
Structural, electronic, mechanical, and dynamical properties of graphene oxides: A first principles...
10
Structural, electronic, mechanical, and dynamical properties of graphene oxides: A first principles study Shweta D. Dabhi, Sanjay D. Gupta, and Prafulla K. Jha Citation: Journal of Applied Physics 115, 203517 (2014); doi: 10.1063/1.4878938 View online: http://dx.doi.org/10.1063/1.4878938 View Table of Contents: http://scitation.aip.org/content/aip/journal/jap/115/20?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Structural, electronic and elastic properties of Fe-doped YN: DFT study AIP Conf. Proc. 1536, 1085 (2013); 10.1063/1.4810612 A DFT study on structural, vibrational properties, and quasiparticle band structure of solid nitromethane J. Chem. Phys. 138, 184705 (2013); 10.1063/1.4803479 DFT investigations of structural and electronic properties of gallium arsenide (GaAs) AIP Conf. Proc. 1482, 64 (2012); 10.1063/1.4757439 First-principles investigations of Zn (Cd) doping effects on the electronic structure and magnetic properties of CoFe2O4 J. Appl. Phys. 109, 07A502 (2011); 10.1063/1.3535442 Full potential linearized augmented plane wave investigations of structural and electronic properties of pyrochlore systems J. Appl. Phys. 96, 6482 (2004); 10.1063/1.1789272 [This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP: 14.139.122.162 On: Sat, 31 May 2014 07:27:19
Transcript of Structural, electronic, mechanical, and dynamical properties of graphene oxides: A first principles...

Structural, electronic, mechanical, and dynamical properties of graphene oxides: A firstprinciples studyShweta D. Dabhi, Sanjay D. Gupta, and Prafulla K. Jha
Citation: Journal of Applied Physics 115, 203517 (2014); doi: 10.1063/1.4878938 View online: http://dx.doi.org/10.1063/1.4878938 View Table of Contents: http://scitation.aip.org/content/aip/journal/jap/115/20?ver=pdfcov Published by the AIP Publishing Articles you may be interested in Structural, electronic and elastic properties of Fe-doped YN: DFT study AIP Conf. Proc. 1536, 1085 (2013); 10.1063/1.4810612 A DFT study on structural, vibrational properties, and quasiparticle band structure of solid nitromethane J. Chem. Phys. 138, 184705 (2013); 10.1063/1.4803479 DFT investigations of structural and electronic properties of gallium arsenide (GaAs) AIP Conf. Proc. 1482, 64 (2012); 10.1063/1.4757439 First-principles investigations of Zn (Cd) doping effects on the electronic structure and magnetic properties ofCoFe2O4 J. Appl. Phys. 109, 07A502 (2011); 10.1063/1.3535442 Full potential linearized augmented plane wave investigations of structural and electronic properties of pyrochloresystems J. Appl. Phys. 96, 6482 (2004); 10.1063/1.1789272
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

Structural, electronic, mechanical, and dynamical properties of grapheneoxides: A first principles study
Shweta D. Dabhi,1 Sanjay D. Gupta,2 and Prafulla K. Jha3,a)
1Department of Physics, Maharaja Krishnakumarsinhji Bhavnagar University, Bhavnagar 364001, India2V. B. Institute of Science, Department of Physics, C. U. Shah University, Wadhwan City - 363030,Surendranagar, India3Department of Physics, Faculty of Science, The Maharaja Sayajirao University of Baroda, Vadodara-390002,India
(Received 10 February 2014; accepted 8 May 2014; published online 30 May 2014)
We report the results of a theoretical study on the structural, electronic, mechanical, and vibrational
properties of some graphene oxide models (GDO, a-GMO, z-GMO, ep-GMO and mix-GMO) at
ambient pressure. The calculations are based on the ab-initio plane-wave pseudo potential density
functional theory, within the generalized gradient approximations for the exchange and correlation
functional. The calculated values of lattice parameters, bulk modulus, and its first order pressure
derivative are in good agreement with other reports. A linear response approach to the density
functional theory is used to derive the phonon frequencies. We discuss the contribution of the
phonons in the dynamical stability of graphene oxides and detailed analysis of zone centre phonon
modes in all the above mentioned models. Our study demonstrates a wide range of energy gap
available in the considered models of graphene oxide and hence the possibility of their use in
nanodevices. VC 2014 AIP Publishing LLC. [http://dx.doi.org/10.1063/1.4878938]
I. INTRODUCTION
Graphene, a single layer of honeycomb carbon lattice has
attracted extensive research activities in recent years due to its
exceptional electrical, mechanical, and thermal properties
which makes it to hold great promise for potential applications
in many technological applications ranging from nanoelec-
tronics, sensors, nanocomposites, batteries, supercapacitors to
hydrogen storage.1–8 However, the pristine graphene is a zero
gap material and much effort has been devoted to create an
energy band gap in graphene based materials for device
applications.8–15 The energy gap can be achieved in pristine
graphene through either nano patterning,9–11 application of
gate voltage, or chemical functionalization.13–15 In addition,
the large scale production of pure graphene sheets remains
challenging. The functionalized graphene sheets can be pro-
duced by exfoliation from graphene oxide (GO).16 The GO
which can have different compositions with various oxidation
levels based on synthesis processes and conditions is currently
of particular interest to scientists. The solubility of GO in
water, ethanol, and other liquids not only makes it to be used
as precursor for fabricating large-scale graphene but also
allows the functionalization of the insoluble and infusible pris-
tine graphene sheets, being flexible tuned as an insulator,
semiconductor, or semimetal with diverse possibilities for wir-
ing with bio and organic molecules.17
The current concern, however, is the proper structural
characterization of the GO for its application as the different
experimental conditions, synthesis processes, oxygen contain-
ing groups, the ratio of the reacted and unreacted sp2 and sp3
carbon atoms, and C:O ratio result in diverse geometries.17–21
Therefore, such diversity in the atomic structures of graphene
oxide usually makes the interpretation of the spectroscopic
results more challenging. Recently, the fully oxidized, ordered
graphene monoxide with stoichiometric ratio C: O¼ 1:1 has
been produced by vacuum thermal reduction in the experi-
