Stereospecific interactions are necessary for Alzheimer's disease amyloid-beta toxicity
14
Neurobiology of Aging 32 (2011) 235–248 Stereospecific interactions are necessary for Alzheimer disease amyloid- toxicity Giuseppe D. Ciccotosto a,b,c , Deborah J. Tew a,b,c , Simon C. Drew a,b,c,f , Danielle G. Smith a,b,c,1 , Timothy Johanssen a,b,c , Varsha Lal c , Tong-Lay Lau b,d,2 , Keyla Perez b,c , Cyril C. Curtain f , John D. Wade e , Frances Separovic b,d , Colin L. Masters c , Jeffrey P. Smith a,c,3 , Kevin J. Barnham a,b,c,4 , Roberto Cappai a,b,∗ a Department of Pathology, The University of Melbourne, VIC 3010, Australia b Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, VIC 3010, Australia c Mental Health Research Institute, VIC 3052, Australia d School of Chemistry, The University of Melbourne, VIC 3010, Australia e Florey Neuroscience Institute, The University of Melbourne, VIC 3010, Australia f School of Physics, Monash University, VIC 3800, Australia Received 27 August 2008; received in revised form 12 February 2009; accepted 18 February 2009 Available online 25 March 2009 Abstract Previous studies suggest membrane binding is a key determinant of amyloid (A) neurotoxicity. However, it is unclear whether this interaction is receptor driven. To address this issue, a D-handed enantiomer of A42 (D-A42) was synthesized and its biophysical and neurotoxic properties were compared to the wild-type A42 (L-A42). The results showed D- and L-A42 are chemically equivalent with respect to copper binding, generation of reactive oxygen species and aggregation profiles. Cell binding studies show both peptides bound to cultured cortical neurons. However, only L-A42 was neurotoxic and inhibited long term potentiation indicating L-A42 requires a stereospecific target to mediate toxicity. We identified the lipid phosphatidylserine, as a potential target. Annexin V, which has very high affinity for externalized phosphatidylserine, significantly inhibited L-A42 but not D-A42 binding to the cultured cortical neurons and Abbreviations: AD, Alzheimer’s diseases; APP, amyloid precursor protein; A, amyloid- peptide; ACSF, artificial cerebral spinal fluid; BCA, bicinchoninic acid; CF, 5-carboxyfluorescein; DBU, 1,8-diazabicyclo[5,4,0]-undec-7-ene; DCF, dichlorofluorescein diacetate; DIV, days in vitro; DMF, dimethylformamide; DPH, diphenyl-1,3,5-hexatriene; EM, electron microscopy; EPR, electron paramagnetic resonance; fEPSP, field excitatory post- synaptic potential; HAE, 4-hydroxyalkenals; HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol; HFS, high-frequency stimulation; LDH, lactate dehydrogenase; LTP, long term potentiation; LUV, large unilamellar vesicles; MDA, malondialdehyde; MALDI-TOF MS, matrix-assisted laser desorption/ionization–time of flight mass spectrometry; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; PBS, phosphate buffered saline; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PICUP, photo-induced crosslinking of unmodified proteins; POPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine]; PS-NBD, 1-oleoyl-2-[6-[(7-nitro-2- 1,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phospho-l-serine; ROS, reactive oxygen species; Ru(Bpy), ruthenium bis(bpy); TFA, trifluoroacetic acid. ∗ Corresponding author. Tel.: +61 3 8344 5868; fax: +61 3 9347 6750. E-mail addresses: [email protected] (K.J. Barnham), [email protected] (R. Cappai). 1 Present address: Department of Biology, University of York, York YO10 5YW, United Kingdom. 2 Present address: Department of Biochemistry and Molecular Biology and Zilkha Neurogenetic Institute, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA. 3 Present address: Department of Biology, Colorado State University Pueblo, CO 81001, USA. 4 Tel.: +61 3 8344 2555. 0197-4580/$ – see front matter © 2009 Elsevier Inc. All rights reserved. doi:10.1016/j.neurobiolaging.2009.02.018
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Stereospecific interactions are necessary for Alzheimer's disease amyloid-beta toxicity

A
inrtsa
bdslflbP1a
U
0d
Neurobiology of Aging 32 (2011) 235–248
Stereospecific interactions are necessary for Alzheimerdisease amyloid-� toxicity
Giuseppe D. Ciccotosto a,b,c, Deborah J. Tew a,b,c, Simon C. Drew a,b,c,f,Danielle G. Smith a,b,c,1, Timothy Johanssen a,b,c, Varsha Lal c,
Tong-Lay Lau b,d,2, Keyla Perez b,c, Cyril C. Curtain f, John D. Wade e,Frances Separovic b,d, Colin L. Masters c, Jeffrey P. Smith a,c,3,
Kevin J. Barnham a,b,c,4, Roberto Cappai a,b,∗a Department of Pathology, The University of Melbourne, VIC 3010, Australia
b Bio21 Molecular Science and Biotechnology Institute, The University of Melbourne, VIC 3010, Australiac Mental Health Research Institute, VIC 3052, Australia
d School of Chemistry, The University of Melbourne, VIC 3010, Australiae Florey Neuroscience Institute, The University of Melbourne, VIC 3010, Australia
f School of Physics, Monash University, VIC 3800, Australia
Received 27 August 2008; received in revised form 12 February 2009; accepted 18 February 2009Available online 25 March 2009
bstract
Previous studies suggest membrane binding is a key determinant of amyloid � (A�) neurotoxicity. However, it is unclear whether thisnteraction is receptor driven. To address this issue, a D-handed enantiomer of A�42 (D-A�42) was synthesized and its biophysical andeurotoxic properties were compared to the wild-type A�42 (L-A�42). The results showed D- and L-A�42 are chemically equivalent withespect to copper binding, generation of reactive oxygen species and aggregation profiles. Cell binding studies show both peptides boundo cultured cortical neurons. However, only L-A�42 was neurotoxic and inhibited long term potentiation indicating L-A�42 requires a
osphatidylserine, as a potential target. Annexin V, which has very high
tereospecific target to mediate toxicity. We identified the lipid ph ffinity for externalized phosphatidylserine, significantly inhibited L-A�42 but not D-A�42 binding to the cultured cortical neurons andAbbreviations: AD, Alzheimer’s diseases; APP, amyloid precursor protein; A�, amyloid-� peptide; ACSF, artificial cerebral spinal fluid; BCA,icinchoninic acid; CF, 5-carboxyfluorescein; DBU, 1,8-diazabicyclo[5,4,0]-undec-7-ene; DCF, dichlorofluorescein diacetate; DIV, days in vitro; DMF,imethylformamide; DPH, diphenyl-1,3,5-hexatriene; EM, electron microscopy; EPR, electron paramagnetic resonance; fEPSP, field excitatory post-ynaptic potential; HAE, 4-hydroxyalkenals; HFIP, 1,1,1,3,3,3-hexafluoro-2-propanol; HFS, high-frequency stimulation; LDH, lactate dehydrogenase; LTP,ong term potentiation; LUV, large unilamellar vesicles; MDA, malondialdehyde; MALDI-TOF MS, matrix-assisted laser desorption/ionization–time ofight mass spectrometry; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt; PBS, phosphateuffered saline; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine; PICUP, photo-induced crosslinking of unmodified proteins;OPC, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; POPS, 1-palmitoyl-2-oleoyl-sn-glycero-3-[phospho-l-serine]; PS-NBD, 1-oleoyl-2-[6-[(7-nitro-2-,3-benzoxadiazol-4-yl)amino]hexanoyl]-sn-glycero-3-phospho-l-serine; ROS, reactive oxygen species; Ru(Bpy), ruthenium bis(bpy); TFA, trifluoroaceticcid.∗ Corresponding author. Tel.: +61 3 8344 5868; fax: +61 3 9347 6750.
E-mail addresses: [email protected] (K.J. Barnham), [email protected] (R. Cappai).1 Present address: Department of Biology, University of York, York YO10 5YW, United Kingdom.2 Present address: Department of Biochemistry and Molecular Biology and Zilkha Neurogenetic Institute, Keck School of Medicine,niversity of Southern California, Los Angeles, CA 90033, USA.3 Present address: Department of Biology, Colorado State University Pueblo, CO 81001, USA.4 Tel.: +61 3 8344 2555.
197-4580/$ – see front matter © 2009 Elsevier Inc. All rights reserved.oi:10.1016/j.neurobiolaging.2009.02.018

2
sp©
K
1
dtkpdcubot2rmefa
alpebp(pabclmi2bPAfliwV
Aeoii
36 G.D. Ciccotosto et al. / Neurobiology of Aging 32 (2011) 235–248
ignificantly rescued L-A�42 neurotoxicity. This suggests that A� mediated toxicity in Alzheimer disease is dependent upon A� binding tohosphatidylserine on neuronal cells.
2009 Elsevier Inc. All rights reserved.
eywords: Cortex; Neurotoxicity; Chiral; Annexin V; Long term potentiation; Membrane phospholipid
rLeabtirtwteaDtela
2
2
csoFtGt1fttwAa1ocws
. Introduction
Alzheimer disease (AD) is the major cause of age-relatedementia. AD is characterized by the abnormal accumula-ion of the amyloid-� peptide (A�) into insoluble aggregatesnown as plaques. A� is derived from the amyloid precursorrotein (APP) (Kang et al., 1987), with A�40 and A�42 theominant peptide species (Masters et al., 1985). The pre-ise mechanism of A�-induced neuronal toxicity remainsnresolved. Since A� contains part of the putative transmem-rane domain of the APP (Kang et al., 1987) the mechanismf A� mediated neurotoxicity may involve interactions withhe cell membrane (Verdier and Penke, 2004; Verdier et al.,004). We previously reported that A� neurotoxicity cor-elated with an increased affinity for the neuronal plasmaembrane (Ciccotosto et al., 2004; Smith et al., 2006; Tickler
t al., 2005). Potential molecular targets on the cell membraneor A� includes lipids, proteoglycans, and proteins (Verdiernd Penke, 2004).
