
Increased Adenovirus Type 5 Mediated Transgene Expression Due to RhoB Down-Regulation
Upload
independentCategory
view
3download
0
HUMAN GENE THERAPY 18:589–602 (July 2007)© Mary Ann Liebert, Inc.DOI: 10.1089/hum.2007.002
Survivin-Driven and Fiber-Modified Oncolytic Adenovirus Exhibits Potent Antitumor Activity
in Established Intracranial Glioma
ILYA V. ULASOV,1 ZENG B. ZHU,2 MATTHEW A. TYLER,1 YU HAN,1 ANGEL A. RIVERA,2
ANDREY KHRAMTSOV,3 DAVID T. CURIEL,2 and MACIEJ S. LESNIAK1
ABSTRACT
The poor prognosis of patients with malignant gliomas necessitates the development of novel therapies. Vi-rotherapy, using genetically engineered adenovectors that selectively replicate in and kill neoplastic cells, rep-resents one such strategy. In this study, we examined several oncolytic vectors with modified transcriptionaland transductional control of viral replication. First, we incorporated the survivin promoter (S) to drive E1Agene expression. We then modified the adenovirus serotype 5 (Ad5) fiber protein via genetic knob switchingor incorporation of peptide ligands to target the following glioma-associated receptors: the Ad3 attachmentprotein, or CD46, �v�3/�v�5 integrins, or heparan sulfate proteoglycans. The three conditionally replicativeadenoviruses, CRAd-S-5/3, CRAd-S-RGD, and CRAd-S-pk7, were then examined in vitro with respect to trans-duction efficiency and tissue specificity. The most promising virus was then tested in vivo for evidence of tu-mor growth inhibition. CRAd-S-pk7 provided the highest level of viral replication and tumor oncolysis inglioma cell lines. At the same time, we observed minimal viral replication and toxicity in normal human brain.Injection of CRAd-S-pk7 inhibited xenograft tumor growth by more than 300% (p � 0.001). Sixty-seven per-cent of treated mice with intracranial tumors were long-term survivors (�110 days; p � 0.005). Analysis oftumor tissue indicated increased adenoviral infectivity, decreased mitotic activity, and enhanced tumor apop-tosis. These findings demonstrate the effectiveness of CRAd-S-pk7 and provide the rationale for further de-velopment of this novel oncolytic virus for glioma gene therapy.
589
INTRODUCTION
GLIOBLASTOMA MULTIFORME (GBM) represents the most ag-gressive form of primary brain cancer and despite surgery,
radiotherapy, or chemotherapy, the median survival rate doesnot exceed 1 year (Lesniak et al., 2001; Lesniak and Brem,2004; Hsieh and Lesniak, 2005; Lesniak, 2005). The invasivenature of GBM, the presence of local micrometastasis, and theinability to effectively penetrate the blood–brain barrier havesuccessfully prevented the development of effective therapies.The use of conditionally replicative adenoviral vectors (CRAds)represents a novel strategy that can be used to treat GBM. Be-
cause these tumors rarely metastasize outside of the central ner-vous system (CNS) and recur in proximity to the original site,direct delivery of an oncolytic virus offers the potential to ef-fectively target these tumors.
Adenoviral vectors are especially suitable in this treatmentstrategy and studies have established the efficacy and safety ofsuch vectors in the CNS. For example, Ad5-�24, an oncolyticvector with pRb binding-deficient E1A, has been tested in vivoin human glioma xenografts in nude mice and found to induceinhibition of tumor growth (Fueyo et al., 2000). Two other vari-ations of Ad5-�24, Ad5�24-hyCD (a vector expressing the cy-tosine deaminase gene, hyCD) and Ad5�24-RGD (an adeno-
1Division of Neurosurgery, Department of Surgery, University of Chicago, Chicago, IL 60637.2Division of Human Gene Therapy, Departments of Medicine, Pathology, and Surgery, and Gene Therapy Center, University of Alabama at
Birmingham, Birmingham, AL 35294.3Department of Pathology, University of Chicago, Chicago, IL 60637.

viral vector with an insertion of the Arg-Gly-Asp motif into thefiber knob of the �24 vector), have been shown to further ex-tend the survival of mice with experimental brain tumors (Lam-fers et al., 2002; Chiocca et al., 2004; Conrad et al., 2005).Most recently, a phase I trial using ONYX-015, an E1B gene-attenuated adenovirus, has been completed and showed that in-jection of up to 1010 plaque-forming units (PFU) was well tol-erated in patients with malignant glioma (Chiocca et al., 2004).Clinical efficacy was also demonstrated in GBM patients treatedwith adenoviral vectors delivering herpes simplex virus thymi-dine kinase (HSV-TK) into the tumor resection cavity, with sig-nificant prolongation of survival in the treatment group (Im-monen et al., 2004). Taken together, studies such as this clearlyprovide the scientific rationale for further development of tar-geted adenoviral gene therapies for GBM.
The therapeutic efficacy of a CRAd relies on the ability ofthe vector to successfully target, transduce, and replicate inGBM. One approach that allows an adenovirus to specificallyreplicate in tumor tissue takes advantage of a tumor-specificpromoter (TSP), which then responds to the specific cellularcues of tumor cells to mediate its replication. An attractive pro-moter element for glioma is survivin (S), a member of the in-hibitor of apoptosis protein (IAP) family. Survivin expressionin gliomas is associated with poor prognosis, increased rates ofrecurrence, and resistance to chemo- and radiotherapy(Chakravarti et al., 2002, 2004; Kajiwara et al., 2003; Yamadaet al., 2003). We have shown that the incorporation of this pro-moter into the adenoviral E1A region is responsible for en-hanced viral replication and an enhanced oncolytic effect in ma-lignant glioma (Van Houdt et al., 2006; Ulasov et al., 2007b).
Although the survivin promoter enhances the killing effectof glioma, there remains a need for a gene delivery componentthat mediates the effective binding and internalization of aCRAd to tumor cells. The wild-type human adenovirus (Ad5)has demonstrated relatively poor transduction in human tumors(Asaoka et al., 2000; Seki et al., 2002; Kanerva et al., 2004;Tango et al., 2004). The accepted rationale for this poor trans-duction is that tumor cells have limited surface expression ofthe Ad5 primary receptor, the coxsackievirus–adenovirus re-ceptor (CAR) (Cripe et al., 2001). Therefore, in an attempt toincrease tumor transduction efficiency, several fiber modifica-tions have been made to increase adenoviral tropism. For ex-ample, we have previously shown that a chimeric Ad5/3 vec-tor, which contains the shaft of Ad5 and knob of Ad3,effectively targets CD46 or the CD80/86 cellular receptor andexhibits increased transduction of malignant glioma comparedwith wild-type Ad5 (Ulasov et al., 2006, 2007a). We have alsoshown that incorporation of an RGD peptide motif into theAd5/3 fiber knob enhances the efficiency of transduction of ma-lignant glioma (Tyler et al., 2006). An alternative approach toenhancing adenoviral tropism has been the introduction ofpolylysine residues into the adenovirus fiber knob. Theseresidues selectively bind heparan sulfate proteoglycans(HSPGs), which are overexpressed in glioma (Qiao et al.,2003). Staba and coworkers used a replication-deficient type 5adenovirus with a pk7 fiber modification and showed that thepk7 modification endowed the virus with an enhanced abilityto transduce glioma (Staba et al., 2000). Our previous findingsas well as those of Staba and coworkers have given us reason-able cause to examine the use of conditionally replicating ade-
noviruses that contain the preceding fiber modifications in thecontext of malignant glioma.
In this study, we hypothesized that transcriptional and trans-ductional control of viral replication would enhance the oncolyticeffect of a virus against malignant glioma. To test this hypothe-sis, we constructed CRAd-S-5/3, CRAd-S-RGD, and CRAd-S-pk7, which bind to CD46, �v�3/�v�5, and HSPG, respectively.We then examined the targeting and oncolytic efficiency of ourCRAds in vitro and in vivo. Our results clearly demonstrate thatCRAd-S-pk7 has enhanced tumor targeting and exhibits enhancedtumor cell killing and should be considered for further preclini-cal development and testing in human clinical trials.
MATERIALS AND METHODS
Cells and cell culture
The human malignant glioma cell lines U-251 MG, U-118MG, U-87 MG, and A172, human lung carcinoma cell lineA549, and human kidney HEK293 cells were purchased fromthe American Type Culture Collection (Manassas, VA). Kings-1 and no.10 glioma cell lines were purchased from the Japan-ese Collection of Research Bioresources (JCRB) Cell Bank (Osaka, Japan). Primary human tissue samples were obtainedfrom patients undergoing intracranial surgery according to aprotocol approved by the Institutional Review Board of the Uni-versity of Chicago (Chicago, IL). All cells were maintained inDulbecco’s modified Eagle’s medium (DMEM) supplementedwith 10% fetal bovine serum (Mediatech, Herndon, VA) andincubated in 37°C and 5% CO2.
