
Stability of Pt-Modified Cu (111) in the Presence of Oxygen and Its Implication on the Overall...
10
Stability of Pt-Modified Cu(111) in the Presence of Oxygen and Its Implication on the Overall Electronic Structure Henrik O ̈ berg, † Toyli Anniyev, ‡,§,∥ Aleksandra Vojvodic, ⊥,# Sarp Kaya, ‡,§ Hirohito Ogasawara, § Daniel Friebel, ‡,§ Daniel J. Miller, ‡,§ Dennis Nordlund, § Uwe Bergmann, ▽ Mathias P. Ljungberg, † Frank Abild-Pedersen, ⊥ Anders Nilsson, †,‡,§ and Lars G. M. Pettersson* ,† † Department of Physics, AlbaNova University Center, Stockholm University, S-106 91 Stockholm, Sweden ‡ Stanford Institute for Materials and Energy Sciences, § Stanford Synchrotron Radiation Lightsource, ⊥ SUNCAT Center for Interface Science and Catalysis, and ▽ Linac Coherent Light Source, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, Menlo Park, California 94025, United States ∥ Department of Physics and # Department of Chemical Engineering, Stanford University, Stanford, California 94305, United States * S Supporting Information ABSTRACT: The electronic structure and stability of Cu(111)-hosted Pt overlayers with and without the presence of atomic oxygen have been studied by means of core-level spectroscopy and density functional theory (DFT). Because of lattice mismatch, Pt(111) overlayers grown on Cu(111) are compressively strained, and hard X-ray photoelectron spectroscopy together with Pt L 3 -edge X-ray absorption spectroscopy (XAS) reveals a pronounced downshift of the Pt d-band owing to the increased overlap of the d-orbitals, an effect also reproduced theoretically. Exposure to oxygen severely alters the surface composition; the O−Cu binding energy largely exceeds that of O−Pt, and DFT calculations predict surface segregation of Cu atoms. Comparing the adsorbate electronic structure for O on unstrained Pt(111) with that of O on Pt- modified Cu(111) using O K-edge XAS and X-ray emission spectroscopy salient differences are observed and calculations show that Cu-segregation to the topmost layer is required to reproduce the measured spectra. It is proposed that O is binding in a hollow site constituted by at least two Cu atoms and that up to 75% of the Pt atoms migrate below the surface. ■ INTRODUCTION An important goal of catalyst design is optimizing the turnover frequency while minimizing the loading of costly precious metals. In fuel cell catalysis, for example, there is a pressing need to reduce the Pt loading of the electrocatalyst and enhance the rate of the oxygen reduction reaction (ORR) taking place at the cathode. Several recent efforts to develop efficient bimetallic catalysts have focused on preparation of “Pt- skin” structures from Pt bimetallic alloys through Pt surface segregation. Indeed, a number of Pt-skin structures have been reported to have ORR activities superior to pure Pt. 1−5 Strained Pt overlayer structures have also been prepared by preferential dissolution (dealloying) of the electrochemically less-noble elements of bimetallic alloys. 6,7 Significant experimental 8−11 and theoretical 12−15 efforts have been dedicated to understanding the reactivity of these materials; of particular interest is how substrate-induced effects modify the electronic and geometric properties of overlayers. 16 For instance, surface strain, which arises from the difference of the lattice parameter of the overlayer from its bulk value, plays a decisive role in the reactivity of these surfaces. 14 Density functional theory (DFT) calculations showed that the adsorption properties of strained overlayers can be modified by changing the nearest-neighbor distances. 14 The modified properties were related to the shift in energy of the d-band center of the surface layer, which was suggested as a single effective measure of the surface−adsorbate interaction energy controlling the kinetics. 17 When discussing Pt skin structures, one crucial question is whether the composition of the exposed surface is stable under reaction conditions. The choice of elements should be such that the core atoms remain in the core instead of segregating to the surface. Surface segregation of the core atoms may be driven by temperature, adsorbates, and electrochemical potential con- ditions. Ma and Balbuena evaluated the surface segregation trend of Pt 3 M (M = Fe, Co, and Ni) alloys in the presence of adsorbed atomic oxygen based on first-principles calculations. 18 They concluded that neither Pt- nor M-segregation is favored on the (111) surface at 1/4 ML oxygen coverage but rather the nonsegregated structures remain the most thermodynamically stable. On the other hand, Ramirez-Caballero et al. investigated surface segregation of Pt/M(111) skin structures under vacuum Received: January 21, 2013 Revised: June 20, 2013 Published: July 18, 2013 Article pubs.acs.org/JPCC © 2013 American Chemical Society 16371 dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−16380
-
Upload
independent -
Category
Documents
-
view
4 -
download
0
Transcript of Stability of Pt-Modified Cu (111) in the Presence of Oxygen and Its Implication on the Overall...
Stability of Pt-Modified Cu(111) in the Presence of Oxygen and ItsImplication on the Overall Electronic StructureHenrik Oberg,† Toyli Anniyev,‡,§,∥ Aleksandra Vojvodic,⊥,# Sarp Kaya,‡,§ Hirohito Ogasawara,§
Daniel Friebel,‡,§ Daniel J. Miller,‡,§ Dennis Nordlund,§ Uwe Bergmann,▽ Mathias P. Ljungberg,†
Frank Abild-Pedersen,⊥ Anders Nilsson,†,‡,§ and Lars G. M. Pettersson*,†
†Department of Physics, AlbaNova University Center, Stockholm University, S-106 91 Stockholm, Sweden‡Stanford Institute for Materials and Energy Sciences, §Stanford Synchrotron Radiation Lightsource, ⊥SUNCAT Center for InterfaceScience and Catalysis, and ▽Linac Coherent Light Source, SLAC National Accelerator Laboratory, 2575 Sand Hill Road, MenloPark, California 94025, United States∥Department of Physics and #Department of Chemical Engineering, Stanford University, Stanford, California 94305, United States
*S Supporting Information
ABSTRACT: The electronic structure and stability of Cu(111)-hostedPt overlayers with and without the presence of atomic oxygen have beenstudied by means of core-level spectroscopy and density functional theory(DFT). Because of lattice mismatch, Pt(111) overlayers grown onCu(111) are compressively strained, and hard X-ray photoelectronspectroscopy together with Pt L3-edge X-ray absorption spectroscopy(XAS) reveals a pronounced downshift of the Pt d-band owing to theincreased overlap of the d-orbitals, an effect also reproduced theoretically.Exposure to oxygen severely alters the surface composition; the O−Cubinding energy largely exceeds that of O−Pt, and DFT calculationspredict surface segregation of Cu atoms. Comparing the adsorbateelectronic structure for O on unstrained Pt(111) with that of O on Pt-modified Cu(111) using O K-edge XAS and X-ray emission spectroscopy salient differences are observed and calculations showthat Cu-segregation to the topmost layer is required to reproduce the measured spectra. It is proposed that O is binding in ahollow site constituted by at least two Cu atoms and that up to 75% of the Pt atoms migrate below the surface.
■ INTRODUCTION
An important goal of catalyst design is optimizing the turnoverfrequency while minimizing the loading of costly preciousmetals. In fuel cell catalysis, for example, there is a pressingneed to reduce the Pt loading of the electrocatalyst andenhance the rate of the oxygen reduction reaction (ORR)taking place at the cathode. Several recent efforts to developefficient bimetallic catalysts have focused on preparation of “Pt-skin” structures from Pt bimetallic alloys through Pt surfacesegregation. Indeed, a number of Pt-skin structures have beenreported to have ORR activities superior to pure Pt.1−5 StrainedPt overlayer structures have also been prepared by preferentialdissolution (dealloying) of the electrochemically less-nobleelements of bimetallic alloys.6,7
Significant experimental8−11 and theoretical12−15 efforts havebeen dedicated to understanding the reactivity of thesematerials; of particular interest is how substrate-induced effectsmodify the electronic and geometric properties of overlayers.16
For instance, surface strain, which arises from the difference ofthe lattice parameter of the overlayer from its bulk value, plays adecisive role in the reactivity of these surfaces.14 Densityfunctional theory (DFT) calculations showed that theadsorption properties of strained overlayers can be modified
by changing the nearest-neighbor distances.14 The modifiedproperties were related to the shift in energy of the d-bandcenter of the surface layer, which was suggested as a singleeffective measure of the surface−adsorbate interaction energycontrolling the kinetics.17
When discussing Pt skin structures, one crucial question iswhether the composition of the exposed surface is stable underreaction conditions. The choice of elements should be such thatthe core atoms remain in the core instead of segregating to thesurface. Surface segregation of the core atoms may be driven bytemperature, adsorbates, and electrochemical potential con-ditions. Ma and Balbuena evaluated the surface segregationtrend of Pt3M (M = Fe, Co, and Ni) alloys in the presence ofadsorbed atomic oxygen based on first-principles calculations.18
They concluded that neither Pt- nor M-segregation is favoredon the (111) surface at 1/4 ML oxygen coverage but rather thenonsegregated structures remain the most thermodynamicallystable. On the other hand, Ramirez-Caballero et al. investigatedsurface segregation of Pt/M(111) skin structures under vacuum
Received: January 21, 2013Revised: June 20, 2013Published: July 18, 2013
Article
pubs.acs.org/JPCC
© 2013 American Chemical Society 16371 dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−16380

and with adsorbed oxygen.19 They predicted that 3d transitionmetal cores in these alloys are unstable both under vacuum andwith 0.25 ML oxygen and tend to segregate to the surface.Menning et al. studied the Pt-M3d-Pt structures in the presenceof oxygen in a combined theoretical and experimental studyand found that the M3d subsurface layer tends to segregate tothe surface with 0.5 ML adsorbed oxygen.20 Volker et al. alsoreported oxygen adsorption-induced surface segregation of Cuatoms in the AuCu bimetallic polycrystalline system.21
In the present work, we consider Pt overlayers epitaxiallygrown on Cu(111) as a model for Pt skin and investigate thevalence electronic structure of the overlayers and ofchemisorbed oxygen on these overlayers in an atom-specificway. By combining synchrotron-based X-ray spectroscopies [OK-edge X-ray absorption and emission spectroscopies (XASand XES, respectively), hard X-ray photoelectron spectroscopy(HAXPES), and Pt L3-edge high-energy-resolution fluorescencedetection X-ray absorption spectroscopy (HERFD XAS)] withDFT calculations, we are able to correlate the bondingcharacteristics of adsorbed O to the electronic structure ofthe surface alloy and also quantify the stability of the differentcompositions at the surface with respect to segregation.