ment.22 The most commonly agreed conclusion that is
achieved is that the graphene can be oxidized by the formation
of epoxy bond [C-O-C] using oxygen to form graphene
oxide.14,23–26 However, till date this proposed model of gra-
phene oxide lacks to predict the atomic structure of GO by
comparing to highly accurate synchrotron based X-ray absorp-
tion near edge measurement.27 The epoxy pair configuration
has been used for low energy structural model for stoichiomet-
ric graphene oxides in theoretical calculations.22,28 Recently,
Zhang et al.17 proposed ether type configuration as the ground
state of the stoichiometric graphene oxide. They observed that
the zigzag graphene monoxide (z-GMO) and armchair gra-
phene monoxide (a-GMO) have lower energy and are more
stable than the epoxy graphene monoxide (ep-GMO) and mix
graphene monoxide (mix-GMO) reported earlier by Xiang
et al.28 In addition, the molecular dynamics study show that
the graphene dioxide (GDO) which is not yet synthesized is
not stable at high temperature17 in contrast to the z-GMO
which is stable up to 2000K. Very recently, Kumar et al.29
have performed the experimental and theoretical investigation
on this material. The experimental finding reveals that the
large scale GO sheets with preserved oxygen content and bet-
ter properties can be synthesized using thermal annealing. The
atomistic simulations based on molecular dynamics simula-
tion and density functional theory together with experimental
results demonstrate that the GO structures undergo a phase
transformation into graphitic and oxidized domains by tem-
perature driven oxygen diffusion. McAllister30 et al. have
reported a detailed analysis of the expansion mechanism of
a)Author to whom correspondence should be addressed. Electronic mail:
[email protected]. Tel.: þ91-265-2795339.
0021-8979/2014/115(20)/203517/9/$30.00 VC 2014 AIP Publishing LLC115, 203517-1
JOURNAL OF APPLIED PHYSICS 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

GO to produce functionalized graphene sheets. Despite the
studies partly summarized above, relatively very little is
known about the materials characteristics in different configu-
rations of graphene oxide. Therefore, we employ quantum-
mechanical calculations in order to shed more light on struc-
tural, energetic, electronic and vibrational properties of this
important class of materials. In the present work, we perform
systematic first principles density functional theory calcula-
tions to investigate the structural, stability, electronic, me-
chanical, and vibrational properties of z-GMO, GDO,
a-GMO, ep-GMO, and mix-GMO. The paper is organized as
follows. After introduction, the computational details section
describes our computational method, provides computational
parameters and details to the determination of materials char-
acteristics followed by the results and discussion in Sec. III.
Finally, in Sec. IV, we conclude our results.
II. COMPUTATIONAL DETAILS
The first principles calculations in the present study were
performed by Quantum Espresso code31 within the plane
wave basis set and Perdew-Burke-Ernzerhof (PBE) plane
wave pseudo potential method.32 The plane wave cutoff
energy is set to 550 eV. The supercells with a vacuum larger
than 15 A are adapted to avoid the interaction with its periodic
images to simulate the isolated nano graphene monoxide. The
special k-points generated by a 24� 24� 1 Monkhorst-Pack
grid in the Brillouin zone (BZ), where the integration is car-
ried out without smearing techniques with shift from origin.
The final plane wave basis set corresponds to the energies and
stress differences as 0.01 meV and 0.01 GPa respectively per
unit cell. A Broyden–Fletcher–Goldfarb–Shanno (BFGS)
algorithm was used to relax electrons, ions, and cell parame-
ters. The second order elastic constants were calculated using
the numerical differentiation of physical stress of a graphene
oxide structures as a function of imposed strain. To calculate
the second order elastic constants, the ground state relaxed ge-
ometry has been considered in terms of both cell parameters
and atomic positions, where the relaxed equilibrium configu-
ration is used as reference system. Finally, using each
deformed structure, the internal degrees of freedom are again
relaxed. The second order derivatives of the stress have been
calculated from the five sets of deformed crystal structures
created using the knowledge of the space group symmetry.
The phonon calculations were performed within the frame-
work of density functional perturbation theory (DFPT).33 The
advantage of DFPT over the conventional method is that it
allows the calculations for the phonon at any wave vector
only using unit cell. The dynamical matrices were calculated
on 4 � 4 � 1 q-point grid sampling.
III. RESULTS AND DISCUSSION
The electronic properties of the oxidized layer of gra-
phene sheet namely graphene oxide depend on the detailed
chemical structure.34–38 A single functional group of epoxide
on graphene can induce significant local distortion with a
new bond formed between carbon (C) and oxygen (O) atoms,
the bonding characteristic of the connecting C atoms can be
changed from planar sp2 to distorted sp3 hybridization. Our
theoretical investigation concentrates on the following key
issues: (1) an arrangement of epoxy functional group in fully
oxidized graphene sheet and (2) the effect of epoxy arrange-
ment on electronic properties on the graphene sheets.
The atomic structure of graphene oxide has been investi-
gated based on the molecular ratio of C: O as 1:1 obtained
from experimental measurement of Mattson et al.22 The pos-
sible crystalline geometrical structures are considered for the
graphene oxides in XY, YZ, and XZ planes together with
perspective view have been shown in the Figs. 1(a)–1(e) for
GDO, a-GMO, z-GMO, ep-GMO, and mix-GMO, respec-
tively. After geometric relaxation, the obtained unit cell pa-
rameters, fractional coordinates, and total energy of different
graphene oxide models such as z-GMO, GDO, a-GMO,
FIG. 1. (a) Atomic structure of GDO along the xy, yz, xz planes with per-
spective view. (b) Atomic structure of a-GMO along the xy, yz, xz planes
with perspective view. (c) Atomic structure of z-GMO along the xy, yz, xz
planes with perspective view. (d) Atomic structure of ep-GMO along the xy,
yz, xz planes with perspective view. (e) Atomic structure of mix-GMO along
the xy, yz, xz planes with perspective view.