Adenoviral constructs
The following four CRAds were generated by our group onthe basis of wild-type adenovirus (AdWT): AdWT-S, CRAd-Survivin-RGD, CRAd-Survivin-5/3, and CRAd-Survivin-pk7(Zhu et al., 2006) (Fig. 1). The CRAd-Survivin constructs havethe following characteristics: (1) CRAd-Survivin constructscontain the human survivin promoter, termed CRAd-S, to driveE1 expression. The survivin-controlled E1 expression cassettewas placed in the original E1 region of the Ad gene as previ-ously described (Van Houdt et al., 2006). The native E1 pro-moter was deleted to avoid nonspecific viral replication; (2) re-combinant adenoviruses were created on the basis ofhomologous recombination in 911 cells between a shuttle vec-tor, pScs/PA/S, which carries a human survivin promoter, anda pVK700-based wild-type adenoviral 5 backbone containingan RGD motif incorporated into the HI loop of the adenoviralknob 5 protein (CRAd-S-RGD) (Dmitriev et al., 1998), a sub-stitution of fiber knob for adenovirus knob 3 type (CRAd-S-5/3) (Tekant et al., 2005), or a polylysine modification of thefiber knob (CRAd-S-pk7) (Wu et al., 2004); (3) the E3 genewas retained in the Ad genome to elevate oncolysis of the CRAdagents; and (4) a poly(A) signal was inserted between the in-verted terminal repeat (ITR) and the survivin promoter to stopthe nonspecific transcriptional activity of the ITR, and to retainthe tumor specificity of the survivin promoter. Viruses were se-lected from single plaques on 911 cells, expanded in A549 cells,and then purified by double CsCl gradient ultracentrifugation(Graham et al., 1977).
ULASOV ET AL.590

In vitro gene transfer
Viral cytopathic assays that were performed in this studyhave been previously described. Briefly, U-87 MG, Kings-1,U-251 MG, U-118 MG, A172, and no.10 cells (5 � 104 cellsper well) were either mock infected or infected with condi-tionally replicative Ad vectors (CRAd-S-5/3, CRAd-S-RGD, orCRAd-S-pk7), AdWT, or replication-deficient virus (reAd) at100, 10, or 1 VP/cell in 0.5 ml of DMEM with 2% FBS. After1 hr infection, the medium was removed and fresh growthmedium was added. Ten days later, the medium was aspiratedand the cells were stained with crystal violet. Pictures weretaken with a Sony digital camera.
Cell viability assay
U-118 MG cells were seeded in a 96-well plate (1 � 104
cells per well) and infected 24 hr later with 100 VP/cell. Theplates were incubated at 37°C and supplemented with 5% CO2.At various time points, the medium was removed and freshmedium containing 20 �l of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium(MTS) solution (Promega, Madison, WI) was added to eachwell. The cells were then incubated for 4 hr and absorbancewas recorded at 490 nm with an enzyme-linked immunosorbentassay (ELISA) plate reader.
Quantitative analysis of viral transcription and replication
U-118 MG human glioma cells (5 � 104 cells per well) weregrown to 60% confluence and seeded onto 24-well plates with1 ml of F12–Dulbecco’s modified Eagle’s medium. The nextday, medium was aspirated and the cells were infected withAdWT-S, CRAd-S-pk7, or AdWT adenoviral vectors at 100
VP/cell, and incubated at 37°C in a humidified atmosphere for1 hr. The cells were rinsed with phosphate-buffered saline(PBS) and 10% growth medium was added. For replicationanalysis, on days 1, 2, 5, and 8 aliquots of medium and de-tached cells were subjected to DNA isolation and quantitativepolymerase chain reaction (PCR). DNA was isolated from cellsaccording to a standard protocol, using a DNeasy tissue kit (Qi-agen, Valencia, CA) and a quantitative real-time PCR assay forE1 and E4 genes was performed. For quantification of E4mRNA expression, cell pellets were collected on days 1 and 3.Isolated RNA was converted into cDNA with the SuperScriptII system (Invitrogen, Carlsbad, CA) and subjected to quantifi-cation. The sequences of specific primers used for E4 amplifi-cation were as follows: sense, 5�-GGAGTGCGCCGAGAC-AAC-3�; antisense, 5�-ACTACGTCCGGCGTTCCAT-3�. Thesequences for E1A were previously described (Rein et al.,2005). The PCR was performed with glyceraldehyde-3-phos-phate dehydrogenase (GAPDH)-specific primers (TaqManGAPDH control reagent; Applied Biosystems, Foster City, CA)to create an internal standard. Quantification using SYBR GreenPCR master mix (Applied Biosystems) was performed accord-ing to vendor recommendations. Data were reported as the ra-tio of E4 copy number per human GAPDH copy.
Evaluation of virus replication and toxicity in normalhuman brain
Primary brain slices were prepared as previously described(Kirby et al., 2004) and cultured for 1 hr at 37°C. The sliceswere then infected with 500 VP per slice of AdWT or CRAd-S-pk7, or were mock infected. Four hours later, the slices wererinsed several times with PBS and fresh growth medium wasadded to the culture. One portion of infected slices was har-vested on days 1 and 3 for further progeny titration on HEK293
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 591
FIG. 1. Structure of conditionally replicative adenoviral vectors used in this study.

cells (Fueyo et al., 2000); the second portion was evaluated forlactate dehydrogenase (LDH) release, using a cell membraneintegrity kit (Promega).
In vivo xenograft models of U-87 MG glioma
Five- to 6-week-old female athymic mice (n � 6 per group,nu/nu; Charles River Laboratories, Wilmington, MA) were usedfor in vivo studies. Animals were cared for according to proto-cols approved by the Animal Use Committee of the Universityof Chicago. U-87 MG cells were cultured under standard con-ditions until they reached 70% confluence, were detached with0.05% trypsin solution, washed with serum-enriched medium,and then centrifuged at 1200 rpm for 5 min at 4°C. U-87 MGxenografts were established by preparing 1 � 106 cells in RPMI(Mediatech) and then injecting them subcutaneously into theflank region. When tumors reached 0.7 cm3, mice bearing tu-mors were divided in three groups: two groups received intra-tumoral CRAd-S-pk7 or AdWT at 1 � 1011 VP/mouse, pre-pared in RPMI; mice in the control group were mock infectedwith RPMI alone. Tumor volume was measured every 2 daysand determined by three-dimensional caliper measurements.
Human glioma xenografts were also generated in the brainsof athymic nude mice (n � 6 per group) according to a proto-col previously described by our group (Hsu et al., 2005; Les-niak et al., 2005). Briefly, 1 � 106 U-87 MG glioma cells wereinoculated intracranially into nu/nu BALB/c mice (day 0).Seven days after tumor inoculation, mice were randomized intothree groups, one receiving CRAd-S-pk7, one receiving AdWT,and one receiving RPMI. A total of 5 � 109 VP in a total vol-ume of 5 �l of RPMI per mouse was injected.
Histology and immunohistochemistry
Tumors obtained from treated and control animals were im-mediately fixed in formalin (3.7% formaldehyde–PBS) and sub-sequently embedded in paraffin. Five-millimeter-thick sectionswere deparaffinized and stained with hematoxylin and eosin(H&E). Tumor sections underwent immunohistochemical analy-sis with goat anti-hexon antibodies (1:500 dilution; ViroStat,Portland, ME) recognizing adenoviral hexon, rabbit monoclonalanti Ki-67 antibodies (M7187, 1:50 dilution; Dako, Glostrup,Denmark), and mouse anti-human CD31 (BD BiosciencesPharmingen, San Diego, CA) and were processed with Histostain(Zymed; Invitrogen) and the EnVision� system (Dako) accord-ing to the manufacturer’s instructions (Ulasov et al., 2006b).
To quantify cells positively stained for hexon, CD31, andKi-67 markers, an automated system was used to assess thequantity and morphology of stained cells (ACIS; Clarient, Al-iso Viejo, CA). Eleven �20 fields were randomly selected andthe computer software quantified the number of positive cellsin each field.
Quantitative analysis of nuclear and cytoplasmicintensities by automated cellular imaging system
The intensity of Ki-67 and hexon staining was measured withthe ACIS as described previously (van Gijssel et al., 2004). Thedensity of CD31 formation was evaluated by counting the num-ber of cells staining for endothelial antigen CD31 in 20 high-power fields and deriving an average value.
Western blot analysis
Protein extracts were prepared from xenograft cells 24 daysafter infection with conditionally replicative adenoviral vectorsor after mock infection. Protein concentrations were determinedby Bradford assay (Bio-Rad, Hercules, CA). Lysates were an-alyzed by Western blot analysis using 10% sodium dodecyl sul-fate (SDS) gels. Lanes were loaded with 50 �g of protein andelectrophoresed for 2 hr at 90 V. Gels were transferred topolyvinylidene difluoride (PVDF) membrane, blocked with 3%nonfat dry milk, and incubated with primary antibodiesovernight at 4°C. Primary antibodies included mouse mono-clonal antibody IgG to �-actin (1:2000 dilution; Abcam, Cam-bridge, MA) and rabbit polyclonal antibody to caspase-3(1:1000 dilution; Cell Signaling Technology, Danvers, MA).Horseradish peroxidase (HRP)-conjugated secondary antibod-ies, including goat anti-mouse (ab5870, 1:2500 dilution; Ab-cam) and goat anti-rabbit (1858415, 1:5000 dilution; PierceBiotechnology, Rockford, IL), were added for 1 hr and proteinsignals were detected with Immobilon Western substrate (Mil-lipore, Billerica, MA). Pictures were taken with a Kodak 440system.
Statistical analysis
Statistical comparison for all experimental conditions was performed by Student t test. Survival was plotted as a Kaplan–Meier survival curve and statistical significance wasdetermined by Kruskal–Wallis nonparametric analysis of vari-ance followed by the nonparametric analog of the New-man–Keuls multiple comparison test. p � 0.05 was consideredstatistically significant.