The plasma membrane lipid composition is heterogeneousnd asymmetrical, a process that is essential for normal cellu-ar function (Bretscher, 1972). The choline-containing lipids,hosphatidylcholine (PC) and sphingomyelin, are primarilynriched in the extracellular outer cell surface of the mem-rane while the amine-containing glycerophospholipids,hosphatidyl-ethanolamine (PE) and phosphatidylserinePS) are primarily located on the cytoplasmic surface of thelasma membrane (reviewed in Daleke, 2003; Pomorski etl., 2004). The role of externalized PS in neuronal mem-ranes is typically used as an identification marker forells that are undergoing apoptosis. Recent evidence high-ighted that a specific neuronal cell population with increased
embrane exposure of externalized PS correlated best withncreased membrane A� binding (Simakova and Arispe,007). Annexin V peptide is a Ca2+ dependent phospholipid-inding protein that has a very high affinity for externalizedS (Mukhopadhyay and Cho, 1996; White et al., 2001).� can induce PS flipping to the extracellular surface sinceuorescently tagged annexin V binding was dramatically
ncreased following neuronal A� treatment, an effect thatas inhibited by co-incubation of A� peptides with annexinantibodies (Lee et al., 2002; White et al., 2001).Numerous hypotheses have been put forward to explain
� toxicity (Cappai and Barnham, 2007b) including the gen-
ration of reactive oxygen species (ROS), the self-assemblyf aggregates that bind and form holes in membranes or A�nteracting with receptors, such as RAGE. To test if A� tox-city and cell membrane binding was receptor mediated i.e.(mDc
equired a stereospecific interaction, we synthesized D- and-handed versions of A�42 because in the absence of a chiralnvironment, both the variants would behave identically ands such if a receptor was not required for toxicity then it woulde expected that both peptides would be equally toxic. Whenhe biological properties of L- and D-A�42 were examined,t was observed that unlike L-A�42, D-A�42 was not neu-otoxic to primary cortical cultures and did not inhibit longerm potentiation (LTP) in mouse hippocampal slices. Whene examined the membrane binding characteristics of the
wo peptides, it was observed that while both L- and D-A�42nantiomers bound to neurons in culture to a similar extent,nnexin V could only inhibit the binding of L-A�42 and not-A�42 variant. Based on these observations, we propose
hat A� membrane binding occurs via PS and therefore thexternalized PS is functioning as a specific early molecu-ar target for A� interaction ultimately leading to cell deathnd/or inhibition of long term potentiation.
. Materials and methods
.1. Peptide synthesis
Peptide synthesis was performed using solid state Fmochemistry, in a continuous flow synthesizer; the synthe-is of the L-handed (levorotatory) peptide was carriedut using Fmoc-L-Ala-PEG-PS resin (Applied Biosystems,oster City, CA) whilst for D-handed (dextrorotatory) pep-
ide, TentaGel S Trt D-Ala-Fmoc resin (RAPP PolymermbH, Tubingen, Germany) was used. Both the syn-
hesis and deprotection steps were undertaken using 2%,8-diazabicyclo[5,4,0]-undec-7-ene (DBU) in dimethyl-ormamide (DMF) from the C-terminus to Ser 8 andhen changed to 20% piperidine in DMF until the N-erminus. All the DBU steps were performed for 5 minhist the 20% piperidine reactions were extended to 10 min.ll the amino acids were coupled to the resin using3-fold excess where 0.5 M 2-(1H-benzotriazole-1-yl)-
,1,3,3-tetramethyluronium hexafluorophosphate and 0.5 Mf N,N-diisopropylethylamine were used as activators. Eachoupling was performed for 1 h after which the resin wasashed with DMF. Upon synthesis completion, the resin was
welled in DMF and either biotin or 5-carboxyfluorescein
CF) was coupled overnight using the standard couplingethods described previously. The resin was washed withMF and methanol, lyophilized and then the peptide wasleaved from the resin by stirring in a solution of 1% water,

biology
0faaT1tawN
2
hsa2isb1u1sobo2
2
ttt
2(
baerbacpbAatlrb
as(tRsstl(wTo
2s
FatuwEnfqfa8
2
bBorDStwo
2
adags
G.D. Ciccotosto et al. / Neuro
.5% triisopropylsilane in trifluoroacetic acid (TFA) for 3 hollowed by HPLC purification. HPLC was performed usingpreparative C8 Vydac column, the column temperature sett 60 ◦C, Buffer A = 0.1% TFA in water and Buffer B = 0.1%FA 10% isopropanol in acetonitrile and a gradient mix of0–90% B for 30 min was set. The peptide-containing frac-ions were collected, lyophilized and later re-analyzed bynalytical HPLC and MALDI-TOF MS. Unlabeled L-A�42as purchased from W.M. Keck Laboratory (Yale University,ew Haven, CT).
.2. Peptide preparation
Dry peptide was weighed and dissolved in 1,1,1,3,3,3-exafluoro-2-propanol (HFIP). It was then dispensed intomall amounts and dried using a speed-vac and then storedt −80 ◦C until use. Aliquots to be used were dissolved in0% final volume of 20 mM NaOH, vortexed and then son-cated in an ice chilled water bath for 15 min. The peptideolution was diluted in water (70% final volume) followedy the addition of 10× PBS (2.7 mM KCl, 1.5 mM KH2PO4,38 mM NaCl, 8 mM Na2HPO4, at pH 7.4) (10% final vol-me). The solution was then spun in a benchtop centrifuge at6,000 × g for 5 min (to remove amorphous aggregates). Theupernatant was stored on ice until used; typically within 1 hf its preparation. Peptide concentrations were determinedy using the calculated molar extinction coefficient valuesf 75,887 M−1cm for A�42 using the absorbance value of14 nm.
.3. CD spectroscopy
CD spectra were obtained at 37 ◦C using a Jasco 810 spec-ropolarimeter. Far-UV CD spectra were obtained from 190o 260 nm with a 1 mm path length. The baseline acquired inhe absence of peptide was subtracted.
.4. Photo-induced crosslinking of unmodified proteinsPICUP) analysis
PICUP enables the formation of covalent bondsetween closely interacting polypeptide chains withoutny prior modifications and without using spacers (Bitant al., 2001). The PICUP interaction is induced byapid visible light photolysis of a Ru(Bpy), Tris(2,2′-ipyridyl)dichlororuthenium(II), complex in the presence ofn electron acceptor. PICUP thus produces a population ofrosslinked oligomerization states. Analysis of the “frozen”roducts reveals the state of aggregation that was present justefore the crosslinking. To 25 �L of freshly prepared 40 �M� peptide in a microfuge tube, 6.25 �L of 1 mM Ru(Bpy)
nd 6.25 �L of 20 mM ammonium persulfate were added in
he dark room. Samples were gently mixed then exposed to aight source for a predetermined length of time (0.5 s) and theeaction quenched by adding 38.5 �L of 2 times SDS loadinguffer-containing �-mercapto-ethanol. Samples were mixed,iscw
of Aging 32 (2011) 235–248 237
nd run on a 16% tris-tricine gel and bands identified byilver staining procedures using manufacturer’s instructionsBio-Rad). Gels were air dried and scanned using a Perfec-ion 1640 scanner (Epson, Australia) in transmissive mode,GB color, and 600 dot per inch resolution. The scans were
aved in TIFF format and imported into Image J analysisoftware (ver 1.41c, NIH). The color images were convertedo 256 greyscale and the image colors inverted. A rectangu-ar box was used to highlight separate bands in each columnrepresenting each Nmer) and the relative density intensitiesere measured in each box and then background corrected.he data are expressed as a percentage of the sum of theligomeric bands measured for each PICUP experiment.
.5. Electron paramagnetic resonance (EPR)pectroscopy
D-A�42 was freshly prepared by making a 200 �M stock.rom a 10 mM stock, 0.5 molar equivalents CuCl2 wasdded and the samples immediately transferred to quartz EPRubes (Wilmad-Labglass, Buena, NJ) and snap frozen in liq-id nitrogen. Continuous wave EPR spectra were obtainedith a Bruker ESP380E spectrometer equipped with anR4131 VT digital temperature controller with a gaseousitrogen flow and a rectangular TE012 cavity. The microwaverequency was measured with an EIP microwave 548A fre-uency counter. Experimental conditions were: microwaverequency, 9.43 GHz; microwave power, 10 mW; modulationmplitude, 4 G; modulation frequency, 100 kHz; sweep time,4 s; and time constant, 42 ms.
.6. Dityrosine formation
The formation of dityrosine adducts by A� has previouslyeen described (Atwood et al., 2004; Smith et al., 2006).riefly, A� peptide was prepared at 10 �M in the presencef 100 �M ascorbate. After 2 h of incubation at 37 ◦C, theeaction was quenched by the addition of EDTA (250 �M).ityrosine content was quantified using a PerkinElmer Lifeciences LS55 fluorescence spectrophotometer with an exci-
ation wavelength of 320 nm and scanning the emissionavelengths from 350 to 500 nm. Maximum light signal wasbserved and recorded at 418 nm.
.7. Aggregation studies using thioflavin T fluorescence
The use of thioflavin T to monitor the presence of �-sheetmyloidogenic structures of A� peptide has previously beenescribed (Tew et al., 2008). Briefly, A� peptide was preparedt 20 �M in the presence of 40 �M thioflavin T and aggre-ation was promoted by shaking in a Biorad Thermoshakeret at 37 ◦C at 1200 rpm. The thioflavin T fluorescence read-
ngs were measured using a Varian Cary Eclipse fluorescencepectrophotometer with a thermostatically controlled multi-ell cuvette holder, at 37 ◦C. The excitation and emissionavelengths were set at 444 and 480 nm, respectively, with
2 biology
aaw
2
weamoaptrpuwmNtC
2
Aa5fir1ipC5(apttSTi1sMdit
2
s
2tmadpmalibmt(gTrcts
2
ipapfimfiworBAiwwcttccga(iav
38 G.D. Ciccotosto et al. / Neuro
slit width of 5 nm and path length of 10 mm used. Thecquired fluorescence data were normalized using Prism soft-are V4.0.