■ EXPERIMENTAL DETAILS
Preparation of Pt(111), Cu(111), Pt/Cu(111), O−Pt(111), and O−Pt/Cu(111). The Pt(111) and Cu(111)crystals were cleaned in an ultrahigh-vacuum (UHV) chamberwith a base pressure better than 1 × 10−10 Torr by cycles ofNe+ ion bombardment at room temperature and annealing to1000 K and 850 K, respectively. The cleaning cycles wererepeated until the contamination levels of C and O were belowthe detection limit of XPS (<0.1%), and the quality of thesurface order was confirmed by low energy electron diffraction(LEED). Heating was achieved using electron bombardmentfrom a tungsten filament situated behind the samples. Thetemperature was measured by chromel−alumel thermocouplesthat were inserted into holes in the sides of the crystals.Pt was evaporated onto Cu(111) using a home-built
evaporator comprising a resistively heated Pt filament with0.25 mm diameter at room temperature. The Pt coverage andgrowth mode were deduced from the ratio of the Pt 4f and Cu2p XPS integrated peak intensities, and Frank−Van der Merwelayer-by-layer growth mode was confirmed (see SupportingInformation for details).22 All Pt coverages up to 5 MLreported in the present work were estimated to within an errorof 0.1 ML. The lateral strain in the deposited Pt layers (∼4%and 3.5% compressive strain for the 1.8 and 2.9 ML thickepitaxially grown overlayers, respectively) was determined usingLEED and was found to be in excellent agreement with aprevious measurement (see Supporting Information).23 Theatomic oxygen layers on Pt(111) and Pt/Cu(111) wereprepared by dosing O2 at a temperature of 375 K to avoidCO contamination. O2 was dosed until saturation coverage wasreached, which according to O 1s XPS is similar for the strainedPt overlayers and for Pt(111) (∼0.25 ML).24 LEED patternsafter exposing the Pt films to oxygen showed neitherdestruction of the pattern due to massive reconstruction norany additional superstructure indicating an ordered oxygenlayer. We also investigated the effect of the temperature onthese systems in the absence of oxygen and found that Pt/Cuintermixing takes place above 450 K and thus did not go above375 K in the surface treatments.
Measurement Details. O K-edge XAS, XES, and XPSmeasurements were performed at beamline 13-2 at the StanfordSynchrotron Radiation Lightsource (SSRL) using an ellipticallypolarized undulator. The sample could be rotated around thepropagation axis of the incoming beam, whose incidence anglewas fixed at 5° with respect to the plane of the sample surface.The resolution of all XPS spectra is better than 0.2 eV. OxygenK-edge XAS spectra were obtained with an overall photon-energy resolution better than 0.1 eV by collecting O KVVAuger electrons. Unoccupied O 2p-states oriented parallel(2pxy) and perpendicular (2pz) to the surface were selectivelyprobed by rotating the sample with respect to the incidentphoton E-vector. The spectra were normalized to the incidentphoton flux, which was measured using the total photocurrentfrom a gold grid located after the refocusing mirror.Additionally, all XAS spectra have been corrected for thebackground contribution of the clean metal substrate.Oxygen K-edge XES spectra were recorded using a grazing
incidence grating spectrometer with a multichannel-platedetector. A nickel-coated spherical−elliptical grating with agroove density of 1100 lines/mm, a curvature of 5 m, and alength of 10 cm was used. With the grazing-incidence angle onthe grating fixed at 5°, XES spectra with an energy resolution of0.7 eV were obtained. The excitation energy was set to the firstabsorption resonance as determined by XAS. Utilizing thealignment of the E-vector of the emitted light with the O 2porbital,25 emission from O 2pxy and 2pxy + 2pz orbitals wasselectively obtained by collecting X-rays in normal and grazingemission, respectively. The 2pz component was extracted bysubtracting ∼50% of the spectra measured in normal emission(O 2pxy only) from those collected in grazing emission(normalized to the same area).25
The HAXPES spectra were obtained at beamline BL47XU atthe SPring-8 synchrotron radiation facility with a totalresolution of 0.25 eV; the incidence angle was set to 0.5° toincrease the surface sensitivity. Pt L3-edge HERFD XASmeasurements were carried out at wiggler beamline 6-2 atSSRL using the Si(111) double-crystal monochromator. The X-ray emission spectrometer, operated in a Rowland circlegeometry, was set up to use the (660) Bragg reflection fromthree spherically bent (R = 1 m) Ge crystals26 and aligned tothe center of the Pt Lα1 emission line; the combined resolutionof the monochromator and analyzers was 1.9 eV, as determinedby the elastic peak full width at half-maximum (fwhm).
■ THEORETICAL DETAILSThe theoretical analysis was performed using GPAW,27,28
which is a real-space-grid-based all-electron DFT codeimplemented in the projector-augmented wave (PAW) formal-ism.29 All calculations were performed using the RPBEexchange-correlation (XC) functional,30 a revision to the PBEXC-functional,31 and a grid spacing of 0.2 Å. Monkhorst−Pack32 k-point sampling was performed using a (4 × 4 × 1)grid for the total energy calculations and with a (10 × 10 × 1)grid for the d-density of states (d-DOS) calculations. Aprevious study of the O/Pt(111) system confirmed that Obinding energies are converged for this choice of parameters.33
The metal surfaces were represented by four-layers-thick(√3 × 2) or (2 × 2) super cells and optimized by relaxingthe two topmost layers while keeping the bottom two layersfixed. In addition, in the adsorption calculations the oxygenatoms were also allowed to relax. Oxygen adsorption energiesare defined as Eads = E[O/slab] − E[slab] − 1/2E[O2]; because
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016372
the high-spin ground state of the O2 molecule is poorlydescribed in DFT,34 E[O2] is calculated from the formationreaction of water from H2 and O2 using the gas-phase energy ofH2O and H2.
35 The PBE lattice parameters of Pt and Cu wereused in the calculations, but a comparison of the PBE andRPBE DOS does not show any significant differences. The d-DOS was calculated as the sum of the local density of states ofthe atomic d-orbitals of the surface Pt and Cu atoms, while thed-band centers were obtained as the first moment of the d-DOS.All XAS spectra were calculated using the half-core−hole
transition-potential approach36,37 as implemented in GPAW38
with a PAW setup containing a half core−hole at the excitedoxygen. The XAS cross-section was obtained using theHaydock recursion scheme39 with 2000 Lanczos vectors, thusavoiding explicitly constructing the unoccupied states. Thespectra were then Gaussian broadened with a fwhm of 0.4 eVfor better comparison with experiment. Because we haveperiodic boundary conditions, large cell sizes (∼15 × 15 × 30Å3) were employed to eliminate core−hole interactionsbetween the cells.The XES spectra were calculated using a code developed by
Ljungberg et al.40 in which vibrational interference effects areincluded through the Kramers−Heisenberg (KH) formula. Thecross-section for nonresonant XES is written in the KHformulation as
∑ ∑σ ωω
′ ∝⟨ | ′ | ⟩⟨ | ′ | ⟩′ − − + Γ
D DE E
( )f n n i
( ) if n
FN NI
n f
2
where, e.g., DFN is DFN(R) = ⟨F|D|N⟩ and lower case lettersdenote vibrational states on the initial (I), intermediate (N),and final (F) electronic potential energy surfaces (PES). In thisapproach we consider the one-dimensional oxygen-surfacestretch mode where the oxygen distance to the surface mapsout the PES’s with a step size of 0.025 Å. The PES’s areconstituted by 60 steps and calculated using the GPAW code.For O adsorbed on Pt(111) and unsegregated Pt/Cu(111), thePES’s are obtained from performing single-point calculations ofeach step while for the segregated systems, the surface isallowed to relax in each step (the O-surface distance and thebottom two layers are kept fixed) to obtain a more accuratedescription of the PES’s; transition moments at each step werecalculated using the ground-state orbitals.41,42 The lifetimewidth, Γ, of the core−hole was chosen as the experimental, 0.18eV,43 although the spectral features are rather insensitive tosmall changes in the lifetime broadening. All calculated spectrawere shifted to match the onset of the experimental spectrabecause Ef was found to vary slightly among the structures,causing uncertainty in the binding energy scale.