RESULTS
CRAd-S-pk7 effectively kills glioma cells in vitro
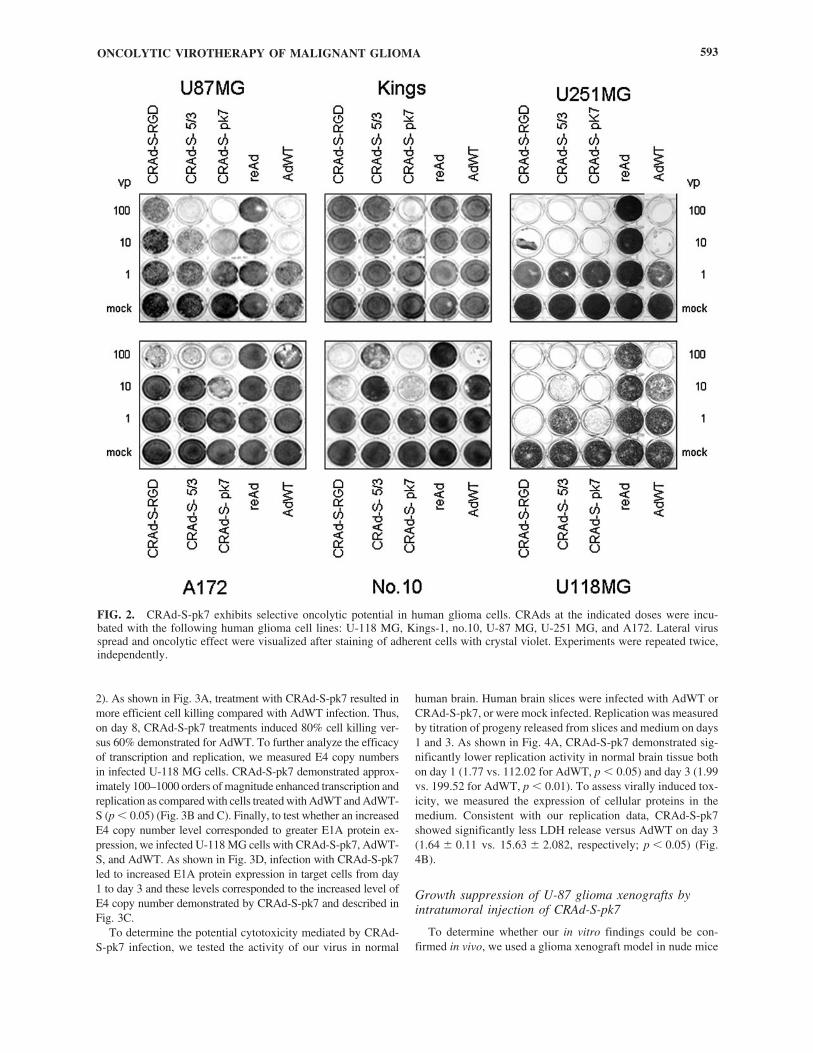
To assess the oncolytic effect of our CRAds, U-87 MG,Kings-1, U-251 MG, A172, no.10, and U-118 MG glioma celllines were exposed to CRAd-S-pk7, CRAd-S-5/3, CRAd-S-RGD, AdWT, or replication-deficient reAd virus at 1, 10, and100 VP/cell. Cytotoxic effect was then assessed via crystal vi-olet staining. Of the tested vectors, CRAd-S-pk7 demonstrateda dose-dependent cytolytic effect in all human glioma cell lines(Fig. 2). The virus induced cell killing at doses as low as 1VP/cell in U-118 MG cells and at 10 VP/cell in U-87 MG andU-251 MG cells. Kings-1, no.10, and A172 cells displayedlower cytotoxicity levels (�10-fold less) than did U-118 MGcells. Of note, the oncolytic effect of CRAd-S-pk7 was supe-rior to AdWT in five of the six tested cell lines. No cytotoxiceffect was observed for the control, replication-defective reAdvector.
CRAd-S-pk7 efficiently and selectively replicates inglioma cells
The in vitro results suggested that a pk7 knob modification im-proves the oncolytic capability of the survivin-modified CRAd invitro. To evaluate the dynamics of cell killing detected by crys-tal violet, we performed an MTS analysis of U-118 MG cells,which were most susceptible at low viral concentrations (see Fig.
ULASOV ET AL.592
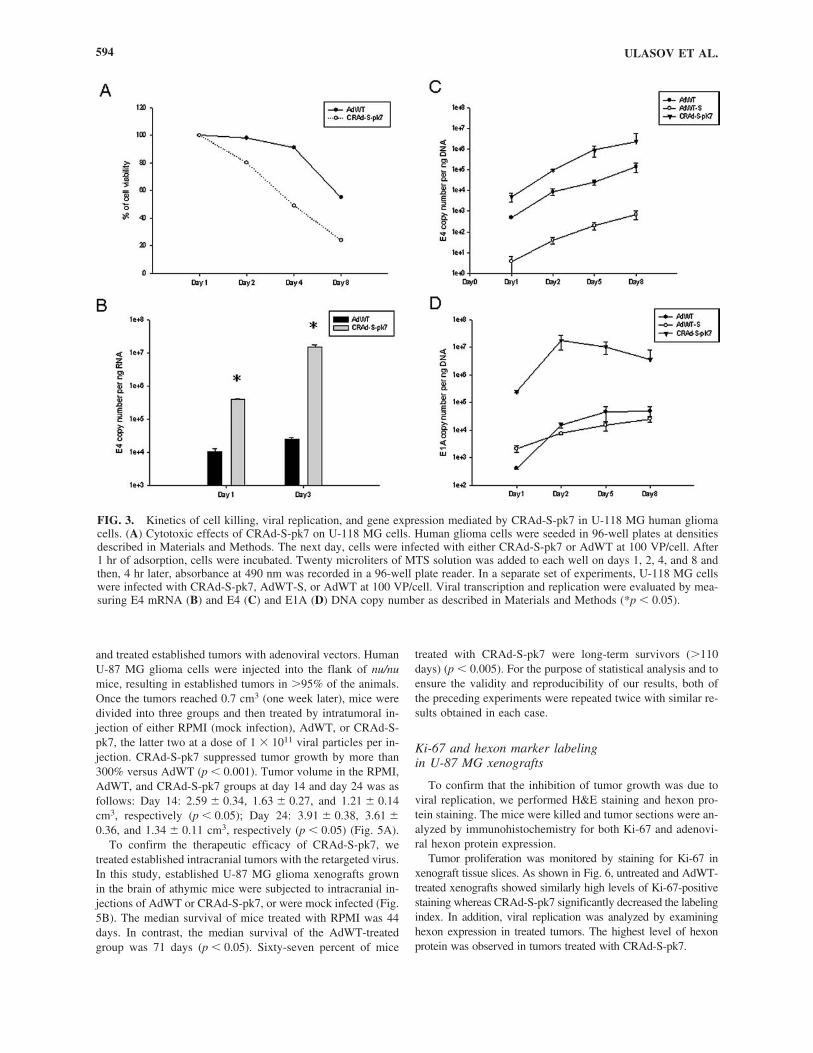
2). As shown in Fig. 3A, treatment with CRAd-S-pk7 resulted inmore efficient cell killing compared with AdWT infection. Thus,on day 8, CRAd-S-pk7 treatments induced 80% cell killing ver-sus 60% demonstrated for AdWT. To further analyze the efficacyof transcription and replication, we measured E4 copy numbersin infected U-118 MG cells. CRAd-S-pk7 demonstrated approx-imately 100–1000 orders of magnitude enhanced transcription andreplication as compared with cells treated with AdWT and AdWT-S (p � 0.05) (Fig. 3B and C). Finally, to test whether an increasedE4 copy number level corresponded to greater E1A protein ex-pression, we infected U-118 MG cells with CRAd-S-pk7, AdWT-S, and AdWT. As shown in Fig. 3D, infection with CRAd-S-pk7led to increased E1A protein expression in target cells from day1 to day 3 and these levels corresponded to the increased level ofE4 copy number demonstrated by CRAd-S-pk7 and described inFig. 3C.
To determine the potential cytotoxicity mediated by CRAd-S-pk7 infection, we tested the activity of our virus in normal
human brain. Human brain slices were infected with AdWT orCRAd-S-pk7, or were mock infected. Replication was measuredby titration of progeny released from slices and medium on days1 and 3. As shown in Fig. 4A, CRAd-S-pk7 demonstrated sig-nificantly lower replication activity in normal brain tissue bothon day 1 (1.77 vs. 112.02 for AdWT, p � 0.05) and day 3 (1.99vs. 199.52 for AdWT, p � 0.01). To assess virally induced tox-icity, we measured the expression of cellular proteins in themedium. Consistent with our replication data, CRAd-S-pk7showed significantly less LDH release versus AdWT on day 3(1.64 � 0.11 vs. 15.63 � 2.082, respectively; p � 0.05) (Fig.4B).