203517-2 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

ep-GMO and mix-GMO are shown in Table I. There is in
general a good agreement with previous available data.17
The z-GMO structure can be viewed as a zigzag carbon
chains linked by oxygen atoms, having C-C-C and C-O-C
zigzag chains in the X- and Y-directions, respectively. Here,
all the carbon atoms are 4-fold co-ordinated possessing two
bonds with its neighboring and C and O atoms. The arm
chair graphene oxide has orthorhombic crystalline structure
in Pmma(#51) space group with 8 atoms per unit cell (Table
I). In the case of a-GMO, arm chair carbon atoms having
chain formation linked to O atoms and form a stable crystal-
line phase. Our calculations indicate that these possible low
energy phases will probably exist in any above considered
structures. Table I clearly brings out that the z-GMO phase is
energetically more stable with very small energy difference
relative to each other, similar to the results of Zhang et al.17 In
Table II, we present the bond lengths and bond angles in the
different configurations of graphene oxide.
To understand the electronic properties of graphene ox-
ide models, we have calculated the electronic dispersion
TABLE I. Structural parameters and total energy of GDO, a-GMO, z-GMO, ep-GMO, and mix-GMO, respectively.
Structure
Lattice parameters (A)
Space group Atomic position
Total energy
per CO unit(eV)a b c
GDO 2.34 2.34 15 P�4m2 (115) C 0.5000 0.5000 0.5000 �510.52743276
O 0.5000 0.0000 0.4447
O 0.0000 0.5000 0.5552
a-GMO 5.02 2.27 15 Pmma (51) C 0.4189 0.0000 0.4554 �587.895606772
C 0.5811 0.0000 0.5446
C 0.0811 0.0000 0.4554
C 0.9189 0.0000 0.5446
O 0.0105 0.5000 0.4047
O 0.9895 0.5000 0.5953
O 0.4895 0.5000 0.4047
O 0.5105 0.5000 0.5953
z-GMO 2.65 2.27 15 Pmma (51) C 0.7500 0.0000 0.4701 -587.95222743
C 0.2500 0.0000 0.5298
O 0.2500 0.5000 0.5856
O 0.7500 0.5000 0.4143
ep-GMO 5.68 2.64 15 Cmmm (65) C 0.3261 0.0000 0.5000 �587.66005036
C 0.6739 0.0000 0.5000
C 0.8259 0.5000 0.5000
C 0.1741 0.5000 0.5000
O 0.5000 0.0000 0.5696
O 0.5000 0.0000 0.4304
O 0.0000 0.5000 0.5696
O 0.0000 0.5000 0.4304
mix-GMO 9.42 5.27 15 Cm (8) C 0.0484 0.1414 0.4399 �587.61688915
C 0.0484 0.8586 0.4399
C 0.5484 0.6414 0.4399
C 0.5484 0.3586 0.4399
C 0.9137 0.3100 0.4328
C 0.9137 0.6900 0.4328
C 0.4137 0.8100 0.4328
C 0.4137 0.1900 0.4328
C 0.2854 0.6429 0.4080
C 0.2854 0.3571 0.4080
C 0.7854 0.1429 0.4080
C 0.7854 0.8571 0.4080
O 0.1592 0.2357 0.3895
O 0.1592 0.7643 0.3895
O 0.6592 0.7357 0.3895
O 0.6592 0.2643 0.3895
O 0.8968 0.5000 0.5019
O 0.3968 0.0000 0.5019
O 0.9289 0.5000 0.3646
O 0.4289 0.0000 0.3646
O 0.3251 0.5000 0.3308
O 0.8251 0.0000 0.3308
O 0.5735 0.5000 0.5204
O 0.0735 0.0000 0.5204
203517-3 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19
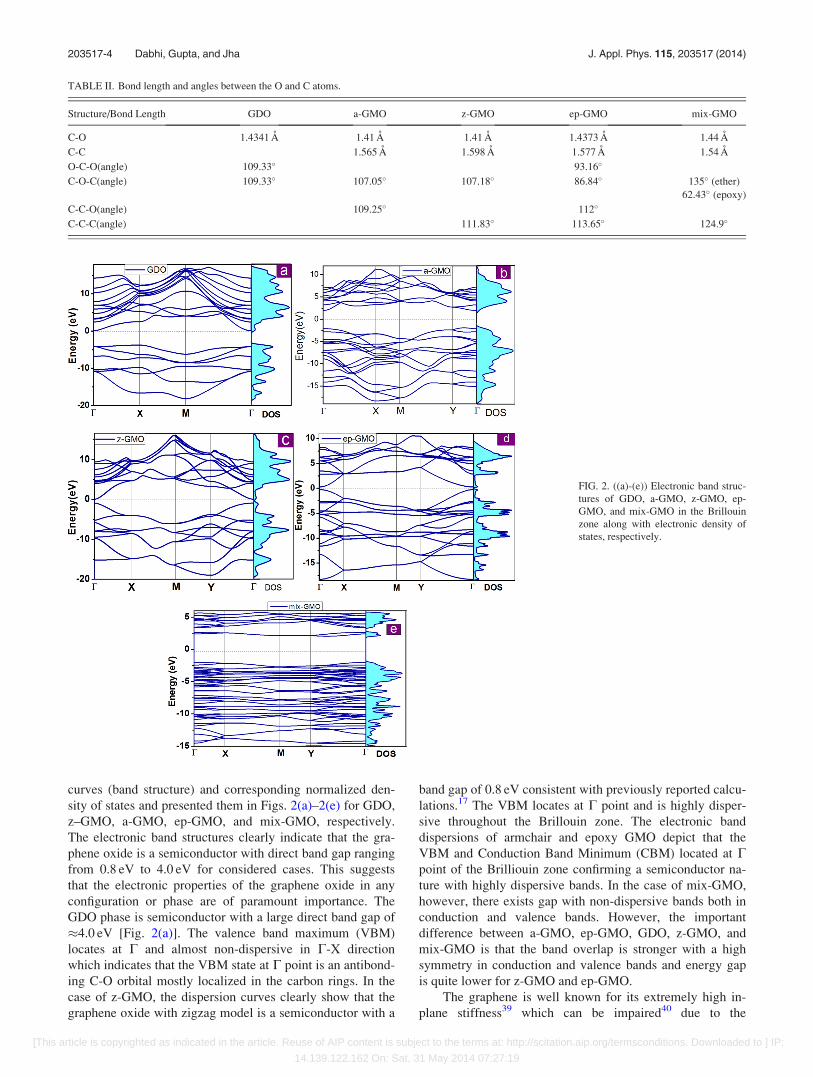
curves (band structure) and corresponding normalized den-
sity of states and presented them in Figs. 2(a)–2(e) for GDO,
z–GMO, a-GMO, ep-GMO, and mix-GMO, respectively.
