asserted that, since the metalatoms contains six electrons in the tg orbitals, whichare of the proper symmetry to overlap with the £ orbitalof the ligand molecules, three pi molecular orbitals areformed. This is in keeping with the conclusion by
148 149Orgel, ' that pi bonding must be assumed in tris-
llf8L. E. Orgel, J. Chem. Phys.. 2$, 1819 (1955)*E. Orgel, "Some Applications of Crystal Field
Theory to Problems in Transition Metal Chemistry," Report to the Xth Solvay Council, Brussels, May, 1956.
(o-phenanthroline)-iron(ll) iodide to account for it3 diamagnetism. The infrared spectra of these complexes support this assertion of pi bonding between metal atom and ligand molecules. It was observed that the position of the C=N stretching vibration in the spectra of the spin-paired iron(ll) complexes, relative to that of the ligand, the spin-free iron(ll) complex, and the analogous spin-free complexes of cobalt(ll) and nickel(ll), indicated a decrease in the bond order of the C»N bond. This decrease in bond order is easily understood in terms of multiple bonding between the metal atom and ligand molecules*
The data, which were obtained from the ultraviolet and visible spectra of these complexes, are not In disagreement with this conclusion* In fact, the charge

transfer bands, observed In the visible spectra of the spin-paired iron(ll) complexes, are consistent with the assumption of pi bonding. As explained earlier, the charge transfer bands indicate a transition of the t^g electrons of the metal into molecular orbitals extending over (delocalized on) the ligand molecules, i.e., an effective oxidation of the iron(ll) ion occurs. Therefore, it is significant that the spectra of the spin-free co- balt(ll) complexes do not exhibit this type of behavior, despite the fact that cobalt(ill) complexes, with ligands of the dipyridyl type, are more stable than those of iron (ill) (with respect to reduction). In other words, it is the pi electron system of the complex containing a divalent metal which is of primary importance, rather than the ease of oxidation of the metal ion, in giving rise to absorptions of this class. The magnetic properties indicate that this strong interaction exists In the iron(ll) complexes but, that it does not exist to such an extent in the spin-free cobalt(II) complexes.
The cobait(ll) complexes which have been prepared with BdH, PAH, PdAdH, and PAT are also of two general types,(1) [Co (AA)3]I2 or [Co(AAA)2 3l2 , and (2) [Co(AA)2Cl2 3* The conductance data Indicate that all of these complexes are octahedral, despite the fact that here, as in the case of the two-to-one iron(ll) complex, solvation of the complex,

[Co(BdH)gClg] occurs resulting in the substitution of solvent molecules (methanol) for halide ion. The magnitude of the molar conductance of this complex (101 ohm"1) indicates that, on an average, slightly more than one chloride is displaced per formula weight (the conductance of a uni-univalent electrolyte such as sodium chloride is approximately 95 ohm"1). This is supported, in several instances, by the magnetic moments of the complexes. All of the complexes of cobalt(ll) with bidentate ligand are strongly paramagnetic, indicating three unpaired electrons however, [Co(PdAdH)2 ]l2 was observed to have a magnetic moment of 2.9 B.M., which is intermediate between the spin only magnetic moments expected for one and three unpaired electrons. The data obtained from a study of the temperature dependence of the magnetic susceptibility of this complex indicate that an equilibrium mixture of spin-free and spin-paired states exists (approximately 70 per centspin-paired state at 300° K.). The basic structure of
7both the spin-paired and spin-free d ion is octahedral; however, one would expect each structure to be distorted toward a tetragonal configuration, the effect being more pronounced in the spin-paired case (here the orbital degeneracy is in the antibonding e^* orbitals) than in the spin-free case (the orbital degeneracy being in the nonbonding tgg orbitals).

192The Infrared spectral data support this conclusion.
The position of the C=N stretching frequency In the spectra of the spin-free complexes resembles that of the nickel(Il) complexes while the spectrum of essentially spin-paired LCo(PdAdH)2 ]l2 resembles that of the spin- paired iron(ll) complexes. The magnetic moments of the BdH and PAH complexes, 4.3 B.M. and 4.6 B.M., respectively, are somewhat lower than the observed values for spin-free octahedral complexes (4.8-5*2 B.M.)• This may indicate a behavior similar to that postulated in case of [CofPdAdHjgl l£* This is reasonable since the other ligands of this series would be expected to have similar ligand (electrostatic) fields.
Lions and M a r t i n * ^ have recently reported the
150F. Lions and K. V. Martin, J. Am. Chem. Soc.,80, 3858 (1958); 19, 2733 (1958).
preparations and characterization of cobalt(II) complexes of 2,6-pyrldindial-bis(benzylimine) and 3>6-dithIaoctan-1, 8-dial bis-d-pyridylhydrazone. They observed that these complexes have magnetic moments of 3*8 B.M. and 2 . 5 B.M., respectively. It was concluded that these complexes are not and cannot be octahedral for two reasons, (l) The octahedral configuration requires the use of dgsp3 hybridized atomic orbitals which, in case a d^, Is Impractical, due to the high promotion energy required to make

193two 3d orbitals available for hybridization (promotion of one electron to the 5 j3 level) . (2) Octahedral cobalt(II)complexes customarily have magnetic moments between 1.7 and 2.0 B.M.; therefore, Lions and Martin imply that these two complexes are five coordinate (tetragonal pyramidal)• It appears much better to consider these complexes to be six-coordinate and tetragonal bipyramidal (or simply tetragonal) and to explain the moments on the basis of an equilibrium mixture of doublet and quartet states as has been demonstrated in the case of ( C o ( P d A d H ) at>ove*It might also be pointed out that our octahedral configuration of a d7 ion does not require promotion of an electron to the 5 JL level, either in terms of molecular orbital theory (the one unpaired electron is antibonding) or in terms of crystal field theory (the one unpaired electron occupies the e* orbitals)• In fact, even a more modern valence bond picture does not require promotion of
Aone electron (dsp -pd hybridized bonds)*One other complex which appears tobelong to this
class of cobalt(ll) compounds (bis-[pyrldlndialbis(methyl- imine)]-cobalt(II) Iodide), was prepared by Figgins,1^
Figgins, loc. cit *
who reported a magnetic moment of 2.34 B>H. It is probably significant that the ligands Involved In these complexes of cobalt(ll) are either trldentate or sexldentate

19^and that they are all well-suited for pi bonding. A study of the variation of the magnetic moments of this series of complexes as a function of temperature should be quite revealing.
As in the case of the iron(ll) and cobalt(ll) complexes of BdH, PAH, PdAdH, and PAT, nickel(ll) forms complexes of the general type, (l) [Ni(AA)^]l2 or [Ni(AAA)2 ]l2,and [Ni(AA)_Xpj. In addition, a continuous variations 2 cstudy of the system, biacetyldihydrazone-nickel(ll), indicates the existence of a one-to-one species In solution which may be formulated as [Ni(BdH) (HgO)5 * These observations are completely in accord with the dipyridyl and o-phenanthroline complexes of nickel(ll). It is also significant that the two-to-one complex of nickel(ll)with dipyridyl, which is usually prepared by thermal
152degredation of the three-to-one complex, has been
152p. Basolo and F. P. Dwyer, loc. clt.
prepared as a hydrate ([Ni(dipyridyl)2cl2 ^*3H20) in this laboratory, using precisely the same preparative method used to prepare the two-to-one complex of BdH. The three water molecules are quickly lost upon heating at 110°C. for a short period of time. Both of these two-to-one complexes disproportionate in water to give the three-to- one and a one-to-one complex. The conductance data on

195methanol solutions indicate that the complexes of both types are octahedral despite the fact that solvation does occur in the methanol solution thus giving rise to a conductance which is larger than that which is expected for a non-electrolyte. The magnetic moments of these spin- free complexes support the assertion that the configuration is octahedral, lying in the general range of values
Oobserved for octahedral complexes with a d configuration (approximately 3 B.M.).
The spectral data indicate that there is considerable interaction between the ligand molecules and the metal atom although it is never as profound as that observed in the spectra of the corresponding iron(ll) complexes. The suggestion of strong interaction between ligand molecules and metal atom, even in spin-free complexes, is supported by the covalent character of tris-(o-phenanthroline)- nickel(ll) iodide, for example, which has been resolved into optically active antipodes This
153p. p. Dwyer, loc. cit.t . Morgan and F. H. Burstall, loc. cit.
155A. Werner, loc. cit.■^F. P. Dwyer and E. C. Gyarfas, loc. cit.
optically active complex racemizes more slowly than the corresponding spin-paired lron(ll) complex. One possible explanation of this phenomenon is presented below.

