
Spatio-temporal patterns of suspended and attached bacterial ...
Upload
independentCategory
view
3download
0
CPCHFT 13 (11) 2617–2804 (2012) · ISSN 1439-4235 · Vol. 13 · No. 11 · August 6, 2012
Concepts: Plasmonic Trapping with a Gold Nanopillar(K. B. Crozier and K. Wang)
Original Contributions: Surface-Attached Proteins Characterizedby FTIR Difference Spectroscopy (K. Gerwert, C. Kötting et al.),
Directionality of Dihydrogen Bonds (A. Lledos, E. S. Shubina et al.)
11/2012
www.chemphyschem.org
A Journal of

Cover Picture
Philipp Pinkerneil, Jçrn G�ldenhaupt, Klaus Gerwert*, andCarsten Kçtting*
ATR-FTIR spectroscopy monitors protein reactions and interactions of monolayers at theatomic level. On p. 2649 K. Gerwert, C. Kçtting et al. describe a method to immobilizeproteins by means of a polyhistidine-tag and nitrilotriacetic-acid-modified lipids. Be-cause polyhistidine tags are often used for protein purification, modified proteins areeasy to access. This makes the method a universal technique for many soluble proteins.The setup allows the investigation of the secondary structure, orientation, reactionmechanisms and protein–ligand or protein–protein interactions.

DOI: 10.1002/cphc.201200358
Surface-Attached Polyhistidine-Tag Proteins Characterized by FTIRDifference Spectroscopy
Philipp Pinkerneil, Jçrn G�ldenhaupt, Klaus Gerwert,* and Carsten Kçtting*[a]
FTIR difference spectroscopy can reveal the molecular detailsof protein reactions.[1–6] The absorption spectrum of a proteinprovides global information, for example, on protein secondarystructure. In order to reveal molecular reaction mechanisms,difference spectroscopy has to be applied. It allows monitoringof functional groups that are involved in conformationalchanges, chemical reactions, H-bond changes, ligand bindingor protein–protein interactions. In these difference spectra, thesmall signal of the reaction is selected out of the usuallya factor of 103 larger, but quiescent, background absorbanceof the whole sample. Therefore identical conditions must bemaintained between the measurements of two different statesof the protein. The reaction can be triggered without movingthe sample, by a laser flash. Although this is a powerfulmethod for photoactive proteins like bacteriorhodopsin (bR)[7]
or the bacterial reaction center (RC),[8] the function of mostproteins cannot be triggered by light. One possible solution toovercome this problem is the use of caged compounds,[9, 10]
which are photolabile precursers of active compounds. For ex-ample, caged GTP can be used for the investigation of GTPas-es.[11]
A more general method to perform difference spectroscopyis the use of immobilized proteins on attenuated total reflec-tion (ATR) crystals.[6, 12, 13] Difference spectra can then be ob-tained by simple buffer exchange, for example, with a ligandversus without. The group of Vogel modified Ge and ZnSe in-ternal reflection elements (IREs) by gold or chemical vapordeposition of SiO2 and subsequently attached a linker mole-cule that reversibly binds peptides by a polyhistidine tag.[14]
During the last years SEIRA (surface-enhanced infrared absorp-tion) spectroscopy of protein monolayers covalently immobi-lized on gold-coated silicon was established.[15] Due to thegold layer this technique is superior for potential-induced dif-ference spectroscopy. However, the strong near-field of therough gold surface might hamper quantitative analysis, be-cause the enhancement factor varies dramatically. We andothers recently established ATR-FTIR difference spectroscopy of
monolayers using a multiple reflection germanium ATR crystalas the internal reflection element (IRE).[13, 16, 17] Due to the highrefractive index, suitable chemical properties, and the goodtransmission in the mid-infrared region, germanium is probablythe best IRE material in the mid-infrared region. Instead of thesurface-enhanced effect in SEIRA, multiple reflection can beused and give similar signal to noise ratios.[17] Further, germani-um surfaces can easily be treated to become hydrophilic andthe spreading of lipid vesicles results a stable lipid bilayer.[16]
Here, we report a method to immobilize proteins by means ofa polyhistidine tag and lipids modified by nitrilotriacetic acid(NTA). Because polyhistidine tags are often used for proteinpurification,[18] modified proteins are easy to access, whichmakes our method a universal technique for the investigationof almost any soluble protein.
