
Transcription regulator LMO4 interferes with neuritogenesis in human SH-SY5Y neuroblastoma cells

Journal of Neuroimmunology 233 (2011) 18–28
Contents lists available at ScienceDirect
Journal of Neuroimmunology
j ourna l homepage: www.e lsev ie r.com/ locate / jneuro im
Novel survivin mutant protects differentiated SK-N-SH human neuroblastoma cellsfrom activated T-cell neurotoxicity
Sara Baratchi, Rupinder K. Kanwar, Jagat R. Kanwar ⁎Laboratory of Immunology and Molecular Biomedical Research, Centre for Biotechnology and Interdisciplinary Biosciences (BioDeakin), Institute for Technology Research and Innovation(ITRI), Deakin University, Waurn Ponds, Victoria 3217, Australia
⁎ Corresponding author. Tel.: +61 3 52271148; fax: +E-mail address: [email protected] (J.R. Ka
0165-5728/$ – see front matter. Crown Copyright © 20doi:10.1016/j.jneuroim.2010.10.036
a b s t r a c t
a r t i c l e i n f oArticle history:Received 19 July 2010Received in revised form 27 October 2010Accepted 29 October 2010
Keywords:SurR9-C84ANeuroprotectionMultiple sclerosisNeuro-degeneration
Currently, there are no known treatments for protection of axonal loss associated with neuroinflammatorydiseases such as multiple sclerosis (MS). Survivin is a member of the inhibitors of the apoptosis (IAP) family ofproteins that its neuroprotective effects have not been studied. We demonstrate here that SurR9-C84A, asurvivin mutant, exhibits a neuroprotective role against the cytotoxic effects of activated T-cell infiltrates,such as granzyme B (GrB). The activated T-cell supernatants induce toxicity on differentiated SK-N-SH cells,which is associated with the loss of Ca2+ homeostasis, the increased population of dead cells, mitochondrialmembrane depolarisation, and the accelerated expression of cyclinD1, caspase3 and Fas, as observed for mostapoptotic cells. Alternatively, the pre-treatment with SurR9-C84A reduces the population of dead cells bybalancing the cytosolic Ca2+ homeostasis, decreasing the level of mitochondrial depolarisation, and alsoreducing the expression of cyclinD1 and caspase3. Our findings suggest that SurR9-C84A has aneuroprotective effect against the cytotoxins existing in activated T-cell supernatants including GrB.
61 3 52273402.nwar).
10 Published by Elsevier B.V. All rights res
Crown Copyright © 2010 Published by Elsevier B.V. All rights reserved.
1. Introduction
Multiple sclerosis (MS) is a debilitating long term disability that isassociated with severe axonal damage during its early stages, as wellas inflammatory responses (Dhib-Jalbut et al., 2006; Kanwar et al.,2009). Due to clinical variability and unpredictable nature of MS,prescribing treatments for specific individuals is complicated, and sofar no therapy has been found with the capability of halting diseaseprogression (Cook, 2003; Kanwar, 2005; Vartanian et al., 2004). Tosuccessfully stop the progression or reverse MS, a treatment mustsimultaneously target a combination of inflammatory and degenerativefactors (Kanwar, 2005; Kanwar et al., 2000, 2004a). The approvedtreatments forMS include the applicationof conventional agents such asinterferon-β, glatiramer acetate and mitoxantrone, as well as thetreatments still under trial such as monoclonal antibodies (i.e.,Atalizumab, Alemtuzumab and Rituximab) and oral drugs, which alltarget inflammatory components of MS pathogenesis (Gold andVoskuhl, 2009). Until now, no direct neuroprotective treatment hasbeen available, especially because administering direct neuroprotectivetherapies needs to penetrate the blood brain barrier (Baratchi et al.,2009; Kanwar et al., 2009).
Targeting members of the apoptosis family of proteins andevaluating their neuroprotective effects have been the subject ofmuch attention (Charriaut-Marlangue, 2004). The apoptosis pathway
is negatively regulated by Bcl-2 and the IAP family of proteins. Theneuroprotective role of IAPs in degenerating neurons has been studiedby different groups (Hefti, 1997; Simons et al., 1999; Xu et al., 1999).Among the differentmembers of IAPs, NIAP andXIAP are shown to haveessential roles in the intracellular signalling of glial cell line-derivedneurotrophic factor (GDNF) (Perrelet et al., 2002). In addition,adenoviral delivery of XIAP into axotomised retinal ganglion cells cansignificantly decrease neuronal death (Kato et al., 2000; Perrelet et al.,2004).
Survivin is another member of the IAP family, which has beenshown to have a role in early brain development (Jiang et al., 2005),and serves a bifunctional role during mitosis and inhibition ofapoptosis (Altieri, 2008; Baratchi et al., 2010a; Kanwar et al., 2010).Survivin consists of two defined domains, including the amino-terminal BIR domain that is composed of an extensive dimerisationinterface (Verdecia et al., 2000). This interface is associated with XIAPand allows the formation of survivin–XIAP complex, which increasesthe stability of XIAP against ubiquitination/proteasomal destructionand the synergistic inhibition of apoptosis (Dohi et al., 2004).However, using wild-type IAPs for the purpose of human clinicaltrial raises concerns due to their role in cancer formation (Kanwaret al., 2004b, 2010). Nevertheless, developing IAP mutants capable ofincreasing the cell proliferation will provide a new therapeuticwindow for neuronal repair and proliferation therapy.
Different forms of survivin mutants (such as C84A, Δ106 andT34A) have been used for the purpose of targeting survivin over-expression in cancer cells (Altieri, 2003b; Cheung et al., 2006; Kanwaret al., 2001; Khan et al., 2009; Li et al., 1998; Pisarev et al., 2003).
erved.

19S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
Among the various survivin mutants, the baculovirus IAP repeat motif(C84A) was initially produced by Li et al. (1998), and has shown pro-apoptotic effects on human cancer cells. Furthermore, we previouslyfound that SurR9-C84A has protective effects against retinoic acid-induced cell toxicity (Baratchi et al., 2010b).
In the present study, we used the SK-N-SH cell line as a culturemodel of retinoic acid-induced neuronal differentiation (Hong et al.,2003; Maris and Matthay, 1999; Wainwright et al., 2001). We showfor the first time, the neuroprotective effect of SurR9-C84A againstcytotoxic elements existing in activated T-cell supernatants such asGrB. Because GrB is a powerful pro-apoptotic member of granzymesfamily and is a very important mediator of damage in progressive MSand other neuroinflammatory disorders (Hoftberger et al., 2004;Neumann et al., 2002), we examined the importance of GrB releasedfrom the activated T-cells in our in-vitro system and compared theprotective effect of SurR9-C84A with GrB inhibitor.
2. Material and method
2.1. Cell line and culture conditions
Human SK-N-SH, obtained from the American Type CultureCollection (ATCC) were grown as a monolayer in the Dulbecco'sMinimumEssentialMedium (DMEM)media supplementedwith 10% ofheat-inactivated fetal bovine serum (FBS), penicillin (20 units/ml) andstreptomycin (20 mg/ml) at 37 °C in a saturated humid atmospherewith 5% CO2. As the cells became confluent, they were split aftertreatment with Trypsin–EDTA. The seeding density varied according tothe experiment. To initiate the differentiation in SK-N-SH, cells weregrown in the DMEM media containing the 20 μM retinoic acid (RA)(Sigma-Aldrich) under dark conditions with replacement of theconditionedmedia every 48–72 h for twoweeks. Cells were consideredto be differentiated if they had at least one process longer than the cellbody regarded as neurite (Chang et al., 2005).
Jurkat E-6.1 cells, a human T lymphocyte cell line, were obtainedfrom the ATCC. This T-cell line was grown in RPMI 1640 mediumsupplemented with 10% fetal bovine serum, 100 μg/ml streptomycinand100 IU/mlpenicillin in ahumidified5%CO2 chamber at37 °C. T-cellswere stimulated using 10 ng/ml PMA (Sigma) and 500 ng/ml ionomy-cine (Sigma) for 72 h in culture. Culture supernatant was then collectedand incubated (1:10 dilution) with differentiated neuronal cells.
Caspase and GrB inhibition was performed using 50 μM ofirreversible broad-range caspase inhibitor Boc-D-FMK and GrBinhibitor I, Z-AAD-CMK (Calbiochem, San Diego, CA, USA) 1 h beforeeach treatment. The best inhibitory concentration was explored byperforming MTT assays.
2.2. Construction of a BIR motif mutant of survivin expressing vector andprotein purification
A cell-permeable (9 arginine residues) form of survivin mutant(SurR9-C84A) was constructed comprising 9 N-terminal arginine resi-dues (R9, cell-permeable peptide carrier) fused to the C84A survivinmutant, as reported previously (Baratchi et al., 2010b). Briefly, the cDNAof a pcDNA3 expression plasmid encoding survivin mutant was excisedand amplified using the sense primer 5′-GGGGATCCATGCGACGACGAC-GACGACGACGACGACGAGGAGCTCCGGCGCTGCCCCAG-3′ (encodes R9)and the antisense primer 5′-GGGATCCTTAGGCAGCCAGC-3′ (Cheunget al., 2006). The resulting PCR product was subcloned into pGEM-T(Promega Corp., Madison, WI), liberated by digestion with BamHI, andcloned into the expression vector pGEX-2T (Pharmacia Biotech, Piscat-away, NJ) to give the vector pGEX-2T/SurR9-C84A. The integrity of thepGEX-2T/SurR9-C84A vector was confirmed by DNA sequence analysis(Cheung et al., 2006). The recombinant SurR9-C84Awas transfected intothe E. coli BL21 strain. The transformants bearing SurR9-C84A plasmidwere inoculated in 1000 ml flask with 500 ml LB medium containing
100 mg/l ampicillin. When the culture OD600nm reached 0.7 at 25 °C, theexpression strains were induced with 0.7 mM isopropylthiogalactoside(IPTG). As the protein was GST tagged, it was purified with glutathioneagarose (GE-heath care) according to the procedure suggested by themanufacturer.