.8. Transmission electron microscopy
Transmission electron microscope images of A� peptidesere taken using previously published methods (Ciccotosto
t al., 2004). Briefly, A� peptides were prepared at 20 �Mnd aggregation was promoted by shaking in a Biorad Ther-oshaker set at 37 ◦C at 1200 rpm for 24 h. 3.5 �l aliquot
f the aggregated A� peptide samples were absorbed ontocarbon coated Formvar film mounted on 300 mesh cop-
er grids (ProSciTech, Qld, Australia). Prior to adsorption,he grids were rendered hydrophilic by glow discharge in aeduced atmosphere of air for 10 s. After 30 s adsorption, sam-les were blotted and negatively stained with 1.5% aqueousranyl acetate (ProSciTech, Qld, Australia). Investigationsere undertaken using a FEI Company Tecnai G2 TF30 trans-ission electron microscope (FEI Company, Eindhoven, Theetherlands) operated at 200 kV. Images were acquired digi-
ally with a Gatan US1000 2kX2k CCD Camera (Pleasanton,A, USA).
.9. H2O2 assay
2′,7′-Dichlorofluorescin diacetate (DCFDA, Sigma–ldrich) was dissolved in 100% dimethyl sulfoxide (5 mM;
rgon purged for 1 h at 20 ◦C) de-acetylated with 50% (v/v)0 mM NaOH for 30 min then neutralized to pH 7.5 to anal concentration of 1 mM (Ciccotosto et al., 2004). Theeactions were carried out in dPBS (treated with chelex-00 and pH adjusted to 7.5 using concentrated NaH2PO4),n a 96 well plate (250 �L/well) containing freshly pre-ared synthetic peptide (200 nM), copper-glycine (150 nMuCl2 and 900 nM glycine) and ascorbate (0, 1.0, 2.5,.0, 7.5, 10.0 �M) in the presence of de-acylated DCFDA100 �M) and horse-radish peroxidase (0.1 �M) incubatedt 25 ◦C. There was a 4:3 molar excess of peptide to cop-er to ensure that there would be no free copper ions inhe solution. The reaction was carried out at 200 nM A�42o reflect the soluble concentration of A� in the brain.tudies were performed on the day of reagent preparation.he fluorescence signal specific for H2O2 was decreased
n parallel samples co-incubated with catalase (40 units/ml;00 nM). Fluorescence readings were recorded on Varioskanpectrofluorometer (Thermo Electron Corporation, Waltham,A) (excitation 485 nm and emission 535 nm) against a stan-
ard curve of reagent grade H2O2 (Univar, NSW, Australia)n PBS for 200 min. All experiments were performed inriplicate.
.10. Primary neuronal cultures
Mouse cortical neuronal cultures were prepared underterile conditions as described previously (Barnham et al.,
2
(
of Aging 32 (2011) 235–248
003; Ciccotosto et al., 2004) and approved by a local insti-utional ethics committee. Briefly, embryonic day 14 C57Bl6
ouse cortices were removed, dissected free of meninges,nd dissociated in 0.025% (w/v) trypsin in Krebs buffer. Theissociated cells were triturated using a filter-plugged fineipette tip, pelleted, resuspended in plating medium (mini-um Eagle’s medium, 10% fetal calf serum, 5% horse serum)
nd counted. Cortical neuronal cells were plated into poly-d-ysine coated 48 well plates at a density of 150,000 cells/welln plating medium. All cultures were maintained in an incu-ator set at 37 ◦C with 5% CO2. After 2 h the platingedium was replaced with fresh neurobasal medium con-
aining B27 supplements, geneticin, and 0.5 mM glutamineall tissue culture reagents were purchased from Invitro-en (Mount Waverly, Australia) unless otherwise stated).his method resulted in cultures highly enriched for neu-
ons (>95% purity) with minimal astrocyte and microglialontamination as determined by immunostaining of cul-ure preparations using specific marker antibodies (data nothown).
.11. Cell viability and cell death assays
The neuronal cells were allowed to mature for 6 daysn culture before commencing treatment using freshly pre-ared neurobasal medium plus B27 supplements minusntioxidants. For the treatment of neuronal cultures, freshlyrepared soluble A� stock solutions were diluted to thenal concentration (0, 5, 10 or 20 �M) in neurobasaledium. The mixtures were then added to neuronal cells
or up to 4 days in vitro (DIV). Cell survival was mon-tored by phase contrast microscopy, and cell viabilityas quantitated using the MTS assay as described previ-usly (Ciccotosto et al., 2004). Briefly, the medium waseplaced with fresh neurobasal medium supplemented with27 lacking antioxidants, and 10% (v/v) MTS (Promegalexandria, NSW, Australia) was added to each well and
ncubated for 3 h at 37 ◦C in a 5% CO2 incubator. Platesere gently shaken, and a 150 �l aliquot from each wellas transferred to separate wells of a 96 well plate. The
olor change of each well was determined by measuringhe absorbance at 490 nm using a Wallac Victor Mul-ireader, and background readings of MTS incubated inell-free medium were subtracted from each value beforealculations. Cell death was monitored by lactate dehydro-enase (LDH) assay from culture supernatants free of serumnd cell debris using an LDH Cytotoxicity Detection KitRoche Diagnostics, Castle Hill, NSW, Australia) accord-ng to manufacturer’s instructions. The data were normalizednd calculated as a percentage of untreated vehicle controlalues.
.12. Long term potentiation experiments
LTP experiments were performed as previously describedBarnham et al., 2008). Briefly, 14- to 40-day-old C57Bl6

biology
mteiCa2Nwbhtoabfysiwtrs
psdl12RenB3dosl8vS5ptb1t
Iedtmt3
at
2
pwAwshstoflIi
2
2warwait(tcupa4dB8mtgPiitowBA
G.D. Ciccotosto et al. / Neuro
ice were decapitated under anesthesia by halothane inhala-ion. These procedures were approved by local institutionalthics committees. Brains were rapidly removed and chilledn ice cold artificial cerebral spinal fluid (ACSF) with reducedaCl2 (0.75 mM) supplemented with 8 mM MgCl2 and 2 mMscorbic acid. ACSF contained 24 mM NaCl, 2.5 mM KCl,mM MgSO4, 2 mM CaCl2, 10 mM d-glucose, 1.25 mMaH2PO4, 26 mM NaHCO3, and was bubbled vigorouslyith 95% O2/5% CO2 (pH 7.38, HCl/NaHCO3). The fore-rain and cerebellum were removed by transverse cuts andemisected brains were sliced horizontally into 350 �m sec-ions taken between 1.75 and 4.6 mm from the dorsal surfacef the brain using a Vibratome 3000. Slices were incubatedt 34 ◦C for 30 min in ACSF supplemented with 2 mM ascor-ate and then chilled to room temperature (∼24 ◦C) and heldor 1.5–5 h prior to electrophysiological recording. For anal-sis of A�42 inhibiting LTP, brain slices were placed in aingle well of a 24 well plate containing 1 �M A� peptiden 2 mL of ACSF media. The brain slices were pretreatedith A�42 or vehicle for 30 min and then transferred to
he recording chamber. ACSF was circulated through theecording chamber at about 5 mL/min in a 50 mL closedystem.
Field potential recordings were made in the hippocam-us by stimulating and recording orthodromically in thetratum radiatum of the CA1 region with an inter-electrodeistance near 350 �m. The stimulating electrode was a bipo-ar concentric electrode and the recording electrode was.5 mm borosilicate glass (Harvard Apparatus) filled withM NaCl and pulled to a resistance of approximately 5 M�.ecordings were made with a microelectrode amplifier (NPIlectronic GmbH, Tamm, Germany) in bridge-mode, con-ected to a 20× preamplifier with 10 kHz low-pass eight-poleessel filtration (Krohn-Hite Corporation, MD, USA; model381 filter/amplifier). Signals were digitized with a Digi-ata 1322A A/D converter (Axon Instruments) and storedn the hard drive of a personal computer using pClamp 8.2oftware (Axon Instruments, Australia). Slices were stimu-ated at 0.033 Hz with a 0.1 ms square pulse using a Masterstimulator (A.M.P.I. Instruments, Jerusalem, Israel) with a
ariable current stimulus isolation unit (Getting Instruments,an Diego, CA, USA). Voltage–time data were collected at0 kHz. Baseline stimulation intensity was that required toroduce 20–30% of the maximum slope of the field excita-ory post-synaptic potential (fEPSP) and was calibrated at theeginning of each experiment. Tetanic stimulation was a 1 s00 Hz pulse delivered in substitution for the test stimulus athe test intensity.
The data were analyzed offline using pClamp 8.2 (Axonnstruments) and Microsoft Excel software. The slope ofach fEPSP was determined by linear regression of theata points following the pre-synaptic fiber volley with
he fitted data range chosen by visual inspection. Nor-alized fEPSP slopes were expressed as the percent ofhe average slope of the baseline data points between0 and 20 min prior to tetanus. LTP was quantified by
aqiS
of Aging 32 (2011) 235–248 239
veraging the data for each slice from 55 to 60 min post-etanus.
.13. Fluorescence histochemistry
Primary cortical neurons (37,500 cells) were seeded onoly-d-lysine coated 12 mm glass coverslips placed in fourell plates. After 6 DIV, cells were treated with 5 �M CF-L-�42 and CF-D-A�42 peptides for 20 h at 37 ◦C. Cells wereashed with phosphate buffered saline (PBS), then 0.1 M
odium carbonate pH 10.0 and fixed in 4% paraformalde-yde for 20 min, rinsed in PBS and finally mounted onto glasslides using mounting media (PermaFluor, Thermo Elec-ron Corporation, Pittsburgh, PA) and left in the dark to dryvernight. Cells were then photographed using a Biorad con-ocal attached to a Zeiss microscope using a 63× oil objectiveens and laser beam excitation/emission filters of 405/480 nm.dentical exposure settings were used for both peptides andmages were processed using confocal assistant software.
