
Modernization of Golgi staining techniques for high ... - Nature

THE JOURNAL OF COMPARATIVE NEUROLOGY 352~92-105 (1995)
Localization of Amyloid P Component in Human Brain: Vascular Staining Patterns
and Association With Alzheimer’s Disease Lesions
LYNN s. PERLMUTTER, ERNESTO BARRON, MARTHA MYERS, DAVID SAPERIA, AND HELENA CHANG CHUI
Department of Neurology, University of Southern California School of Medicine, Los Angeles, California 90033 (L.S.P., E.B., M.M.); Alzheimer’s Disease Diagnostic and Treatment Center,
Rancho Los Amigos Medical Center, Downey, California 90242 (D.S., H.C.C.)
ABSTRACT Amyloid P component is a normal serum protein that is highly conserved across phylogeny.
Although it resembles the classic acute-phase reactant C-reactive protein, and is considered to be a normal extracellular matrix component, its physiologic role in humans is unknown. Amyloid P component is also colocalized with accumulations of all recognized forms of amyloid. The present study uses light and electron microscopy to compare the cerebral localization of amyloid P component in cases with (n = 19) and without (n = 15) Alzheimer’s disease (AD). In non-AD cases, amyloid P component was predominantly localized to the cerebrovasculature. Perivascular staining was observed in most cases, more so in the white than in the gray matter. In AD cases, amyloid P component was localized to all three characteristic histopathologic lesions, namely, neurofibrillary tangles, senile plaques, and amyloid angiopathy. Furthermore, in cases with prominent staining of gray matter parenchymal lesions, intravascular staining was decreased. Given the fixation and processing methods used, amyloid P component was never seen to be localized to the cerebrovascular basement membrane. These data argue against amyloid P component’s postulated role as the anchor for vascular p-amyloid deposition. Because there is no evidence for intrinsic amyloid P component production in brain, its perivascular and parenchymal distributions suggest either compromise of the blood-brain barrier or transport across vascular endothelium. o 199.5 Wiley-Liss, Inc.
Indexing terms: blood-brain barrier, microglia, neurofibrillary tangles, senile plaques, vascular basement membrane
Amyloid P component is a normal serum protein synthe- sized in the liver but apparently not in the brain (Kalaria et al., 1991b). I t has been localized to elastic microfibrils (Breathnach et al., 1981b), glomerular basement mem- branes (Dyck et al., 1980a,b; Breathnach et al., 1981a), and all forms of amyloid (Pepys et al., 1979b; Castafio and Frangione, 1988). Its physiological role in humans is as yet unknown. Amyloid P component is highly conserved across phylogeny, from invertebrates to primates (Baltz et al., 1982). It resembles the classic acute-phase reactant C- reactive protein, in both configuration and amino acid sequence (Pepys et al., 1978). Whereas amyloid P compo- nent is an acute-phase reactant in mice (Pepys et al., 1979a), in humans it is not significantly elevated in the serum during acute inflammation (Pepys et al., 1978).
Amyloid P component binds in a calcium-mediated manner to several ligands, including chondroitin, dermatan, and heparan sulfates (Hamazaki, 1987; Schwalbe et al., 1991); fibronectin (Wright et al., 1983; Rostagno et al., 1986); complement C4-binding protein (Schwalbe et al., 1990; Kalaria and Kroon, 1992); and amyloid fibrils (Pepys et al., 197913). In addition, it binds to macrophages in a receptor- mediated manner (Siripont et al., 1988), where it increases bactericidal activity (Siripont et al., 19881, activates C3b and C3bi receptors to promote phagocytosis (Wright et al.,
Accepted July 5, 1994. Address reprint requests to Lynn S. Perlmutter at her present address,
Miles Inc. Pharmaceuticals Division, Institute for Dementia Research, 400 Morgan Lane, West Haven, CT 06516.
O 1995 WILEY-LISS, INC.

AMYLOID P COMPONENT IN HUMAN BRAIN 93
TABLE 1. Patient Characteristics and Amyloid P Component Immunoreactive Vascular Staining Patterns’ ~
White matter vasculature Grey matter vasculature
Case no. DX Age PM hrs Sex Intravasc Perivasc Intravasc Perivasc 1 2 3 4 5 6 7 8
10 11 12 13 14 15 16 17 18 19
Mean SD
1 2 3 4 5 6 7 8 9
10 11 12 13 14 15 Mean SD
AD AD AD AD AD AD AD AD AD AD AD AD AD AD AD AD AD AD AD
MID MID PSP PSP PSP Tangle Pick‘s ALS OBS OBS CBD Heart disease Heart disease Cancer Gunshot
60 77 76 88 63 82 64 84 78 76 76 75 83 88 68 78 64 75 72
75.1 8.3
73 83 70 78 77 85 47 85 70 90 60 41 83 82 39 70.9 16.6
4 5 5 5 5 4 4 6 7 4 5 4 5 5 7 6 9 5 3.5
5.2 1.3
4 6 8 4 3 7 4 4 4 4
10 12.5 14 4
11
6.6 3.6
F M M F F F M F F F M F M M M F M F M
M F F F F F M F F F F M M M M
++ ++ ++ +
++ ++ ++ + + +
++ ++ ++ ++ ++ +
++
-
-
++ ++ ++ ++ ++ ++ ++ +
++ ++ ++ +
++ ++
-
++ ++ ++ ++ ++ ++ +
-
-
- -
++ ++ ++ ++ ++ +
++ -
++ ++ ++ ++ +
++ ++ ++ ++ ++ ++ +
-
- -
++ ++ + + - -
++ + + - - -
++ - - + + + +
++ + +
++ ++ ++ ++ ++ ++ ++ +
-
- ++ ++
‘MID, multi-infarct dementia; PSP, progressive supranuclear palsy; Tangle, numerous cortical NFT, hut few plaques; Pick’s, Pick’s disease; ALS, amyotrophic lateral sclerosis; OBS, organic brain syndrome (dementia of unknown etiology); CBD, cortimbasal degeneration. + + , Present; +, variable; - , absent.
19831, and induces enhanced production of interleukin- 1 (Sarlo and Mortensen, 1985). Thus, although its precise function remains obscure, its strict evolutionary conserva- tion and binding characteristics suggest an important immunologic role.
The distribution of amyloid P component is altered in several pathologic states. Distinctive, abnormal staining patterns are seen in the glomerular basement membranes of patients with diabetes, systemic vasculitis, and several types of nephritis (Dyck et al., 1980a; Melvin et al., 1986; Yang et al., 1992). In addition, it is detected within a number of lesions associated with chronic neurodegenera- tive disease, including Pick and Lewy bodies (Kalaria et al., 1991a). Amyloid P component is a globular component of the amyloid accumulations that occur in all types of sys- temic and local amyloidosis examined including renal, cutaneous, cerebral amyloid angiopathy, and Creutzfeldt- Jakob disease (Westermark et al., 1975; Breathnach et al., 1981a; Rowe et al., 1984; Coria et al., 1988; Yang and Gallo, 1990; Kalaria et al., 1991a). Recently, amyloid P component has been associated with all of the amyloidotic lesions of Alzheimer’s disease (AD)-cerebrovascular amyloid, senile plaques, and neurofibrillary tangles (NFT; Coria et al., 1988; Duong et al., 1989; Kalaria and Grahovac, 1990; Akiyama et al., 1991; Kalaria et al., 1991a). These data suggest that amyloid P component may act as a “pathologi- cal chaperone” (Wisniewski and Frangione, 1992;
Wisniewski et al., 1993), i.e., mediate the formation of P-pleated amyloid fibrils from polypeptide fragments.
The present study (briefly reported elsewhere; Saperia et al., 1988; Perlmutter et al., 1992) was designed to examine further the distribution of amyloid P component in AD and non-AD brain at both the light and electron microscopic levels. We report that 1) in all cases, amyloid P component immunoreactivity was found within the capillary lumen or endothelial cells but was not seen on the basement mem- brane of the cerebral microvasculature; 2) diffuse perivascu- lar deposits of amyloid P component immunoreactivity were equally likely to occur in AD and control cases; 3) in AD cases, intracellular (I)-NFT and extracellular (El-NFT as well as compact and diffuse plaque-like accumulations were always stained; 4) the increased staining of AD lesions in the gray matter was accompanied by decreased intravas- cular labeling but not with increased perivascular accumu- lations. It is as yet unknown how amyloid P component enters the brain: a specific intracerebral transport mecha- nism or blood-brain barrier compromise may be responsible for its parenchymal localization.
MATERIALS AND METHODS Subjects
Samples were obtained from 19 patients with pathologi- cally confirmed AD [CERAD criteria (Mirra et al., 1990);
94 L.S. PERLMUTTER ET AL.
3

AMYLOID P COMPONENT IN HUMAN BRAIN
mean age = 75.1 * 8.3 years; postmortem interval = 5.2 ? 1.3 hours] and 15 non-AD controls (mean age = 70.9 +. 16.6 years; postmortem interval = 6.6 f 3.6 hours). Diagnosis, age, hours postmortem (PM hours), and sex for each case is detailed in Table 1. The tissue specimens were obtained from the Southern California Alzheimer’s Research Consor- tium and the National Neurological Research Bank.
