
The role of KRT5+ progenitors in chronic otitis media - UCL ...

Repository of the Max Delbrück Center for Molecular Medicine (MDC) Berlin (Germany) http://edoc.mdc-berlin.de/9394/
Sirt1 contributes critically to the redox-dependent fate of neural progenitors
Timour Prozorovski, Ulf Schulze-Topphoff, Robert Glumm, Jan Baumgart, Friederike Schröter, Olaf Ninnemann, Elise Siegert, Ivo Bendix, Oliver Brüstle, Robert Nitsch, Frauke Zipp, and Orhan Aktas
Published in final edited form as: Nature Cell Biology. 2008 Apr; 10(4): 385-394 | doi: 10.1038/ncb1700 Nature Publishing Group (U.K.) ►

Final Draft
Sirt1 contributes critically to the redox-dependent fate of neural progenitors Timour Prozorovski1,a, Ulf Schulze-Topphoff1,a, Robert Glumm1, Jan Baumgart2, Friederike Schröter1, Olaf Ninnemann2, Elise Siegert1, Ivo Bendix1, Oliver Brüstle3, Robert Nitsch2, Frauke Zipp1,a, and Orhan Aktas1,a
1 Cecilie Vogt Clinic for Neurology in the Helios Klinikum Berlin-Buch, Charité – Universitätsmedizin Berlin, and Max Delbrück Center for Molecular Medicine, Charitéplatz 1, 10117 Berlin, Germany 2 Institute for Cell and Neurobiology, Charité – Universitätsmedizin Berlin, Germany 3 Institute of Reconstructive Neurobiology, University of Bonn Medical Center and Hertie Foundation, 53105 Bonn, Germany a These authors contributed equally to this work.
ABSTRACT | Repair processes that are activated in response to neuronal injury, be it inflammatory, ischaemic, metabolic, traumatic or other cause, are characterized by a failure to replenish neurons and by astrogliosis. The underlying molecular pathways, however, are poorly understood. Here, we show that subtle alterations of the redox state, found in different brain pathologies, regulate the fate of mouse neural progenitor cells (NPCs) through the histone deacetylase (HDAC) Sirt1. Mild oxidation or direct activation of Sirt1 suppressed proliferation of NPCs and directed their differentiation towards the astroglial lineage at the expense of the neuronal lineage, whereas reducing conditions had the opposite effect. Under oxidative conditions in vitro and in vivo, Sirt1 was upregulated in NPCs, bound to the transcription factor Hes1 and subsequently inhibited pro-neuronal Mash1. In utero shRNA-mediated knockdown of Sirt1 in NPCs prevented oxidation-mediated suppression of neurogenesis and caused upregulation of Mash1 in vivo. Our results provide evidence for an as yet unknown metabolic master switch that determines the fate of neural progenitors.
Introduction In the adult mammalian brain, multipotent and self-renewing NPCs have the capacity to generate new neurons, astrocytes and oligodendrocytes. Neural progenitors may thus function as a regenerative tool to compensate for damage in the central nervous system (CNS) [1,2]. However, many pathological conditions in the brain are characterized by the emergence of more astrocytes (through a process known as reactive astrogliosis) [3] than neurons. Thus, replacement of damaged neurons remains insufficient and may account for fixed clinical deficits. Inflammation, which generates an oxidative milieu and is present in a variety of neurological diseases [4], has been reported recently to inhibit neurogenesis in the brain [5]. Changes in the intracellular redox state have been observed during the self-renewal and differentiation processes of dividing O-2A progenitor cells [6]; however, the molecular mechanisms underlying this phenomenon have yet to be defined. We therefore systematically assessed the effects of redox modulation on neural stem cells both in vitro and in vivo, as well as in the animal model of multiple sclerosis, experimental autoimmune encephalomyelitis (EAE).
Sirt1, a mammalian homologue of Sir2, the yeast HDAC [7], is a possible candidate for redox modulation because its activity is regulated by nicotinamide adenine dinucleotide (NAD+) and it is therefore, sensitive to the redox state and cellular metabolism. Indeed, Sirt1 has been identified recently as a redox sensor in mouse embryonic fibroblasts and undifferentiated muscle cells, in which it controls proliferation, cell cycle arrest and differentiation [8-10]. Moreover, independently generated Sirt1-deficient mice have been reported to exhibit severe neural defects, including exencephaly [11] and disturbed neuroretinal morphogenesis [12]. As Sirt1 lacks a DNA-binding domain, it has to be recruited to target promoters by sequence-specific transcription factors to induce chromatin remodelling and subsequently regulate gene expression [13]. Sirt1 and dSir2 (the Sirt1-related protein
in Drosophila melanogaster) function together with Hes1 as transcriptional co-repressors [13,14]. In the developing brain, Hes1 prevents premature neuronal differentiation by repressing the activation of the pro-neuronal basic helix-loop-helix (bHLH) transcription factor Mash1 (ref. 15). Here, we report that subtle (non-toxic) alterations of the redox state are crucial in regulating proliferation and differentiation of neural progenitors in a Sirt1–Hes1-dependent fashion involving regulation of Mash1.
Results Redox state regulates self-renewal of NPCs
Mouse cortical NPCs can be isolated and cultured from the embryonic stage onwards [16]. They mostly (~95%) express nestin (Fig.1a), a marker typically found in multipotent, uncommitted stem cells [1], although few of these cells express the lineage-specific differentiation markers β-III-tubulin (for neurons) or glial fibrillary acidic protein (GFAP, for astrocytes). In the presence of basic fibroblast growth factor (bFGF), NPCs proliferate, showing the classical stem-cell property of self-renewal [17]. NPCs were plated as monolayers under proliferation-sustaining conditions (that is, in the presence of bFGF) and exposed to pro-oxidative buthionine sulfoximine (BSO), an inhibitor of glutathione synthetase that causes intracellular accumulation of reactive oxygen species [6]. Low, non-toxic BSO concentrations (Supplementary Information, Fig.S1) caused a reduction in total cell number and decreased the proportion of proliferating Ki67(+) cells by 30% (Fig.1b, P < 0.05, compared with vehicle controls). Incubation of NPCs with pro-oxidative diethyl-dithiocarbamate (DETC), which suppresses superoxide dismutase and causes the accumulation of superoxide anions, had a similar effect (Fig.1b). Blocking other anti-oxidative enzymes, such as glutathione peroxidase (using mercaptosuccinic acid) and catalase (using amino-1, 2, 4-triazole), similarly reduced NPC proliferation (data not shown). Thus, regardless of the specific anti-oxidative enzyme systems targeted, a net induction of oxidative
MDC Repository | http://edoc.mdc-berlin.de/9394/ 1

Prozorovski T et al.
stress inhibits NPC proliferation. Application of the reducing compounds lipoic acid or N-acetylcysteine (NAC), however, had the opposite effect: we observed an increase of 39% or 22%, respectively, in the number of Ki67(+) cells (Fig.1b, P < 0.05). Ki67 immunolabelling was limited exclusively to nestin(+) cells, indicating that the redox effects on proliferation are in fact restricted to the undifferentiated proportion of the NPC population (Fig.1a). The findings based on Ki67 detection were confirmed by analysing 5-bromodeoxyuridine (BrdU) incorporation (Fig.1b). The constant number of condensed nuclei (Hoechst dye 33258 staining) and the results from independent cell-death detection assays (Supplementary Information, Fig.S1) again confirmed the absence of toxic bystander-effects.