Our novel measurement platform is shown in Figure 1. First,a solid supported lipid bilayer (SSLB) is formed by vesiclespreading on top of an ATR crystal. We used small unilamellarvesicles consisting of a mixture of the natural 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and the head-group-modified lipid 1,2-di-(9Z-octadecenoyl)-sn-glycero-3-[(N-(5-amino-1-carboxypentyl)iminodiaceticacid)succinyl] (NTA-DOGS). The spreading of these vesicles resulted in a bilayercontaining NTA groups, which enables protein immobilizationvia polyhistidine tags. Because the process of bilayer formationcan be done within the spectrometer by using a flow-throughsystem, we could easily monitor this process by the infrared
Figure 1. Schematic representation of our technique. A SSLB containingNTA-modified lipids is attached on top of a germanium ATR crystal by hydro-philic interactions. Any His-tag-modified protein can be immobilized and in-vestigated by FTIR spectroscopy.
[a] P. Pinkerneil,+ Dr. J. G�ldenhaupt,+ Prof. Dr. K. Gerwert, Dr. habil. C. KçttingLehrstuhl f�r BiophysikRuhr-Universit�t Bochum44780 Bochum (Germany)E-mail : [email protected]
[+] both authors contributed equally to this work.
Supporting information for this article is available on the WWW underhttp://dx.doi.org/10.1002/cphc.201200358.
Re-use of this article is permitted in accordance with the Terms and Con-ditions set out at http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)1439-7641/homepage/2267_onlineopen.html.
ChemPhysChem 2012, 13, 2649 – 2653 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim 2649

absorptions of the lipids, and obtained the kinetics and the dif-ference spectrum of the immobilization. From these data, sur-face concentration, lipid phase, and order parameters could becalculated, which then allowed the quantification of the bilayerquality. Larger vesicles did not spread completely and led toabsorptions higher than expected for one bilayer (SupportingInformation, Figure S1). On the other hand, bilayers preparedby small vesicles were stable and could be used for several
days. We verified that the layer was complete (Supporting In-formation, Figure 2).
Figure 2 A shows the time course of the lipid immobilizationprocess with our optimized conditions. The adsorption of thevesicles was monitored by the absorbance of the asymmetricCH2 stretching vibration at 2924 cm�1. After 10 min, a stableabsorption of 20 mOD was reached as expected for a single bi-layer[17] with a surface concentration similar to Langmuir–
Figure 2. Formation of the lipid bilayer. A) Typical time course of vesicle spreading monitored by the CH2 stretching vibration of the lipid. B) Difference spec-trum of the bilayer formation. Bands facing upwards are due to the bilayer, bands facing downwards are due to the displaced water.
Figure 3. Protein immobilization. A) Typical time course of protein immobilization. We observed specific binding by the NTA groups, because no binding wasobserved without Ni2 + . B) Immobilization of N-Ras 1-180 by the hexa- and deca-His tags. C) The protein can be removed by imidazole. D) IC50 of the displace-ment by imidazole.