.14. Peptide binding to cells
Isolated mouse primary cortical cells were plated into4 well plates at a seeding density of 600,000 cells perell and allowed to mature for 6 DIV. 5 �M of L-A�42
nd biotin labeled D-A�42 peptides were added to neu-onal cells in culture for 20 h. The cortical neurons in theells were briefly washed in ice cold 0.1 M sodium carbon-
te pH 10.0 (this step removes non-specific peptide bindingn wells) and the cells scraped up from the wells and pro-eins extracted from the cells in 200 �L of extraction buffer20 mM Tris, pH 7.4, 1 mM EDTA, 0.25 M sucrose/0.1% Tri-on X-100). The amount of protein content in each sampleell extract was determined by BCA assay according to man-facturer’s instructions (Pierce, Rockford, IL). Total A�42eptide bound to cell membrane extracts was determined bydding 30 �g of protein sample cell extract per well using a8 well slot blot apparatus. Immunoblots were allowed to airry then re-wet in PBS, boiled for 5 min in PBS and cooled.lots were blocked for 1 h in TBST (10 mM Tris–HCl pH.0, 150 mM NaCl, 0.1% Tween 20) containing 5% skimilk. The immunoblots were then probed for 2 h at room
emperature with an anti-A� monoclonal antibody (WO2, tar-eting A� residues 5–8, diluted 1/50 from culture medium inBS/0.1% Tween 20), then washed 3 times in 10 min intervals
n TBST, then probed with a peroxidase sheep anti-mousemmunoglobulin-diluted 1/5000 in TBST for 1 h at roomemperature and washed several times again. This antibodynly detects the L-A�42 peptide. To detect D-A�42, the blotsere probed with streptavidin-HRP secondary antibody only.lots were developed with ECL reagent (Amersham, Sydney,ustralia) for 1 min as per the manufacturer’s instructions
nd the chemiluminescent signals from immunoblots wereuantitated using a charged-coupled device camera imag-ng system and GeneTool analysis software (GeneGnome,yngene, Cambridge, UK).

2 biology
2
dBp31haP(1rdatwTpauaff1fl1rfcwfpb
2
lc
3
3L
cctoavs
pri
ttDafi(w
iiaAgubde
(AeHaa
bsiBaof(
ucflepirTtatoafi
40 G.D. Ciccotosto et al. / Neuro
.15. Flip-flop fluorescence assay
The flip-flop assays were performed using a previouslyescribed method (Lau et al., 2006) with some modifications.riefly, synthetic 1-palmitoyl-2-oleoyl-sn-glycero-3-hosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero--[phospho-l-serine] (POPS) and fluorescently labeled-oleoyl-2-[6-[(7-nitro-2-1,3-benzoxadiazol-4-yl)amino]-exanoyl]-sn-glycero-3-phospho-l-serine (PS-NBD) werell purchased from Avanti Polar Lipids Inc. (Alabaster, AL).OPC (2.75 mg/2 mL), POPS (2.75 mg/2 mL) and PS-NBD2 mg/mL) were dissolved in chloroform, then mixed in a:1:0.0125 ratio in a small conical flask. The solvent wasemoved under a stream of N2 gas, followed by vacuumrying. To the resultant multi-lamellar film, PBS (pH 7.4)nd glass beads (Sigma, Australia) were added and the flaskransferred to a shaker at 37 ◦C for 1 h. The lipid suspensionas decanted and freeze–thawed in liquid nitrogen 5 times.he vesicles were then passed 13 times through a 100 nmolycarbonate membrane (Whatman) using a mini-extruderpparatus (Avanti Polar Lipids, Alabaster, AL) to yield largenilamellar vesicles (LUVs). This LUV mix was added to500 �L cuvette and placed in a Varian fluorimeter for
urther analysis. The fluorimeter settings used were 475 nmor excitation and 535 nm for emission, 5 nm slit width,min cycle time and set temperature 15 ◦C. Initial maximaluorescence readings then were recorded at 5 min, 50 �L ofM Na2SO4 was added to the cuvettes to quench externally
esiding PS-NBD and readings were allowed to stabilizeor another 20 min. The lipid mix was transferred to a tubeontaining HFIP treated A�42 peptide (100 �g) and mixedell by vortexing for 10 s then transferred back to the cuvette
or further measurements. Following the addition of A�42eptides, the fluorescence intensity data were normalizedetween t = 0 and t = 25 min and reploted.
.16. Statistical analyses
Results are shown as mean ± S.E.M. The data were ana-yzed by two-tailed Student’s t-test. Values of p < 0.05 wereonsidered significant.
. Results
.1. Biophysical properties of D-Aβ42 are the same as-Aβ42
To test if the A� toxicity and cell binding was stereospe-ific we synthesized D- and L-handed versions of A�42. Weompared the biophysical, metal binding and redox proper-ies of the D- and L-A�42 peptides. The secondary structure
f both soluble forms of the L- and D-handed A�42 wasssessed by CD spectroscopy and are in agreement with pre-ious reports (Wiesehan et al., 2003) showing that the CDpectra of D-A�42 is a mirror inverse profile of the L-handedatDs
of Aging 32 (2011) 235–248
eptide (Fig. 1A). Both soluble variants show predominantlyandom coil structures with the maximal rotation at approx-mately 200 nm for both structures.
To determine if there were differences in the aggrega-ion profile at earlier time points we used PICUP to studyhe oligomerization of A� (Bitan et al., 2001); both L- and-A�42 formed oligomers ranging from monomers to hex-
mers as well as an approximately 56 kDa species but nobrillar structures were seen in the loading lanes in the gelsFig. 1B). The abundance of the different oligomeric speciesas similar between the L- and D-A�42 peptides.Recent EXAFS data suggest A� interacts with Cu2+
n distorted six-coordinated (3N3O) geometry in PBS 7.4,ncluding three histidines, glutamic or/and aspartic acid andxial water (Streltsov et al., 2008). The EPR spectrum of D-�42 at pH 7.4 displayed spin Hamiltonian parameters of|| ∼ 2.27 and A|| (63Cu) ∼ 180 × 10−4 cm−1 (Fig. 1C). Notnexpectedly, there are no differences in EPR parametersetween the L- and D-handed A� indicating similar Cu coor-ination spheres (Karr and Szalai, 2007; Kowalik-Jankowskat al., 2003; Syme et al., 2004).
The A�:Cu complex can catalytically generate H2O2Huang et al., 1999) and we tested the ability of L- and D-�42 to produce H2O2 using the DCF assay (Ciccotosto
t al., 2004; Huang et al., 1999). The peptides generated2O2 with similar Km values (4.3 ± 1.0 and 4.2 ± 0.9 �M)
nd Vmax values (19.1 ± 2.2 and 17.5 ± 1.8 nM/min) for L-nd D-A�42 peptides, respectively (n = 3).
Tyrosine 10 is a key residue in the production of ROSy the A�:Cu complex (Barnham et al., 2004) and dityro-ine adducts have been suggested to play an important rolen A� aggregation and neurotoxicity (Atwood et al., 2004;arnham et al., 2004). Dityrosine adducts of A� are formeds a by-product when the peptide is incubated in the presencef Cu2+ and excess reductant such as ascorbate. Both peptideormed such adducts when incubated with Cu and ascorbateFig. 1D).
The aggregation of L- and D-handed A�42 was detectedsing thioflavin T fluorescence assay whereby the fluores-ence level of thioflavin T is known to increase when theuorophore binds to amyloidogenic �-sheet structures (Tewt al., 2008). As expected both the L- and D-handed A�42eptide variants displayed identical aggregation character-stics showing an initial lag phase then rapid aggregationeaching maximal fluorescence at about 120 min (Fig. 1E).he results shown are of a single experiment, representa-
ive of a number of similar aggregation experiments. Theggregation curve observed is diagnostic of amyloid forma-ion, typically involves a lag phase during which nucleationccurs, followed by a period of rapid aggregation (Andreund Timasheff, 1986; Harper and Lansbury, 1997). To con-rm D-handed A�42 forms fibril structures, images of the
ggregated peptides were taken using a transmission elec-ron microscope (Fig. 1F). The images clearly show that the-handed A�42 can form fibrils that are similar in size andhape to the L-handed A�42 peptide.
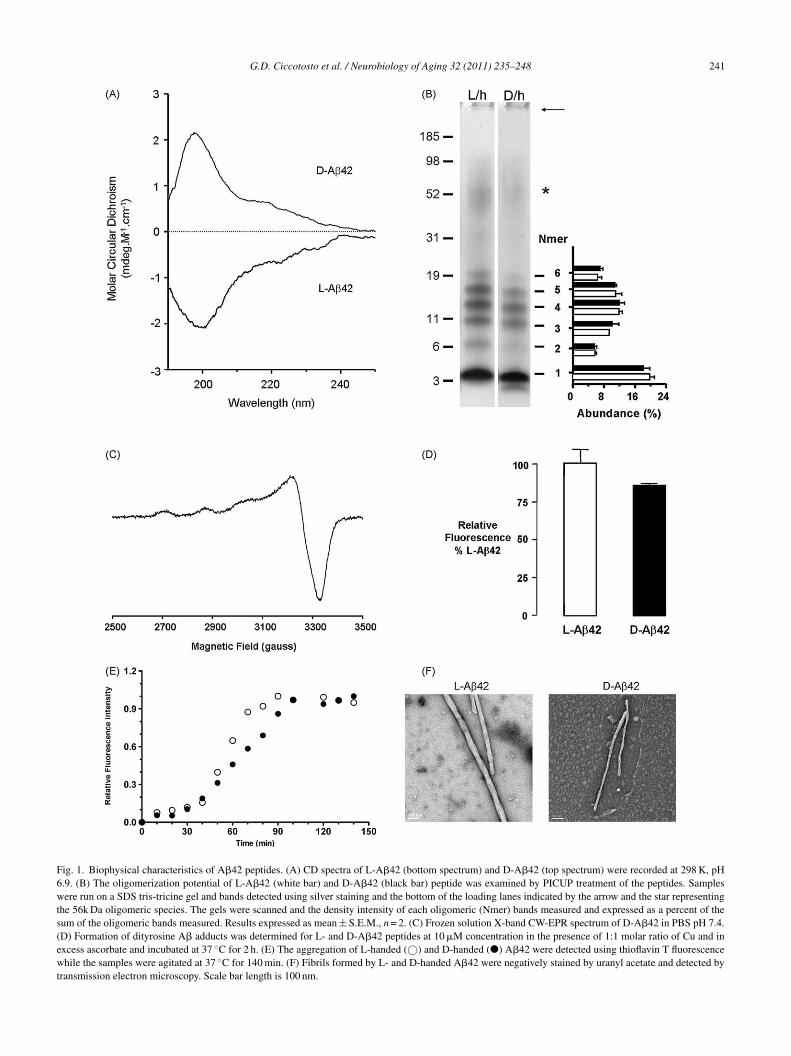
G.D. Ciccotosto et al. / Neurobiology of Aging 32 (2011) 235–248 241
Fig. 1. Biophysical characteristics of A�42 peptides. (A) CD spectra of L-A�42 (bottom spectrum) and D-A�42 (top spectrum) were recorded at 298 K, pH6.9. (B) The oligomerization potential of L-A�42 (white bar) and D-A�42 (black bar) peptide was examined by PICUP treatment of the peptides. Sampleswere run on a SDS tris-tricine gel and bands detected using silver staining and the bottom of the loading lanes indicated by the arrow and the star representingthe 56k Da oligomeric species. The gels were scanned and the density intensity of each oligomeric (Nmer) bands measured and expressed as a percent of thesum of the oligomeric bands measured. Results expressed as mean ± S.E.M., n = 2. (C) Frozen solution X-band CW-EPR spectrum of D-A�42 in PBS pH 7.4.(D) Formation of dityrosine A� adducts was determined for L- and D-A�42 peptides at 10 �M concentration in the presence of 1:1 molar ratio of Cu and inexcess ascorbate and incubated at 37 ◦C for 2 h. (E) The aggregation of L-handed (©) and D-handed (�) A�42 were detected using thioflavin T fluorescencewhile the samples were agitated at 37 ◦C for 140 min. (F) Fibrils formed by L- and D-handed A�42 were negatively stained by uranyl acetate and detected bytransmission electron microscopy. Scale bar length is 100 nm.