Redox state regulates differentiation of NPCs Withdrawal of bFGF caused spontaneous differentiation of NPCs into astrocytes, neurons and oligodendrocytes, reflecting their multipotentiality [17]. We therefore investigated whether the redox system also affects the multipotentiality of NPCs. Cells were first cultured in the presence of bFGF and redox modulators for two days and then bFGF was withdrawn. After 7 days under pro-oxidative conditions, the proportion of astrocytes (GFAP(+)) increased by 40% (BSO) or 35% (DETC; Fig.1c,d, P < 0.05), whereas the proportion of neurons (NeuN(+)) decreased by 40% (BSO) and 26% (DETC; P < 0.05). Reducing conditions had the opposite effect: lipoic acid increased the proportion of NeuN(+) cells by 70% and decreased the proportion of GFAP(+) cells by 17% (P < 0.05); NAC produced similar effects (Fig.1d). Nestin was downregulated in all cultures, indicating that their differentiation capacity was not altered. Furthermore, no differences were found in oligodendrocyte frequency (Supplementary Information, Fig.S2). We then isolated and tracked single NPC clones grown under adherent cell culture conditions. In the presence of bFGF, each clone expanded into a distinct colony consisting of 10–50 cells. With BSO challenge, the number of cells derived from one single clone was reduced (Fig.1e), whereas the number of apoptotic cells was unaffected (data not shown). When bFGF was withdrawn, cells from a given colony differentiated spontaneously into either neurons or astrocytes. Most (>90%) of these colonies were mixed, consisting of both neurons and astrocytes, indicating that the clone from which they were derived was bipotent. We also observed monopotent (that is, purely neuronal or astroglial) colonies, suggesting that the initial clone was already committed. Pro-oxidative conditions did not change the proportion of bipotent-to-monopotent colonies (Fig.1f). However, BSO altered the cellular composition of bipotent colonies, reducing the number of neurons by 46% and increasing the number of astrocytes by 22% (Fig.1g, P < 0.05). The same results were obtained when NPC clones were expanded under free-floating conditions.
Oxidation-induced Sirt1 contributes to neural-fate decision In NPCs exposed to pro-oxidative conditions, the expression of Sirt1 was markedly upregulated (Table 1), whereas levels of the neural transcription factors NeuroD,
Hes1, Hes5, Olig1 and Olig2, which are known to influence the development of neural stem cells [18], were not altered. We assessed the expression levels of other members of the HDAC family as they also function in cellular chromatin remodelling and may, therefore, contribute to neural specification. Pro-oxidative conditions slightly dereased the expression of HDAC2 and HDAC3 in proliferating, but not in differentiating NPCs (Supplementary Information, Table S1). In contrast, Sirt1 expression levels remained upregulated in proliferating and differentiating NPCs. We therefore targeted Sirt1 in NPCs. The Sirt1 activator, resveratrol [19], was added to freshly plated, proliferating NPCs for two days before differentiation. Resveratrol increased the proportion of astrocytes and reduced the proportion of neurons in a Sirt1-dependent fashion (Fig.1h), thus mimicking the effects of mild oxidative conditions. To confirm the direct contribution of Sirt1 to redox-mediated differentiation processes, we silenced Sirt1 in NPCs before BSO challenge, using two short interfering (si) RNAs designed against different exons, and indeed, oxidation-mediated effects were prevented (Fig.1i).
Sirt1 cooperates with Hes1, deacetylates H3K9 and inhibits Mash1 To analyse the downstream effects of Sirt1 upregulation, we performed immunoprecipitation experiments using an anti-Sirt1 antibody. These analyses revealed that, in total NPC lysates, mild pro-oxidative conditions activated the Sirt1–Hes1 complex (Fig.2a) and downregulated acetylation of the histone residue H3K9 (Fig.2b), a putative target of Sirt1. Acetylation of H4K16, another possible substrate of Sirt1 (ref. 20), as well as acetylation of whole H3 or H4, remained unchanged. As seen in Fig.2c, both mild pro-oxidative conditions (inducing Sirt1) and direct Sirt1 activation (using resveratrol) markedly decreased Mash1 gene expression under proliferative and spontaneously differentiating conditions. To determine whether the Sirt1–Hes1 complex mediates Mash1 repression during a pro-oxidative shift, we depleted Sirt1 and Hes1 with siRNAs designed against different exons (Supplementary Information, Fig.S3). Indeed, oxidation-mediated Mash1 repression did not occur when Hes1 or Sirt1 were silenced (Fig.2d) or in Sirt1-/- progenitor cells [12] (Fig.2e).
Oxidation mediates H3K9 deacetylation and Sirt1 association at the Mash1 promoter Consequently, we hypothesized that, under conditions of oxidative challenge, Sirt1 cooperates with Hes1 as part of the co-repressor complex that inhibits Mash1 transcription. Generally, Hes1 regulates the expression of Mash1 by binding to three class-C hairy binding sites (CACGCG) [21]. We performed chromatin immunoprecipitation (ChIP) analysis of the two proximal C sites in differentiating NPCs and found that, under pro-oxidative conditions, Sirt1 was markedly upregulated in the Mash1 promoter region (Fig.2f; for quantification see Supplementary Information, Fig.S4a). Additionally, in view of a link between Mash1 activation and H3K9 acetylation [22], and reduced acetyl-H3K9 levels in total NPC lysates under oxidative conditions (Fig.2b), we investigated H3K9 acetylation directly at the Mash1 promoter. Vehicle-treated cells had an abundance of acetyl-H3K9 at the
MDC Repository | http://edoc.mdc-berlin.de/9394/ 2

Prozorovski T et al.
Mash1 promoter; however, oxidative conditions markedly reduced H3K9 acetylation (Fig.2f). We next investigated whether oxidation-mediated Sirt1 deacetylase activity may be a global phenomenon, affecting the promoters of other potentially redox-dependent Sirt1 targets, such as uncoupling protein 2 (UCP2; ref. 23) or cellular inhibitor of apoptosis 2 (cIAP2; ref. 24). We found that mild (non-toxic) oxidation caused slight increases in UCP2 and cIAP2 expression in NPCs (Fig.2g), whereas ChIP analysis did not reveal any changes in Sirt1 association or H3K9 acetylation at the corresponding promoters (Fig.2h). In contrast to the marked upregulation of Sirt1 at the Mash1 promoter, the level of Hes1 bound to the same sites was not affected (Fig.2f). This suggests that association of Sirt1 with Hes1 does not affect Hes1 binding to specific sites in the Mash1 promoter under oxidative conditions.
Sirt1 stabilizes the TLE1-containing complex to repress Mash1 activation To perform in-depth analysis of oxidation-mediated regulation of Mash1 transcription, we exposed differentiating NPCs to platelet-derived growth factor (PDGF), which has been shown previously to induce substantial Mash1 expression and thus to facilitate the analysis of cofactors at this promoter [25]. Here, induction of Mash1 expression was found to be regulated by poly(ADP–ribose) polymerase-1 (PARP-1), leading to the dismissal of the groucho (Gro) Transducin Like Enhancer of split 1 (TLE1) co-repressor and to assembly of the histone acetyltransferase-containing co-activator complex, including CREB-binding protein (CBP), at the Mash1 promoter (Fig.3a) [25]. It should be noted that in the presence of PDGF, Hes1 blockade alone does not cause upregulation of Mash1 in vitro [25]. However, parallel to our findings in NPCs after withdrawal of only bFGF, Sirt1 deacetylase activity was required for Mash1 repression in the presence of PDGF: oxidative conditions did not repress Mash1 expression when Sirt1 was silenced (Supplementary Information, Fig.S5a,b) or after treatment with the selective Sirt1 inhibitors sirtinol [26] and splitomicin [27] (Fig.3b). Mechanistically, under oxidative conditions, the Mash1 promoter was occupied by high levels of the co-repressors TLE1 and Sirt1, but low levels of the co-activator CBP. Consistent with the spontaneous NPC differentiation model (Fig.2f), Hes1 and PARP-1 — regardless of the redox conditions — showed no relevant regulation at the Mash1 promoter (Fig.3a; for quantification, see Supplementary Information, Fig.S4b). Oxidation does not seem to affect their binding activity, but rather, operates through the change of cofactors. Indeed, silencing of the co-repressor TLE1 alone (Supplementary Information, Fig.S5c,d) partially abrogated the effects of mild oxidation on Mash1 expression (Fig.3c). These data suggest that, in NPCs exposed to pro-oxidative conditions, Sirt1 stabilizes the TLE1-containing repressor complex to repress Mash1 activation.
