
T he functional role of the JAK-STAT pathway in post-infarction remodeling
10
Cardiovascular Research 57 (2003) 129–138 www.elsevier.com / locate / cardiores The functional role of the JAK–STAT pathway in post-infarction remodeling a a,c b a c Hala El-Adawi , Lili Deng , Anthony Tramontano , Steven Smith , Eduardo Mascareno , a b a,b, * Kalyan Ganguly , Ricardo Castillo , Nabil El-Sherif a New York Harbor VA Healthcare System, Brooklyn Campus, Brooklyn, NY 11209, USA b Department of Medicine, State University of New York, Downstate Medical Center, Box 1199, 450 Clarkson Avenue, Brooklyn, NY 11203, USA c Department of Anatomy and Cell Biology, State University of New York, Downstate Medical Center, Brooklyn, NY 11203, USA Received 18 April 2002; accepted 19 July 2002 Abstract Objectives: Recently, the Janus kinase / signal transducer and activator of transcription (JAK–STAT) signaling pathway was found to be prominently associated with activation of the autocrine loop of the heart tissue-localized renin angiotensin system (RAS). We investigated if the JAK–STAT pathway is activated in the post-myocardial infarction (MI) non-ischemic myocardium (NIM), destined to undergo remodeling and whether blockade of the pathway in vivo can modify early post-MI remodeling. Methods: We investigated the time course of tyrosine phosphorylation of JAK–STAT and gp130 proteins in the NIM of post-MI rat heart as well as the binding activity of STAT proteins to the St-domain of the angiotensinogen gene promoter. We further compared the effects of in vivo blockade of RAS by the AT receptor (AT R) blocker losartan with the in vivo blockade of JAK–STAT pathway by the specific JAK2 blocker tyrphostin 1 1 AG490 on certain aspects of early post-MI remodeling. Results: We showed that JAK2, STATs 1, 3, 5a and 6 and gp130 proteins are tyrosine phosphorylated as early as 5–30 min post-MI and that STATs 1, 3, and 5a remain activated up to 7 days post-MI. Gel mobility shift assay showed a strong binding activity of STAT proteins to the St-domain of angiotensinogen gene promoter in 1-day post-MI NIM. The binding was significantly reduced in rat hearts previously treated with losartan or tyrphostin AG490. Supershift experiments identified STATs 3 and 5a as specifically interacting with the St-domain. Both AT R and JAK2 blockade resulted in significant amelioration of the 1 increase of protein phosphatase 1 activity and decrease in basal level of p16-phospholamban that may underlie early diastolic dysfunction, as well as partial amelioration of early downregulation of Kv4.2 gene expression that may underlie increased arrhythmogenicity of 3-day post-MI heart. On the other hand, while blockade of AT R significantly ameliorated apoptotic changes in 1-day post-MI border zone, 1 blockade of JAK2 increased apoptosis. Conclusions: The study provides compelling evidence in favor of the linkage of the JAK–STAT pathway with the angiotensin II autocrine loop and uncovers a mechanism by which selective activation of a set of STAT proteins underlies mobilization of the gene activation program intrinsic to post-MI remodeling. It also suggests that drugs that inhibit JAK–STAT phosphorylation may provide a new approach to modify post-MI remodeling. This needs to be confirmed in long term in vivo studies in the post-MI heart. 2002 European Society of Cardiology. Published by Elsevier Science B.V. All rights reserved. Keywords: Apoptosis; Infarction; Remodeling; Renin angiotensin system; Signal transduction 1. Introduction post-MI remodeling [1,2]. More recently, the Janus kinase / signal transducer and activator of transcription (JAK– A plethora of experimental and clinical data show that STAT) signaling pathway was found to be prominently the renin-angiotensin system (RAS) plays a major role in associated with activation of the autocrine loop of the heart tissue-localized RAS [3]. Angiotensin II (ANGII) uses a signal pathway in cardiac myocytes in which the promoter of the gene encoding its prohormone, angiotensinogen *Corresponding author. Tel.: 11-718-270-4147; fax: 11-718-630- 3740. E-mail address: [email protected] (N. El-Sherif). Time for primary review 26 days. 0008-6363 / 02 / $ – see front matter 2002 European Society of Cardiology. Published by Elsevier Science B.V. All rights reserved. PII: S0008-6363(02)00614-4 by guest on August 3, 2016 Downloaded from
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of T he functional role of the JAK-STAT pathway in post-infarction remodeling

Cardiovascular Research 57 (2003) 129–138www.elsevier.com/ locate/cardiores
T he functional role of the JAK–STAT pathway in post-infarctionremodeling
a a,c b a cHala El-Adawi , Lili Deng , Anthony Tramontano , Steven Smith , Eduardo Mascareno ,a b a,b,*Kalyan Ganguly , Ricardo Castillo , Nabil El-Sherif
aNew York Harbor VA Healthcare System, Brooklyn Campus, Brooklyn, NY 11209,USAbDepartment of Medicine, State University of New York, Downstate Medical Center, Box 1199, 450Clarkson Avenue, Brooklyn, NY 11203,USA
cDepartment of Anatomy and Cell Biology, State University of New York, Downstate Medical Center, Brooklyn, NY 11203,USA
Received 18 April 2002; accepted 19 July 2002
Abstract
Objectives: Recently, the Janus kinase/signal transducer and activator of transcription (JAK–STAT) signaling pathway was found tobe prominently associated with activation of the autocrine loop of the heart tissue-localized renin angiotensin system (RAS). Weinvestigated if the JAK–STAT pathway is activated in the post-myocardial infarction (MI) non-ischemic myocardium (NIM), destined toundergo remodeling and whether blockade of the pathway in vivo can modify early post-MI remodeling.Methods: We investigated thetime course of tyrosine phosphorylation of JAK–STAT and gp130 proteins in the NIM of post-MI rat heart as well as the binding activityof STAT proteins to the St-domain of the angiotensinogen gene promoter. We further compared the effects of in vivo blockade of RAS bythe AT receptor (AT R) blocker losartan with the in vivo blockade of JAK–STAT pathway by the specific JAK2 blocker tyrphostin1 1
AG490 on certain aspects of early post-MI remodeling.Results: We showed that JAK2, STATs 1, 3, 5a and 6 and gp130 proteins aretyrosine phosphorylated as early as 5–30 min post-MI and that STATs 1, 3, and 5a remain activated up to 7 days post-MI. Gel mobilityshift assay showed a strong binding activity of STAT proteins to the St-domain of angiotensinogen gene promoter in 1-day post-MI NIM.The binding was significantly reduced in rat hearts previously treated with losartan or tyrphostin AG490. Supershift experiments identifiedSTATs 3 and 5a as specifically interacting with the St-domain. Both AT R and JAK2 blockade resulted in significant amelioration of the1
increase of protein phosphatase 1 activity and decrease in basal level of p16-phospholamban that may underlie early diastolic dysfunction,as well as partial amelioration of early downregulation of Kv4.2 gene expression that may underlie increased arrhythmogenicity of 3-daypost-MI heart. On the other hand, while blockade of AT R significantly ameliorated apoptotic changes in 1-day post-MI border zone,1
blockade of JAK2 increased apoptosis.Conclusions: The study provides compelling evidence in favor of the linkage of the JAK–STATpathway with the angiotensin II autocrine loop and uncovers a mechanism by which selective activation of a set of STAT proteinsunderlies mobilization of the gene activation program intrinsic to post-MI remodeling. It also suggests that drugs that inhibit JAK–STATphosphorylation may provide a new approach to modify post-MI remodeling. This needs to be confirmed in long term in vivo studies inthe post-MI heart. 2002 European Society of Cardiology. Published by Elsevier Science B.V. All rights reserved.