196Kinetic studies of both the rate of racemization
and rate of dissociation (or ligand exchange) have been made on the systems, iron(ll)-dipyridyl, Iron(ll)-o-phenanthroline, nlckel(ll)-dipyridyl, and nickel(ll)-o-
157phenanthroline. It was observed that the rate constant
*57p. p. Dwyer, loc. cit.
for the dissociative process, in the nickel(ll) complexes, is identical to that of the racemization process, indicating that the mechanism of racemization is a dissociative one. In contrast, the rate constant for the racemization process in the iron(ll) complexes, is approximately ten times as large as the rate constant for the dissociative process* From these data, it must be concluded that the racemization process involves an intramolecular rearrangement. This type of process, which does not necessitate the rupture of any bonds, was first postualted to occur in the case of the o-phenanthroline complex by Basolo, Hayes, and Neumann. J The two optical Isomers which are
Basolo, J. c. Hayes, and H. M. Neumann, J. Am. Chem. Soc., 7£, 5102 (1953b Jo , 3807 (195*0-
of the same energy are separated by a potential energy barrier with respect to Inversion of configuration. In order to achieve racemization by means of an inversion, an energy of activation is required, i.e., the vibrational

197energy of the molecule must be sufficient to lift the
molecule to the high energy state which Is Intermediate
to the two normal states, both of which are optically
active. At the top of this energy barrier, there Is an
equal probability of formation for both configurations
provided that they are of equal energy. The net result
of this process is racemization.The vibrational energy required to pass over the
potential energy barrier may be obtained from an external
source, e.g., thermal excitation; however, in the case ofthe o-phenanthroline complex of lron(ll) for example,this energy may arise internally. As discussed previously,
159Orgel concludes from crystal field calculations that the
159l . E. Orgel, "Some Applications of Crystal Field Theory to Problems in Transition Metal Chemistry, ” Report to the Xth Solvay Council, Brussels, May, 1956.
tris-(o-phenanthroline) complex of iron(ll) lies very close to the cross over point between the spin-paired and spin-free states. If the energy of separation is on the order of kT, a fractional population of the excited state will result. Since transitions from one spin-state to the other involve a shortening and lengthening of the metal- ligand bands, i.e., vibration along the metal-llgand band txls, molecular vibrations may arise which will effectively decrease the potential energy barrier of inversion and

198hence, to racemization. As a result, more molecules would attain the vibrational energy required to cause this intramolecular rearrangement to occur in a given period of time. In spin-paired complexes which are far removed from the spin-free excited state, little or no intramolecular rearrangement can occur, e.g., in cobalt(lll) complexes.The smaller rate constant which is observed in the tris- (biacetyldehydrazone)-iron(ll) ion may be attributed to a ligand field which is larger than that of o-phenanthroline. Utilizing the same concept for the spin-free nickel(ll) complexes it is seen that no internal vibrations can occur as a result of transitions between spin-free and spin- paired states; therefore, the rate of racemization is primarily a function of bond lability, i.e., the rate of dissociation.
As a consequence of the apparent stability of these nickel(ll) complexes and since one of the corresponding iron(ll) complexes has been resolved (which racemizes more slowly than either the analogous dipyridyl or o- phenanthroline iron(ll) complex), one might expect that tris-(biacetyldihydrazone)-nickel(ll) chloride can be resolved into the optically active forms. Furthermore, on the basis of the analogous system, one might expect this optically active nickel(Il) complex to racemize more slowly than the corresponding iron(ll) complex.

199An orange-colored (normal-) complex and a green-
colored (iso-) complex of PdAdH have been prepared. The ultraviolet and infrared spectra of these two isomers are identical. The visible region of the spectra is identical with respect to high probability transitions; however, the d-d transition frequencies have not been studied. These might provide an indication of the nature of these two species. Certainly, it seems quite improbable that they are geometric isomers of the face-edge type or coordination isomers (coordination through one or more amine groups in one case and through the imine nitrogen atoms in the other), since this type of isomerization should result in a difference in all three spectral regions. The temperature dependence of the magnetic moments of these two forms was determined; however, this provided no basis of distinction. It is significant, however, that the moments of both of the complexes appear to decrease as the temperature is lowered, a break occuring In the curve constructed by plotting l/T against the molar susceptibility near liquid nitrogen temperatures. This temperature dependence may be of the type observed in the complex, [Co(PdAdH)g]l2 , which was discussed previously. At present, no explanation can be given to account for these two apparent forms of bis-(pyridindialdihydrazone)-nickel(Il) iodide•

200As a consequence of the many similarities which
exist between the complexes of iron(ll), cobalt(ll) and nickel(ll) with ligands of the aromatic beterocycllc diamine, and acyclic diimine type on the one hand, and with biacetyldehydrazone, pyridinalhydrazone, pyridindialdihy- drazone and pyridinal-p-tolylimine on the other, it is concluded that this latter class of complexes resemble those of the dipyridyl class more closely than those of the dioxime type.

III. THE COMPLEXES OF NICKEL(ll) AND DIMETHYLGLYOXIME, BIACETYLOXIMEMETHOXIME, BIACETYLHYDRAZONEOXIME,
AND BIACETYLHYDRAZONEMETHOXIME
A* Introduction
It was shown conclusively, in Part II of this dissertation, that the complexes of lron(ll), coblat(ll), and nickel(Il) which are formed with the ligands, biacetylidi- hydrazone, pyridinalhydrazone, pyrldindialdihydrazone, and pyridinal-p-tolylimine, more closely resemble the complexes of these metals with ligands of the aromatic heterocyclic- diamines, the -dlimines, and ligands containing functional groups of both types (pyridlnalmethylimine) than the complexes which these metals form with the oC -dioxime type of ligands. Although the question pertaining to the classification of the hydrazone complexes which were prepared, Is answered, the question of why these hydrazone complexes should differ so greatly In character from the complexes of the dioximes remains unanswered. As pointed out earlier, one might expect that the amine group of these hydrazone complexes would appear to bear closer resemblance to the OH group of the oxime function than to an alkyl group of an imine, for example. It had been anticipated that these hydrazone complexes might have properties which were intermediate to the dipyridyl complexes on the one
201

202hand and to the o( -dioxlmes, on the other. In order to determine the extent of these differences and, perhaps, to determine some of the factors responsible for this divergent character, the series of complexes described In this section has been studied.
The one obvious difference between dimethylglyox- lme and biacetyldihydrazone, for example, is the much greater acidic character of the hydrogen on the oxime function. In fact, the majority of the investigations which have previously been carried out on the reactions of the of-dioxlmes with various metals were made under conditions conducive to the ionization of a proton from an oxime function.
For the purposes of this investigation, it was decided to use only one metal, nickel(ll). The reason for this choice lies In the fact that, of the three metal ions, nickel(ll), cobalt(ll), and iron(ll), only nickel (II) forms well-characterized complexes with both the o( -dioximes and the acyclic dihydrazones and dlimines.
In this investigation, the methods of preparation and the physical and chemical properties of the complexes formed by nickel(ll) with the ligands dimethylglyoxime (H-DMG), biacetyloximemethoxime (H-BOM), biacetylhydra- zoneoxime (H-BHO), and biacetylhydrazonemethoxime (BHM), are reported. The methods used to characterize these

203complexes are essentially the same as those described In Part II of this dissertation. Before proceeding to the Experimental section, a more detailed characterization of the complexes of nickel(Il) with the dioxlmes will be given.
The first o( -dioxime complex of nickel(ll) was prepared by Tschugaeff* who reacted dimethylglyoxime with
1L. Tschugaeff, Z. anorg. Chem., 46, 144 (l9°5).
nickel(ll) to produce the familiar red, water insoluble, neutral complex which was shown subsequently, to be diamagnetic. On the basis of geometric isomerism, which was first proposed by Hantzsch and Werner, symmetrical diox- imes were shown to exist in the three isomeric forms shown below. Of these three forms, only the anti- form reacts
C - C C - C^ \\ / / %N N N N
/ \ / /HO OH HO HOanti- amphl-
C - C * ^
N N\ /OH HO
522-
Figure 40

204with nickel(ll) ion to give the product obtained by Tschaugaeff. Proof of the existence of tautomeric forms of the dioxlmes by Brady and Mehta^ led Pfeiffer^ to
^0. L. Brady and R. P. Mehta, J. Chem. Soc., 125.2297 (1924).
3P. Pfeiffer, Ber., M , l8ll (1930); P. Pfeiffer and J. Richarz, Ber., b l , 1^3 (1928).
postulate a five membered chelate ring for this complex,the bonding occurring through the unsaturated nitrogenatoms rather than through the oxygens as had been postu-
4lated originally.
^A. Werner and P. Pfeiffer, "Neuere Anschauungen im Gebiete der anorganischen Chemie," Fifth Edition,
• Friedrich Vieweg and Sohn, Brunswick, 1923> PP* 164, 302.
5Pfeiffer demonstrated that one of the oxime groups
5P. Pfeiffer, Ber., 6£, l8ll (1930).
could be replaced with an imlno or methylimino group without substantially altering the nature of the complex formed. He also prepared the neutral oxlmemethoxime complexes which were reported to have similar properties to the
-dioxlmes.
6p. Pfeiffer, loc. cit.