2.3. Cell cycle analysis
RA differentiated and undifferentiated SK-N-SH cells were har-vested, fixed and permeabilized over night in ice-cold 70% ethanol(Sigma). The cells were washed twice with PBS. RNA was digestedwith RNase A (Sigma). The DNA was stained with Propidium Iodide(PI) (10 μg/ml). Fluorescence was recorded in BD FACS Canto™ IIanalyser. Instrument settings were adjusted to move the G0/G1 peakto 200 relative fluorescence units. A total of 10,000 events perconditionwere recorded. Data was evaluated using the BD FACS CantoSoftware.
2.4. Neurotoxicity assay
To evaluate the level of cell toxicity, MTT and Trypan blue exclusionassays were conducted. The colorimetric MTT (3-(4, 5-dimethylthiazol-2yl)-2, 5-diphenyltetrazoliumbromide) assay was conducted with differ-entiated SK-N-SH cells. For differentiation, 5×103 cells were seeded in 96well plates and incubated with RA for a period of 10 days. Differentiatedcellswerepre-treatedwith/without SurR9-C84A ingrowthmedia for 24 hfollowed by treatment with 10% (final concentration of 18 pg/ml)activated Jurkat supernatant for 24 h prior to MTT assay. For Trypanblue exclusion, cells were seeded at 105/ml and differentiated for 10 daysin a 6-well plate. Cells were pre-treated with 75 μg/ml SurR9-C84A incontrol media for 24 h and treated with activated T-cell supernatants foranother 24 h. The cells were then stained with Trypan blue for 5 min,washed with PBS, pH 7.4, and fixed with 4% paraformaldehyde. Trypanblue positive and negative cells were counted in randomly selected fieldsfrom at least three different quadrants of the culture well.
Approximately 200 cells were counted in each well. Eachexperiment was conducted in triplicate wells. Mean (±SEM) valueswere calculated from at least three independent experiments.
2.5. Immuno-depletion of GrB
For immuno-depletion of GrB from the activated T-cell supernatants,the supernatant derived from the activated T-cells was incubated withpreswollenproteinG sepharose (Sigma-Aldrich) for 2 h to eliminate thenon-specific bindings. Themixturewas centrifuged and the supernatantwas incubated with anti-GrB or isotype-match control antibody (Cellsignalling) overnight at 4 °C. The incubated mixture was furtherincubated with protein G sepharose for 2 h at 4 °C and filtered throughthe column.
2.6. Quantitative RT-PCR for analysis of mRNA expression
RNA was extracted from the total cell lysate using the GenElute™Mammalian Total RNA Miniprep kit (Sigma-Aldrich) according to theprocedure suggestedby themanufacturer. The cDNAwasprepared from5 μg of total extracted RNA and SUPERSCRIPT III reverse-transcriptase(Invitrogen), oligo-dT as primer and nuclease-free water, followed byquantitative real time PCR in triplicate. The reaction mixture of all genetypes (primer sequence in Table 1) was subjected to 36 PCRamplification cycles of 60 s at 94 °C, 60 s at 60 °C, and 90 s at 72 °C.
2.7. Immunocytochemistry (ICC) and Western blotting (WB)
Cells (105 cells/well) were grown in 8well chamber slides for 24 h.Following the treatment, cells were rinsed with PBS, fixed for 15 minwith freshly prepared 4% paraformaldehyde/PBS and permeabilized

Table 1Primer sequences of applied genes.
Name Forward primer sequences Reverse primer sequence Ref
Fas GAAGGACATGGCTTAGAAGTG ACTTAGTGTCATGACTCCAGC Roh et al. (2002)Caspase3 CTCGGTCTGGTACAGATGTCGATG GGTTAACCCGGGTAAGAATGTGCA Deligezer et al. (2006)Cyclin D1 GAACAAACAGATCATCCGCAA CCCTTCTGGTATCAAAATGC Imanishi et al. (2001)β-Actin CTCACCGAGCGCGGCTACA CTCCTGCTTGCTGATCCACAT Li et al. (2009)
20 S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
for 10 min in 0.2% Triton X100/PBS. After blocking with 2% normal goatserum for20 min, cellswere incubated for 1 h at room temperaturewithantibodies against rabbit anti-β-Tubulin III (1/400, Sigma-Aldrich) andmouse anti-Neurofilament 200 (1/500, Sigma-Aldrich). FITC labelledgoat anti-mouse (1/160, Sigma-Aldrich) and goat anti-rabbit (1/100,Sigma-Aldrich) were used as secondary antibodies. TUNEL stainingwasperformed using in situ cell death detection kit (Roche) according to themanufacturer's instructions.
Slides weremountedwithmountingmedia (Vector Labs, USA). ForWestern blot analysis, 50 μg of dividing or differentiated cell lysateprotein was used. Cell lysate was prepared using the RIPA buffer(150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate,0.1% SDS and 50 mMTris, PH 8.0) containing protease inhibitor tablets(Roche Applied Science). The total protein content was measuredusing theBradford assay. Sampleswere electrophoresedand transferredto the PVDF membrane. Primary antibodies used were mouse anti-survivin (D8) (1/2000, SANTA CRUZ) and goat anti-β-actin (1/1000,Santa Cruz), rabbit anti-caspase3 (1/1000, Cell signalling), mouse anti-cyclinD1 (1/2000, Cell signalling), rabbit anti-Fas (1/1000, Cellsignalling) and rabbit anti-GrB (1/1000, Cell signalling). Secondaryantibodies were peroxidase conjugated anti-mouse (1/2000, Cellsignalling), peroxidase conjugated anti-goat (1/10,000, Sigma-Aldrich)and peroxidase conjugated anti-rabbit (1/2000, cell signalling). Theblots were developed using the chemiluminescence assay system (ECLkit; Amersham Biosciences).
2.8. Measurement of cytosolic calcium ([Ca2+]i)
[Ca2+]i was determined by the Fura-2/acetoxymethyl ester (AM)(Sigma) using the methods similar to those previously reported (Hirstet al., 1999; Kanwar et al., 1995). Cells were incubated with 3 μM Fura-2/AM for 30 min at 37 °C in the dark and washed with Krebs/HEPES buffer(143.3 mMNa+, 4.7 mMK+, 2.5 mMCa2+, 1.3 mMMg2+, 125.6 mMCl−,25 mM HCO3
−, 1.2 mM H2PO4−, 1.2 mM SO4
2−, 11.7 mM glucose, and10 mMHEPES, pH7.4) to remove the extra cellular fura-2/AM. The loadedcells were further incubated at room temperature for de-esterification ofthe Fura-2/AM. Loaded cells were treated with activated T-cell super-natants. Readings were performed at 37 °C using “FlexStation III”microplate reader containing the mixer. Cells were alternativelyexcited at 340 and 380 nm by a monochrometer, and emission wasrecorder at 510 nm using Bucher Biotec software (Bucher BiotecAG, Switzerland). [Ca2+]i was estimated by translating thefluorescence ratio of 340/380 nm to [Ca2+] using the Grynkiewicz
method as: Ca2 +� �
i nMð Þ = Kd × R−RminRmax−R × Q, where Kd=225 nM
(for Ca2+ binding to Fura-2 at 37 °C), R=340/380 ratio at differenttimes, Rmax=380/340 ratio under Ca2+saturating conditions,Rmin=340/380 nm ratio under Ca2+ free conditions and Q is the ratioof Fmin/Fmax at 380 nm. The calibration values were determined in thepresence of 1 mM Ca2+ (Rmax) or 10 mM EGTA (Rmin).
2.9. Determination of the intrinsic mitochondrial membrane potential(Δψm)
Mitochondrial membrane potential variation was determined usingthe MitoLight Mitochondrial Apoptosis Detection Kit (Chemicon)
according to the manufacturer's instruction. MitoLight is a lipophiliccationic dye, which stains live cell mitochondria according to theirmembrane potentials. In healthy cells, the dye accumulates andaggregates in the mitochondria and fluoresces red (Em=585–590 nm) while in apoptotic cells, the dye remains in the cytosol andfluoresces green (Em=527–530 nm).
Briefly, cells were washed with PBS. Diluted mitolight reagent wasadded to the cells and incubated at 37 °C in 5% CO2 for 20 min. Cellswere washed with PBS and the fluorescence was measured atλexcitation=485 nm and λemission=580 nm with a spectrofluorome-ter. The ratio of red (Em=585–590 nm) to green (Em=527–530 nm) fluorescence reflects the Δψm.
2.10. Image and data analysis on cell cultures
Analysis and photography of fluorescent immunostained cellswere carried out using an inverted Leica SP5 confocal microscope.Results are shown as the average±SEM of data extracted from two tothree experiments, unless stated otherwise.
2.11. Data interpretation and statistical analysis
Results are expressed as means±SEM of at least three experi-ments unless otherwise stated. For the statistical evaluation ofnumerical data, one way ANOVA and Student's t-test were used tocompute the differences. In all cases, the number of asterisks indicatesthe level of significance as **Pb0.01 and *Pb0.05, and no asteriskindicates PN0.05.
3. Results
3.1. Neuronal cell differentiation and cell cycle analysis
Cell differentiation was induced in SK-N-SH cells with 20 μM ofretinoic acid (RA) for a period of 10 days. This treatment restricted thecell division and changed the morphology, as characterised by theneurite extension (Fig. 1A–B).