Keywords: Apoptosis; Infarction; Remodeling; Renin angiotensin system; Signal transduction
1 . Introduction post-MI remodeling [1,2]. More recently, the Janus kinase/signal transducer and activator of transcription (JAK–
A plethora of experimental and clinical data show that STAT) signaling pathway was found to be prominentlythe renin-angiotensin system (RAS) plays a major role in associated with activation of the autocrine loop of the heart
tissue-localized RAS [3]. Angiotensin II (ANGII) uses asignal pathway in cardiac myocytes in which the promoterof the gene encoding its prohormone, angiotensinogen*Corresponding author. Tel.:11-718-270-4147; fax:11-718-630-
3740.E-mail address: [email protected](N. El-Sherif). Time for primary review 26 days.
0008-6363/02/$ – see front matter 2002 European Society of Cardiology. Published by Elsevier Science B.V. All rights reserved.PI I : S0008-6363( 02 )00614-4
by guest on August 3, 2016
Dow
nloaded from

130 H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138
(ANG), serves as the target site for the STAT proteins. The inhibitor tyrphostin AG490 was administered intraperitone-JAK–STAT pathway is also a major signal transduction ally in a total dose of 5 mg/kg/day, starting 2 h prior topathway of the cytokine superfamily through the gp130- LAD ligation [11].dependent pathway [4]. Recently, the JAK–STAT pathwaywas shown to be activated in ischemic myocardium [5–7].However, its role in the remodeling of the post-MI non- 2 .3. Hemodynamic measurementsischemic myocardium (NIM) remains largely unexplored.The present study will show that the JAK–STAT pathway Hemodynamic measurements were obtained before theis activated early in the post-MI NIM. Further, the heart was removed from sham-operated rats (n55), 3-dayfunctional role of the activated pathway was contrasted post-MI rats (n55), and 3-day post-MI rats pretreated withwith that of RAS by investigating the effects of specific losartan (n55) or tyrphostin (n55). A 2-F mi-blockade of either pathways in vivo on selected functional cromanometer-tipped catheter (model SPR-407 Miller In-aspects of early post-MI remodeling. strument, Houston, TX) was inserted through the right
carotid artery into the aorta and subsequently advanced tothe LV. LV systolic pressure (LVSP), LV end-diastolic
2 . Methods pressure (LVEDP), peak positive and peak negative firstderivative of LV pressure (1dP/dt and 2dP/dt) were
2 .1. Experimental model calculated as previously reported by averaging 10 consecu-tive cycles [12].
The post-MI rat model: 2-month-old Sprague–Dawleyrats weighing 200 to 250 g underwent left anteriordescending artery (LAD) ligation or sham operation, as 2 .4. Tyrosine phosphorylation studiespreviously described [8]. The LAD was ligated|2–3 mmfrom its origin, which resulted in a moderate size infarct Monoclonal or polyclonal antibodies to JAK2, STAT(31–46% of left ventricle, LV). Animals were sacrificed at proteins 1, 3, 5a and 6, gp130, and anti-phosphotyrosinevarious time intervals after LAD ligation. Sham-operated were obtained from Upstate Biotechnology, Santa Cruzanimals underwent thoracotomy without LAD ligation and Biotechnology, and Sigma. Tissues were harvested fromserved as controls. For studies conducted up to 3 days sham-operated LV, and from the post-MI non-ischemic LVpost-MI the ischemic/ infarcted zone and the non-ischemic at 5, 30, 120 min and 1, 3, and 7 days post-MI. To preparezone were identified by means of injection of 1% Evans cell extracts, cells were washed in PBS and then extractedblue dye into the inferior vena cava before sacrifice. The in lysis buffer containing (in mmol / l) Tris–HCl (pH 7.4)viable area was stained blue, while the infarcted area 20, NaCl 100, EDTA 5, NaF 50, Na P O 10, Na VO 1,3 2 7 3 4
remained pale. After 3 days post-MI, the infarct tissue was phenylmethylsulfonyl fluoride 1 and 1.0% Triton X-100,identified visually or by examination of Van-Giesen stained 10% glycerol, 0.1% SDS, 1.0% deoxycholic acid, 10transverse LV sections. With the help of microdissection, mg/ml aprotinin, and 10mg/ml leupeptin. The lysatestissue samples representing the NIM were obtained from were centrifuged at 10 0003g for 15 min. Protein con-the free LV wall excluding the ischemic/ infarcted area plus centration was determined by the Bio-Rad protein assay.the border zone. The border zone was defined as the 1-mm Cell lysates were incubated with 1mg/ml of the respectivearea next to the ischemic/ infarcted area. Infarct size was antibodies overnight at 48C. Immunocomplexes weredetermined planimetrically as the ratio of infarct tissue or collected by incubating with 50ml of protein A- orscar to the length of the entire LV endocardial circumfer- G-Sepharose for 2 h. Immunoprecipitates were washedence as described previously [9]. It has been shown that four times with ice-cold lysis buffer. The pellets wereinfarct size significantly affects post-MI remodeling [9]. re-suspended in 23 sample buffer containing 50 mmol / lData were collected only from rats with moderate (31– Tris (pH 6.8), 2% SDS, 2%b-mercaptoethanol, 2%46%) or large (,46%) infarcts. The investigation con- glycerol, and bromophenol blue. The samples were sub-formed with theGuide for the Care and Use of Laboratory jected to SDS–PAGE and were transferred to reinforcedAnimals (NIH publication no. 85-23, revised 1996). nitrocellulose membranes (Schleicher & Schuell). The
membranes were blocked with 5% BSA in Tris-buffered2 .2. Drug protocol saline–Tween solution (20 mmol / l Tris–HCl (pH 7.41),
150 mmol / l NaCl, and 0.05% Tween 20) for 2 h at roomOne group of rats received the AT R antagonist losar- temperature. Blots were immunolabeled overnight at 48C1
tan. The drug was administered to rats 1 week prior to with anti-phosphotyrosine antibody. Proteins were visual-LAD ligation in a dose of 10 mg/kg/day [10]. The drug ized by enhanced chemiluminescence (Amersham). Thewas added to the drinking water, with careful monitoring blots were stripped and reprobed with the same antibodiesof water consumption and body weight to ensure proper used for their immunoprecipitation, to ensure equal loadingdrug dosage. In the second group of rats the JAK2 of the proteins.