95
Antibodies Amyloid P component. A commercial polyclonal anti-
body (DAKO-Patts, Santa Barbara, CA) raised in rabbit against human amyloid P component was used. Specificity has been confirmed by crossed immunoelectrophoresis and immunoblot analysis in which the antibody has been shown to recognize an approximately 26 kD protein (Iseki et al., 1988; Kalaria et al., 1991a). This antibody has previously been used to detect human cerebral amyloid both within tissue and in vitro (Coria et al., 1988; Iseki et al., 1988; Kalariaet al., 1991a).
A commercially available monoclonal antibody, subclass IgGl from tissue culture supernatant (Chemicon International, Temecula, CAI, was raised against purified bovine glomerular HSPG and characterized by Kemeny et al. (1988). Direct and competitive inhibition enzyme-linked immunosorbent as- says, immunoblotting, and immunoinhibition immunofluo- rescence were used to demonstrate the specificity of the antibody for the protein core of basement membrane HSPG (Kemeny et al., 1988). We and others have previously demonstrated that HSPG labels E-NFT as well as amyloid accumulations (Snow et al., 1990; Perlmutter et al., 1991).
This monoclonal antibody (a generous gift of Dr. Dale Schenk, Athena Neurosciences) was raised against p-amyloid 1-28 and specifically recog- nizes 1-16 of p-amyloid (i.e., 653-668 of the (3-amyloid protein precursor). It also recognizes both the secreted and full-length forms of P-amyloid protein precursor 695 and 751, although it reacts most strongly with the p-amyloid 1-40 fragment. When used without pretreatment, only a subset of plaques are labeled; the present studies used a formic acid (88%) pretreatment for 3 minutes, allowing detection of diffuse as well as compact plaques and cerebral amyloid angiopathy (Hyman et al., 1991).
This well-characterized monoclonal antibody (Pa- pasozomenos, 1989), also of the IgGl subclass, was a generous gift of Dr. Lester Binder. It has proven useful for labeling the highly phosphorylated form of tau found in the paired helical filaments that comprise the NFT (Papasozo- menos, 1989).
Heparan sulfate proteoglycan (HSPG).
P-amyloid 1-28 (AP 1-28).
Tau-2.
Tissue preparation The leptomeninges were stripped at the time of autopsy.
Tissue blocks (approximately 1.5 x 1.0 x 1.0 cm) from several neocortical regions affected by the lesions of AD (Brodmann areas 7, 9, 17, 22), the hippocampal formation, and cerebellum were immersed in 2% paraformaldehyde, 0.2% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) for at least 2 weeks. Blocks were rinsed, sectioned with a vibratome (50 pm), and treated in 1% HzOz in 10% ethanol for 30 minutes. Sections used for AP 1-28 staining were further incubated in formic acid (88%) for 3 minutes. Most sections were next preincubated in 10% normal goat serum before being placed in amyloid P component (1:4,000), HSPG (l:lO), A@ 1-28 (5 pg/ml), or tau-2 (1:500) primary antibody for 60 hours at 4°C. To ensure that the amyloid P component that was detected in the tissue sections was not derived from the exogenous normal serum, the blocking step was omitted for a subset of sections. The antibodies were visualized with the biotin-avidin-peroxidase technique according to the instructions in the kit (Vectastain; Vector, Burlingame, CA). Diaminobenzidine, either with or with- out nickel ammonium sulfate enhancement (as per instruc- tions in the Vectastain kit), was used as chromagen. The antibody was omitted from adjacent sections as an immuno- cytochemical control.
Some of the immunostained sections were microdissected and prepared for electron microscopic analysis: after treat- ment with osmium tetroxide/potassium ferracyanide, they were infiltrated with “Epon”/araldite resin, embedded, thin sectioned, and lightly contrasted with Reynold’s lead stain. Remaining immunostained sections were mounted on gelatinized slides, counterstained with thioflavine S (fluorescent marker for amyloid), and coverslipped.
Analyses The localization of amyloid P component, HSPG, p-amy-
loid, and tau-2 in selected brain regions was qualitatively examined at the light and electron microscopic levels. Particular attention was paid to the distribution of the vascular staining pattern of amyloid P component, and the degree (“+ +” = present; “+” = variable; “-” = absent) of intravascular and perivascular staining in the gray and white matter was coded by an experimenter blind to diagnostic category. For statistical purposes, “+ +” was coded as 2, “+” as 1, and “-” as 0. Correlations between vascular staining pattern and age or postmortem time were computed. Three hypotheses were proposed based on the results of qualitative analyses. The first two concerned the degree of intravascular or perivascular staining in the gray
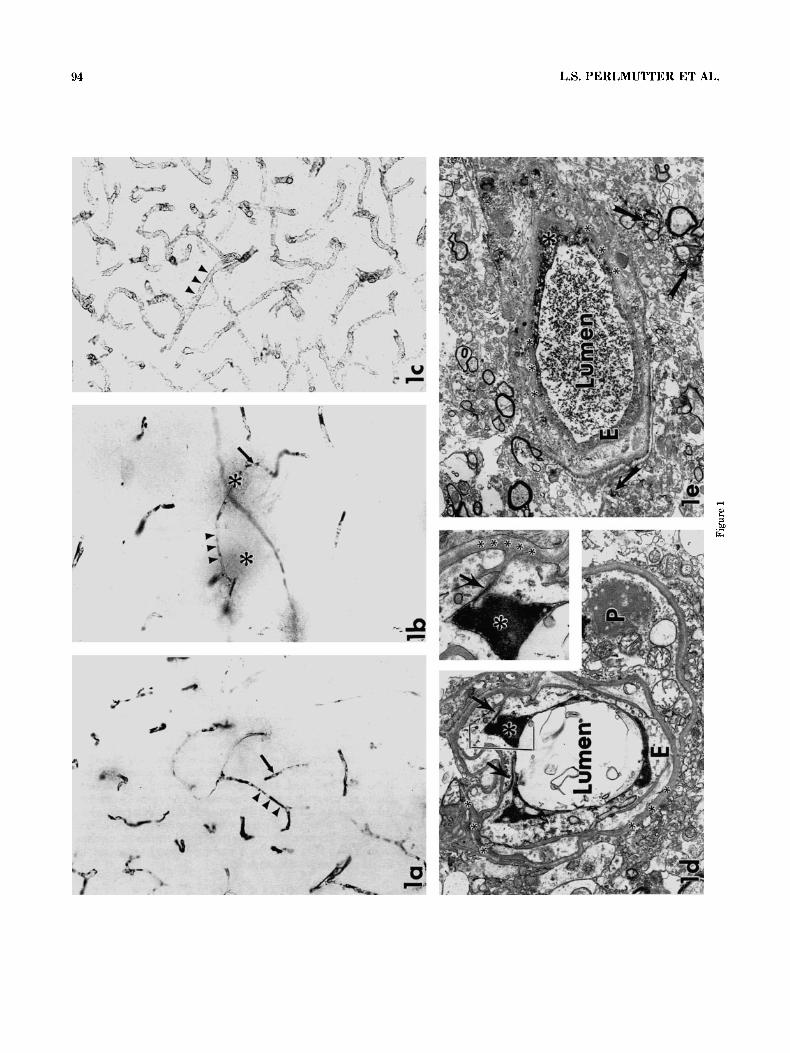
Fig. 1. Amyloid P component immunoreactivity labeled the vascula- ture in most subjects with individual variation in regional intensity and distribution. a,b: Staining was clearly localized to the plasma within the vascular lumen (arrowheads). Intraluminal blood cells were not immunoreactive (arrows). Diffuse perivascular staining was also com- mon (asterisk in b). Both intravascular and diffuse perivascular stain- ing appeared to be more consistently present in the white matter than the gray. c: In contrast to the intravascular localization of amyloid P component, immunoreaction product for the vascular basement mem- brane component, heparan sulfate proteoglycan (HSPG), decorated the outer surfaces of the microvasculature (arrowheads). d,e: In electron microscopic analyses, amyloid P component immunoreaction product
was never seen to decorate the vascular basement membrane (small asterisk). Rather, amyloid P component immunoreactivity was local- ized to the capillary lumen or endothelial cell cytosol (large asterisk). The endothelial cells (E) appeared grossly normal, with no vacuoliza- tion or swelling, and at times intact tight junctions could be seen (d, inset in d; arrows). In the inset, note the lumenal immunoreaction product (large asterisk) abutting an intact tight junction (arrow). Diffuse perivascular amyloid P component-immunoreactive deposits ie; arrows) were not associated with obvious morphological evidence of breakdown of the blood-brain barrier. P, pericyte. Magnification = ~ 2 0 6 in a<, ~ 9 , 9 0 0 in d, ~ 3 1 , 2 0 0 in inset, X5,250 in e.

96
vs. white matter and were tested using paired t-tests (SAS, 1985). The third hypothesis was tested using a logistic regression model (SAS, 19901, where the degree of intra-or perivascular staining in the gray or white matter was used as the predictor of diagnostic category (AD vs. non-AD).
L.S. PERLMUTTER ET AL.