2650 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 2649 – 2653

Blodgett films.[19] The absorption difference spectrum of bilayerformation is defined as the difference between the infrared ab-sorptions before and after this bilayer formation (Figure 2 B).Whereas the water displaced by the lipids produced negativepeaks, all absorption bands of the lipids result in positivepeaks in this difference spectrum. Besides the absorption ofthe asymmetric CH2 stretching vibration mentioned above,these positive peaks include the absorptions of the symmetricCH2 stretching vibration at 2853 cm�1, (asymmetric CH3 at2958 cm�1 and symmetric at 2873 cm�1), of the carbonylgroups at 1737 cm�1, of the carboxylgroups of NTA at1411 cm�1 and of the phosphates at 1230 cm�1 and 1087 cm�1.The band positions are typical for a lipid membrane in theliquid disordered phase. We further evaluated the quality ofthe bilayer by means of order parameters, which are a measureof the orientational distribution of the dipole moments of theabsorption. These values (S = 0.2 for the lipid alkane chain andS =�0.23 for the carbonyl ab-sorption) are in line with litera-ture values from NMR[20] andIR.[17]
In the next step, any polyhisti-dine-tagged protein can be im-mobilized. Again, this processcan be directly monitored by theabsorption bands of the ad-sorbed species. In this case, weused the amide II absorption ofthe protein (Figure 3 A). The pro-tein only adsorbed if Ni2 + waspresent in the buffer, leading toa saturation of the surfacewithin approximately 45 min,which indicated specific bindingby the His tag. We optimized theconditions using N-Ras 1-180with a C-terminal polyhistidinetag. In this case, the membraneanchoring via the polyhistidinetag mimicked the natural lipidanchor, which is also C-terminaland known to be less struc-tured.[21] We used either a hexa-His tag or a deca-His tag (Fig-ure 3 B). The deca-His tag led toconsiderably more adsorbedprotein, approximately fourtimes that of the hexa-His tagunder identical conditions with15 % NTA groups. Using 25 %NTA-modified lipids and thedeca-His tag, we obtaineda signal of 14 mOD for the ami-de II band (Figure 3 A). This isclose to the maximum absorp-tion of 16 mOD obtained forlipid-anchored N-Ras, indicating
a surface coverage of about 70 % for the Ras protein. The half-life time under our conditions was about 12 h.
The protein immobilization could be reversed by washingwith imidazole (Figure 3 C), which provides additional proof forspecific binding by the His tag. We used different concentra-tions of imidazole and found an IC50 of 22.5 mm for the deca-His tag and 25 mol % NTA-DOGS (Figure 3 D). This is betweenthe values obtained for mono- and bis-NTA-modified polyethy-leneglycol brushes.[22] The reason for the stable immobilizationis probably that the NTA groups are laterally mobile, whichallows multivalent binding. This leads to a much stronger bind-ing and prohibits the protein from coming off.[23]
Using this setup, several types of experiments are possiblethat can provide a variety of information. Again we exemplifythese experiments using the GTPase Ras. By taking the spec-trum before and after the protein immobilization process, theabsorption spectrum of the protein was derived (Figure 4 A).
Figure 4. Applications of our technique. A) The FTIR spectrum of the protein reveals global features such as sec-ondary structure by decomposition of the amide I band. B) By using polarized infrared light (Epp and Evp), informa-tion on the orientation can be gained because absorptions with a transition dipole moment parallel to the per-pendicular of the ATR crystal (mk) interact more strongly with Epp and result a positive peak in the dichroic differ-ence spectrum D*, whereas absorptions with a transition dipole moment parallel to the membrane (m? ) interactmore strongly with Evp and result a negative peak in D*. In D* of N-Ras1-166 a positive peak at 1661 cm�1 indi-cates that the majority of the transition dipole moments of the helical parts of the protein are perpendicular tothe membrane. The transition dipole moment of helices is in the helix direction, resulting in an orientation asshown in Figure 1. A quantitative description for the calculation of orientations from D* is published elsewhere.[26]
ChemPhysChem 2012, 13, 2649 – 2653 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 2651

From this, for example, the secondary structure ofthe protein can be obtained by decomposition of theamide I band.[16] For these kinds of measurements,D2O should be used to prevent disturbance by theabsorption signal of water. In our decomposition wefound 33.4 % a-helix, 24.3 % b-sheet, 26.8 % b-turnand 15.4 % random coil. This agrees within the exper-imental error with both the X-ray structure[24] andearlier FTIR measurement with lipid-anchored Ras.[16]
A similar amide I band was also obtained for K-Ras insolution.[25] The slight increase in b-sheet and b-turnindicated that the peptide bonds of the additionalten NTA-bound histidine residues absorbed in thisregion.