Sirt1 and Mash1 expression patterns in germinal brain regions To determine the relevance of our findings in vivo, we first performed an immunohistological analysis of Mash1 and Sirt1 patterning in the postnatal day 2 (P2) central
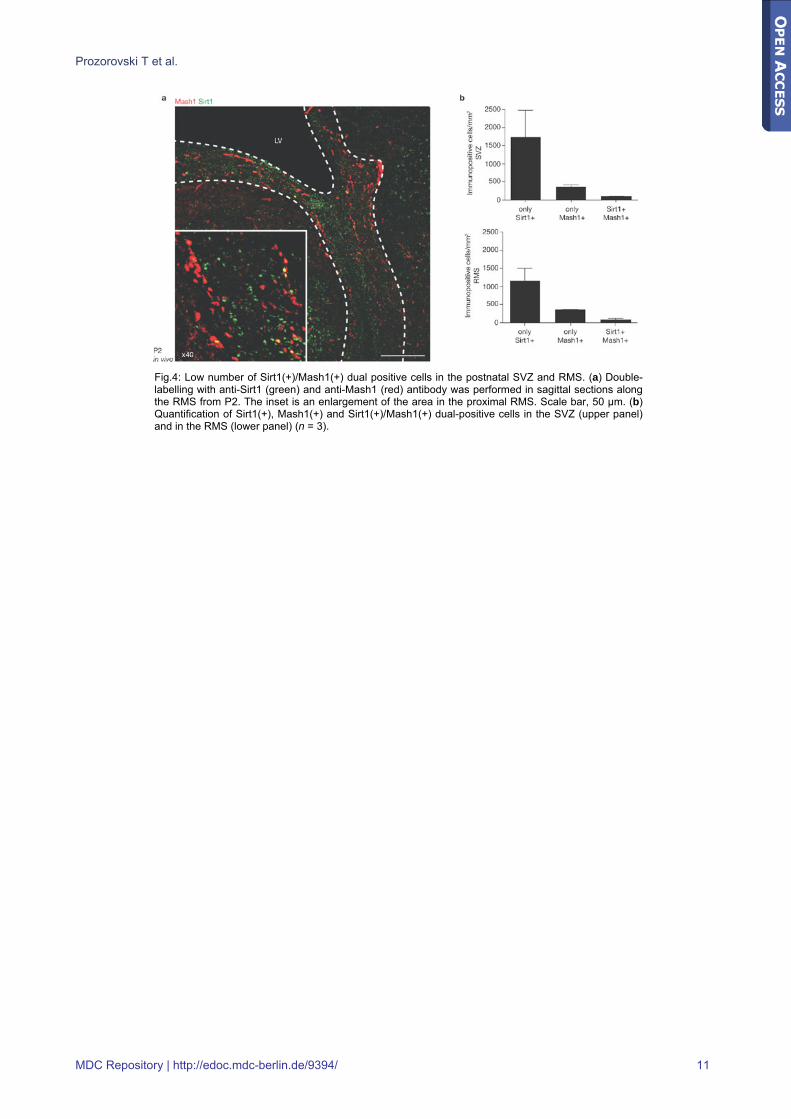
nervous system (Fig.4a). At this stage, Mash1 was detected in progenitors residing in germinal brain regions [28,29] (that is, in the subventricular zone (SVZ), along the rostral migratory stream (RMS) and in the centre of the olfactory bulb) and was shown to contribute to the specification of both neurons and oligodendrocytes [30]. We also found numerous Sirt1(+) cells in the SVZ of the lateral ventricle and along the RMS (Fig.4a). It is important to note that despite the close proximity of Sirt1(+) and Mash1(+) cells, very few cells were positive for both markers (Fig.4b), suggesting that they have different roles in neural-fate decision.
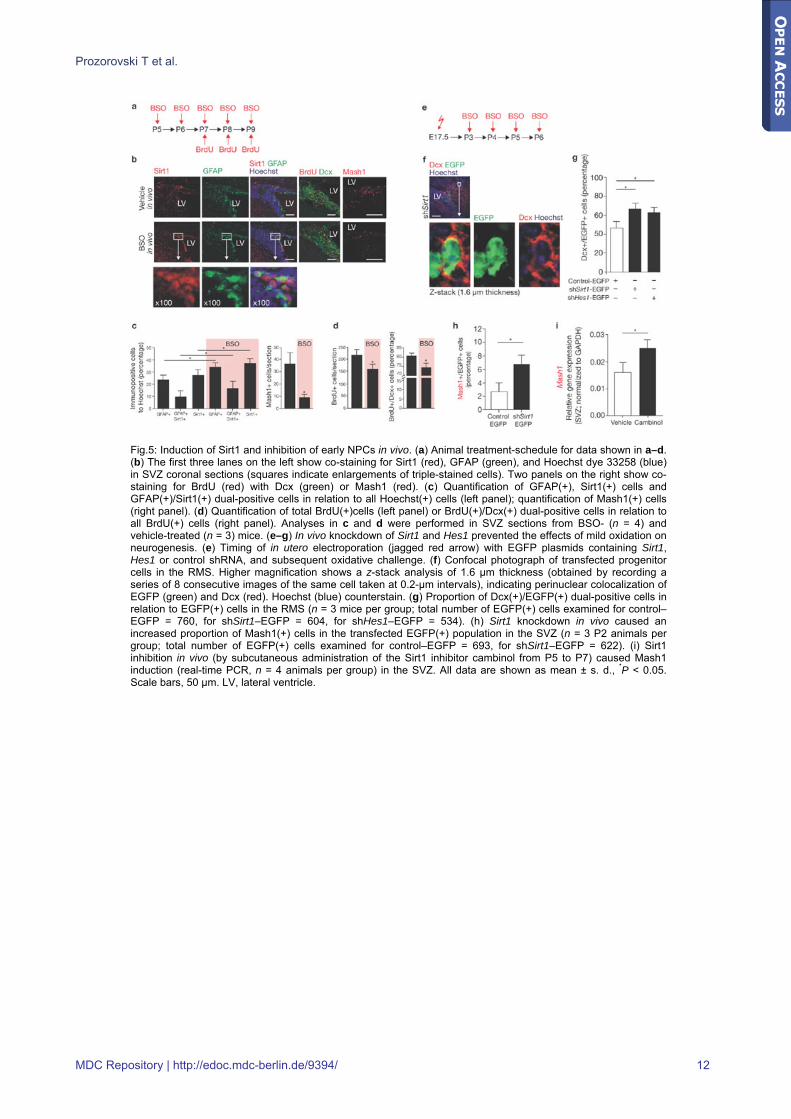
In vivo oxidative challenge inhibits neurogenesis in a Sirt1-dependent fashion Next we investigated whether the fate of NPCs in vivo is also modulated by a pro-oxidative shift. Mouse pups received BSO by daily subcutaneous injections, starting from P5 until P9, and were pulsed with BrdU between P7 and P9 before analysis of the SVZ (Fig.5a). Expression of Sirt1 was increased in the SVZ and mainly confined to GFAP(+) cells; however, the number of Mash1(+) cells decreased (Fig.5b,c). At the same time, the number of proliferating cells, identified by BrdU staining, decreased (Fig.5d). As precursor cells migrate from the SVZ through the RMS, these regions were stained with the neuroblast marker doublecortin (Dcx). BSO-treatment reduced the proportion of cells double-labelled for BrdU and Dcx (Fig.5d) but did not cause a specific depletion of neuroblasts, as reflected by unaltered numbers of TUNEL(+), Dcx(+) cells. To investigate the direct contribution of Sirt1 and Hes1 to oxidation-mediated effects in vivo, we performed knockdown of these genes using in utero electroporation. Animals were electroporated at embryonic day (E) 17.5 with enhanced green fluorescent protein (EGFP) plasmids containing Sirt1, Hes1 or control short-hairpin (sh) RNA (Fig.5e); transfected cells could be tracked up to P7 (Fig.5f) [31]. As expected, Sirt1 or Hes1 knockdown resulted in a higher proportion of Dcx(+) cells in mice subjected to oxidation from P3 to P6 (Fig.5g), indicating increased neurogenesis in vivo. It must be noted, however, that in Hes1-knockdown mice, the absolute number of EGFP(+) cells was reduced by approximately 80%. Although Hes1 is already known to be crucial for inhibiting neural differentiation during development [15], our in vivo knockdown strategy demonstrates the hitherto unknown contribution of Sirt1 to neural-fate decision in the oxidative milieu. Moreover, depletion of Sirt1 in animals electroporated in utero at E15 caused an increase in the proportion of Mash1(+) cells in the EGFP(+) population at P2, indicating increased neurogenesis (Fig.5h). Consistently, subcutaneous treatment of pups from P5–P7 with cambinol, a small-molecule Sirt1 inhibitor [32], caused upregulation of Mash1 in the SVZ (Fig.5i).