Growth suppression of U-87 glioma xenografts byintratumoral injection of CRAd-S-pk7
To determine whether our in vitro findings could be con-firmed in vivo, we used a glioma xenograft model in nude mice
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 593
FIG. 2. CRAd-S-pk7 exhibits selective oncolytic potential in human glioma cells. CRAds at the indicated doses were incu-bated with the following human glioma cell lines: U-118 MG, Kings-1, no.10, U-87 MG, U-251 MG, and A172. Lateral virusspread and oncolytic effect were visualized after staining of adherent cells with crystal violet. Experiments were repeated twice,independently.
and treated established tumors with adenoviral vectors. HumanU-87 MG glioma cells were injected into the flank of nu/numice, resulting in established tumors in 95% of the animals.Once the tumors reached 0.7 cm3 (one week later), mice weredivided into three groups and then treated by intratumoral in-jection of either RPMI (mock infection), AdWT, or CRAd-S-pk7, the latter two at a dose of 1 � 1011 viral particles per in-jection. CRAd-S-pk7 suppressed tumor growth by more than300% versus AdWT (p � 0.001). Tumor volume in the RPMI,AdWT, and CRAd-S-pk7 groups at day 14 and day 24 was asfollows: Day 14: 2.59 � 0.34, 1.63 � 0.27, and 1.21 � 0.14cm3, respectively (p � 0.05); Day 24: 3.91 � 0.38, 3.61 �0.36, and 1.34 � 0.11 cm3, respectively (p � 0.05) (Fig. 5A).
To confirm the therapeutic efficacy of CRAd-S-pk7, wetreated established intracranial tumors with the retargeted virus.In this study, established U-87 MG glioma xenografts grownin the brain of athymic mice were subjected to intracranial in-jections of AdWT or CRAd-S-pk7, or were mock infected (Fig.5B). The median survival of mice treated with RPMI was 44days. In contrast, the median survival of the AdWT-treatedgroup was 71 days (p � 0.05). Sixty-seven percent of mice
treated with CRAd-S-pk7 were long-term survivors (110days) (p � 0.005). For the purpose of statistical analysis and toensure the validity and reproducibility of our results, both ofthe preceding experiments were repeated twice with similar re-sults obtained in each case.
Ki-67 and hexon marker labeling in U-87 MG xenografts
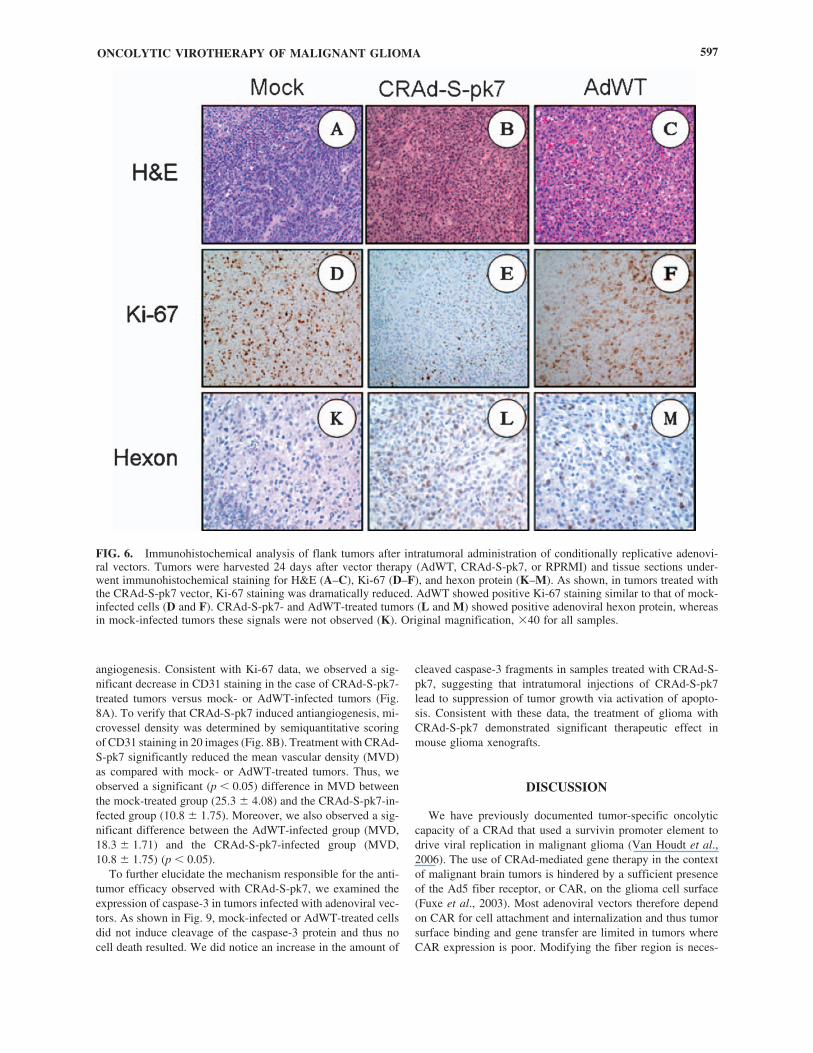
To confirm that the inhibition of tumor growth was due toviral replication, we performed H&E staining and hexon pro-tein staining. The mice were killed and tumor sections were an-alyzed by immunohistochemistry for both Ki-67 and adenovi-ral hexon protein expression.
Tumor proliferation was monitored by staining for Ki-67 inxenograft tissue slices. As shown in Fig. 6, untreated and AdWT-treated xenografts showed similarly high levels of Ki-67-positivestaining whereas CRAd-S-pk7 significantly decreased the labelingindex. In addition, viral replication was analyzed by examininghexon expression in treated tumors. The highest level of hexonprotein was observed in tumors treated with CRAd-S-pk7.
ULASOV ET AL.594
FIG. 3. Kinetics of cell killing, viral replication, and gene expression mediated by CRAd-S-pk7 in U-118 MG human gliomacells. (A) Cytotoxic effects of CRAd-S-pk7 on U-118 MG cells. Human glioma cells were seeded in 96-well plates at densitiesdescribed in Materials and Methods. The next day, cells were infected with either CRAd-S-pk7 or AdWT at 100 VP/cell. After1 hr of adsorption, cells were incubated. Twenty microliters of MTS solution was added to each well on days 1, 2, 4, and 8 andthen, 4 hr later, absorbance at 490 nm was recorded in a 96-well plate reader. In a separate set of experiments, U-118 MG cellswere infected with CRAd-S-pk7, AdWT-S, or AdWT at 100 VP/cell. Viral transcription and replication were evaluated by mea-suring E4 mRNA (B) and E4 (C) and E1A (D) DNA copy number as described in Materials and Methods (*p � 0.05).

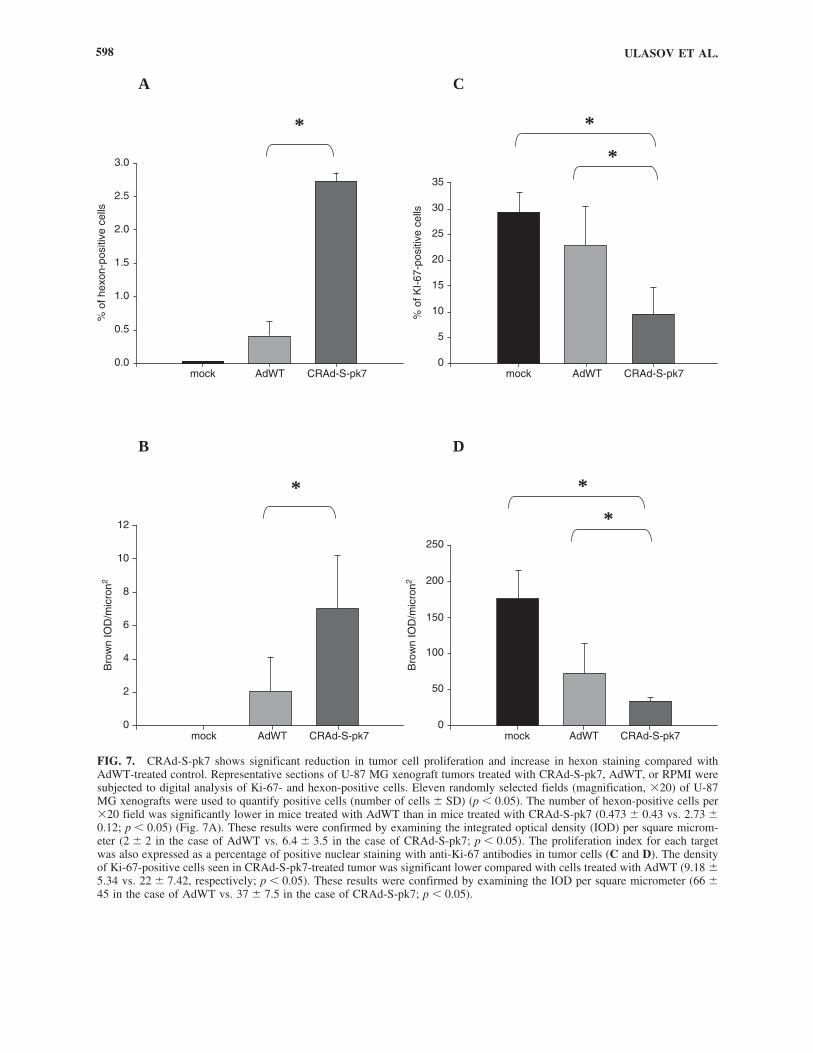
To objectively characterize the delayed growth effect in-duced by CRAd-S-pk7, we measured hexon and Ki-67 label-ing indexes (Fig. 7). We used digital analysis software (ACIS)to quantify the positively stained cells as described in Materi-als and Methods. The number of hexon-positive cells per �20field was significantly lower in mice treated with AdWT thanin mice treated with CRAd-S-pk7 (0.473 � 0.43 vs. 2.73 �0.12, respectively; p � 0.05) (Fig. 7A). These results were con-firmed by examining integrated optical density (IOD) per squaremicrometer (2 � 2 in the case of AdWT vs. 6.4 � 3.5 in thecase of CRAd-S-pk7; p � 0.05) (Fig. 7B). The proliferation in-dex for each target was also expressed as a percentage of pos-
itive nuclear staining with anti-Ki-67 antibodies in tumor cells(Fig. 7C and D). The density of Ki-67-positive cells seen inCRAd-S-pk7-treated tumor was significantly lower comparedwith cells treated with AdWT (9.18 � 5.34 vs. 22 � 7.42; p �0.05). These results were confirmed by examining the IOD persquare micrometer (66 � 45 in the case of AdWT vs. 37 � 7.5in the case of CRAd-S-pk7, p � 0.05).