■ RESULTS AND DISCUSSION
Electronic Structure of Pt Overlayers on Cu(111). Toinvestigate how compressive strain and ligand interactionsinfluence the electronic structure of Pt overlayers, we
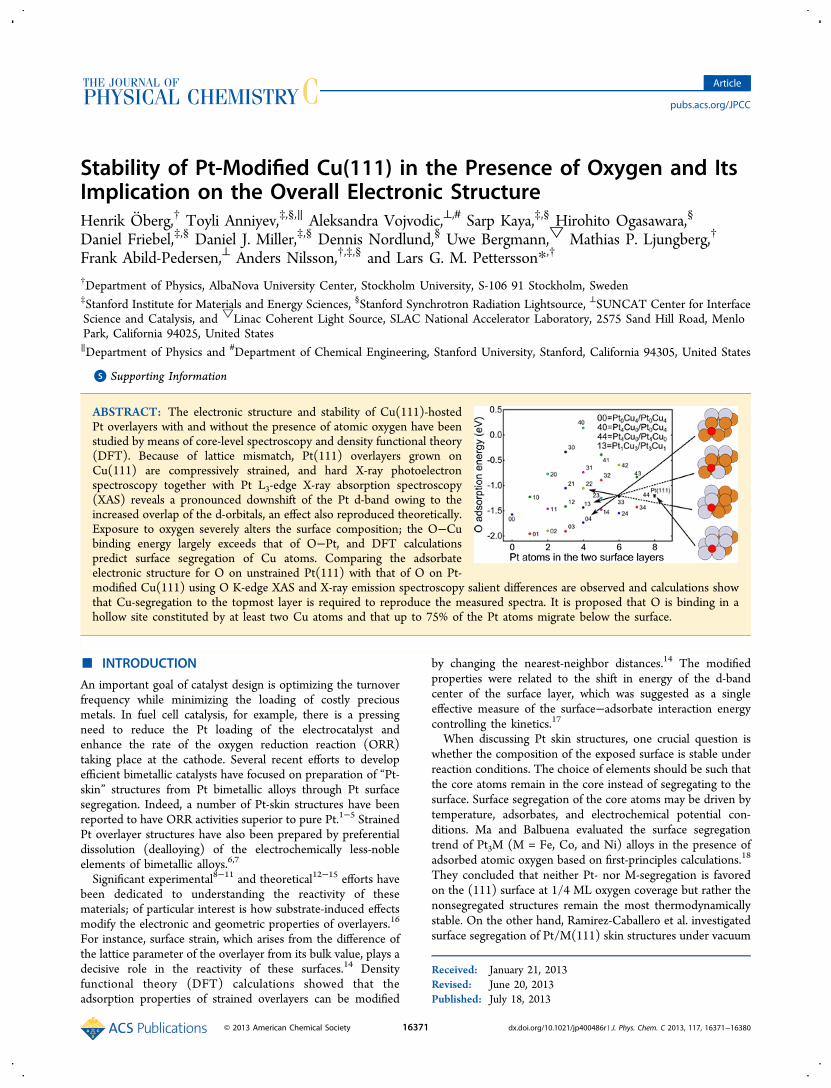
Figure 1. (a) HAXPES and Pt L3-edge XAS spectra for polycrystalline Pt foil and 1.8 and 2.9 ML Pt/Cu(111). The HAXPES spectra werebackground subtracted and normalized to the orbital occupancy obtained from DFT calculations. The Pt L3-edge XAS spectra (taken with theHERFD method) were normalized to the intensity 10 eV above the absorption edge. (b) Calculated DOS projected onto the d-states of pure andstrained surface layer of Pt(111) and of Pt overlayers on Cu(111). (c) Side views of 1 and 2 ML Pt overlayers on Cu(111.).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016373

performed valence band HAXPES and Pt L3-edge HERFD XASmeasurements on Pt foil and 1.8 and 2.9 ML Pt/Cu(111)samples (Figure 1).Whereas soft X-ray valence band photoemission spectra from
thin Pt/Cu(111) samples (Pt thickness <5 ML) exhibitsubstrate-related features despite surface-sensitive measure-ment,44 HAXPES can probe the partial Pt d-DOS selectivelybecause the favorable ratio of the Pt 5d and Cu 3d crosssections at 8 keV (∼29.3) significantly suppresses the signalfrom the Cu(111) substrate.45 The HAXPES spectra of 1.8 and2.9 ML Pt/Cu(111) (Figure 1) are thus dominated by the Pt5d band with negligible contributions from the Cu substrate.Moreover, because the penetration depth of 8 keV photons at agrazing-incidence angle of 0.5° is ∼22.6 Å (∼10 Pt layers), theHAXPES spectrum of Pt(111) is expected to probe the Pt 5dDOS of bulk Pt; we can assume, then, that Pt(111) and Pt foilwould yield identical HAXPES spectra. We have therefore usedHAXPES spectra to deduce strain- and substrate-inducedchanges in the width (Wd) and center position (εd) of the Pt 5dband for the different Pt thicknesses of Pt/Cu(111).Pt L3-edge XAS is an element-specific technique that probes
the Pt-projected unoccupied d-DOS and therefore comple-ments soft X-ray XPS and HAXPES measurements of thevalence band. The near-edge region of Pt L3-edge spectraexhibits a characteristic “white-line” (WL) or absorptionmaximum due to 2p→5d transitions. Moreover, by acquiringXAS spectra using the HERFD method,26,46−48 we were able tosignificantly reduce the effect of the Pt 2p core−hole lifetimebroadening and thus better resolve variations in the WLintensity. Variations in the WL intensity thus indicate changesin unoccupied 5d states and consequently changes in the d-band filling arising from chemical interactions between the Ptoverlayer and the Cu(111) substrate (ligand effect). Figure 1aclearly demonstrates that the thinner Pt overlayers experiencestronger electronic structure modifications, which could not bestraightforwardly resolved into strain and ligand effects becauseboth of them exist simultaneously in the experiment.To be able to separate the substrate and strain effects in the
electronic structure when compressing the Pt overlayers, weperformed calculations on a set of model systems, namely purePt(111) slabs with optimized (a = 3.98 Å) and reduced latticeconstants (a = 3.78 Å, corresponding to a similar strain as forthe epitaxially grown Pt in the experiment), as well as 1 and 2ML of Pt on Cu(111) slabs (see Figure 1). Compressinglaterally the pure Pt substrate isolates effects in the electronicstructure induced by pure strain, while the model containingoverlayers of Pt on Cu(111) accounts for both strain and ligandeffects, i.e., also hybridization between the Pt overlayer and theCu(111) substrate. In Figure 1b, we show the computed DOSprojected onto the d-orbitals of the top layer Pt atoms in allfour models.The combined experimental and theoretical results shown in
Figure 1 exhibit a clear trend for the Pt overlayers compared tothe Pt foil: The WL intensity in the Pt L3-edge XAS isdecreased, the DOS at the Fermi level (EF) is reduced, and theintensity of the shoulder at 5 eV is increased. All these effectsare consistent with modifications of the Pt d-band. The reducedintensity of the WL is also an indication of a somewhatincreased Pt 5d-band occupation. Indeed, the d-band centersbased on the experimental distribution of occupied states of the1.8 and 2.9 ML Pt/Cu(111) samples are found to be at 3.69and 3.58 eV, respectively, as compared to 3.29 eV for Pt foil.
This picture is consistent with the d-DOS obtained from thecalculations where pure compressive strain broadens the Pt d-band significantly, resulting in an overall downshift of the bandto preserve the filling (ligand effects are not included here).49
The d-band center, εd, for structure-relaxed Pt(111) is 2.45 eVcompared to 2.75 eV for that of compressed Pt(111) (asmentioned, HAXPES probes the occupied states of the Pt 5d-band, hence the discrepancy in the absolute position of theexperimental and calculated εd, where the latter is calculated asthe first moment of the d-DOS integrated over the entire d-band). In the HAXPES spectrum we observe a pronouncedpeak with a singularity building up at EF which is not present inthe calculated DOS of the Pt surface atoms. However, if weproject the DOS on all Pt atoms in the cell (not shown in thefigure), hence taking the bulk contribution into account, thepeak arises and the bandwidth increases to that of theexperiment. We choose to show only the former to focus onthe surface contribution and facilitate comparison between thedifferent structure models. The singularity property of thepronounced peak can be attributed to the dynamics in the XESprocess often causing this kind of behavior near threshold;50
the calculated DOS is obtained from ground-state orbitals in asingle-particle approach and will therefore not capture theeffect. Because the corresponding states of the overlayersystems lie further below EF compared to that for Pt(111),the singularity toward EF is absent in the experiment for these.Adding ligand effects, i.e., having a hybridization between the
Pt overlayer and the Cu(111) substrate, broadens thecomputed Pt d-band further and consequently εd shifts downeven further. For the 1 ML thick Pt overlayer, there is anadditional feature at ∼6 eV in the Pt d-band. As a second layerof Pt is added, that is, we consider the 2 ML of Pt on Cu(111),this peak is smeared out and moves toward lower bindingenergy, i.e., becomes more Pt like, in good agreement with theHAXPES spectrum. Moreover, εd for 1 and 2 ML of Pt onCu(111) is 3.14 and 3.03 eV, respectively, which correspondsto a shift similar in magnitude to that derived from theexperiment. The combined effect of compressive strain and thesubstrate−overlayer interaction is therefore a downward shift ofthe center of the Pt 5d-band and fewer unoccupied d-statesabove EF in correspondence to the observed reduction in WLintensity.The d-band model readily rationalizes these results.
Assuming an approximately constant d-band occupation forthe 2.9 ML overlayer similar to pure Pt(111), which isrigorously correct if only strain effects are present,13 then εd andWd are coupled such that the d-band broadens as its centermoves away from EF.