by guest on August 3, 2016
Dow
nloaded from

H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138 131
2 .5. St-domain binding studies Chomczyniski and Sacchi of homogenization in acidguanidinium thiocyanate followed by phenol–chloroform
Tissues harvested from sham-operated LV and 1-day-old extraction and ethanol precipitation [14]. The amount ofpost-MI non-infarcted LV were dissociated into cells and RNA recovered in each sample was determined spectro-the nuclei prepared by dounce homogenization in the photometrically at a wavelength of 260 nm and theappropriate lysis buffer. Nuclei were collected by centrifu- integrity of each sample confirmed by analysis on agation, treated with 0.5 M NaCl for 1 h at 48C, and denaturing agarose gel. Quantitative evaluation was per-re-centrifuged and aliquots of the nuclear lysates were formed using scanning densitometric analysis. For com-frozen in liquid N . The nuclear lysates were used in a gel parisons between groups, the arbitrary densitometric units2
mobility shift assay (GMSA) using as probe the St-domain were normalized to the value of the cyclophilin gene.of the ANG gene promoter. The St-domain is the promoterelement that binds STAT proteins (the nucleotide sequence2 .9. Measurement of caspase-3 activityis: 59-GGGTTCCTGGAAGGG-39) [3]. All radioactivelylabeled probes were incubated with nuclear lysate at 48C The activity of caspase-3 was determined in the 1-dayfor 30 min. Protein–probe complexes were electrophoresed post-MI border zone with the caspase-3 cellular activityin 8% polyacrylamide gel and visualized by autoradiog- assay kit (Biomol Research, PA, USA) according to theraphy. To identify which of the activated STAT proteins manufacturer’s instructions. The activity was measured byspecifically bind to the St-domain of the ANG gene means of detection of chromophorep-nitroanilide afterpromoter supershift experiments were conducted as fol- cleavage from the labeled substrate Asp–Glu–Val–Asplows: prior to addition of St-domain probes, antibodies to (DEVD)–p-nitroanilide. Tissue samples (10 mg) werethe STAT proteins 1, 3, 5a and 6 were added to the nuclear solubilized by using a Polytron homogenizer with a celllysates and incubated on ice for 4 h, then the St-domain lysis buffer (50 mM HEPES, pH 7.4, 0.1% CHAPS, 1 mMprobe was added and incubated as outlined above. DTT, 0.1 mM EDTA). Equal amounts of protein lysates
were then reacted with 50mM DEVD–p-nitroanilide for 12 .6. Protein phosphatase 1 activity h at 378C. The activity was determined in a microtiter
plate-reader at 405 nm and the results were calibrated withAssays were performed using the manufacturer’s proto- known concentrations ofp-nitroanilide.
col (protein phosphatase assay system; Life Technologies,Rockville, MD, USA). Phosphatase activity was measured 2 .10. Apoptosis assayat 308C using P-labeled phosphorylasea as substrate innoninfarcted LV myocardium obtained from sham-operated To visualize apoptotic nuclei in cardiac myocytes in theanimals, 3-day post-MI animals and 3-day post-MI animals 1-day post-MI border zone, in situ labeling of fragmentedpretreated with losartan or tyrphostin. DNA was performed using In Situ Death Detection Kit,
Fluorescein (Roche Molecular Biochemicals). Sections for2 .7. Phospholamban phosphorylation formalin fixed and paraffin-embedded rat left ventricular
myocardium were placed on charged slides. Sections wereTo compare the level of phosphorylated phospholamban dewaxed with microwave irrigation (370 W for 5 min in
and total phospholamban in the same four groups of 200 ml of 0.1 M citrate buffer, pH 6). Slides were thenanimals, the antibody p-16 that recognized phosphorylated cooled rapidly by immediately adding 80 ml of distilledSer16 in phospholamban, which is known to be the main water (20–258C). Slides were immersed for 30 min atphosphorylation site for protein kinase A, was used. After room temperature in 0.1 M Tris–HCl, containing 3% BSA,a second antibody and luminescent detection, the mem- pH 7.5 then rinsed twice with PBS at RT. Cell permeabili-brane was stripped and relabeled with antibody A1, which zation was achieved by rinsing slides in 0.1% Tritonrecognized total phospholamban [12]. X-100, 0.1% sodium citrate for 2 min on ice (48C) after
which slides were then rinsed twice with PBS. TUNEL12 .8. K channel gene expression reaction mixture (100ml) (terminal deoxynucleotidyl
transferase from calf thymus mixed with nucleotide mix-Details of the preparation of RNA from ventricular ture in reaction buffer) was added to each slide after which
myocardium and RNase protection assay have been re- a coverslip was applied. Slides were then incubated for 30ported previously [13]. Noninfarcted LV myocardium from min at 378C in a humidified chamber, washed three timesthe LV free wall was dissected from sham-operated in PBS for 5 min, and evaluated under a fluorescenceanimals, 3-day post-MI animals and 3-day post-MI animals microscope. Normal myocardial tissue sections (Sham)pretreated with losartan or tyrphostin. The tissues were were pretreated with Dnase I (IU/ML) dissolved in 50 mMrinsed in saline to remove excess blood, snap frozen in Tris–HCl, pH 7.5, 1 mM MgCl , 1 mg/ml BSA for 102