To monitor the cell cycle stage, SK-N-SH cells were induced todifferentiate, and the cell cycle was analysed by fluorescence-activated cell sorting (FACS) using PI staining. According to theseresults, more than 70% of undifferentiated SK-N-SH cells were at theG2/M phase, whereas after differentiation they arrested in G0/G1(Fig. 1C–D). These results were further confirmed by RT-PCR andWestern blotting (Fig. 1E–F). We found that survivin was over-expressed in undifferentiated SK-N-SH cells, whereas its expressiondeclined dramatically (4 fold±0.1; Pb0.001) at bothmRNAandproteinlevels, which correlated with the cell cycle arrest in G0/G1 phase afterdifferentiation. However, survivin was also expressed in G2/M.
3.2. Activated T-cells release GrB
First, to determine whether activated Jurkat cells releases solubleneurotoxicants, we treated the differentiated neuronal cells withsupernatant derived from PMA/ionomycine activated T-cells orunstimulated T-cells, and studied the cell viability by either MTT orTrypan blue exclusion assays. The activated T-cell supernatant
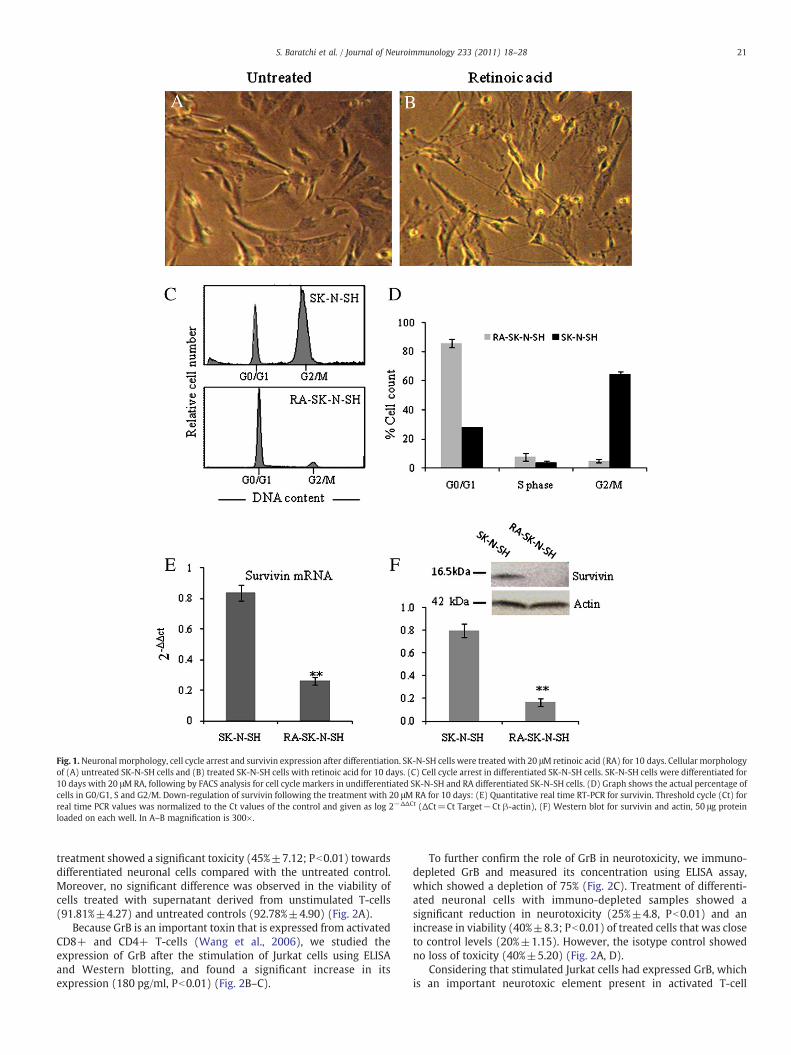
Fig. 1.Neuronal morphology, cell cycle arrest and survivin expression after differentiation. SK-N-SH cells were treated with 20 μM retinoic acid (RA) for 10 days. Cellular morphologyof (A) untreated SK-N-SH cells and (B) treated SK-N-SH cells with retinoic acid for 10 days. (C) Cell cycle arrest in differentiated SK-N-SH cells. SK-N-SH cells were differentiated for10 days with 20 μMRA, following by FACS analysis for cell cycle markers in undifferentiated SK-N-SH and RA differentiated SK-N-SH cells. (D) Graph shows the actual percentage ofcells in G0/G1, S and G2/M. Down-regulation of survivin following the treatment with 20 μM RA for 10 days: (E) Quantitative real time RT-PCR for survivin. Threshold cycle (Ct) forreal time PCR values was normalized to the Ct values of the control and given as log 2−ΔΔCt (ΔCt=Ct Target−Ct β-actin), (F) Western blot for survivin and actin, 50 μg proteinloaded on each well. In A–B magnification is 300×.
21S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
treatment showed a significant toxicity (45%±7.12; Pb0.01) towardsdifferentiated neuronal cells compared with the untreated control.Moreover, no significant difference was observed in the viability ofcells treated with supernatant derived from unstimulated T-cells(91.81%±4.27) and untreated controls (92.78%±4.90) (Fig. 2A).
Because GrB is an important toxin that is expressed from activatedCD8+ and CD4+ T-cells (Wang et al., 2006), we studied theexpression of GrB after the stimulation of Jurkat cells using ELISAand Western blotting, and found a significant increase in itsexpression (180 pg/ml, Pb0.01) (Fig. 2B–C).
To further confirm the role of GrB in neurotoxicity, we immuno-depleted GrB and measured its concentration using ELISA assay,which showed a depletion of 75% (Fig. 2C). Treatment of differenti-ated neuronal cells with immuno-depleted samples showed asignificant reduction in neurotoxicity (25%±4.8, Pb0.01) and anincrease in viability (40%±8.3; Pb0.01) of treated cells that was closeto control levels (20%±1.15). However, the isotype control showedno loss of toxicity (40%±5.20) (Fig. 2A, D).
Considering that stimulated Jurkat cells had expressed GrB, whichis an important neurotoxic element present in activated T-cell
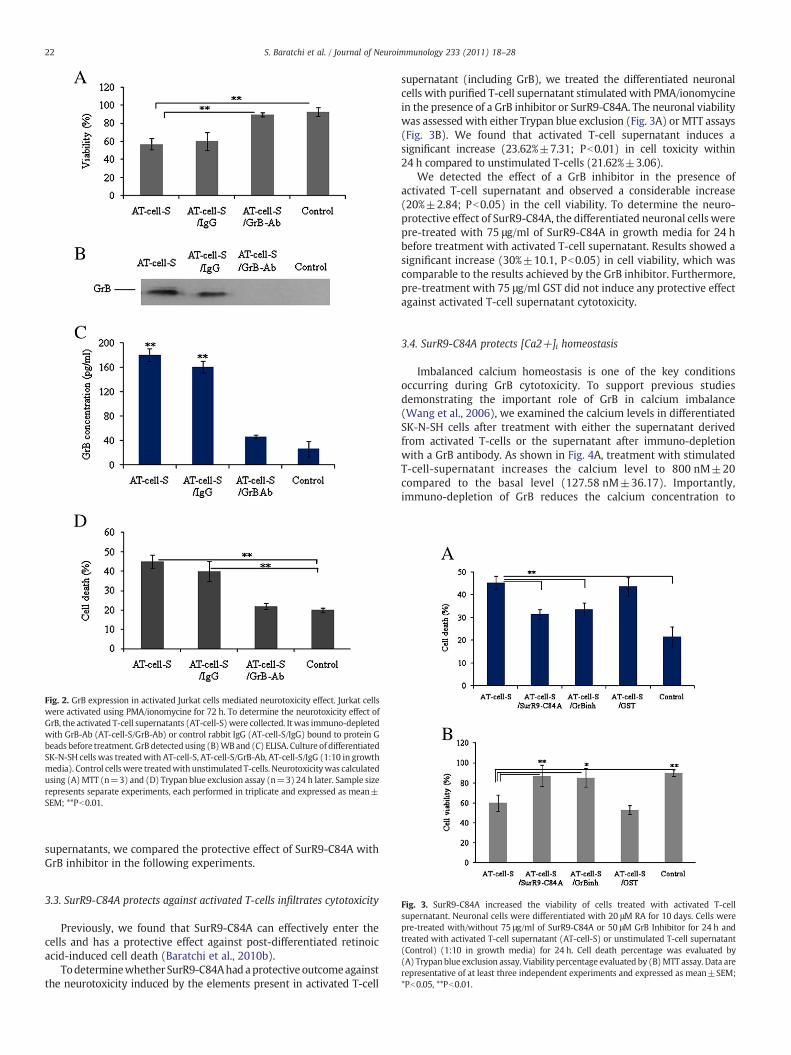
Fig. 2. GrB expression in activated Jurkat cells mediated neurotoxicity effect. Jurkat cellswere activated using PMA/ionomycine for 72 h. To determine the neurotoxicity effect ofGrB, the activated T-cell supernatants (AT-cell-S)were collected. Itwas immuno-depletedwith GrB-Ab (AT-cell-S/GrB-Ab) or control rabbit IgG (AT-cell-S/IgG) bound to protein Gbeads before treatment. GrB detectedusing (B)WB and (C) ELISA. Culture of differentiatedSK-N-SH cells was treatedwith AT-cell-S, AT-cell-S/GrB-Ab, AT-cell-S/IgG (1:10 in growthmedia). Control cellswere treatedwith unstimulated T-cells. Neurotoxicitywas calculatedusing (A)MTT (n=3) and (D) Trypan blue exclusion assay (n=3) 24 h later. Sample sizerepresents separate experiments, each performed in triplicate and expressed as mean±SEM; **Pb0.01.
