
Synthesis and properties of mononuclear and binuclear molybdenum complexes derived from...
9
Available online at www.sciencedirect.com Spectrochimica Acta Part A 69 (2008) 706–714 Synthesis and properties of mononuclear and binuclear molybdenum complexes derived from bis(2-hydroxy-1-naphthaldehyde)oxaloyldihydrazone Ram A. Lal a,∗ , Debajani Basumatary a , Syamal Adhikari b , Arvind Kumar c a Department of Chemistry, North-Eastern Hill University, Shillong 793022, Meghalaya, India b Department of Chemistry, Tripura University, Suryamaninagar 799130, Tripura, India c Institute of Chemistry, Academia Sinica, 128 Academia Road, Sec.2, Nankang, Taipei, 115, Taiwan, ROC Received 17 January 2007; received in revised form 9 May 2007; accepted 9 May 2007 Abstract The monomer molybdenum(VI) complex [MoO 2 (napoxlhH 2 )]·2H 2 O (1) has been synthesized from the reaction of MoO 2 (acac) 2 with bis(2- hydroxy-1-naphthaldehyde)oxaloyldihydrazone (napoxlhH 4 ) in 1:1 molar ratio in ethanol under reflux. This complex on reaction with pyridine/3- picoline/4-picoline yielded the dimer molybdenum(VI) complexes [Mo 2 O 4 (napoxlhH 2 ) 2 (A) 2 ]·2H 2 O (A = py (2), 3-pic (3), 4-pic (4)), whereas reaction with isonicotinoylhydrazine (inhH 3 ) and salicyloylhydrazine (sylshH 3 ) lead to the reduction of the metal centre yielding monomeric molybdenum(V) complexes [Mo(napoxlhH 2 )(hzid)]·2H 2 O (where hzidH 3 = inhH 3 (5) and sylshH 3 (6)). The complexes have been characterized by elemental analyses, molecular weight determinations, molar conductance data, magnetic moment data, electronic, IR, ESR and 1 H NMR spectroscopic studies. The complexes (5) and (6) are paramagnetic to the extent of one unpaired electron. The electronic spectra of the complexes are dominated by strong charge transfer bands. In all of the complexes, the principal dihydrazone ligand has been suggested to coordinate to the metal centres in the anti-cis-configuration. The complexes (1), (5) and (6) are suggested to have six-coordinate octahedral stereochemistry around molybdenum(VI) and molybdenum(V) metal centres, respectively, while the complexes (2)–(4) are suggested to have eight coordinate dodecahedral stereochemistry around molybdenum(VI) metal centre. © 2007 Elsevier B.V. All rights reserved. Keywords: Mononuclear and binuclear; Molybdenum complexes; Bis(2-hydroxy-1-naphthaldehyde)oxaloyldihydrazone; Magnetic moment; Spectroscopic studies 1. Introduction Owing to their biological relevance for molybdoenzymes [1] and also due to the use of high oxidation state molybde- num species in, for example, olefin metathesis catalysis [2], the coordination chemistry of mononuclear molybdenum(IV), (V) and (VI) complexes continues to attract a great deal of attention. The function of molybdenum core depends on the functionalities present in the ligands [3]. Extended X-ray absorp- tion fine structure (EXAFS) spectroscopic studies have also implicated the presence of a sulphur atom, besides oxygen and nitrogen at the active sites of molybdoenzymes. Further, recently it has been shown that the NNN functionalities gener- ∗ Corresponding author. Tel.: +91 364 2722616; fax: +91 364 2550486. E-mail address: lal [email protected] (R.A. Lal). ated by a pyrazolylborate ligand [4] also activate the MoO 2 2+ unit towards oxo-transfer. However, the possibility of acti- vation of MoO 2 2+ unit towards oxo-transfer by other NNO or NOO functionalities cannot be eliminated [5]. All these observations have kindled renewed interest in the coordination chemistry of molybdenum. The chemical information gained in studying the molybdenum coordination complexes, may be transferable to enzyme structure/functions questions not read- ily solvable by studying the enzyme themselves. Further, the metal complexes derived from polyfunctional ligands exhibit interesting properties as electrocatalysts [6], as models of bio- logical systems [7] and as precursors for electrically conducting polymers [8]. Further, interest has been shown, very recently, in the synthesis of molybdenum complexes of macrocylic lig- ands with a view to make an assessment of the role that the ligand backbone and side chains of macrocycle may play in determining the redox and spectroscopic properties of a 1386-1425/$ – see front matter © 2007 Elsevier B.V. All rights reserved. doi:10.1016/j.saa.2007.05.023
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Synthesis and properties of mononuclear and binuclear molybdenum complexes derived from...

A
hprmbsatad©
K
1
[nt(aftiar
1d
Available online at www.sciencedirect.com
Spectrochimica Acta Part A 69 (2008) 706–714
Synthesis and properties of mononuclear and binuclear molybdenumcomplexes derived from
bis(2-hydroxy-1-naphthaldehyde)oxaloyldihydrazone
Ram A. Lal a,∗, Debajani Basumatary a, Syamal Adhikari b, Arvind Kumar c
a Department of Chemistry, North-Eastern Hill University, Shillong 793022, Meghalaya, Indiab Department of Chemistry, Tripura University, Suryamaninagar 799130, Tripura, India
c Institute of Chemistry, Academia Sinica, 128 Academia Road, Sec.2, Nankang, Taipei, 115, Taiwan, ROC
Received 17 January 2007; received in revised form 9 May 2007; accepted 9 May 2007
bstract
The monomer molybdenum(VI) complex [MoO2(napoxlhH2)]·2H2O (1) has been synthesized from the reaction of MoO2(acac)2 with bis(2-ydroxy-1-naphthaldehyde)oxaloyldihydrazone (napoxlhH4) in 1:1 molar ratio in ethanol under reflux. This complex on reaction with pyridine/3-icoline/4-picoline yielded the dimer molybdenum(VI) complexes [Mo2O4(napoxlhH2)2(A)2]·2H2O (A = py (2), 3-pic (3), 4-pic (4)), whereaseaction with isonicotinoylhydrazine (inhH3) and salicyloylhydrazine (sylshH3) lead to the reduction of the metal centre yielding monomericolybdenum(V) complexes [Mo(napoxlhH2)(hzid)]·2H2O (where hzidH3 = inhH3 (5) and sylshH3 (6)). The complexes have been characterized
y elemental analyses, molecular weight determinations, molar conductance data, magnetic moment data, electronic, IR, ESR and 1H NMRpectroscopic studies. The complexes (5) and (6) are paramagnetic to the extent of one unpaired electron. The electronic spectra of the complexesre dominated by strong charge transfer bands. In all of the complexes, the principal dihydrazone ligand has been suggested to coordinate to
he metal centres in the anti-cis-configuration. The complexes (1), (5) and (6) are suggested to have six-coordinate octahedral stereochemistryround molybdenum(VI) and molybdenum(V) metal centres, respectively, while the complexes (2)–(4) are suggested to have eight coordinateodecahedral stereochemistry around molybdenum(VI) metal centre. 2007 Elsevier B.V. All rights reserved.y-1-n
auvoociti
eywords: Mononuclear and binuclear; Molybdenum complexes; Bis(2-hydrox
. Introduction
Owing to their biological relevance for molybdoenzymes1] and also due to the use of high oxidation state molybde-um species in, for example, olefin metathesis catalysis [2],he coordination chemistry of mononuclear molybdenum(IV),V) and (VI) complexes continues to attract a great deal ofttention. The function of molybdenum core depends on theunctionalities present in the ligands [3]. Extended X-ray absorp-ion fine structure (EXAFS) spectroscopic studies have also
mplicated the presence of a sulphur atom, besides oxygennd nitrogen at the active sites of molybdoenzymes. Further,ecently it has been shown that the NNN functionalities gener-∗ Corresponding author. Tel.: +91 364 2722616; fax: +91 364 2550486.E-mail address: lal [email protected] (R.A. Lal).
