
Fanconi syndrome in adults
11
Fanconi Syndrome in Adults A Manifestation of a Latent Form of Myeloma JORGE E. MALDONADO, M.D., Ph.D. JORGE A. VELOSA, M.D. ROBERT A. KYLE, M.D. RICHARD D. WAGONER, M.D. KEITH E. HOLLEY, M.D. ROBERT M. SALASSA, M.D. Rochester, Minnesota From the Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55901. This investigation was supported in part by Research Grants CA- 11911D, CA-11911F and CA-11911GG from the National Institutes of Health, U.S. Public Health Service, and by the Pearl Goldberg Fund. Requests for reprints should be addressed to Dr. Jorge E. Mafdonado, Mayo Clinic, Roches- ter, Minnesota 55901. Manuscript accepted June 4. 1974. From a review of 17 cases of Fanconi syndrome with Bence Jones proteinuria and myeloma or amyloidosis, including three new cases reported here in detail, there emerges a well defined set of characteristics. In most cases, the diagnosis of Fanconi syndrome preceded the development of myeloma or amyloidosis. Myeloma preceding the development of Fanconi syndrome has not been reported. All the patients had Bence Jones proteinuria, but in some it could be detected only by electrophoresis or immu- noelectrophoresis. In the seven cases in which the Bence Jones protein was typed, it was of kappa type. There were no serum protein monoclonal abnormalities. In the bone marrow and renal samples of half of the patients, crystalline cytoplasmic inclusion bodies were present in lymphoplasmacytic elements and renal tubular cells. It is proposed that patients with Fanconi syndrome and Bence Jones proteinuria have a distinct type of plasma cell disorder or variant of the monoclonal gammopathies, characterized by a slow progression of the tumor and by an early phase dominated by the metabolic complications of the renal proximal tubular dys- function. Adult patients with Fanconi syndrome should be careful- ly investigated for the presence of Bence Jones protein and a plasmacytic dyscrasia should be excluded. Renal Fanconi syndrome in adults has been reported in association with myeloma, amyloidosis and Bence Jones proteinuria in at least 14 cases [l-13]. In the three cases we are reporting here, all the patients presented with the Fanconi syndrome and Bence Jones proteinuria in the absence of diagnosable myeloma or amyloidosis. In one case, the patient is alive and clinically well 5 years after the diagnosis of Fanconi syndrome was made: in another, amyloidosis developed 16 l/2 years after the diagnosis of Fanconi syndrome was first considered; and in the third, the patient died of myeloma 6 years after the diagnosis of Fanconi syndrome was made. From our experience and from a review of the literature, well de- fined characteristics emerge for this group of patients, and we think that they represent a distinct variant of the monoclonal gam- mopathies. CASE REPORTS Case 1. This 66 year old Caucasian man was first seen at the Mayo Clinic in July 1956 with proctitis. The results of physical examination were otherwise unremarkable. Pertinent laboratory studies are summarized in Tables I and II. Proteinuria (positive Bence Jones reaction) and glycosuria were incidental findings. There was no family history of diabetes mellitus, 354 March 1975 The American Journal of Mediclne Volume 58
-
Upload
independent -
Category
Documents
-
view
7 -
download
0
Transcript of Fanconi syndrome in adults

Fanconi Syndrome in Adults
A Manifestation of a Latent Form of Myeloma
JORGE E. MALDONADO, M.D., Ph.D.
JORGE A. VELOSA, M.D.
ROBERT A. KYLE, M.D.
RICHARD D. WAGONER, M.D.
KEITH E. HOLLEY, M.D.
ROBERT M. SALASSA, M.D.
Rochester, Minnesota
From the Mayo Clinic and Mayo Foundation, Rochester, Minnesota 55901. This investigation was supported in part by Research Grants CA- 11911D, CA-11911F and CA-11911GG from the National Institutes of Health, U.S. Public Health Service, and by the Pearl Goldberg Fund. Requests for reprints should be addressed to Dr. Jorge E. Mafdonado, Mayo Clinic, Roches- ter, Minnesota 55901. Manuscript accepted June 4. 1974.
From a review of 17 cases of Fanconi syndrome with Bence Jones proteinuria and myeloma or amyloidosis, including three new cases reported here in detail, there emerges a well defined set of characteristics. In most cases, the diagnosis of Fanconi syndrome preceded the development of myeloma or amyloidosis. Myeloma preceding the development of Fanconi syndrome has not been reported. All the patients had Bence Jones proteinuria, but in some it could be detected only by electrophoresis or immu- noelectrophoresis. In the seven cases in which the Bence Jones protein was typed, it was of kappa type. There were no serum protein monoclonal abnormalities. In the bone marrow and renal samples of half of the patients, crystalline cytoplasmic inclusion bodies were present in lymphoplasmacytic elements and renal tubular cells.
It is proposed that patients with Fanconi syndrome and Bence Jones proteinuria have a distinct type of plasma cell disorder or variant of the monoclonal gammopathies, characterized by a slow progression of the tumor and by an early phase dominated by the metabolic complications of the renal proximal tubular dys- function. Adult patients with Fanconi syndrome should be careful- ly investigated for the presence of Bence Jones protein and a plasmacytic dyscrasia should be excluded.
Renal Fanconi syndrome in adults has been reported in association with myeloma, amyloidosis and Bence Jones proteinuria in at least 14 cases [l-13]. In the three cases we are reporting here, all the patients presented with the Fanconi syndrome and Bence Jones proteinuria in the absence of diagnosable myeloma or amyloidosis. In one case, the patient is alive and clinically well 5 years after the diagnosis of Fanconi syndrome was made: in another, amyloidosis developed 16 l/2 years after the diagnosis of Fanconi syndrome was first considered; and in the third, the patient died of myeloma 6 years after the diagnosis of Fanconi syndrome was made.
From our experience and from a review of the literature, well de- fined characteristics emerge for this group of patients, and we think that they represent a distinct variant of the monoclonal gam- mopathies.