Using polarized infrared light, dichroic measure-ments are possible. From this type of experiment, theorientation of functional groups relative to the mem-brane can be obtained. As shown in Figure 4 B(inset), an absorbing group with the transition dipolemoment perpendicular to the membrane will absorbparallel polarized light more strongly (absorbanceApp) than vertically polarized light (absorbance Avp).This results in a positive peak in the dichroic differ-ence spectrum D*, which is calculated by D* =
App�Riso·Avp (for details, see the Supporting Informa-tion).[17] The positive peak in the amide I region, rep-resenting the helical content, shows that N-Ras1-166was orientated as shown in Figure 4 B, with mosthelices perpendicular to the membrane. Thus, the ori-entation was similar to membrane-bound lipidatedN-Ras.[26] This indicates that in this case, the artificialbinding not only immobilized the protein efficientlybut even mimicked the natural anchor to a certaindegree. Interestingly, N-Ras 1-180 does not show thisorientation (Supporting Information, Figure S3). Thelonger linker enabled an isotropic distribution.
Furthermore, difference spectra can be obtainedby utilizing the immobilization of the protein ina flow-through system (Figure 5 A). Thus, protein re-actions could be induced in different ways, includingchanges in pH, addition or exchange of ligands, orsmall molecules. Here, we show the action of BeF3
�
on Ras·GDP. BeF3� mimics the g-phosphate and led
to a conformational change of the GTPase from its“off” into its “on” state.[27] The observed difference spectrum isvery similar to those obtained from Ras in solution and fromlipidated membrane-bound Ras.[28] For example, the Thr35marker band in the “on” state at 1684 cm�1 (in D2O) is well-re-solved. The reaction can be repeated many times to obtainsignal-to-noise ratios from monolayers that are similar to thesignal to noise from bulk experiments. Thus, reaction mecha-nisms can be investigated at the atomic level.
Another possibility is the use of the system in a surface-plas-mon-resonance-type experiment, but gaining additional chem-ical information of infrared spectroscopy. Figure 5 B shows theassociation and dissociation of the effector protein NORE1A(199–358)[29] to Ras using solutions with varying NORE1A con-
centrations. From these experiments, the KD of the two pro-teins could be calculated. We obtained a value of 45 nm,which is in good agreement with the literature value of80 nm.[29] In addition, the infrared absorption spectrum of thesecond protein was obtained. Again, difference spectra of pro-tein–protein interactions in various conditions, such as withand without small molecules, are possible.
In conclusion, we established a universal label-free methodfor the spectroscopic investigation of polyhistidine-tagged pro-teins. FTIR difference spectra can reveal reaction mechanismsof proteins at the atomic level. Usually, this method is limitedto proteins with photoactivatable groups or redox active pro-teins. Our technique overcomes this limitation and allows FTIR
Figure 5. A) By simple buffer exchange, difference spectra are obtained. Here, the inter-action of Ras·GDP with a small molecule (BeF3
�) is shown. This molecule mimics the g-phosphate and switches Ras into its “on” conformation as seen by the absorption of themarker band for the “on” state, the carbonyl vibration of Thr35.[28] Repetitive changes be-tween buffer with and without BeF3
� enable repetitive switching of the protein. B) Pro-tein–protein interactions can be studied in a manner similar to surface plasmon reso-nance, but with the additional chemical information of the FTIR spectrum. Here, the as-sociation and the dissociation of NORE1A from Ras is shown. The raise can be fitted bya single exponential function. The plot of the time constant kobs against the NORE1A con-centration (inset) revealed the KD.
2652 www.chemphyschem.org � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim ChemPhysChem 2012, 13, 2649 – 2653

difference spectroscopic investigations of any soluble or mem-brane attached protein by a simple buffer exchange. Further,information on secondary structure, molecular orientation,small-molecule or protein–protein interactions can be ob-tained.
Acknowledgements
We thank Christian Herrmann for the NORE1A plasmid. We ac-knowledge the Deutsche Forschungsgemeinschaft (SFB 642) forfinancial support.
Keywords: bilayers · difference spectroscopy · irspectroscopy · proteins · reaction mechanisms
[1] W. M�ntele, Trends Biochem. Sci. 1993, 18, 197.[2] K. Gerwert, Biol. Chem. 1999, 380, 931.[3] H. Kandori, Recent Res. Dev. Phys. Chem. 2001, 5, 161.[4] C. Kçtting, K. Gerwert, ChemPhysChem 2005, 6, 881.[5] A. Barth, P. I. Haris, Biological and Biomedical Infrared Spectroscopy,
Vol. 2, IOS, Amsterdam, 2009.[6] K. Ataka, T. Kottke, J. Heberle, Angew. Chem. 2010, 122, 5544; Angew.