14,51 The observed broadening of the Pt5d-band has a direct physical interpretation, since compressivestrain reduces the distance between nearest-neighbor surfaceatoms and thus increases the d−d orbital overlap. Pt−Cuhybridization, leading to increased Pt 5d-band filling, is alsoexpected to shift εd away from the EF, because the energy of thePt 5d-band is known to be lowered by alloying Pt with Cu.52
The observed downshift of the d-band moreover accords withcalculations of a pseudomorphic Pt monolayer on Cu(111)performed by Ruban et al.15
Similar spectroscopic evidence of Pt d-band downshift wasreported in our previous work for a core−shell strained Pt−Cunanoparticle catalyst, which exhibited exceptional activitytoward ORR.52 In that study it was shown, through acombination of HAXPES and Pt L2-edge XAS, how the Pt5d-band experienced a downshift due to strain and alloying
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016374
effects. In particular, a reduced d-DOS at the EF and reducedWL intensity were observed for the catalyst as compared tobulk Pt. The similarity of the spectroscopic features observedfor Pt−Cu nanoparticles and Pt/Cu(111) (features at 1 eV andaround 5 eV in HAXPES) reaffirms the validity of using thelatter system as a model for the clean surface of the real catalyst,that is, a catalyst in a nongaseous environment. However, whenconsidering oxygen adsorbed on the surface of Pt/Cu(111),discussed in the following section, severe adsorbate-inducedeffects are observed which further affect the electronic structureand cause segregation of Cu atoms to the surface.Stability of Pt Overlayers on Cu(111) upon O
Adsorption. The surface characteristics can change dramati-cally in the presence of a gaseous environment. Adsorbates can,for example, give rise to surface relaxation and surfacereconstruction and even induce segregation. When it comesto surface stability of multicomponent systems, e.g., layered oralloyed systems in the presence of adsorbates, there is a trade-off between the energy cost due to exchange of top surface layeratoms with the ones from the second surface layer or furtherdown in the bulk, and the energy gain due to adsorbate−surfacebond formation at the rearranged surface compared to theoriginal structure. If the latter is larger, the result is adsorbate-induced segregation. Previous studies have shown that atomicoxygen binds about 0.5 eV more strongly to Cu(111) than toPt(111),53 which indicates that O adsorption might lead tosegregation in a Pt−Cu binary system.To elucidate whether the existence of Cu in the topmost Pt
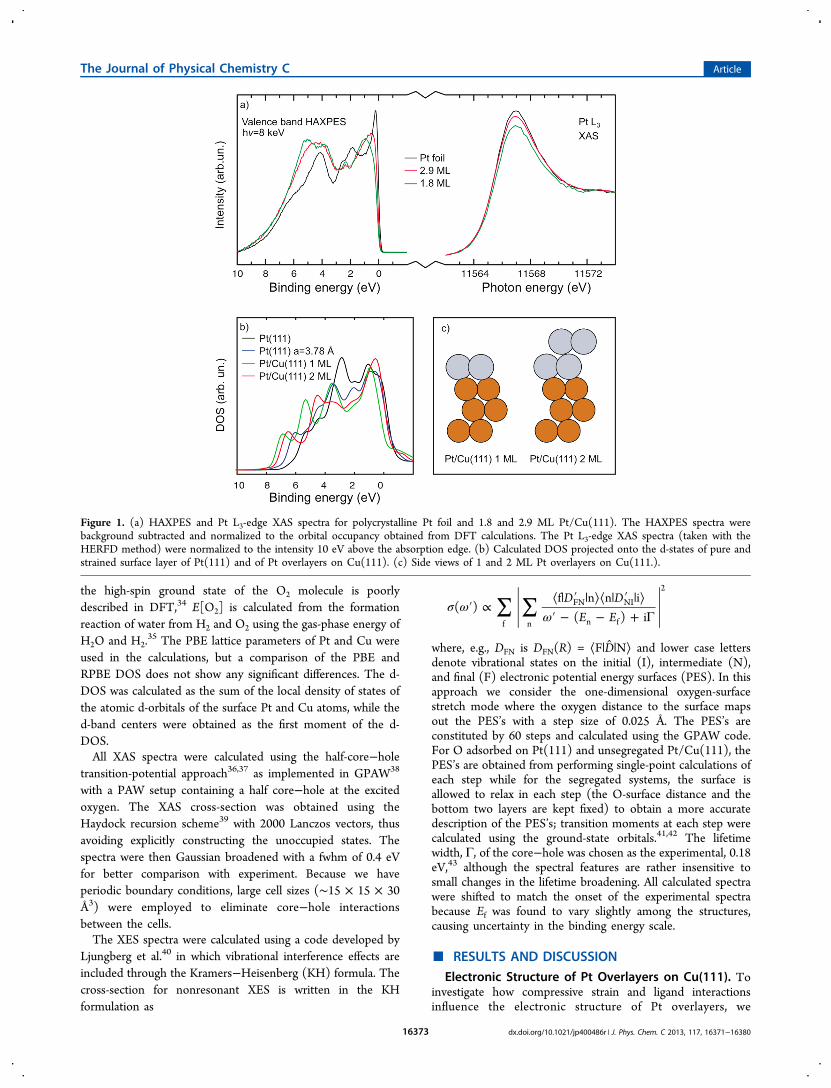
layers is favorable, we performed calculations of O adsorptionon PtxCu4−x/PtyCu4−y/Cu(111) surfaces with x and y = 1, 2, 3,or 4. In particular, a pure Cu(111) surface corresponds to x = y= 0 (denoted as xy = 00), a 1 ML Pt overlayer on the Cu(111)surface corresponds to x = 4 and y = 0 (denoted as xy = 40),and a surface with a 1:3 Pt:Cu ratio in the topmost surface layer(denoted as xy = 13) and a 3:1 Pt:Cu ratio in the secondsurface layer corresponds to x = 1 and y = 3 (denoted as xy =31). Because there are several surface compositions that havethe same chemical formula, we assign the letters A, B, C, and/orD to the four atom positions in the top surface layer and theletters E, F, G, and/or H to the different positions in the secondsurface layer to indicate where the Pt atoms are located. Thebinding energy of oxygen adsorbed at a 0.25 ML coverage ineither fcc or hcp sites was computed for all 256 possible surfacecombinations.Figure 2a shows the most stable O adsorption energies for
every combination of x and y for all considered PtxCu4−x/PtyCu4−y/Cu(111) surfaces. For all surfaces, we found thatadsorption in the fcc site is preferred over the hcp site. We findthat O binds more strongly on the Cu(111) surface (Eads =−1.57 eV) than on either the Pt(111) surface (Eads = −1.10eV), which is in agreement with previous studies,17 or the 1 MLPt/Cu(111) surface (Eads = 0.18 eV, note that this slightlypositive Eads means that the O adsorption relative to O2 in gasphase is not favorable), or the 2 ML Pt/Cu(111) surface (Eads =−1.22 eV). The O adsorption energy as a function of thenumber of Pt atoms in the surfaces forms a fan-like shape. Asimilar observation was found in a study of CO adsorption onCu−Pt surface alloys.53 We find that O adsorbs most stronglyon the Pt0Cu4/Pt1Cu3/Cu(111) surface (denoted as 01 inFigure 2), that is, the Cu(111) surface with one Pt atom in thesecond layer. For a given xy structure, we find that the strongestO-surface bond is at the surface with the minimal/maximal
amount of Pt/Cu atoms, which shows that the energy gain inthe O−Cu bond formation is large.The strongest O adsorption energy on a surface with a Pt
content corresponding to the 1 ML Pt overlayer in theexperiment (a total of four Pt in the two first surface layers) isthe Pt0Cu4/Pt4Cu0/Cu(111) (denoted as 04 in Figure 2); inother words, a Pt layer sandwiched as the second layer of theCu(111) surface and not the 1 ML Pt-overlayer surface(denoted as 40 in Figure 2). However, this finding is notenough to conclude if the 04 Pt−Cu near surface alloy will bethe favorable surface upon O adsorption. Therefore, we alsocalculate the O-induced segregation energies.Figure 2b shows the difference in O-induced segregation
energy as one Pt atom at a time segregates from the topmostsurface layer to the second surface layer, i.e., the energydifference between structure xy and structure (x − 1)(y + 1)(i.e. structures that have the same value of x + y); the numberof O, Pt, and Cu atoms remains constant in the comparison butone top-layer Pt atom and one second-layer Cu areinterchanged. We have chosen to analyze differences in Oinduced segregation energies because we are interested insystems with the same amount of Pt atoms and hence theresults are independent of the Pt and Cu reference energies. IfEO[xy] − EO[(x − 1)(y + 1)] > 0, the (x − 1)(y + 1) structureis more stable than the xy structure. If, on the other hand,EO[xy] − EO[(x − 1)(y + 1)] < 0, the xy structure is the morestable. Let us first focus on the structures with four Pt atoms inthe two surface layers. We find that the effect of O on thesegregation results in the Pt1Cu3/Pt3Cu1/Cu(111) surface (13in Figure 2, a surface with 1:3 ratio between Pt and Cu in thetopmost layer and a 3:1 ratio in the second layer) and thePt2Cu2/Pt2Cu2/Cu(111) surface (22 in Figure 2, a surface withhalf Pt and Cu in both the topmost and second layer) beingpreferred structures and not the 40 or the 04 structure. Two of