CASE REPORTS
Case 1. This 66 year old Caucasian man was first seen at the Mayo Clinic in July 1956 with proctitis. The results of physical examination were otherwise unremarkable. Pertinent laboratory studies are summarized in Tables I and II. Proteinuria (positive Bence Jones reaction) and glycosuria were incidental findings. There was no family history of diabetes mellitus,
354 March 1975 The American Journal of Mediclne Volume 58

FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
TABLE I Hematologic Laboratory Values
Hemo-
globin
Urinary Protein
Quantity Serum Protein Electrophoresis (g/dl)
Time (g/dl) BJP* (g/24 hr) EPt Albumin Alpha1 Alpha2 Beta Gamma Bone Marrow1
19561957 13.2-14.5 + 1964 13.0 +
1967 12.5 + 1969 13.5 +
1973 9.5
195'7 13.7 + 0.455 Alpha2
9/64 9.3-8.7 + 11.0 Alpha2
11/64-12/64 . . . . . . . . . . .
1969-1970 15.1 +
1972 15.0 +
1973 15.0 +
0.135 Beta
3.7 Beta
4.7 Beta
3.7 Beta
Beta
(kappa)
2.4
3.1
1.7
Beta
Beta
Beta
(kappa)
* BJP = Bence Jones reaction.
f EP = electrophoretic spike.
$ NDx = not diagnostic.
9 In this instance, g/dl.
2.97
3.70
3.42 2.94
2.91
3.60
3.65
. . .
3.36
4.03
4.22
and a glucose tolerance test was normal. Serum protein electrophoresis revealed no monoclonal spike; gamma globulin was slightly decreased.
The patient returned in October 1957 with polyuria. Uri- nary protein electrophoresis showed a beta-gamma spike. Heavy aminoaciduria was demonstrated. Bone marrow was normal. Roentgenograms of the chest, pelvis and head were within normal limits.
Although the diagnosis of Fanconi syndrome was sus- pected during the 1957 visit, it was not fully documented until February 1964 (Table II), when he returned com- plaining of generalized weakness and pain in the heels, pelvic girdle, spinal column and rib cage. There was de- mineralization of the spinal column, ribs and hips, and a compression fracture of the body of the ninth thoracic ver- tebra, but no osteolytic lesions were seen. A 24 hour urine specimen contained 3.7 g of protein which showed a beta spike. The bone marrow was still nondiagnostic. Treat- ment with vitamin 0 (100,000 units, at first daily and then every other day) relieved the skeletal pain, but the regimen had to be temporarily discontinued because of symptomat- ic hypercalcemia. Neutra-Phase (Willen Drug Co., Balti-
Case 1 0.25 0.48 0.31 0.74
0.33 0.76 0.37 0.67
0.40 0.62
Case 2
0.34 0.76
0.30 0.79
. . . . . .
Case 3
0.35 0.79
0.18 0.50
0.31 0.49
0.73 0.73 0.80 0.62
0.76 0.62 1.02 0.69
0.51 0.46
1.10 1.25
0.79 1.16
. . .
0.87
0.68
0.75
. . .
0.79
0.68
0.49
NDx; 3% plasma cells
NDx; 5.5% lymphocytoid plasma
cells
Not done
NDx; 9% lymphocytoid plasma cells
containing cytoplasmic crystalline
inclusion bodies
Autopsy specimen NDx; systemic
amyloidosis demonstrated (bone
marrow involved)
NDx; diffuse increase in lympho-
cytes (32%) Diffuse increase in lymphocytes
(39%) Diagnostic of myeloma (before
death and at autopsy)
NDx; 10.5% lymphoid plasma cells with cytoplasmic crystalline
inclusion bodies
NDx; 12’% lymphoid plasma cells with inclusion bodies
NDx; 9% lymphoid plasma cells with
inclusion bodies
more, Md.) was added to the program but was taken only once or twice a week.
Except for an episode of vesical neck obstruction in Oc- tober 1967, the patient did well. Therapy with vitamin D and Neutra-Phos was continued.
The patient’s final visit to the Mayo Clinic was in Febru- ary 1969. In the interim, his physician had added calcium carbonate and oral potassium to the regimen to correct the acidosis (which had worsened) and the hypokalemia that had ensued. The skeletal pain had recurred. The labo- ratory results were unchanged from the previous visit ex- cept for an increase in serum creatinine. The pelvic and spinal roentgenograms showed persistent demineraliza- tion. A bone marrow aspirate revealed focal areas of in- creased plasma cells with lymphoid features (these cells constituted 9 per cent of the nucleated marrow elements) but was not considered diagnostic of myeloma. Retrospec- tive analysis disclosed the presence of rectangular, rhom- boid or round, mildly basophilic, cytoplasmic inclusion bod- ies in the lymphoplasmacytic elements. The vitamin D and Neutra-Phos regimen was continued. Deterioration of the patient’s general status occurred over the next 2 years,