Mechanism of tumor growth suppression
To assess the effect of adenoviral replication on tumor ves-sel density, tumor sections were stained for CD31, a marker of
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 595
FIG. 4. Evaluation of CRAd-mediated toxicity in normal human brain slices. Human brain tissue slices were infected with Advectors at 500 VP/cell. Twenty-four or 72 hr postinfection, progeny (A) were isolated from medium and tissue slices and titratedin HEK293 cells. Alternatively, LDH release (B) was measured in slice medium. Results are presented as plaque-forming unitsper milliliter (A) and as percent toxicity (B). Compared with the positive control (AdWT), CRAd-S-pk7 demonstrated signifi-cantly lower replication activity in normal brain tissue both on day 1 (1.77 vs. 112.02, p � 0.05) and day 3 (1.99 vs. 199.52,p � 0.01). LDH data corroborate replication data and revealed more LDH released from AdWT-infected cells than from CRAd-S-pk7-infected cells (15.63 � 2.082 vs. 1.64 � 0.11, p � 0.05).
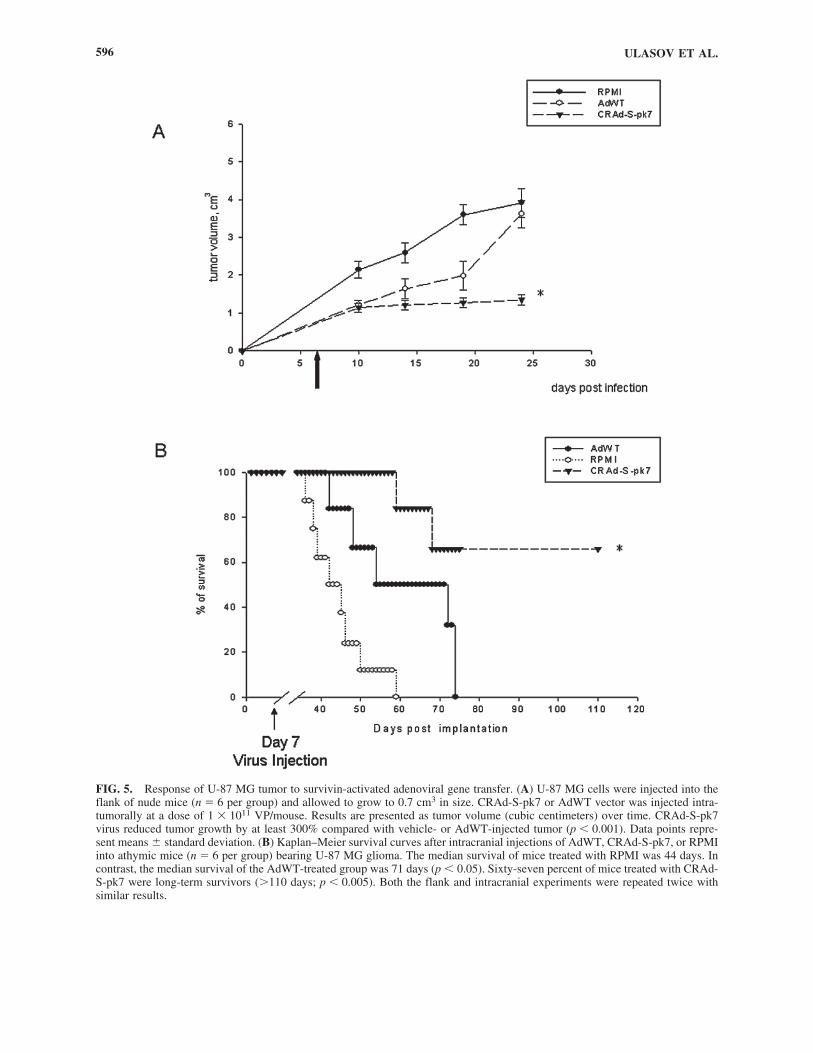
FIG. 5. Response of U-87 MG tumor to survivin-activated adenoviral gene transfer. (A) U-87 MG cells were injected into theflank of nude mice (n � 6 per group) and allowed to grow to 0.7 cm3 in size. CRAd-S-pk7 or AdWT vector was injected intra-tumorally at a dose of 1 � 1011 VP/mouse. Results are presented as tumor volume (cubic centimeters) over time. CRAd-S-pk7virus reduced tumor growth by at least 300% compared with vehicle- or AdWT-injected tumor (p � 0.001). Data points repre-sent means � standard deviation. (B) Kaplan–Meier survival curves after intracranial injections of AdWT, CRAd-S-pk7, or RPMIinto athymic mice (n � 6 per group) bearing U-87 MG glioma. The median survival of mice treated with RPMI was 44 days. Incontrast, the median survival of the AdWT-treated group was 71 days (p � 0.05). Sixty-seven percent of mice treated with CRAd-S-pk7 were long-term survivors (110 days; p � 0.005). Both the flank and intracranial experiments were repeated twice withsimilar results.
ULASOV ET AL.596
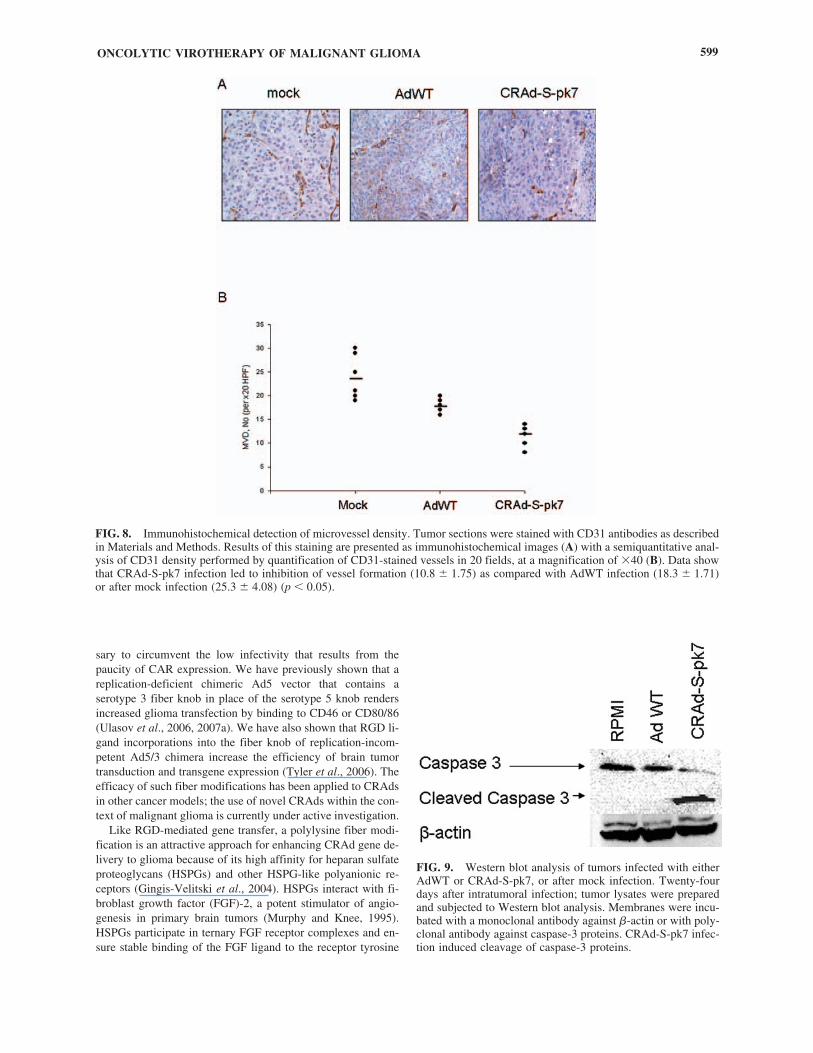
angiogenesis. Consistent with Ki-67 data, we observed a sig-nificant decrease in CD31 staining in the case of CRAd-S-pk7-treated tumors versus mock- or AdWT-infected tumors (Fig.8A). To verify that CRAd-S-pk7 induced antiangiogenesis, mi-crovessel density was determined by semiquantitative scoringof CD31 staining in 20 images (Fig. 8B). Treatment with CRAd-S-pk7 significantly reduced the mean vascular density (MVD)as compared with mock- or AdWT-treated tumors. Thus, weobserved a significant (p � 0.05) difference in MVD betweenthe mock-treated group (25.3 � 4.08) and the CRAd-S-pk7-in-fected group (10.8 � 1.75). Moreover, we also observed a sig-nificant difference between the AdWT-infected group (MVD,18.3 � 1.71) and the CRAd-S-pk7-infected group (MVD,10.8 � 1.75) (p � 0.05).