22 S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
supernatants, we compared the protective effect of SurR9-C84A withGrB inhibitor in the following experiments.
Fig. 3. SurR9-C84A increased the viability of cells treated with activated T-cellsupernatant. Neuronal cells were differentiated with 20 μM RA for 10 days. Cells werepre-treated with/without 75 μg/ml of SurR9-C84A or 50 μM GrB Inhibitor for 24 h andtreated with activated T-cell supernatant (AT-cell-S) or unstimulated T-cell supernatant(Control) (1:10 in growth media) for 24 h. Cell death percentage was evaluated by(A) Trypan blue exclusion assay. Viability percentage evaluated by (B)MTT assay. Data arerepresentative of at least three independent experiments and expressed as mean±SEM;*Pb0.05, **Pb0.01.
3.3. SurR9-C84A protects against activated T-cells infiltrates cytotoxicity
Previously, we found that SurR9-C84A can effectively enter thecells and has a protective effect against post-differentiated retinoicacid-induced cell death (Baratchi et al., 2010b).
Todeterminewhether SurR9-C84Ahadaprotective outcomeagainstthe neurotoxicity induced by the elements present in activated T-cell
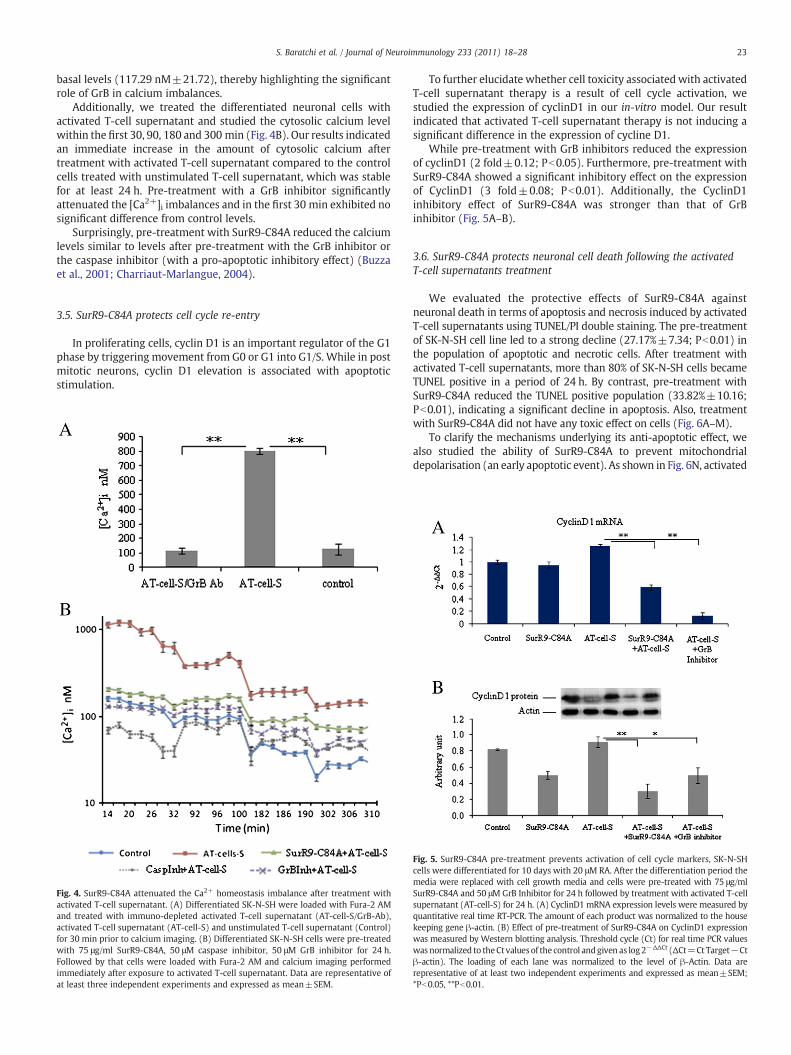
supernatant (including GrB), we treated the differentiated neuronalcells with purified T-cell supernatant stimulated with PMA/ionomycinein the presence of a GrB inhibitor or SurR9-C84A. The neuronal viabilitywas assessed with either Trypan blue exclusion (Fig. 3A) or MTT assays(Fig. 3B). We found that activated T-cell supernatant induces asignificant increase (23.62%±7.31; Pb0.01) in cell toxicity within24 h compared to unstimulated T-cells (21.62%±3.06).
We detected the effect of a GrB inhibitor in the presence ofactivated T-cell supernatant and observed a considerable increase(20%±2.84; Pb0.05) in the cell viability. To determine the neuro-protective effect of SurR9-C84A, the differentiated neuronal cells werepre-treated with 75 μg/ml of SurR9-C84A in growth media for 24 hbefore treatment with activated T-cell supernatant. Results showed asignificant increase (30%±10.1, Pb0.05) in cell viability, which wascomparable to the results achieved by the GrB inhibitor. Furthermore,pre-treatment with 75 μg/ml GST did not induce any protective effectagainst activated T-cell supernatant cytotoxicity.
3.4. SurR9-C84A protects [Ca2+]i homeostasis
Imbalanced calcium homeostasis is one of the key conditionsoccurring during GrB cytotoxicity. To support previous studiesdemonstrating the important role of GrB in calcium imbalance(Wang et al., 2006), we examined the calcium levels in differentiatedSK-N-SH cells after treatment with either the supernatant derivedfrom activated T-cells or the supernatant after immuno-depletionwith a GrB antibody. As shown in Fig. 4A, treatment with stimulatedT-cell-supernatant increases the calcium level to 800 nM±20compared to the basal level (127.58 nM±36.17). Importantly,immuno-depletion of GrB reduces the calcium concentration to
23S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
basal levels (117.29 nM±21.72), thereby highlighting the significantrole of GrB in calcium imbalances.
Additionally, we treated the differentiated neuronal cells withactivated T-cell supernatant and studied the cytosolic calcium levelwithin the first 30, 90, 180 and 300 min (Fig. 4B). Our results indicatedan immediate increase in the amount of cytosolic calcium aftertreatment with activated T-cell supernatant compared to the controlcells treated with unstimulated T-cell supernatant, which was stablefor at least 24 h. Pre-treatment with a GrB inhibitor significantlyattenuated the [Ca2+]i imbalances and in the first 30 min exhibited nosignificant difference from control levels.
Surprisingly, pre-treatment with SurR9-C84A reduced the calciumlevels similar to levels after pre-treatment with the GrB inhibitor orthe caspase inhibitor (with a pro-apoptotic inhibitory effect) (Buzzaet al., 2001; Charriaut-Marlangue, 2004).
3.5. SurR9-C84A protects cell cycle re-entry
In proliferating cells, cyclin D1 is an important regulator of the G1phase by triggering movement from G0 or G1 into G1/S. While in postmitotic neurons, cyclin D1 elevation is associated with apoptoticstimulation.
Fig. 4. SurR9-C84A attenuated the Ca2+ homeostasis imbalance after treatment withactivated T-cell supernatant. (A) Differentiated SK-N-SH were loaded with Fura-2 AMand treated with immuno-depleted activated T-cell supernatant (AT-cell-S/GrB-Ab),activated T-cell supernatant (AT-cell-S) and unstimulated T-cell supernatant (Control)for 30 min prior to calcium imaging. (B) Differentiated SK-N-SH cells were pre-treatedwith 75 μg/ml SurR9-C84A, 50 μM caspase inhibitor, 50 μM GrB inhibitor for 24 h.Followed by that cells were loaded with Fura-2 AM and calcium imaging performedimmediately after exposure to activated T-cell supernatant. Data are representative ofat least three independent experiments and expressed as mean±SEM.
To further elucidate whether cell toxicity associated with activatedT-cell supernatant therapy is a result of cell cycle activation, westudied the expression of cyclinD1 in our in-vitro model. Our resultindicated that activated T-cell supernatant therapy is not inducing asignificant difference in the expression of cycline D1.
While pre-treatment with GrB inhibitors reduced the expressionof cyclinD1 (2 fold±0.12; Pb0.05). Furthermore, pre-treatment withSurR9-C84A showed a significant inhibitory effect on the expressionof CyclinD1 (3 fold±0.08; Pb0.01). Additionally, the CyclinD1inhibitory effect of SurR9-C84A was stronger than that of GrBinhibitor (Fig. 5A–B).
3.6. SurR9-C84A protects neuronal cell death following the activatedT-cell supernatants treatment
We evaluated the protective effects of SurR9-C84A againstneuronal death in terms of apoptosis and necrosis induced by activatedT-cell supernatants using TUNEL/PI double staining. The pre-treatmentof SK-N-SH cell line led to a strong decline (27.17%±7.34; Pb0.01) inthe population of apoptotic and necrotic cells. After treatment withactivated T-cell supernatants, more than 80% of SK-N-SH cells becameTUNEL positive in a period of 24 h. By contrast, pre-treatment withSurR9-C84A reduced the TUNEL positive population (33.82%±10.16;Pb0.01), indicating a significant decline in apoptosis. Also, treatmentwith SurR9-C84A did not have any toxic effect on cells (Fig. 6A–M).