March 1975 The American Journal of Medlclne Volume 58 355
FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
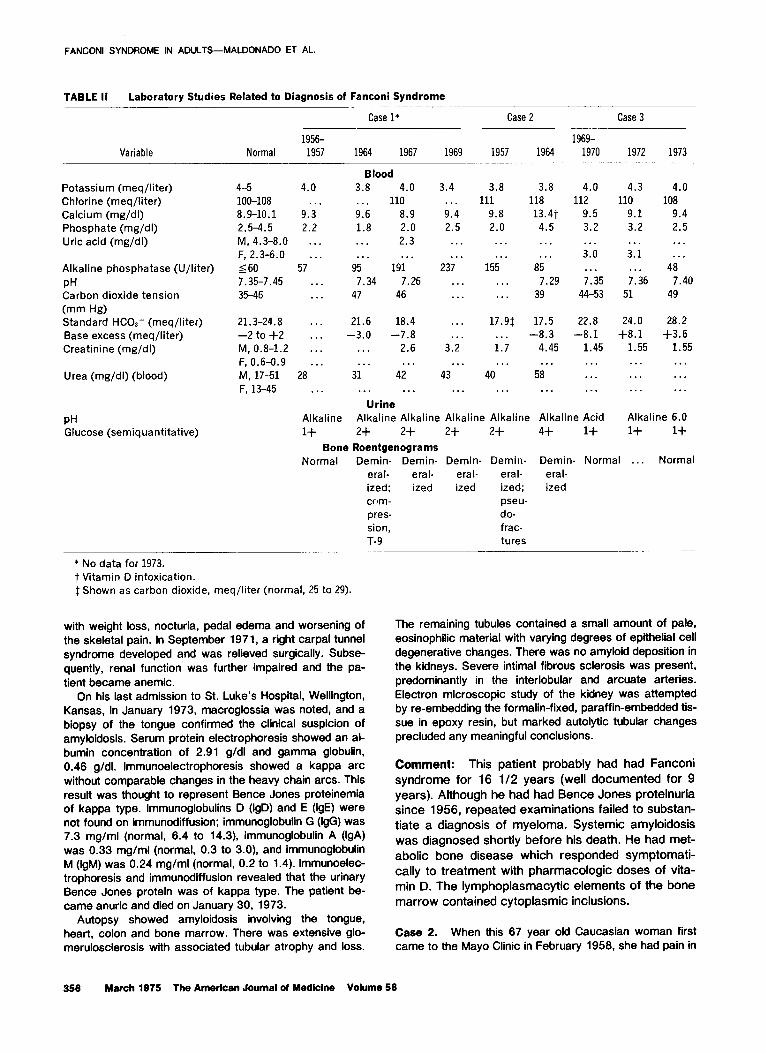
TABLE II Laboratory Studies Related to Diagnosis of Fanconi Syndrome
Variable Normal 1956- 1957
Case l* Case 2 Case 3 - _
1969- 1964 1967 1969 1957 1964 1970 1972 1973
Blood 3.8 4.0 . . . 110 9.6 8.9 1.8 2.0 . . . 2.3 . . . . . .
95 191 7.34 7.26
47 46
Potassium (meq/liter) Chlorine (meq/liter) Calcium (mg/dI) Phosphate (mg/dl) Uric acid (mg/dl)
Alkaline phosphatase (U/liter) PH Carbon dioxide tension (mm Hg) Standard HCOI- (meq/liter) Base excess (meq/liter) Creatinine (mg/dl)
Urea (mg/di) (blood)
4-5 4.0 loo-108 . . . 8.9-10.1 9.3 2.5-4.5 2.2 M, 4.3-8.0 . . . F, 2.3-6.0 . . .
560 57 7.35-7.45 . . . 35-46 . . .
3.4 . . . 9.4 2.5
3.8 3.8 4.0 4.3 4.0 111 118 112 110 108
9.8 13.4t 9.5 9.1 9.4 2.0 4.5 3.2 3.2 2.5
. . . 3.0 . . .
7.35 44-53
. . . 3.1 . . .
7.36 51
. . .
. . . 237
. . . . . .
155 . . .
85 7.29
39
. . . 48
7.40 49
. . *
. . .
21.3-24.8 . . . -2 to +2 .*. M, 0.8-1.2 . . .
F, 0.6-0.9 . . . M, 17-51 28 F, 13-45 . . .
21.6 18.4 -3.0 -7.8
. . . 2.6
..* . . . 31 42
17.9$ . . . 1.7 . . .
40
17.5 22.8 24.0 28.2 -8.3 -8.1 +8.1 +3.6
4.45 1.45 1.55 1.55 . . .
58 . . . . . .
. . .
. . . . . . . . . . . .
. . . 3.2
. . . Urine
Alkaline Alkaline Alkaline Alkaline Alkaline Alkaline Acid Alkaline 6.0
1+ 2+ 2+ 2+ 2+ 4+ 1+ 1+ 1+
Bone Roentgenograms Normal Demin- Demin- Demin- Demin- Demin- Normal . . . Normal
eral- eral- eral- eral- eral- ized; ized ized ized; ized com- pseu- pres- do- sion, frac- T-9 tures
_
PH Glucose (semiquantitative)
* No data for 1973. t Vitamin D intoxication 1 Shown as carbon dioxide, meq/liter (normal, 25 to 29).
with weight loss, nocturia, pedal edema and worsening of the skeletal pain. In September 1971, a right carpal tunnel syndrome developed and was relieved surgically. Subse- quently, renal function’ was further impaired and the pa- tient became anemic.
On his last admission to St. Luke’s Hospital, Wellington, Kansas, in January 1973, macroglossia was noted, and a biopsy of the tongue confirmed the clinical suspicion of amylokfosis. Serum protein electrophoresis showed an al- bumin concentration of 2.91 g/dl and gamma globulin, 0.46 g/dl. lmmunoelectrophoresis showed a kappa arc without comparable changes in the heavy chain arcs. This result was thought to represent Bence Jones proteinemia of kappa type. lmmunoglobulins D (IgD) and E (IgE) were not found on immunodiffusion; immunoglobulin G (IgG) was 7.3 mg/ml (normal, 6.4 to 14.3), immunoglobulin A (IgA) was 0.33 mg/ml (normal, 0.3 to 3.0), and immunoglobulin M (IgM) was 0.24 mg/ml (normal, 0.2 to 1.4). Immunoelec- trophoresis and immunodiffusion revealed that the urinary Bence Jones protein was of kappa type. The patient be- came anuric and died on January 30, 1973.
Autopsy showed amyloidosis involving the tongue, heart, colon and bone marrow. There was extensive glo- merulosclerosis with associated tubular atrophy and loss.
The remaining tubules contained a small amount of pale, eosinophilic material with varying degrees of epithelial cell degenerative changes. There was no amyloid deposition in the kidneys. Severe intimal fibrous sclerosis was present, predominantly in the interlobular and arcuate arteries. Electron microscopic study of the kidney was attempted by re-embedding the formalin-fixed, paraffin-embedded tis- sue in epoxy resin, but marked autolytic tubular changes precluded any meaningful conclusions.