liquid nitrogen, and stored at270 8C. Total RNA was min at 258C to induce DNA strand breaks to serve as aextracted from the LV using the standard protocol of positive control. Biotin-dUTP was omitted in negative
by guest on August 3, 2016
Dow
nloaded from

132 H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138
controls. The sections were examined by light microscopy shown in studies of acute lymphoblastic leukemia [11] andand an apoptotic index was calculated as the percentage of was later confirmed in rat cardiomyocyte culture [15]. Wepositive myocytes observed in 10-high power fields per tested the specificity of tyrphostin in vivo on two other keysection. kinases that are activated in the 1-day post-MI NIM and
found that tyrphostin at 5 mg/kg/day has no effect on2 .11. Measurement of apoptosis-related proteins activated JNK and Akt but specifically inhibited JAK2
phosphorylation (Fig. 1E).The expression of the apoptosis-related proteins Bax and Fig. 2 shows the results of GMSA. Fig. 2A shows a
Bcl-xL in the 1-day post-MI border zone was examined by strong St-domain/STAT binding activity in 1-day oldWestern blot analysis. Equal amounts of protein (30mg) post-MI NIM. Binding was significantly reduced in post-were separated by 20% SDS–PAGE gel and immuno- MI rat heart previously treated with either the AT R1
blotted with anti-Bax and anti-Bcl-2 (Upstate Biotechnol- blocker, losartan or the JAK2 blocker, tyrphostin. Spe-ogy). cificity of binding by STAT proteins to the wild-type
St-domain was demonstrated by competing out specific2 .12. Statistical analysis STAT–St-domain binding by co-incubating with a 1003
excess of non-radioactive St-domain probe. A mutatedStatistical analysis was performed by means of Student’s form of the probe was also used as negative control to
t-test and one-way ANOVA followed by the Bonferroni further ensure specificity of binding by STAT proteins.procedure for multiple group comparisons. A value of Fig. 2B illustrates a supershift experiment to identify theP,0.05 was considered significant. STAT proteins that specifically interact with the St-domain
and shows that the STAT–DNA complexes were disruptedsignificantly by anti-STAT 3 and anti-STAT 5a.
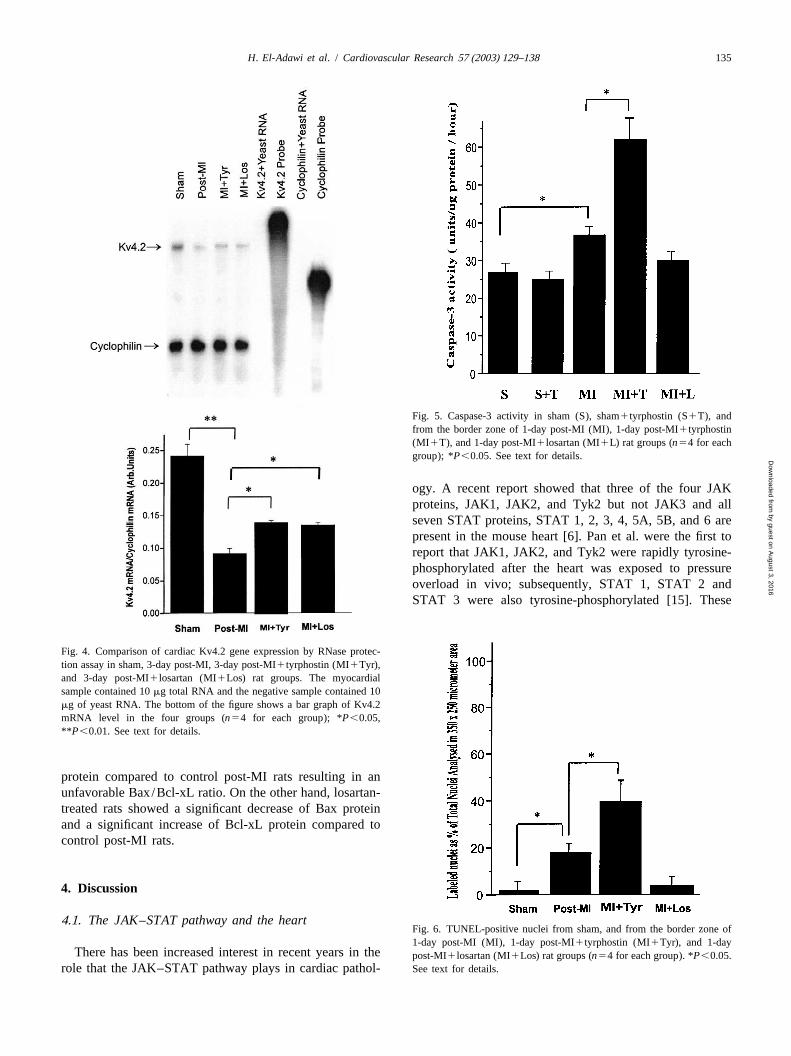
3 . Results Fig. 3A shows that protein phosphatase 1 activitysignificantly increased in the NIM 3-day post-MI by 32%
Table 1 shows the effects of pretreatment with losartan compared to sham. Three-day post-MI rats treated withor tyrphostin on hemodynamic parameters in 3-day post- losartan or tyrphostin showed significantly less increase ofMI rats. Compared to sham-operated animals, 3-day post- protein phosphatase 1 activity (15% and 12%, respectively,MI rats had significantly lower LVSP, higher LVEDP, and P,0.05). Fig. 3B compares the level of p16-phospholam-decreased1 dP/dt and 2 dP/dt. Losartan-treated 3-day ban and total phospholamban in sham, 3-day post-MI ratspost-MI rats had a significantly lower LVEDP and higher and 3-day post-MI rats treated with losartan or tyrphostin.2dP/dt compared to control post-MI rats. The1dP/dt There was a 52% decrease in the basal level of p16-was also higher than non-treated animals but the difference phospholamban in the NIM of 3-day post-MI rat. In 3-daydid not reach statistical significance. The tyrphostin-treated post-MI rats treated with losartan or tyrphostin, the de-rats showed similar hemodynamic changes to the losartan- crease in basal level of p16-phospholamban was signifi-treated group. Thus both drugs seem to improve the early cantly ameliorated (52% vs. 15% vs. 10% in sham,diastolic dysfunction in the 3-day post-MI rat. losartan group, and tyrphostin group, respectively,P,
Fig. 1A shows that phosphorylation of JAK2 in the NIM 0.01).was detected as early as 30 min post-LAD ligation and Fig. 4 shows the Kv4.2 mRNA level in sham, 3-daypeaked between 2 h and 1-day post-MI. Phosphorylation of post-MI rats, 3-day post-MI rats treated with losartan, andSTAT proteins 1, 3, 5a and 6 and gp130 protein followed 3-day post-MI rats that received tyrphostin. The mRNAclosely the same time course of JAK2 phosphorylation. level of Kv4.2 was significantly decreased by 61% in theFig. 1B–D shows that STATs 1, 3 and 5a phosphorylation NIM of 3-day post-MI rats compared to sham (P,0.01).remained significantly elevated up to 7 days post-MI. The decrease in Kv4.3 mRNA level was significantlySTAT 6 and gp130 phosphorylation returned to control ameliorated in rats treated with losartan (38%) or tyrphos-levels in the 3-day post-MI samples (not shown). tin (39%),P,0.05.