RESULTS The control experiments confirmed the specificity of the
immunostaining: No labeling was ever seen when primary antibody was omitted, and amyloid P component staining was identical with and without the normal goat serum blocking step (data not shown). Amyloid P component immunoreactivity labeled the vasculature in most subjects, with individual variation in regional intensity and distribu- tion. Larger vessels were only sometimes stained, whereas capillaries were often labeled. The immunoreaction product was localized intravascularly (Fig. l a ) as well as in a diffuse perivascuIar pattern (Fig. lb). Both intravascular and diffuse perivascular staining appeared to be more consis- tently present in the white matter than the gray. These empirical observations were formally tested as hypotheses 1 and 2 (see below). Unlike the uniform immunostaining of the outer wall of the capillary bed seen with the antibody against the vascular basement membrane component HSPG (Fig. lc), amyloid P component immunolabeled the plasma within the vascular lumen (Fig. la,b). Intraluminal blood cells were not immunoreactive (Fig. la,b). Electron micros- copy confirmed that amyloid P component immunoreactiv- ity was localized to the capillary lumen or endothelial cell cytosol (Fig. ld,e). The endothelial cells appeared grossly normal, with no vacuolization or swelling, and, at times, an intact tight junction could be seen (Fig. Id). In no case for either the non-AD or AD cases did amyloid P component immunoreaction product decorate the vascular basement membrane (Fig. ld,e). Finally, the diffuse perivascular location of amyloid P component immunoreaction product was not associated with obvious morphological evidence of breakdown of the blood-brain barrier (Fig. le).
Qualitative analyses: Non-AD In non-AD cases, immunostaining for amyloid P compo-
nent was most often restricted to the vasculature (Fig. la,b). Extravascular staining of the cytosol of neurons and sometimes glia was often accompanied by focal regions of attenuated vascular staining in both non-AD (Fig. 2a,b) as well as AD (Fig. 3a) samples. In the few non-AD cases with limited numbers of plaques or tangles, these lesions were amyloid P component immunoreactive as well (data not shown).
Qualitative analyses: AD As was noted above, increased extravascular staining (in
this case, tangles and plaques) was associated with de- creased vascular staining (Fig. 3a). Overall, it appeared that AD cases could be discriminated by the lack of gray matter intravascular staining; this observation gave rise to hypoth- esis 3, which was quantitatively tested (see below). Amyloid P component immunoreaction product darkly stained all of the characteristic lesions of AD including NFT (Fig. 3b), plaque-like accumulations (Fig 3b), and cerebral amyloid angiopathy (Fig. 3c,d). The degree of colocalization of amyloid P component and amyloid could not be determined in the present analysis, as the amyloid P component
immunoreaction product reduced subsequent staining of amyloid deposits with thioflavine S. In general, however, two to four times as many plaque-like accumulations were seen in sections immunostained for amyloid P component than in adjacent sections stained with thioflavine S alone.
At the light microscopic level, amyloid P component immunolabeling of neurons was similar to that seen with thioflavine S (Fig. 4a,b) and was clearly distinct from the diffuse cytosolic pattern seen in some non-AD cases (Fig. 2a,b). At the ultrastructural level, amyloid P component labeled cytosol and some filaments of I-NFT-containing neurons (Fig. 4c). Although some NFT remained unstained (Fig. 4c), E-NFT were consistently darkly labeled (Fig. 4d). A close association between amyloid P component-immuno- reactive fibrils of an E-NFT and a microglial cell was seen (Fig. 4e). At high magnification, the fibrils of the E-NFT appeared to be attached to the microglial cell (Fig. 4e, inset). Microglia are ultrastructurally identified as cells with dense body-filled cytoplasm and a characteristic pattern of chroma- tin clumping beneath the nuclear envelope (Peters et al., 1991).
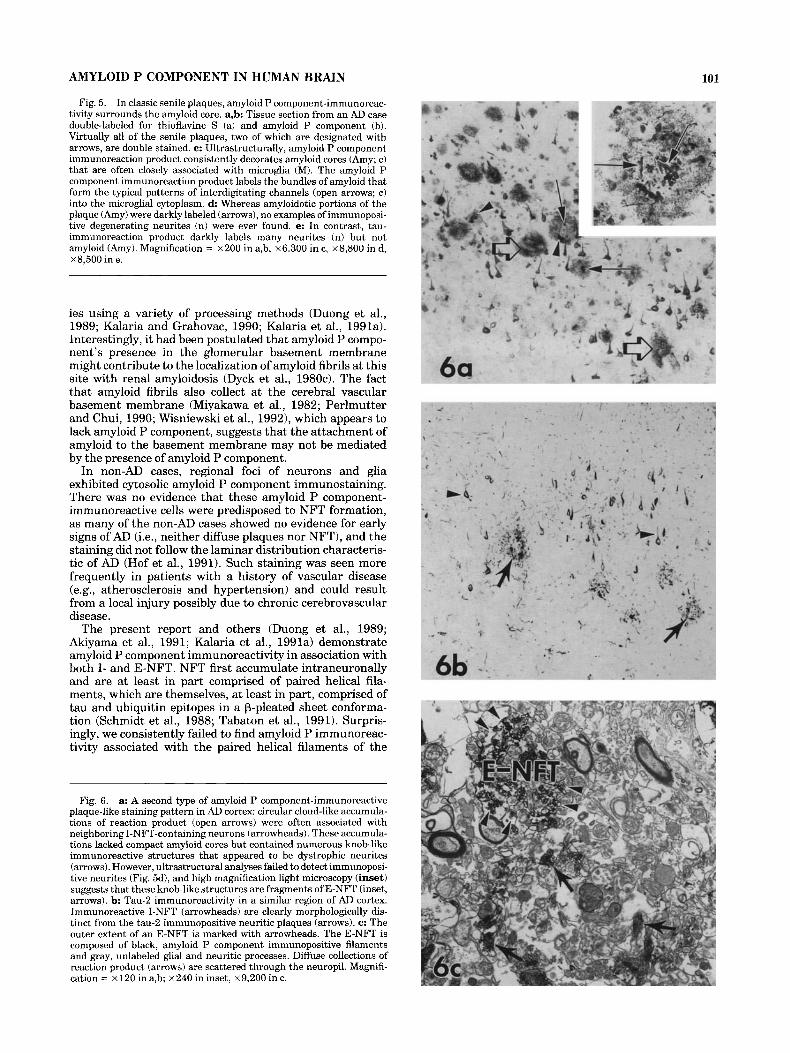
Plaque-like accumulations of amyloid P component- immunostaining were highly variable in size and configura- tion. In classic senile plaques, the amyloid P component immunoreactivity surrounded an amyloid core (Fig. 5a,b). Ultrastructurally, amyloid P component immunoreaction product consistently decorated the amyloid fibrils, includ- ing the compact amyloid cores that were closely associated with microglia (Fig. 5c). Although amyloidotic portions of the plaque were darkly labeled, no immunolabeled degener- ating neurites were seen (Fig. 5d).
A second type of amyloid P component-immunoreactive plaque-like staining pattern consisted of circular cloud-like accumulations often associated with neighboring I-NFT- containing neurons. These accumulations lacked compact amyloid cores but contained numerous knob-like immuno- reactive structures that appeared to be dystrophic neurites (Fig. 6a). However, high magnification light microscopy (Fig. 6a, inset) as well as comparisons with tau-2 immunola- beled degenerating neurites (Fig. 6b) revealed that these knob-like structures were not neurites. Instead, ultrastruc- tural analyses (Fig. 6c) suggest that these structures were E-NFT surrounded by diffuse amyloid P component- immunoreactive accumulations.