milpiati
386-1425/$ – see front matter © 2007 Elsevier B.V. All rights reserved.oi:10.1016/j.saa.2007.05.023
aphthaldehyde)oxaloyldihydrazone; Magnetic moment; Spectroscopic studies
ted by a pyrazolylborate ligand [4] also activate the MoO22+
nit towards oxo-transfer. However, the possibility of acti-ation of MoO2
2+ unit towards oxo-transfer by other NNOr NOO functionalities cannot be eliminated [5]. All thesebservations have kindled renewed interest in the coordinationhemistry of molybdenum. The chemical information gainedn studying the molybdenum coordination complexes, may beransferable to enzyme structure/functions questions not read-ly solvable by studying the enzyme themselves. Further, theetal complexes derived from polyfunctional ligands exhibit
nteresting properties as electrocatalysts [6], as models of bio-ogical systems [7] and as precursors for electrically conductingolymers [8]. Further, interest has been shown, very recently,
n the synthesis of molybdenum complexes of macrocylic lig-nds with a view to make an assessment of the role thathe ligand backbone and side chains of macrocycle may playn determining the redox and spectroscopic properties of a
a Act
mcrmmtrtopk
prodoowtM1rt
2
dhElO(nhwmlmF
ldbpbta8ttatppd
tSTEEtppXmFt
2[
sto
fil
2[4
4rmrwa
ti
2[[
b
4rorcwith ether and air dried over anhydrous CaCl2 and collected.
R.A. Lal et al. / Spectrochimic
olybdenum centre [5]. Acyl, aroyl and pyridoyl-hydrazonesontaining amide, azomethine and phenol functions are recentlyecognized as polyfunctional ligands [9] which can react withetal ions either in keto form and enol form. Although, oxo-olybdenum(VI), (V), (IV) complexes of several bi-, tri- and
etra dentate nitrogen, sulphur and oxygen donor system areeported [5,6,10–12], a careful survey of literature has failedo locate any study on mononuclear molybdenum complexesf dihydrazone derived from condensation of acyl-, aroyl-, andyridoyl-dihydrazines with o-hydroxy aromatic aldehydes andetones [9].
Further, although some first row transition metal ion andolynuclear molybdenum complexes of dihydrazones have beeneported [9,12], yet the mononuclear molybdenum complexesf the ligand bis(2-hydroxy-1-naphthaldehyde)oxaloyl-ihydrazone are virtually absent in the literature to the bestf our knowledge. Accordingly, our interest in the chemistryf oxometal cations ligated to nitrogen and oxygen donorsith NO2 and N2O4 chromophores [13–15] has prompted us
o pursue the present work which reports the mononuclearo(VI) complex, derived from the title ligand bis(2-hydroxy-
-naphthaldehyde)oxaloyldihydrazone (napoxlhH4) and itseactivity study with proton and electron donor reagents andhe characterization of the resulting compounds.
. Experimental
Ammonium paramolybdate tetrahydrate, acetyl acetone,iethyl oxalate, hydrazine hydrate, 2-hydroxy-1-naphthalde-yde, isonicotinoylhydrazine, pyridine and acetonitrile were-Merck grade reagents. MoO2(acac)2 [16] and salicy-
oylhydrazine [17] were prepared by literature methods.xaloyldihydrazine was prepared by reacting ethyloxalate
1 mol) with hydrazine hydrate (2 mols). Bis(2-hydroxy-1-aphthaldehyde)oxaloyldihydrazone was prepared by refluxingot dilute ethanol solution of oxaloyldihydrazone (1 mol)ith 2-hydroxy-1-naphthaldehyde (2 mol) by the literatureethod [21,28]. The yellow precipitate obtained was crystal-
ized from ethanol, dried in an electric oven at ca. 70 ◦C,.p. > 300 ◦C(dec). Found: C, 68.00; H, 4.25; N, 13.32; calcd.or C24H18N4O4, C, 67.61; H, 4.25; N, 13.15.
Molybdenum in the complexes was determined by standarditerature method [18]. Carbon, hydrogen and nitrogen wereetermined by microanalysis. Water molecules were determinedy heating the sample at ca. 110, 130, 180 ◦C, respectively, whileyridine, 3-picoline and 4-picoline molecules were determinedy heating the sample at 220 ◦C. Thermogravimetric studies ofhe complex was carried out manually by heating the samplest a particular temperature for 1/2 h in the temperature range0–250 ◦C at an interval of 5 ◦C in hot air oven and estimatinghe weight loss. The vapours were passed through a trap con-aining anhydrous copper sulphate, a CHCl3 solution containing
drop of 5 N NaOH solution. Molecular weight determina-
ions were carried out in DMSO (spectral grade) by freezingoint depression method. The molar conductance of the com-lexes at 10−3 M dilution in DMSO was measured using airect reading conductivity meter – 303 with a dip conduc-lmo
a Part A 69 (2008) 706–714 707
ivity cell. IR spectra were recorded on a Perkin-Elmer – 983pectrophotometer in the range 4000–180 cm−1 in KBr disk.he 1H NMR spectra of the complexes were recorded on anM-390, 90 MHz Spectrophotometer using DMSO-d6 solution.lectronic spectra were recorded on a DMR-21 Spectropho-
ometer in solid state. The ESR spectra of the compounds inowdered form at room temperature and liquid nitrogen tem-erature were recorded at X-band frequency on a Varian E-112/Q-band spectrometer, DPPH was used as an internal fieldarker. The magnetic susceptibilities were determined by thearaday method at room temperature using Hg[Co(NCS)4] as
he calibrant.
.1. Preparation of the complexesMoO2(napoxlhH2)]·2H2O (1)
To an ethanol solution of MoO2(acac)2 was added ligandolution in ethanol maintaining the molar ratio at 1:1. The reac-ion mixture was refluxed for 2 h and cooled which yielded anrange red precipitate.
This was filtered and washed with ethanol, hot benzene andnally with ether and air dried over anhydrous CaCl2 and col-
ected.
.2. Preparation of the complexesMo2O4(napoxlhH2)2(A)2].2H2O (A = py(2), 3-pic(3),-pic(4))
The complex [MoO2(napoxlhH2)]·2H2O (1) (1.5 gm,0 cm3) was suspended in ethanol accompanied by gentle stir-ing for 15 min at 70 ◦C. To this suspension, pyridine was addedaintaining the molar ratio 1:16. The reaction mixture was
efluxed for 1 h. This precipitated the greenish yellow complexhich was filtered, washed with ethanol and finally with ether
nd air dried over anhydrous CaCl2.The complexes (3) and (4) were also prepared by essen-
ially following the above method using 3-picoline or 4-picolinenstead of pyridine.