To clarify the mechanisms underlying its anti-apoptotic effect, wealso studied the ability of SurR9-C84A to prevent mitochondrialdepolarisation (an early apoptotic event). As shown in Fig. 6N, activated
Fig. 5. SurR9-C84A pre-treatment prevents activation of cell cycle markers, SK-N-SHcells were differentiated for 10 days with 20 μM RA. After the differentiation period themedia were replaced with cell growth media and cells were pre-treated with 75 μg/mlSurR9-C84A and 50 μM GrB Inhibitor for 24 h followed by treatment with activated T-cellsupernatant (AT-cell-S) for 24 h. (A) CyclinD1 mRNA expression levels were measured byquantitative real time RT-PCR. The amount of each product was normalized to the housekeeping gene β-actin. (B) Effect of pre-treatment of SurR9-C84A on CyclinD1 expressionwas measured by Western blotting analysis. Threshold cycle (Ct) for real time PCR valueswas normalized to theCt valuesof the control andgiven as log2−ΔΔCt (ΔCt=Ct Target−Ctβ-actin). The loading of each lane was normalized to the level of β-Actin. Data arerepresentative of at least two independent experiments and expressed as mean±SEM;*Pb0.05, **Pb0.01.
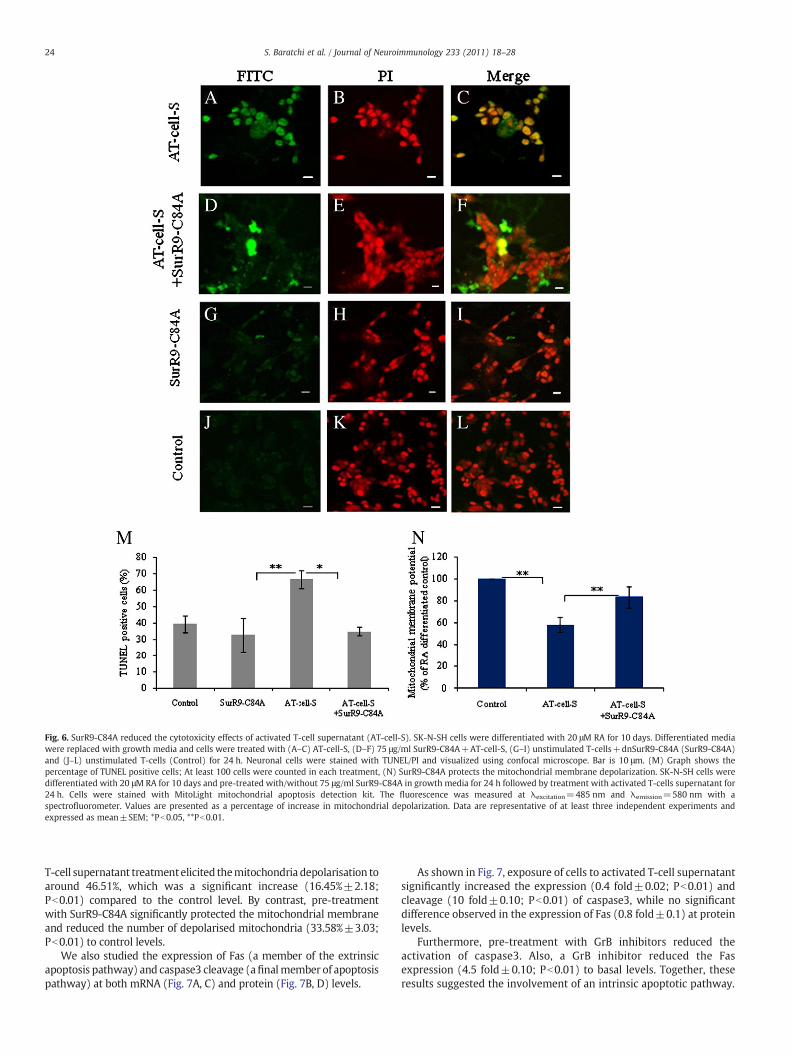
Fig. 6. SurR9-C84A reduced the cytotoxicity effects of activated T-cell supernatant (AT-cell-S). SK-N-SH cells were differentiated with 20 μM RA for 10 days. Differentiated mediawere replaced with growth media and cells were treated with (A–C) AT-cell-S, (D–F) 75 μg/ml SurR9-C84A+AT-cell-S, (G–I) unstimulated T-cells+dnSurR9-C84A (SurR9-C84A)and (J–L) unstimulated T-cells (Control) for 24 h. Neuronal cells were stained with TUNEL/PI and visualized using confocal microscope. Bar is 10 μm. (M) Graph shows thepercentage of TUNEL positive cells; At least 100 cells were counted in each treatment, (N) SurR9-C84A protects the mitochondrial membrane depolarization. SK-N-SH cells weredifferentiated with 20 μM RA for 10 days and pre-treated with/without 75 μg/ml SurR9-C84A in growth media for 24 h followed by treatment with activated T-cells supernatant for24 h. Cells were stained with MitoLight mitochondrial apoptosis detection kit. The fluorescence was measured at λexcitation=485 nm and λemission=580 nm with aspectrofluorometer. Values are presented as a percentage of increase in mitochondrial depolarization. Data are representative of at least three independent experiments andexpressed as mean±SEM; *Pb0.05, **Pb0.01.
24 S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
T-cell supernatant treatmentelicited themitochondria depolarisation toaround 46.51%, which was a significant increase (16.45%±2.18;Pb0.01) compared to the control level. By contrast, pre-treatmentwith SurR9-C84A significantly protected the mitochondrial membraneand reduced the number of depolarised mitochondria (33.58%±3.03;Pb0.01) to control levels.
We also studied the expression of Fas (a member of the extrinsicapoptosis pathway) and caspase3 cleavage (afinalmember of apoptosispathway) at both mRNA (Fig. 7A, C) and protein (Fig. 7B, D) levels.
As shown in Fig. 7, exposure of cells to activated T-cell supernatantsignificantly increased the expression (0.4 fold±0.02; Pb0.01) andcleavage (10 fold±0.10; Pb0.01) of caspase3, while no significantdifference observed in the expression of Fas (0.8 fold±0.1) at proteinlevels.
Furthermore, pre-treatment with GrB inhibitors reduced theactivation of caspase3. Also, a GrB inhibitor reduced the Fasexpression (4.5 fold±0.10; Pb0.01) to basal levels. Together, theseresults suggested the involvement of an intrinsic apoptotic pathway.
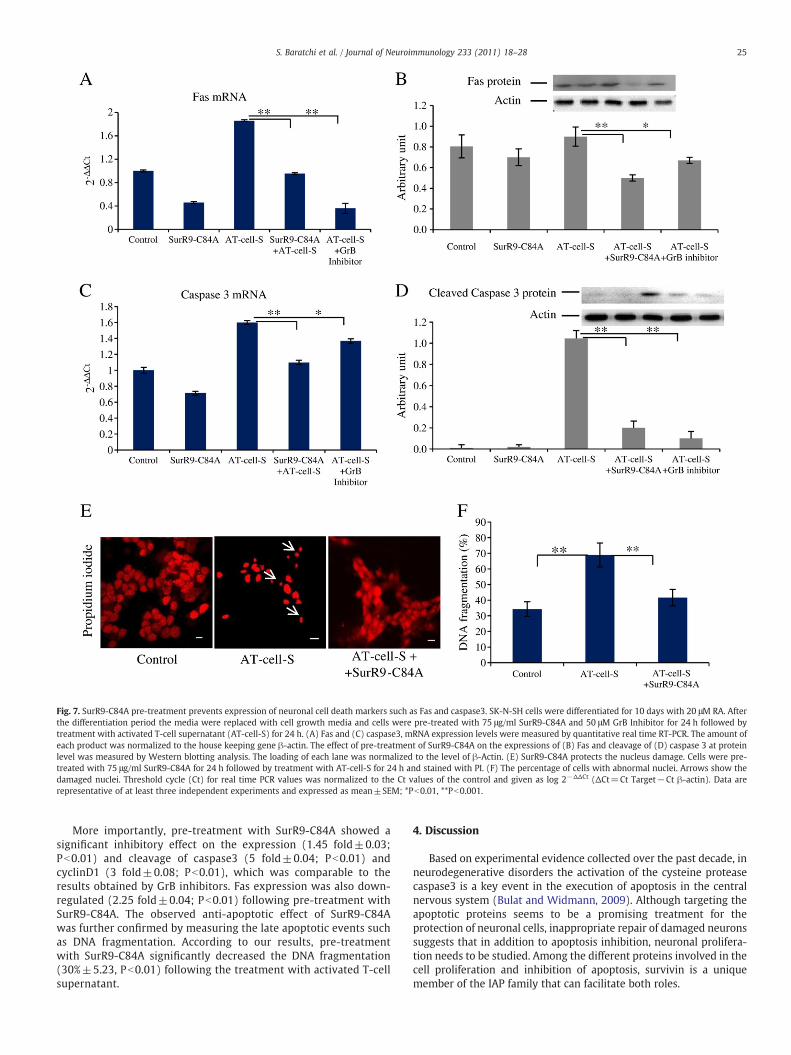
Fig. 7. SurR9-C84A pre-treatment prevents expression of neuronal cell death markers such as Fas and caspase3. SK-N-SH cells were differentiated for 10 days with 20 μM RA. Afterthe differentiation period the media were replaced with cell growth media and cells were pre-treated with 75 μg/ml SurR9-C84A and 50 μM GrB Inhibitor for 24 h followed bytreatment with activated T-cell supernatant (AT-cell-S) for 24 h. (A) Fas and (C) caspase3, mRNA expression levels were measured by quantitative real time RT-PCR. The amount ofeach product was normalized to the house keeping gene β-actin. The effect of pre-treatment of SurR9-C84A on the expressions of (B) Fas and cleavage of (D) caspase 3 at proteinlevel was measured by Western blotting analysis. The loading of each lane was normalized to the level of β-Actin. (E) SurR9-C84A protects the nucleus damage. Cells were pre-treated with 75 μg/ml SurR9-C84A for 24 h followed by treatment with AT-cell-S for 24 h and stained with PI. (F) The percentage of cells with abnormal nuclei. Arrows show thedamaged nuclei. Threshold cycle (Ct) for real time PCR values was normalized to the Ct values of the control and given as log 2−ΔΔCt (ΔCt=Ct Target−Ct β-actin). Data arerepresentative of at least three independent experiments and expressed as mean±SEM; *Pb0.01, **Pb0.001.