Comment: This patient probably had had Fanconi syndrome for 16 l/2 years (well documented for 9 years). Although he had had Bence Jones proteinuria since 1956, repeated examinations failed to substan- tiate a diagnosis of myeloma. Systemic amyloidosis was diagnosed shortly before his death. He had met- abolic bone disease which responded symptomati- cally to treatment with pharmacologic doses of vita- min D. The lymphoplasmacytic elements of the bone marrow contained cytoplasmic inclusions.
Case 2. When this 67 year old Caucasian woman first came to the Mayo Clinic in February 1956, she had pain in
356 March 1975 The American Journal of Medicine Volume 58

the hips, legs and ribs, and weakness of the lower ex- tremities. She had had glycosuria in 1950 and proteinuria in 195 1. The major physical finding was diminished muscle strength in both legs. Laboratory results are summarized in Tables I and II.
Roentgenograms of the pelvis, spinal column and ribs showed demineralization. There were pseudofractures in the pelvis, and the roentgenographic picture was interpret- ed as osteomalacia. A diagnosis of Fanconi syndrome was corroborated by the laboratory findings (Table II). The uri- nary excretion of alpha-amino acid nitrogen was in- creased. She had grade 2 proteinuria and a positive Bence Jones reaction. Protein electrophoresis showed that most of the protein in the urine migrated as an alpha* globulin. The serum protein electrophoretic pattern and the sedi- mentation rate were normal. A bone marrow aspirate was not diagnostic and contained 32 per cent lymphocytes and 2 per cent plasmacytes. Treatment with 50,000 units of vi- tamin D daily and oral potassium was begun.
The patient’s symptoms were alleviated, and she was not seen at the Mayo Clinic again until September 1964, when she returned with generalized weakness and inter- mittent pedal edema. During the interval between visits, her renal function had deteriorated markedly. However, bone roentgenograms showed cessation of the demineral- ization and disappearance of the pseudofractures. Pancy- topenia was found. The sedimentation rate was 113 mm in 1 hour (Westergren). Bence Jones proteinuria persisted, and electrophoresis of urinary protein again showed an alpha2 globulin narrow-based spike with 11 g of protein/24 hours. A bone marrow aspirate showed 39 per cent lym- phocytes and 1 per cent plasma cells but was thought to be nondiagnostic.
In November 1964, easy bruising and gingival bleeding developed. When the patient was seen elsewhere, the bone marrow aspirate was diagnostic of multiple myelo- ma. Therapy with melphalan (Alkeranm) and prednisone was started. Severe epistaxis and gastrointestinal bleed- ing developed, and the patient died on December 12, 1964.
Autopsy confirmed the diagnosis of myeloma. Some of the myeloma cells contained ovoid and rhomboid inclusion bodies. There was glomerulosclerosis involving from 15 to 20 per Cent of the glomeruli, and generalized atrophy and loss of the proximal convoluted tubular elements. Numer- ous small, granular, eosinophilic casts were present, and several tubules contained small amounts of calcium. Ap- proximately 20 per cent of the glomeruli were fibrotic. Electron microscopy of the renal tissue obtained at autop- sy was not satisfactory.
Comment: This woman had had proteinuria and glycosuria for 14 and 13 years, respectively, and documented Fanconi syndrome for 5 years before she died of myeloma. Although she had had Bence Jones proteinuria for 5 years (and perhaps longer) and her marrow was suggestive of lymphoprolifera- tion, it was not possible to make a diagnosis of mye- loma until the end of her illness. There were cyto- plasmic inclusion bodies in the myeloma cells in the
FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
bone marrow obtained at autopsy. She had the type of metabolic bone disease seen in Fanconi syn- drome, which responded to large doses of vita- min D.
Case 3. This 50 year old Caucasian man came to the Mayo Clinic in January 1969 for evaluation of glycosuria that had been discovered in the preceding year. He had had proteinuria since 1964. His physical examination gave unremarkable results. The pertinent laboratory data are summarized in Tables I and II.
A diagnosis of Fanconi syndrome was made based on the presence of glycosuria, phosphaturia (tubular reab- sorption of phosphorus, 68.8 per cent; normal, 85 to 95 per cent), aminoaciduria (cystine, ornithine and lysine pri- marily, and tyrosine, valine and phenylalanine to a lesser extent), and hypouricemia. He did not have an acidification defect when challenged with an ammonium chloride load, and renal function was slightly below normal as assessed by inulin clearance. The patient denied exposure to heavy metals, and the urine contained no excess of lead or arse- nic. A 24 hour urine specimen contained 2.4 g of protein that migrated as a dense beta globulin band. The Bence Jones heat test was positive. The serum protein electro- phoretic pattern was normal. Those bones examined roentgenologically were normal. A kidney biopsy specimen showed mild nonspecific changes (minimal segmental mesangial matrix increase).
When the patient returned in January 1970, the pre- viously noted abnormalities and the Bence Jones protein- uria (2.1 g/24 hours) still were present, but the serum pro- tein electrophoretic pattern remained normal. A bone mar- row aspirate contained 10.5 per cent plasma cells. Some cells had lymphoid features and many contained needle- like cytoplasmic inclusion bodies that stained faintly blue with Wright’s stain. The bone marrow was not diagnostic.
The patient was seen again in January 1971. He had re- mained asymptomatic. Renal function remained stable. A repeat bone marrow aspirate showed 12 per cent plasma cells, again with crystalline inclusion bodies. In February 1972, the situation was essentially unchanged. A meta- static bone survey was negative.
