
Sympathoadrenomedullary activity in the neuroleptic malignant syndrome
10
334 BIOL PSYCHIATRY 1992;32:334-343 Sympathoadrenomedullary Activity in the Neuroleptic Malignant Syndrome Ronald J. Gurrera and Jorge A. Romero Many clinical features of the neuroleptic malignant syndrome suggest that sympathetic nervous system hyperactivity is involved in the pathophysiology of this disorder. Only a few studies have examined levels of catecholamines or their metabolites in patients with NMS; results so far have been inconclusive. In the present study urinary catecholamine metabolites obtained during the course of NMS were studied with respect to frequently reported signs and symptoms of NMS. The principal findings are that (!) elevated urinary catecholamines and metabolites are a frequent but inconstant feature of NM$; (2) it is likely that sympathetic nervous system hyperactivity contributes to the picture offulminant NMS; and (3) the role of the adrenal medulla in producing excess catecholamines during NMS is uncertain. Introduction The pathophysiology of neuroleptic malignant syndrome (NMS) remains unknown, but the prominence of autonomic dysregulation has led some investigators to postulate a role for the sympathetic nervous system in this disorder. Increased sympathetic activity was first demonstrated by Feibel and Schiffer ( 1981), who found increased plasma and urinary levels of norepinephrine (NE) and epinephrine (E) in a case of NMS. Subsequent reports of sympathoadrenomedullary (SAM) activity in NMS have been inconsistent. Some have confirmed increased levels of NE and E in urine and blood during the active phase of NMS (Hashimoto et al 1984; Ansseau et ai 1986), but others have not (Pernstein 1979; Tollefson 1982; Scarlett et al 1983; Singh et al 1989). In one study, eight patients with NMS were compared with unmatched healthy volunteers; CSF NE was elevated during the active phase, but normal after recovery, whereas CSF MHPG did not differ from controls (Nisijima and Ishiguro 1990). Data regarding other neurotransmitters in NMS are more equivocal. CSF HVA may be decreased (Nisijima and lshiguro 1990) or increased (Ansseau et al 1986) during the acute phase. Urinary 5HIAA was normal in the only two NMS cases in which it was measured (Feibel and Schiffer 1981, Bernstein 1979). CSF 5HIAA may be normal (Ans- seau et al 1986) or decreased (Nisijima and lshiguro 1990). From the Psychiatry Service (RJG) and Neurology Service (JAR), Department of Veterans' Affairs Medical Center, Brockton, Massachusetts and the Departments of Psychiatry (RJG) and Neurology (JAR), Harvard Medical School, Boston MA. This work was supported by OVA Medical Research funds, it was undertaken and completed at the Brockton DVA Medical Center, Brockton. Massachusetts. Address reprint requests to Dr. Ronald J. Gurrera, Brockton DVAMC, Psychiatry (I 16A), Building 60, 940 Belmont Street, Brockton, MA 02401. Received April 9, 1991, revised February 21, 1992. © 1992 Society of Biological Psychiatry 0006-3223/92/$05.00
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Sympathoadrenomedullary activity in the neuroleptic malignant syndrome

334 BIOL PSYCHIATRY 1992;32:334-343
Sympathoadrenomedullary Activity in the Neuroleptic Malignant Syndrome
Ronald J. Gurrera and Jorge A. Romero
Many clinical features of the neuroleptic malignant syndrome suggest that sympathetic nervous system hyperactivity is involved in the pathophysiology of this disorder. Only a few studies have examined levels of catecholamines or their metabolites in patients with NMS; results so far have been inconclusive. In the present study urinary catecholamine metabolites obtained during the course of NMS were studied with respect to frequently reported signs and symptoms of NMS. The principal findings are that (!) elevated urinary catecholamines and metabolites are a frequent but inconstant feature of NM$; (2) it is likely that sympathetic nervous system hyperactivity contributes to the picture of fulminant NMS; and (3) the role of the adrenal medulla in producing excess catecholamines during NMS is uncertain.
Introduction
The pathophysiology of neuroleptic malignant syndrome (NMS) remains unknown, but the prominence of autonomic dysregulation has led some investigators to postulate a role for the sympathetic nervous system in this disorder. Increased sympathetic activity was first demonstrated by Feibel and Schiffer ( 1981), who found increased plasma and urinary levels of norepinephrine (NE) and epinephrine (E) in a case of NMS. Subsequent reports of sympathoadrenomedullary (SAM) activity in NMS have been inconsistent. Some have confirmed increased levels of NE and E in urine and blood during the active phase of NMS (Hashimoto et al 1984; Ansseau et ai 1986), but others have not (Pernstein 1979; Tollefson 1982; Scarlett et al 1983; Singh et al 1989). In one study, eight patients with NMS were compared with unmatched healthy volunteers; CSF NE was elevated during the active phase, but normal after recovery, whereas CSF MHPG did not differ from controls (Nisijima and Ishiguro 1990).