The specificity of tyrphostin in a dose of 5mmol / l /day Fig. 5 shows caspase-3 activity in sham-operated LV, inas selective inhibitor of JAK2 phosphorylation was first the border zone of 1-day post-MI rat heart and 1-day
Table 1Effects of pretreatment with losartan (L) or tyrphostin (T) on hemodynamic parameters in 3-day post myocardial infarction (MI) rat
Sham MI MI1L MI 1T
LVSP, mmHg 113612 10368* 9969 1026101 1LVEDP, mmHg 2.660.4 7.161.9* 3.160.5 3.260.4
1dP/dt, mmHg/s 79106341 59676306* 67126502 655464561 1
2dP/dt, mmHg/s 66406610 54596360* 62126390 618463151*P ,0.05, sham vs. MI. P,0.05, MI vs. MI1L or MI1T.
by guest on August 3, 2016
Dow
nloaded from
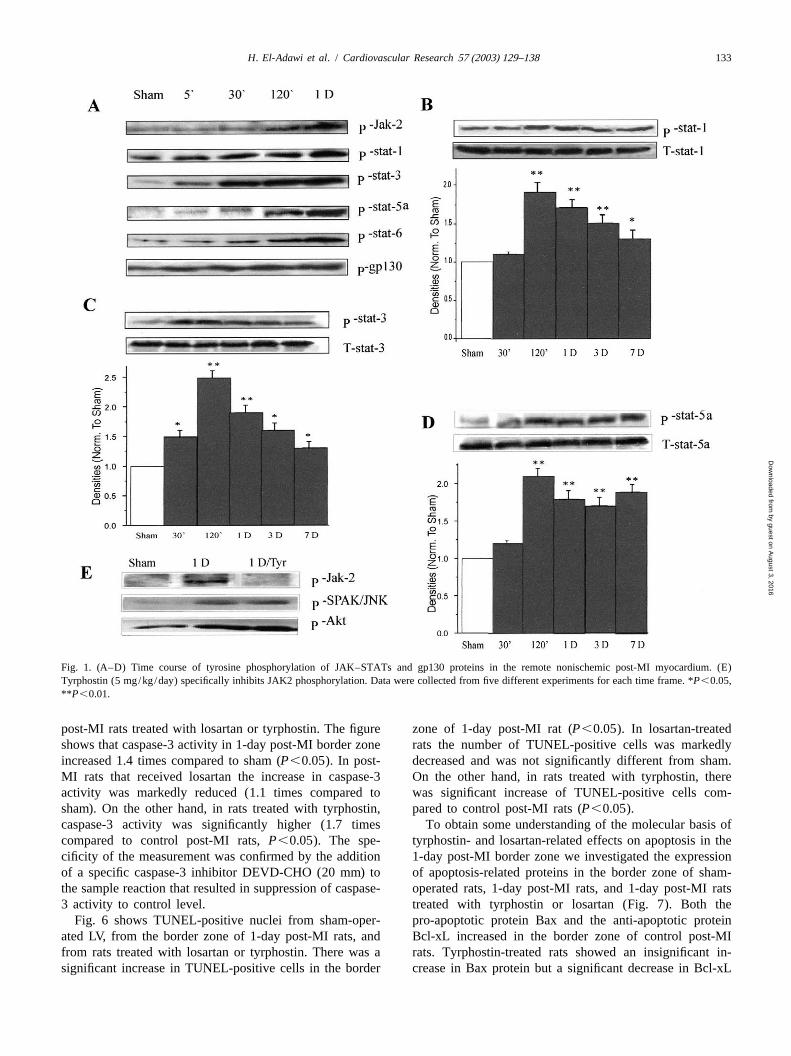
H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138 133
Fig. 1. (A–D) Time course of tyrosine phosphorylation of JAK–STATs and gp130 proteins in the remote nonischemic post-MI myocardium. (E)Tyrphostin (5 mg/kg/day) specifically inhibits JAK2 phosphorylation. Data were collected from five different experiments for each time frame. *P,0.05,**P ,0.01.