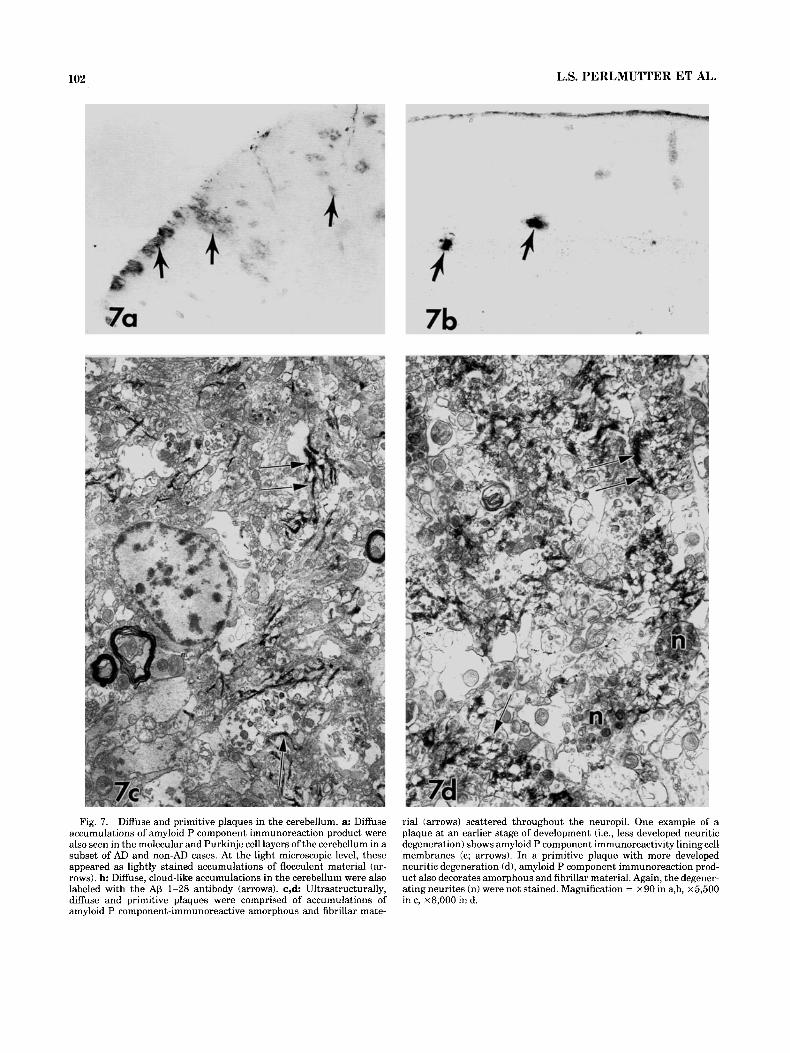
Diffuse clouds of amyloid P component immunoreaction product were particularly conspicuous in upper neocortical layers and varied markedly in size (Fig. 3b). Diffuse accumu- lations were also seen in the molecular and Purkinje cell layers of the cerebellum in a subset of AD and non-AD cases. At the light microscopic level, these appeared as lightly stained accumulations of flocculent material (Fig. 7a). Similar cerebellar diffuse accumulations were labeled with the AP 1-28 antibody (Fig. 7b). Ultrastructurally, diffuse and primitive plaques were comprised of accumula- tions of amyloid P component-immunoreactive amorphous and fibrillar material scattered throughout the neuropil, at times lining the cell membrane of longitudinally sectioned processes (Fig. 7c). In primitive plaques (i.e., plaques containing amyloid fibrils, some dystrophic neurites, no compact amyloid core; Yamaguchi et al., 1988), the amyloid fibrils but, again, not the neurites were consistently immu- nolabeled for amyloid P component (Fig. 7d).
AMYLOID P COMPONENT IN HUMAN BRAIN
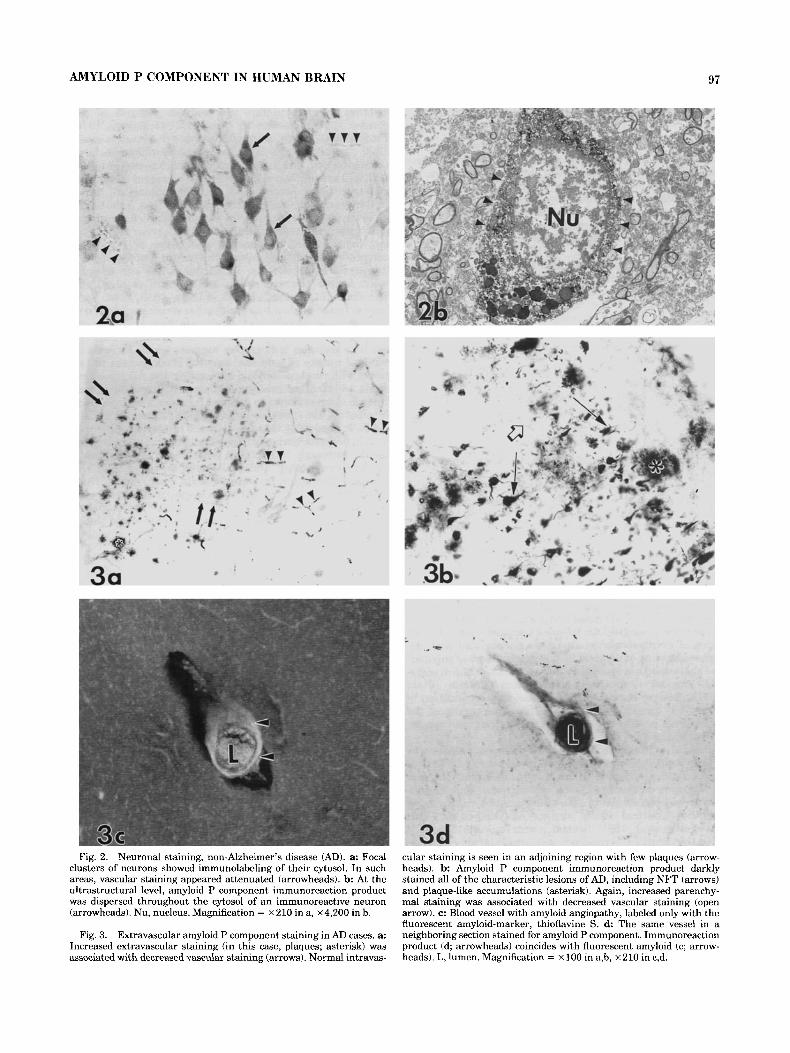
Fig. 2. Neuronal staining, non-Alzheimer’s disease (AD). a: Focal clusters of neurons showed immunolabeling of their cytosol. In such areas, vascular staining appeared attenuated (arrowheads). b At the ultrastructural level, amyloid P component immunoreaction product was dispersed throughout the cytosol of an immunoreactive neuron (arrowheads). Nu, nucleus. Magnification = x 210 in a, ~ 4 , 2 0 0 in b.
Fig. 3. Extravascular amyloid P component staining in AD cases. a: Increased extravascular staining (in this case, plaques; asterisk) was associated with decreased vascular staining (arrows). Normal intravas-
97
cular staining is seen in an adjoining region with few plaques (arrow- heads). b Amyloid P component immunoreaction product darkly stained all of the characteristic lesions of AD, including NFT (arrows) and plaque-like accumulations (asterisk). Again, increased parenchy- mal staining was associated with decreased vascular staining (open arrow). c: Blood vessel with amyloid angiopathy, labeled only with the fluorescent amyloid-marker, thioflavine S. d The same vessel in a neighboring section stained for amyloid P component. Immunoreaction product (d; arrowheads) coincides with fluorescent amyloid (c; arrow- heads). L, lumen. Magnification = x 100 in a,b, ~ 2 1 0 in c,d.

98 L.S. PERLMUTTER ET AL.
Amyloid P component staining of the vasculature
To test whether age or postmortem delay affected the vascular staining pattern as well as to test the three empirically-derived hypotheses, a semi-quantitative scoring of the degree of gray and white matter intravascular and perivascular staining (Table 1) was used. First, there were no significant correlations between vascular staining pat- tern and either age or postmortem time (Table 2). Second, each of the three hypotheses was statistically significant (Table 2).
Hypothesis 1. Across all cases (AD and non-AD), there was more intravascular staining in the white than the gray matter (paired t test: t (33) = 3.48, P < 0.002).
Hypothesis 2. Across all cases (AD and non-AD), there was more perivascular staining in the white than the gray matter (paired t test: t (33) = 3.19, P < 0.005).
Hypothesis 3. A greater degree of intravascular gray matter staining was predictive of a diagnosis of non-AD (logistic regression: Wald x2 = 4.96, P = 0.03). This variable remained a significant predictor (P < 0.05) even when the effects of the other three variables were first taken into account.
DISCUSSION Amyloid P component immunoreactivity exhibited sev-
eral distinct patterns. Although these patterns were not significantly correlated with age or postmortem delay, information concerning agonal state was not available, and, therefore, its effects cannot be evaluated. Intravascular staining was seen in most subjects with individual variation in regional intensity and distribution. Overall, intravascu- lar staining was less frequent in the gray than the white matter, especially in AD cases. Diffuse perivascular staining was seen in all cases and was more frequent in the white than the gray matter. Finally, two patterns of extravascular staining were seen: focal regions of cytosolic staining in both non-AD and AD cases and widespread labeling of all three types of amyloidotic lesions associated with AD. Both types of extravascular staining were associated with de- creased labeling of adjacent blood vessels. Because polymer- ase chain-reaction analyses show no evidence of intracere- bral amyloid P component production (Kalaria et al., 1991b), the perivascular and extravascular localization of amyloid P component suggests either compromise of the blood-brain barrier or increased transport across vascular endothelial cells.
Several findings support the possibility of selective or intermittent leakage of the blood-brain barrier in normal aging. First, a variety of serum proteins including albumin, prealbumin, immunoglobulins, complement, and fibrino- gen have been found in the brain parenchyma in some cognitively normal elderly persons (Alafuzoff et al., 1987; Rozemuller et al., 1988). Second, normal aging is associated with thinning of the capillary wall, which has been attrib- uted to loss of pericytes and endothelial cytoplasm (Stewart et al., 1987). Such thinning occurs predominantly in the white matter, and could predispose to transient leaks in the blood-brain barrier (Stewart et al., 1987) leading to the perivascular extravasation of amyloid P component re- ported here. Alternatively, perivascular localization may result from blood-brain barrier compromise during the agonal state or postmortem period.