Data regarding other neurotransmitters in NMS are more equivocal. CSF HVA may be decreased (Nisijima and lshiguro 1990) or increased (Ansseau et al 1986) during the acute phase. Urinary 5HIAA was normal in the only two NMS cases in which it was measured (Feibel and Schiffer 1981, Bernstein 1979). CSF 5HIAA may be normal (Ans- seau et al 1986) or decreased (Nisijima and lshiguro 1990).
From the Psychiatry Service (RJG) and Neurology Service (JAR), Department of Veterans' Affairs Medical Center, Brockton, Massachusetts and the Departments of Psychiatry (RJG) and Neurology (JAR), Harvard Medical School, Boston MA.
This work was supported by OVA Medical Research funds, it was undertaken and completed at the Brockton DVA Medical Center, Brockton. Massachusetts.
Address reprint requests to Dr. Ronald J. Gurrera, Brockton DVAMC, Psychiatry (I 16A), Building 60, 940 Belmont Street, Brockton, MA 02401.
Received April 9, 1991, revised February 21, 1992.
© 1992 Society of Biological Psychiatry 0006-3223/92/$05.00

Sympathoadrenomedullary Activity in NMS BIOL PSYCHIATRY 335 1992:32:334--343
We examine a series of patients who met diagnostic criteria for NMS and for whom urinary sympathoadrenomeduUary activity data were available. This study is part of a larger retrospective study of NMS.
Methods
Subject Selection
Subjects were drawn from inpatient referrals to a neurology consultation service between 1982 and 1988. During this period it was customary for all patients on the medical and psychiatry services to be referred to the neurology service when NMS was suspected. After exclusion of 18 patients due to insufficient medical records or confounding medical illness or treatment, a total of 65 suspected NMS episodes in 45 subjects remained. Urinary SAM data were available for 13 (28.9%) subjects, corresponding to 15 (23.1%) suspected episodes. Twelve episodes met published diagnostic criteria for NMS (Levenson 1985) and three were partial syndromes.
Data Collection Urinary SAM data (catecholamines, total metanephrines, and vanillylmandelic acid [VMA]) were recorded by subject and episode; multiple collections related to the same episodes were recorded individually. Values for the following variables were recorded if they were obtained or observed at the time of urine collection: temperature, heart rate, respiratory rate, blood pressure, CPK, WBC, agitation, confusion, rigidity, and tremors. In recording data, preference was given to data obtained on the same day the urine collection was initiated. If data for a variable were unavailable on that day, the following day was used. If data were unavailable on both days, a missing value for that variable was recorded. Thus, data for all variables listed above were obtained within 36 hr after the 24-hr urine collection had begun. In addition, the following signs and symptoms were recorded if they occurred at any time during the course of the episode: thirst, diaphoresis, pallor, and flushing.
Data Analysis Pearson correlation coefficients were computed to examine relationships between urinary SAM measures and continuous variables. When more than one determination had been made, the midpoint of the highest and lowest values for that day was used: this occurred only for temperature, heart rate, respiratory rate, and blood pressure. Log-transformed CPK values (IogCPK) were used for all calculations to minimize the effect of extreme values. For discontinuous variables, SAM measures were dichotomized into elevated and normal groups based on clinical laboratory guidelines and Fisher's exact test was applied.
Missing values were excluded pairwise for all comparisons due to small sample size. Data were analyzed twice, first using the entire sample, then using a subgroup of "acute NMS" SAM collections. The latter were operationally defined as follows: (1) the cor- responding episode met Levenson's diagnostic criteria for NMS and (2) SAM collection was within 15 days of the peak date* for that episode. The second analysis was performed
*Prior to data analysis each episode of NMS was carefully reviewed in order to assign a "peak" date for that episode (i.e.. the day on which the clinical syndrome as a whole reached its maximal intensity). Peak date was determined by the clinical judgment of a physician, based on consideration of vital signs and progress notes: laboratory results were considered ;¢ the reviewer (RG) was unable to assign a peak date based on clinical information alone.