Sirt1 expression patterns and modulation in EAE Finally, we investigated Sirt1 expression in autoimmune demyelination, in which oxidative stress is known to occur [33,34]. Brain pathology in mice with EAE, induced by transfer of encephalitogenic T cells into naive recipients [35], showed a lack of Sirt1 expression in non-inflamed brain regions (Fig.6a). In contrast, areas affected by
MDC Repository | http://edoc.mdc-berlin.de/9394/ 3

Prozorovski T et al.
typical perivascular leukocyte infiltration and reactive astrogliosis showed marked Sirt1 upregulation in GFAP(+) cells with astroglial morphology (Fig.6b–d), but very low Mash1 expression (Fig.6e). In these cells, Sirt1 expression was mainly intranuclear, suggesting its involvement in chromatin silencing (Fig.6c). Moreover, application of the Sirt1 activator resveratrol to EAE animals revealed an increase of GFAP(+), BrdU(+) early progenitors in lesions (Fig.6f), suggesting enhanced gliogenesis through increased Sirt1 deacetylase activity.
Discussion Our studies show that subtle (non-toxic) redox alterations, present in a variety of CNS diseases [4], critically affect the self-renewal capacity of NPCs. They also extend previous observations derived from O-2A progenitor cells in vitro [6] and show that oxidative conditions favour astrocyte expansion at the expense of neurogenesis, whereas reducing conditions have the opposite effect. The oxidation-mediated increase of astrocytes does not seem to be linked to either an enhanced proliferation of astrocyte precursors or to restriction of the multipotentiality of NPCs. Rather, oxidation directs differentiation of uncommitted NPCs towards the astroglial lineage by altering their 'default' development programme. Moreover, our data suggest that Sirt1 (a class III NAD+-dependent HDAC with a crucial role in various cellular processes that comprise energy metabolism, transcriptional silencing and DNA repair) [36] is the mediator of the observed redox effects, as mild oxidation in NPCs led most notably to increased Sirt1 deacetylase activity at the Mash1 promoter. On the basis of our observations, oxidation does not induce a global histone deacetylation, as the acetylation status of whole histone H3 or whole histone H4 was not reduced under these conditions. On the contrary, oxidation reduced acetylation of the H3K9, but not of the H4K16 residue, both known to be regulated by Sirt1 (ref. 20). This effect was reversed when we applied the functional Sirt1 blocker splitomicin. In contrast to these observations, trichostatin A, which inhibits class I and class II HDACs, induced a global change in acetylation (in this case an increase). There was no modulation of other potentially redox-dependent Sirt1 targets, such as UCP2 or cIAP2. We may conclude from these data that the observed loss of H3K9 acetylation at the Mash1 promoter is not part of a general phenomenon in the context of oxidation-mediated Sirt1 activation, but rather, that oxidation results in a stronger association of Sirt1 to the Mash1 promoter, leading to a targeted and local deacetylation of H3K9 and subsequently to Mash1 inhibition. As Mash1 has a crucial role in neurogenesis [18], Sirt1 must be considered to contribute to NPC fate in vivo.
Indeed, analysis of the developing brain revealed a predominantly non-coincident distribution of Sirt1 and Mash1, suggesting that they have opposing roles in commitment and differentiation of neural progenitors. Furthermore, to clarify the roles of Sirt1 and Hes1 under oxidative conditions in vivo, we performed gene knockdown using in utero electroporation. This technique allows a selective and transient manipulation of gene function in vivo in a subset of cells in otherwise normal tissue [31,37]. As reported previously, knockout of Sirt1 or Hes1 is lethal [11,12,15]. Combined with the fluorescence tagging of the successfully silenced cells, the selective and transient knockdown of these genes enabled in vivo
clonal analysis and the confirmation of our in vitro findings.
In conclusion, a pro-oxidative shift induces Sirt1 expression in neural progenitors both in vitro and in vivo. This in turn promotes astrogenesis and inhibits neurogenesis through the interaction of Sirt1 with Hes1 and subsequent repression of Mash1. By providing an explanation for downregulation of neurogenesis in disease, our data suggest new targets for the regulation of neural cell-fate in neurological disorders. Targeting these mechanisms may minimize undesirable aspects of reactive astrogliosis and improve the success of therapeutic neural stem-cell implantation.
Methods Cell culture
Cerebral cortical NPCs were obtained from C57BL/6 mice embryos (Charles River), according to standard procedures [38]. Cells isolated from E17.5 cortices (as described in the Supplementary Information Methods) were seeded onto uncoated T75 tissue culture flasks in serum-free neurobasal medium (NBM) supplemented with 2% B27 and 1% GlutaMAX-I, and then induced to proliferate as free-floating neurospheres by addition of recombinant bFGF (10 ng ml-1; ref. 39). Clonal analysis and proliferating/differentiating conditions of NPCs are described in the Supplementary Information Methods. For experiments shown in Fig.2e, Sirt1-/- (knockout of the catalytic domain of Sirt1) [12] mice or wild-type littermates were used. For genotyping, a mix of primers was used, as listed in Supplementary Information, Table S2.
Immunocytochemistry
Immunocytochemical analysis of proliferating and differentiating cultures was performed, as described in the Supplementary Information Methods.
Immunoprecipitation and western blotting
NPCs were cultured for 48 h in the presence of bFGF (10 ng ml-1), collected and treated as described in the Supplementary Information Methods, to perform Sirt1 immunoprecipitation and subsequent western blotting for Sirt1 and Hes1. To determine the acetylation level of histones H3 and H4, total histones were isolated by acidic extraction (see Supplementary Information Methods). Resulting precipitates (equivalent to 20 μg protein) were fractionated by 15% SDS–PAGE and incubated overnight at 4 °C with anti-acetyl-H3K9 (1:1000), anti-acetyl-H4K16 (1:1000), anti-acetyl-H3 (1:1000), anti-acetyl-H4 (1:1000), anti-H3 (1:500) and anti-H4 (1:500) antibodies (Biomol). Protein bands were detected using secondary antibody coupled to horseradish peroxidase (Dako) and the ECL-plus system (Amersham Biosciences).
Chromatin immunoprecipitation (ChIP)
NPCs were grown for 48 h in the presence of bFGF (10 ng ml-1) on poly-D-lysine-coated 6-well plates. Cells were washed and fresh NBM without bFGF was added for 24 h to induce differentiation. BSO (5 μM) was continuously present in the culture. Cells were treated for 10 min with
MDC Repository | http://edoc.mdc-berlin.de/9394/ 4

Prozorovski T et al.