25S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
More importantly, pre-treatment with SurR9-C84A showed asignificant inhibitory effect on the expression (1.45 fold±0.03;Pb0.01) and cleavage of caspase3 (5 fold±0.04; Pb0.01) andcyclinD1 (3 fold±0.08; Pb0.01), which was comparable to theresults obtained by GrB inhibitors. Fas expression was also down-regulated (2.25 fold±0.04; Pb0.01) following pre-treatment withSurR9-C84A. The observed anti-apoptotic effect of SurR9-C84Awas further confirmed by measuring the late apoptotic events suchas DNA fragmentation. According to our results, pre-treatmentwith SurR9-C84A significantly decreased the DNA fragmentation(30%±5.23, Pb0.01) following the treatment with activated T-cellsupernatant.
4. Discussion
Based on experimental evidence collected over the past decade, inneurodegenerative disorders the activation of the cysteine proteasecaspase3 is a key event in the execution of apoptosis in the centralnervous system (Bulat and Widmann, 2009). Although targeting theapoptotic proteins seems to be a promising treatment for theprotection of neuronal cells, inappropriate repair of damaged neuronssuggests that in addition to apoptosis inhibition, neuronal prolifera-tion needs to be studied. Among the different proteins involved in thecell proliferation and inhibition of apoptosis, survivin is a uniquemember of the IAP family that can facilitate both roles.

26 S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
We have reported for the first time, the neuroprotective effect of acell-permeable form of a survivin mutant (SurR9-C84A), in which thecysteine at position 84 in the zinc-coordination site has beensubstituted with alanine and was amino-terminally fused during thesynthesis with a non-arginine (R9) peptide to render its cellpermeable (Cheung et al., 2006). Using R9 peptide is advantageousover other carriers, such as polylysine or ornithine, because it exhibitsa greater cellular uptake (Mitchell et al., 2000), and it is superior toviral systems because it is less toxic and will not induce a significantimmune response (Morris et al., 2000). More importantly, R9activated proteins has the ability to cross the blood brain barrier,which can provide a safe and non-invasive ability for the future of in-vivo studies (Kumar et al., 2007).
SurR9-C84Awas employed to investigate the survival mechanismsin differentiated neural cells after the induction of apoptosis byactivated T-cell supernatants, which has not been studied previously.The SurR9-C84A protein has been shown to bind to polymerisedmicrotubules and localise to the microtubule organising centre ofinterphase cells in a manner indistinguishable from wild-typesurvivin (Li et al., 1998). Although the effect of SurR9-C84A on cellsover-expressing survivin has been studied in a few different cancercell lines, its protective ability in neuronal cell lines (with low levels ofsurvivin expression) has not been investigated. In terms of cellproliferation, the results obtained from the SurR9-C84A treatment onSK-N-SH cells during different stages of differentiation led to thefollowing results. The proliferation of cells was reduced at day 0,increased during days 4–6, and remained unchanged at day 10 withrespect to untreated control cells.
Release of GrB from activated T-cells at the early stage of MS andits neurotoxic effect have been reported previously (Malmestromet al., 2008). These cytotoxic effects appear to be due to the activationof the apoptosis pathway and disruption of calcium homeostasis(Wang et al., 2006). An important mechanism for counteractingneuronal cytotoxicity might be through targeting the family membersof apoptotic proteins or cell proliferation markers.
In this context, our results showed that after differentiation of SK-N-SH cells with RA, their cell cycle arrested in G0/G1 and survivinexpression was consequently reduced, which was expected becausesurvivin is mostly expressed in G2/M according to the previousfindings (Altieri, 2003a). Activated T-cells play an important roleduring the pathogenesis of MS. They induce neuronal damage throughdirect cell-to-cell contact (Giuliani et al., 2003; Nitsch et al., 2004) orin a contact independent manner by releasing soluble factors such asGrB (Tomkinson et al., 1989). Both CD4+ and CD8+ forms of T-cellsare able to release GrB (Aguiló et al., 2010; Tomkinson et al., 1989). Inthis study, the inflammatory environment of the CNS during MS wasreproduced through the activation of Jurkat cells as a model of T-cells.According to our results, supernatants derived from PMA/inomycineactivated T-cells contained GrB. Also, pre-treatment of neuronal cellswith GrB depleted supernatant increased the viability of neuronalcells to near normal conditions.
GrB is the most powerful pro-apoptotic member of the granzymefamily of proteins. Once liberated from T-cells, it enters the target cellthrough endocytosis and mediates the cell death by activation of theapoptosis pathway through cleavage of aspartic acid residues andactivation of caspases8 and 10, as well as effector caspases3 and 7(Wang et al., 2006). Its role in mediating neuronal death during focalcerebral ischemia (Chaitanya et al., 2010) and MS (Malmestrom et al.,2008) has been previously reported.
In the present study, the treatment of differentiated neuronal cellswith activated T-cell supernatant had a significant toxic effect, whichwas GrB dependent. However, the immuno-depletion of GrB or usinga GrB Inhibitor (Z-AAD-CMK) reduced the cytotoxicity. The GrBinhibitor forms a tetrahedral complex with the GrB active site andsubsequently decreases its cytotoxic effects by reducing the cleavageof caspase3 and [Ca2+]i overload (Shi et al., 1992).
It has been shown that survivin has a role in the control of mitosisand cell cycle by its association with aurora B and INCENP at thecentromer/kinetochore, which forms the chromosomal passengercomplex (Bolton et al., 2002). Survivin is also required for neuronalsurvival during brain development and in the postnatal period (Jianget al., 2005). Thus, SurR9-C84A is a survivinmutant that we previouslyfound for its neuroprotective effect against post differentiationretinoic acid-induced cell death (Baratchi et al., 2010b).
According to our current results, pre-treatment of cells withSurR9-C84A increased the resistance of cells to toxic effects inducedby activated T-cell supernatant. Interestingly, the time course ofSurR9-C84A-induced neuroprotection showed a shorter treatmentwas needed for SK-N-SH cells to induce cytoprotective effects againstGrB. These findings are important because SurR9-C84A can play asignificant role in the detoxification of GrB, which is an importanttoxin released by activated T-cells during the progress of inflammation.In this regard, our results demonstrated that differentiated SK-N-SHcells treated with SurR9-C84A, in an experimental condition similar toan inflammatory environment, could remarkably increase resistance toneuronal death in terms of apoptosis and necrosis elicited by activatedT-cell supernatant.
Several lines of evidence have suggested the involvement ofimbalanced calcium homeostasis in various neurodegenerative dis-orders. To confirm the protective effect of SurR9-C84A, we evaluated itseffect on calcium homeostasis in the cells treated with activated T-cellsupernatants. As the protective effect of caspase inhibitors and GrBinhibitors has been demonstrated previously (Ray, 2006; Yang et al.,1998),we compared theirprotectiveeffect oncalciumhomeostasiswithSurR9-C84A. Here, we report that cultured differentiated neuronstreatedwith activated T-cell supernatant can increase the cytosolic Ca2+
within the first few hours of injury, which occurred before orconcurrently with the activation of caspase3, and before the cell deathwas detectable by PI staining. Our results further indicated that pre-treatment of cells with SurR9-C84A reduces cytosolic calcium levels,which was similar to the effect of caspase inhibitors and GrB inhibitors(Fig. 8).
A large body of evidence suggests the absence of cell division and thecontinued arrest of cell cycle as the core features of neuronal identity,and failure to do so causes neuronal degeneration (Herrup and Yang,2007). Moreover, there are a number of reports indicating theexpression of cell cycle markers including cyclinD1 in neurons frompatients suffering from neurodegenerative disease (Busser et al., 1998;Nagy et al., 1997; Smith et al., 1999; Yang et al., 2001). For instance,cyclinD1 expression is elevated in dying neurons under differentapoptotic conditions, and the inhibition of cyclin D-dependent kinasesboosts the survival of primary cortical neurons (Sumrejkanchanakijet al., 2003). Here, we found the down-regulation of cyclinD1 followedby SurR9-C84A therapy. This effect is important, since it reduces thetransition from G0/G1 to S phase and thereby protecting the cell cyclearrest. Since exit from the cell cycle is the main reason for apoptosisactivation in post mitotic neurons, the results suggest that SurR9-C84Ais protecting the differentiated SK-N-SH cells from apoptosis byinducing the cell cycle arrest.
More importantly, for the first time, we showed that the anti-apoptotic effect of SurR9-C84A was through its ability to preventmitochondrial depolarisationwith the activation of apoptotic signallingmediated by caspase3 and Fas,which are responsible for the breakdownof DNA into the oligosomes (an irreversible late stage of apoptosis)(Fig. 8).