Chem. Int. Ed. 2010, 49, 5416.[7] F. Garczarek, K. Gerwert, Nature 2006, 439, 109.[8] R. Brudler, R. Rammelsberg, T. T. Woo, E. D. Getzoff, K. Gerwert, Nat.
Struct. Biol. 2001, 8, 265.[9] V. Cepus, C. Ulbrich, C. Allin, A. Troullier, K. Gerwert, Methods Enzymol.
1998, 291, 223.[10] J. E. T. Corrie in Dynamic Studies in Biology (Eds. : M. Goeldner, R. Givens),
Wiley-VCH, Weinheim, 2005, p. 1.[11] C. Kçtting, A. Kallenbach, Y. Suveyzdis, A. Wittinghofer, K. Gerwert, Proc.
Natl. Acad. Sci. USA 2008, 105, 6260.
[12] E. Goormaghtigh, V. Raussens, J. M. Ruysschaert, Biochim. Biophys. ActaRev. Biomembr. 1999, 1422, 105.
[13] N. Hassler, D. Baurecht, G. Reiter, U. P. Fringeli, J. Phys. Chem. C 2011,115, 1064.
[14] P. Rigler, W.-P. Ulrich, P. Hoffmann, M. Mayer, H. Vogel, ChemPhysChem2003, 4, 268.
[15] K. Ataka, J. Heberle, Biopolymers 2006, 82, 415.[16] J. G�ldenhaupt, Y. Adig�zel, J. Kuhlmann, H. Waldmann, C. Kçtting, K.
Gerwert, FEBS J. 2008, 275, 5910.[17] C. Kçtting, J. G�ldenhaupt, K. Gerwert, Chem. Phys. 2012, 396, 72.[18] J. A. Bornhorst, J. J. Falke, Applications of Chimeric Genes and Hybrid Pro-
teins, Part A, Methods in Enzymology, Vol. 326, Elsevier, Amsterdam,2000, p. 245.
[19] S. A. Tatulian, Biochemistry 2003, 42, 11898.[20] J. Seelig, A. Seelig, Q. Rev. Biophys. 1980, 13, 19.[21] R. Thapar, J. G. Williams, S. L. Campbell, J. Mol. Biol. 2004, 343, 1391.[22] S. Lata, A. Reichel, R. Brock, R. Tampe, J. Piehler, J. Am. Chem. Soc. 2005,
127, 10205.[23] M. Bhagawati, S. Lata, R. Tampe, J. Piehler, J. Am. Chem. Soc. 2010, 132,
5932.[24] M. V. Milburn, L. Tong, A. M. Devos, A. Brunger, Z. Yamaizumi, S. Nishi-
mura, S. H. Kim, Science 1990, 247, 939.[25] K. Weise, S. Kapoor, C. Denter, J. Nikolaus, N. Opitz, S. Koch, G. Triola, A.
Herrmann, H. Waldmann, R. Winter, J. Am. Chem. Soc. 2011, 133, 880.[26] J. G�ldenhaupt, T. Rudack, P. Bachler, G. Triola, H. Waldmann, C. Kçtting,
K. Gerwert, unpublished results.[27] J. F. Diaz, A. Sillen, Y. Engelborghs, J. Biol. Chem. 1997, 272, 23138.[28] C. Kçtting, A. Kallenbach, Y. Suveyzdis, C. Eichholz, K. Gerwert, ChemBio-
Chem 2007, 8, 781.[29] B. Stieglitz, C. Bee, D. Schwarz, O. Yildiz, A. Moshnikova, A. Khokhlatch-
ev, C. Herrmann, EMBO J. 2008, 27, 1995.