2057Brady and Muers' proposed the hydrogen bridged
70 . L. Brady and M. M. Muers, J* Chem. Soc., 1930,1599.
structure of these dioxime complexes to account for the fact that no isomeric forms had been observed. The planar configuration of these four coordinate nickel(ll) complexes was proved, chemically, by the preparation of com-
Oplexes with unsymmetrically substituted - dioximes.
8S. Sugden, J. Chem. Soc., 1932, 246; S. Sugden andH. J. Cavell, J. Chem.~Soc., 1^35, o*l.
-
Paneth and Thilo observed that hydrogen chloride
% . Paneth and E. Thilo, Z. anorg. Chem., 147,196 (1925).
reacted with bis-(dimethylglyoxime) nickel(ll) to form a blue-colored product which they formulated as [NifH-DMG^] Clg. This observation was substantiated by Sharpe and Wakefield^ who, on the basis of conductance data in
^A. G. Sharpe and D. B. Wakefield, J. Chem. Soc., 1251. *96.
acetone and of the reaction with silver nitrate, concluded that the complex Is octahedral and should be formulated as [NiCH-DMG^Clg] • They report that this product Is decomposed by all solvents except acetone, however, work in this

206laboratory has demonstrated that it is possible to prepare
this complex in ethanol to which a few drops of concen
trated hydrochloric acid have been added and that it can
be recrystallized from glacial acetic acid without decom
position. Sharpe and Wakefield al3o report that this
product can be acetylated with acetyl chloride, forming a
light green product; however, this product has been shown
by J i c h a t o consist of [Ni(H-DMG)Cl^J mixed with the
^ D . C. Jicha, unpublished results.
uncoordinated, acylated ligand.Rather recently, the structure of bis-(dimethylgly-
oxime) nickel(ll) has been determined by X-ray diffraction 12techniques. Godycki and Rundle concluded from their
^ L . E. Godycki and R. E. Rundle, Acta Cryst., 6 9 ,^87 (1953).
study that the planar molecules are so arranged that the nickel atoms are only 3*24 A. apart, indicating weak metal- metal bonding. The separation between the oxygen atoms of the two coordinated ions (2.44 A.) is the shortest known separation of hydrogen-bonded oxygen atoms. Godycki and Rundle also suggest that the very low solubility of the nickel(ll) complex is associated with the postulated metal-metal bonding. There are a number of instances in which it is observed that the introduction of larger groups

207on the -dioxime molecule appears to prevent the metal- metal Interaction, thus resulting In an Increase in solubility . -3
-3a . g . Sharpe and D. B. Wakefield, loc * cit.
B. Experimental
1. Preparations and Analytical DataDichloro-bls-(dimethyIglyoxlme)-nlckel(ll).- One
and nineteen-hundreths grams of nickel(ll) chloride 6- hydrate (0.005 mole) was dissolved In 30 ml. of absolute alcohol which contained 0 . 5 ml. of concentrated hydrochloric acid. To this solution was added, with stirring, a solution which had been prepared by dissolving l.lo g. of dimethylglyoxime (0 ,0 1 mole) in 5° ml. of boiling ethanol. During the addition of the dimethylglyoxime solution, the mixture became deep blue In color. The volume of the solution was reduced to 30 ml. by evaporation with a stream of dry air. The blue product, which had separated from the solution, was filtered and washed several times with cold, absolute alcohol. This product was recrystallized from a small amount of glacial acetic acid by warming gently to speed dissolution and then cooling; yield 80 per cent. Anal. Calcd. for [NiCc^HgNgOg^C^^: C, 26.55; H, 4.46; N, 15.48. Found: C, 2 6 .3 6; H, 4.44;N, 15.33.

Blacetyloxlmemethoxlme.- Eleven and fifty-one hundreths grams of 2,3-butanedione-2-methoxime (0.10 mole) wa3 added to 5^ ml* of absolute alcohol. To this was added, with stirring, a solution which had been prepared by dissolving 6.95 g. of hydroxylaminehydrochloride (0.10 mole) in 5° ml. of absolute alcohol. To this mixture was added 5*61 g. of potassium hydroxide (0.10 mole) to exactly neutralize the hydrochloride. Approximately 75 ml. of water was added to the reaction mixture and the alcohol was distilled from the mixture. As the alcohol was being distilled from the mixture, a white, crystalline product separated. The product was filtered with suction, washed with several portions of cold water, and dried in vacuo over 91*5 per cent. Anal. Calcd. forC5H10N2°2: Cf ^.14; H, 7.?4; N, 21.53. Found: C, 46.05H, 7.91; N, 21.15.
Dichloro-biB-(blacetylhydrazonemethoxlme-nlckel(ll) Six and ninety-five hundreths grams of hydroxylamine hydrochloride (0.10 mole) was dissolved in 100 ml.of absolute alcohol which contained 4.00 g. of sodium hydroxide (0.10 mole). A white solid (NaCl) began to separate immediately. This product was removed by filtering. The filtrate was added, with stirring, to a solution which had been prepared by dissolving 11.89 g* of nickel(ll) chloride 6-hydrate in 100 ml. of absolute alcohol. To

209this solution was added 11 .51 g* of 2 ,3-butanedione-2- methoxime (0.10 mole). No significant color change was observed during the course of this reaction. The total volume of the solution was reduced to approximately 100
ml. and the green solid which had separated was filtered with suction. The dark green, crystalline product was washed with several small portions of absolute alcohol, and dried in vacuo over 77*6 yer cent. Anal.Calcd. for [Nl(C5Hl0Ng02) Clg]: C, 30.80; H, 5.17; N,14.37. Found: C, 29.84; H. 5-35; N, 1397*
Blacetylhydrazoneoxime.- Twenty milliliter of absolute alcohol was added to 2 0 .2 0 g. of 2 ,3-butanedione- 2-oxime (0.20 mole). To the resulting solution was added 6.42 g. of anhydrous hydrazine (0 .2 1 mole), with stirring. Upon stirring and cooling a very light-colored, crystalline product separated. This product was filtered and recrystallized from the minimum amount of absolute ethanol; yield 66*5 per cent. Anal. Calcd. for C^H^N^O: C, 41.73;H, 7.88; N, 36.50. Found: C, 41.88, 41.95; H, 8 .0 5, 7 .8 9;N, 36.50.
Dlchloro-bls-(blacetylhydrazoneoxlme)-nickel(II).- Two and thirty-eight hundreths grams of nickel(ll) chloride 6-hydrate (0 . 0 1 mole) was dissolved in 50 ml. of absolute alcohol. To this solution was added another solution which contained 2 .3O g. of blacetylhydrazoneoxime.

210Immediately, a silver-blue product began to separate. The product was filtered with suction, washed with several small portions of absolute alcohol, and dried in vacuo at room temperature over ^2^5 yield 83*5 per cent. Anal. Calcd. for [NiCC^H^OjgClg]: C, 26.70; H, 5.04; N,23.36. Found: C, 26.00; H, 5*01; N, 23.28.
Bls-(blacetylhydrazoneoxime)-nickel(ll).- Two and thirty-eight hundreths grams of nickel(ll) chloride 6- hydrate (0.01 mole) was dissolved in 20 ml. of water. To this solution was added 0.3 g. of ammonium chloride and approximately 2 ml. of concentrated ammonium hydroxide.To the resulting solution (PH''%"8), 2.3O g* of blacetylhydrazoneoxime (0.02 mole) was added with stirring. Immediately the solution became a brown-red in color. After stirring for approximately thirty second^ a magenta- colored, paste like product was formed. This product was transferred to a suction filter and washed with a larige volume of water (until the wash water gave no chloride ion test). The magenta-colored product was dried in vacuo over P205; yield 76*7 per cent. Anal. Calcd. for [Ni(C4HgN30)2]: C, 33*^8; H, 5*62; N, 29.29. Found:C, 33*39; H. 5*78; N, 2 9.0 7.
Tris-(blacetylhydrazoneoxime)-nlckel(ll) Perchlorate 2-Hydrate.- A small amount of the blue-gray dichloro-bis- (blacetylhydrazoneoxime)-nickel(ll) was dissolved in water,

211resulting in a pink-colored solution. Upon adding approximately 0.5 g. of sodium perchlorate to the solution, a red, crystalline product separated and was filtered with suction. These crystals were washed in several small portions of cold water, and dried in vacuo over Anal. Calcd. for [ N i ^ H ^ O ) (C104)2*2H20: C, 22.55;H, 4.89; N, 20.10. Found: C, 22.92; H, 4.99; N, 20.10.
Blacetylhydrazonemethoxlme.- Eleven and fifty-one hundreths grams of 2,3-butanedione-2-methoxime (0.10 mole) was placed in 10 ml.of absolute ethanol. To this solution was added 3*21 g. of hydrazine (0.10 mole] which had been diluted with 20 ml. of absolute alcohol. Upon cooling in an lce-bath, a white crystalline product formed, which tended to entrain the solvent. This crude product was filtered and recrystallized from absolute ethanol; yield 92*9 per cent. Anal. Calcd. for 46.49; H, 8.59; N, 32.53* Pound: C, 46.50; H, 8.51;N, 32.56.
Dlc hloro-b1s-(blac e tylhydra z0neme thoxime)-nickel(I I ) One and twenty-nine hundreths grams of biacetyl- hydrazonemethoxime (0.10 mole) was dissolved in 10 ml.of absolute ethanol. To this was added a solution which had been prepared by dissolving 1.19 g* of nlckel(ll) chloride 6-hydrate in 30 ml. of absolute ethanol. The color of the solution became blue-green and a light silver-green

212product separated and was filtered, washed with cold absolute ethanol, and dried in vacuo over P2O5 ; yield 59 per cent. Anal. Calcd. for [NitC^H^N^Oj^C]^]: C, 30*93;H, 5.72; N, 21.66. Found: C, 31*03, 31*08; H, 5*81,5.60; N, 21.66, 2 1 .5 8.
2. Physical MeasurementsThe techniques used in this series of investiga
tions are essentially those which were described in the Experimental section, Part II, of this dissertation.
C . Results
1. Conductivity MeasurementsThe molar conductances have been determined for
four nickel(ll) complexes and are reported in Table 21. There are two distinct types of complexes involved here, both of which are formulated as nonlonic. In one type, a coordinated halide Ion satisfies the electrovalence of the nickel(ll) ion, whereas, in the second type, the bi- dentate ligand has lost a proton from an oxime function giving the ligand a formal, negative charge. The first type of complex Is postulated to be six coordinate and octahedral, the second, four coordinate and square planar.
Absolute methanol was used as the solvent in these determinations since these complexes appeared to be conveniently soluble in it and in order to facilitate com
parison with the data of Table 4.