242 G.D. Ciccotosto et al. / Neurobiology
Fig. 2. Neuro- and synapto-toxicity of L- and D-A�42. (A) Primary corticalneurons were grown at low density (1.25 × 105 cells/cm2) for 6 days, andthe viability of these cells following peptide treatment was determined bymeasuring the inhibition of MTS reduction. Cortical neurons were treatedwith 5, 10, 20 �M peptide for 96 h in serum free media. (B) Brain slicesfrom C57bl6 mice were treated with 1 �M L-A�42 (©), D-A�42 (�) orvehicle ( ) for 30 min in CSF-like media then the brain slice was allowed toequilibrate for a further 30 min, and LTP induced using an HFS stimulationprotocol (see Section 2) at time t = 0 (arrow) with 0.5 min recordings for60 min taken using a recording electrode to obtain the fEPSP slope ofbaseline. (C) The data points between 50.5 and 60.5 min were averaged foreach treatment group and expressed as the percent above baseline. Resultsare expressed as mean ± S.E.M. Statistical comparisons between vehicle and
3i
trpfc1n
imtaso3htfaapcpoa
3t
iaDttCcdadis(aoca
t#
r
of Aging 32 (2011) 235–248
.2. The D-Aβ42 lacks neurotoxicity and does notnhibit LTP
Having confirmed that the biophysical characteristics ofhe two peptides were alike, we then tested if A� toxicity waseceptor mediated by measuring their neurotoxic activity inrimary mouse cortical cultures. Treating the cortical culturesor 4 days with L-A�42 (0, 5, 10 or 20 �M) induced signifi-ant reduction in cell viability in a dose dependent manner at0 and 20 �M (Fig. 2A). In contrast, D-A�42 displayed noeurotoxicity at any concentration tested (Fig. 2A).
Synaptotoxicity was then assessed by examining the abil-ty of the A� peptides to inhibit LTP in the CA1 region of
ouse hippocampal slices. Brain slices were initially pre-reated for 30 min with 1 �M A� peptide in ACSF mediand then calibrated on the recording apparatus to produce atable baseline at the stimulus strength producing 20–30%f the maximum slope of the fEPSP. After a minimum of0 min of stable baseline recording, LTP was induced byigh-frequency stimulation (HFS). The levels of LTP, relativeo the baseline, were measured as a percentage increase in theEPSP slopes averaged from 50.5 to 60.5 min post-tetanus,nd are presented in Fig. 2B and C. The vehicle control gaven fEPSP of 140 and as expected, the L-A�42 peptide com-letely prevented LTP (Fig. 2C) with an fEPSP of 100. Inontrast, treatment of the hippocampal slices with D-A�42eptide did not affect the levels of LTP, yielding an fEPSPf 140. Therefore, D-A�42 is inactive as an inhibitor of LTPnd as inducer of neurotoxicity.
.3. Cell binding experiments show that D-Aβ42 bindso neurons in culture
A� neurotoxicity is associated with neuronal cell bind-ng (Ciccotosto et al., 2004; Smith et al., 2006; Tickler etl., 2005); therefore, we examined whether the inability of-A�42 to induce neurotoxicity and inhibit LTP was due
o altered cell binding characteristics. Carboxyfluoresceinagged L- and D-A�42 peptides (termed CF-L-A�42 andF-D-A�42, respectively) were added to primary corticalultures and the amount of peptide bound to the cells wasetermined by fluorescence confocal microscopy (Fig. 3And B). To ensure that the CF tag on L-A�42 peptideid not alter A� toxic activity, we treated cortical neuronsn culture with 10 �M CF-L-A�42 for 96 h and found aimilar decrease in cell viability as observed for L-A�4283.9 ± 3.4 and 81.8 ± 5.7% cell viability for CF-L-A�42
nd L-A�42, respectively). Subtoxic concentrations (5 �M)f CF-L-A�42 and CF-D-A�42 were used and the fluores-ence intensity was similar for both L- and D-A�42 peptidesnd both peptides bound to the neuronal cell body, axonsreatment groups using Student’s t-test. *p < 0.05, **p < 0.01 versus vehicle,p < 0.01 versus D-A�42. Cell toxicity assays were done in triplicate andepeated at least 3 times. LTP experiments were repeated 12 times.
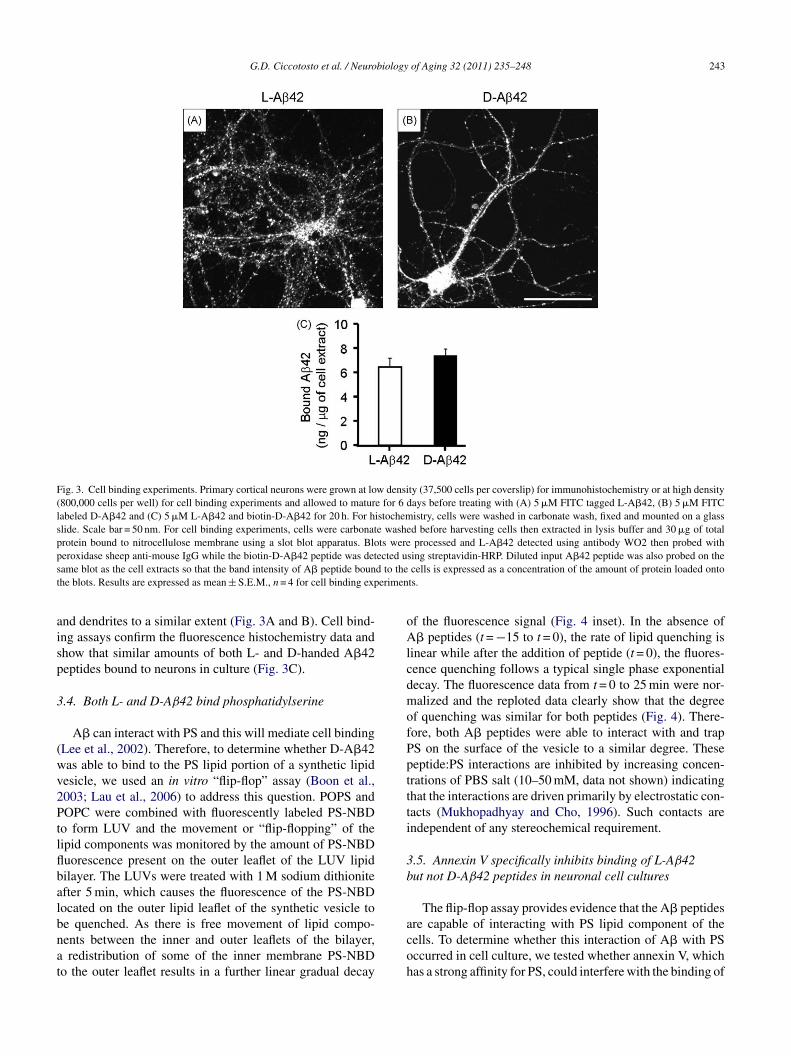
G.D. Ciccotosto et al. / Neurobiology of Aging 32 (2011) 235–248 243
Fig. 3. Cell binding experiments. Primary cortical neurons were grown at low density (37,500 cells per coverslip) for immunohistochemistry or at high density(800,000 cells per well) for cell binding experiments and allowed to mature for 6 days before treating with (A) 5 �M FITC tagged L-A�42, (B) 5 �M FITClabeled D-A�42 and (C) 5 �M L-A�42 and biotin-D-A�42 for 20 h. For histochemistry, cells were washed in carbonate wash, fixed and mounted on a glassslide. Scale bar = 50 nm. For cell binding experiments, cells were carbonate washed before harvesting cells then extracted in lysis buffer and 30 �g of totalprotein bound to nitrocellulose membrane using a slot blot apparatus. Blots were processed and L-A�42 detected using antibody WO2 then probed withp ected us d to thet erimen
aisp
3
(wv2Ptlflbalbnat
oAlcdmofPpttti
3b
eroxidase sheep anti-mouse IgG while the biotin-D-A�42 peptide was detame blot as the cell extracts so that the band intensity of A� peptide bounhe blots. Results are expressed as mean ± S.E.M., n = 4 for cell binding exp
nd dendrites to a similar extent (Fig. 3A and B). Cell bind-ng assays confirm the fluorescence histochemistry data andhow that similar amounts of both L- and D-handed A�42eptides bound to neurons in culture (Fig. 3C).