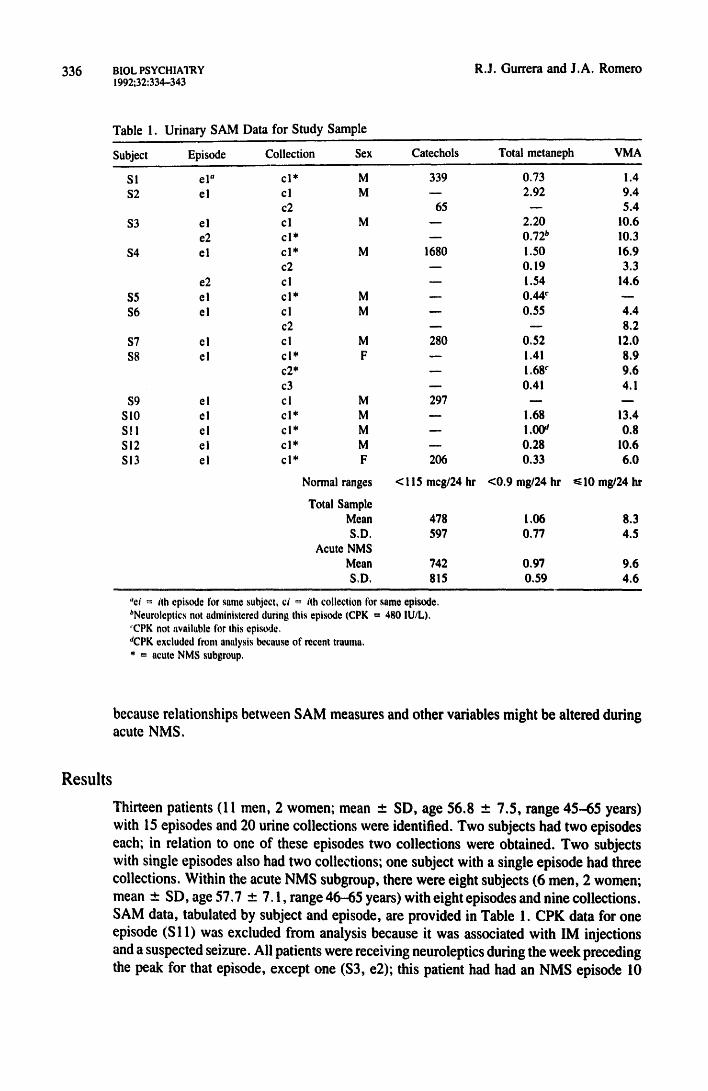
336 BIOL PSYCHIATRY 1992;32:334--343
R.J. Gurrera and J.A. Romero
Table 1. Ur ina ry S A M Data for S t u d y Sample
Subject Episode Collection Sex Catechols Total metaneph VMA
SI el" e l* M 339 0.73 1.4 $2 e l cl M - - 2.92 9.4
c2 65 - - 5.4
$3 e l e l M - - 2.20 10.6 e2 e l * - - 0.72 b 10.3
$4 el e l * M 1680 1.50 16.9 c2 -- 0.19 3.3
e2 cl - - 1.54 14.6 $5 el c l * M - - 0.44 c - -
$6 el cl M - - 0.55 4.4 c2 - - - - 8.2
$7 el cl M 280 0.52 12.0
$8 el c l* F - - 1.41 8.9 c 2 ' - - ! .68 c 9.6
c3 - - 0.41 4.1
$9 e I c I M 297 - - - - S l0 el e l * M - - 1.68 13.4 S l l el c l * M - - 1.00 a 0.8 SI2 el e l * M - - 0.28 10.6 SI3 el e l * F 206 0.33 6.0
Normal ranges <115 meg/24 hr <0 .9 rag/24 hr ~<10 mg/24 hr
Total Sample Mean
S.D. Acute NMS
Mean
S.D.
478 1,06 8 . 3
597 0,77 4.5
742 0.97 9.6 815 0,59 4,6
"ei = itll episode Ibr same subject, ci ~ ith collection for same episode, bNeuroleptics not administered during this episode (CPK = 480 IU/L), 'CPK not available tbr this episode. 'eCPK excluded front analysis because of recent trauma, * = acute NMS subgroup,
because relationships between SAM measures and other variables might be altered during acute NMS.
Results
Thirteen patients (11 men, 2 women; mean _+ SD, age 56.8 _ 7.5, range 45--65 years) with 15 episodes and 20 urine collections were identified. Two subjects had two episodes each; in relation to one of these episodes two collections were obtained. Two subjects with single episodes also had two collections; one subject with a single episode had three collections. Within the acute NMS subgroup, there were eight subjects (6 men, 2 women; mean ± SD, age 57,7 ± 7.1, range 46-65 years) with eight episodes and nine collections. SAM data, tabulated by subject and episode, are provided in Table 1. CPK data for one episode (S 11) was excluded from analysis because it was associated with IM injections and a suspected seizure. All patients were receiving neuroleptics during the week preceding the peak for that episode, except one ($3, e2); this patient had had an NMS episode 10

BIOL PSYCHIATRY 1992:32:334--343
337
20
-15
-10
- 5
0
1.5
0.6
Catecholemines & Metenephrines (mg124h)
60
I I B O
B V V
0
I
VMA (mg124h)
0
0
I I I I I I I I I I I I
600
CPK (IU/L)
I I I I I
6000
Sympathoadrenomedullary Activity in NMS
Series '1 a Series2 o Series3
Sedee 1 . , clitecholemlnee Sodee 2 u total metlmephdnoe
Sedos 3 - VMA
Figure 1. IogCPK vs. Urinary SAM Metabolites
weeks earlier (el), while receiving neuroleptics. In all cases, treatment of NMS was limited to neuroleptic discontinuation and supportive measures.