213TABLE 21
MOLAR CONDUCTANCES OF THE NONIONIC COMPLEXES OF NICKEL(II) WITH H-BOM, H-BHO AND BHM
Compound/ i ohm-
Mohm"*
Cation
[n i (h -b o m )2c i 2 ] 109.8 (58)*[Ni(BHO)2 ] 7.0[Ni(H-BHO)2Cl2 3 8.0[Ni(BHM)2Cl2 3 13.
* This value is calculated on the basis that one chloride Ion has been replaced by methanol.
The low molar conductance values of the last three complexes leaves no doubt that these are non-electrolytes. One might have anticipated, however, that the conductance of the four coordinate complexes of nickel(ll) with blacetylhydrazoneoxime would be lower since it is formulated analogously to the dimethylglyoxime complex. The fact that it does not have a smaller conductivity may be associated with the difference In solubility In methanol. [Ni(DMG)2 ] is so slightly soluble in methanol that It is impossible to determine its conductance, with the equipment available, whereas the complex, [Ni(BHO)2 3, appears to be rather soluble.
The conductance of [NlCBOM^C^]* 109*8 ohm"*, appears to resemble more closely the two-to-one nonionic

complexes listed In Table 4, Section II, than those ofH-BHO and BHM. It appears that slightly more than onechloride has been replaced per formula weight by reaction
14with the solvent. Sharpe and Wakefield report that
•^A. G. Sharpe and D. B. Wakefield, loc. cit.
[Nl(H-DMG)2C12 J is intermediate in this respect, the solvation reaction (which produces ions) proceeding at a measurable rate in acetone.
2. Magnetic MeasurementsThe magnetic susceptibilities of three ligands
and four complexes have been determined in this series of compounds. They are listed in Tables 22 and 23*
TABLE 22MOLAR SUSCEPTIBILITIES OF LIGANDS AND ANIONS
*y 5Compound m X 10
H-DMG* - 1*8.96H-BHO - 4 6 . 4 6 t 3 * 7 5H-BOM - 4 7 . 9 2 ± 1 8 . 5 1
BHM - 6 7 * 6 4 t 8.60Chloride** - 23.4 - 1.3Iodide** - 50.6 ± 1.6Water** - 1 3 . 0
* Calculated using Pascal*s Constants.** P. W. Selwood, Magnetochemistry," Second Edition,
Interscience Publishers, Inc., New York, 1956, p. 36.

215TABLE 23
MOLAR SUSCEPTIBILITIES OF COMPLEXES
Compound "XM X 106 ef f
[Ni(DMG)2 3 --- 0[n i (b h o )2 3 - 215 0[Ni (H-DMG) 2C’l2 3* --- 3 .0 7
[Ni(H-BHO)2Cl2 3 3890 3 .1 2
[N1(H-B0M)2C12 3 3980 3 .1 8
[ni(bhm)2ci2 3 4170 3 .2 4
[Ni(H-BHO)3 3l2** 3100 2.37
* A. G. Sharpe and D. B. Wakefield, loc. clt.** The sample utilized in this measurement was
later found to be slightly impure.
The magnetic data indicate that the nonionic complex of nickel(ll) with H-BHO, [Ni(BH0)2 ], is spin-paired and planar (Just as the analogous H-DMG complex of nlckel(ll) is spin-paired), while the dlchloro, two-to- one complexes of all the ligands which were studied are spin-free and octahedral.
3* Ultraviolet and Visible SpectraThe ultraviolet spectra of the two ligands which
do not contain hydrazine functions (H-BOM and H-DMG) and the spin-free complexes of these two ligands ([Ni(H-B0M)2 Clg] and Ni(H-DMG)2Cl2 3) are characterized by a single

216strong absorption (Figures 4l, 42, 43 and 44). These absorptions lie between 227 nyx and 235 (Table 24).
In contrast, the spectrum of H-BHO, a molecule containing both an oxime and a hydrazone group, is characterized by two major absorptions, the lower frequency band occurring at 252 vcijU and the higher frequency band occurring as a shoulder at 241 m^ (Figure 4l)• The molar extinction coefficients of the two bands are roughly equal (11000 and 13000 respectively). Although the 259
mjx absorption of BHM does not actually have a well- defined shoulder on the high frequency side, there is a marked increase in broadness (Figure 44)• In the spectrum of the spin-free complex of BHM, [Ni(BHM)gClg], this characteristic Is not so obvious. The spectrum of the spin-free complex of H-BHO, [Ni(H-BHO)2 ^ 2 * re^lect;s an Interaction which appears to be quite different from that present in the analogous BHM complex, two absorptions occurring instead of one (Figures 42 and 44)• The stronger absorption Is at 244 tn/4 while the second, which is not well-resolved from the higher frequency absorption, occurs at 280 mJLA. . The spectra of the two spin-paired complexes, [Ni(DMQ)g] and [NifBHOjg] are rather unique by comparison with the spectra of the spin-free complexes of their respective ligands and, by comparison, to each other. Whereas the major absorptions in the spectra of

Perc
ent
Tro
nsm
itto
nce
217
D im e th y lg ly o x im e
B io c e ty lh y d ro z o n e o x im e20
4 0
6 0
8 0
K)02 2 9 2 5 3 2 7 7 3 0 1 3 2 5
W ave L ength in M ill im icrons
Figure 41. Ultraviolet Spectra of DimethyIglyoxlraeand Biacetylhydrazoneoxime•

Tro
nsm
itton
ce
218
-------------- [N i (H-0MG)2 CI2 ]I cm. celt, 3 x lO '9 M
------------[N i(D M G )tJ «I cm. cell, I x lO"9 M
-------------- [N i(H -B H O )2CI2 3I cm. cell, 2 x I0-9 M
.......... [Ni(BHO)jjI cm. cell, 2 x K T9 M
001% so lu tio n of 12N HCl in absolute ethanol was used as sol vent.
** Chloroform solution.
3 4 9 3 7 3
Wave Length in M illim icrons
Figure 42. Ultraviolet Spectra of the Nickel(ll)Complexes of Dimethylglyoxime andBiaeetylhydrazoneoxlme.

Tro
nsm
itto
nce
• [N i(O M G )e ]I cm . c e ll , 5 * K
• [NifH-DMOgCI*]i cm c e ll , 3 x l(
■ [N i (H - B H 0 ) f C lj ]I cm ce ll, I x 10
[N i( B H 0 )* J20
i-5■» C hloroform s o lu t io n
4 0
6 0
8 0
1006 6 06 0 04 2 0 5 4 03 6 0 4 8 0
Wove Length in Millimicrons
Figure 43. Visible Spectra of the Nickel(Il) Complexes of Dimethylglyoxime and Biacetylhydrazoneoxime.

Tro
nsm
itto
nce
220
B iocetylox imemethox i me
20[Ni {H -B 0M)z C(2 ]
i cm. cell, 2 x I0"9 M B iacetyl hydro zonemethoxime
4 0
6 0
8 0
3 49 373277 301 325
Wbve Length in Millimicrons
229 253
Figure 44. Ultraviolet Spectra of Biacetyloximemethoxime,Biacetylhydrazonemethoxime, [Ni(BHM)PCl0],and [Ni(H-B0M)2Cl2l. d

IABIi2*
ULTRAVIOLET AND VISBB AM M ONS OF N l(ll) COBP1EKS AND U 0 DS
Compound A r m A fmi A w a x A f m A £max
H-DMQ 227 16000 m m m aaa • •0 m m m ••• aaa
[NlfB-DNOljClj] 229 32000 ■ am m m m • art • rtrt rt rt • m m m M5 25
[Nl(DHO)j] ••• m u m ■ •• m m m1
262 8A00 337 A0O0 *25 1300
fi-BOM ••• u u u 235 16000 aaa ■ art • aa aaa • •a
[N1(H-B0H)2C 1 1 aaa u m u 235 32000 • •• aaa « o 10
H-BHO m m m m m m Oil 11000 252 13000 • a* m m m m m m
[NllN-BHOljCljj m m m m m m m 18000 280 97OO • •• • •• • aa aaa
[ll(BHO)j] 229 15000 m m m aa« • «• • art 337 3500 555 253
BHM • •a aaa m m m 259 15000 • aa ai« m m m m m m
[ N l W j C l j ] aaa aaa m m m •aa 259 31000 • aa •aa m m m aaa