.4. Both L- and D-Aβ42 bind phosphatidylserine
A� can interact with PS and this will mediate cell bindingLee et al., 2002). Therefore, to determine whether D-A�42as able to bind to the PS lipid portion of a synthetic lipidesicle, we used an in vitro “flip-flop” assay (Boon et al.,003; Lau et al., 2006) to address this question. POPS andOPC were combined with fluorescently labeled PS-NBD
o form LUV and the movement or “flip-flopping” of theipid components was monitored by the amount of PS-NBDuorescence present on the outer leaflet of the LUV lipidilayer. The LUVs were treated with 1 M sodium dithionitefter 5 min, which causes the fluorescence of the PS-NBDocated on the outer lipid leaflet of the synthetic vesicle to
e quenched. As there is free movement of lipid compo-ents between the inner and outer leaflets of the bilayer,redistribution of some of the inner membrane PS-NBDo the outer leaflet results in a further linear gradual decay
acoh
sing streptavidin-HRP. Diluted input A�42 peptide was also probed on thecells is expressed as a concentration of the amount of protein loaded ontots.
f the fluorescence signal (Fig. 4 inset). In the absence of� peptides (t = −15 to t = 0), the rate of lipid quenching is
inear while after the addition of peptide (t = 0), the fluores-ence quenching follows a typical single phase exponentialecay. The fluorescence data from t = 0 to 25 min were nor-alized and the reploted data clearly show that the degree
f quenching was similar for both peptides (Fig. 4). There-ore, both A� peptides were able to interact with and trapS on the surface of the vesicle to a similar degree. Theseeptide:PS interactions are inhibited by increasing concen-rations of PBS salt (10–50 mM, data not shown) indicatinghat the interactions are driven primarily by electrostatic con-acts (Mukhopadhyay and Cho, 1996). Such contacts arendependent of any stereochemical requirement.
.5. Annexin V specifically inhibits binding of L-Aβ42ut not D-Aβ42 peptides in neuronal cell cultures
The flip-flop assay provides evidence that the A� peptides
re capable of interacting with PS lipid component of theells. To determine whether this interaction of A� with PSccurred in cell culture, we tested whether annexin V, whichas a strong affinity for PS, could interfere with the binding of
244 G.D. Ciccotosto et al. / Neurobiology of Aging 32 (2011) 235–248
Fig. 4. Flip-flop of PS lipids. The fluorescence intensity of PS-NBD wasmeasured following the addition of L-handed (�) and D-handed (*) A�42peptides. POPC, POPS, and PS-NBD were mixed to create LUVs. Excesssodium dithionite was added to the LUV resulting in quenching of the PS-NBD head groups located on the outer membrane leaflet. (A) The constantflip-flopping of the lipid head groups results in further linear quenching ofthe PS-NBD fluorescence as a function of time (inset), but addition of A�42(denoted by arrow, t = 0) stabilizes the PS head group to the outer leafletoqot
AwAL
FwmaFtlaptpbtmgV
Fig. 6. Annexin V inhibits A� neurotoxicity. Primary cortical neurons weregrown at low density (125,000 cells/cm2) for 5 days, and the cell death fol-lowing a neurotoxic insult was determined by LDH assay. Cortical neuronswere treated with annexin V in the absence or presence of 10 �M L-A�42 (�)for 72 h in serum free media. Results are normalized and the data expressedas a percentage of the untreated vehicle ( ) controls. Results are expressedama
tsi
f the bilayer, causing a single phase exponential decay of fluorescenceuenching. (B) The raw fluorescence intensity values following the additionf A�42 peptide are normalized and replotted. Results are representative ofwo experiments.
� peptides (Fig. 5). The annexin V protein and A� peptidesere added to the cells at the same time. The amount of� membrane binding in culture was similar between for- and D-handed A�42 using the slot blot assay (Fig. 5) and
ig. 5. Annexin V inhibits cell binding of L-A�42. Primary cortical neuronsere grown at high density (800,000 cells per well) for cell binding experi-ents and allowed to mature for 6 days before treating with 5 �M L-A�42
nd biotin tagged D-A�42 for 20 h alone or in the presence of annexin V.or cell binding experiments, cells were carbonate washed before harvesting
hen extracted in lysis buffer and 30 �g of total protein bound to nitrocellu-ose membrane using a slot blot apparatus. Blots were processed for L-A�42nd detected using monoclonal antibody WO2 (Ciccotosto et al., 2004) thenrobed with peroxidase sheep anti-mouse IgG while the biotin-D-A�42 pep-ide was detected using streptavidin-HRP directly. Diluted input for A�
eptides were also probed on the same blot as the cell extracts so that theand intensity of A� peptides bound to the cells is expressed as a concentra-ion of the amount of protein loaded onto the blots. Results are expressed as
ean ± S.E.M., n = 4. Statistical comparisons between annexin V treatmentroups was analyzed using Student’s t-test, **p < 0.01 versus 0 nM annexin.
atanA
3A
wVats(braTso
4
abtt
s mean ± S.E.M., n = 7. Statistical comparisons between annexin V treat-ent groups were analyzed using Student’s t-test, *p < 0.05 versus 0 nM
nnexin V.
hese results are in agreement with CF-A�42 labeled peptideshown in Fig. 3A and B. Cell binding experiments were set upn the presence of 10 nM annexin V. The results showed thatnnexin V significantly inhibited the binding of L-A�42 tohe cortical neurons in culture (p < 0.001) (Fig. 5). In contrast,nnexin V did not block the binding of the D-A�42 to theeurons in culture indicating that, unlike L-A�42, the D-�42 peptide did not bind to PS in the neurons.
.6. Annexin V specifically inhibits the neurotoxicity ofβ peptides in neuronal cell cultures
To determine whether this interaction of L-A�42 with PSas necessary for neurotoxicity, we tested whether annexinprotein, could inhibit the neurotoxicity of L-A�42. The
nnexin V protein and A� peptides were added to the cells athe same time. The addition of annexin V peptide at 10 nMignificantly inhibited the toxicity of L-A�42 (p < 0.05)Fig. 6). The annexin V peptide specifically targeted andound to PS and not A� since we found that pretreating neu-ons for 30 min with annexin V before adding A� peptidelso resulted in the inhibition of toxicity (data not shown).he inhibitory effects of annexin V for L-A�42 toxicity werepecific since annexin V alone had no effect on the cell deathf untreated neurons.
. Discussion
The mechanism of A� toxicity remains unresolved and
fundamental question is whether A� toxicity is mediatedy interactions with a protein receptor. Potential recep-ors on the plasma membrane of glia and neurons includehe alpha-7 nicotinic acetylcholine receptor, receptor for

biology
aprl(2mtedtRmsafpstpocbd
tecrTospDpAfiimse2ha(aat
ae2oPcb
thoootU5ctibrtuwtrwwottPLirtirctpiaaa
basPibtttAimaL
G.D. Ciccotosto et al. / Neuro
dvanced glycosylation end-products, amyloid precursorrotein, the N-methyl-d-aspartate receptor, the P75 neu-otrophin receptor, the scavenger receptors, CD36 andow-density lipoprotein receptor-related protein membersfor detailed review on this area see (Cappai and Barnham,007a; Verdier et al., 2004). These proteins can bindonomeric A�, non-fibrillar or fibrillar A�. Other fac-
ors implicating A� toxicity include membrane perturbationither through the formation of pore-like structures or throughetergent like effects (Arispe, 2004; Demuro et al., 2005), andhe interaction of A� with redox active metals to generateOS (Bush, 2003). To ascertain whether these non-receptorediated effects were sufficient for A� toxicity we synthe-
ized D-handed A�42 peptide and investigated its neurotoxicctivity. Stereoisomers, the left (L) and right (D) handedorms of amino acids, have the same exact chemical com-osition and are mirror images of each other and have theame physical and chemical characteristics but are struc-urally (stereochemically) different. As a result, it would beredicted that the physico-chemical characteristic propertiesf the A� peptides should occur independently of peptidehirality and that the toxic activity of L- and D-A� coulde correlated to either a sterospecific or non-stereospecificependent phenomena.
The D-A�42 peptide was not toxic and hence unableo reduce cell viability or inhibit LTP, two important prop-rties associated with neuro/synaptotoxicity of L-A�42. Inontrast, Cribbs et al. (1997) showed that D-A�42 was neu-otoxic in their cell culture model (Cribbs et al., 1997).he reasons for the discrepancy may be due to a numberf different experimental parameters used between studiesuch as cell type used (mouse cortical versus rat hippocam-al), culture methods (6 DIV and 96 h treatment versus 2IV and 24 h treatment), culture conditions (NB/B27 sup-lements versus DMEM/N2 supplements), preparation of� peptide (soluble oligomeric species versus aggregatedbrillar structures) and method for measuring cell viabil-
ty (MTS assay versus trypan blue exclusion assay). Ourodel for measuring A� neurotoxicity has provided con-
istent and reproducible results over many years (Barnhamt al., 2003, 2004; Ciccotosto et al., 2004; Smith et al.,006, 2007; Tickler et al., 2005). Therefore, while D-A�42as a number of the biophysical characteristics commonlyssociated with A� toxicity, such as peptide aggregationinto oligomers or fibrils), Cu coordination, and the gener-tion of ROS, these specific characteristics of this peptidere not sufficient for the toxicity associated with this pep-ide.
We and others have shown that A� neurotoxicity is medi-ted via interactions with the plasma membrane (Ciccotostot al., 2004; Kourie, 2001; Lau et al., 2006; Muller et al.,001; Simakova and Arispe, 2007; Wang et al., 2001). A loss
f lipid asymmetry was observed in AD tissue where PC andE levels are reduced (Nitsch and Frotscher, 1992) and theholesterol to phospholipid ratio decreased by 30% in ADrains (Mason et al., 1992). Castegna et al. (2004) reported(b1V
of Aging 32 (2011) 235–248 245
hat two highly reactive products of lipid peroxidation, 4-ydroxynonena and acrolein, both elevated in AD brain, mayxidatively modify the flippase enzyme resulting in the lossf phospholipid asymmetry and subsequent externalizationf PS to the outer cell membrane, an effect that would con-ribute to the neuronal cell loss in AD (Castegna et al., 2004).sing radiolabeled annexin V, brain images from 4 out ofAD patients showed multifocal cortical annexin V uptakes
ompared to the control group (Lampl et al., 2006) suggestinghat there is increased surface expression of PS on neuronsn AD patients. Therefore, it was interesting to observe thatoth L- and D-A�42 could bind to the cell surface of neu-onal cells in culture (Fig. 3). PS has been implicated in A�oxicity (Lee et al., 2002; Simakova and Arispe, 2007) andsing an in vitro assay we showed that both L- and D-A�42ere able to interact with PS to a similar degree via elec-
rostatic interactions (Fig. 4). Simakova and Arispe (2007),ecently reported that the cell-selective neurotoxicity of A�as associated with cell membrane A� binding, an effecthich was correlated with increased cell membrane exposuref PS (Simakova and Arispe, 2007). We now present evidencehat membrane exposed PS is a molecular target for A� sincehe addition of annexin V, which is a high affinity ligand forS, to our cortical cultures caused significant inhibition of-A�42 binding (Fig. 5) and significant inhibition of toxic-
ty (Fig. 6). In addition, using SELDI-TOF MS, we recentlyeported that the annexin V specifically inhibited dimer andrimer oligomeric species and but not monomeric A� fromnteracting with PS membranes (Hung et al., 2008). Theseesults are consistent with previous reports that annexin V atoncentrations greater than 30 nM could block A�40 cyto-oxicity in PC12 cells (Lee et al., 2002). In contrast, a recentaper showed that annexin V did not affect the surface bind-ng of A�42 to PC12 cells by flow cytometry (Bateman etl., 2007), but in that case the cells were treated with 2.7 nMnnexin V, at which concentration it had no affect in ourssays either.