In January 1973, the patient was still asymptomatic. The hemogram, creatinine concentration and bone survey remained normal. The urine contained a beta globulin (1.7 g/24 hours) that was identified as kappa type light chain. lmmunoelectrophoresis failed to detect a monoclonal serum protein. The bone marrow contained 9 per cent plasma cells, many with crystalline inclusion bodies. Total gamma globulin was decreased. The serum immunoglobu- lin values were IgG 6.2 mg/ml, IgA 0.65 mg/ml and IgM 0.46 mg/ml. Urinary amino acid excretion was normal in several first-degree relatives. Electron Mlcrosc~plc Studies. Bone marrow: Sam- ples for electron microscopy were obtained in 1971, 1972 and 1973. After aspiration, the marrow particles were promptly fixed in 3 per cent glutaraldehyde in 0.1 M phos- phate buffer (pH 7.3), postfixed in 2 per cent osmic acid, dehydrated and embedded in epoxy. The ultra thin sections were double-stained with lead citrate and uranyl acetate and viewed with a Hitachi HU-12 electron microscope.
March 1975 The American Journal of Medicine Volume 58 357

FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
Figure 7. Complete section of one plasma cell and partial section of another, showing multiple electrorxfzs- mic inclusion bodies. Some inclusion bodies have geometric configuration; others are of irregular shape and variable densi- ty. N = nucleus. Original magnification X 16,500.
358 March 1975 The American Journal of Medicine Volume 58

FANCONI SYNDROME IN ADULTS-MALDONAOO ET AL.
March 1975 The American Journal of Medicine Volume 59 359

FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
The number of cells with plasmacytic features was mogeneous or with a linear periodicity. In some inclusion
higher in the electron microscopic preparations than in the bodies, a homogeneous, moderately dense background
smears. A striking feature was the presence of cytoplas- was mixed with areas .of high density. Other structures re-
mic inclusion bodies of increased but variable electron sulted from the irregular aggregation of dense, rounded or
density (Figures 1 and 2). These inclusion bodies appeared “lumpy” structures of electron-dense material.
most often as needle- or rod-shaped structures, either ho- Some of the cytoplasmic inclusion bodies showed a
TABLE 111 Reported Cases of Fanconi Syndrome with Myeloma or “Premyeloma”
source
Patient’s Sex and
&a (yr)* Year
Sirota, Hamerman [l]
Dragsted, Hjorth [2]
Engle, Wallis [3]
Muntendam 14)
Short, Smith [5]
Gomes da Costa et al. [6]
Dedmon et al. [7]
Costanza, Smaller [B]
Harrison, Blainey [9]
Horn et al. [lo]
. . . 1960
M,51 19541
1955
F,67 1960
M.40 19632
1963
1970
M,46 19618
1963
1964
1965 1969
1973
F,59 1964$
Headley et al. (111 F,55
Lee et al. (121 F,34
Finkel et al. [13] F,57
M,54 19441
1947 1951
M,49 19441
1949 1951
1952
1953
M,38 1952
1952
F,54 19521
1953
1955
M,65 1957
1965
1966
19611
1963
1964
1963$ 1966
19581 1961 1964 1967 1968 1970
Summary of History
Findings
Glycosuria; proteinuria
Dx Fanconi syndrome
Dx myeloma
Proteinuria
Proteinuria; glycosuria
Osteomalacia
Dx Fanconi syndrome
Dx myeloma
Proteinuria
Dx Fanconi syndrome and myeloma
Proteinuria; ankle and foot pain
Glycosuria Dx Fanconi syndrome and myeloma
Glycosuria; Bence Jones proteinuria; Dx
Fanconi syndrome and myeloma;
death; autopsy, amyloidosis
Dx Fanconi syndrome and myeloma
Glycosuria
Dx Fanconi syndrome and myeloma
Dx Fanconi syndrome and myeloma
Thirst, polyuria, muscle weakness
Dx Fanconi syndrome
Myeloma Tiredness, lassitude, “muscle pains”
“Bone pains”
Polyuria
Dx Fanconi syndrome
Nephrocalcinosis Sudden death
Back pain, waddling gait; Dx Fanconi
syndrome . . .
Dx myeloma
Nocturia
Glycosuria; proteinuria, etc. (?Fanconi
syndrome)
Dx myeloma
Glycosuria Glycosuria, proteinuria, etc.; Dx Fanconi
syndrome and “variant light chain
disease”
Proteinuria Proteinuria, glycosuria, etc. Dx Fanconi syndrome
BJPt
-
+
NM
+
+ +
+ + (ep)
+ K (iep)
+ K (ied
+ Cw)
K (iep)
+
K (iw)
. . . Epistaxis; heavy proteinuria (6-9 g/24 hr) Death; autopsy, amyloidosis
+ (ep) K (rep)
*Age at time of diagnosis of myeloma or Fanconi syndrome or at death. t BJP = Bence Jones protein; NM = not mentioned; ep = electrophoresis; iep = immunoelectrophoresis. $ “Premyeloma” + myeloma. 5 “Premyeloma.”
360 March 1975 The American Journal of Medlclne Volume 58

FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
clear-cut lattice arrangement with a linear periodicity of 85 to 105 A (Figure 2). The over-all appearance was honey- combed and, at high magnification, there was a suggestion that the lines were formed by fibers with a central hole producing an irregular “ropy” appearance.
The larger, needle-like inclusion bodies reached 5 or 8 pm in length but, interestingly, often had a geometric con- figuration with a well defined hexagonal, pentagonal or rectangular shape. Furthermore, of interest but of un- known significance, in the center of these inclusion bodies there often were “empty” spaces of similar geometric shape.
The cytoplasmic inclusion bodies were invariably sur- rounded by agranular or smooth membranes and were present in different areas of the cytoplasm. There were no inclusion bodies inside the sacs of the rough endoplasmic reticulum, which was most often of the flattened lamellar type [ 141. The predominant plasma cells were of well dif- ferentiated, mature type. Kldney: The kidney tissue was obtained by needle biopsy in January 1989 but was processed in standard fashion for light microscopy only; for electron microscopy, the sample was re-embedded in epoxy and sectioned, stained and viewed as described for the bone marrow. For orientation, thicker (0.5 to 1.0 pm) sections were examined for selec- tion of areas in which cytoplasmic inclusion bodies were seen. Although preservation of the tissues was of substan- dard quality for electron microscopy, well defined electron- dense inclusion bodies were seen in the cytoplasm of tu- bular cells. Some of the larger, needle-like inclusion bodies had a remarkable similarity to those seen in the bone mar- row. In addition, there were smaller, rounded, comma- shaped or ovoid inclusion bodies.