The status of the blood-brain barrier in AD has also been the subject of intense scrutiny. Structural alterations of the
cerebral microvasculature are common features of the disease: increased tortuosity (Hassler, 1965; Ravens, 1978; Fischer et al., 1990), attenuated vessels (Perlmutter et al., 1990), possible gaps in endothelial cell tight junctions (Stewart et al., 1992), and increased thickness, reduplica- tion, and vacuolization of the vascular basement membrane (Ravens, 1978; Mancardi et al., 1980; Scheibel, 1984; Perlmutter et al., 1990; Perlmutter and Chui, 1990) have all been reported. Although these structural changes sug- gest the possibility of altered blood-brain barrier permeabil- ity, there is still no conclusive evidence of functional compromise. Although a variety of serum proteins includ- ing prealbumin, albumin, immunoglobulins and comple- ment factors (Powers et al., 1981; Mann et al., 1982; Shirahama et al., 1982; Ishii and Haga, 1983, 1984) have been detected in plaque amyloid, other serum proteins such as C-reactive protein and serum amyloid A are absent (Duong et al., 1989; Kalaria et al., 1991a). Although positron emission tomography suggests a mild increase in cerebral uptake of iodinated-amyloid P component in pa- tients with cerebral amyloid angiopathy and AD (Hawkins et al., 19911, other imaging and permeability studies pro- vide no clear evidence to support functional leakage (Ala- fuzoff et al., 1987; Schlageter et al., 1987; Rozemuller et al., 1988). It is possible that the presence of amyloid P compo- nent in brain parenchyma may reflect changes in transendo- thelial transport rather than alterations in blood-brain barrier permeability. Indeed, specific impairment of the glucose transporter in isolated brain microvessels from lesion-affected areas of AD brain suggests that transport may be affected (Kalaria and Harik, 1989). Future studies are obviously needed to determine the status of the blood- brain barrier and the mechanisms for amyloid P component entry into the brain.
Amyloid P component has been immunocytochemically localized to glomerular and alveolar basement membrane, arterioles, bronchioles and sarcolemma of cardiac and smooth muscle (Dyck et al., 1980a,b; Melvin et al., 1986), as well as to elastic tissue fibers in the dermis (Breathnach et al., 1983). Recently, minute connective tissue structures called “pentosomes,” which are usually found in the vicin- ity of microfibrils in connective tissue, have been shown to be amyloid P component subunits (Inoue, 1991). Amyloid P component has not been localized to all types of basement membrane, however, and is lacking from renal tubular (Dyck et al., 198Oc), dermal-epidermal (Breathnach et al., 19831, and dermal papillary vascular basement membranes (Breathnach et al., 1983). The present study demonstrates that cerebrovascular basement membranes also lack amy- loid P component, at least under the fixation and staining conditions used here. Nor was immunolabeling of the cerebral vascular basement membrane evident in the photo- micrographs presented in previous light-microscopic stud-
Fig. 4. a: Thioflavine S labeling of I-NFT (arrows). b Amyloid P component immunolabeling of I-NFT was also fibrillar (arrows), simi- lar to that seen with thioflavine S. c: At the ultrastructural level, amyloid P component labeled both filaments (arrowheads) and cytosol (asterisk) of an I-NFT-containing neuron; some NFT remain unstained (arrows). d: E-NFT were consistently darkly labeled (arrowheads). e: A close association between amyloid P component-immunoreactive fibrils of an E-NFT and a microglial cell (MI was seen (arrows). At high magnification (inset), the fibrils appeared to be attached to the microglial cell (arrows). Nu, nucleus. Magnification = ~ 1 4 5 in a,b, x6,600inc, x3,900ind, x7,500ine, ~14 ,250in inse t .
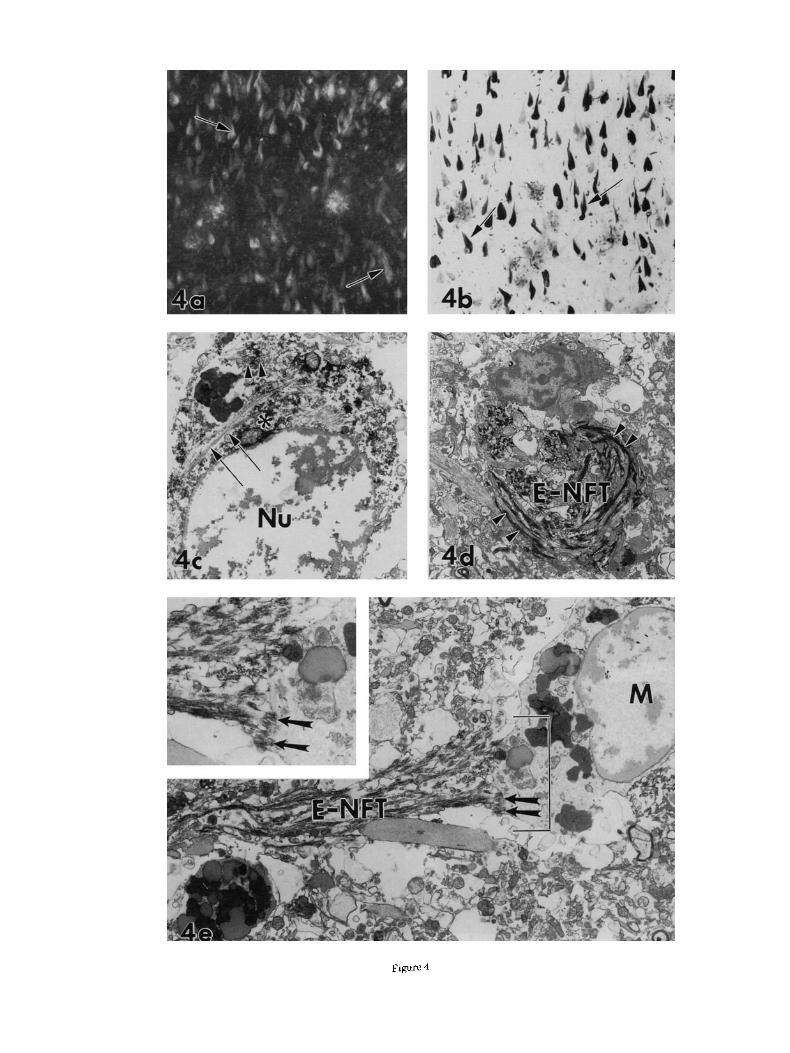
Figure 4
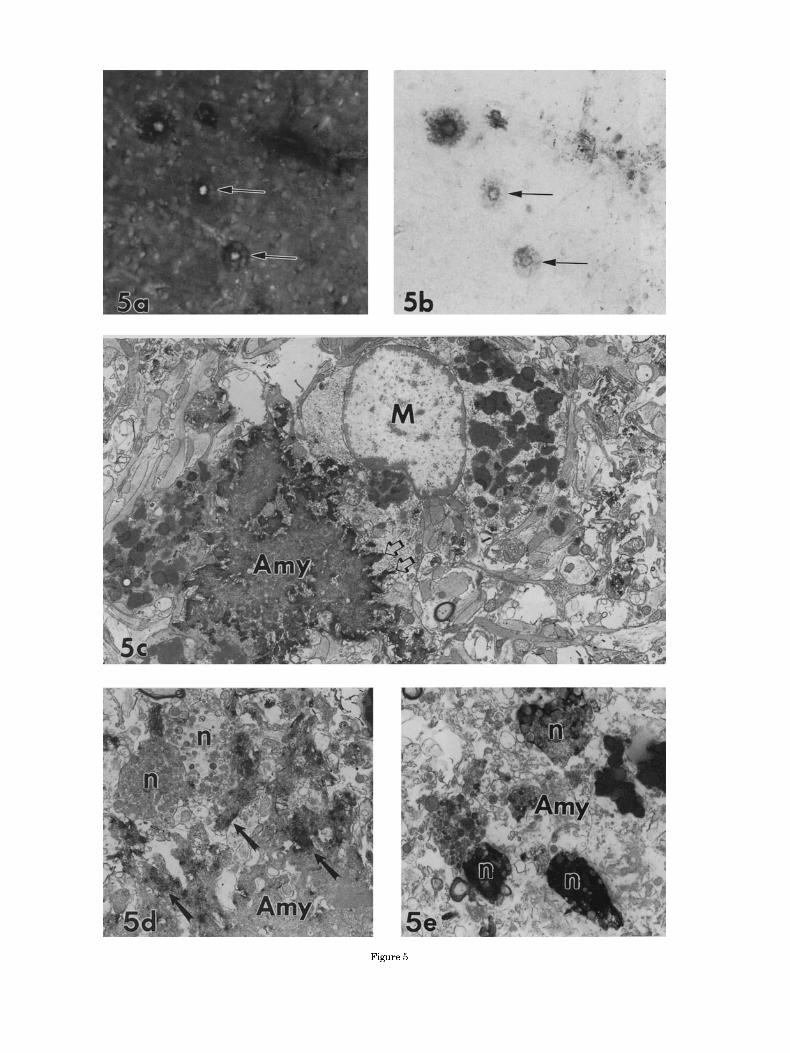
Figure 5
AMYLOID P COMPONENT IN HUMAN BRAIN 101
Fig. 5. In classic senile plaques, amyloid P component-immunoreac- tivity surrounds the amyloid core. a,b Tissue section from an AD case double-labeled for thioflavine S (a) and amyloid P component (b). Virtually all of the senile plaques, two of which are designated with arrows, are double stained. c: Ultrastructurally, amyloid P component immunoreaction product consistently decorates amyloid cores (Amy; c) that are often closely associated with microglia (MI. The amyloid P component immunoreaction product labels the bundles of amyloid that form the typical patterns of interdigitating channels (open arrows; c) into the microglial cytoplasm. d: Whereas amyloidotic portions of the plaque (Amy) were darkly labeled (arrows), no examples of immunoposi- tive degenerating neurites (n) were ever found. e: In contrast, tau- immunoreaction product darkly labels many neurites (n) but not amyloid (Amy). Magnification = x200 in a,b, ~ 6 , 3 0 0 in c, ~8 ,800 in d, ~ 8 , 5 0 0 in e.