Figure 2. (a) Calculated most stable O adsorption energy onPtxCu4−x/PtyCu4−y/Cu(111) for all possible x and y between 1 and 4.(b) Calculated O induced segregation energies for systems with thesame amount Pt atoms, i.e., same x + y values. (c) Top view of therelaxed structures of O adsorbed on PtxCu4−x/PtyCu4−y/Cu(111) for afew representative surfaces.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016375
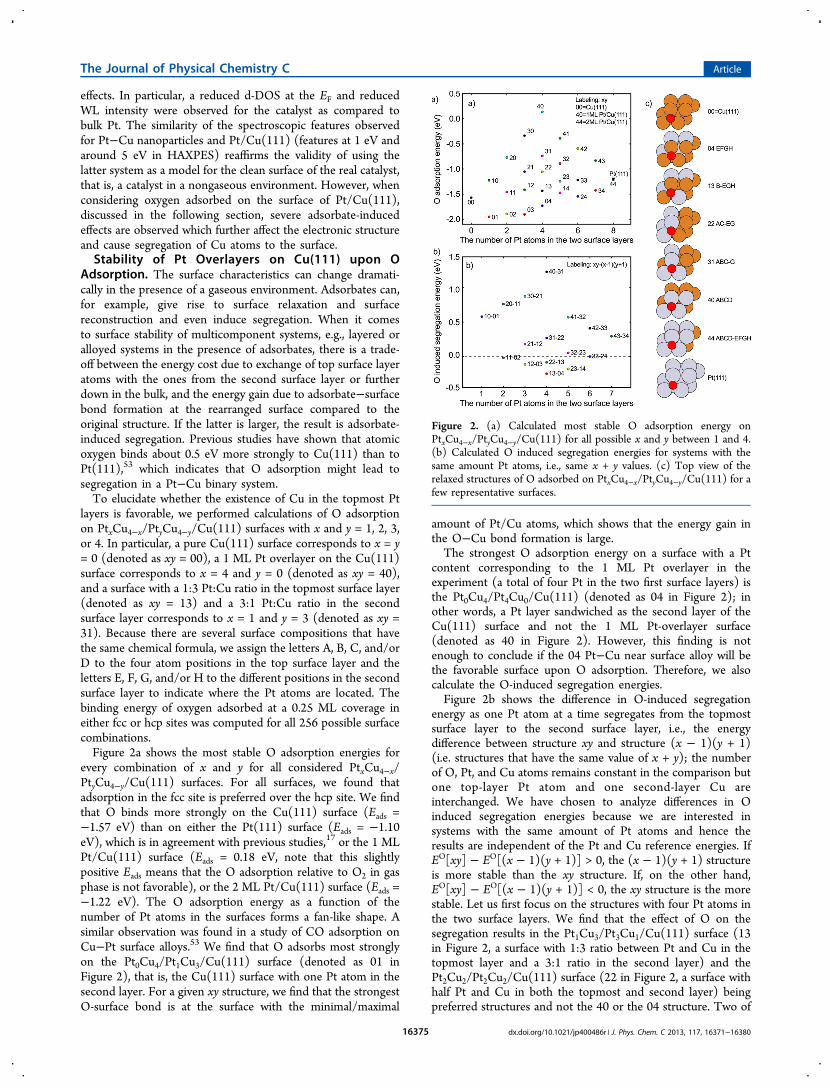
the Pt1Cu3/Pt3Cu1/Cu(111) surfaces, namely, the B-EGH andthe C-EGH surfaces with O adsorbed in the fcc site and bindingto two Cu and one Pt surface atoms are the most stablestructures by 0.05 eV compared to the other Pt1Cu3/Pt3Cu1/Cu(111) structures (see Figure 3 for structure models). Wechoose to model the spectra for the Pt1Cu3/Pt3Cu1/Cu(111)structures because the difference in total energy between Oadsorbed on the 22 and 13 structures is small (within theaccuracy of our calculations); hence, one could expect that theycoexist at finite temperatures. We have analyzed the DOS of thePt2Cu2/Pt2Cu2/Cu(111) structure, and it shows propertiessimilar to that for Pt1Cu3/Pt3Cu1/Cu(111) (see next section).We have performed an analysis of the thermodynamics of the
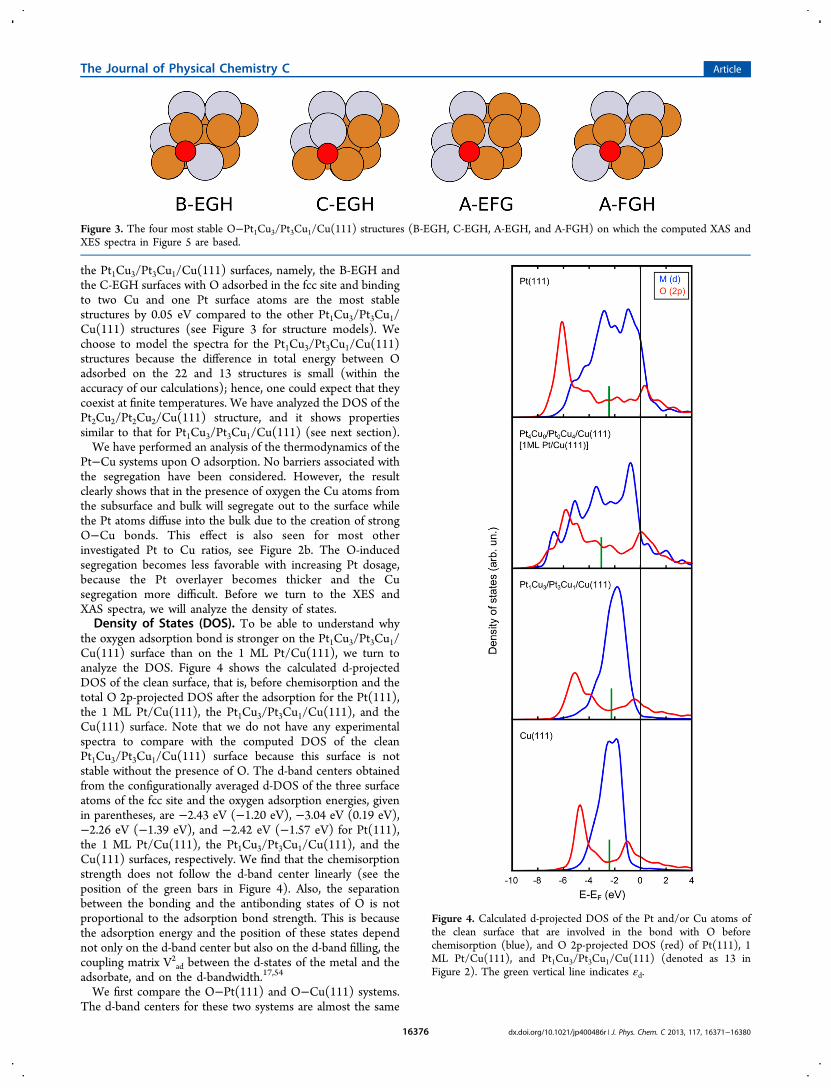
Pt−Cu systems upon O adsorption. No barriers associated withthe segregation have been considered. However, the resultclearly shows that in the presence of oxygen the Cu atoms fromthe subsurface and bulk will segregate out to the surface whilethe Pt atoms diffuse into the bulk due to the creation of strongO−Cu bonds. This effect is also seen for most otherinvestigated Pt to Cu ratios, see Figure 2b. The O-inducedsegregation becomes less favorable with increasing Pt dosage,because the Pt overlayer becomes thicker and the Cusegregation more difficult. Before we turn to the XES andXAS spectra, we will analyze the density of states.Density of States (DOS). To be able to understand why
the oxygen adsorption bond is stronger on the Pt1Cu3/Pt3Cu1/Cu(111) surface than on the 1 ML Pt/Cu(111), we turn toanalyze the DOS. Figure 4 shows the calculated d-projectedDOS of the clean surface, that is, before chemisorption and thetotal O 2p-projected DOS after the adsorption for the Pt(111),the 1 ML Pt/Cu(111), the Pt1Cu3/Pt3Cu1/Cu(111), and theCu(111) surface. Note that we do not have any experimentalspectra to compare with the computed DOS of the cleanPt1Cu3/Pt3Cu1/Cu(111) surface because this surface is notstable without the presence of O. The d-band centers obtainedfrom the configurationally averaged d-DOS of the three surfaceatoms of the fcc site and the oxygen adsorption energies, givenin parentheses, are −2.43 eV (−1.20 eV), −3.04 eV (0.19 eV),−2.26 eV (−1.39 eV), and −2.42 eV (−1.57 eV) for Pt(111),the 1 ML Pt/Cu(111), the Pt1Cu3/Pt3Cu1/Cu(111), and theCu(111) surfaces, respectively. We find that the chemisorptionstrength does not follow the d-band center linearly (see theposition of the green bars in Figure 4). Also, the separationbetween the bonding and the antibonding states of O is notproportional to the adsorption bond strength. This is becausethe adsorption energy and the position of these states dependnot only on the d-band center but also on the d-band filling, thecoupling matrix V2
ad between the d-states of the metal and theadsorbate, and on the d-bandwidth.17,54
We first compare the O−Pt(111) and O−Cu(111) systems.The d-band centers for these two systems are almost the same
Figure 3. The four most stable O−Pt1Cu3/Pt3Cu1/Cu(111) structures (B-EGH, C-EGH, A-EGH, and A-FGH) on which the computed XAS andXES spectra in Figure 5 are based.
Figure 4. Calculated d-projected DOS of the Pt and/or Cu atoms ofthe clean surface that are involved in the bond with O beforechemisorption (blue), and O 2p-projected DOS (red) of Pt(111), 1ML Pt/Cu(111), and Pt1Cu3/Pt3Cu1/Cu(111) (denoted as 13 inFigure 2). The green vertical line indicates εd.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016376
but there is a large difference in the O adsorption energy andthe separation between the bonding and antibonding states.This is because V2
ad for Pt is almost four times stronger than forCu, resulting in a more pronounced Pauli repulsion for the Ptsystem.17,54 In addition, because the Pt(111) band is muchbroader, i.e., the d-bandwidth is larger, the separation betweenthe bonding and the antibonding states is larger and the peaksare broader than for Cu(111).We find that the Pt1Cu3/Pt3Cu1/Cu(111) surface has a d-
band that mostly resembles that of the Cu(111) surface, in bothits center and width, while the d-band of 1 ML Pt/Cu(111)more resembles that of the Pt(111) surface. Therefore, the O2p-projected DOS for the Pt1Cu3/Pt3Cu1/Cu(111) system hasnarrower bonding and antibonding peaks, and the separationbetween them is smaller than the corresponding O 2p DOSpeaks for the 1 ML Pt/Cu(111) system. Note that, despite thepeak with antibonding character being more downshifted in thePt1Cu3/Pt3Cu1/Cu(111) system than in the 1 ML Pt/Cu(111)system, the oxygen chemisorption energy is stronger on thePt1Cu3/Pt3Cu1/Cu(111) surface. Again, this is because there isan interaction between the O and the Cu surface atoms that isstronger due to the weaker Pauli repulsion between O and Cucompared to that between O and Pt. In the following sectionwe will focus on the electronic structure through a comparisonof theoretical and experimental XES and XAS spectra.