.3. Preparation of the complexesMo(napoxlhH2)(inh)]·2H2O (5) andMo-(napoxlhH2)(sylsh)]·2H2O (6)
These complexes were prepared under dinitrogen atmospherey the following general procedure.
The complex [MoO2(napoxlhH2)]·2H2O (1) (1.5 gm,0 cm3) was suspended in ethanol accompanied by gentle stir-ing for 15 min at 70 ◦C. To this suspension ethanolic solutionf inhH3, was added maintaining the molar ratio at 1:6. Theeaction mixture was refluxed for 4 h. This precipitated the redomplex which was filtered, washed with ethanol and finally
The complex [Mo(napoxlhH2)(sylsh)]·2H2O was also iso-ated in a similar manner by employing sylshH3 instead of inhH3aintaining the molar ratio 1:6. This precipitated the reddish
range complex.
7 ica Acta Part A 69 (2008) 706–714
3
tct(((ar
ctT2n
3
bcmslwtcivcfitm
3
ttd(rsiorci
3
sss
ight
,de
com
posi
tion
tem
pera
ture
,pe
rcen
tage
yiel
d,an
alyt
ical
,m
agne
ticm
omen
t,m
olar
cond
ucta
nce
and
elec
tron
icsp
ectr
alda
tafo
rM
oco
mpl
exes
ofB
is(2
-hyd
roxy
-1-n
apht
hald
ehyd
e)az
one
d/co
mpl
exM
olec
ular
wei
ght
expt
.(th
eo)
D.P
(◦C
)Y
ield
(%)
Ana
lysi
s:fo
und
(Cal
cd)%
μef
f(B
.M.)
Mol
arco
nduc
tanc
e(S
cm−1
mol
−1)
Ele
ctro
nic
spec
tral
band
s(n
m)
Mo
CH
N
2(n
apox
lhH
2)]
·2H2O
Ora
nge
red
620
±25
(588
)>
300
7516
.75
(16.
33)
50.3
7(4
8.98
)3.
35(3
.40)
9.31
(9.5
2)D
ia2.
532
0,37
0,44
0,55
0O
4(n
apox
lhH
2) 2
(py)
2]·2
H2O
ish
Yel
low
1240
±60
(129
8)>
300
9014
.48
(14.
80)
27.3
0(2
6.81
)3.
60(3
.54)
10.4
8(1
0.79
)D
ia2.
036
5,44
0,56
0
O4(n
apox
lhH
2) 2
(3-p
ic) 2
]·2H
2O
nish
yello
w13
05±
65(1
326)
>30
092
14.1
0(1
4.80
)54
.72
(54.
30)
3.72
(3.7
7)10
.81
(10.
56)
Dia
2.9
360,
450,
580
O4
(nap
oxlh
H2) 2
(4-p
ic) 2
]·2H
2O
ish
yello
w12
40±
60(1
298)
>30
089
14.9
5(1
4.48
)53
.85
(54.
30)
3.82
(3.7
7)10
.31
(10.
56)
Dia
3.1
355,
445,
575
napo
xlhH
2)(
inh)
]·2H
2O
Dar
kre
d70
0±
30(6
90)
>30
095
8.89
5(9
.24)
52.6
2(5
2.17
)3.
96(3
.91)
14.6
2(1
4.20
)1.
692.
734
0,36
0,37
1,45
0,60
0,71
0na
poxl
hH2)(
syls
h)]·2
H2O
ish
oran
ge69
0±
32(7
05)
>30
090
9.63
(9.8
7)52
.42
(52.
77)
3.92
(3.9
7)11
.50
(11.
91)
1.70
3.5
365,
480,
560,
750
08 R.A. Lal et al. / Spectrochim
. Results and discussion
The complexes isolated in the present study together withheir colour, decomposition point, analytical data and molaronductance are set out in Table 1. Based on the analytical data,he complexes are suggested to have the compositions [MoO2napoxlhH2)]·2H2O (1), [Mo2O4(napoxlhH2)2(A)2]·2H2OA = py(2), 3-pic(3), 4-pic(4)), [Mo(napoxlhH2)(inh)]·2H2O5) and [Mo(napoxlhH2)(sylsh)]·2H2O (6). These complexesre orange red, yellowish brown, red and reddish orange,espectively. These complexes are air stable.
The experimental values of the molecular weight for theomplexes (1), (5) and (6) suggested their monomer struc-ure while a dimeric structure for the complexes (2) and (4).hese complexes have molar conductance values in the region.0–3.5 ohm−1 cm2 mol−1 in DMSO indicating that they areon-electrolyte in this solvent [19].
.1. Thermal studies
All of the complexes melt with decomposition above 300 ◦Cut complex (1) decomposes without melting. All of theomplexes show loss of weight corresponding to two waterolecules at 110 ◦C indicating that they are present in the lattice
tructure of the complexes. These complexes do not show anyoss of weight at 180 ◦C ruling out the possibility of presence ofater molecules in the coordination sphere of the complexes. On
he other hand, the complexes (2)–(4) show weight loss at 220 ◦Corresponding to two pyridine/3-picoline/4-picoline moleculesndicating that they are coordinated to the metal centre. Theapours evolved in this temperature range 220–230 ◦C in theomplex (2) turn the CHCl3 and 5 M NaOH solution red con-rming that they originate from pyridine molecules indicating
hat they are present in the first coordination sphere around theetal centre.
.2. Magnetic moment
The μB values for the complexes (1)–(4) are zero indicatinghat they are diamagnetic. These facts conspicuously suggest thathese complexes contain molybdenum in +6 oxidation state with0 electronic configuration. On the other hand, the compounds5) and (6) are paramagnetic to the extent of 1.63 and 1.70 BM,espectively. The μB values for the complexes are close to thepin-only value for a formally d1 system. This feature can benterpreted as being indicative of the effective quenching of therbital angular momentum by a low symmetry ligand field sur-ounding the metal centre. The μB values in the present Mo(V)omplexes dismiss the possibility of any significant metal-metalnteractions.