In the sample as a whole, 5/6 (83%) NMS episodes had elevated catecholamines, 7/14 (50%) had elevated total metanephrines, and 7/13 (54%) had elevated VMA. Metaneph- rines were correlated with catecholamines (r = 0.9"/, p = 0 . 0 3 , , = 4) and showed a trend toward correlation with VMA (r = 0.44,p = 0.09, n = 16). Urinary catecholamines were significantly correlated with systolic blood pressure (r = 0.95, p = 0.05) although sample size was small (n = 4) due to missing catecholamine values. Heart rate showed a nearly significant correlation with VMA (r = 0.49, p = 0.06, n = 15), but this was due l,~tgely to a single value; when this was excluded, the correlation became nonsig- nificant (r = 0.38, p = 0.19, n = 14). Nine of nine patients without agitation at the time of SAM collection had normal VMA, but 3/4 agitated patients had elevated VMA (p = 0.01, two-tailed Fisher's exact test). No CPK data were available for non-acute episodes.
Within the acute NMS subgroup, 3/3 (100%) NMS episodes had elevated catechol- amines, 3/8 (38%) had elevated metanephrines, and 4/'/ (5./%) had elevated VMA. LogCPK was positively correlated with catecholamines (r = 1.00, p = 0.02, n = 3), metanephrines (r = 0.73, p = 0.06, n = 7)and VMA (r = 0.94, p = 0.002, n = 7). These correlations were unchanged when the patient who had not received neuroleptics recently was excluded (catecholamines: r = 1.00, n = 3, p = 0.02; metanephrines: r = 0../3, n = 6, p < 0.1; VMA: r = 0.95, n = 6, p = 0.004). Figure 1 illustrates the overall positive relationship between CPK and SAM activity in the acute NMS subgroup. Five of five patients without documented thirst during their NMS episode had

338 BIOL PSYCHIATRY 1992~32:334--343
R.J. Gurrera and J.A. Romero
normal metanephrines, whereas 3/3 with evidence of thirst had elevated metanephrines (p - 0.02, two-tailed Fisher's exact test). There was a nonsignificant association of flushing with elevated metanephrines (p = O. 11, two-tailed Fisher's exact test).
The acute NMS subgroup did not differ from the rest of the sample in mean age, height, weight, or SAM measures ([t] -< 1.11, p -> 0.28), or psychiatric diagnosis (p = 0.16).
Discussion
These data should be interpreted cautiously because of small sample size and the method of subject selection. Urinary catecholamine data were not obtained randomly but reflected a medical assessment that a pheochromocytoma needed to be ruled out. Because of this, our data may overestimate the incidence of elevated SAM activity in NMS. Some urine collections may have been incomplete, and there were missing values due to the retro- spective nature of this study. These factors would tend to underestimate the frequency of abnormal SAM activity in NMS, and to obscure relationships between some variables.
The major findings of this study are that increased sympathoadrenomeduilary activity is present in some cases of NMS and that it appears to be associated with specific features of the syndrome. In particular, there is a strong association between elevated CPK and increased SAM activity, especially VMA, during the acute phase of NMS. Also, increased levels of total metanephrines are associated with thirst in the acute NMS subgroup and elevated VMA excretion is associated with agitation in the sample as a whole. The latter finding did not retain significance in the acute NMS subgroup Oecause of small sample size (seven of nine nonagitated subjects were excluded from this subgroup).