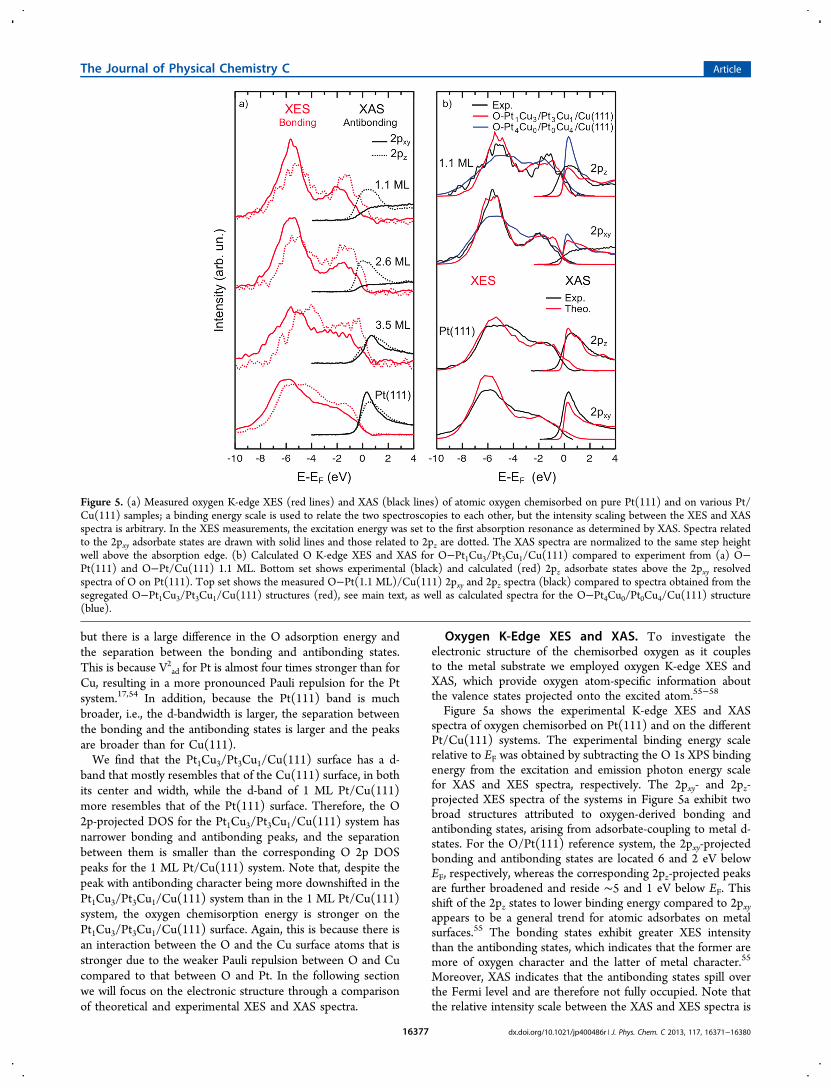
Oxygen K-Edge XES and XAS. To investigate theelectronic structure of the chemisorbed oxygen as it couplesto the metal substrate we employed oxygen K-edge XES andXAS, which provide oxygen atom-specific information aboutthe valence states projected onto the excited atom.55−58
Figure 5a shows the experimental K-edge XES and XASspectra of oxygen chemisorbed on Pt(111) and on the differentPt/Cu(111) systems. The experimental binding energy scalerelative to EF was obtained by subtracting the O 1s XPS bindingenergy from the excitation and emission photon energy scalefor XAS and XES spectra, respectively. The 2pxy- and 2pz-projected XES spectra of the systems in Figure 5a exhibit twobroad structures attributed to oxygen-derived bonding andantibonding states, arising from adsorbate-coupling to metal d-states. For the O/Pt(111) reference system, the 2pxy-projectedbonding and antibonding states are located 6 and 2 eV belowEF, respectively, whereas the corresponding 2pz-projected peaksare further broadened and reside ∼5 and 1 eV below EF. Thisshift of the 2pz states to lower binding energy compared to 2pxyappears to be a general trend for atomic adsorbates on metalsurfaces.55 The bonding states exhibit greater XES intensitythan the antibonding states, which indicates that the former aremore of oxygen character and the latter of metal character.55
Moreover, XAS indicates that the antibonding states spill overthe Fermi level and are therefore not fully occupied. Note thatthe relative intensity scale between the XAS and XES spectra is
Figure 5. (a) Measured oxygen K-edge XES (red lines) and XAS (black lines) of atomic oxygen chemisorbed on pure Pt(111) and on various Pt/Cu(111) samples; a binding energy scale is used to relate the two spectroscopies to each other, but the intensity scaling between the XES and XASspectra is arbitrary. In the XES measurements, the excitation energy was set to the first absorption resonance as determined by XAS. Spectra relatedto the 2pxy adsorbate states are drawn with solid lines and those related to 2pz are dotted. The XAS spectra are normalized to the same step heightwell above the absorption edge. (b) Calculated O K-edge XES and XAS for O−Pt1Cu3/Pt3Cu1/Cu(111) compared to experiment from (a) O−Pt(111) and O−Pt/Cu(111) 1.1 ML. Bottom set shows experimental (black) and calculated (red) 2pz adsorbate states above the 2pxy resolvedspectra of O on Pt(111). Top set shows the measured O−Pt(1.1 ML)/Cu(111) 2pxy and 2pz spectra (black) compared to spectra obtained from thesegregated O−Pt1Cu3/Pt3Cu1/Cu(111) structures (red), see main text, as well as calculated spectra for the O−Pt4Cu0/Pt0Cu4/Cu(111) structure(blue).
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016377

arbitrary such that the relative population of antibonding andbonding states cannot be concluded from the figure in a simpleway.The Fermi level (i.e., the zero of the energy scale) does not
coincide with the XAS peak positions for 1.1 and 2.6 ML Pt/Cu(111) but crosses the leading edges of the Pt(111) and 3.5ML Pt/Cu(111) spectra. It is known that the intensity and theenergy onset of the first absorption resonance could beinfluenced by the core−hole; the dynamic response of theexcited (or ionized) system could lead to modifications in XASspectra near the Fermi level. The measured XAS spectra maynot, therefore, directly reflect the unoccupied oxygen-projectedp-DOS; this effect was observed previously for O/Ni(100) andN/Ni(100)59 and was found to be stronger for adsorbatesystems with low DOS above EF. The shift of the XAS peakposition closer to EF with decreasing Pt thickness is thusconsistent with the expected increase in occupation of the O 2pstates.Turning to Figure 5b, we compare the symmetry-resolved
theoretical and experimental spectra for the O−Pt(111) system,bottom set, and the theoretical estimate to the 1.1 ML O−Pt/Cu(111) system, top set. To model the XES and XAS spectraof the PtCu alloy structures, i.e., the O−Pt1Cu3/Pt3Cu1/Cu(111) structures, which we predicted to be the most stablesystems upon O adsorption, we first calculated the spectra forthe four model systems with the lowest total energy, denoted B-EGH, C-EGH, A-EGH, and A-FGH (see Figure 3; forclarification of the notations, see the section Stability of PtOverlayers on Cu(111) upon O Adsorption). In these systemsthe O adsorbate is adsorbed in the fcc site, bonding to two Cusurface atoms and one Pt surface atom. The spectra were thenweighted together according to the Boltzmann distribution oftheir total energy differences; the obtained weights are 0.388(B-EGH), 0.388 (C-EGH), 0.127 (A-EFG), and 0.096 (A-FGH). Also, the spectra of O−Pt4Cu0/Pt0Cu4/Cu(111), i.e.,exposing O only to Pt, are shown as a reference.In both XES and XAS, the overall agreement between theory
and experiment is very good. Focusing on the XES for the O−Pt1Cu3/Pt3Cu1/Cu(111) system, we note in the 2pxy projectionan excellent overlap between the O−Pt1Cu3/Pt3Cu1/Cu(111)spectrum and the experiment while the computed spectrum ofthe O−Pt4Cu0/Pt0Cu4/Cu(111) system has significantly broad-er features and resembles that of O on Pt(111). A similarobservation is found for the 2pz resolved spectra where theseparation between the bonding and the antibonding peaksalmost disappears for the O−Pt4Cu0/Pt0Cu4/Cu(111) system,i.e., if segregation is not considered, indicating an exaggeratedinvolvement of Pt in the adsorbate−substrate bond comparedto the O−Pt1Cu3/Pt3Cu1/Cu(111) system.Turning to the unoccupied states in the XAS, the salient
feature near EF for the 2pz states in the experiment is wellreproduced by the spectrum representing the O−Pt1Cu3/Pt3Cu1/Cu(111) system, giving additional strength to theargument of Cu segregation, while for O−Pt4Cu0/Pt0Cu4/Cu(111), the peak is too pronounced, as also expected from theDOS analysis in the previous section where it was shown thatincreasing O coordination to Cu pulls the p-band further belowEF. In the 2pxy projection of the XAS, the experimentalspectrum has no distinct onset while for the O−Pt4Cu0/Pt0Cu4/Cu(111) system we observe a sharp feature comparableto that of O on Pt(111). For the O−Pt1Cu3/Pt3Cu1/Cu(111)spectrum, the peak intensity decreases as the representation ofPt in the 3-fold hollow to which O binds is reduced, and the
agreement with the experimental spectrum is slightly improved.The asymmetry of the binding hollow site of the O (to 2 Cuand 1 Pt) would possibly suggest a Franck−Condon effect, butan investigation using a linear coupling model60 givesinsignificant effects on the spectrum; this could possibly bedue to the small size of the surface unit cell restricting possibledistortions in the core-excited state. Despite the discrepancy inthe 2pxy projection of the XAS, the overall agreement betweenthe theoretical and experimental spectra is very good.Hence, we conclude that Cu segregation into the top layer is
required to reproduce the features in the experimental spectraand, comparing with the energetics in the previous section onwhich the choice of structures was made, we deem it likely thatthe magnitude of Cu segregation to the surface is up to 75%and that O favors bonding to a hollow site constituted by twoCu atoms and one Pt atom in the first layer.61
■ CONCLUSIONSHAXPES and Pt L3-edge XAS together with DFT calculationsshow that the d-band center of the Pt skin layers on Cu shiftsdown compared to that of pure Pt, as indicated by a broadeningof the Pt d-band and intensity reduction near the Fermi edge.As the influence of oxygen adsorption is considered, Cusegregation to the surface becomes favorable and the stability ofthe Pt skin structures change. Calculated adsorption energiesand relative stabilities predict a magnitude of up to 75% of Cumigration to the topmost surface layer. The calculated O K-edge XES and XAS spectra, selectively probing the O-derivedbonding and antibonding states in the adsorbate−substratesystem, show excellent agreement with the experiment. Thenarrowing in the O 2p-states and smaller separation betweenthe bonding and antibonding peaks as the adsorbate−metalorbital overlap changes are attributed to bond formationbetween O and Cu present in the topmost surface layer. Ourresults demonstrate the importance of looking beyond theproperties of the substrate itself in the pursuit of catalystcandidates; the stability, hence also the reactivity, of skincatalysts may under reaction conditions decrease significantly.In this respect it should be noted that electrochemicallydealloyed catalysts, such as those reported in ref 44, constitute aspecial case where the segregated component goes into solutionand does not remain at the surface.