222H-DMG and H-BOM and in the spectra of their spin-free com plexes occur at relatively high frequencies, in contrast, the strongest absorption in the spectrum of [Ni(DMG)2 3 occurs at 262 m jj, , a lower frequency than any other major absorption in this series. In the spectra of H-FHO and BHM, and of their spin-free complexes, the major absorption tends to remain at relatively low frequencies; however, in the spectrum of the spin-paired complex of H-BHO the major absorption occurs at 229 in/* , a shift of 15 m^u from the position of this absorption in the spin-free complex. These two-spin paired complexes do have one point of similarity in their spectra, a high intensity band at 337 m/x (the maxima are 4000 and 350° respectively).
It is Interesting to note that the extinction coefficient of the absorption band which Is characteristic of these ligands Is unaltered by complexing in a number of cases. For example, see Figure 44 which contains the spectra of two ligands and their spin-free nickel(ll) complexes.
Obviously, the absorptions which are most pronounced in these spectra must be associated with the pi electron system of the conjugated methine group. The fact that this band usually occurs at lower frequencies in the spectrum of the compounds which contain the

223hydrazone group may Indicate that the amine groups are partly responsible for their shift of position to lower frequencies. As might be predicted, the methyl ether derivative appears to be somewhat Intermediate in Its positions with respect to the oximes and hydrazones.
This entire picture is consistent with a greater Interaction or delocalization of electrons In the hydrazone type of ligand than in the oxime or methoxime type.It will be recalled that the spectrum of BdH has a single strong absorption near 258 mja . . That of the hydrazone- oxime, H-BHO, has a strong band at 252 m w. and a shoulder at 2h l mjuL , the indication being that replacement of one hydrazone group by an oxime function gives rise to two absorptions, one mainly associated with the oxime function and the second, with the hydrazone group. The displacement of these bands from the positions observed in the dioxime and the dihydrazone may be explained by assuming that replacement of a hydrazone group in BdH by an oxime group decreases the total Interaction possible within the molecule, giving rise to a shift to higher frequencies for the band which must be associated with the pi system encompassing the remaining hydrazone group. On the other hand, the absorption primarily associated with the oxime group, occurs in this system at a lower frequency than those In the dioxime as a result of enhanced conjugation
arising from the adjacent hydrazone group.

The visible spectra of these complexes reveal exactly the same type of behavior which was observed in the visible spectra of the hydrazone complexes described In Part II of this dissertation. The spectra of the two spin-paired complexes, [Nl(DMG)2 ) and lNi(BHO)2 3, have absorptions which appear to be charge transfer in nature and which occur at 425 m ju. and 555 , respectively(Figure 42). However, the intensities of these bands are much lower than the charge transfer bands in the spin- paired iron(ll) complexes (Tables 13 and 14). The spectrum of [Nl(DMG)2 ] exhibits another band at 480 mjn which appears to be too strong to be derived from a cl-d transition, but almost too weak to be a charge transfer absorption. The spectra of the spin-free complexes, [Ni(H-DMG)2 Cl2 ] and [Ni(H-BOM)2C12 ], exhibit absorptions which must, from molar extinction coefficients and position, involve d-d transitions ( ^ s 405 m yU , & s 25 and * 410,£, = 10, respectively, Figures 43 and 45- The spectrum of [Nl(H-BHO)2Clg3 also gives an indication of a d-d transition In this general region; however, the concentration of the complex was too low to detect the band center.
The positions of charge transfer bands in the visible region of the spectrum and the shifts of the ultraviolet absorption bands appear to be In agreement with the suggestion that ei bonding Is a significant

T ro
nsm
itto
nca
225
20 .2
4 0 -
6 0
8 0 "
7206 6 06 0 05 4 04 8 0
Wav* Length in Millimicrons
3 6 0 4 2 0
Figure 45. Visible Spectra of [Ni(H-BOM)2Cl2 ] andCNi(BHM)2Clg].

226factor in determining the nature of the bonds in the neutral, planar spin-paired complexes, [Ni(AA)23. No such charge transfer band is observed in the spectra of the octahedral complexes of the general formula [Ni(AA)2
X^]. The visible spectra of these complexes are characterized by the presence of absorptions ascribable to d-d transitions of the type usually associated with octahedral ions. In keeping with the conclusion that electronic interaction between the ligands and the metal ion are relatively uncomplicated, in these octahedral complexes, the ultraviolet spectra differ very slightly from those of the free ligands. Qualitatively, this distinction between the effect on the ultraviolet spectra resulting from the formation of planar ions and that associated with the formation of octahedral ions was pointed out by Mellor and Craig who observed a similar effect in a variety of systems.
D. Discussion
It was pointed out In the Introduction to this dissertation and again, in considerable detail, in the Introduction to Parts II and III, that the complexes which the periodic group VIII metal ions, iron(ll), cobalt(ll), and nickel(ll), commonly form with ligands of the aromatic heterocyclic diamine class are quite

different from the best characterized complexes formed by these metal ions with ligands of the o( -dioxime type. Part II of this dissertation described studies concerned with a variation in structural parameter of a series of ligands which has been shown to be closely related to the dipyridyl type ligands. The properties of the complexes formed by iron(ll), cobalt(Il), and nickel(ll) with biacetyldlhydrazone, (BdH), have been considered. These complexes might be expected to have properties intermediate between those of the o( -dioximes and those of aromatic heterocyclic diamines. Ligands of mixed type were prepared, e.g., PAH and PdAdH. These Investigations demonstrated that such hydrazone complexes more nearly resembled those of the dipyridyl class rather than those of the ©(-dioxime class despite the similarity of the NH2 part of the hydrazone to the OH group of the oxime.In this section (Part III), the variation In structures of the several ligands studied is so designed that these molecules are still more closely related to the dioxime class, specifically, an oxime or a methyloxime group is present In each case. Pfeiffer first investigated the effects associated with similar changes In structural parameters* He replaced one oxime function with groups such as an Imlme or methyl imine without significant alteration of the properties of the neutral two-to-one

223nickel(ll) complexes formed by the ligands. The present
Investigation is concerned with dimethylglyoxime (H-DMG,
structure IV), biacetyloxlmemethoxlme (H-BOM, structure
Xl), blacetylhydrazoneoxime (H-30M, structure XII), and
biacetylhydrasonemethoxime (BHM, structure XIIl)•
The basic differences in these ligands may be seen by comparing their structures. If, as seems to be the case, only the oxime hydrogen atom is acidic, then only H-DMG, H-BOM, and H-BHO contain ionizable protons.
The molar conductances, magnetic susceptibilities,and ultraviolet and visible spectra of the nickel(ll) compounds of these ligands have been determined and the results of these measurements have been utilized in the characterization of these substances. The infrared spectra have also been recorded and are contained in
CE
XI XII
X II I

229Appendix IV. It will be recalled that three general types of complexes have been characterized with respect to the dipyridyl and oC-dioxime complexes of iron(ll), cobalt(ll), and nickel(ll). They are:
(1) [n i (a a )3]++(2) [M1(AA)2X2 ](3) [Nl(AA) 2 3•
The dipyridyl complexes include types (l) and (2), while the -dioxime complexes embrace types (2) and (3)* In the desire to make these groups overlap through the formation of complexes of all three types with one or more ligands, an attempt was made to prepare the complex cation tris-(dimethylglyoxime)-nickel(ll). For this purpose, solvents were chosen which would tend to inhibit ionization of a proton from one of the oxime groups, e.g., glacial acetic acid, due to the acid character of the solvent or which would be relatively stable toward reaction with any other acid included in the system for the purpose of inhibiting the ionization of the oxime function, e.g., ethanol. Different starting materials were used over a range of concentrations; however, all of these attempts resulted in the formation of complexes of types (2) and (3) or in a complex of the type [Ni(H-DMG) Clg]. Attempts to prepare the analogous complexes of H-BOM and BHM were not successful; however, this may be attributed to greater steric requirements of the molecules.

230It is significant, however, that the ligand,
biacetylhydrazoneoxime (H-BHO), which would be expected to be intermediate in character between H-DMG and BdH, does form complexes of all three types. It is also significant that the three-to-one complex of H-BHO is obtained in the pure state with much difficulty.
The complexes which are of immediate concern, type (2), are observed to undergo different reactions with water than those in which a dipyridyl type ligand is present. In the second case (dipyridyl type), the complexes [Ni(AA)2X2 ] disproportionate forming [Ni(AA)^]+f whereas those in which the ligand Is of the -dioxime type lose a proton and revent to type (3) complexes [Ni(AA)2 J. The reaction of the H-BHO complex, which Is Intermediate in type, may be controlled by adjusting the pH of the solution.
Low
[ N i ( B H O ) 2 3
The conductance (in ethanol) and magnetic moment data indicate that the -dioxime complexes of type (2), [NifAA^X^J, are octahedral in structure, as are the analogous complexes with ligands of the dipyridyl class. There is a difference, however, with respect to the conductance data. The type (2) complexes of the hydrazones

undergo rapid solvation in ethanol, thus giving rise to conductivities very near those predicted for uni-univalent electrolytes, the complexes [N i (H-BOM) ] and[Ni(H-BHO)gClg3 undergo comparatively little solvation in the time required for mixing of the solutions and measurement (Table 2l). The rapid solvation reactions of [Ni(BdH)2Cl2 ] and [Ni(BHM)2C12 J may be attributed to repulsions between the methyl and amine groups in a trans- dlchloro configuration of the octahedral complex. This stress in the system will be relieved if the chloride ions occupy £is-positions; however, a els arrangement of two negative groups (Cl") is electrostatically unfavorable.If the system is labile, replacement of a chloride ion by a solvent molecule occurs, which then lead to the formation of a more stable comple having a cis-structure in which only one negatively charged group is present.