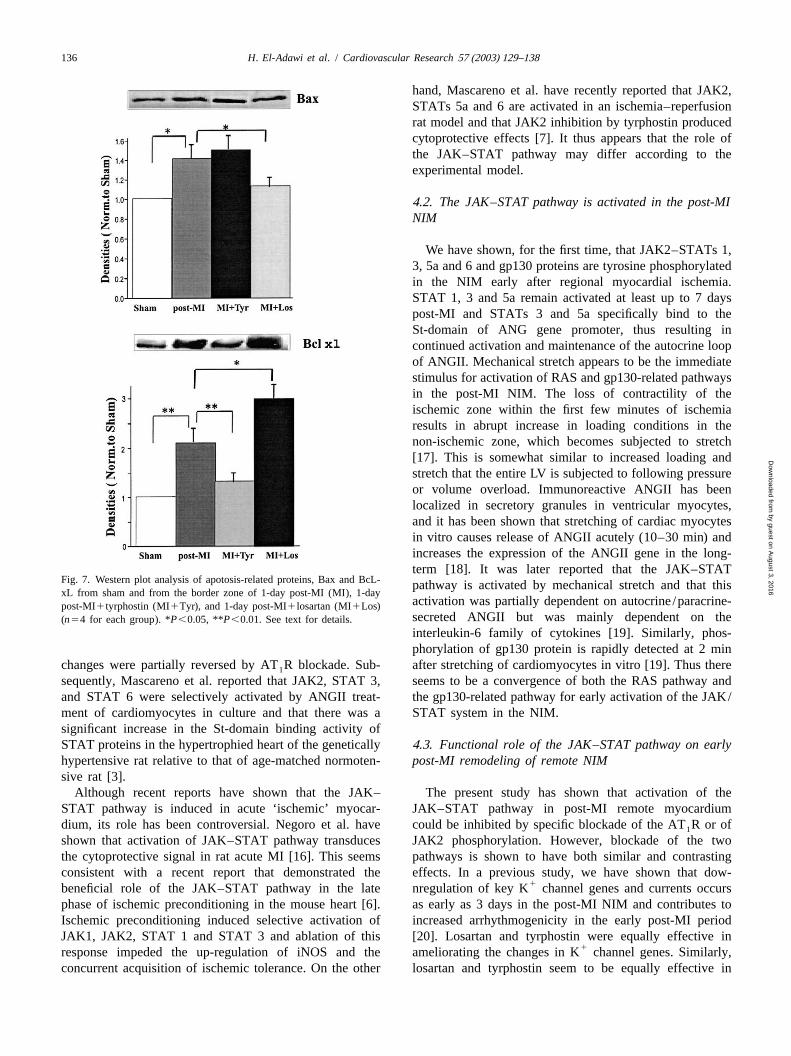
post-MI rats treated with losartan or tyrphostin. The figure zone of 1-day post-MI rat (P,0.05). In losartan-treatedshows that caspase-3 activity in 1-day post-MI border zone rats the number of TUNEL-positive cells was markedlyincreased 1.4 times compared to sham (P,0.05). In post- decreased and was not significantly different from sham.MI rats that received losartan the increase in caspase-3 On the other hand, in rats treated with tyrphostin, thereactivity was markedly reduced (1.1 times compared to was significant increase of TUNEL-positive cells com-sham). On the other hand, in rats treated with tyrphostin, pared to control post-MI rats (P,0.05).caspase-3 activity was significantly higher (1.7 times To obtain some understanding of the molecular basis ofcompared to control post-MI rats,P,0.05). The spe- tyrphostin- and losartan-related effects on apoptosis in thecificity of the measurement was confirmed by the addition 1-day post-MI border zone we investigated the expressionof a specific caspase-3 inhibitor DEVD-CHO (20 mm) to of apoptosis-related proteins in the border zone of sham-the sample reaction that resulted in suppression of caspase- operated rats, 1-day post-MI rats, and 1-day post-MI rats3 activity to control level. treated with tyrphostin or losartan (Fig. 7). Both the
Fig. 6 shows TUNEL-positive nuclei from sham-oper- pro-apoptotic protein Bax and the anti-apoptotic proteinated LV, from the border zone of 1-day post-MI rats, and Bcl-xL increased in the border zone of control post-MIfrom rats treated with losartan or tyrphostin. There was a rats. Tyrphostin-treated rats showed an insignificant in-significant increase in TUNEL-positive cells in the border crease in Bax protein but a significant decrease in Bcl-xL
by guest on August 3, 2016
Dow
nloaded from
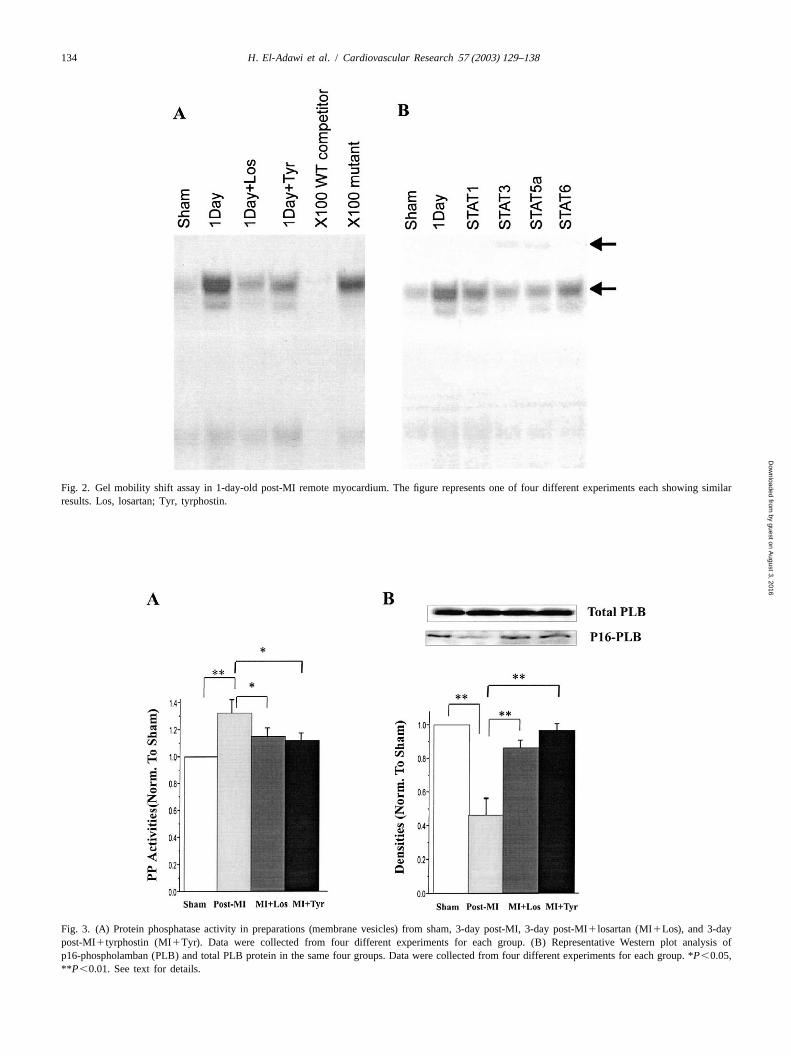
134 H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138
Fig. 2. Gel mobility shift assay in 1-day-old post-MI remote myocardium. The figure represents one of four different experiments each showing similarresults. Los, losartan; Tyr, tyrphostin.