ies using a variety of processing methods (Duong et al., 1989; Kalaria and Grahovac, 1990; Kalaria et al., 1991a). Interestingly, it had been postulated that amyloid P compo- nent’s presence in the glomerular basement membrane might contribute to the localization of amyloid fibrils at this site with renal amyloidosis (Dyck et al., 1980~). The fact that amyloid fibrils also collect at the cerebral vascular basement membrane (Miyakawa et al., 1982; Perlmutter and Chui, 1990; Wisniewski et al., 1992), which appears to lack amyloid P component, suggests that the attachment of amyloid to the basement membrane may not be mediated by the presence of amyloid P component.
In non-AD cases, regional foci of neurons and glia exhibited cytosolic amyloid P component immunostaining. There was no evidence that these amyloid P component- immunoreactive cells were predisposed to NFT formation, as many of the non-AD cases showed no evidence for early signs of AD (i.e., neither diffuse plaques nor NFT), and the staining did not follow the laminar distribution characteris- tic of AD (Hof et al., 1991). Such staining was seen more frequently in patients with a history of vascular disease (e.g., atherosclerosis and hypertension) and could result from a local injury possibly due to chronic cerebrovascular disease.
The present report and others (Duong et al., 1989; Akiyama et al., 1991; Kalaria et al., 1991a) demonstrate amyloid P component immunoreactivity in association with both I- and E-NFT. NFT first accumulate intraneuronally and are at least in part comprised of paired helical fila- ments, which are themselves, at least in part, comprised of tau and ubiquitin epitopes in a @-pleated sheet conforma- tion (Schmidt et al., 1988; Tabaton et al., 1991). Surpris- ingly, we consistently failed to find amyloid P immunoreac- tivity associated with the paired helical filaments of the
Fig. 6. a: A second type of amyloid P component-immunoreactive plaque-like staining pattern in AD cortex: circular cloud-like accumula- tions of reaction product (open arrows) were often associated with neighboring I-NFT-containing neurons (arrowheads). These accumula- tions lacked compact amyloid cores but contained numerous knob-like immunoreactive structures that appeared to be dystrophic neurites (arrows). However, ultrastructural analyses failed to detect immunoposi- tive neurites (Fig. 5d), and high magnification light microscopy (inset) suggests that these knob-like structures are fragments of E-NFT (inset, arrows). b: Tau-2 immunoreactivity in a similar region of AD cortex. Immunoreactive I-NFT (arrowheads) are clearly morphologically dis- tinct from the tau-2 immunopositive neuritic plaques (arrows). c: The outer extent of an E-NFT is marked with arrowheads. The E-NFT is composed of black, amyloid P component immunopositive filaments and gray, unlabeled glial and neuritic processes. Diffuse collections of reaction product (arrows) are scattered through the neuropil. Magnifi- cation = x 120 in a,b; ~ 2 4 0 in inset, x 9,200 in c.
102
Fig. 7. Diffuse and primitive plaques in the cerebellum. a: Diffuse accumulations of amyloid P component immunoreaction product were also seen in the molecular and Purkinje cell layers of the cerebellum in a subset of AD and non-AD cases. At the light microscopic level, these appeared as lightly stained accumulations of flocculent material (ar- rows). b: Diffuse, cloud-like accumulations in the cerebellum were also labeled with the A0 1-28 antibody (arrows). c,d Ultrastructurally, diffuse and primitive plaques were comprised of accumulations of amyloid P component-immunoreactive amorphous and fibrillar mate-
L.S. PERLMUTTER ET AL.
rial (arrows) scattered throughout the neuropil. One example of a plaque at an earlier stage of development (i.e., less developed neuritic degeneration) shows amyloid P component immunoreactivity lining cell membranes (c; arrows). In a primitive plaque with more developed neuritic degeneration (d), amyloid P component immunoreaction prod- uct also decorates amorphous and fihrillar material. Again, the degener- ating neurites (n) were not stained. Magnification = x90 in a,b, x 5,500 in c, ~ 8 , 0 0 0 in d.

AMYLOID P COMPONENT IN HUMAN BRAIN 103
TABLE 2. Statistical Analyses
Intravascular Perivascular
White matter Gray matter White matter Gray matter
Age -0.21 (ns!2 -0.23 (ns! -0.09 (ns! -0.17 (nsl postmortem delay Postmortem delay -.014 (ns! -0.20 (ns! -0.06 (nsl 0.08 (ns!
vs gray matter
Correlations’ between degree of vascular staining and age1
Paired t tests for intravasculariperivascular staining in white Mean 1.62 1.15 1.41 0.79 SD 0.64 0.81 0.84 0.93 Mean difference 0.47 0.62 SD of mean difference 0.79 1.13 t 3.48 3.19 Statistical probability P < 0.002 P < 0.005
Logistic regression using degree of staining in intravasculari Wald x 2 0.18 4.96 0.17 0.57 perivascular whiteigray matter as predictors of diagnostic Statistical probability P = 0.68 P = 0.03 P = 0.68 P = 0 4 5 category (AD vs. non-AD)
‘Criterion value (P < 0.051 of Pearson r = 0.349. %s, not significant.
plaque neurites. There are several possible reasons for this. First is the sensitivity of the technique and the possibility that the immunoreagents cannot penetrate into the neu- rites. However, the light micrograph of tau-2 labeling in Figure 6b of this paper and our published electron micro- graph of tau-2 immunolabeled neurites (Perlmutter et al., 1991) argue against this idea. A second possible explanation takes into consideration the differences between paired helical filaments and NFT. Amyloid P component may be associated with both, but the epitope may not be available in the former. On the other hand, amyloid P component might become associated with NFT as it forms from the individual paired helical filament segments. Further work is necessary to solve this enigma.
Evidence suggests that neurons do not produce amyloid P component (Kalaria et al., 1991b); thus, the appearance of amyloid P component immunoreactivity in I-NFT and in the cytosol of some neurons in both non-AD and AD cases suggests that injured cells may have a take-up mechanism for extracellular amyloid P component. This possibility requires further examination as well.
As NFT become extracellular, they are infiltrated by astrocytes, microglial processes, and degenerating neurites (Schmidt et al., 1988; Cras et al., 1991; Yamaguchi e t al., 1991a,b). They lose immunoreactivity for certain epitopes (Schmidt et al., 1988; Tabaton et al., 1991) but acquire immunoreactivity for P-protein (Hyman et al., 1989; Taba- ton et al., 1991; Yamaguchi et al., 1991b; Perry et al., 1992). In the present report, we describe diffuse, cloud-like amy- loid P component accumulations associated with I-NFT as well as with E-NFT (Fig. 6a,c). The role of these extracellu- lar accumulations in disease pathogenesis is unknown. Finally, similarities in the cellular and biochemical composi- tion of E-NFT and senile plaques have suggested to some investigators that E-NFT may represent the nidus for the development of a subset of plaques (Perry et al., 1991; Pappolla et al., 1992). The associated accumulation of amyloid P component may play a role in the progression of these pathologic events as well.
ACKNOWLEDGMENTS We thank Scott Lyness, Chris Zarow, and Galen Buckwal-
ter for their statistical assistance. This research was sup- ported by NIH grant AG-07624 and Sankyo Co. Ltd. Research Institute. Tissue specimens were obtained from the Southern California Alzheimer’s Research Consortium (NIH lP50-AG05142) and the National Neurological Re- search Bank, VAMC Wadsworth Division (Los Angeles, CA 90073; sponsored by NINCDS/NIMH, NMSS, HD Founda- tion, Comprehensive Epilepsy Program, Tourette’s Syn-
drome Association, Dystonia Research Foundation, and the U.S. Veteran’s Administration).
LITERATURE CITED Akiyama, H., T. Yamada, T. Kawamata, and P.L. McGeer (1991) Association
of amyloid P component with complement proteins in neurologically diseased brain tissue. Brain Res. 548:349-352.
Alafuzoff, I., R., Adolfsson, I. Grundke-Iqbal, and B. Winblad (1987) Blood-brain barrier in Alzheimer dementia and in nondemented elderly. Acta Neuropathol. 73:160-166.