Comment: This patient had had proteinuria for 9 years. A diagnosis of Fanconi syndrome was made in 1969. Despite the Bence Jones proteinuria, a diagno- sis of myeloma. has not been established, although the patient is suspected of having a plasmacytic dys- crasia. Lymphoplasmacytic elements in the bone marrow and tubular cells in the kidney contained sim- ilar crystalline cytoplasmic inclusion bodies. As yet, metabolic bone disease has not developed, but the serum phosphorus level has decreased.
COMMENTS
The most pertinent information from the 14 cases of the association of Fanconi syndrome with Bence Jones proteinuria and myeloma or amyloidosis re- ported [l-13] in the literature is summarized in Table Ill.
Typical or highly suggestive features of Fanconi syndrome (a group of abnormalities resulting from dysfunction of the proximal renal tubules and includ- ing glycosuria, aminoaciduria, phosphaturia, acidosis and frequently osteomalacia) preceded the diagnosis of myeloma or amyloidosis in 11 patients ([ 1,2,4,7, 10-131; Case 1, [9]; and our Cases ,l and 2). The
phase of premyeloma has lasted as long as 16 l/2 years. In three patients [3,5,8], the two diagnoses were made simultaneously. In one case, the clinical information is incomplete [6]. Of the two patients de- scribed in 1967 by Harrison and Blainey [9], in one (Case l), myeloma developed in 1970 after 7 l/2 years of Fanconi syndrome, and in the other nephrocalcinosis developed and the patient died sud- denly in 1973 of unknown causes (personal commu- nication): this second patient remained clinically in a “premyelomatous” state until his death. One of our patients (Case 3) thus far can only be suspected of harboring a plasmacytic dyscrasia. Based on several of the cases reported in the literature and on the evo- lution in our Cases 1 and 2, in this patient a clearly di- agnostic condition, such as myeloma or amyloidosis, probably will develop. It is of great interest that, de- spite much experience with myeloma, no cases have been reported in which Fanconi syndrome developed after myeloma.
Multiple myeloma can cause nonspecific deminer- alization alone. However, in addition to demineraliza- tion, there frequently are characteristic osteolytic le- sions. In other patients the demineralization is the re- sult of osteomalacia, and typical pseudofractures may be present. Osteomalacia occurs when there is dysfunction of the proximal renal tubules (renal tubu- lar acidosis type II) with inappropriate renal excretion of phosphorus and resulting hypophosphatemia. The serum alkaline phosphatase value is high and, in con- trast to many other patients with myeloma, the serum calcium level is normal. A roentgenologic pic- ture of osteomalacia with associated pseudo- fractures was found in six patients ([1,2,7,12,13] and our Case 2). In an additional patient the laborato- ry values, but not the roentgenograms, were consis- tent with osteomalacia (Case 1, [9]); in another pa- tient, who had no pseudofractures, osteomalacia was proved by biopsy (Case 2, [9]). Demineralization alone was found in two patients [4,11]. Osteolytic le- sions were found in three patients [5,8, lo], all of whom by then had fully developed myeloma. In our Case 3, roentgenographic evidence of osteomalacia has not yet developed, and the serum alkaline phos- phatase value has remained normal: however, serum phosphorus is decreasing and clinical osteomalacia may develop in the future.
With the exception of one of the two patients (Case 2) described by Harrison and Blainey [9], and who had an initial “normal” bone marrow, all the other patients, including ours,, had a plasmacytic dys- crasia at one time or another. This dyscrasia may have consisted originally of mild plasmacytosis or lymphoplasmacytosis, but with the eventual progres- sion of the disease to myeloma or to amyloidosis. In
March 1975 The American Journal of Medlclne Volume 58 381

FANCONI SYNDROME IN ADULTS-MALDONAOO ET AL.
at least two cases ([ 121 and our Case 3), the in- crease in plasmacytic elements was greater in the samples examined with the electron microscope than in the bone marrow smears examined by light microscopy.
Of particular interest was the finding of cytoplas- mic inclusion bodies in the lymphoplasmacytic ele- ments. These were specifically reported in six cases [3,7,8,1 l-131 and were present in our three cases. In several other cases, cytoplasmic crystals were not mentioned and may have been present but unrecog- nized. These inclusion bodies are described as rec- tangular, rhomboid or rounded, and usually show a slightly basophilic staining. In three cases ([ 12,131 and our Case 3), electron microscopy has been per- formed. The ultrastructural appearance of the lym- phoplasmacytic elements containing the inclusion bodies is characteristic, with frequent clear-cut crys- talline structures with a linear lattice. Perhaps the most salient feature is the fact that in all three cases the inclusion bodies appeared to be surrounded by smooth membranes. A variety of electron-dense cy- toplasmic inclusion bodies have been described in the plasma-myeloma cell [ 14-171, but by and large they are surrounded by rough or granular endoplas- mic reticulum in contrast to the definite and consis- tent smooth membranes seen in the patients dis- cussed here.
Bessis [ 171 published electron micrographs of cy- toplasmic crystalline inclusion bodies surrounded by smooth membranes in what he described as an “ex- ceptional” type of variant of plasma-myeloma cell, but he gave no clinical information about the case. In 1949, Neumann [ 181 reported a case of myeloma with Bence Jones proteinuria in which there were rhomboid crystals in the myeloma cells and in the renal tubules. These two cases probably belong in the category of patients we have included in our study. Likewise, Ito et al. [19] reported a case of kappa light chain disease (myeloma) with plasmacyt- ic and renal tubular cytoplasmic inclusions exactly as in our Case 3 and proposed a pathologic model for the crystalline protein. The scanty clinical information provided in the paper does not permit a diagnosis of Fanconi syndrome.