Elevated catecholamine levels can cause focal myocardial necrosis in patients with pheochromocytoma (Bloom 1987), in rats infused with large amounts of epinephrine (E) and norepinephrine (NE) (Van Vliet et al 1966), and in dogs infused with inotropic amines (Sandusky et al 1990). Elevated CPK and CPK-MB fraction occurs in children treated with intravenous isoproterenol for severe as~.hma (Maguire et al 1986). Pheochromocytoma can produce focal myositis in skeletal muscle, accompanied by elevated CPK, even in the absence of myocarditis (Bhatnagar et al 1986). The muscle necrosis that occurs in these settings is apparently due to a direct effect of catecholamines on calcium influx into myocytes (Bloom 1987), although lesions produced by excess catecholamines can be indistinguishable from those associated with severe atherosclerosis (Van Vliet et al 1966). This is relevant because sympathetic innervation of skeletal muscle controls va- soconstrictor activity of myocytes rather than individual skeletal motor units (WaUin 1984). In our sample only 3/14 (21%) episodes with CPK data had elevated MB fractions (range 5.2-6.6%) at any time during the course of the episode, suggesting that some of the CPK might have been myocardial in origin but that a more diffuse process involving skeletal muscle was also present.
CPK may increase following motor activity, but not in proportion to it; marathon running is consistently associated with elevated CPK, but moderate exercise is not (Haibach and Hosler 1985). CPK elevations have also been reported in children with neuro!eptic-induced dystonia (Lui 1979). Agitation and rigidity were unrelated to CPK in our sample.
According to Kopin (1985) E is the major catecholamine released from the adrenal medulla into the bloodstream, whereupon it acts hormonally at distant receptor sites; in contrast, NE is the neurotransmitter released from sympathetic nerve endings, and it acts

Sympathoadrenomedullary Activity in NMS BIOL PSYCHIATRY 339 1992;32:334-343
within or close to the neuroeffector junction within which it is released. Plasma NE levels reflect the total overflow of this transmitter at sympathetic neuroeffector junctions, and in resting humans there is a good correlation between plasma NE levels and electrical activity of sympathetic nerves supplying skeletal muscle (Wallin 1984).
VMA is the major urinary excretion product of E and NE in humans; metanephrine (M) and normetanephrine (NM) are important intermediates in its formation. The per- centage of total NE metabolites represented by NM is an index of sympathetic activation, whereas M reflects adrenal medullary activity. Over sufficiently long intervals of relatively constant release there is good agreement between urinary excretion and plasma levels of catecholamines (Kopin, 1985).
The VMA-IogCPK correlation observed here is consistent with a localized and specific effect of increased sympathetic nervous system activity on muscle tissue in acute NMS. If the elevated VMA observed here were due to circulating E, as in a state of adreno- medullary hyperactivity, it would be expected to correlate at least as strongly with other physiologic variables such as heart rate and blood pressure. This reasoning is supported by the observation that pheochromocytomas produce many of the signs and symptoms associated with NMS (hypertension, tachycardia, tachypnea, pallor, flushing, diaphoresis, and tremor), but elevated CPK is not among these (Finton et al 1984). A 0n~v~ toxic effect of neuroleptics on muscle tissue, possibly mediated by disordered calcium metab- olism due to inhibition of calmodulin (Weiss et al 1982), cannot be excluded on the basis of our data. Such a process, if present, might have rendered muscle cells more vulnerable to the effects of excess catecholamines.
Total metanephrines (NM + M) are reported in this study because fractionation was generally not done. However, fractionated total metanephrines were available for one patient (S8), who was found to have elevated NM (1.10 mcg, normal < 0.9 mcg) but not M (0.31 mcg, normal < 0.40 mcg). This lends additional support to the speculation that the VMA-IogCPK correlation occurs in acute NMS patients with sympathetic nervous system, but not adrenal medullary, hyperactivity.
Research has linked increased catecholamine secretion to a variety of conditions in- cluding immobilization stress (Kvetnansky et al 1985), isometric exercise (Ziegler et ai 1985), and hyperactivity (Post et al 1977). None of our patients was in restraints near the time of urine collection, and there was no statistical association betweee rigidity and SAM activity. However, there was a significant association between agitation and VMA in our sample. In a group of affective disorder patients, Swann et al (1991) found that mixed sympathoadrenomedullary activation was correlated with affective and behavioral symptoms, but that sympathetic nervous system activity alone was more strongly cor- related with psychomotor agitation. Catecholamine-like cardiomyopathy has been linked to agitation in the setting of NMS (Schibuk and Schachter 1986).
It has been observed that NMS resembles malignant hyperthermia (MH) in terms of muscle tissue contractile responses (Downey et ai 1984) and microscopic evidence for a myopathic process (Tollefson 1982; Downey et al 1984; Jones and Dawson 1989). In a recent study (Haggendal et al 1990), pigs susceptible to MH were given halothane, and plasma E and NE were followed. The time of onset for various MH signs varied greatly, but metabolic events significantly preceded signs of increased sympathetic activity, leading the authors to conclude that increased sympathetic activity does not initiate MH but strongly contributes to the fulminant MH syndrome.