IV. SUMMARY
Iron(ll), cobalt(ll), and nickel(ll) are known to form well-characterized complexes with ligands of the aromatic heterocyclic diamine type, e.g., 2,2‘bipyridine and o-phenanthroline, the oC-diimine type, e.g., biacetylbis- methylimine, and those of mixed type, e.g., pyridinalmethy- limine. The six coordinate iron(ll) complexes of these ligands are invariably spin-paired while the analogous co- balt(ll) and nickel(ll) complexes are spin-free. In general, it may be stated that the physical and chemical properties of these complexes present a very logical and consistent picture. This might have been anticipated, since all of these ligand molecules contain the same conjugated methine group; therefore, the same chelate ring is present in all of the complexes formed by these ligand molecules.If one considers the properties of the complexes formed by these dipositive metals with ligands of the -dioxime type, e.g., dimethylglyoxime, which also contains this same conjugated methine structural unit, the self consistent picture tends to break down. Iron(Il) has been found to form a four coordinate complex with dimethylglyoxime, while cobalt(ll) and nickel(ll) prefer to form neutral, spin-paired, four coordinate complexes with dimethylglyoxime. The main difference between the ligands represented by dimethylglyoxime, on the one hand, and dipyridyl, on the other, is that the
232

233latter type have an alkyl group or part of an aryl group attached to the unsaturated nitrogen atom while the former have a hydroxyl group attached to the unsaturated nitrogen atom.
Biacetyldihydrazone, pyridinalhydrazone, and pyrid- indialdlhydrazone which also contain the conjugated methine structural unit, were studied during the course of this investigation in order to determine which, of the two general types of ligands, they more closely resembled (dipyridyl or dimethylglyoxime type)• It was anticipated that the NHg group, characteristic of these hydrazone ligands, would resemble, more closely, the OH group, characteristic of the -dioximes, than the alkyl group of the aromatic heterocyclic diamine and -dilmine ligands. However, the complexes which were prepared, using Iron(ll), cobalt(ll), and nickel(ll), were found to resemble the complexes formed by these metals with the dipyridyl class of ligands rather than the complexes of these metals with the dimethylglyoxime class of ligands. Conductance data indicate that all of the complexes formed are six coordinate.
The magnetic data indicate that the six coordinate iron(ll) complexes are spin-paired. One iron(ll) complex, tPe(BdH)gJig* was resolved into optically active isomers which racemized more slowly than either the analogous dipyridyl or o-phenanthroline complex, indicating that the biacetyldihydrazone complex may be the most stable. Infrared studies of the iron(ll) complexes, which were formed

with these hydrazone ligands, indicate that the interaction between the unsaturated nitrogen (donor) atoms and the metal ion is greatest in the iron(ll) complexes. This increased interaction in the iron(ll) complexes is not inconsistent with the postulate of multiple bonding between donor atom and metal atom. The visible and ultraviolet spectra of the iron(ll) complexes are entirely consistent with this suggestion. It has been suggested that multiple bonding is responsible for the stability of the analogous dipyridyl and o-phenanthroline complexes of iron(ll)•
All of the cobalt(ll) complexes of the hydrazone ligands were determined to be highly paramagnetic and spin- free, with the exception of LCo(PdAdH)2 ]l2, which has a magnetic moment of 2-9 B.M., Intermediate between the moment expected for a spin-paired and a spin-free cobalt(ll) complex. This seemingly anomalous magnetic moment can be explained on the basis of an equilibrium mixture of the two spin states. The spectral data which were obtained also support this conclusion.
Nickel(ll) forms spin-free six coordinate complexes with the hydrazone ligands. There were formed, also, six coordinate complexes of the type [Ni(AA)2X g 3 * which react with water to form the six coordinate (trls-bidentate) complexes, discussed above.
In a continued attempt to find ligands which were
Intermediate between the ^-dioximes, on the one hand, and

235the aromatic heterocyclic diamines and the o( -dilmines, on the other, three additional ligands were studied. They were biacetylhydrazoneoxime, biacetylhydrazonemethoxime, and biacetyloximemethoxirae. These ligands are intermediate between the oC-dioxime type and the hydrazone type (which were shown to be similar to the dipyridyl type of ligand)•
Only the nickel(II) complexes of these last three ligands were investigated, since, of the three metals used In the first part of this investigation, only nickel(ll) forms we11-characterized complexes with both types of ligands. With these two classes of ligands, nickel(ll) was found to form three types of complexes, (l) rNi(AA)^] ,(2) fNi(AA)2X g ], and (3) fNi(AA)2 ]. The dipyridyl class of ligands form types (l) and (2), the latter dispropor- tlonating in water to yield type (1) complexes, whereas, the dimethylglyoxime class of ligands form types (2) and(3). In this case, type (2) complexes react with water to form type (3) complexes. Of the three ligands which ap peared to be Intermediate between the c^-dloxlmes and the hydrazones, only biacetylhydrazoneoxime formed complexes of all three types. This was accomplished by varying the reaction conditions to either promote or inhibit Ionization of a proton from the oxime group. The experimental evidence indicates that [Ni(H-BH0 )2ClgJ may be reacted in aqueous solution to form either the type (l) or type (3) complex. Therefore, biacetylhydrazoneoxime, alone, appears to be

236truly Intermediate between ligands of the dipyridyl type and the dimethylglyoxime type.
It is concluded from this study that the complexes of the hydrazone containing ligands more closely resemble the dipyridyl type of complex than the dimethylglyoxime type of complex for two reasons, (l) the inability of the NHg group to lose a proton in aqueous solution thus forming a neutral complex, and (2) the apparent enhancement of the basic character of the hydrazone containing ligand due to the presence of the NH^ group.

APPENDIX I237
MAGNETIC SUSCEPTIBILITY MEASUREMENTS
The molar susceptibilities of the ligand molecules were determined using water as the standard while those of the complexes were determined using ferrous ammonium sulfate 6-hydrate as the standard* The gram susceptibilities of these standards are given by Selwood * -0*720;
A pj s = T + 1 ) • The samples were placed in a
■ P. W. Selwood, loc * cit *
pyrax sample tube (approximately 5 mm* inside diameter) which had been marked, after calibration, to denote a volume of 3 ml* (this corresponded to approximately 17 cm. of tube length)• All measurements were made at 300 K. - 2 and at a current of 8 amperes*
Provided that the sample extends from a region in which the magnetic field is homogeneous to one in which the field is negligible, the gram susceptibility of a sample is given by the expression
2gA = W TTp
(where £ is the gravitationat constant, and auj is the apparent change in weight of the Bample when placed in a magnetic field, H is the field strength, A is the cross