Received: April 25, 2012
Published online on June 15, 2012
ChemPhysChem 2012, 13, 2649 – 2653 � 2012 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim www.chemphyschem.org 2653

Supporting Information� Copyright Wiley-VCH Verlag GmbH & Co. KGaA, 69451 Weinheim, 2012
Surface-Attached Polyhistidine-Tag Proteins Characterized by FTIRDifference Spectroscopy
Philipp Pinkerneil, Jçrn G�ldenhaupt, Klaus Gerwert,* and Carsten Kçtting*[a]
cphc_201200358_sm_miscellaneous_information.pdf
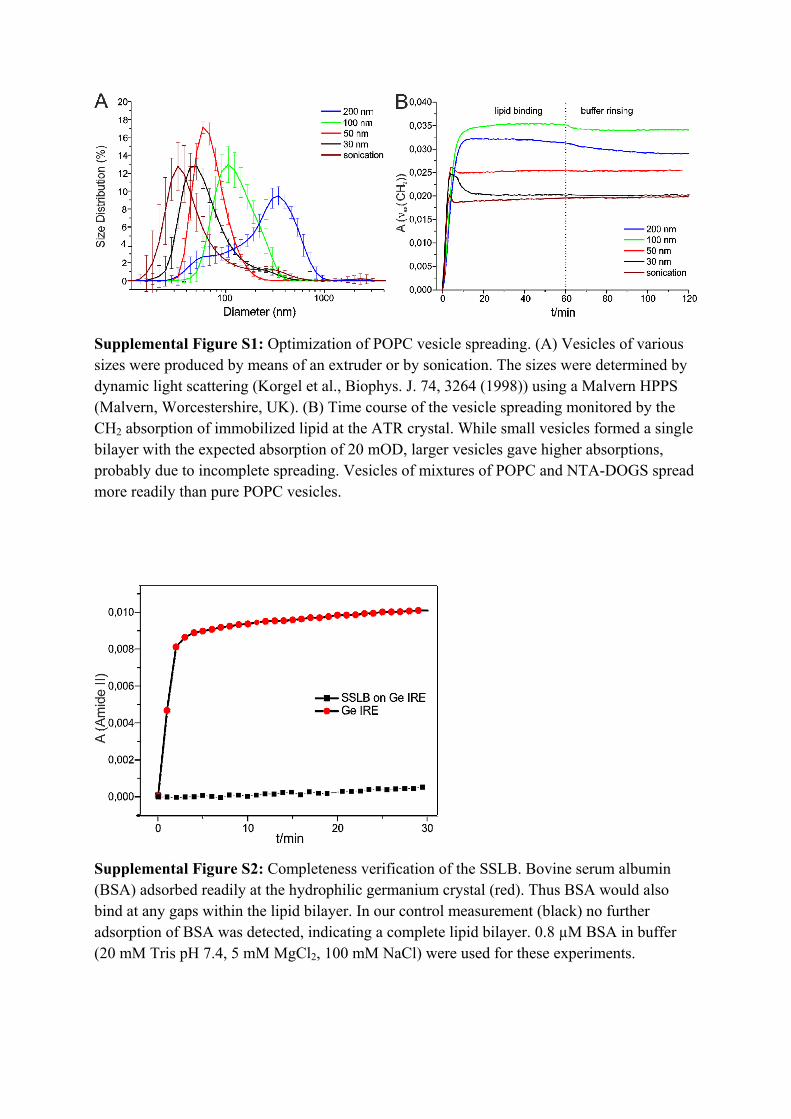
Supplemental Figure S1: Optimization of POPC vesicle spreading. (A) Vesicles of various sizes were produced by means of an extruder or by sonication. The sizes were determined by dynamic light scattering (Korgel et al., Biophys. J. 74, 3264 (1998)) using a Malvern HPPS (Malvern, Worcestershire, UK). (B) Time course of the vesicle spreading monitored by the CH2 absorption of immobilized lipid at the ATR crystal. While small vesicles formed a single bilayer with the expected absorption of 20 mOD, larger vesicles gave higher absorptions, probably due to incomplete spreading. Vesicles of mixtures of POPC and NTA-DOGS spread more readily than pure POPC vesicles.
Supplemental Figure S2: Completeness verification of the SSLB. Bovine serum albumin (BSA) adsorbed readily at the hydrophilic germanium crystal (red). Thus BSA would also bind at any gaps within the lipid bilayer. In our control measurement (black) no further adsorption of BSA was detected, indicating a complete lipid bilayer. 0.8 µM BSA in buffer (20 mM Tris pH 7.4, 5 mM MgCl2, 100 mM NaCl) were used for these experiments.