In 12 cases the renal changes have been studied in specimens obtained by biopsy or at autopsy ([2,3,5,7,8,9,12,13] and in our three cases). In the majority there were degenerative changes in the tu- bular cells, particularly of the proximal convoluted tu- bules. In eight cases ([3,5,7.8,12,13]; Case 1, [9]; and our Case 3), crystalline inclusion bodies were present in the tubular cells. In three cases (112, 131 and our Case 3), electron microscopic studies were made; there was a remarkable similarity between the
plasmacytic and the renal tubular inclusion bodies, as noted in our Case 3.
Essentially all the patients have had Bence Jones proteinuria which, in our cases and in several others [ 10,12,13], has been part of the premyelomatous phase of the disease. Furthermore, kappa type was present in all seven cases in which Bence Jones pro- tein was typed ([9,10.13,14] and our Cases 1 and 3). Electrophoresis of the urine was sometimes nec- essary for the demonstration of Bence Jones protein. Urine protein electrophoresis should be part of the study of an adult with Fanconi syndrome. Except for Bence Jones proteinemia in the terminal phase in one of our patients (Case l), there were no monoclo- nal serum spikes in any case.
Three of the patients ([5,13] and our Case 1) have had systemic amyloidosis as the terminal phase of their illnesses but, despite the renal tubular dys- function, the kidneys have been free of amyloid infil- tration in these cases.
In vitro, the Bence Jones protein is toxic to the renal tubular cell [20,21]. It therefore is tempting to postulate that tubular reabsorption of the light chain leads to damage of the renal tubule and to its dys- function, with the secondary development of the Fan- coni syndrome. This has been proposed by other in- vestigators [8,9]. it is necessary, however, to explain first the abnormal production and the urinary excre- tion of the Bence Jones protein. The most plausible explanation is that the group of patients we present have a basic plasma cell disorder of slow growth rate that first expresses itself by the excretion of light chain, and this in turn leads to damage of the renal tubule and to the Fanconi syndrome. Many years may be necessary for the full development of myelo- ma or amyloidosis and, in the interim, the clinical pic- ture is dominated by the metabolic manifestations secondary to the renal tubular disease.
Recently, lsobe and Osserman [22] emphasized the striking association of Bence Jones proteinuria and amyloidosis and again proposed that, in the pathogenesis of amyloidosis, the initial abnormalities are abnormal plasma cells and the production of Bence Jones protein. In many instances, the devel- opment of clinically overt amyloidosis may take a long time to occur, such as in Case 1 of this report.
In two patients described by our group, Bence Jones proteinuria has been present for many years without evidence of the development of myeloma, amyloidosis or other malignant disease [23]. These patients do not have impaired renal function: they have no features of the Fanconi syndrome. It is evi- dent then that, even when present for a prolonged time, Bence Jones proteinuria is not always nephro- toxic. Additional unknown factors, perhaps the chem-
362 March 1975 The American Journal of Medicine Volume 58

ical composition of the Bence Jones protein, may have a role in renal tubular damage and dysfunction.
Renal tubular dysfunction, usually distal, has been reported in disorders accompanied by polyclonal hy- pergammaglobulinemia but without monoclonal Bence Jones proteinuria [24-281.
If the cytoplasmic inclusion bodies of the marrow plasma cells are of protein nature-a likely possibil- ity-they may have some relatively unique proper- ties. The fact that they are surrounded by smooth membranes suggests that they are within elements probably derived from the Golgi apparatus. This suggests an intracellular metabolic synthetic block or disturbance at this level. Second, these inclusion bodies have peculiar ultrastructural features and form crystalline lattices with special configurational arrangements. Finally, the inclusion bodies in the renal tubular cells are morphologically similar. In ad- dition, all the patients typed thus far have had kappa light chains.
ACKNOWLEDGMENTS
We are grateful for the clinical information provided on Case 1 by Dr. T. J. Luellen, Wichita, Kansas. We thank Dr. Shelby Rose, St. Luke’s Hospital, Welling- ton, Kansas, who kindly made available to us the bi- opsy specimens of the tongue in Case 1 and the au- topsy protocol and tissues as well as urine and serum samples obtained shortly before the patient’s death.
We are also grateful to Dr. H. G. Langford, Univer- sity of Mississippi Medical Center, Jackson, for mak- ing clinical records and bone marrow and autopsy material in Case 2 available to us, and to Dr. J. C.
FANCONI SYNDROME IN ADULTS-MALDONADO ET AL.
Hunt, Mayo Clinic, for his valuable assistance in the study of the patient in Case 3.
ADDENDUM
Since this manuscript was accepted for publication, we have seen another patient with both myeloma and Fanconi syndrome. This patient, a 59 year old man, had Bence Jones proteinuria and features sug- gestive of acquired adult Fanconi syndrome 3 years before the diagnosis of multiple myeloma was estab- lished. There was no monoclonal serum peak. In 24 hours he excreted 3.7 g of protein which gave a pos- itive Bence Jones reaction and had a monoclonal configuration by electrophoresis. This monoclonal urinary protein was kappa type. Many of the abnor- mal plasma cells in the bone marrow were of lym- phoid type and contained small rounded hyalin inclu- sion bodies. This patient shared many of the features of the other patients we described herein.
One of our patients (Case 3) was seen in October 1974; he was clinically well without evidence of bone disease. The urinary protein excretion was 3.9 g/24 hours.
Recently, Clyne et al. [29] were able to induce renal proximal tubular lesions in rats by the intraperi- toneal injection of kappa type Bence Jones proteins. Electron microscopy revealed crystallized inclusion bodies in the proximal tubular cells; by immunofluo- rescence, kappa chains were demonstrated in these cells. It is suggested that the Bence Jones protein is filtered in the glomerulus and reabsorbed in the renal tubule. This experimental study supports the hypothe- sis that, in the cases of Fanconi syndrome reviewed in our paper, a similar phenomenon occurred.