To further elucidate the mechanism responsible for the anti-tumor efficacy observed with CRAd-S-pk7, we examined theexpression of caspase-3 in tumors infected with adenoviral vec-tors. As shown in Fig. 9, mock-infected or AdWT-treated cellsdid not induce cleavage of the caspase-3 protein and thus nocell death resulted. We did notice an increase in the amount of
cleaved caspase-3 fragments in samples treated with CRAd-S-pk7, suggesting that intratumoral injections of CRAd-S-pk7lead to suppression of tumor growth via activation of apopto-sis. Consistent with these data, the treatment of glioma withCRAd-S-pk7 demonstrated significant therapeutic effect inmouse glioma xenografts.
DISCUSSION
We have previously documented tumor-specific oncolyticcapacity of a CRAd that used a survivin promoter element todrive viral replication in malignant glioma (Van Houdt et al.,2006). The use of CRAd-mediated gene therapy in the contextof malignant brain tumors is hindered by a sufficient presenceof the Ad5 fiber receptor, or CAR, on the glioma cell surface(Fuxe et al., 2003). Most adenoviral vectors therefore dependon CAR for cell attachment and internalization and thus tumorsurface binding and gene transfer are limited in tumors whereCAR expression is poor. Modifying the fiber region is neces-
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 597
FIG. 6. Immunohistochemical analysis of flank tumors after intratumoral administration of conditionally replicative adenovi-ral vectors. Tumors were harvested 24 days after vector therapy (AdWT, CRAd-S-pk7, or RPRMI) and tissue sections under-went immunohistochemical staining for H&E (A–C), Ki-67 (D–F), and hexon protein (K–M). As shown, in tumors treated withthe CRAd-S-pk7 vector, Ki-67 staining was dramatically reduced. AdWT showed positive Ki-67 staining similar to that of mock-infected cells (D and F). CRAd-S-pk7- and AdWT-treated tumors (L and M) showed positive adenoviral hexon protein, whereasin mock-infected tumors these signals were not observed (K). Original magnification, �40 for all samples.
ULASOV ET AL.598
FIG. 7. CRAd-S-pk7 shows significant reduction in tumor cell proliferation and increase in hexon staining compared withAdWT-treated control. Representative sections of U-87 MG xenograft tumors treated with CRAd-S-pk7, AdWT, or RPMI weresubjected to digital analysis of Ki-67- and hexon-positive cells. Eleven randomly selected fields (magnification, �20) of U-87MG xenografts were used to quantify positive cells (number of cells � SD) (p � 0.05). The number of hexon-positive cells per�20 field was significantly lower in mice treated with AdWT than in mice treated with CRAd-S-pk7 (0.473 � 0.43 vs. 2.73 �0.12; p � 0.05) (Fig. 7A). These results were confirmed by examining the integrated optical density (IOD) per square microm-eter (2 � 2 in the case of AdWT vs. 6.4 � 3.5 in the case of CRAd-S-pk7; p � 0.05). The proliferation index for each targetwas also expressed as a percentage of positive nuclear staining with anti-Ki-67 antibodies in tumor cells (C and D). The densityof Ki-67-positive cells seen in CRAd-S-pk7-treated tumor was significant lower compared with cells treated with AdWT (9.18 �5.34 vs. 22 � 7.42, respectively; p � 0.05). These results were confirmed by examining the IOD per square micrometer (66 �45 in the case of AdWT vs. 37 � 7.5 in the case of CRAd-S-pk7; p � 0.05).
mock AdWT CRAd-S-pk70.0
*
0.5
1.0
1.5
2.0
2.5
3.0
% o
f hex
on-p
ositi
ve c
ells
A
B
mock AdWT CRAd-S-pk70
*
5
10
15
20
25
30
35
% o
f KI-
67-p
ositi
ve c
ells
C
*
mock AdWT CRAd-S-pk70
*
50
100
150
200
250
Bro
wn
IOD
/mic
ron2
*
mock AdWT CRAd-S-pk70
2
4
6
8
10
12
Bro
wn
IOD
/mic
ron2
*
D
sary to circumvent the low infectivity that results from thepaucity of CAR expression. We have previously shown that areplication-deficient chimeric Ad5 vector that contains aserotype 3 fiber knob in place of the serotype 5 knob rendersincreased glioma transfection by binding to CD46 or CD80/86(Ulasov et al., 2006, 2007a). We have also shown that RGD li-gand incorporations into the fiber knob of replication-incom-petent Ad5/3 chimera increase the efficiency of brain tumortransduction and transgene expression (Tyler et al., 2006). Theefficacy of such fiber modifications has been applied to CRAdsin other cancer models; the use of novel CRAds within the con-text of malignant glioma is currently under active investigation.
Like RGD-mediated gene transfer, a polylysine fiber modi-fication is an attractive approach for enhancing CRAd gene de-livery to glioma because of its high affinity for heparan sulfateproteoglycans (HSPGs) and other HSPG-like polyanionic re-ceptors (Gingis-Velitski et al., 2004). HSPGs interact with fi-broblast growth factor (FGF)-2, a potent stimulator of angio-genesis in primary brain tumors (Murphy and Knee, 1995).HSPGs participate in ternary FGF receptor complexes and en-sure stable binding of the FGF ligand to the receptor tyrosine
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 599
FIG. 8. Immunohistochemical detection of microvessel density. Tumor sections were stained with CD31 antibodies as describedin Materials and Methods. Results of this staining are presented as immunohistochemical images (A) with a semiquantitative anal-ysis of CD31 density performed by quantification of CD31-stained vessels in 20 fields, at a magnification of �40 (B). Data showthat CRAd-S-pk7 infection led to inhibition of vessel formation (10.8 � 1.75) as compared with AdWT infection (18.3 � 1.71)or after mock infection (25.3 � 4.08) (p � 0.05).
FIG. 9. Western blot analysis of tumors infected with eitherAdWT or CRAd-S-pk7, or after mock infection. Twenty-fourdays after intratumoral infection; tumor lysates were preparedand subjected to Western blot analysis. Membranes were incu-bated with a monoclonal antibody against �-actin or with poly-clonal antibody against caspase-3 proteins. CRAd-S-pk7 infec-tion induced cleavage of caspase-3 proteins.

kinase (Alanko et al., 1994). Certain HSPGs have been shownto be overexpressed exclusively in glioma vessel endothelialcells, contributing to the aggressive proliferation of glioblas-toma (Qiao et al., 2003).
Replication-deficient adenoviruses with a pk7 fiber modifi-cation have exhibited enhanced transfection in passaged gliomacell lines (Staba et al., 2000). Moreover, Wickham and col-leagues have previously shown that a replication-defective ad-enovirus with a pk7 modification can effectively target vascu-lar smooth muscle (Wickham et al., 1997) and thereforemodulate the antiangiogenic properties important for tumorcontrol. Because of these findings, a pk7 fiber modificationseemed suitable for achieving more efficient CRAd gene trans-fer to neoplastic areas, further enhancing the tumor-regressingcapabilities of the CRAd. To identify which fiber modificationendows the survivin promoter-driven CRAd with the best in-fectivity profile, we compared gene transfer capabilities ofCRAd-S-5/3, CRAd-S-RGD, CRAd-S-pk7, and AdWT.
In this study, we found that transcriptional and transductionalmodulation, in the form of survivin promoter and pk7 fibermodification, represents the most effective approach for gliomavirotherapy. These findings were confirmed both in vitro andin vivo. Cytotoxic assays using passaged glioblastoma multi-forme cells clearly indicate that CRAd-S-pk7 is more efficientthan CRAd-S-RGD, CRAd-S-5/3, and AdWT in cell transduc-tion and killing. Furthermore, E1A and E4 copy number assaysdemonstrate that the increased cytotoxic effect is a result of tu-mor-specific promoter and pk7 fiber modification.
CRAd-S-pk7 demonstrated a superior ability to delay tumorprogression compared with the wild-type adenovirus and mockinfections. This result has implications as to how pk7 directsattachment to the glioma periphery via HSPG expression (Stabaet al., 2000). HSPG–FGF-2 signaling complexes aggregate atthe tumor periphery to promote angiogenesis and endothelialcell differentiation. The availability of the HSPG signaling com-plexes at the tumor margin renders the outer neoplastic regionamenable to CRAd-S-pk7 infection and oncolysis. Thus,CRAd-S-pk7 may inhibit tumor migration via binding to “pro-proliferation” signaling systems and subsequently inducing on-colysis via its survivin-promoted replication.
Although the success obtained with CRAd-S-pk7 in con-trolling and prolonging the survival of mice with intracranialtumors is encouraging, one of the limitations of this study isthe use of oncolytic vectors in immunodeficient mice. How-ever, there is no fully permissive animal model in which braintumor virotherapy and the immune response can be examinedat the same time. This limitation, however, has not impeded thedevelopment of oncolytic vectors for brain tumor therapy. Asan example, the ONYX-015 oncolytic adenovirus has com-pleted a phase I clinical trial (Chiocca et al., 2004). This viruswas shown to exhibit significant oncolytic efficacy in preclin-ical studies of xenografts in nude mice (Shinoura et al., 1999;Geoerger et al., 2002, 2003); when tested in humans, injectionsof up to 1010 PFU were safe. Of the 24 patients tested in thisphase I study, only 2 seroconverted from negative to positivefor adenovirus antibodies. Other oncolytic adenovectors, suchas Ad-�24-RGD, have also completed promising preclinicalstudies in nude mice, and are about to enter a human clinicaltrial for glioma (Fueyo et al., 2000; Suzuki et al., 2001; Lam-fers et al., 2002; Portella et al., 2002). Moreover, studies com-
paring replication-defective versus oncolytic viruses haveshown that although preexisting antibodies significantly atten-uate the activity of replication-defective adenovirus, they ex-hibit no effect on the activity of replicating adenovirus (Tsai etal., 2004). These are significant findings with important impli-cations for glioma virotherapy because of the relatively im-munocompromised state of these patients, who are typically re-ceiving chronic steroid medication and have been treated withradiation and chemotherapy, and for injection of the virus withinthe brain, an organ that is relatively immunoprivileged. Of note,data obtained with immunodeficient mice have been critical toestablishing the rationale for employment of tropism-modifiedadenoviral vectors by relevant federal regulatory bodies.