■ ASSOCIATED CONTENT*S Supporting InformationThe estimation of the layer-by-layer growth of Pt overlayers andthe intrinsic strain accompanied is demonstrated. This materialis available free of charge via the Internet at http://pubs.acs.org.
■ AUTHOR INFORMATIONCorresponding Author*E-mail: [email protected] ContributionsThe manuscript was written through contributions from allauthors. All authors have given approval to the final version ofthe manuscript. H.O., T.A., and A.V. contributed equally.NotesThe authors declare no competing financial interest.
■ ACKNOWLEDGMENTSThis work is supported by the Department of Energy, Office ofBasic Energy Sciences, Division of Materials Sciences and
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016378

Engineering, under contract DE-AC02-76SF00515, and theSwedish Research Council (VR). This research was partlycarried out at the Stanford Synchrotron Radiation Lightsource,a National User Facility operated by Stanford University onbehalf of the U.S. Department of Energy, Office of Basic EnergySciences. Portions of this research were performed at SPring-8with the approval of Japan Synchrotron Radiation ResearchInstitute as Nanotechnology Support Project of the Ministry ofEducation, Culture, Sports, Science and Technology (proposalno. 2009B1751/BL-47XU). The DFT spectrum calculationswere performed on resources provided by the Swedish NationalInfrastructure for Computing (SNIC) at the HPC2N center.F.A.P. and A.V. gratefully acknowledge support from the U.S.Department of Energy under contract number DE-AC02-76SF00515. D.F. is grateful to the Alexander von HumboldtFoundation for a Feodor Lynen fellowship.
■ REFERENCES(1) Toda, T.; Igarashi, H.; Uchida, H.; Watanabe, M. Enhancementof the Electroreduction of Oxygen on Pt Alloys with Fe, Ni, and Co. J.Electrochem. Soc. 1999, 146, 3750−3756.(2) Stamenkovic, V. R.; Mun, B. S.; Arenz, M.; Mayrhofer, K. J. J.;Lucas, C. A.; Wang, G. F.; Ross, P. N.; Markovic, N. M. Trends inElectrocatalysis on Extended and Nanoscale Pt-Bimetallic AlloySurfaces. Nat. Mater. 2007, 6, 241−247.(3) Stamenkovic, V. R.; Fowler, B.; Mun, B. S.; Wang, G. F.; Ross, P.N.; Lucas, C. A.; Markovic, N. M. Improved Oxygen ReductionActivity on Pt3Ni(111) via Increased Surface Site Availability. Science2007, 315, 493−497.(4) Chen, S.; Ferreira, P. J.; Sheng, W. C.; Yabuuchi, N.; Allard, L. F.;Shao-Horn, Y. Enhanced Activity for Oxygen Reduction Reaction on“Pt3Co” Nanoparticles: Direct Evidence of Percolated and Sandwich-Segregation Structures. J. Am. Chem. Soc. 2008, 130, 13818−13819.(5) Chen, S.; Sheng, W. C.; Yabuuchi, N.; Ferreira, P. J.; Allard, L. F.;Shao-Horn, Y. Origin of Oxygen Reduction Reaction Activity on“Pt3Co” Nanoparticles: Atomically Resolved Chemical Compositionsand Structures. J. Phys. Chem. C 2009, 113, 1109−1125.(6) Koh, S.; Strasser, P. Electrocatalysis on Bimetallic Surfaces:Modifying Catalytic Reactivity for Oxygen Reduction by VoltammetricSurface Dealloying. J. Am. Chem. Soc. 2007, 129, 12624−12625.(7) Yang, R. Z.; Leisch, J.; Strasser, P.; Toney, M. F. Structure ofDealloyed PtCu3 Thin Films and Catalytic Activity for OxygenReduction. Chem. Mater. 2010, 22, 4712−4720.(8) Pedersen, M. Ø.; Helveg, S.; Ruban, A.; Stensgaard, I.; Lægsgaard,E.; Nørskov, J. K.; Besenbacher, F. How a Gold Substrate Can Increasethe Reactivity of a Pt Overlayer. Surf. Sci. 1999, 426, 395−409.(9) Schlapka, A.; Lischka, M.; Groß, A.; Kasberger, U.; Jakob, P.Surface Strain versus Substrate Interaction in Heteroepitaxial MetalLayers: Pt on Ru(0001). Phys. Rev. Lett. 2003, 91, 016101.(10) Yu, W.; Porosoff, M. D.; Chen, J. G. Review of Pt-BasedBimetallic Catalysis: From Model Surfaces to Supported Catalysts.Chem. Rev. 2012, 112, 5780−5817.(11) Zhang, J. L.; Vukmirovic, M. B.; Xu, Y.; Mavrikakis, M.; Adzic,R. R. Controlling the Catalytic Activity of Platinum-MonolayerElectrocatalysts for Oxygen Reduction with Different Substrates.Angew. Chem., Int. Ed. 2005, 44, 2132−2135.(12) Hammer, B.; Morikawa, Y.; Nørskov, J. K. CO Chemisorptionat Metal Surfaces and Overlayers. Phys. Rev. Lett. 1996, 76, 2141−2144.(13) Kitchin, J. R.; Nørskov, J. K.; Barteau, M. A.; Chen, J. G. Role ofStrain and Ligand Effects in the Modification of the Electronic andChemical Properties of Bimetallic Surfaces. Phys. Rev. Lett. 2004, 93,156801.(14) Mavrikakis, M.; Hammer, B.; Nørskov, J. K. Effect of Strain onthe Reactivity of Metal Surfaces. Phys. Rev. Lett. 1998, 81, 2819−2822.
(15) Ruban, A.; Hammer, B.; Stoltze, P.; Skriver, H. L.; Nørskov, J. K.Surface Electronic Structure and Reactivity of Transition and NobleMetals. J. Mol. Catal. A: Chem. 1997, 115, 421−429.(16) Anniyev, T.; Kaya, S.; Rajasekaran, S.; Ogasawara, H.; Nordlund,D.; Nilsson, A. Tuning the Metal−Adsorbate Chemical Bond throughthe Ligand Effect on Platinum Subsurface Alloys. Angew. Chem., Int.Ed. 2012, 51, 7724−7728.(17) Hammer, B.; Nørskov, J. K. Theoretical Surface Science andCatalysis - Calculations and Concepts. In Advances in Catalysis;Academic Press Inc: San Diego, 2000; Vol. 45, pp 71.(18) Ma, Y. G.; Balbuena, P. B. Pt Surface Segregation in BimetallicPt3M Alloys: A Density Functional Theory Study. Surf. Sci. 2008, 602,107−113.(19) Ramirez-Caballero, G. E.; Ma, Y.; Callejas-Tovar, R.; Balbuena,P. B. Surface Segregation and Stability of Core-Shell Alloy Catalysts forOxygen Reduction in Acid Medium. Phys. Chem. Chem. Phys. 2010, 12,2209−2218.(20) Menning, C. A.; Hwu, H. H.; Chen, J. G. Experimental andTheoretical Investigation of the Stability of Pt-3d-Pt(111) BimetallicSurfaces under Oxygen Environment. J. Phys. Chem. B 2006, 110,15471−15477.(21) Volker, E.; Williams, F. J.; Calvo, E. J.; Jacob, T.; Schiffrin, D. J.O2-Induced Cu Surface Segregation in Au-Cu Alloys Studied by Angle-Resolved XPS and DFT Modelling. Phys. Chem. Chem. Phys. 2012, 14,7448−7455.(22) Belkhou, R.; Barrett, N. T.; Guillot, C.; Barbier, A.; Eugene, J.;Carriere, B.; Naumovic, D.; Osterwalder, J. Growth of Pt/Cu(111)Characterized by Auger-Electron Spectroscopy, Core Level Photo-emission and X-Ray Photoelectron Diffraction. Appl. Surf. Sci. 1993,65−6, 63−70.(23) Fusy, J.; Menaucourt, J.; Alnot, M.; Huguet, C.; Ehrhardt, J. J.Growth and Reactivity of Evaporated Platinum Films on Cu(III): AStudy by AES, RHEED and Adsorption of Carbon Monoxide andXenon. Appl. Surf. Sci. 1996, 93, 211−220.(24) Puglia, C.; Nilsson, A.; Hernnas, B.; Karis, O.; Bennich, P.;Martensson, N. Physisorbed, Chemisorbed and Dissociated O2 onPt(111) Studied by Different Core-Level Spectroscopy Methods. Surf.Sci. 1995, 342, 119−133.(25) Nilsson, A. X-ray Emission Studies of Adsorbates. J. ElectronSpectrosc. Relat. Phenom. 1998, 93, 143−152.(26) Glatzel, P.; Bergmann, U. High Resolution 1s Core Hole X-raySpectroscopy in 3d Transition Metal Complexes - Electronic andStructural Information. Coord. Chem. Rev. 2005, 249, 65−95.(27) Mortensen, J. J.; Hansen, L. B.; Jacobsen, K. W. Real-Space GridImplementation of the Projector Augmented Wave Method. Phys. Rev.B 2005, 71, 035109.