In conclusion, the present study shows that activated T-cells releaseGrB, which had a significant neurotoxic effect by disrupting the calciumhomeostasis and increasing the expression of cyclinD1 and Fas, therebyleading to mitochondrial membrane depolarisation and activation ofcaspase3 indifferentiatedSK-N-SHcells.More importantly,we found thatSurR9-C84A protects neuronal cells against activated T-cell supernatantsand reduces the cytotoxic signals. The precise mechanism by which this

Fig. 8. The proposed mechanism for neuroprotective effect of SurR9-C84A. The activated T-cells deteriorate the neural cells by releasing GrB, leading to (i) activating caspase3,increases cytosolic calcium concentration and calcium over load in Mitochondria, and (ii) release of calcium from endoplasmic reticulum (ER), both contributing to mitochondrialmembrane depolarization, increasing caspase3 activation and DNA cleavage. By contrast, SurR9-C84A plays a triple role in protecting from cell damaged by (i) inhibiting theactivation of caspase3, (ii) reducing the cytosolic calcium concentration, and (iii) decreasing mitochondrial membrane depolarization.
27S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
survivin mutant protects the post mitotic neurons is not completelyunderstood. Since SurR9-C84A reduces the cyclinD1expression, it is likelythat it has more nuclear functions and protects the cell cycle re-entry indifferentiated neurons. The results suggest that inducing the cell cyclearrest might help the calcium homeostasis; protect the mitochondrialmembrane potential and in-activation of apoptosis (Fig. 8). Whether thisprotective effect is through the dimerization with wild-type survivin oraltering the expression of Smac/Diablo needs further investigation.
SurR9-C84A can be applied in neuroprotective strategies to protectdifferentiated neural cells from cell cycle re-entry and apoptosis.Additionally, one important advantage of this mutant over the wild-type survivin is that it does form tumour due to its pro-apoptoticeffect in cancer cells.
Because the over-expression of survivin has been reported instimulated T-cells derived from patients with active MS, the neuropro-tective ability of SurR9-C84A has the potential to be employed for futureneurodegenerative therapies, and may also be further evaluated fortargeting stimulated T-cells for the treatment of neurodegenerativediseases such as MS (Sharief et al., 2002) and other brain injuries.
Acknowledgement
We gratefully acknowledge the financial support from Institute forTechnology Research and Innovation (ITRI), Deakin University.
References
Aguiló, J.I., Anel, A., Catalán, E., Sebastián, A., Acín-Pérez, R., Naval, J., Wallich, R., Simon,M.M., Pardo, J., 2010. Granzyme B of cytotoxic T cells induces extramitochondrialreactive oxygen species production via caspase-dependent NADPH oxidaseactivation. Immunol. Cell Biol. 88, 545–554.
Altieri, D.C., 2003a. Survivin versatile modulation of cell division and apoptosis incancer. Oncogene 22, 8581–8589.
Altieri, D.C., 2003b. Validating survivin as a cancer therapeutic target. Nat. Rev. Cancer3, 46–54.
Altieri, D.C., 2008. Opinion — survivin, cancer networks and pathway-directed drugdiscovery. Nat. Rev. Cancer 8, 61–70.
Baratchi, S., Kanwar, R.K., Khoshmanesh, K., Vasu, P., Ashok, C., Hittu, M., Parratt, A.,Krishnakumar, S., Sun, X.Y., Sahoo, S.K., Kanwar, J.R., 2009. Promises of nanotechnol-ogy for drug delivery to brain in neurodegenerative diseases. Curr. Nanosci. 5, 15–25.
Baratchi, S., Kanwar, R.K., Kanwar, J.R., 2010a. Survivin: a target from brain cancer toneurodegenerative disease. Crit. Rev. Biochem. Mol. Biol. 45, 535–554.
Baratchi, S., Kanwar, R.K., Chun, H.A.C., Kanwar, J.R., 2010b. Proliferective and protectiveeffects of SurR9-C84A on differentiated neuronal cells. J. Neuroimmunol. 227,120–132.
Bolton, M.A., Lan, W.J., Powers, S.E., McCleland, M.L., Kuang, J., Stukenberg, P.T., 2002.Aurora B kinase exists in a complexwith survivin and INCENP and its kinase activity isstimulatedbysurvivinbindingand ina complex inase activity isphosphorylation.Mol.Biol. Cell 13, 3064–3077.
Bulat, N., Widmann, C., 2009. Caspase substrates and neurodegenerative diseases. BrainRes. Bull. 80, 251–267.
Busser, J., Geldmacher, D.S., Herrup, K., 1998. Ectopic cell cycle proteins predict the sitesof neuronal cell death in Alzheimer's disease brain. J. Neurosci. 18, 2801–2807.
Buzza,M.S., Hirst, C.E., Bird, C.H., Hosking, P.,McKendrick, J., Bird, P.I., 2001. The granzymeBinhibitor PI-9, is present inendothelial andmesothelial cells suggesting that it protectsbystander cells during immune responses. Cell. Immunol. 210, 21–29.
Chaitanya, G.V., Schwaninger, M., Alexander, J.S., Babu, P.P., 2010. Granzyme-b isinvolved in mediating post-ischemic neuronal death during focal cerebral ischemiain rat model. Neuroscience 165, 1203–1216.
Chang, P.A., Chen, R.,Wu,Y.J., 2005. Reduction of neuropathy target esterasedoesnot affectneuronal differentiation, but moderate expression induces neuronal differentiation inhuman neuroblastoma (SK-N-SH) cell line. Mol. Brain Res. 141, 30–38.
Charriaut-Marlangue, C., 2004. Apoptosis: A Target for Neuroprotection. AdisInternational Ltd, pp. 185–190.
Cheung, C.H.A., Kanwar, J., Krissansen, G.W., 2006. A cell-permeable dominant-negativesurvivin protein as a tool to understand how survivin maintains tumour cellsurvival. Ejc Suppl. 4, 488.
Cook, S.D., 2003. Advancing treatment with interferon beta-1b (Betaferon(R)/Betaseron(R)) in the next decade: thinking beyond the standard dose. J. Neurol.250, 15–20.

28 S. Baratchi et al. / Journal of Neuroimmunology 233 (2011) 18–28
Deligezer, U., Erten, N., Akisik, E.E., Dalay, N., 2006. Circulating fragmented nucleosomalDNA and caspase-3 mRNA in patients with lymphoma and myeloma. Experimentaland Molecular Pathology 80, 72–76.
Dhib-Jalbut, S., Arnold, D.L., Cleveland,D.W., Fisher,M., Friedlander, R.M.,Mouradian,M.M.,Przedborski, S., Trapp, B.D., Wyss-Coray, T., Yong, V.W., 2006. Neurodegenerationand neuroprotection in multiple sclerosis and other neurodegenerative diseases.J. Neuroimmunol. 176, 198–215.
Dohi, T., Okada, K., Xia, F., Wilford, C.E., Samuel, T., Welsh, K., Marusawa, H., Zou, H.,Armstrong, R., Matsuzawa, S., Salvesen, G.S., Reed, J.C., Altieri, D.C., 2004. An IAP–IAPcomplex inhibits apoptosis. J. Biol. Chem. 279, 34087–34090.
Giuliani, F., Goodyer, C.G., Antel, J.P., Yong, V.W., 2003. Vulnerability of human neuronsto T cell-mediated cytotoxicity. J. Immunol. 171, 368–379.
Gold, S.M., Voskuhl, R.R., 2009. Estrogen treatment in multiple sclerosis. J. Neurol. Sci.286, 99–103.
Hefti, F., 1997. Pharmacology of neurotrophic factors. Annu. Rev. Pharmacol. Toxicol. 37,239–267.
Herrup, K., Yang, Y., 2007. Cell cycle regulation in the postmitotic neuron: oxymoron ornew biology? Nat. Rev. Neurosci. 8, 368–378.
Hirst, R.A., Harrison, C., Hirota, K., Lambert, D.G., 1999. Measurement of [Ca2+](i) inwhole cell suspensions using fura-2. Calcium Signal. Protoc. 114, 31–39.
Hoftberger, R., Aboul-Enein, F., Brueck, W., Lucchinetti, C., Rodriguez, M., Schmidbauer,M., Jellinger, K., Lassmann, H., 2004. Expression of major histocompatibilitycomplex Class I molecules on the different cell types in multiple sclerosis lesions.Brain Pathol. 14, 43–50.
Hong, M.S., Hong, S.J., Barhoumi, R., Burghardt, R.C., Donnelly, K.C., Wild, J.R., Venkatraj, V.,Tiffany-Castiglioni, E., 2003. Neurotoxicity induced in differentiated SK-N-SH-SY5Yhuman neuroblastoma cells by organophosphorus compounds. Toxicol. Appl.Pharmacol. 186, 110–118.
Imanishi, Y., Hosokawa, Y., Yoshimoto, K., Schipani, E.,Mallya, S., Papanikolaou, A., Kifor, O.,Tokura, T., Sablosky, M., Ledgard, F., Gronowicz, G., Wang, T.C., Schmidt, E.V., Hall, C.,Brown, E., Bronson, R., Arnold, A., 2001. Primary hyperparathyroidism caused byparathyroid-targeted overexpression of cyclin D1 in transgenic mice. Journal ofClinical Investigation 107, 1093–1102.
Jiang, Y.Y., de Bruin, A., Caldas, H., Fangusaro, J., Hayes, J., Conway, E.M., Robinson, M.L.,Altura, R.A., 2005. Essential role for survivin in early brain development. J. Neurosci.25, 6962–6970.
Kanwar, J.R., 2005. Anti-inflammatory immunotherapy for multiple sclerosis/experi-mental autoimmune encephalomyelitis (EAE) disease. Curr. Med. Chem. 12,2947–2962.
Kanwar, R.K., Ganguly, N.K., Kumar, L., Rakesh, J., Panigrahi, D.,Walia, B.N.S., 1995. Calciumand protein kinase C play an important role in Campylobacter jejuni-induced changesin Na+ and Cl− transport in rat ileum in vitro. Biochim. Biophys. Acta Mol. Basis Dis.1270, 179–192.