In conclusion, our results indicate that transcriptional and trans-ductional control of adenoviral replication can greatly enhance thevirotherapy of malignant glioma. Our novel adenovirus, CRAd-S-pk7, exhibited enhanced tropism, replication, and oncolytic ca-pacity in the setting of malignant glioma. At the same time, weobserved minimal, if any, toxicity to normal brain. The enhancedoncolytic effect mediated by pk7 warrants further studies of thisnovel CRAd in future preclinical and clinical studies.
ACKNOWLEDGMENTS
The authors thank Baogen Lu and Hong Ju Wu (Gene Ther-apy Center, University of Alabama) for help with propagationof CRAds. The authors thank Maria Tretiakova (Department ofPathology, University of Chicago) for excellent technical as-sistance with automated image analysis system and software.This study was supported by a grant from the American Col-lege of Surgeons (M.L.) and the National Institutes of Neuro-logical Disorders and Stroke (K08 NS046430; M.L.).
AUTHOR DISCLOSURE STATEMENT
No competing financial interests exist.
REFERENCES
ALANKO, T., TIENARI, J., LEHTONEN, E., and SAKSELA, O.(1994). Development of FGF-dependency in human embryonic car-cinoma cells after retinoic acid-induced differentiation. Dev. Biol.161, 141–153.
ASAOKA, K., TADA, M., SAWAMURA, Y., IKEDA, J., and ABE,H. (2000). Dependence of efficient adenoviral gene delivery in ma-lignant glioma cells on the expression levels of the coxsackievirusand adenovirus receptor. J. Neurosurg. 92, 1002–1008.
CHAKRAVARTI, A., NOLL, E., BLACK, P.M., FINKELSTEIN,D.F., FINKELSTEIN, D.M., DYSON, N.J., and LOEFFLER, J.S.(2002). Quantitatively determined survivin expression levels are ofprognostic value in human gliomas. J. Clin. Oncol. 20, 1063–1068.
CHAKRAVARTI, A., ZHAI, G.G., ZHANG, M., MALHOTRA, R.,LATHAM, D.E., DELANEY, M.A., ROBE, P., NESTLER, U.,SONG, Q., and LOEFFLER, J. (2004). Survivin enhances radiationresistance in primary human glioblastoma cells via caspase-inde-pendent mechanisms. Oncogene 23, 7494–7506.
CHIOCCA, E.A., ABBED, K.M., TATTER, S., LOUIS, D.N.,HOCHBERG, F.H., BARKER, F., KRACHER, J., GROSSMAN,
ULASOV ET AL.600

S.A., FISHER, J.D., CARSON, K., ROSENBLUM, M.,MIKKELSEN, T., OLSON, J., MARKERT, J., ROSENFELD, S.,NABORS, L.B., BREM, S., PHUPHANICH, S., FREEMAN, S.,KAPLAN, R., and ZWIEBEL, J. (2004). A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenu-ated adenovirus, ONYX-015, into the peritumoral region of recur-rent malignant gliomas, in the adjuvant setting. Mol. Ther. 10,958–966.
CONRAD, C., MILLER, C.R., JI, Y., GOMEZ-MANZANO, C.,BHARARA, S., MCMURRAY, J.S., LANG, F.F., WONG, F.,SAWAYA, R., YUNG, W.K., and FUEYO, J. (2005). �24-hyCDadenovirus suppresses glioma growth in vivo by combining oncoly-sis and chemosensitization. Cancer Gene Ther. 12, 284–294.
CRIPE, T.P., DUNPHY, E.J., HOLUB, A.D., SAINI, A., VASI, N.H.,MAHLLER, Y.Y., COLLINS, M.H., SNYDER, J.D., KRASNYKH,V., CURIEL, D.T., WICKHAM, T.J., DEGREGORI, J., BERGEL-SON, J.M., and CURRIER, M.A. (2001). Fiber knob modificationsovercome low, heterogeneous expression of the coxsackievirus-ade-novirus receptor that limits adenovirus gene transfer and oncolysisfor human rhabdomyosarcoma cells. Cancer Res. 61, 2953–2960.
DMITRIEV, I., KRASNYKH, V., MILLER, C.R., WANG, M.,KASHENTSEVA, E., MIKHEEVA, G., BELOUSOVA, N., andCURIEL, D.T. (1998). An adenovirus vector with genetically mod-ified fibers demonstrates expanded tropism via utilization of a cox-sackievirus and adenovirus receptor-independent cell entry mecha-nism. J. Virol. 72, 9706–9713.
FUEYO, J., GOMEZ-MANZANO, C., ALEMANY, R., LEE, P.S.,MCDONNELL, T.J., MITLIANGA, P., SHI, Y.X., LEVIN, V.A.,YUNG, W.K., and KYRITSIS, A.P. (2000). A mutant oncolytic ad-enovirus targeting the Rb pathway produces anti-glioma effect invivo. Oncogene 19, 2–12.
FUXE, J., LIU, L., MALIN, S., PHILIPSON, L., COLLINS, V.P., andPETTERSSON, R.F. (2003). Expression of the coxsackie and ade-novirus receptor in human astrocytic tumors and xenografts. Int. J.Cancer 103, 723–729.
GEOERGER, B., GRILL, J., OPOLON, P., MORIZET, J., AUBERT,G., TERRIER-LACOMBE, M.J., BRESSAC DE-PAILLERETS, B.,BARROIS, M., FEUNTEUN, J., KIRN, D.H., and VASSAL, G.(2002). Oncolytic activity of the E1B-55 kDa-deleted adenovirusONYX-015 is independent of cellular p53 status in human malignantglioma xenografts. Cancer Res. 62, 764–772.
GEOERGER, B., GRILL, J., OPOLON, P., MORIZET, J., AUBERT,G., LECLUSE, Y., VAN BEUSECHEM, V.W., GERRITSEN, W.R.,KIRN, D.H., and VASSAL, G. (2003). Potentiation of radiation ther-apy by the oncolytic adenovirus dl1520 (ONYX-015) in human ma-lignant glioma xenografts. Br. J. Cancer 89, 577–584.
GINGIS-VELITSKI, S., ZETSER, A., KAPLAN, V., BEN-ZAKEN,O., COHEN, E., LEVY-ADAM, F., BASHENKO, Y., FLUGEL-MAN, M.Y., VLODAVSKY, I., and ILAN, N. (2004). Heparanaseuptake is mediated by cell membrane heparan sulfate proteoglycans.J. Biol. Chem. 279, 44084–44092.
GRAHAM, F.L., SMILEY, J., RUSSELL, W.C., and NAIRN, R.(1977). Characteristics of a human cell line transformed by DNAfrom human adenovirus type 5. J. Gen. Virol. 36, 59–74.
HSIEH, J.C., and LESNIAK, M.S. (2005). Surgical management ofhigh-grade gliomas. Expert Rev. Neurother. 5, S33–S39.
HSU, W., LESNIAK, M.S., TYLER, B., and BREM, H. (2005). Lo-cal delivery of interleukin-2 and Adriamycin is synergistic in thetreatment of experimental malignant glioma. J. Neurooncol. 74,135–140.
IMMONEN, A., VAPALAHTI, M., TYYNELA, K., HURSKAINEN,H., SANDMAIR, A., VANNINEN, R., LANGFORD, G., MUR-RAY, N., and YLA-HERTTUALA, S. (2004). AdvHSV-tk gene ther-apy with intravenous ganciclovir improves survival in human ma-lignant glioma: A randomised, controlled study. Mol. Ther. 10,967–972.
KAJIWARA, Y., YAMASAKI, F., HAMA, S., YAHARA, K., YOSH-IOKA, H., SUGIYAMA, K., ARITA, K., and KURISU, K. (2003).Expression of survivin in astrocytic tumors: Correlation with malig-nant grade and prognosis. Cancer 97, 1077–1083.
KANERVA, A., BAUERSCHMITZ, G.J., YAMAMOTO, M., LAM,J.T., ALVAREZ, R.D., SIEGAL, G.P., CURIEL, D.T., and HEM-MINKI, A. (2004). A cyclooxygenase-2 promoter-based condition-ally replicating adenovirus with enhanced infectivity for treatment ofovarian adenocarcinoma. Gene Ther. 11, 552–559.
KIRBY, T.O., RIVERA, A., REIN, D., WANG, M., ULASOV, I.,BREIDENBACH, M., KATARAM, M., CONTRERAS, J.L.,KRUMDIECK, C., YAMAMOTO, M., ROTS, M.G., HAISMA,H.J., ALVAREZ, R.D., MAHASRESHTI, P.J., and CURIEL, D.T.(2004). A novel ex vivo model system for evaluation of condition-ally replicative adenoviruses therapeutic efficacy and toxicity. Clin.Cancer Res. 10, 8697–8703.