1% formaldehyde at room temperature. The crosslinking reaction was quenched using glycine (125 mM), cells were washed several times in ice-cold PBS and lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl at pH 8.0 with protease inhibitors). The lysate was ultrasonicated under conditions yielding fragments ranging from 200–600 bp and then diluted in immunoprecipitation buffer (1% Triton X-100, 150 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl at pH 8.0). The lysate (300 μg) was used for a 12-h immunoprecipitation with 3 μg of specific antibody at 4 °C. ChIPs were performed with the following antibodies: rabbit polyclonal anti-Sirt1 (1:200), rabbit polyclonal anti-Hes1 (1:200), rabbit polyclonal anti-TLE1 (1:200), rabbit polyclonal anti-CBP (1:200), rabbit polyclonal anti-PARP1 (1:200; all Santa Cruz), rabbit polyclonal anti-acetyl-histone H3K9 (1:1000; Biomol) and IgG from rabbit serum (1 μg ml-1; Sigma). Complexes were collected for 2 h by using recombinant protein A–agarose beads (Sigma) pre-absorbed with salmon-sperm DNA (500 μg ml-1) and bovine serum albumin (100 μg ml-1). After the washing and elution steps, samples were incubated with RNAse (500 μg ml-1) for 15 min and Proteinase K (500 μg ml-1) for 1 h. Formaldehyde crosslinking was reversed by adding NaCl (200 mM) with a 12-h incubation at 65 °C. Samples (100 μl) were purified with GFX PCR purification kit columns (Amersham Pharmacia) and used as a template in PCRs to detect the Mash1 promoter region between the -268 to -71 bps upstream from ATG start codon and placed directly downstream from CACGCG (-287 to -282) and CACGCG (-276 to -271). A high-affinity binding for Hes1 has been described for these two class-C sites [21]. For the mouse cIAP2 promoter, the NF-κB-dependent regulatory region between the -467 to -255 upstream from ATG start codon was selected for primer design. This region, which contains two canonical NF-κB DNA-binding elements [40], was chosen for its homology with human cIAP2 (described previously to be regulated in a Sirt1-dependent manner [24]). For the UCP2 promoter, the regulatory region between the -1462 to -1,237 bps upstream from the ATG codon was chosen for its homology with rat UCP2, which was described previously to be regulated in a Sirt1-dependent manner [23]. Control reactions using primers amplifying promoter regions in the mouse β-actin gene detected no PCR product. All primer sequences are described in the Supplementary Information Methods. Quantification of ChIP analysis was performed by real-time PCR (see Supplementary Information Methods).
EAE induction, Sirt1 modulation and BSO treatment
SJL/J mice were killed by intraperitoneal injection of ketamine (Ketavet, Upjohn) and xylazine (Rompun, Bayer) 12 days after induction of EAE, as described previously [35]. At this stage, mice had developed disease, as they showed hind-limb paresis and tail plegia. Animal care was in accordance with institutional guidelines. To examine the effects of Sirt1 activation in EAE, mice were treated with either resveratrol (50 mg kg-1) or vehicle solution (Cremophore EL and ethanol; Sigma) from day 6 to day 12 after immunization. BrdU (60 mg kg-1; Sigma) was administered by daily intraperitoneal injections on the last two days before animals were killed. To determine the effects of oxidation alone (that is, in the absence of inflammation), BSO (2.5 mmol kg-1; prepared from 1M stock in isotonic saline at pH 7.4) or vehicle alone was administered subcutaneously to mouse pups
twice daily, as described previously [41], according to the treatment schedules shown in Fig.5a or 5e. Pups were then killed, perfused transcardially with saline and 4% paraformaldehyde, and brains were embedded in TissueTek. Immunohistochemistry analysis and confocal microscopy imaging were performed as described in the Supplementary Information Methods. The Sirt1 inhibitor cambinol (100 mg kg-1) or vehicle solution alone (Cremophor EL and ethanol) was administered subcutaneously to mouse P5 pups twice daily. Pups were killed after two days of treatment, SVZ tissue was isolated from 350-mm frontal sections, as described previously [42], and Mash1 gene expression was analysed by real-time PCR analysis.
siRNA transfection
Transfections were performed with 1μg ml-1 cationic lipid β-Argfectin35-plus (Atugen). Synthetic Silencer Sirt1 siRNA (30 nM; Ambion, siRNA ID#174220) specific for exon 5, Sirt1 siRNA (30 nM; Ambion, siRNA ID#174219) specific for exon 2, Hes1 siRNA (30 nM; Ambion, siRNA ID#100393) specific for exon 2, Hes1 siRNA (30 nM; Ambion, siRNA ID#158034) specific for exon 5, TLE1 siRNA (30 nM; Ambion, siRNA ID#187335) specific for exon 20 and TLE1 siRNA (30 nM; Ambion, siRNA ID#187336) specific for exon 9 were used. For details of siRNA transfection and shRNA cloning, see Supplementary Information Methods.
In utero electroporation
The in utero electroporation experiments were carried out as described previously [31], in accordance with a protocol approved by the local Ethics Committee. We used time-pregnant C57BL/6 mice at E15.5–E17.5 (post coitum). After anaesthesia with ketamine (10 mg ml-1) and xylazine (1 mg ml-1), the uterine horns were exposed. DNA solution (1–1.5 μl per embryo) coloured with Fast Green was injected through the uterine wall into the lateral ventricle of two of the pups by pulled-glass capillaries (World Precision Instruments). Electric pulses were delivered to embryos by holding the injected brain through the uterine wall with forceps-type electrodes (CUY650P5; Unique Medical Imada) connected to a square-pulse generator (CUY21 EDIT; Nepagene). Five pulses (38 V, 50 ms) were applied at 950-ms intervals. The uterine horns were carefully replaced into the abdominal cavity before the muscle wall and the abdomen were sutured.
Statistical analysis
Statistical analysis was performed using Student's t-test. All data are shown as mean with standard deviations (s.d.). Probabilities of P < 0.05 were considered significant.
Acknowledgements We thank Nancy Nowakowski and Sophia Bardehle for technical assistance, Alistair Noon and Andrew Mason for reading the manuscript as native English speakers and Christine Kutschbach for editorial assistance. We thank Frederick W. Alt and Yuko Fujiwara (Children's Hospital, Harvard Medical School and Howard Hughes Medical Institute, Boston) for providing Sirt1-/- mouse mating pairs,
MDC Repository | http://edoc.mdc-berlin.de/9394/ 5

Prozorovski T et al.
and Atugen AG (Berlin, Germany) for providing transfection lipids. This work was supported by grants from the Institute for Multiple Sclerosis Research Göttingen and the Gemeinnützige Hertie-Stiftung (to F. Z. and O. B.), the Deutsche Forschungsgemeinschaft (SFB-TRR 43 to R. N., F. Z. and O. A., as well as SFB 665 to R.N. and SFB 650 to F. Z.) and the Bundesministerium fr Bildung und Forschung (to F.Z.).
Competing interests statement The authors declare no competing financial interests.
Corresponding Author Frauke Zipp, e-mail: [email protected]
References 1. McKay, R. Stem cells in the central nervous system.
Science 276, 66–71 (1997).
2. Wernig, M. & Brustle, O. Fifty ways to make a neuron: shifts in stem cell hierarchy and their implications for neuropathology and CNS repair. J. Neuropathol. Exp. Neurol. 61, 101–110 (2002).
3. Ridet, J. L., Malhotra, S. K., Privat, A. & Gage, F. H. Reactive astrocytes: cellular and molecular cues to biological function. Trends Neurosci. 20, 570–577 (1997).
4. Zipp, F. & Aktas, O. The brain as a target of inflammation: common pathways link inflammatory and neurodegenerative diseases. Trends Neurosci. 29, 518–527 (2006).
5. Monje, M. L., Toda, H. & Palmer, T. D. Inflammatory blockade restores adult hippocampal neurogenesis. Science 302, 1760–1765 (2003).
6. Smith, J., Ladi, E., Mayer-Proschel, M. & Noble, M. Redox state is a central modulator of the balance between self-renewal and differentiation in a dividing glial precursor cell. Proc. Natl Acad. Sci. USA 97, 10032–10037 (2000).
7. Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
8. Chua, K. F. et al. Mammalian SIRT1 limits replicative life span in response to chronic genotoxic stress. Cell Metab. 2, 67–76 (2005).
9. Brunet, A. et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015 (2004).
10. Fulco, M. et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol. Cell 12, 51–62 (2003).
11. McBurney, M. W. et al. The mammalian SIR2α protein has a role in embryogenesis and gametogenesis. Mol. Cell Biol. 23, 38–54 (2003).
12. Cheng, H.L. et al. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl Acad. Sci. USA 100, 10794–10799 (2003).
13. Rosenberg, M. I. & Parkhurst, S. M. Drosophila Sir2 is required for heterochromatic silencing and by euchromatic Hairy/E(Spl) bHLH repressors in segmentation and sex determination. Cell 109, 447–458 (2002).