.3. ESR spectra
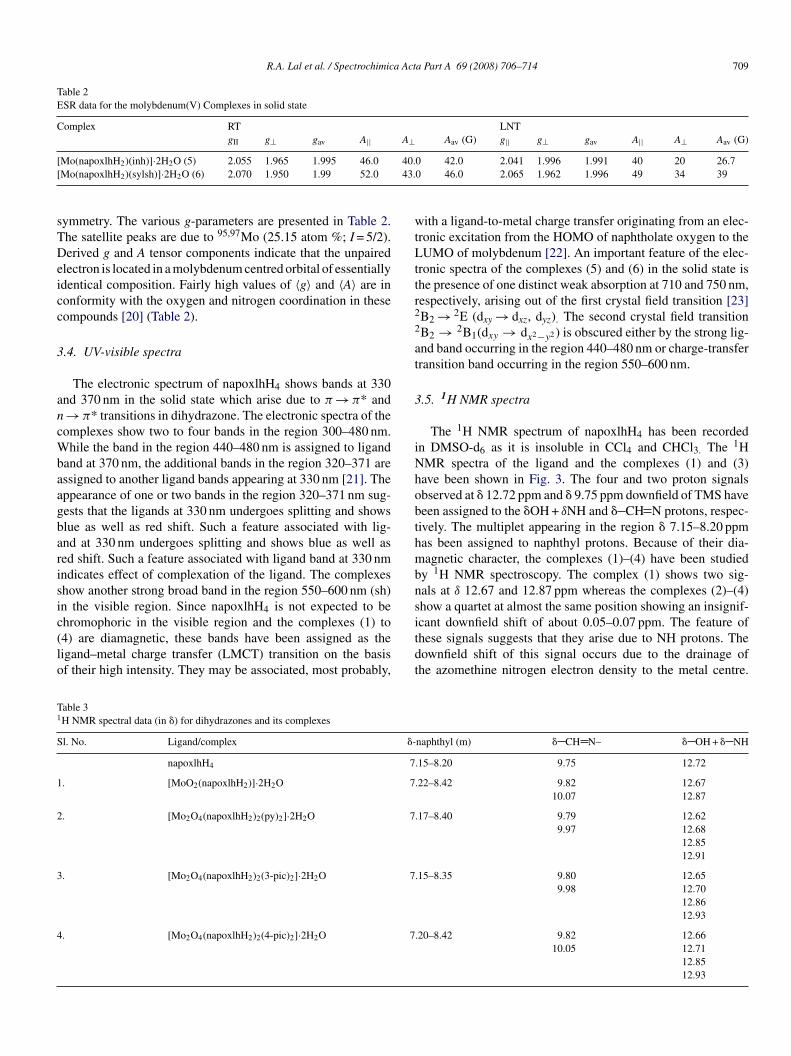
The complexes (5) and (6) have been characterized by ESRpectroscopy. The ESR spectrum of the complex (6) has beenhown in Fig. 2. The X-band ESR spectra of the complexes in theolid state at RT and LNT can be explained by assuming axial Ta
ble
1M
olec
ular
we
oxal
oyld
ihyd
r
SI.n
o.L
igan
1.[M
oO2.
[Mo 2
Gre
en3.
[Mo 2
Bro
w4.
[Mo 2
Gre
en5.
[Mo(
6.[M
o(R
edd
R.A. Lal et al. / Spectrochimica Acta Part A 69 (2008) 706–714 709
Table 2ESR data for the molybdenum(V) Complexes in solid state
Complex RT LNTgII g⊥ gav A|| A⊥ Aav (G) g|| g⊥ gav A|| A⊥ Aav (G)
[ 40.[ 43.
sTDeicc
3
ancWbaagbarisic(lo
wtLttr2
2
at
3
iNhobthmbns
T1
S
1
2
3
4
Mo(napoxlhH2)(inh)]·2H2O (5) 2.055 1.965 1.995 46.0Mo(napoxlhH2)(sylsh)]·2H2O (6) 2.070 1.950 1.99 52.0
ymmetry. The various g-parameters are presented in Table 2.he satellite peaks are due to 95,97Mo (25.15 atom %; I = 5/2).erived g and A tensor components indicate that the unpaired
lectron is located in a molybdenum centred orbital of essentiallydentical composition. Fairly high values of 〈g〉 and 〈A〉 are inonformity with the oxygen and nitrogen coordination in theseompounds [20] (Table 2).
.4. UV-visible spectra
The electronic spectrum of napoxlhH4 shows bands at 330nd 370 nm in the solid state which arise due to π → π* and→ π* transitions in dihydrazone. The electronic spectra of theomplexes show two to four bands in the region 300–480 nm.hile the band in the region 440–480 nm is assigned to ligand
and at 370 nm, the additional bands in the region 320–371 aressigned to another ligand bands appearing at 330 nm [21]. Theppearance of one or two bands in the region 320–371 nm sug-ests that the ligands at 330 nm undergoes splitting and showslue as well as red shift. Such a feature associated with lig-nd at 330 nm undergoes splitting and shows blue as well ased shift. Such a feature associated with ligand band at 330 nmndicates effect of complexation of the ligand. The complexeshow another strong broad band in the region 550–600 nm (sh)n the visible region. Since napoxlhH4 is not expected to be
hromophoric in the visible region and the complexes (1) to4) are diamagnetic, these bands have been assigned as theigand–metal charge transfer (LMCT) transition on the basisf their high intensity. They may be associated, most probably,itdt
able 3H NMR spectral data (in �) for dihydrazones and its complexes
l. No. Ligand/complex �-
napoxlhH4 7
. [MoO2(napoxlhH2)]·2H2O 7
. [Mo2O4(napoxlhH2)2(py)2]·2H2O 7
. [Mo2O4(napoxlhH2)2(3-pic)2]·2H2O 7
. [Mo2O4(napoxlhH2)2(4-pic)2]·2H2O 7
0 42.0 2.041 1.996 1.991 40 20 26.70 46.0 2.065 1.962 1.996 49 34 39
ith a ligand-to-metal charge transfer originating from an elec-ronic excitation from the HOMO of naphtholate oxygen to theUMO of molybdenum [22]. An important feature of the elec-
ronic spectra of the complexes (5) and (6) in the solid state ishe presence of one distinct weak absorption at 710 and 750 nm,espectively, arising out of the first crystal field transition [23]B2 → 2E (dxy → dxz, dyz). The second crystal field transitionB2 → 2B1(dxy → dx2−y2 ) is obscured either by the strong lig-nd band occurring in the region 440–480 nm or charge-transferransition band occurring in the region 550–600 nm.