LAMFERS, M.L., GRILL, J., DIRVEN, C.M., VAN BEUSECHEM,V.W., GEOERGER, B., VAN DEN BERG, J., ALEMANY, R.,FUEYO, J., CURIEL, D.T., VASSAL, G., PINEDO, H.M., VAN-DERTOP, W.P., and GERRITSEN, W.R. (2002). Potential of theconditionally replicative adenovirus Ad5-�24RGD in the treatmentof malignant gliomas and its enhanced effect with radiotherapy. Can-cer Res. 62, 5736–5742.
LESNIAK, M.S. (2005). Novel advances in drug delivery to brain can-cer. Technol. Cancer Res. Treat. 4, 417–428.
LESNIAK, M.S., and BREM, H. (2004). Targeted therapy for brain tu-mours. Nat. Rev. Drug Discov. 3, 499–508.
LESNIAK, M.S., LANGER, R., and BREM, H. (2001). Drug deliveryto tumors of the central nervous system. Curr. Neurol. Neurosci. Rep.1, 210–216.
LESNIAK, M.S., UPADHYAY, U., GOODWIN, R., TYLER, B., andBREM, H. (2005). Local delivery of doxorubicin for the treatmentof malignant brain tumors in rats. Anticancer Res. 25, 3825–3831.
MURPHY, P.R., and KNEE, R.S. (1995). Basic fibroblast growth fac-tor binding and processing by human glioma cells. Mol. Cell. En-docrinol. 114, 193–203.
PORTELLA, G., SCALA, S., VITAGLIANO, D., VECCHIO, G., andFUSCO, A. (2002). ONYX-015, an E1B gene-defective adenovirus,induces cell death in human anaplastic thyroid carcinoma cell lines.J. Clin. Endocrinol. Metab. 87, 2525–2531.
QIAO, D., MEYER, K., MUNDHENKE, C., DREW, S.A., andFRIEDL, A. (2003). Heparan sulfate proteoglycans as regulators offibroblast growth factor-2 signaling in brain endothelial cells: Spe-cific role for glypican-1 in glioma angiogenesis. J. Biol. Chem. 278,16045–16053.
REIN, D.T., BREIDENBACH, M., KIRBY, T.O., HAN, T., SIEGAL,G.P., BAUERSCHMITZ, G.J., WANG, M., NETTELBECK, D.M.,TSURUTA, Y., YAMAMOTO, M., DALL, P., HEMMINKI, A., andCURIEL, D.T. (2005). A fiber-modified, secretory leukoprotease in-hibitor promoter-based conditionally replicating adenovirus for treat-ment of ovarian cancer. Clin. Cancer Res. 11, 1327–1335.
SEKI, T., DMITRIEV, I., SUZUKI, K., KASHENTSEVA, E.,TAKAYAMA, K., ROTS, M., UIL, T., WU, H., WANG, M., andCURIEL, D.T. (2002). Fiber shaft extension in combination with HIloop ligands augments infectivity for CAR-negative tumor targetsbut does not enhance hepatotropism in vivo. Gene Ther. 9,1101–1108.
SHINOURA, N., YOSHIDA, Y., TSUNODA, R., OHASHI, M.,ZHANG, W., ASAI, A., KIRINO, T., and HAMADA, H. (1999).Highly augmented cytopathic effect of a fiber-mutant E1B-defectiveadenovirus for gene therapy of gliomas. Cancer Res. 59, 3411–3416.
STABA, M.J., WICKHAM, T.J., KOVESDI, I., and HALLAHAN,D.E. (2000). Modifications of the fiber in adenovirus vectors increasetropism for malignant glioma models. Cancer Gene Ther. 7, 13–19.
SUZUKI, K., FUEYO, J., KRASNYKH, V., REYNOLDS, P.N.,CURIEL, D.T., and ALEMANY, R. (2001). A conditionally replica-
ONCOLYTIC VIROTHERAPY OF MALIGNANT GLIOMA 601

tive adenovirus with enhanced infectivity shows improved oncolyticpotency. Clin. Cancer Res. 7, 120–126.
TANGO, Y., TAKI, M., SHIRAKIYA, Y., OHTANI, S., TOKU-NAGA, N., TSUNEMITSU, Y., KAGAWA, S., TANI, T.,TANAKA, N., and FUJIWARA, T. (2004). Late resistance to ade-noviral p53-mediated apoptosis caused by decreased expression ofcoxsackie–adenovirus receptors in human lung cancer cells. CancerSci. 95, 459–463.
TEKANT, Y., DAVYDOVA, J., RAMIREZ, P.J., CURIEL, D.T.,VICKERS, S.M., and YAMAMOTO, M. (2005). Oncolytic adeno-viral therapy in gallbladder carcinoma. Surgery 137, 527–535.
TSAI, V., JOHNSON, D.E., RAHMAN, A., WEN, S.F., LAFACE, D.,PHILOPENA, J., NERY, J., ZEPEDA, M., MANEVAL, D.C., DE-MERS, G.W., and RALSTON, R. (2004). Impact of human neutral-izing antibodies on antitumor efficacy of an oncolytic adenovirus ina murine model. Clin. Cancer Res. 10, 7199–7206.
TYLER, M.A., ULASOV, I.V., BOROVJAGIN, A., SONABEND,A.M., KHRAMTSOV, A., HAN, Y., DENT, P., FISHER, P.B.,CURIEL, D.T., and LESNIAK, M.S. (2006). Enhanced transductionof malignant glioma with a double targeted Ad5/3-RGD fiber-mod-ified adenovirus. Mol. Cancer Ther. 5, 2408–2416.
ULASOV, I.V., TYLER, M.A., ZHENG, S., HAN, Y., and LESNIAK,M.S. (2006). CD46 represents a target for adenoviral gene therapyof malignant glioma. Hum. Gene Ther. 17, 556–564.
ULASOV, I.V., TYLER, M.A., ZHENG, S., HAN, Y., and LESNIAK,M.S. (2007a). Targeting adenovirus to CD80/CD86 increases genetransfer efficiency to malignant glioma. J. Neurosurg. (in press).
ULASOV, I.V., RIVERA, A.A., SONABEND, A.M., RIVERA, L.B.,WANG, M., ZHU, Z.B., and LESNIAK, M.S. (2007b). Comparativeevaluation of survivin, midkine, and CXCR4 promoters for tran-scriptional targeting of glioma gene therapy. Cancer Biol. Ther. (inpress).
VAN GIJSSEL, H.E., SCHILD, L.J., WATT, D.L., ROTH, M.J.,WANG, G.Q., DAWSEY, S.M., ALBERT, P.S., QIAO, Y.L., TAY-LOR, P.R., DONG, Z.W., and POIRIER, M.C. (2004). Polycyclicaromatic hydrocarbon–DNA adducts determined by semiquantitativeimmunohistochemistry in human esophageal biopsies taken in 1985.Mutat. Res. 547, 55–62.
VAN HOUDT, W.J., HAVIV, Y.S., LU, B., WANG, M., RIVERA,A.A., ULASOV, I.V., LAMFERS, M.L., REIN, D., LESNIAK, M.S.,SIEGAL, G.P., DIRVEN, C.M., CURIEL, D.T., and ZHU, Z.B.(2006). The human survivin promoter: A novel transcriptional tar-geting strategy for treatment of glioma. J. Neurosurg. 104, 583–592.
WICKHAM, T.J., TZENG, E., SHEARS, L.L., 2nd, ROELVINK, P.W.,LI, Y., LEE, G.M., BROUGH, D.E., LIZONOVA, A., and KOVESDI,I. (1997). Increased in vitro and in vivo gene transfer by adenovirusvectors containing chimeric fiber proteins. J. Virol. 71, 8221–8229.
WU, H., HAN, T., LAM, J.T., LEATH, C.A., DMITRIEV, I.,KASHENTSEVA, E., BARNES, M.N., ALVAREZ, R.D., andCURIEL, D.T. (2004). Preclinical evaluation of a class of infectiv-ity-enhanced adenoviral vectors in ovarian cancer gene therapy. GeneTher. 11, 874–878.
YAMADA, Y., KUROIWA, T., NAKAGAWA, T., KAJIMOTO, Y.,DOHI, T., AZUMA, H., TSUJI, M., KAMI, K., and MIYATAKE,S. (2003). Transcriptional expression of survivin and its splice vari-ants in brain tumors in humans. J. Neurosurg. 99, 738–745.
ZHU, Z.B., CHEN, Y., MAKHIJA, S.K., LU, B., WANG, M.,RIVERA, A.A., YAMAMOTO, M., WANG, S., SIEGAL, G.P.,CURIEL, D.T., and MCDONALD, J.M. (2006). Survivin promoter-based conditionally replicative adenoviruses target cholangiocarci-noma. Int. J. Oncol. 29, 1319–1329.
Address reprint requests to:Dr. Maciej S. Lesniak
Division of NeurosurgeryUniversity of Chicago
5841 S. Maryland Avenue, MC 3026Chicago, IL 60637
E-mail: [email protected]
Received for publication January 4, 2007; accepted after revi-sion May 27, 2007.
Published online: July 11, 2007.
ULASOV ET AL.602
Copyright © 2022 FDOKUMEN




