14. Takata, T. & Ishikawa, F. Human Sir2-related protein SIRT1 associates with the bHLH repressors HES1 and HEY2 and is involved in HES1- and HEY2-mediated transcriptional repression. Biochem. Biophys. Res. Commun. 301, 250–257 (2003).
15. Ishibashi, M. et al. Targeted disruption of mammalian Hairy and Enhancer of split homolog-1 (HES-1) leads to up-regulation of neural helix-loop-helix factors, premature neurogenesis, and severe neural tube defects. Genes Dev. 9, 3136–3148 (1995).
16. Reynolds, B. A., Tetzlaff, W. & Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 12, 4565–4574 (1992).
17. Vescovi, A. L., Reynolds, B. A., Fraser, D. D. & Weiss, S. bFGF regulates the proliferative fate of unipotent (neuronal) and bipotent (neuronal/astroglial) EGF-generated CNS progenitor cells. Neuron 11, 951–966 (1993).
18. Bertrand, N., Castro, D. S. & Guillemot, F. Proneural genes and the specification of neural cell types. Nature Rev. Neurosci. 3, 517–530 (2002).
19. Howitz, K.T. et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425, 191–196 (2003).
20. Vaquero, A. et al. Human SirT1 interacts with histone H1 and promotes formation of facultative heterochromatin. Mol. Cell 16, 93–105 (2004).
21. Chen, H. et al. Conservation of the Drosophila lateral inhibition pathway in human lung cancer: a hairy-related protein (HES-1) directly represses achaete-scute homolog-1 expression. Proc. Natl Acad. Sci. USA 94, 5355–5360 (1997).
22. Williams, R. E. et al. Neural induction promotes large-scale chromatin reorganisation of the Mash1 locus. J. Cell Sci. 119, 132–140 (2006).
23. Bordone, L. et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic β cells. PLoS Biol. 4, e31 (2006).
24. Yeung, F. et al. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 (2004).
25. Ju, B. G. et al. Activating the PARP-1 sensor component of the groucho/ TLE1 co-repressor complex mediates a CaMKinase IIδ-dependent neurogenic gene activation pathway. Cell 119, 815–829 (2004).
26. Grozinger, C. M., Chao, E. D., Blackwell, H. E., Moazed, D. & Schreiber, S. L. Identification of a class of small molecule inhibitors of the sirtuin family of NAD-dependent deacetylases by phenotypic screening. J. Biol. Chem. 276, 38837–38843 (2001).
27. Bedalov, A., Gatbonton, T., Irvine, W. P., Gottschling, D. E. & Simon, J. A. Identification of a small molecule inhibitor of Sir2p. Proc. Natl Acad. Sci. USA 98, 15113–15118 (2001).
28. Alvarez-Buylla, A. & Lim, D. A. For the long run: maintaining germinal niches in the adult brain. Neuron 41, 683–686 (2004).
29. Gotz, M. & Huttner, W. B. The cell biology of neurogenesis. Nature Rev. Mol. Cell Biol. 6, 777–788 (2005).
30. Parras, C. M. et al. Mash1 specifies neurons and oligodendrocytes in the postnatal brain. EMBO J. 23, 4495–4505 (2004).
31. Brandt, N. et al. The neural EGF family member CALEB/NGC mediates dendritic tree and spine complexity. EMBO J. 26, 2371–2386 (2007).
MDC Repository | http://edoc.mdc-berlin.de/9394/ 6

Prozorovski T et al.
32. Heltweg, B. et al. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 66, 4368–4377 (2006).
33. Steinman, L. Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85, 299–302 (1996).
34. Frohman, E. M., Racke, M. K. & Raine, C. S. Multiple sclerosis — the plaque and its pathogenesis. N. Engl. J. Med. 354, 942–955 (2006).
35. Aktas, O. et al. Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 46, 421–432 (2005).
36. Leibiger, I. B. & Berggren, P. O. Sirt1: a metabolic master switch that modulates lifespan. Nature Med. 12, 34–36 (2006).
37. Bai, J. et al. RNAi reveals doublecortin is required for radial migration in rat neocortex. Nature Neurosci. 6, 1277–1283 (2003).
38. Ciccolini, F. & Svendsen, C. N. Fibroblast growth factor 2 (FGF-2) promotes acquisition of epidermal growth factor (EGF) responsiveness in mouse striatal precursor cells: identification of neural precursors responding to both EGF and FGF-2. J. Neurosci. 18, 7869–7880 (1998).
39. Reynolds, B. A. & Weiss, S. Clonal and population analyses demonstrate that an EGF-responsive mammalian embryonic CNS precursor is a stem cell. Dev. Biol. 175, 1–13 (1996).
40. Croxton, R. et al. Daxx represses expression of a subset of antiapoptotic genes regulated by nuclear factor-κB. Cancer Res. 66, 9026–9035 (2006).
41. Merad-Saidoune, M. et al. Overproduction of Cu/Zn-superoxide dismutase or Bcl-2 prevents the brain mitochondrial respiratory dysfunction induced by glutathione depletion. Exp. Neurol. 158, 428–436 (1999).
42. Lois, C. & varez-Buylla, A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc. Natl Acad. Sci. USA 90, 2074–2077 (1993).
MDC Repository | http://edoc.mdc-berlin.de/9394/ 7

Prozorovski T et al.
Fig.1: Impact of redox and Sirt1 modulation on self-renewal and multipotentiality of NPCs. (a, b) Opposite effects of pro-oxidative (BSO, DETC) or reducing (lipoic acid (LA), NAC) redox modulators on self-renewal after two days of bFGF treatment. Ki67(+) immunolabelling (red) was limited exclusively to nestin(+) cells (green). Nuclei were counterstained with Hoechst dye 33258. Proliferation was quantified by counting Ki67(+) or BrdU(+) cells. (c, d) Opposite effects of redox modulators on multipotentiality. (c) Cells were allowed to differentiate in the presence of redox modulators and stained for GFAP (green; astrocytes) and NeuN (red; neurons). (d) Quantification of differentiation. (e–g) Characterization of colonies derived from single NPCs under adherent culture conditions (clonal analysis). (e) Average number of cells per individual colony. (f) Percentages of clones which gave rise to either mixed colonies; that is, consisting of neuronal (β-III-tubulin(+)) and astroglial (GFAP(+)) cells (bipotent), or to purely neuronal or purely astroglial colonies. (g) Proportion of astrocytes and neurons within mixed (bipotent) clones. (h) Effect of selective Sirt1 modulation on NPC differentiation. NPCs were exposed to the Sirt1 activator, resveratrol, under unmodified redox conditions. The direct contribution of Sirt1 was investigated by depletion of Sirt1 by RNAi. (i) NPCs were first transfected with two different Sirt1 siRNAs (siRNA1, siRNA2) or nonsense siRNA, then exposed to oxidative conditions. NPC differentiation was quantified by identifying astrocytes (GFAP(+)) and neurons (β -III-tubulin(+)). All data are mean ± s. d. of three independent experiments, *P < 0.05, compared with vehicle controls. Scale bars, 20 μm.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 8
Prozorovski T et al.