Kanwar, J.R., Harrison, J.E.B., Wang, D.M., Leung, E., Mueller, W., Wagner, N., Krissansen,G.W., 2000. Beta 7 integrins contribute to demyelinating disease of the centralnervous system. J. Neuroimmunol. 103, 146–152.
Kanwar, J.R., Shen, W.P., Kanwar, R.K., Berg, R.W., Krissansen, G.W., 2001. Effects ofsurvivin antagonists on growth of established tumors and B7-1 immunogenetherapy. J. Natl Cancer Inst. 93, 1541–1552.
Kanwar, J.R., Kanwar, R.K., Krissansen, G.W., 2004a. Simultaneous neuroprotection andblockade of inflammation reverses autoimmune encephalomyelitis. Brain 127,1313–1331.
Kanwar, J.R., Shen, W.P., Kanwar, R.K., Sun, X.Y., Berg, R.W., Krissansen, G.W., 2004b.Survivin antagonists and antisense HIF-1 alpha stimulate the generation of tumor-specific CTLs: may be beneficial for the treatment of large lymphomas. Cancer GeneTher. 11, 853.
Kanwar, J.R., Kanwar, R.K., Burrow, H., Baratchi, S., 2009. Recent advances on the roles ofNO in cancer and chronic inflammatory disorders. Curr. Med. Chem. 16, 2373–2394.
Kanwar, R.K., Cheung, C.H.A., Chang, J.Y., Kanwar, J.R., 2010. Recent advances in anti-survivin treatments for cancer. Curr. Med. Chem. 17.
Kato, A.C., Ferri, A., Monnier, D., Liston, P., MacKenzie, A., Perrelet, D., 2000. Adenoviralgene transfer of HIAP1, HIAP2, XIAP and NAIP can rescue motoneurons afterneonatal sciatic nerve axotomy. Eur. J. Neurosci. 12, 229.
Khan, S., Aspe, J.R., Asumen, M.G., Almaguel, F., Odumosu, O., Acevedo-Martinez, S., DeLeon, M., Langridge, W.H.R., Wall, N.R., 2009. Extracellular, cell-permeable survivininhibits apoptosis while promoting proliferative and metastatic potential. Br. J.Cancer 100, 1073–1086.
Kumar, P., Wu, H.Q., McBride, J.L., Jung, K.E., Kim, M.H., Davidson, B.L., Lee, S.K., Shankar,P., Manjunath, N., 2007. Transvascular delivery of small interfering RNA to thecentral nervous system. Nature 448, 39–43.
Li, F.Z., Ambrosini, G., Chu, E.Y., Plescia, J., Tognin, S., Marchisio, P.C., Altieri, D.C., 1998.Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 396,580–584.
Li, L., Feng, T.T., Lian, Y.Y., Zhang, G.F., Garen, A., Song, X., 2009. Role of humannoncoding RNAs in the control of tumorigenesis. Proceedings of the NationalAcademy of Sciences of the United States of America 106, 12956–12961.
Malmestrom, C., Lycke, J., Haghighi, S., Andersen, O., Carlsson, L., Wadenvik, H., Olsson,B., 2008. Relapses in multiple sclerosis are associated with increased CD8(+) T-cellmediated cytotoxicity in CSF. J. Neuroimmunol. 196, 159–165.
Maris, J.M., Matthay, K.K., 1999. Molecular biology of neuroblastoma. J. Clin. Oncol. 17,2264–2279.
Mitchell, D.J., Kim, D.T., Steinman, L., Fathman, C.G., Rothbard, J.B., 2000. Polyarginineenters cells more efficiently than other polycationic homopolymers. J. Pept. Res. 56,318–325.
Morris, M.C., Chaloin, L., Heitz, F., Divita, G., 2000. Translocating peptides and proteinsand their use for gene delivery. Curr. Opin. Biotechnol. 11, 461–466.
Nagy, Z., Esiri, M.M., Cato, A.M., Smith, A.D., 1997. Cell cycle markers in thehippocampus in Alzheimer's disease. Acta Neuropathol. 94, 6–15.
Neumann, H., Medana, I.M., Bauer, J., Lassmann, H., 2002. Cytotoxic T lymphocytes inautoimmune and degenerative CNS diseases. Trends Neurosci. 25, 313–319.
Nitsch, R., Pohl, E.E., Smorodchenko, A., Infante-Duarte, C., Aktas, O., Zipp, F., 2004.Direct impact of T cells on neurons revealed by two-photon microscopy in livingbrain tissue. J. Neurosci. 24, 2458–2464.
Perrelet, D., Ferri, A., Liston, P., Muzzin, P., Korneluk, R.G., Kato, A.C., 2002. IAPs areessential for GDNF-mediated neuroprotective effects in injured motor neurons invivo. Nat. Cell Biol. 4, 175–179.
Perrelet, D., Perrin, F.E., Liston, P., Korneluk, R.G., MacKenzie, A., Ferrer-Alcon, M., Kato,A.C., 2004. Motoneuron resistance to apoptotic cell death in vivo correlates with theratio between X-linked inhibitor of apoptosis proteins (XIAPs) and its inhibitor,XIAP-associated factor 1. J. Neurosci. 24, 3777–3785.
Pisarev, V., Yu, B., Salup, R., Sherman, S., Altieri, D.C., Gabrilovich, D.I., 2003. Full-lengthdominant-negative survivin for cancer immunotherapy. Clin. Cancer Res. 9,6523–6533.
Ray, S.K., 2006. Currently evaluated calpain and caspase inhibitors for neuroprotectionin experimental brain ischemia. Curr. Med. Chem. 13, 3425–3440.
Roh, C.R., Lee, J.W., Kang, B.H., Yang, S.H., Kim, B.G., Bae, D.S., Kim, J.H., Lee, J.H., 2002.Differential expressions of Fas and Fas ligand in human placenta. Journal of KoreanMedical Science 17, 213–216.
Sharief, M.K., Noori, M.A., Douglas, M.R., Semra, Y.K., 2002. Upregulated survivinexpression in activated T lymphocytes correlates with disease activity in multiplesclerosis. Eur. J. Neurol. 9, 503–510.
Shi, L.F., Kam, C.M., Powers, J.C., Aebersold, R., Greenberg, A.H., 1992. Purification ofthree cytotoxic lymphocyte granule serine proteases that induce apoptosis throughdistinct substrate and target cell interactions. J. Exp. Med. 176, 1521–1529.
Simons, M., Beinroth, S., Gleichmann, M., Liston, P., Korneluk, R.G., MacKenzie, A.E., Bahr,M., Klockgether, T., Robertson,G.S.,Weller,M., Schulz, J.B., 1999. Adenovirus-mediatedgene transfer of inhibitors of apoptosis proteins delays apoptosis in cerebellar granuleneurons. J. Neurochem. 72, 292–301.
Smith, M.Z., Nagy, Z., Esiri, M.M., 1999. Cell cycle-related protein expression in vasculardementia and Alzheimer's disease. Neurosci. Lett. 271, 45–48.
Sumrejkanchanakij, P., Tamamori-Adachi, M., Matsunaga, Y., Eto, K., Ikeda, M.A., 2003.Role of cyclin D1 cytoplasmic sequestration in the survival of postmitotic neurons.Oncogene 22, 8723–8730.
Tomkinson, B.E., Brown, M.C., Ip, S.H., Carrabis, S., Sullivan, J.L., 1989. Soluble CD8during T-cell activation. J. Immunol. 142, 2230–2236.
Vartanian, T., Sorensen, P.S., Rice, G., 2004. Impact of neutralizing antibodies on theclinical efficacy of interferon beta in multiple sclerosis. J. Neurol. 251, 25–30.
Verdecia, M.A., Huang, H.K., Dutil, E., Kaiser, D.A., Hunter, T., Noel, J.P., 2000. Structure ofthe human anti-apoptotic protein survivin reveals a dimeric arrangement. Nat.Struct. Biol. 7, 602–608.
Wainwright, L.J., Lasorella, A., Iavarone, A., 2001. Distinct mechanisms of cell cyclearrest control the decision between differentiation and senescence in humanneuroblastoma cells. Proc. Natl Acad. Sci. USA 98, 9396–9400.
Wang, T., Allie, R., Conant, K., Haughey,N., Turchan-Chelowo, J., Hahn, K., Rosen, A., Steiner,J., Keswani, S., Jones, M., Calabresi, P.A., Nath, A., 2006. Granzyme B mediatesneurotoxicity through a G-protein-coupled receptor. FASEB J. 20, 1209–1214.
Xu, D.G., Bureau, Y., McIntyre, D.C., Nicholson, D.W., Liston, P., Zhu, Y.X., Fong, W.G.,Crocker, S.J., Korneluk, R.G., Robertson, G.S., 1999. Attenuation of ischemia-inducedcellular and behavioral deficits by X chromosome-linked inhibitor of apoptosisprotein overexpression in the rat hippocampus. J. Neurosci. 19, 5026–5033.
Yang, X.H., Stennicke, H.R., Wang, B.K., Green, D.R., Janicke, R.U., Srinivasan, A., Seth, P.,Salvesen, G.S., Froelich, C.J., 1998. Granzyme Bmimics apical caspases— descriptionof a unified pathway for trans-activation of executioner caspase-3 and -7. J. Biol.Chem. 273, 34278–34283.
Yang, Y., Geldmacher, D.S., Herrup, K., 2001. DNA replication precedes neuronal celldeath in Alzheimer's disease. J. Neurosci. 21, 2661–2668.
Copyright © 2022 FDOKUMEN




