The electronic band structures clearly indicate that the gra-
phene oxide is a semiconductor with direct band gap ranging
from 0.8 eV to 4.0 eV for considered cases. This suggests
that the electronic properties of the graphene oxide in any
configuration or phase are of paramount importance. The
GDO phase is semiconductor with a large direct band gap of
�4.0 eV [Fig. 2(a)]. The valence band maximum (VBM)
locates at C and almost non-dispersive in C-X direction
which indicates that the VBM state at C point is an antibond-
ing C-O orbital mostly localized in the carbon rings. In the
case of z-GMO, the dispersion curves clearly show that the
graphene oxide with zigzag model is a semiconductor with a
band gap of 0.8 eV consistent with previously reported calcu-
lations.17 The VBM locates at C point and is highly disper-
sive throughout the Brillouin zone. The electronic band
dispersions of armchair and epoxy GMO depict that the
VBM and Conduction Band Minimum (CBM) located at Cpoint of the Brilliouin zone confirming a semiconductor na-
ture with highly dispersive bands. In the case of mix-GMO,
however, there exists gap with non-dispersive bands both in
conduction and valence bands. However, the important
difference between a-GMO, ep-GMO, GDO, z-GMO, and
mix-GMO is that the band overlap is stronger with a high
symmetry in conduction and valence bands and energy gap
is quite lower for z-GMO and ep-GMO.
The graphene is well known for its extremely high in-
plane stiffness39 which can be impaired40 due to the
TABLE II. Bond length and angles between the O and C atoms.
Structure/Bond Length GDO a-GMO z-GMO ep-GMO mix-GMO
C-O 1.4341 A 1.41 A 1.41 A 1.4373 A 1.44 A
C-C 1.565 A 1.598 A 1.577 A 1.54 A
O-C-O(angle) 109.33� 93.16�
C-O-C(angle) 109.33� 107.05� 107.18� 86.84� 135� (ether)
62.43� (epoxy)
C-C-O(angle) 109.25� 112�
C-C-C(angle) 111.83� 113.65� 124.9�
FIG. 2. ((a)-(e)) Electronic band struc-
tures of GDO, a-GMO, z-GMO, ep-
GMO, and mix-GMO in the Brillouin
zone along with electronic density of
states, respectively.
203517-4 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

oxidation. Motivated with this fact and aim to understand the
effect of oxidation on the mechanical properties of graphene
oxide, we have calculated the elastic properties of the above
mentioned graphene oxide structures. The second order elas-
tic constants are calculated using both cell parameters and
atomic positions of optimized geometries and the numerical
differentiation of physical stress in a plane as a function of
the imposed strain. The calculated elastic constants of the
above mentioned graphene oxide structures are shown in
Table III. It is found that the GDO has the maximum value
327 GPa nm along the x-direction, indicating that the in-
plane stiffness in the case of GDO type graphene oxide is
highly isotropic along the x-direction, whereas in the case of
a-GMO and z-GMO it greatly weakens. However, the
y-direction elastic stiffness (C22) is maximum for a-GMO
and minimum for ep-GMO. As far as arm chair and zigzag
GMO are concerned, both x- and y-directions elastic stiff-
ness are close which result almost in the same value of Bulk
modulus and Young’s modulus. While comparing the iso-
tropic mechanical properties of the conventional pristine gra-
phene sheet and its hydrogenated or fluorinated structures41
the anisotropic elastic property exhibited in z-GMO and
a-GMO are quite unique having anisotropy factor(AF) value
� 86% and 83%, respectively. The evaluated directional ani-
sotropy in z-GMO and a-GMO can be attributed to the dif-
ference in strength between the O-bridged ether type
configuration and zigzag and arm chair carbon chains. The
simple hexagonal graphene has isotropic symmetry (there-
fore, is elastically isotropic); however, in the present gra-
phene oxide models GDO, a-GMO, z-GMO, ep-GMO, and
mix-GMO modeled by oxidation of honeycomb graphene
lattice. Each conformer characterized by a specific oxygen
sub lattice alternate on the both sides of the carbon lattice in
the x-y plane. The GDO, a-GMO, z-GMO, and ep-GMO
conformers show trigonal and orthorhombic symmetry,
respectively. This shows anisotropic symmetry and causes
an anisotropic linear elastic behavior. Due to this anisotropic
symmetry case in above said GMO models, two deforma-
tions have been applied: (i) An axial deformation along the
arm chair direction and (ii) a shear deformation. Further, we
observe that the difference between C11 and C12 for all aniso-
tropic GMO models is very large. The value of C44
measuring the resistance to shear deformation, consistent in
GDO, a-GMO, z-GMO while drastically increases in the
ep-GMO. Interestingly, a negative C12 is unexpected and
worth for further investigation. However, very small value of
C12 than C11 in all cases leads to the positive shear constant
implying mechanical stability of all structures at 0K. It is
clear from Table III that the oxidation of graphene remark-
ably affects the mechanical properties of graphene and hence
the structural anisotropy. The maximum value of in-plane
stiffness, C11 for GDO signifies that this type of graphene
oxide is more isotropic in x-direction. However, this is less
isotropic than graphene in x-direction. The implications of
the C11, C12, C13, C22, C23, C33, C44 become more significant
due to shape anisotropy meshes with crystal structure anisot-
ropy in 2D nanostructures. The modeled GMO with aniso-
tropic crystal symmetry lead to nontrivial physical
consequences in terms of elastic and vibrational properties.