.5. 1H NMR spectra
The 1H NMR spectrum of napoxlhH4 has been recordedn DMSO-d6 as it is insoluble in CCl4 and CHCl3. The 1HMR spectra of the ligand and the complexes (1) and (3)ave been shown in Fig. 3. The four and two proton signalsbserved at � 12.72 ppm and � 9.75 ppm downfield of TMS haveeen assigned to the �OH + δNH and � CH N protons, respec-ively. The multiplet appearing in the region � 7.15–8.20 ppmas been assigned to naphthyl protons. Because of their dia-agnetic character, the complexes (1)–(4) have been studied
y 1H NMR spectroscopy. The complex (1) shows two sig-als at δ 12.67 and 12.87 ppm whereas the complexes (2)–(4)how a quartet at almost the same position showing an insignif-
cant downfield shift of about 0.05–0.07 ppm. The feature ofhese signals suggests that they arise due to NH protons. Theownfield shift of this signal occurs due to the drainage ofhe azomethine nitrogen electron density to the metal centre.naphthyl (m) � CH N– � OH + � NH
.15–8.20 9.75 12.72
.22–8.42 9.82 12.6710.07 12.87
.17–8.40 9.79 12.629.97 12.68
12.8512.91
.15–8.35 9.80 12.659.98 12.70
12.8612.93
.20–8.42 9.82 12.6610.05 12.71
12.8512.93
710R
.A.L
aletal./Spectrochimica
Acta
PartA 69 (2008) 706–714
Table 4Characteristic IR bands (cm−1) for Bis(2-hydroxy-1-naphthaldehyde)oxaloyldihydrazone and its molybdenum complexes
SI. no. Ligand/complex ν(OH) + ν(NH) ν(C O) ν(C N) Amide(II) +ν(C O) ν(C O) ν(M O) ν(M O)(phenolic) ν(M O)(carbonyl) Other bands
napoxlhH4 3500-3300(sbr); 3382(s),3332s(s)
1655(vs) 1616(s) 1528(s) 1278(w) – – – –
1. [MoO2(napoxlhH2)]·2H2O 3550-3000(sbr); 3462(s),3181(s)
1674(m); 1660(s) 1616(s); 1593(vs) 1545(vs) 1283(s) 944(vs); 916(vs) 590(s) 557(m) – –
2. [Mo2O4(napoxlhH2)2(py)2]·H2O 3550-3000(sbr); 3616(s),3500(s) 3167(s), 3103(s)
1677(s); 1658(s) 1618(vs); 1598(s) 1531(s) 1283(m) 952(s); 907(m) 592(w) 556(w) – –
3. [Mo2O4(napoxlhH2)(3-pic)2]·2H2O 3550-3000(sbr); 3400(s);3217(s)
1685(m); 1660(s) 1618(s); 1600(s) 1540(vs) 1284(w) 945(vs); 920(m) 590(s) 550(m) – –
4. [Mo2O4(napoxlhH2)(4-pic)2]·2H2O 3600-3000(sbr); 3450(s);3270(s)
1680(s); 1662(s) 1616(s); 1596(s) 1535(s) 1285(s) 952(vs); 910(m) 585(m) 540(s) – –
5. [Mo(napoxlhH2)(inh)]·2H2O 3550-3000(sbr); 3411(s) 1674(m); 1662(s) 1614(s); 1595(vs) 1532(s) 1273(w) – 589(m) 531(w) 444(w) 1506(s) 1003(s)6. [Mo(napoxlhH2)(sylsh)]·2H2O 3615(m); 3300-
3000(sbr); 3217(s)1675(m); 1660(s) 1614(s); 1597(s) 1533(s) 1276(s) – 591(m) 530(m) 486(w) 1482(s)
Fig.1.(A
)Staggered-configurfiguration.
The
�C
HN
signalo
two
resonancesand
appT
hissignal
shows
dowT
hesplitting
ofthis
signcoordination
ofdihydrazone
throughthe
azomethine
nitrogenatom
sto
them
etalcentre.The
complexes
(1)–(4)showtw
osig-
nalsin
theregion
�9.79–10.02
ppmdue
toazom
ethineprotons
while
thecom
plex(1)
shows
two
signalsand
thecom
plexes
Fig.2.
X-band
roomtem
peraturesolid
stateE
SRspectrum
of[M
o(napoxlhH2 )(syslh)]·2H
2 O(6),frequency
9.02G
Hz.
ation.(B)A
nti-cisconfiguratio
bservedat
�9.75
ppmears
inthe
region�
9.nfield
shiftof
about0
alinto
two
signalsshow
n.(c)Syn-ciscon-
alsosplits
into79–10.07
ppm.
.13–0.19ppm
.s
theeffect
of
R.A. Lal et al. / Spectrochimica Acta Part A 69 (2008) 706–714 711
oO2
(tadt
�pdtiCgiohetacedt
pocta
pbstmpg[
3prrtasWia4ocotp
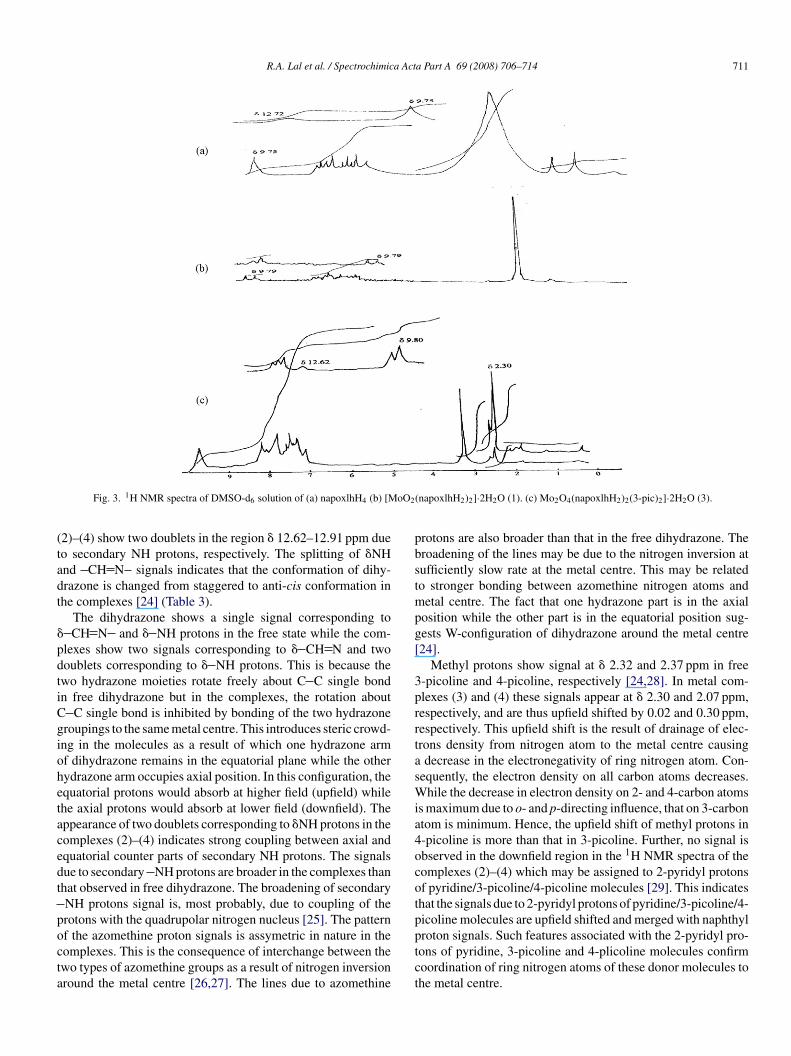
Fig. 3. 1H NMR spectra of DMSO-d6 solution of (a) napoxlhH4 (b) [M
2)–(4) show two doublets in the region � 12.62–12.91 ppm dueo secondary NH protons, respectively. The splitting of �NHnd CH N signals indicates that the conformation of dihy-razone is changed from staggered to anti-cis conformation inhe complexes [24] (Table 3).