Fig. 3. (A) Protein phosphatase activity in preparations (membrane vesicles) from sham, 3-day post-MI, 3-day post-MI1losartan (MI1Los), and 3-daypost-MI1tyrphostin (MI1Tyr). Data were collected from four different experiments for each group. (B) Representative Western plot analysis ofp16-phospholamban (PLB) and total PLB protein in the same four groups. Data were collected from four different experiments for each group. *P,0.05,**P ,0.01. See text for details.
by guest on August 3, 2016
Dow
nloaded from
H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138 135
Fig. 5. Caspase-3 activity in sham (S), sham1tyrphostin (S1T), andfrom the border zone of 1-day post-MI (MI), 1-day post-MI1tyrphostin(MI1T), and 1-day post-MI1losartan (MI1L) rat groups (n54 for eachgroup); *P,0.05. See text for details.
ogy. A recent report showed that three of the four JAKproteins, JAK1, JAK2, and Tyk2 but not JAK3 and allseven STAT proteins, STAT 1, 2, 3, 4, 5A, 5B, and 6 arepresent in the mouse heart [6]. Pan et al. were the first toreport that JAK1, JAK2, and Tyk2 were rapidly tyrosine-phosphorylated after the heart was exposed to pressureoverload in vivo; subsequently, STAT 1, STAT 2 andSTAT 3 were also tyrosine-phosphorylated [15]. These
Fig. 4. Comparison of cardiac Kv4.2 gene expression by RNase protec-tion assay in sham, 3-day post-MI, 3-day post-MI1tyrphostin (MI1Tyr),and 3-day post-MI1losartan (MI1Los) rat groups. The myocardialsample contained 10mg total RNA and the negative sample contained 10mg of yeast RNA. The bottom of the figure shows a bar graph of Kv4.2mRNA level in the four groups (n54 for each group); *P,0.05,**P ,0.01. See text for details.
protein compared to control post-MI rats resulting in anunfavorable Bax/Bcl-xL ratio. On the other hand, losartan-treated rats showed a significant decrease of Bax proteinand a significant increase of Bcl-xL protein compared tocontrol post-MI rats.
4 . Discussion
4 .1. The JAK–STAT pathway and the heartFig. 6. TUNEL-positive nuclei from sham, and from the border zone of1-day post-MI (MI), 1-day post-MI1tyrphostin (MI1Tyr), and 1-day
There has been increased interest in recent years in thepost-MI1losartan (MI1Los) rat groups (n54 for each group). *P,0.05.role that the JAK–STAT pathway plays in cardiac pathol- See text for details.
by guest on August 3, 2016
Dow
nloaded from
136 H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138
hand, Mascareno et al. have recently reported that JAK2,STATs 5a and 6 are activated in an ischemia–reperfusionrat model and that JAK2 inhibition by tyrphostin producedcytoprotective effects [7]. It thus appears that the role ofthe JAK–STAT pathway may differ according to theexperimental model.
4 .2. The JAK–STAT pathway is activated in the post-MINIM
We have shown, for the first time, that JAK2–STATs 1,3, 5a and 6 and gp130 proteins are tyrosine phosphorylatedin the NIM early after regional myocardial ischemia.STAT 1, 3 and 5a remain activated at least up to 7 dayspost-MI and STATs 3 and 5a specifically bind to theSt-domain of ANG gene promoter, thus resulting incontinued activation and maintenance of the autocrine loopof ANGII. Mechanical stretch appears to be the immediatestimulus for activation of RAS and gp130-related pathwaysin the post-MI NIM. The loss of contractility of theischemic zone within the first few minutes of ischemiaresults in abrupt increase in loading conditions in thenon-ischemic zone, which becomes subjected to stretch[17]. This is somewhat similar to increased loading andstretch that the entire LV is subjected to following pressureor volume overload. Immunoreactive ANGII has beenlocalized in secretory granules in ventricular myocytes,and it has been shown that stretching of cardiac myocytesin vitro causes release of ANGII acutely (10–30 min) andincreases the expression of the ANGII gene in the long-term [18]. It was later reported that the JAK–STAT
Fig. 7. Western plot analysis of apotosis-related proteins, Bax and BcL- pathway is activated by mechanical stretch and that thisxL from sham and from the border zone of 1-day post-MI (MI), 1-day
activation was partially dependent on autocrine/paracrine-post-MI1tyrphostin (MI1Tyr), and 1-day post-MI1losartan (MI1Los)secreted ANGII but was mainly dependent on the(n54 for each group). *P,0.05, **P,0.01. See text for details.interleukin-6 family of cytokines [19]. Similarly, phos-phorylation of gp130 protein is rapidly detected at 2 min
changes were partially reversed by AT R blockade. Sub- after stretching of cardiomyocytes in vitro [19]. Thus there1
sequently, Mascareno et al. reported that JAK2, STAT 3, seems to be a convergence of both the RAS pathway andand STAT 6 were selectively activated by ANGII treat- the gp130-related pathway for early activation of the JAK/ment of cardiomyocytes in culture and that there was a STAT system in the NIM.significant increase in the St-domain binding activity ofSTAT proteins in the hypertrophied heart of the genetically 4 .3. Functional role of the JAK–STAT pathway on earlyhypertensive rat relative to that of age-matched normoten- post-MI remodeling of remote NIMsive rat [3].
Although recent reports have shown that the JAK– The present study has shown that activation of theSTAT pathway is induced in acute ‘ischemic’ myocar- JAK–STAT pathway in post-MI remote myocardiumdium, its role has been controversial. Negoro et al. have could be inhibited by specific blockade of the AT R or of1
shown that activation of JAK–STAT pathway transduces JAK2 phosphorylation. However, blockade of the twothe cytoprotective signal in rat acute MI [16]. This seems pathways is shown to have both similar and contrastingconsistent with a recent report that demonstrated the effects. In a previous study, we have shown that dow-
1beneficial role of the JAK–STAT pathway in the late nregulation of key K channel genes and currents occursphase of ischemic preconditioning in the mouse heart [6]. as early as 3 days in the post-MI NIM and contributes toIschemic preconditioning induced selective activation of increased arrhythmogenicity in the early post-MI periodJAK1, JAK2, STAT 1 and STAT 3 and ablation of this [20]. Losartan and tyrphostin were equally effective in
1response impeded the up-regulation of iNOS and the ameliorating the changes in K channel genes. Similarly,concurrent acquisition of ischemic tolerance. On the other losartan and tyrphostin seem to be equally effective in
by guest on August 3, 2016
Dow
nloaded from

H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138 137
counteracting the early decrease in basal level of p16- term effects of blockade of the JAK–STAT pathway in thephospholamban and both drugs were shown to significantly post-MI heart.ameliorate the early diastolic dysfunction in the 3-daypost-MI rat. We have previously reported that in the 3-week post-MI heart, remodeled hypertrophy of NIM is at A cknowledgementsits maximum and the heart is usually in a compensatedstage with no evidence of heart failure. However, signifi- Supported in part by VA MERIT and REAP grants tocant diastolic dysfunction of the hypertrophied myocar- Nabil El-Sherif, MD.dium was present and was attributed, to a large extent, todecreased basal level of p16-PLB caused by increasedprotein phosphatase 1 activity [12]. The present study R eferencesshows, for the first time, that the increase in proteinphosphatase 1 activity and subsequent decreased level of
[1] Pfeffer MA, Braunwald E. Ventricular remodeling after myocardialp16-phospholamban, the main regulatory protein of infarction; experimental observations and clinical implications.SERCA2, occurs as early as 3 days post-MI in the remote Circulation 1990;81:1161–1172.