Our data suggest a similar, secondary role for increased SAM activity in NMS. SAM measures were positively associated with CPK, but they were unrelated to other cardinal

340 BIOL PSYCHIATRY 1992;32:334-343
R.J. Gurrera and J.A. Romero
features of NMS such as fever and rigidity. Dissociation of elevated CPK and rhabdom- yolysis on the one hand, and fever and rigidity on the other, has been reported in patients with NMS (Conlon 1986; Dhib-Jalbut et al 1987).
A secondary, albeit important, role for SAM hyperactivi:y in NMS may account for inconsistency among reports regarding the efficacy of bromocriptine in the management of this disorder (cf. Rosenberg and Green 1989; Shalev et al 1989). In men (Mannelli et al 1985) and women (Mancini et al 1985) bromocriptine lowers plasma NE. Bromocriptine appears tu inhibit NE release from sympathetic nerve terminals, and this effect is mediated by dopaminergic receptors (Mannelli et al 1985). The antihypertensive effect of bro- mocriptine may also involve tonic inhibitory dopaminergic receptors in the adrenal medulla (Carmichael 1986). If reduction of SAM activity contributes to the therapeutic action of bromocriptine in NMS, then bromocriptine would be less effective when this feature is not present. Our data suggest that SAM hyperactivity is not always present in NMS, which may account for inconsistent observations regarding the efficacy of bromocriptine in this disorder.
The association of elevated total metanephrines with thirst is interesting but difficult to interpret. It is possible that these patients were volume depleted and that increased sympathetic activity and fluid consumption reflected an attempt to regulate blood pressure in a volume-depleted state. Our data do not permit an accurate assessment of volume status for these subjects. In rats allowed to drink freely, vasopressin release is associated with release of NE, but not E, from the adrenal medulla (Carmichael 1986).
Elevated urinary SAM measures do not indicate whether the primary site of abnormality is central or peripheral, but this may not be a useful distinction. Central and peripheral noradrenergic function are anatomically and physiologically related (Potter et al 1985), and may constitute an interactive unit (Maas and Leckman 1983). In one study (Ziegler et al 1977), a strong positive correlation was found between NE levels in blood and CSF. An increase in central NE relative to DA activity has been proposed as a cause of NMS (Schibuk and Schachter 1986), and CSF NE is increased in acute NMS (Nisijima and lshiguro 1990).
Both sympathetic nervous system and adrenal medulla are regulated by preganglionic sympathetic neurons, which are under hypothalamic control (Teychenne et al 1985; Carmichael 1986). In the absence of normal hypothalamic regulation the autonomic reflex loop formed by efferent and afferent sympathetic neurons becomes prone to unrestrained responses to various afferent stimuli (Lake and Ziegler 1985). Both sympathetic nervous system and hypothalamus have been implicated as sites of dysregulation in NMS (Kurlan et al 1984), and autopsy findings have provided some support for this (Horn et al 1988).
The acute NMS patient who was not currently receiving neuroleptics was indistin- guishable from the other acute NMS patients on the basis of SAM data. The occurrence of an NMS-like syndrome following neuroleptic discontinuation has been reported (Co- quet-Le Pape 1989, cited in Keilam 1990), and in that case the clinical picture was also indistinguishable from NMS. This is interesting in light of a recent report by Otani et ai ( 1991 ) in which an apparent familial predisposition to NMS in a mother and two daughters is discussed. The mother and one daughter both had unexplained NMS-like syndromes prior to ever receiving neuroleptics, and subsequently developed neuroleptic-associated NMS later in life; the authors propose that these cases suggest a genetic vulnerability to NMS. We would add that these cases also suggest that vulnerability to NMS can manifest itself in the absence of neuroleptic exposure. Our patient may have had an episode such as this.

Sympathoadrenomedullary Activity in NMS BIOL PSYCHIATRY 1992;32:334-343
341
Summary Urinary measures of sympathoadrenomedullary (SAM) activity were found to be elevated frequently in a series of retrospectively studied NMS episodes. Statistically significant relationships were found between SAM measures and several features of NMS: CPK, agitation, and thirst. The sympathetic nervous system is the most likely source of elevated urinary SAM measures in our sample, but involvement of the adrenal medulla cannot be ruled out. The primary site of sympathetic nervous system dysregulation is unknown, and may involve central (hypothalamus) as well as peripheral (sympathetic reflex arc) structures. Systematic collection of urinary SAM measures, together with fractionation of urinary total metanephrines, in patients with acute NMS, should provide useful in- formation regarding the role and origin of SAM hyperactivity in NMS.
References Ansseau M, Reynolds CF Ill, Kupfer DJ, et al (1986): Central dopaminergic and noradrenergic
receptor blockade in a patient with neuroleptic malignant syndrome. J Clin Psychiatry 47:320- 321.