The dihydrazone shows a single signal corresponding toCH N and � NH protons in the free state while the com-
lexes show two signals corresponding to � CH N and twooublets corresponding to � NH protons. This is because thewo hydrazone moieties rotate freely about C C single bondn free dihydrazone but in the complexes, the rotation about
C single bond is inhibited by bonding of the two hydrazoneroupings to the same metal centre. This introduces steric crowd-ng in the molecules as a result of which one hydrazone armf dihydrazone remains in the equatorial plane while the otherydrazone arm occupies axial position. In this configuration, thequatorial protons would absorb at higher field (upfield) whilehe axial protons would absorb at lower field (downfield). Theppearance of two doublets corresponding to �NH protons in theomplexes (2)–(4) indicates strong coupling between axial andquatorial counter parts of secondary NH protons. The signalsue to secondary NH protons are broader in the complexes thanhat observed in free dihydrazone. The broadening of secondaryNH protons signal is, most probably, due to coupling of therotons with the quadrupolar nitrogen nucleus [25]. The pattern
f the azomethine proton signals is assymetric in nature in theomplexes. This is the consequence of interchange between thewo types of azomethine groups as a result of nitrogen inversionround the metal centre [26,27]. The lines due to azomethineptct
(napoxlhH2)2]·2H2O (1). (c) Mo2O4(napoxlhH2)2(3-pic)2]·2H2O (3).
rotons are also broader than that in the free dihydrazone. Theroadening of the lines may be due to the nitrogen inversion atufficiently slow rate at the metal centre. This may be relatedo stronger bonding between azomethine nitrogen atoms andetal centre. The fact that one hydrazone part is in the axial
osition while the other part is in the equatorial position sug-ests W-configuration of dihydrazone around the metal centre24].
Methyl protons show signal at � 2.32 and 2.37 ppm in free-picoline and 4-picoline, respectively [24,28]. In metal com-lexes (3) and (4) these signals appear at � 2.30 and 2.07 ppm,espectively, and are thus upfield shifted by 0.02 and 0.30 ppm,espectively. This upfield shift is the result of drainage of elec-rons density from nitrogen atom to the metal centre causingdecrease in the electronegativity of ring nitrogen atom. Con-
equently, the electron density on all carbon atoms decreases.hile the decrease in electron density on 2- and 4-carbon atoms
s maximum due to o- and p-directing influence, that on 3-carbontom is minimum. Hence, the upfield shift of methyl protons in-picoline is more than that in 3-picoline. Further, no signal isbserved in the downfield region in the 1H NMR spectra of theomplexes (2)–(4) which may be assigned to 2-pyridyl protonsf pyridine/3-picoline/4-picoline molecules [29]. This indicateshat the signals due to 2-pyridyl protons of pyridine/3-picoline/4-icoline molecules are upfield shifted and merged with naphthyl
roton signals. Such features associated with the 2-pyridyl pro-ons of pyridine, 3-picoline and 4-plicoline molecules confirmoordination of ring nitrogen atoms of these donor molecules tohe metal centre.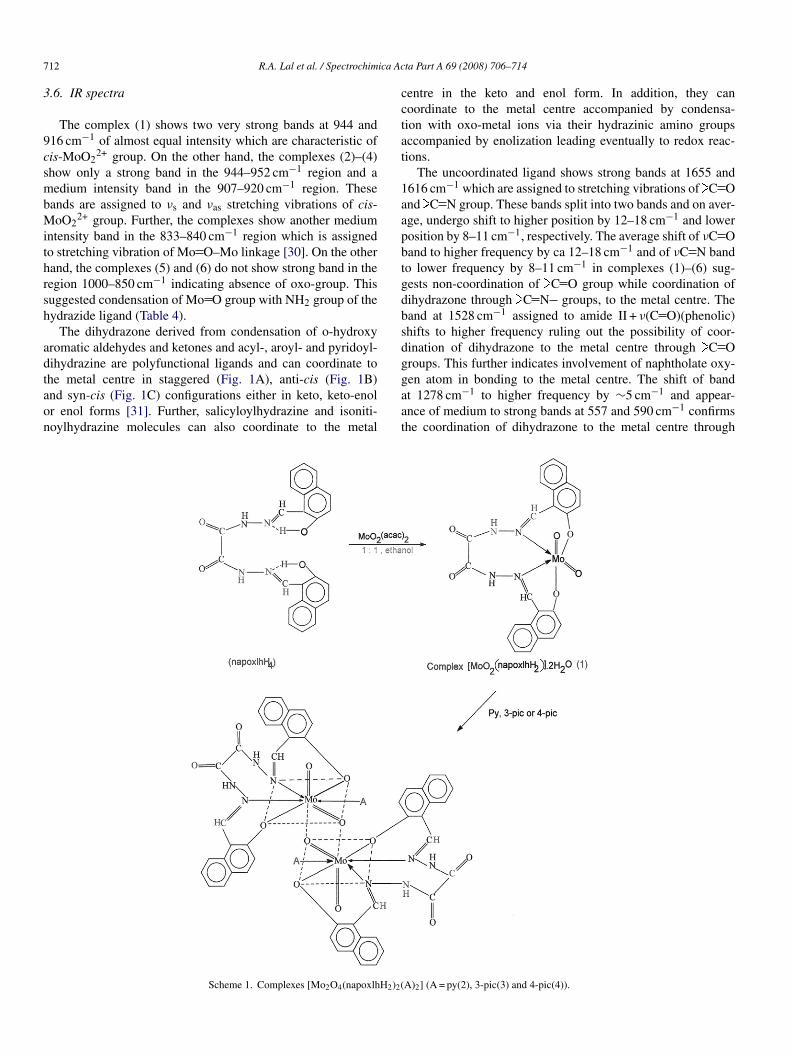
7 ica Ac
3
9csmbMithrsh
adtaon
cctat
1aapbtgdbsdg
12 R.A. Lal et al. / Spectrochim
.6. IR spectra
The complex (1) shows two very strong bands at 944 and16 cm−1 of almost equal intensity which are characteristic ofis-MoO2
2+ group. On the other hand, the complexes (2)–(4)how only a strong band in the 944–952 cm−1 region and aedium intensity band in the 907–920 cm−1 region. These
ands are assigned to νs and νas stretching vibrations of cis-oO2
2+ group. Further, the complexes show another mediumntensity band in the 833–840 cm−1 region which is assignedo stretching vibration of Mo O–Mo linkage [30]. On the otherand, the complexes (5) and (6) do not show strong band in theegion 1000–850 cm−1 indicating absence of oxo-group. Thisuggested condensation of Mo O group with NH2 group of theydrazide ligand (Table 4).
The dihydrazone derived from condensation of o-hydroxyromatic aldehydes and ketones and acyl-, aroyl- and pyridoyl-ihydrazine are polyfunctional ligands and can coordinate to
he metal centre in staggered (Fig. 1A), anti-cis (Fig. 1B)nd syn-cis (Fig. 1C) configurations either in keto, keto-enolr enol forms [31]. Further, salicyloylhydrazine and isoniti-oylhydrazine molecules can also coordinate to the metalgaat
Scheme 1. Complexes [Mo2O4(napoxlhH2)2(
ta Part A 69 (2008) 706–714
entre in the keto and enol form. In addition, they canoordinate to the metal centre accompanied by condensa-ion with oxo-metal ions via their hydrazinic amino groupsccompanied by enolization leading eventually to redox reac-ions.