The Table III shows directional elastic anomaly in terms
C12, C13, and C33 value of modeled GMO which can be
attributed to a variation in Young’s modulus for GMO com-
pared to its value in the case of graphene.
To understand the changes that occur in the vibrational
spectrum of graphene oxide resulting from the complete oxi-
dation of graphene, the full phonon calculations have been
performed for above considered configurations of graphene
oxide. The phonon calculations are important for variety of
properties and phenomena of a material and hence are inte-
gral part of material’s research including graphene and
related materials. The study of vibrational properties of gra-
phene using experiment, mainly the Raman spectroscopy or
theoretical calculations, is to determine the number and ori-
entation of layers, electron-phonon interaction, thermal con-
duction, etc.8,42–51 Besides, the phonon dispersion curves of
a material also confirm its dynamical stability. Therefore, the
lattice dynamics is a reliable test for an optimized structure
through the variation of frequency of phonon modes with
wave vector, k in BZ. If frequency of any phonon is imagi-
nary in BZ, the structure is said to be dynamically unstable.
If a phonon mode has imaginary frequency, it does not gen-
erate restoring force to execute the oscillations and the sys-
tem is forced to deviate from its structural configurations. To
understand the behavior of each mode and dynamical stabil-
ity of different structures of graphene oxide, we have calcu-
lated the phonon dispersion curves of graphene oxide
structures and presented them in Figs. 3(a)–3(e). The present
figure clearly brings out the differences in the phonon disper-
sion curves of different graphene oxide structures. However,
we note that the phonon dispersion curves of all considered
structures contain phonon modes with positive frequency
with the exception of ep-GMO where lowest acoustical
branch intersects not exactly at zone centre but slightly away
like the out of plane (ZA) mode of graphene.8,42–51 This indi-
cates that the modes are the genuine local minimum and ca-
pable of existing in real. Our results are more or less
consistent with the other available calculations. In the case
of mix-GMO, the range of frequency increases up to �1400
cm�1 in contrast to the usual range of �1200–1250 cm�1
TABLE III. Elastic stiffness with associated mechanical properties for
GDO, a-GMO, z-GMO, ep-GMO, and mix-GMO.
Structure/Elasic Constant GDO a-GMO z-GMO ep-GMO mix-GMO
C11 (GPa nm) 327 199 198 273 140
C12 (GPa nm) �1.65 �1.2 �0.9 �0.15 12
C13 (GPa nm) �7.2 31.05 �1.35 10.05 �7.7
C22 (GPa nm) 329 454 453 198 190
C23 (GPa nm) �7.2 17.6 11.8 8.85 �6.3
C33 (GPa nm) �20.25 �40.8 �27.6 �18.15 �2.7
C44 (GPa nm) 1.8 1.05 0.9 11.55 �4.4
C55 (GPa nm) 1.8 30.9 30.9 �12.3 28.95
C66 (GPa nm) 36.75 70.05 69.9 55.5 77.85
Voigt Bulk Modulus
(GPa nm)
44.8 78.5 78.2 54.5 34.2
Voigt Young Modulus
(GPa nm)
123 140 139 96.2 90
AF % 83.1 83.4 85.9 �195 247
203517-5 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19
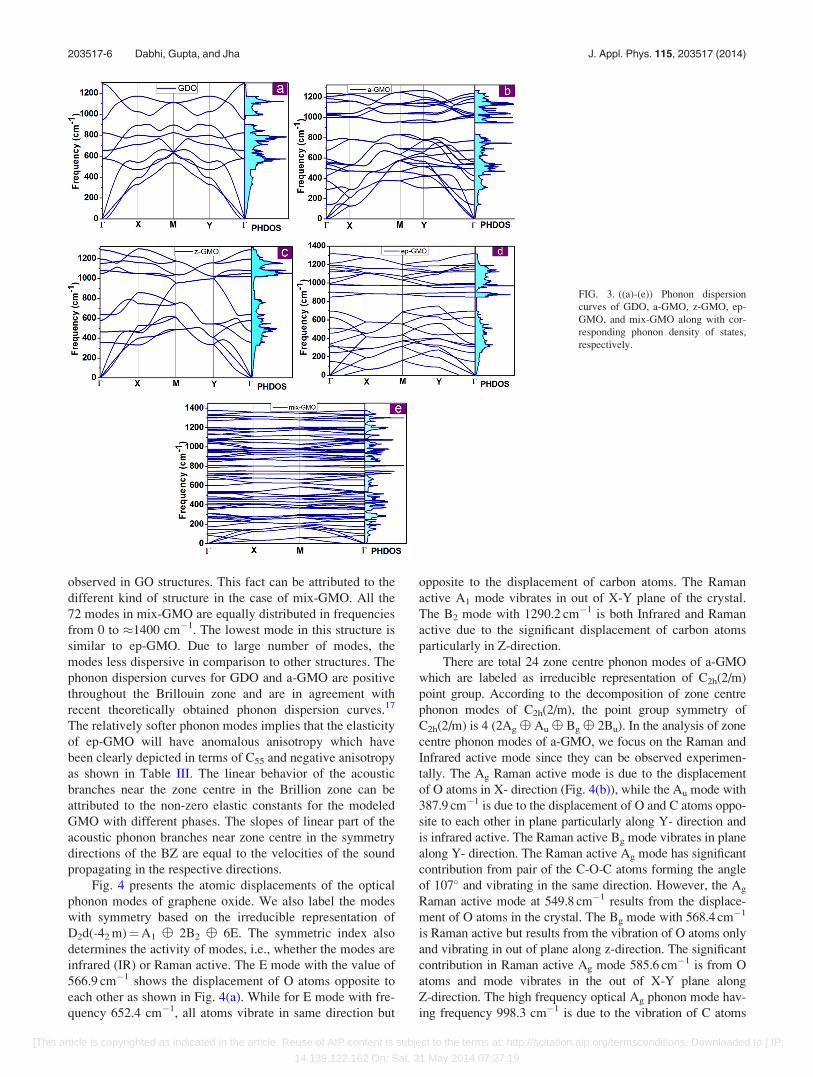
observed in GO structures. This fact can be attributed to the
different kind of structure in the case of mix-GMO. All the
72 modes in mix-GMO are equally distributed in frequencies
from 0 to �1400 cm�1. The lowest mode in this structure is
similar to ep-GMO. Due to large number of modes, the
modes less dispersive in comparison to other structures. The
phonon dispersion curves for GDO and a-GMO are positive
throughout the Brillouin zone and are in agreement with
recent theoretically obtained phonon dispersion curves.17
The relatively softer phonon modes implies that the elasticity
of ep-GMO will have anomalous anisotropy which have
been clearly depicted in terms of C55 and negative anisotropy
as shown in Table III. The linear behavior of the acoustic
branches near the zone centre in the Brillion zone can be
attributed to the non-zero elastic constants for the modeled
GMO with different phases. The slopes of linear part of the
acoustic phonon branches near zone centre in the symmetry
directions of the BZ are equal to the velocities of the sound
propagating in the respective directions.