(28) Enkovaara, J.; Rostgaard, C.; Mortensen, J. J.; Chen, J.; Dułak,M.; Ferrighi, L.; Gavnholt, J.; Glinsvad, C.; Haikola, V.; Hansen, H. A.;et al. Electronic Structure Calculations with GPAW: A Real-SpaceImplementation of the Projector Augmented-Wave Method. J. Phys.:Condens. Matter. 2010, 22, 253202.(29) Blochl, P. E. Projector Augmented-Wave Method. Phys. Rev. B1994, 50, 17953−17979.(30) Hammer, B.; Hansen, L. B.; Nørskov, J. K. ImprovedAdsorption Energetics within Density-Functional Theory UsingRevised Perdew-Burke-Ernzerhof Functionals. Phys. Rev. B 1999, 59,7413−7421.(31) Perdew, J. P.; Burke, K.; Ernzerhof, M. Generalized GradientApproximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865−3868.(32) Monkhorst, H. J.; Pack, J. D. Special Points for Brillouin-ZoneIntegrations. Phys. Rev. B 1976, 13, 5188−5192.(33) Miller, D. J.; Oberg, H.; Naslund, L. Å.; Anniyev, T.; Ogasawara,H.; Pettersson, L. G. M.; Nilsson, A. Low O2 Dissociation Barrier onPt(111) Due to Adsorbate-Adsorbate Interactions. J. Chem. Phys.2010, 133, 224701.(34) Kurth, S.; Perdew, J. P.; Blaha, P. Molecular and Solid-StateTests of Density Functional Approximations: LSD, GGAs, and Meta-GGAs. Int. J. Quantum Chem. 1999, 75, 889−909.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016379

(35) Nørskov, J. K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.;Kitchin, J. R.; Bligaard, T.; Jonsson, H. Origin of the Overpotential forOxygen Reduction at a Fuel-Cell Cathode. J. Phys. Chem. B 2004, 108,17886−17892.(36) Leetmaa, M.; Ljungberg, M. P.; Lyubartsev, A.; Nilsson, A.;Pettersson, L. G. M. Theoretical Approximations to X-ray AbsorptionSpectroscopy of Liquid Water and Ice. J. Electron Spectrosc. Relat.Phenom. 2010, 177, 135−157.(37) Triguero, L.; Pettersson, L. G. M.; Ågren, H. Calculations ofNear-Edge X-ray-Absorption Spectra of Gas-Phase and ChemisorbedMolecules by Means of Density-Functional and Transition-PotentialTheory. Phys. Rev. B 1998, 58, 8097−8110.(38) Ljungberg, M. P.; Mortensen, J. J.; Pettersson, L. G. M. AnImplementation of Core Level Spectroscopies in a Real SpaceProjector Augmented Wave Density Functional Theory Code. J.Electron Spectrosc. Relat. Phenom. 2011, 184, 427−439.(39) Haydock, R.; Heine, V.; Kelly, M. J. Electronic Structure Basedon Local Atomic Environment for Tight-Binding Bands. J. Phys. C:Solid State Phys. 1972, 5, 2845−2858.(40) Ljungberg, M. P.; Pettersson, L. G. M.; Nilsson, A. VibrationalInterference Effects in X-ray Emission of a Model Water Dimer:Implications for the Interpretation of the Liquid Spectrum. J. Chem.Phys. 2011, 134, 044513.(41) Fohlisch, A.; Hasselstrom, J.; Bennich, P.; Wassdahl, N.; Karis,O.; Nilsson, A.; Triguero, L.; Nyberg, M.; Pettersson, L. G. M.Ground-State Interpretation of X-ray Emission Spectroscopy onAdsorbates: CO Adsorbed on Cu(100). Phys. Rev. B 2000, 61,16229−16240.(42) Triguero, L.; Pettersson, L. G. M.; Ågren, H. Calculations of X-ray Emission Spectra of Molecules and Surface Adsorbates by Meansof Density Functional Theory. J. Phys. Chem. A 1998, 102, 10599−10607.(43) Neeb, M.; Rubensson, J. E.; Biermann, M.; Eberhardt, W.Coherent Excitation of Vibrational Wave Functions Observed in CoreHole Decay Spectra of O2, N2 and CO. J. Electron Spectrosc. Relat.Phenom. 1994, 67, 261−274.(44) Strasser, P.; Koh, S.; Anniyev, T.; Greeley, J.; More, K.; Yu, C.F.; Liu, Z. C.; Kaya, S.; Nordlund, D.; Ogasawara, H.; et al. Lattice-Strain Control of the Activity in Dealloyed Core-Shell Fuel CellCatalysts. Nat. Chem. 2010, 2, 454−460.(45) Scofield, J. H. Theoretical Photoionization Cross Sections from 1 to1500 keV; Lawrence Livermore National Laboratory: Livermore, CA,1973.(46) Friebel, D.; Miller, D. J.; O’Grady, C. P.; Anniyev, T.; Bargar, J.;Bergmann, U.; Ogasawara, H.; Wikfeldt, K. T.; Pettersson, L. G. M.;Nilsson, A. In Situ X-ray Probing Reveals Fingerprints of SurfacePlatinum Oxide. Phys. Chem. Chem. Phys. 2011, 13, 262−266.(47) Hamalainen, K.; Siddons, D. P.; Hastings, J. B.; Berman, L. E.Elimination of the Inner-Shell Lifetime Broadening in X-rayAbsorption Spectroscopy. Phys. Rev. Lett. 1991, 67, 2850−2853.(48) de Groot, F. M. F.; Krisch, M. H.; Vogel, J. Spectral Sharpeningof the Pt L Edges by High-Resolution X-ray Emission. Phys. Rev. B2002, 66, 195112.(49) Hammer, B.; Nørskov, J. K. Electronic Factors Determining theReactivity of Metal Surfaces. Surf. Sci. 1995, 343, 211−220.(50) von Barth, U.; Grossmann, G. Dynamical Effects in X-raySpectra and the Final-State Rule. Phys. Rev. B 1982, 25, 5150−5179.(51) Bligaard, T.; Nørskov, J. K. Ligand Effects in HeterogeneousCatalysis and Electrochemistry. Electrochim. Acta 2007, 52, 5512−5516.(52) Anniyev, T.; Ogasawara, H.; Ljungberg, M. P.; Wikfeldt, K. T.;MacNaughton, J. B.; Naslund, L. Å.; Bergmann, U.; Koh, S.; Strasser,P.; Pettersson, L. G. M.; et al. Complementarity between High-EnergyPhotoelectron and L-Edge Spectroscopy for Probing the ElectronicStructure of 5d Transition Metal Catalysts. Phys. Chem. Chem. Phys.2010, 12, 5694−5700.(53) Andersson, K. J.; Calle-Vallejo, F.; Rossmeisl, J.; Chorkendorff,I. Adsorption-Driven Surface Segregation of the Less Reactive AlloyComponent. J. Am. Chem. Soc. 2009, 131, 2404−2407.
(54) Abild-Pedersen, F.; Nilsson, A.; Nørskov, J. K. Comment on“Using Photoelectron Spectroscopy and Quantum Mechanics toDetermine d-Band Energies of Metals for Catalytic Applications. J.Phys. Chem. C 2013, 117, 6914−6915.(55) Nilsson, A.; Pettersson, L. G. M. Chemical Bonding on SurfacesProbed by X-ray Emission Spectroscopy and Density FunctionalTheory. Surf. Sci. Rep. 2004, 55, 49−167.(56) Nilsson, A.; Pettersson, L. G. M. Adsorbate Electronic Structureand Bonding on Metal Surfaces. In Chemical Bonding at Surfaces andInterfaces; Nilsson, A., Pettersson, L. G. M., Nørskov, J. K., Eds.;Elsevier: Amsterdam, 2008; p 57.(57) Nilsson, A.; Wassdahl, N.; Weinelt, M.; Karis, O.; Wiell, T.;Bennich, P.; Hasselstrom, J.; Fohlisch, A.; Stohr, J.; Samant, M. LocalProbing of the Surface Chemical Bond Using X-ray EmissionSpectroscopy. Appl. Phys. A: Mater. Sci. Process. 1997, 65, 147−154.(58) Wassdahl, N.; Nilsson, A.; Wiell, T.; Tillborg, H.; Duda, L. C.;Guo, J. H.; Martensson, N.; Nordgren, J.; Andersen, J. N.; Nyholm, R.Soft X-ray Emission Studies of Adsorbates. Phys. Rev. Lett. 1992, 69,812−815.(59) Zdansky, E. O. F.; Nilsson, A.; Tillborg, H.; Bjorneholm, O.;Martensson, N.; Andersen, J. N.; Nyholm, R. Electronic Structure ofAtomic Adsorbates from X-ray Absorption Spectroscopy - ThresholdEffects and Higher Excited States. Phys. Rev. B 1993, 48, 2632−2641.(60) Kolczewski, C.; Puttner, R.; Plashkevych, O.; Ågren, H.;Staemmler, V.; Martins, M.; Snell, G.; Schlachter, A. S.; Sant’Anna, M.;Kaindl, G. Detailed Study of Pyridine at the C 1s and N 1s IonizationThresholds: The Influence of the Vibrational Fine Structure. J. Chem.Phys. 2001, 115, 6426−6437.(61) Oezaslan, M.; Hasche, F.; Strasser, P. PtCu3, PtCu and Pt3CuAlloy Nanoparticle Electrocatalysts for Oxygen Reduction Reaction inAlkaline and Acidic Media. J. Electrochem. Soc. 2012, 159, B444−B454.
The Journal of Physical Chemistry C Article
dx.doi.org/10.1021/jp400486r | J. Phys. Chem. C 2013, 117, 16371−1638016380




