[2] Lijnen P, Petrov V. Renin–angiotensin system, hypertrophy and geneNIM and seems to be dissociated from the hypertrophicexpression in cardiac myocytes. J Mol Cell Cardiol 1999;31:949–response per se.970.On the other hand, blockade of AT R and JAK21 [3] Mascareno E, Dhar EM, Siddiqui MAQ. Signal transduction and
phosphorylation produced contrasting effects on apoptotic activator of transcription (STAT) protein-dependent activation ofchanges in the 1-day post-MI border zone. While there is ANG promoter. A cellular signal for hypertrophy in cardiac muscle.no evidence that apoptosis in the border zone contributes Proc Natl Acad Sci USA 1998;95:5590–5594.
[4] Heim MH. The Jak–STAT pathway; cytokine signaling from thesignificantly to post-MI remodeling, continued apoptosis inreceptor to the nucleus. J Recept Signal Transduct 1999;19:75–120.the remote myocardium with the resulting loss of car-
[5] Omura T, Yoshiyama M, Ishikura F et al. Myocardial ischemiadiomyocytes may influence the remodeling process activates the Jak–STAT pathway through angiotensin II signaling in[21,22]. Little data are available on the complex interplay vivo myocardium of rats. J Mol Cell Cardiol 2000;32:572–581.of hypertrophic, pro-apoptotic and anti-apoptotic signals [6] Xuan YT, Guo Y, Han H et al. An essential role of the Jak–STAT
pathway in ischemic preconditioning. Proc Natl Acad Scion post-MI remodeling.2000;98:9050–9055.
[7] Mascareno E, El-Shafei M, Maulik N et al. Jak–STAT signaling isassociated with cardiac dysfunction during ischemia and reperfu-4 .4. Study limitationsion. Circulation 2001;104:325–329.
[8] Qin D, Zang ZH, Caref EB et al. Cellular and ionic basis ofThe present study does not address the specific molecu- arrhythmias in post-infarction remodeled ventricular myocardium.
lar mechanisms and signaling pathways involved in the Circ Res 1996;79:461–473.1downregulation of K gene expression, the increase in [9] Pfeffer MA, Pfeffer JM, Fishbein MC et al. Myocardial infarct size
and ventricular function in rats. Circ Res 1979;44:503–512.protein phosphatase 1 activity, or apoptosis in the post-MI[10] Jain M, Liao R, Ngoy S et al. Angiotensin II receptor blockadeheart as it relates to either the ANGII–AT R pathway or1 attenuates the deleterious effects of exercise training on post-MI
the JAK–STAT pathway. These areas of research should ventricular remodeling in rats. Cardiovasc Res 2000;46:66–72.be of considerable interest. [11] Meydan N, Grunberger T, Dadi H et al. Inhibition of acute
lymphoblastic leukemia by a Jak-2 inhibitor. Nature 1996;379:645–648.
[12] Huang B, Wang S, Qin D, Boutjdir M, El-Sherif N. Diminished5 . Conclusions basal phosphorylation level of phospholamban in the post-infarction
remodeled rat ventricle. Role ofb-adrenergic pathway, G protein,i
phosphodiesterase, and phosphatases. Circ Res 1999;85:848–855.We have shown that the JAK–STAT pathway is acti-[13] Gidh-Jain M, Huang B, Jain P, El-Sherif N. Differential expression
vated in the post-MI remote NIM and that the activation 1of voltage-gated K channel genes in left ventricular remodeledcould be blocked in vivo by a specific inhibitor of JAK2 myocardium after experimental myocardial infarction. Circ Resphosphorylation. Blockade of the JAK–STAT pathway has 1996;79:669–675.
[14] Chomczynski P, Sacchi N. Single-step method of RNA isolation bybeneficial functional consequences on early post-MI re-acid guanidinium thiocyanate–phenol–chloroform extraction. Analmodeling equivalent to that of the AT R blockade. How-1 Biochem 1987;162:156–159.
ever, there are contrasting effects of blockade of the two [15] Pan J, Fukuda K, Kodama H et al. Role of angiotensin II inpathways on at least one aspect of early post-MI remodel- activation of the JAK/STAT pathway induced by acute pressureing, i.e. apoptotic changes in 1-day post-MI border zone. overload in the rat heart. Circ Res 1997;81:611–617.
[16] Negoro S, Kunisada K, Tone E et al. Activation of JAK/STATThe similar /contrasting effects of blocking of eitherpathway transduces cytoprotective signal in rat acute myocardialpathways on the various aspects of long term post-MIinfarction. Cardiovasc Res 2000;47:797–805.
remodeling including hypertrophy, diastolic and systolic [17] Sutton MG, St J, Sharpe N. Left ventricular remodeling afterdysfunction, and survival remain unknown. The present myocardial infarction. Pathophysiology and therapy. Circulationfindings indicate the need of in vivo studies of the long- 2000;101:2981–2988.
by guest on August 3, 2016
Dow
nloaded from

138 H. El-Adawi et al. / Cardiovascular Research 57 (2003) 129–138
[18] Sadoshima J, Izumo S. Molecular characterization of angiotensin genes and currents in the postinfarction heart. J Cardiovasc Elec-II-induced hypertrophy of cardiac myocytes and hyperplasia of trophysiol 2000;11:1252–1261.cardiac fibroblasts. Critical role of the AT receptor subtype. Circ [21] Cheng W, Kajstura J, Nitahara JA et al. Programmed myocyte cell1
Res 1993;73:413–423. death affects the viable myocardium after infarction in rats. Exp Cell[19] Pan J, Fukada K, Saito M et al. Mechanical stretch activates the Res 1996;226:316–327.
Jak–STAT pathway in rat cardiomyocytes. Circ Res 1999;84:1127– [22] Palojoki E, Sarasate A, Eriksson A. Cardiomyocyte apoptosis and1130. ventricular remodeling after myocardial infarction in rats. Am J
1[20] Huang B, Qin D, El-Sherif N. Early down-regulation of K channel Physiol 2001;280:H2726–2731.
by guest on August 3, 2016
Dow
nloaded from




