Fig. 4 presents the atomic displacements of the optical
phonon modes of graphene oxide. We also label the modes
with symmetry based on the irreducible representation of
D2d(-42 m)¼A1 � 2B2 � 6E. The symmetric index also
determines the activity of modes, i.e., whether the modes are
infrared (IR) or Raman active. The E mode with the value of
566.9 cm�1 shows the displacement of O atoms opposite to
each other as shown in Fig. 4(a). While for E mode with fre-
quency 652.4 cm�1, all atoms vibrate in same direction but
opposite to the displacement of carbon atoms. The Raman
active A1 mode vibrates in out of X-Y plane of the crystal.
The B2 mode with 1290.2 cm�1 is both Infrared and Raman
active due to the significant displacement of carbon atoms
particularly in Z-direction.
There are total 24 zone centre phonon modes of a-GMO
which are labeled as irreducible representation of C2h(2/m)
point group. According to the decomposition of zone centre
phonon modes of C2h(2/m), the point group symmetry of
C2h(2/m) is 4 (2Ag � Au � Bg � 2Bu). In the analysis of zone
centre phonon modes of a-GMO, we focus on the Raman and
Infrared active mode since they can be observed experimen-
tally. The Ag Raman active mode is due to the displacement
of O atoms in X- direction (Fig. 4(b)), while the Au mode with
387.9 cm�1 is due to the displacement of O and C atoms oppo-
site to each other in plane particularly along Y- direction and
is infrared active. The Raman active Bg mode vibrates in plane
along Y- direction. The Raman active Ag mode has significant
contribution from pair of the C-O-C atoms forming the angle
of 107� and vibrating in the same direction. However, the Ag
Raman active mode at 549.8 cm�1 results from the displace-
ment of O atoms in the crystal. The Bg mode with 568.4 cm�1
is Raman active but results from the vibration of O atoms only
and vibrating in out of plane along z-direction. The significant
contribution in Raman active Ag mode 585.6 cm�1 is from O
atoms and mode vibrates in the out of X-Y plane along
Z-direction. The high frequency optical Ag phonon mode hav-
ing frequency 998.3 cm�1 is due to the vibration of C atoms
FIG. 3. ((a)-(e)) Phonon dispersion
curves of GDO, a-GMO, z-GMO, ep-
GMO, and mix-GMO along with cor-
responding phonon density of states,
respectively.
203517-6 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

in X-Y plane and Raman active, while Bg mode with fre-
quency 1009.5 cm�1 is due to the displacement of C atoms in
X-Y plane. The Raman active Ag mode at 1023.2 cm�1
vibrates in X-Y plane. Both Ag modes with frequency 1205.3
cm�1 and 1233.2 cm�1 vibrates in and out of plane, respec-
tively, and both are Raman active.
According to the decomposition of the twelve zone
centre phonon mode of D2h(mmm) point group symmetry,
the phonon modes can be written as D2h(mmm)¼ 2 (Ag �
B1u � B2u � B3u � B2g � B3g) for z-GMO. The Raman
active B2g mode is due to the displacement of O and C atoms
in X-Y plane particularly in X- direction. However, the
Raman active modes B3g, B2g, and Ag with values 585.95,
1051.29, and 1188.59 cm�1 are due to the displacement of
both O and C atoms opposite to each other along
Z-direction. The Ag Raman active modes with frequency
FIG. 4. ((a)-(e)) Zone center phonon modes along with phonon frequencies in GDO, a-GMO, z-GMO, ep-GMO and mix-GMO structure, respectively.
203517-7 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

638.43 and 1282.24 cm�1 are due to the simultaneous dis-
placement of O and C atoms out of X-Y plane. The B3u and
B1u infrared active modes at 464.52 cm�1 and 1072.93 cm�1
are due to the displacement of O and C atoms opposite to
each other in X-Y plane particularly in Y- and X-direction,
respectively, as shown in Fig. 4(c).