The uncoordinated ligand shows strong bands at 1655 and616 cm−1 which are assigned to stretching vibrations of C Ond C N group. These bands split into two bands and on aver-ge, undergo shift to higher position by 12–18 cm−1 and lowerosition by 8–11 cm−1, respectively. The average shift of νC Oand to higher frequency by ca 12–18 cm−1 and of νC N bando lower frequency by 8–11 cm−1 in complexes (1)–(6) sug-ests non-coordination of C O group while coordination ofihydrazone through C N groups, to the metal centre. Theand at 1528 cm−1 assigned to amide II + ν(C O)(phenolic)hifts to higher frequency ruling out the possibility of coor-ination of dihydrazone to the metal centre through C Oroups. This further indicates involvement of naphtholate oxy-
en atom in bonding to the metal centre. The shift of bandt 1278 cm−1 to higher frequency by ∼5 cm−1 and appear-nce of medium to strong bands at 557 and 590 cm−1 confirmshe coordination of dihydrazone to the metal centre throughA)2] (A = py(2), 3-pic(3) and 4-pic(4)).
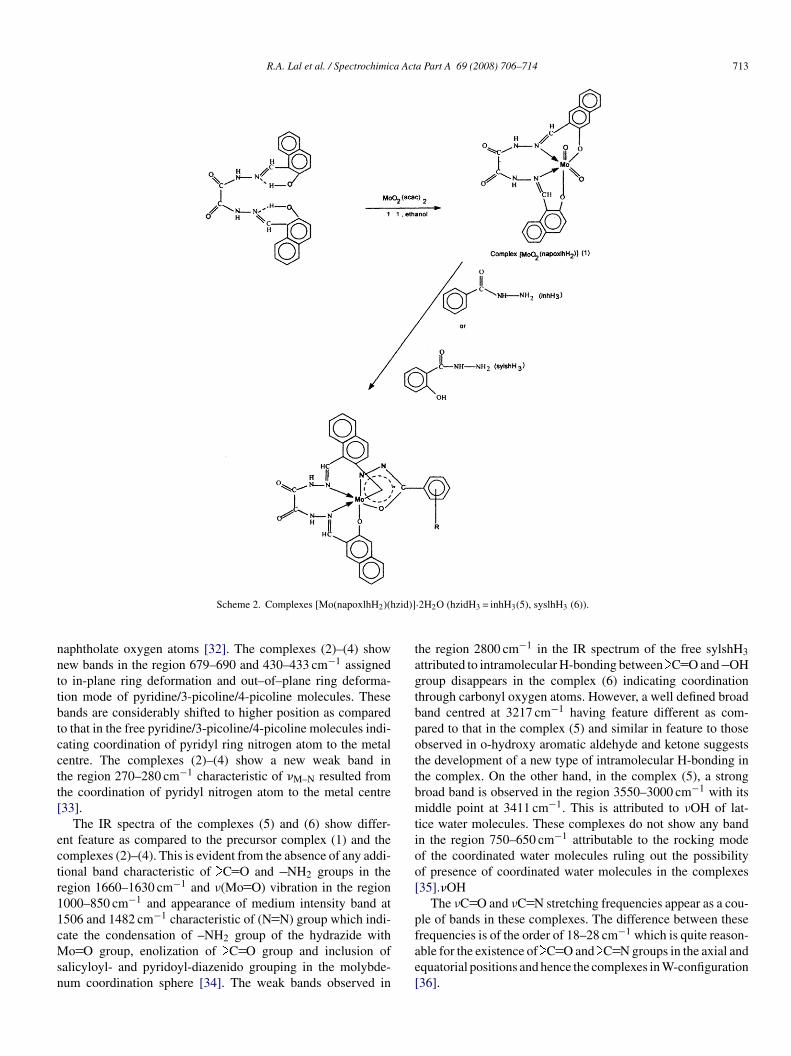
R.A. Lal et al. / Spectrochimica Acta Part A 69 (2008) 706–714 713
zid)]
nnttbtcctt[
ectr11cMsn
tagtbpottbmtioo[
p
Scheme 2. Complexes [Mo(napoxlhH2)(h
aphtholate oxygen atoms [32]. The complexes (2)–(4) showew bands in the region 679–690 and 430–433 cm−1 assignedo in-plane ring deformation and out–of–plane ring deforma-ion mode of pyridine/3-picoline/4-picoline molecules. Theseands are considerably shifted to higher position as comparedo that in the free pyridine/3-picoline/4-picoline molecules indi-ating coordination of pyridyl ring nitrogen atom to the metalentre. The complexes (2)–(4) show a new weak band inhe region 270–280 cm−1 characteristic of �M–N resulted fromhe coordination of pyridyl nitrogen atom to the metal centre33].
The IR spectra of the complexes (5) and (6) show differ-nt feature as compared to the precursor complex (1) and theomplexes (2)–(4). This is evident from the absence of any addi-ional band characteristic of C O and NH2 groups in theegion 1660–1630 cm−1 and ν(Mo O) vibration in the region000–850 cm−1 and appearance of medium intensity band at506 and 1482 cm−1 characteristic of (N N) group which indi-
ate the condensation of –NH2 group of the hydrazide witho O group, enolization of C O group and inclusion ofalicyloyl- and pyridoyl-diazenido grouping in the molybde-um coordination sphere [34]. The weak bands observed in
fae[
·2H2O (hzidH3 = inhH3(5), syslhH3 (6)).
he region 2800 cm−1 in the IR spectrum of the free sylshH3ttributed to intramolecular H-bonding between C O and OHroup disappears in the complex (6) indicating coordinationhrough carbonyl oxygen atoms. However, a well defined broadand centred at 3217 cm−1 having feature different as com-ared to that in the complex (5) and similar in feature to thosebserved in o-hydroxy aromatic aldehyde and ketone suggestshe development of a new type of intramolecular H-bonding inhe complex. On the other hand, in the complex (5), a strongroad band is observed in the region 3550–3000 cm−1 with itsiddle point at 3411 cm−1. This is attributed to νOH of lat-
ice water molecules. These complexes do not show any bandn the region 750–650 cm−1 attributable to the rocking modef the coordinated water molecules ruling out the possibilityf presence of coordinated water molecules in the complexes35].�OH
The νC O and νC N stretching frequencies appear as a cou-le of bands in these complexes. The difference between these
requencies is of the order of 18–28 cm−1 which is quite reason-ble for the existence of C O and C N groups in the axial andquatorial positions and hence the complexes in W-configuration36].
7 ica Ac
4
dccmmepatufccwc
cpifr
A
frUa
R
[[
[
[
[
[
[[[
[[
[
[
[
[
[
[[[[[[[[[
[
[36] H. Adams, N.A. Bailey, M.J.S. Dwyer, D.E. Fenton, P.C. Hellier, P.D.