Baltz, M.L., F.C. De Beer, S. Feinstein, E.A. Munn, C.P. Milstein, T.C. Fletcher, J.F. March, J. Taylor, C. Bruton, J.R. Clamp, A.J.S. Davies, and M.B. Pepys (1982) Phylogenetic aspects of C-reactive protein and related proteins. Ann. NY Acad. Sci. 389:49-75.
Breathnach, S.M., B. Bhogal, R.F. Dyck, F.C. De Beer, M.M. Black, and M.B. Pepys (1981a) Immunohistochemical demonstration of amyloid P compo- nent in skin of normal subjects and patients with cutaneous amyloidosis. Br. J. Dermatol. 105r115-124.
Breathnach, S.M., S.M. Melrose, B. Bhogal, F.C. De Beer, R.F. Dyck, G. Tennent, M.M. Black, and M.B. Pepys (1981b) Amyloid P component is located on elastic fibre microfibrils in normal human tissue. Nature 293.652454.
Breathnach, S.M., S.M. Melrose, B. Bhogal, F.C. De Beer, M.M. Black, and M.B. Pepys (1983) Immunohistochemical studies of amyloid P compo- nent distribution in normal human skin. J. Invest. Dermatol. 80536-90.
Castatio, E.M., and B. Frangione (1988) Biology of disease. Human amyloido- sis, Alzheimer disease and related disorders. Lab. Invest. 58:122-132.
Coria, F., E.M. Castafio, F. Prelli, M. Larrondo-Lillo, S.G. van Duinen, M.L. Shelanski, and B. Frangione (1988) Isolation and characterization of amyloid P component from Alzheimer’s disease and other types of cerebral amyloidosis. Lab. Invest. 58t454-457.
Cras, P., M. Kawai, S. Siedlak, and G. Perry (1991) Microglia are associated with the extracellular neurofibrillary tangles of Alzheimer disease. Brain Res. 558:312-3 14.
Duong, T., E.C. Pommier, and A.B. Scheibel (1989) Immunodetection of human serum amyloid P component in Alzheimer’s disease. Acta Neuropathol. 78:429437.
Dyck, R.F., D.J. Evans, C.M. Lockwood, A.J. Rees, D. Turner, and M.B. Pepys (1980a) Amyloid P-component in human glomerular basement membrane. Lancet 2.606-609,
Dyck, R.F., M. Kershaw, N. McHugh, and M.B. Pepys (1980b) Immunohisto- chemical staining of normal and pathological human tissues with antibody to serum amyloid P-component (SAP). In G.G. Glenner, P. Pinho e Costa, and F. de Freitas (eds): Amyloid and Arnyloidosis. Amsterdam: Excerpta Medica, pp. 50-54.
Dyck, R.F., C.M. Lockwood, M. Kershaw, N. McHugh, V.C. Duance, M.L. Baltz, and M.B. Pepys (1980~) Amyloid P-component is a constituent of normal human glomerular basement membrane. J. Exp. Med. 152:1162- 1174.
Fischer, V.W., A. Siddiqi, and Y. Yusufaly (1990) Altered angioarchitecture in selected areas of brains with Alzheimer’s disease. Acta Neuropathol. 79:672-679.
Hamazaki, H. (1987) Ca2+-mediated association of human serum amyloid P component with heparan sulfate and dermatan sulfate. J. Biol. Chem. 262:1456-1460.
Hassler, 0. (1965) Vascular changes in senile brains I. A micro-angiographic study. Acta Neuropathol. 540-53.

104 L.S. PERLMUTTER ET AL.
Hawkins, P.N., P. Tyrrell, T. Jones, M.N. Rossor, P. Roques, M. Myers, R. Lamhrecht, S. Richardson, G. Gudmundsson, and M.B. Pepys (1991) Metabolic and scintigraphic studies with radiolahelled serum amyloid P component in amyloidosis: Apphcations to cerebral deposits and Alzhei- mer’s disease with positron emission tomography. Bull. Clin. Neurosci. 56:178-190.
Hof, P.R., D.P. Perl, A.J. Loerzel, and J.H. Morrison (1991) Neurofihrillary tangle distribution in the cerebral cortex of Parkinsonism-dementia cases from Guam: Differences with Alzheimer’s disease. Brain Res. 564:306-313.
Hyman, B.T., G.W. Van Hoesen, K. Beyreuther, and C.L. Masters (1989) A4 amyloid protein immunoreactivity is present in Alzheimer’s disease neurofihrillary tangles. Neurosci. Lett. 101:352-355.
Hyman, B.T., R.B. Tanzi, K. Marzloff, R. Barhour, and D.B. Schenk (1991) Kunitz protease inhibitor containing amyloid beta protein precursor immunoreactivity in Alzheimer’s disease. J. Neuropathol. Exp. Neurol. 51: 76-83.
Inoue, S. (1991) Pentosome-a new connective tissue component-is a subunit of amyloid P. Cell Tissue Res. 263:431438.
Iscki, E., N. Amano, T. Matsuishi, S. Yokoi, N. Arai, and S. Yagishita (1988) A case of familial, atypical Alzheimer’s disease: Immunohistochemical study of amyloid P-component. Neuropathol. Appl. Neurol. 14:169-174.
Ishii, T., and S. Haga (1983) Immuno-electron microscopic localization of immunoglobulins in amyloid fibrils of senile plaques. Acta Neuropathol. 61: 76-80.
Ishii, T., and S. Haga (1984) Immuno-electron-microscopic localization of complements in amyloid fibrils of senile plaques. Acta Neuropathol. 63296-300.
Kalaria, R.N., and I. Grahovac (1990) Serum amyloid P immunoreactivity in hippocampal tangles, plaques and vessels: Implications for leakage across the blood-brain barrier in Alzheimer’s disease. Brain Res. 51 6:349- 353.
Kalaria, R.N., and S.I. Harik (1989) Reduced glucose transporter a t the blood-brain barrier and cerebral cortex in Alzheimer’s disease. J. Neuro- chem. 5324:1083-1088.
Kalaria, R.N., and S.N. Kroon (1992) Complement inhibitor C4-binding protein in amyloid deposits containing serum amyloid P in Alzheimer’s disease. Biochem. Biophys. Rcs. Commun. 186r461466.
Kalaria, R.N., P.G. Galloway, and G. Perry (1991a) Widespread serum amyloid P immunoreactivity in cortical amyloid deposits and the neurofi- brillary pathology of Alzheimer’s disease and other degenerative disor- ders. Neuropathol. Appl. Neurobiol. 17:189-201.
Kalaria, R.N., T.E. Golde, M.L. Cohen, and S.G. Younkin (1991h) Serum amyloid P in Alzheimer’s disease. Implications for dysfunction of the blood-brain barrier. Ann. NY Acad. Sci. 640: 145-148.
Kemeny, E., H.M. Fillit, S. Damle, R. Mahabir, N.A. Kefalides, J.D. Gregory, T. Antonovych, S. Sabnis, and J.B. Zabriskie (1988) Monoclonal antibod- ies to heparan sulfate proteoglycan: Development and application to the study of normal tissue and pathologic human kidney biopsies. Connect. Tissue Res. 18~9-25.
Mancardi, G.L., F. Perdelli, C. Rivano, A. Leonardi, and 0. Bugiani (1980) Thickening of the basement membrane of cortical capillaries in Alzhei- mer’s disease. Acta Neuropathol. 49.79-83.
Mann, D.M.A., J.S. Davies, P.O. Yatcs, and J. Hawkes (1982) Immunohisto- chemical staining of senile plaques. Neuropathol. Appl. Neurobiol. 8:55-61.
Melvin, T., Y. Kim, and A.F. Michael (1986) Amyloid P component is not present in the glomerular basement membrane in Alport-type hereditary nephritis. Am. J. Pathol. 125:460464.
Mirra, S.S., D.W. McKeel, B.J. Crain, J.P. Hughes, G. van Belle, and A. Heyman (1990) Quality assurance in the neuropathology assessment of Alzheimer’s disease: A multi-center study of the Consortium to Estah- lish a Registry for Alzheimer’s Disease (CERAD). J. Neuropathol. Exp. Neurol. 49r336
Miyakawa, T., A. Shimoji, R. Kuramoto, and Y. Higuchi (1982) The relationship between senile plaques and cerebral hlood vessels in Alzhei- mer’s disease and senile dementia. Virchows Arch Cell Pathol. 40:121- 129.
Papasozomenos, S.C. (1989) Tau protein immunoreactivity in dementia of the Alzheimer type. Lab. Invest. 50:123-137.
Pappolla, M.A., R.A. Omar, K. Sambamurti, J.P. Anderson, and N.K. Rohakis (1992) The genesis of the senile plaque: Further evidence in support of its neuronal origin. Am. J. Pathol. 141:1151-1159.
Pepys, M.B., A.C. Dash, R.E. Markham, H.C. Thomas, and B.D. Williams (1978) Comparative clinical study of protein SAP (amyloid P component) and C-reactive protein in serum. Clin. Exp. Immunol. 32119-124.
Pepys, M.B., M. Baltz, K. Gomer, A.J.S. Davies, and M. Doenhoff (1979a) Serum amyloid P-component is an acute-phase reactant in the mouse. Nature 278:259-261.