There are twenty four zone centre phonon modes in the
case of ep-GMO and are labeled with the irreducible repre-
sentations of D2h(mmm) point group. According to decom-
position of this point group symmetry, the phonon modes
can be written as D2h(mmm) ¼ 2 (2Ag � 2B1u � 2B2u �
2B3u � B1g � 2B2g � B3g). The Ag, B1g, B2g, and B3g pho-
non modes are Raman active while B1u, B2u and B3u are
infrared active. From the analysis of displacement of atoms
in Fig. 4(d), the modes B2u, two B3g, B2u, B3u, B2g, Ag, and
Ag, with value of 8.79, 3.75, �1.25, 312.7, 626.4, 696.4,
848.7, and 899.8 cm�1, respectively, occur due to the dis-
placement of O atoms in the crystal. However, the phonon
modes B1g, B1u, B2u, B2g, B1g, Ag, B1u, B2u, B3u, and Ag,
with corresponding values of 151.0, 244.4, 331.8, 984.4, and
992.5 cm�1 are due to the combined displacement of C and
O atoms as shown in Fig. 4(d).
The Fig. 4(e) presents the displacement pattern of all the
phonon modes in mix-GMO. We have focused particularly
on modes only close to 1200 cm�1 and 1400 cm�1 as all the
structures have phonon modes around 1200 cm�1 and the
phonon modes at �1400 cm�1 exist only in mix-GMO. It is
clearly demonstrated that the stretching modes are in and out
of phase in X-Y plane.
It is important to shed some light on the stability of the
different oxidized graphene structures, particularly, in com-
parison to earlier theoretical and experimental evidences.
Several atomic models including the well celebrated Lerf-
Klinowski model have been proposed with different
functional groups.18,19,38 The Lerf-Klinowski model for
functional group involves theoretical modeling of hydroxy,
epoxy, and carbonyl in terms of structural and energetically
favorable stability using the experimental NMR data.37 The
oxidized structures in the present study are quasihexagonal
lattice with parallel-aligned epoxy pairs (ep-GMO), the
O-driven unzipping of clamped epoxide which lowers the
energy through release of local strain and modeled as zigzag
carbon chains graphene monoxide (z-GMO), and arm chair
graphene monoxide (a-GMO), graphene monoxide contain-
ing ether group, normal epoxy and epoxy pair (mix-GMO),
and graphene dioxide (GDO). All the modeled GO studied in
the present manuscripts are chemically homogeneous and or-
dered structures developed using quantum mechanical many
body approach within the framework of periodic boundary
conditions using density functional theory. The current theo-
retical quantum mechanical approach within density func-
tional theory is quite successful in finding the global
minimum, which has been revealed in the present manuscript
by investigating structural, electronic, mechanical, and dy-
namical stability. In the present study, all the considered
structures of GO are fully oxidized structures and in are
good agreement with earlier experimental finding.23
Furthermore, the recent work of Kumar et al.29 also supports
our theoretical findings for the fully oxidized graphene
monoxides. As separated GO belongs from the same confor-
mal, our work is the extension of the detailed study of fully
oxidized GO in the possible conformal after the separation
process.
IV. CONCLUSION
In summary, we have applied density functional theory to
elucidate the structural, electronic, elastic, and phonon proper-
ties of different models of graphene oxide. Our calculated lat-
tice parameters, elastic stiffness and phonon frequencies are in
good agreement with available theoretical data. The calculated
zero temperature phonon dispersion curves and elastic stiff-
ness values confirm the dynamical and mechanical stability of
the graphene oxide in GDO, a-GMO and mix-GMO struc-
tures. However, the electronic band structure and total energy
calculation supports the semiconducting nature for all above
considered structures and reveals the electronic stability of the
z-GMO and ep-GMO structures of the graphene monoxides.
The band structures reveal that the considered graphene oxide
structures have band gap from 0.8 to 4 eV depending on the
structures of graphene oxide. The wide range of band gap
available with graphene oxide suggests that they have poten-
tial applications in nanodevices.
ACKNOWLEDGMENTS
Computations carried out on the computer cluster
PAWAN at the Department of Physics, Maharaja
Krishnakumasinhji Bhavnagar University funded by the
Department of Science and Technology, Government of
India, are highly appreciated for this work. Part of the calcu-
lations was performed at HPC facility IUAC, New Delhi.
One of us (S.D.) highly appreciates the financial support in
terms of Inspire Research Fellowship by the Department of
Science and Technology, Government of India.
1J. Wu, H. A. Becerril, Z. Bao, Z. Liu, Y. Chen, and P. Peumans, Appl.
Phys. Lett. 92, 263302 (2008).2K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, and
S. V. Dubonos, Science 306, 666 (2004).3K. A. Ritter and J. W. Lyding, Nature Mater. 8, 235 (2009).4C. Berger, Z. Song, X. Li, X. Wu, N. Brown, and C. Naud, Science 312,
1191 (2006).5M. I. Katsnelson, K. S. Novoselov, and A. K. Geim, Nat. Phys. 2, 620
(2006).6F. Miao, S. Wijeratne, Y. Zhang, U. C. Coskun, W. Bao, and C. N. Lau,
Science 317, 1530 (2007).7A. K. Geim and K. S. Novoselov, Nature Mater. 6, 183 (2007).8S. K. Gupta, H. R. Soni, and P. K. Jha, AIP Adv. 3, 032117 (2013).9T. Ohta, A. Bostwick, T. Seyller, K. Horn, and E. Rotenberg, Science 313,
951 (2006).10M. Y. Han, B. Ozyilmaz, Y. Zhang, and P. Kim, Phys. Rev. Lett. 98,
206805 (2007).11G. Giovannetti, P. A. Khomyakov, G. Brocks, P. J. Kelly, and J. Brink,
Phys. Rev. B 76, 073103 (2007).12X. Li, X. Wang, L. Zhang, S. Lee, and H. Dai, Science 319, 1229 (2008).13X. Wu, M. Sprinkle, X. Li, F. Ming, C. Berger, and W. A. Heer, Phys.