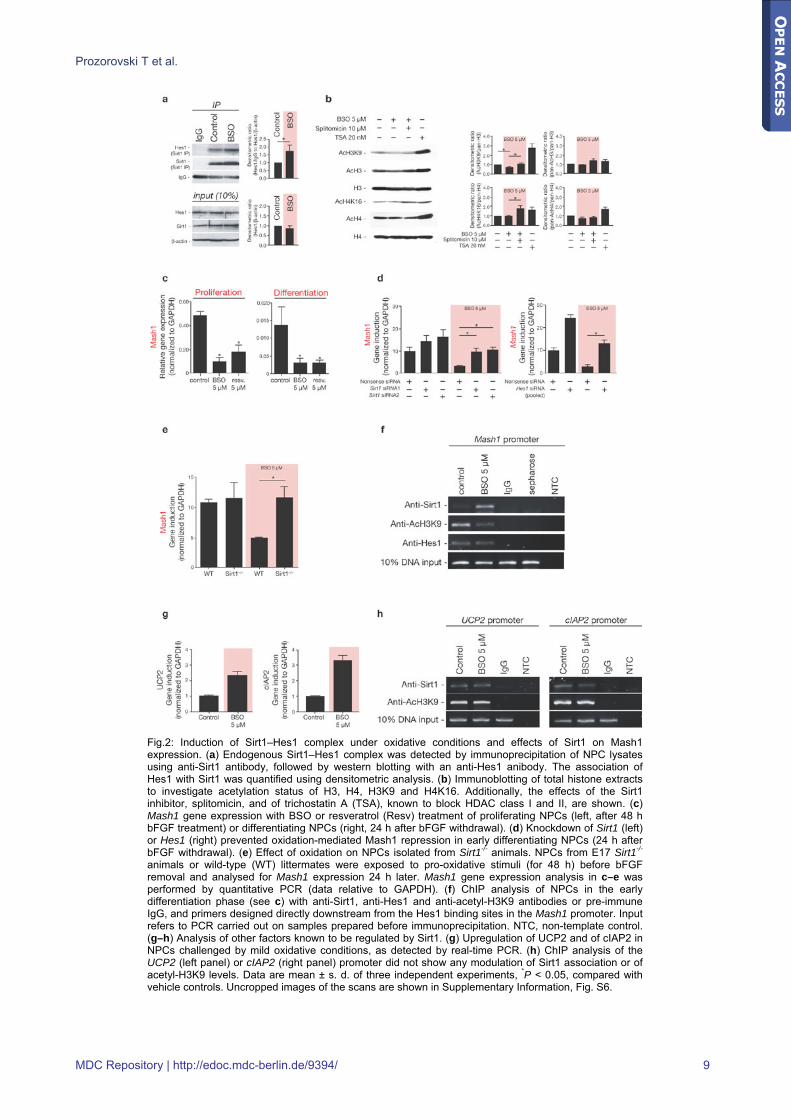
Fig.2: Induction of Sirt1–Hes1 complex under oxidative conditions and effects of Sirt1 on Mash1 expression. (a) Endogenous Sirt1–Hes1 complex was detected by immunoprecipitation of NPC lysates using anti-Sirt1 antibody, followed by western blotting with an anti-Hes1 anibody. The association of Hes1 with Sirt1 was quantified using densitometric analysis. (b) Immunoblotting of total histone extracts to investigate acetylation status of H3, H4, H3K9 and H4K16. Additionally, the effects of the Sirt1 inhibitor, splitomicin, and of trichostatin A (TSA), known to block HDAC class I and II, are shown. (c) Mash1 gene expression with BSO or resveratrol (Resv) treatment of proliferating NPCs (left, after 48 h bFGF treatment) or differentiating NPCs (right, 24 h after bFGF withdrawal). (d) Knockdown of Sirt1 (left) or Hes1 (right) prevented oxidation-mediated Mash1 repression in early differentiating NPCs (24 h after bFGF withdrawal). (e) Effect of oxidation on NPCs isolated from Sirt1-/- animals. NPCs from E17 Sirt1-/- animals or wild-type (WT) littermates were exposed to pro-oxidative stimuli (for 48 h) before bFGF removal and analysed for Mash1 expression 24 h later. Mash1 gene expression analysis in c–e was performed by quantitative PCR (data relative to GAPDH). (f) ChIP analysis of NPCs in the early differentiation phase (see c) with anti-Sirt1, anti-Hes1 and anti-acetyl-H3K9 antibodies or pre-immune IgG, and primers designed directly downstream from the Hes1 binding sites in the Mash1 promoter. Input refers to PCR carried out on samples prepared before immunoprecipitation. NTC, non-template control. (g–h) Analysis of other factors known to be regulated by Sirt1. (g) Upregulation of UCP2 and of cIAP2 in NPCs challenged by mild oxidative conditions, as detected by real-time PCR. (h) ChIP analysis of the UCP2 (left panel) or cIAP2 (right panel) promoter did not show any modulation of Sirt1 association or of acetyl-H3K9 levels. Data are mean ± s. d. of three independent experiments, *P < 0.05, compared with vehicle controls. Uncropped images of the scans are shown in Supplementary Information, Fig. S6.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 9
Prozorovski T et al.
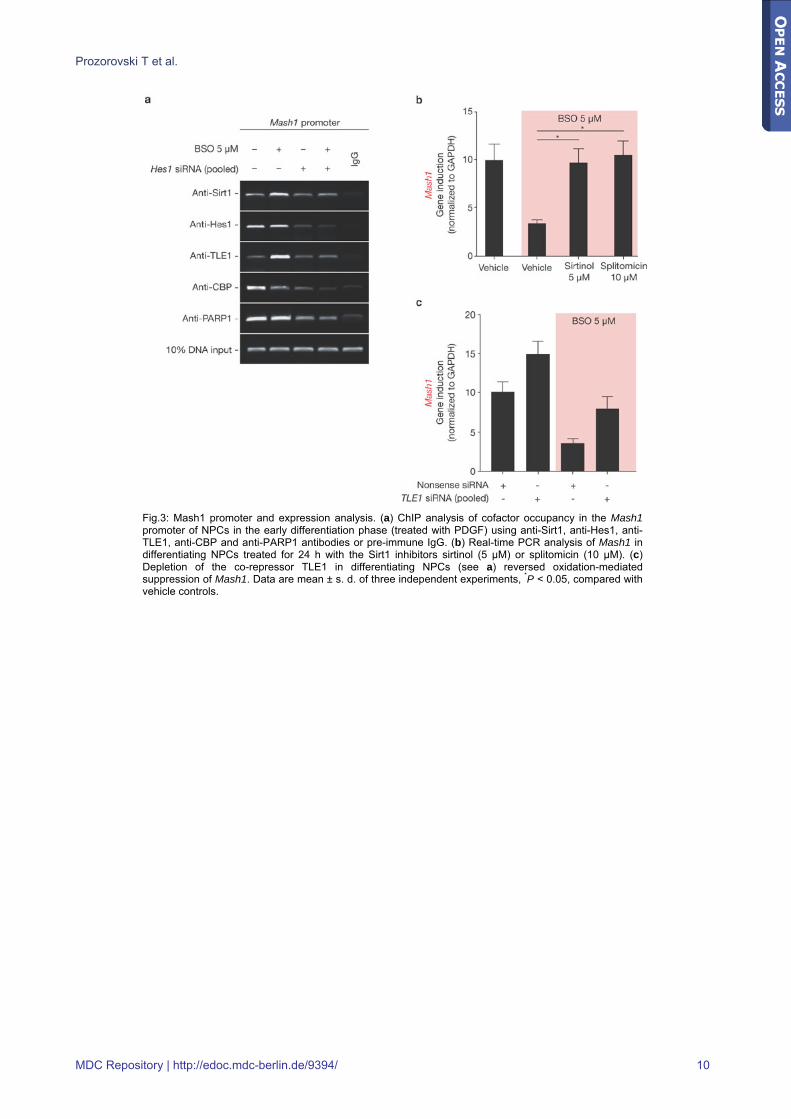
Fig.3: Mash1 promoter and expression analysis. (a) ChIP analysis of cofactor occupancy in the Mash1 promoter of NPCs in the early differentiation phase (treated with PDGF) using anti-Sirt1, anti-Hes1, anti-TLE1, anti-CBP and anti-PARP1 antibodies or pre-immune IgG. (b) Real-time PCR analysis of Mash1 in differentiating NPCs treated for 24 h with the Sirt1 inhibitors sirtinol (5 μM) or splitomicin (10 μM). (c) Depletion of the co-repressor TLE1 in differentiating NPCs (see a) reversed oxidation-mediated suppression of Mash1. Data are mean ± s. d. of three independent experiments, *P < 0.05, compared with vehicle controls.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 10
Prozorovski T et al.
Fig.4: Low number of Sirt1(+)/Mash1(+) dual positive cells in the postnatal SVZ and RMS. (a) Double-labelling with anti-Sirt1 (green) and anti-Mash1 (red) antibody was performed in sagittal sections along the RMS from P2. The inset is an enlargement of the area in the proximal RMS. Scale bar, 50 μm. (b) Quantification of Sirt1(+), Mash1(+) and Sirt1(+)/Mash1(+) dual-positive cells in the SVZ (upper panel) and in the RMS (lower panel) (n = 3).