REFERENCES
7.
6.
Sirota JH. Hamerman D: Renal function studies in an adult subject with the Fanconi syndrome. Am J Med 16: 138, 1954.
Dragsted PJ. Hjorth N: The association of the Fanconi syn- drome with malignant disease. Dan Med Bull 3: 177, 1956.
Engle RL Jr, Wallis LA: Multiple myeloma and the adult Fan- coni syndrome. I. Report of a case with crystal-like de- posits in the tumor cells and in the epithelial cells of the kidney. Am J Med 22: 5, 1957.
Muntendam DH: Muftipele myelomatosis en het syndroom van Da Toni-Fanconl. Ned Tljdschr Geneeskd 102: 1890, 1958.
Short IA, Smlth JP: Myelomatosis associated with glycosur- ia and aminoaclduria. Scott Med J 4: 89, 1959.
Comes da Costa SF, Relvas MESA, Halpern MJ, Da Silva JAF: Contribucies para o estudo da patologia quimica de urn case de mieloma associado ao sindroma de Fan- coni. Gaz Med Portuguesa 13: 583, 1960.
Dedmon RE, West JH, Schwartz TB: The adutt Fanconi syn- drome: report of two cases, one with multiple myeloma. Med Clin North Am 47: 191, 1983.
Costanza DJ, Smoller M: Multiple myeloma with the Fanco-
9.
10.
11.
12.
13.
14.
15.
16.
ni syndrome: study of a case, with electron microscopy of the kidney. Am J Med 34: 125, 1963.
Harrison JF, Blainey JD: Adult Fanconi syndrome with monoclonal abnormality of lmmunoglobulin light chain. J Clin Pathol20: 42, 1967.
Horn ME, Knapp MS, Page FT, Walker WHC: Adult Fanconi syndrome and multiple myelomatosis. J Clin Pathol 22: 414, 1969.
Headley RN, King JS Jr, Cooper MR, Felts JH: Multiple mye- loma presenting as adult Fanconi syndrome. Clin Chem 18: 293, 1972.
Lee DBN, Drinkard JP, Rosen VJ. Gonick HC: The adult Fanconi syndrome: observations on etiology, morpholo- gy, renal function and mineral metabolism in three pa- tients. Medicine (Batt) 51: 107. 1972.
Finkel PN, Kronenberg K, Pesce AJ, Pollack VE. Pirani CL: Adult Fanconi syndrome, amylokfosis and marked n-light chain protelnuria. Nephron 10: 1, 1973.
Mafdonado JE, Brown AL Jr, Bayrd ED, Pease CL: Ultra- structure of the myeloma cell. Cancer 19: 1613, 1968.
Zucker-Franklin D: Structural features of cells associated with the paraproteinemias. Semin Hematol 1: 185, 1984.
Maldonado JE. Brown AL Jr, Bayrd ED, Pease CL: Cyto-
March 1975 The Amerkan Journal of Medlclne Volume 58 383

FANCONI SYNDROME IN ADULTS-MALOONADO ET AL.
plasmic and intranuclear inclusion electrondense bodies in the myeloma cell: light and electron microscopy ob- servations. Arch Pathol81: 484, 1966.
17. Bessis M: Living Blood Cells and Their Ultrastructure. New York, Springer-Verlag, 1973.
18. Neumann V: Multiple plasma-cell myeloma with crystalline deposits in the tumour cells and in the kidneys. J Pathol Biol61: 165, 1949.
19. lto S, Goshima K. Ninomi M, Horikoshi N, Nomura S, Sugi- ura K, Yamazaki K, Hirabayashi N, Nishi Y: Electron mi- croscopic studies of the crystalline inclusions in the mye- loma cells and kidney of K-Bence Jones protein type myeloma. Acta Haematol Jap 33: 598, 1970.
20. Preuss HG, Hammack WJ, Murdaugh HV: The effect of Bence Jones protein on the in vitro function of rabbit renal cortex. Nephron 5: 210, 1967.
21. Weiss FR: Role of Bence Jones and other urinary proteins in renal dysfunction (abstract). J Lab Clin Invest 76: 1048, 1970.
22. lsobe T, Osserman EF: Patterns of amyloidosis and their association wtth plasma-cell dyscrasia, monoclonal im- munoglobulins and BenceJones proteins. N Engl J Med
290: 473, 1974. 23. Kyle RA, Maldonado JE, Bayrd ED: Mlopathlc Bence Jones
proteinurla-a distinct entity? Am J Med 55: 222, 1973.
24. Wilson ID, Williams RC Jr, Tobfan L Jr: Renal tubular acido- sis: three cases with lmmunoglobulin abnormalities in the patients and their kindreds. Am J Med 43: 356, 1967.
25. Shearn MA, Tu WH: Latent renal tubular acidosis in Sjnr_ gren’s syndrome. Ann Rheum Dis 27: 27, 1968.
26. Runeberg L. L&devirta J, Collan Y, Jokinen EJ: Renal tubu- lar dysfunction and hypergammaglobulinaemla: electro- lyte balance, electron microscopic hypergammaglobu- linaemia. Acta Med Stand 189: 341, 1971,.
27. Walker BR, Alexanoer F, Tannenbaum PJ: Fanconi syn- drome with renal tubular acidosis and light chain protein- uria. Nephron 8:103, 1971.
28. Kamm DE, Fischer MS: Proximal renal tubular acidosis and the Fanconi syndrome in a patient with hypergamma- globulinemia. Nephron 9: 208, 1972.
29. Clyne DH, Brendstrup L, First MR. et al.: Renal effects of in- traperitoneal kappa chain Injection: induction of crystals in renal tubular cells. Lab Invest 31: 131, 1974.
364 March 1975 The American Journal of Medlclne Volume 58




