14 R.A. Lal et al. / Spectrochim
. Conclusion
In the present study, we have prepared some monomeric andimeric molybdenum(V) and molybdenum(VI) complexes andharacterized them on the basis of data obtained from physico-hemical and spectroscopic studies. The reaction of monomericolybdenum(VI) complex with proton donor reagents givesonomeric molybdenum(V) complexes while reaction with
lectron donor reagents gives dimeric molybdenum(VI) com-lexes. The dihydrazone coordinates to the metal centre throughzomethine nitrogen atoms and naphtholate oxygen atoms as aetradentate N2O2 donor. The C O and NH groups remainncoordinated. The dihydrazone exists in the staggered con-ormation in the free state while it is isomerized to anti-cisonformation in the complexes. The molybdenum centre has sixoordinate octahedral geometry in the complexes (1), (5) and (6)hile it has eight coordinate dodecahedral geometry [37] in the
omplexes (2)–(4). In the complexes (2)–(4) the molybdenum
entres are bonded as . In all of the complexes, therincipal dihydrazone ligand is coordinated to the metal centren keto form in the anti-cis-configuration. Tentative structuresor the complexes have been proposed in Schemes 1 and 2,espectively.
cknowledgements
The authors are thankful to the Head, RSIC, CDRI, Lucknowor C, H, N analyses and to the Head RSIC, as IIT, Madras forecording esr spectra. One of the authors (RAL) is thankful to theniversity Grants Commission, New Delhi, India for financial
ssistance through a research grant.
eferences
[1] J.M. Tunney, J. McMaster, C.D. Garner, in: J.A. McCleverty, T.J. Meyer(Eds.), Comprehensive Coordination Chemistry II, vol. 8, Elsevier, Ams-terdam, 2003, p. 459.
[2] A.H. Hoveyda, R.R. Schrock, in: E.N. Jacobson, A. Pfaltz, H. Yamamoto(Eds.), Comprehensive Asymmetric Catalysis, vol. 1, Springer, Heielberg,2004, p. 207.
[3] K.S. Burger, G. Haselhorst, S. Stotzel, T. Weyermuller, K. Wieghardt, B.Nuber, J. Chem. Soc., Dalton’s Trans. (1993) 1987.
[4] C.S.J. Chang, D. Collison, F.E. Mabbs, J.H. Enermark, Inorg. Chem. 29(1990) 2261;C.S.J. Chang, J.H. Enemark, Inorg. Chem. 30 (1991) 683.
[5] S.P. Gramer, R. Wahe, K.V. Rajagopalan, J. Am. Chem. Soc. 103 (1981)7721.
[6] C.L. Bailey, R.D. Bereman, D.P. Rillema, R. Nowak, Inorg. Chem. 25(1986) 933.
[7] V. Gottfried, A. Weiss, Z. Dori, J. Am. Chem. Soc. 102 (1980) 3942.[8] M. Hunziker, G. Rihs, Inorg. Chim. Acta. 102 (1985) 39.[9] R.L. Dutta, Md.M. Hossain, J. Scient. Ind. Res. 44 (1985) 635;
[
ta Part A 69 (2008) 706–714
A. Bacchi, L.P. Battaglia, M. Carcelli, C. Pellizi, G. Pellizi, C. Solinas,M.A. Zoroddi, J. Chem. Soc. Dalton Trans. (1993) 775;A. Bonardi, S. Ianelli, C. Pellizi, G. Pellizi, C. Solinas, Inorg. Chim. Acta.232 (1995) 211;M.P. Degaonkar, V.G. Puranik, S.S. Tavale, S. Gopinath, Bull. Chem. Japan67 (1994) 1797.
10] J.M. Berg, R.H. Holm, J. Am. Chem. Soc. 107 (1985) 917.11] M. Chaudhuri, J. Chem. Soc., Dalton’s Trans. (1985) 115;
M. Chaudhuri, Inorg. Chem. 24 (1985) 3011.12] S. Purohit, A.P. Koley, L.S. Prasad, P.T. Manohran, S. Ghosh, Inorg. Chem.
28 (1989) 3735.13] R.A. Lal, Indian J. Chem. 25A (1986) 979;
R.A. Lal, M.N. Singh, R.K. Thapa, Indian J. Chem. 26a (1987) 883.14] R.A. Lal, S. Das, R.K. Thapa, Inorg. Chim. Acta. 132 (1987) 129;
R.A. Lal, S. Adhikari, A. Pal, A.N. Siva, A. Kumar, J. Chem. Res. (S) 122(1997).
15] R.A. Lal, Polyhedron 8 (1989) 2527;M. Husain, S.S. Bhattacharjee, K.B. Singh, R.A. Lal, Polyhedron 10 (1991)779.
16] G.J.J. Chen, J.W. McDonald, W.E. Newton, Inorg. Chem. 15 (1976) 2612.17] G. Struve, P. Raclehansen, J. Prakt. Chem. 50 (1943) 239.18] A.I. Vogel, Text book of Quantitative Inorganic Analysis, Longman, Lon-
don, 1973.19] J.W. Geary, Coord. Chem. Rev. 7 (1971) 81.20] W.E. Cleladjun, K.M. Barnhardt, K. Yamonnonchi, D. Collision,
F.E. Mabbs, R.B. Ortega, J.H. Enemark, Inorg. Chem. 26 (1987)1017.
21] R.A. Lal, L.M. Mukherjee, A.N. Siva, A. Pal, S. Adhikari, K.K. Narang,M.K. Singh, Polyhedron 12 (1993) 2351.
22] D.G. McCollum, L. Hall, C. White, R. Ostrandor, A.L. Rheingold, J. Whe-lan, B. Bosnich, Inorg. Chem. 33 (1994) 924;E.I. Olomon, K.W. Penfield, D.E. Wilcox, Struct. Bonding (Berl.) 53 (1983)1.
23] C.D. Garner, T.H. Hiller, F.E. Hiller, F.E. Mabbs, Chem. Phys. Lett. 32(1975) 224.
24] L.M. Jackman, S. Sternhell, Application of Nuclear Magnetic ResonanceSpectroscopy in Organic Chemistry, chapter 3, vol. 10, second ed., Perga-mon Press, Amsterdam, 1978.
25] E.A.B. Elsorth, D.W.H. Ramkin, S. Cradock, Structural Methods in Inor-ganic Chemistry, chapter 2, first ed., Blackwell Scientific Publications,London, 1987.
26] J.L. Sudmier, C.N. Reilley, Anal. Chem. 36 (1964) 1689–1704.27] Y.F. Fujiwara, C.N. Reilly, Anal. Chem. 40 (1968) 890.28] J.C.N. Ma, E.W. Warnhoff, Can. J. Chem. 43 (1961) 143.29] T.K. Wu, B.P. Daily, J. Chem. Phys. 41 (1965) 1849.30] O.A. Rajan, A. Chakraborty, Inorg. Chem. 20 (1981) 660.31] R.A. Lal, S. Adhikari, Indian J. Chem. 35A (1996) 607.32] G.C. Percy, Spectrochim. Acta 32 (1976) 1287.33] A.B.P. Lever, B.S. Ramaswamy, Can. J. Chem. 51 (1973) 1582.34] H. Kang, S. Liu, S.N. Shaikh, T. Nicholson, J. Zubieta, Inorg. Chem. 28
(1989) 920;M.D. Fitzroy, J.M. Frederikson, K.S. Murray, M.R. Snow, Inorg. Chem. 24(1985) 3265.
35] A.C. Fabretti, C.G. Franchini, C.P. Preti, G. Tosi, Can. J. Chem. 19A (1980)137.
Hewpsted, J.M. Latour, J. Chem. Soc., Dalton Trans. (1993) 1207.37] C.G. Young, in: J.A. McCleverty, T.J. Meyer, A.G. Wedd (Eds.), Compre-
hensive Coordination Chemistry II, vol. 4, Elsevier Pergamon, Amsterdam,2004, p. 415.




