233sectional area of the sample, and is the apparent density) . Stratton^ has determined that the magnetic field
^W. J. Stratton, loc. cit.
is homogeneous in a region extending from the center of the pole pieces to a point approximately 0 .5 cm. in any direction and that the field becomes negligible beyond 8 cm (see Figures 46 and 47)• The restrictions imposed upon the placement and length of the sample have been met in these determinations.
Since the field strength and sample tube positioning are constant throughout this series of measurements, it fol lows that
Since the gram susceptibility of each standard is known and since the apparent density and change in weight can be measured, the equation may be simplified by making the substitution
Therefore, It is seen that the gram susceptibility of the complex or ligand is given by the expression
sample standardsample
~)C standard x fi standardstandard
sample$ xA sample
P sample
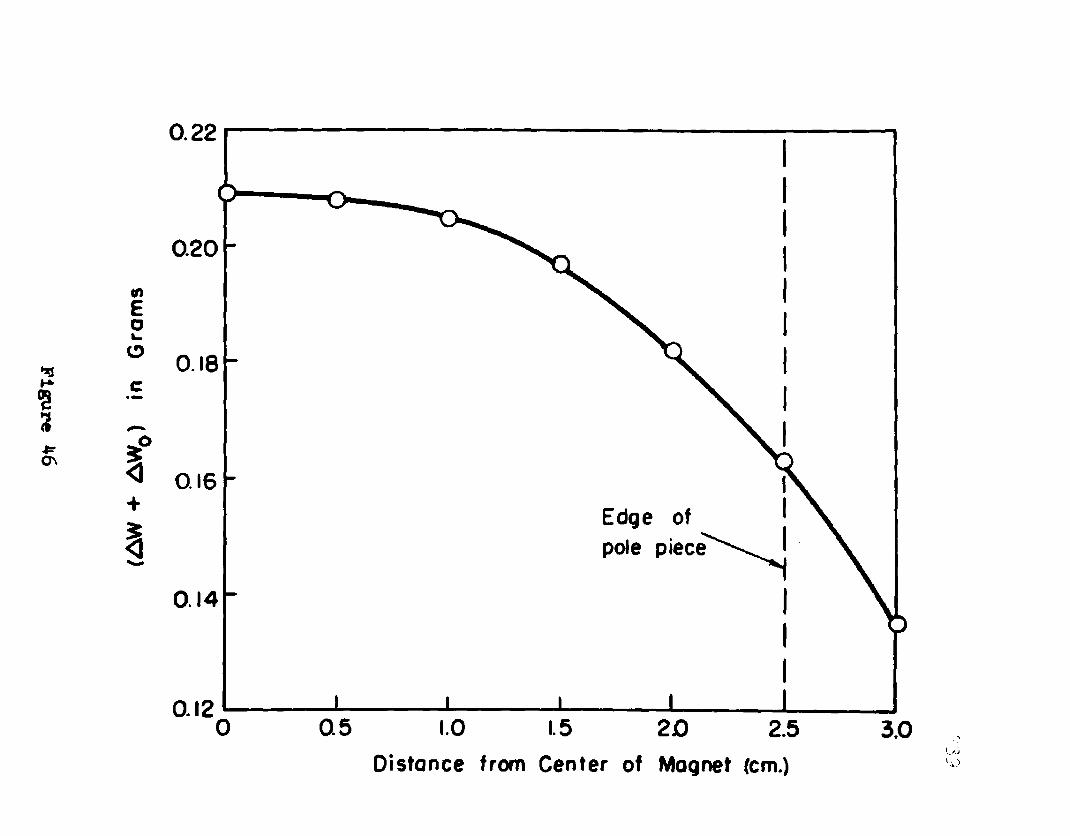
0.22
0.20 “
(A
Eot.o
s
*=■o\
0.10 -
$%
Q I 6 -
0.14 ‘
Edge of pole piece
0.12 1 1 1Q5 1.0 1.5 20 2.5
D istance from Center of Magnet (cm.)3.0
VO

lif
9JLT
&T&
(AW
+
AW
0)
in G
ram
s
0 2 0 “
016
012
008
0.04
t.
L ength of S am p le (cm.)

241The molar susceptibility, " X - M is given by the expression
7 , M = 7 x M (the molecular weight)
5 represents the average value of several determinations of Q under the same conditions.
TABLE 25
CALIBRATION OF FIE ID WITH WATER
Apparent Density A w Q x 106
0.9963 -0.0046 155.9O .9962 -0.0047 1 5 2 .6
0.9971 -0.0047 152.7Average 153-7 - 1.4

242
TABLE 26CALIBRATION OF FIEID WITH FERROUS AMMONIUM
SULFATE 6 -HYDRATE
Apparent Density ^ w Q x 106
0.9964 0.1865 1 68 .3
I.O35O 0.1937 1 6 8 .3
0.9378 0.1755 168.31 .0 1 1 0 0.1972 161.5O .9885 O .1825 1 7 0 .6
O .9883 O .1823 1 7 0 .8, + Average 167*9 - 2

MAGNETIC SUSCEPTIBILITYTABLE 27 DATA FOR SOLID SAMPLES OP LIGANDS *
Compound r A w y * 106M
1 ( x 106 M (ave)
Biacetyldihydrazone 0 .6 3 1 0 -0 .0 0 2 0 -48.70.6843 -0 .0 0 2 1 -47.2O .4 0 5 9 -0 .0 0 1 6 -6 9 .20.4045 -0.0027 -117.1 -70.6 - 23
PyridlndiaIdihydrazone 0 .3 6 0 7 -0.0019 -132.2O .3683 -0.0014 -95.3 -113*S - 19
PyrIdina1-p-to ly limine 0.4410 -0 .0 0 0 7 -47.9 +O .5 6 6 1 -0.0014 -74.6 -6 1 .3 - 13
* 5 = -t-53 x 10"6
ro■*=■OJ

244TABLE 28
MAGNETIC SUSCEPTIBILITY DATA FOR SOLID SAMPLESOF COMPLEXES *
JQ y xio6 y xl0bCompound / ^ w A M A m (ave)
[Fe(BdH)3 3l2 0 .7 2 0 80.6647
-0.0014-0.0014
-213-231 -2 2 2
[Co(BdH)2Cl2 3 0.53490 .3 6 6 9
0 .0 8 6 6O.O598
97429806 9774
tCo(BdH)3 ]l2 0.78840 .8 1 1 6
0.04950.0511
69136932 6923
[Ni(BdH) 2C12 ] 0.52460.4131O .5 1 1 4
O.O354O.O2590 .0 3 1 6
405937693715 3848
[Ni(BdH)3 JCl2 0 .3 9 8 1O .4 3 6 3
0.01840 .0 2 0 3
36663691 3679
[Fe(PAH) 3 3l2 0 .5 7 6 50.5242-0 .0 0 0 7-0.0007
-137-151 -144
[c o (p a h )3 ]i 2 O.6 5 5OO .5 5 8 2
0.04860.0414
84328425 8429
iNi(PAH)3 3l2 0.64090.6625
0.02100 .0 2 1 8
37203740 3730
[Fe(PdAdH)2 3l2 O .5 1 3 40 .5 2 7 5
-0 .0 0 1 0-0.0008
-21-1 6 -19
[Co(FdAdH)2 3l2 0.4913O.51OI 0.01490.0147
32563093 3175
Normal-[Ni(PdAdH)2 3l2 0 .5 0 6 10.5357
0.01910.0199 4051
3990 4021[Fe(PAT)2Clg 3•2HgO O .2 5 2 3
0.26570.2688
0 .0 3 0 7O.O336O.O334
113471179811586 11577

245TABLE 28 (Contd.)
/O yv Y xlO° y xlO6 Compound ( /- M /-M (ave)
(Fe(PAT)3]l2 *2H20 0.37930.4431
-0.0006-0,0014
-248-496 -372
[Co (PAT)^]I2 *3H20 0.48360.4970
0.02970.0329
984010610 10225
[n i (p a t)2c i2 3*2h 2o 0.40340.4610
0.01910.0218
44164410 4413
[Nl(PAT)2(CIO^)2 3 *H20 0.60180.6465
0 .0 2 2 30 .0 2 3 2
41594028 4094tNl(PAT)33l2 *2H20 0.5247
0 .5 2 1 10.01360.0133
40774020 4028
* 5 = 167*9 x lO-6

APPENDIX II246
MAGNETIC SUSCEPTIBILITY MEASUREMENTS
The molar susceptibilities of the complexes contained herein were also determined by the Gouy method (as were those contained in Appendix I)• The balance used In this series of determinations was an Ainsworth semi-micro balance of 0 .0 0 5 mg. sensitivity rather than the balance used In the first series of determinations, which was a Christian-Becker analytical balance of 0.1 mg sensitivity. In addition, the field strength used for this series of determinations was slightly smaller than that used previously, due to a small increase in gap-width between the pole pieces of the magnet.
TABLE 29CALIBRATION OP MAGNETIC FIEID WITH PERROUS
AMMONIUM SULFATE 6-HYDRATE
Apparent Density ^ w Q x 106
0.92662 0.17077 171.2481.00178 0.18456 171.3060.99832 O .1 7998 175.0581.04634 0.19135 172.560
Average 172.543 - 1 .2 6 6

247
TABLE 30MAGNETIC SUSCEPTIBILITY DATA FOR SOLID
SAMPLES OF COMPLEXES*
Compound P A w X M x l ° 6
[Fe(BdH)3 ]l2 O.703O2 -0.00135 -2 1 6 .1
[Fe(PAH)3]l2 0.68742 -0.00027 -45*6[Fe(PdAdH)2 ]l2 O .5 5 1 2 5 -O.OOO74 -147*3[Fe(PAT)3]l2 «2H20 0.36540 -O.OOO75 -331.0[Co(PdAdH)2 Jl2 0.55357 0.01752 3490Normal-[Ni(PdAdH)2 ]l2 0.45377 0.01795 4361Iso-[Ni(PdAdH)2 ]l2 0.52239
0.312980.017260.01268
Average364240413842
* 5 a 1 7 2 .5 x 10“6

APPENDIX III
TEMPERATURE DEPENDENCE OP MAGNETIC SUSCEPTIBILITY
The molar susceptibilities of the complexes contained herein were also determined by the Guoy method (as were those contained In Appendixes I and II)• The equipment which was described in Appendix II was used in this series of determinations.
TABLE 31TEMPERATURE DEPENDENCE OF THE MOLAR SUSCEPTIBILITIES OF
SOME SPIN-PAIRED IRON(II) COMPLEXES*
Compound/°
A w v “ 6 T. °K.
[Fe(BdH)o]lo 0 .7 0 3 0 2 -O.OO135 -2 1 6 .0 6 300J c -0.00127 -2 0 3 .2 8 80
[Fe(PAH) J l 0 0.68742 -0.00027 -45*64 300-0 .0 0 0 0 3 -5 .0 5 2320 .0 0 0 1 0 1 6 .9 0 2060 .0 0 0 1 7 28.74 1830 .0 0062 104.74 80
[Fe(PdAdH)0 Jl0 0.55125 -0 .0 0074 -147*31 300C C. -0 .0 0 0 6 3 -125.43 232-0 .0 0 0 5 0 -99.54 206-0 .0 0 0 3 8 -75.63 183-0.00046 -91.59 80
[Fe(PAT)3 ]l2 *2H20 O .36 5*1-0 -0.00075 -330.98 300-0.00077 -339.77 232-O.OOO56 -2 4 7 .0 7 186-0.00053 -233*89 80
* 5 = 172.5 X 10"6