Supplemental Figure S3: Absorption spectra using polarized light (App and Avp) of N-Ras 1-180 and the calculated dichroic spectrum D*. No significant peak was observed in D* in the amide I region indicating an isotropic orientation of the protein.
Experimental Methods
N-Ras1–166His6, N-Ras 1-166His10 , N-Ras 1-180His6 and N-Ras 1-180His10 were expressed
as described for N-Ras without the His-tag.[1] The purification was carried out with
immobilized metal ion affinity chromatography (IMAC) and size exclusion chromatography.
For the experiments shown in Figure 4 N-Ras 1-166His10 were used and for the experiments
shown in Figure 5, N-Ras 1-180His10 were used. NORE1A (199-358) was expressed and
purified as a GST fusion protein according to the literature.[2] The GST-tag was removed by
cleavage with thrombin.
The measurements were performed with a vertical ATR multireflection unit, (Specac,
Orpington, UK) mounted in a Vertex 80V spectrometer (BrukerOptics, Ettlingen, Germany).
The internal reflection element was a 52 mm · 20 mm · 2 mm trapezoidal germanium ATR
plate with an aperture angle of 45°. Only one side of the IRE was used, which resulted in 13
active reflections. Measurements were done at 293 K with a resolution of 2 cm1 and a
scanner speed of 80 kHz using double sided forward backward data acquisition. Between 50
and 200 scans were averaged. Fourier transformation was done with a zero filling factor of 4,
Mertz phase correction and Blackmann-Harris three-term apodization. Absorbances are
calculated from the intensity I relative to the reference intensity I0 (before the immobilization
or reaction) according to A = log(I/I0). By means of a polarizing filter, we obtained
absorption spectra for parallel polarized light (App) and vertical polarized light (Avp). The
dichroic difference spectrum D* is calculated by D*=AppRiso·Avp. Riso corrects for the

different electric field strength for parallel and vertical polarized light. It was shown that
under our experimental conditions Riso = 1.72.[3]
POPC and NTA-DOGS were purchased from Avanti Polar Lipids (Alabaster, Alabama,
USA). Vesicle formation bilayer preparation was done in the same manner as described for
pure POPC.[3] The lipid mixture for the SSLB formation had a concentration of 410 µM (up
to 25 mol% NTA-DOGS). The kinetics of the vesicle spreading was monitored by the
asymmetric CH2 stretching vibration between 2925 cm-1 and 2922 cm-1.
Protein immobilization was achieved with a solution of 1 µM N-Ras in buffer (20 mM Tris
pH 7.4, 5 mM MgCl2, 100 mM NaCl, 0.1 mM NiCl2, 1 mM TCEP, 0.1 mM GDP). The
kinetics of the protein immobilization were monitored by the amide II mean absorption
between 1550 cm-1 and 1545 cm-1. Secondary structure analysis was done in D2O with the
same procedure and parameter set as used for lipidated Ras.[4] We used 4 mM BeF2 and 16
mM NaF (1:6 molar ratio of beryllium to fluoride) for the generation of BeF3to mimic the -
phosphate in the protein activity tests. To determine the dissociation constant of the
NORE1A·N-Ras complex, the apparent association rates (kobs) were plotted against the
concentration of NORE1A. The slope of the resulting regression line gives kon and the
intercept with the y-axis the koff. The dissociation constant KD is the quotient of these two rate
constants.
[1] J. Tucker, G. Sczakiel, J. Feuerstein, J. John, R. S. Goody, A. Wittinghofer, EMBO J. 1986, 5, 1351.
[2] B. Stieglitz, C. Bee, D. Schwarz, O. Yildiz, A. Moshnikova, A. Khokhlatchev, C. Herrmann, EMBO J. 2008, 27, 1995.
[3] C. Kötting, J. Güldenhaupt, K. Gerwert, Chem. Phys. 2012, 396, 72. [4] J. Güldenhaupt, Y. Adigüzel, J. Kuhlmann, H. Waldmann, C. Kötting, K. Gerwert,
Febs Journal 2008, 275, 5910.
Copyright © 2022 FDOKUMEN




