An important finding from the present study is that theinding of D-A�42 to the cell surface was not inhibited bynnexin V (Fig. 5) in contrast to our in vitro data whichhowed that both L- and D-A�42 are able to interact withS with similar affinities. The inability of annexin V to
nhibit D-A�42 binding to cells suggests that D-A�42 isinding fortuitously via another higher affinity ligand onhe cell surface. Since D-A�42 is not a native molecule,his interaction is not physiologically relevant. An alterna-ive explanation for the difference in effect of annexin V on� binding to neurons may be explained if annexin V directly
nteracted with L-handed A�42 peptide. Using surface plas-on resonance technology, which enables rapid and sensitive
nalysis of protein/protein interactions, we found that neither-handed nor D-handed A�42 peptide bound to annexin V
Supplementary Fig. 1). Although A� has been shown toind to membrane bound proteins such as RAGE (Yan et al.,996), p75NTR (Yaar et al., 2002), and others (reviewed byerdier and Penke, 2004) we would speculate that annexin

246 G.D. Ciccotosto et al. / Neurobiology
Table 1Summary of properties of L- and D-handed A� peptides in various assays.
L-handed A�42 D-handed A�42
Biophysical and biochemical propertiesSecondary structure Predominantly
random coiledPredominantlyrandom coiled
Chiral ChiralFibril formation Yes YesOligomer formation Yes YesBinding to Cu2+ Yes YesBinding to synthetic lipids Yes YesFlip-flop of PS Yes YesGeneration of H2O2 Yes YesDityrosine adduct formation Yes Yes
Biological propertiesNeurotoxicity Yes NoInhibition of LTP Yes NoCell binding Yes YesAnnexin V effect on toxicity Blocks toxicity Not determined as
D-hand is nottoxic
Annexin V blocks binding Yes No
Vntfndna
iftssnr(2aWmimwAap
C
i
A
RRtMf
A
f2
R
A
A
A
A
B
B
B
B
B
B
B
Annexin V binds to A� No No
blocks the binding of A� to neurons by a similar mecha-ism proposed by Andree et al. (1992) where they showedhat annexin V forms ordered clusters on the membrane sur-ace and completely inhibited prothrombinase binding butot other procoagulants (Andree et al., 1992). Therefore, thisata strongly support the model that cell binding, per se, isot sufficient for A� toxicity and that it must be mediated viaspecific interaction with PS.
In summary, the present data indicate that the biophys-cal and biochemical properties that have been describedor A� and summarized in Table 1 are insufficient to behe sole determinant responsible for A� neurotoxic andynaptotoxic activity because all of these parameters wereimilar for the D- and L-A�42 peptides, yet D-A�42 wason-toxic. A body of data indicate that A� mediated neu-otoxicity requires it to bind to neuronal cell membranesBarnham et al., 2004; Ciccotosto et al., 2004; Gong et al.,003; Lambert et al., 1998; Shankar et al., 2007; Simakovand Arispe, 2007; Smith et al., 2006; Tickler et al., 2005).
hile others have highlighted that the presence of PS on theembrane surface correlated well with increased A� bind-
ng (Simakova and Arispe, 2007), we now show that A�embrane binding is mediated through a direct interactionith PS and that is a pivotal event on the critical pathway to� toxicity. This raises the possibility that strategies aimed
t inhibiting A�/PS interactions would have therapeuticotential.
onflict of interest statement
The authors declare that there are no conflicts of interestn associated with this paper.
B
C
of Aging 32 (2011) 235–248
cknowledgements
This work was funded by National Health and Medicalesearch Council of Australia (NHMRC) and the Australianesearch Council (ARC). The authors acknowledge the Elec-
ron Microscopy Unit of Bio21 Institute, The University ofelbourne, for assistance with the electron microscopy per-
ormed in the course of this research.
ppendix A. Supplementary data
Supplementary data associated with this article can beound, in the online version, at doi:10.1016/j.neurobiolaging.009.02.018.
eferences
ndree, H.A., Stuart, M.C., Hermens, W.T., Reutelingsperger, C.P., Hemker,H.C., Frederik, P.M., Willems, G.M., 1992. Clustering of lipid-boundannexin V may explain its anticoagulant effect. J. Biol. Chem. 267,17907–17912.
ndreu, J.M., Timasheff, S.N., 1986. The measurement of cooperative pro-tein self-assembly by turbidity and other techniques. Methods Enzymol.130, 47–59.
rispe, N., 2004. Architecture of the Alzheimer’s A�P ion channel pore. J.Membr. Biol. 197, 33–48.
twood, C.S., Perry, G., Zeng, H., Kato, Y., Jones, W.D., Ling, K.Q., Huang,X., Moir, R.D., Wang, D., Sayre, L.M., Smith, M.A., Chen, S.G., Bush,A.I., 2004. Copper mediates dityrosine cross-linking of Alzheimer’samyloid-�. Biochemistry 43, 560–568.
arnham, K.J., Ciccotosto, G.D., Tickler, A.K., Ali, F.E., Smith, D.G.,Williamson, N.A., Lam, Y.H., Carrington, D., Tew, D., Kocak, G., Voli-takis, I., Separovic, F., Barrow, C.J., Wade, J.D., Masters, C.L., Cherny,R.A., Curtain, C.C., Bush, A.I., Cappai, R., 2003. Neurotoxic, redox-competent Alzheimer’s beta-amyloid is released from lipid membraneby methionine oxidation. J. Biol. Chem. 278, 42959–42965.
arnham, K.J., Haeffner, F., Ciccotosto, G.D., Curtain, C.C., Tew, D.,Mavros, C., Beyreuther, K., Carrington, D., Masters, C.L., Cherny, R.A.,Cappai, R., Bush, A.I., 2004. Tyrosine gated electron transfer is key tothe toxic mechanism of Alzheimer’s disease beta-amyloid. FASEB J. 18,1427–1429.
arnham, K.J., Kenche, V.B., Ciccotosto, G.D., Smith, D.P., Tew, D.J.,Liu, X., Perez, K., Cranston, G.A., Johanssen, T.J., Volitakis, I., Bush,A.I., Masters, C.L., White, A.R., Smith, J.P., Cherny, R.A., Cappai, R.,2008. Platinum-based inhibitors of amyloid-beta as therapeutic agentsfor Alzheimer’s disease. Proc. Natl. Acad. Sci. U.S.A. 105, 6813–6818.
ateman, D.A., McLaurin, J., Chakrabartty, A., 2007. Requirement ofaggregation propensity of Alzheimer amyloid peptides for neuronal cellsurface binding. BMC Neurosci. 8, 29.
itan, G., Lomakin, A., Teplow, D.B., 2001. Amyloid �-protein oligomeriza-tion: prenucleation interactions revealed by photo-induced cross-linkingof unmodified proteins. J. Biol. Chem. 276, 35176–35184.
oon, J.M., Lambert, T.N., Sisson, A.L., Davis, A.P., Smith, B.D., 2003.Facilitated phosphatidylserine (PS) flip-flop and thrombin activationusing a synthetic PS scramblase. J. Am. Chem. Soc. 125, 8195–8201.
retscher, M.S., 1972. Asymmetrical lipid bilayer structure for biologicalmembranes. Nat. New Biol. 236, 11–12.
ush, A.I., 2003. The metallobiology of Alzheimer’s disease. Trends Neu-rosci. 26, 207–214.
appai, R., Barnham, K.J., 2007a. Delineating the Mechanism ofAlzheimer’s disease A� peptide neurotoxicity. Neurochem. Res. 33,526–532.

biology
C
C
C
C
D
D
G
H
H
H
K
K
K
K
L
L
L
L
M
M
M
M
N
P
S
S
S
S
S
S
T
T
G.D. Ciccotosto et al. / Neuro
appai, R., Barnham, K.J., 2007b. Molecular determinants of Alzheimer’sdisease A� peptide neurotoxicity. Future Neurol. 4, 397–409.
astegna, A., Lauderback, C.M., Mohmmad-Abdul, H., Butterfield, D.A.,2004. Modulation of phospholipid asymmetry in synaptosomal mem-branes by the lipid peroxidation products, 4-hydroxynonenal andacrolein: implications for Alzheimer’s disease. Brain Res. 1004,193–197.
iccotosto, G.D., Tew, D., Curtain, C.C., Smith, D., Carrington, D., Mas-ters, C.L., Bush, A.I., Cherny, R.A., Cappai, R., Barnham, K.J., 2004.Enhanced toxicity and cellular binding of a modified amyloid betapeptide with a methionine to valine substitution. J. Biol. Chem. 279,42528–42534.
ribbs, D.H., Pike, C.J., Weinstein, S.L., Velazquez, P., Cotman, C.W., 1997.All-D-enantiomers of beta-amyloid exhibit similar biological propertiesto all-L-beta-amyloids. J. Biol. Chem. 272, 7431–7436.
aleke, D.L., 2003. Regulation of transbilayer plasma membrane phospho-lipid asymmetry. J. Lipid Res. 44, 233–242.
emuro, A., Mina, E., Kayed, R., Milton, S.C., Parker, I., Glabe, C.G.,2005. Calcium dysregulation and membrane disruption as a ubiquitousneurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 280,17294–17300.
ong, Y., Chang, L., Viola, K.L., Lacor, P.N., Lambert, M.P., Finch, C.E.,Krafft, G.A., Klein, W.L., 2003. Alzheimer’s disease-affected brain:presence of oligomeric A� ligands (ADDLs) suggests a molecularbasis for reversible memory loss. Proc. Natl. Acad. Sci. U.S.A. 100,10417–10422.