MDC Repository | http://edoc.mdc-berlin.de/9394/ 11
Prozorovski T et al.
Fig.5: Induction of Sirt1 and inhibition of early NPCs in vivo. (a) Animal treatment-schedule for data shown in a–d. (b) The first three lanes on the left show co-staining for Sirt1 (red), GFAP (green), and Hoechst dye 33258 (blue) in SVZ coronal sections (squares indicate enlargements of triple-stained cells). Two panels on the right show co-staining for BrdU (red) with Dcx (green) or Mash1 (red). (c) Quantification of GFAP(+), Sirt1(+) cells and GFAP(+)/Sirt1(+) dual-positive cells in relation to all Hoechst(+) cells (left panel); quantification of Mash1(+) cells (right panel). (d) Quantification of total BrdU(+)cells (left panel) or BrdU(+)/Dcx(+) dual-positive cells in relation to all BrdU(+) cells (right panel). Analyses in c and d were performed in SVZ sections from BSO- (n = 4) and vehicle-treated (n = 3) mice. (e–g) In vivo knockdown of Sirt1 and Hes1 prevented the effects of mild oxidation on neurogenesis. (e) Timing of in utero electroporation (jagged red arrow) with EGFP plasmids containing Sirt1, Hes1 or control shRNA, and subsequent oxidative challenge. (f) Confocal photograph of transfected progenitor cells in the RMS. Higher magnification shows a z-stack analysis of 1.6 μm thickness (obtained by recording a series of 8 consecutive images of the same cell taken at 0.2-μm intervals), indicating perinuclear colocalization of EGFP (green) and Dcx (red). Hoechst (blue) counterstain. (g) Proportion of Dcx(+)/EGFP(+) dual-positive cells in relation to EGFP(+) cells in the RMS (n = 3 mice per group; total number of EGFP(+) cells examined for control–EGFP = 760, for shSirt1–EGFP = 604, for shHes1–EGFP = 534). (h) Sirt1 knockdown in vivo caused an increased proportion of Mash1(+) cells in the transfected EGFP(+) population in the SVZ (n = 3 P2 animals per group; total number of EGFP(+) cells examined for control–EGFP = 693, for shSirt1–EGFP = 622). (i) Sirt1 inhibition in vivo (by subcutaneous administration of the Sirt1 inhibitor cambinol from P5 to P7) caused Mash1 induction (real-time PCR, n = 4 animals per group) in the SVZ. All data are shown as mean ± s. d., *P < 0.05. Scale bars, 50 μm. LV, lateral ventricle.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 12
Prozorovski T et al.
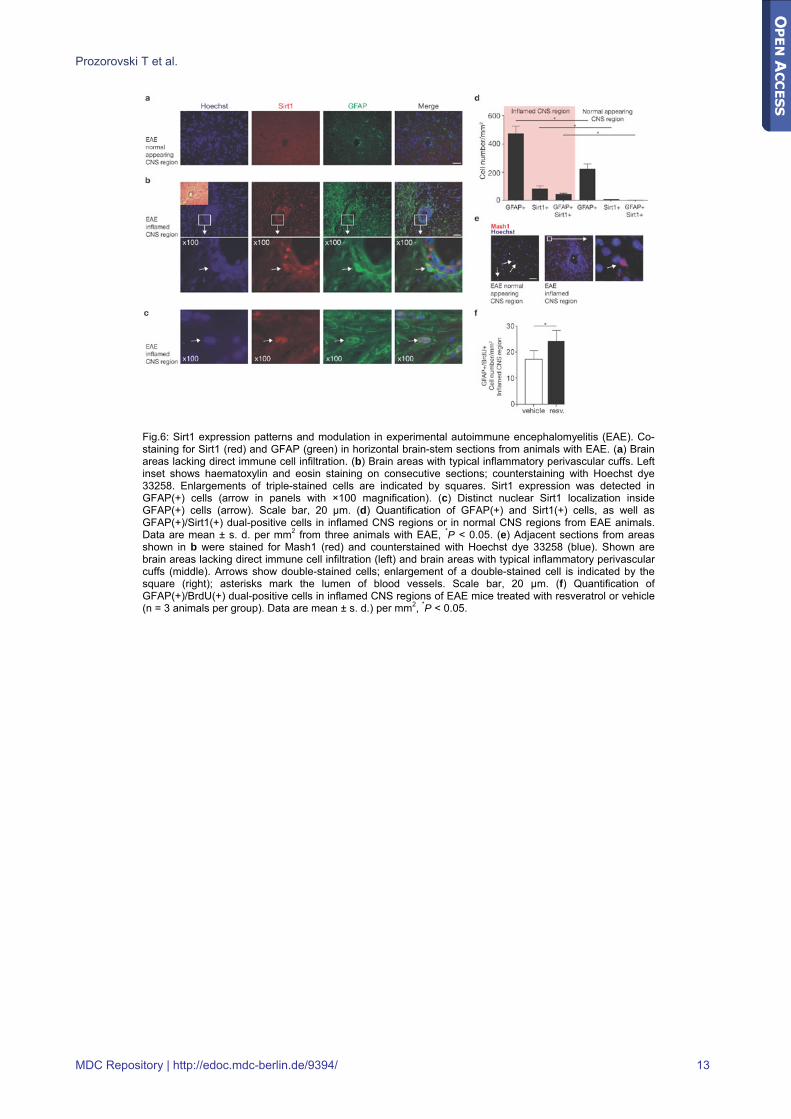
Fig.6: Sirt1 expression patterns and modulation in experimental autoimmune encephalomyelitis (EAE). Co-staining for Sirt1 (red) and GFAP (green) in horizontal brain-stem sections from animals with EAE. (a) Brain areas lacking direct immune cell infiltration. (b) Brain areas with typical inflammatory perivascular cuffs. Left inset shows haematoxylin and eosin staining on consecutive sections; counterstaining with Hoechst dye 33258. Enlargements of triple-stained cells are indicated by squares. Sirt1 expression was detected in GFAP(+) cells (arrow in panels with ×100 magnification). (c) Distinct nuclear Sirt1 localization inside GFAP(+) cells (arrow). Scale bar, 20 μm. (d) Quantification of GFAP(+) and Sirt1(+) cells, as well as GFAP(+)/Sirt1(+) dual-positive cells in inflamed CNS regions or in normal CNS regions from EAE animals. Data are mean ± s. d. per mm2 from three animals with EAE, *P < 0.05. (e) Adjacent sections from areas shown in b were stained for Mash1 (red) and counterstained with Hoechst dye 33258 (blue). Shown are brain areas lacking direct immune cell infiltration (left) and brain areas with typical inflammatory perivascular cuffs (middle). Arrows show double-stained cells; enlargement of a double-stained cell is indicated by the square (right); asterisks mark the lumen of blood vessels. Scale bar, 20 μm. (f) Quantification of GFAP(+)/BrdU(+) dual-positive cells in inflamed CNS regions of EAE mice treated with resveratrol or vehicle (n = 3 animals per group). Data are mean ± s. d.) per mm2, *P < 0.05.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 13

Prozorovski T et al.
Table1: Impact of the redox state on transcriptional regulators
Gene name NCBI GeneID BSO1 LA2
NeuroD 18012 -1.01 ± 0.16 1.47 ± 0.23
Hes1 15205 -1.96 ± 0.34 2.43 ± 0.40
Hes5 15208 1.27 ± 0.22 -1.08 ± 0.19
Olig1 50914 -1.39 ± 0.16 1.43 ± 0.18
Olig2 50913 -1.88 ± 0.21 1.36 ± 0.39
Sirt1 93759 6.50 ± 1. 36 1.46 ± 0.37
Displayed are relative fold changes (means ± s. d.) of quantitative real-time PCR. Values are normalized to expression levels under vehicle treatment. 11BSO, 1 μM; 22LA, 5 μM.
MDC Repository | http://edoc.mdc-berlin.de/9394/ 14
Copyright © 2022 FDOKUMEN




