TABLE 32TEMPERATURE DEPENDENCE OF THE MOLAR SUSCEPTIBILITY
OF [Co (PdAdH)2 3l2*
Compound jO A w ^ x 10^ T.°K.
[Co(PdAdH)2 ]l2 O .55357 0.020140.017700.017520 .0 15010.014740.014730.02633
4234372134902990293629345245
37330030023220618380
* 5 = 1 7 2 .5 x 10"6
TABLE 33TEMPERATURE DEPENDENCE OF THE
THE NICKEL(II) COMPLEXES OFMOLAR SUS CE PTIBI LI TIES OF PYRIDINDIAIDIHYDRAZONE
Compound ^ x 10° 0T. K.
Normal-[Ni(PdAdH)2 ]l2 0.^5377 0.017950.02234
0.024230 .0 2 5 3 60.03380
46875753621264878537
30023220619180
Iso-[Nl(PdAdH)2 ]l2 0.52239 0.017260.021370.023160.024830.03315
36424510488752406995
30023220619980
* 5 = 172.5 x 10“6

250APPENDIX IV
The Infrared spectra of the nlckel(ll) complexes which were prepared during the course of this investigation with the ligands dimethylglyoxime, biacetylhydrazone- oxlme, blacetyloximemethoxlme and biacetylhydrazonemethoxime have been determined using the techniques discussed in Part II of this dissertation (Figures 48, 49, 50, 51, and 52)• The major peaks which are characteristic of these spectra, are listed in Tables 34, 35* 36, 37, and 3 8; however, the assignments for these absorptions have not been made* In general, the infrared spectra of these complexes appear to be somewhat more complex than the spectra of the analogous biacetyldihydrazone complexes, particularly in the double bond region (near 1600 cm”*). In order to make any definite assignments In this region, the complexes which contain the hydrazone group should be deuterated, thereby removing the NHg deformation which was observed to occur near 1640 cm"* in the complexes of biacetyldihydrazone. Also, from considerations of the number of peaks In this region of the spectra, It appears that there may be both an asymmetric and a symmetric C=N stretching vibration In the 15OO-165O region of the spectra. This was not observed In the spectra of the complexes which were discussed in Part II of this dissertation, with the possible exception of the spin-paired iron(ll) complexes

251
too
•o
•o
to -Oimi
*00IOOOtoo
to
•o -J V ^ V
to [NMMWtg] INKH-0M%C%]
I I L8000-Lj. 1 X
4000 •00
Figure 48. Infrared Spectra of Dlmethylglyoxime,[N1(H-DMG)2CI2 ], and [Nl(DMG)2 J.

ioo r
8 0
6 0
*- 4 0
7 0 08009001000ooo2000
8 0
60
20B iat«tylh>6fw an*o»im «
7 0 08009 0 010008 0 020004000 3000
Figure 49. Infrared Spectra of 2,3-butanedione-2-oxime and Biacetylhydrazoneoxlme.

Transmittance
LOO
80r
6 0
4 0
20 tW ilH-BHOJ.CI,--------- [Ni<BHO)( ]
TOOaoo900KX30190020004000 3000UMV* num ber* , cm ^
100
80
60 f\40-
20
TOO8009 0 01000<90020004000 3000Wow* num ber*, c m '1
Figure 50. Infrared Spectra of the Nickel(ll) Complexesof Biacetylhydrazoneoxlme.

25^
too
•0
40
to0
4000 aooo ._L 4- •000 900 TOO
1001
«o>
60-
t-
20.
4000 3000 2000 900 900 700
too
00
I40
£0
8000 1500 TOO
Figure 51* Infrared Spectra of 2,3-butanedIone-2-methoxime,Biacetyloximemethoxime, and [NlfH-BOMjgC^].

255
100
ao8I 60 ru
Biecetylhydreio"W'Wlh*ime
TOO600900COO190020004000 3000 I, cm '
100
00
40
20TOOBOO2000
Figure 52. Infrared Spectra of Biacetylhydrazonemethoximeand [Ni(BHM)2Cl2].

256
TABLE 34INFRARED ABSORPTION BANDS (cm-1) FOR DIMETHYLGLYOXIME
AND ITS NICKEL(II) COMPLEXES
Dimethylglyoxime [Ni(DMG) [N1(H-DMG)2C12
3226 s (b) 3401 w 3135 vs1420 s (b) 1572 s 1669 m1350 s 1408 m 1403 s1140 m 1368 m 1372 s (c^)971 vs (b) 1236 s 1353 3 (d2)873 vs (b) 1100 s 1282 s741 m (vb) 988 m 1053 vs (b)705 m (b) 893 w M 947 m (V '
744 w (b) 807 m702 s (b)

257
TABLE 35INFRARED ABSORPTION BANDS (cm"1) FOR 2,3-BUTANEDIONE-2-
OXIME AND BIACETYLHYDRAZONEOXIME
2,3-butanedione-2-oxime Biac e tylhydrazoneo xlme
3290 s (d) 3401 s1675 vs 322a 3
1447 s (b) 1645 S
1366 s 1613 S
1312 m 1577 S
1133 s (b) 1435 S
1020 vs (d) 1368 3
983 m 1151 S
936 m 1121 S
799 m (b) 1007 vs (b)771 m 978 s
919 vs692 m (b)

258
TABLE 36INFRARED ABSORPTION BANDS (cm-1) FOR [Ni(H-BHO)pClP ],
[NI(BH0)2 3, AND LNi(H-BHO)3 ]l2*
[n i (h -b h o )2c i 2 ] [n i (b h o )2 3 [n i (h -b h o )3 ]i2*
3401 vs 3226 vs 3250 vs (b)3322 3 2703 s3215 s I656 m1597 s 1567 s 1576 s1546 s 1484 s 1536 m1449 3 1379 m (b) 1390 m (b)1374 S 1242 vs 1342 s
1335 3 1170 vs 1230 m1206 m 1103 s 1115 s1127 m 999 m (b) 1094 m
1053 vs 906 m (b) 1015 vs (b)952 m 833 m 960 vs (b)798 m 728 3 930 3
692 m 772 3
755 s (b)i 660 3
* Sample was Impure.

259
TABLE 37INFRARED SPECTRA OF 2,3-BUTANEDIONE-2-METHOXIME. BIACETYIRYDRAZONEMETHOXIME, AND [Nl(H-BOM)2C12 J
2,3-butanedione-2- Biacetylhydrazone-methoxime methoxlme [Ni(H-BOM)
2849 m 3185 vs (b) 3145 m1642 vw
1701 vs 1580 w 2907 vs1610 m 1422 vs (b) I007 vw1425 m 1362 vs 1548 vw1359 3 1284 vs 1460 vs1300 m lOlo vs (b) 1377 s1119 s 1068 s1047 vs (b) 973 vs (b) 921 m917 s (b) 885 vs (b) 812 m780 m 735 m (b)
690 m

260
TABLE 38INFRARED SPECTRA OF BIACETYLHYDRAZONEMETHOXIME
AND [Ni(BHM)2Cl2 ]
Biacetylhydrazonemethoxime [Ni(BHM)2Cl2 3
3 3 7 8 s 3311 s (nr)3236 s 3236 s (nr)2 9 4 1 s 3175 s (nr)2 8 2 5 m 1593 s1645 s 1529 m1587 s 1437 m (b)1462 s (dj) 1370 m1441 (sh » <*2 ) 1205 m1362 vs 1116 m1139 s 1050 vs (b)1114 s 912 m1178 vs (b) 795 m958 m 680 w904 vs796 vs (b)744 vs (b)670 m

with ligands containing the pyridine nucleus. The absence of any well-characterized peaks in the double bond region of the spectrum of dimethylglyoxime presents an additional complexing factor. The C-CH^ deformations which are present in these spectra are observed to lie in the region 1462 - 1350 cm"* which is in agreement with the spectra of the biacetyldihydrazone complexes (1464 - 1353 cm"*}.

2o2AUTOBIOGRAPHY
I, Robert Carl Stoufer, was born in Ashland, Ohio,
November 3, 1930* I received my secondary school educa
tion at Ashland High School, and my undergraduate training
at Otterbein College, which granted me the Bachelor of
Arts and Bachelor of Science degrees in 1952. I served
in the United States Army from 1952 to 1955* I enrolled
at The Ohio State University in 1955* where I studied in
the Department of Chemistry. I completed the requirements
for the degree Doctor of Philosophy under the direction
of Dr. Dary]eH. Busch, Professor of Inorganic Chemistry.




