arper, J.D., Lansbury Jr., P.T., 1997. Models of amyloid seeding inAlzheimer’s disease and scrapie: mechanistic truths and physiologi-cal consequences of the time-dependent solubility of amyloid proteins.Annu. Rev. Biochem. 66, 385–407.
uang, X., Cuajungco, M.P., Atwood, C.S., Hartshorn, M.A., Tyndall, J.D.,Hanson, G.R., Stokes, K.C., Leopold, M., Multhaup, G., Goldstein, L.E.,Scarpa, R.C., Saunders, A.J., Lim, J., Moir, R.D., Glabe, C., Bowden,E.F., Masters, C.L., Fairlie, D.P., Tanzi, R.E., Bush, A.I., 1999. Cu(II)potentiation of Alzheimer A� neurotoxicity: correlation with cell-freehydrogen peroxide production and metal reduction. J. Biol. Chem. 274,37111–37116.
ung, L.W., Ciccotosto, G.D., Giannakis, E., Tew, D.J., Perez, K., Masters,C.L., Cappai, R., Wade, J.D., Barnham, K.J., 2008. Amyloid-beta pep-tide (A�) neurotoxicity is modulated by the rate of peptide aggregation:Abeta dimers and trimers correlate with neurotoxicity. J. Neurosci. 28,11950–11958.
ang, J., Lemaire, H., Unterbeck, A., Salbaum, J.M., Masters, C.L.,Grzeschik, K., Multhaup, G., Beyreuther, K., Müller-Hill, B., 1987.The precursor of Alzheimer’s disease amyloid A4 protein resemblesa cell-surface receptor. Nature 325, 733–736.
arr, J.W., Szalai, V.A., 2007. Role of aspartate-1 in Cu(II) binding to theamyloid-beta peptide of Alzheimer’s disease. J. Am. Chem. Soc. 129,3796–3797.
ourie, J.I., 2001. Mechanisms of amyloid beta protein-induced modi-fication in ion transport systems: implications for neurodegenerativediseases. Cell. Mol. Neurobiol. 21, 173–213.
owalik-Jankowska, T., Ruta, M., Wisniewska, K., Lankiewicz, L., 2003.Coordination abilities of the 1–16 and 1–28 fragments of beta-amyloidpeptide towards copper(II) ions: a combined potentiometric and spec-troscopic study. J. Inorg. Biochem. 95, 270–282.
ambert, M.P., Barlow, A.K., Chromy, B.A., Edwards, C., Freed, R.,Liosatos, M., Morgan, T.E., Rozovsky, I., Trommer, B., Viola, K.L.,Wals, P., Zhang, C., Finch, C.E., Krafft, G.A., Klein, W.L., 1998.Diffusible, nonfibrillar ligands derived from A�1–42 are potent cen-tral nervous system neurotoxins. Proc. Natl. Acad. Sci. U.S.A. 95,6448–6453.
ampl, Y., Lorberboym, M., Blankenberg, F.G., Sadeh, M., Gilad, R., 2006.Annexin V SPECT imaging of phosphatidylserine expression in patientswith dementia. Neurology 66, 1253–1254.
au, T.L., Ambroggio, E.E., Tew, D.J., Cappai, R., Masters, C.L., Fidelio,G.D., Barnham, K.J., Separovic, F., 2006. Amyloid-beta peptide disrup-
V
of Aging 32 (2011) 235–248 247
tion of lipid membranes and the effect of metal ions. J. Mol. Biol. 356,759–770.
ee, G., Pollard, H.B., Arispe, N., 2002. Annexin 5 and apolipoprotein E2protect against Alzheimer’s amyloid-�-peptide cytotoxicity by competi-tive inhibition at a common phosphatidylserine interaction site. Peptides23, 1249–1263.
ason, R.P., Shoemaker, W.J., Shajenko, L., Chambers, T.E., Herbette,L.G., 1992. Evidence for changes in the Alzheimer’s disease brain cor-tical membrane structure mediated by cholesterol. Neurobiol. Aging 13,413–419.
asters, C.L., Multhaup, G., Simms, G., Pottgiesser, J., Martins, R.N.,Beyreuther, K., 1985. Neuronal origin of a cerebral amyloid: neu-rofibrillary tangles of Alzheimer’s disease contain the same proteinas the amyloid of plaque cores and blood vessels. EMBO J. 4,2757–2763.
ukhopadhyay, S., Cho, W., 1996. Interactions of annexin V with phospho-lipid monolayers. Biochim. Biophys. Acta 1279, 58–62.
uller, W.E., Kirsch, C., Eckert, G.P., 2001. Membrane-disordering effectsof beta-amyloid peptides. Biochem. Soc. Trans. 29, 617–623.
itsch, R., Frotscher, M., 1992. Reduction of posttraumatic transneuronal“early gene” activation and dendritic atrophy by the N-methyl-d-aspartate receptor antagonist MK-801. Proc. Natl. Acad. Sci. U.S.A.89, 5197–5200.
omorski, T., Holthuis, J.C.M., Herrmann, A., van Meer, G., 2004. Trackingdown lipid flippases and their biological functions. J. Cell. Sci. 117,805–813.
hankar, G.M., Bloodgood, B.L., Townsend, M., Walsh, D.M., Selkoe, D.J.,Sabatini, B.L., 2007. Natural oligomers of the Alzheimer amyloid-betaprotein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27,2866–2875.
imakova, O., Arispe, N.J., 2007. The cell-selective neurotoxicity of theAlzheimer’s A� peptide is determined by surface phosphatidylserine andcytosolic ATP levels. Membrane binding is required for Abeta toxicity.J. Neurosci. 27, 13719–13729.
mith, D.P., Ciccotosto, G.D., Tew, D.J., Fodero-Tavoletti, M.T., Johanssen,T., Masters, C.L., Barnham, K.J., Cappai, R., 2007. Concentra-tion dependent Cu(2+) induced aggregation and dityrosine formationof the Alzheimer’s disease amyloid-beta peptide. Biochemistry 46,2881–2891.
mith, D.P., Smith, D.G., Curtain, C.C., Boas, J.F., Pilbrow, J.R., Ciccotosto,G.D., Lau, T.L., Tew, D.J., Perez, K., Wade, J.D., Bush, A.I., Drew, S.C.,Separovic, F., Masters, C.L., Cappai, R., Barnham, K.J., 2006. Copper-mediated amyloid-beta toxicity is associated with an intermolecularhistidine bridge. J. Biol. Chem. 281, 15145–15154.
treltsov, V.A., Titmuss, S.J., Epa, V.C., Barnham, K.J., Masters, C.L.,Varghese, J.N., 2008. The structure of the amyloid {beta}-peptide highaffinity copper II binding site in Alzheimer’s disease. Biophys. J. 95,3447–3456.
yme, C.D., Nadal, R.C., Rigby, S.E., Viles, J.H., 2004. Copper binding tothe amyloid-beta (A�) peptide associated with Alzheimer’s disease: fold-ing, coordination geometry, pH dependence, stoichiometry, and affinityof A�-(1–28): insights from a range of complementary spectroscopictechniques. J. Biol. Chem. 279, 18169–18177.
ew, D.J., Bottomley, S.P., Smith, D.P., Ciccotosto, G.D., Babon, J., Hinds,M.G., Masters, C.L., Cappai, R., Barnham, K.J., 2008. Stabilizationof neurotoxic soluble beta-sheet-rich conformations of the Alzheimer’sdisease amyloid-beta peptide. Biophys. J. 94, 2752–2766.
ickler, A.K., Smith, D.G., Ciccotosto, G.D., Tew, D.J., Curtain, C.C., Car-rington, D., Masters, C.L., Bush, A.I., Cherny, R.A., Cappai, R., Wade,J.D., Barnham, K.J., 2005. Methylation of the imidazole side chains ofthe Alzheimer disease amyloid-beta peptide results in abolition of super-
oxide dismutase-like structures and inhibition of neurotoxicity. J. Biol.Chem. 280, 13355–13363.erdier, Y., Penke, B., 2004. Binding sites of amyloid beta-peptide in cellplasma membrane and implications for Alzheimer’s disease. Curr. Pro-tein Peptide Sci. 5, 19–31.

2 biology
V
W
W
W
Y
48 G.D. Ciccotosto et al. / Neuro
erdier, Y., Zarandi, M., Penke, B., 2004. Amyloid beta-peptide interac-tions with neuronal and glial cell plasma membrane: binding sites andimplications for Alzheimer’s disease. J. Peptide Sci. 10, 229–248.
ang, S.S., Rymer, D.L., Good, T.A., 2001. Reduction in cholesterol andsialic acid content protects cells from the toxic effects of beta-amyloidpeptides. J. Biol. Chem. 276, 42027–42034.
hite, A.R., Guirguis, R., Brazier, M.W., Jobling, M.F., Hill, A.F.,
Beyreuther, K., Barrow, C.J., Masters, C.L., Collins, S.J., Cappai, R.,2001. Sublethal concentrations of prion peptide PrP106-126 or theamyloid beta peptide of Alzheimer’s disease activates expression ofproapoptotic markers in primary cortical neurons. Neurobiol. Dis. 8,299–316.Y
of Aging 32 (2011) 235–248
iesehan, K., Buder, K., Linke, R.P., Patt, S., Stoldt, M., Unger, E., Schmitt,B., Bucci, E., Willbold, D., 2003. Selection of D-amino-acid peptides thatbind to Alzheimer’s disease amyloid peptide A�1-42 by mirror imagephage display. Chembiochem. 4, 748–753.
aar, M., Zhai, S., Fine, R.E., Eisenhauer, P.B., Arble, B.L., Stewart, K.B.,Gilchrest, B.A., 2002. Amyloid � binds trimers as well as monomersof the 75-kDa neurotrophin receptor and activates receptor signaling. J.
Biol. Chem. 277, 7720–7725.an, S.D., Chen, X., Fu, J., Chen, M., Zhu, H., Roher, A., Slattery, T.,Zhao, L., Nagashima, M., Morser, J., Migheli, A., Nawroth, P., Stern, D.,Schmidt, A.M., 1996. RAGE and amyloid-beta peptide neurotoxicity inAlzheimer’s disease. Nature 382, 685–691.