Pepys, M.B., R.F. Dyck, F.C. De Beer, M. Skinner, and AS. Cohen (197913) Binding of serum amyloid P-component ( S A P ) by amyloid fibrils. Clin. Exp. Immunol. 38284-293.
Perlmutter, L.S., and H.C. Chui (1990) Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review. Brain Res. Bull. 24:677-686.
Perlmutter, L.S., H.C. Chui, D. Saperia, and J. Athanikar (1990) Microangi- opathy and the colocalization of heparan sulfate proteoglycan with amyloid in senile plaques of Alzheimer’s disease. Brain Res. 508~13-19.
Perlmutter, L.S., E. Barron, D. Saperia, and H.C. Chui (1991) Association between vascular basement membrane components and the lesions of Alzheimer’s disease. J. Neurosci. Res. 30:673-681.
Perlmutter, L.S., M.A. Myers, E. Barron, D. Saperia, and H.C. Chui (1992) Amyloid P component and Alzheimer amyloid lesions. Soc. Neurosci. Ahstr. 18:1442
Perry, G., P. Cras, M. Kawai, P. Mulvihill, and M. Tabaton (1991) Transfor- mation of neurofihrillary tangles in the extracellular space. Bull. Clin. Neurosci. 56:107-119.
Perry, G., P. Cras, S.L. Siedlak, M. Tahaton, and M. Kawai (1992) Beta- protein immunoreactivity is found in the majority of neurofihrilllary tangles of Alzheimer’s disease. Am. J. Pathol. 140:283-290.
Peters, A,, S.L. Palay, and A. DeF. Webster (1991) The Fine Structure of the Nervous System. New York: Oxford University Press, pp. 304-311.
Powers, J.M., W.W. Schlaepfer, M.C. Willingham, and B.J. Hall (1981) An immunoperoxidase study of senile cerebral amyloidosis with pathoge- netic considerations. J. Neuropathol. Exp. Neurol. 40:592-612.
Ravens, J.R. (1978) Vascular changes in the human senile hrain. Adv. Neurol. 20:487-501.
Rostagno, A., B. Frangione, E. Pearlstein, and A. Garcia-Pardo (1986) Fibronectin binds to amyloid P component. Localization of the binding site to the 31,000 dalton c-terminal domain. Biochem. Biophys. Res. Commun. 140:12-20.
Rowe, L.F., 0. Jensson, P.D. Lewis, J.M. Candy, G.A. Tennent, and M.B. Pepys (1984) Immunohistochemical demonstration of amyloid P compo- nent in cerehro-vascular amyloidosis. Neuropathol. Appl. Neurohiol. 10.53-6 1.
Rozemuller, J.M., P. Eikelenhoom, W. Kamphorst, and F.C. Stam (1988) Lack of evidence for dysfunction of the blood-brain harrier in Alzhei- mer’s disease: An immunohistochemical study. Neurohiol. Aging 9:383- 391.
Saperia, D., L.S. Perlmutter, J. Athanikar, and H.C. Chui (1988) Amyloid P component in dementia. Soc. Neurosci. Abstr. 14:638
Sarlo, K.T., and R.F. Mortensen (1985) Enhanced interleukin 1 (IL-1) production mediated by mouse serum amyloid P component. Cell Immunol. 93:398
SAS Institute Inc. (1985) SAS Users Guide: Basics, Version 5 Addition. Cary, NC: SAS Institute Inc.
SAS Institute Inc. (1990) SAS Technical Report P-200, SASiSTAT Software: CALIS and LOGISTIC Procedures, Release 6.04. Cary, NC: SAS Insti- tute Inc.
Scheihel, A.B. (1984) Changes in brain capillary structure in aging and dementia. In J. Wertheimer and M. Marois (eds): Senile Dementia: Outlook for the Future. New York: Alan R. Liss, Inc., pp. 137-149.
Schlageter, N.L., R.E. Carson, and S.I. Rapoport (1987) Examination of blood-brain harrier permeability in dementia of the Alzheimer type with [68GalEDTA and positron emission tomography. J. Cereh. Blood Flow Metah. 7:l-8.
Schmidt, M.L., R.E. Gur, R.C. Gur, and J.Q. Trojanowski (1988) Intraneuro- nal and extracellular neurofihrillary tangles exhibit mutually exclusive cytoskeletal antigens. Ann. Neurol. 23:184-189.
Schwalbe, R.A., B. Dahlback, and G.L. Nelsestuen (1990) Independent association of serum amyloid P component, protein S, and complement C4b with complement C4b-binding protein and subsequent association of the complex with membranes. J. Biol. Chem. 265:21749-21757.
Schwalbe, R.A., B. Dahlhack, and G.L. Nelsestuen (1991) Heparin influence on the complex of serum amyloid P component and complement C4h-binding protein. J. Biol. Chem. 266:12896-12901.
Shirahama, T., M. Skinner, P. Westermark, A. Rubinow, AS. Cohen, A. Brun, and T.L. Kemper (1982) Senile cerebral amyloid prealhumin as a common constituent in the neuritic plaque, in the neurofibrillary tangle, and the microangiopathic lesion. Am. J. Pathol. 107:41-50.
Siripont, J., J.M. Teho, and R.F. Mortensen (1988) Receptor-mediated

AMYLOID P COMPONENT IN HUMAN BRAIN 105
binding of the acute-phase reactant mouse serum amyloid P-component (SAP) to macrophages. Cell Immunol. 11 7.939-252.
Snow, A.D., H. Mar, D. Nochlin, R.T. Sekiguchi, K. Kimata, Y. Koike, and T.N. Wight (1990) Earlyaccumulation of heparan sulfate in neurons and in the beta-amyloid protein containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 137:1253-1270.
Stewart, P.A., M. Magliocco, K. Hayakawa, C.L. Farrell, R.F. DelMaestro, J. Girvin, J.C.E. Kaufmann, H.V. Vinters, and J. Gilbert (1987) Aquantita- tive analysis of blood-brain barrier ultrastructure in the aging human. Microvasc. Res. 33.5270-282.
Stewart, P.A., K. Hayakawa, M.-A. Akers, and H.V. Vinters (1992) A morphometric study of the blood-brain barrier in Alzheimer’s disease. Lab. Invest. 67:734-742.
Tabaton, M., S. Cammarata, G. Mancardi, V. Manetto, L. Autilio-Gambetti, G Perry, and P. Gambetti (1991) Ultrastructural localization of p-amy- loid, 7, and ubiquitin epitopes in extracellular neurofibrillary tangles. Proc. Natl. Acad. Sci. USA88:2098-2102.
Westermark, P., M. Skinner, and AS. Cohen (1975) The P-component of amyloid of human islets of langerhans. Seand. J. Immunol. 4:95-97.
Wisniewski, H.M., J. Wegiel, K.C. Wang, and B. Lach (1992) Ultrastructural studies of the cells forming amyloid in the cortical vessel wall in Alzheimer’s disease. Acta Neuropathol. 84:117-127.
Wisniewski, T., and B. Frangione (1992) Apolipoprotein E: A pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci. Lett. 135235-238.
Wisniewski, T., A. Golabek, E. Matsubara, J. Ghiso, and B. Frangione (1993) Apolipoprotein E: Binding to soluble Alzheimer’s p-amyloid. Biochem. Biophys. Res. Commun. 192359-365.
Wright, S.D., L.S. Craigmyle, and S.C. Silverstein (1983) Fibronectin and serum amyloid P component stimulate C3b- and C3hi-mediated phagocy- tosis in cultured human monocytes. J. Exp. Med. 158:1338-1343.
Yamaguchi, H., S. Hirai, M. Morimatsu, M. Shoji, and Y. Ihara (1988) A variety of cerebral amyloid deposits in the brains of the Alzheimer-type dementia demonstrated by beta immunoprotein staining. Acta Neuro- pathol. 76:541-549.
Yamaguchi, H., Y. Nakazato, T. Kawarabayashi, K. Ishiguro, Y. Ihara, M. Morimatsu, and S. Hirai (1991a) Extracellular neurofibrillary tangles associated with degenerating neurites and neuropil threads in Alzheimer- type dementia. Acta Neuropathol. 813503-609.
Yamaguchi, H., Y. Nakazato, M. Shoji, K. Okamoto, Y. Ihara, M. Morimatsu, and S. Hirai (1991b) Secondary deposition of beta amyloid within extracellular neurofibrillary tangles in Alzheimer-type dementia. Am. J. Pathol. 138:699-705.
Yang, G.C.H., and G.R. Gallo (1990) Protein A-Gold immunoelectron microscopic study of amyloid fibrils, granular deposits, and fibrillar luminal aggregates in renal amyloidosis. Am. J. Pathol. 137;1223-1231.
Yang, G.C.H., R. Nieto, I. Stachura, and G.R. Gallo (1992) Ultrastructural immunohistochemical localization of polyclonal IgG, C3, and amyloid P component on the Congo red-negative amyloid-like fibrils of fibrillary glomerulopathy. Am. J. Pathol. 141:409419.
Copyright © 2022 FDOKUMEN




