Bemstein RA (1979): Malignant neuroleptic syndrome: an atypical case. Psychosomatics 20:840- 846.
Bhatnagar D, Carey P, Pollard A (1986): Focal myositis and elevated creatine kinase levels in a patient with phaeochromocytoma. Postgrad Med J 62:197-198.
Bloom S (1987): Catecholamine cardiomyopathy (letter). N Engl J Med 317:900. Carmichael SW (1986): The Adrenal Medulla, 1st edition. NYC, Cambridge University Press. Conlon P (1986): The spectrum concept of neuroleptic toxicity (letter). Am J Psychiatry 143:811, Dhib-Jalbut S, Hesselbrock R, Mouradian MM, Means ED (1987): Bromocriptine treatment of
neuroleptic malignant syndrome. J Clin Psychiatry 48:69-73. Downey GP, Rosenberg M, Caroff S, et al (1984): Neuroleptic malignant syndrome: patient with
unique clinical and physiologic features. Am J Med 77:338-340. Feibel JH, Schiffer RB (198 !): SympathoadrenomeduUary hyperactivity in the neuroleptic malignant
syndrome: a case report. Am J Psychiatry 138:1115-1116. Finton CK, Chernow B, Keiser HR (1984): Pheochromocytoma: clinical considerations. In Ziegler
MG, Lake CR (eds), Norepinephrine [Frontiers of Clinical Neuroscience, vol 2], Chapter 32. Baltimore, Williams & Wilkins, pp 486-493.
Haggendal J, Jonsson L, Carlsten J (1990): The role of sympathetic activity in initiating malignant hyperthermia. Acta Anaesthesiol Scand 34:677-682.
Haibach H, Hosler MW (1985): Serum creatine kinase in marathon runners. Experientia (Basel) 41:39-40.
Hashimoto F, Sherman CB, Jeffery WH (1984): Neuroleptic malignant syndrome and dopaminergic. blockade. Arch Intern Med 144:629-630.
Horn E, Lach B, Lapierre Y, Hrdina P (I 988): Hypothalamic pathology in the neuroleptic malignant syndrome. Am J Psychiatry 145:617-620.
Jones EM, Dawson A (1989): Neuroleptic malignant syndrome: a case report with post-mortem brain and muscle pathology. J Neurol Neurosurg Psychiatry 52:1006-1009.
Kellam AMP (1990): The (frequently) neuroleptic (potentially) malignant syndrome. Br J Psychiatry 157:169-173.
Kopin IJ (1985): Biochemical evaluation of sympatho-adrenal medullary activity--an overview. In Ben-Jonathan N, Bahr JM, Weiner RI (eds), Catecholamines as hormone regulators [Serono Symposia Publications, vol 18]. New York, Raven Press, pp 175-188.

342 BIOL PSYCHIATRY ! 992;32:334-343
R.J. Gurrera and J.A. Romero
Kurlan R, Hamill R, Shoulson I (1984): Neuroleptic malignant syndrome: review. Clinical Neu- ropharmacology 7:109-120.
Kvetnansky R, Torda T, Dominiak P, Vigas M, Nemeth S, Grobecker H (1985): Stress induced changes in the adrenergic system. In Ben-Jonathan N, Bahr JM, Weiner RI (eds), Catecholamines as Hormone Regulators [Serene Symposia Publications, vol 18]. New York, Raven Press, pp 237-257.
Lake CR, Ziegler MG (1985): Techniques for the assessment and interpretion of catecholamine measurements in neuropsychiatric patients. In Lake ER, Ziegler MG (eds), The Catecholamines in Psychiatric andNeurologic Disorders, I st edition, Chapter 1. Boston, Butterworth Publishers, pp 1-34.
Levenson JL (1985): Neuroleptic malignant syndrome. Am J Psychiatly 142:i 137-1145.
Lui WY (1979): Phenothiazine-induced dystonia associated with an increase in serum creatine phosphokinase. Arch Dis Child 54:150-151.
Maas JW, Leckman JF ( 1983): Relationships between central nervous system noradrenergic function and plasma and urinary MHPG and other norepinephrine metabolites. In Maas JW (ed), MHPG: Basic Mechanisms and Psychopathology, Chapter 3. Orlando, FL, Academic Press, Inc., pp 33-43.
Maguire JF, Geha RS, Umetsu DT (1986): Myocardial specific creatine phosphokinase isoenzyme elevation in children with asthma treated with intravenous isoproterenol. J Allergy Clin immunol 78:631-636.