Rev. Lett. 101, 026801 (2008).14I. Jung, D. A. Dikin, R. D. Piner, and R. S. Ruoff, Nano Lett. 8, 4283
(2008).15D. C. Elias, R. R. Nair, T. M. G. Mohiuddin, S. V. Morozov, P. Blake,
M. P. Halsall, A. C. Ferrari, D. W. Boukhvalov, M. I. Katsnelson, A. K.
Geim, and K. S. Novoselov, Science 323, 610 (2009).16J. Yan, L. Xian, and M. Y. Chou, Phys. Rev. Lett. 103, 086802 (2009).
203517-8 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19

17S. Zhang, J. Zhou, Q. Wang, and P. Jena, J. Phys. Chem. C 117, 1064 (2013).18A. Lerf, H. Heb, T. Riedl, M. Forster, and J. Klinowski, Solid State Ionics
101, 857 (1997).19H. He, J. Klinowski, M. Forster, and A. Lerf, Chem. Phys. Lett. 287, 53
(1998).20K. A. Mkhoyan, A. W. Contryman, J. Silcox, D. A. Stewart, G. Eda, C.
Mattevi, S. Miller, and M. Chhowalla, Nano Lett. 9, 1058 (2009).21T. Szab�o, O. Berkesi, P. Forg�o, K. Josepovits, Y. Sanakis, D. Petridis, and
I. D�ek�any, Chem. Mater. 18, 2740 (2006).22E. C. Mattson, H. Pu, S. Cui, M. A. Schofield, S. Rhim, G. Lu, M. J.
Nasse, R. S. Ruoff, M. Weinert, M. Gajdardziska-Josifovska, J. Chen, and
C. J. Hirschmugl, ACS Nano 5, 9710 (2011).23H. C. Schniepp, J. L. Li, M. J. McAllister, H. Sai, M. Herrera-Alonso,
D. H. Adamson, R. K. Prud’homme, R. Car, D. A. Saville, and I. A.
Aksay, J. Phys. Chem. B 110, 8535 (2006).24M. Jana, S. Saha, P. Khanra, N. C. Murmu, S. K. Srivastava, T. Kuila, and
J. H. Lee, Mater. Sci. Eng., B 186, 33 (2014).25Z. Xu and K. Xue, Nanotechnology 21, 045704 (2010).26S. Saxena, T. A. Tyson, S. Shukla, E. Negusse, H. Chen, and J. Bai, Appl.
Phys. Lett. 99, 013104 (2011).27S. Saxena, T. A. Tyson, and E. Negusse, J. Phys. Chem. Lett. 1, 3433 (2010).28H. J. Xiang, S. H. Wei, and X. G. Gong, Phys. Rev. B 82, 035416 (2010).29P. V. Kumar, N. M. Bardhan, S. Tongay, J. Wu, A. M. Belcher, and J. C.
Grossman, Nat. Chem. 6, 151 (2013).30M. J. McAllister, J. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J.
Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud’homme, and
I. A. Aksay, Chem. Mater. 19, 4396 (2007).31P. Giannozzi et al., J. Phys. Condens. Matter 21, 395502 (2009); URL:
http://www.quantum-espresso.org.32J. P. Perdew, K. Burke, and M. Ernzerhof, Phys. Rev. Lett. 77, 3865 (1996).
33S. Baroni, S. Gironcoli, A. D. Corso, and P. Giannozzi, Rev. Mod. Phys.
73, 515 (2001).34T. Nakajima, A. Mabuchi, and R. Hagiwara, Carbon 26, 357 (1988).35M. Mermoux, Y. Chabre, and A. Rousseau, Carbon 29, 469 (1991).36T. Nakajima and Y. Matsuo, Carbon 32, 469 (1994).37A. Lerf, H. He, M. Forster, and J. Klinowski, J. Phys. Chem. B 102, 4477
(1998).38H. He, T. Riedl, A. Lerf, and J. Klinowski, J. Phys. Chem. 100, 19954
(1996).39C. Lee, X. Wei, J. W. Kysar, and J. Hone, Science 321, 385 (2008).40R. J. W. E. Lahaye, H. K. Jeong, C. Y. Park, and Y. H. Lee, Phys. Rev. B
79, 125435 (2009).41H. Sahin, M. Topsakal, and S. Ciraci, Phys. Rev. B 83, 115432 (2011).42A. Das, S. Pisana, B. Chakraborty, S. Piscanec, S. K. Saha, U. V.
Waghmare, K. S. Novoselov, H. R. Krishnamurthy, A. K. Geim, A. C.
Ferrari, and A. K. Sood, Nat. Nanotechnol. 3, 210 (2008).43S. K. Saha, R. Ch. Chandrakanth, H. R. Krishnamurthy, and U. V.
Waghmare, Phys. Rev. B 80, 155414 (2009).44S. Piscanec, M. Lazzeri, F. Mauri, A. C. Ferrari, and J. Robertson, Phys.
Rev. Lett. 93, 185503 (2004).45A. Gr€uneis, R. Saito, T. Kimura, L. G. Cancado, M. A. Pimenta, A. Jorio,
A. G. Souza Filho, G. Dresselhaus, and M. S. Dresselhaus, Phys. Rev. B
65, 155405 (2002).46H. Wang, Y. Wang, X. Cao, M. Feng, and G. Lan, J. Raman Spectrosc. 40,
1791 (2009).47N. Mounet and N. Marzari, Phys. Rev. B 71, 205214 (2005).48J. Yan, W. Y. Ruan, and M. Y. Chou, Phys. Rev. B 77, 125401 (2008).49K. H. Michel and B. Verberck, Phys. Rev. B 78, 085424 (2008).50L. Wirtz and A. Rubio, Solid State Commun. 131, 141 (2004).51J. Jiang, H. Tang, B. Wang, and Z. Su, Phys. Rev. B 77, 235421 (2008).
203517-9 Dabhi, Gupta, and Jha J. Appl. Phys. 115, 203517 (2014)
[This article is copyrighted as indicated in the article. Reuse of AIP content is subject to the terms at: http://scitation.aip.org/termsconditions. Downloaded to ] IP:
14.139.122.162 On: Sat, 31 May 2014 07:27:19




