Mancini A, Barontini M, Kleiman A, Armando I, Levin G, Molocznik I (1985): Effect of bro- mocriptine on plasma catecholamines in normal subjects and subjects with PRL secreting tumors. In Ben-Jonathan N, Bahr JM, Weiner RI (eds), Catecholamines as Hormone Regulators [Serene Symposia Publications, vol 18]. New York, Raven Press, p 372.
Mannelli M, Cuomo S, DeFeo ML, Maggi M, Piazzini M, Guazzelli R (1985): Dopaminergic inhibition of norepinephrine release. In Ben4onathan N, Bahr JM, Weiner RI (eds), Cate- cholamines as Hormone Regulators [Serene Symposia Publications, vol 18]. New York, Raven Press, p 362.
Nisijima K, lshiguro T (1990): Neuroleptic malignant syndrome: a study of CSF monoamiae metabolism. Bioi Psychiatry 27:280-288.
Otani K, Horiuchi M, Kondo T, Kaneko S, Fukushima Y (1991): Is the predisposition to neuroleptic malignant syndrome genetically transmitted? Br J Psychiatry 158:850-853.
Post RM, Stoddard FJ, Gillin JC, Buchsbaum MS, Runkle DC, Black KE, Bunney WE Jr (1977): Alterations in motor activity, sleep, and biochemistry in a cycling manic-depressive patient. Arch Gen Psychiatry 34:470-477.
Potter WZ, Ross RJ, Zavadil AP ill (1985): Norepinephrine in the affective disorders: classic biochemical approaches, in Lake ER, Ziegler MG (eds), The Catecholamines in Psychiatric and Neurologic Dist, rders, 1st edition, Chapter I I. Boston, Butterworth Publishers, pp 213- 233.
Rosenberg MR, Green M (1989): Neuroleptic malignant syndrome: review of response to therapy. Arch Intern Med 149:!927-1931.
Sandusky GE, Means JR, Todd GC (1990): Comparative cardiovascular toxicity in dogs given inotropic agents by continuous intravenous infusion. Toxicol Pathol 18:268-278.
Scarlett JD, Zimmerman R, Berkovic SF (1983): Neuroleptic malignant syndrome. Aust NZ J Med 13:70-73.
Schibuk M, Schachter D (1986): A role for catecholamines in the pathogenesis of neuroleptic malignant syndrome. Can J Psychiat~ 31:66--69.
Shalev A, Hermesh H, Munitz H (1989): Mortality from neuroleptic malignant syndrome. J Clin Psychiatry 50:18-25.
Singh SP, Giridhar C, Avasthi A (1989): Neuroleptic malignant syndrome with trifluperidol. 8r J Psychiatry 155:561-563.

Sympathoadrenomedullary Activity in NMS BIOL PSYCHIATRY 343 1992;32:334--343
Swann AC, Secunda SK, Koslow SH, Katz MM, Bowden CL, Maas JW, Davis JM, Robins E (1991): Mania: sympathoadrenal function and clinical state. Psychiatry Res 37:195-205.
Teychenne PF, Feuerstein G, Lake CR, Ziegler MG (1985): Central catecholamine systems: in- teraction with neurotransmitters in normal subjects and in patients with selected neurologic diseases. In Lake ER, Ziegler MG (eds), The Catecholamines in Psychiatric and Neurologic Disorders, Ist edition, Chapter 5. Boston, Butterworth Publishers, pp 91-I 19.
Tollefson G (1982): A case of neuroleptic malignant syndrome: in vitro muscle comparison with malignant hyperthermia. J Clin Psychopharmacol 2:266-270.
Van Vliet PD, Burchell HB, Titus JL (1966): Focal myocarditis associated with pheochromocytoma. N Engl J Med 274:1102-I 108.
Wallin BG (1984): Sympathetic activity in human extremity nerves and its relationship to plasma NE. In Ziegler MG, Lake CR (eds), Norepinephrine [Frontiers of Clinical Neuroscience, vol 2], Chapter 28. Baltimore, Williams & Wilkins, pp 431-438.
Weiss B, Prozialeck WC, Wallace TL (1982): Interaction of drugs with calmodulin: biochemical, pharmacological and clinical implications. Biochemical Pharmacology 31:2217-2226.
Ziegler MG, Lake CR, Wood JH, Brooks BR, Ebert MH (1977): Relationship between norepi- nephrine in blood and cerebrospinal fluid in the presence of a blood-cerebrospinal fluid barrier for norepinephrine. J Neurochem 28:677-679.
Ziegler MG, Milano A J, Hull E (1985): The catecholaminergic response to stress and exercise. In Lake ER, Ziegler MG (eds), The Catecholamines in Psychiatric and Neurologic Disorders, I st edition, Chapter 2. Boston, Butterworth Publishers, pp 37-53.




















